§ 27. СВЯЗИ-ЛОКАЛИЗАЦИЯ, ОТДЕЛЬНЫЕ ПАРЫ, ИОННЫЕ ОСТОВЫ
Изложенная до сих пор теория основывалась на молекулярно-орбитальном описании, в котором использовались функции  для молекулярно-орбитальных пар, даже если мы имели дело с большими насыщенными молекулами с сильно локализованными электронами. В такой теории оболочки разделяются на внутренние и внешние. Однако для насыщенных молекул мы еще должны рассмотреть, каким образом возникают локализованные связи и отдельные пары, почему энергии связи совершенно не зависят от молекулярного окружения и какие силы действуют между связями. К счастью, нет необходимости заново начинать рассмотрение таких систем с помощью другой теории. Простое преобразование переводит молекулярные орбитали, на которых основывается многоэлектронная теория, в локализованные орбитали.
для молекулярно-орбитальных пар, даже если мы имели дело с большими насыщенными молекулами с сильно локализованными электронами. В такой теории оболочки разделяются на внутренние и внешние. Однако для насыщенных молекул мы еще должны рассмотреть, каким образом возникают локализованные связи и отдельные пары, почему энергии связи совершенно не зависят от молекулярного окружения и какие силы действуют между связями. К счастью, нет необходимости заново начинать рассмотрение таких систем с помощью другой теории. Простое преобразование переводит молекулярные орбитали, на которых основывается многоэлектронная теория, в локализованные орбитали.
В этой задаче полная функция и полная энергия также имеют орбитальную и корреляционную части.
Орбитальная часть была получена Леннардом-Джонсом [42, 46, 47], показавшим, что в определителе  построенном на хартри-фоковских молекулярных орбиталях самосогласованного поля, электроны уже локализованы относительно друг друга (например, в
построенном на хартри-фоковских молекулярных орбиталях самосогласованного поля, электроны уже локализованы относительно друг друга (например, в  как упоминалось в § 7. Это распределение электронов относительно друг друга лучше всего описывать с помощью унитарного «эквивалентного» или «локализованного» орбитального преобразования
как упоминалось в § 7. Это распределение электронов относительно друг друга лучше всего описывать с помощью унитарного «эквивалентного» или «локализованного» орбитального преобразования  которое не меняет
которое не меняет 

где  хартри-фоковские молекулярные орбитали самосогласованного поля со спином,
хартри-фоковские молекулярные орбитали самосогласованного поля со спином,  -новые локализованные спин-орбитали,
-новые локализованные спин-орбитали,  матрица, эрмитово сопряженная матрице
матрица, эрмитово сопряженная матрице 
Отметим, что выражение (1416) в действительности содержит два независимых преобразования: одно для нечетных  т. е.
т. е.  -спин-орбиталей, а другое для четных
-спин-орбиталей, а другое для четных  -спин-орбиталей.
-спин-орбиталей.
Если взять

то  [выражение
[выражение  и
и  [выражение (13)] сохраняют свой вид
[выражение (13)] сохраняют свой вид

однако формула (12) больше не применима. Вместо нее имеем

где  недиагональные хартри-фоковские орбитальные энергии, а
недиагональные хартри-фоковские орбитальные энергии, а  — диагональная орбитальная энергия;
— диагональная орбитальная энергия;  те же самые величины, что и раньше [определяются формулами (4а) и (8), но
те же самые величины, что и раньше [определяются формулами (4а) и (8), но  заменено на
заменено на 
В  эквивалентные
эквивалентные  -орбитали [46—48] определяются при помощи условий симметрии. Для других молекул с меньшей симметрией, например для молекул
-орбитали [46—48] определяются при помощи условий симметрии. Для других молекул с меньшей симметрией, например для молекул  преобразование
преобразование  можно определить как преобразование, минимизирующее величины обменных членов [49, 50, 57] в (144), или же с помощью аналогичных критериев [51].
можно определить как преобразование, минимизирующее величины обменных членов [49, 50, 57] в (144), или же с помощью аналогичных критериев [51].
Локализованные орбитали были подробно изучены Поплом [43], Линнетом [44] и Холлом [50]. Совсем недавно Рюденберг и Эдмистон развили методику для их расчета, основанную на критерии «минимального обмена» [49, 50].
Холл [50] скоррелировал потенциалы ионизации углеводородов от этана до декана, полагая неизменными локализованные орбитали и энергии 
Потенциалы ионизации в небольшой степени подвержены воздействию корреляции электронов («теорема Купмена» [1]). Однако корреляция весьма важна для теплот образования и конфигурационных изменений [2].
Харли, Леннард-Джонс и Попл [23] ввели корреляцию в локализованных группах с помощью подобранной для этого случая волновой функции

и произвольного условия (1). Ограничения этого приближения [24—26] обсуждаются после формулы (1) в § 4 и в ряде работ [3, 10, 23, 27—31] (см. примечание 29 в работе [10]).
Помимо остальных ограничений, выражение (146) не позволяет переходить от локальных свойств в основном состоянии к свойствам делокализованных электронов, например при рассмотрении электронных спектров.
К счастью, нет необходимости использовать различные теории для локализованных групп и для делокализованных электронов. Оба случая охватываются одной и той же «многоэлектронной теорией». Необходимо лишь выяснить, каким образом преобразуются [104] функции  и Есотг при действии того же самого оператора
и Есотг при действии того же самого оператора  который преобразует функцию
который преобразует функцию  Зная это, можно переходить от делокализованного описания к локализованному, и наоборот [12].
Зная это, можно переходить от делокализованного описания к локализованному, и наоборот [12].
А. Преобразование точной энергии
Точная энергия определяется выражением (60) через молекулярные спин-орбитали  Чтобы усмотреть, каким образом преобразуется корреляционная энергия, если преобразование, заданное (141а), применено к хартри-фоковским молекулярным орбиталям, подставим
Чтобы усмотреть, каким образом преобразуется корреляционная энергия, если преобразование, заданное (141а), применено к хартри-фоковским молекулярным орбиталям, подставим

в выражение (60)

где

Меняя порядок суммирования и замечая, что оператор одинаков для всех электронов, получаем

Кроме того, имеем

где  все еще молекулярные спин-орбитали,
все еще молекулярные спин-орбитали,  корреляционные волновые функции между локализованными спин-орбиталями. Они также локализованы. Данная функция
корреляционные волновые функции между локализованными спин-орбиталями. Они также локализованы. Данная функция  если
если  отличаются лишь своим спином, является корреляционной волновой функцией для этой связи, отдельной пары или пары внутренней оболочки, подобной
отличаются лишь своим спином, является корреляционной волновой функцией для этой связи, отдельной пары или пары внутренней оболочки, подобной  . С другой стороны, если
. С другой стороны, если  относятся к локализованным орбиталям с различными пространственными частями (межоболочечная корреляция), соответствующая функция
относятся к локализованным орбиталям с различными пространственными частями (межоболочечная корреляция), соответствующая функция  определяет вандерваальсово притяжение между локализованными орбиталями отдельных групп.
определяет вандерваальсово притяжение между локализованными орбиталями отдельных групп.
Когда в распоряжении имеются молекулярно-орбитальные парные функции скажем, в виде разложений по методу конфигурационного взаимодействия или в виде зависимости от  формула (149) дает немедленно корреляции в локализованном описании. Например, формула (149) могла бы быть применена к результатам Несбета [48] для молекулы
формула (149) дает немедленно корреляции в локализованном описании. Например, формула (149) могла бы быть применена к результатам Несбета [48] для молекулы  (метод хартри-фоковских молекулярных орбиталей, дополненный методом конфигурационного взаимодействия). Тем самым можно было бы извлечь из этих результатов корреляционную энергию
(метод хартри-фоковских молекулярных орбиталей, дополненный методом конфигурационного взаимодействия). Тем самым можно было бы извлечь из этих результатов корреляционную энергию  -связи и «вандерваальсово притяжение» между двумя различными
-связи и «вандерваальсово притяжение» между двумя различными  -связями.
-связями.
Выражение (149) показывает, что энергия  насыщенной молекулы в точности равна сумме корреляционных энергий связей, отдельных пар и ионного остова и внутримолекулярного вандерваальсова притяжения между этими группами. Результат этот точен настолько же, насколько справедливо само уравнение Шредингера, однако пока он чисто формальный. Точные функции
насыщенной молекулы в точности равна сумме корреляционных энергий связей, отдельных пар и ионного остова и внутримолекулярного вандерваальсова притяжения между этими группами. Результат этот точен настолько же, насколько справедливо само уравнение Шредингера, однако пока он чисто формальный. Точные функции  которые входят в выражение (148), сопоставляются с точной функцией
которые входят в выражение (148), сопоставляются с точной функцией  [выражение (20)] таким же образом, как это было сделано для
[выражение (20)] таким же образом, как это было сделано для  [выражение
[выражение  в принципе они могут зависеть от всех других членов функции
в принципе они могут зависеть от всех других членов функции
Остаются следующие вопросы:
1) каким образом можно непосредственно получить функции  (поскольку это было бы более удобно, нежели сначала находить функции
(поскольку это было бы более удобно, нежели сначала находить функции 
2) Какие подразумеваются эффекты окружения в  и локализованных корреляционных энергиях?
и локализованных корреляционных энергиях?

Б. Преобразования приближенных выражений для энергии
Выражения для энергии, даваемые теорией возмущений и многоэлектронной теорией, имеют один и тот же вид (60).
Если учесть парную функцию первого порядка  удовлетворяющую (70), то соотношение (69) даст нам следующее выражение для энергии с точностью до второго порядка:
удовлетворяющую (70), то соотношение (69) даст нам следующее выражение для энергии с точностью до второго порядка:

В многоэлектронной теории функция  переходит в
переходит в  [выражение (101)], как только каждая из энергий
[выражение (101)], как только каждая из энергий  [выражение (86)] минимизирована так, что пары «во всех порядках» удовлетворяют уравнению (100), при этом (85) вновь принимает вид
[выражение (86)] минимизирована так, что пары «во всех порядках» удовлетворяют уравнению (100), при этом (85) вновь принимает вид

Отметим, что выражение (60), (152а) и (153а) выглядят одинаково. Однако в каждом из них парная корреляционная функция  различна. Выражение (60) получено из точной функции а в выражениях (152а) и (153а) подразумевается, что пары являются несвязанными между собой и соответствующие парные корреляционные функции не зависят явно от
различна. Выражение (60) получено из точной функции а в выражениях (152а) и (153а) подразумевается, что пары являются несвязанными между собой и соответствующие парные корреляционные функции не зависят явно от  или от
или от  и т. д.
и т. д.
Так как формы этих выражений одинаковы, то преобразования (147) и (149) с равным основанием применимы  и (153а). Результирующие выражения, подобны (148), но содержат теперь либо корреляционную функцию первого порядка (а либо
и (153а). Результирующие выражения, подобны (148), но содержат теперь либо корреляционную функцию первого порядка (а либо  порядки) парные корреляционные функции
порядки) парные корреляционные функции  «многоэлектронной теории». Имеем
«многоэлектронной теории». Имеем

и

Различие между  в выражении (1536) и
в выражении (1536) и  в выражении (148) аналогично тому, которое имеет место между функцией
в выражении (148) аналогично тому, которое имеет место между функцией  и функцией полученной из полной точной функции
и функцией полученной из полной точной функции 
В. Преобразования вариационных энергий
Выражения (152а) и (153а) в предыдущем разделе справедливы только для таких парных корреляционных функций, которые удовлетворяют отвечающим им эффективным уравнениям Шредингера (70) или (100а). Чтобы получить парные корреляционные функции  в случае, когда исходят из молекулярных орбиталей, необходимо минимизировать вариационные выражения вида (86). Выражения (1526) и (1536) имеют силу лишь для оптимальных функций
в случае, когда исходят из молекулярных орбиталей, необходимо минимизировать вариационные выражения вида (86). Выражения (1526) и (1536) имеют силу лишь для оптимальных функций  Чтобы найти их непосредственно, необходимо располагать вариационным принципом для каждого
Чтобы найти их непосредственно, необходимо располагать вариационным принципом для каждого 
Пригодность концепции «энергии связи» указывает на то, что, когда рассматривается большая молекула, гораздо легче получить непосредственно те или иные  для корреляций локализованного и вандерваальсова типа, чем получить вначале
для корреляций локализованного и вандерваальсова типа, чем получить вначале  Это обстоятельство имеет большую практическую ценность. Чтобы рассчитать, к примеру, a priori энергию
Это обстоятельство имеет большую практическую ценность. Чтобы рассчитать, к примеру, a priori энергию
 -саязи, не нужно получать молекулярные орбитали, скажем, из рассмотрения всей молекулы, протеина или полностью проводить вначале расчеты по методу Хартри — Фока, дополненному методом конфигурационного взаимодействия»
-саязи, не нужно получать молекулярные орбитали, скажем, из рассмотрения всей молекулы, протеина или полностью проводить вначале расчеты по методу Хартри — Фока, дополненному методом конфигурационного взаимодействия»
Чтобы понять, каким образом  можно непосредственно подсчитать, обратимся вначале к теории возмущений. Вариационная форма для выражения (152а) [см. (63), (64) и
можно непосредственно подсчитать, обратимся вначале к теории возмущений. Вариационная форма для выражения (152а) [см. (63), (64) и  имеет вид
имеет вид

Функции  являются пробными функциями для получения парных корреляционных функций первого порядка в молекулярном орбитальном описании. Они стремятся к
являются пробными функциями для получения парных корреляционных функций первого порядка в молекулярном орбитальном описании. Они стремятся к  когда
когда 
Первая сумма в  та же самая, что и в (152а); она преобразуется путем подстановки (147) и (149). Величина
та же самая, что и в (152а); она преобразуется путем подстановки (147) и (149). Величина  содержит [см. выражения (7) и (12) и примечание на стр. 116] одну и ту же часть для всех электронов:
содержит [см. выражения (7) и (12) и примечание на стр. 116] одну и ту же часть для всех электронов:

Следовательно обращение соотношения (149) дает

Отметим, что изменено лишь обозначение переменных суммирования в  Во всех других
Во всех других
отношениях величины  совершенно аналогичны, они обе включают в себя потенциал
совершенно аналогичны, они обе включают в себя потенциал  всей молекулы, определяемый формулой (8) с заменой спин-орбиталей молекулярными орбиталями.
всей молекулы, определяемый формулой (8) с заменой спин-орбиталей молекулярными орбиталями.
Величина  содержит часть энергии
содержит часть энергии  которую можно вычислить при помощи молекулярных орбиталей; она различается для каждой пары орбиталей; так, используя преобразование (149) дважды (и слева и справа), получаем
которую можно вычислить при помощи молекулярных орбиталей; она различается для каждой пары орбиталей; так, используя преобразование (149) дважды (и слева и справа), получаем

Тильда над  для простоты опущена. В итоге выражение (154) дает
для простоты опущена. В итоге выражение (154) дает

В выражении (156) мы использовали

и 

где  матрица орбитальных энергий, вычисляемых при помощи молекулярных орбиталей,
матрица орбитальных энергий, вычисляемых при помощи молекулярных орбиталей,  матрица орбитальных энергий локализованных состояний [см. (143) и (145)]. В выражении (157б)
матрица орбитальных энергий локализованных состояний [см. (143) и (145)]. В выражении (157б)

Теперь в соответствии с методикой вариационного приближения, использующего теорию возмущений, необходимо было бы минимизировать каждую
функцию  взяв пробные функции и потребовав, чтобы
взяв пробные функции и потребовав, чтобы

где  все еще полные молекулярные орбитали. Эта процедура и эффекты окружения, которые внесены орбиталями
все еще полные молекулярные орбитали. Эта процедура и эффекты окружения, которые внесены орбиталями  и полной хартри-фоковской функцией
и полной хартри-фоковской функцией  в потенциалы
в потенциалы  в (1576), будут обсуждаться в следующем разделе.
в (1576), будут обсуждаться в следующем разделе.
Члены  легко оцениваются. Они намного меньше, чем член
легко оцениваются. Они намного меньше, чем член  так что применимо вариационное приближение с использованием теории возмущений (см. § 14). Те же самые величины
так что применимо вариационное приближение с использованием теории возмущений (см. § 14). Те же самые величины  которые найдены из (1576), можно использовать, чтобы подсчитать
которые найдены из (1576), можно использовать, чтобы подсчитать 
Холл [50] получил эмпирические значения для недиагональных хартри-фоковских энергий  от углеводородов метана до декана. Применяя выражение (149) к несбетовой [48] функции для
от углеводородов метана до декана. Применяя выражение (149) к несбетовой [48] функции для  представляющей собой хартри-фоковскую молекулярную орбиталь, исправленную с помощью метода конфигурационного взаимодействия, получаем [105] грубую оценку для При
представляющей собой хартри-фоковскую молекулярную орбиталь, исправленную с помощью метода конфигурационного взаимодействия, получаем [105] грубую оценку для При  величина
величина  в результате для метана получаем
в результате для метана получаем

а для полной корреляционной энергии мы имели

Величина  является эмпирической оценкой для полной корреляционной энергии
является эмпирической оценкой для полной корреляционной энергии  она не равна величине, полученной Несбетом [48] и составляющей
она не равна величине, полученной Несбетом [48] и составляющей 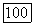
Если ограничиваться 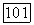 то функцию
то функцию 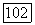 можно аппроксимировать функцией полученной минимизацией (1576),
можно аппроксимировать функцией полученной минимизацией (1576),
Рассмотрим теперь полные верхние пределы, даваемые для 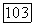 волновыми функциями вида (69а) и (76). Энергия [выражение (65)], полученная с помощью
волновыми функциями вида (69а) и (76). Энергия [выражение (65)], полученная с помощью 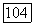 содержит
содержит 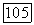 Выражение (65), записанное более детально, имеет ту же самую форму, что и
Выражение (65), записанное более детально, имеет ту же самую форму, что и 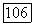 [соотношение (78)], однако оно содержит функции
[соотношение (78)], однако оно содержит функции 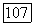 полученные минимизацией меньшей доли от
полученные минимизацией меньшей доли от 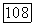 [выражение (154в)]. Далее, сравнение (65), (154в) и (78) показывает, что
[выражение (154в)]. Далее, сравнение (65), (154в) и (78) показывает, что 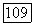 включает поправки третьего порядка к парным функциям, а также трехэлектронные члены
включает поправки третьего порядка к парным функциям, а также трехэлектронные члены

где 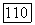 определяется формулами (79) — (81), однако там вместо функций
определяется формулами (79) — (81), однако там вместо функций 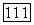 надо подставить
надо подставить 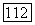
Полная функция 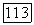 не меняется при преобразовании к локализованным орбиталям [выражение (141)], поскольку величины, входящие в (64), все еще инвариантны при таком преобразовании. Таким образом, значения
не меняется при преобразовании к локализованным орбиталям [выражение (141)], поскольку величины, входящие в (64), все еще инвариантны при таком преобразовании. Таким образом, значения 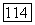 по отдельности остаются неизменными. С другой стороны, ожидается возрастание вклада пар в
по отдельности остаются неизменными. С другой стороны, ожидается возрастание вклада пар в 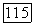 до сравнению с трехэлектронными корреляциями
до сравнению с трехэлектронными корреляциями 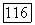 Локализованные пары (которые, однако, не обязательно совпадают с вандерваальсовыми членами) должны быть в большей мере расцепленными (независимыми друг от друга) по сравнению с тем, что имело место для парных корреляционных функций, основанных на молекулярных орбиталях. Тот же аргумент [10] применяют к
Локализованные пары (которые, однако, не обязательно совпадают с вандерваальсовыми членами) должны быть в большей мере расцепленными (независимыми друг от друга) по сравнению с тем, что имело место для парных корреляционных функций, основанных на молекулярных орбиталях. Тот же аргумент [10] применяют к 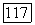 [выражение (82)]. Из этого следует, что лучшая энергия и волновая функция должны бы получаться путем минимизации большей части энергии, содержащей вариационные энергии локализованных пар во «всех порядках» (т. е. включая
[выражение (82)]. Из этого следует, что лучшая энергия и волновая функция должны бы получаться путем минимизации большей части энергии, содержащей вариационные энергии локализованных пар во «всех порядках» (т. е. включая 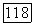 а не до второго порядка, как в (1576). Полученные в результате этого функции
а не до второго порядка, как в (1576). Полученные в результате этого функции 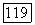 должны бы были дать меньшие значения для остаточных членов в
должны бы были дать меньшие значения для остаточных членов в 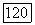
Подробности таких преобразований, применяемых в полной форме (82), пока еще в стадии изучения [105]. По аналогии с (86) и из рассмотрения влияний окружения на связь, влияния ионных остовов и т. д. можно предположить, что вариационное выражение для локализованных пар во «всех порядках» дается приведенным ниже выражением (162). В любом случае может оказаться достаточным учет только 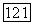 так что можно работать с выражением (157).
так что можно работать с выражением (157).
Г. Влияние молекулярного окружения на связь
Экспериментальные энергии связи мало меняются от молекулы к молекуле, например в насыщенных углеводородах эти изменения лежат в пределах 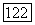 . Возникает вопрос о том, совместимы ли эффекты молекулярного окружения, подразумеваемые в данных выше выражениях с такого рода постоянством энергий связи.
. Возникает вопрос о том, совместимы ли эффекты молекулярного окружения, подразумеваемые в данных выше выражениях с такого рода постоянством энергий связи.
Проблему молекулярного окружения можно подразделить на части: молекулярное окружение в хартри-фоковском приближении и корреляционная часть.
Локализованные орбитали из (145) подвержены действию потенциала всего хартри-фоковского «фона» 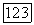 т. е. потенциала молекулы; кроме того, на них оказывает влияние принцип исключения, приводящий к тому, что они выбираются ортогональными ко всем другим
т. е. потенциала молекулы; кроме того, на них оказывает влияние принцип исключения, приводящий к тому, что они выбираются ортогональными ко всем другим 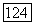 а также приводящий к обменным членам. Потенциал
а также приводящий к обменным членам. Потенциал 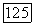 включающий притяжения ядер, вызывает поляризацию определенных групп у отрицательных ионов. Обмен и ортогональность приводят к несвязывающим отталкиваниям [106, 107]. И эти два последних эффекта и потенциал
включающий притяжения ядер, вызывает поляризацию определенных групп у отрицательных ионов. Обмен и ортогональность приводят к несвязывающим отталкиваниям [106, 107]. И эти два последних эффекта и потенциал 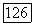 вследствие экранировки ядер имеют малый радиус действия. Таким образом, на орбитали
вследствие экранировки ядер имеют малый радиус действия. Таким образом, на орбитали 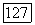 в основном воздействует их непосредственное окружение. Эта орбитальная часть проблемы была исследована [42—44, 49, 50], хотя, по-видимому, необходимо провести еще значительную работу, чтобы рассмотреть, каким образом локализованы
в основном воздействует их непосредственное окружение. Эта орбитальная часть проблемы была исследована [42—44, 49, 50], хотя, по-видимому, необходимо провести еще значительную работу, чтобы рассмотреть, каким образом локализованы 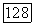 и насколько они постоянны.
и насколько они постоянны.
Корреляционная часть функции связи, назовем ее 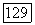 имеют те же самые пространственные части), также подвержена действию полного молекулярного хартри-фоковского
имеют те же самые пространственные части), также подвержена действию полного молекулярного хартри-фоковского 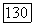 хотя
хотя 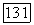 в (1576) являются почти несвязанными друг с другом.
в (1576) являются почти несвязанными друг с другом.
Как и в случае атома (см. § 24), влияние окружения на 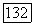 через
через 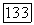 проявляется двояким образом:
проявляется двояким образом:
1. Потенциал 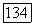 входящий в формулы
входящий в формулы

включает в себя кулоновский и обменный потенциалы всех 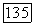 молекулярных орбиталей. Однако, применяя (141а), потенциал
молекулярных орбиталей. Однако, применяя (141а), потенциал  можно записать через кулоновский и обменный потенциалы для
можно записать через кулоновский и обменный потенциалы для 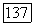 Далее видно, что действие
Далее видно, что действие 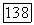 например
например 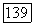 -связи
-связи 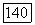 в в этане, экранировано кулоновской частью потенциала
в в этане, экранировано кулоновской частью потенциала 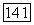 этой связи. Этот экранированный потенциал обращается в нуль на расстоянии до
этой связи. Этот экранированный потенциал обращается в нуль на расстоянии до 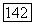 -свя-зей других метальных групп. Несомненно,
-свя-зей других метальных групп. Несомненно, 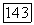 -связь весьма слабо экранирована
-связь весьма слабо экранирована 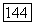 электронами от заряда своего собственного атома углерода; однако в § 24 мы упоминали, насколько нечувствительны «динамические» корреляции к изменениям заряда ядра. Судя по Не,
электронами от заряда своего собственного атома углерода; однако в § 24 мы упоминали, насколько нечувствительны «динамические» корреляции к изменениям заряда ядра. Судя по Не, 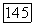 (см. табл. 4) и значениям корреляции
(см. табл. 4) и значениям корреляции 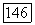 , следует ожидать, что влияние этих эффектов окружения
, следует ожидать, что влияние этих эффектов окружения 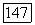 -типа, скажем, на
-типа, скажем, на 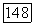 -связь будет не больше
-связь будет не больше 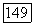 т. е. в пределах точности экспериментальных энергий связи.
т. е. в пределах точности экспериментальных энергий связи.
2. Функции ортогональны к полным молекулярным орбиталям 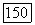 [выражение (157г)]. Согласно (141а), это ведет к следующим результатам:
[выражение (157г)]. Согласно (141а), это ведет к следующим результатам:

Таким образом, как и парная корреляционная функция (§ 10), функция 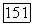 ортогональна не только к своим собственным
ортогональна не только к своим собственным 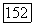 (аналогично влиянию
(аналогично влиянию 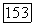 но также ко всем другим функциям
но также ко всем другим функциям 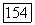 Последний «эффект запрета» теперь представляет другой тип влияния окружения. Можно ожидать, что влияние его на
Последний «эффект запрета» теперь представляет другой тип влияния окружения. Можно ожидать, что влияние его на 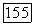 -связи меньше
-связи меньше 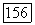 Даже в атоме лития (см. табл. 4), где электроны расположены концентрически и, следовательно, сильнее запрет,
Даже в атоме лития (см. табл. 4), где электроны расположены концентрически и, следовательно, сильнее запрет, 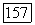 -ортогональность изменяет 812 для
-ортогональность изменяет 812 для 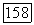 лишь примерно на
лишь примерно на 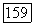 Случай остова
Случай остова 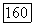 конфигурацией (см. § 25) аналогичен. Соотношения (1416) переводят эту конфигурацию в
конфигурацией (см. § 25) аналогичен. Соотношения (1416) переводят эту конфигурацию в 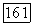 как в случае
как в случае 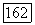 рассмотренном ниже. Энергии
рассмотренном ниже. Энергии 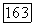 этих локализованных остовов должны быть подобны
этих локализованных остовов должны быть подобны 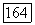 для
для 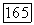 и располагаться, вероятно, внутри интервала
и располагаться, вероятно, внутри интервала 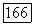
Из этих рассмотрений, по-видимому, следует, что влияние окружения во всяком случае, пока речь идет о корреляционной части, находится в пределах точности экспериментальных энергий связи. Корреляционные энергии отдельных пар и ионных остовов также, по-видимому, не меняются в пределах указан ной выше точности 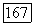
Д. Расчет энергии связей
Так как эффекты окружения весьма малы, корреляционные энергии связей или отдельных пар можно подсчитать порознь, как это делалось, например, для молекулы 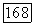
При расчетах с точностью до второго порядка минимизируем (1576), однако в выражении для 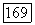 заменяем
заменяем 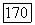 соответствующими частями из этой связи. Таким образом, вместо (
соответствующими частями из этой связи. Таким образом, вместо (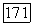 можно написать
можно написать

где 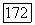 различаются лишь своими спинами.
различаются лишь своими спинами.
По аналогии с уравнением (86) можно предположить, что во всех порядках

где функция 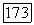 включает в себя лишь потенциалы, вычисленные с помощью
включает в себя лишь потенциалы, вычисленные с помощью 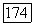 Однако, чтобы подтвердить это выражение, если не удовлетворится (1576), которое сейчас представляется достаточно обоснованным, необходимо дальнейшее исследование.
Однако, чтобы подтвердить это выражение, если не удовлетворится (1576), которое сейчас представляется достаточно обоснованным, необходимо дальнейшее исследование.
Пробные функции могут зависеть от координат 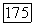 аналогично функции Джеймса — Кулиджа [108, 92] для
аналогично функции Джеймса — Кулиджа [108, 92] для 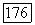 При использовании таких функций часть
При использовании таких функций часть 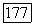 расщепляется вначале в соответствии с
расщепляется вначале в соответствии с 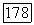
Чтобы рассчитать a priori энергию связи, необходимо обратиться к хартри-фоковской части именно этой связи. Метод Хартри — Фока для локализованных групп находится пока еще в стадии развития [52, 53].
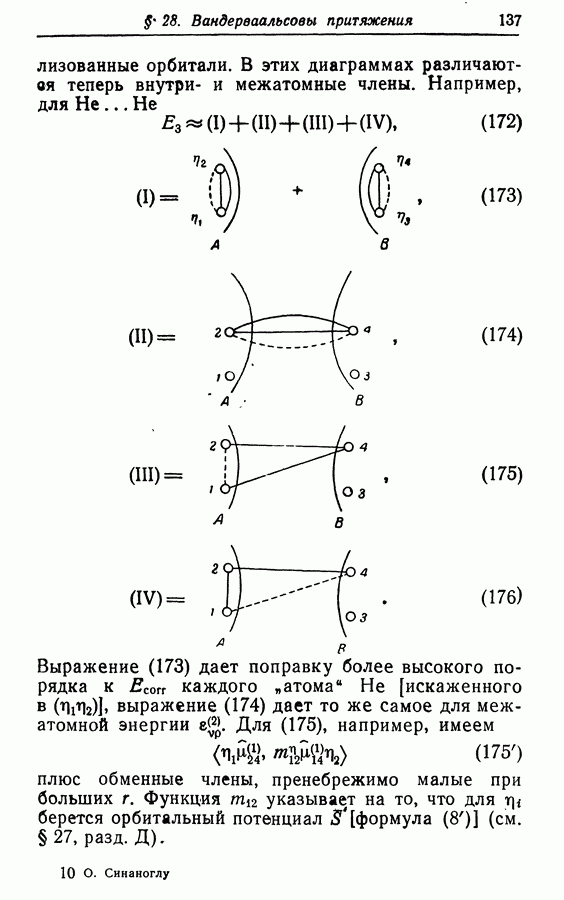
Как следует из (12), (143) и (144), вариационная форма Ел, 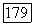 имеет вид
имеет вид

а пробные орбитали 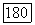 подчинены следующему требованию:
подчинены следующему требованию:

Снова комбинируя члены экранировки и пренебрегая взаимодействиями между группами, получаем















































































































































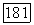 отличаются лишь спинами, и
отличаются лишь спинами, и 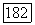 оставшиеся части именно для этой связи.
оставшиеся части именно для этой связи.
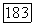 к внутренним оболочкам и выбирая для
к внутренним оболочкам и выбирая для 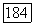 потенциал остова
потенциал остова 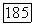 углеродного иона. Этот потенциал затем можно улучшить, используя функцию
углеродного иона. Этот потенциал затем можно улучшить, используя функцию 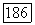 из, скажем, связанной орбитали
из, скажем, связанной орбитали 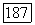 (например в
(например в  или же для расчета
или же для расчета 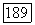 -связи в
-связи в 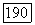 можно взять
можно взять 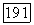 часть из
часть из 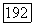 Необходимо рассмотреть, насколько практически выполнимы и хорошо определены подобные действия. Здесь они упомянуты лишь как возможные.
Необходимо рассмотреть, насколько практически выполнимы и хорошо определены подобные действия. Здесь они упомянуты лишь как возможные.

