§ 28. ВАНДЕРВААЛЬСОВЫ ПРИТЯЖЕНИЯ
Общепринято рассматривать вопрос о вандерва-альсовых притяжениях между двумя атомами, например Не  Не, с помощью теории Гайтлера — Лондона. Притяжение при минимальном расстоянии между атомами отчасти обусловлено искажением гайтлер-лондоновских атомных орбиталей (эффектом «орбитальной средней поляризации») [3], а при больших мёжъядерных расстояниях
Не, с помощью теории Гайтлера — Лондона. Притяжение при минимальном расстоянии между атомами отчасти обусловлено искажением гайтлер-лондоновских атомных орбиталей (эффектом «орбитальной средней поляризации») [3], а при больших мёжъядерных расстояниях  оно возникает в основном из-за одновременных возбуждений в обоих
оно возникает в основном из-за одновременных возбуждений в обоих
атомах (т. е. из-за обычных лондоновских дисперсионных сил). Эти два эффекта сравнивались [109] для  в триплетном состоянии при различных значениях
в триплетном состоянии при различных значениях 
Эффект искажения для системы взаимодействующих атомов автоматически учитывается в хартри-фоковских расчетах, основывающихся на молекулярных орбиталях самосогласованного поля. Расчет Ранзила [110] с помощью «наилучшим образом ограниченных» хартри-фоковских молекулярных орбиталей самосогласованного поля хорошо передавал взаимодействие Не  Не, если межатомное расстояние бралось равным приблизительно
Не, если межатомное расстояние бралось равным приблизительно  (т. е. было минимальным)
(т. е. было минимальным)  однако он приводил к слишком малому притяжению для
однако он приводил к слишком малому притяжению для  больших, чем примерно 1,2. Притяжение при больших
больших, чем примерно 1,2. Притяжение при больших  является «межоболочечным» корреляционным эффектом. Этот эффект до сих пор рассматривали с помощью теории лондоновских дисперсионных сил. Его считали ответственным за несвязывающие притяжения между различными частями насыщенной молекулы для того, чтобы получить теплоты образования и изомеризации [2].
является «межоболочечным» корреляционным эффектом. Этот эффект до сих пор рассматривали с помощью теории лондоновских дисперсионных сил. Его считали ответственным за несвязывающие притяжения между различными частями насыщенной молекулы для того, чтобы получить теплоты образования и изомеризации [2].
Теория Лондона использует второй порядок теории возмущений в ее обычном (рэлей-шредингеров-ском) варианте, т. е. в виде бесконечной суммы по полному базисному набору. Этот набор для системы из двух взаимодействующих молекул  состоит из всех произведений собственных функций для систем
состоит из всех произведений собственных функций для систем  по отдельности. Таким образом, в теории Лондона предполагается не только отсутствие перекрывания между основными состояниями атомов, но также и отсутствие перекрывания между любыми виртуальными атомными возбужденными состояниями [3].
по отдельности. Таким образом, в теории Лондона предполагается не только отсутствие перекрывания между основными состояниями атомов, но также и отсутствие перекрывания между любыми виртуальными атомными возбужденными состояниями [3].
Рассмотрим, например, два атома водорода, имеющие одинаковые спины и расположенные на
расстоянии  [12]. При таком разделении атомов перекрывание между орбиталями основных
[12]. При таком разделении атомов перекрывание между орбиталями основных  -состояний фактически пренебрежимо мало. Однако Боровский радиус состояния пропорционален
-состояний фактически пренебрежимо мало. Однако Боровский радиус состояния пропорционален  так что даже следующее виртуальное возбужденное
так что даже следующее виртуальное возбужденное  -состояние, которое должно бы давать вклад в лондоновские силы, обнаруживает большое перекрывание. Следовательно, непосредственное рассмотрение не подтверждает применимость лондоновской формулы даже для вполне разумных межъядерных расстояний
-состояние, которое должно бы давать вклад в лондоновские силы, обнаруживает большое перекрывание. Следовательно, непосредственное рассмотрение не подтверждает применимость лондоновской формулы даже для вполне разумных межъядерных расстояний  Таким образом, использование этой формулы применительно к внутренним оболочкам молекулы также требует подтверждения [3, 104].
Таким образом, использование этой формулы применительно к внутренним оболочкам молекулы также требует подтверждения [3, 104].
Экспериментальное значение коэффициента при члене с  меньше (более отрицательно) [2], чем его лондоновское значение на множитель 2,2 для
меньше (более отрицательно) [2], чем его лондоновское значение на множитель 2,2 для  но оно отличается лишь на множитель 1,2 для Не и 1,09 для
но оно отличается лишь на множитель 1,2 для Не и 1,09 для  Донаф и Питцер [2] приписали это корреляциям более высокого порядка, нежели второй, которые становятся существенными в больших атомах (см. ниже).
Донаф и Питцер [2] приписали это корреляциям более высокого порядка, нежели второй, которые становятся существенными в больших атомах (см. ниже).
Мак Вини [30] распространил применение групповых функций [выражение (146)] на межмолекулярные силы, добавляя к волновым функциям, рассчитанным во втором порядке, набор многоэлектронных функций  для каждого атома
для каждого атома  соответственно. Функции, отвечающие атому А, предполагались ортогональными в смысле уравнения (1) не только друг к другу, но также и к функциям, отвечающим атому В. Нет рецептов для получения этих функций. Таким образом, слишком жесткое требование (1) относительно ортогональности между группами функций
соответственно. Функции, отвечающие атому А, предполагались ортогональными в смысле уравнения (1) не только друг к другу, но также и к функциям, отвечающим атому В. Нет рецептов для получения этих функций. Таким образом, слишком жесткое требование (1) относительно ортогональности между группами функций  эквивалентно [3] постулату отсутствия перекрывания в теории Лондона. Эти ограничения изучались применительно к взаимодействию Не... Не [29].
эквивалентно [3] постулату отсутствия перекрывания в теории Лондона. Эти ограничения изучались применительно к взаимодействию Не... Не [29].
Многоэлектронная теория [3, 104] дает вандер-ваальсовы притяжения для всех  как для межмолекулярных, так и для внутримолекулярных случаев, без предположений относительно перекрывания. Нет необходимости различать исходные приближения или же строить различные теории: одну для больших
как для межмолекулярных, так и для внутримолекулярных случаев, без предположений относительно перекрывания. Нет необходимости различать исходные приближения или же строить различные теории: одну для больших 
другую для  близких
близких  Многоэлектронная теория хорошо покрывает трудную [111] промежуточную область.
Многоэлектронная теория хорошо покрывает трудную [111] промежуточную область.
А. Внутримолекулярные притяжения
Члены  относящимися к пространственно различным локализованным спин-орбиталям, существенны для межгрупповых корреляций. Они описывают обобщенные притяжения Лондона — ван дер Ваальса. Однако
относящимися к пространственно различным локализованным спин-орбиталям, существенны для межгрупповых корреляций. Они описывают обобщенные притяжения Лондона — ван дер Ваальса. Однако  представлено в замкнутом виде, не разложенном по атомгаым базисным наборам.
представлено в замкнутом виде, не разложенном по атомгаым базисным наборам.
Соотношения (148) и (149) определяют вандер-ваальсовы притяжения между локализованными группами при использовании любой функции  (такой, как хартри-фоковская, или из метода конфигурационного взаимодействия) в молекулярно-орбитальном описании. На этом пути, т. е. при переходе к локализованному описанию, можно получить, например, корреляционную энергию между двумя
(такой, как хартри-фоковская, или из метода конфигурационного взаимодействия) в молекулярно-орбитальном описании. На этом пути, т. е. при переходе к локализованному описанию, можно получить, например, корреляционную энергию между двумя  -связями в
-связями в 
Членов второго порядка  [выражение (1526)] оказывается достаточно, чтобы рассчитать, скажем, энергию изомеризации с притяжениями между связями, например
[выражение (1526)] оказывается достаточно, чтобы рассчитать, скажем, энергию изомеризации с притяжениями между связями, например  Каждая группа имеет всего несколько электронов, как в случае взаимодействия Не
Каждая группа имеет всего несколько электронов, как в случае взаимодействия Не  Не, и поправки высших порядков, упоминавшиеся выше [2], например, для
Не, и поправки высших порядков, упоминавшиеся выше [2], например, для  здесь не нужны. Использование лондоновских сил в углеводородах [2] можно обосновать [3] с помощью выражения (1526) по крайней мере до того, как мы коснемся проблемы перекрывания базисных функций и эффектов запрета.
здесь не нужны. Использование лондоновских сил в углеводородах [2] можно обосновать [3] с помощью выражения (1526) по крайней мере до того, как мы коснемся проблемы перекрывания базисных функций и эффектов запрета.
Вандерваальсовы члены можно получить отдельно без проведения расчета для полной молекулы. Межгрупповые пары нужно было бы минимизировать в (157б), используя пробные функции  Связь между различными
Связь между различными  которые нужны, чтобы
которые нужны, чтобы
связать с определенной одной или несколькими парными функциями  Например, в
Например, в 

соответственно

когда  Это обусловлено тем, что в выражении (141) а спин-орбитали преобразуются независимо от
Это обусловлено тем, что в выражении (141) а спин-орбитали преобразуются независимо от  спин-орбиталей. Особенно прост Не
спин-орбиталей. Особенно прост Не  Не случай, в котором не будет внутриатомных пар
Не случай, в котором не будет внутриатомных пар  Расчет функций
Расчет функций  и 824 в таком случае совпадает с расчетом корреляционной энергии
и 824 в таком случае совпадает с расчетом корреляционной энергии  в
в  -состоянии [109]. При больших
-состоянии [109]. При больших  пренебрегая разницей между оса и
пренебрегая разницей между оса и  парами, имеем
парами, имеем

Использование  исключает проблему перекрывания.
исключает проблему перекрывания.
Уже применение  дает вандерваальсовы притяжения Не
дает вандерваальсовы притяжения Не  или же
или же  с достаточной точностью. Это можно показать, сравнивая результаты, полученные с помощью лондоновской формы
с достаточной точностью. Это можно показать, сравнивая результаты, полученные с помощью лондоновской формы  и экспериментальных данных [2]. Однако для больших атомов
и экспериментальных данных [2]. Однако для больших атомов  использование только
использование только  приводит [2] к ошибочному множителю порядка 2,2. Чтобы объяснить множитель 2, мы рассмотрим ниже отношения между результатами многоэлектронной теории и результатами Донафа и Питцера [2]. Это покажет также, каким образом можно подсчитать поправки при всех
приводит [2] к ошибочному множителю порядка 2,2. Чтобы объяснить множитель 2, мы рассмотрим ниже отношения между результатами многоэлектронной теории и результатами Донафа и Питцера [2]. Это покажет также, каким образом можно подсчитать поправки при всех  не обращаясь к аппроксимациям лондоновского типа [2].
не обращаясь к аппроксимациям лондоновского типа [2].
Ошибки, по-видимому, возникают не из-за каких-либо поправок, которые обычно появляются для каждого парного члена  в (167) подобно поправкам от непрерывного спектра к формуле Лондона. Они были бы в таком случае пропорциональны
в (167) подобно поправкам от непрерывного спектра к формуле Лондона. Они были бы в таком случае пропорциональны  и давали бы аналогичные поправочные множители как для Не и
и давали бы аналогичные поправочные множители как для Не и  так и для
так и для 
Полная энергия, даваемая многоэлектронной теорией, включает корреляции  [соотношения (78) —
[соотношения (78) —
(80)]. Эти многоэлектронные корреляции пренебрежимо малы по сравнению с полной энергией атома Еоотт или с энергией связи стабильной молекулы (см. § 18). С другой стороны, корреляционная энергия (167) между двумя несвязанными атомами весьма мала, чтобы стать основой нашего подхода. Лондоновское притяжение между двумя атомами неона [2], расположенными на расстоянии 4,0 А  равно всего лишь
равно всего лишь  по сравнению с Есотт
по сравнению с Есотт  отдельного атома неона [1]. В таком случае члены
отдельного атома неона [1]. В таком случае члены  могли бы оказаться сравнимыми с Следовательно, рассмотрение этих многоэлектронных членов
могли бы оказаться сравнимыми с Следовательно, рассмотрение этих многоэлектронных членов  становится особенно существенным в связи с межмолекулярными силами.
становится особенно существенным в связи с межмолекулярными силами.
Предположим, что, использовав соотношения (141) и (145), мы преобразовали (69а):

аналогичные результаты получим и для  [выражения (76) и (77)]. В таком случае энергию можно вновь оценить при помощи диаграмм, как это делалось в § 18 с применением молекулярных орбиталей. За исключением
[выражения (76) и (77)]. В таком случае энергию можно вновь оценить при помощи диаграмм, как это делалось в § 18 с применением молекулярных орбиталей. За исключением  [выражение (157в)], формула для энергии обычно имеет тот же вид; что и (159) и (79) — (81), но в ней
[выражение (157в)], формула для энергии обычно имеет тот же вид; что и (159) и (79) — (81), но в ней  заменены на
заменены на 
С помощью молекулярно-орбитальных пар мы имели бы [пренебрегая (81)]:

Аналогичные диаграммы можно нарисовать для энергии, полученной для функции (170), используя
Аналогично выглядит выражение для энергии, полученное не по теории возмущений, т. е. с помощью 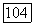 Функции
Функции 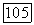 в (172), появляющиеся там согласно
в (172), появляющиеся там согласно 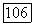 заменяются на
заменяются на 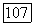 которые минимизируют полную энергию пар [выражение (162), включая (173) или (174), но отнюдь не (1576)].
которые минимизируют полную энергию пар [выражение (162), включая (173) или (174), но отнюдь не (1576)].
Члены, выбранные Донафом и Питцером [2] [см» их уравнения (21) и (22)], соответствуют 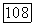 [выражение (167)] и (175) в гайтлер-лондоновском приближении
[выражение (167)] и (175) в гайтлер-лондоновском приближении 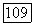 перекрыванием пренебрегают). Их волновая функция не содержит внутриатомные
перекрыванием пренебрегают). Их волновая функция не содержит внутриатомные 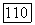 так что не появляются члены
так что не появляются члены  дополнительном диполь-дипольном приближении исчезает член
дополнительном диполь-дипольном приближении исчезает член 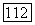 [но не
[но не 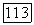 или
или 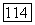
Поправочный множитель [2] 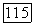 равен 1,2 в Не и 2,2 в
равен 1,2 в Не и 2,2 в 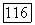 где
где

Число диаграмм типа 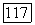 равно
равно

по сравнению с числом пар папь в Поэтому в Не 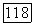 Не
Не

а если взять лишь наиболее поляризуемые 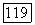 электроны в
электроны в 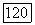 то получим
то получим

Считая, что для отдельных диаграмм приблизительно справедливо то же самое отношение, можно показать, что поправочный множитель примерно в 5 раз больше в тяжелых атомах, нежели в гелии. Отношение наблюдаемых поправок примерно равно
пар, таких, как в (168), некоторые члены из треугольных диаграмм в молекулярно-орбитальноад описании можно непосредственно сопоставить с 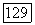 и возможно упростить расчеты этой величины. Необходима дальнейшая работа в этом направлении (см. дополнение автора в конце данной работы).
и возможно упростить расчеты этой величины. Необходима дальнейшая работа в этом направлении (см. дополнение автора в конце данной работы).
Члены 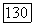 и
и 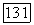 по-видимому, остаются основными членами в (172) при меньших
по-видимому, остаются основными членами в (172) при меньших 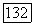 Как член
Как член 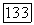 так и член
так и член 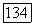 содержат
содержат 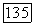 однако Следовательно, можно ожидать, что член
однако Следовательно, можно ожидать, что член  значительно меньше члена
значительно меньше члена 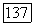 Можно оценить величину члена
Можно оценить величину члена 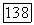 (влияние атомной корреляции на вандерваальсово притяжение). Она, по-видимому, мала, поскольку
(влияние атомной корреляции на вандерваальсово притяжение). Она, по-видимому, мала, поскольку 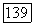 почти ортогональны, а невелико. Величину
почти ортогональны, а невелико. Величину 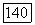 в (157в) также необходимо рассчитать при меньших
в (157в) также необходимо рассчитать при меньших 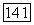 (см. дополнение автора).
(см. дополнение автора).
В заключении этого раздела отметим, что многоэлектронная теория дает возможность рассчитывать непосредственно межмолекулярные притяжения при всех 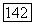 хотя они и представляют собой по абсолютной величине малые корреляционные эффекты.
хотя они и представляют собой по абсолютной величине малые корреляционные эффекты.
Расчеты по методу Хартри — Фока, например, для 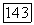 -части составной системы
-части составной системы 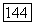 остаются решающей предпосылкой для рассмотрения подобных больших систем. Для дальнодействующей части притяжения необходимы лишь хартри-фоковские функции отдельных атомов; поправки, которые обычно возникают от использования точных атомных функций, по-видимому, несущественны.
остаются решающей предпосылкой для рассмотрения подобных больших систем. Для дальнодействующей части притяжения необходимы лишь хартри-фоковские функции отдельных атомов; поправки, которые обычно возникают от использования точных атомных функций, по-видимому, несущественны.































































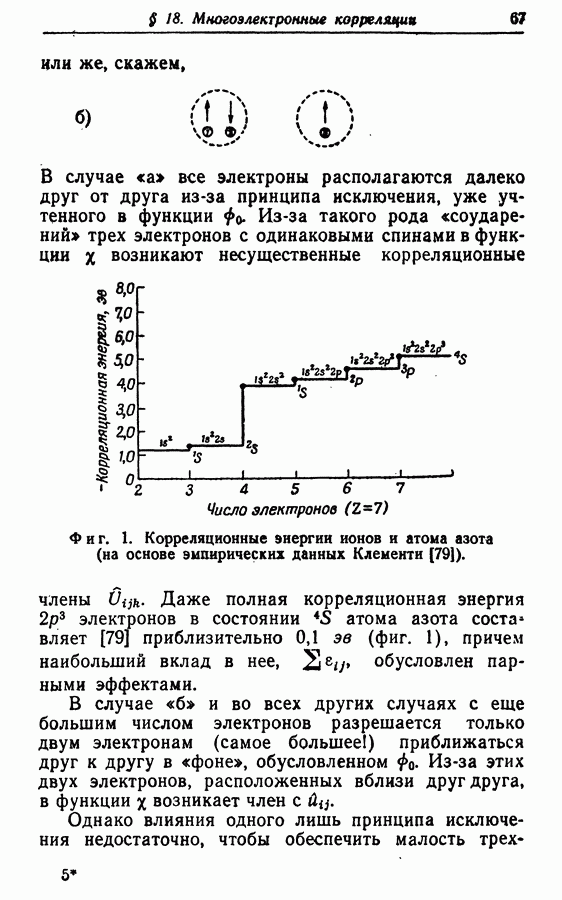






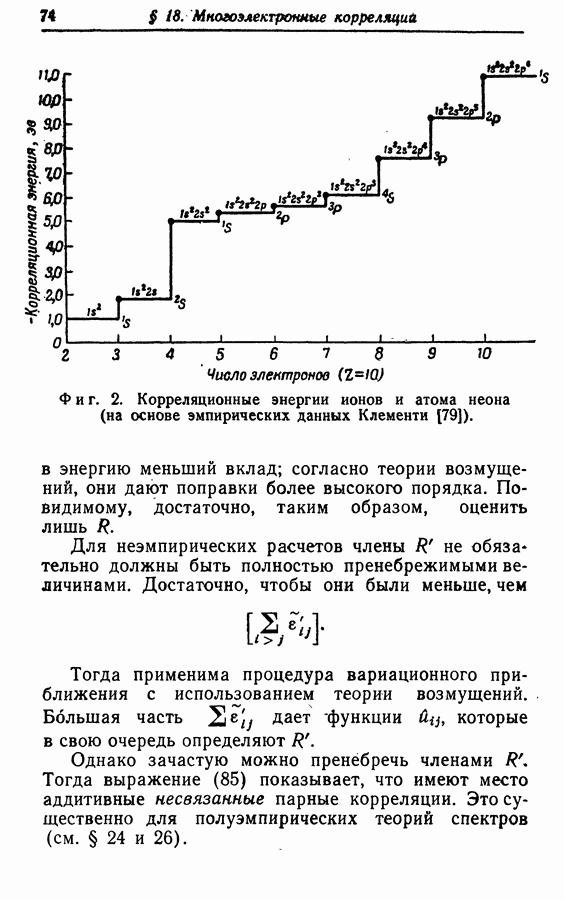



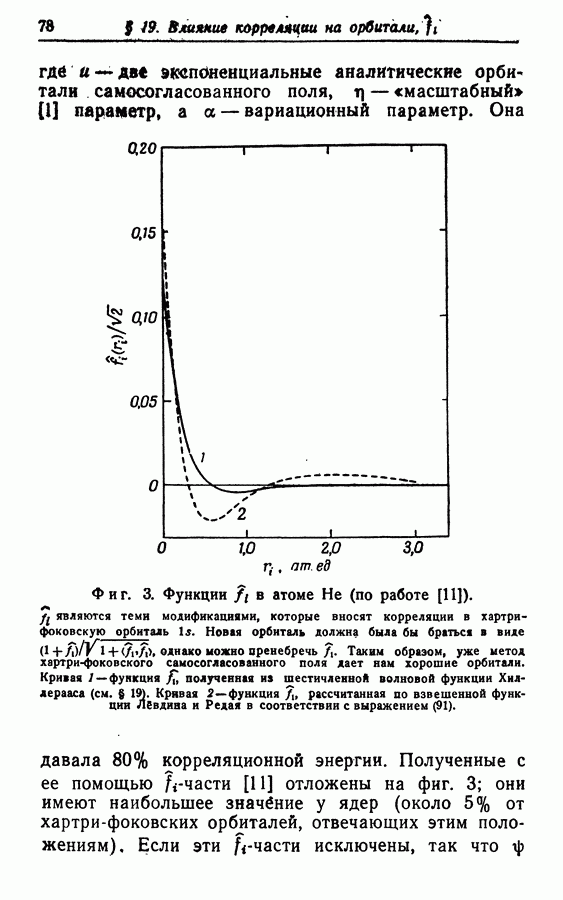





























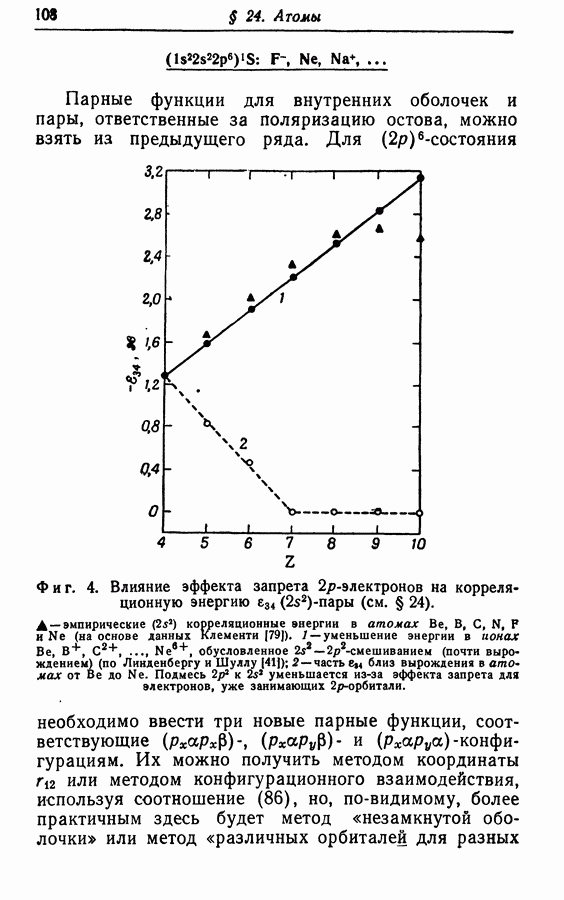









































 обладала соответствующей симметрией (как обсуждалось для атомов в § 23), нуждается в дальнейшем исследовании [105].
обладала соответствующей симметрией (как обсуждалось для атомов в § 23), нуждается в дальнейшем исследовании [105].
 можно сопоставить с локализованными корреляциями
можно сопоставить с локализованными корреляциями  прямо применяя выражение (149). Этим можно обосновать полуэмпирический метод, с помощью которого из легко оцениваемых локализованных корреляций получаются молекулярно-орбитальные парные корреляции. Подобный метод может оказаться полезным в расчете ультрафиолетовых спектральных распределений, основанном на простой теории молекулярных орбиталей, и дать оценки сдвигов, обусловленных корреляциями. Было бы практически интересно рассмотреть
прямо применяя выражение (149). Этим можно обосновать полуэмпирический метод, с помощью которого из легко оцениваемых локализованных корреляций получаются молекулярно-орбитальные парные корреляции. Подобный метод может оказаться полезным в расчете ультрафиолетовых спектральных распределений, основанном на простой теории молекулярных орбиталей, и дать оценки сдвигов, обусловленных корреляциями. Было бы практически интересно рассмотреть  -переходы.
-переходы.
 [110] хартри-фоковская часть объясняет постепенное искажение атомных орбиталей
[110] хартри-фоковская часть объясняет постепенное искажение атомных орбиталей  и замену их эквивалентными орбиталями
и замену их эквивалентными орбиталями  при меньших
при меньших  Ранзиловские [110] хартри-фоковские функции
Ранзиловские [110] хартри-фоковские функции  будучи преобразованы [57] к различным
будучи преобразованы [57] к различным  позволяют получить эти орбитали. Поведение молекулы
позволяют получить эти орбитали. Поведение молекулы  вплоть до диссоциации Несбет рассмотрел с помощью метода Хартри — Фока, непосредственно используя в нем эквивалентные орбитали [5].
вплоть до диссоциации Несбет рассмотрел с помощью метода Хартри — Фока, непосредственно используя в нем эквивалентные орбитали [5].
 пригодно также для корреляционных частей [выражения (77) и (153а)] волновой функции и энергии и приводит к обобщенным лондон-вандерваальсовым членам при всех
пригодно также для корреляционных частей [выражения (77) и (153а)] волновой функции и энергии и приводит к обобщенным лондон-вандерваальсовым членам при всех  Последние являются теми парными функциями в (1526), которые имеют две локализованные орбитали
Последние являются теми парными функциями в (1526), которые имеют две локализованные орбитали  с пространственными частями,
с пространственными частями,

 означает межмолекулярную вандерваальсову часть; атом А содержит
означает межмолекулярную вандерваальсову часть; атом А содержит  электронов, а атом
электронов, а атом  причем
причем  так что в выражении (167) имеется папъ членов. Даже на малых расстояниях
так что в выражении (167) имеется папъ членов. Даже на малых расстояниях  мы можем говорить об индивидуальных «молекулах» или «атомах», если будем рассматривать локализованные орбитали как искаженные формы орбиталей отдельных атомов или молекул.
мы можем говорить об индивидуальных «молекулах» или «атомах», если будем рассматривать локализованные орбитали как искаженные формы орбиталей отдельных атомов или молекул.
 большая часть усилий тратится на получение больших внутриатомных корреляций. При описании с помощью эквивалентных орбиталей рассматриваются как раз межатомные пары [выражение (167)] с применением
большая часть усилий тратится на получение больших внутриатомных корреляций. При описании с помощью эквивалентных орбиталей рассматриваются как раз межатомные пары [выражение (167)] с применением  мало по сравнению с полной величиной
мало по сравнению с полной величиной  и поэтому необходимо рассчитать также
и поэтому необходимо рассчитать также  [выражение (157в)]. Внутриатомные корреляции (как в Не) не чувствительны к детальному виду атомных орбиталей и потенциалам IV Следовательно, даже при меньших
[выражение (157в)]. Внутриатомные корреляции (как в Не) не чувствительны к детальному виду атомных орбиталей и потенциалам IV Следовательно, даже при меньших  атомные орбитали искажаются в орбитали
атомные орбитали искажаются в орбитали  Полагают, что они сократят чисто атомные вклады для
Полагают, что они сократят чисто атомные вклады для 

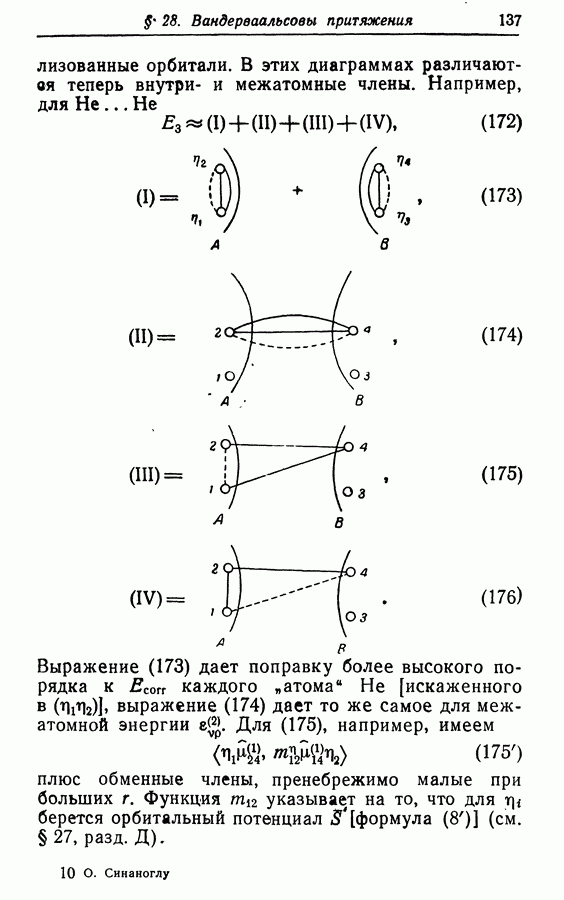
 каждого атома Не [искаженного в
каждого атома Не [искаженного в  выражение (174) дает то же самое для межатомной энергии
выражение (174) дает то же самое для межатомной энергии 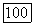 Для (175), например, имеем
Для (175), например, имеем

 Функция
Функция  указывает на то, что для
указывает на то, что для 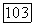 берется орбитальный потенциал 3 [формула (8)] (см. § 27, разд. Д).
берется орбитальный потенциал 3 [формула (8)] (см. § 27, разд. Д).
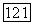 Однако трудно понять, почему этот множитель остается примернр тем же самым в
Однако трудно понять, почему этот множитель остается примернр тем же самым в 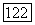
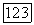 Например, член
Например, член 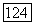 [выражение (175)] можно подсчитать с помощью
[выражение (175)] можно подсчитать с помощью 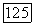 и
и 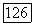 в замкнутом виде.
в замкнутом виде.
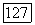 в молекулярно-орбитальном описании. Как упоминалось в § 27 разд. В, относительная величина вклада пар [выражение (173)] по сравнению с частью, даваемой треугольными диаграммами (175) и (176), должна быть больше в
в молекулярно-орбитальном описании. Как упоминалось в § 27 разд. В, относительная величина вклада пар [выражение (173)] по сравнению с частью, даваемой треугольными диаграммами (175) и (176), должна быть больше в 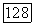 -описании, нежели в молекулярно-орбитальном описании [выражение (171)]. Тем не менее благодаря простым преобразованиям отдельных
-описании, нежели в молекулярно-орбитальном описании [выражение (171)]. Тем не менее благодаря простым преобразованиям отдельных