Пред.
След.
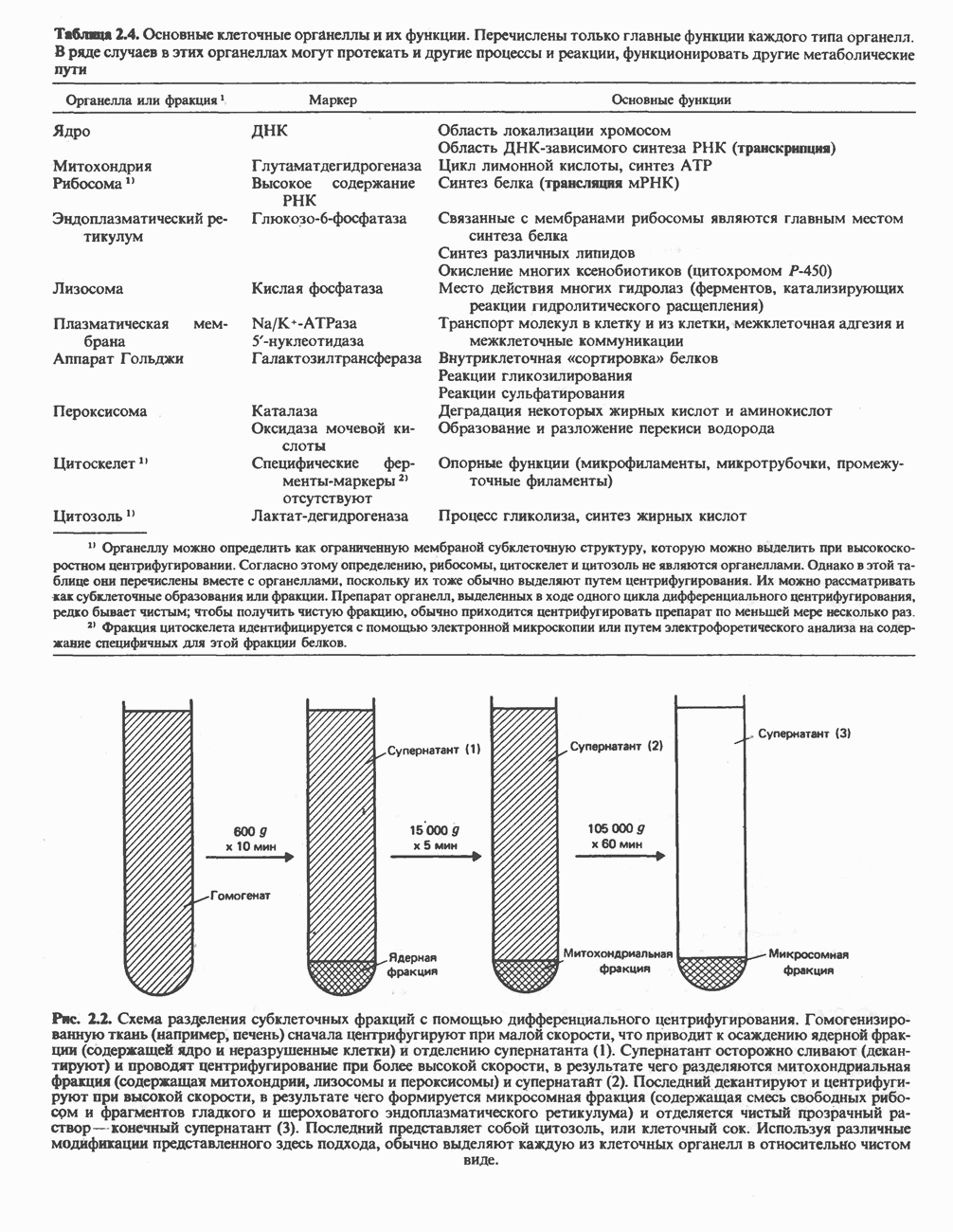
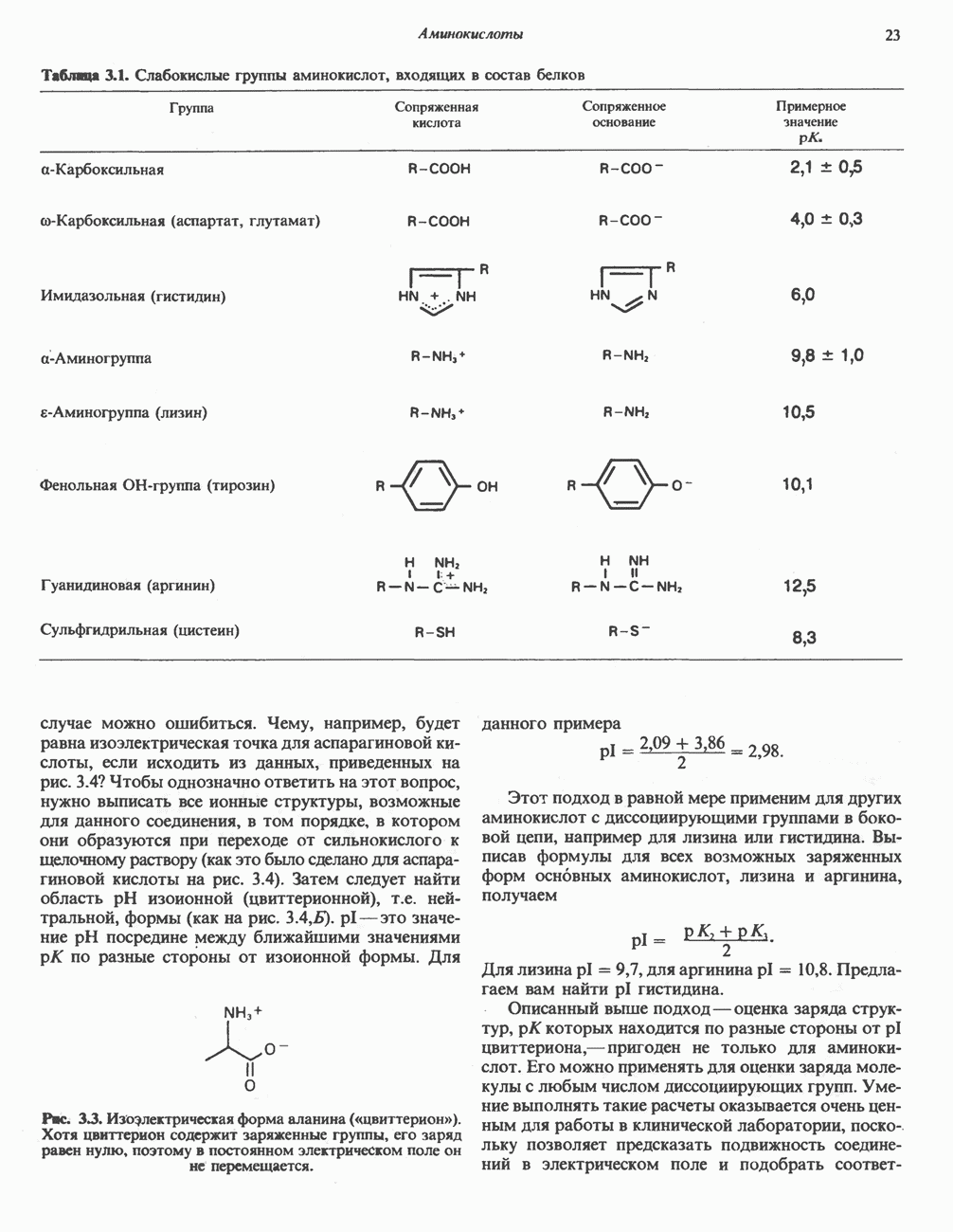
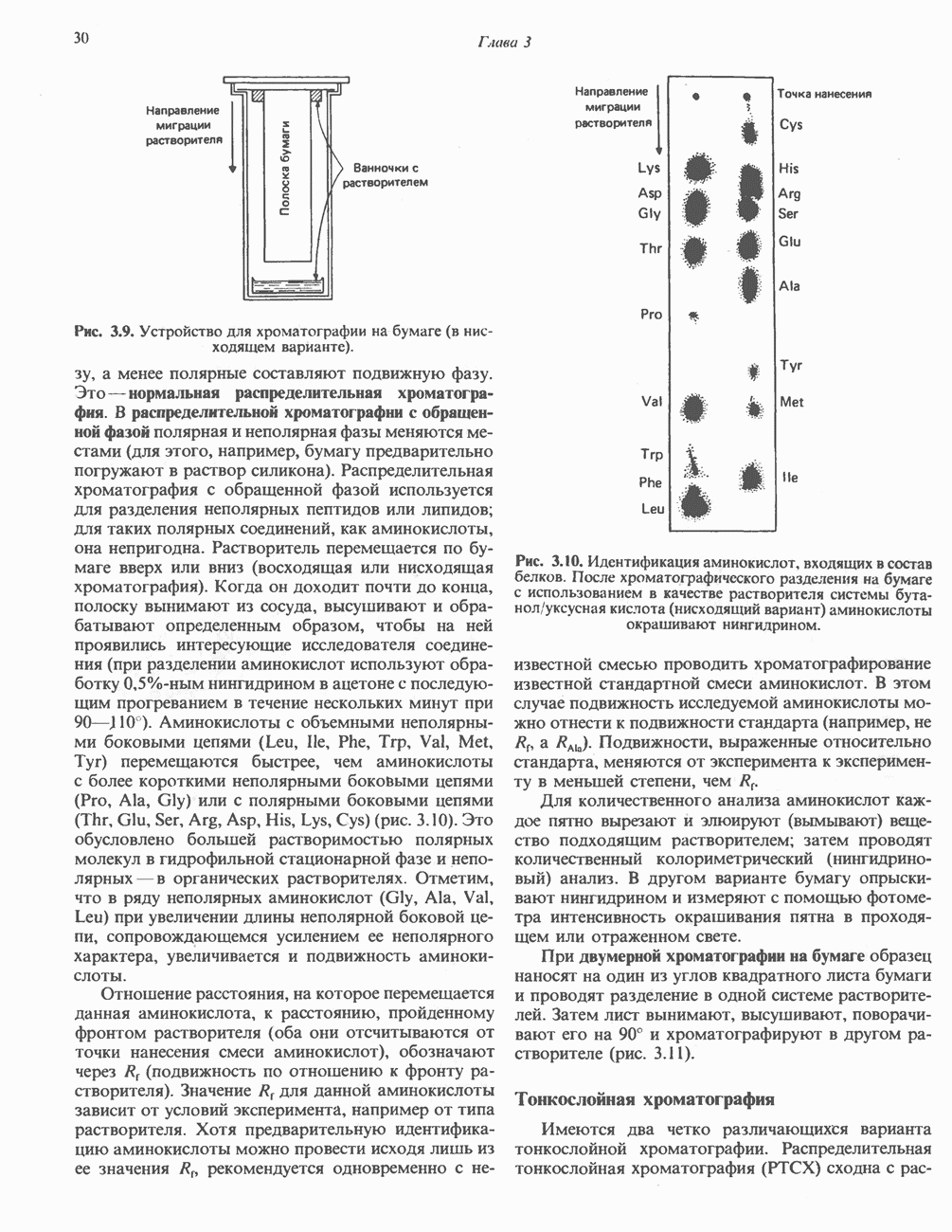
Макеты страниц
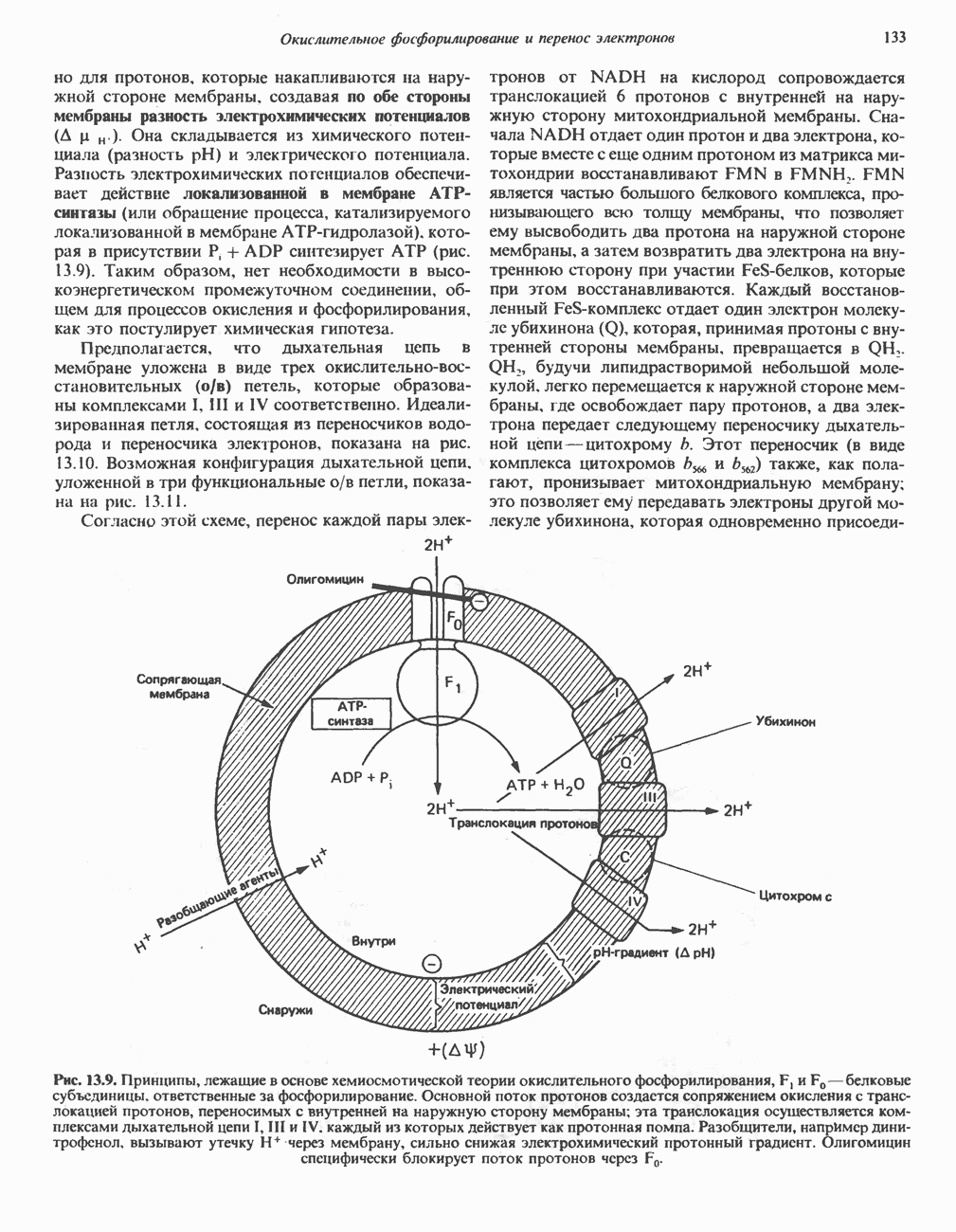
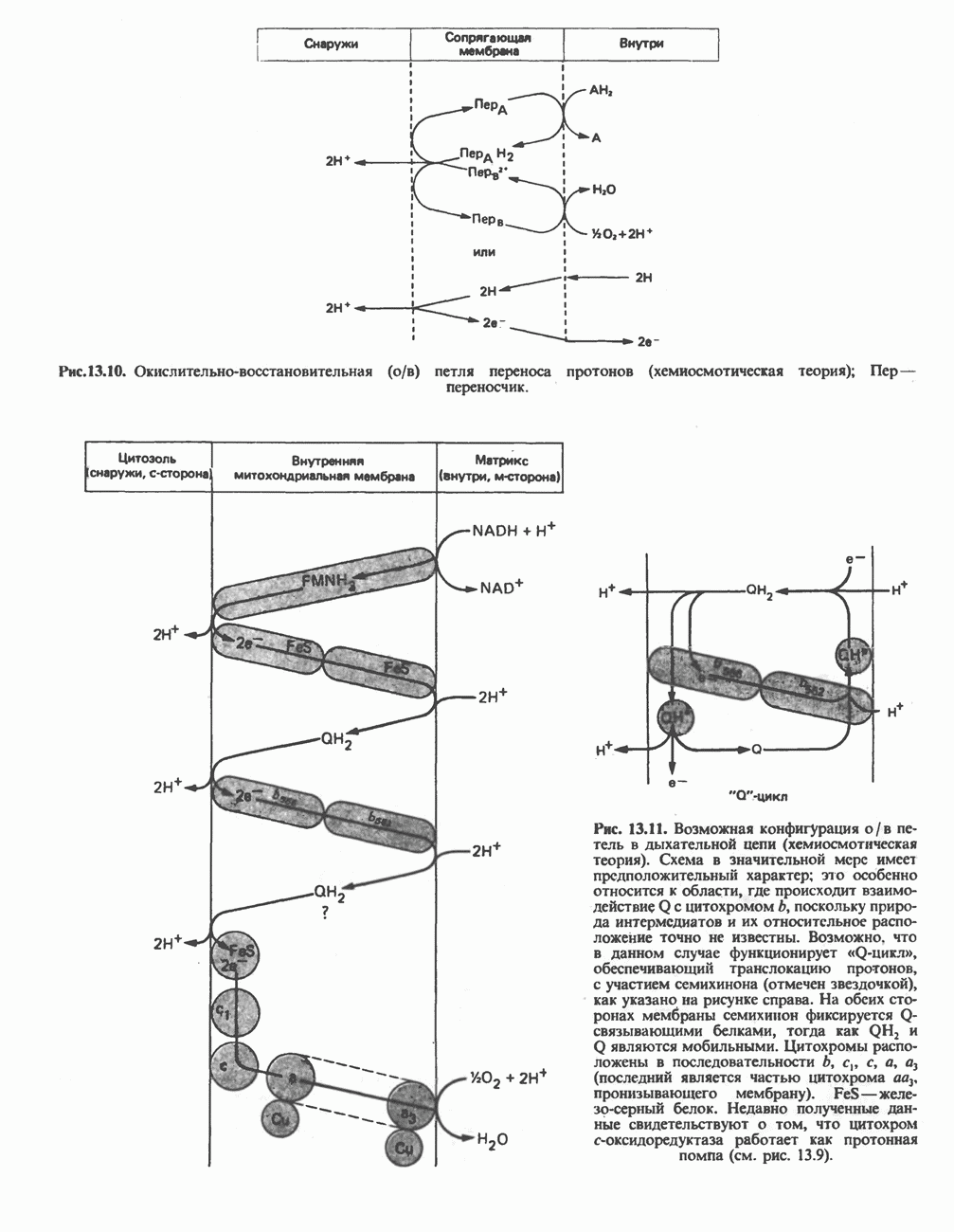
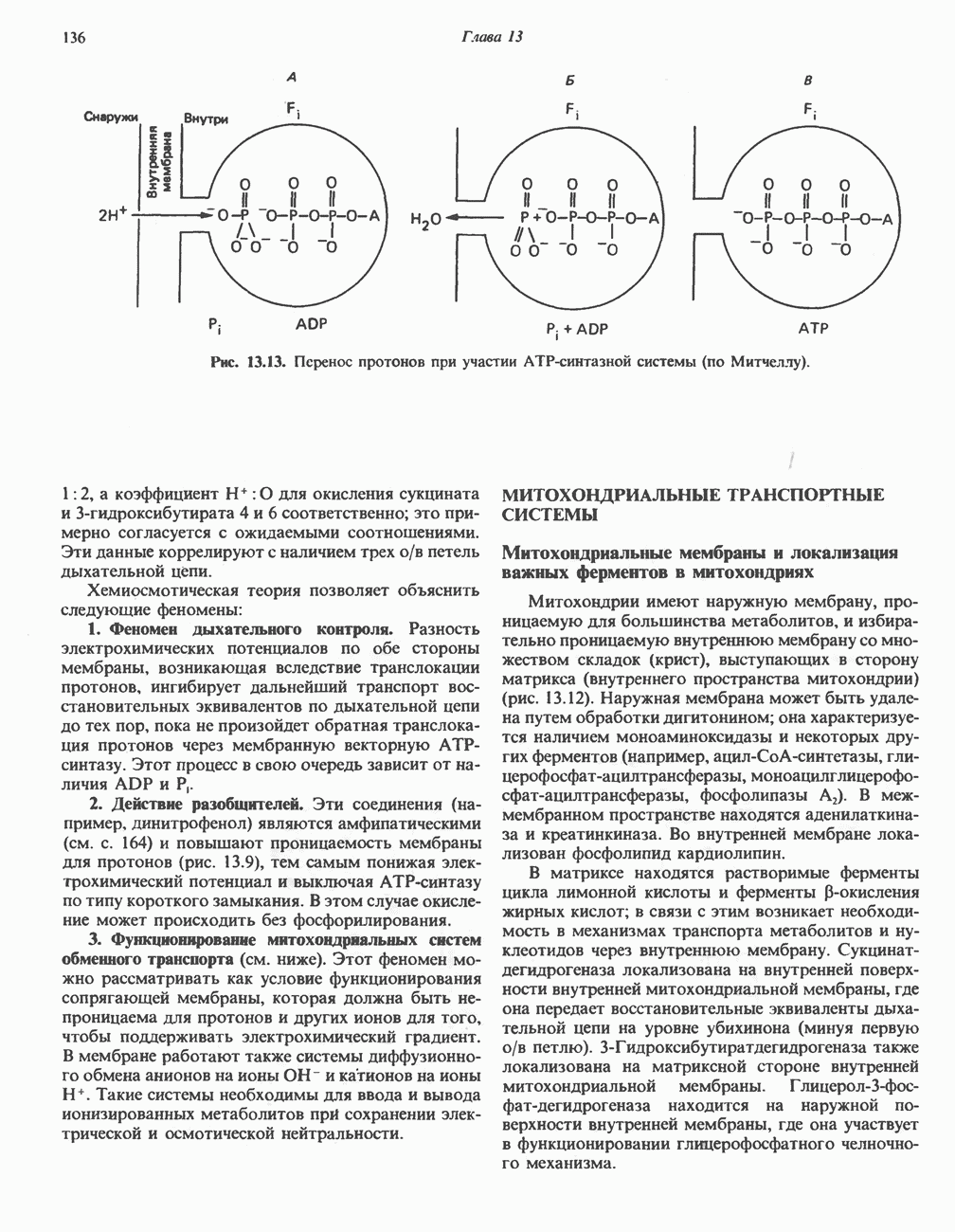
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
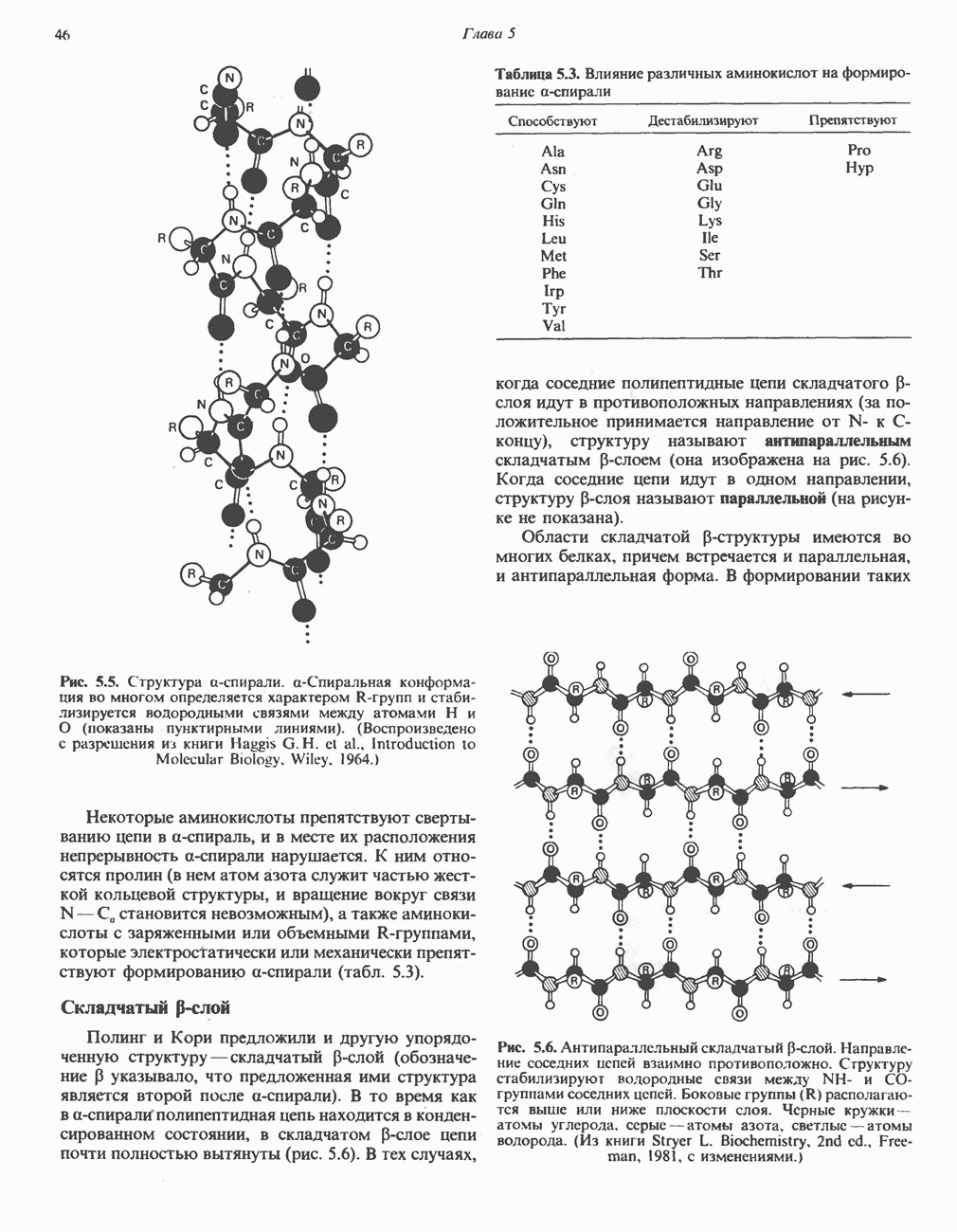
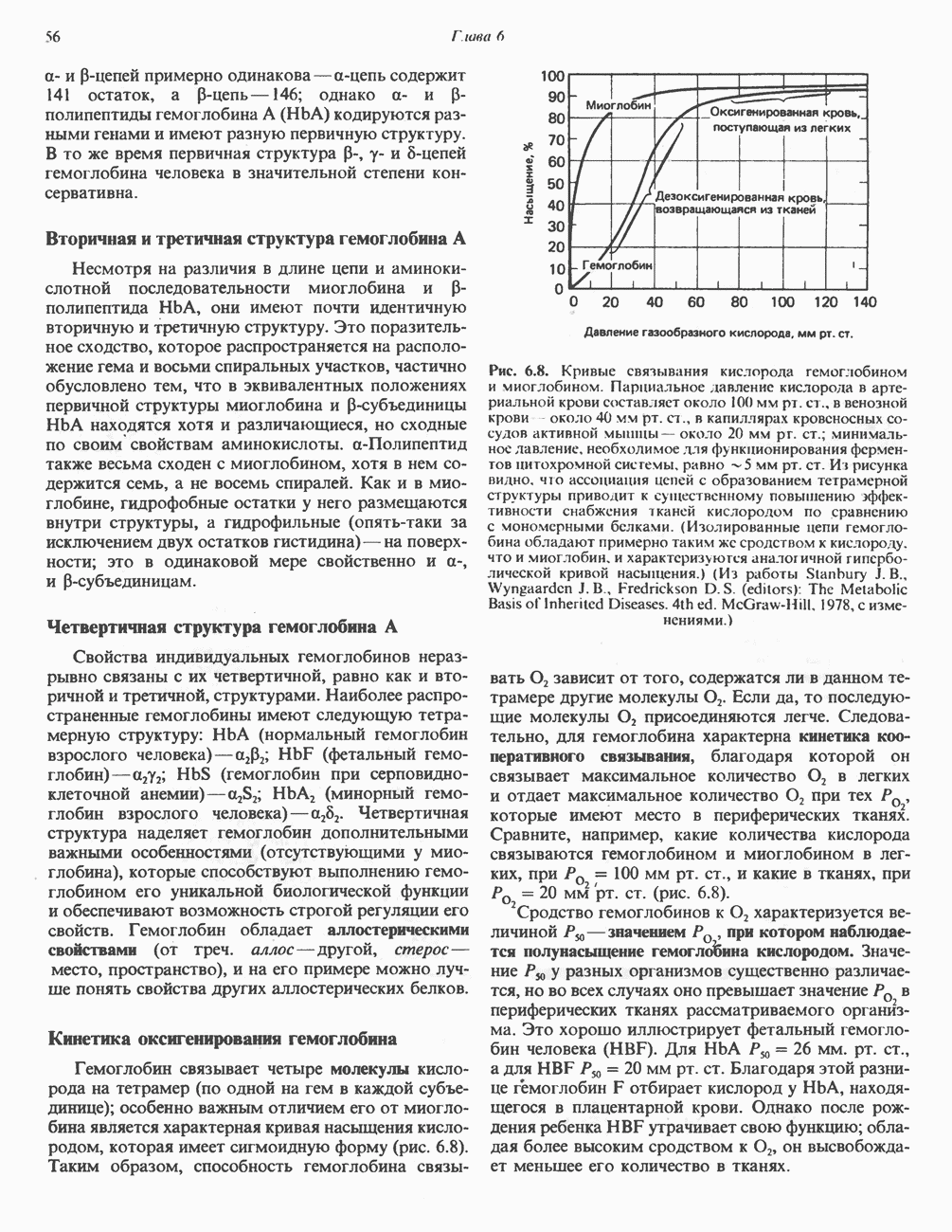
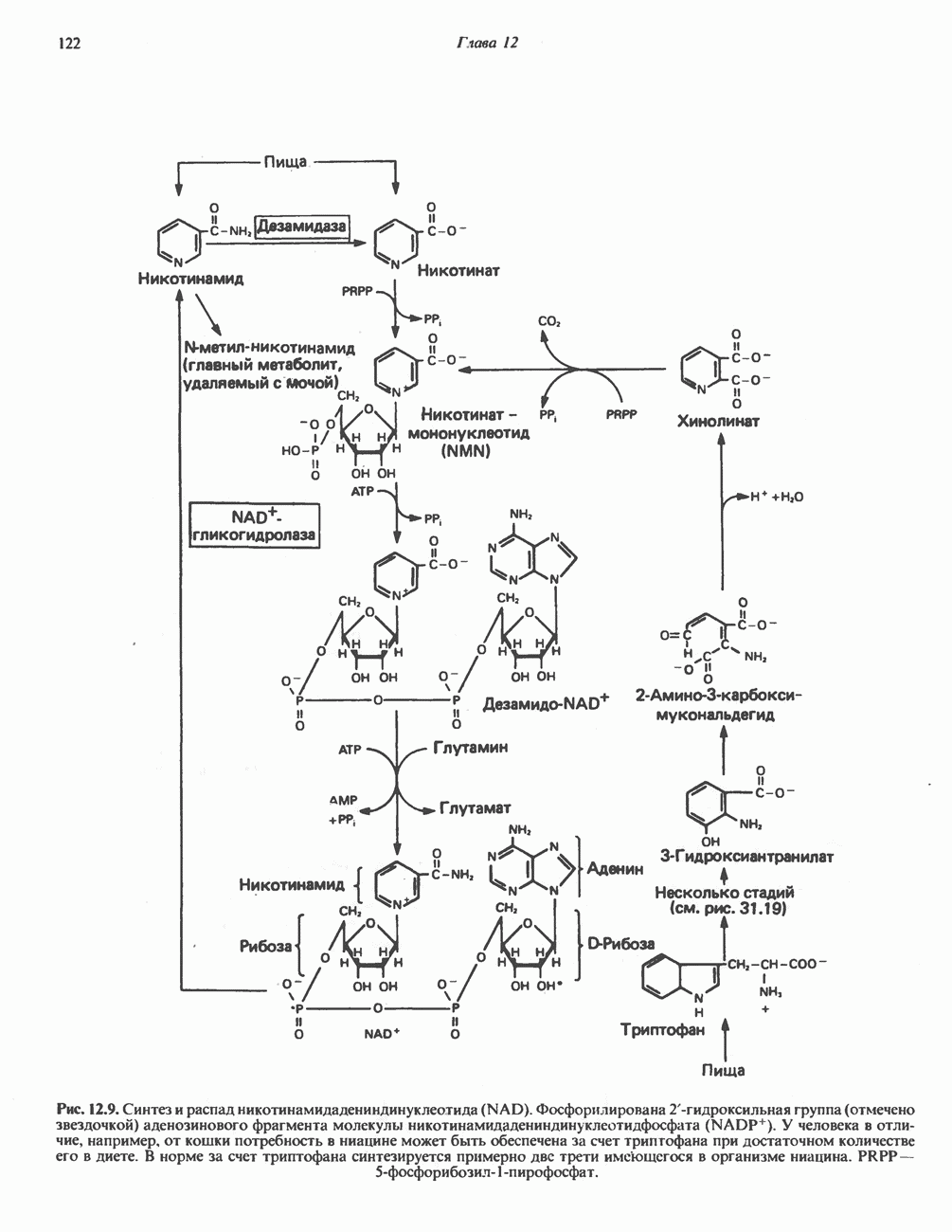
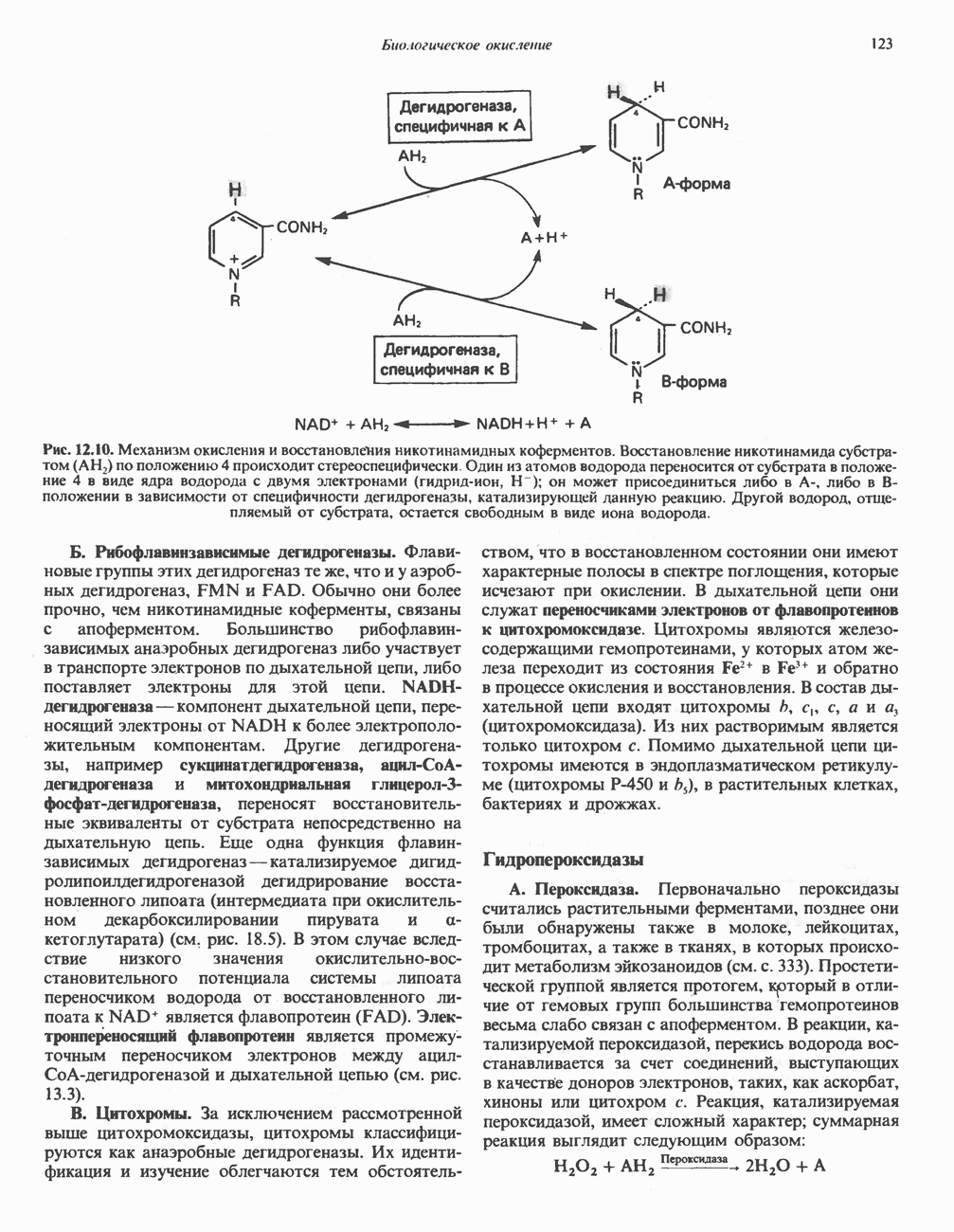
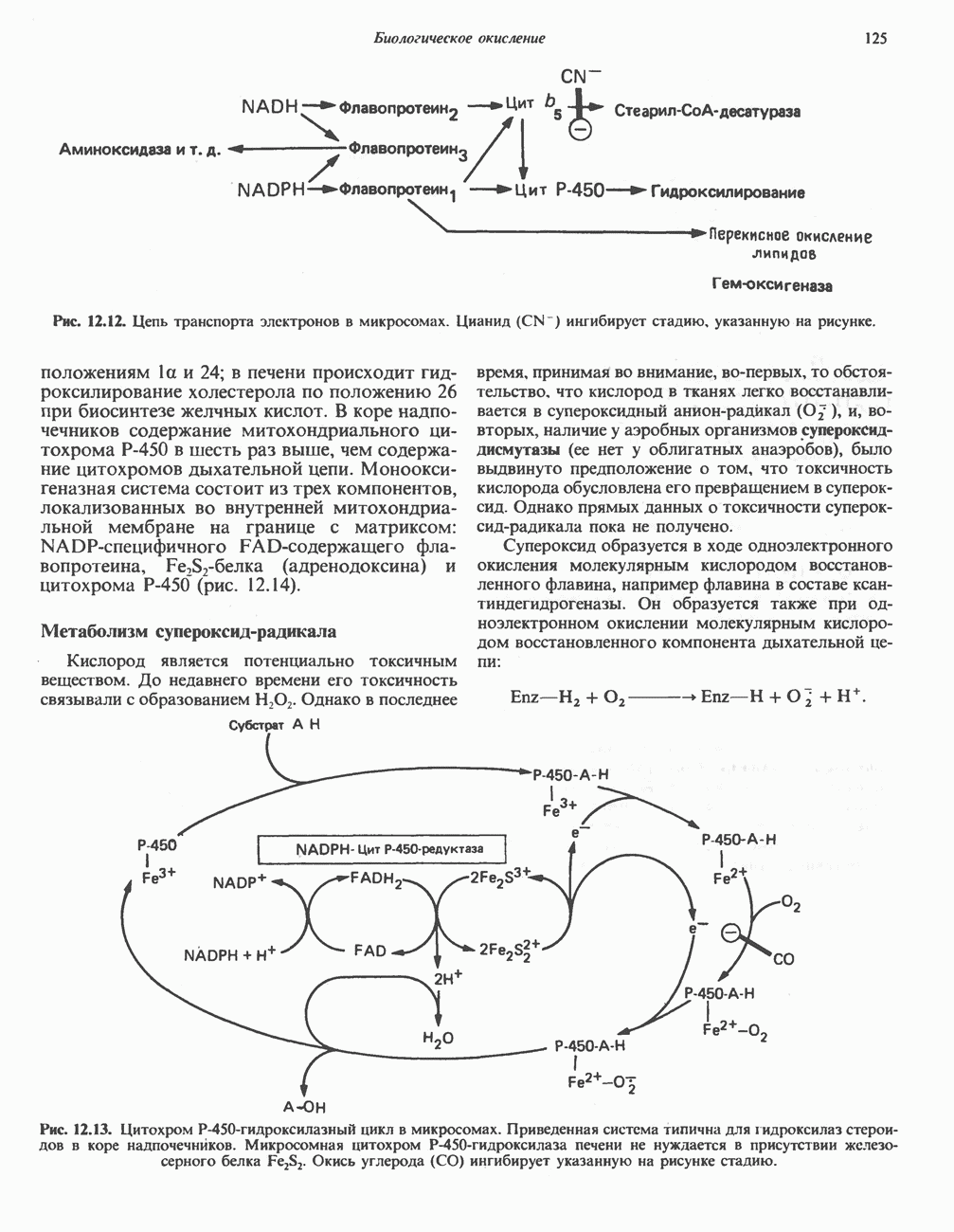
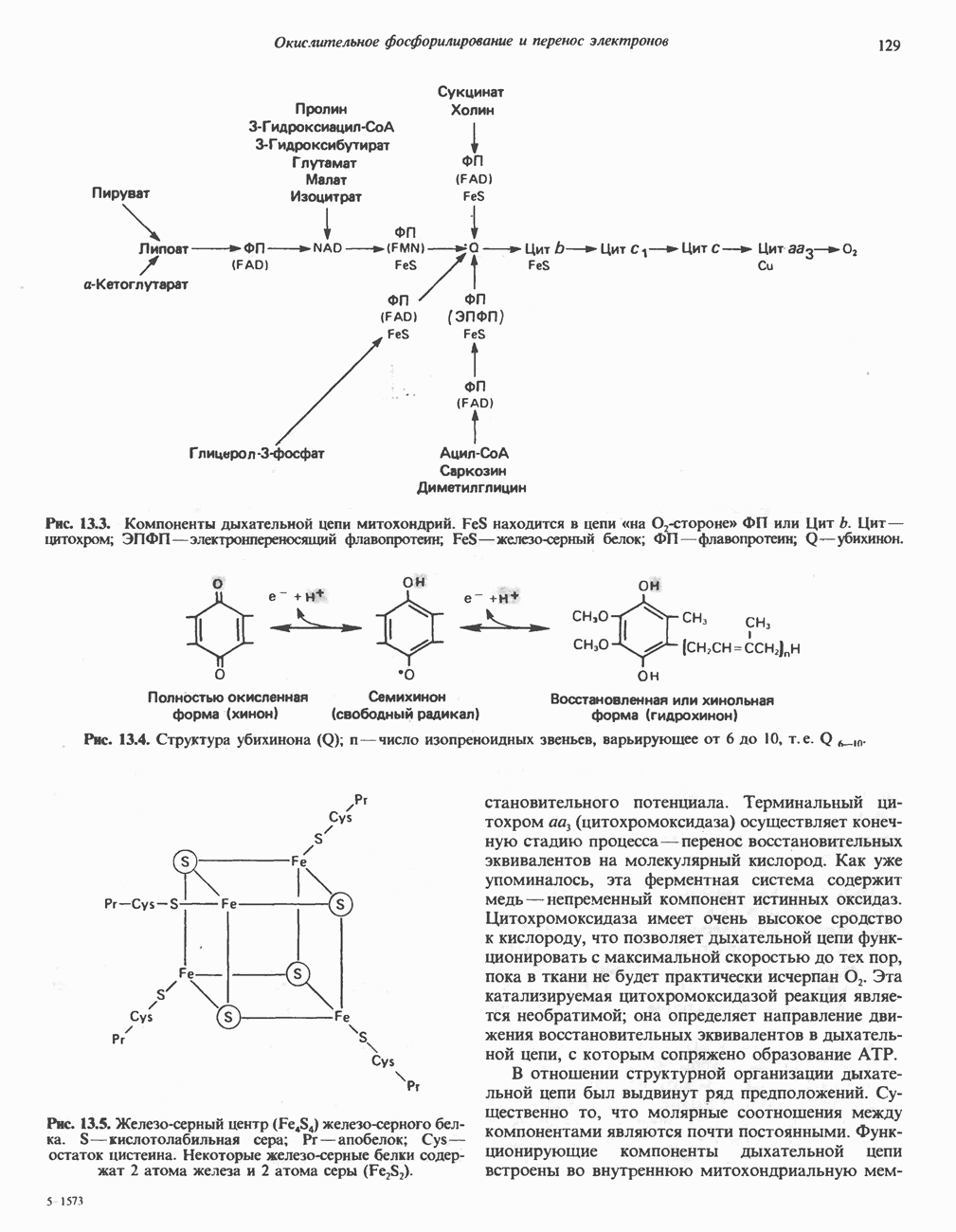
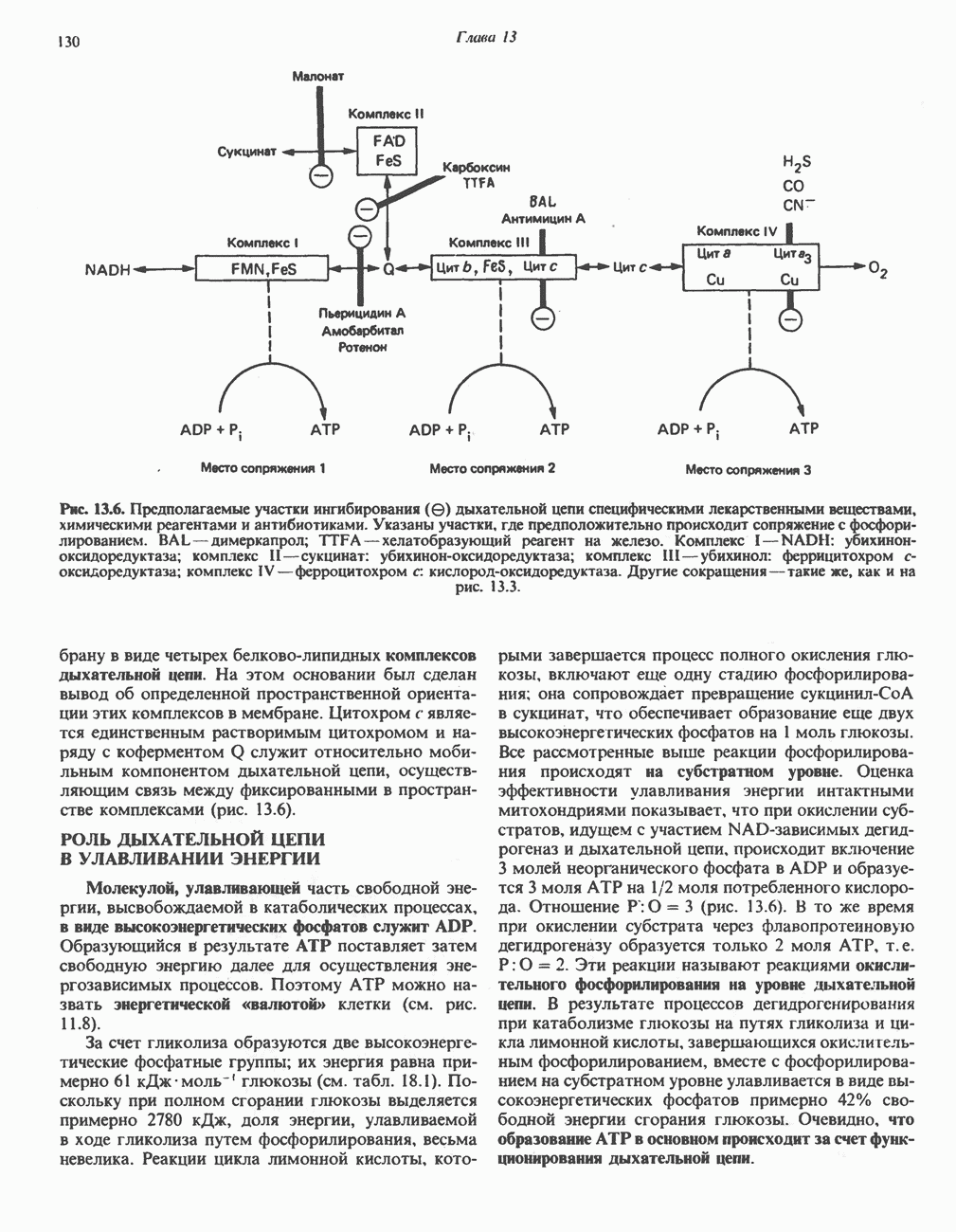
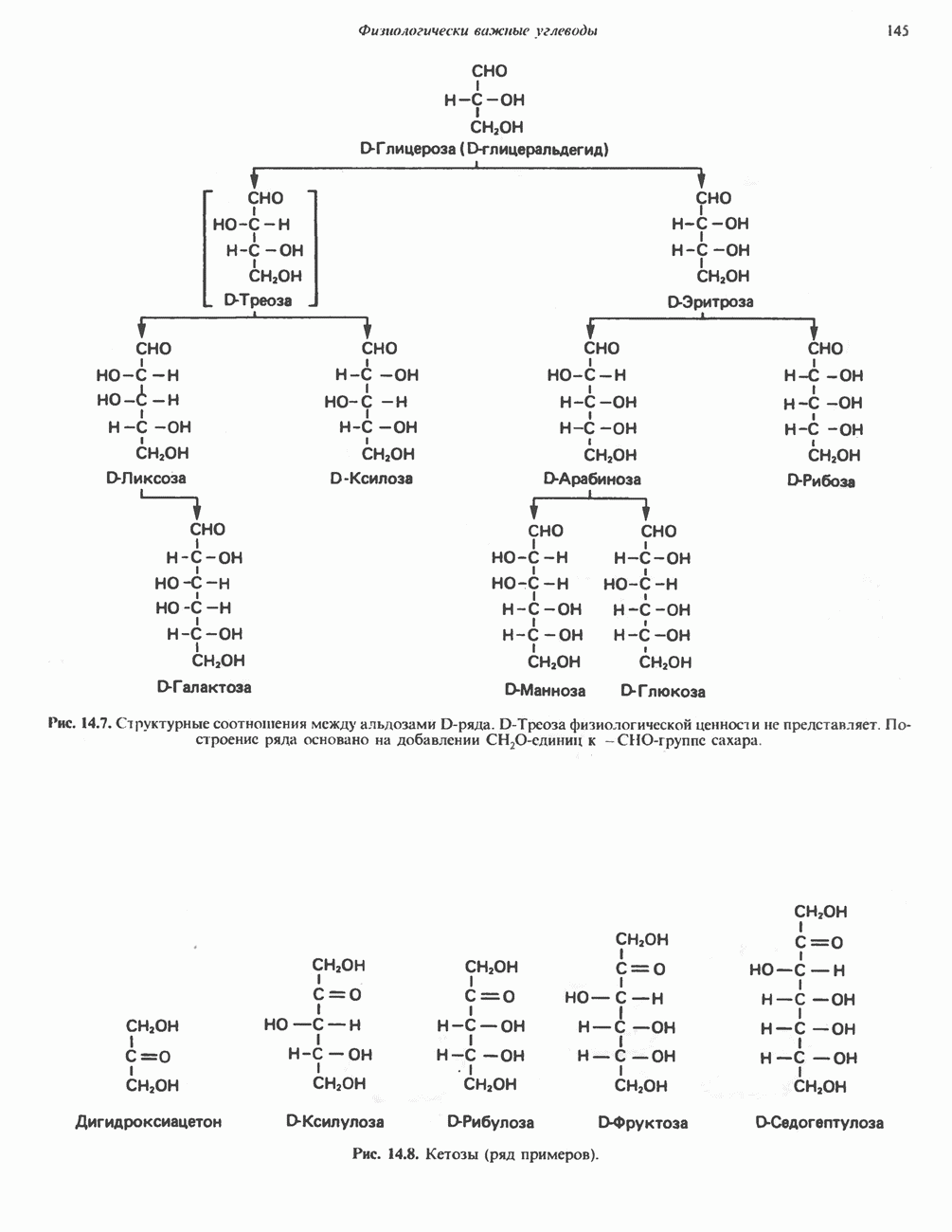
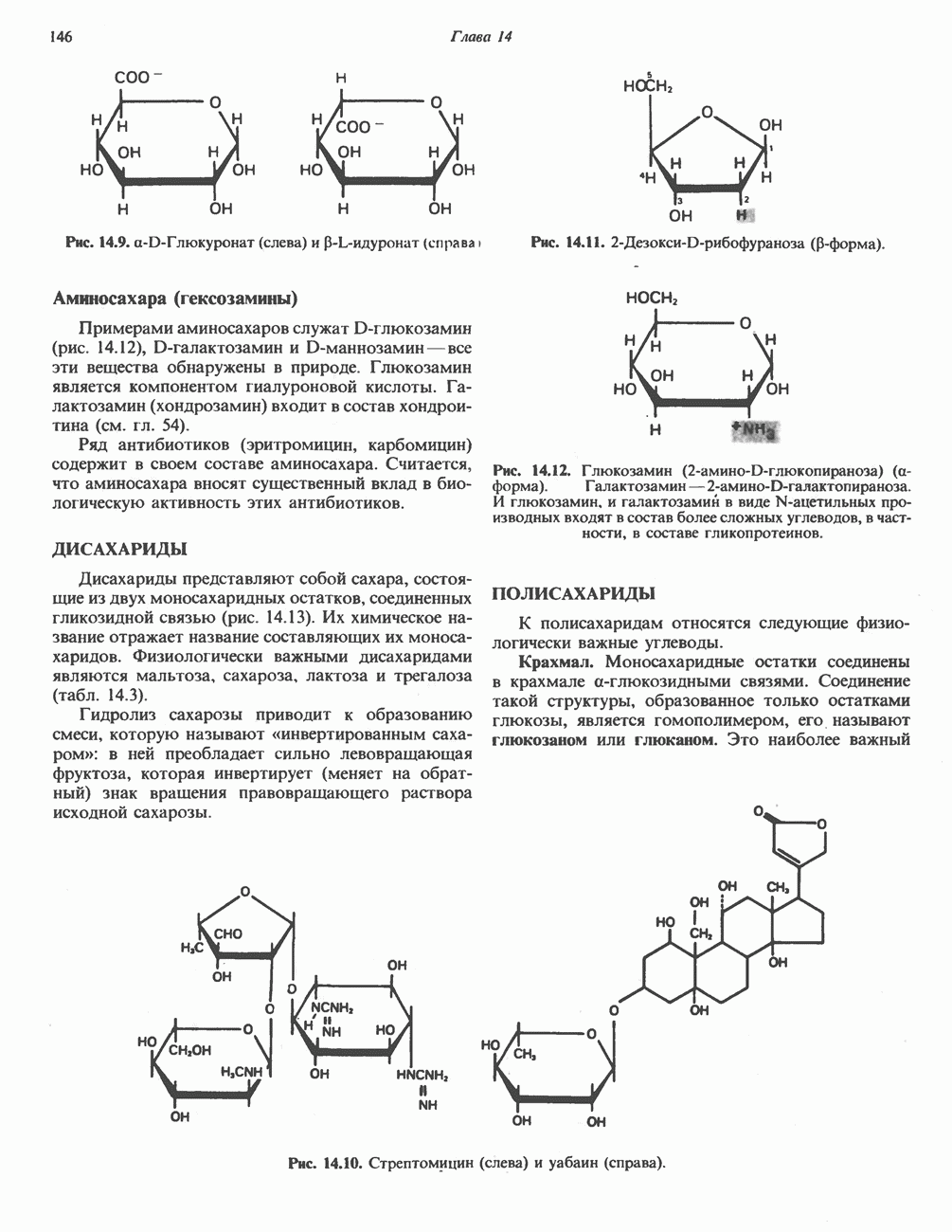
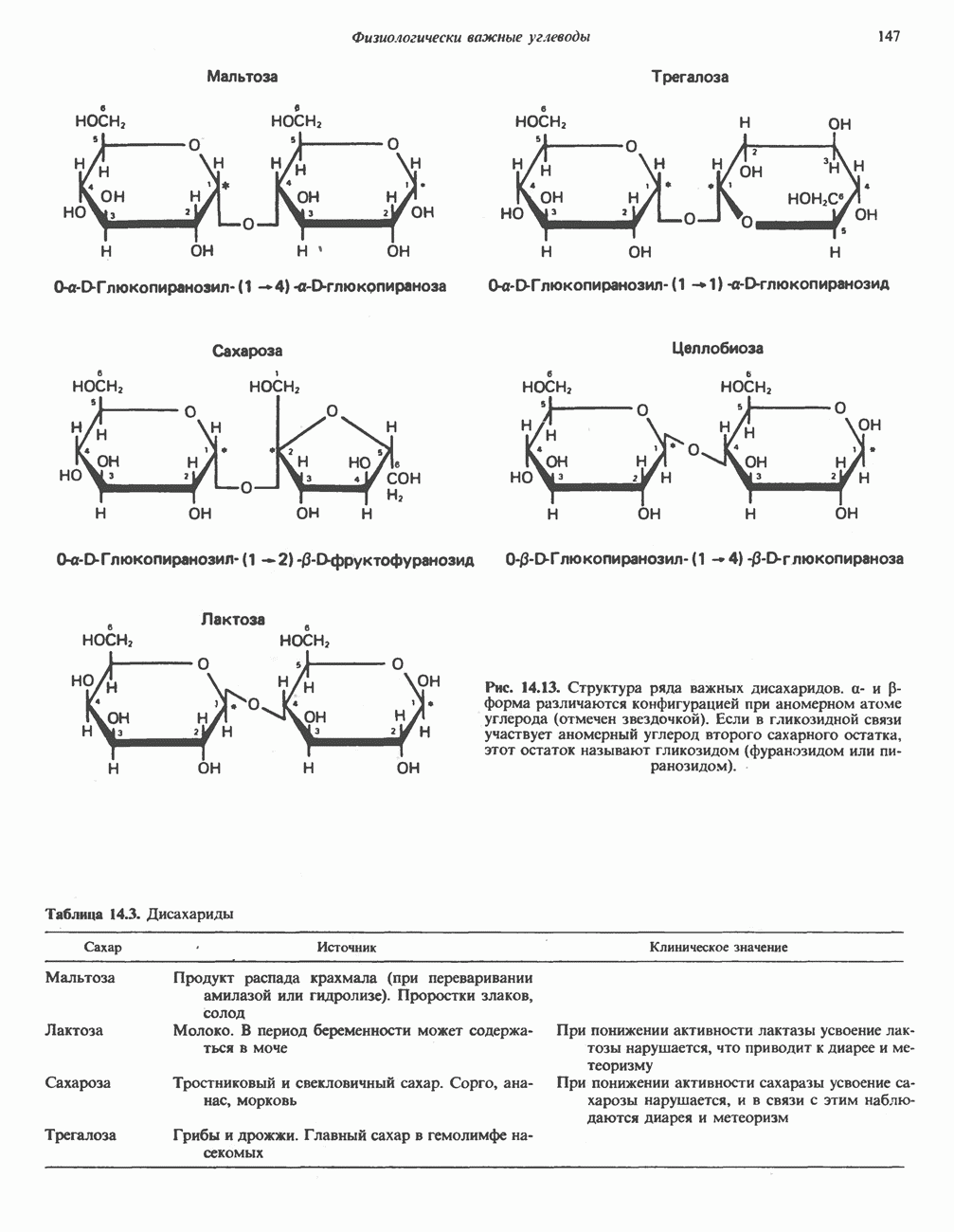
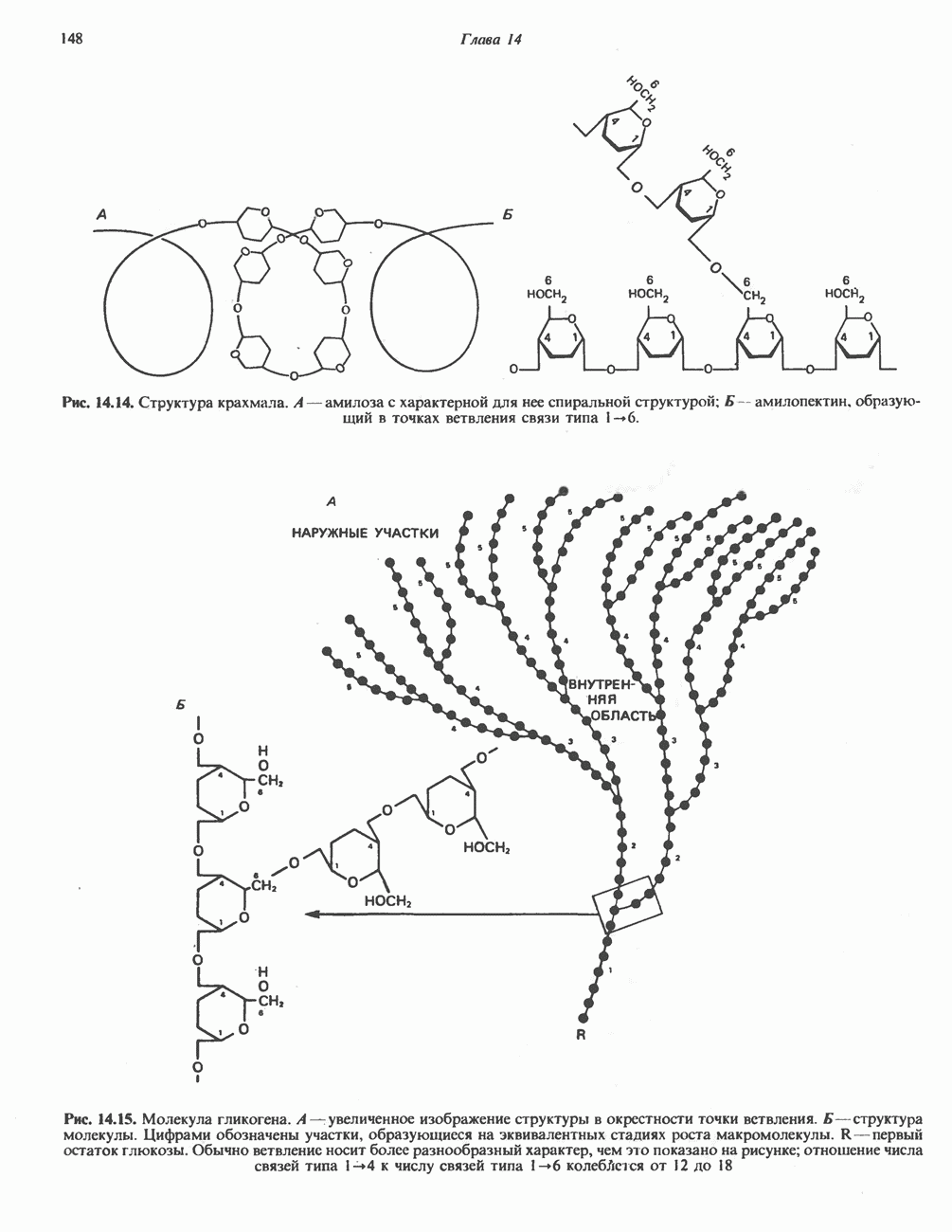
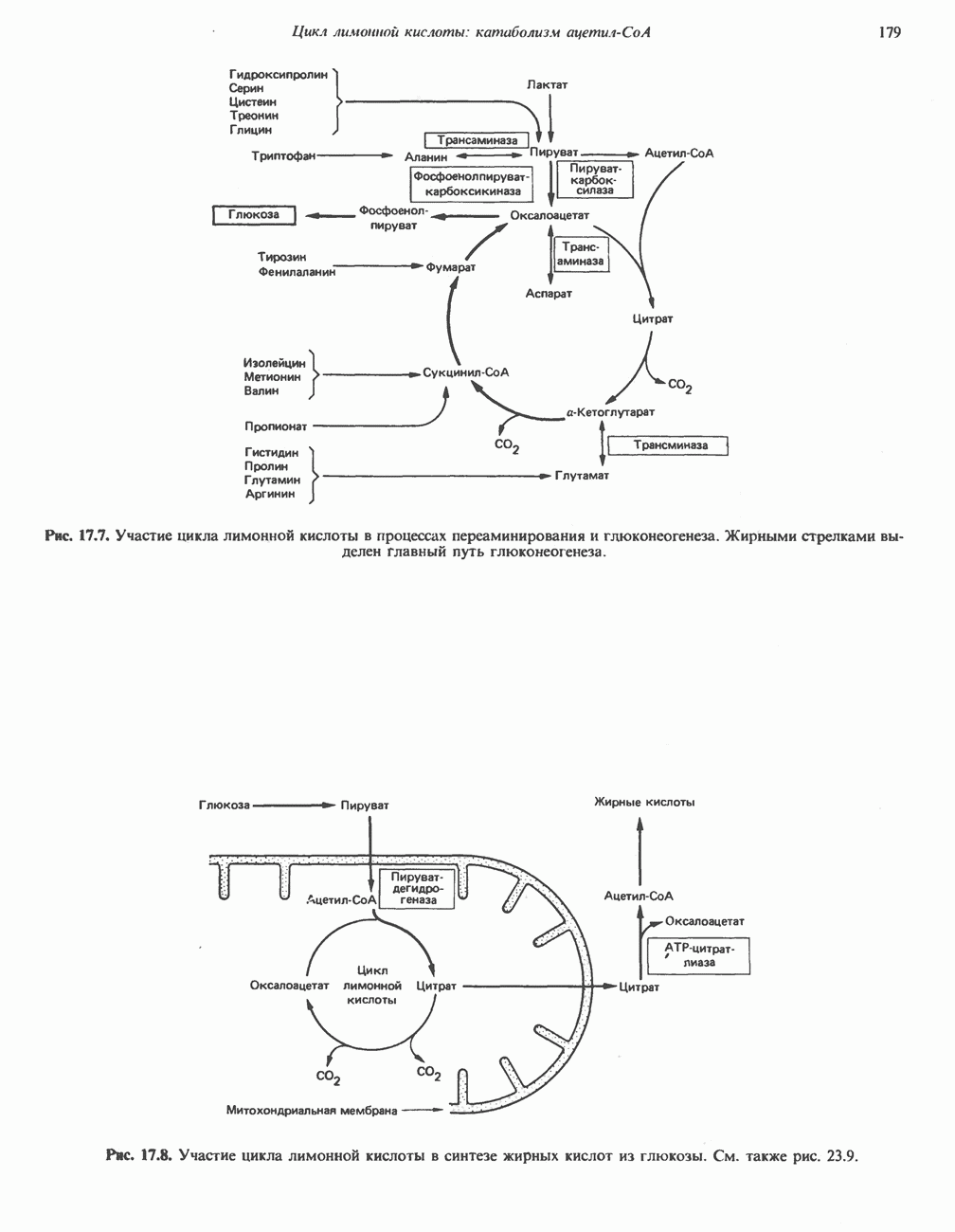
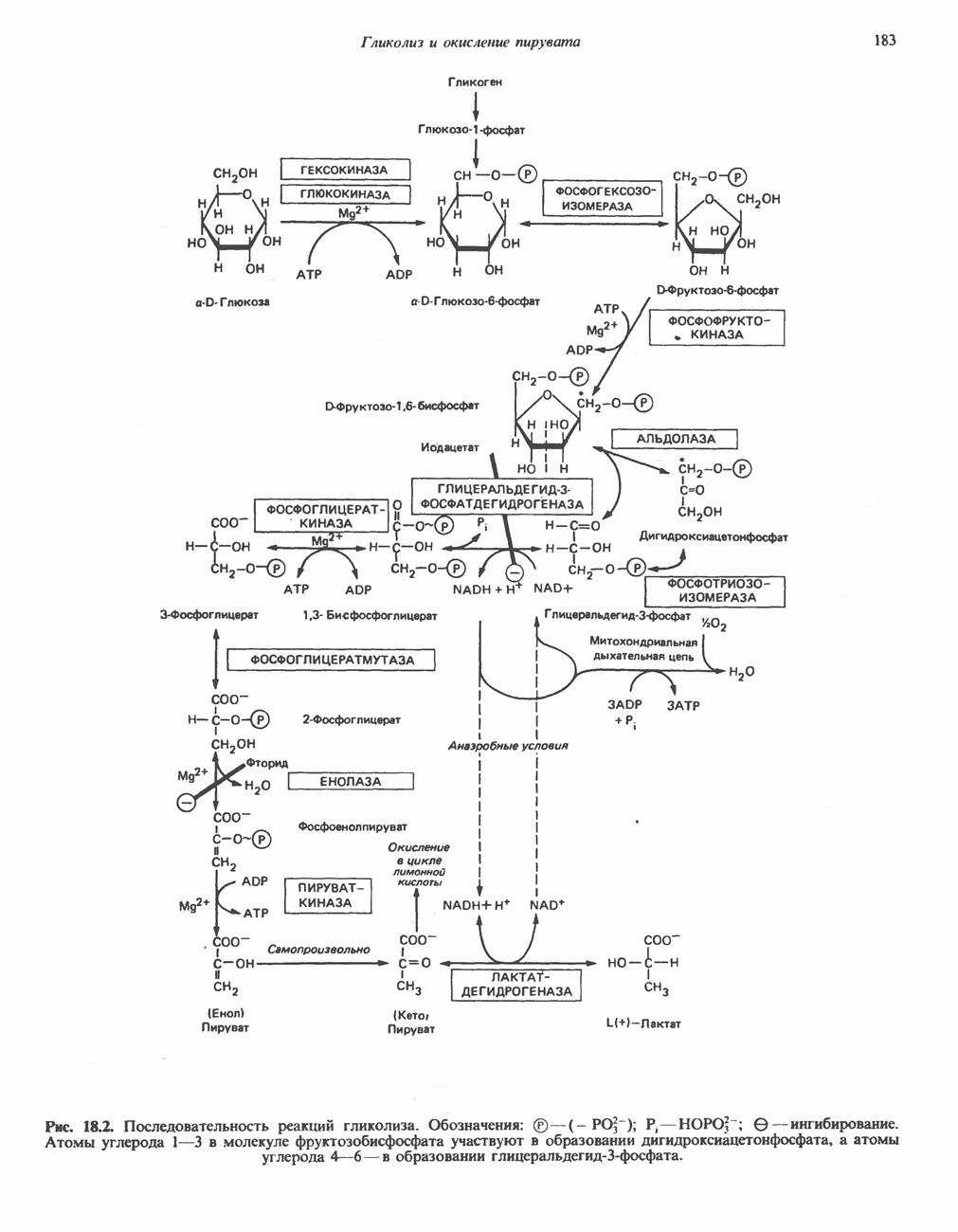
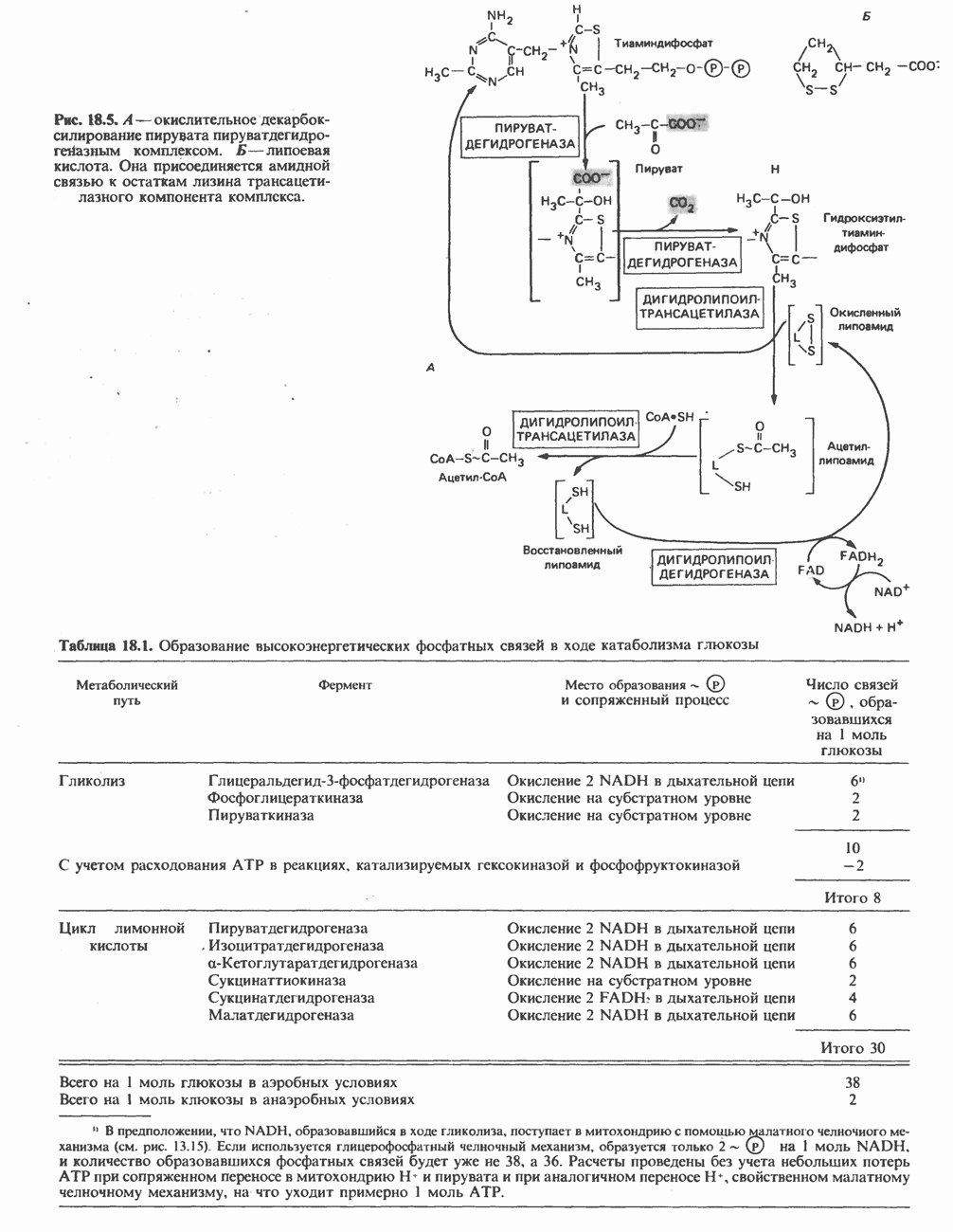
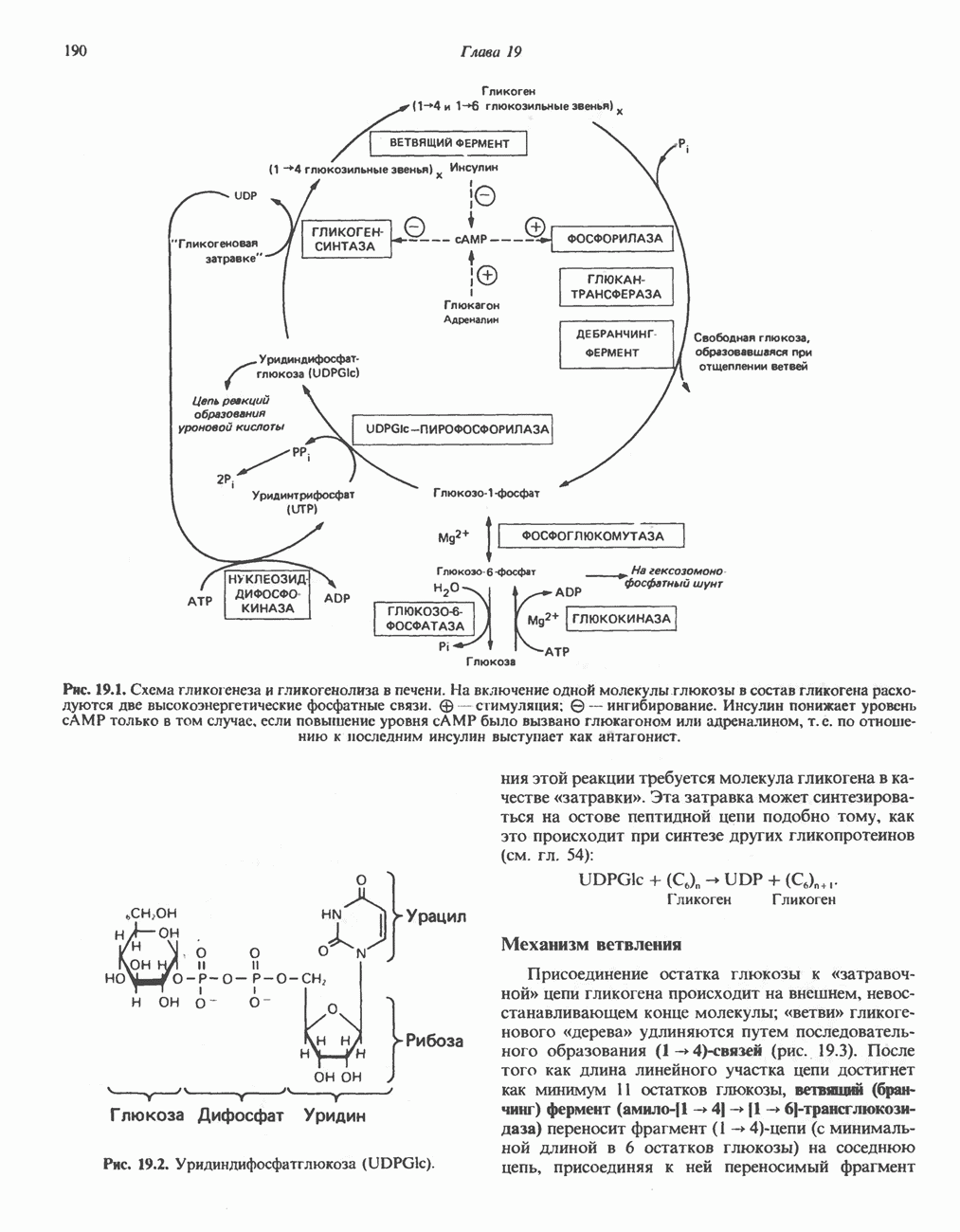
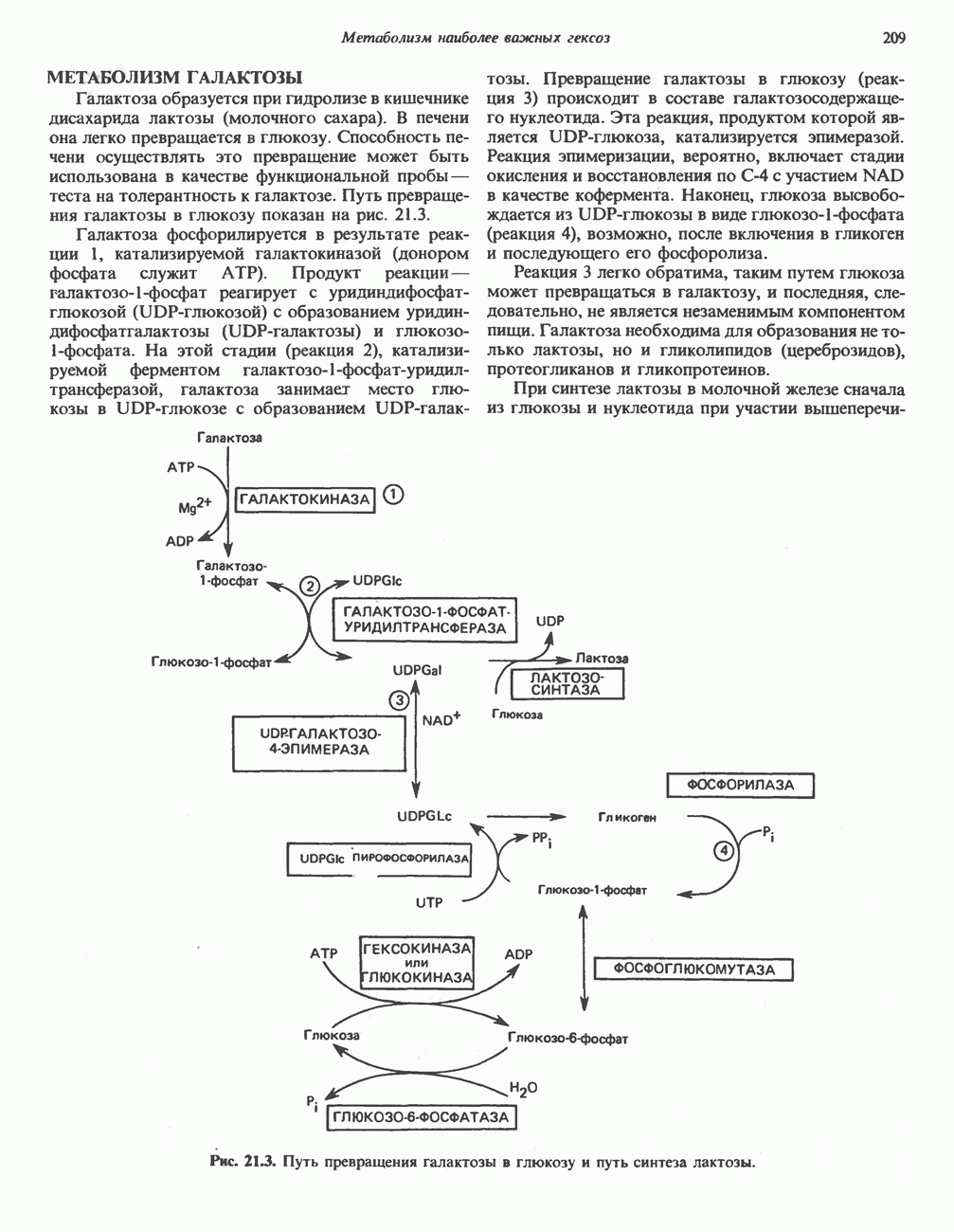
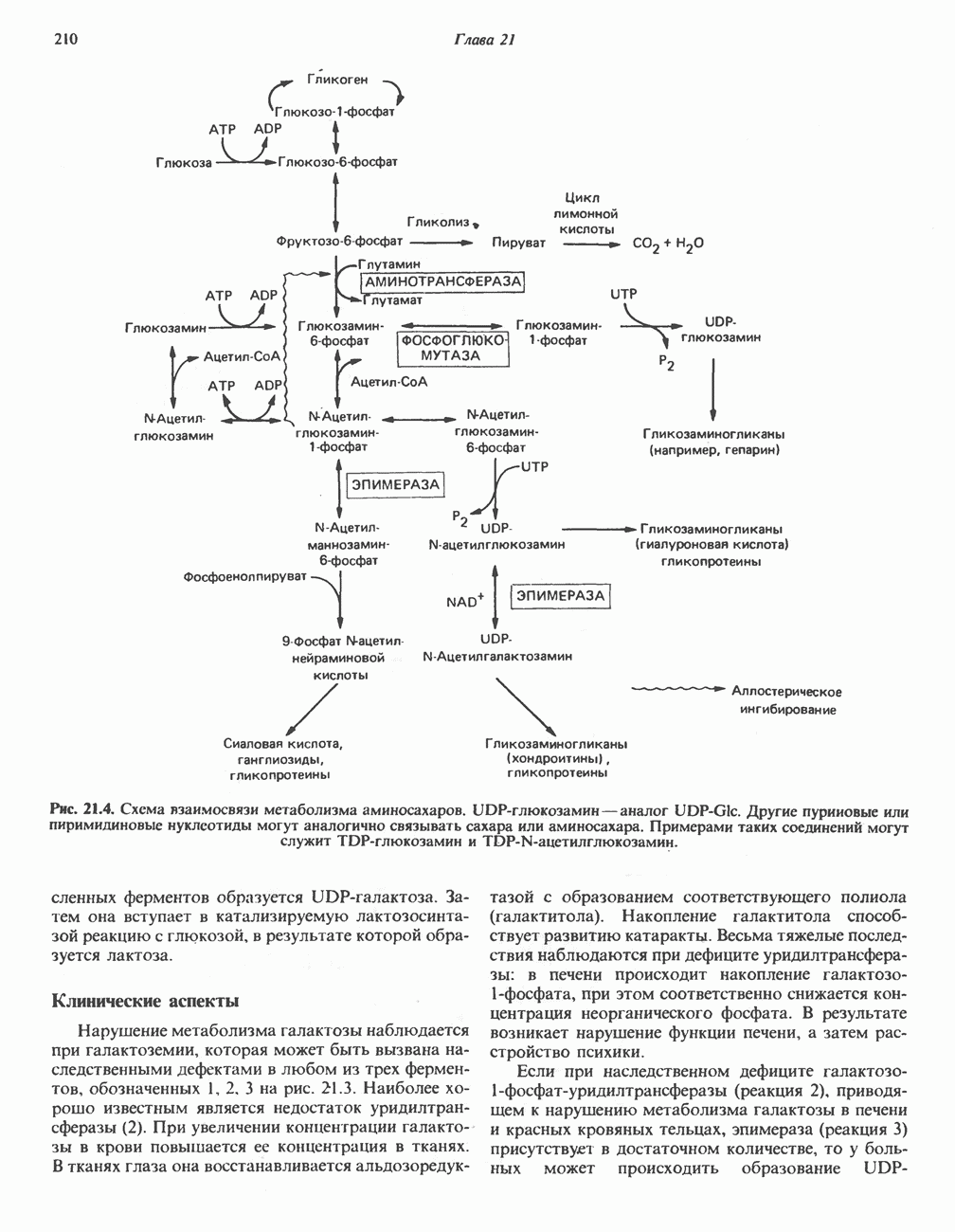
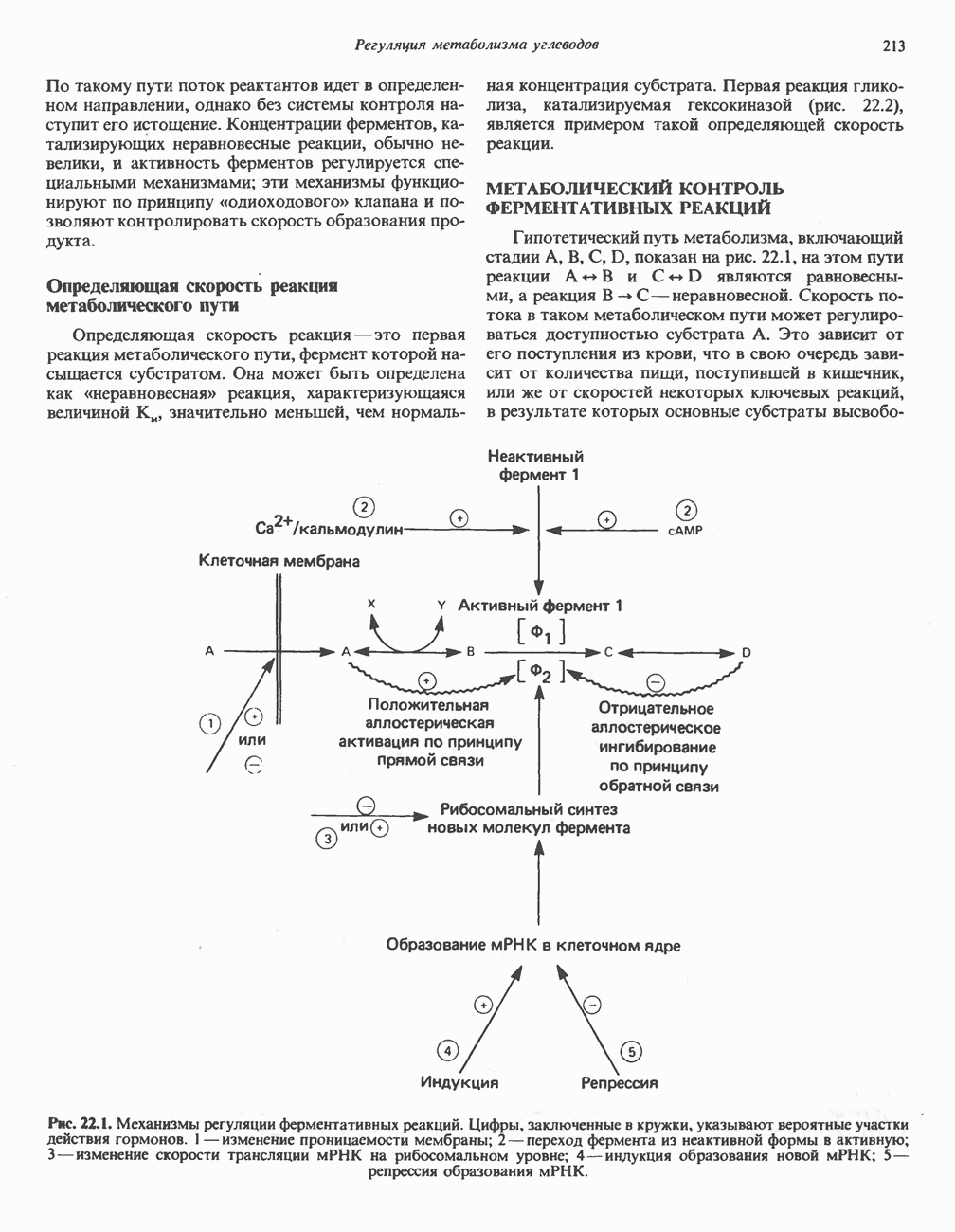
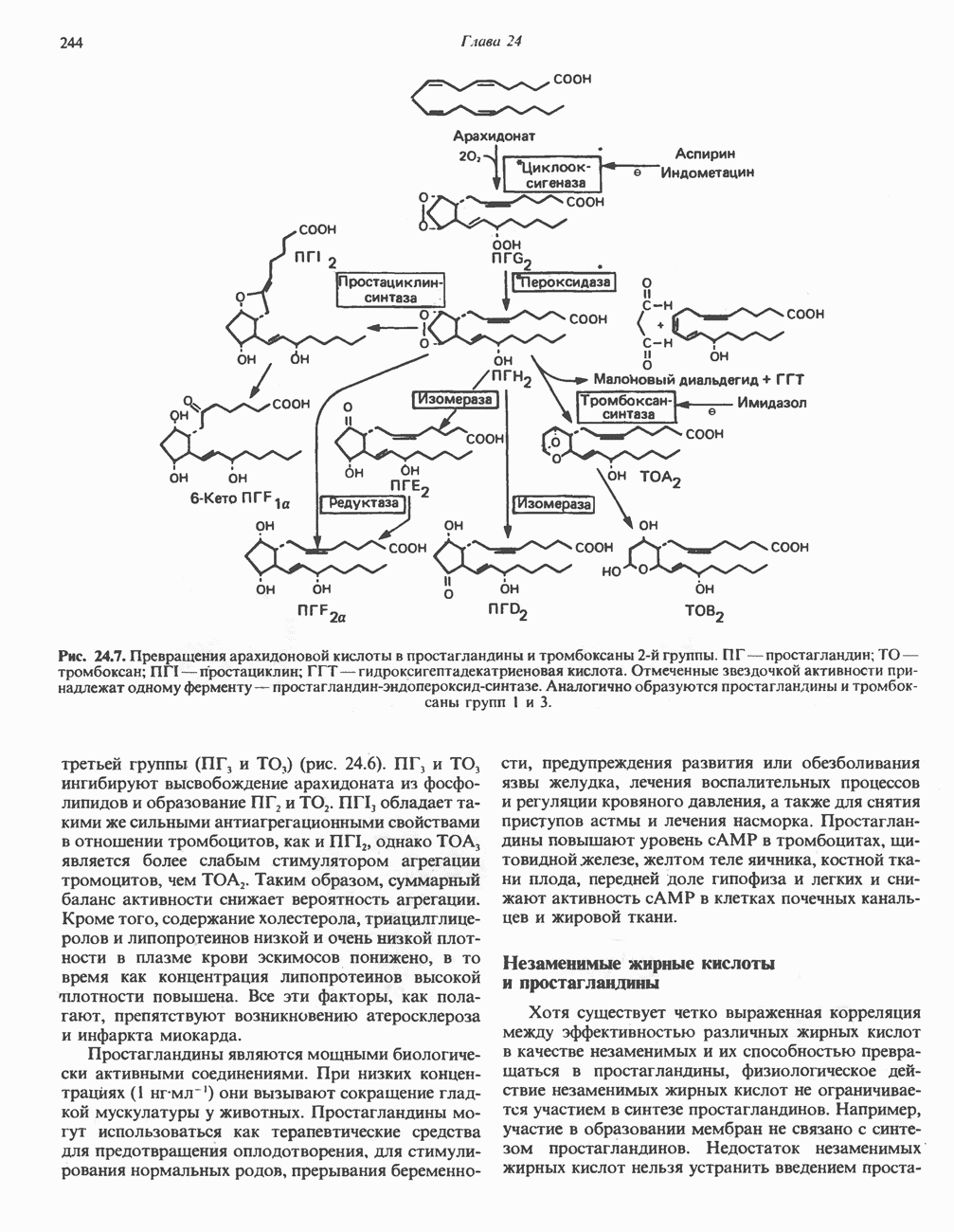
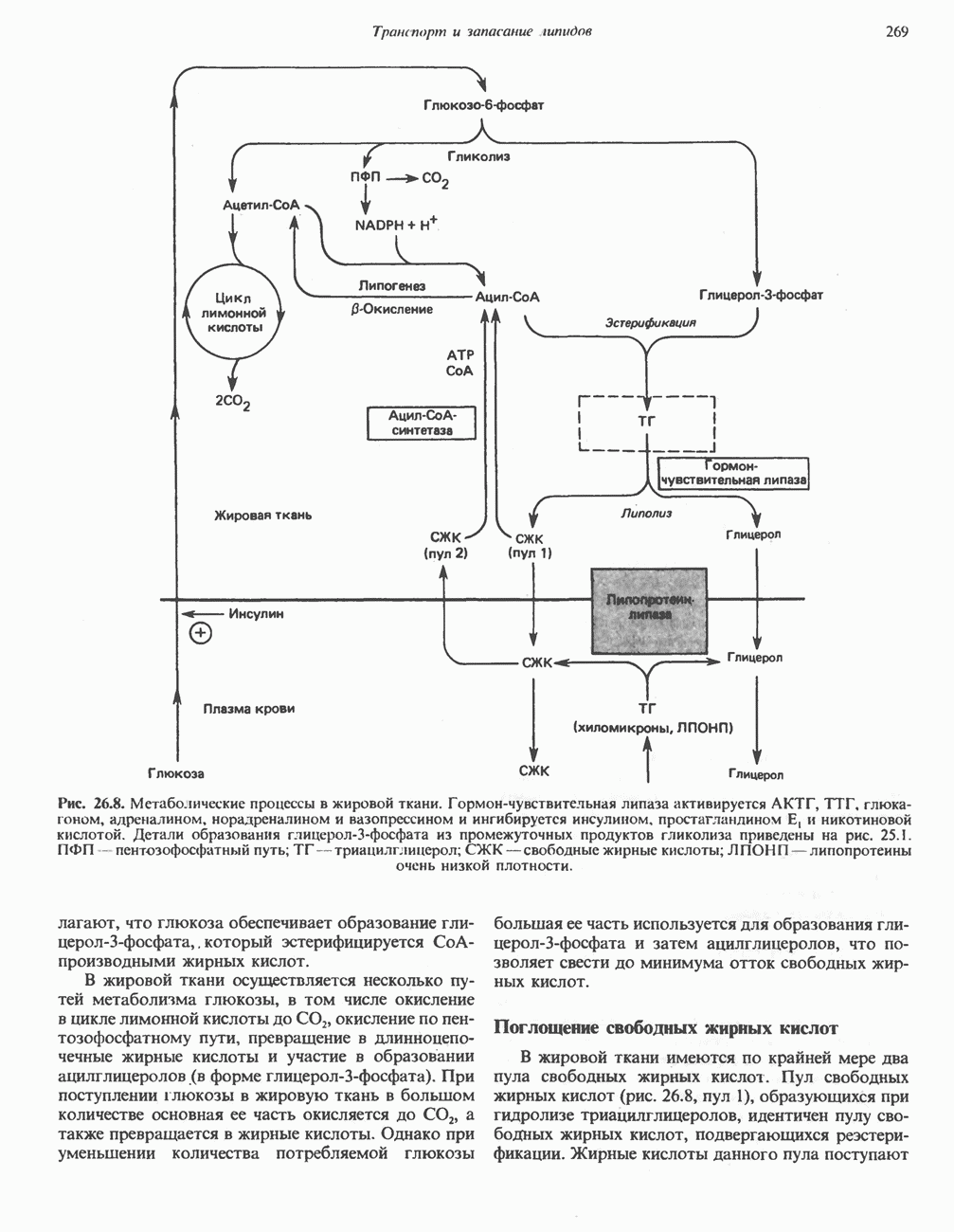
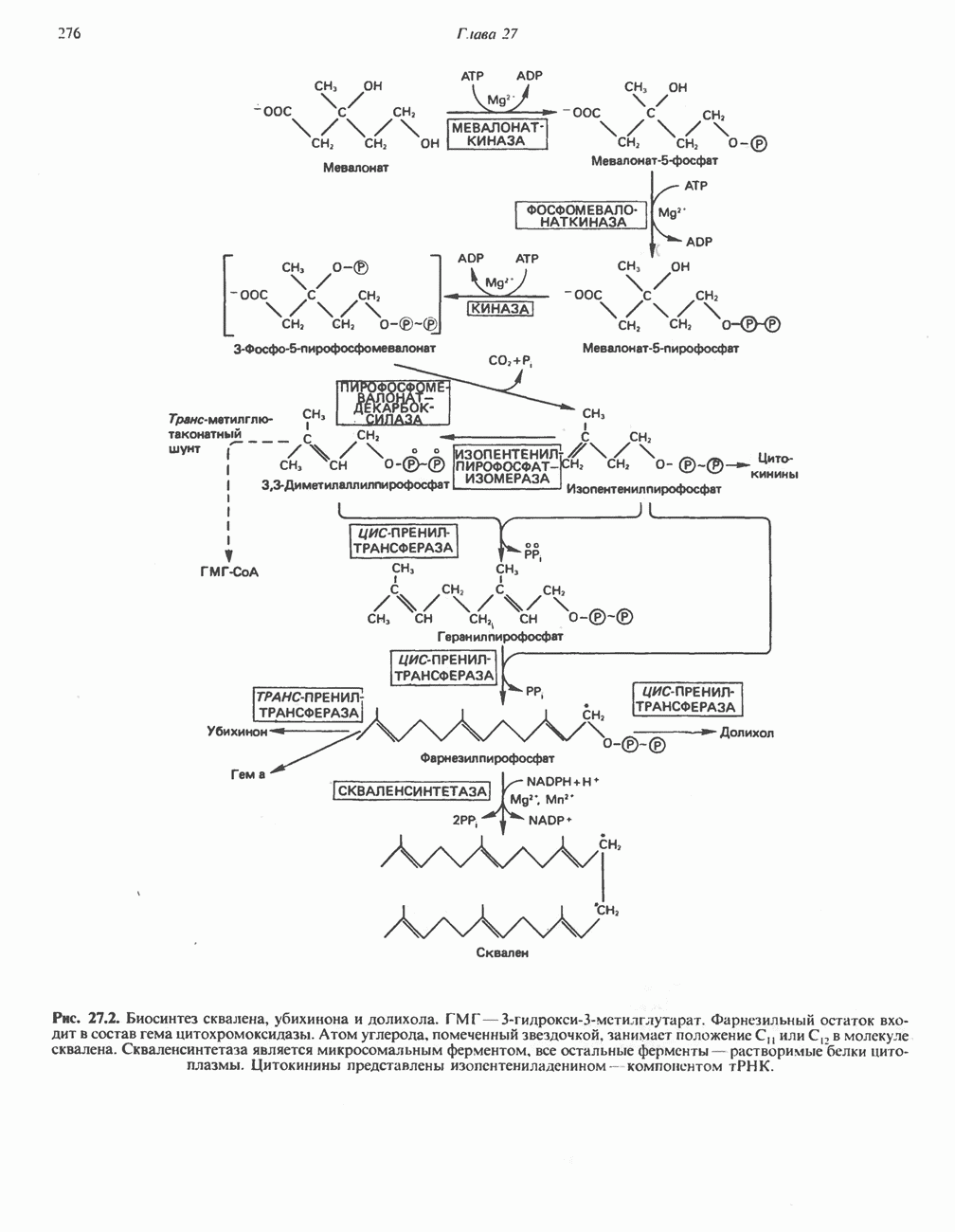
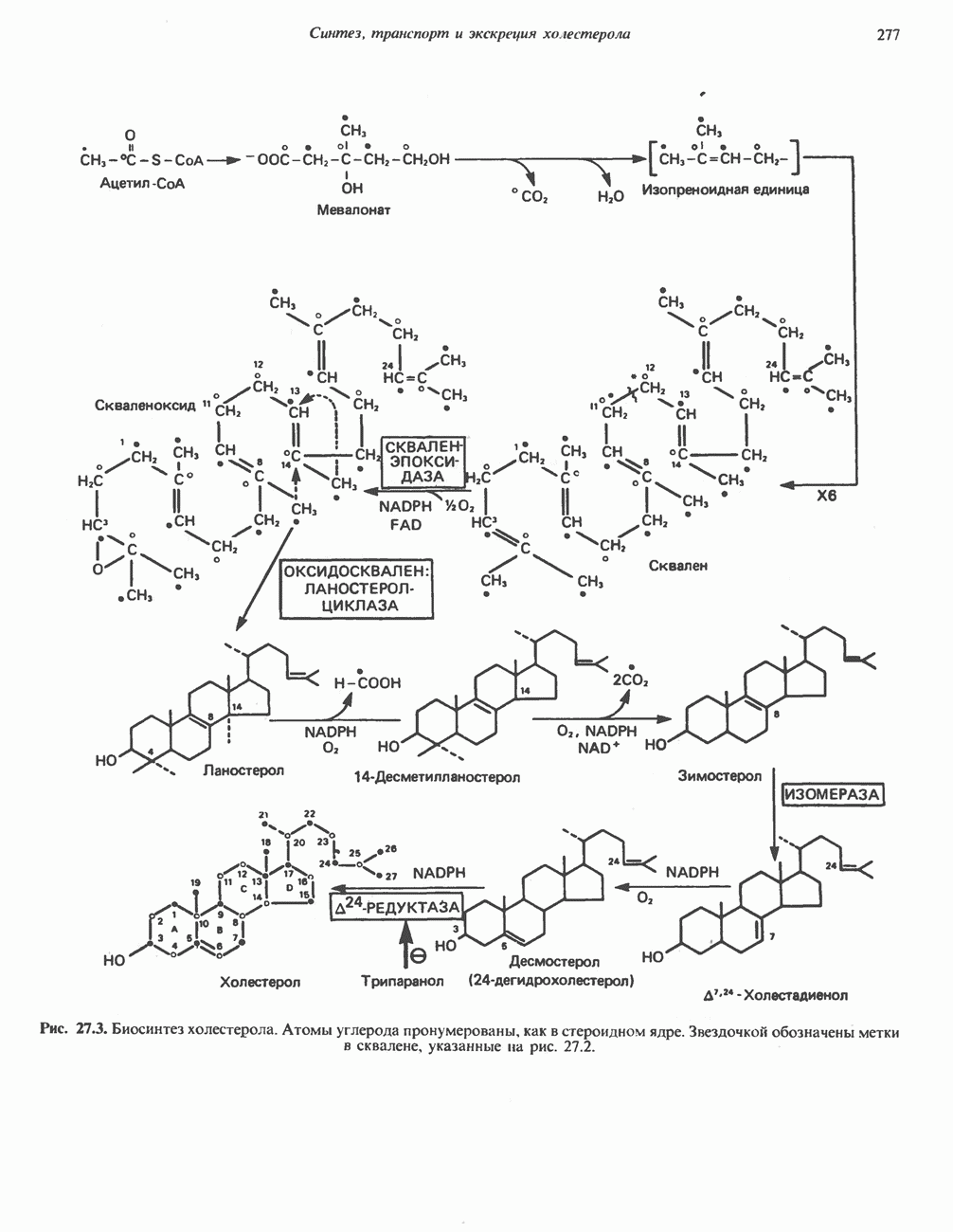
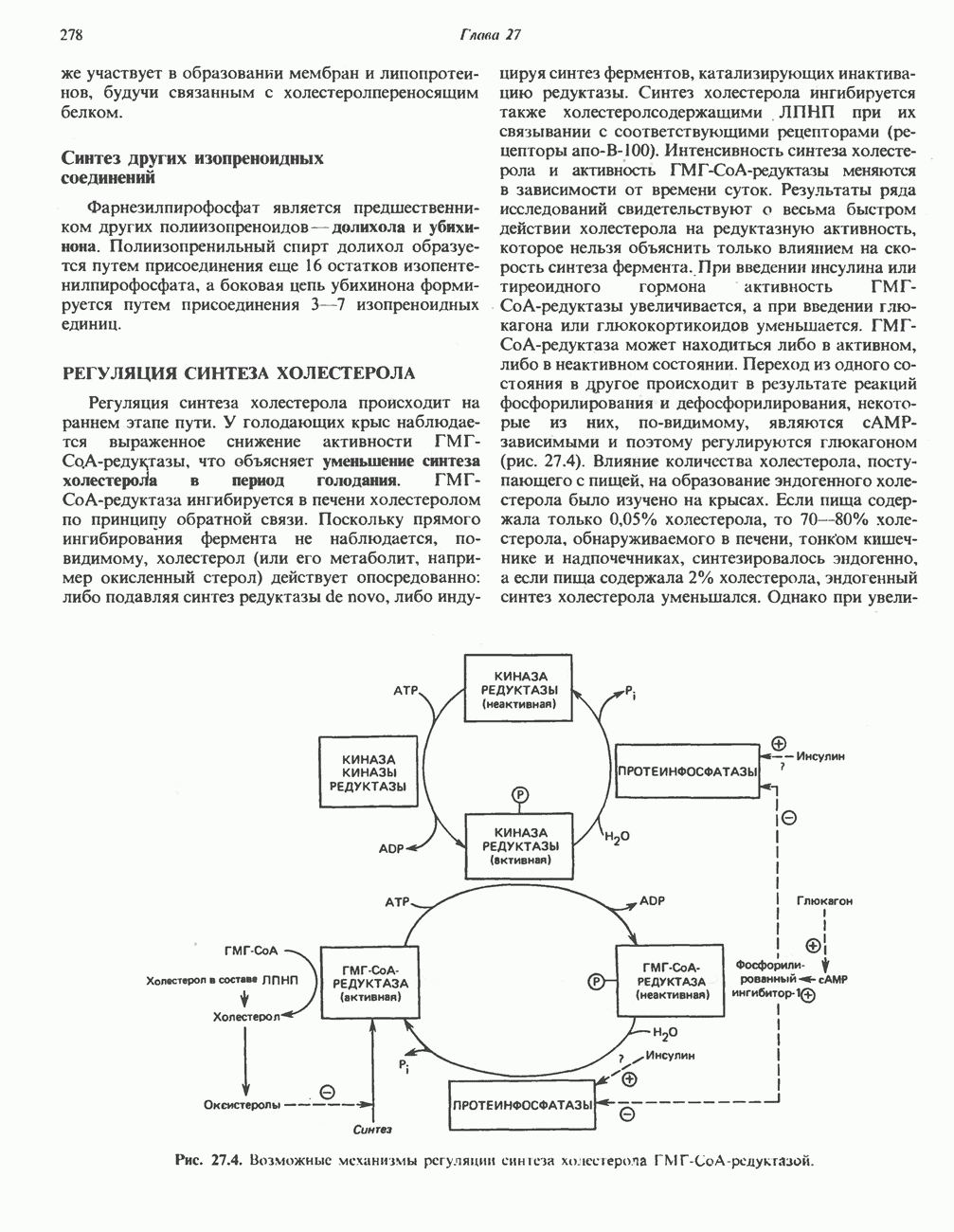
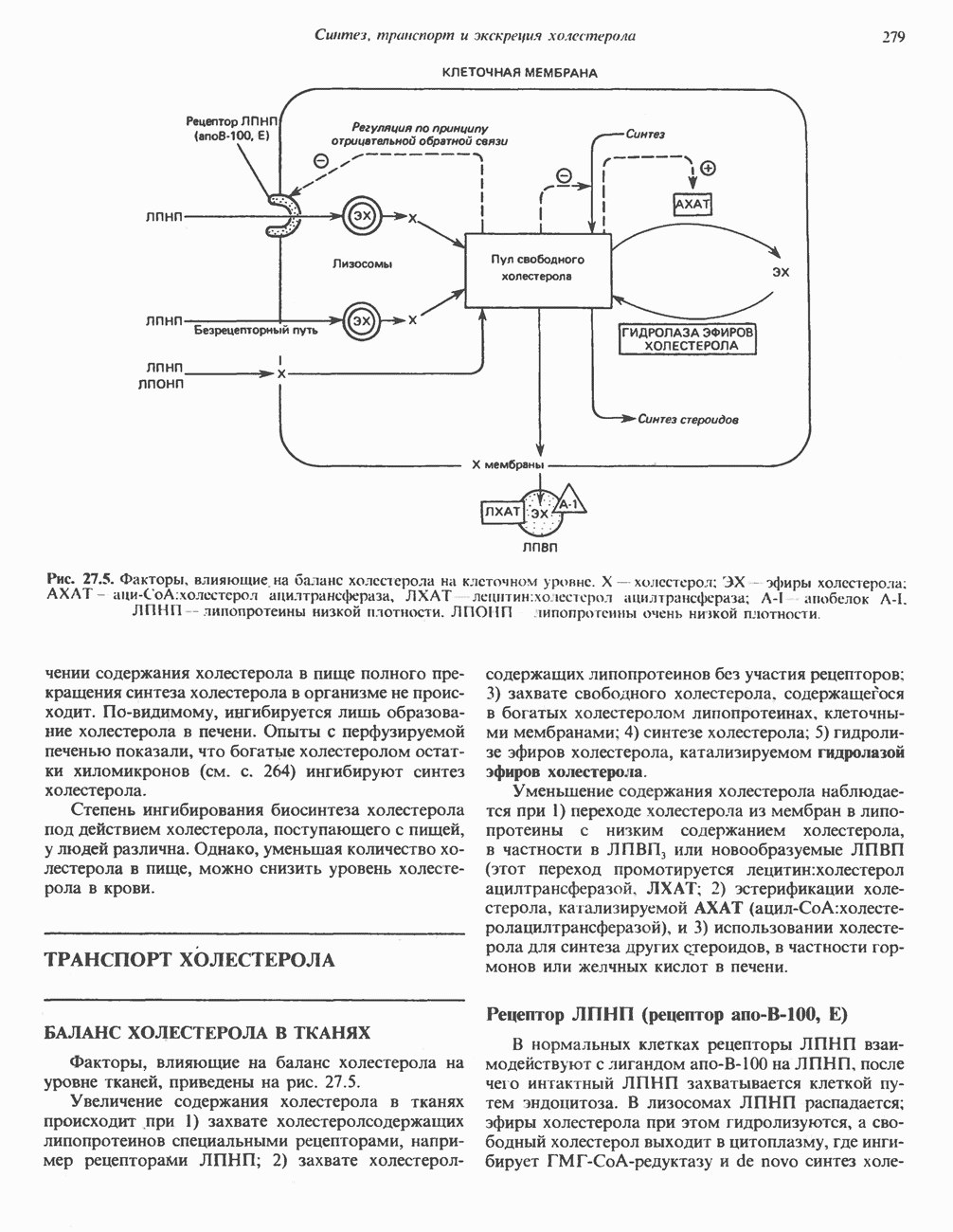
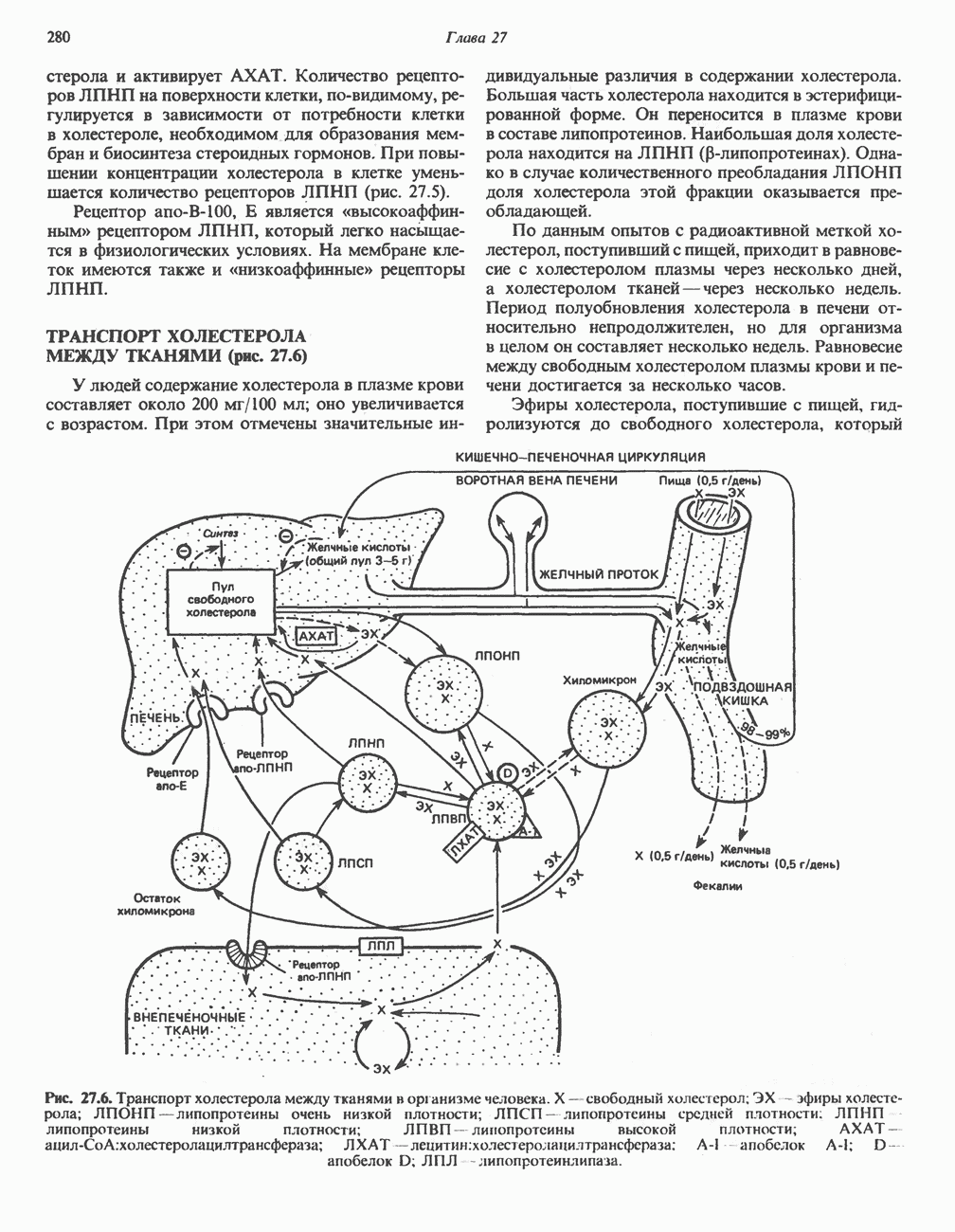
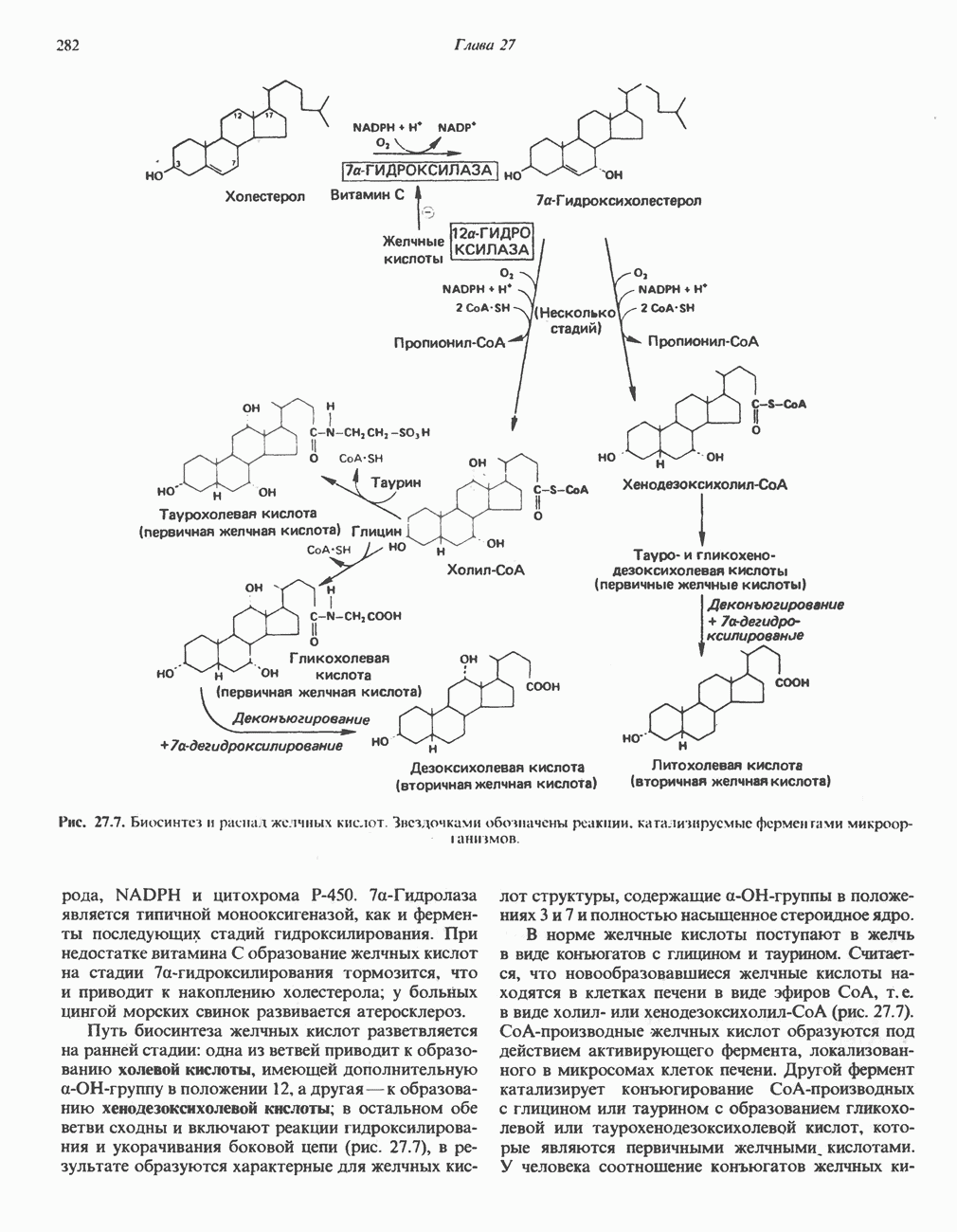
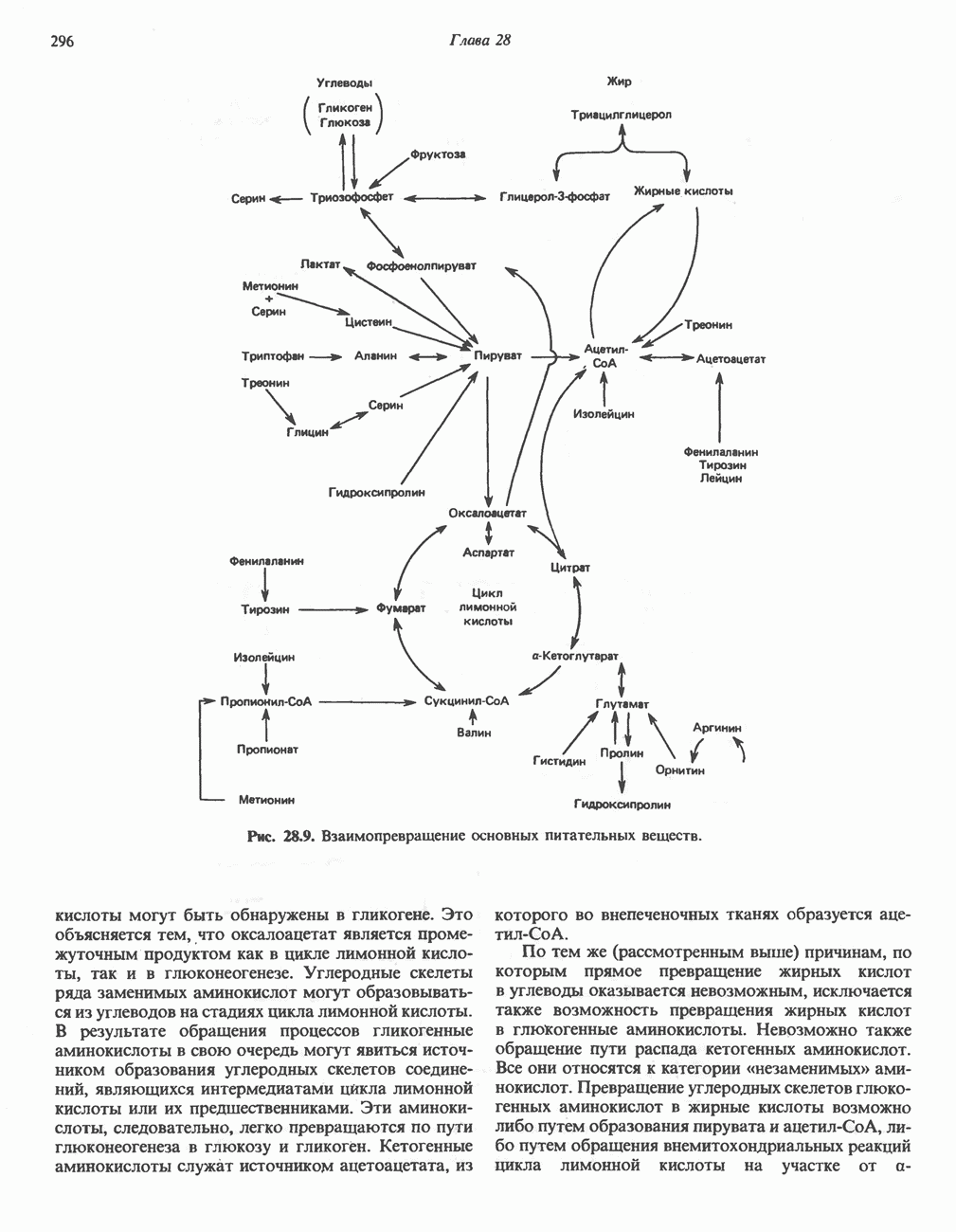
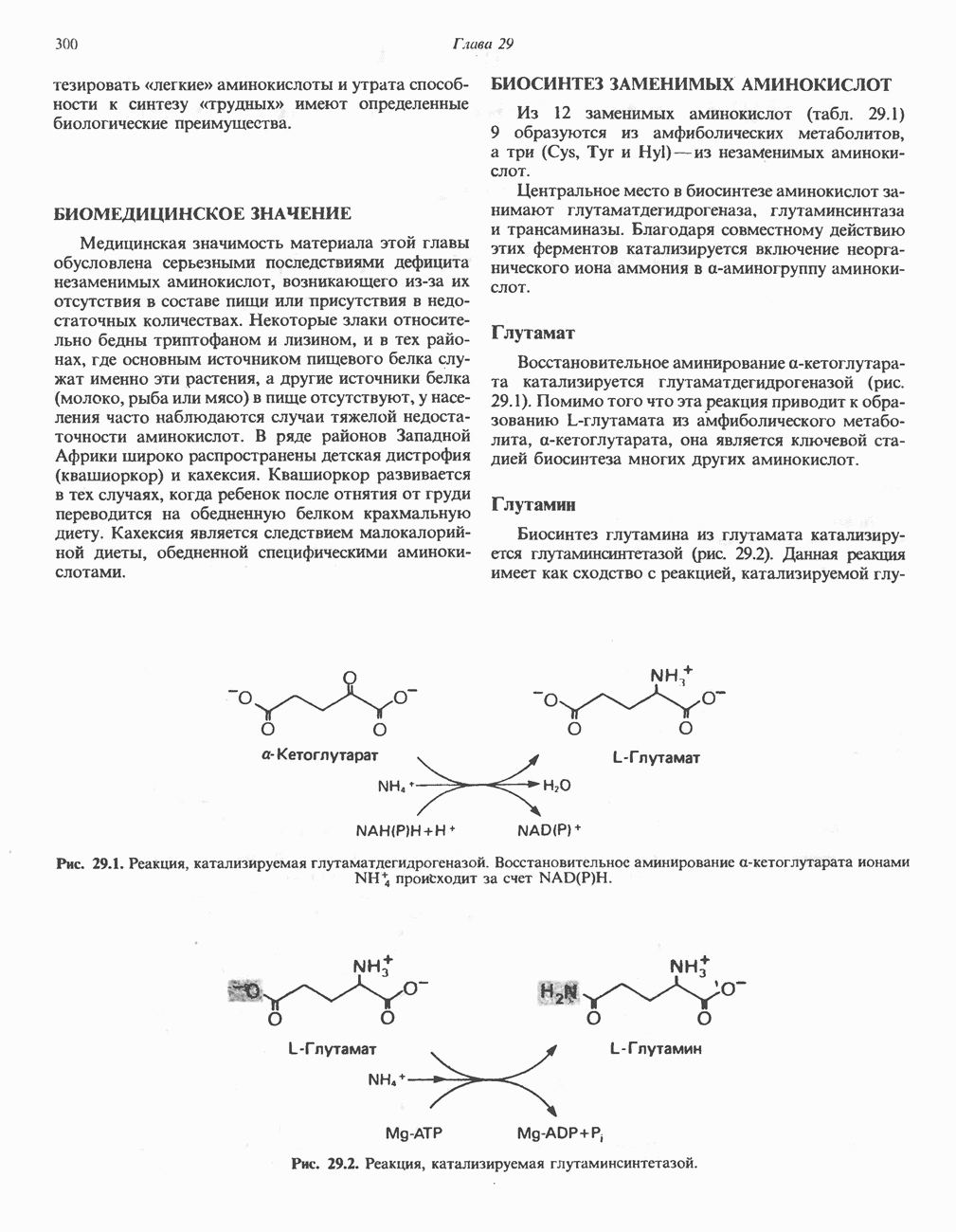
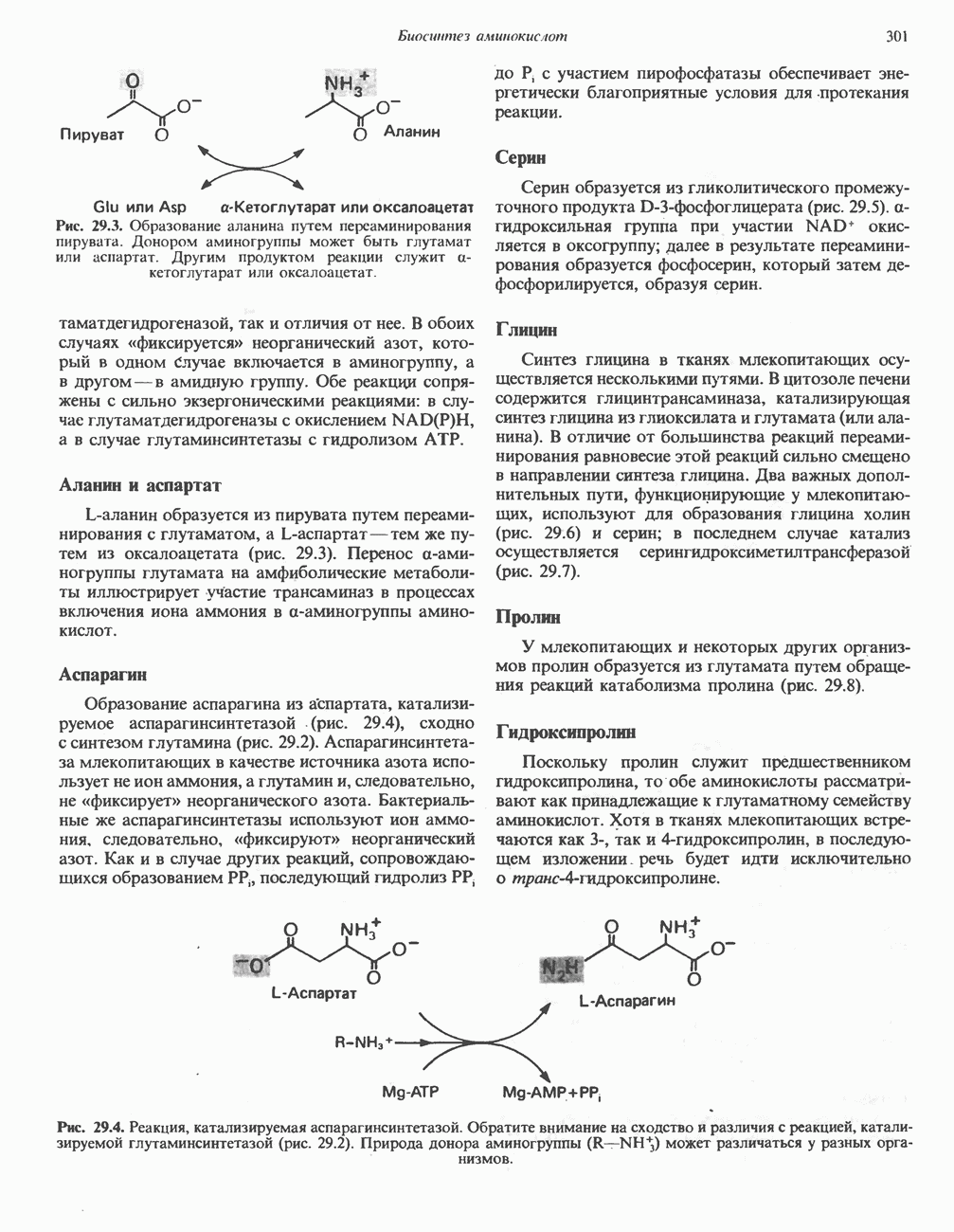
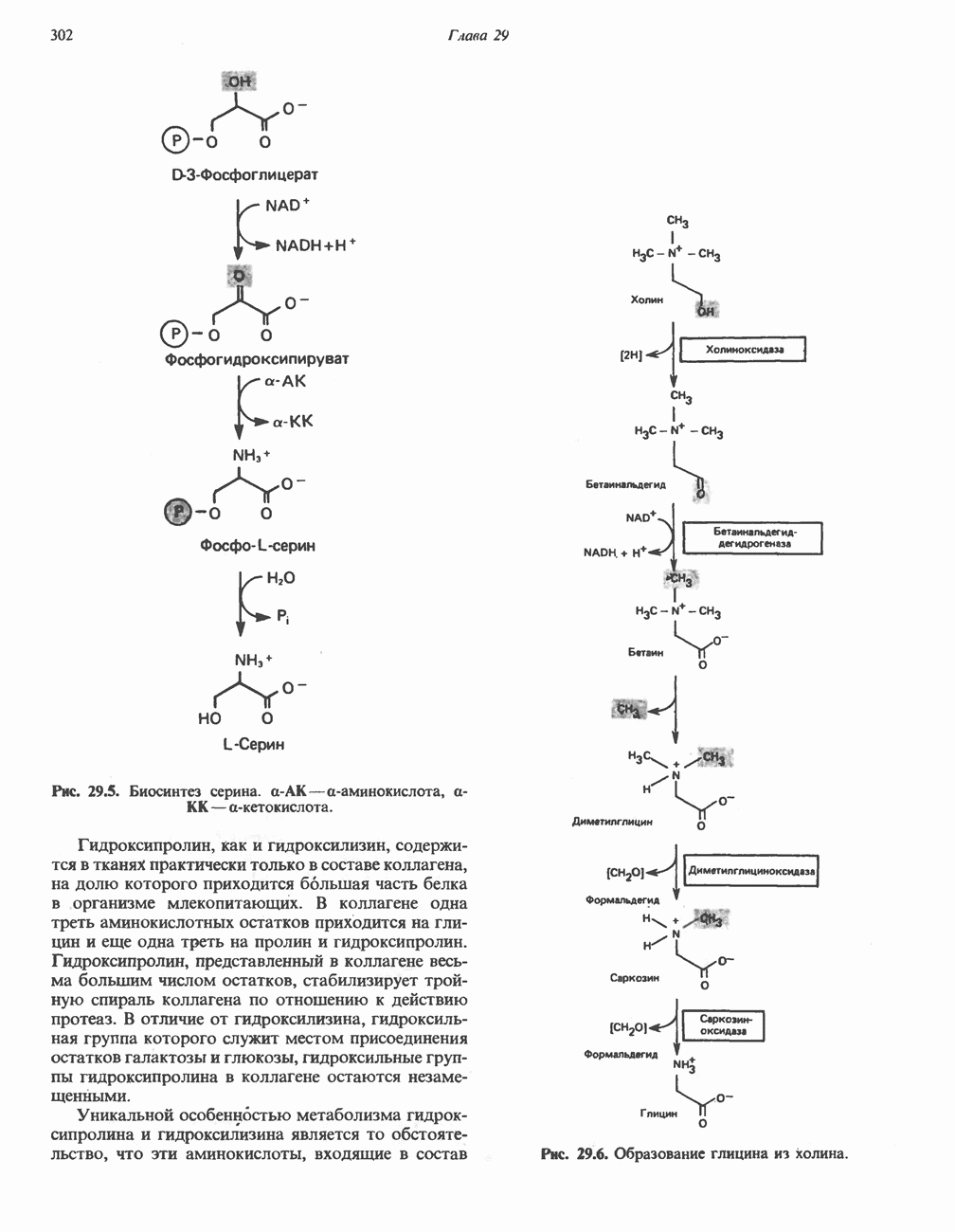
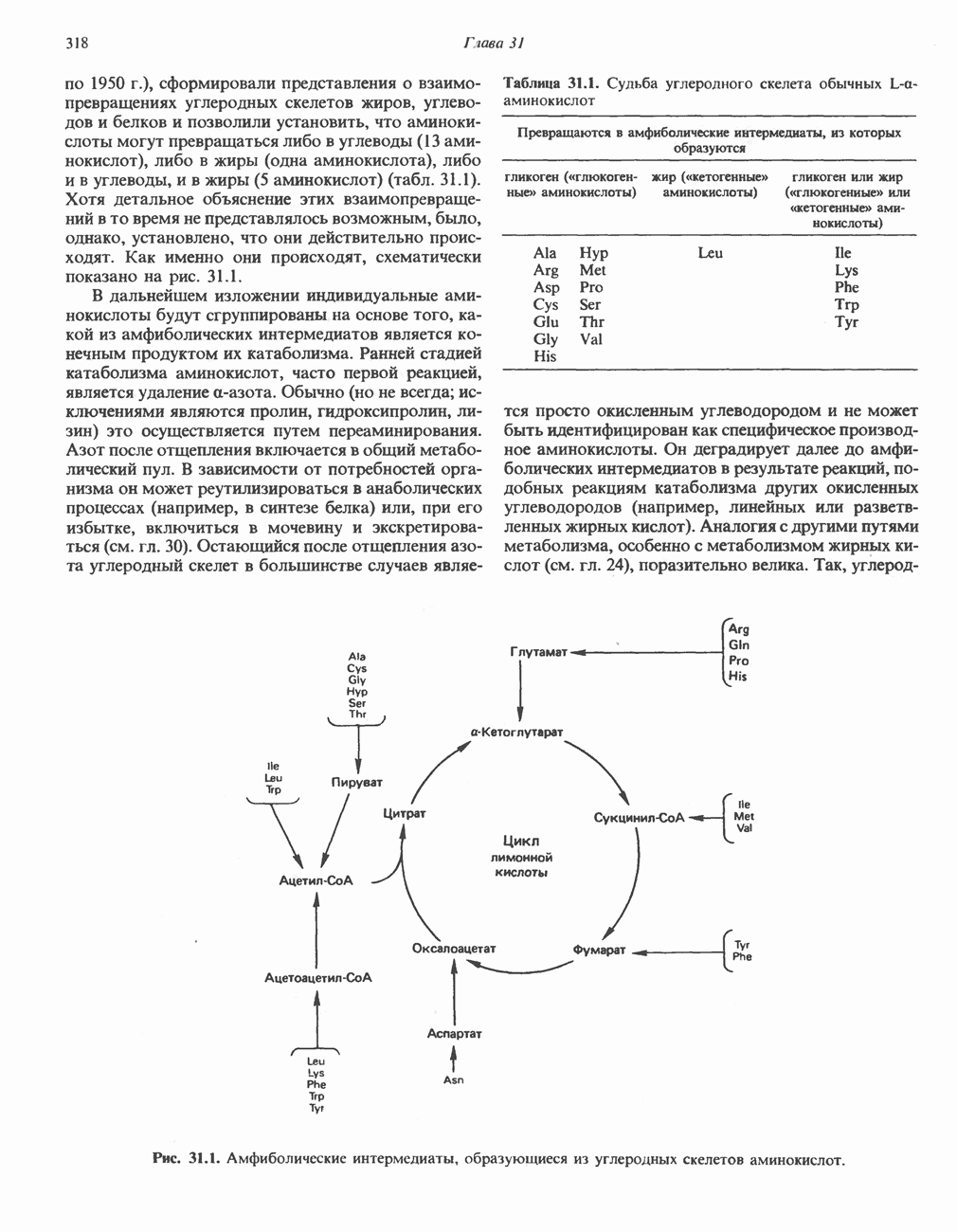
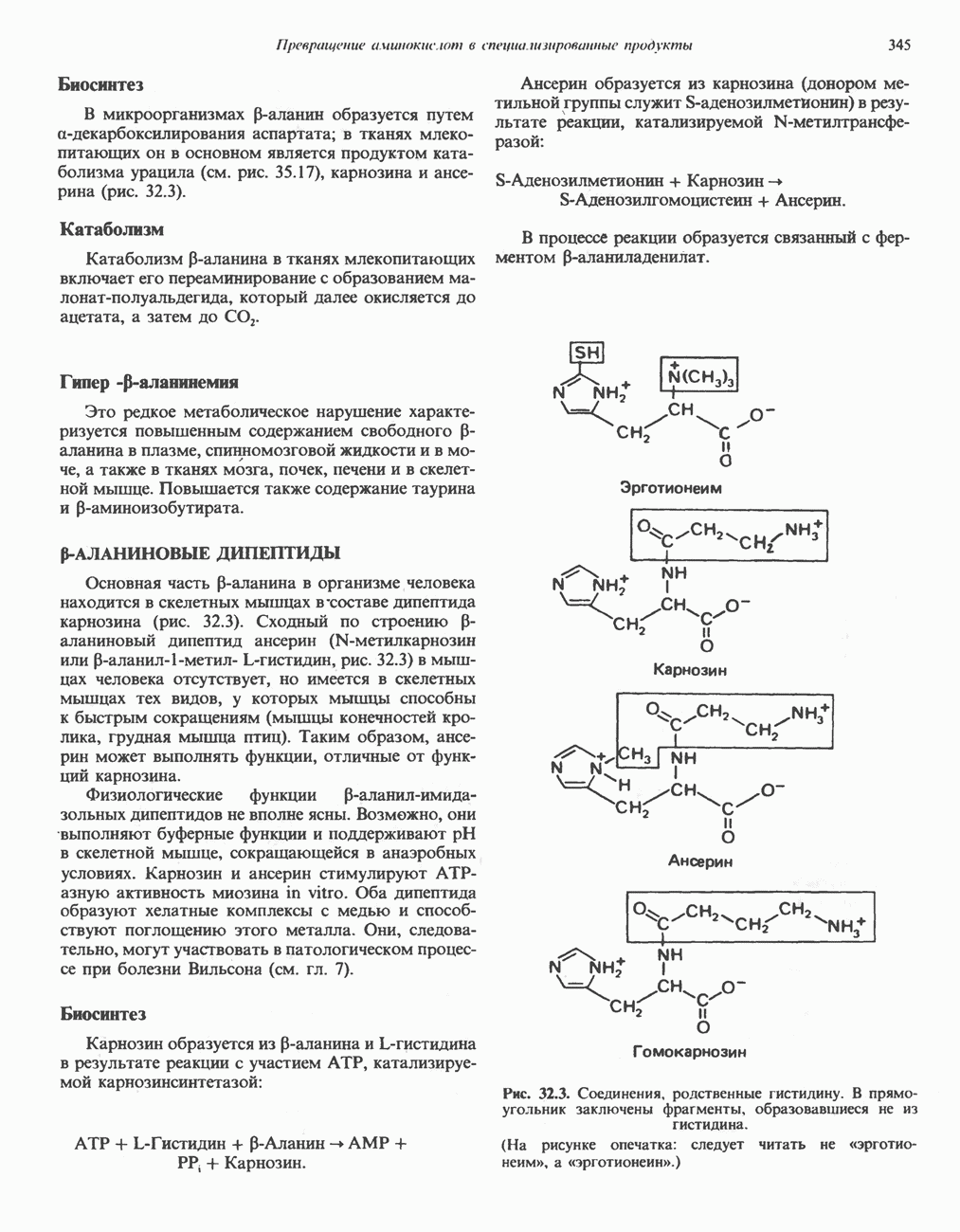
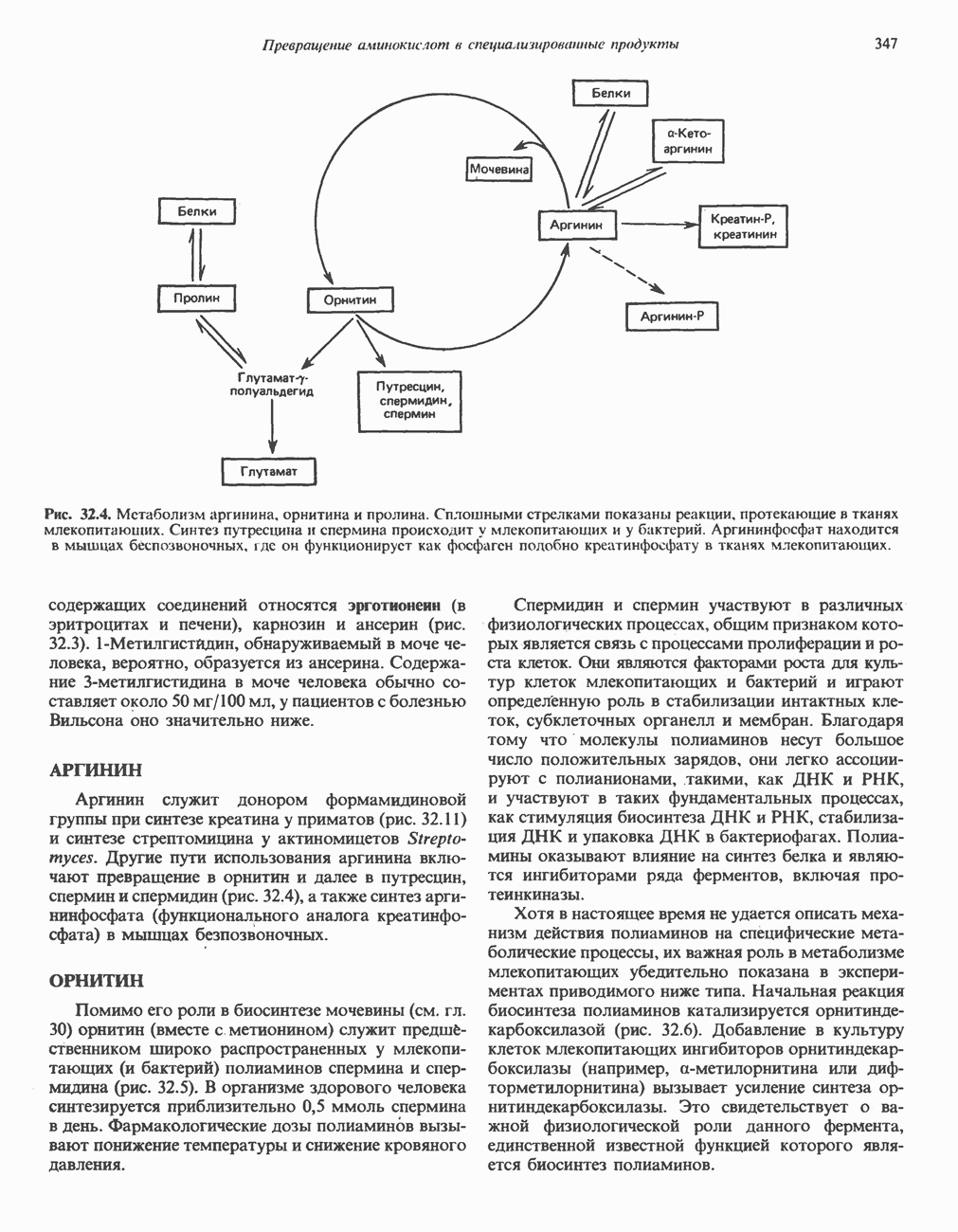
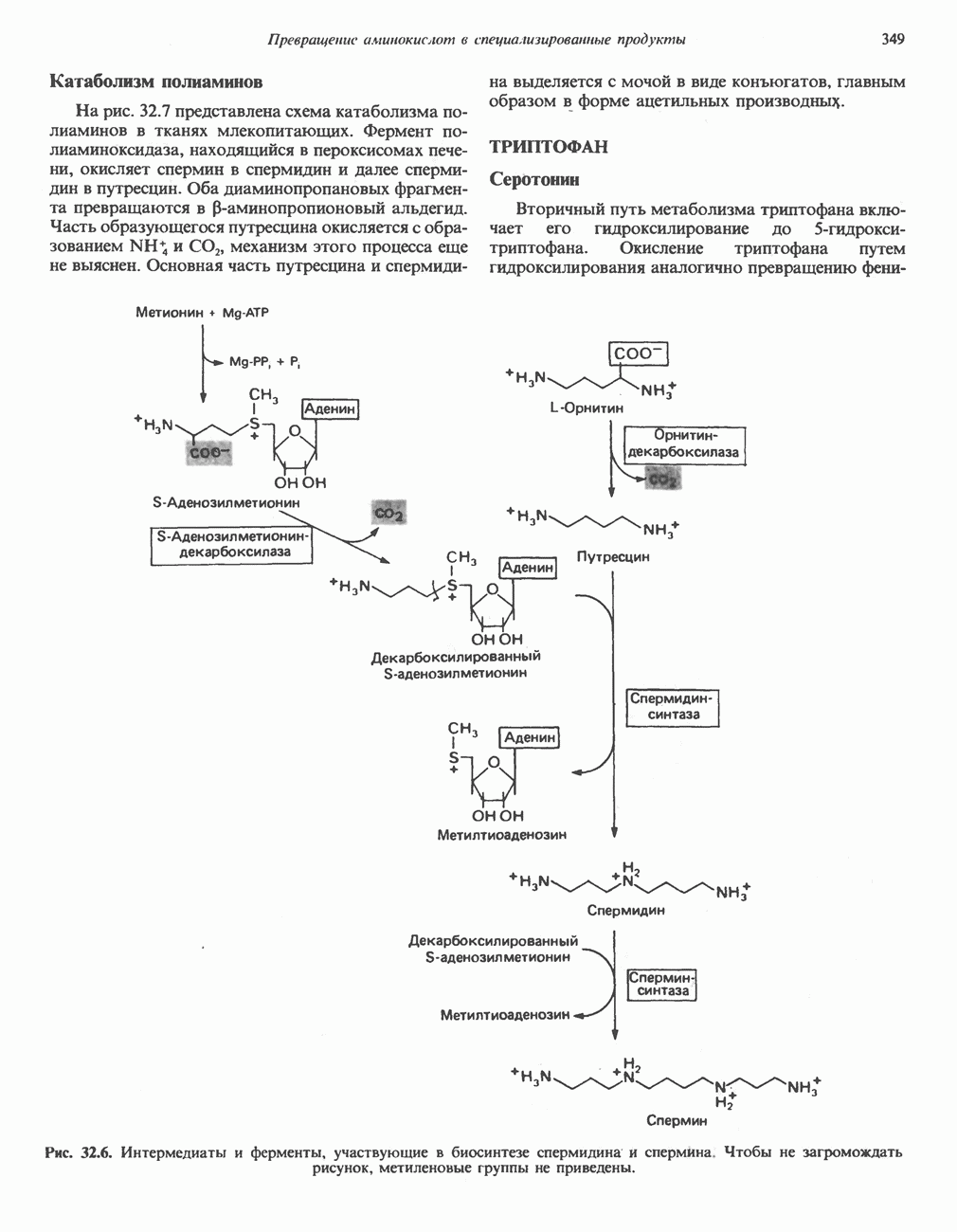
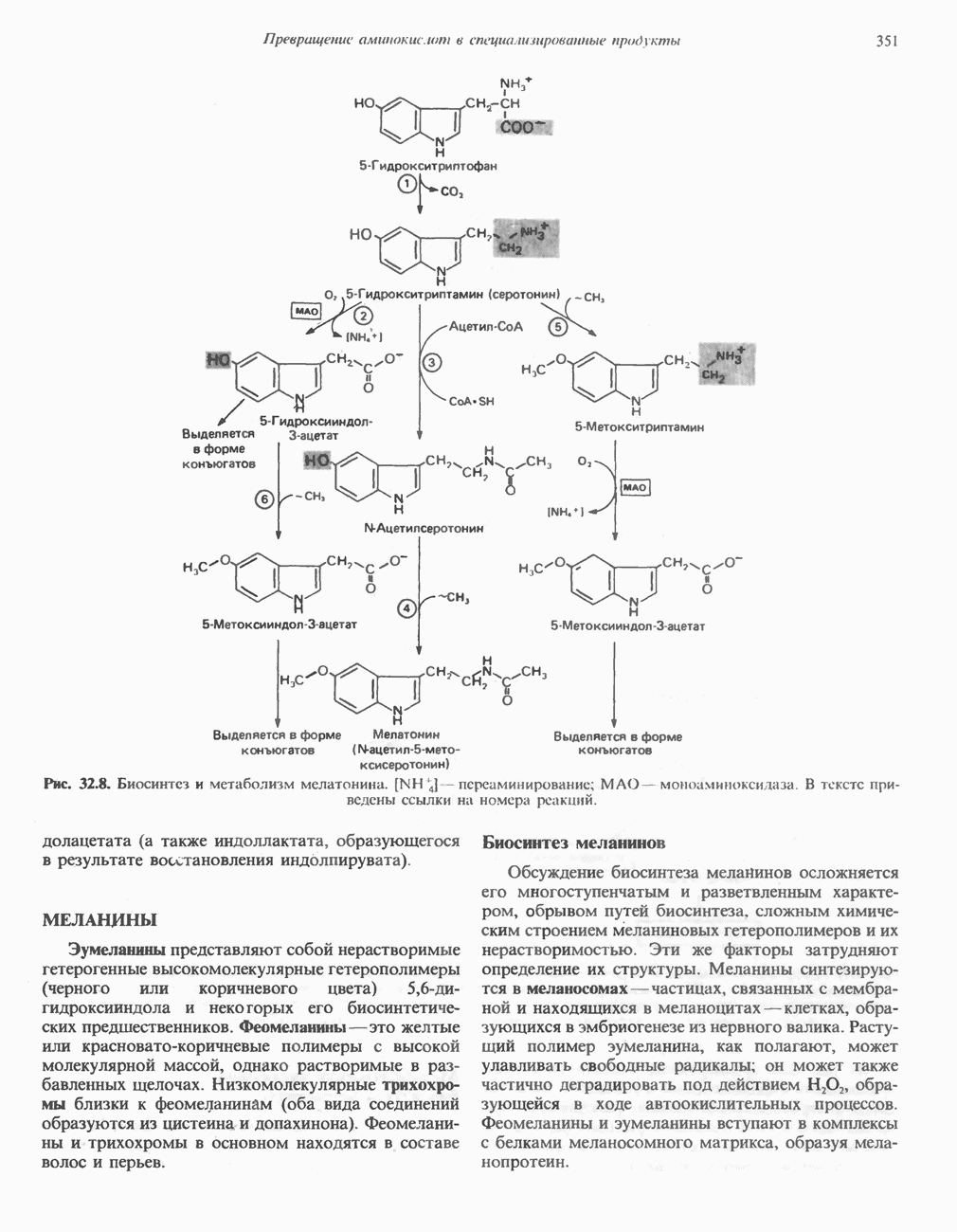
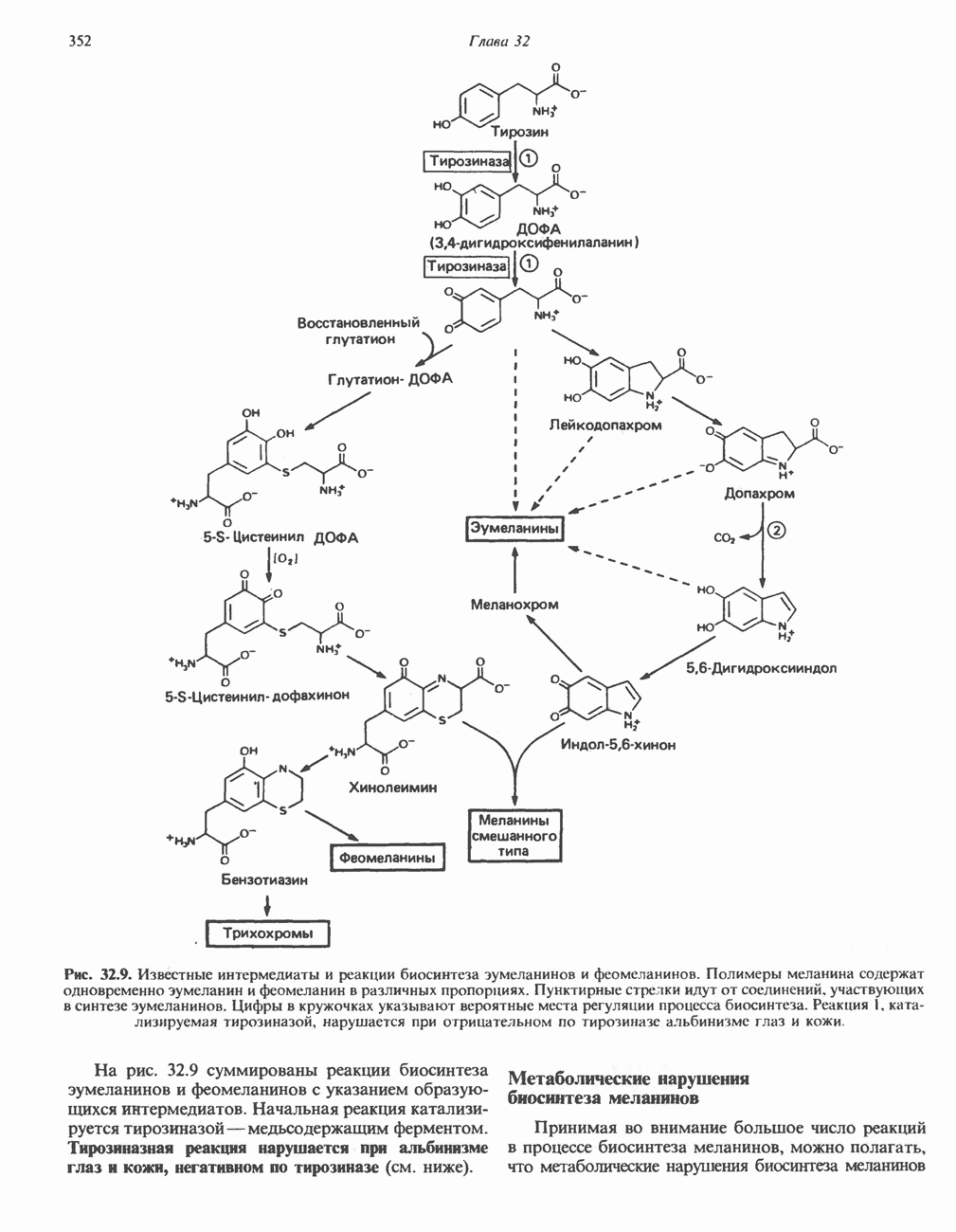
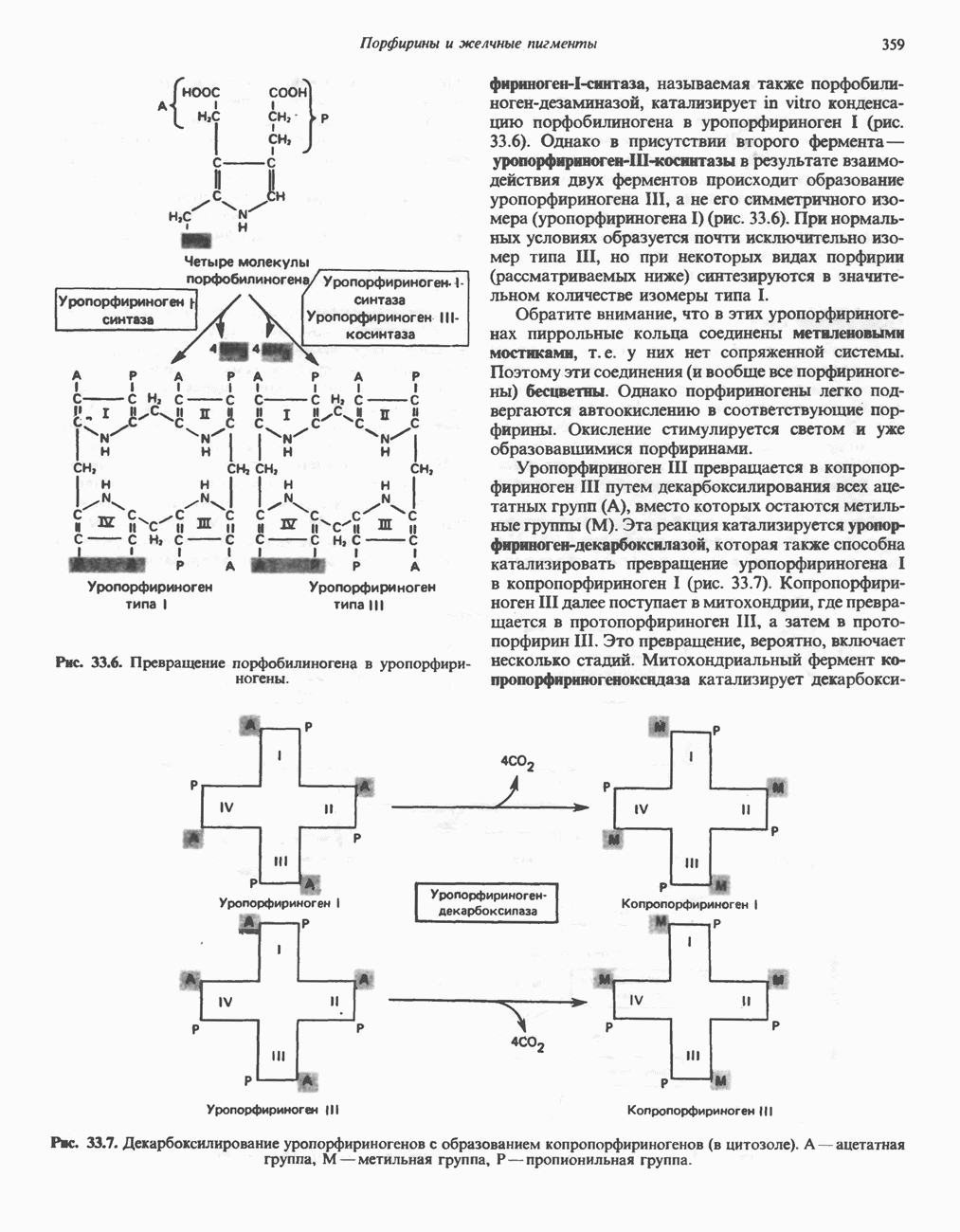
ГЕМОГЛОБИНЫБиологическая функция гемоглобиновГемоглобины—структурно-родственные белки, находящиеся в эритроцитах позвоночных. Они выполняют две важные биологические функции: 1) переносят 2) переносят Первичная структура гемоглобина АВ отличие от миоглобина, который не имеет четвертичной структуры, гемоглобины представляют собой тетрамерные белки, молекулы которых образованы различными типами полипептидных цепей (они обозначаются а- и Вторичная и третичная структура гемоглобина АНесмотря на различия в длине цепи и аминокислотной последовательности миоглобина и р-полипептида Четвертичная структура гемоглобина АСвойства индивидуальных гемоглобинов неразрывно связаны с их четвертичной, равно как и вторичной и третичной, структурами. Наиболее распространенные гемоглобины имеют следующую тетрамерную структуру: Кинетика оксигенирования гемоглобинаГемоглобин связывает четыре молекулы кислорода на тетрамер (по одной на гем в каждой субъединице); особенно важным отличием его от миоглобина является характерная кривая насыщения кислородом, которая имеет сигмоидную форму (рис. 6.8). Таким образом, способность гемоглобина связывать
Рис. 6.8. Кривые связывания кислорода гемоглобином и миоглобином. Парциальное давление кислорода в артериальной крови составляет около 100 мм рт. ст., в венозной крови около 40 мм рт. ст., в капиллярах кровеносных сосудов активной мышцы— около 20 мм рт. ст.; минимальное давление, необходимое для функционирования ферментов цитохромной системы, равно Если да, то последующие молекулы 02 присоединяются легче. Следовательно, для гемоглобина характерна кинетика кооперативного связывания, благодаря которой он связывает максимальное количество 02 в легких и отдает максимальное количество 02 при тех PQ, которые имеют место в периферических тканях. Сравните, например, какие количества кислорода связываются гемоглобином и миоглобином в легких, при Сродство гемоглобинов к Оксигенирование сопровождается значительными конформационными изменениями в гемоглобинеСвязывание О, сопровождается разрывом солевых связей, образованных концевыми карбоксильными группами субъединиц (рис. 6.9). Это облегчает связывание следующих молекул Четвертичная структура частично оксигенированного гемоглобина описывается как Т-состояние (от англ. taut — напряжение); полностью оксигенированному гемоглобину Конформационные изменения в окружении гемогруппыОксигенирование гемоглобина, как и миоглобина, сопровождается структурными изменениями в окружении гемогруппы. При оксигенировании атом железа, который в дезоксигемоглобине выступал на 0,06 нм из плоскости гемового кольца, втягивается в эту плоскость (рис. 6.13). Вслед за атомом железа ближе к гему перемещается и проксимальный гистидин
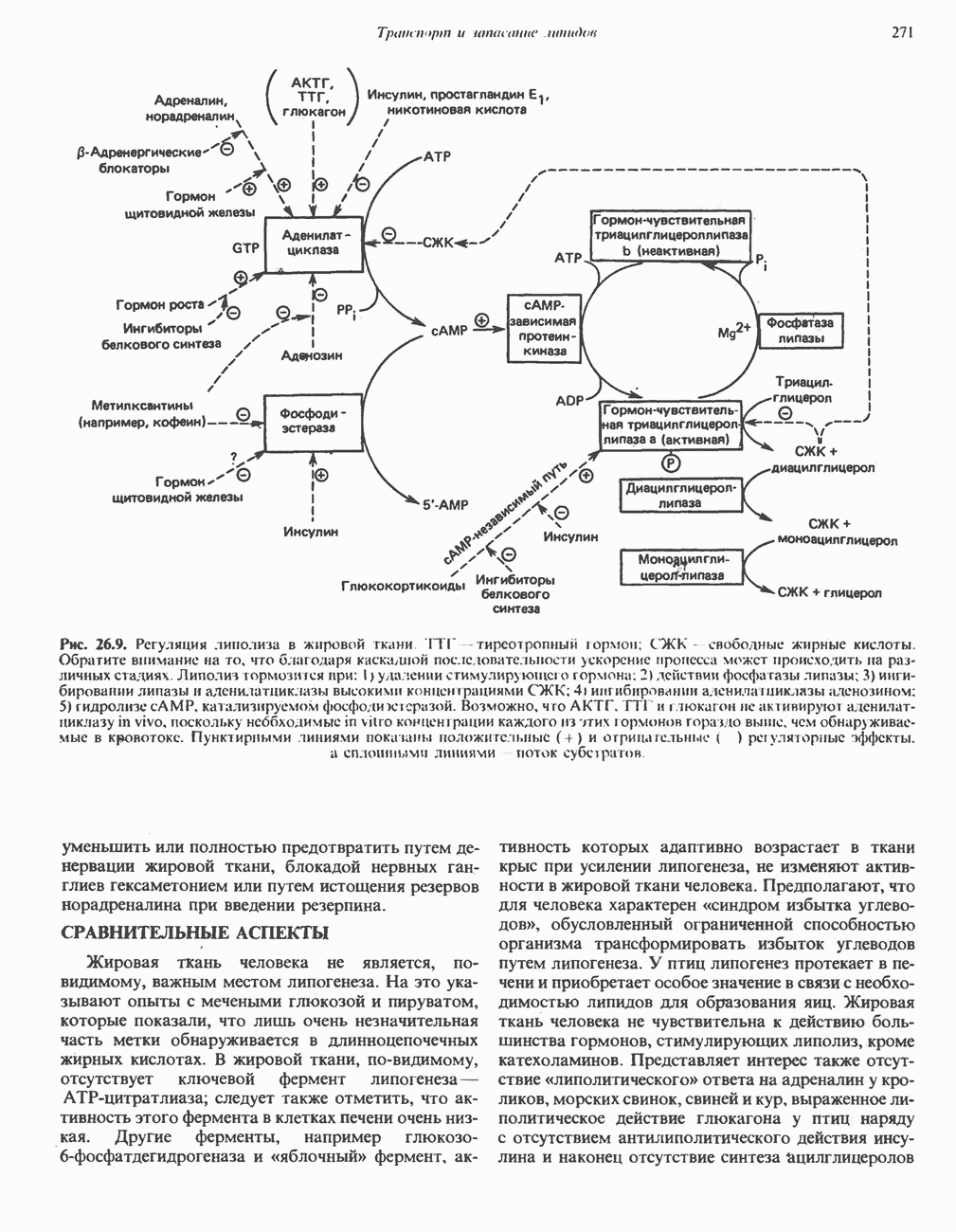
Рис. 6.9. Солевые связи между субъединицами в дезоксигемоглобине. При оксигенировании эти нековалентные связи, обусловленные электростатическими взаимодействиями, разрушаются. (Из книги Stryer L.: Biochemistry, 2nd ed.. Freeman, 1981, с изменениями.) Транспорт двуокиси углеродаГемоглобин не только переносит кислород от легких к периферическим тканям, но и ускоряет транспорт
Рис. 6.10. Переход гемоглобина из Т- в
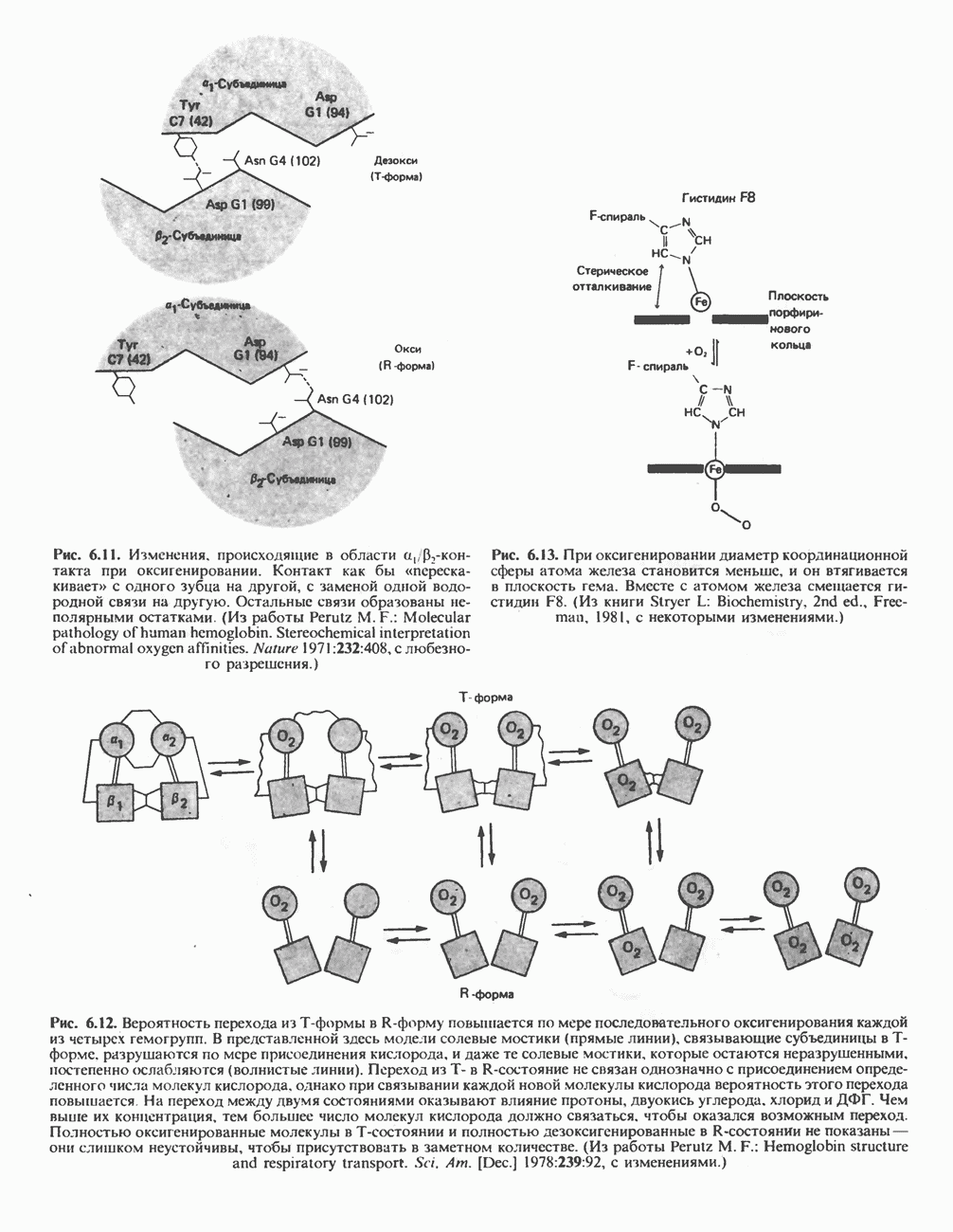
Рис. 6.11. Изменения, происходящие в области
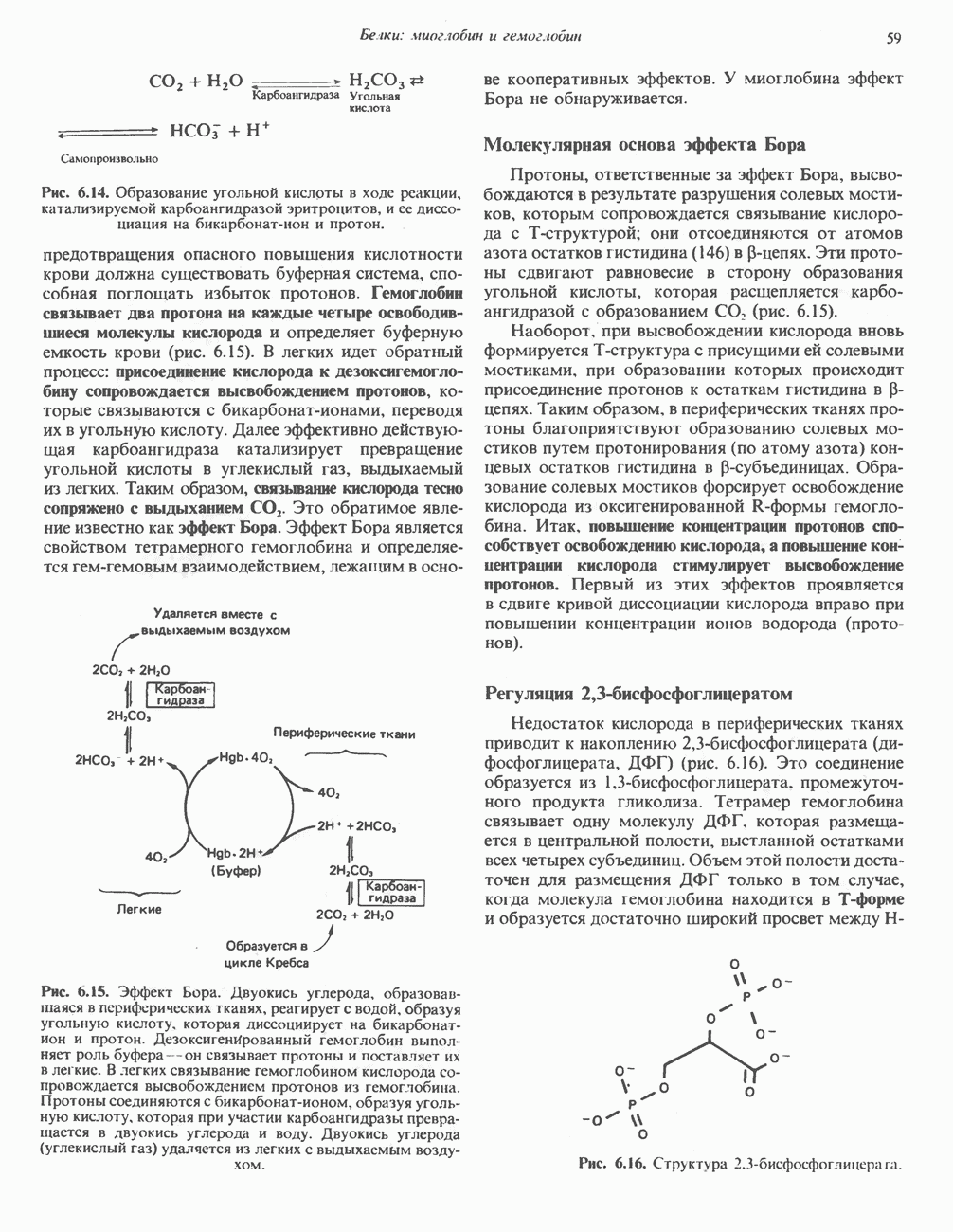
Рис. 6.12. Вероятность перехода из Т-формы в R-форму повышается по мере последовательного оксигенирования каждой из четырех гемогрупп. В представленной здесь модели солевые мостики (прямые линии), связывающие субъединицы в Т-форме. разрушаются по мере присоединения кислорода, и даже те солевые мостики, которые остаются неразрушенными, постепенно ослабляются (волнистые линии). Переход из Т- в R-состояние не связан однозначно с присоединением определенного числа молекул кислорода, однако при связывании каждой новой молекулы кислорода вероятность этого перехода повышается. На переход между двумя состояниями оказывают влияние протоны, двуокись углерода, хлорид и ДФГ. Чем выше их концентрация, тем большее число молекул кислорода должно связаться, чтобы оказался возможным переход. Полностью оксигенированные молекулы в Т-состоянии и полностью дезоксигенированные в R-состоянии не показаны — они слишком неустойчивы, чтобы присутствовать в заметном количестве. (Из работы Perutz М. F.: Hemoglobin structure and respiratory transport. Sci. Am. [Dec.] 1978:239:92, с изменениями.)
Рис. 6.13. При оксигенировании диаметр координационной сферы атома железа становится меньше, и он втягивается в плоскость гема. Вместе с атомом железа смещается гистидин F8. (Из книги Stryer L: Biochemistry, 2nd ed.. Freeman, 1981, с некоторыми изменениями.)
Рис. 6.14. Образование угольной кислоты в ходе реакции, катализируемой карбоангидразой эритроцитов, и ее диссоциация на бикарбонат-ион и протон. предотвращения опасного повышения кислотности крови должна существовать буферная система, способная поглощать избыток протонов. Гемоглобин связывает два протона на каждые четыре освободившиеся молекулы кислорода и определяет буферную емкость крови (рис. 6.15). В легких идет обратный процесс: присоединение кислорода к дезоксигемогло-бину сопровождается высвобождением протонов, которые связываются с бикарбонат-ионами, переводя их в угольную кислоту. Далее эффективно действующая карбоангидраза катализирует превращение угольной кислоты в углекислый газ, выдыхаемый из легких. Таким образом, связывание кислорода тесно сопряжено с выдыханием
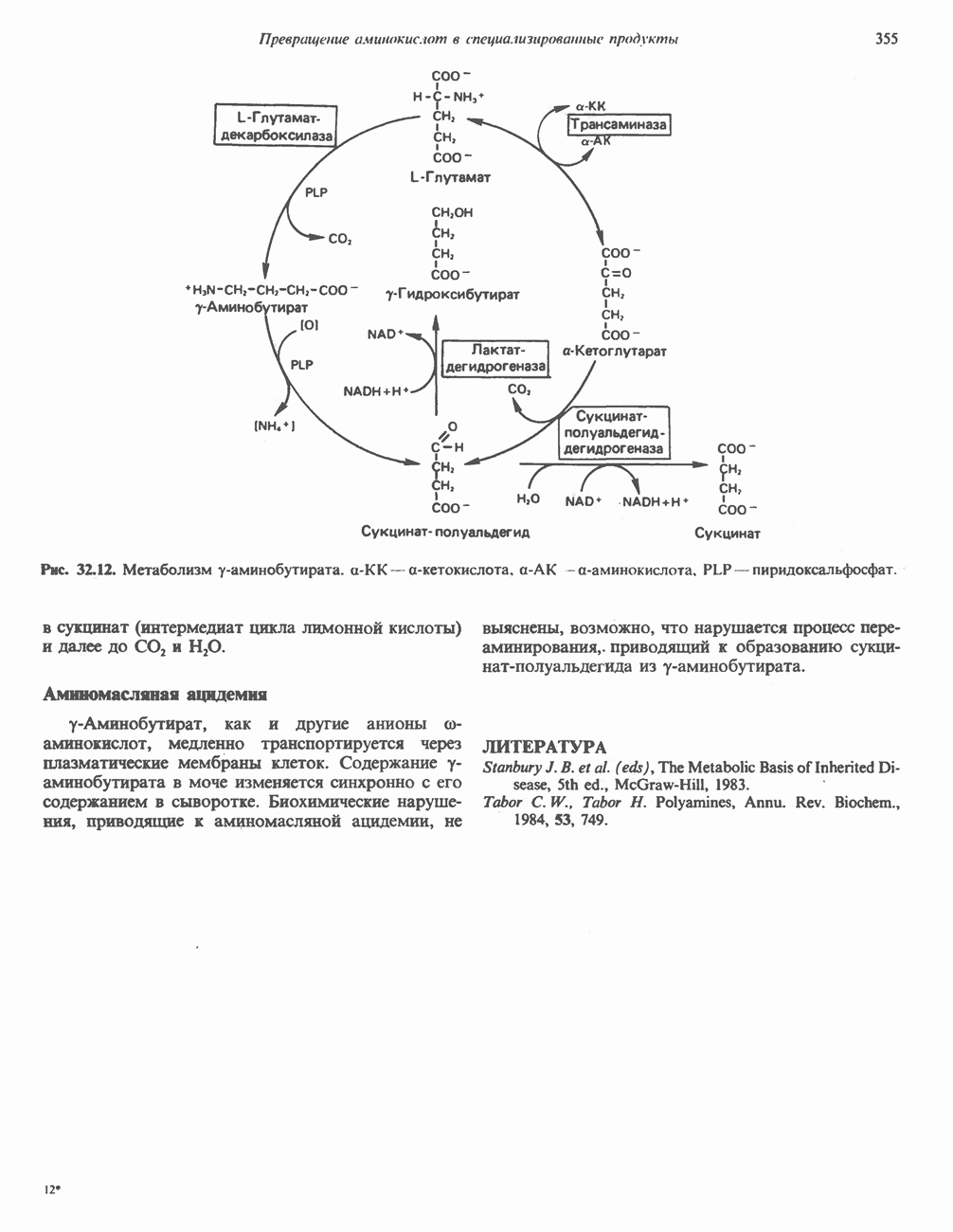
Рис. 6.15. Эффект Бора. Двуокись углерода, образовавшаяся в периферических тканях, реагирует с водой, образуя угольную кислоту, которая диссоциирует на бикарбонат-ион и протон. Дезоксигенированный гемоглобин выполняет роль буфера — он связывает протоны и поставляет их в легкие. В легких связывание гемоглобином кислорода сопровождается высвобождением протонов из гемоглобина. Протоны соединяются с бикарбонат-ионом, образуя угольную кислоту, которая при участии карбоангидразы превращается в двуокись углерода и воду. Двуокись углерода (углекислый газ) удаляется из легких с выдыхаемым воздухом. Молекулярная основа эффекта БораПротоны, ответственные за эффект Бора, высвобождаются в результате разрушения солевых мостиков, которым сопровождается связывание кислорода с Т-структурой; они отсоединяются от атомов азота остатков гистидина (146) в ( Наоборот, при высвобождении кислорода вновь формируется Т-структура с присущими ей солевыми мостиками, при образовании которых происходит присоединение протонов к остаткам гистидина в Р-цепях. Таким образом, в периферических тканях протоны благоприятствуют образованию солевых мостиков путем протонирования (по атому азота) концевых остатков гистидина в ( Регуляция 2,3-бисфосфоглицератомНедостаток кислорода в периферических тканях приводит к накоплению 2,3-бисфосфоглицерата (ди-фосфоглицерата, ДФГ) (рис. 6.16). Это соединение образуется из 1,3-бисфосфоглицерата. промежуточного продукта гликолиза. Тетрамер гемоглобина связывает одну молекулу ДФГ, которая размещается в центральной полости, выстланной остатками всех четырех субъединиц. Объем этой полости достаточен для размещения ДФГ только в том случае, когда молекула гемоглобина находится в Т-форме и образуется достаточно широкий просвет между Н-
Рис. 6.16. Структура 2,3-бисфосфоглицерага. спиралями С фетальным гемоглобином ДФГ связывается менее прочно, чем с гемоглобином взрослого человека, поскольку в его Пусковым механизмом перехода между R- и Т-формами гемоглобина служит перемещение атома железа в плоскость порфиринового кольца или от нее. Источником свободной энергии для этих процессов (около 3000 кал/моль) служат стерические и электростатические факторы. Таким образом, совсем небольшое смещение атома
Рис. 6.17. Механизм связывания ДФГ с дезоксигемоглобином человека. ДФГ взаимодействует с тремя положительно заряженными группами в каждой из Мутантные гемоглобины человека Мутации генов, кодирующих а- и В семействе гемоглобинов М остатки проксимального или дистального гистидина в а- или Р-субъединицах заменены на остатки тирозина. Атом железа в составе гема находится в этом случае в Мутации, приводящие к преимущественному образованию Гемоглобин при серповидноклеточнон анемииВ гемоглобине S остаток
Рис. 6.18. Схема, поясняющая взаимодействие липкого участка гемоглобина S (черный треугольник) с рецептором липкого участка (светлый треугольник) дезоксигемоглобина А и дезоксигемоглобина S. Наличие комплементарных участков на поверхности молекулы дезоксигемоглобина S способствует его полимеризации с образованием волокнистых структур. В присутствии дезоксигемоглобина А процесс полимеризации останавливается, поскольку на поверхности этой молекулы липкого участка нет. (Из книги Stryer L.: Biochemistry, 2nd ed., Freeman, 1981, с некоторыми изменениями.) дезоксигемоглобина S механически деформируют эритроцит, придавая ему серповидную форму, что приводит к лизису клеток и множеству вторичных клинических проявлений. Таким образом, если бы можно было поддерживать гемоглобин S в оксигенированном состоянии или по крайней мере свести к минимуму концентрацию дезоксигенированного гемоглобина S, то нам удалось бы предотвратить полимеризацию дезоксигенированного гемоглобина S и образование «серповидных» клеток. Ясно, что полимеризации подвержена Т-форма гемоглобина S. Интересно отметить (хотя в практическом плане это мало существенно), что ферри-ион метгемоглобина А остается в плоскости порфиринового кольца и тем самым стабилизирует R-форму гемоглобина. То же относится и к гемоглобину при серповидноклеточной анемии: гемоглобин S в ферри-состоянии (метге-моглобин S) не подвержен полимеризации, поскольку он стабилизирован в R-форме. В дезоксигемоглобине А тоже имеется рецепторный участок, способный взаимодействовать с липким участком оксигенированного или дезоксигенированного гемоглобина S (рис. 6.18), но присоединения «липкого» гемоглобина S к дезоксигемоглобину А недостаточно для образования полимера, поскольку сам дезоксигемоглобин А липкого участка не содержит и не может связать следующую молекулу гемоглобина. Следовательно, связывание дезоксигемоглобина А с R- или Т-формой гемоглобина S прерывает полимеризацию. В результате полимеризации дезоксигемоглобина S образуются спиральные фибриллярные структуры. При этом каждая молекула гемоглобина контактирует с четырьмя соседними молекулами (рис. 6.19). Образование подобных трубчатых волокон ответственно за механические нарушения в содержащем
Рис. 6.19. Предполагаемая спиральная структура волокна из агрегированных молекул дезоксигемоглобина S. (Из работы Maugh Т. II: A new understanding of sickle cell emerges. Science 1981:211:265, с разрешения.)
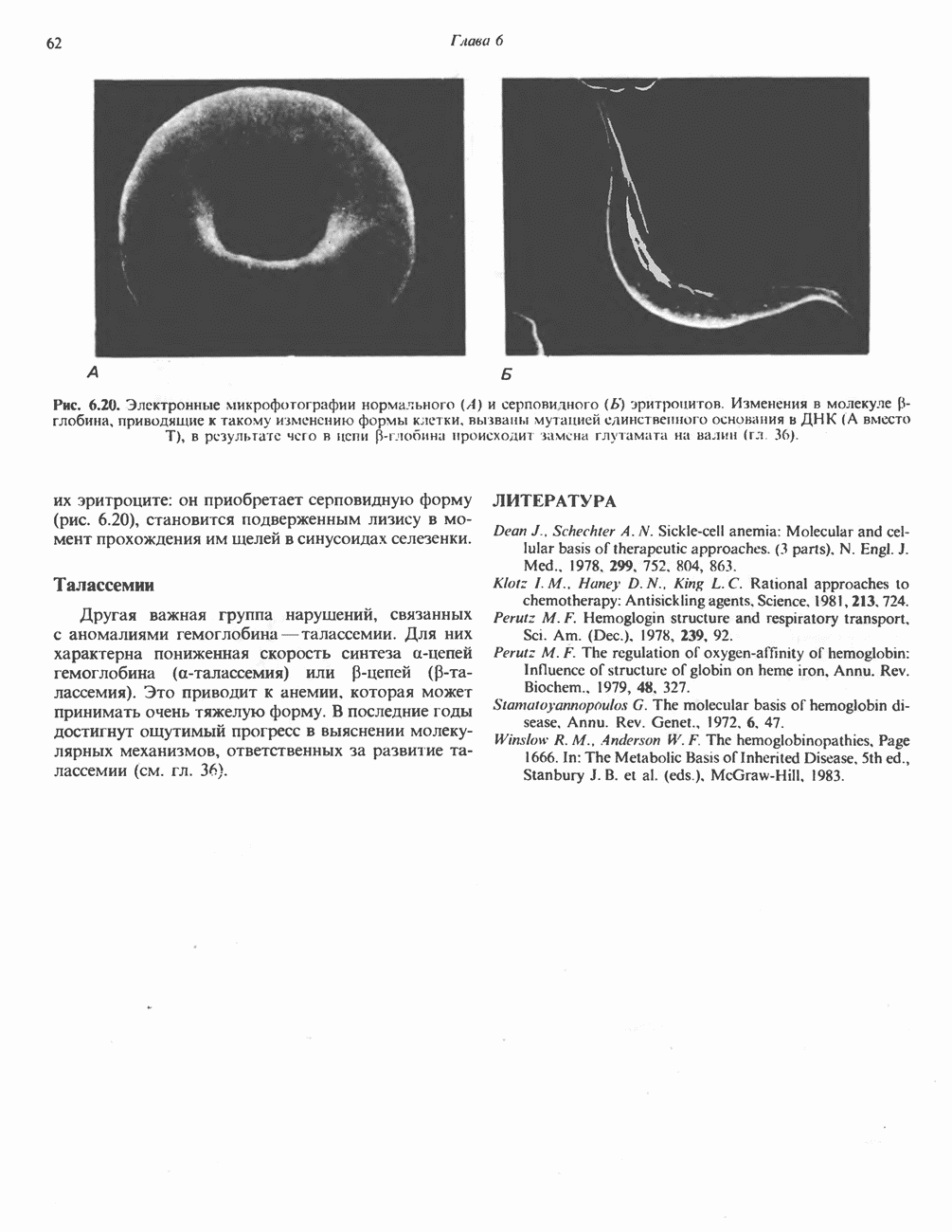
Рис. 6.20. Электронные микрофотографии нормального (А) и серповидного (Б) эритроцитов. Изменения в молекуле р-глобина, приводящие к такому изменению формы клетки, вызваны мутацией единственного основания в ДНК (А вместо Т), в результате чего в цепи Р-глобина происходит замена глутамата на валин (гл. 36). их эритроците: он приобретает серповидную форму (рис. 6.20), становится подверженным лизису в момент прохождения им щелей в синусоидах селезенки. ТалассемииДругая важная группа нарушений, связанных с аномалиями гемоглобина — талассемии. Для них характерна пониженная скорость синтеза а-цепей гемоглобина (а-талассемия) или P-цепей (Р-талассемия). Это приводит к анемии, которая может принимать очень тяжелую форму. В последние годы достигнут ощутимый прогресс в выяснении молекулярных механизмов, ответственных за развитие талассемии (см. гл. 36). ЛИТЕРАТУРАDean J.. Schechter А. N. Sickle-cell anemia: Molecular and cellular basis of therapeutic approaches. (3 parts). N. Engl. J. Med.. 1978. 299. 752, 804, 863. Kloti I. М.. Haney D.N., King L.C. Rational approaches to chemotherapy: Antisickling agents. Science, 1981, 213, 724. Perutz M. F. Hemoglogin structure and respiratory transport, Sci. Am. (Dec.). 1978, 239, 92. Perutz M. F. The regulation of oxygen-affinity of hemoglobin: Influence of structure of globin on heme iron, Annu. Rev. Biochem.. 1979, 48. 327. Stamatoyarmopoulos G. The molecular basis of hemoglobin disease, Annu. Rev. Genet., 1972, 6, 47. Winslow R.M., Anderson W.F. The hemoglobinopathies. Page 1666. In: The Metabolic Basis of Inherited Disease, 5th ed., Stanbury J. B. et al. (eds.), McGraw-Hill, 1983.
|
 |
Оглавление
|























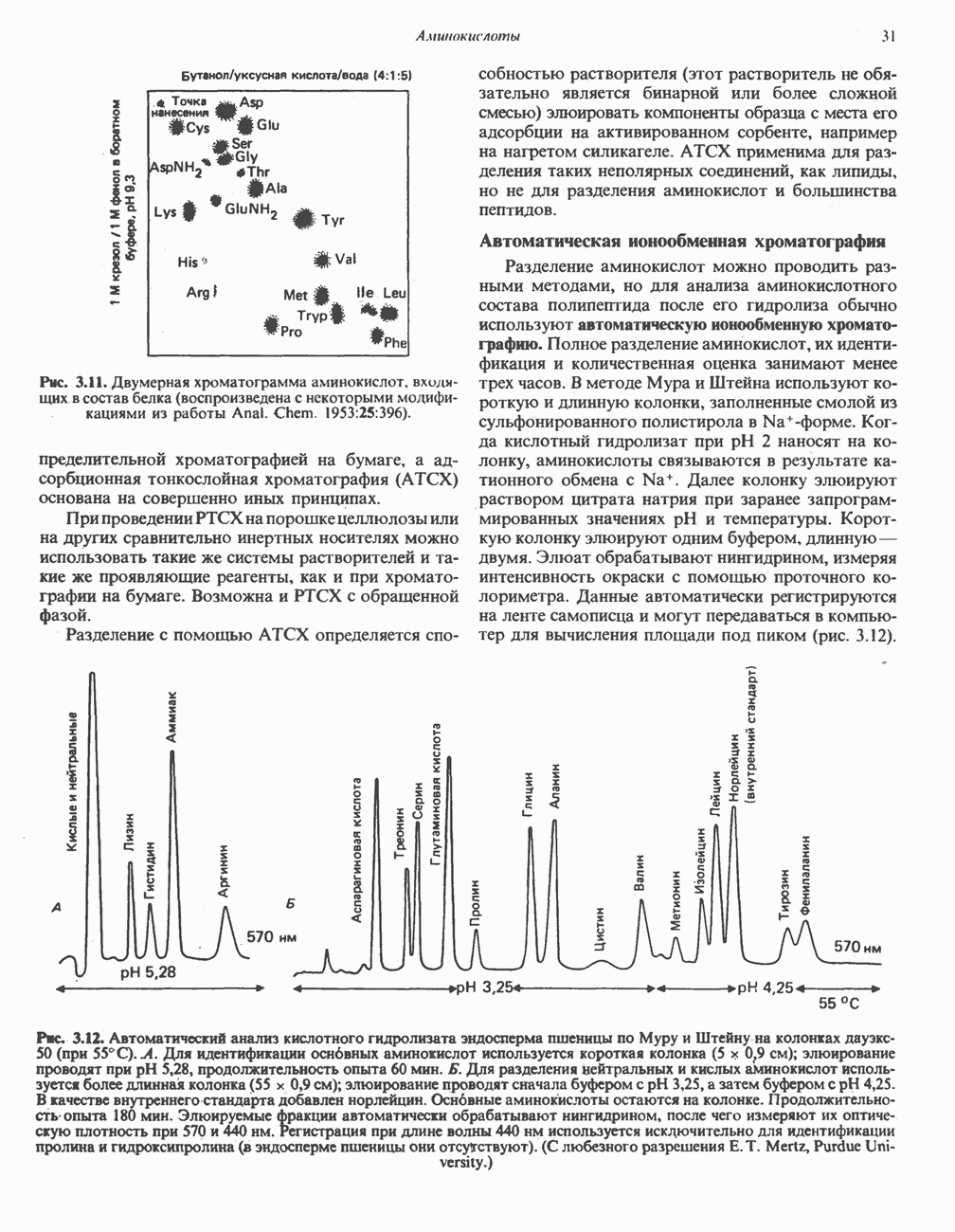


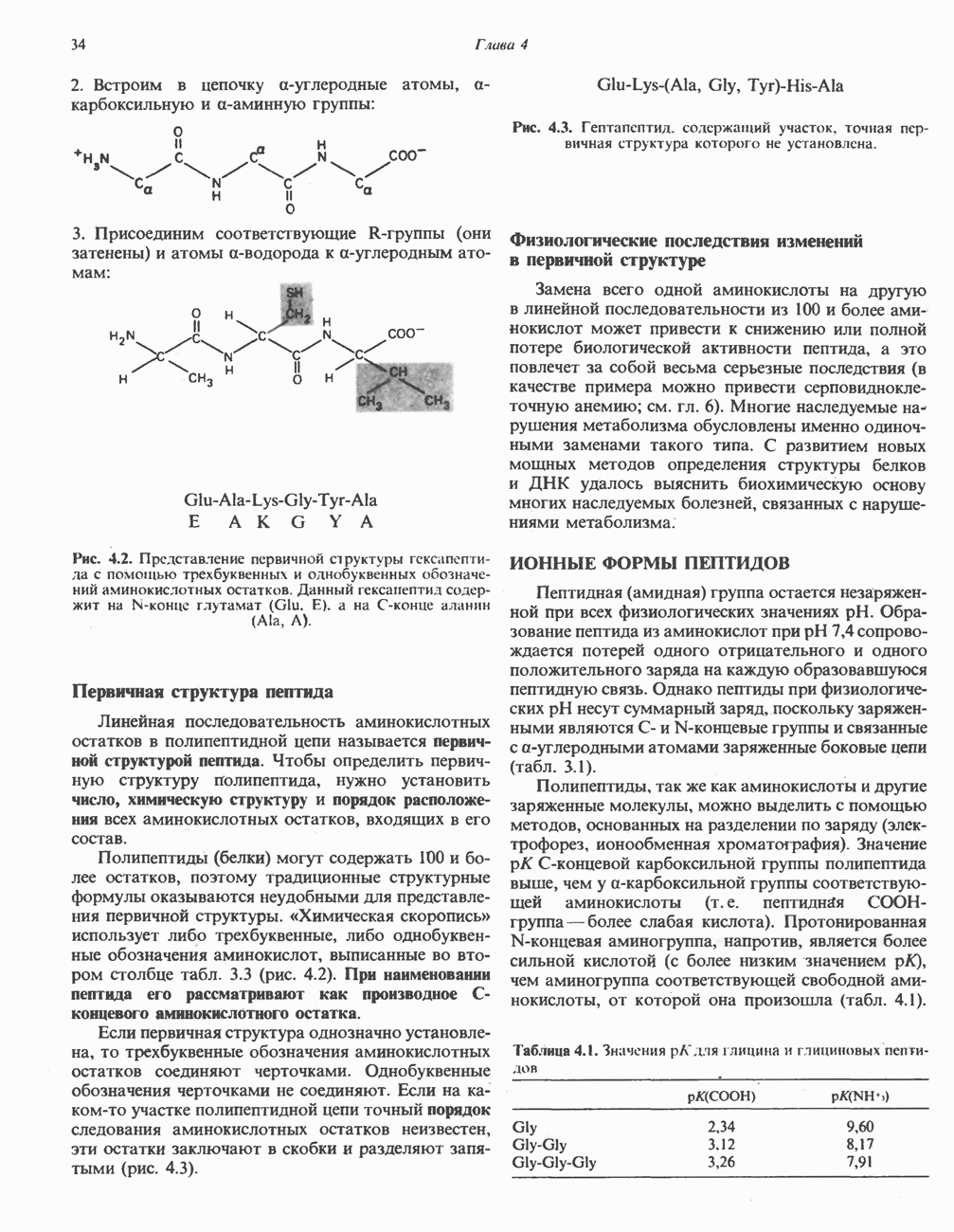


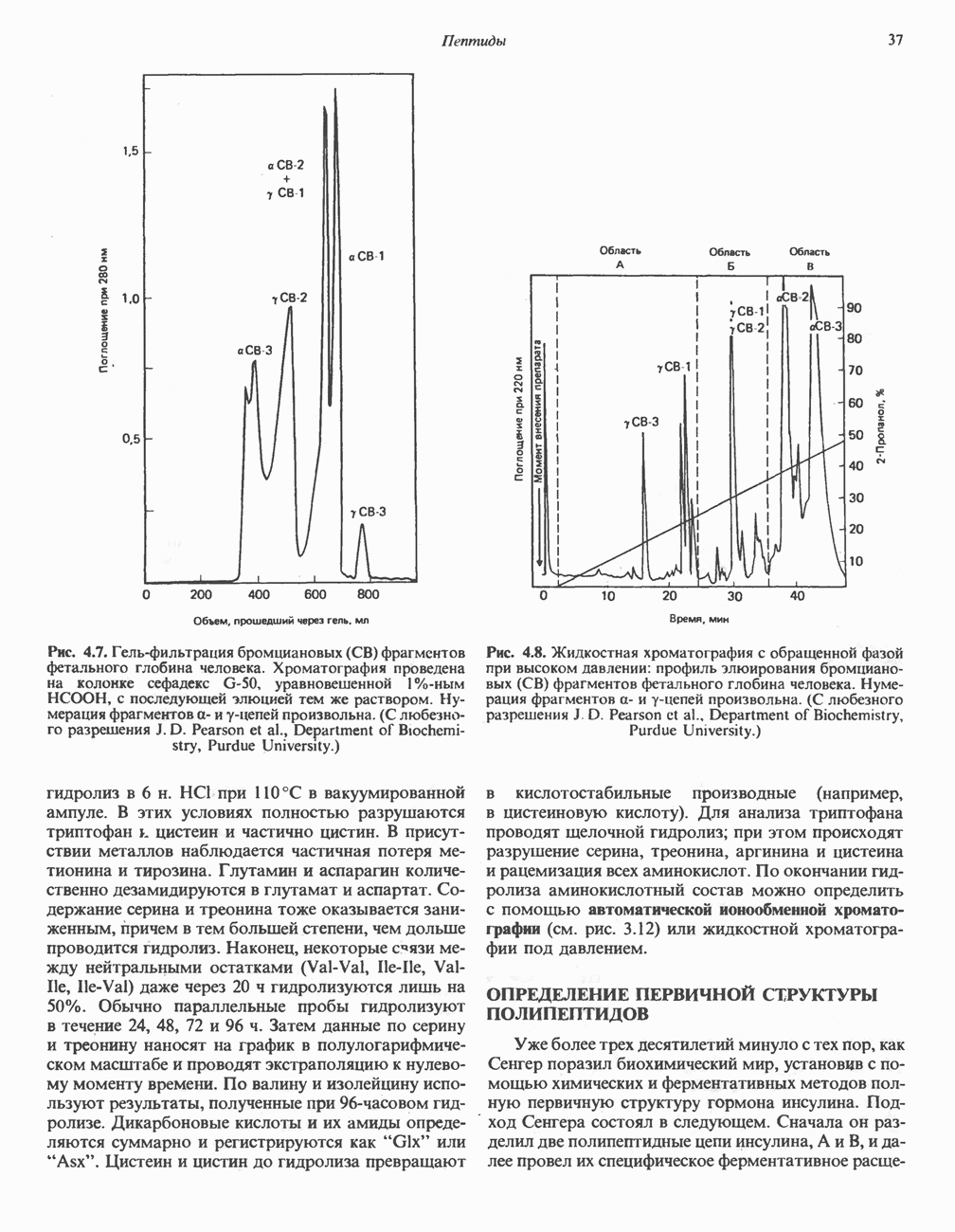


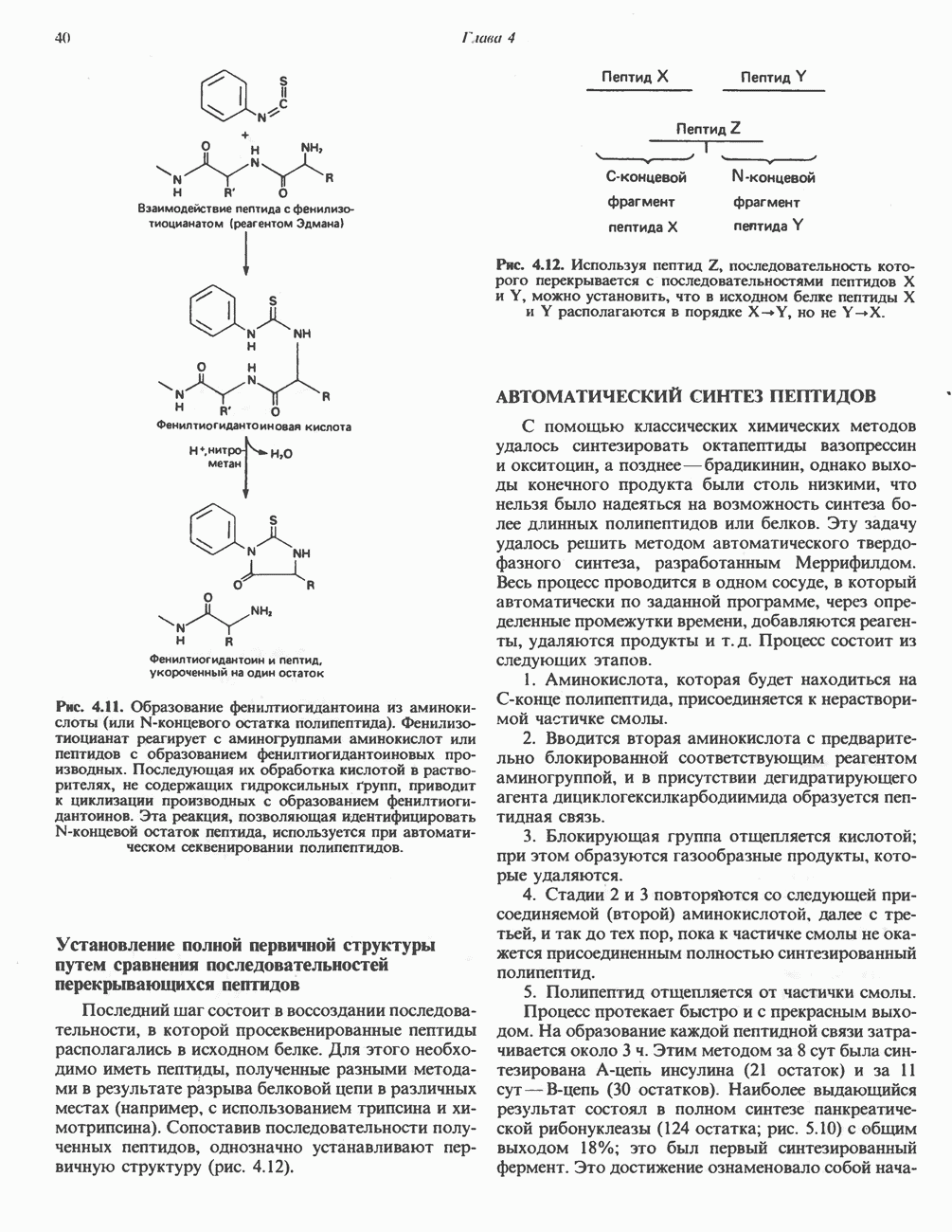
























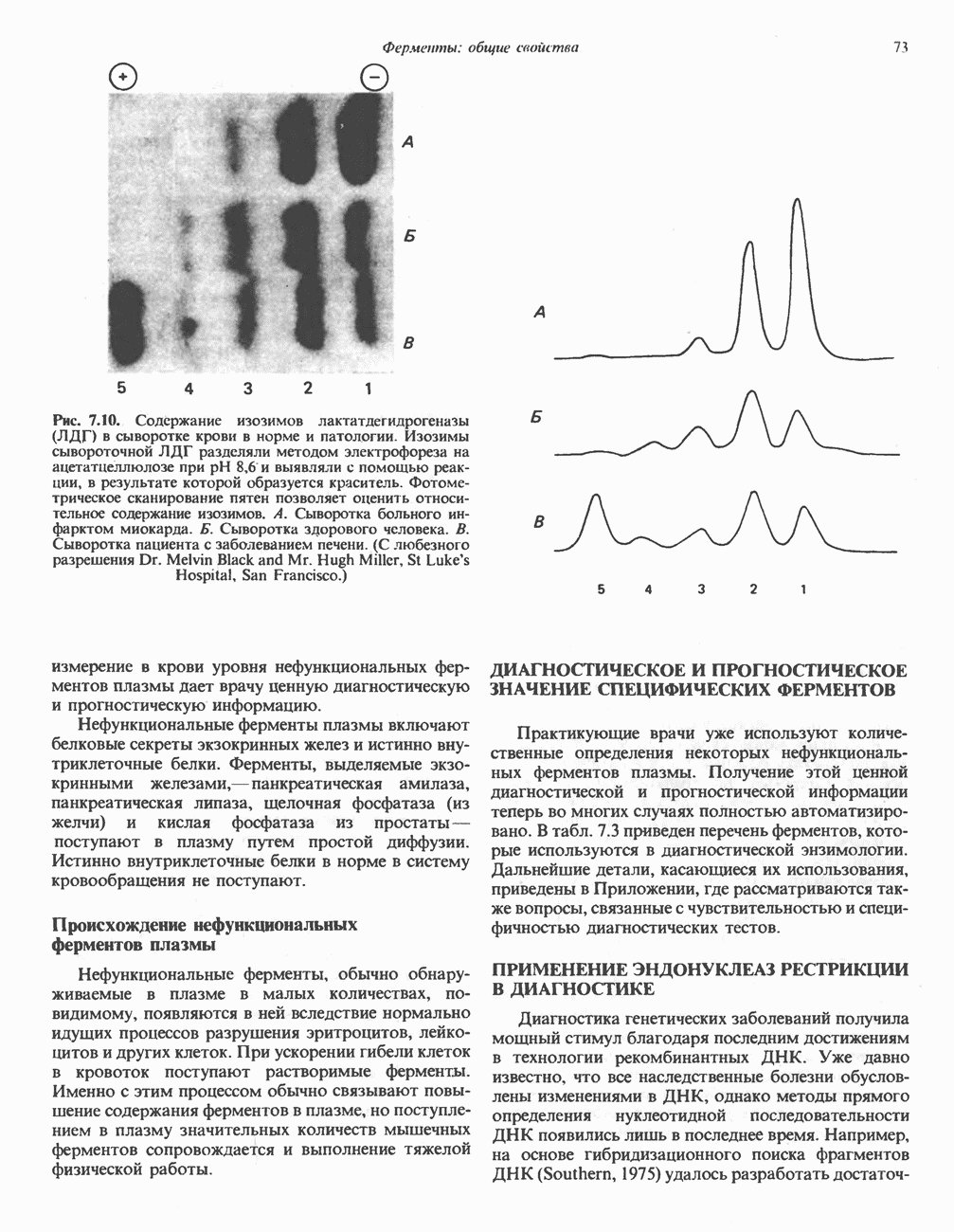



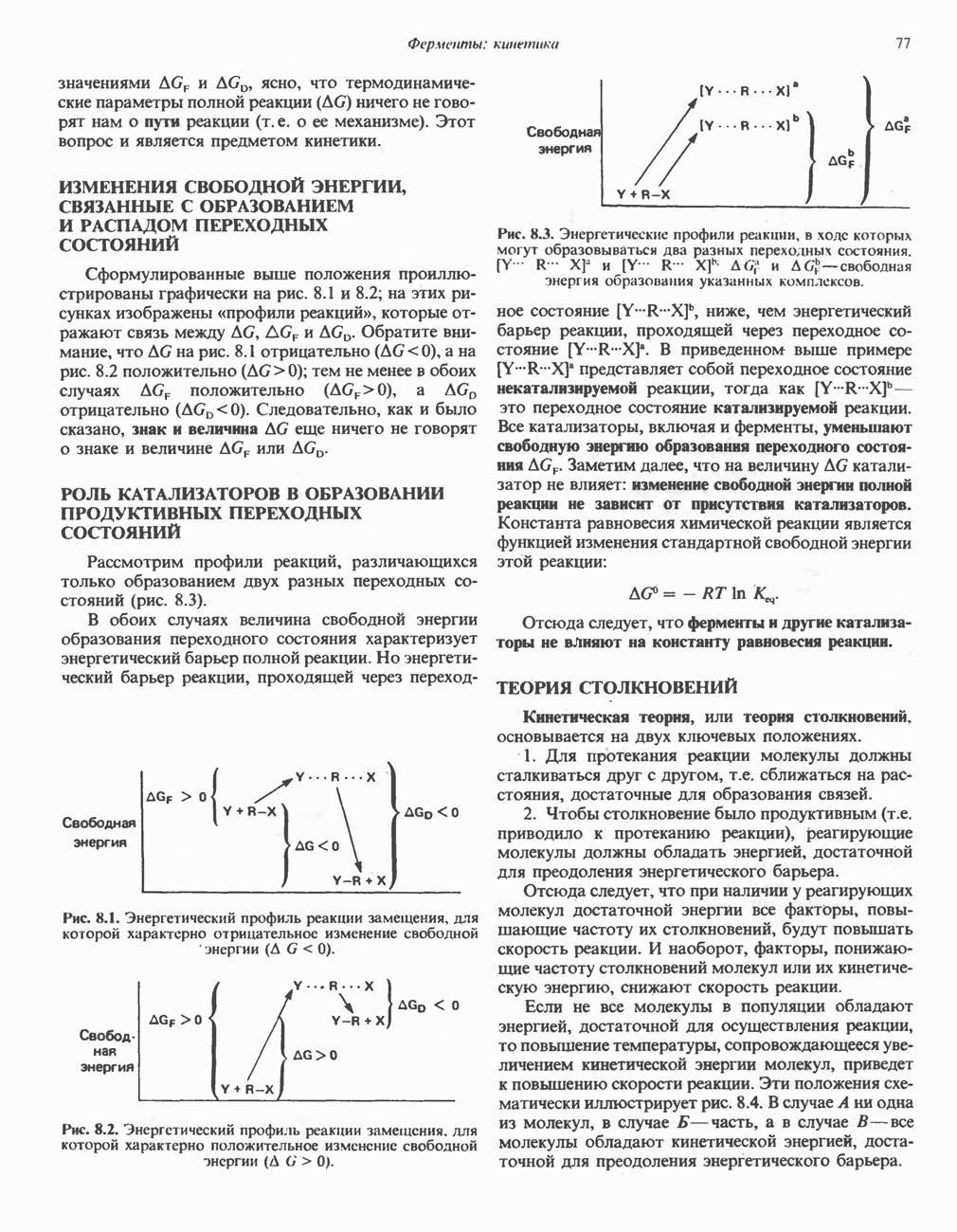


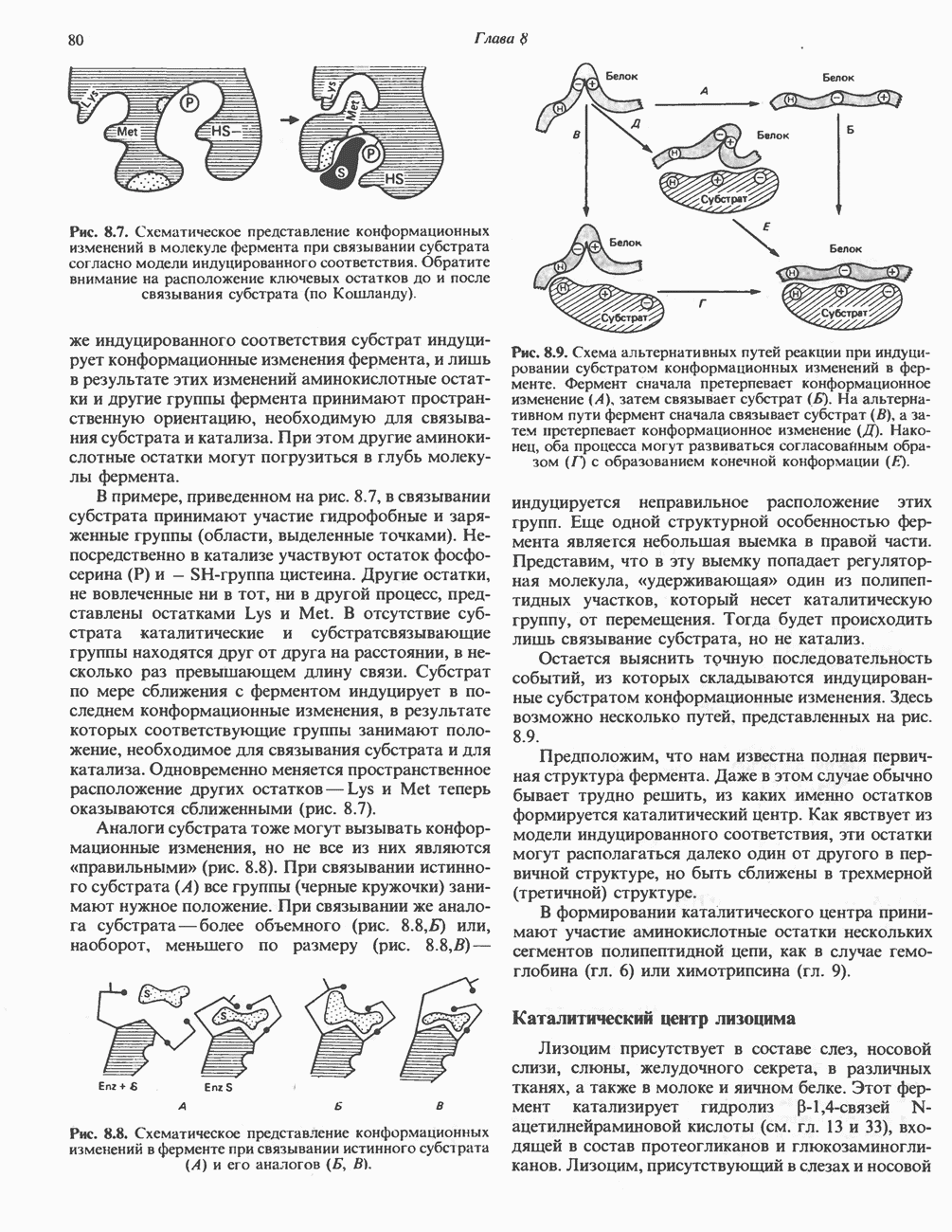

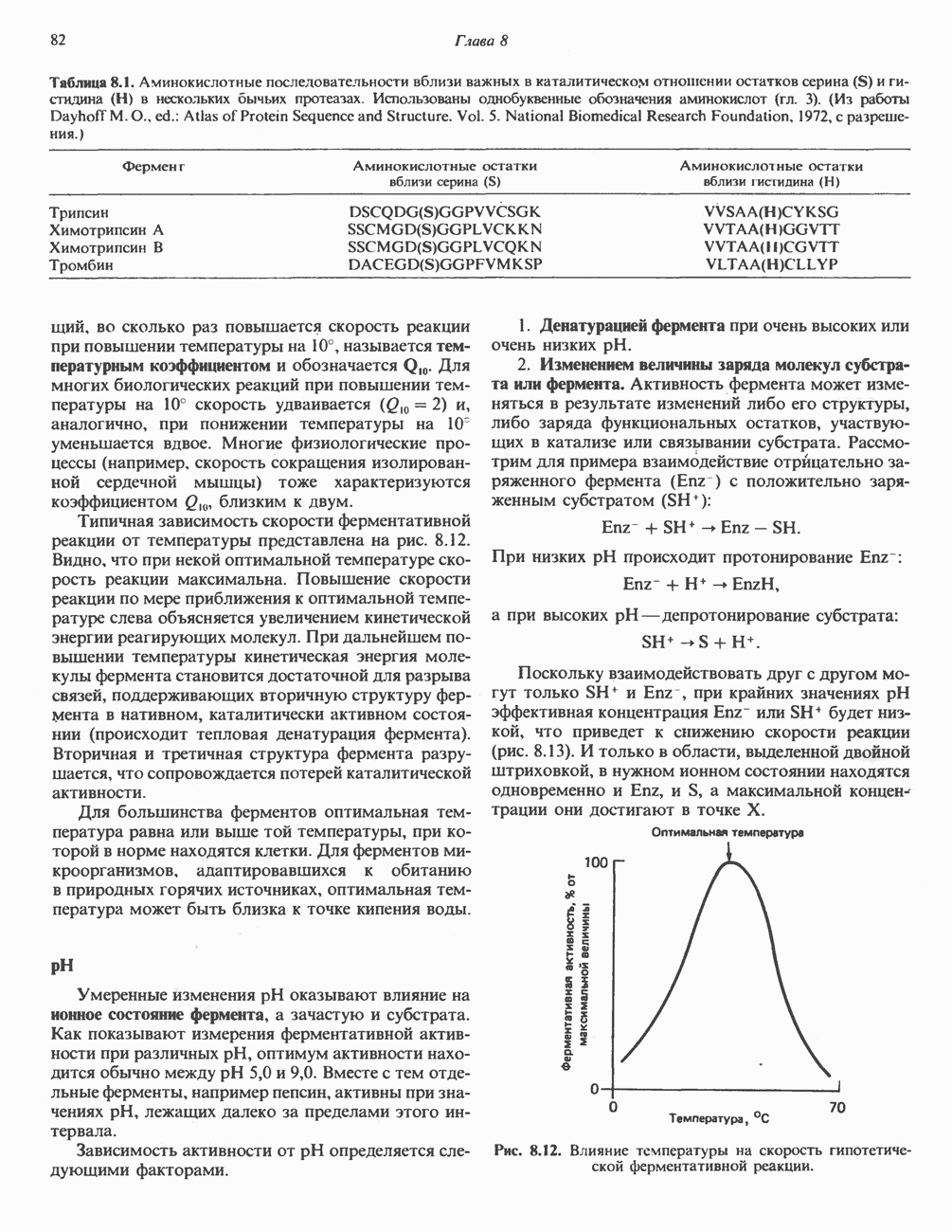






































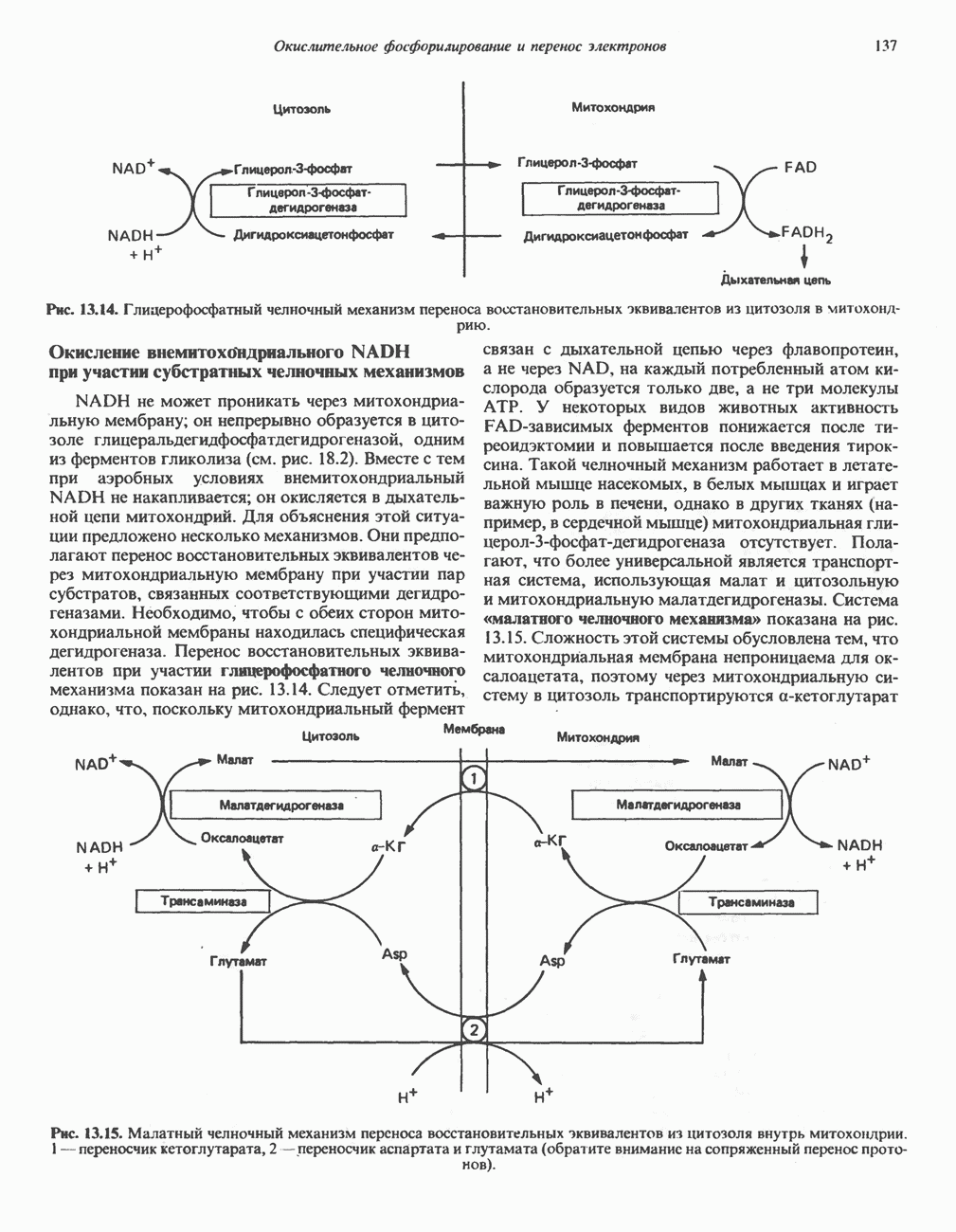



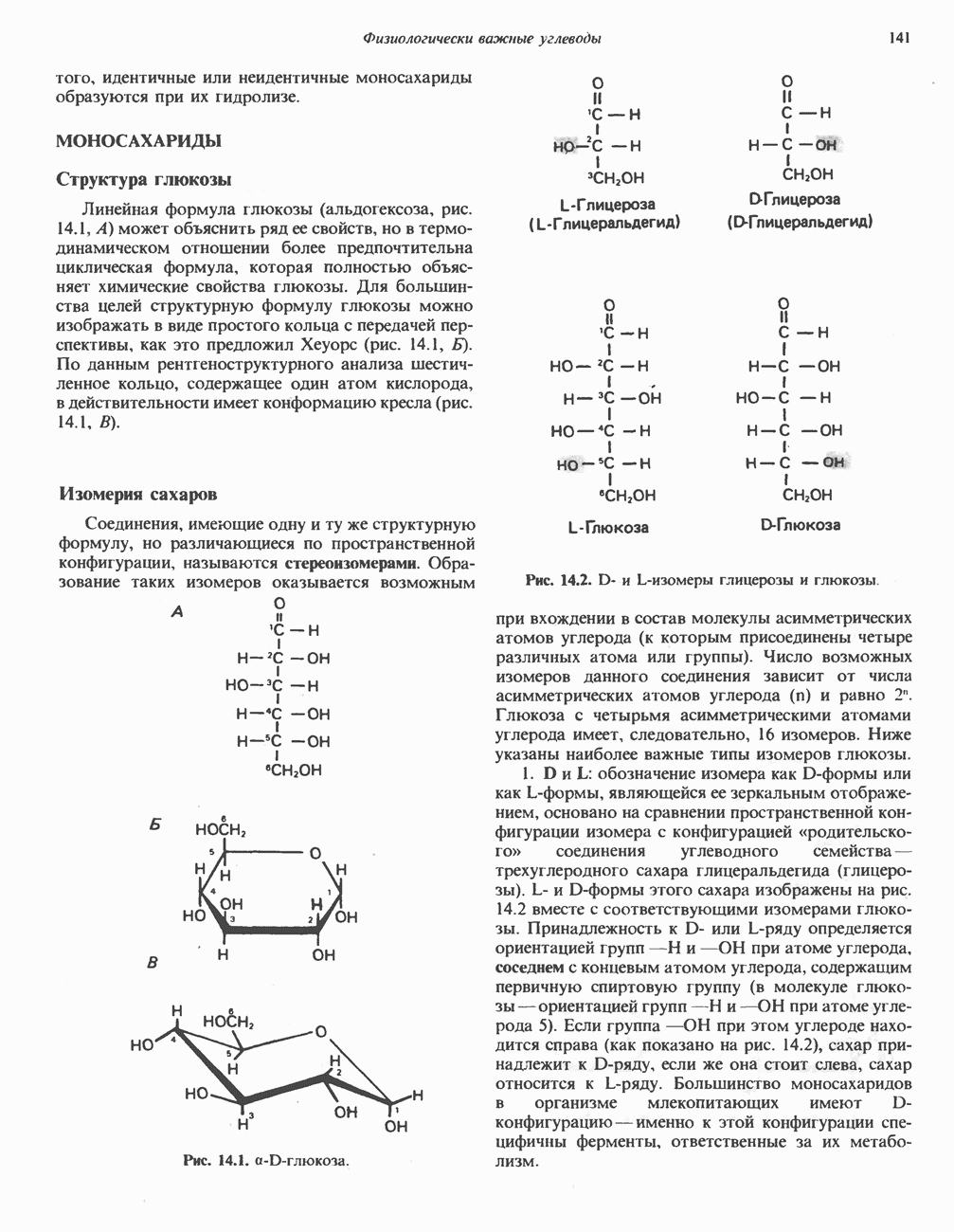
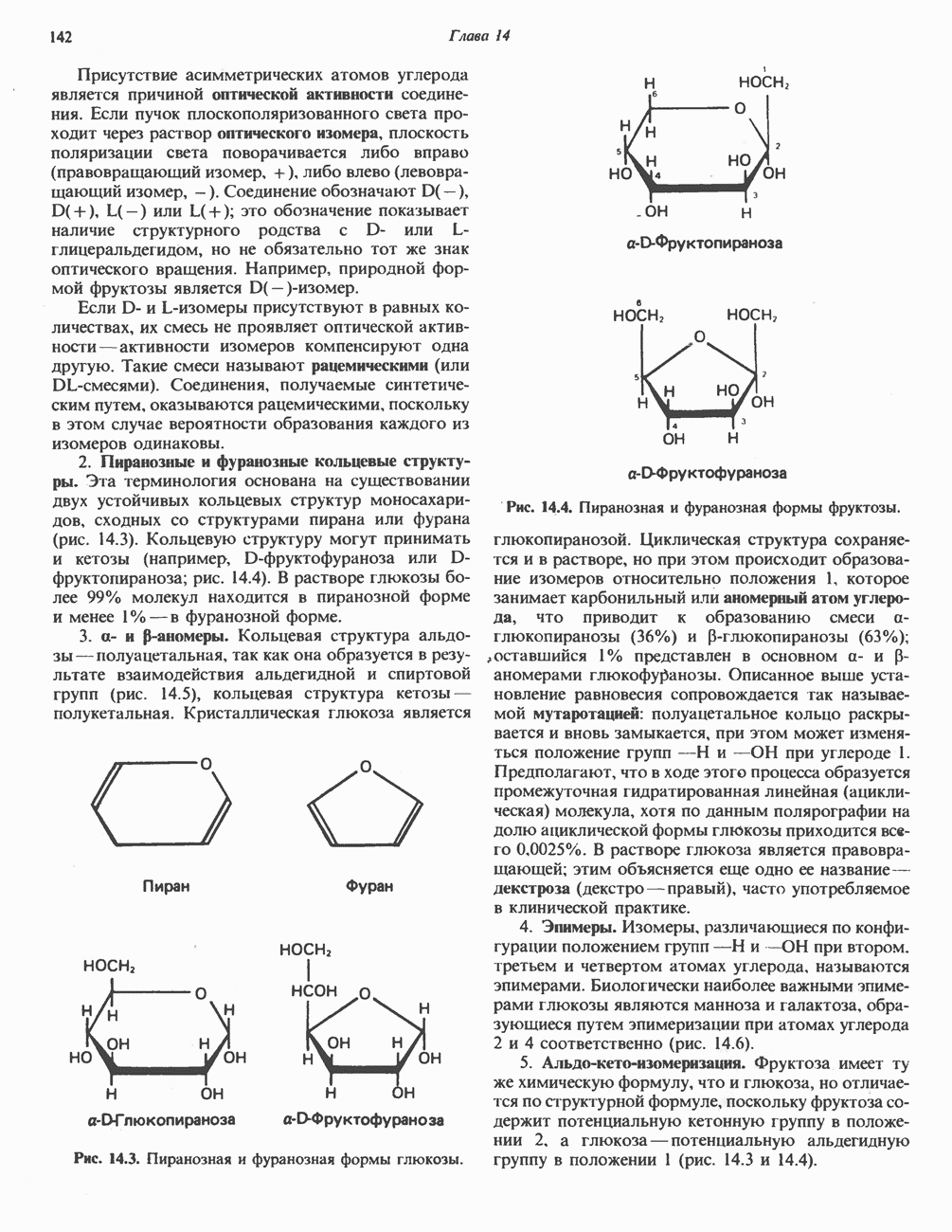
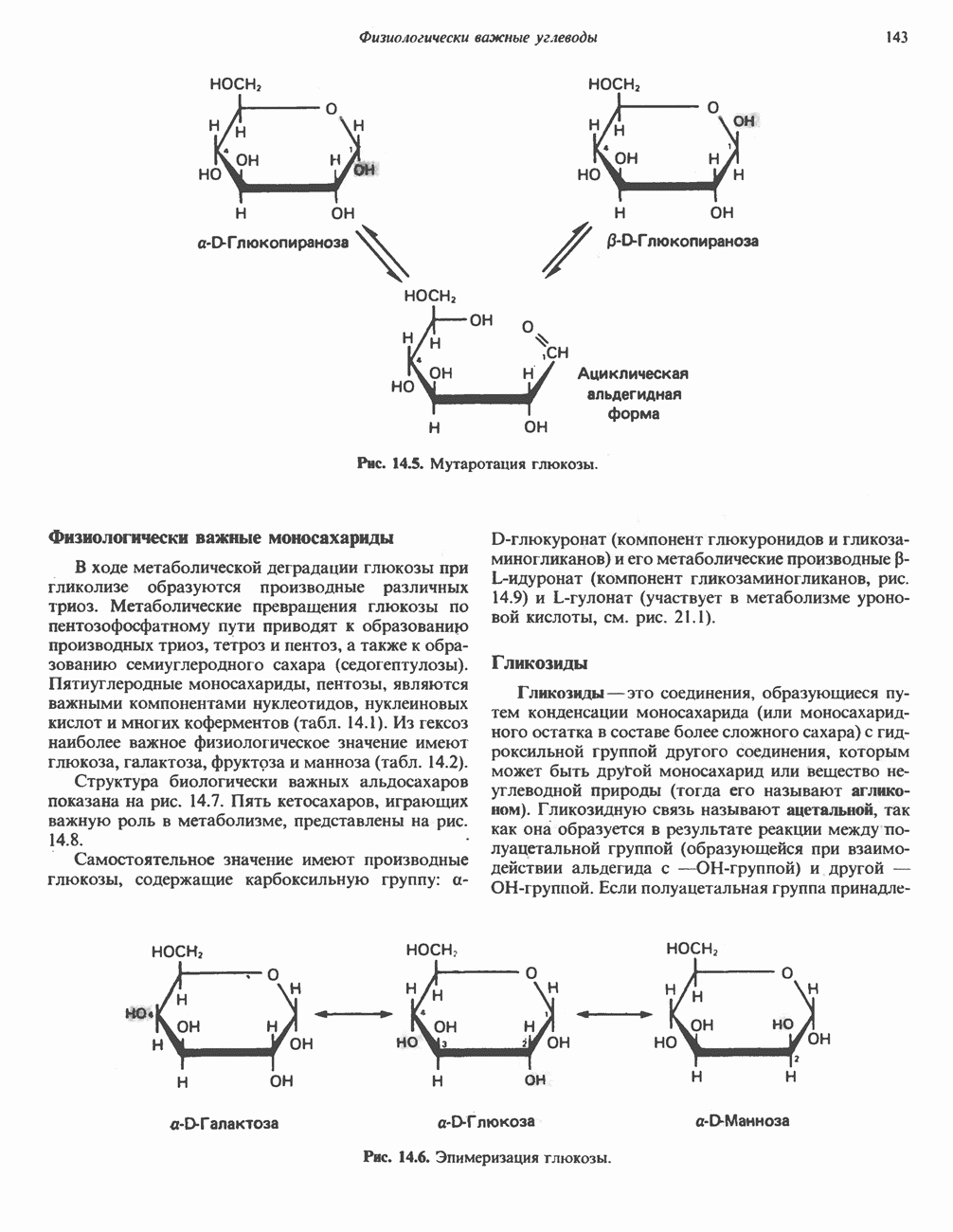














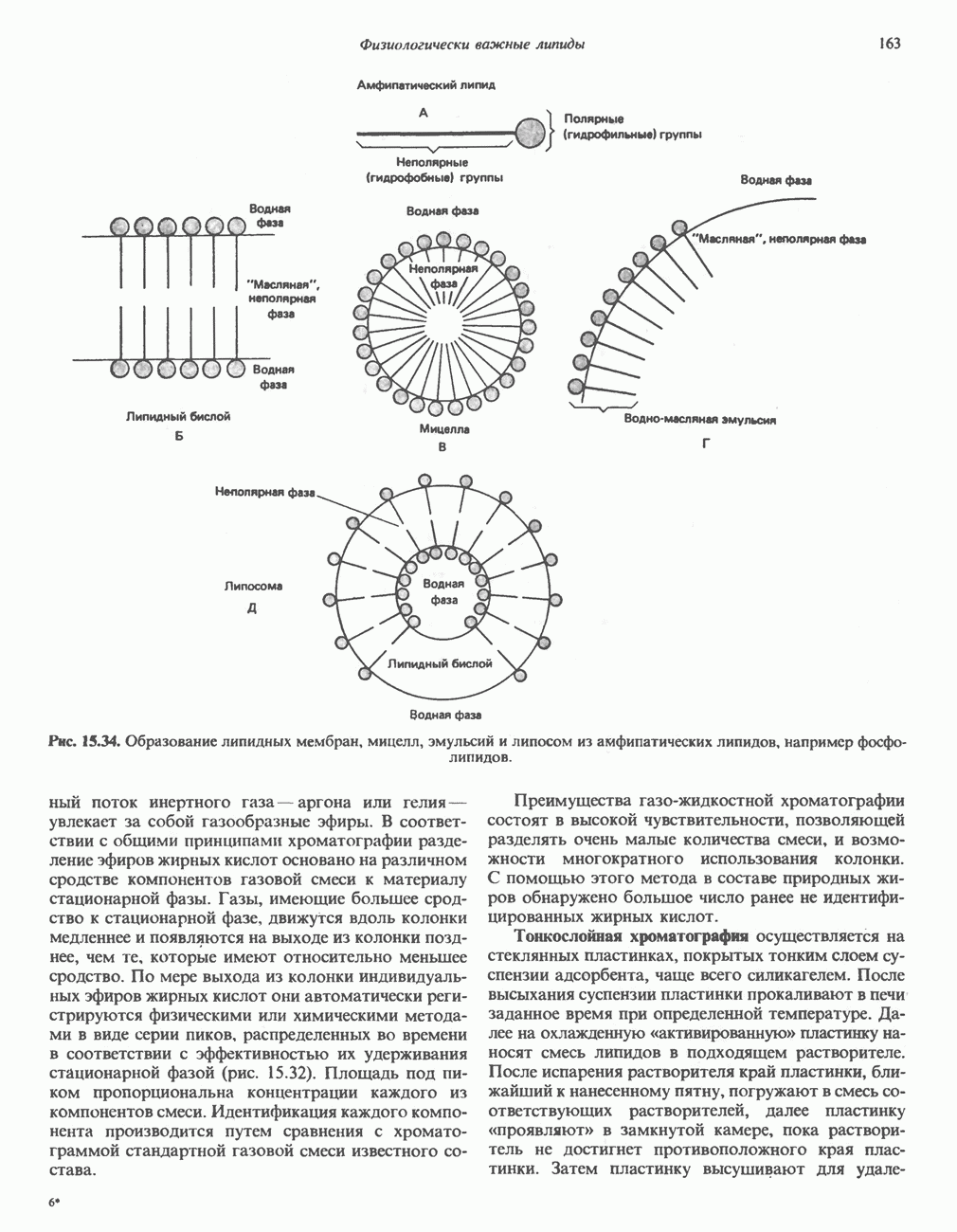


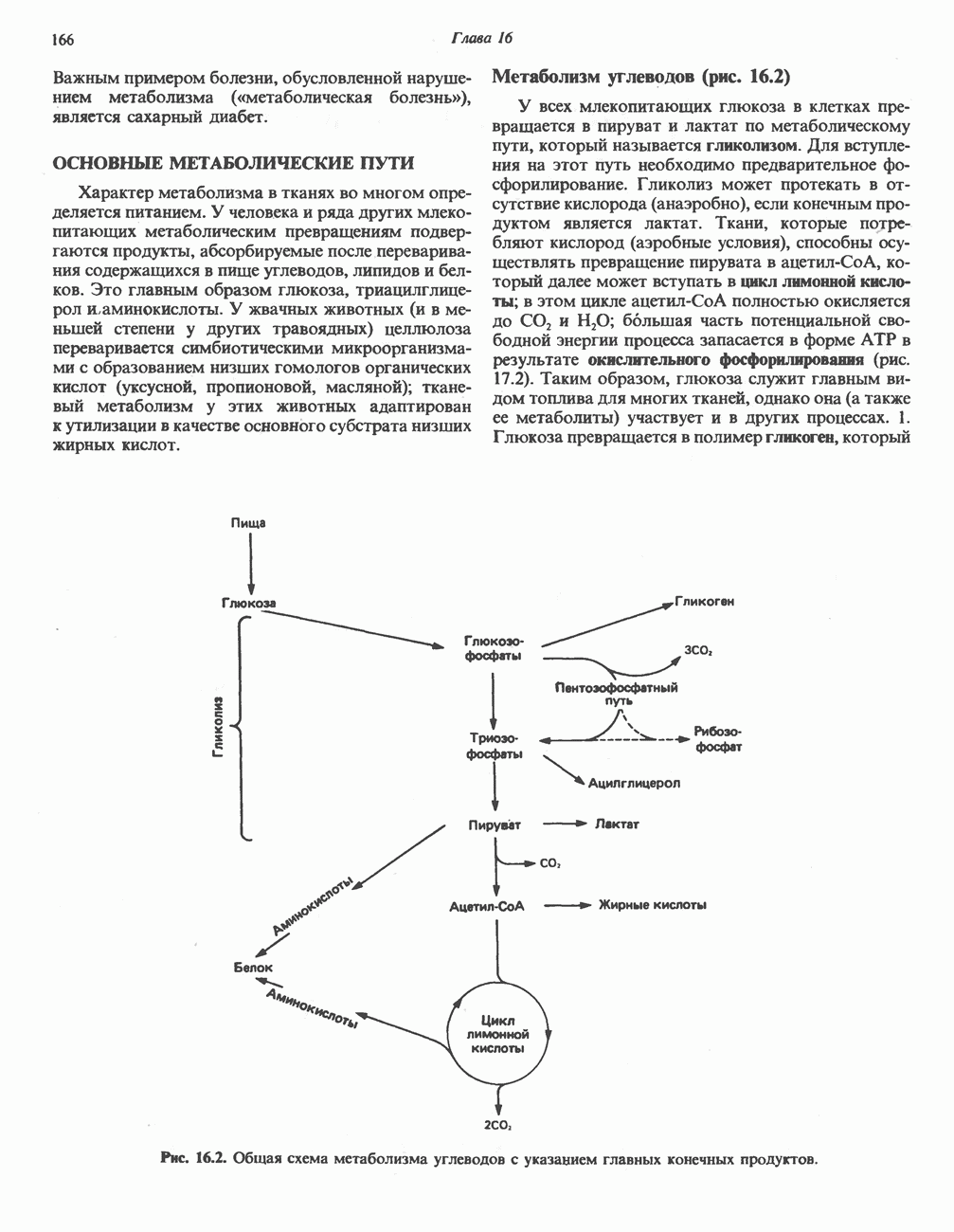


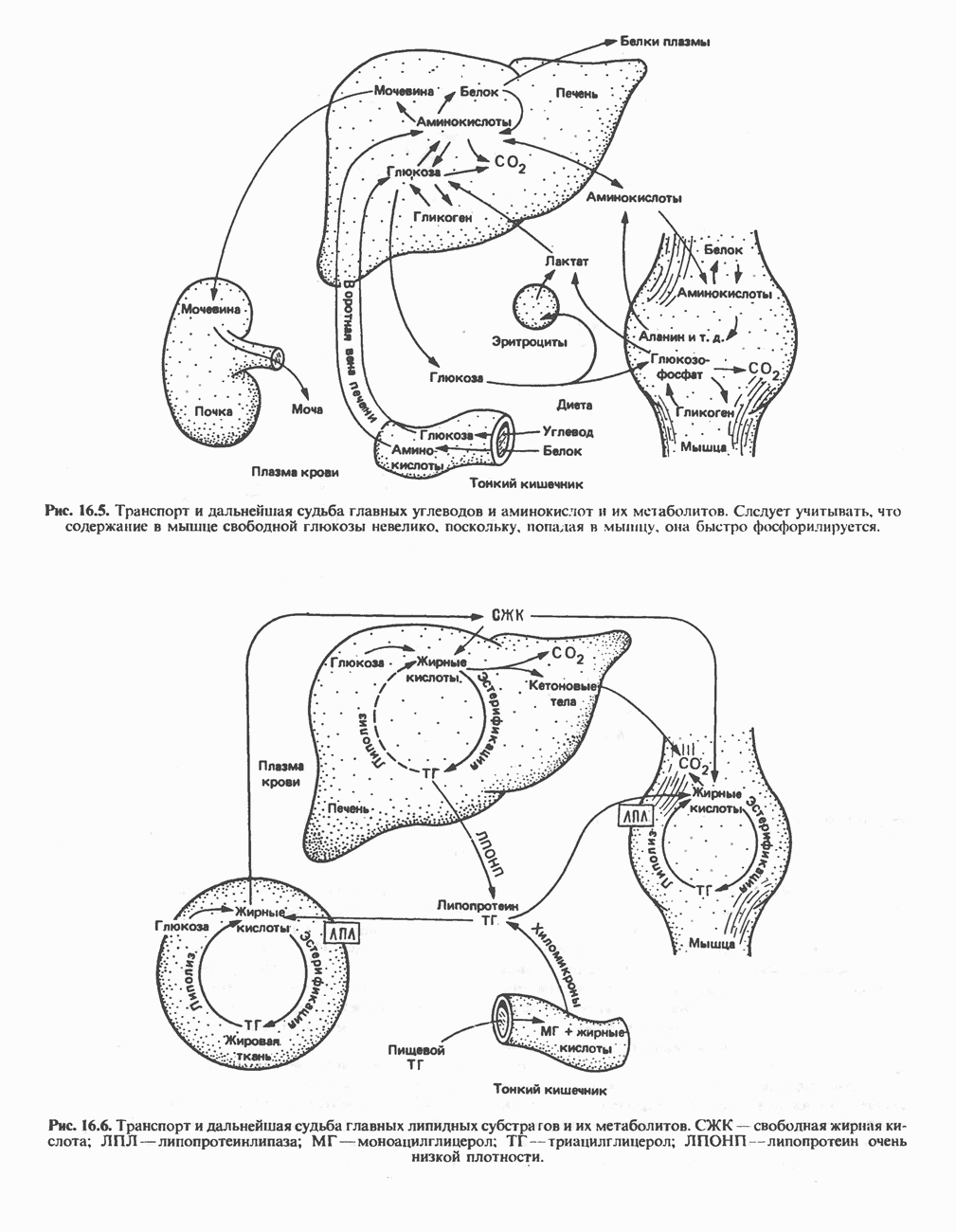
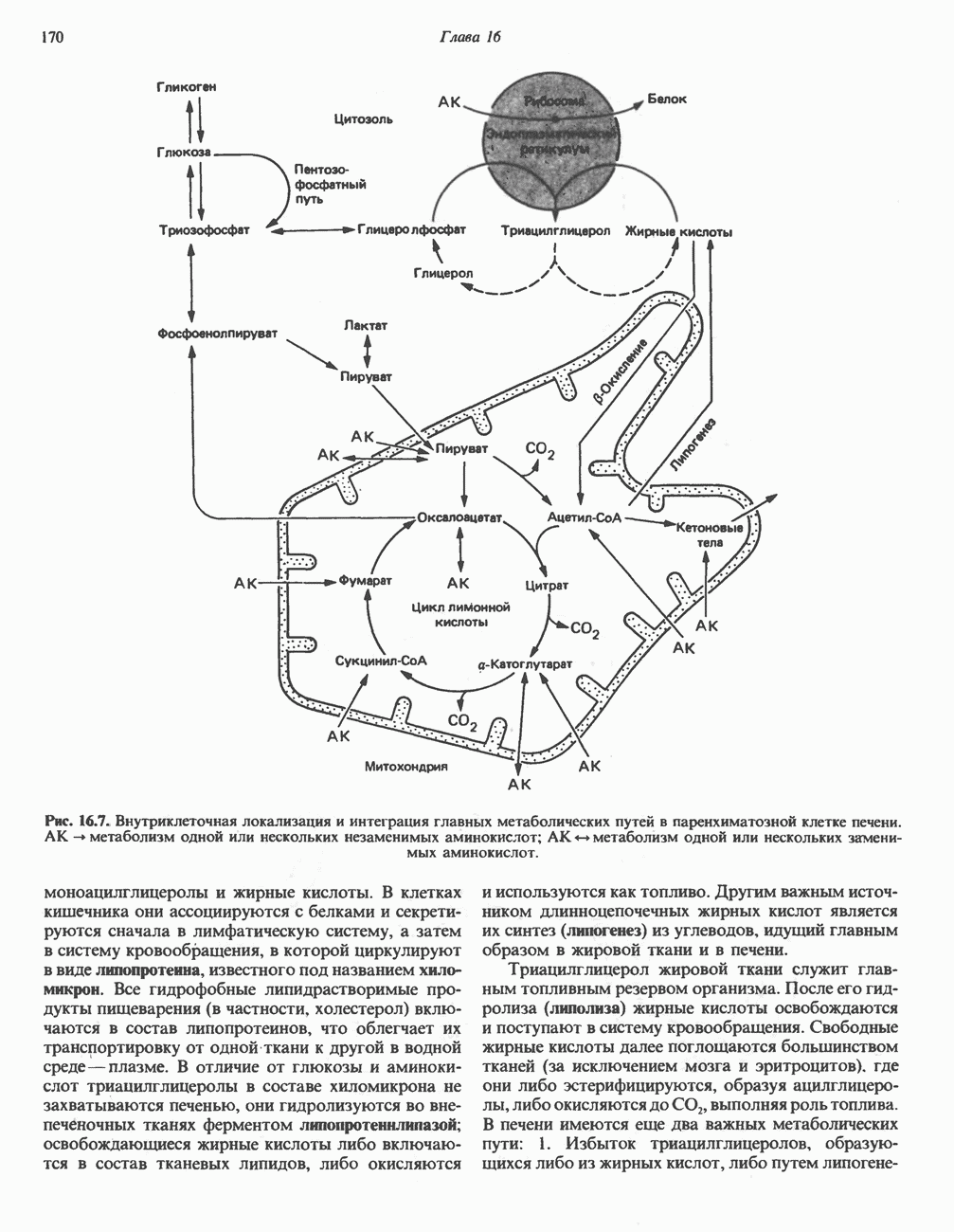


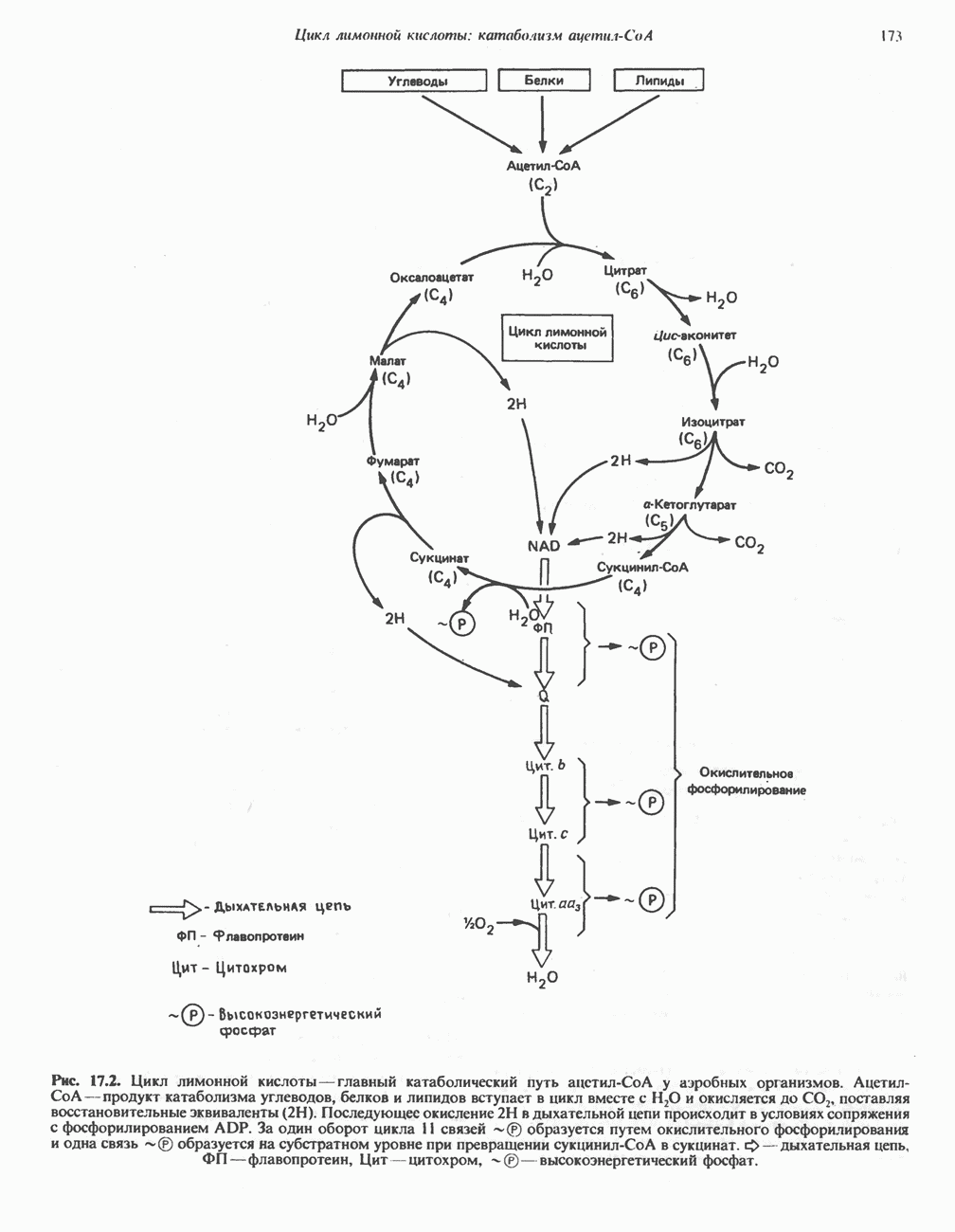

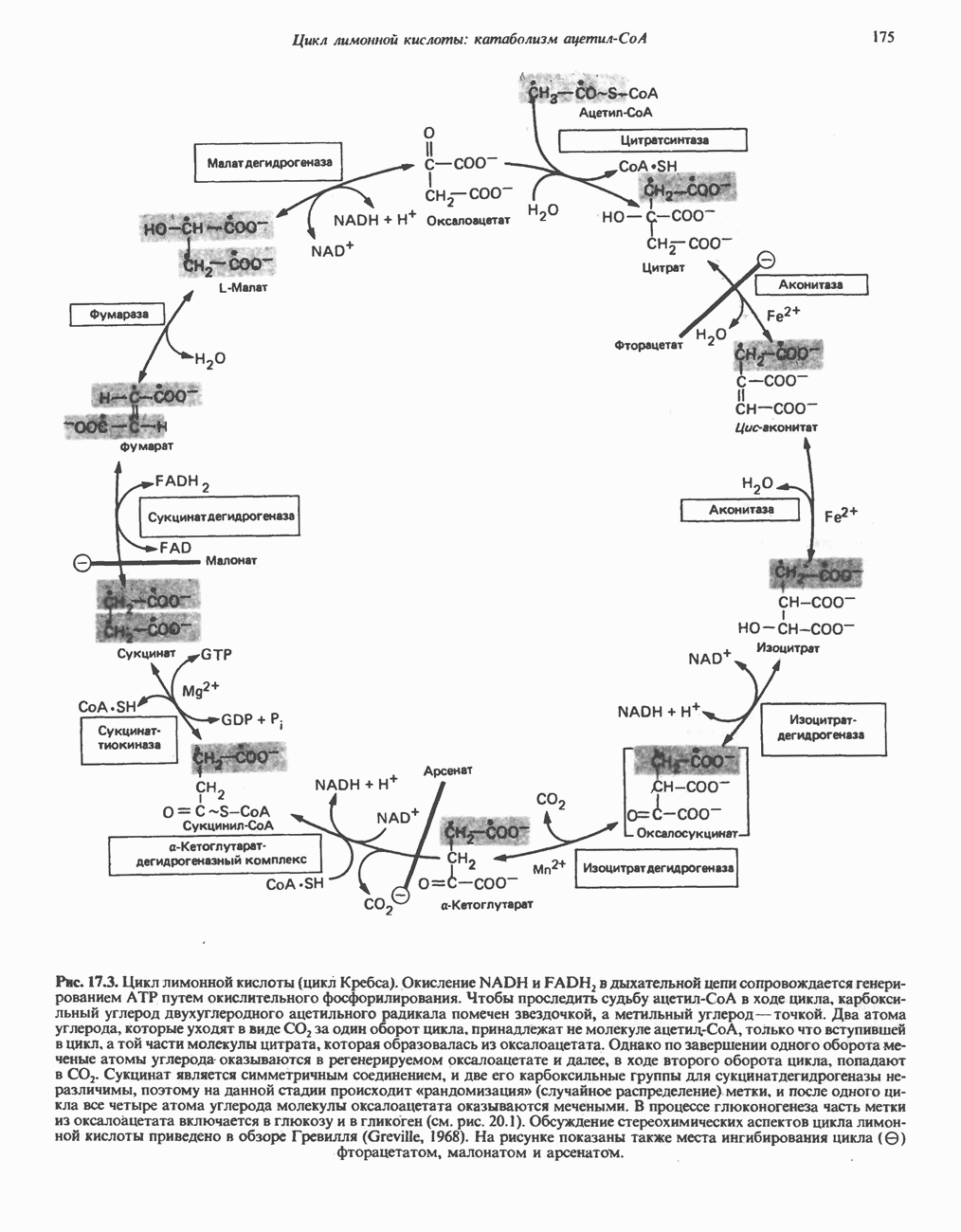


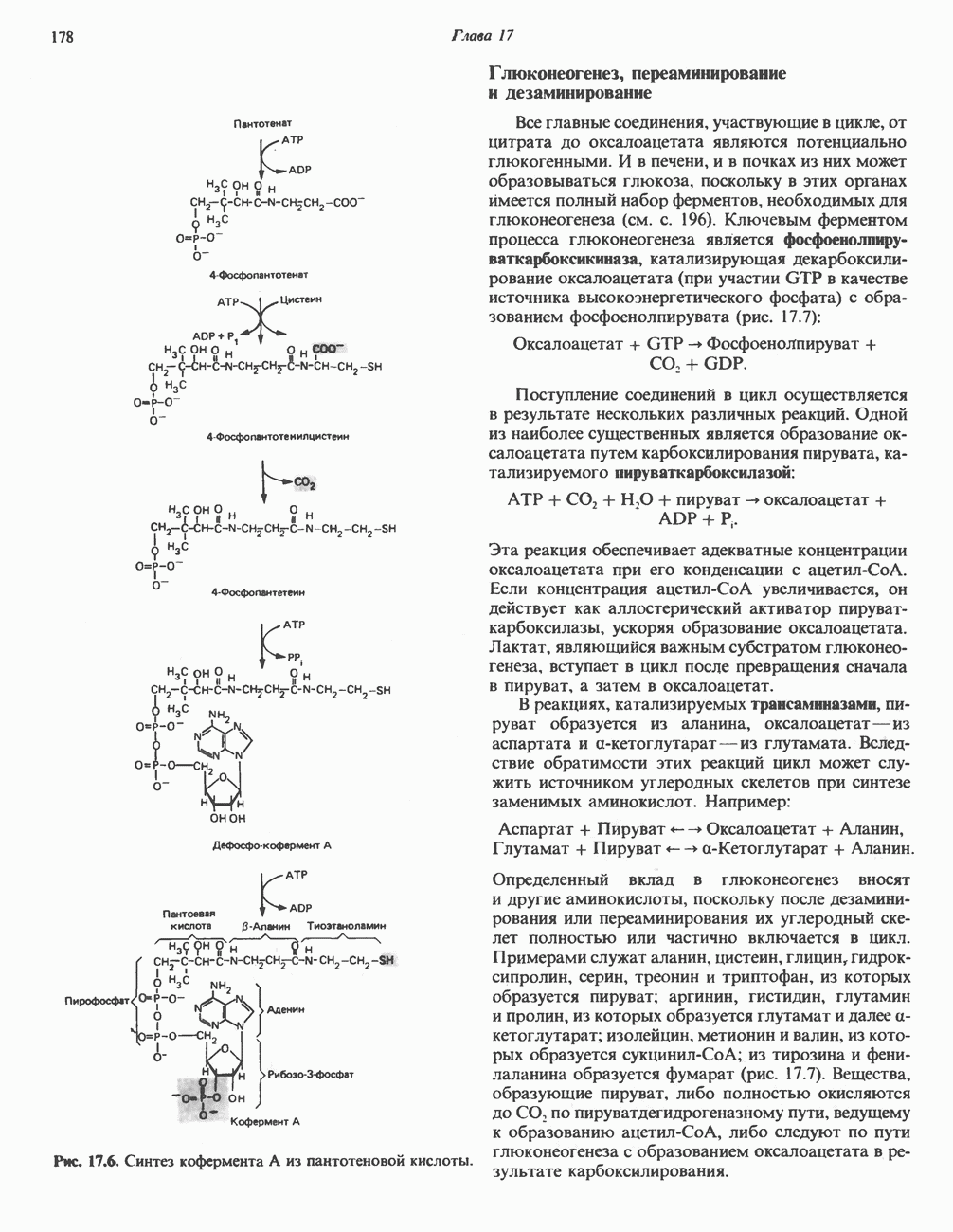








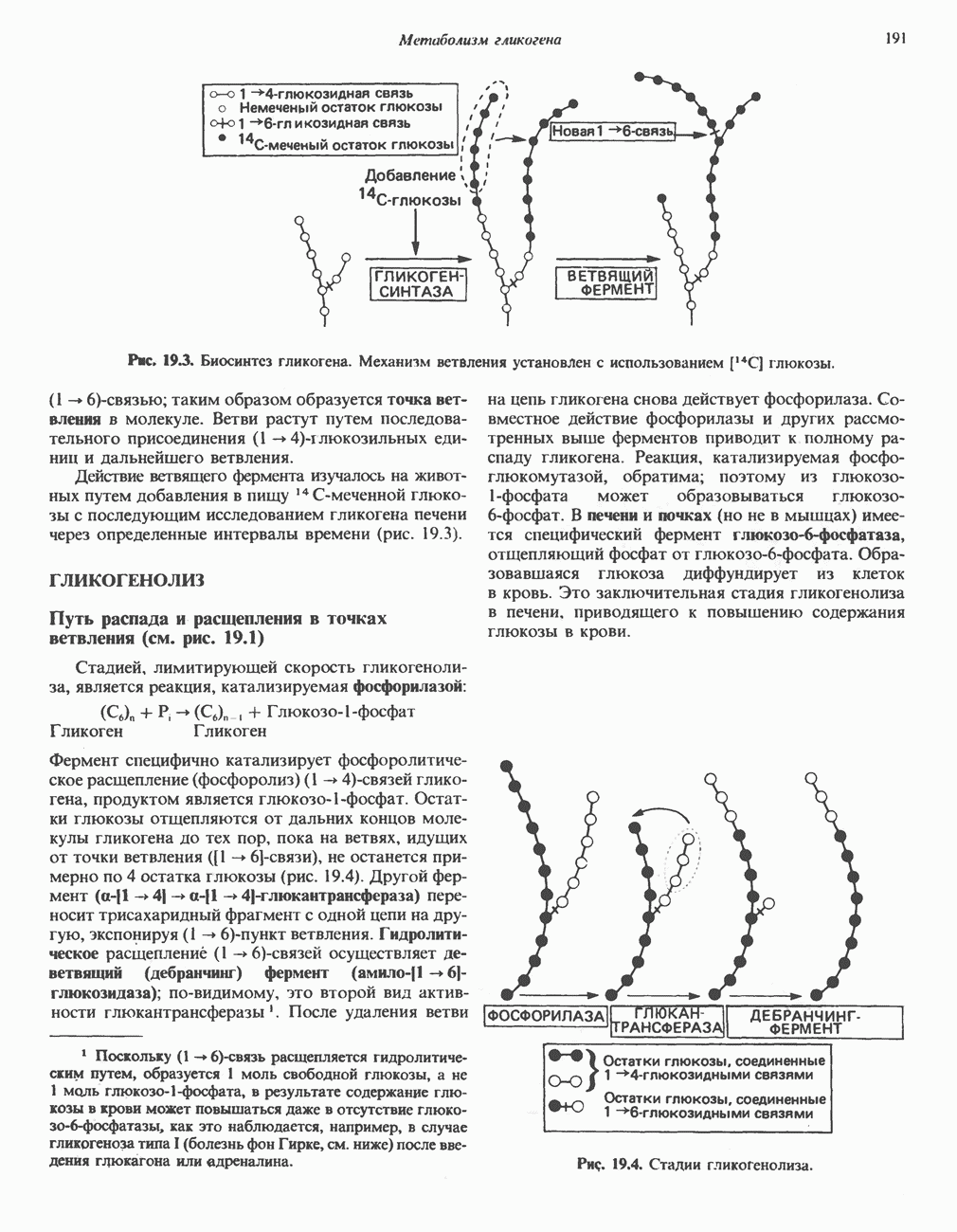
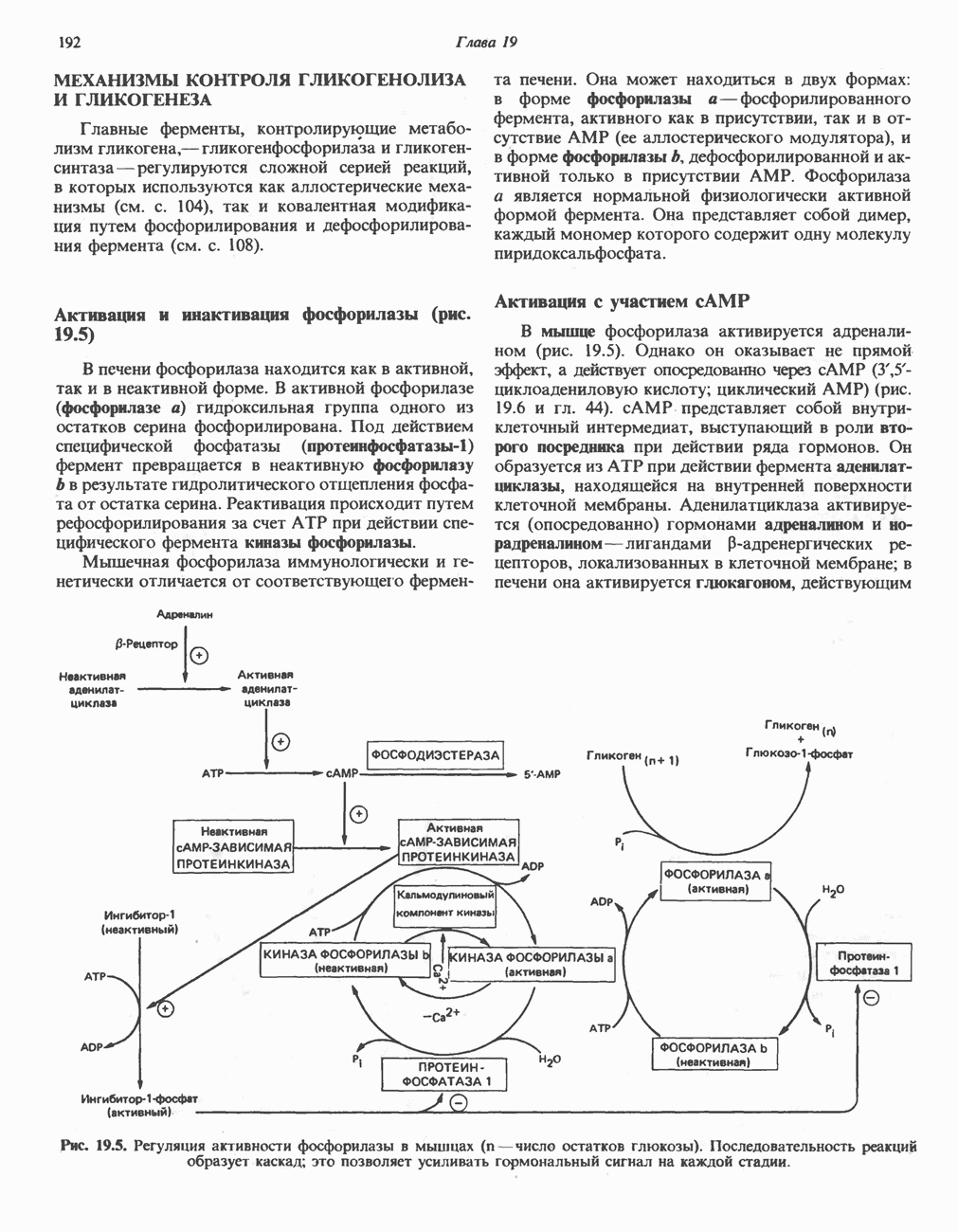

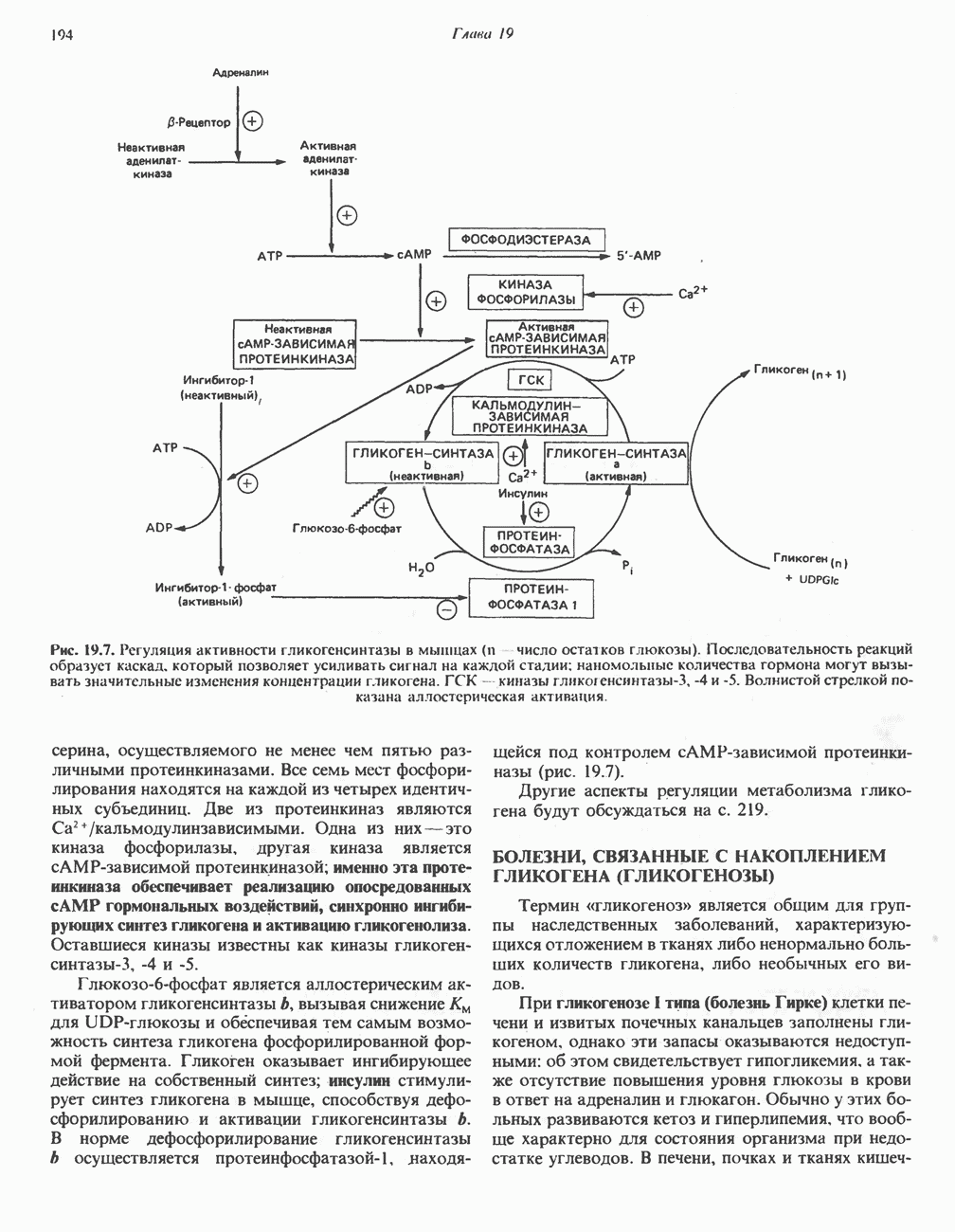


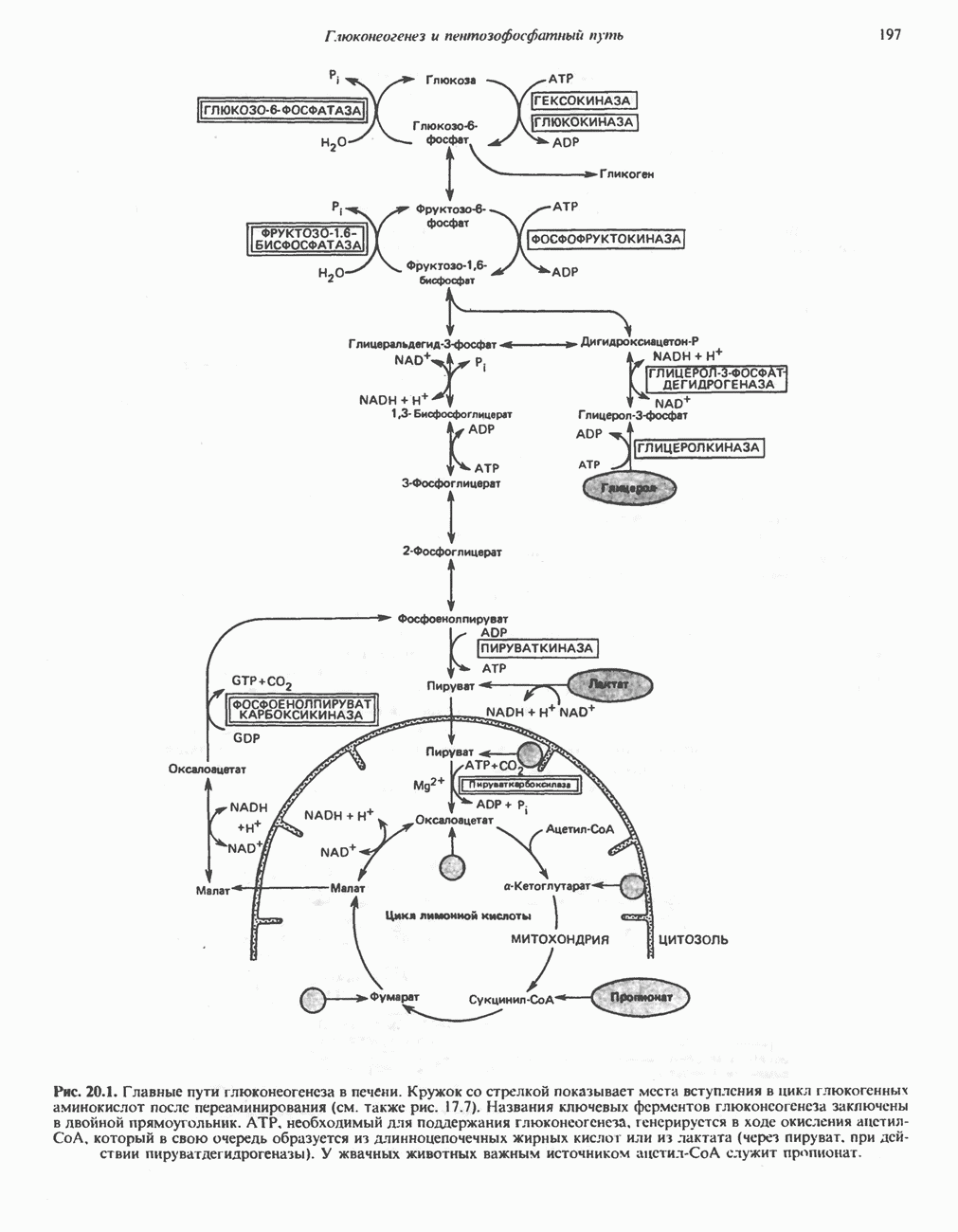



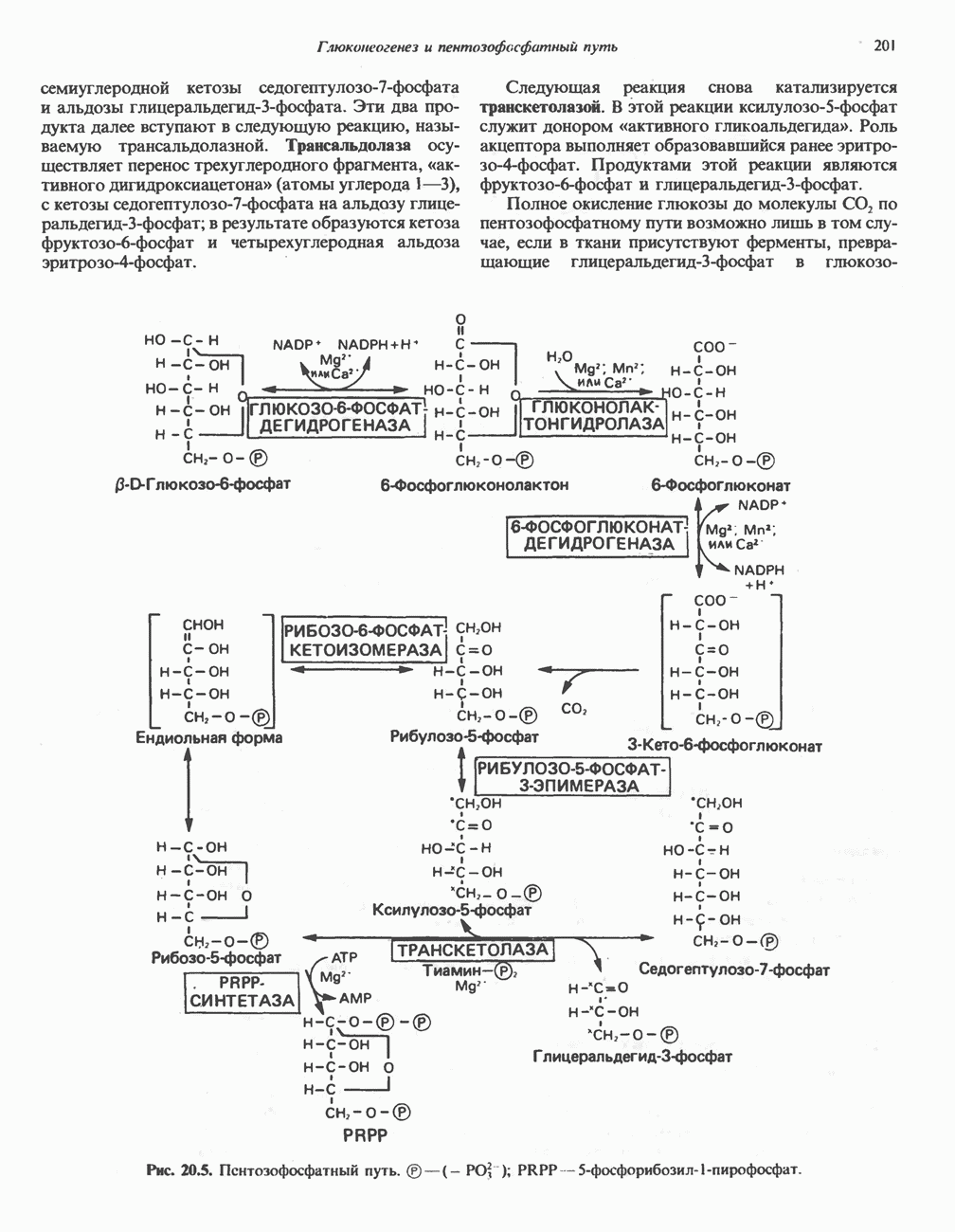

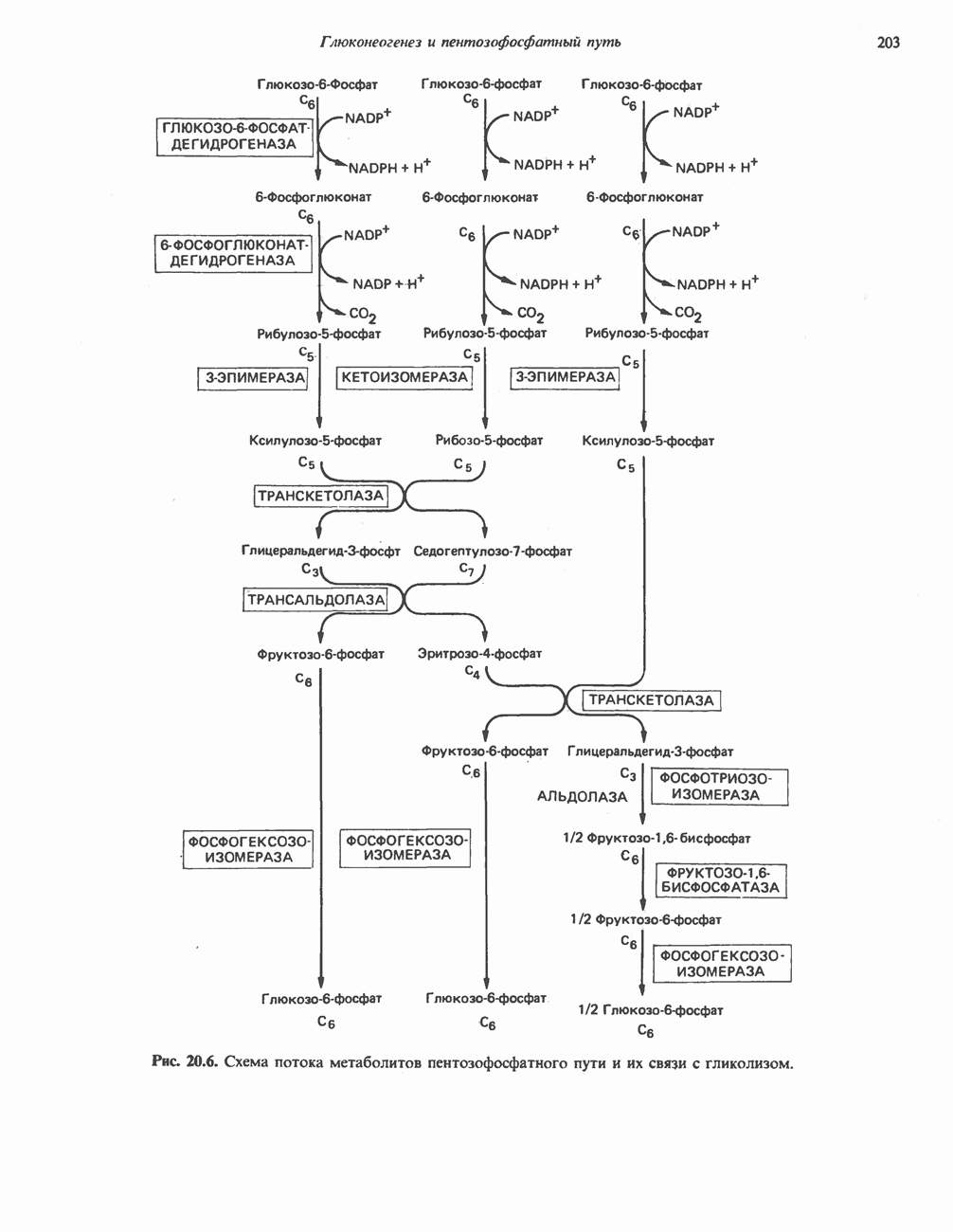


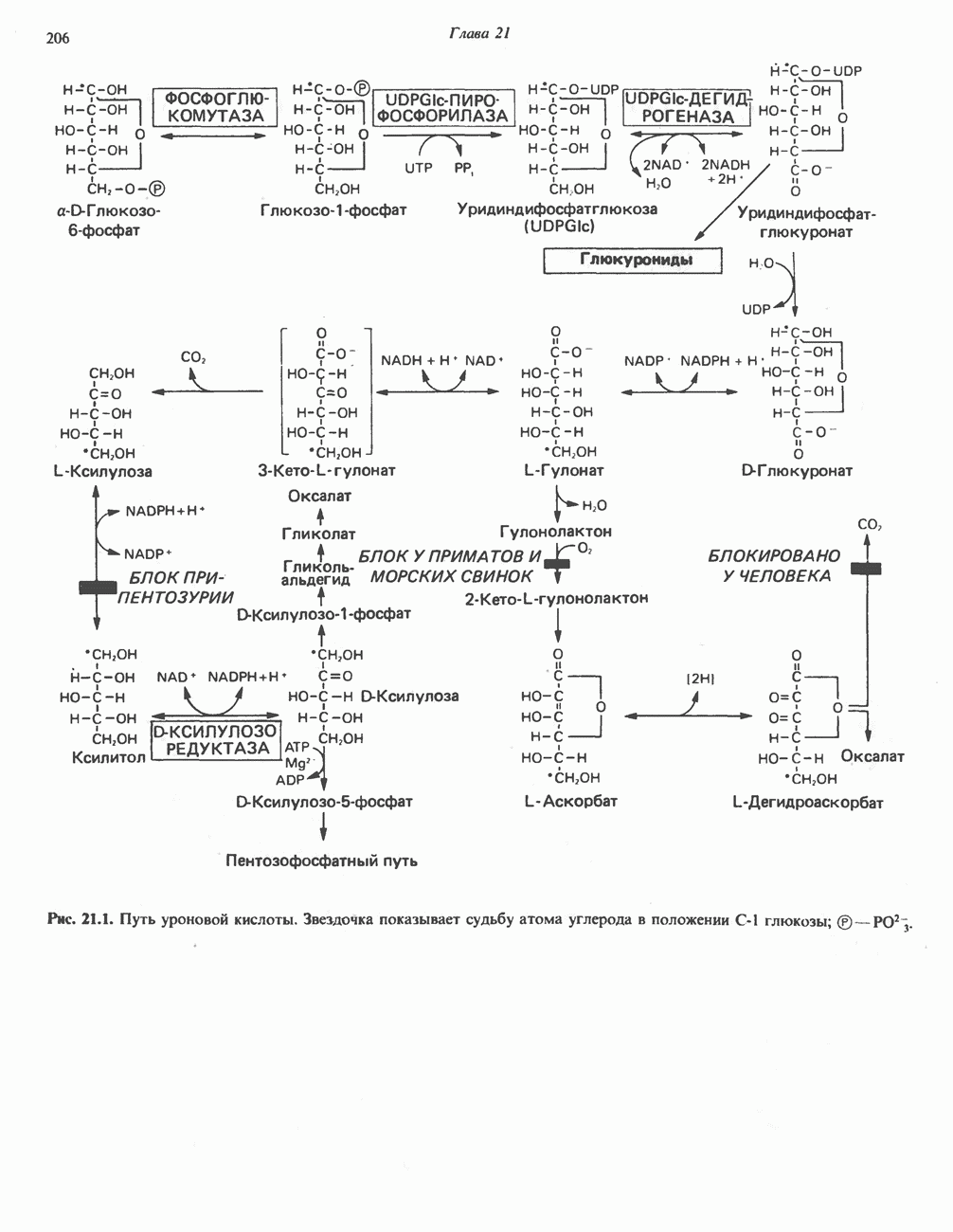

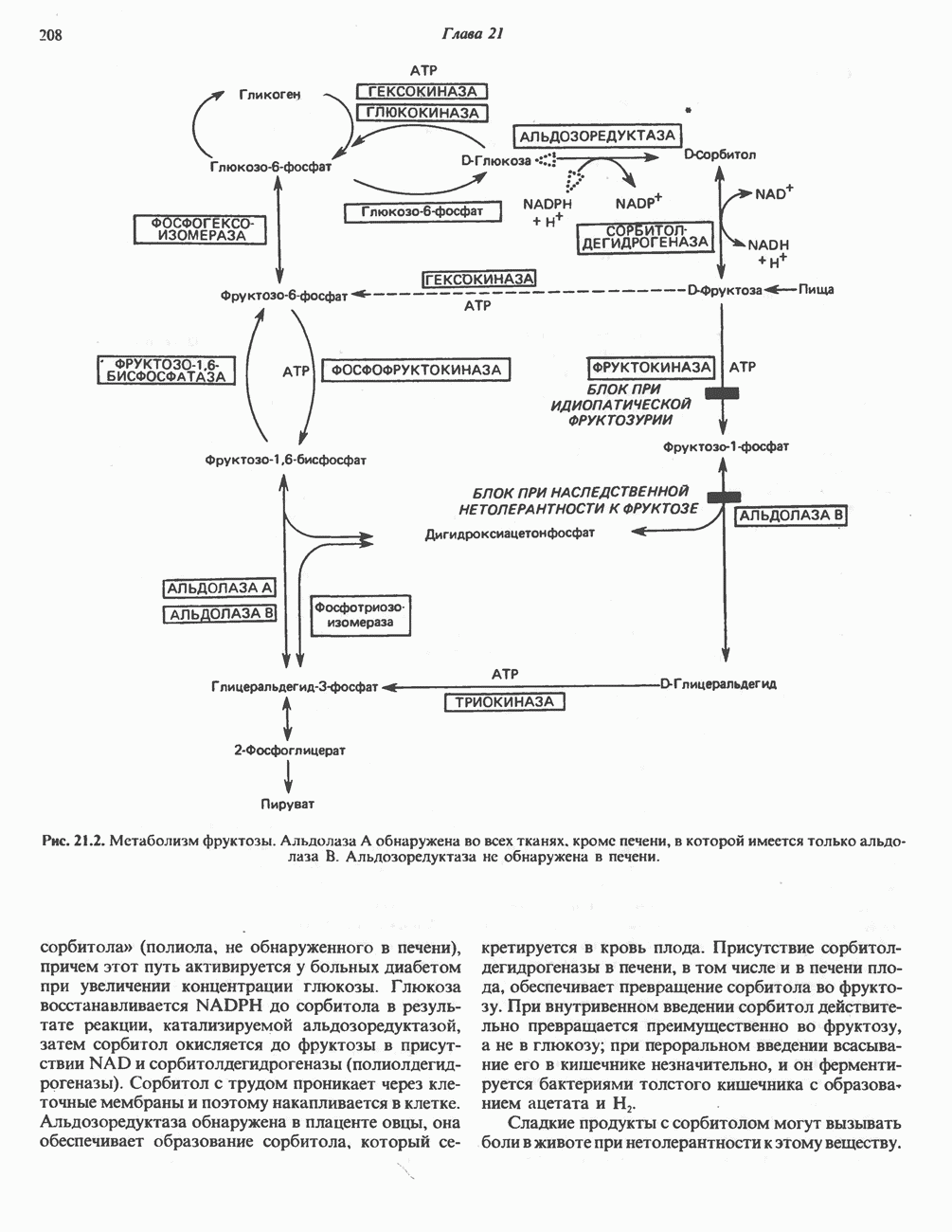



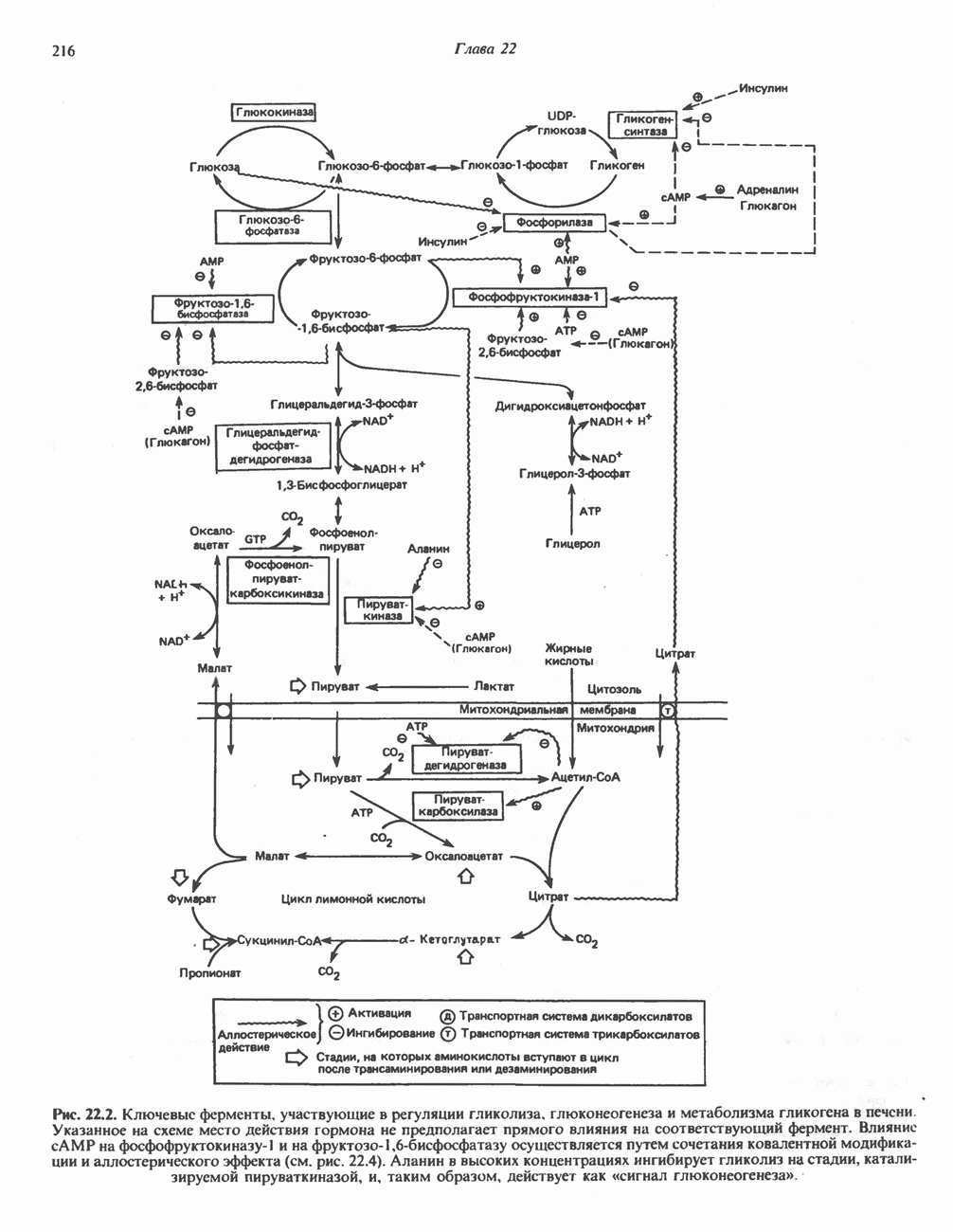
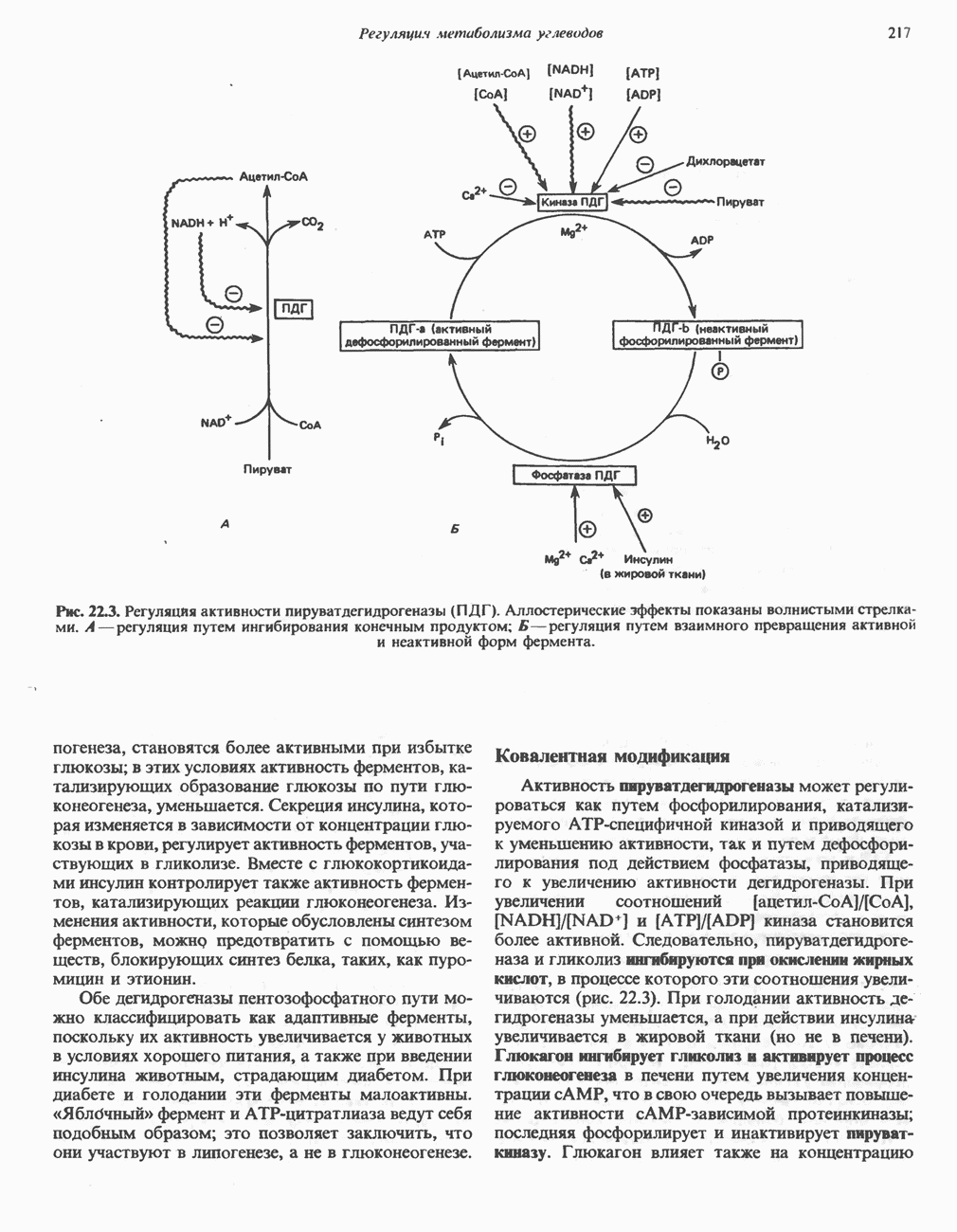

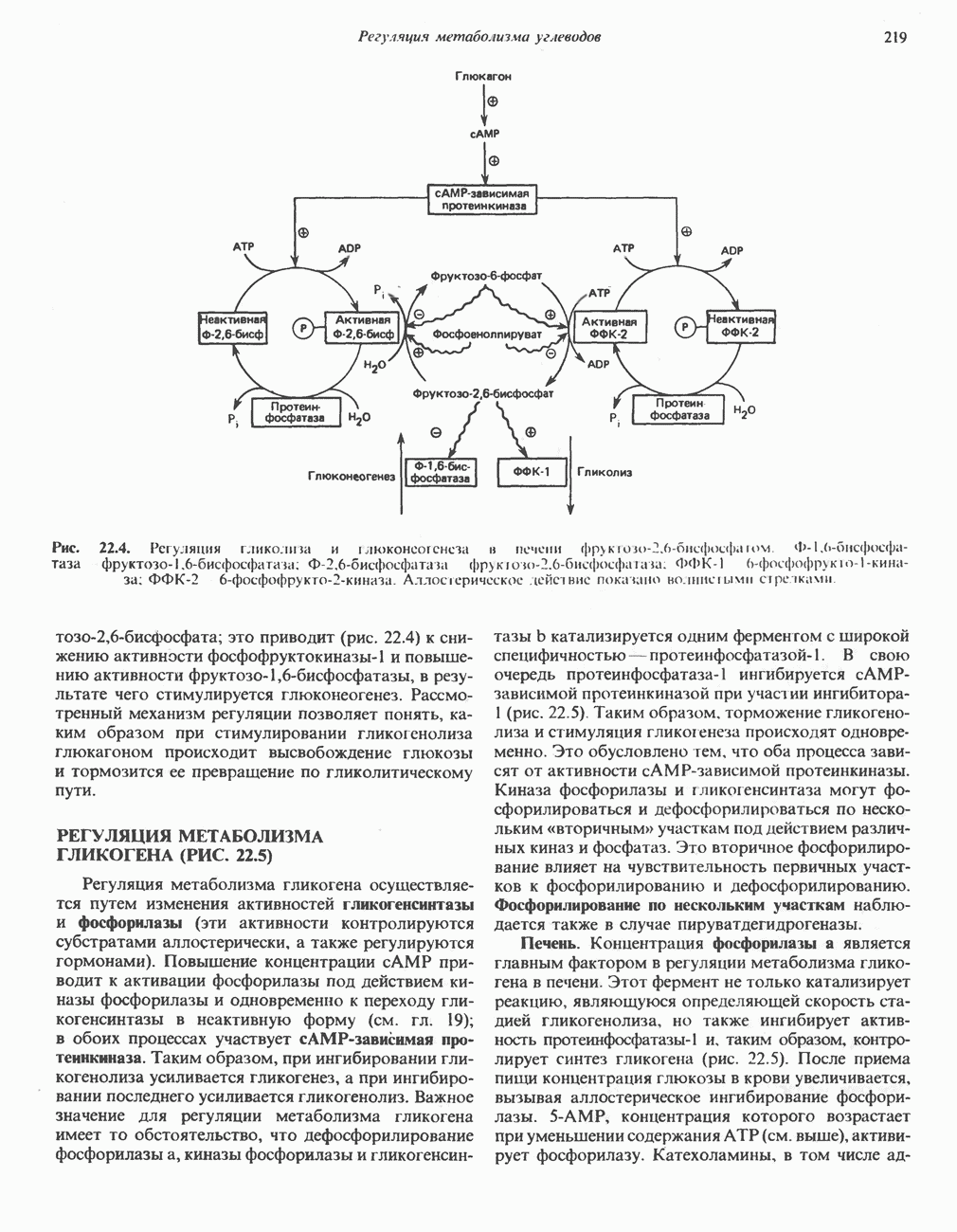
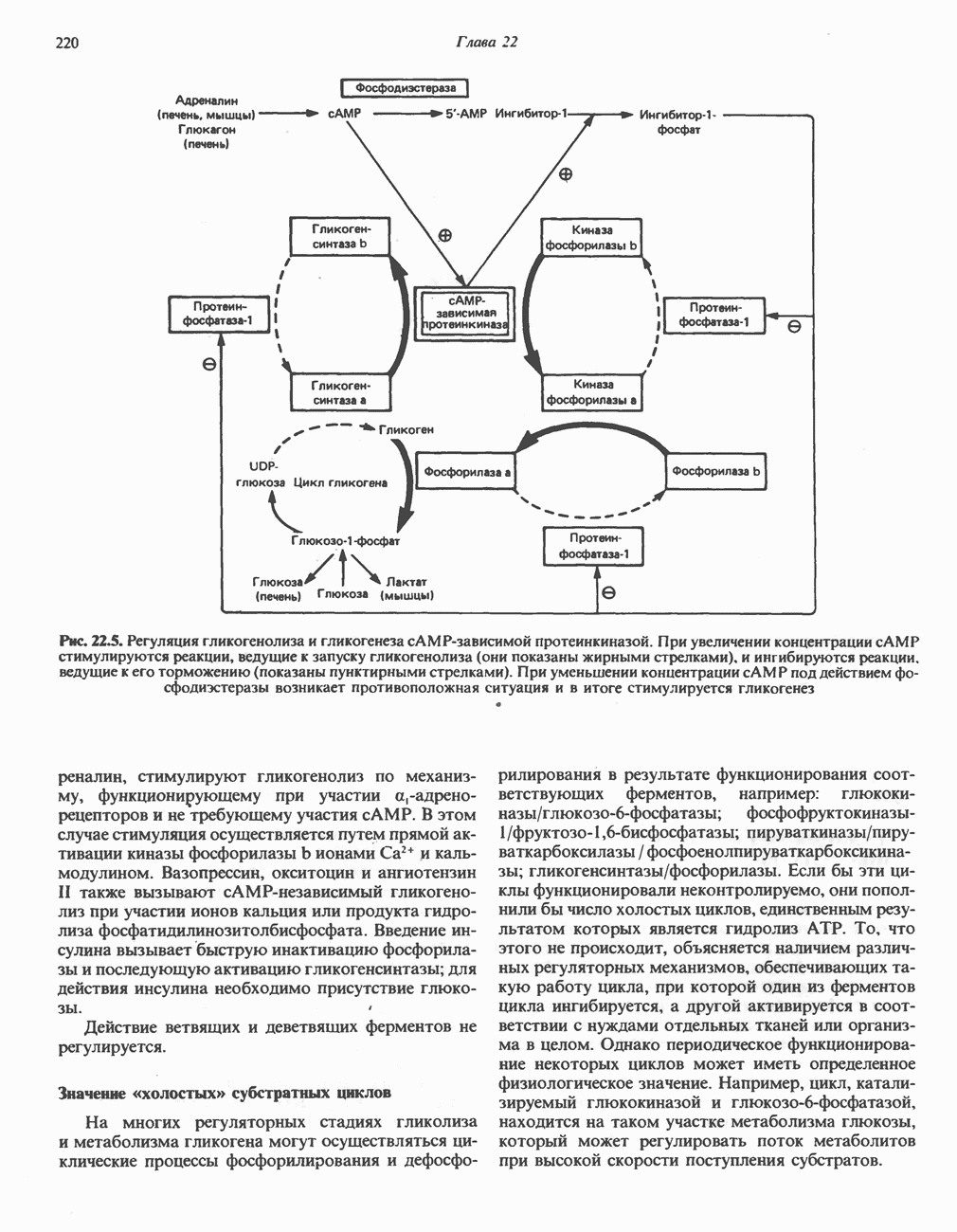
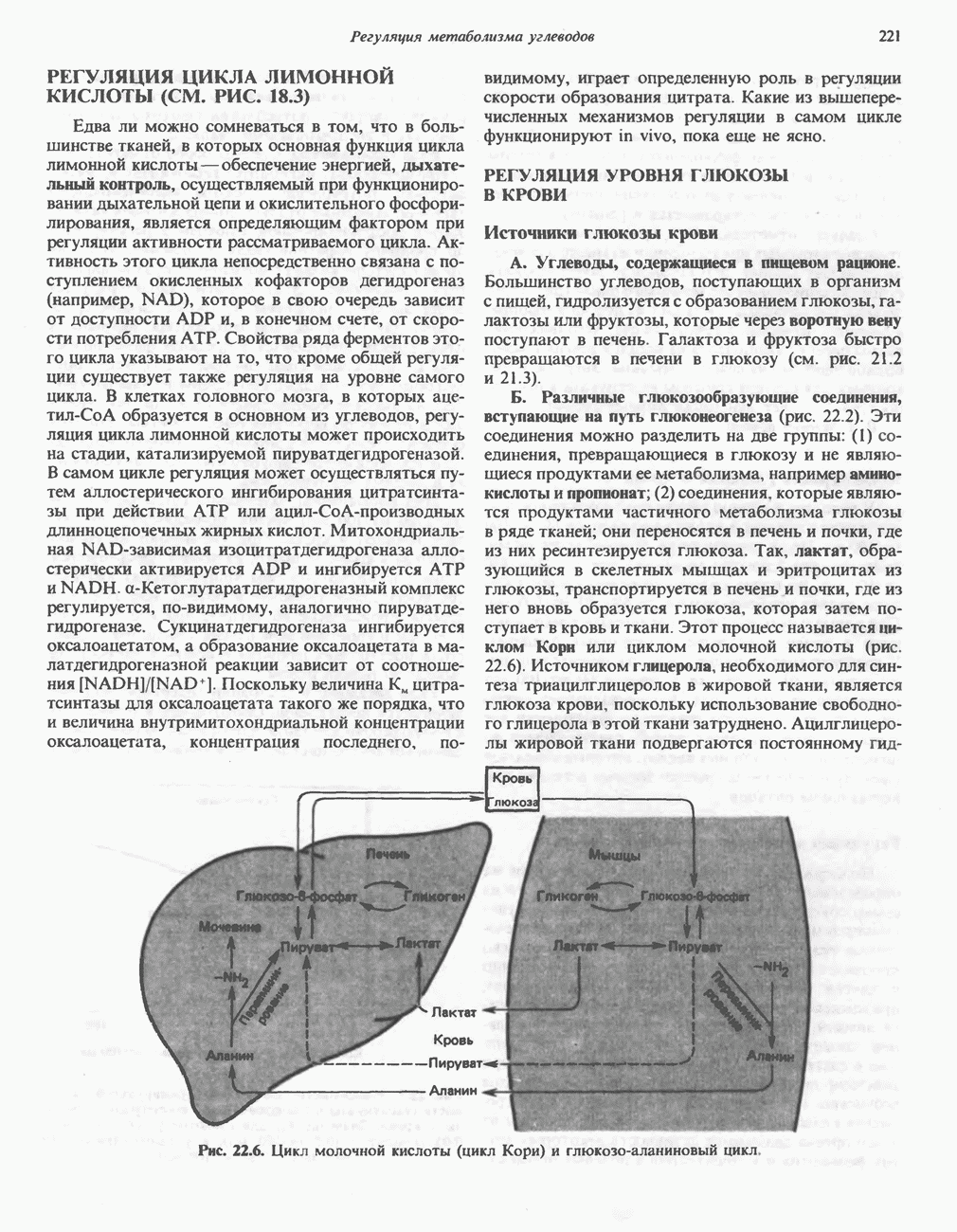





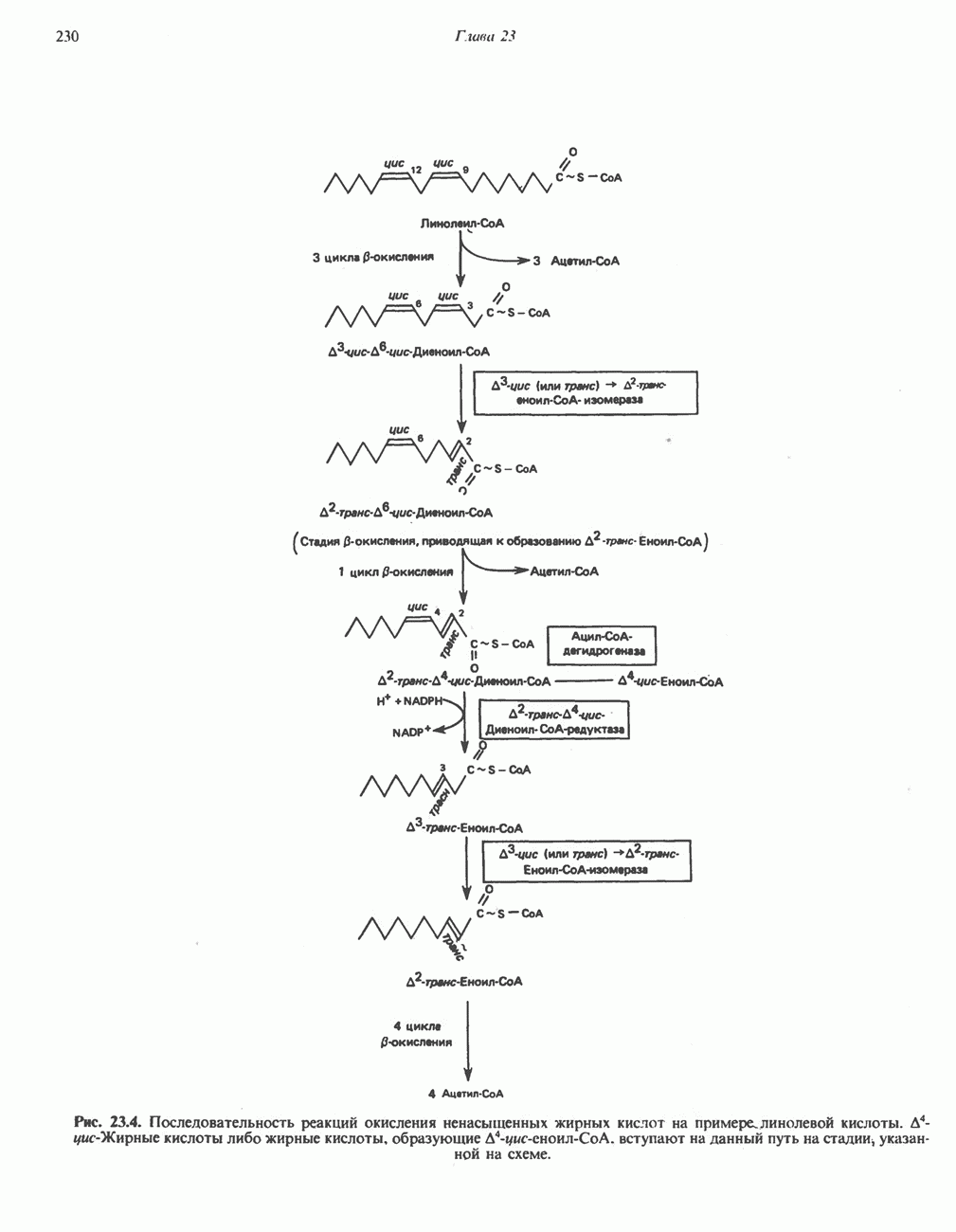

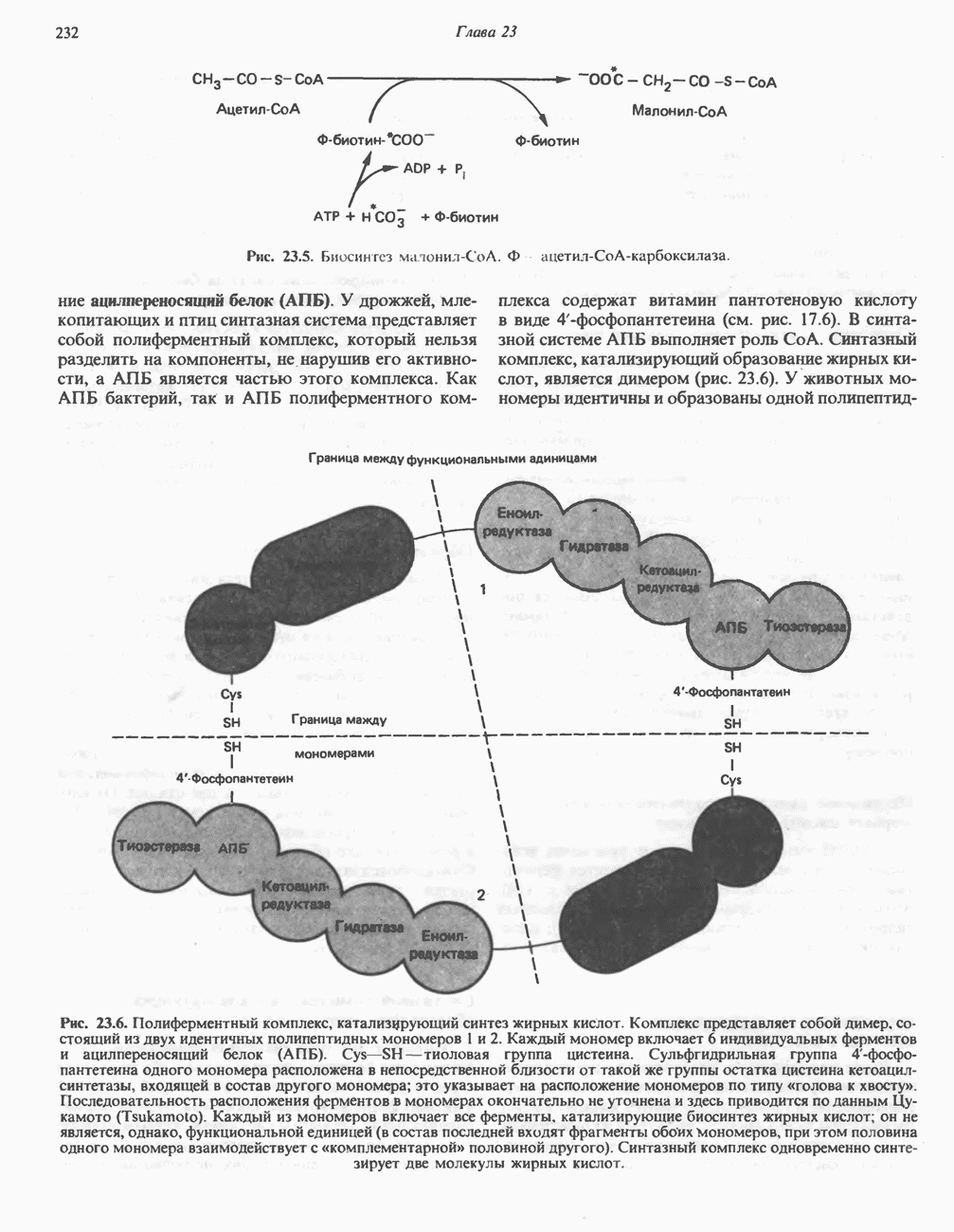
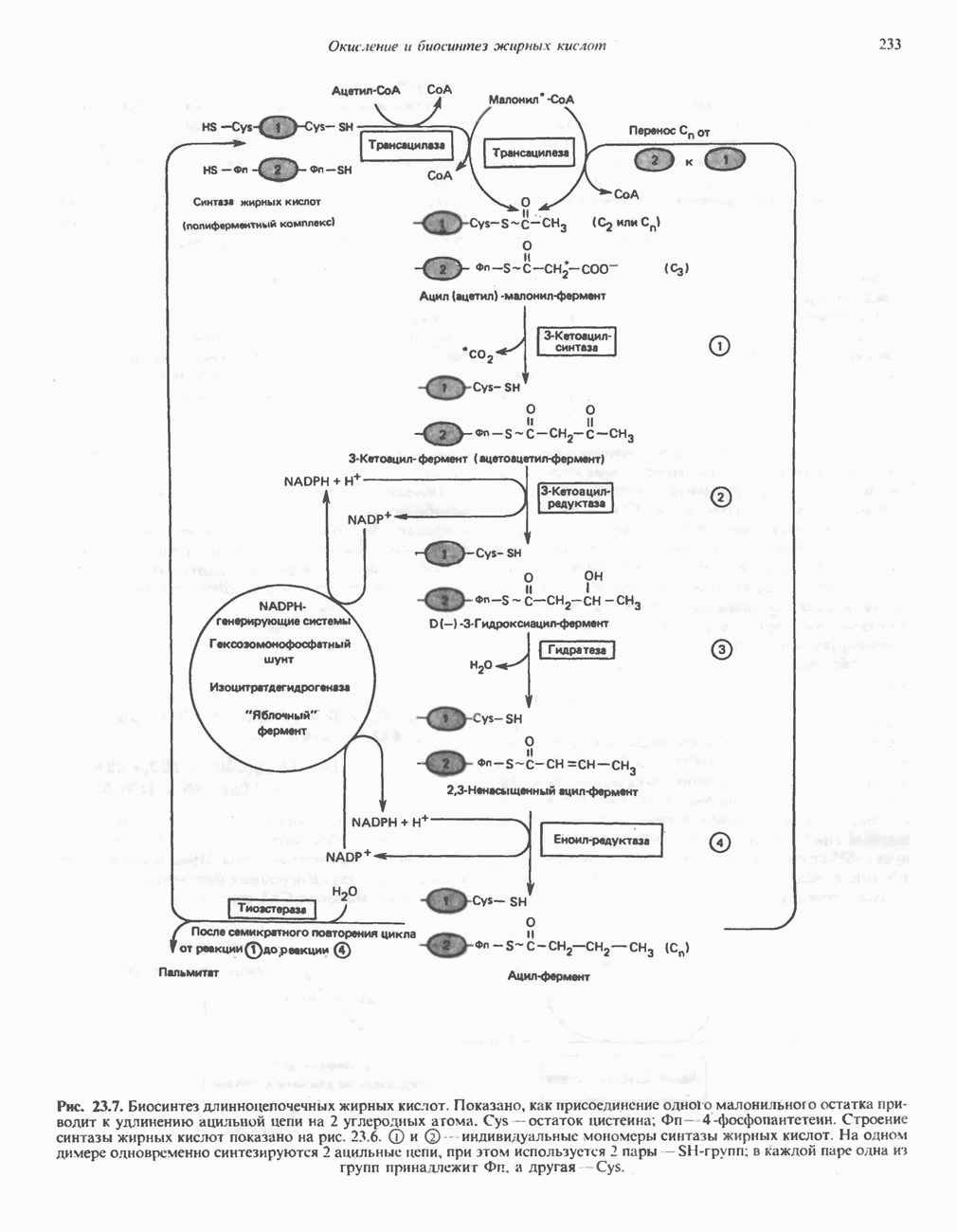

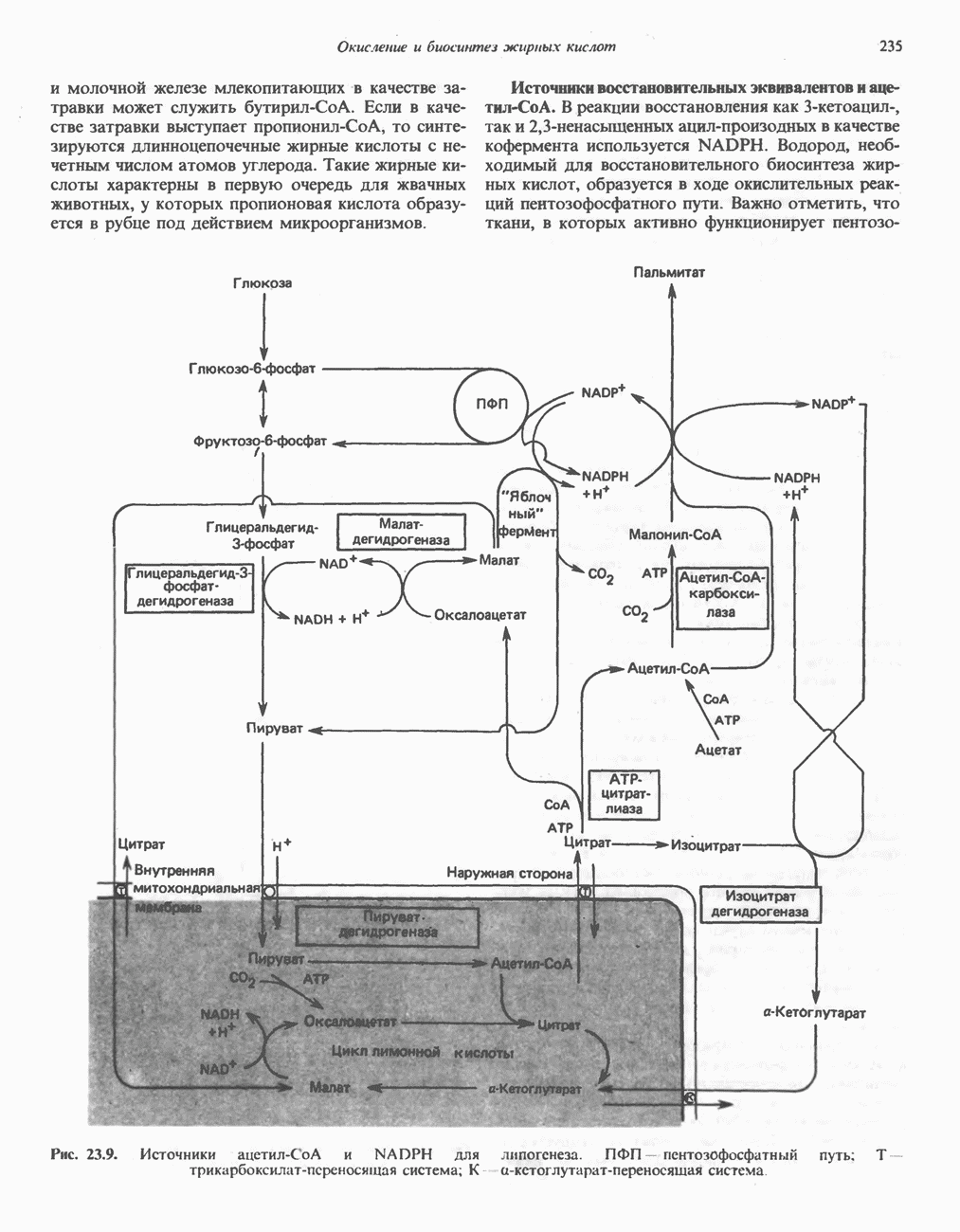



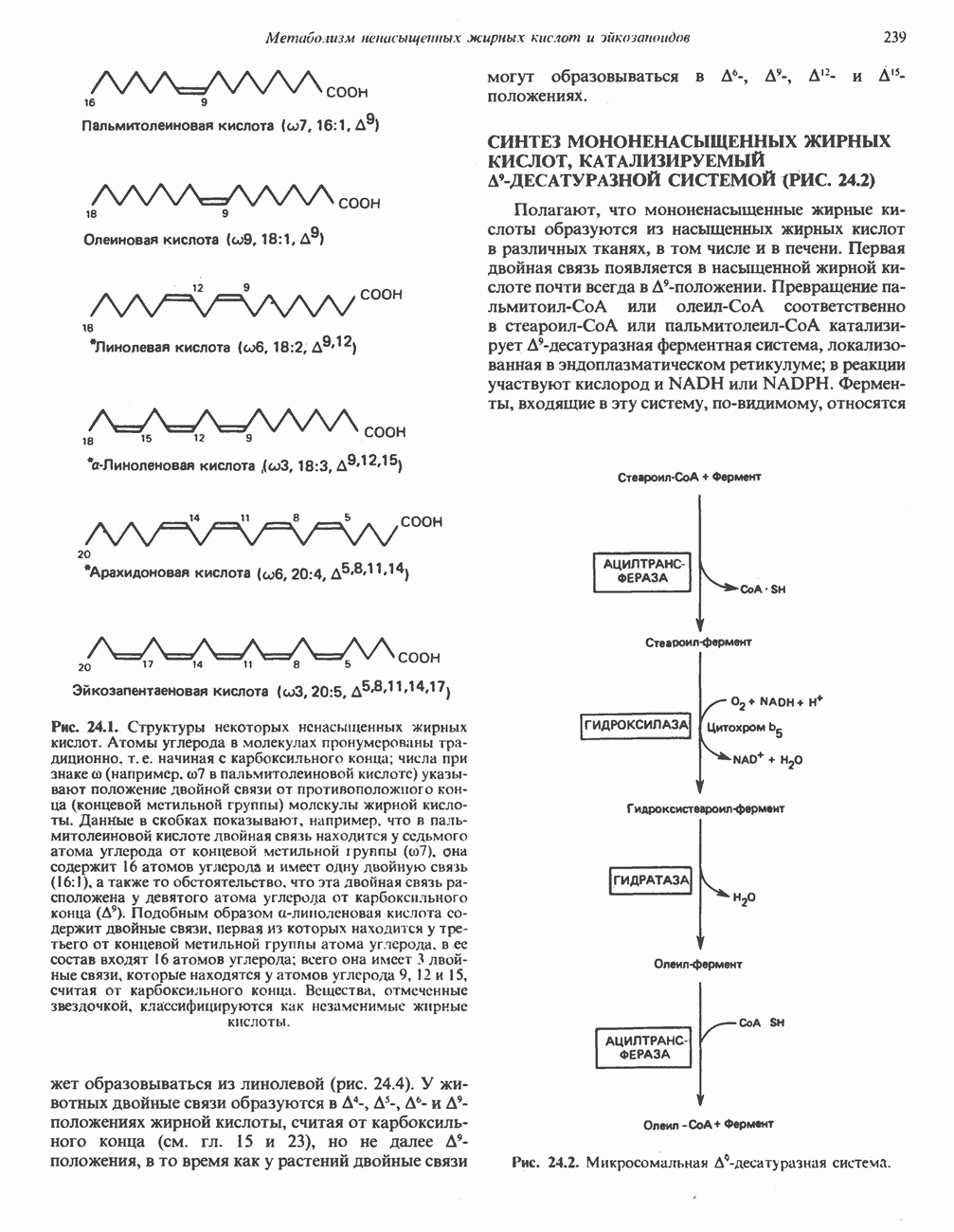
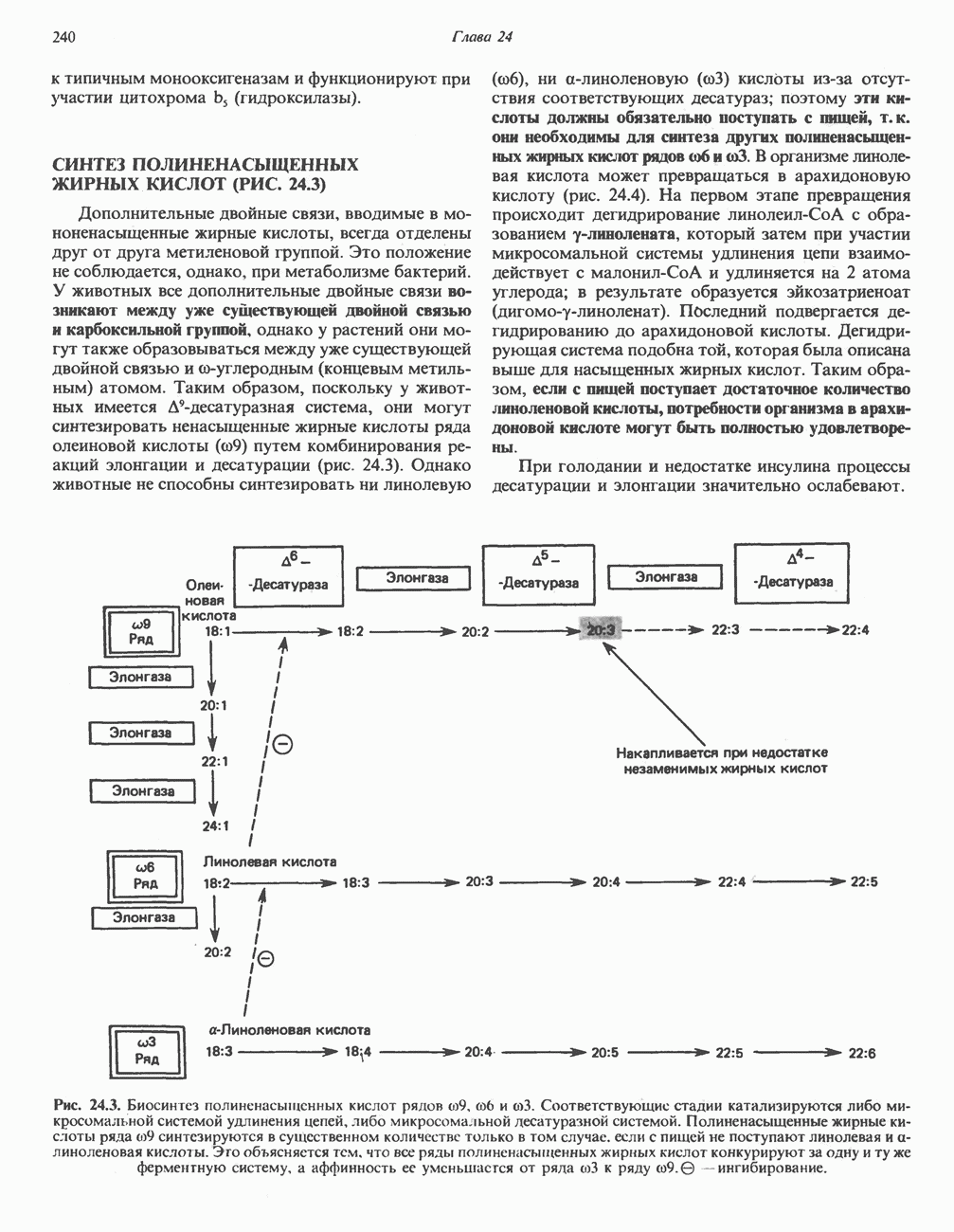
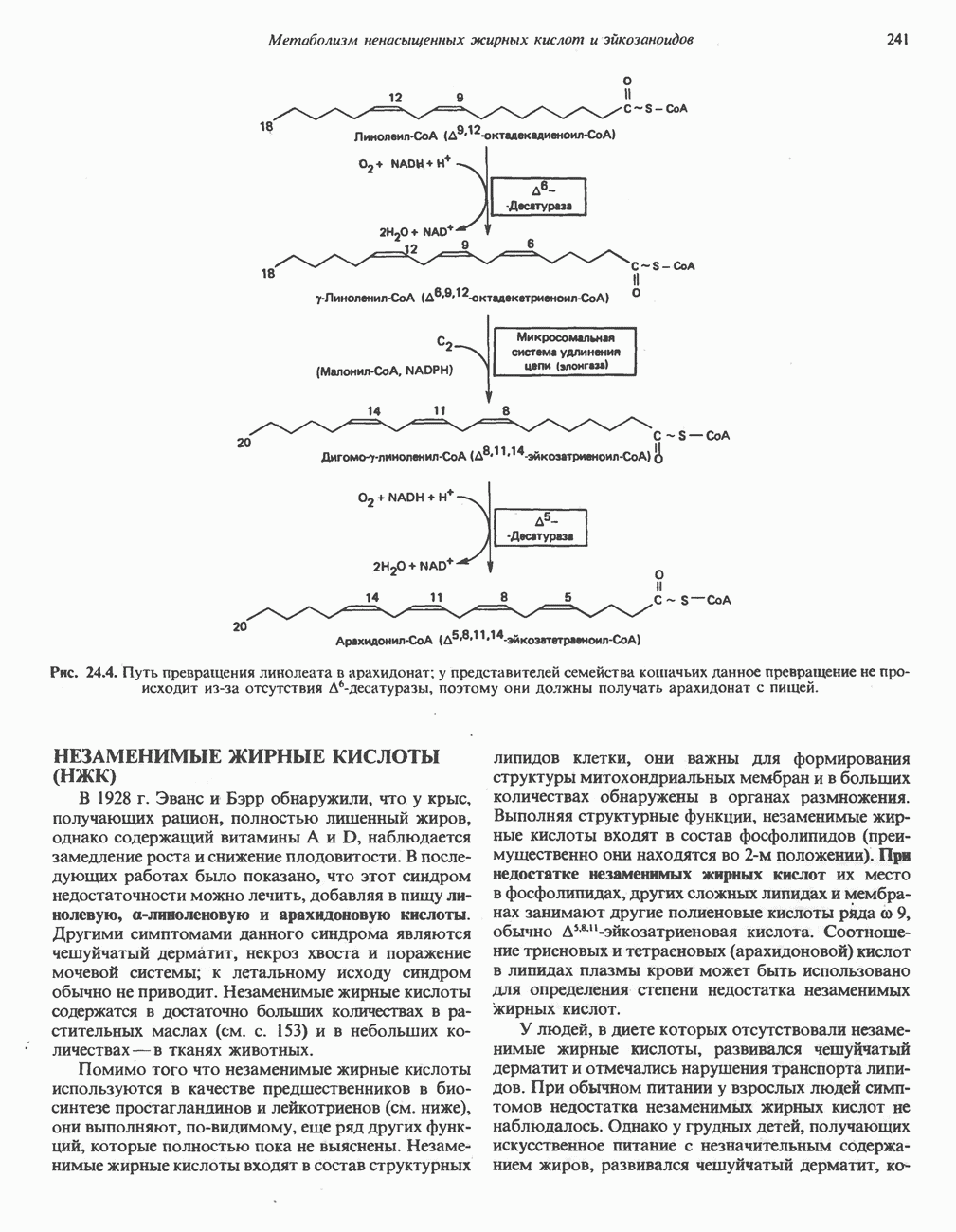

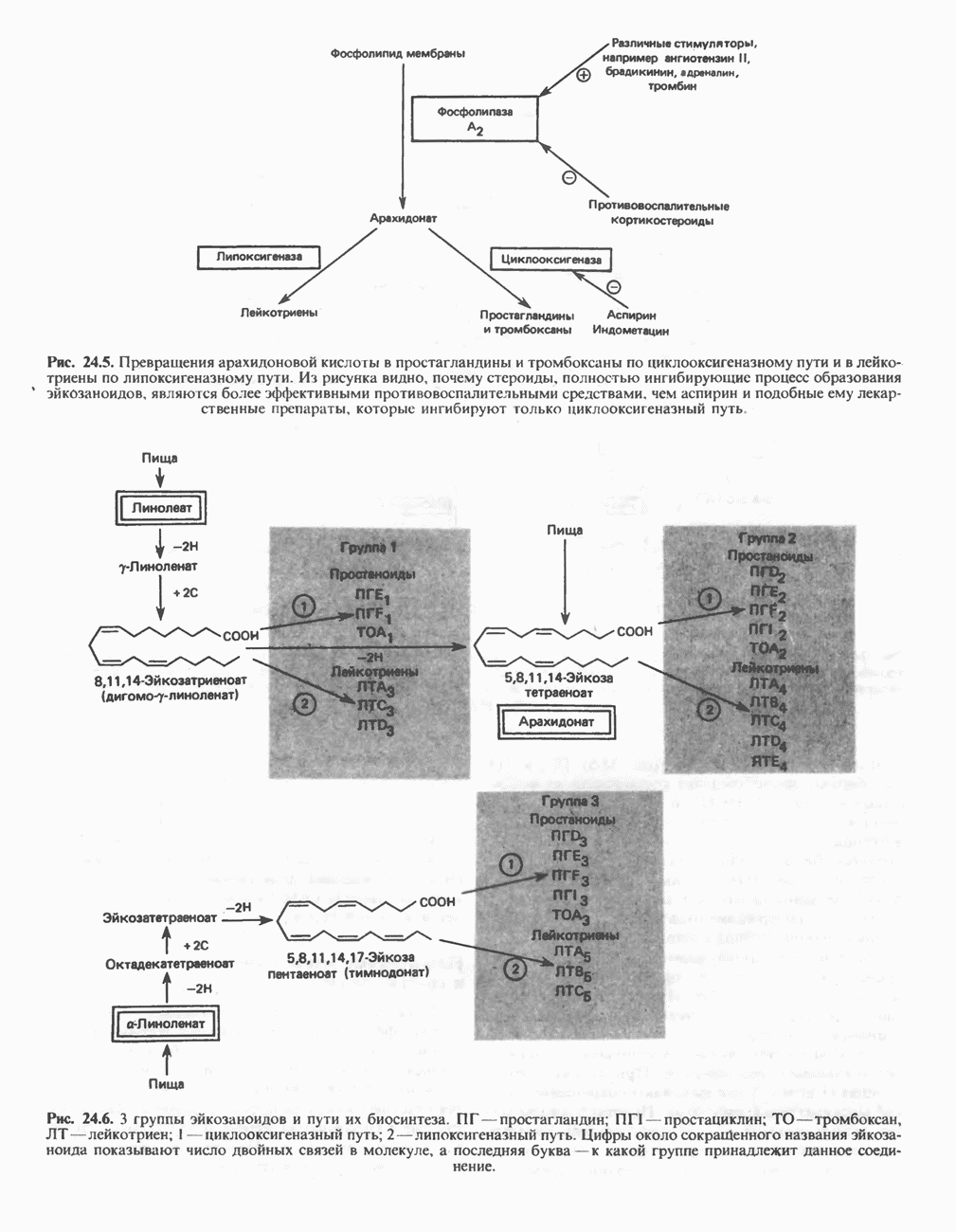
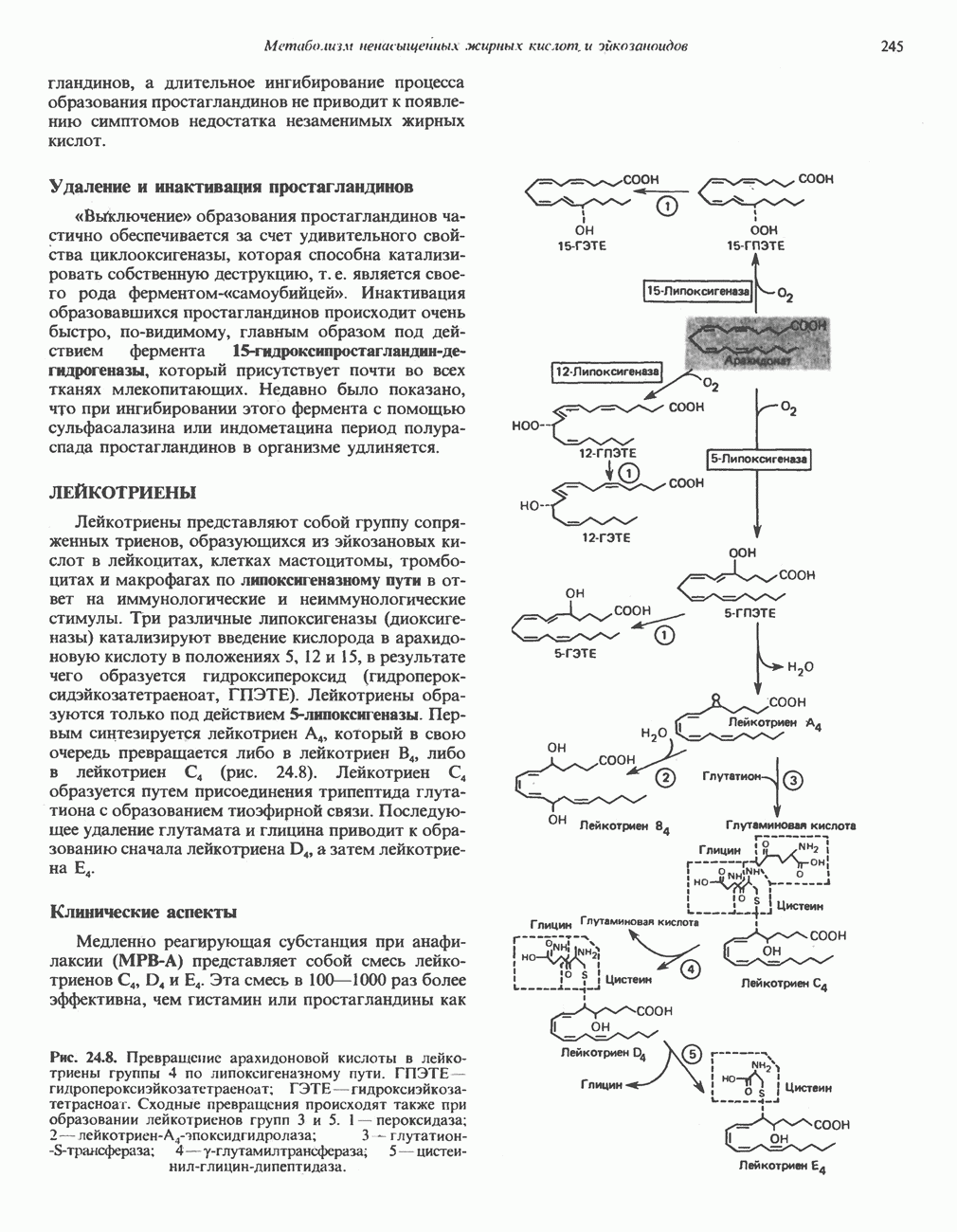





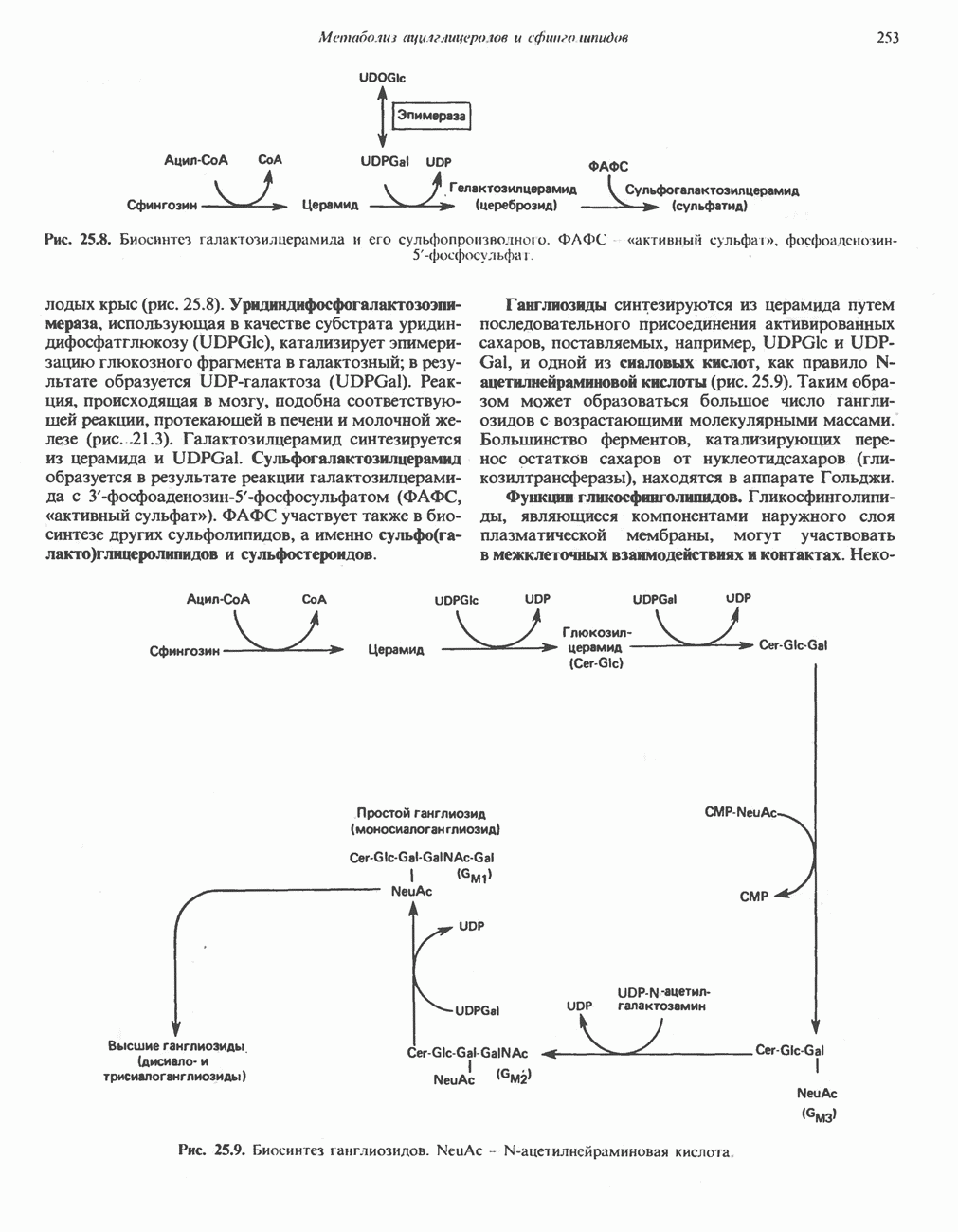
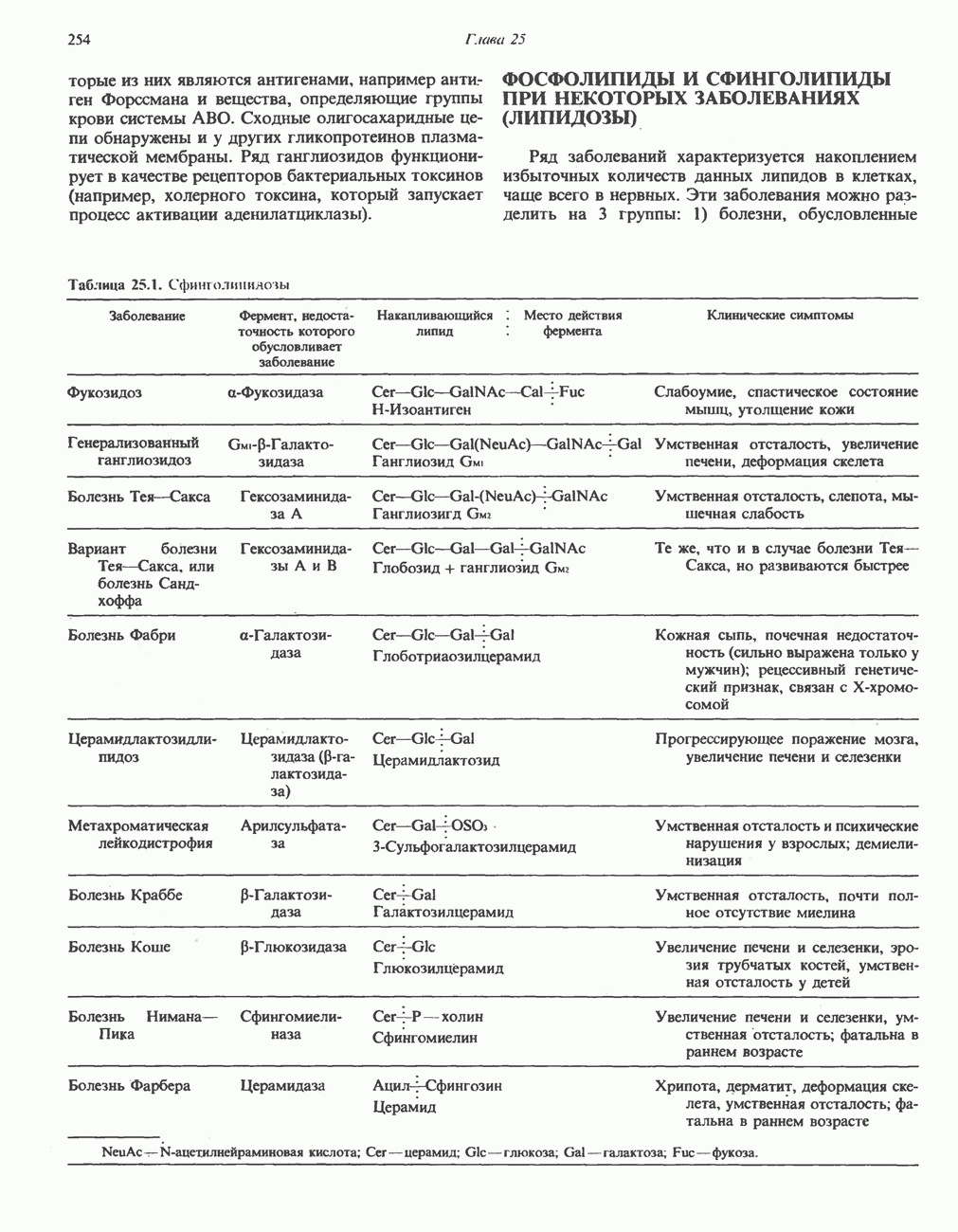


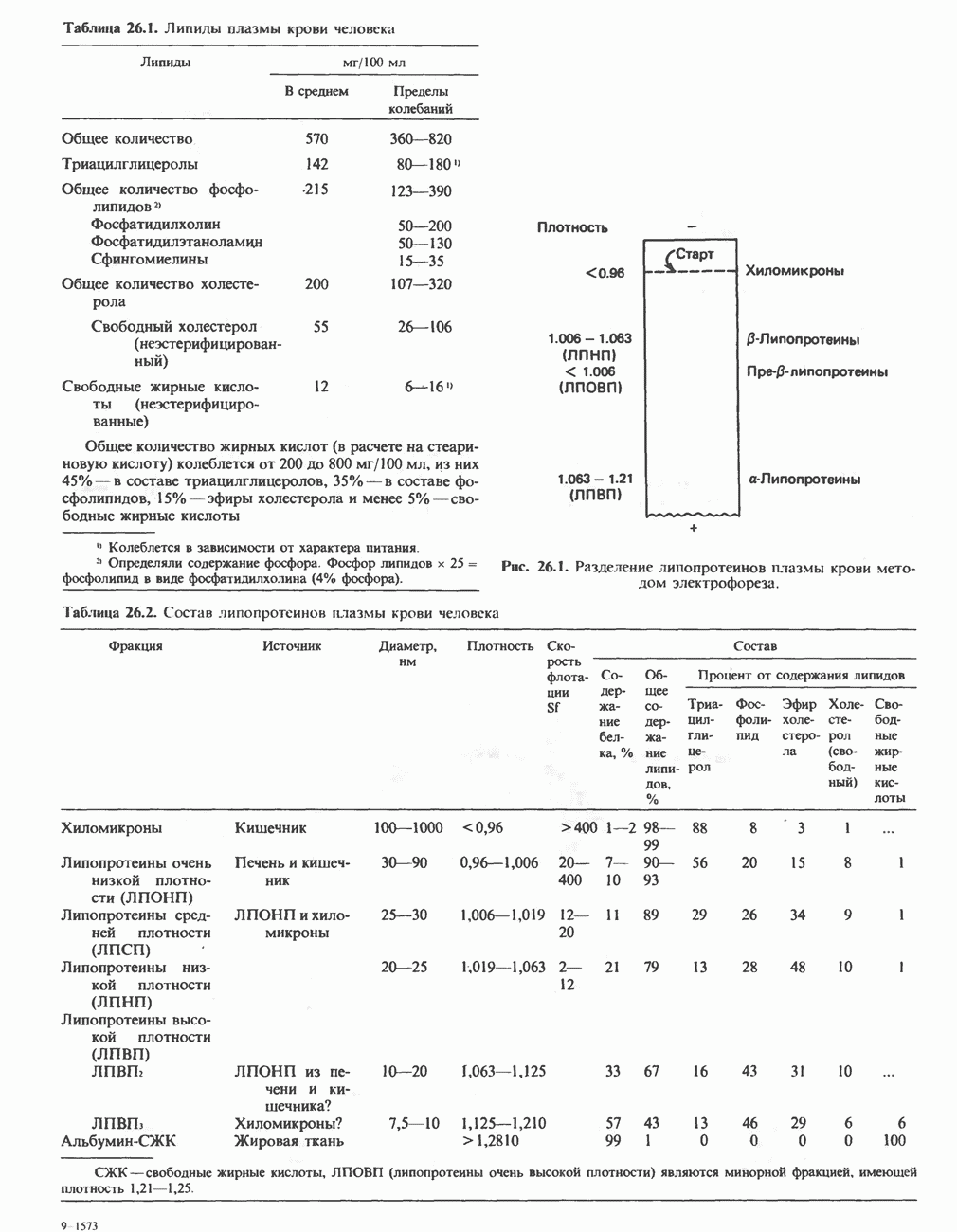
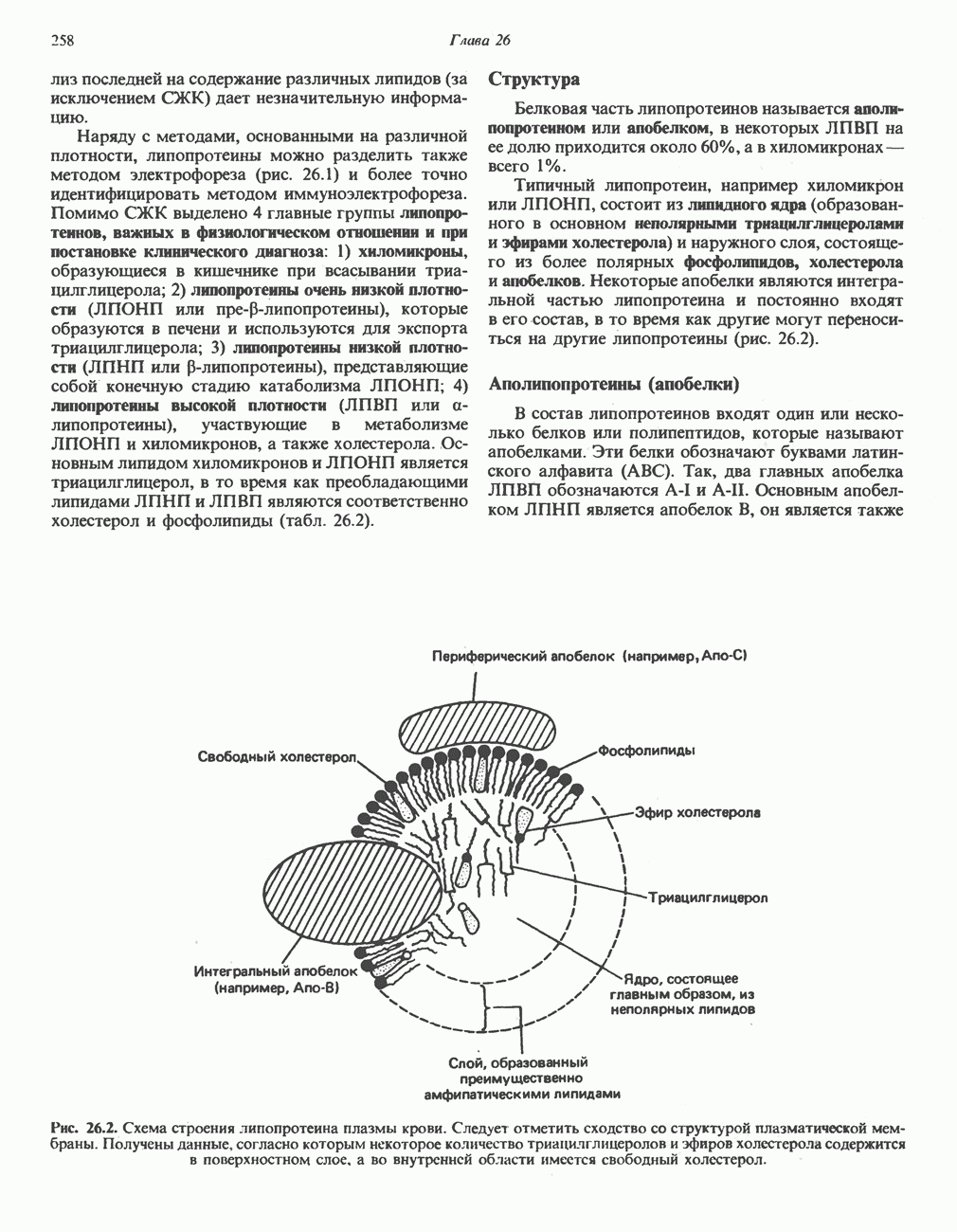

















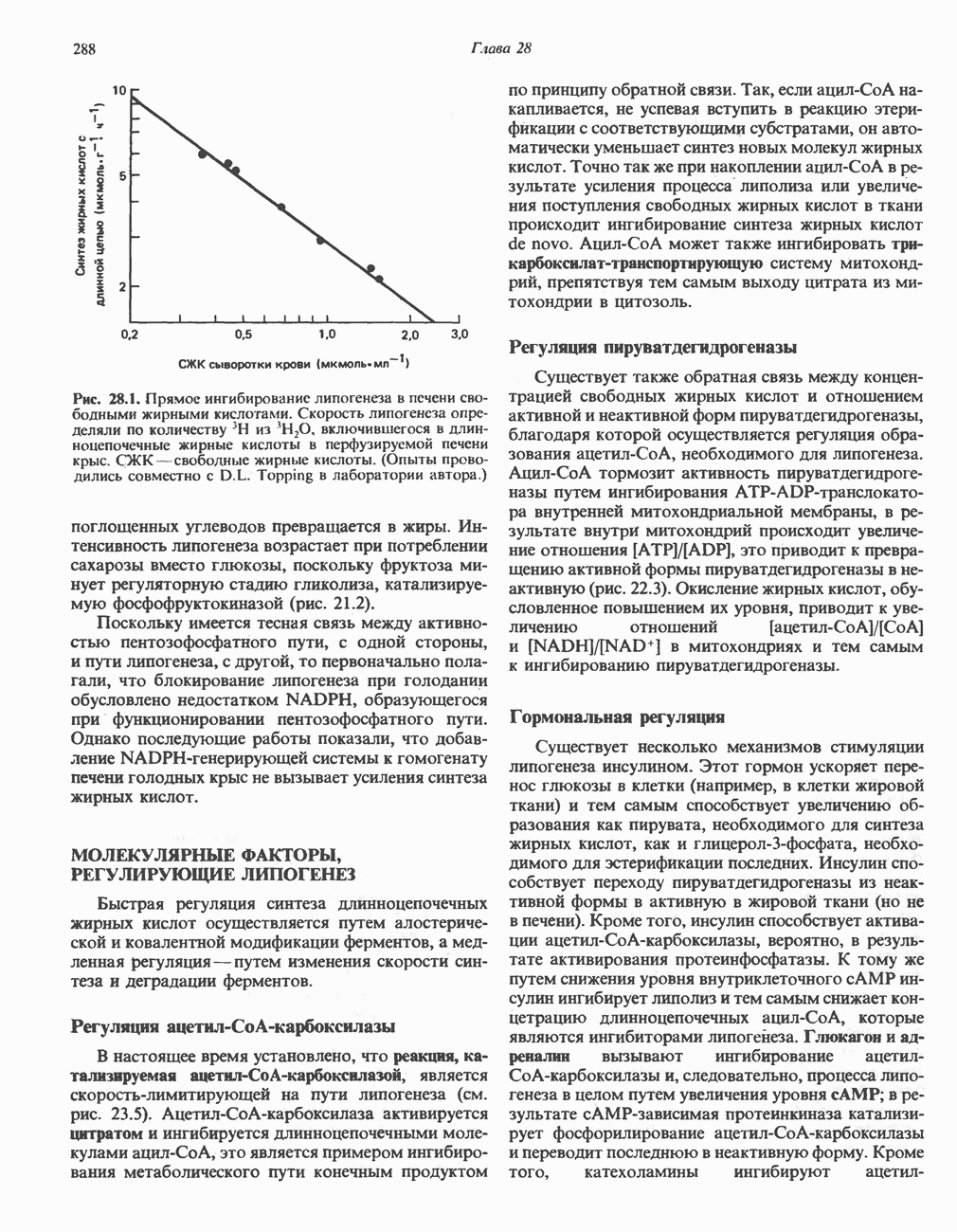

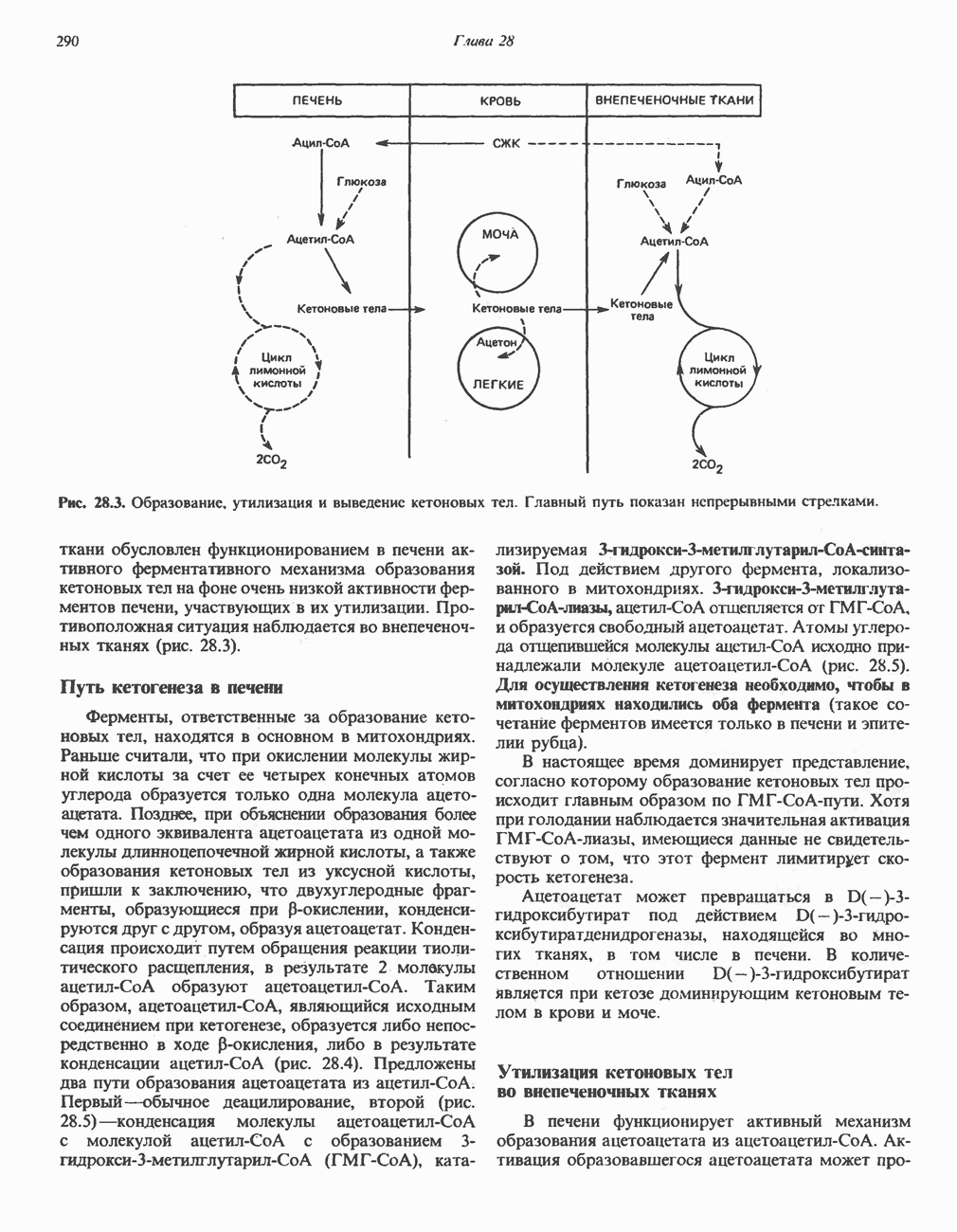
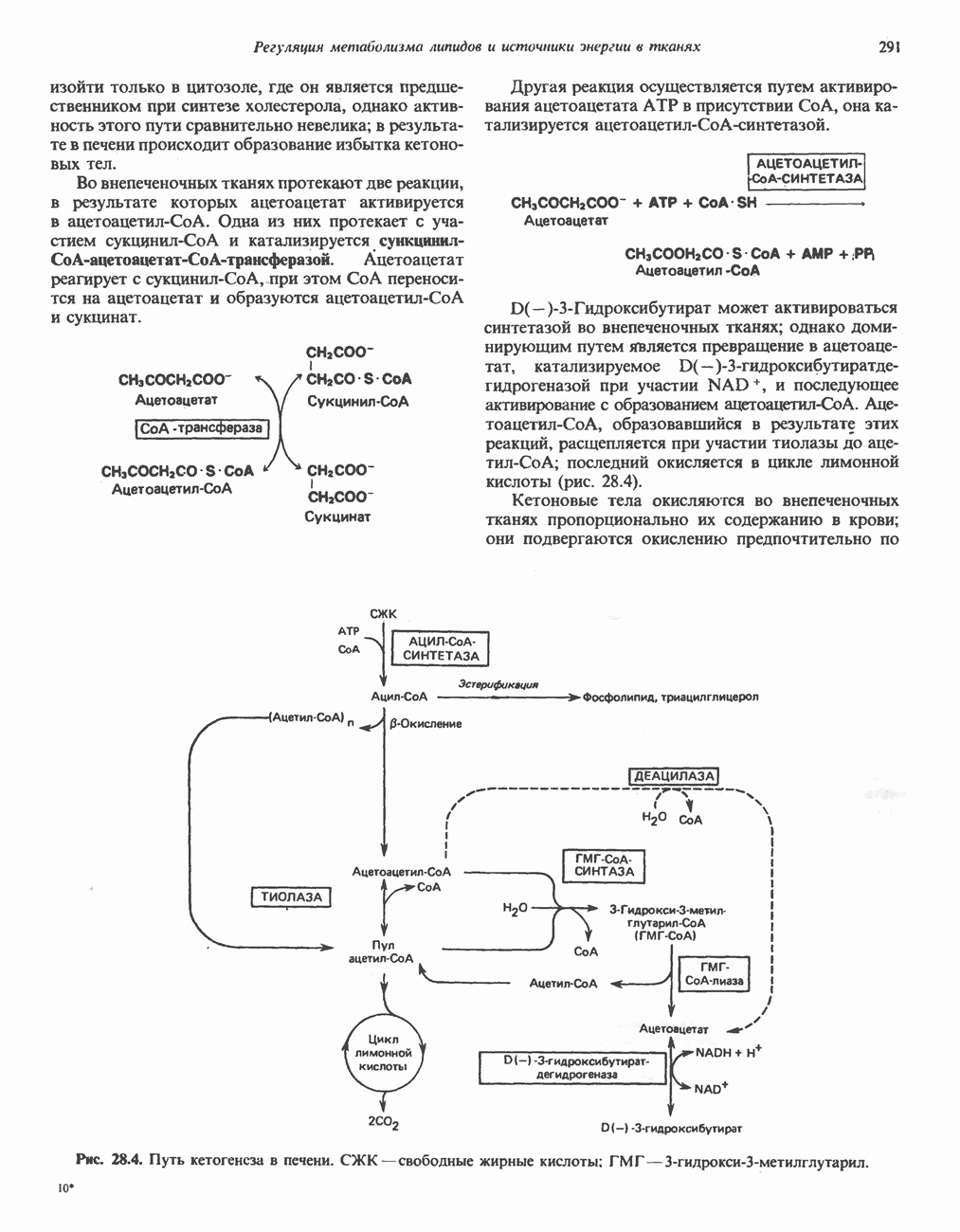



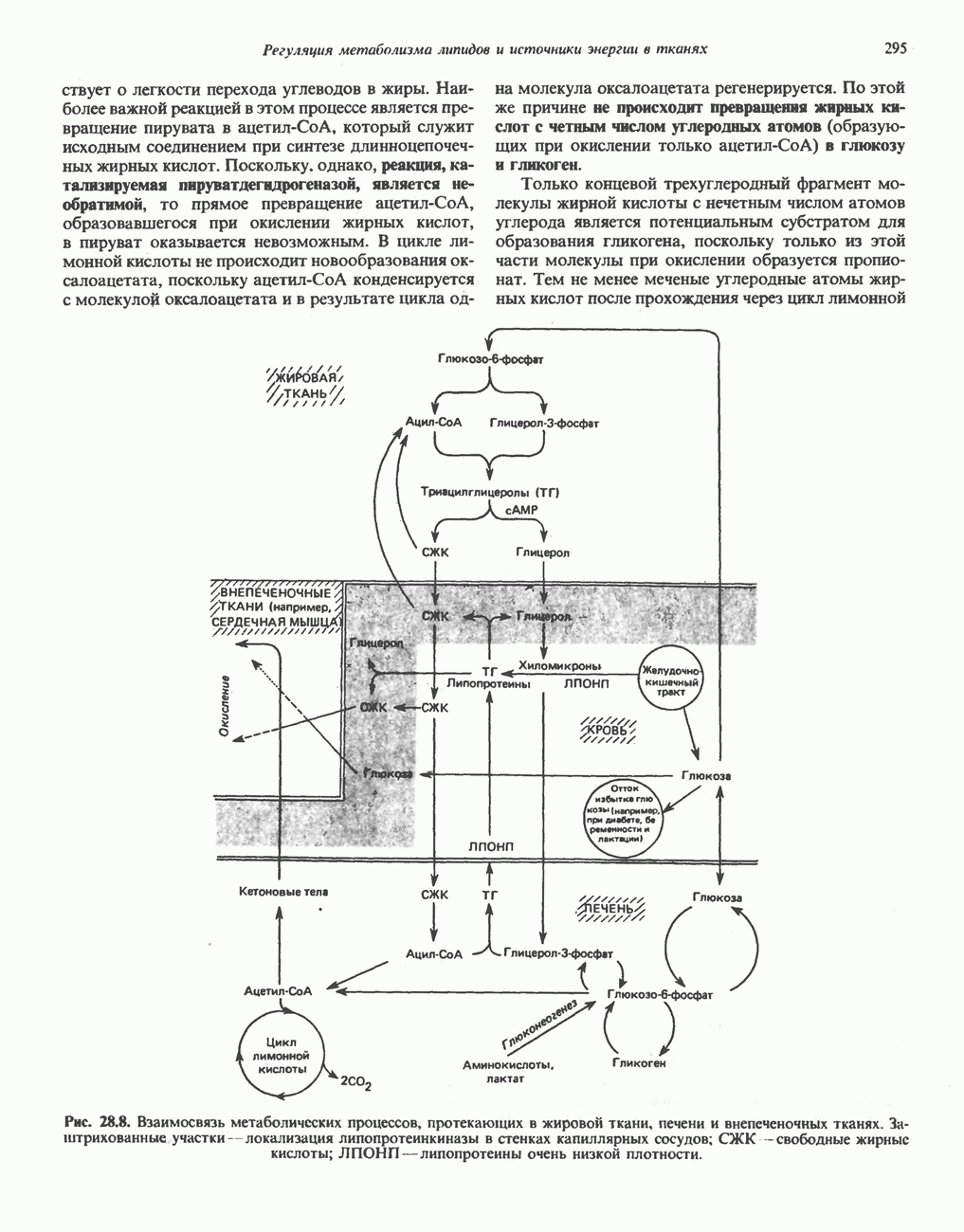



















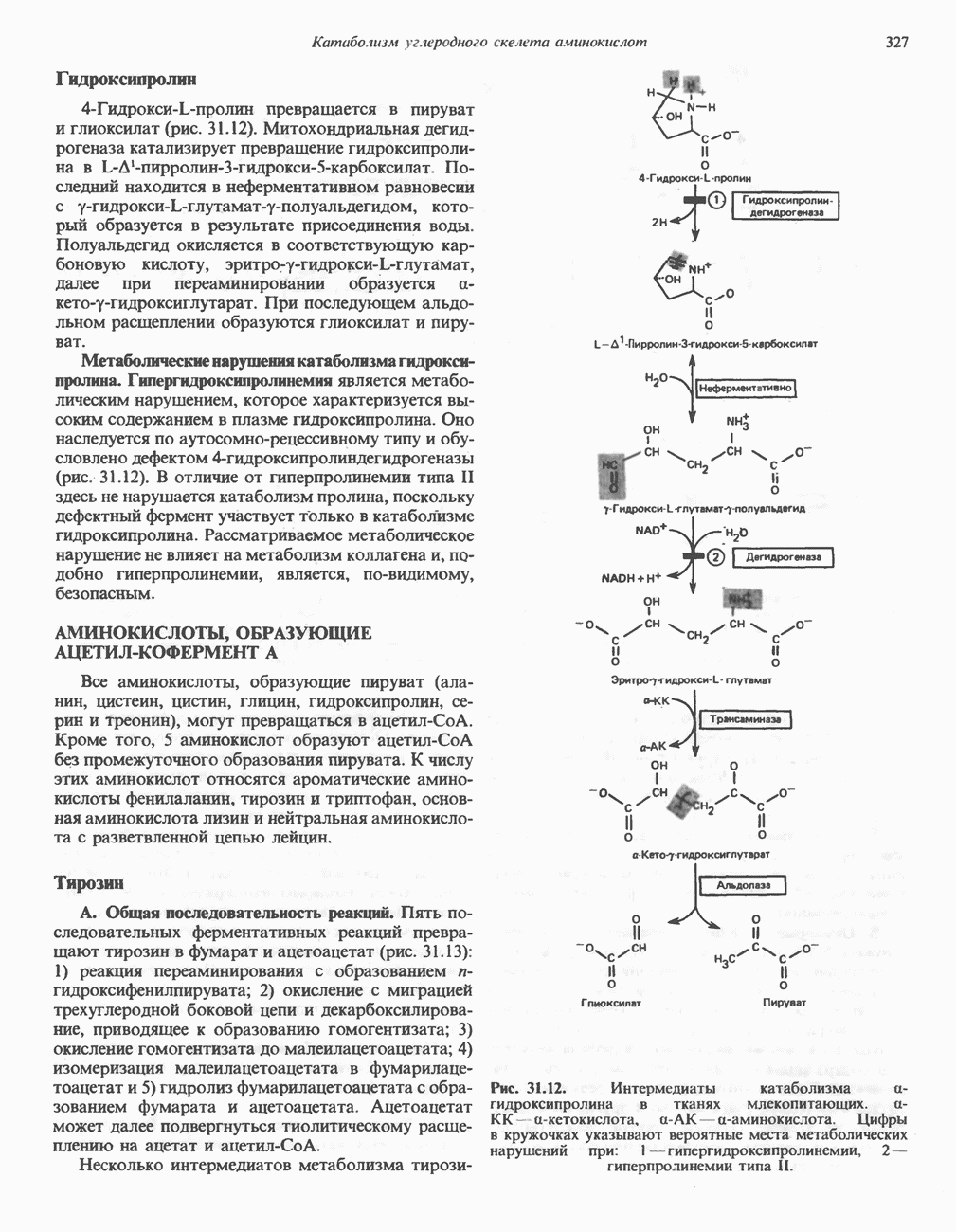

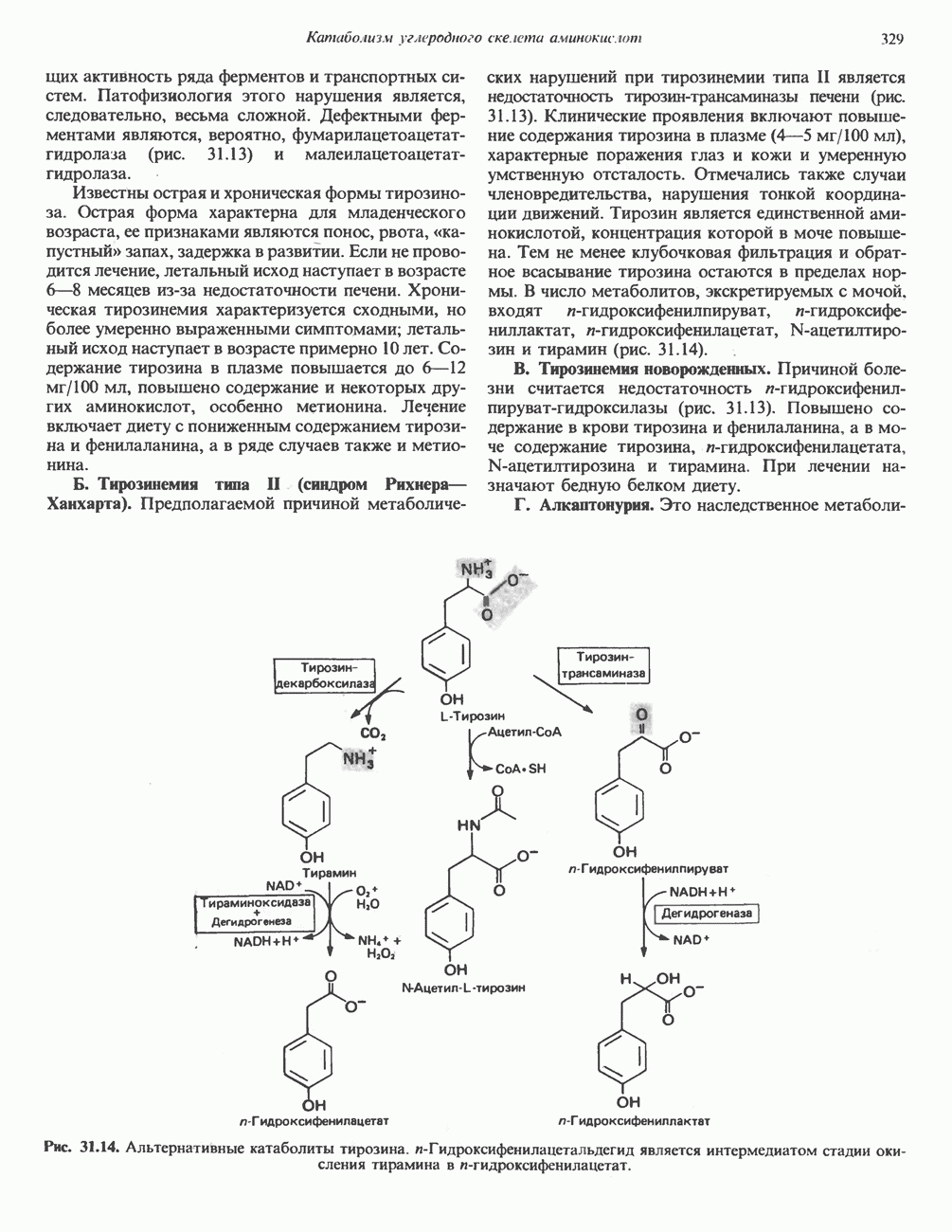



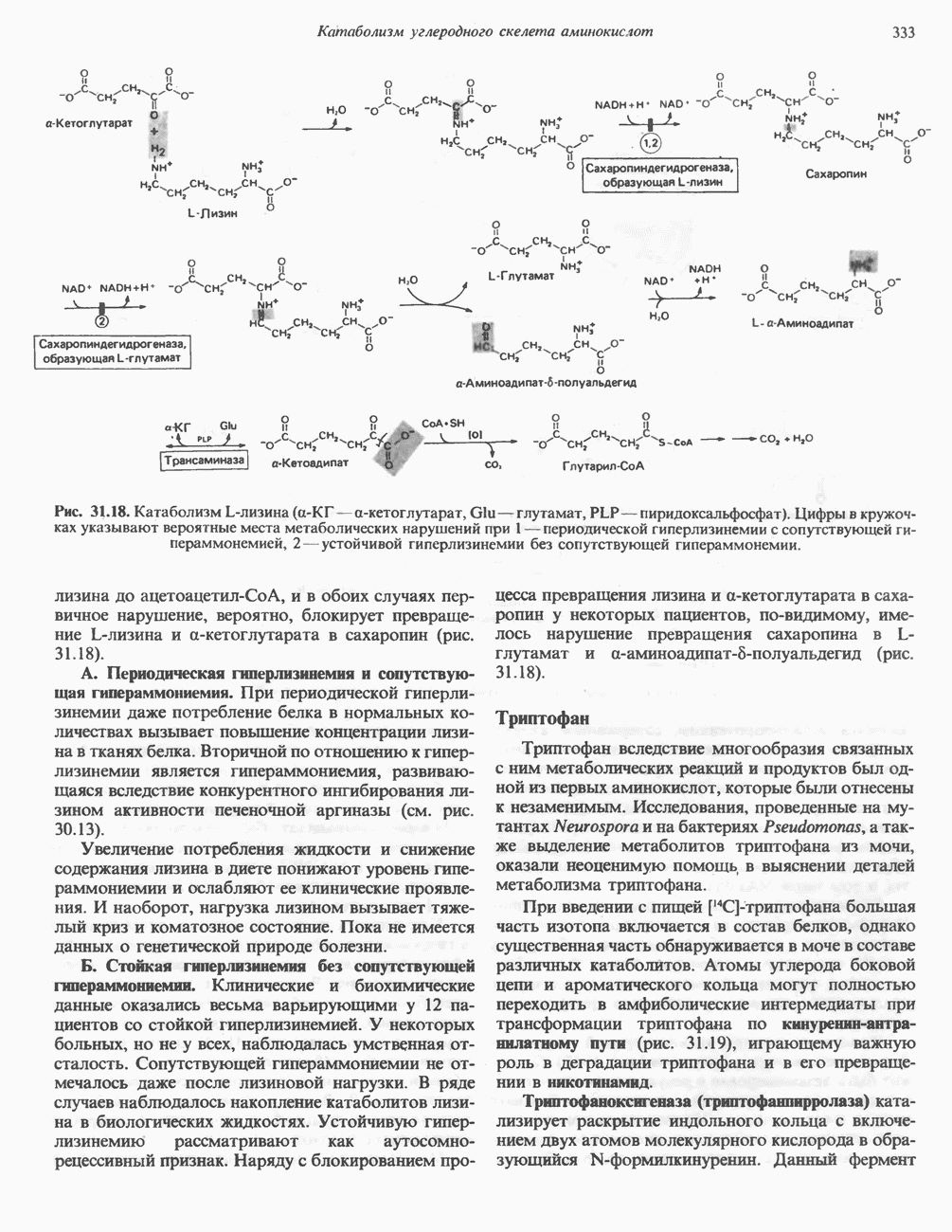
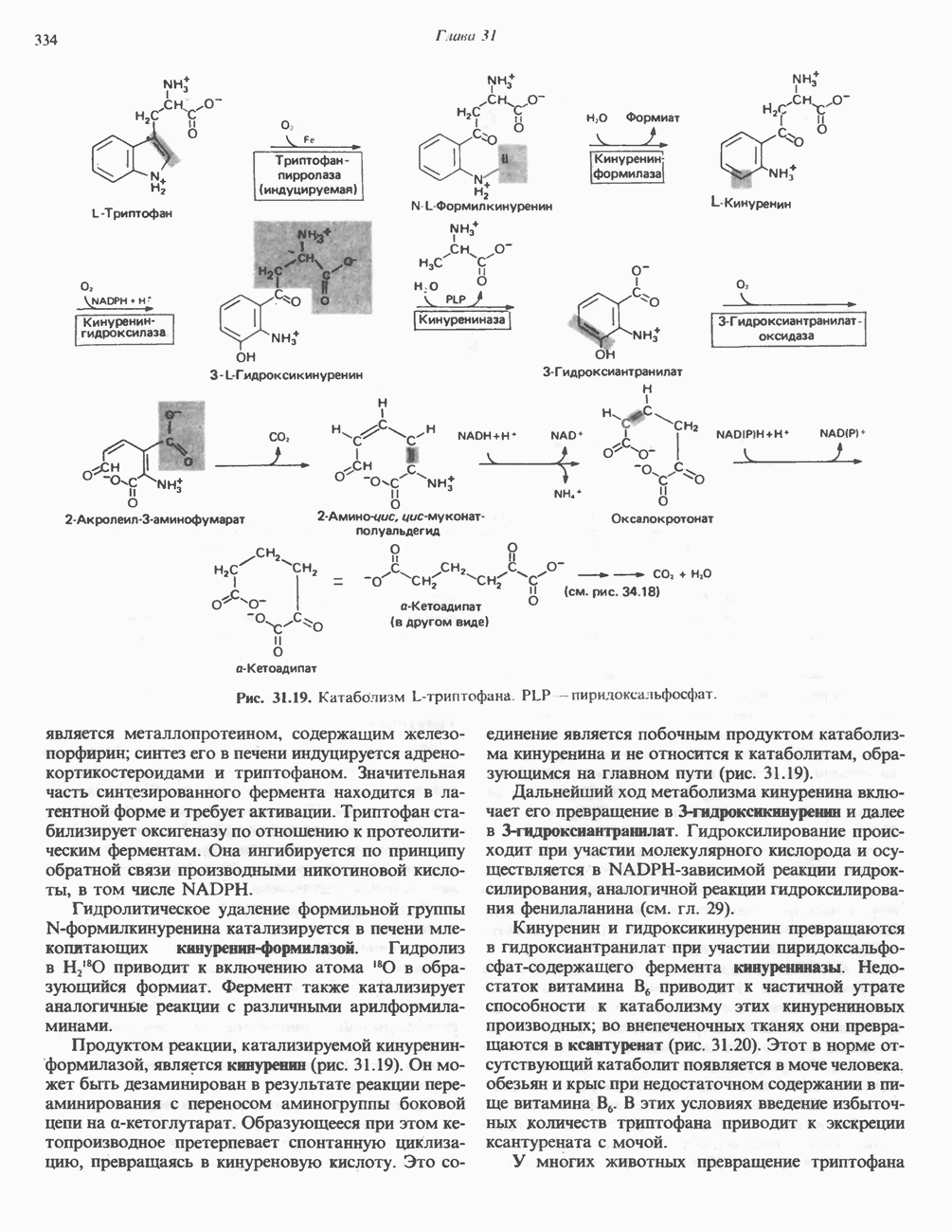























 из легких к периферическим тканям;
из легких к периферическим тканям;
 и протоны от периферических тканей к дыхательным органам для последующего выведения из организма. Сравнительная биохимия гемоглобинов чрезвычайно интересна сама по себе, однако мы здесь сосредоточим внимание только на гемоглобинах человека.
и протоны от периферических тканей к дыхательным органам для последующего выведения из организма. Сравнительная биохимия гемоглобинов чрезвычайно интересна сама по себе, однако мы здесь сосредоточим внимание только на гемоглобинах человека.
 , и т. д.). В состав молекулы входят по две цепи двух разных типов. Длина
, и т. д.). В состав молекулы входят по две цепи двух разных типов. Длина
 -цепей примерно одинакова - a-цепь содержит 141 остаток, а Р-цепь—146; однако а- и (
-цепей примерно одинакова - a-цепь содержит 141 остаток, а Р-цепь—146; однако а- и ( -полипептиды гемоглобина А (НЬА) кодируются разными генами и имеют разную первичную структуру. В то же время первичная структура
-полипептиды гемоглобина А (НЬА) кодируются разными генами и имеют разную первичную структуру. В то же время первичная структура  и
и  -цепей гемоглобина человека в значительной степени консервативна.
-цепей гемоглобина человека в значительной степени консервативна.
 , они имеют почти идентичную вторичную и третичную структуру. Это поразительное сходство, которое распространяется на расположение гема и восьми спиральных участков, частично обусловлено тем, что в эквивалентных положениях первичной структуры миоглобина и Р-субъединицы
, они имеют почти идентичную вторичную и третичную структуру. Это поразительное сходство, которое распространяется на расположение гема и восьми спиральных участков, частично обусловлено тем, что в эквивалентных положениях первичной структуры миоглобина и Р-субъединицы  находятся хотя и различающиеся, но сходные по своим свойствам аминокислоты. а-Полипептид также весьма сходен с миоглобином, хотя в нем содержится семь, а не восемь спиралей. Как и в миоглобине, гидрофобные остатки у него размещаются внутри структуры, а гидрофильные (опять-таки за исключением двух остатков гистидина) - на поверхности; это в одинаковой мере свойственно и
находятся хотя и различающиеся, но сходные по своим свойствам аминокислоты. а-Полипептид также весьма сходен с миоглобином, хотя в нем содержится семь, а не восемь спиралей. Как и в миоглобине, гидрофобные остатки у него размещаются внутри структуры, а гидрофильные (опять-таки за исключением двух остатков гистидина) - на поверхности; это в одинаковой мере свойственно и  и Р-субъединицам.
и Р-субъединицам.
 (нормальный гемоглобин взрослого человека) -
(нормальный гемоглобин взрослого человека) -  (фетальный гемоглобин) -
(фетальный гемоглобин) -  (гемоглобин при серповидноклеточной
(гемоглобин при серповидноклеточной  (минорный гемоглобин взрослого человека) -
(минорный гемоглобин взрослого человека) -  . Четвертичная структура наделяет гемоглобин дополнительными важными особенностями (отсутствующими у миоглобина), которые способствуют выполнению гемоглобином его уникальной биологической функции и обеспечивают возможность строгой регуляции его свойств. Гемоглобин обладает аллостерическими свойствами (от треч. аллос — другой, стерос — место, пространство), и на его примере можно лучше понять свойства других аллостерических белков.
. Четвертичная структура наделяет гемоглобин дополнительными важными особенностями (отсутствующими у миоглобина), которые способствуют выполнению гемоглобином его уникальной биологической функции и обеспечивают возможность строгой регуляции его свойств. Гемоглобин обладает аллостерическими свойствами (от треч. аллос — другой, стерос — место, пространство), и на его примере можно лучше понять свойства других аллостерических белков.
 зависит от того, содержатся ли в данном тетрамере другие молекулы 02.
зависит от того, содержатся ли в данном тетрамере другие молекулы 02.

 мм рт. ст. Из рисунка видно, что ассоциация цепей с образованием теграмерной структуры приводит к существенному повышению эффективности снабжения тканей кислородом по сравнению с мономерными белками. (Изолированные цепи гемоглобина обладают примерно таким же сродством к кислороду, что и миоглобин, и характеризуются аналогичной гиперболической кривой насыщения.) (Из работы Stanbury J. В., Wyngaardcn J.B., Fredrickson D. S. (editors): The Metabolic Basis of Inherited Diseases. 4th ed. McGraw-Hill, 1978, с изменениями.)
мм рт. ст. Из рисунка видно, что ассоциация цепей с образованием теграмерной структуры приводит к существенному повышению эффективности снабжения тканей кислородом по сравнению с мономерными белками. (Изолированные цепи гемоглобина обладают примерно таким же сродством к кислороду, что и миоглобин, и характеризуются аналогичной гиперболической кривой насыщения.) (Из работы Stanbury J. В., Wyngaardcn J.B., Fredrickson D. S. (editors): The Metabolic Basis of Inherited Diseases. 4th ed. McGraw-Hill, 1978, с изменениями.)
 мм рт. ст., и какие в тканях, при
мм рт. ст., и какие в тканях, при  мм рт. ст. (рис. 6.8).
мм рт. ст. (рис. 6.8).
 характеризуется величиной
характеризуется величиной  —значением
—значением  при котором наблюдается полунасыщение гемоглобина кислородом. Значение
при котором наблюдается полунасыщение гемоглобина кислородом. Значение  у разных организмов существенно различается, но во всех случаях оно превышает значение
у разных организмов существенно различается, но во всех случаях оно превышает значение  в периферических тканях рассматриваемого организма. Это хорошо иллюстрирует фетальный гемоглобин человека (HBF). Для
в периферических тканях рассматриваемого организма. Это хорошо иллюстрирует фетальный гемоглобин человека (HBF). Для  мм. рт. ст., а для
мм. рт. ст., а для  мм рт. ст. Благодаря этой разнице гемоглобин F отбирает кислород у
мм рт. ст. Благодаря этой разнице гемоглобин F отбирает кислород у  , находящегося в плацентарной крови. Однако после рождения ребенка HBF утрачивает свою функцию; обладая более высоким сродством к 02, он высвобождает меньшее его количество в тканях.
, находящегося в плацентарной крови. Однако после рождения ребенка HBF утрачивает свою функцию; обладая более высоким сродством к 02, он высвобождает меньшее его количество в тканях.
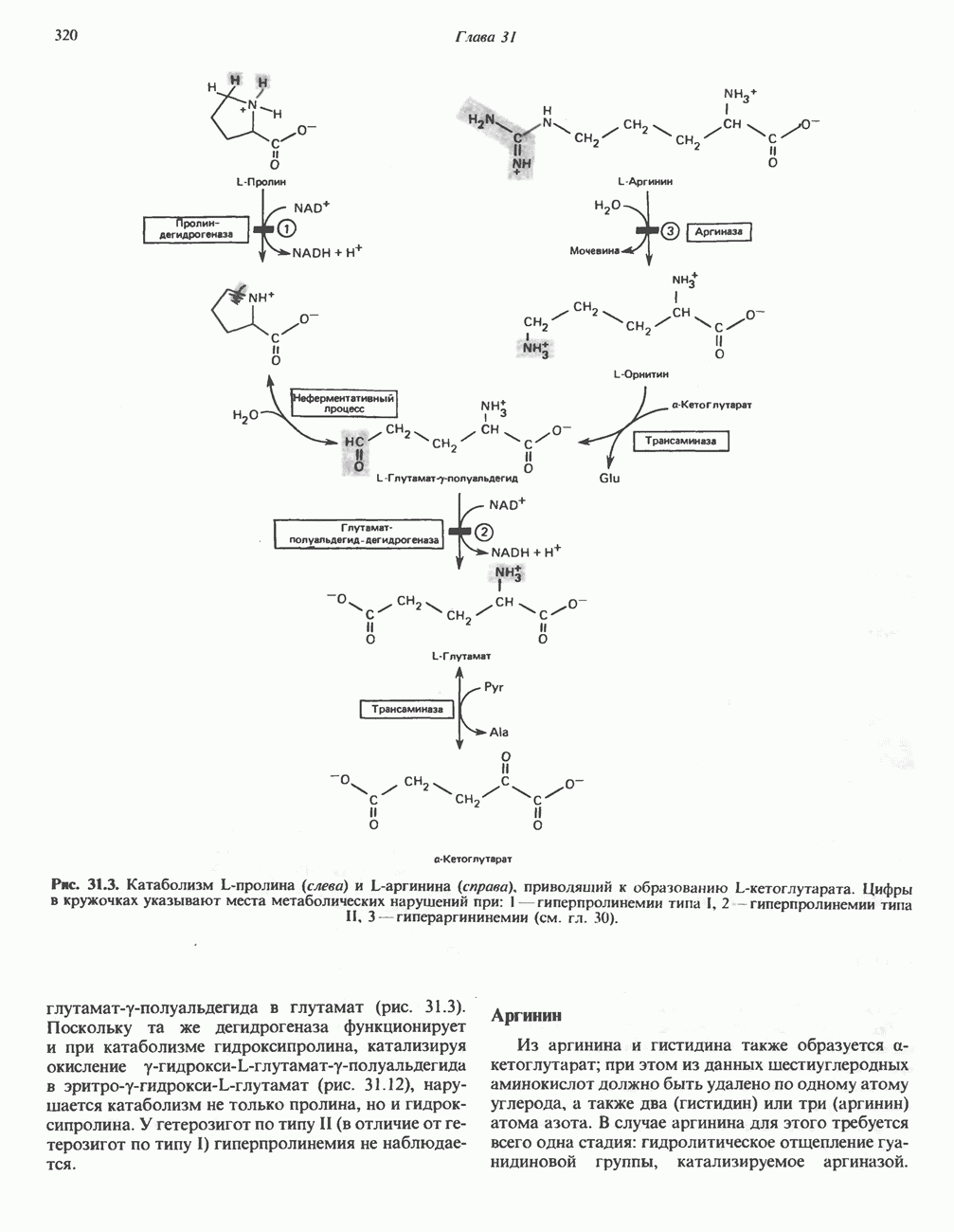
 , поскольку при этом требуется разрыв меньшего числа солевых связей. Указанные изменения заметно влияют на вторичную, третичную и особенно четвертичную структуру гемоглобина. При этом одна
, поскольку при этом требуется разрыв меньшего числа солевых связей. Указанные изменения заметно влияют на вторичную, третичную и особенно четвертичную структуру гемоглобина. При этом одна  -пара субъединиц поворачивается относительно другой
-пара субъединиц поворачивается относительно другой  -пары, что приводит к компактизации тетрамера и повышению сродства гемов к
-пары, что приводит к компактизации тетрамера и повышению сродства гемов к  (рис. 6.10 и 6.11)
(рис. 6.10 и 6.11)
 отвечает R-состояние (relaxed — релаксированное) (рис. 6.12). Термины
отвечает R-состояние (relaxed — релаксированное) (рис. 6.12). Термины  Т-состояния используют для характеристики четвертичной структуры аллостерических ферментов; меньшим сродством к субстрату обладает Т-состояние.
Т-состояния используют для характеристики четвертичной структуры аллостерических ферментов; меньшим сродством к субстрату обладает Т-состояние.
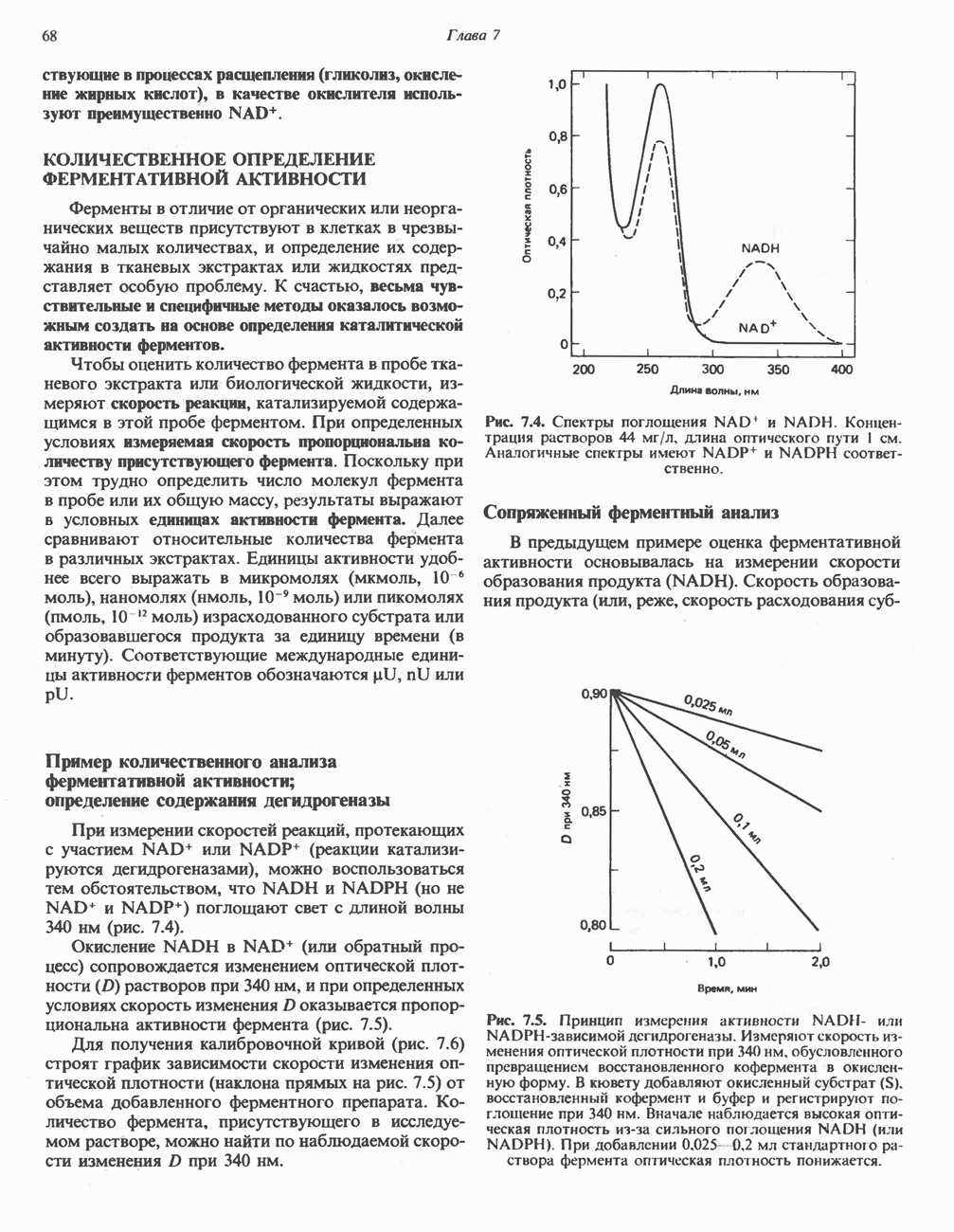
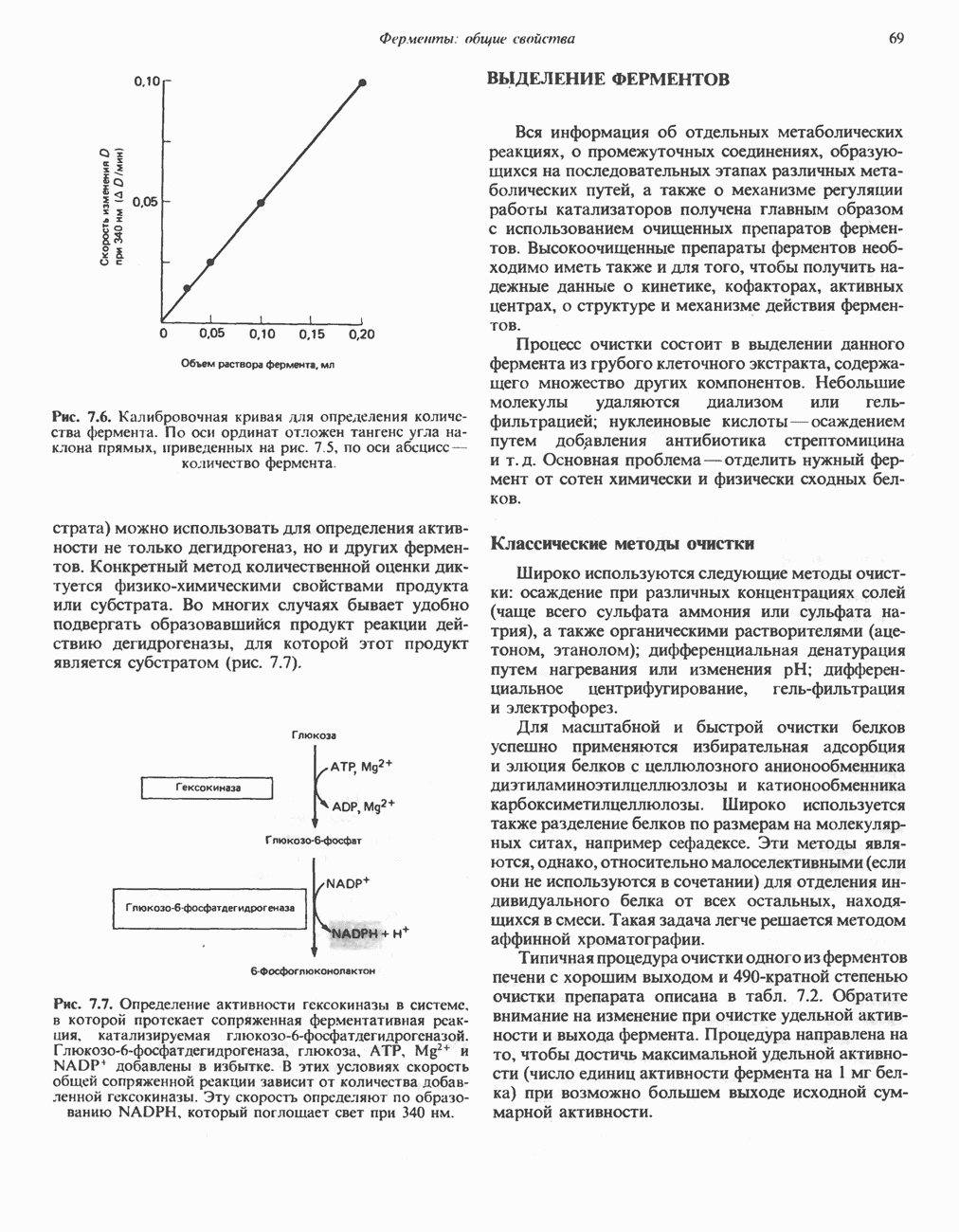
 а также связанные с ним соседние остатки.
а также связанные с ним соседние остатки.

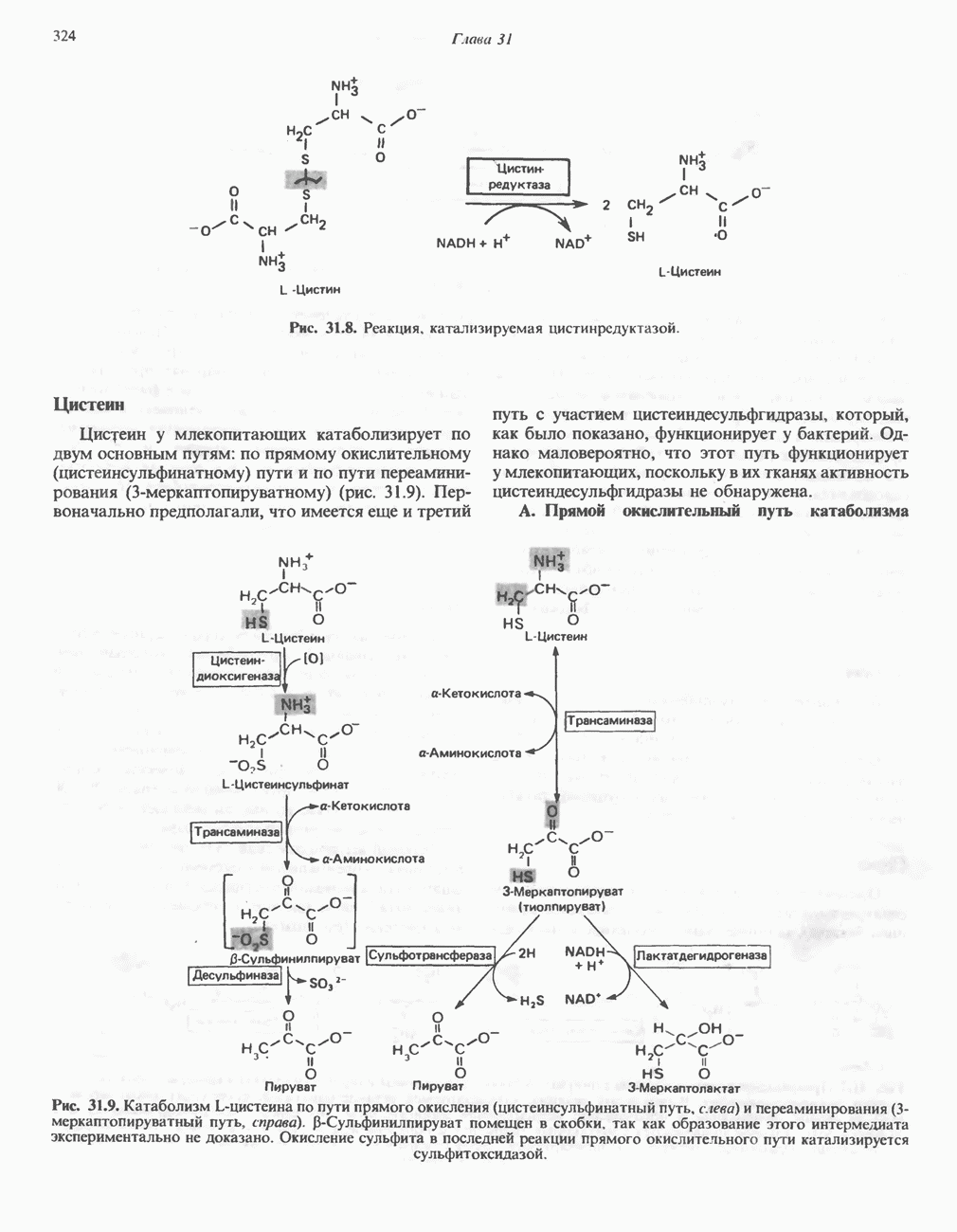
 от тканей к легким. Гемоглобин связывает
от тканей к легким. Гемоглобин связывает  сразу после высвобождения кислорода; примерно
сразу после высвобождения кислорода; примерно  присутствующего в крови, переносится молекулами гемоглобина. Находящаяся в эритроцитах карбоангидраза катализирует превращение поступающего из тканей
присутствующего в крови, переносится молекулами гемоглобина. Находящаяся в эритроцитах карбоангидраза катализирует превращение поступающего из тканей  в угольную кислоту (рис. 6.14). Угольная кислота быстро диссоциирует на бикарбонат-ион и протон, причем равновесие сдвинуто в сторону диссоциации. Для
в угольную кислоту (рис. 6.14). Угольная кислота быстро диссоциирует на бикарбонат-ион и протон, причем равновесие сдвинуто в сторону диссоциации. Для

 -форму сопровождается поворотом одной пары жестко связанных субъединиц
-форму сопровождается поворотом одной пары жестко связанных субъединиц  на 15 относительно другой такой же пары
на 15 относительно другой такой же пары  Ось вращения эксцентрична, т. е. одновременно происходит сдвиг димера
Ось вращения эксцентрична, т. е. одновременно происходит сдвиг димера  ближе к оси тетрамера. На этом рисунке показан поворот и смещение затененной
ближе к оси тетрамера. На этом рисунке показан поворот и смещение затененной  -пары относительно незатененной
-пары относительно незатененной  -пары (последняя считается неподвижной)
-пары (последняя считается неподвижной)

 -контакта при оксигенировании. Контакт как бы «перескакивает» с одного зубца на другой, с заменой одной водородной связи на другую. Остальные связи образованы неполярными остатками. (Из работы Perutz М. F.: Molecular pathology of human hemoglobin. Stereochemical interpretation of abnormal oxygen affinities. Nature 1971:232:408, с любезного разрешения.)
-контакта при оксигенировании. Контакт как бы «перескакивает» с одного зубца на другой, с заменой одной водородной связи на другую. Остальные связи образованы неполярными остатками. (Из работы Perutz М. F.: Molecular pathology of human hemoglobin. Stereochemical interpretation of abnormal oxygen affinities. Nature 1971:232:408, с любезного разрешения.)



 . Это обратимое явление известно как эффект Бора. Эффект Бора является свойством тетрамерного гемоглобина и определяется гем-гемовым взаимодействием, лежащим в основе кооперативных эффектов. У миоглобина эффект Бора не обнаруживается.
. Это обратимое явление известно как эффект Бора. Эффект Бора является свойством тетрамерного гемоглобина и определяется гем-гемовым взаимодействием, лежащим в основе кооперативных эффектов. У миоглобина эффект Бора не обнаруживается.

 -цепях. Эти протоны сдвигают равновесие в сторону образования угольной кислоты, которая расщепляется карбоангидразой с образованием СО; (рис. 6.15).
-цепях. Эти протоны сдвигают равновесие в сторону образования угольной кислоты, которая расщепляется карбоангидразой с образованием СО; (рис. 6.15).
 -субъединицах. Образование солевых мостиков форсирует освобождение кислорода из оксигенированной R-формы гемоглобина. Итак, повышение концентрации протонов способствует освобождению кислорода, а повышение концентрации кислорода стимулирует высвобождение протонов. Первый из этих эффектов проявляется в сдвиге кривой диссоциации кислорода вправо при повышении концентрации ионов водорода (протонов).
-субъединицах. Образование солевых мостиков форсирует освобождение кислорода из оксигенированной R-формы гемоглобина. Итак, повышение концентрации протонов способствует освобождению кислорода, а повышение концентрации кислорода стимулирует высвобождение протонов. Первый из этих эффектов проявляется в сдвиге кривой диссоциации кислорода вправо при повышении концентрации ионов водорода (протонов).

 -цепей. Связывание ДФГ осуществляется путем образования солевых мостиков между атомами кислорода ДФГ и группами, принадлежащими обеим
-цепей. Связывание ДФГ осуществляется путем образования солевых мостиков между атомами кислорода ДФГ и группами, принадлежащими обеим  -цепям: концевыми аминогруппами остатков
-цепям: концевыми аминогруппами остатков  аминогруппами остатков
аминогруппами остатков  и боковыми группами остатков
и боковыми группами остатков  . Таким образом, ДФГ стабилизирует дезоксигениро-ванную Т-форму гемоглобина, образуя поперечные связи между
. Таким образом, ДФГ стабилизирует дезоксигениро-ванную Т-форму гемоглобина, образуя поперечные связи между  -цепями—дополнительные солевые мостики, которые должны быть разрушены при переходе гемоглобина из Т- в R-форму.
-цепями—дополнительные солевые мостики, которые должны быть разрушены при переходе гемоглобина из Т- в R-форму.
 -цепи в положении
-цепи в положении  находится не
находится не  который не может участвовать в формировании солевых мостиков, удерживающих ДФГ в центральной полости. -Поэтому ДФГ в меньшей степени способствуют стабилизации Т-формы фетального гемоглобина и последний обладает более высоким сродством к кислороду по сравнению с гемоглобином взрослого человека.
который не может участвовать в формировании солевых мостиков, удерживающих ДФГ в центральной полости. -Поэтому ДФГ в меньшей степени способствуют стабилизации Т-формы фетального гемоглобина и последний обладает более высоким сродством к кислороду по сравнению с гемоглобином взрослого человека.
 относительно порфиринового кольца вызывает значительные изменения конформации гемоглобина и решающим образом воздействует на его ответную реакцию на сигнал, поступающий из внешней среды.
относительно порфиринового кольца вызывает значительные изменения конформации гемоглобина и решающим образом воздействует на его ответную реакцию на сигнал, поступающий из внешней среды.

 -цепей. (Из работы Amone A.: X-ray diffraction study of bonding of 2,3-diphosphoglycerate to human deoxyhemoglobin. Nature 1972:237:146, с разрешения.)
-цепей. (Из работы Amone A.: X-ray diffraction study of bonding of 2,3-diphosphoglycerate to human deoxyhemoglobin. Nature 1972:237:146, с разрешения.)
 -цепи, могут существенным образом сказываться на их биологической функции. Известно несколько сот мутантных гемоглобинов человека (в большинстве случаев функционально активных), и о некоторых из них, отличающихся сильным изменением биологических функций, речь пойдет ниже. Патологическое состояние, при котором мутация вызывает изменение биологической функции темоглобина, называют гемоглобинопатией.
-цепи, могут существенным образом сказываться на их биологической функции. Известно несколько сот мутантных гемоглобинов человека (в большинстве случаев функционально активных), и о некоторых из них, отличающихся сильным изменением биологических функций, речь пойдет ниже. Патологическое состояние, при котором мутация вызывает изменение биологической функции темоглобина, называют гемоглобинопатией.
 -состоянии, что обусловлено образованием прочного ионного комплекса с фенолятным анионом тирозина. Результатом такой аномалии является метгемоглобинемия, поскольку ферри-гем не способен связывать
-состоянии, что обусловлено образованием прочного ионного комплекса с фенолятным анионом тирозина. Результатом такой аномалии является метгемоглобинемия, поскольку ферри-гем не способен связывать  . В
. В  -цепи гемоглобина М
-цепи гемоглобина М  -равновесие сдвинуто в сторону образования Т-формы. Сродство к кислороду низкое, эффект Бора отсутствует. В
-равновесие сдвинуто в сторону образования Т-формы. Сродство к кислороду низкое, эффект Бора отсутствует. В  -цепях гемоглобинов М может происходить переход между R- и Т-состояниями и, следовательно, наблюдается эффект Бора.
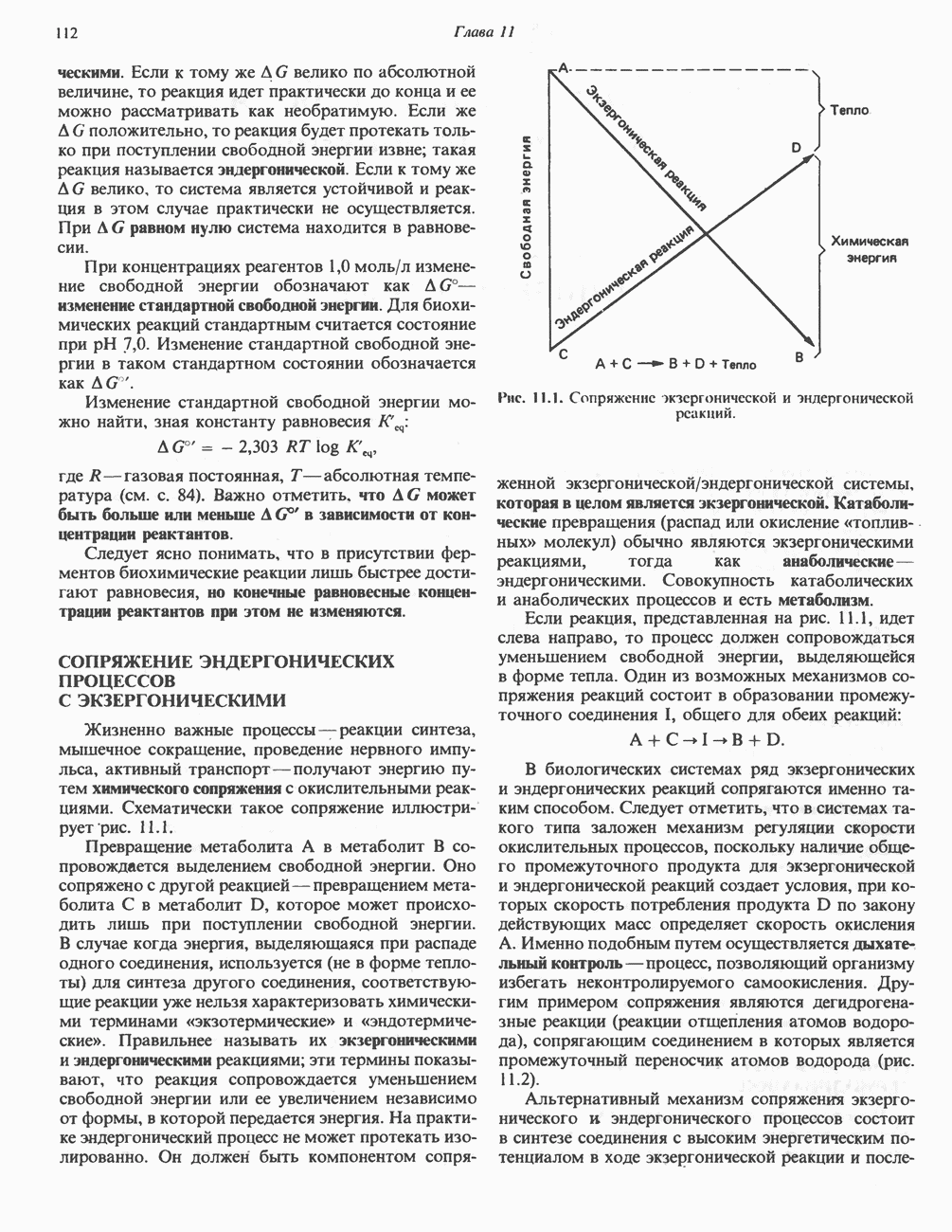
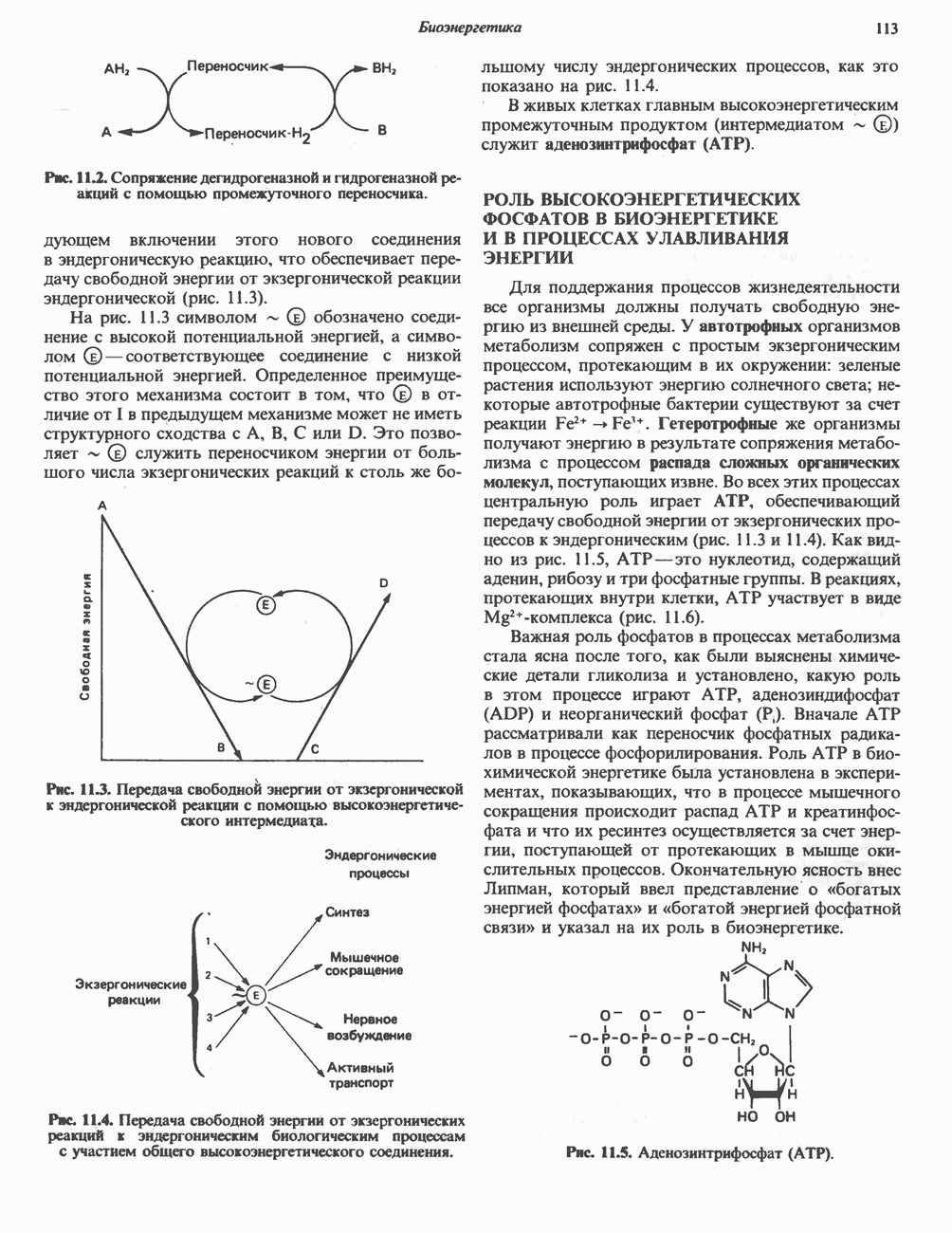
-цепях гемоглобинов М может происходить переход между R- и Т-состояниями и, следовательно, наблюдается эффект Бора.
 -формы (в качестве примера можно привести гемоглобин Чезапик), отличаются тем, что соответствующие гемоглобины обладают повышенным сродством к кислороду. Подобные гемоглобины не способны поставлять достаточное количество кислорода периферическим тканям. Возникает тканевая гипоксия, ведущая к развитию полицитемии (повышению концентрации эритроцитов).
-формы (в качестве примера можно привести гемоглобин Чезапик), отличаются тем, что соответствующие гемоглобины обладают повышенным сродством к кислороду. Подобные гемоглобины не способны поставлять достаточное количество кислорода периферическим тканям. Возникает тканевая гипоксия, ведущая к развитию полицитемии (повышению концентрации эритроцитов).
 замещен на
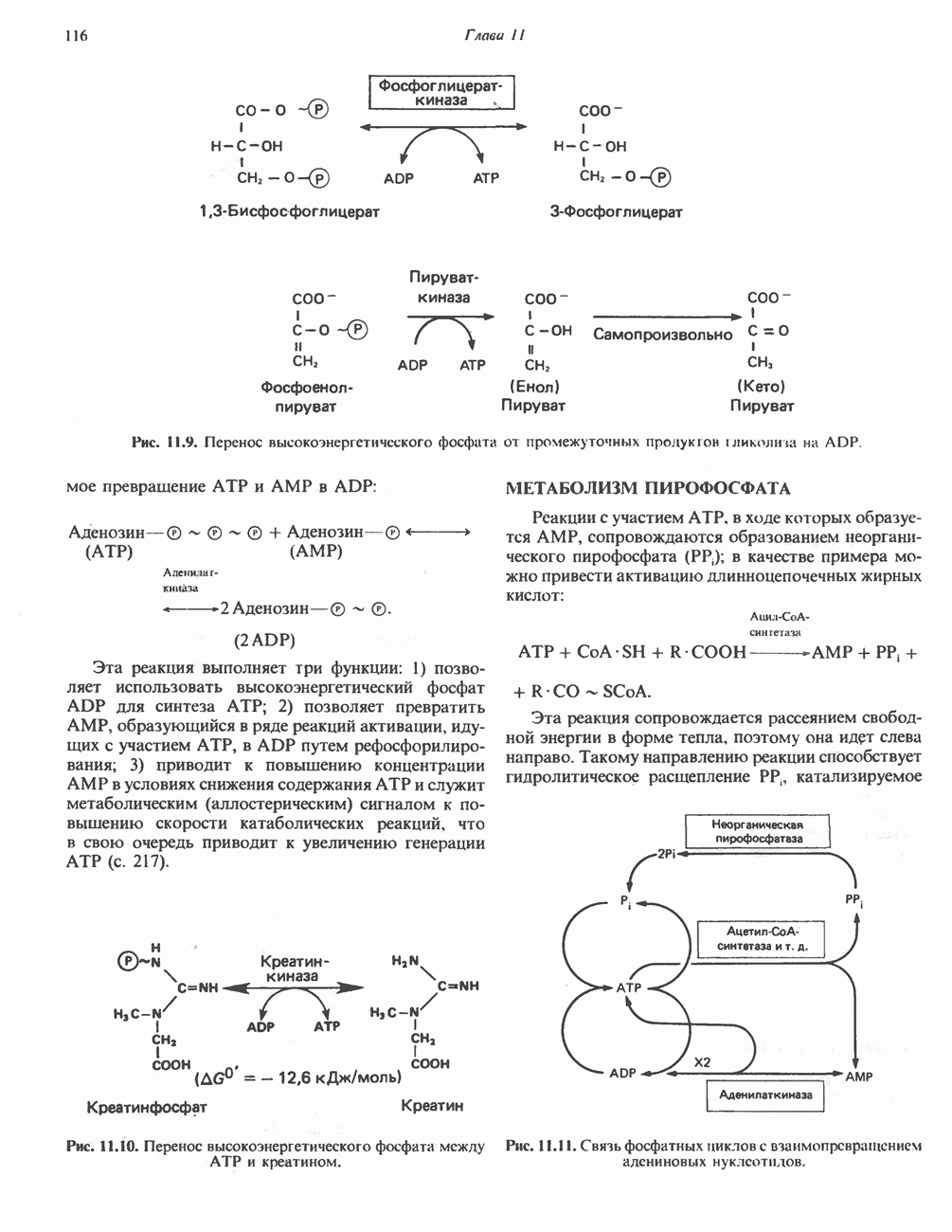
замещен на  Остаток
Остаток  (Glu или Val) располагается на поверхности молекулы гемоглобина и контактирует с водой, и замещение полярного остатка
(Glu или Val) располагается на поверхности молекулы гемоглобина и контактирует с водой, и замещение полярного остатка  на неполярный
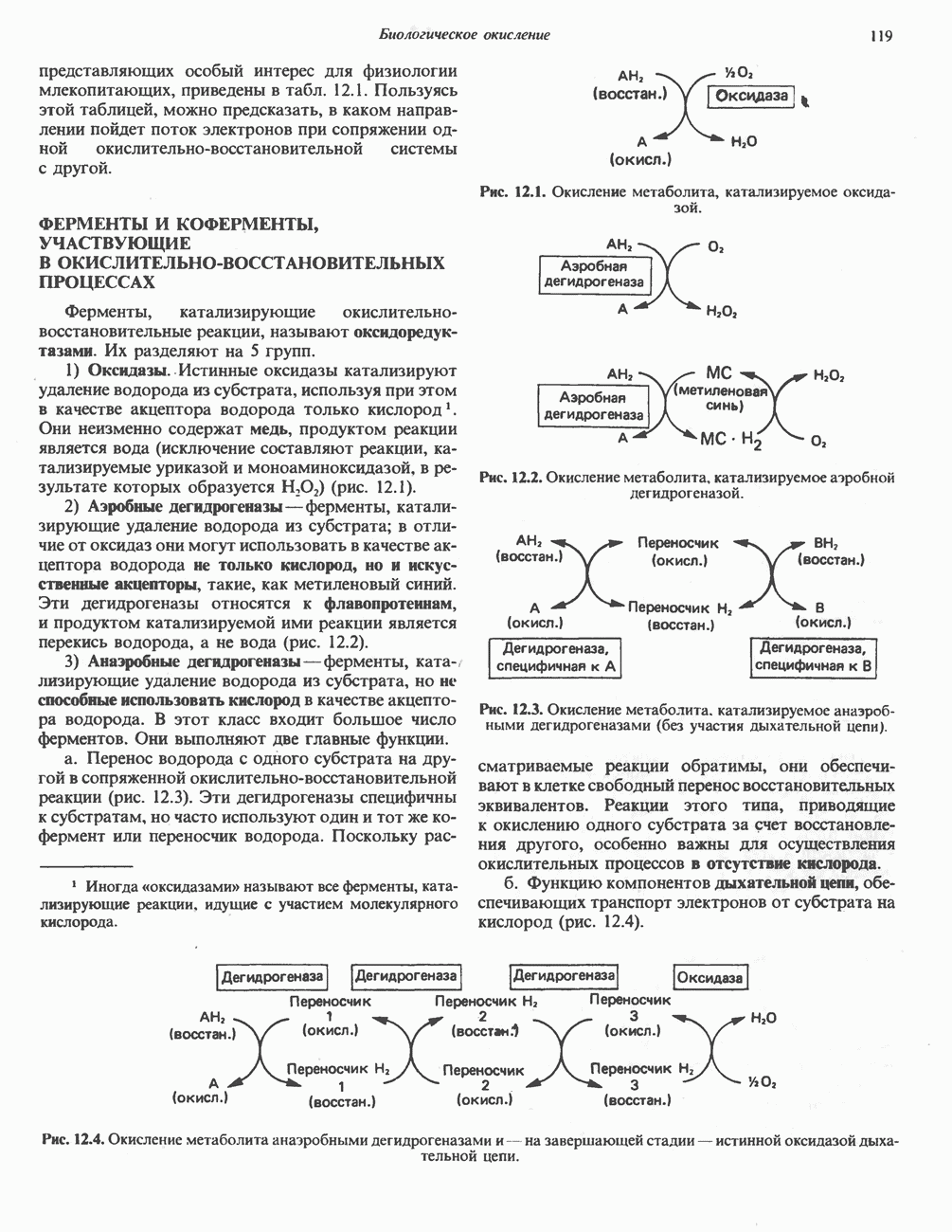
на неполярный  приводит к появлению на поверхности Р-субъединицы «липкого участка». Этот липкий участок присутствует как в оксигенированном, так и в дезоксигенированном гемоглобине S (в гемоглобине А он отсутствует). На поверхности дезоксигенированного гемоглобина существует комплементарный участок, способный прочно связываться с липким участком Р-субъединицы, тогда как в оксигенированном гемоглобине этот участок маскируется другими группами (рис. 6.18). Когда гемоглобин S переходит в дезоксигенированное состояние, его липкий участок связывается с комплементарным участком на другой молекуле дезоксигенированного гемоглобина. Происходит полимеризация дезоксигемоглобина S и его осаждение в виде длинных волокон. Волокна
приводит к появлению на поверхности Р-субъединицы «липкого участка». Этот липкий участок присутствует как в оксигенированном, так и в дезоксигенированном гемоглобине S (в гемоглобине А он отсутствует). На поверхности дезоксигенированного гемоглобина существует комплементарный участок, способный прочно связываться с липким участком Р-субъединицы, тогда как в оксигенированном гемоглобине этот участок маскируется другими группами (рис. 6.18). Когда гемоглобин S переходит в дезоксигенированное состояние, его липкий участок связывается с комплементарным участком на другой молекуле дезоксигенированного гемоглобина. Происходит полимеризация дезоксигемоглобина S и его осаждение в виде длинных волокон. Волокна


