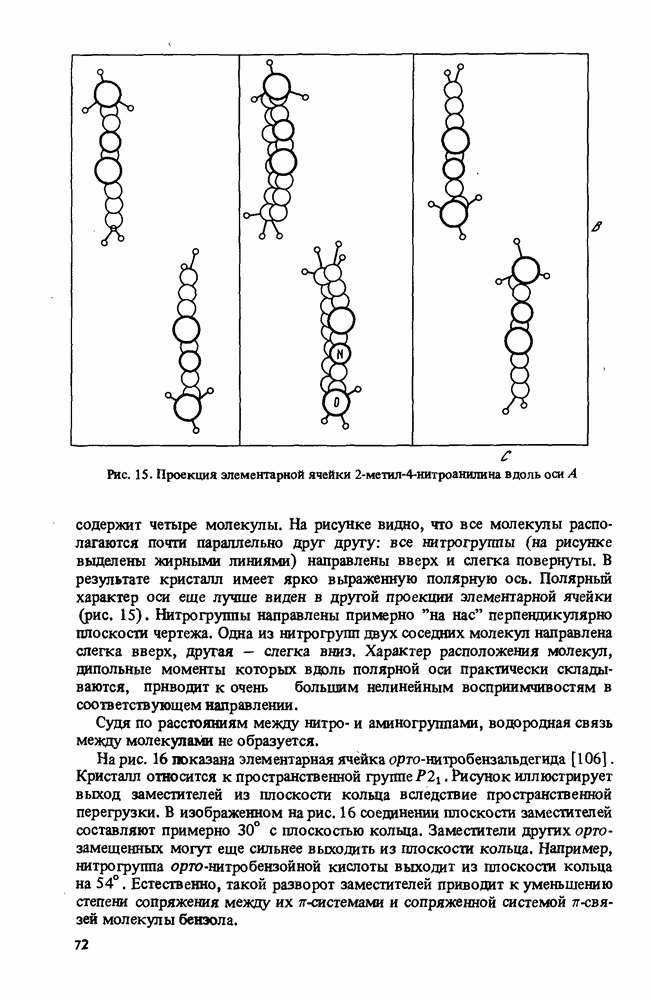
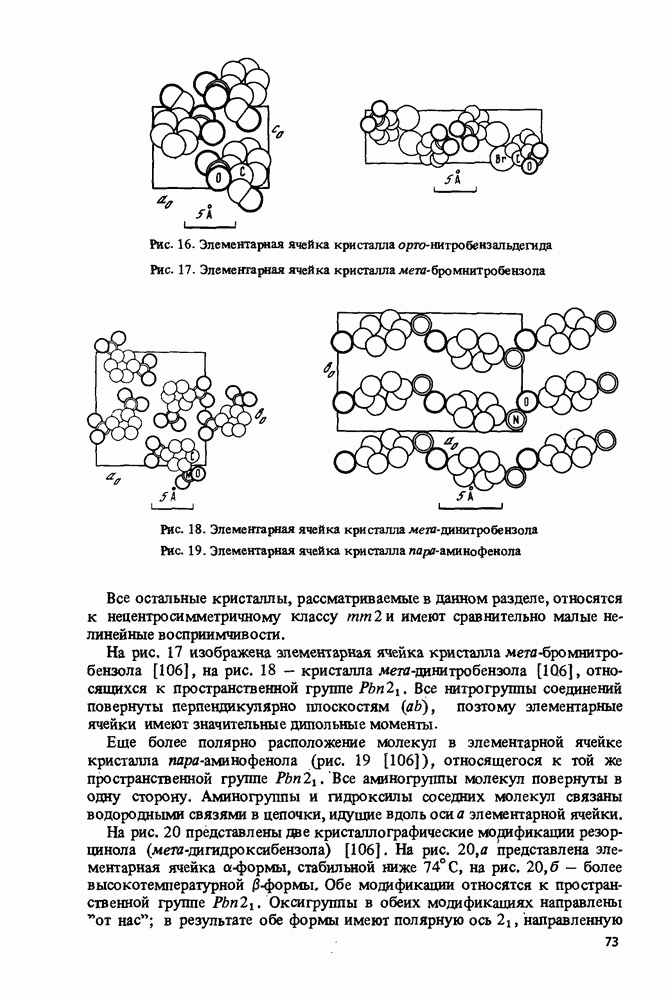
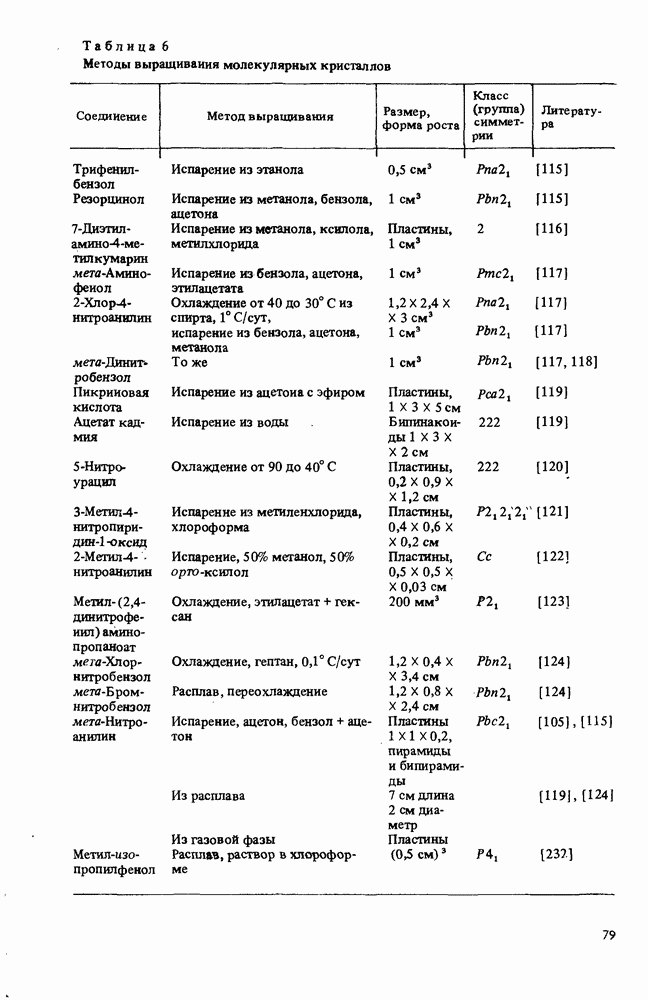
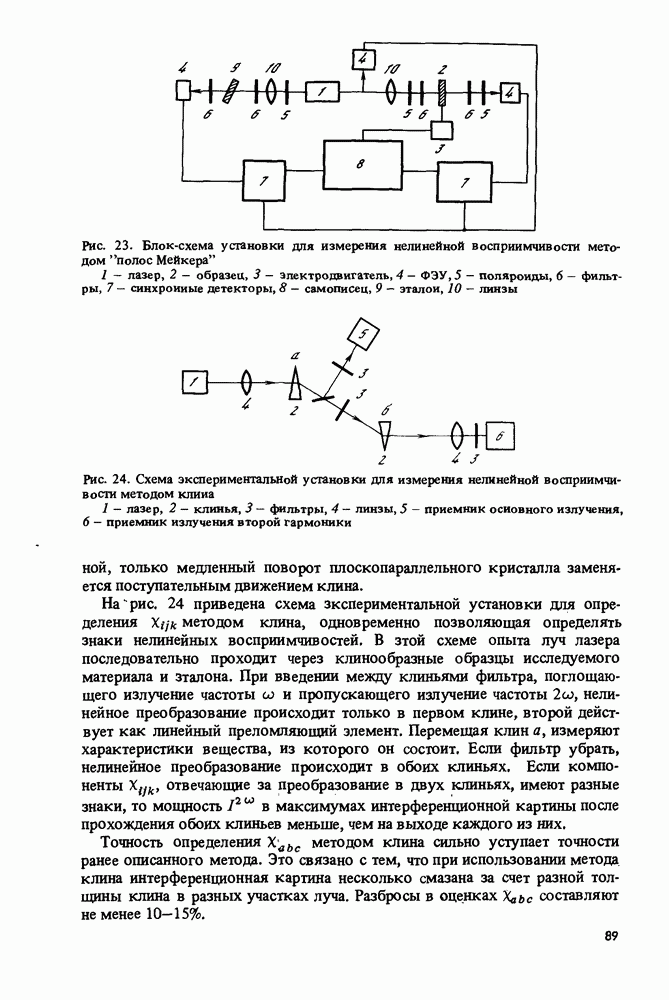
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
2.3. Некоторые методы квантовой химииВ этом разделе кратко описаны принципы расчета параметров молекул, что позволит оценить надежность результатов расчетов, полученных в разные годы, разными методами [69,81]. Для полного квантовомеханического описания молекулы, т. е. системы, состоящей из
зависящую от координат ядер
Гамильтониан
Для решения уравнения Шредингера с гамильтонианом (71) используется целый ряд приближений. Прежде всего задача решается в приближении Борна-Оцценгеймера. т. е. движения электронов и ядер считаются независимыми; полная волновая функция системы считается произведением функций, описывающих электроны и ядра. Электронная волновая функция определяется при фиксированом положении ядер и зависит от координат последних как от параметров. В гамильтониане (71) при этом остаются только электронные члены. Для нахождения электронной волновой функции
всегда больше, чем точное значение энергии системы
где Поэтому для нахождения
Наложение этих условий дает Пробная функция чаще всего выбирается в приближении Хартри-Фока-Рутана. В этом приближении полная волновая функция многозлектронной системы представляется в виде комбинаций волновых функций отдельных электронов
(антисимметризованное произведение и однозлектронных молекулярных новой функции
Спиновый фактор Задача нахождения волновой функции много электронной системы сводится, таким образом, к подбору однозлектронных волновых функций. Для нахождения зтих функций может быть применен вариационный принцип. Однозлектронные молекулярные орбитали представляются в виде функций от некоторых параметров. Например, можно считать, что каждая молекулярная орбиталь
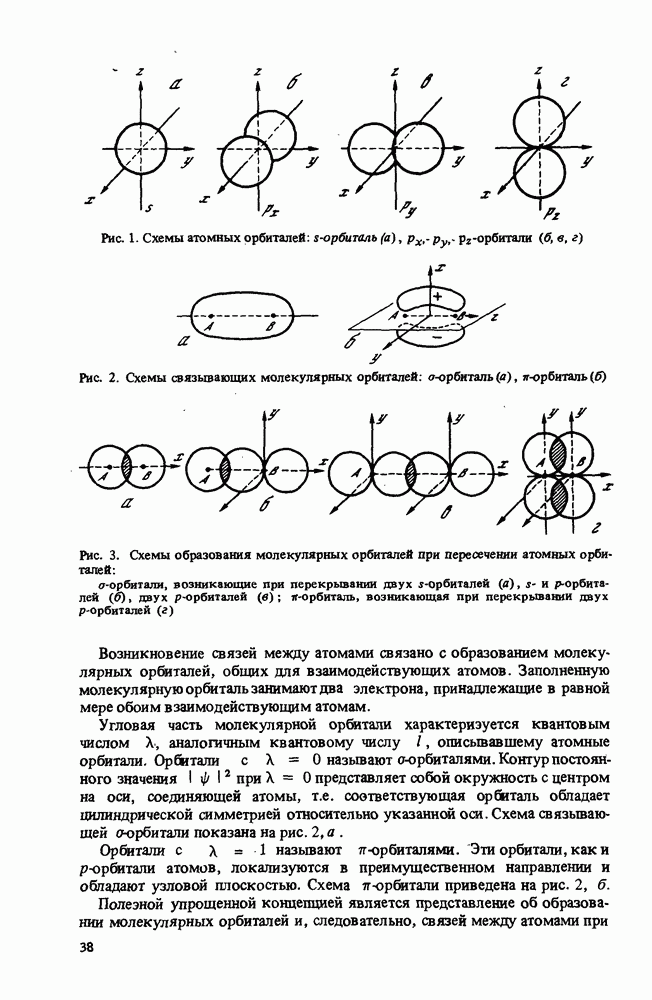
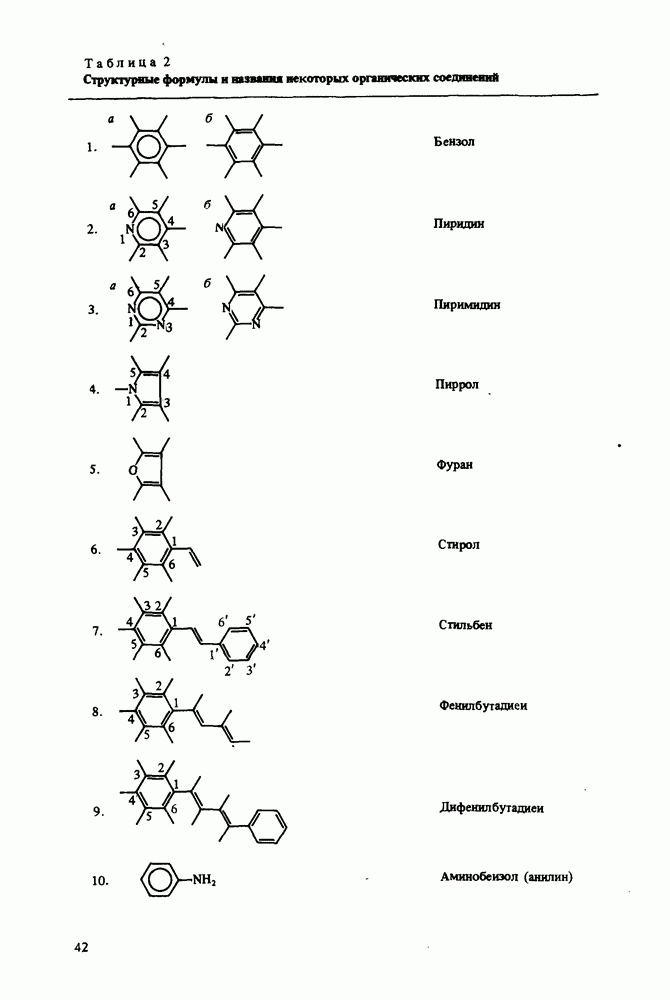
Основную идею этого приближения можно понять на основе следующего рассуждения. Если электрон находится в молекуле вблизи какого-либо атома, то силы, действующие на него, не слишком отличаются от сил, которые действовали бы на него, если бы он входил в изолированный атом. Поэтому МО электрона вблизи атома должна напоминать В качестве
Иногда вариационный принцип используется более тонким образом, с помощью так называемого метода самосогласованного поля (ССП). В этом случае, начиная с некоторого выбранного ряда однозлектронных орбиталей, рассчитывается поле, которое создается в месте Методы МО ЛKAО и ССП применялись в вышеописанном виде в основном до 1970 г. [82]. Главным недостатком метода МО ЛKAO является плохая точность. Как говорилось выше, точность решения зависит от подбора пробных функций. Если МО выбираются в виде (75), а АО в виде (76), то не удается достигнуть хорошего описания достаточно сложных молекул. Если варьировать также параметры АО, то для решения каждой новой задачи весь объем вычислений приходится повторять сначала, даже если рассматриваются сходные молекулы. Что касается метода ССП, то его недостатком является необходимость повторять процесс самосогласования для каждого состояния системы. В связи с вышесказанным в последнее время чаще используют так называемые полузмпирические методы [83, 84]. Полузмпирические методы отличаются от вышеописанных более тщательным выбором пробных функций. Во всех полузмпирических методах используется приближение ЛКАО (75), а Коэффициенты
Здесь
Матричные элементы
и матричные элементы кулоновского взаимодействия между парами электронов
Очевидно, что система уравнений Рутана (77) нелинейна, так как матричные элементы Основной объем вычислений при нахождении коэффициентов Рутана составляет вьиисление интегралов (78)-(80). Поэтому в различных полуэмпирических методах используются приближения, в которых многие из них равны нулю. Кроме того, рассматривают не все электроны системы, а лишь некоторую их совокупность, например валентные электроны (валентное приближение) или электроны, входящие в комбинации АО соседних атомов. Для сопряженных систем рассматривают МО, в которых участвуют лишь АО электронов, входящих в Современные широко используемые полузмпирические методы основаны на валентном приближении. Условно полузмпирические методы делятся на две группы: методы CNDO или ППДП - полного пренебрежения дифференциальным перекрыванием [83] и методы INDO или В методах CNDO из двухзлектронных интегралов рассштриваются только кулоновские вида
В методах INDO, кроме кулоновских, учитываются и одноцентровые обменные интегралы вида
Таким образом, чтобы определить однозлектронные молекулярные орбитали одним из полузмпирических методов, надо найти интегралы перекрывания Например, кулоновские и обменные интегралы описывают суммами вида
Многоцентровые интегралы выражают в виде функций от одноцентровых и интегралов перекрывания Остовные интегралы Некоторые из перечисленных величин, от которых зависят интегралы, находятся из эксперимента. Так, сродство к электрону Параметры Слзйтера-Кондона Таким образом, при использовании полузмпирических методов для расчета параметров новой молекулы нет необходимости повторять все вычисления. Выбирают существенный для данной задачи ансамбль электронов и АО, которые необходимо учитывать. Затем, используя параметризацию одного из полузмпирических методов, рассчитывают необходимые интегралы. После этого решается самосогласованная задача поиска коэффициентов Рутана. Это позволяет определить все МО данной молекулы. Выбор волновой функции всей системы означает размещение выбранного коллектива электронов по получившимся МО. Выбор занятых МО проводится описанным выше методом минимизации энергии системы. Искомая волновая функция системы является слзйтеровским детерминантом (74), составленным из занятых орбиталей. Некоторые МО оказываются незанятыми (виртуальными). Некоторая модификация рассмотренных полузмпирических методов позволяет описать возбужденные состояния молекул. В принципе для описания возбужденных состояний можно использовать виртуальные орбитали, Однако они не вполне подходят для зтой цели, так как их энергия (см. (77)) соответствует взаимодействию электрона с нейтральной молекулой. Фактически электрон возбужденной молекулы взаимодействует с ионом, Виртуальные МО используются для определения параметров возбужденных состояний методом конфигурационных взаимодействий (KB). Возбужденные конфигурации получают, рассматривая, кроме дважды занятых спин-орбиталей, две однократно занятые орбитали. Каждая конфигурация описывается слзйтеровским детерминантом Для учета KB полная волновая функция системы представляется в виде линейной комбинации слзйтеровских детерминантов, описывающих учитываемые конфигурации:
Оптимальные значения коэффициентов (84) находят из решения секулярной задачи
Решение секулярного уравнения (85) дает значения энергий состояний Отметим, что взаимодействуют между собой конфигурации одинаковой симметрии. Расчет возбужденных состояний тем точнее, чем больше рассмотрено различных конфигураций. Если учитываются только конфигурации, соответствующие однократным возбуждениям, то для получения достаточно точного решения следует строить конфигурации, в которых принимают участие все занятые и виртуальные МО. При расчете больших молекул учет только однократных возбуждений дает удовлетворительные результаты, так как конфигураций с одинаковой симметрией достаточно много. Если не удается учесть все такие конфигурации, выбирают те из них, диагональные матричные элементы которых лежат в заданном интервале энергий. В таких случаях удовлетворительно описываются лишь несколько возбужденных состояний с минимальными значениями энергии, При расчете параметров малых молекул обычно недостаточно учитывать лишь однократно возбужденные состояния. В настоящее время для расчета параметров возбужденных состояний молекул чаще всего применяется модификация метода CNDO, называемая методом CNDO/S. По сравнению с методом CNDO изменена параметризация: все параметры модифицированы для лучшего описания возбужденных состояний. Считается, что метод CNDO/S позволяет правильно найти частоты Неоднократно предпринимались попытки обобщения параметризации методов INDO для расчета возбужденных состояний. Однако в целом эти методы не получили столь широкого распространения, как методы типа CNDO. Зная волновые функции основного и возбужденного состояний молекул, можно определить их различные параметры. Например, можно вычислить матричные элементы оператора дипольного момента молекулы:
Вычисление дает дипольный момент основного или возбужденного состояния в зависимости от выбора По разности дипольных моментов основного и возбужденного состояний молекулы можно оценить степень ПЗ соответствующего перехода. Степень ПЗ можно оценить также по изменению электронной плотности вблизи различных атомов. Общая электронная плотность
Наконец, степень ПЗ можно оценить, просто определив распределение электронной плотности в молекуле. Для этого достаточно рассчитать значения Примеры различных способов оценки ПЗ будут приведены в следующем разделе. Квантовохимические методы позволяют рассчитывать гиперполяризуемости молекул. Существует несколько способов таких вычислений. Один из них [85, 86] заключается в вычислении гиперполяризуемости по формуле (52), где В других работах [46,51] многоэлектронный гамильтониан (71) модифицируется с помощью добавления к нему гамильтониана (47), описывающего дипольное взаимодействие молекулы с электрическим полем. Рассматривая гамильтониан (47) как возмущение, находят матричные элементы оператора дипольного момента в различных приближениях. Линейная поляризуемость и гиперполяризуемости находятся аналогично (49):
Наконец, в некоторых работах [87] рассчитывается дипольный момент свободных электронов в потенциальной яме. Затем поляризуемости рассчитываются по формулам (87). Результаты, полученные при расчете гиперполяризуемостей всеми указанными методами, будут обсуждаться в разд. 4.5. Отметим, что в принципе оба описанных метода — использование формулы (52) и формулы (87) — эквивалентны, так как применение теории возмущений к системе, описываемой гамильтонианом (71), должно привести к формуле, аналогичной (52), описывающей смешивание волновых функций основного и возбужденного состояний. Однако в некоторых случаях вычисление поляризуемостей по формулам (87) проще, особенно когда для описания молекулы применяется метод типа INDO [46]. Главной причиной упрощения вычислений в этом случае является уже упоминавшееся отсутствие надежной параметризации методов INDO для расчета возбужденных состояний молекул.
|
 |
Оглавление
|
















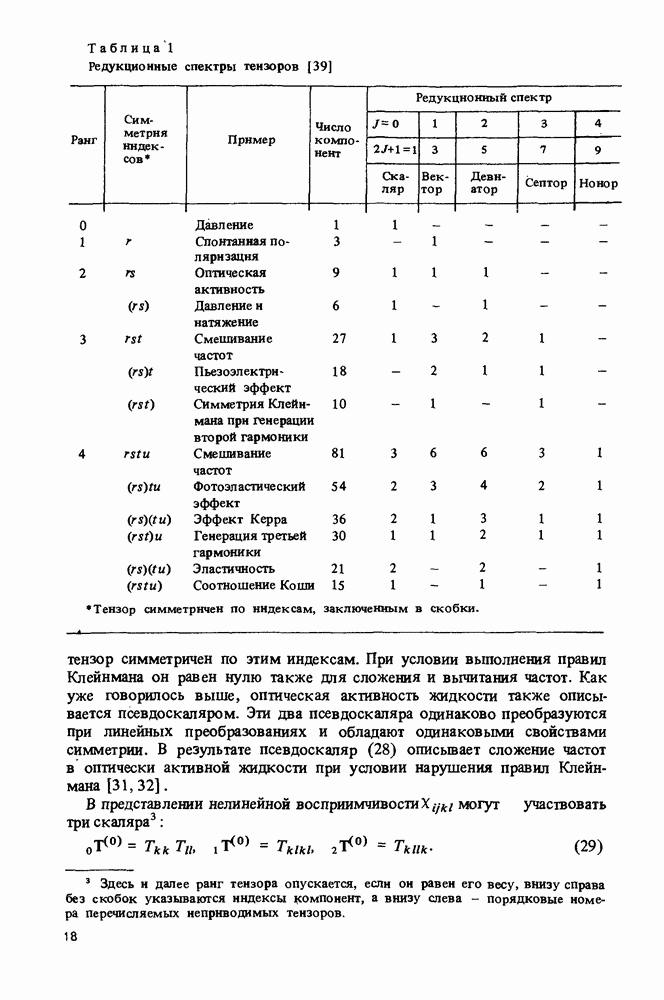













































































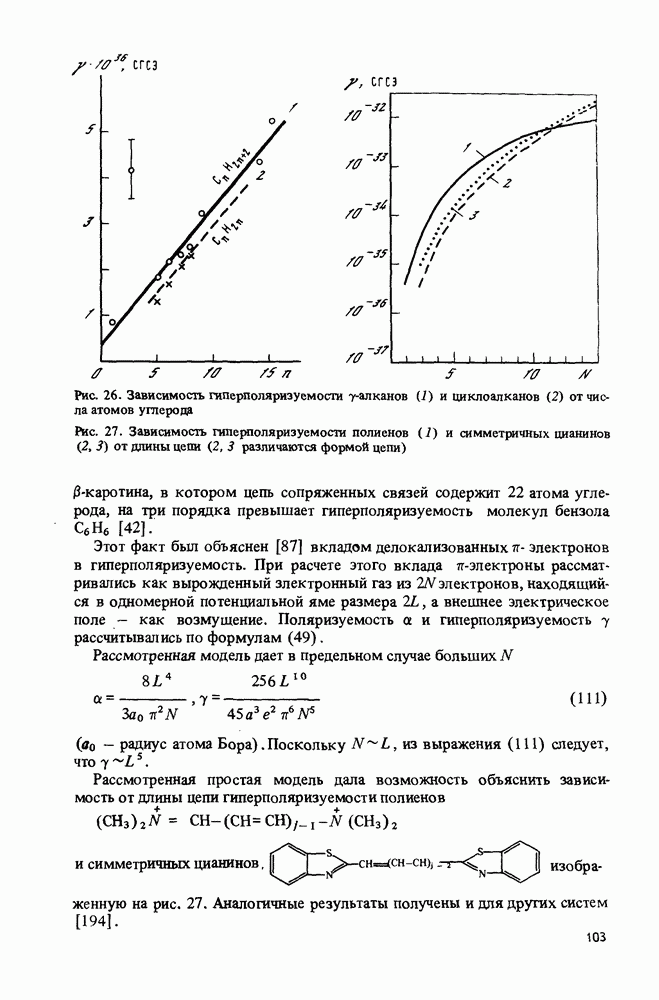























































































 ядер и
ядер и  электронов, достаточно знать ее волновую функцию
электронов, достаточно знать ее волновую функцию

 координат электронов
координат электронов  и спинов электронов и ядер
и спинов электронов и ядер  Эта функция является решением уравнения Шредингера
Эта функция является решением уравнения Шредингера

 в зтом уравнении при использовании атомной системы единиц имеет вид
в зтом уравнении при использовании атомной системы единиц имеет вид

 операторы Лапласа, действующие на координаты электронов и ядер соответственно,
операторы Лапласа, действующие на координаты электронов и ядер соответственно,  расстояние от
расстояние от  электрона до А ядра, между электронами
электрона до А ядра, между электронами  и между ядрами
и между ядрами  заряд ядра А.
заряд ядра А.
 используется вариационный принцип. Согласно этому принципу, если
используется вариационный принцип. Согласно этому принципу, если  любая гладкая и нормированная функция, а
любая гладкая и нормированная функция, а  точный гамильтониан системы, то величина интеграла
точный гамильтониан системы, то величина интеграла

 имеющее вид
имеющее вид

 точная волновая функция, описывающая систему.
точная волновая функция, описывающая систему.
 -функции, описывающей систему, подбирается так называемая пробная функция, зависящая от ряда параметров
-функции, описывающей систему, подбирается так называемая пробная функция, зависящая от ряда параметров  Далее вычисляют интеграл (72) и находят его минимальное значение для каждого из параметров
Далее вычисляют интеграл (72) и находят его минимальное значение для каждого из параметров  Математически это выражается условием
Математически это выражается условием

 уравнений, при решении которых получается
уравнений, при решении которых получается  значений вариационных параметров Вероятность получить таким путем точное значение энергии
значений вариационных параметров Вероятность получить таким путем точное значение энергии  и волновой функции
и волновой функции  зависит от адекватности выбора пробной функции и параметров
зависит от адекватности выбора пробной функции и параметров 
 Поскольку волновая функция не должна меняться при замене электронов (принцип неразличимости частиц в квантовой механике)
Поскольку волновая функция не должна меняться при замене электронов (принцип неразличимости частиц в квантовой механике)  удовлетворительной комбинацией является слзйгеровский детерминант
удовлетворительной комбинацией является слзйгеровский детерминант

 функций, или спин-орбиталей). Каждая спин-орбиталь одного электрона есть произведение координатной волновой функции, или орбитали, и
функций, или спин-орбиталей). Каждая спин-орбиталь одного электрона есть произведение координатной волновой функции, или орбитали, и

 не зависит от координат и имеет два собственных значения:
не зависит от координат и имеет два собственных значения:  Орбитали с разными спин-факторами ортогональны.
Орбитали с разными спин-факторами ортогональны.
 есть линейная комбинация атомных орбиталей
есть линейная комбинация атомных орбиталей 

 электрона в этом атоме.
электрона в этом атоме.
 можно взять, например, орбитали электрона в центрально-симметричном поле (слзйтеровские функции):
можно взять, например, орбитали электрона в центрально-симметричном поле (слзйтеровские функции):

 сферические гармоники, зависящие только от угловых координат электрона
сферические гармоники, зависящие только от угловых координат электрона  радиальная функция, зависящая от типа
радиальная функция, зависящая от типа  (например, для
(например, для  -электронов
-электронов  , для
, для  -электронов
-электронов  ). Затем функции вида (76) подставляются в уравнения (74), (75), определяются интегралы (72), и коэффициенты находятся согласно вариационному принципу
). Затем функции вида (76) подставляются в уравнения (74), (75), определяются интегралы (72), и коэффициенты находятся согласно вариационному принципу 
 электрона; с помощью вариационного метода определяется затем новое выражение для
электрона; с помощью вариационного метода определяется затем новое выражение для  электронной орбитали. Процедура выполняется для каждого электрона системы и приводит к нахождению нового ряда однозлектронных орбиталей. Расчеты повторяются до достижения самосогласования, т. е. до тех пор, пока
электронной орбитали. Процедура выполняется для каждого электрона системы и приводит к нахождению нового ряда однозлектронных орбиталей. Расчеты повторяются до достижения самосогласования, т. е. до тех пор, пока  ряд орбит не совпадает с
ряд орбит не совпадает с  рядом.
рядом.
 часто выбираются в виде слзйтеровских функций (76). Однако коэффициенты
часто выбираются в виде слзйтеровских функций (76). Однако коэффициенты  определяются другими методами. Так, коэффициенты разложения всех МО по
определяются другими методами. Так, коэффициенты разложения всех МО по  находятся одновременно, причем уже на зтом этапе проводится процедура самосогласования. Это значительно увеличивает объем вычислений, несмотря на то что часть параметров не рассчитывается, а берется из эксперимента. Поэтому полуэпирические методы стали доступными в связи с развитием вычислительной техники.
находятся одновременно, причем уже на зтом этапе проводится процедура самосогласования. Это значительно увеличивает объем вычислений, несмотря на то что часть параметров не рассчитывается, а берется из эксперимента. Поэтому полуэпирические методы стали доступными в связи с развитием вычислительной техники.
 в полуэмпирических методах находят с помощью решения уравнений Рутана. Эти уравнения получают, решая секулярную задачу
в полуэмпирических методах находят с помощью решения уравнений Рутана. Эти уравнения получают, решая секулярную задачу

 энергии соответствующих орбиталей,
энергии соответствующих орбиталей,  матричные элементы гамильтониана (71),
матричные элементы гамильтониана (71),  интегралы перекрывания:
интегралы перекрывания:

 можно рассчитать, зная матричные элементы его одноэлектронных (остовных) составляющих:
можно рассчитать, зная матричные элементы его одноэлектронных (остовных) составляющих:


 зависят от
зависят от  Ее решают итерационным методом. При этом выбирают пробные значения
Ее решают итерационным методом. При этом выбирают пробные значения  по ним рассчитывают матричные элементы
по ним рассчитывают матричные элементы  Система (77) решается относительно
Система (77) решается относительно  после чего процесс повторяется, т. е. снова рассчитываются
после чего процесс повторяется, т. е. снова рассчитываются  Процесс повторяют до тех пор, пока не выполнится какой-либо критерий сходимости, например
Процесс повторяют до тех пор, пока не выполнится какой-либо критерий сходимости, например  или
или  (
( — последовательные этапы приближения).
— последовательные этапы приближения).
 -системы. Ограничивают также число членов разложения (75). Например, при рассмотрении молекул с насыщенными связями можно в качестве МО брать лишь
-системы. Ограничивают также число членов разложения (75). Например, при рассмотрении молекул с насыщенными связями можно в качестве МО брать лишь
 -систему.
-систему.
 частичного пренебрежения дифференциальным перекрыванием [84],
частичного пренебрежения дифференциальным перекрыванием [84],


 (см. (78)), остовные интегралы
(см. (78)), остовные интегралы  а также интегралы кулоновского
а также интегралы кулоновского  и обменного
и обменного  взаимодействий. Все
взаимодействий. Все  величины находят полузмпирически, т. е. выражают через некоторые параметры, известные из эксперимента.
величины находят полузмпирически, т. е. выражают через некоторые параметры, известные из эксперимента.

 параметры Слзйтера-Кондона, зависящие от вида атомов,
параметры Слзйтера-Кондона, зависящие от вида атомов,  коэффициенты, зависящие от вида рассматриваемых
коэффициенты, зависящие от вида рассматриваемых 

 выражают через параметры Слзйтера-Кондона, потенциалы ионизации
выражают через параметры Слзйтера-Кондона, потенциалы ионизации  и сродство к электрону
и сродство к электрону 
 измеряется с помощью полярографического восстановления, потенциал ионизации
измеряется с помощью полярографического восстановления, потенциал ионизации  определяется методом электронного удара и фотоионизации.
определяется методом электронного удара и фотоионизации.
 , интегралы перекрывания и коэффициенты разложения
, интегралы перекрывания и коэффициенты разложения  и слегка различны в разных модификациях полузмпирических методов. Все эти величины табулируются для каждой вариации: параметры
и слегка различны в разных модификациях полузмпирических методов. Все эти величины табулируются для каждой вариации: параметры  для каждого сорта атомов второго и третьего периодов таблицы Менделеева, величины
для каждого сорта атомов второго и третьего периодов таблицы Менделеева, величины  для рассматриваемых орбиталей
для рассматриваемых орбиталей 


 и коэффициенты
и коэффициенты  соответствующие этим состояниям.
соответствующие этим состояниям.
 тг-пер входов. Метод дает разумные результаты и при описании основного состояния молекулы.
тг-пер входов. Метод дает разумные результаты и при описании основного состояния молекулы.

 -функций.
-функций.
 вблизи атома
вблизи атома  представляет собой сумму электронных плотностей, вносимых каждым электроном на каждой
представляет собой сумму электронных плотностей, вносимых каждым электроном на каждой 

 число электронов на орбитали
число электронов на орбитали  коэффициент
коэффициент  для
для  (ср. с (75)).
(ср. с (75)).
 для основного и возбужденного состояний для каждой точки пространства.
для основного и возбужденного состояний для каждой точки пространства.
 волновые функции всех состояний молекулы (как основного, так и возбужденных). Очевидно, что указанный метод позволяет оценить вклад различных переходов. Волновые функции молекулы рассчитываются обычно методом CNDO/S.
волновые функции всех состояний молекулы (как основного, так и возбужденных). Очевидно, что указанный метод позволяет оценить вклад различных переходов. Волновые функции молекулы рассчитываются обычно методом CNDO/S.
