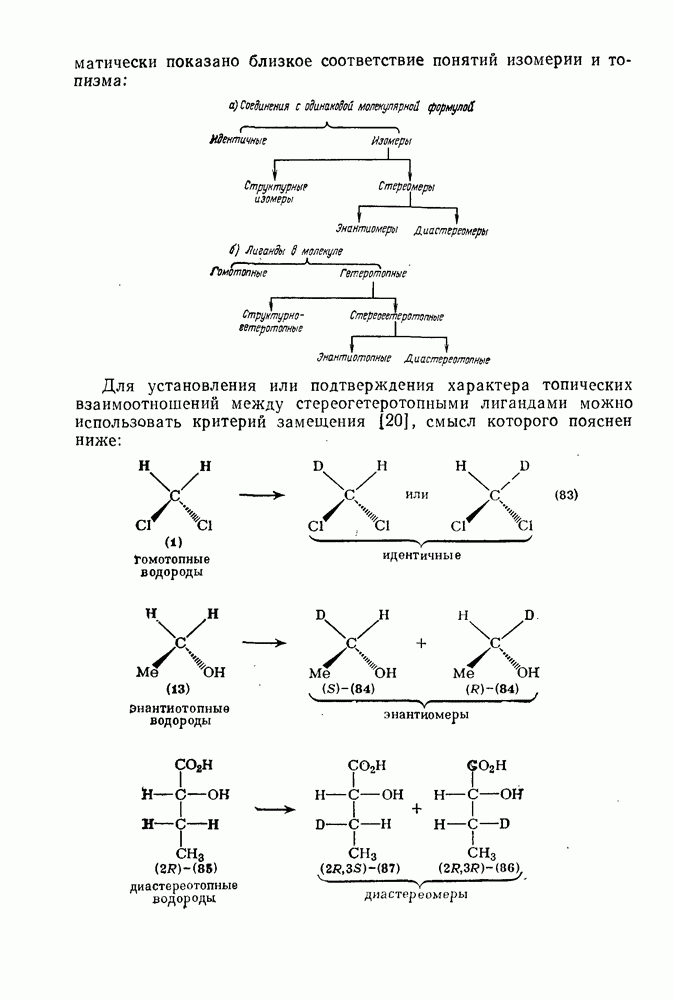
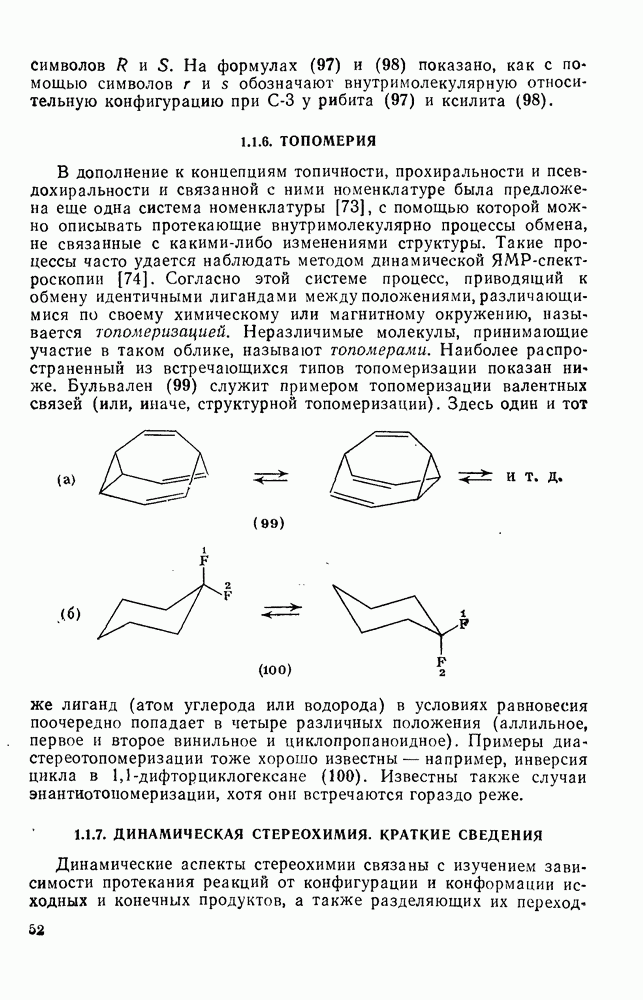
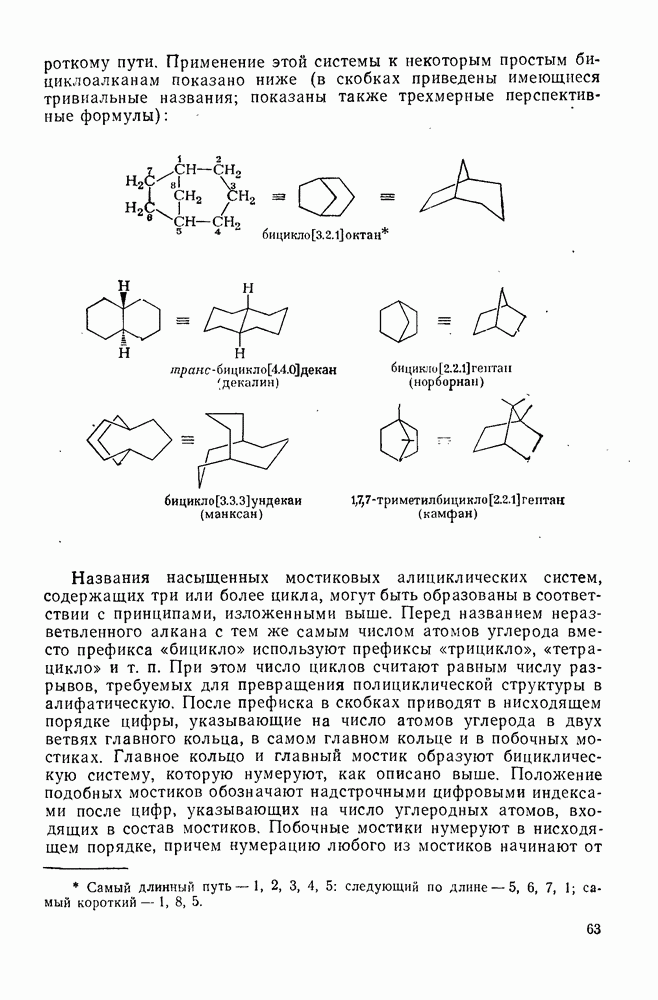
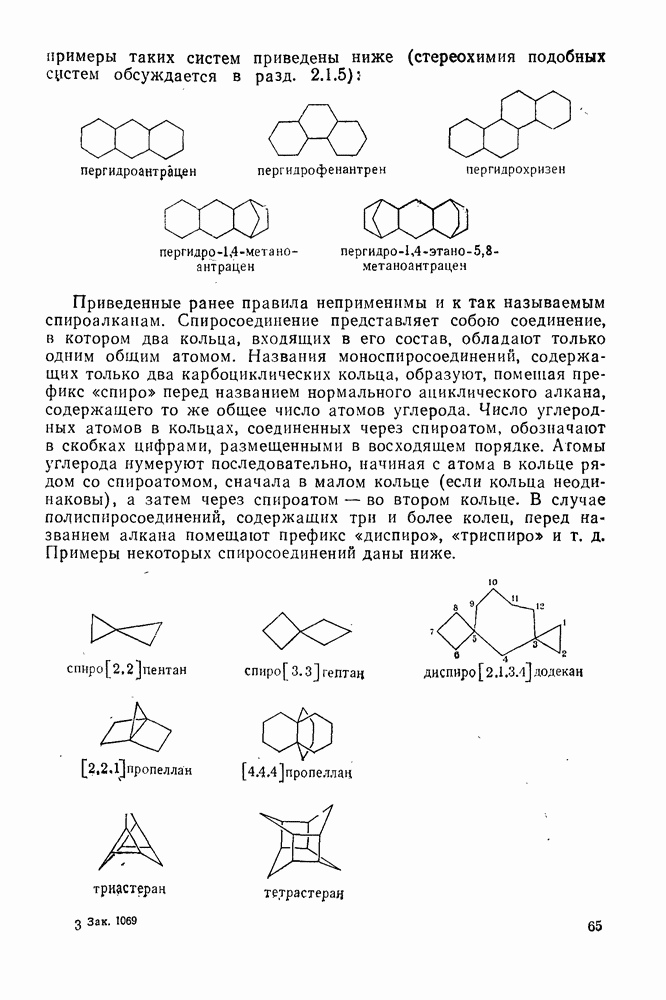
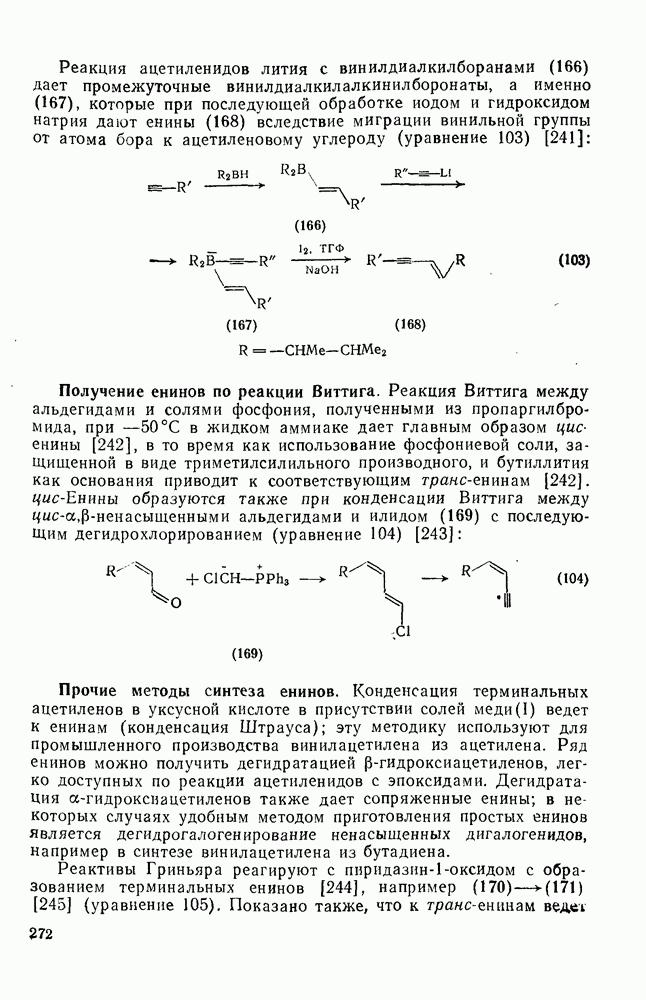
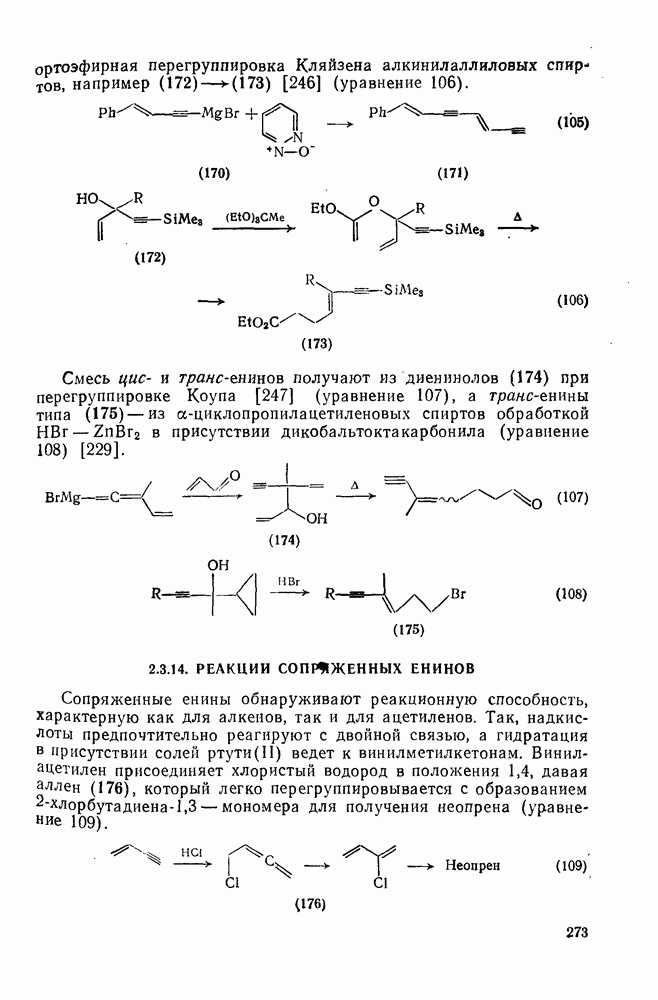
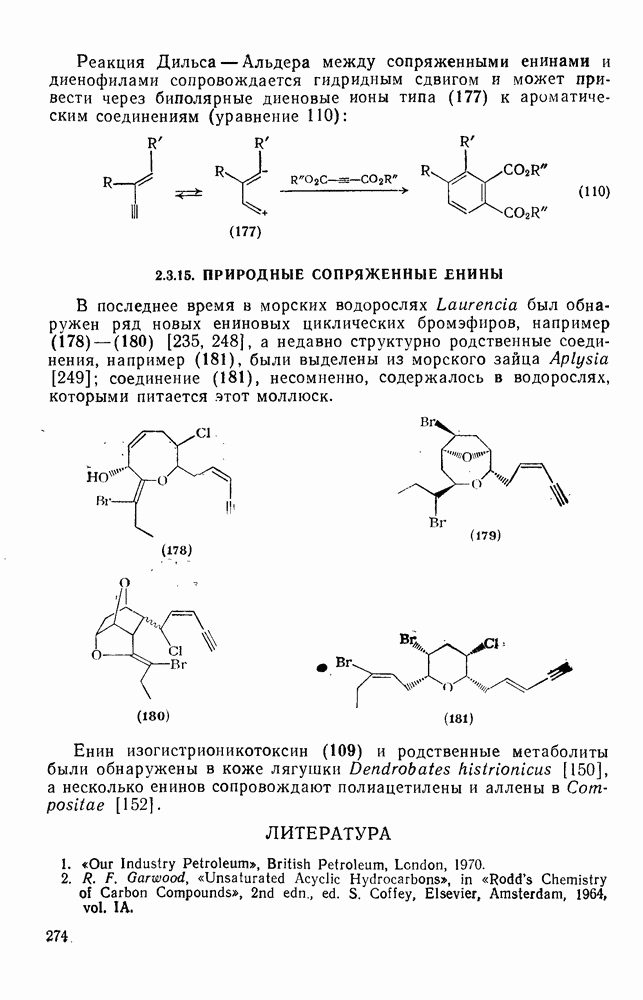
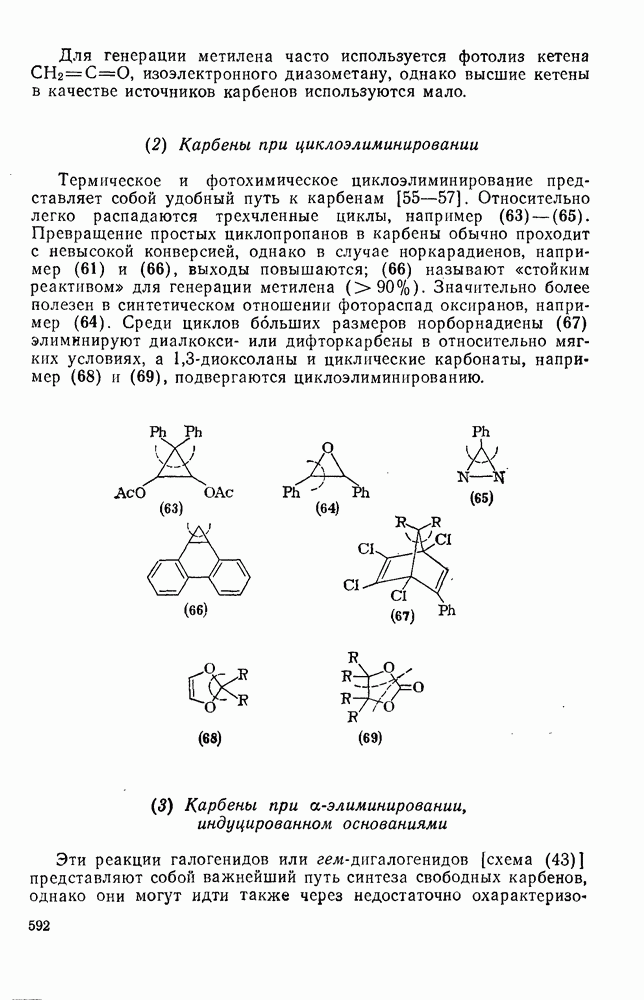
2.8.1.2. Методы генерации и реакции радикалов
Типичные радикальные реакции, например галогенирование ял-канов, присоединение к олефинам, винильная полимеризация, гемолитическое ароматическое замещение, включают последователь ность всех или некоторых элементарных стадий, приведенных ниже [уравнения  ].
].

Диспропорционирование 
 -СНз (13)
-СНз (13)
Радикальные реакции начинаются с разложения какого-нибудь лабильного соединения как первичного источника радикалов (инициирование). Затем следует ряд реакций между радикалами и молекулами (рост цепи) [см., например, уравнения  ], которые обычно приводят к продуктам реакции; заканчиваются радикальные реакции рекомбинацией или диспропорционированием радикалов (обрыв цепи) [см., например, уравнения (12) и (13)]. Для химии радикалов более обычны цепные реакции в сравнении с ионными реакциями: новый радикал, образующийся за счет реакций присоединения или отрыва, часто способен быстро взаимодействовать с молекулой субстрата с образованием другого радикала, который ведет цепь [см. например, схемы (14) и (21)] до тех пор, пока она не оборвется, за счет взаимодействия между радикалами. Развитие (продолжение) цепи обусловлено высокой реакционной способностью радикалов, ведущих цепь, в радикал-молекулярных реакциях. Благодаря этому сохраняется низкая концентрация радикалов, а следовательно, и невысокая скорость радикал-радикальных реакций обрыва цепи.
], которые обычно приводят к продуктам реакции; заканчиваются радикальные реакции рекомбинацией или диспропорционированием радикалов (обрыв цепи) [см., например, уравнения (12) и (13)]. Для химии радикалов более обычны цепные реакции в сравнении с ионными реакциями: новый радикал, образующийся за счет реакций присоединения или отрыва, часто способен быстро взаимодействовать с молекулой субстрата с образованием другого радикала, который ведет цепь [см. например, схемы (14) и (21)] до тех пор, пока она не оборвется, за счет взаимодействия между радикалами. Развитие (продолжение) цепи обусловлено высокой реакционной способностью радикалов, ведущих цепь, в радикал-молекулярных реакциях. Благодаря этому сохраняется низкая концентрация радикалов, а следовательно, и невысокая скорость радикал-радикальных реакций обрыва цепи.

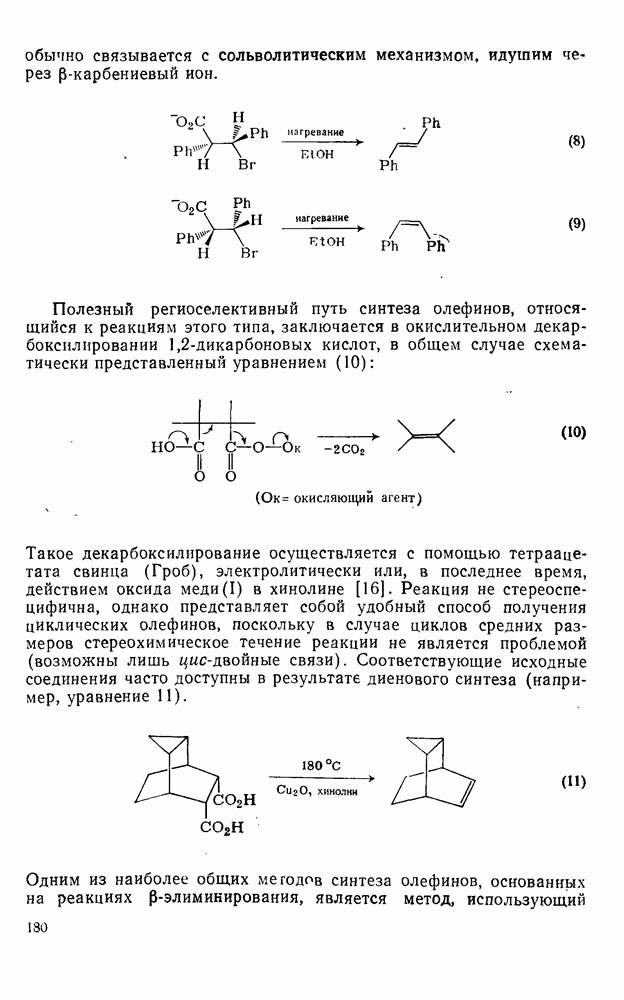
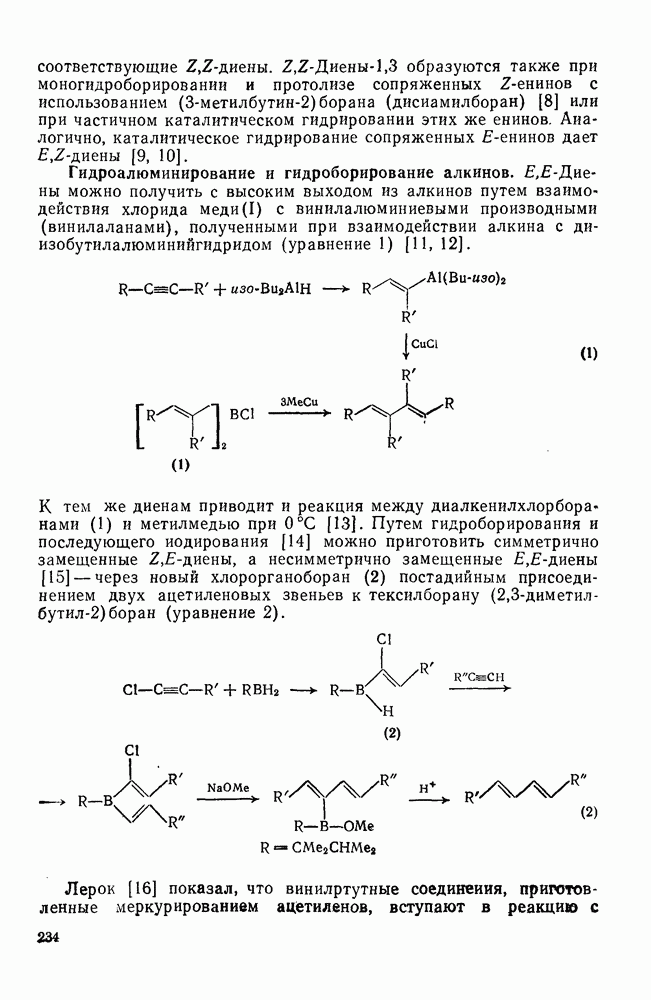
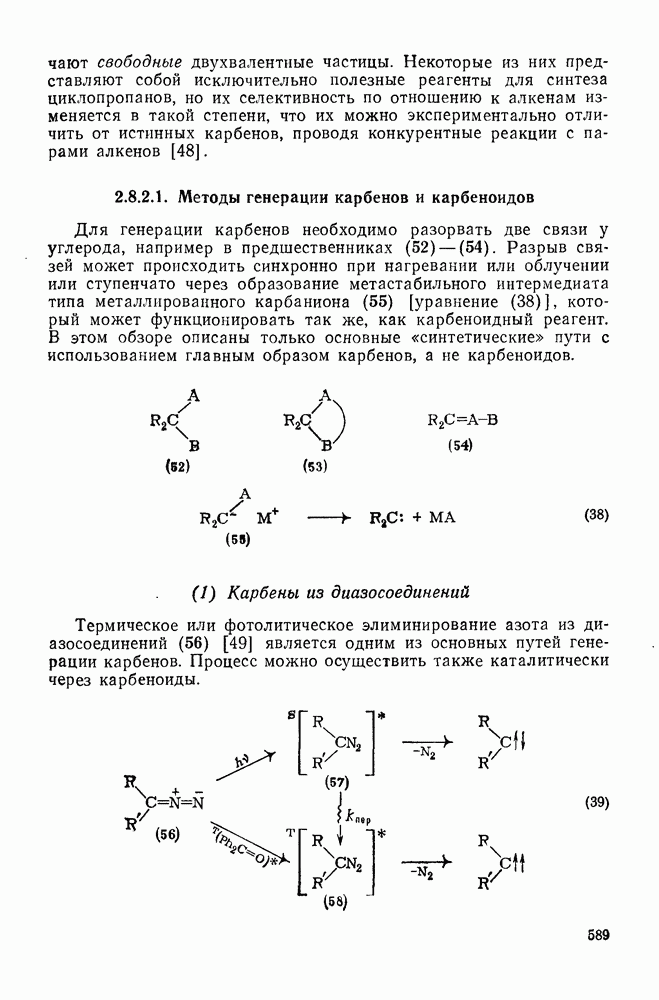
Существует два общих метода генерации определенных радикалов, необходимых для реакции: 1) построение молекулы, содержащей необходимый фрагмент, которая может подвергаться гомолизу или превращаться в радикалы с помощью окислительно-восстановительной реакции; 2) разложение инициатора с образованием реакционноспособных радикалов, которые затем превращают органический субстрат в желаемые радикалы путем отщепления или присоединения.
Во втором методе природа первоначального инициатора не играет существенной роли при условии, что он разлагается при подходящей температуре и образует радикалы, которые обладают достаточной реакционной способностью для участия в дальнейших превращениях.
(1) Первичное образование радикалов
Наиболее обычным методом первичной генерации радикалов является гомолитическое расщепление связей. Большинство связей термически устойчиво в пределах температур, обычно используемых для проведения реакций в растворе  . Однако имеется несколько групп с энергией связи менее
. Однако имеется несколько групп с энергией связи менее  , в первую очередь
, в первую очередь  и азогруппы
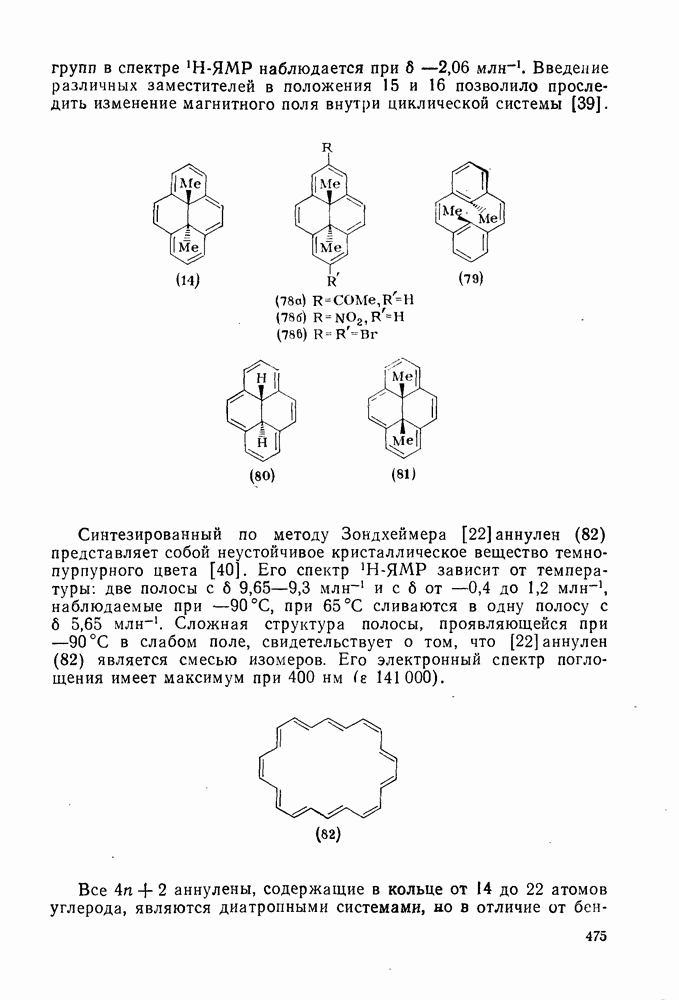
и азогруппы  , которые расщепляются при этих температурах с достаточной скоростью. Поскольку химия пероксидов и азосоединений [15, 16] обсуждается в другом месте, здесь достаточно кратко остановиться на их применении в радикальных реакциях.
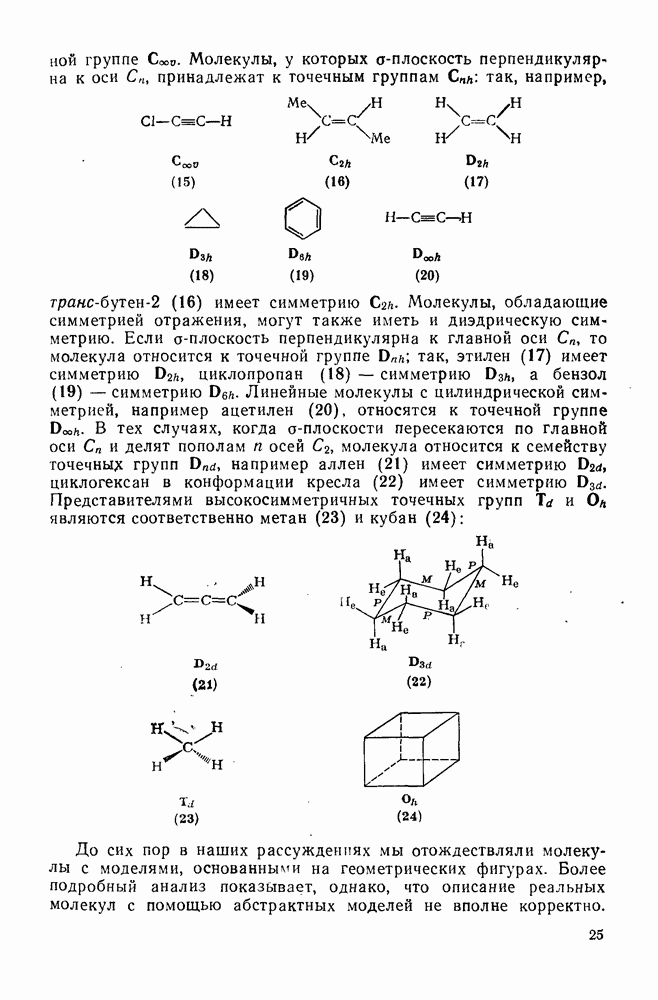
, которые расщепляются при этих температурах с достаточной скоростью. Поскольку химия пероксидов и азосоединений [15, 16] обсуждается в другом месте, здесь достаточно кратко остановиться на их применении в радикальных реакциях.
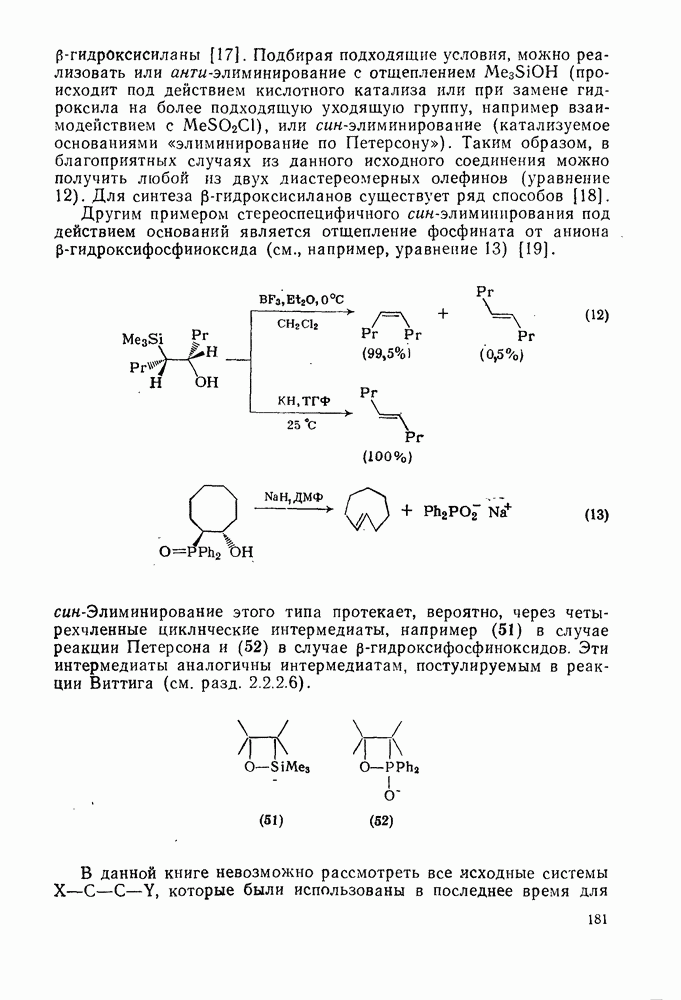
Как полагают, в переходном состоянии термолиза азосоединений связи в значительной степени разрыхлены. Поэтому легкость реакции определяется устойчивостью образующихся радикалов и действительно используется как мера устойчивости радикала [96]. Так, хотя диметилдиимид  является слишком устойчивым, чтобы использовать его в качестве инициатора, дибензильный аналог
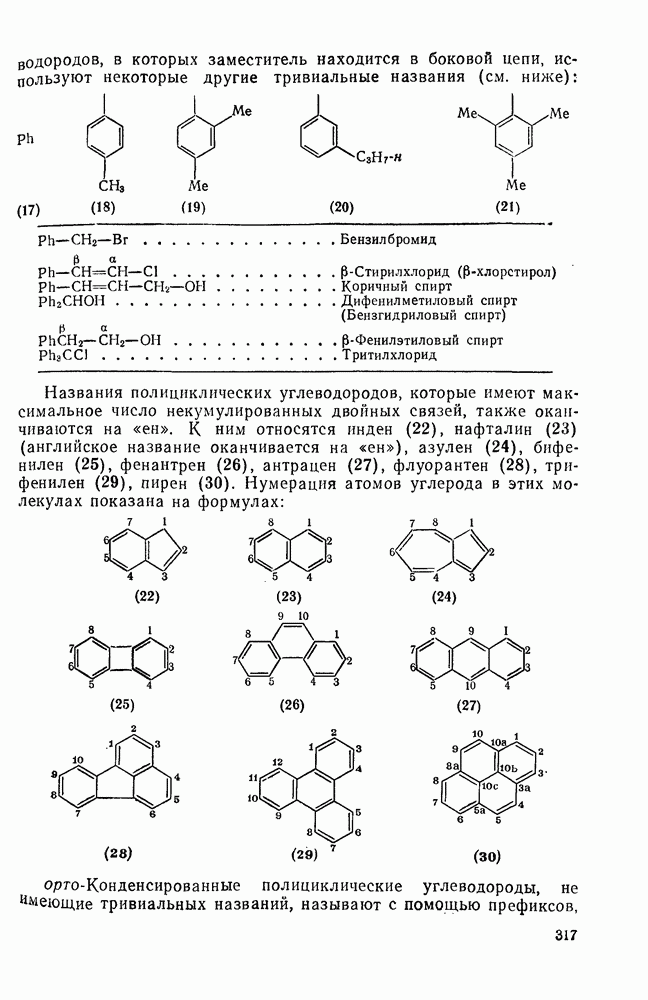
является слишком устойчивым, чтобы использовать его в качестве инициатора, дибензильный аналог  и
и  азобисизобутиронитрил
азобисизобутиронитрил  разлагаются во много сотен раз быстрее. Однако использование таких соединений в качестве инициаторов общего назначения довольно ограничено, поскольку они образуют радикалы, стабилизованные резонансным взаимодействием, и их реакционная способность в реакциях отрыва не очень высока. Стабилизация только одного из образующихся радикалов значительно увеличивает скорость разложения. Например, азобензол устойчив при
разлагаются во много сотен раз быстрее. Однако использование таких соединений в качестве инициаторов общего назначения довольно ограничено, поскольку они образуют радикалы, стабилизованные резонансным взаимодействием, и их реакционная способность в реакциях отрыва не очень высока. Стабилизация только одного из образующихся радикалов значительно увеличивает скорость разложения. Например, азобензол устойчив при  , а фенилазотрифенилме- тан (14) легко разлагается [уравнение (15)] при
, а фенилазотрифенилме- тан (14) легко разлагается [уравнение (15)] при  . Это соединение является удобным источником фенильных радикалов, однако при этом образуются также устойчивые трифенилметильные радикалы, действующие как ингибиторы. Хотя термическое расщепление простых алифатических азосоединений происходит с трудом, их фотолиз является основным путем генерации алкильных радикалов. Радикалы образуются также при фотолизе смешанных алкиларилазосоединений за счет расщепления их неустойчивых цис-изомеров [16], однако фотолиз ароматических азосоединений приводит только к цис/транс-изомеризации.
. Это соединение является удобным источником фенильных радикалов, однако при этом образуются также устойчивые трифенилметильные радикалы, действующие как ингибиторы. Хотя термическое расщепление простых алифатических азосоединений происходит с трудом, их фотолиз является основным путем генерации алкильных радикалов. Радикалы образуются также при фотолизе смешанных алкиларилазосоединений за счет расщепления их неустойчивых цис-изомеров [16], однако фотолиз ароматических азосоединений приводит только к цис/транс-изомеризации.

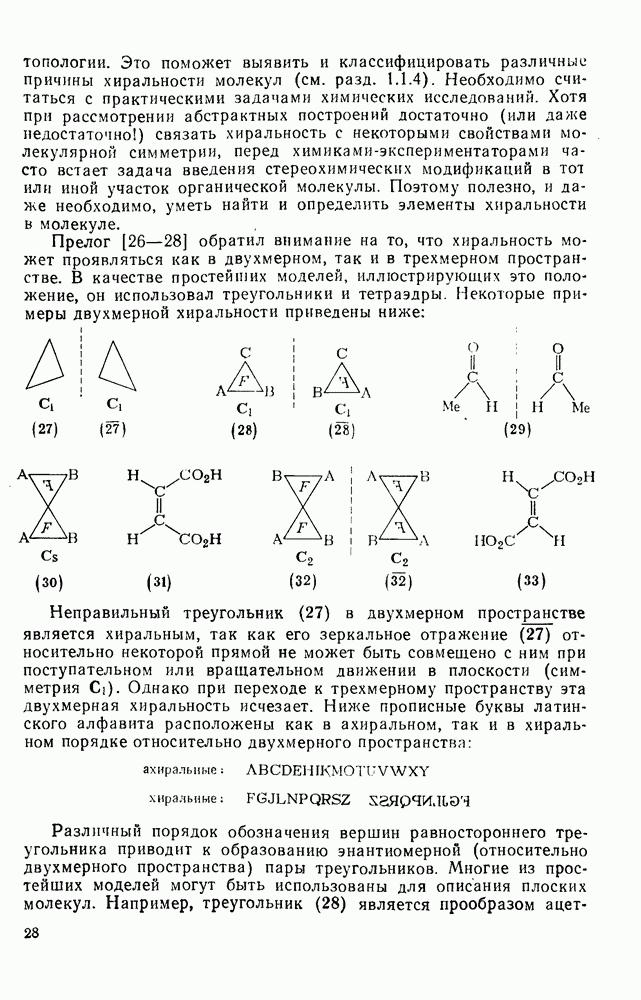
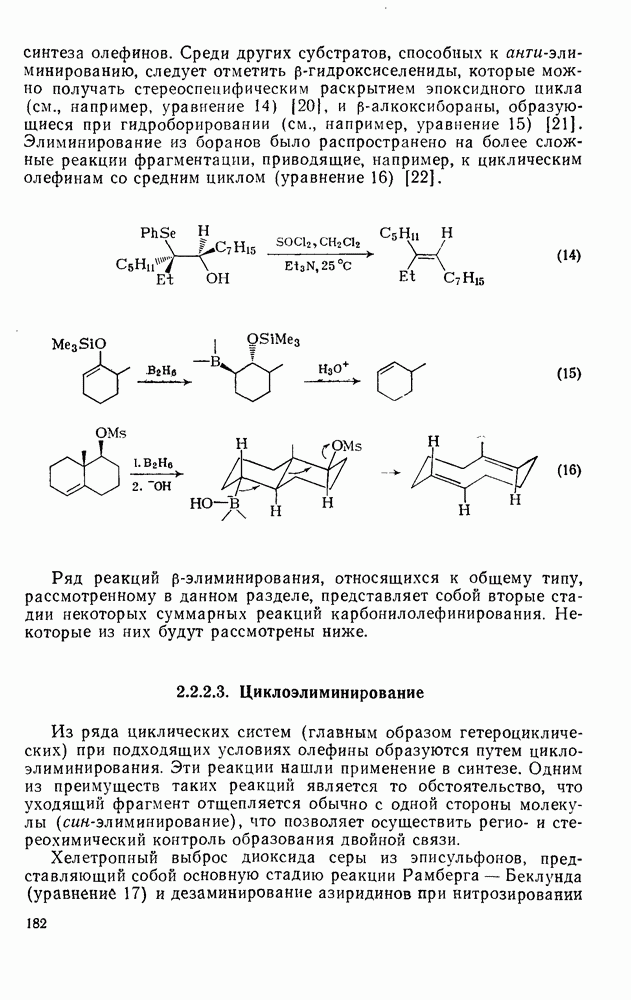
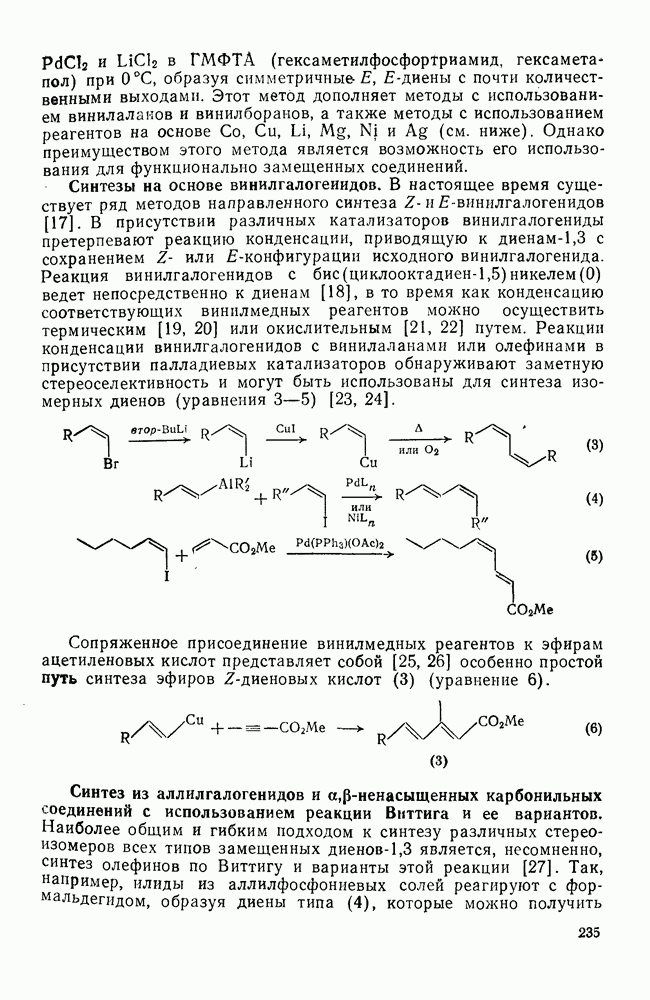
Первичные и вторичные диалкилпероксиды слишком неустойчивы, чтобы широко использоваться в качестве инициаторов, однако третичные алкилпероксиды представляют собой ценный источник реакционноспособных алкоксильных и алкильных радикалов [схема (16)]. Диацилпероксиды образуют радикалы как при термическом, так и при фотохимическом разложении [уравнение  ], однако для генерации вторичных и третичных алкильных радикалов предпочитают использовать фотолиз, поскольку при этом не возникают конкурентные гетеролитические реакции. Если
], однако для генерации вторичных и третичных алкильных радикалов предпочитают использовать фотолиз, поскольку при этом не возникают конкурентные гетеролитические реакции. Если  алкильная группа, то промежуточно образующиеся карбоксильные радикалы очень быстро распадаются [см. уравнение
алкильная группа, то промежуточно образующиеся карбоксильные радикалы очень быстро распадаются [см. уравнение  ], не выходя из клетки растворителя, однако при разложении диароилпероксидов в присутствии реакционноспособных субстратов часто получают продукты превращения ароилоксильных радикалов
], не выходя из клетки растворителя, однако при разложении диароилпероксидов в присутствии реакционноспособных субстратов часто получают продукты превращения ароилоксильных радикалов  . Это уменьшает ценность таких соединений как специфического источника арильных радикалов, но не как инициаторов общего назначения. Аналогично диацилпероксидам разлагаются пероксиэфиры, в особенности удобны пероксалаты и пероксидикарбонаты, поскольку они разлагаются с удовлетворительными скоростями при
. Это уменьшает ценность таких соединений как специфического источника арильных радикалов, но не как инициаторов общего назначения. Аналогично диацилпероксидам разлагаются пероксиэфиры, в особенности удобны пероксалаты и пероксидикарбонаты, поскольку они разлагаются с удовлетворительными скоростями при  , давая более реакционноспособные радикалы, чем азосоединения, например азобисизобутиронигрил, разлагающийся в той же области температур. Термическое разложение пероксидов не всегда имеет первый порядок; во многих системах они расходуются также в бимолекулярных индуцируемых радикалами реакциях.
, давая более реакционноспособные радикалы, чем азосоединения, например азобисизобутиронигрил, разлагающийся в той же области температур. Термическое разложение пероксидов не всегда имеет первый порядок; во многих системах они расходуются также в бимолекулярных индуцируемых радикалами реакциях.

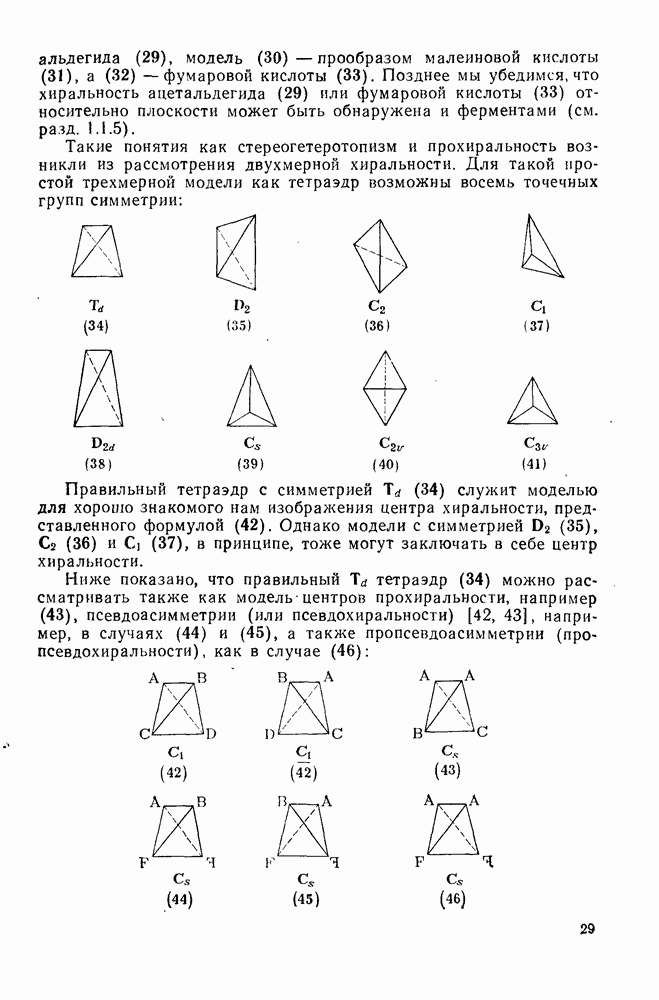
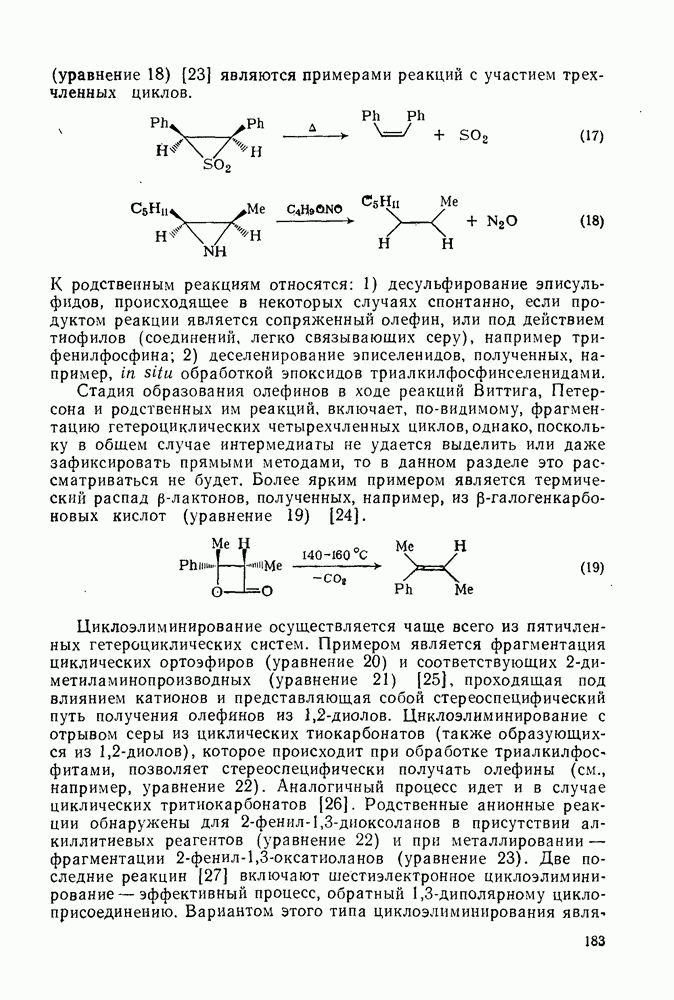
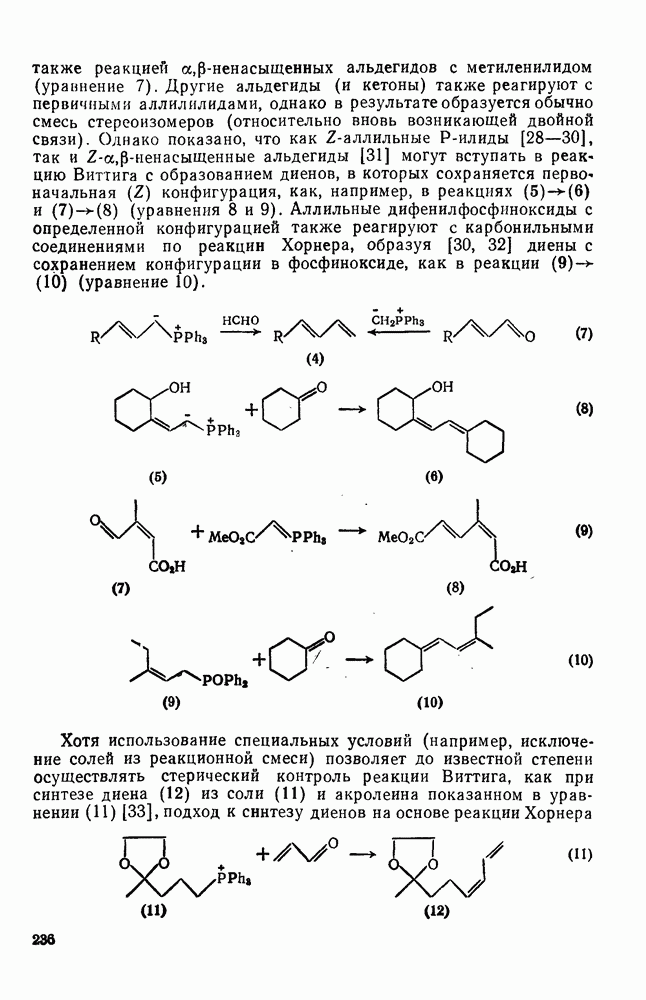
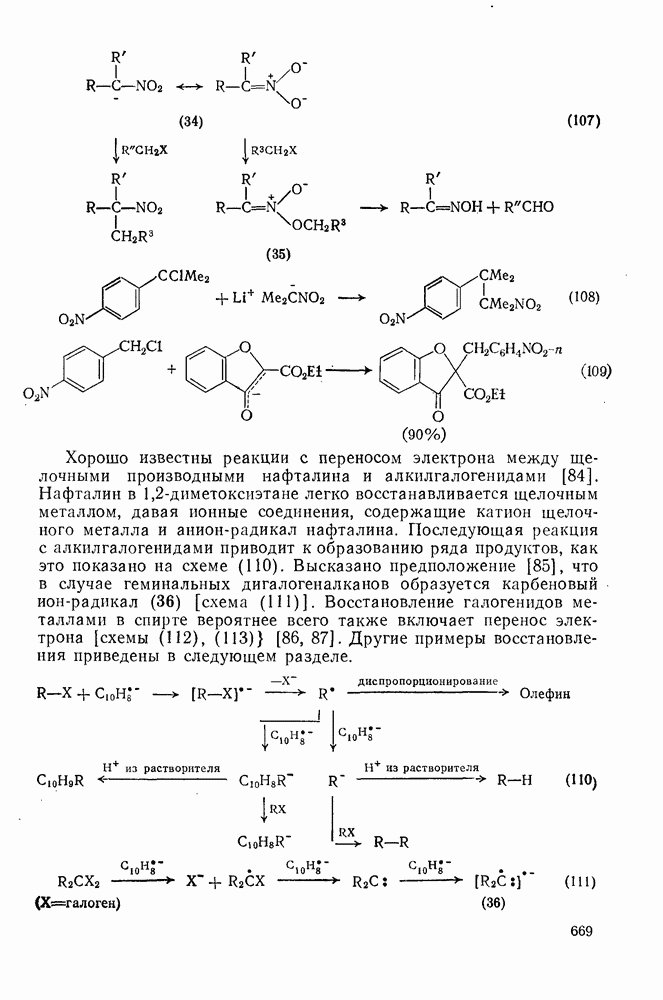
Арильные радикалы генерируют обычно непосредственно; одним из их источников являются диароилпероксиды, но существует также несколько методов, где в качестве исходных веществ используются легко доступные ароматические амины или их производные. Эти методы основаны на одноэлектронном восстановлении ионов диазония  в очень лабильные диазониевые радикалы
в очень лабильные диазониевые радикалы  , которые быстро теряют азот {см., например, уравнение
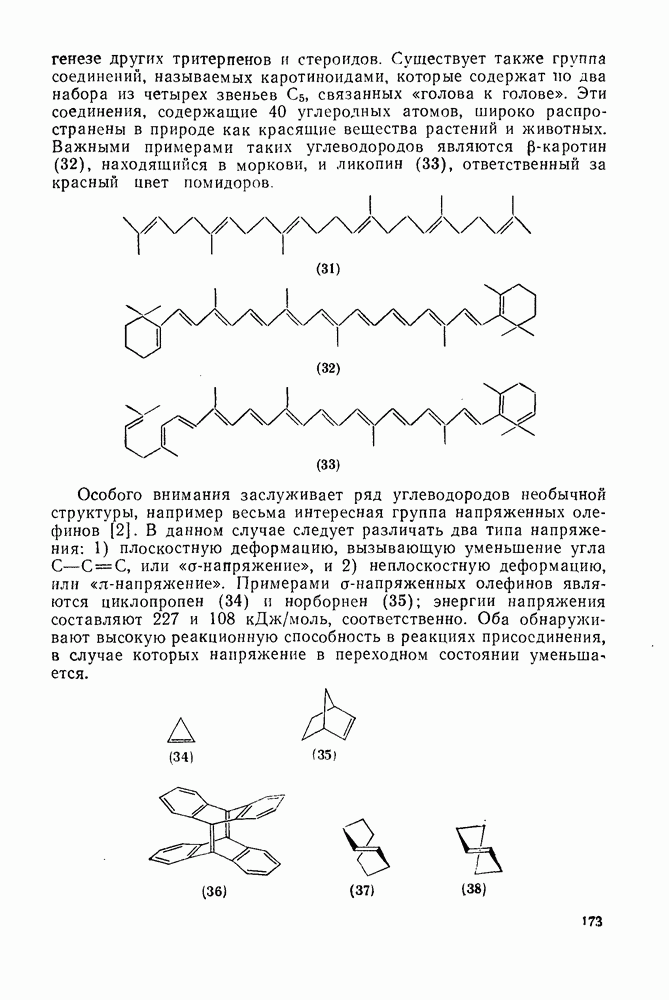
, которые быстро теряют азот {см., например, уравнение  [17]. Реакция Гомберга [уравнение (18)] и разложение ациларилнитрозаминов [уравнение (19)] используются, в частности, для проведения гомолитического ароматического замещения и относятся к цепным реакциям [18]. Апротонное диазотирование ароматических аминов органическими нитритами
[17]. Реакция Гомберга [уравнение (18)] и разложение ациларилнитрозаминов [уравнение (19)] используются, в частности, для проведения гомолитического ароматического замещения и относятся к цепным реакциям [18]. Апротонное диазотирование ароматических аминов органическими нитритами  и реакции арилдиазонийтетрафторборатов с ацетатом калия в присутствии катализатора межфазового переноса —
и реакции арилдиазонийтетрафторборатов с ацетатом калия в присутствии катализатора межфазового переноса —  [19е] — являются наиболее удобными путями генерации различных арильных радикалов. Органические нитриты реагируют также с
[19е] — являются наиболее удобными путями генерации различных арильных радикалов. Органические нитриты реагируют также с  -диарилтриазенами, давая с высоким выходом ароматические радикалы [20].
-диарилтриазенами, давая с высоким выходом ароматические радикалы [20].


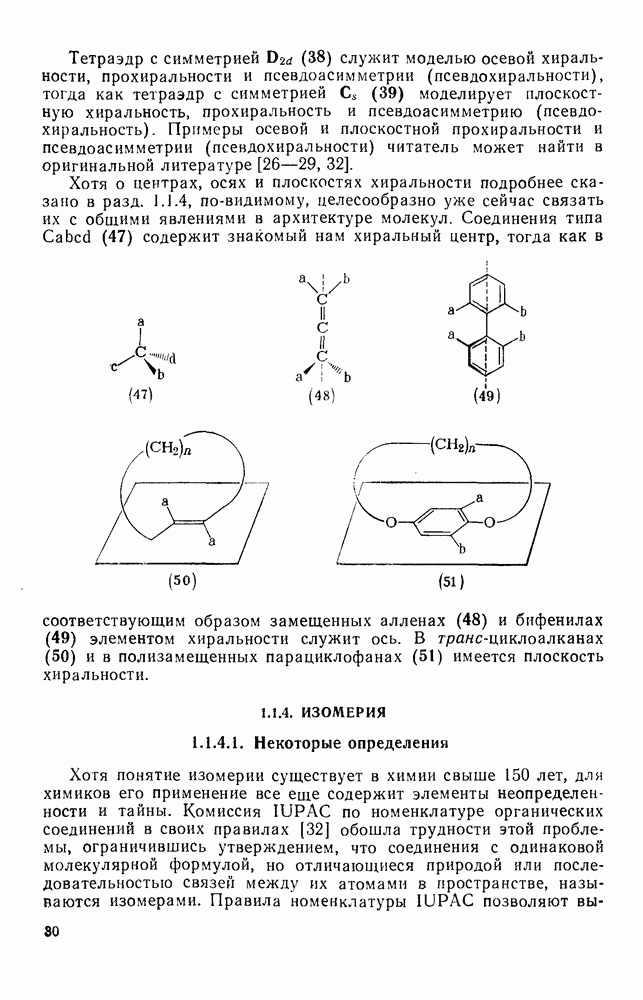
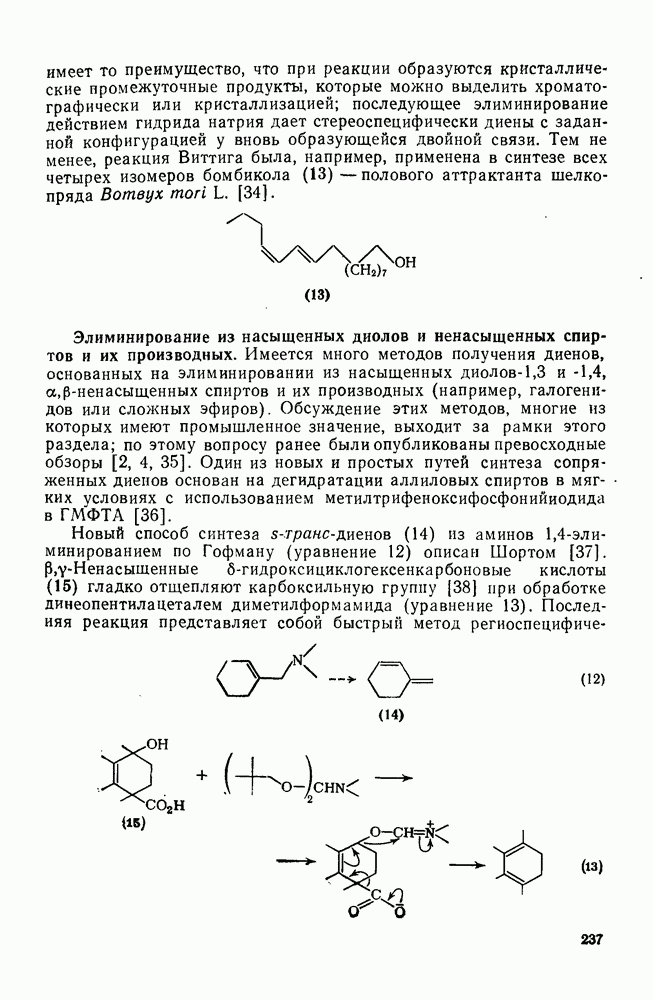
Для генерации радикалов важен также фотолиз галогенов, полигалогенметанов, арилиодидов, алкил- и арилнитритов [21], кетонов и органических соединений ртути. Гомолитическое расщепление многих классов соединений стимулируется облучением лучами высокой энергии, рентгеновскими или  -лучами, а также быстрыми электронами [22].
-лучами, а также быстрыми электронами [22].
(2) Отрыв водорода и других атомов
Элементарной стадией многих свободнорадикальных реакций является образование нового радикала за счет отрыва водорода или других атомов [уравнение (7)]. Отрыв водорода происходит при действии атомов и радикалов различных типов через более или менее линейное переходное состояние. Легкость реакции зависит от реакционной способности участвующего радикала и химического окружения водорода [23]. Важную роль играют сила связи  , полярные факторы и несвязные взаимодействия. Все радикалы обнаруживают одинаковый общий порядок реакционной способности:
, полярные факторы и несвязные взаимодействия. Все радикалы обнаруживают одинаковый общий порядок реакционной способности:  , однако селективность изменяется в широких пределах и зависит главным образом от реакционной способности атакующего радикала (табл. 2.8.1). Радикалы с высокой реакционной способностью, образующие сильные связи с водородом, например
, однако селективность изменяется в широких пределах и зависит главным образом от реакционной способности атакующего радикала (табл. 2.8.1). Радикалы с высокой реакционной способностью, образующие сильные связи с водородом, например  , проявляют очень низкую селективность в то время как радикалы с низкой реакционной способностью, например
, проявляют очень низкую селективность в то время как радикалы с низкой реакционной способностью, например  , проявляют высокую селективность. Следует отметить, что радикалы, образуемые большинством инициаторов (алкильные, арильные и алкоксильные радикалы), обладают только умеренной селективностью. В рамках постулата Хэммонда переходное состояние для реакционноспособных радикалов, например (15), возникает на начальном отрезке координаты реакции и характеризуется незначительным разрыхлением связи
, проявляют высокую селективность. Следует отметить, что радикалы, образуемые большинством инициаторов (алкильные, арильные и алкоксильные радикалы), обладают только умеренной селективностью. В рамках постулата Хэммонда переходное состояние для реакционноспособных радикалов, например (15), возникает на начальном отрезке координаты реакции и характеризуется незначительным разрыхлением связи  , поэтому различия в силе связи
, поэтому различия в силе связи  или в устойчивости первоначального радикала не оказывают большого влияния на скорость процесса.
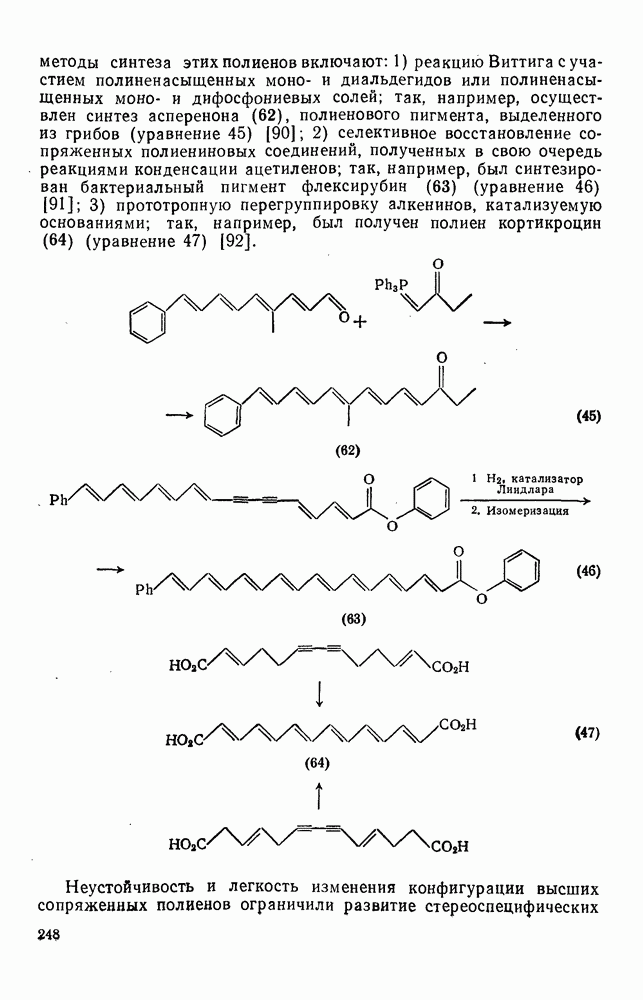
или в устойчивости первоначального радикала не оказывают большого влияния на скорость процесса.
Таблица 2.8.1. Относительные скорости отрыва водорода от алканов [23]

Таблица 2.8.3. Относительная селективность при отрыве водорода из замещенных бутанов в газовой фазе [1а]

Значительное влияние полярного эффекта на скорости реакций отрыва водорода было описано в общих чертах в разд. 2.8.1.1. Например, скорость отрыва водорода от метильной группы в замещенных толуолах (20) уменьшается при введении электроноакцепторных групп Y, которые дестабилизуют переходное состояние (21). Корреляционный анализ по Гаммету этих реакций дает низкие значения  для реакций отрыва водорода радикалами
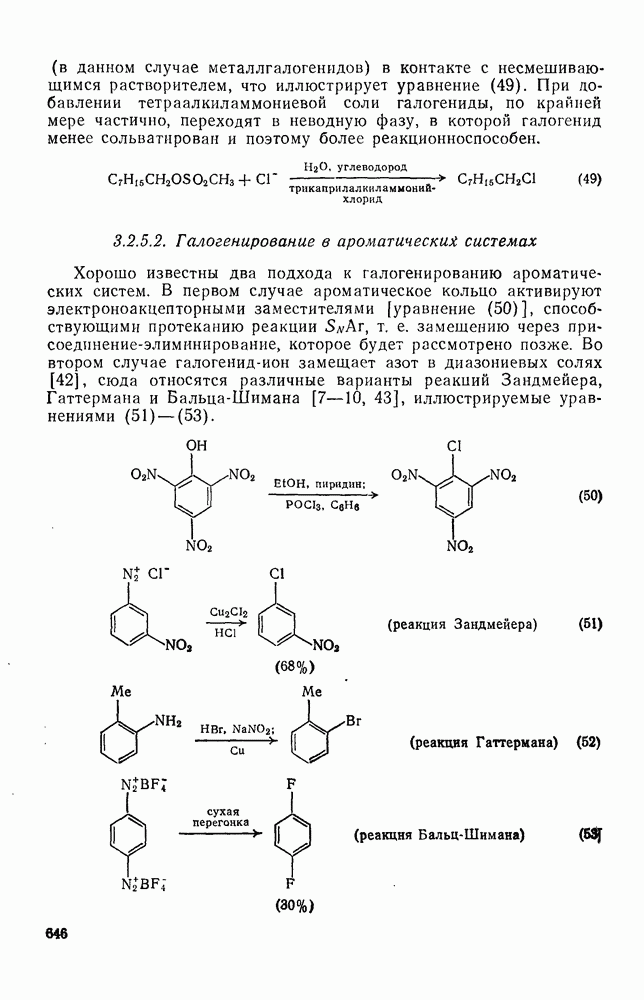
для реакций отрыва водорода радикалами  , что указывает на малую зависимость скорости от полярных эффектов, однако для радикалов с большим сродством к электрону получены более высокие значения
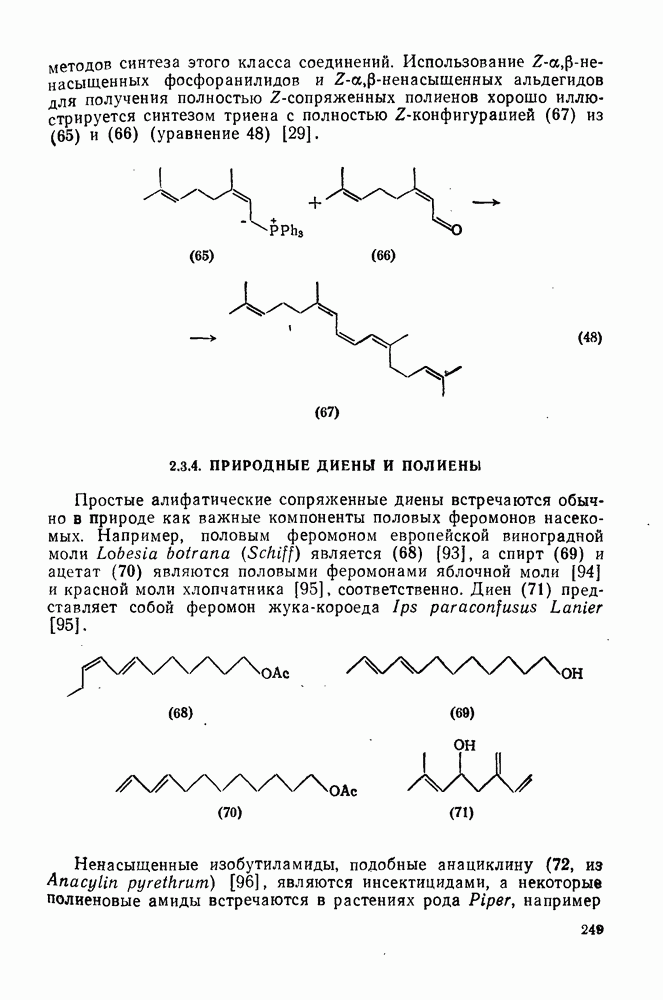
, что указывает на малую зависимость скорости от полярных эффектов, однако для радикалов с большим сродством к электрону получены более высокие значения  , например
, например  . Аналогичное влияние полярных эффектов наблюдается в случае чисто алифатических систем. Обратите внимание, как индуктивные эффекты групп CN и трет-
. Аналогичное влияние полярных эффектов наблюдается в случае чисто алифатических систем. Обратите внимание, как индуктивные эффекты групп CN и трет- (табл. 2.8.3) влияют на реакционную способность
(табл. 2.8.3) влияют на реакционную способность  и
и  -связей
-связей  к отрыву водорода радикалами
к отрыву водорода радикалами  . Результаты, полученные в случае отрыва водорода радикалами
. Результаты, полученные в случае отрыва водорода радикалами  , отличаются от приведенных для
, отличаются от приведенных для  , что объясняется большим разрыхлением связи в переходном состоянии. Повышение реакционной способности
, что объясняется большим разрыхлением связи в переходном состоянии. Повышение реакционной способности  -положения указывает на то, что резонансная стабилизация первоначального радикала
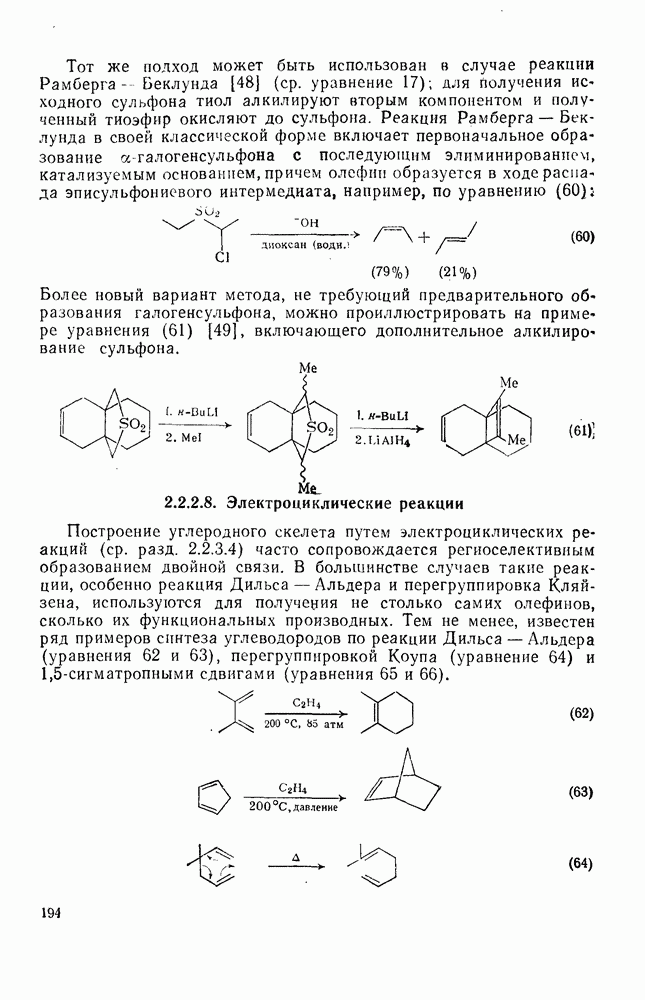
-положения указывает на то, что резонансная стабилизация первоначального радикала  -группой превышает ее дезактивирующий полярный эффект. Однако
-группой превышает ее дезактивирующий полярный эффект. Однако  -положение все еще дезактивируется полярным эффектом [1а].
-положение все еще дезактивируется полярным эффектом [1а].
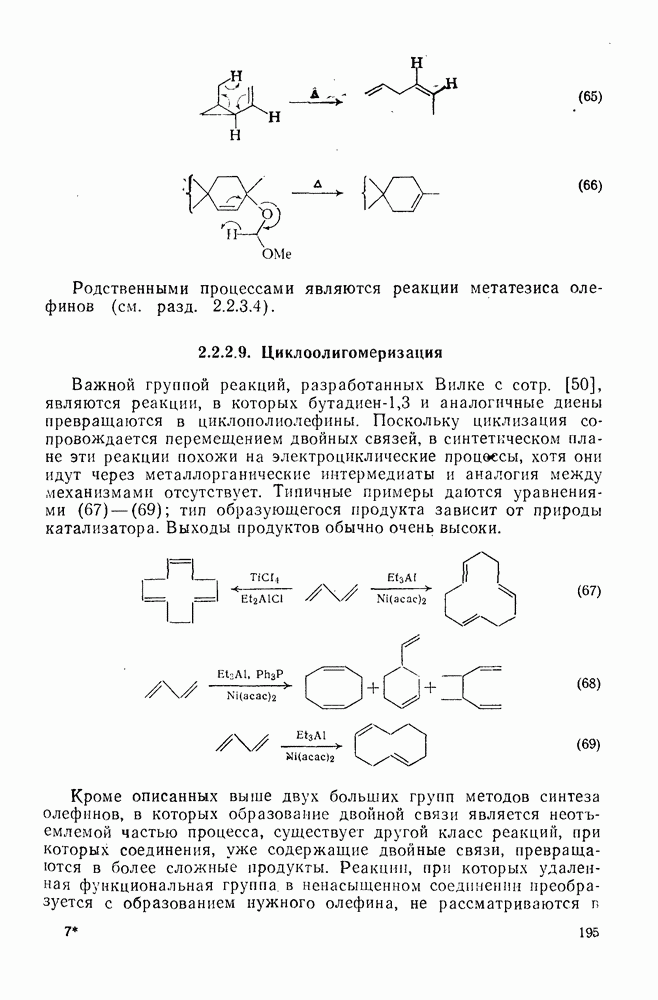
Отрыв галогенов может также осуществляться многими радикалами; реакционная способность падает в ряду:  , отвечающему прочности связи
, отвечающему прочности связи  , за исключением водорода. Полагают, что полярное отталкивание между заполненными орбиталями галогена и приближающимся радикалом замедляет отрыв галогена по сравнению с отрывом водорода и объясняет аномальное положение последнего. Изучение кинетики отрыва иода показало, что переходное состояние поляризовано противоположно тому, как это имеет место в (21). Отрыв галогенов и водорода арильными радикалами, генерированными из арил-аминов, представляет собой удобный вариант реакции Зандмейера [19].
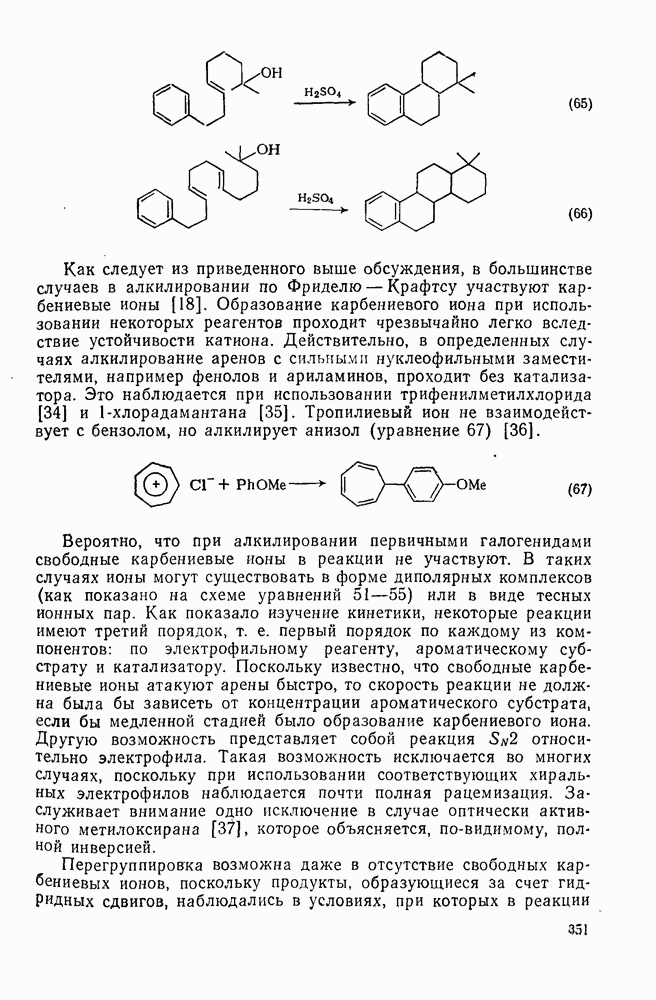
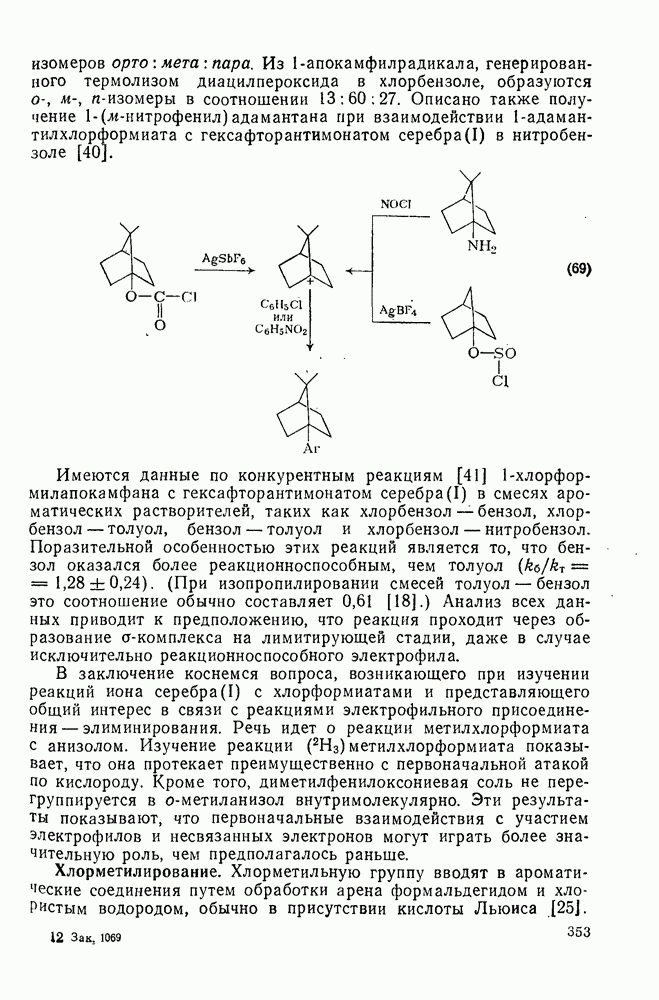
, за исключением водорода. Полагают, что полярное отталкивание между заполненными орбиталями галогена и приближающимся радикалом замедляет отрыв галогена по сравнению с отрывом водорода и объясняет аномальное положение последнего. Изучение кинетики отрыва иода показало, что переходное состояние поляризовано противоположно тому, как это имеет место в (21). Отрыв галогенов и водорода арильными радикалами, генерированными из арил-аминов, представляет собой удобный вариант реакции Зандмейера [19].
Все типы гемолитического замещения были рассмотрены недавно в [25].
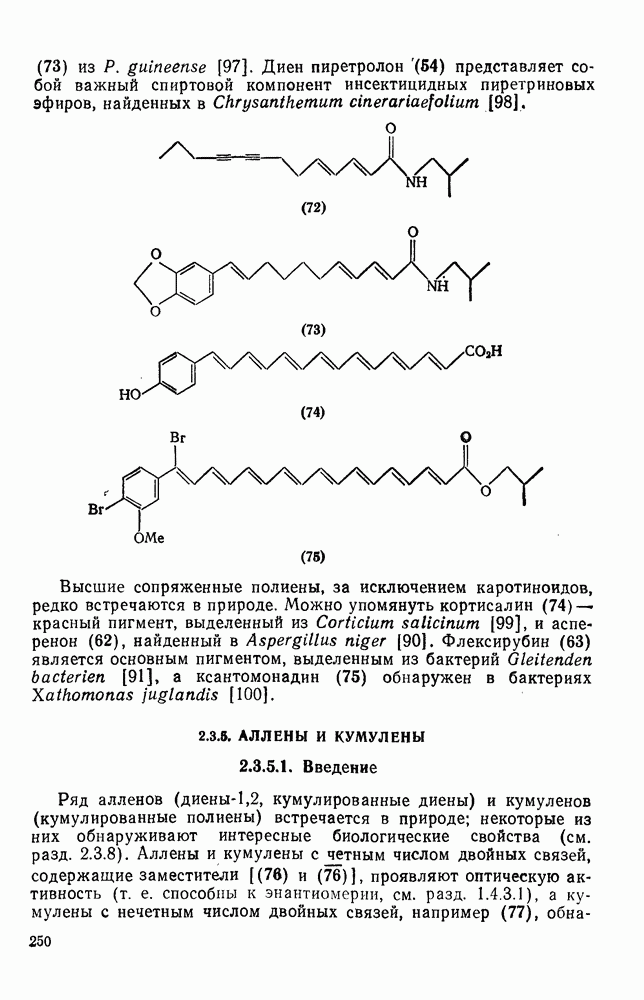
(3) Присоединение
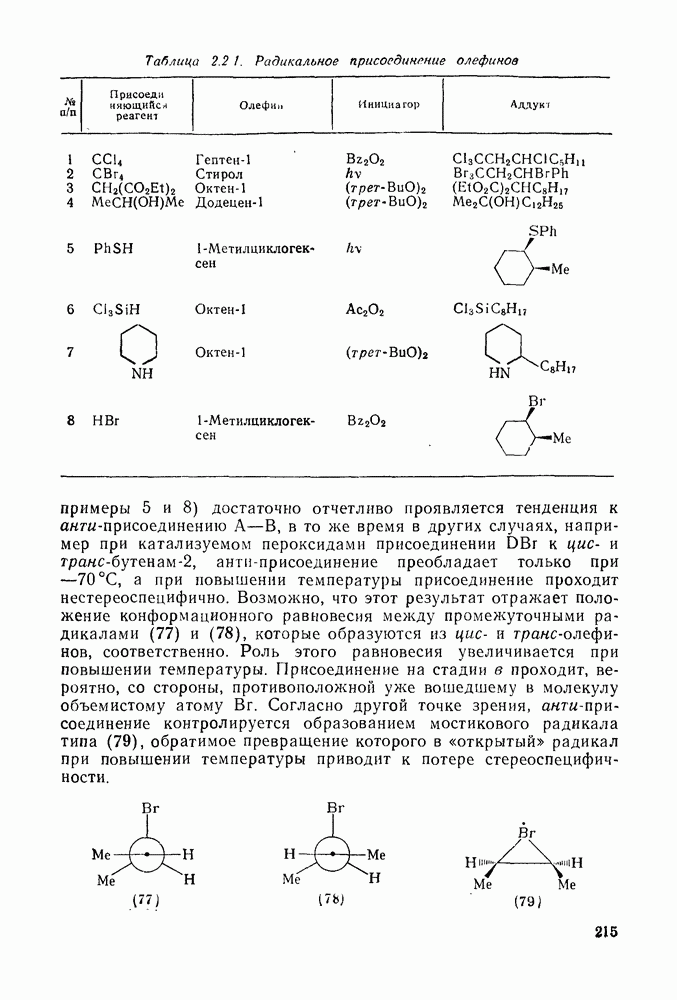
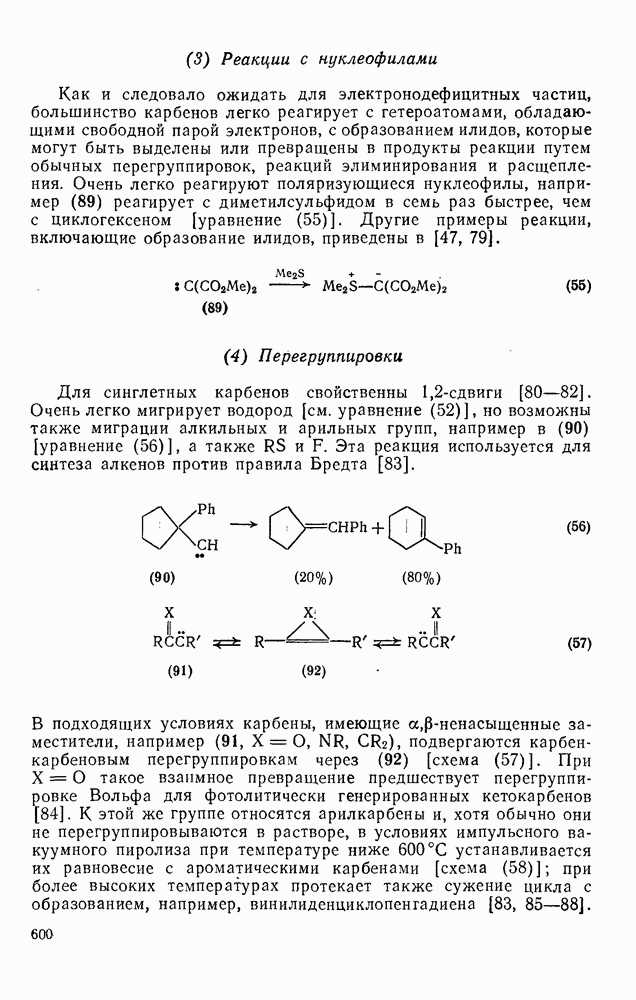
Различные радикалы присоединяются [уравнение (9)] к алке-нам, диенам, ароматическим соединениям, алкинам и к другим соединениям с ненасыщенными связями [26]. Стадия присоединения в реакциях с алкенами обычно является частью цепного процесса [схема  ], приводящего к образованию аддуктов
], приводящего к образованию аддуктов  (24), теломеров, например (25), или высокомолекулярных соединений. Едва ли нужно говорить о важности процессов радикальной полимеризации, но и образование аддуктов 1: 1 также является важной синтетической реакцией [27], применимой к широкому кругу аддендов, например к полигалогенметанам, карбоновым кислотам, эфирам, нитрилам, спиртам, аминам и разнообразным радикалам с радикальным центром на гетероатоме. Преимущественное образование при реакции аддуктов
(24), теломеров, например (25), или высокомолекулярных соединений. Едва ли нужно говорить о важности процессов радикальной полимеризации, но и образование аддуктов 1: 1 также является важной синтетической реакцией [27], применимой к широкому кругу аддендов, например к полигалогенметанам, карбоновым кислотам, эфирам, нитрилам, спиртам, аминам и разнообразным радикалам с радикальным центром на гетероатоме. Преимущественное образование при реакции аддуктов  либо полимеров определяет конкуренция между стадиями (б) и (в) на схеме (21), и хотя это в большой степени зависит от природы реагирующих веществ, все же изменение условий реакции позволяет в значительной мере контролировать направление процесса. Алкены, образующие стабилизованные радикалы (23), которые ведут цепь, дают преимущественно полимеры. Например, стирол
либо полимеров определяет конкуренция между стадиями (б) и (в) на схеме (21), и хотя это в большой степени зависит от природы реагирующих веществ, все же изменение условий реакции позволяет в значительной мере контролировать направление процесса. Алкены, образующие стабилизованные радикалы (23), которые ведут цепь, дают преимущественно полимеры. Например, стирол  легко присоединяет радикалы, однако образующийся при этом резонансно стабилизованный радикал на стадии переноса цепи [стадия (б)] имеет низкую реакционную способность и реагирует предпочтительно с другой молекулой стирола. Такие алкены образуют главным образом полимеры, за исключением тех случаев, когда в адденде имеется достаточно слабая связь, чтобы стадия переноса (б) могла конкурировать со стадией дальнейшего присоединения (в). Наоборот, менее стабилизованные ведущие цепь радикалы генерированные из таких алкенов, как, например,
легко присоединяет радикалы, однако образующийся при этом резонансно стабилизованный радикал на стадии переноса цепи [стадия (б)] имеет низкую реакционную способность и реагирует предпочтительно с другой молекулой стирола. Такие алкены образуют главным образом полимеры, за исключением тех случаев, когда в адденде имеется достаточно слабая связь, чтобы стадия переноса (б) могла конкурировать со стадией дальнейшего присоединения (в). Наоборот, менее стабилизованные ведущие цепь радикалы генерированные из таких алкенов, как, например,  , обладают большей реакционной способностью на стадии (б) и преимущественно дают с высоким выходом аддукты.
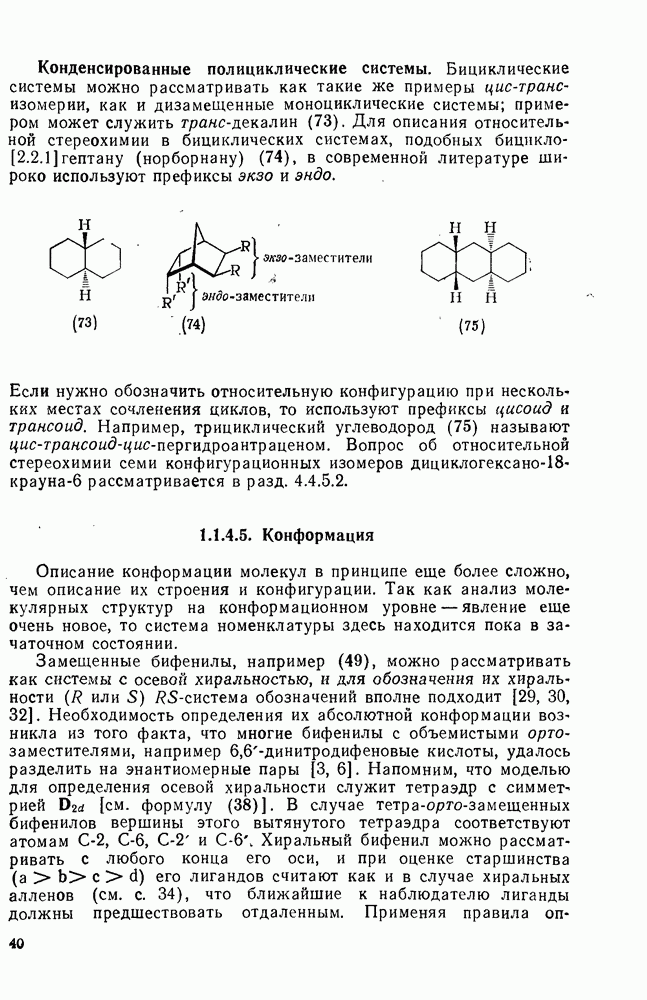
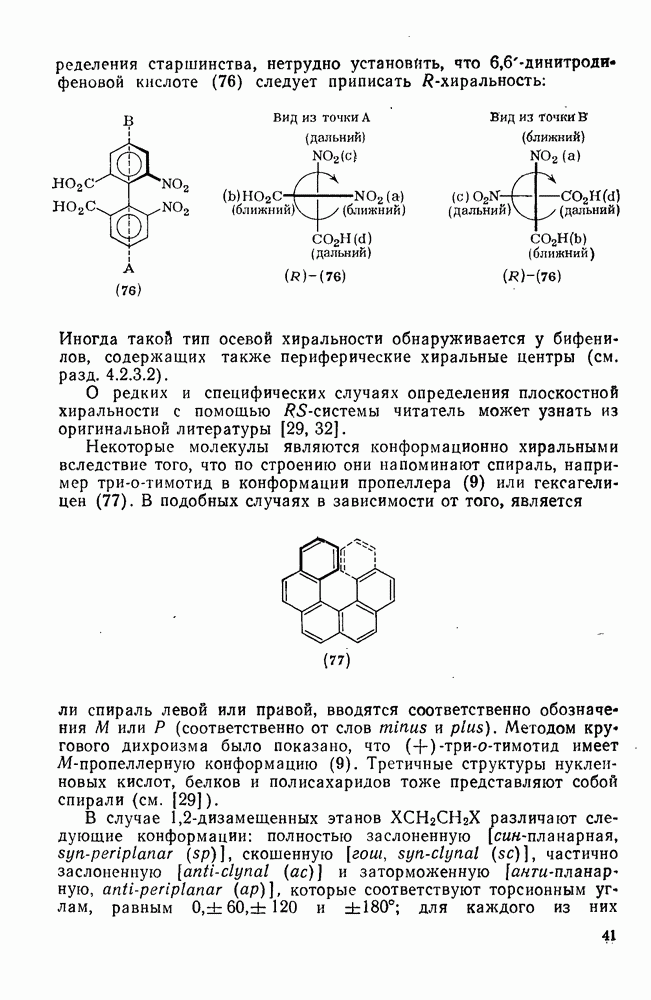
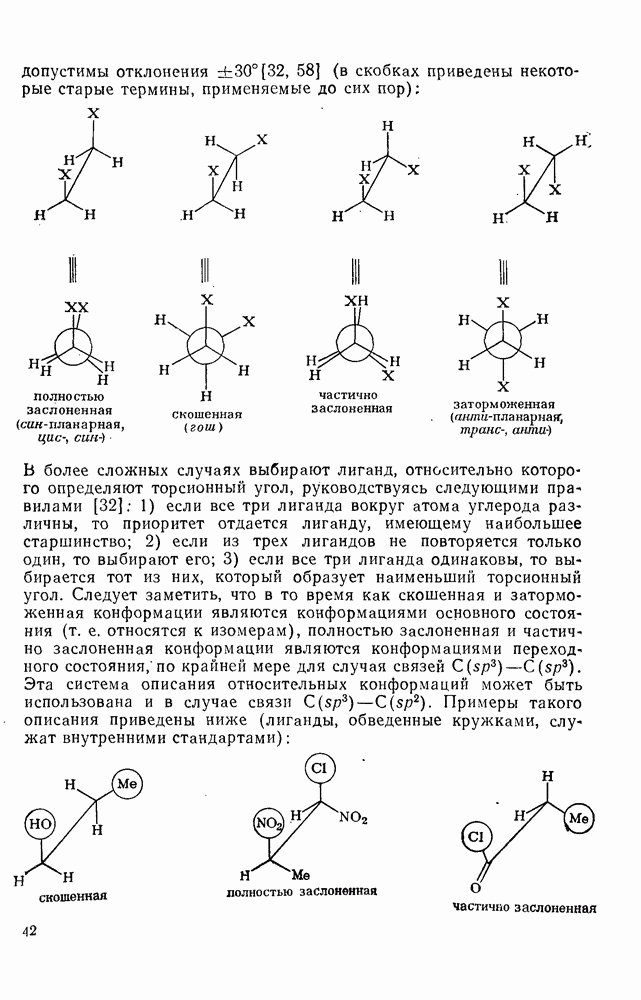
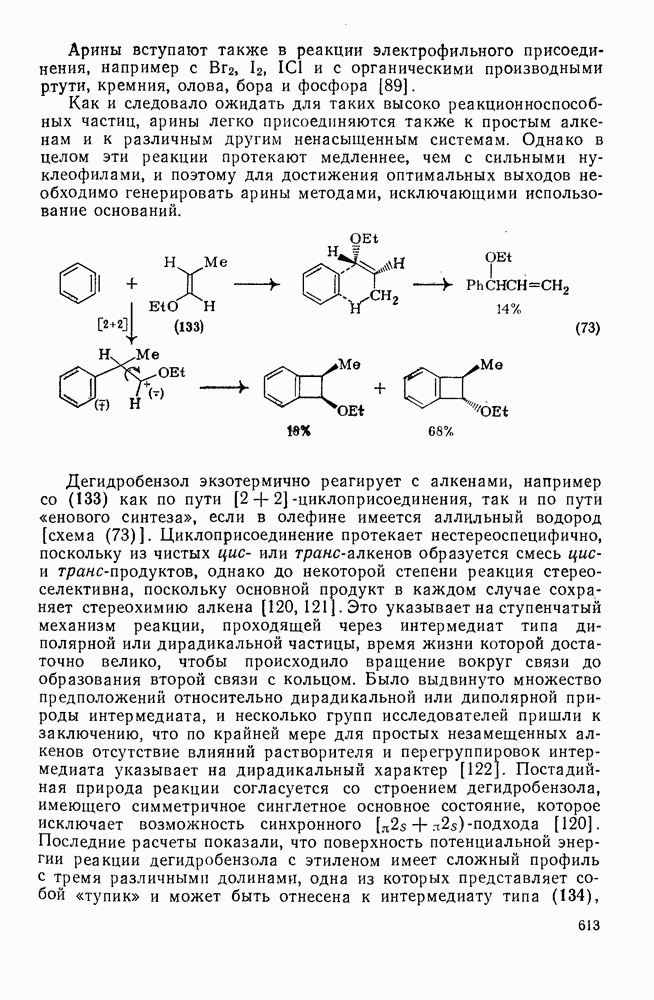
, обладают большей реакционной способностью на стадии (б) и преимущественно дают с высоким выходом аддукты.

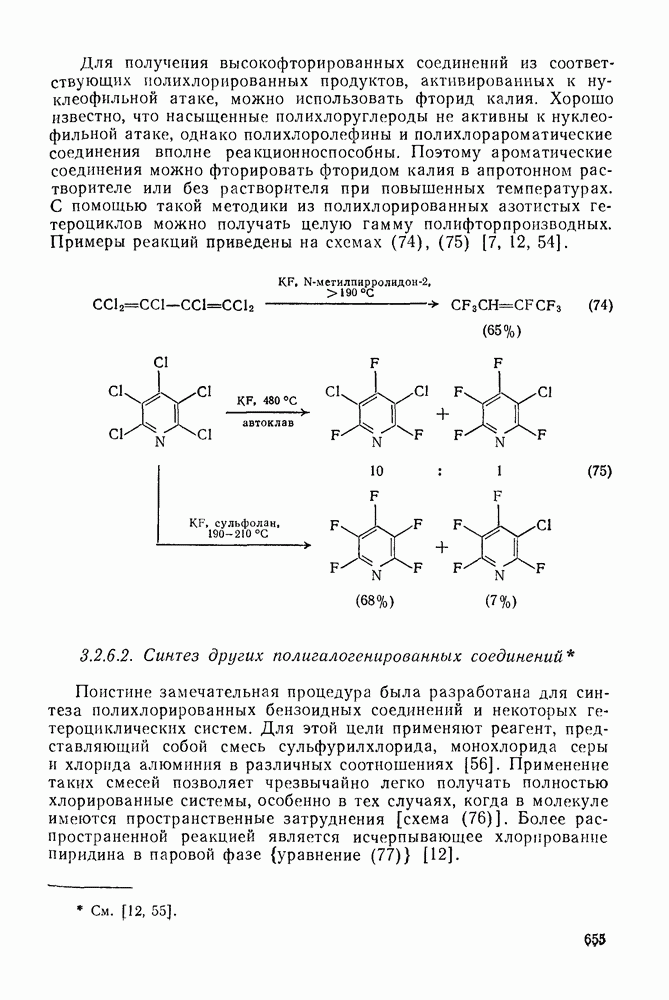
Можно добиться высоких выходов аддуктов, поддерживая высокую концентрацию адденда и низкую концентрацию алкена, что способствует стадии (б) и не благоприятствует стадии (в). Это особенно важно для аддендов, когда на стадии переноса необходимо разорвать достаточно сильную связь. Присоединение простых алкильных радикалов является экзотермической и необратимой при обычных условиях реакцией в растворе. Однако присоединение с образованием более слабых связей, например в реакциях с радикалами  ,
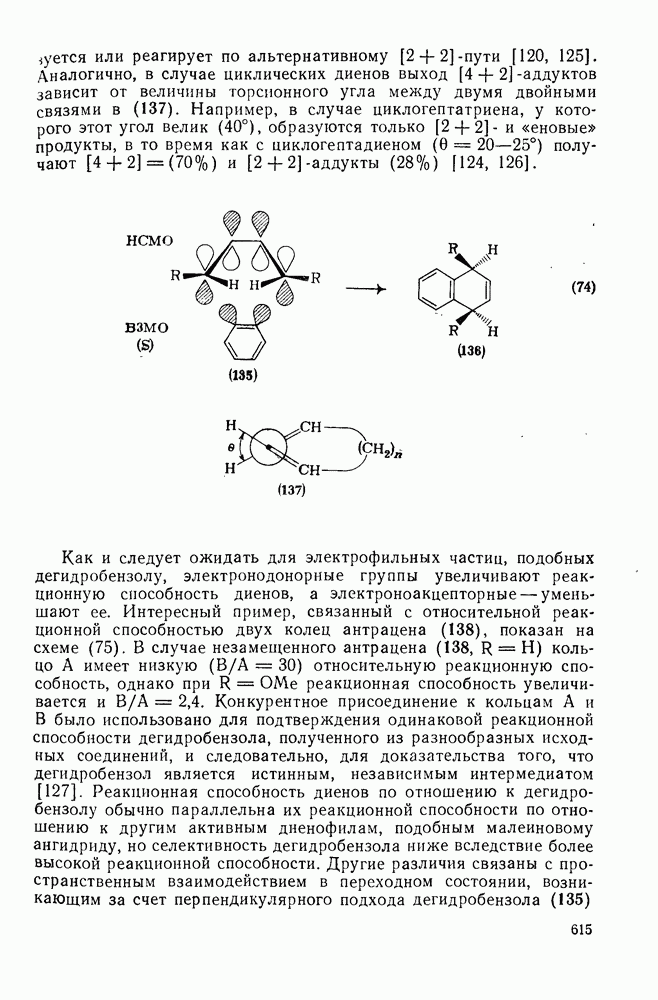
,  и многими гетероатомными радикалами, в значительной степени обратимо. На скорость стадии присоединения (а) влияют как полярные, так и пространственные факторы, например скорость присоединения к замещенным этиленам радикалов
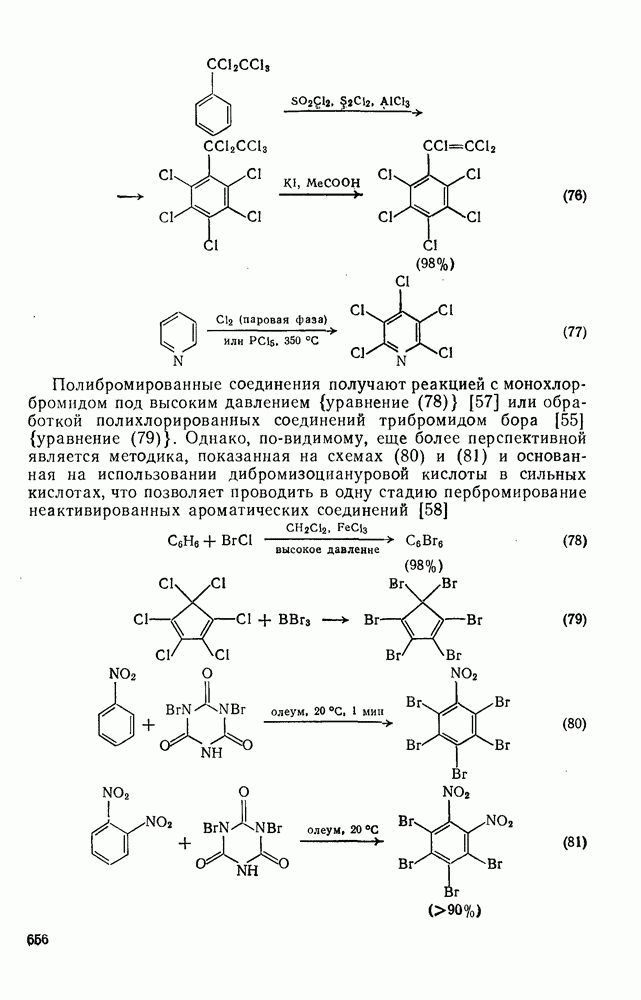
и многими гетероатомными радикалами, в значительной степени обратимо. На скорость стадии присоединения (а) влияют как полярные, так и пространственные факторы, например скорость присоединения к замещенным этиленам радикалов  и
и  , которые имеют электрофильный характер, увеличивается при введении электронодонорных заместителей и уменьшается при наличии электроноакцепторных заместителей. Для незамещенных алкильных радикалов, например
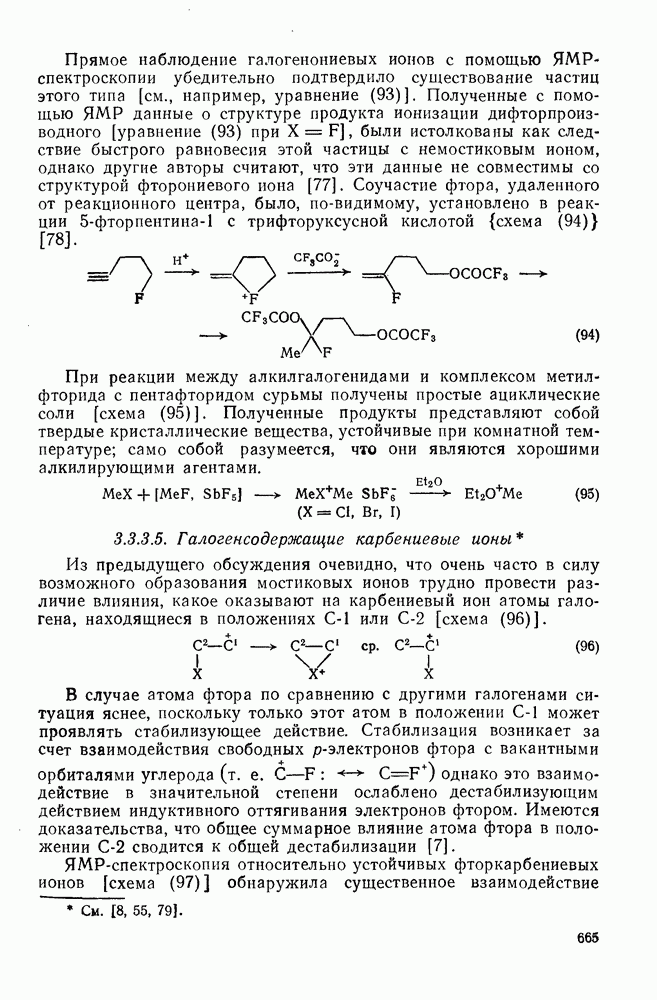
, которые имеют электрофильный характер, увеличивается при введении электронодонорных заместителей и уменьшается при наличии электроноакцепторных заместителей. Для незамещенных алкильных радикалов, например  , которые являются слабо нуклеофильными, наблюдается противоположная зависимость. Так, реакционная способность алкенов резко падает при увеличении числа метальных заместителей за счет как полярных, так и пространственных факторов. Были определены энергии активации присоединения радикалов
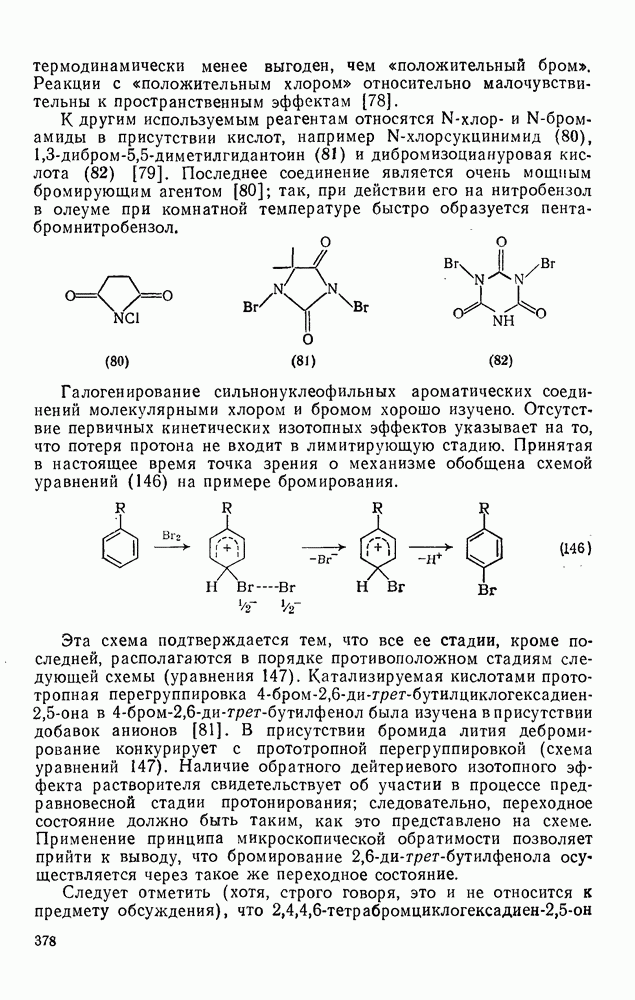
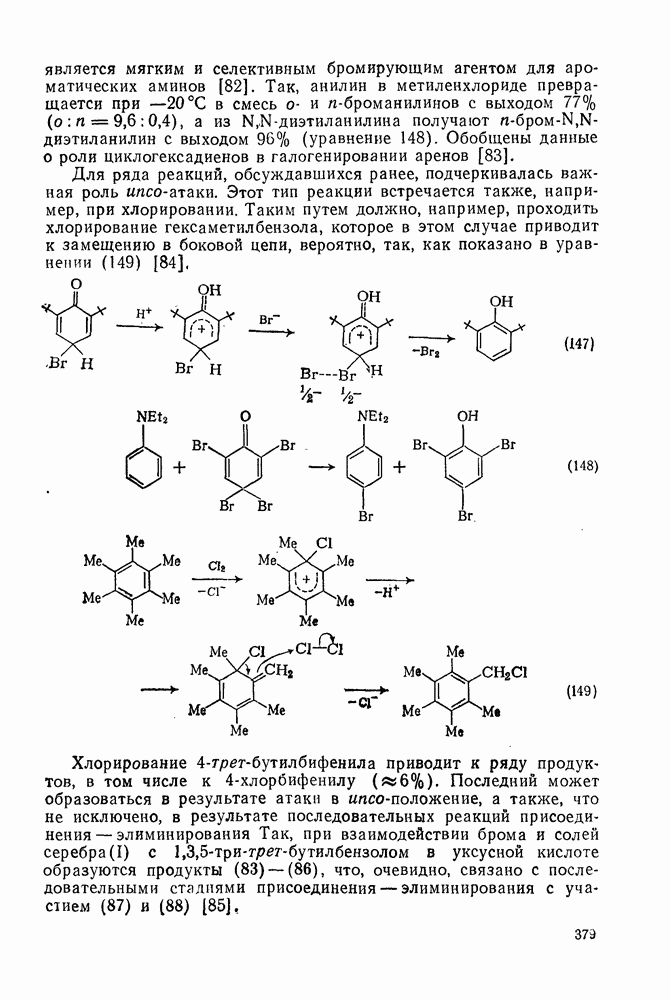
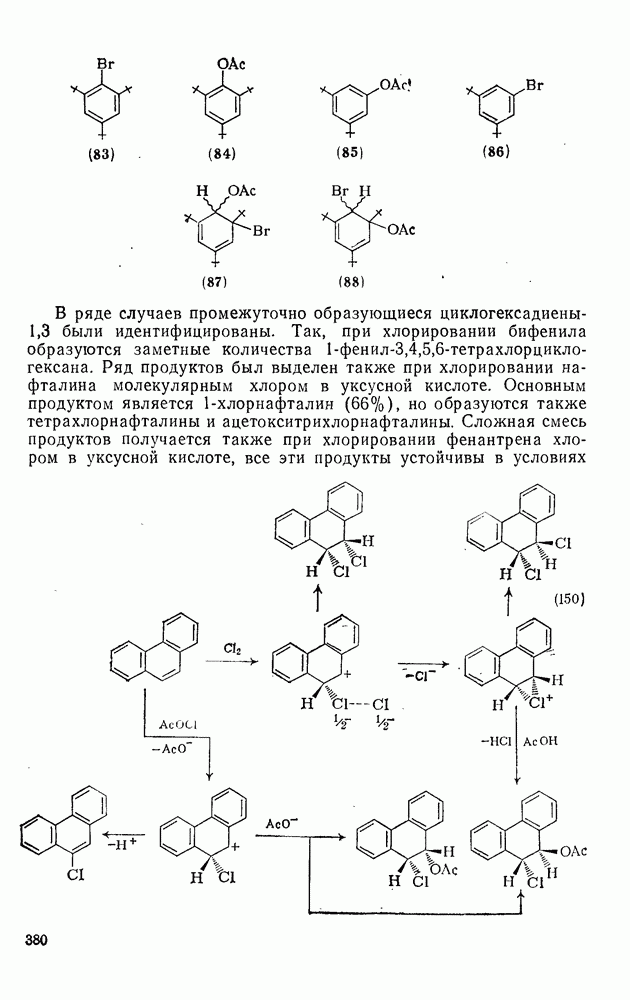
, которые являются слабо нуклеофильными, наблюдается противоположная зависимость. Так, реакционная способность алкенов резко падает при увеличении числа метальных заместителей за счет как полярных, так и пространственных факторов. Были определены энергии активации присоединения радикалов  к обоим винильным углеродным атомам алкенов типа
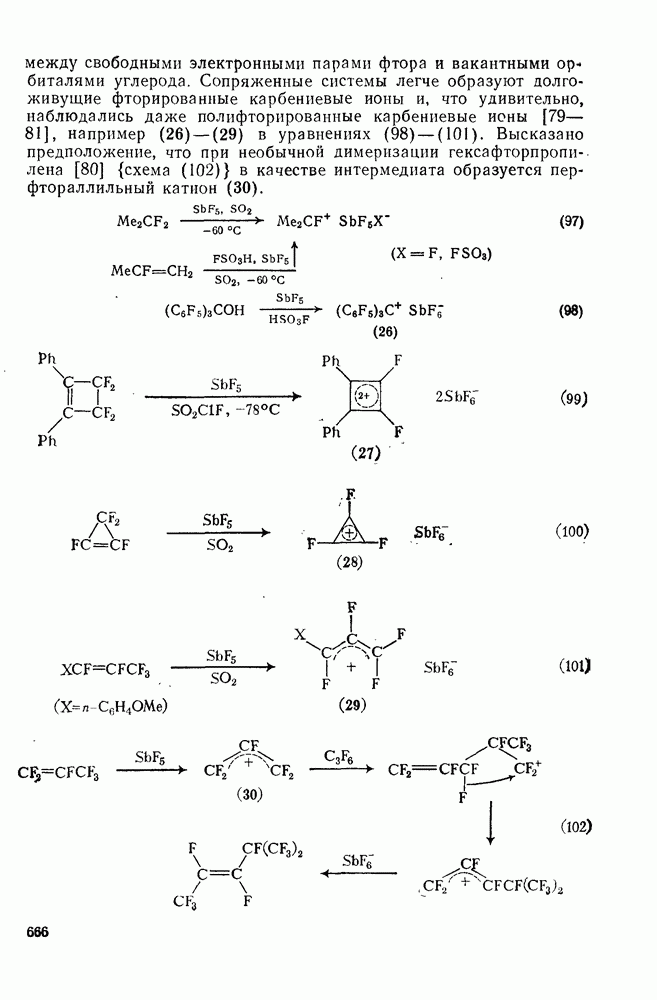
к обоим винильным углеродным атомам алкенов типа  . В случае присоединения к концевой группе
. В случае присоединения к концевой группе  с
с
образованием радикалов  наблюдались лишь небольшие изменения энергии активации в зависимости от природы заместителей X и Y; следовательно, степень резонансной стабилизации образующегося радикала [28], по-видимому, мало влияет на скорость стадии присоединения по крайней мере к этой системе
наблюдались лишь небольшие изменения энергии активации в зависимости от природы заместителей X и Y; следовательно, степень резонансной стабилизации образующегося радикала [28], по-видимому, мало влияет на скорость стадии присоединения по крайней мере к этой системе

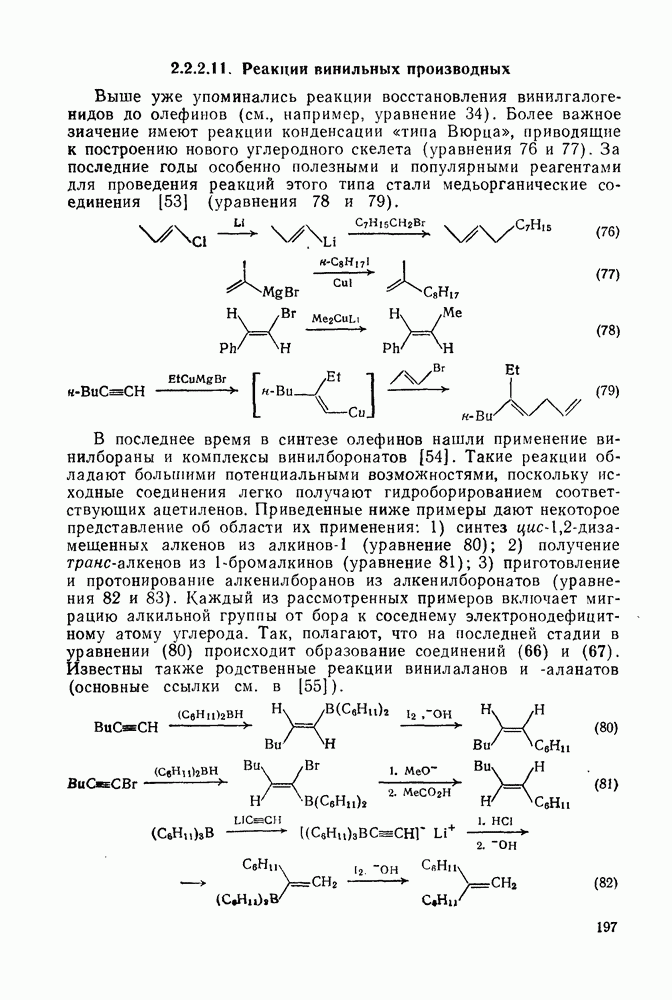
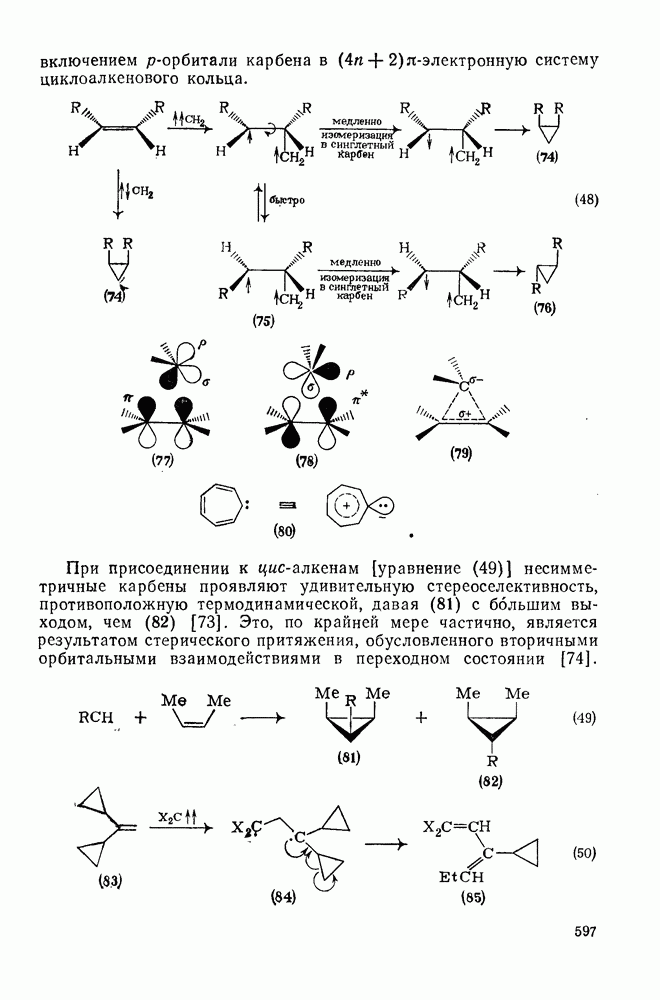
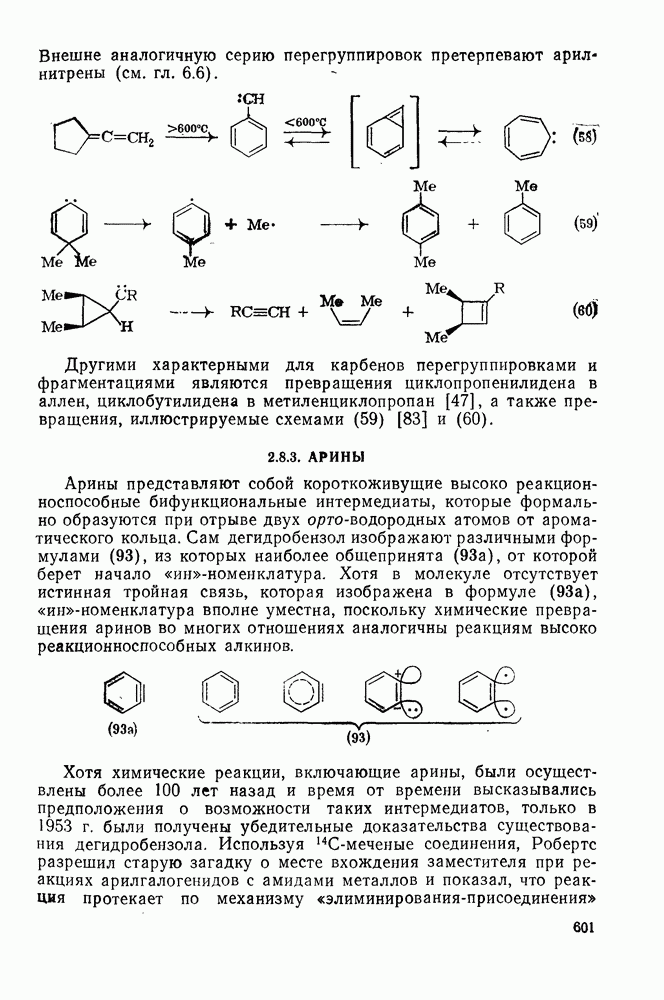
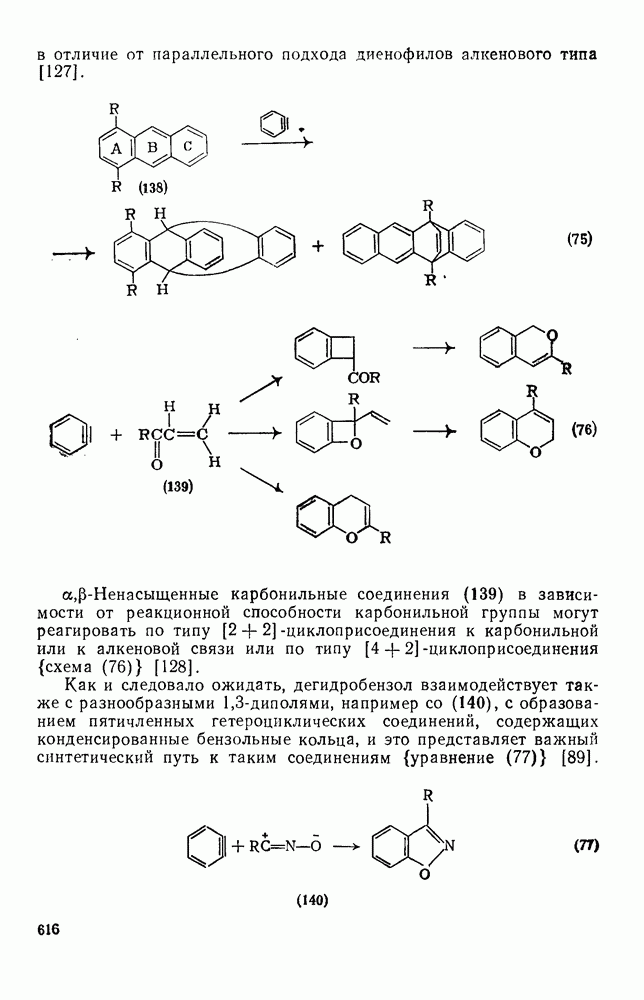
В принципе аддендный радикал может присоединяться к любому концу двойной связи в алкене с образованием радикалов (26) и (27) [схема (22)]. В случае терминальных алкенов  продукты реакции обычно образуются почти исключительно за счет радикала (27); тем самым синтетическая ценность реакции возрастает. Направление присоединения почти всегда можно предсказать, используя простое правило, согласно которому атака радикала направлена преимущественно на наименее замещенный углеродный атом двойной связи. Такое направление реакции объясняют обычно тем, что присоединение проходит через «более устойчивый» из двух возможных радикалов, о чем можно судить, оценивая резонансную делокализацию в обоих радикалах.
продукты реакции обычно образуются почти исключительно за счет радикала (27); тем самым синтетическая ценность реакции возрастает. Направление присоединения почти всегда можно предсказать, используя простое правило, согласно которому атака радикала направлена преимущественно на наименее замещенный углеродный атом двойной связи. Такое направление реакции объясняют обычно тем, что присоединение проходит через «более устойчивый» из двух возможных радикалов, о чем можно судить, оценивая резонансную делокализацию в обоих радикалах.
Теддер полагает, что простая оценка резонансной стабилизации недостаточна; для выбора между двумя направлениями более корректно учитывать прочность образующихся новых связей [29]. В опубликованном недавно обзоре, посвященном проблеме ориентации, Теддер и Уолтон подчеркнули важность и пространственных, и полярных эффектов [30]. Полярные эффекты, например, играют весомую роль в ориентации присоединения к  . Так, электрофильные радикалы, например
. Так, электрофильные радикалы, например  , присоединяются преимущественно к группе
, присоединяются преимущественно к группе  , в случае радикалов
, в случае радикалов  имеет место обратная ориентация и присоединение проходит главным образом к концевой группе
имеет место обратная ориентация и присоединение проходит главным образом к концевой группе  .
.
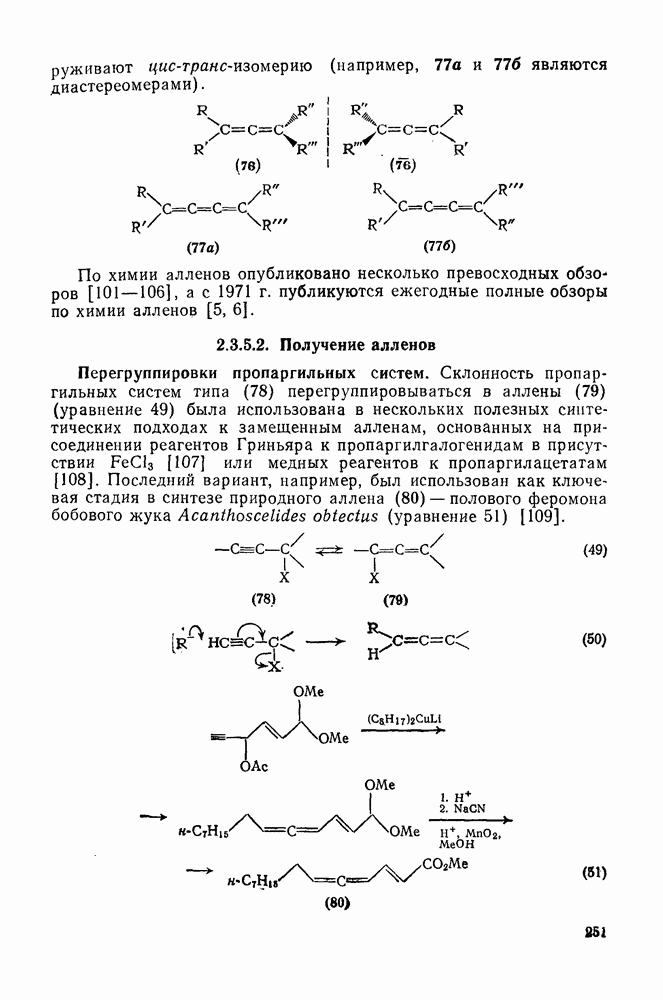
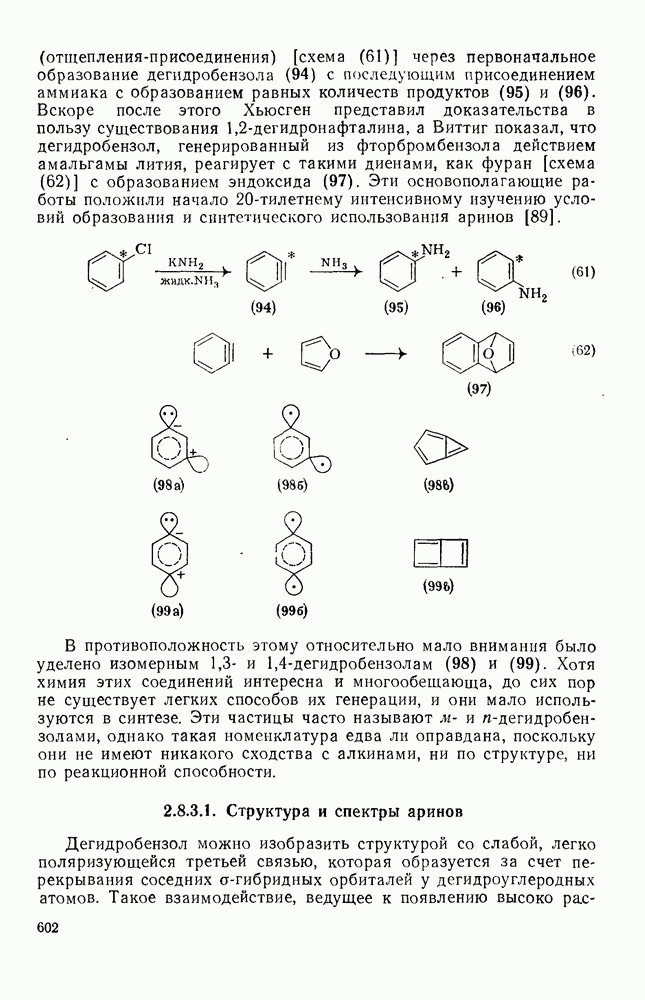
Реакции присоединения радикалов  отличаются тем, что они проходят как стереоспецифическое транс-присоединение. Это часто рассматривают как доказательство в пользу образования мостикового интермедиата (28) [схема (23)] [31]. Присоединение тиолов также проходит предпочтительно по типу транс-присоединения, однако оно менее стереоспецифично. В последние годы большое внимание уделялось изучению внутримолекулярного присоединения [32]. Особенно интересны
отличаются тем, что они проходят как стереоспецифическое транс-присоединение. Это часто рассматривают как доказательство в пользу образования мостикового интермедиата (28) [схема (23)] [31]. Присоединение тиолов также проходит предпочтительно по типу транс-присоединения, однако оно менее стереоспецифично. В последние годы большое внимание уделялось изучению внутримолекулярного присоединения [32]. Особенно интересны  радикалы
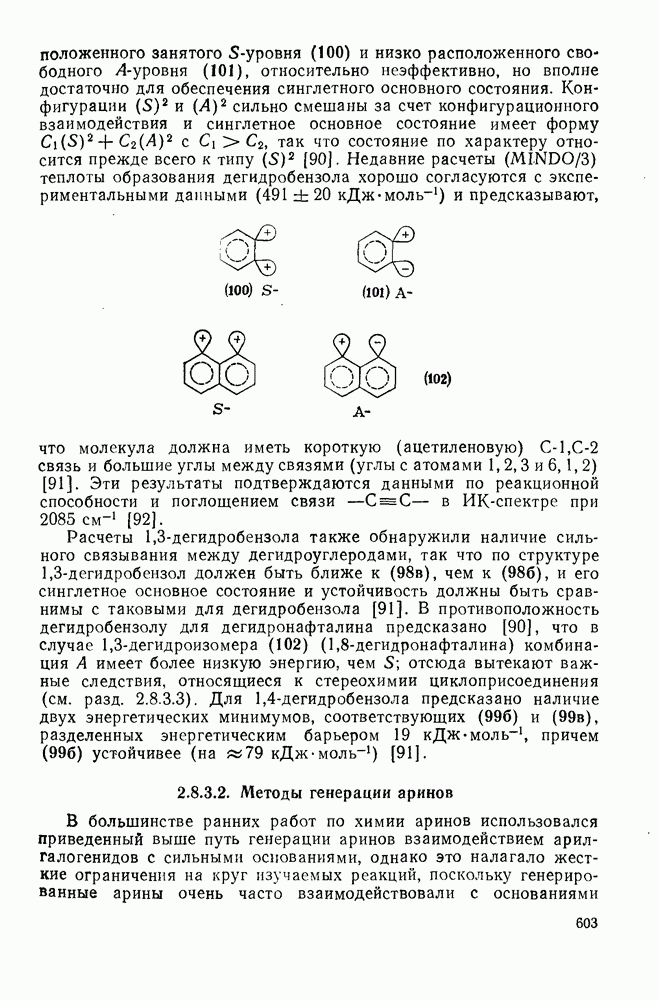
радикалы  , в которых стереоэлектронные факторы направляют циклизацию в сторону образования менее устойчивого радикала (30) [схема (24)]. В случае более стабилизованных радикалов
, в которых стереоэлектронные факторы направляют циклизацию в сторону образования менее устойчивого радикала (30) [схема (24)]. В случае более стабилизованных радикалов  образуются как (30), так и (31).
образуются как (30), так и (31).

Присоединение к сопряженным диенам обычно дает  -аддук-ты, если только нет структурных особенностей, нарушающих сопряжение. В алленах большинство радикалов атакует терминальные положения, однако некоторые радикалы, например
-аддук-ты, если только нет структурных особенностей, нарушающих сопряжение. В алленах большинство радикалов атакует терминальные положения, однако некоторые радикалы, например  , присоединяются к центральному углеродному атому. При радикальном присоединении к ацетиленам образуются реакционноспособные винильные радикалы, которые обычно быстро превращаются в алкены за счет переноса цепи. Присоединение гидробромида протекает стереоспецифично, но в реакциях с большинством других аддендов образуются оба стереоизомера.
, присоединяются к центральному углеродному атому. При радикальном присоединении к ацетиленам образуются реакционноспособные винильные радикалы, которые обычно быстро превращаются в алкены за счет переноса цепи. Присоединение гидробромида протекает стереоспецифично, но в реакциях с большинством других аддендов образуются оба стереоизомера.
Полициклические ароматические углеводороды и некоторые гетероциклические соединения реагируют значительно активнее бензола. Например, антрацен реагирует с фенильными радикалами в 250 раз быстрее, а также взаимодействует с менее реакционноспособными радикалами  , которые к бензолу не присоединяются.
, которые к бензолу не присоединяются.
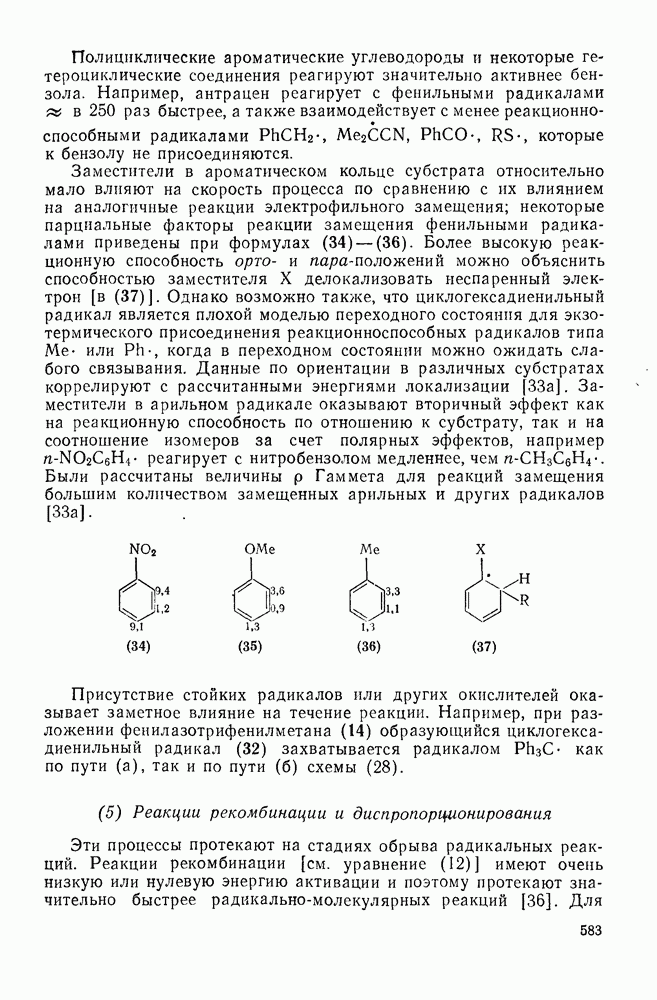
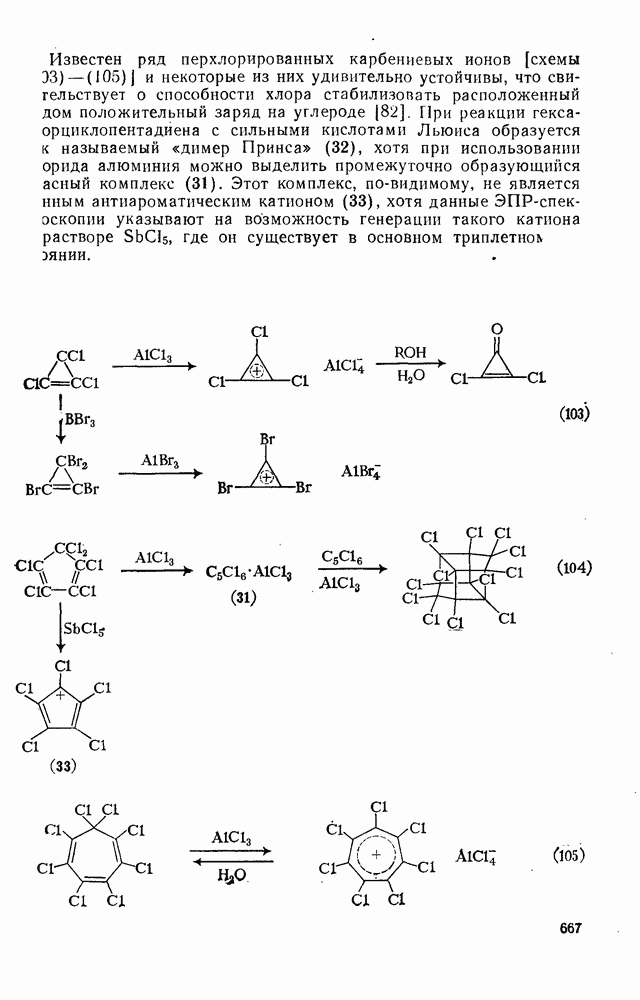
Заместители в ароматическом кольце субстрата относительно мало влияют на скорость процесса по сравнению с их влиянием на аналогичные реакции электрофильного замещения; некоторые парциальные факторы реакции замещения фенильными радикалами приведены при формулах (34) — (36). Более высокую реакционную способность орто- и пара-положений можно объяснить способностью заместителя X делокализовать неспаренный электрон [в (37)]. Однако возможно также, что циклогексадиенильный радикал является плохой моделью переходного состояния для экзотермического присоединения реакционноспособных радикалов типа  или
или  , когда в переходном состоянии можно ожидать слабого связывания. Данные по ориентации в различных субстратах коррелируют с рассчитанными энергиями локализации [33а]. Заместители в арильном радикале оказывают вторичный эффект как на реакционную способность по отношению к субстрату, так и на соотношение изомеров за счет полярных эффектов, например
, когда в переходном состоянии можно ожидать слабого связывания. Данные по ориентации в различных субстратах коррелируют с рассчитанными энергиями локализации [33а]. Заместители в арильном радикале оказывают вторичный эффект как на реакционную способность по отношению к субстрату, так и на соотношение изомеров за счет полярных эффектов, например  реагирует с нитробензолом медленнее, чем
реагирует с нитробензолом медленнее, чем  . Были рассчитаны величины
. Были рассчитаны величины  Гаммета для реакций замещения большим количеством замещенных арильных и других радикалов [33а].
Гаммета для реакций замещения большим количеством замещенных арильных и других радикалов [33а].

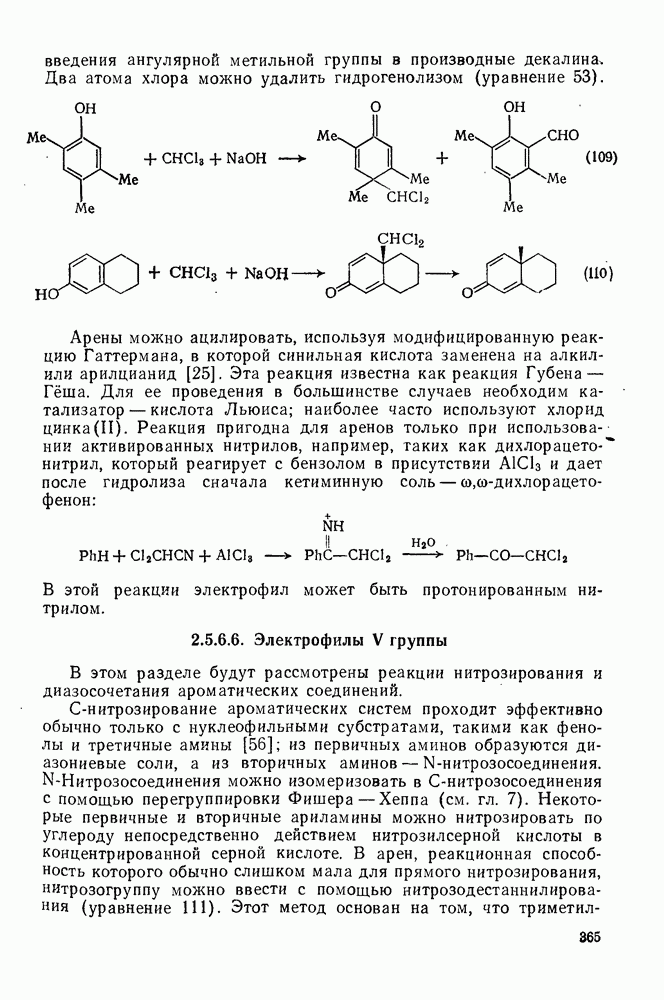
Присутствие стойких радикалов или других окислителей оказывает заметное влияние на течение реакции. Например, при разложении фенилазотрифенилметана (14) образующийся циклогексадиенильный радикал (32) захватывается радикалом  как по пути (а), так и по пути (б) схемы (28).
как по пути (а), так и по пути (б) схемы (28).
(5) Реакции рекомбинации и диспропорционирования
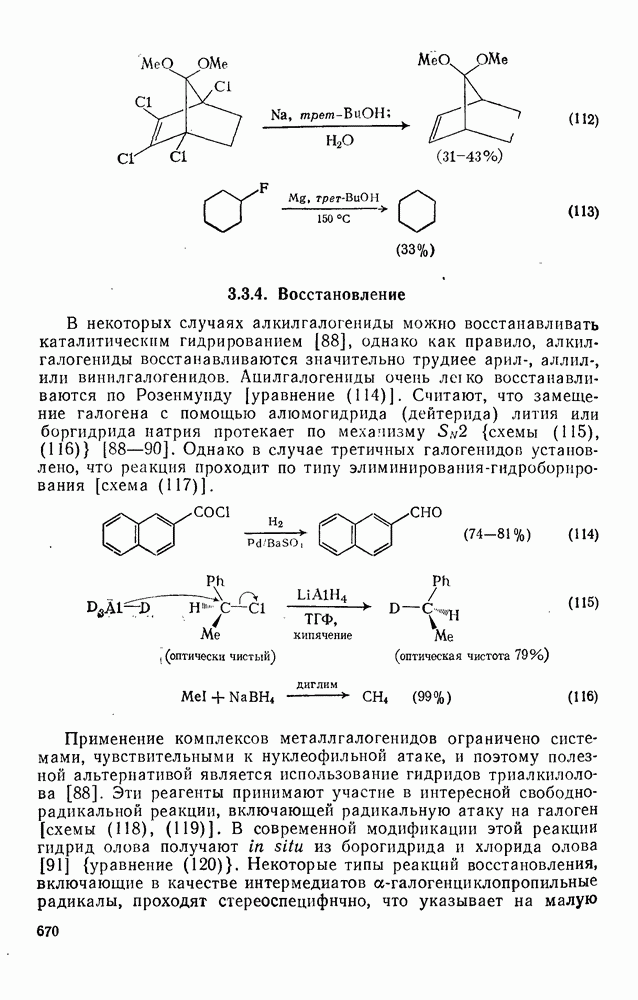
Эти процессы протекают на стадиях обрыва радикальных реакций. Реакции рекомбинации [см. уравнение (12)] имеют очень низкую или нулевую энергию активации и поэтому протекают значительно быстрее радикально-молекулярных реакций [36].
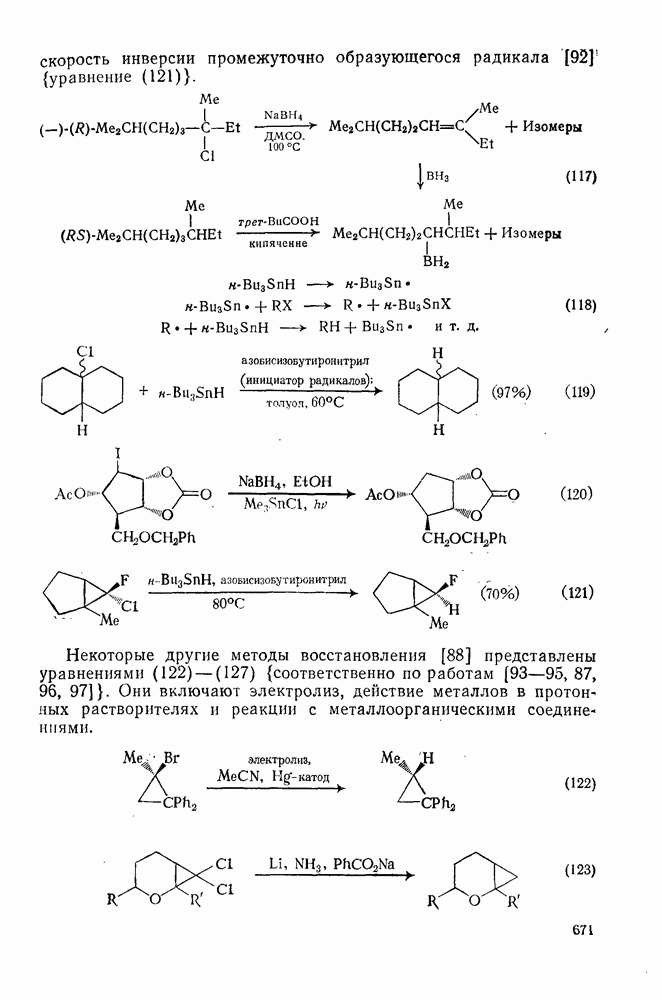
Для реакций в жидкой фазе константа скорости димеризации радика» лов с радикальным центром на атоме углерода обычно составляет  (для сравнения константа скорости радикального присоединения к алкенам равна
(для сравнения константа скорости радикального присоединения к алкенам равна  ) и близка к диффузионно контролируемому пределу. Следует отметить, что скорость рекомбинации резонансно стабилизованных радикалов, например
) и близка к диффузионно контролируемому пределу. Следует отметить, что скорость рекомбинации резонансно стабилизованных радикалов, например  и
и  , не ниже скорости менее стабилизованных частиц, однако димеризация замедляется при наличии пространственных затруднений, таких как в радикалах (1) и (5). Более низкая скорость димеризации
, не ниже скорости менее стабилизованных частиц, однако димеризация замедляется при наличии пространственных затруднений, таких как в радикалах (1) и (5). Более низкая скорость димеризации  по сравнению с
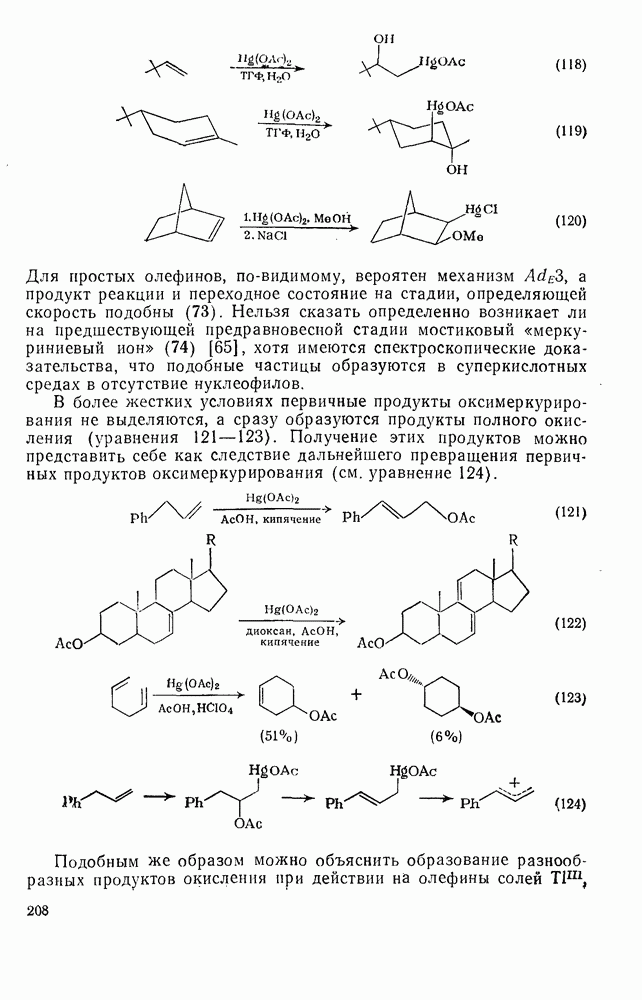
по сравнению с  обусловлена полярным отталкиванием. Скорость реакции диспропорционирования [см. уравнение (13)] аналогична скорости реакции рекомбинации и поэтому не может включать обычный отрыв водорода, поскольку энергия активации в радикал-молекулярных реакциях равна
обусловлена полярным отталкиванием. Скорость реакции диспропорционирования [см. уравнение (13)] аналогична скорости реакции рекомбинации и поэтому не может включать обычный отрыв водорода, поскольку энергия активации в радикал-молекулярных реакциях равна  . Отношения констант скорости реакций диспропорционирования и рекомбинации
. Отношения констант скорости реакций диспропорционирования и рекомбинации  для
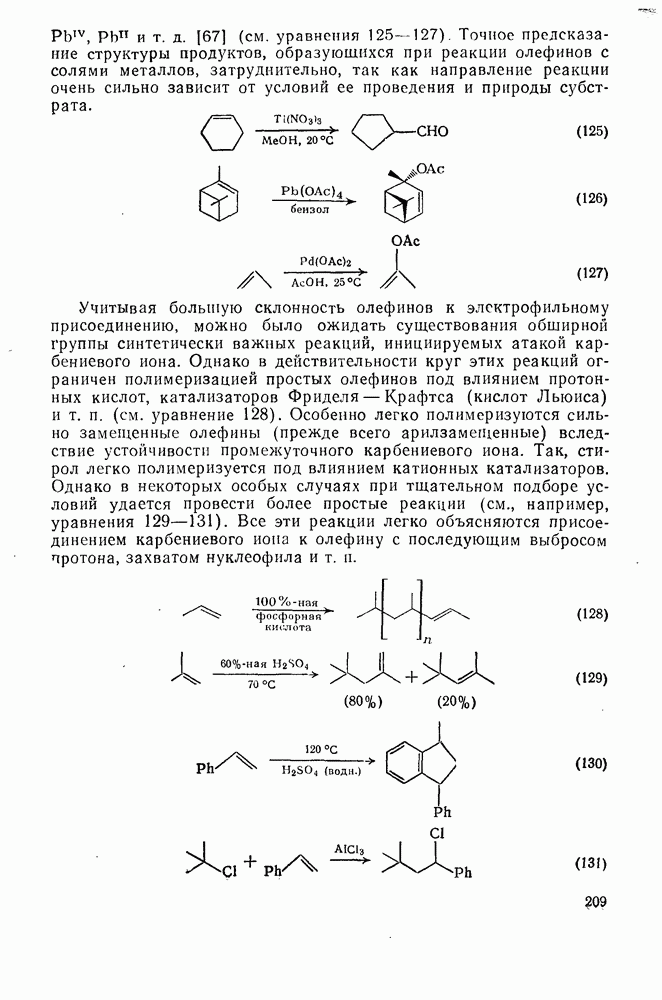
для  -водорода первичных, вторичных и третичных алкильных радикалов составляют соответственно 0,03; 0,1 и 0,4. Это отношение уменьшается в случае резонансно стабилизованных радикалов и, например, для
-водорода первичных, вторичных и третичных алкильных радикалов составляют соответственно 0,03; 0,1 и 0,4. Это отношение уменьшается в случае резонансно стабилизованных радикалов и, например, для  равно 0,005. Кроме процессов, контролируемых диффузией, обе эти реакции могут проходить в клетке растворителя [37]. В тех случаях, когда пары радикалов генерируются в непосредственной близости друг от друга, как, например, при разложении азосоединений или пероксидов, некоторые из них попарно рекомбинируются в клетке растворителя еще до того, как осуществится диффузия и пройдут реакции с молекулами субстрата или другими радикалами. Степень клеточной рекомбинации возрастает с увеличением вязкости растворителя и может в значительной степени уменьшать эффективность инициаторов.
равно 0,005. Кроме процессов, контролируемых диффузией, обе эти реакции могут проходить в клетке растворителя [37]. В тех случаях, когда пары радикалов генерируются в непосредственной близости друг от друга, как, например, при разложении азосоединений или пероксидов, некоторые из них попарно рекомбинируются в клетке растворителя еще до того, как осуществится диффузия и пройдут реакции с молекулами субстрата или другими радикалами. Степень клеточной рекомбинации возрастает с увеличением вязкости растворителя и может в значительной степени уменьшать эффективность инициаторов.
Реакции рекомбинации можно использовать в синтетических целях, например для получения таких димеров, как  и
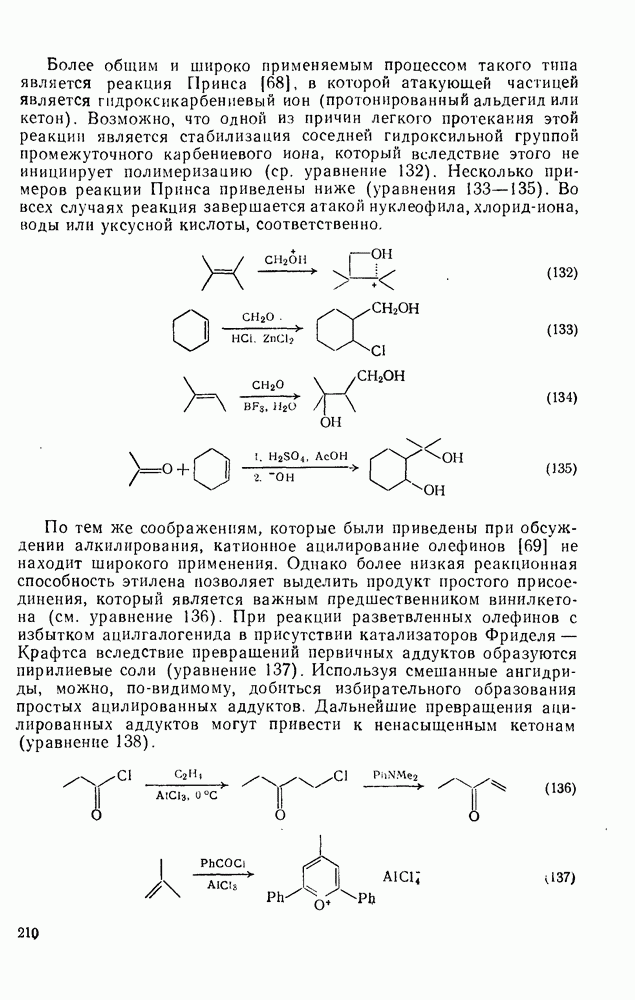
и  , а также при окислительном сочетании фенолов (см. разд. 4.2.3.2). Стойкие радикалы используют также в качестве небольших радикальных ловушек, часто для ингибирования цепных реакций.
, а также при окислительном сочетании фенолов (см. разд. 4.2.3.2). Стойкие радикалы используют также в качестве небольших радикальных ловушек, часто для ингибирования цепных реакций.
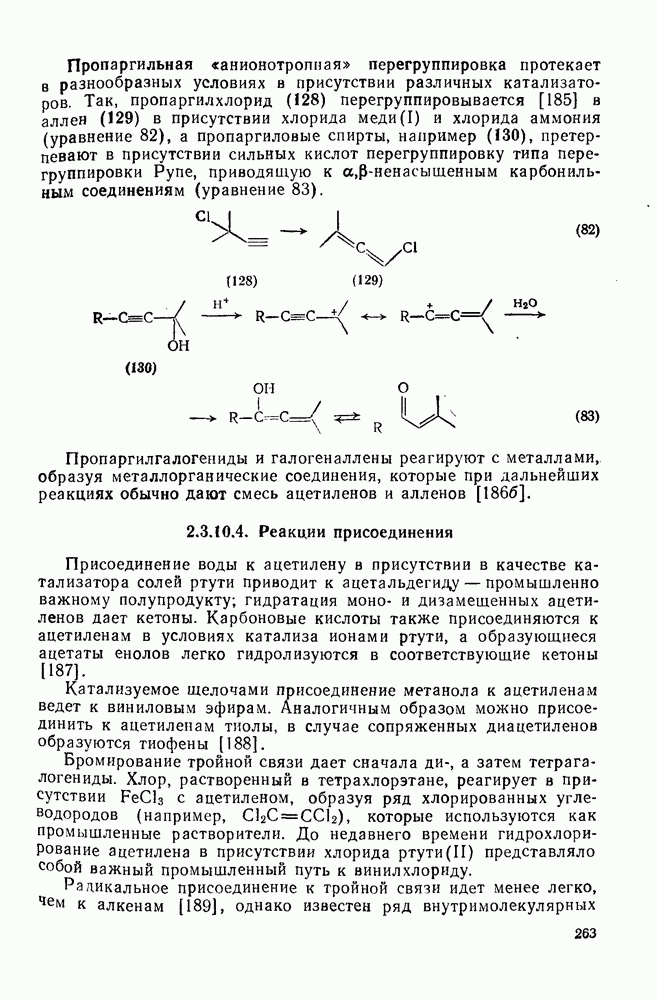
(6) Фрагментации и перегруппировки [32б]
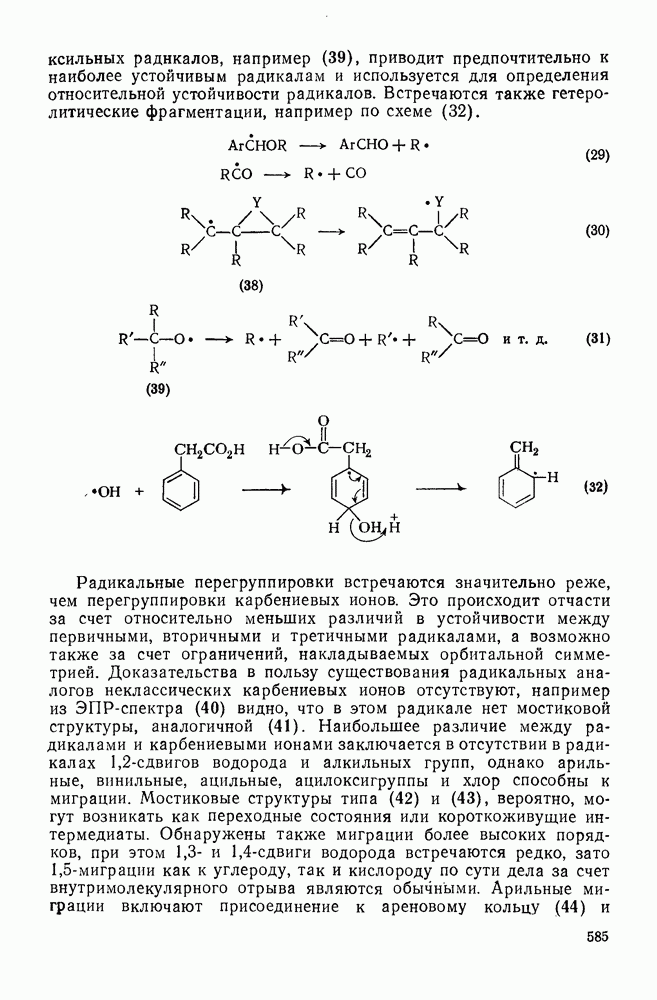
Некоторые примеры гомолитической фрагментации радикалов приведены в уравнениях (10), (16) и (29) — (31). Фрагментация — реакция, обратная радикальному присоединению к алкенам [см. уравнение  ], свойственна радикалам, у которых
], свойственна радикалам, у которых  или
или
 . При
. При  фрагментация возможна только в тех случаях, когда вовлекается слабая или сильно напряженная связь. Имеются примеры внутримолекулярной фрагментации, например циклопро-пил-гомоаллильная перегруппировка
фрагментация возможна только в тех случаях, когда вовлекается слабая или сильно напряженная связь. Имеются примеры внутримолекулярной фрагментации, например циклопро-пил-гомоаллильная перегруппировка  и аналогичное раскрытие кольца оксиранов
и аналогичное раскрытие кольца оксиранов  .
.
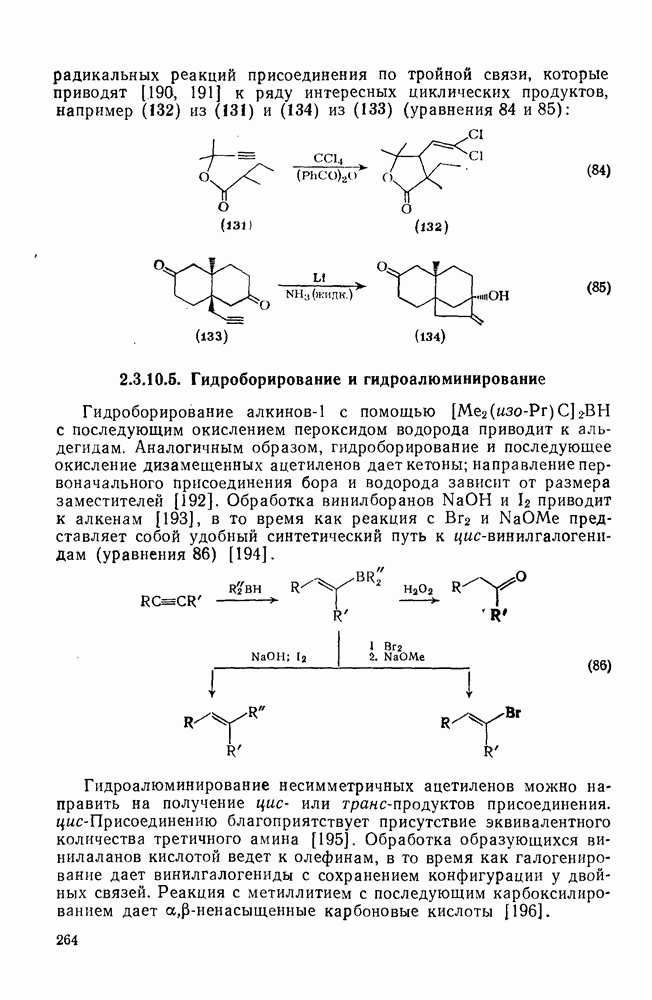
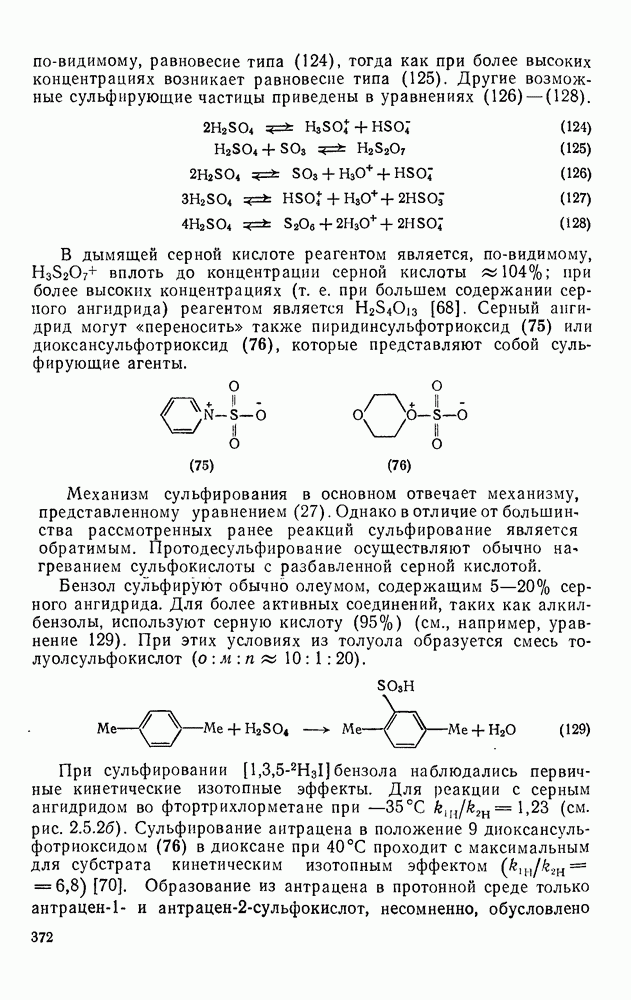
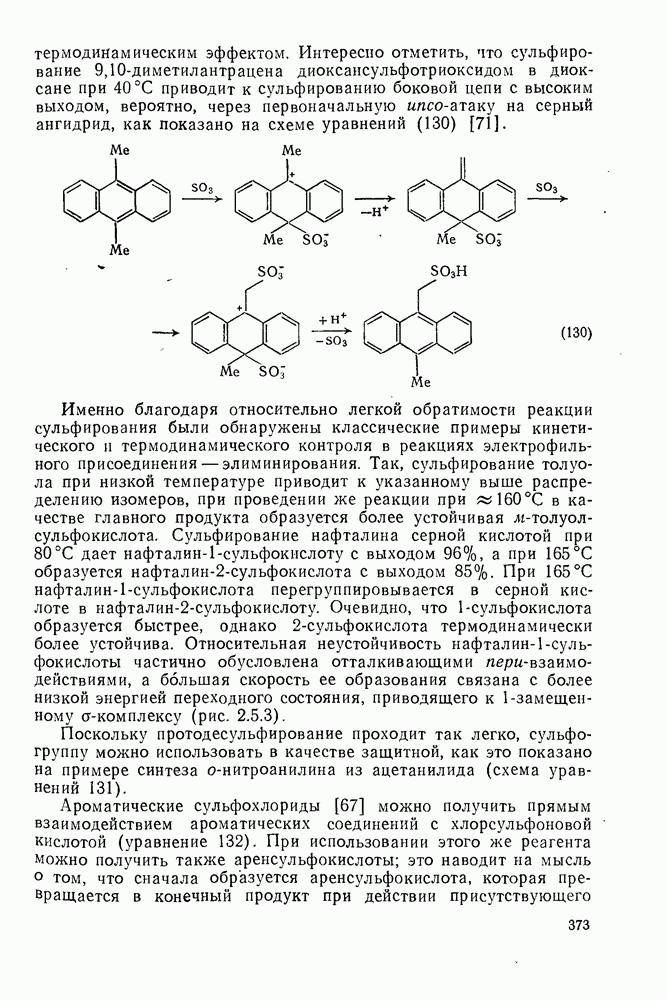
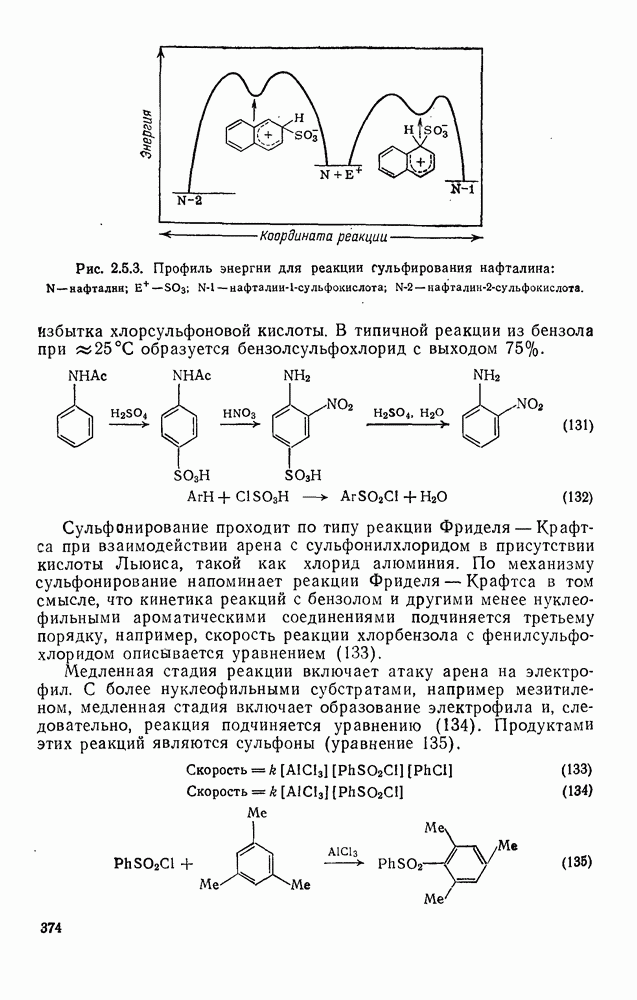
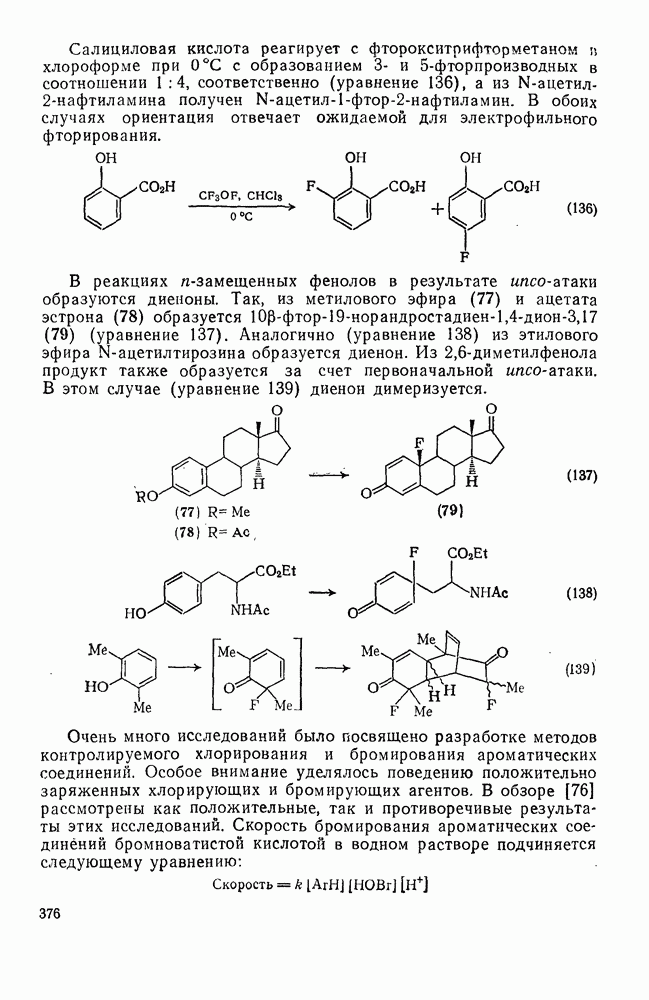
Фрагментация алкоксильных радикалов, например (39), приводит предпочтительно к наиболее устойчивым радикалам и используется для определения относительной устойчивости радикалов. Встречаются также гетеролитические фрагментации, например по схеме (32).

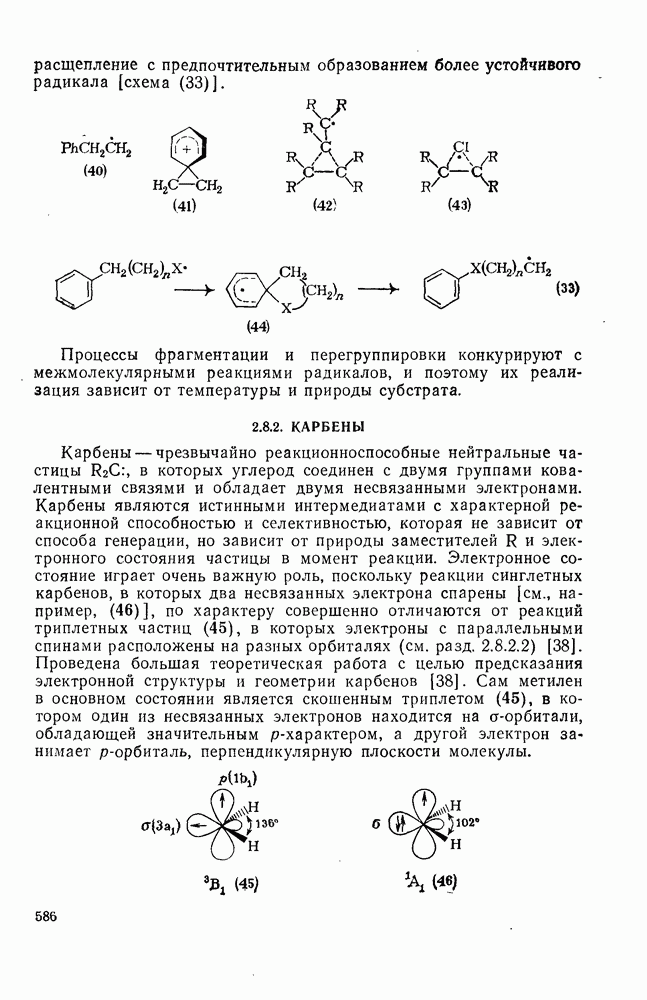
Радикальные перегруппировки встречаются значительно реже, чем перегруппировки карбениевых ионов. Это происходит отчасти за счет относительно меньших различий в устойчивости между первичными, вторичными и третичными радикалами, а возможно также за счет ограничений, накладываемых орбитальной симметрией. Доказательства в пользу существования радикальных аналогов неклассических карбениевых ионов отсутствуют, например из ЭПР-спектра (40) видно, что в этом радикале нет мостиковой структуры, аналогичной (41). Наибольшее различие между радикалами и карбениевыми ионами заключается в отсутствии в радикалах  -сдвигов водорода и алкильных групп, однако арильные, винильные, ацильные, ацилоксигруппы и хлор способны к миграции. Мостиковые структуры типа (42) и (43), вероятно, могут возникать как переходные состояния или короткоживущие интермедиаты. Обнаружены также миграции более высоких порядков, при этом 1,3- и
-сдвигов водорода и алкильных групп, однако арильные, винильные, ацильные, ацилоксигруппы и хлор способны к миграции. Мостиковые структуры типа (42) и (43), вероятно, могут возникать как переходные состояния или короткоживущие интермедиаты. Обнаружены также миграции более высоких порядков, при этом 1,3- и  -сдвиги водорода встречаются редко, зато
-сдвиги водорода встречаются редко, зато  -миграции как к углероду, так и кислороду по сути дела за счет внутримолекулярного отрыва являются обычными.
-миграции как к углероду, так и кислороду по сути дела за счет внутримолекулярного отрыва являются обычными.
Арильные миграции включают присоединение к ареновому кольцу (44) и расщепление с предпочтительным образованием более устойчивого радикала [схема (33)].
 (44)
(44)
Процессы фрагментации и перегруппировки конкурируют с межмолекулярными реакциями радикалов, и поэтому их реализация зависит от температуры и природы субстрата.
























































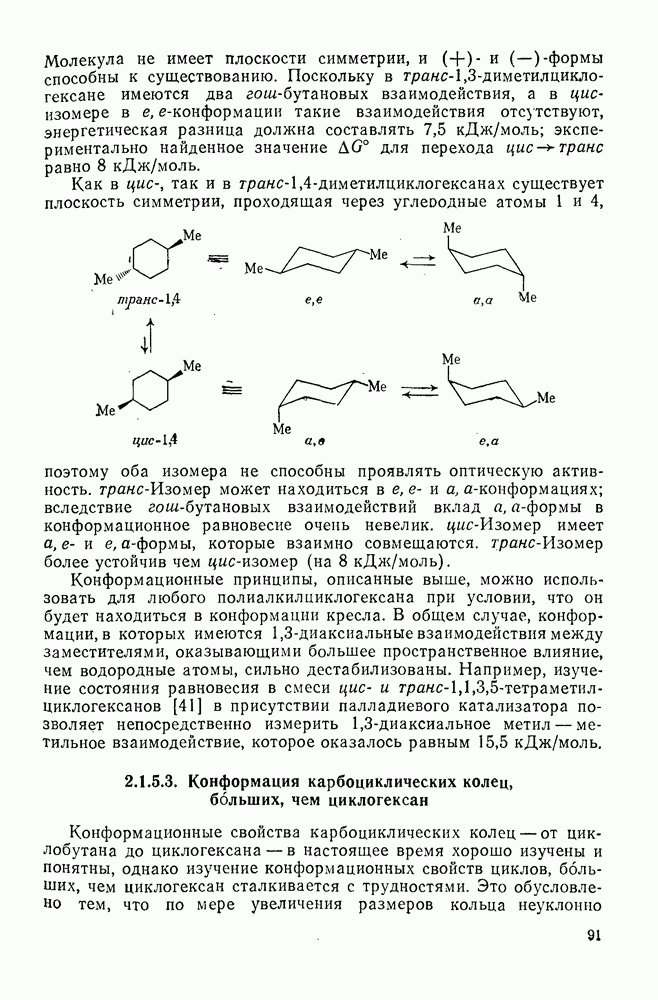




























































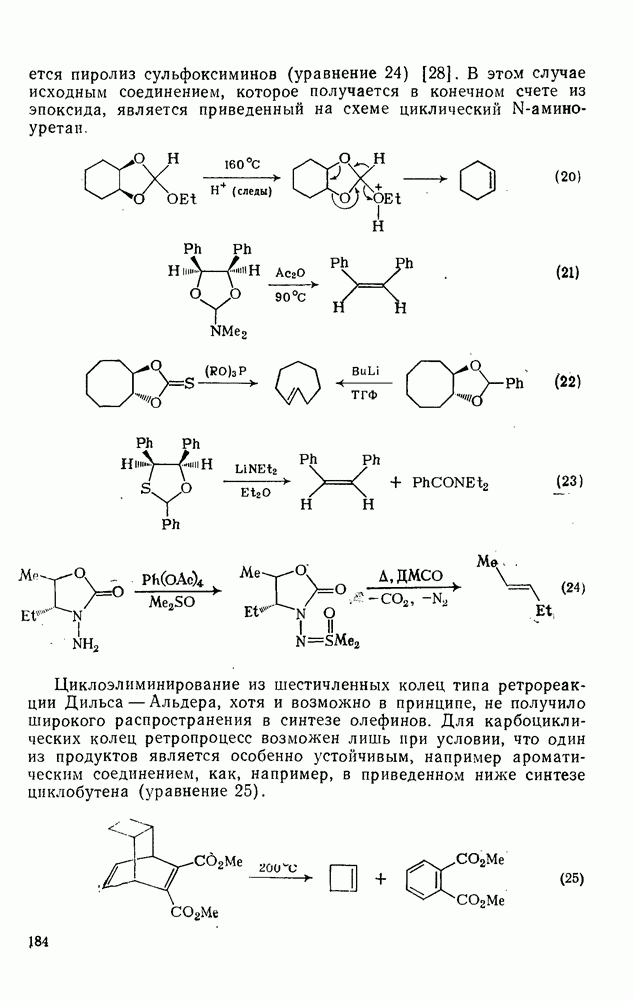
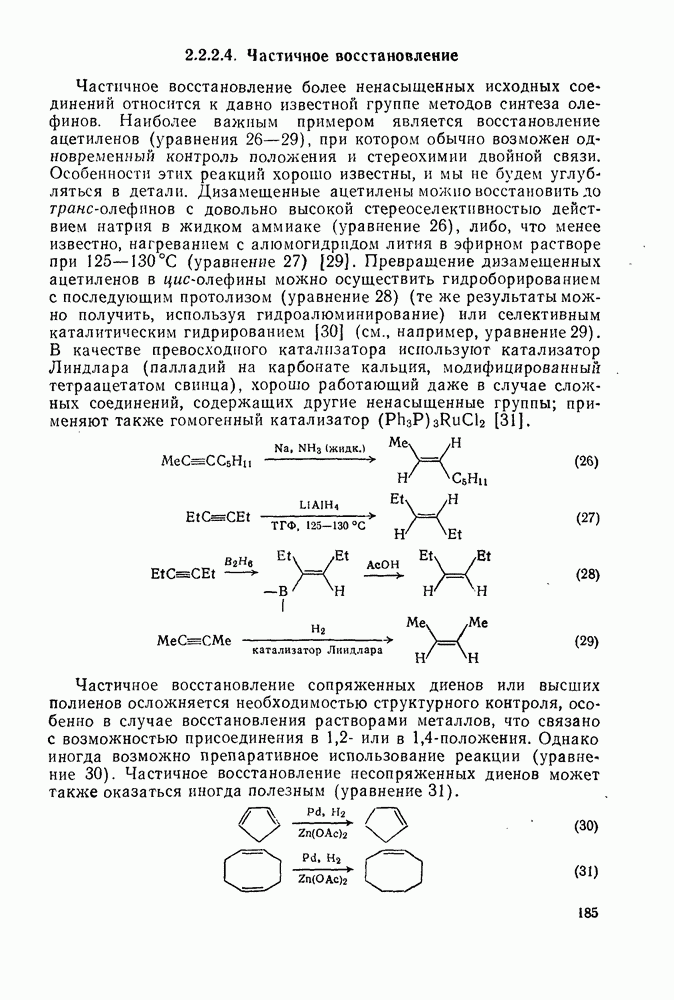
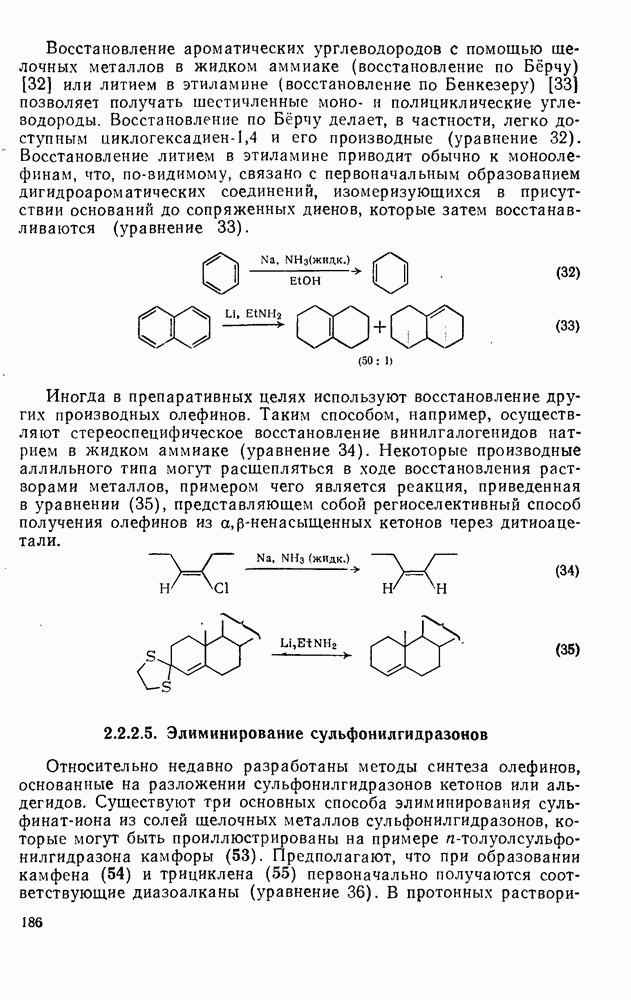
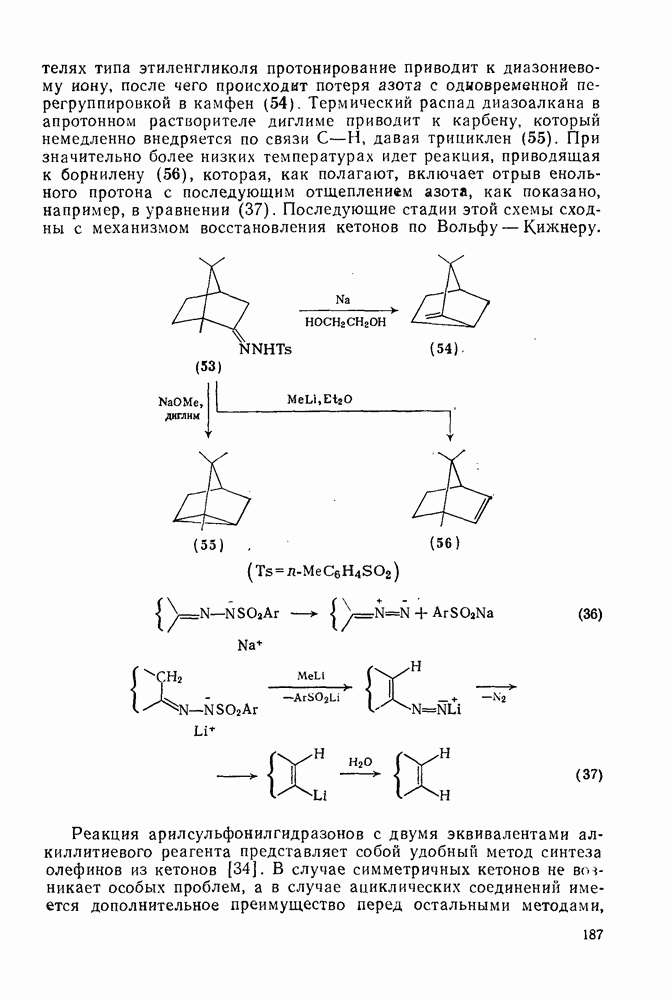























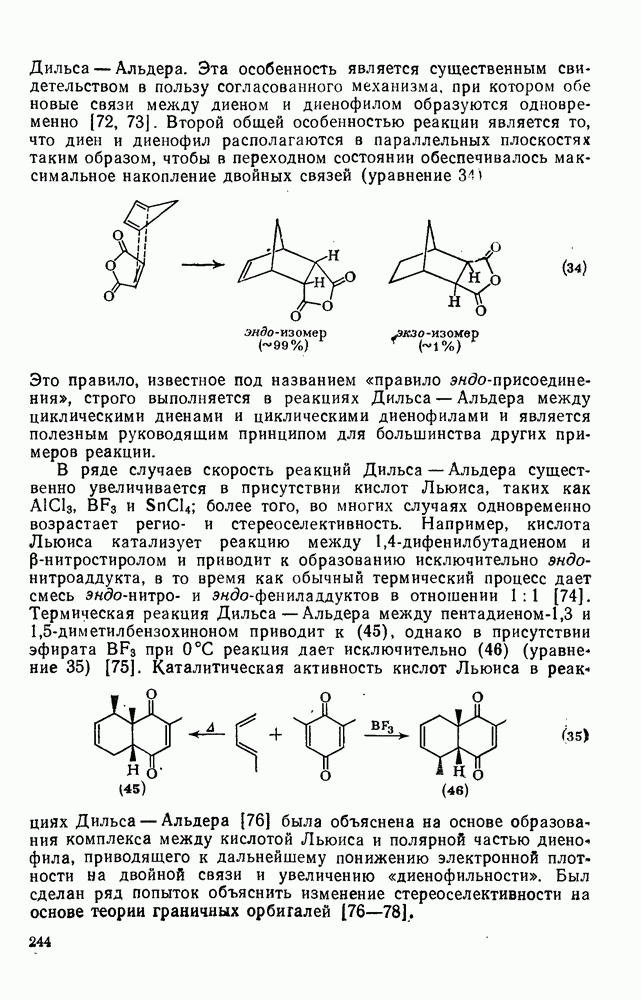
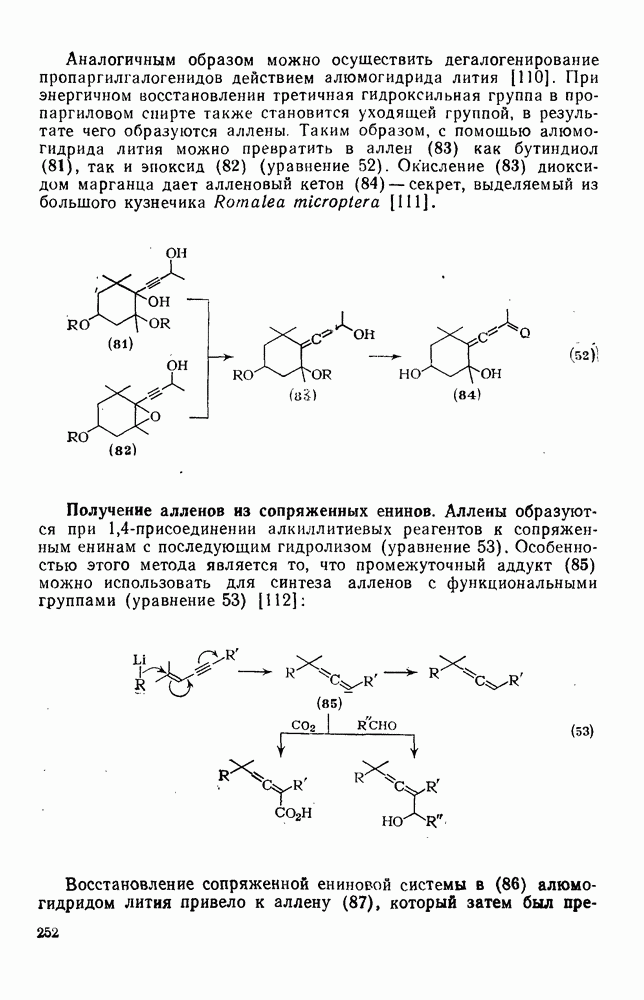

































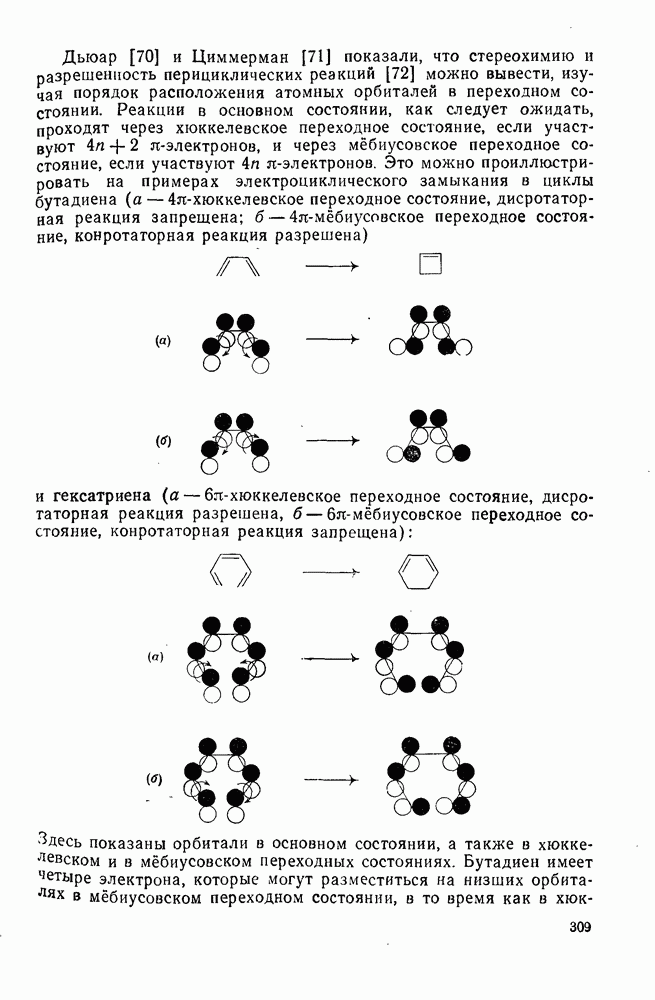





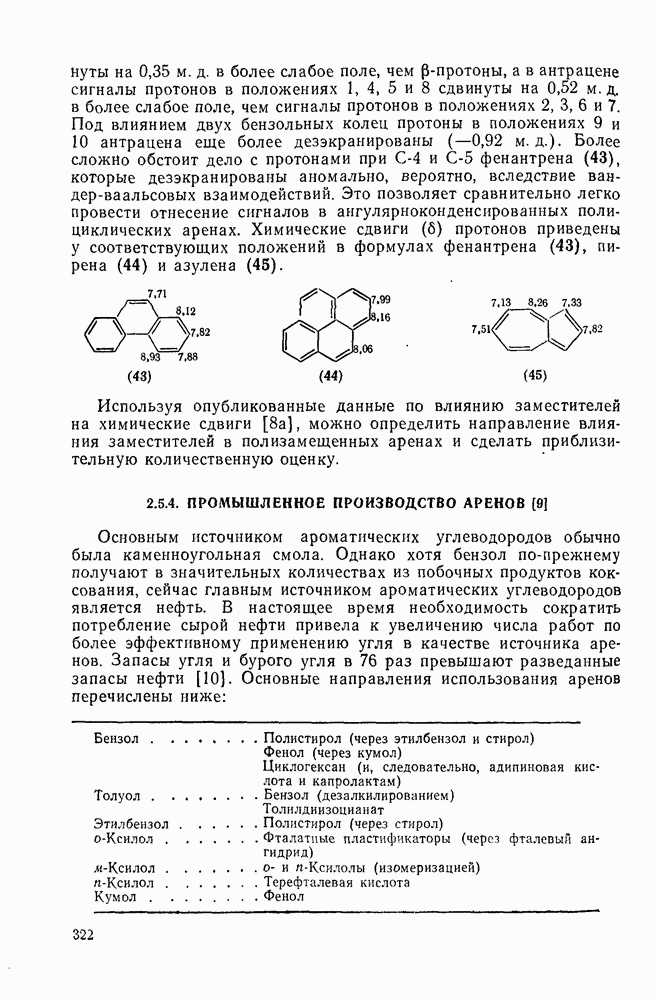





























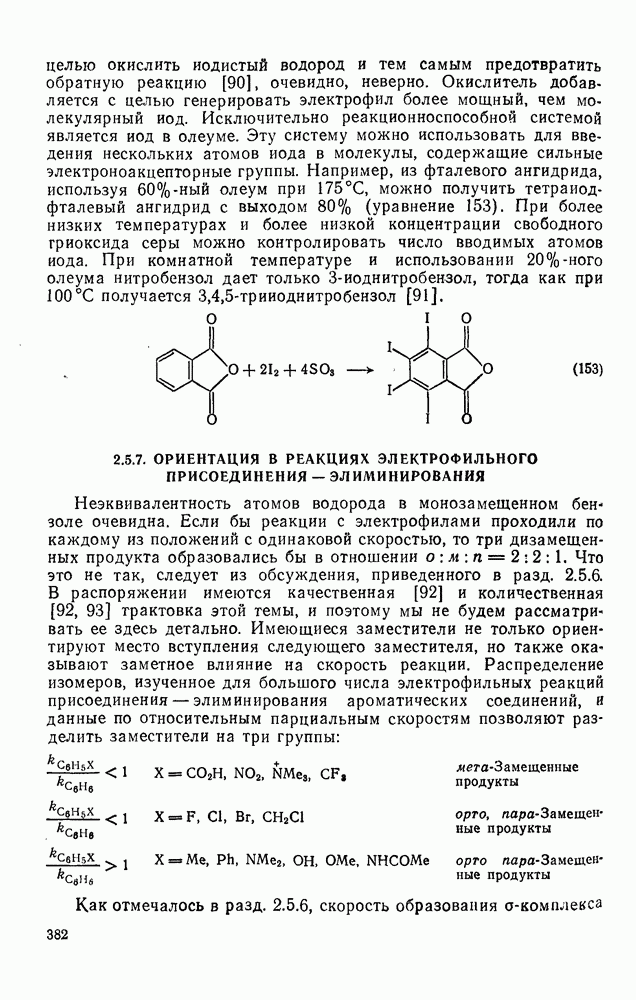





















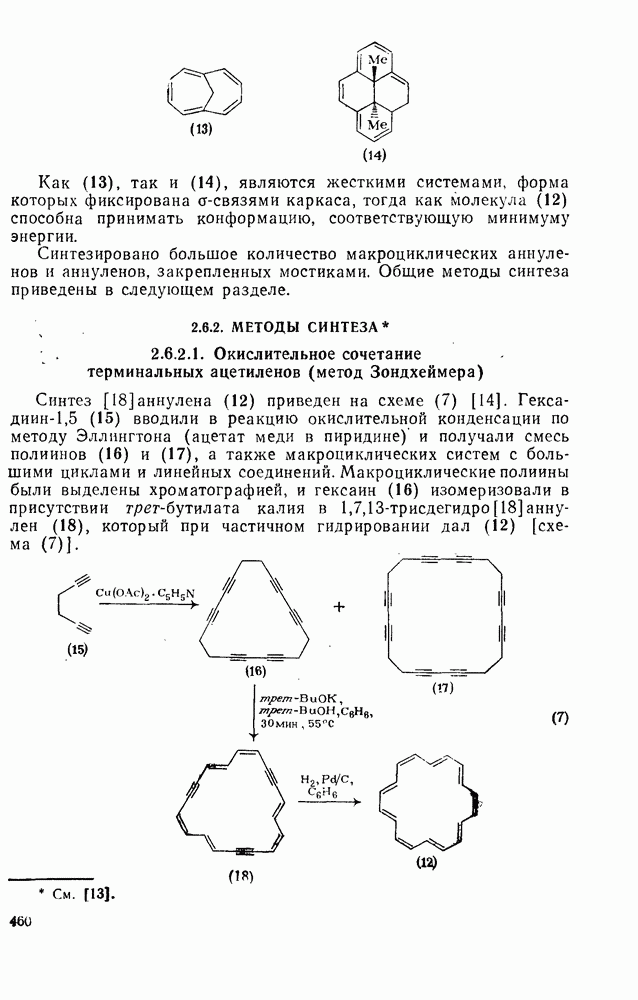
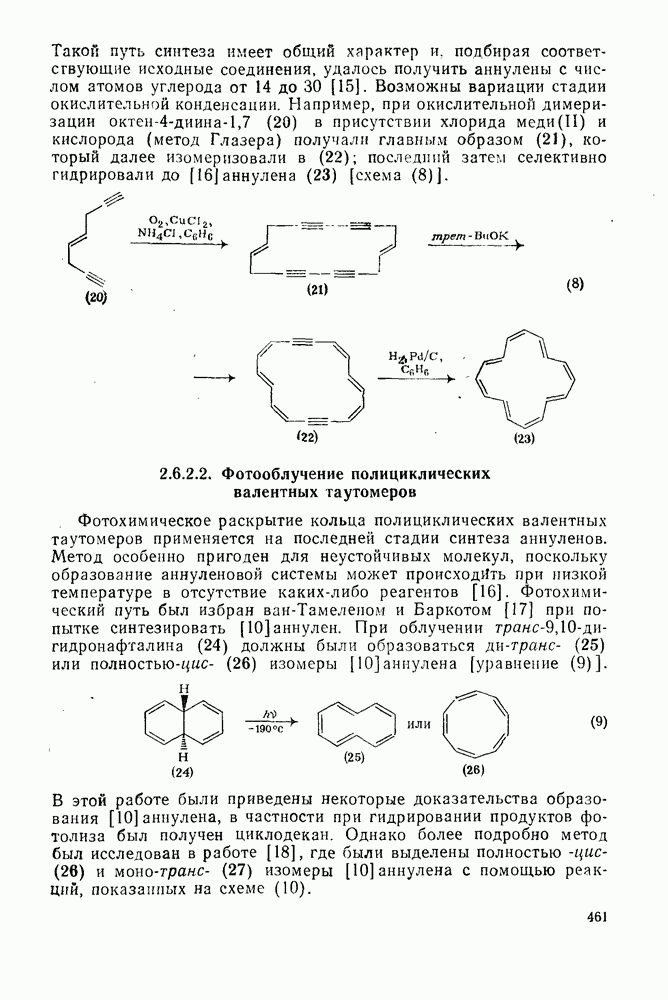
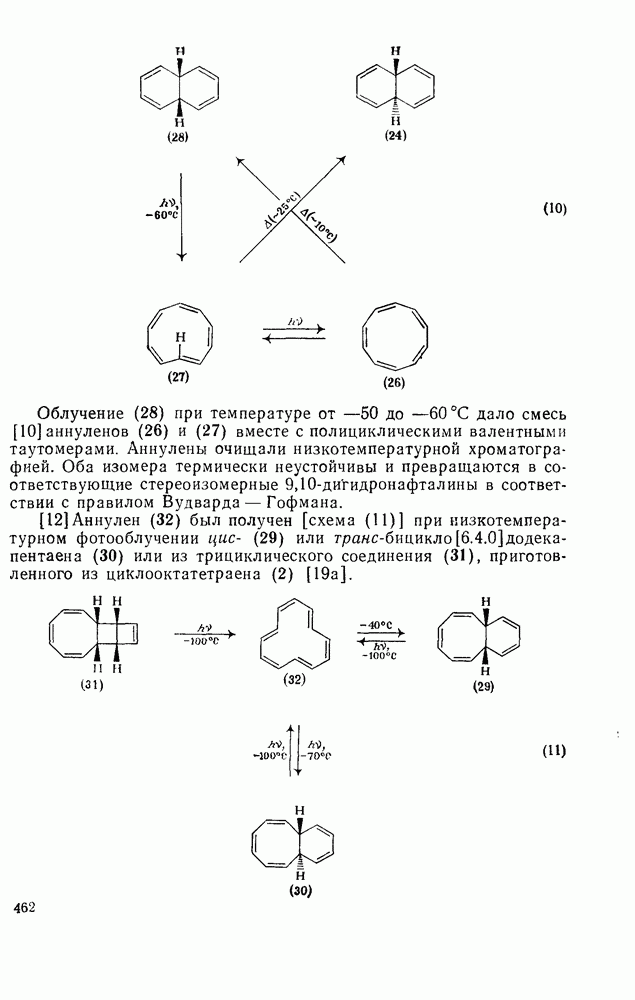
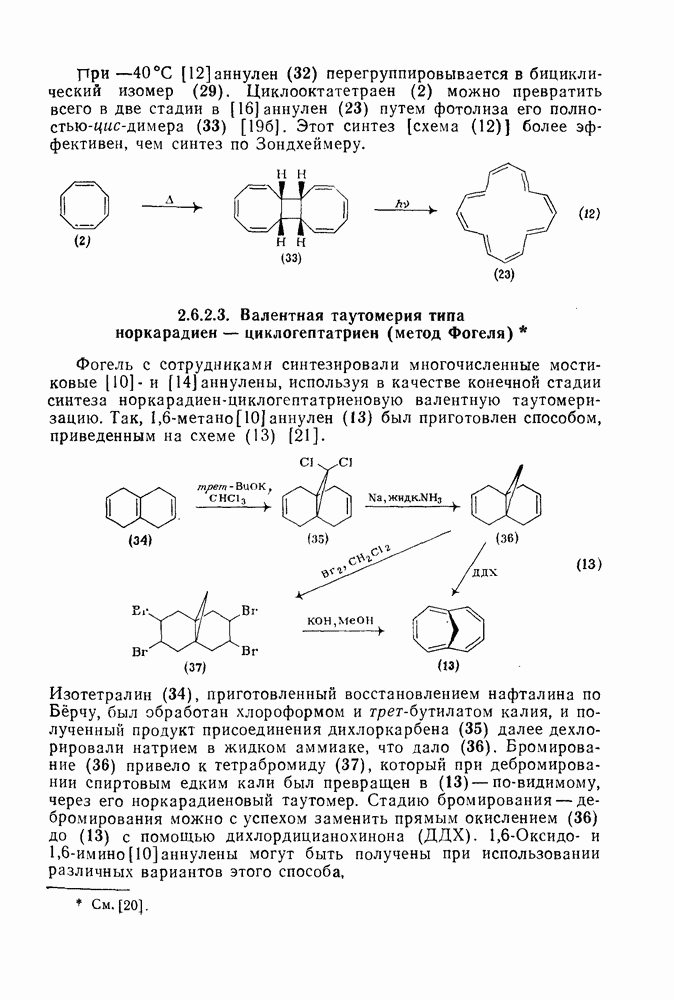




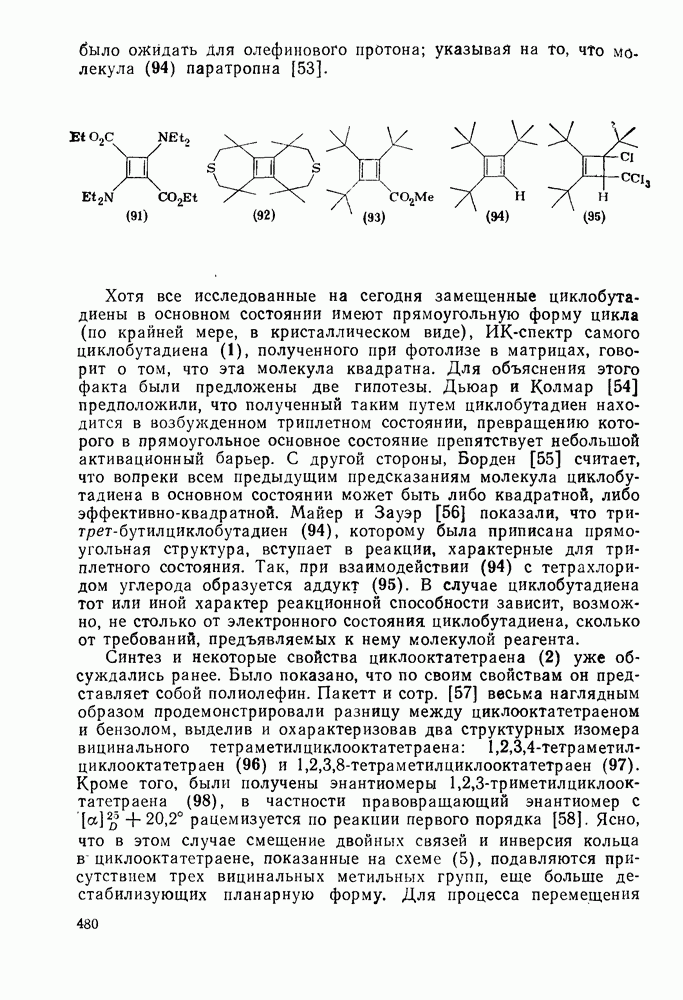



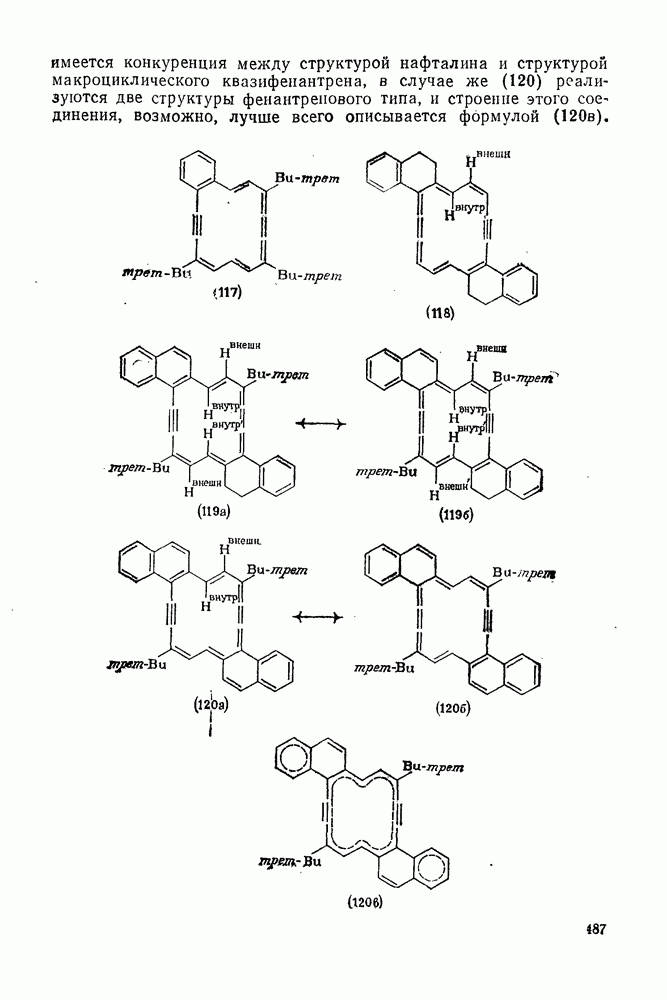
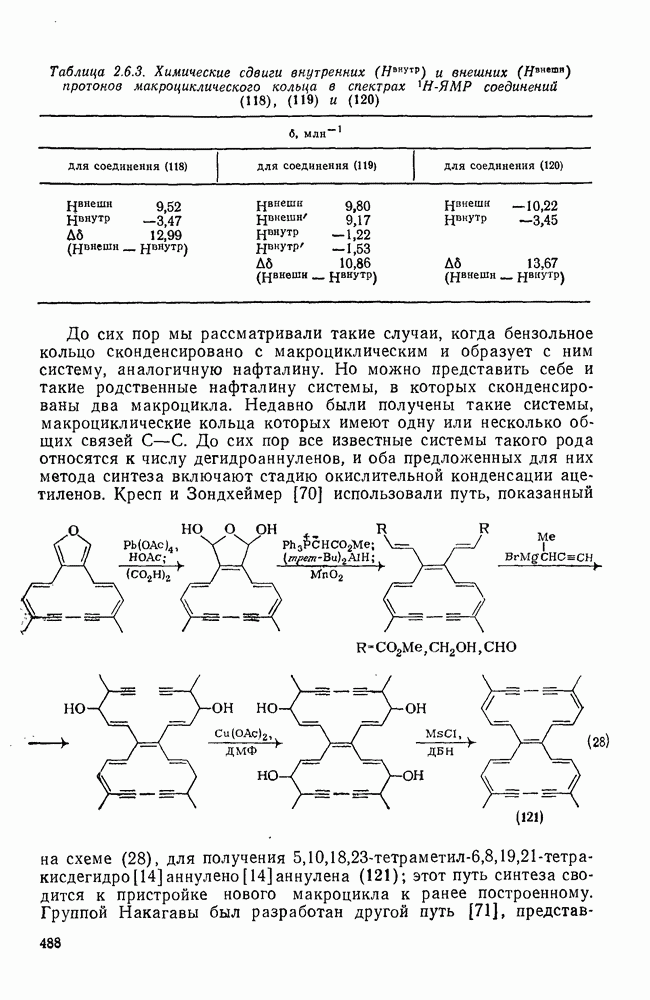
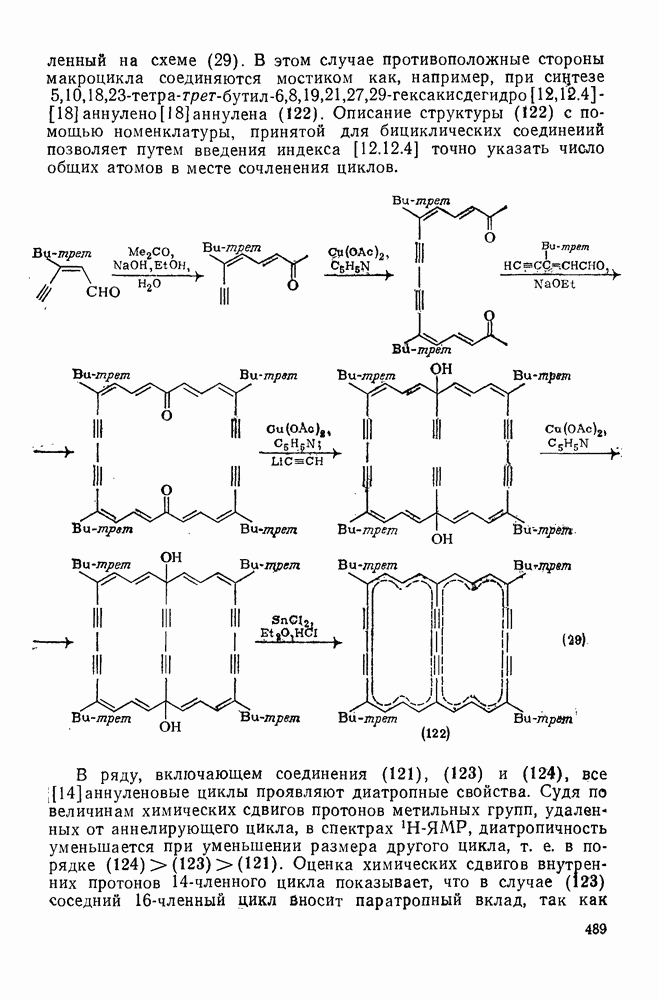
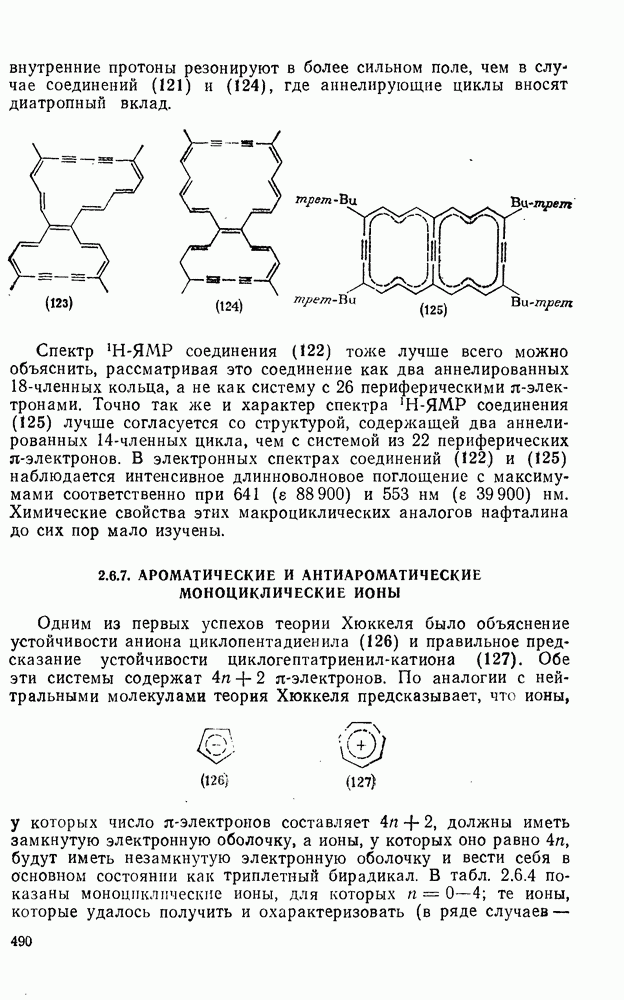

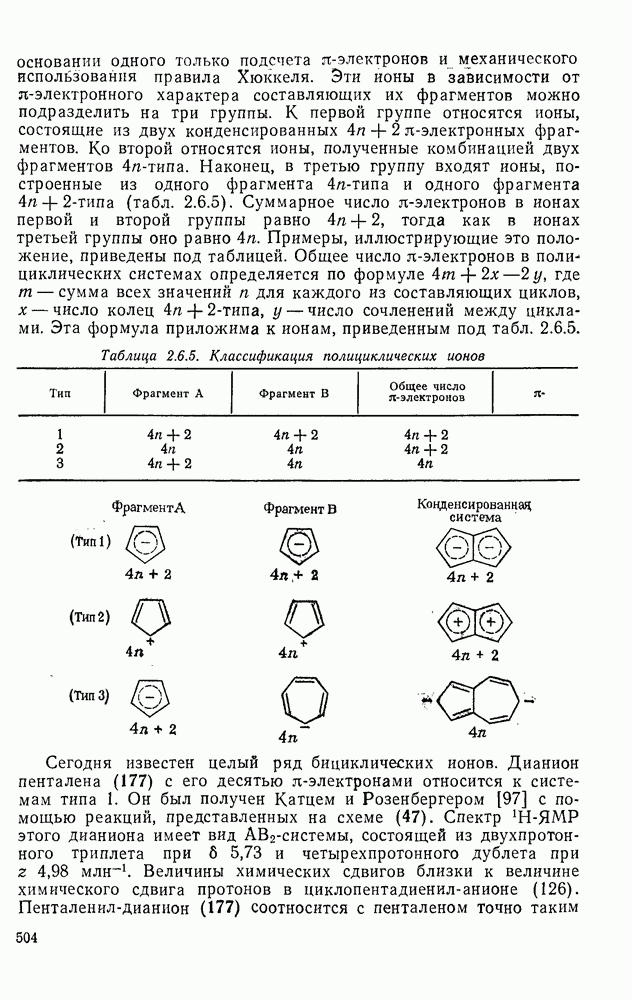













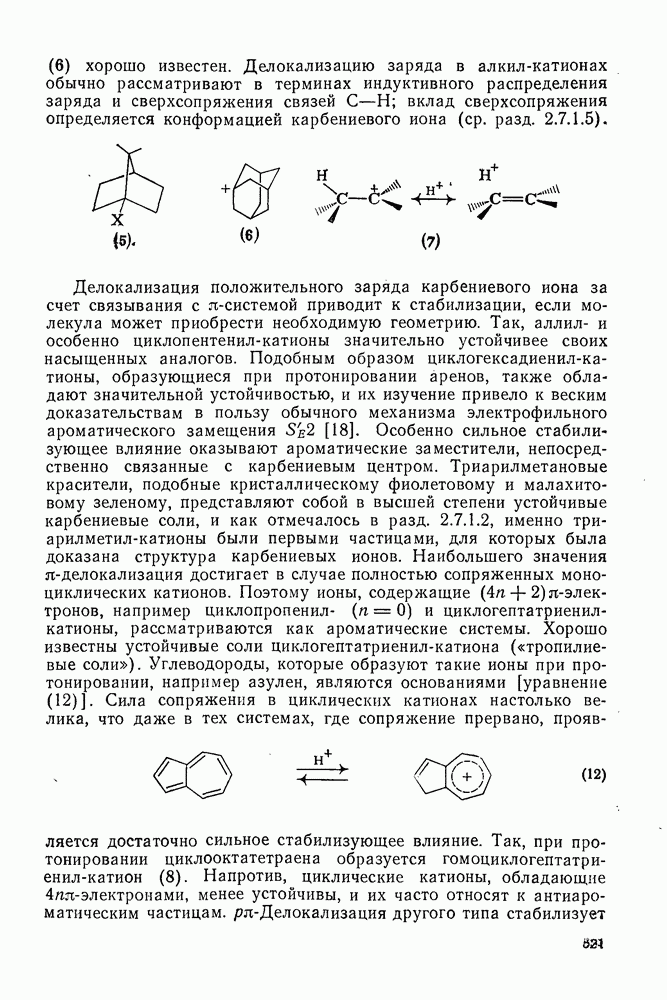





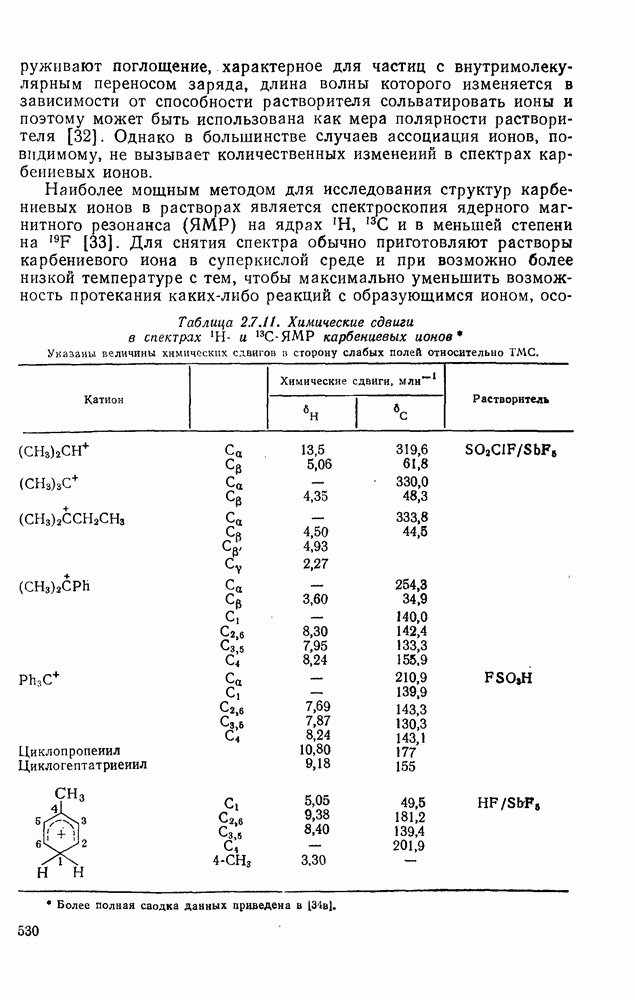
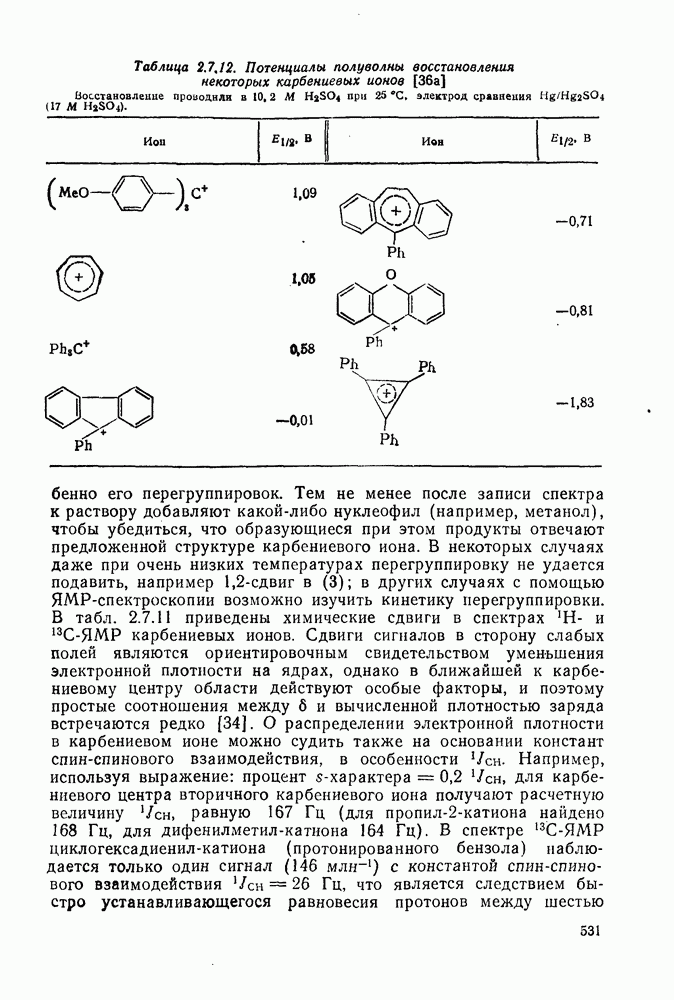

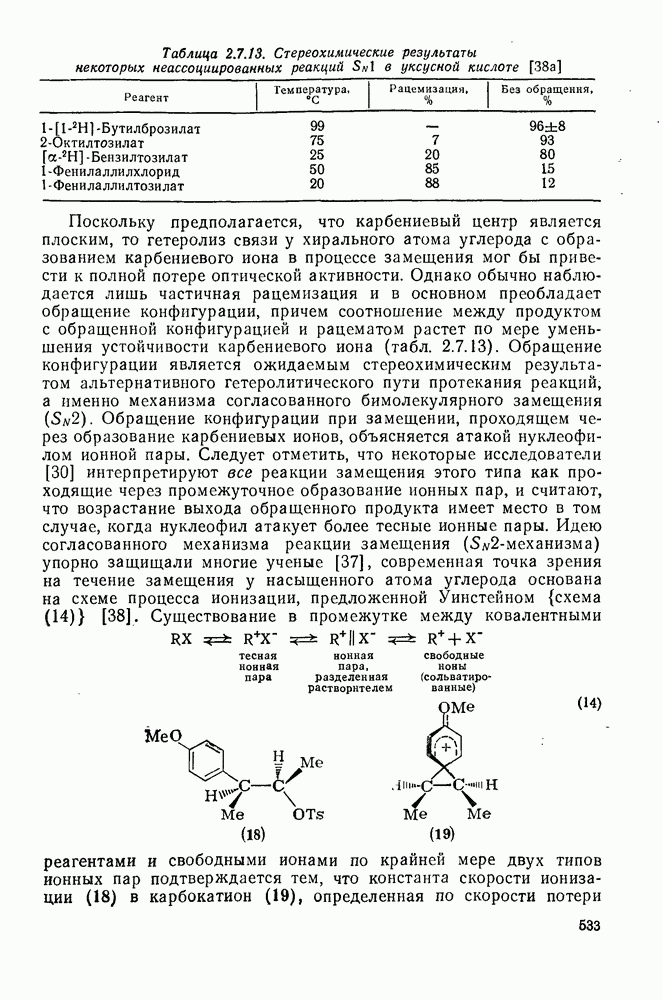
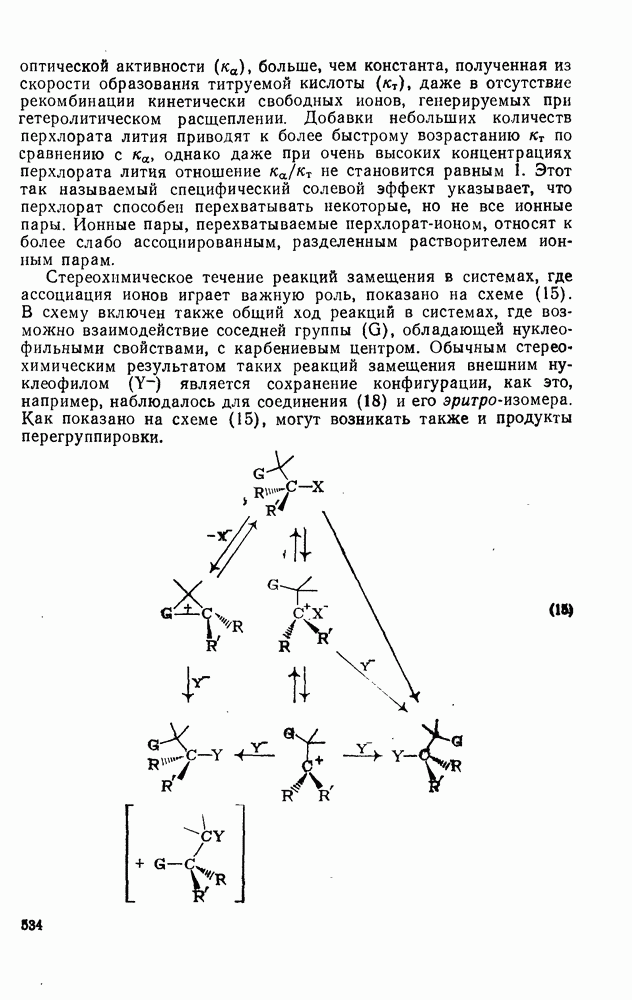











































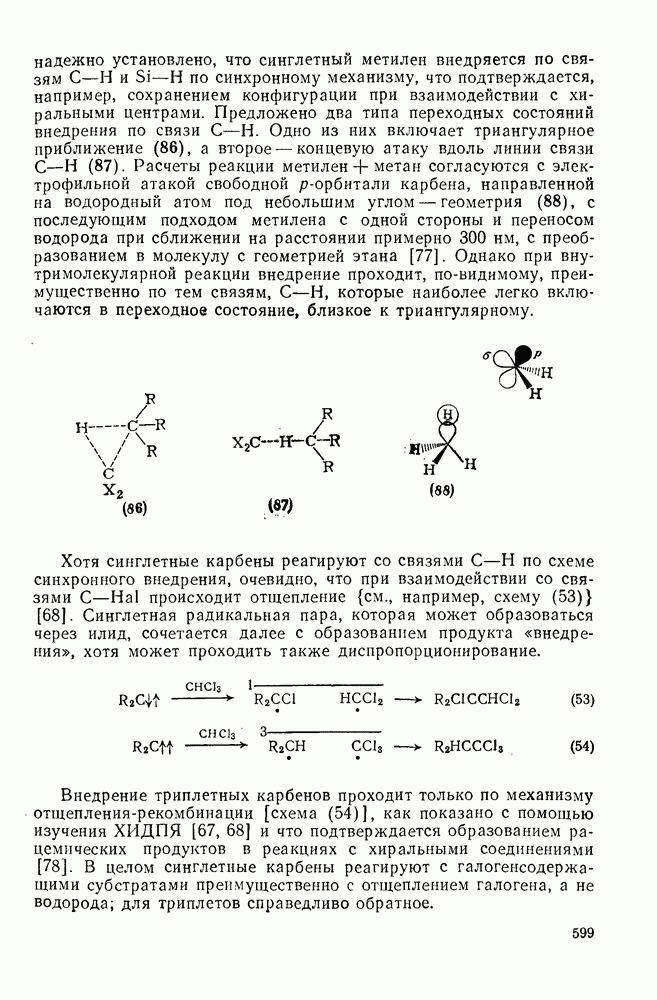




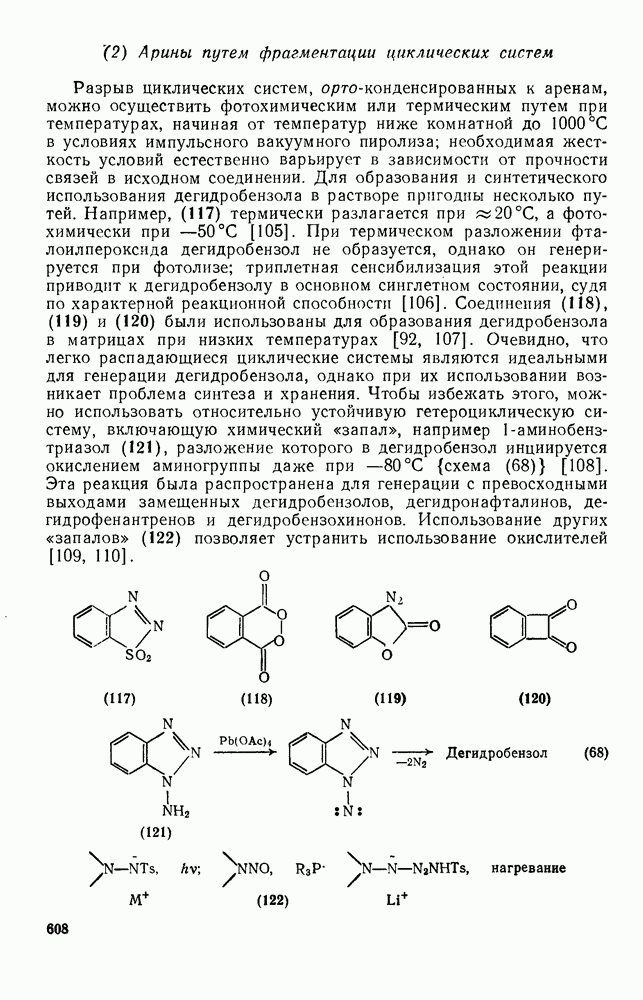














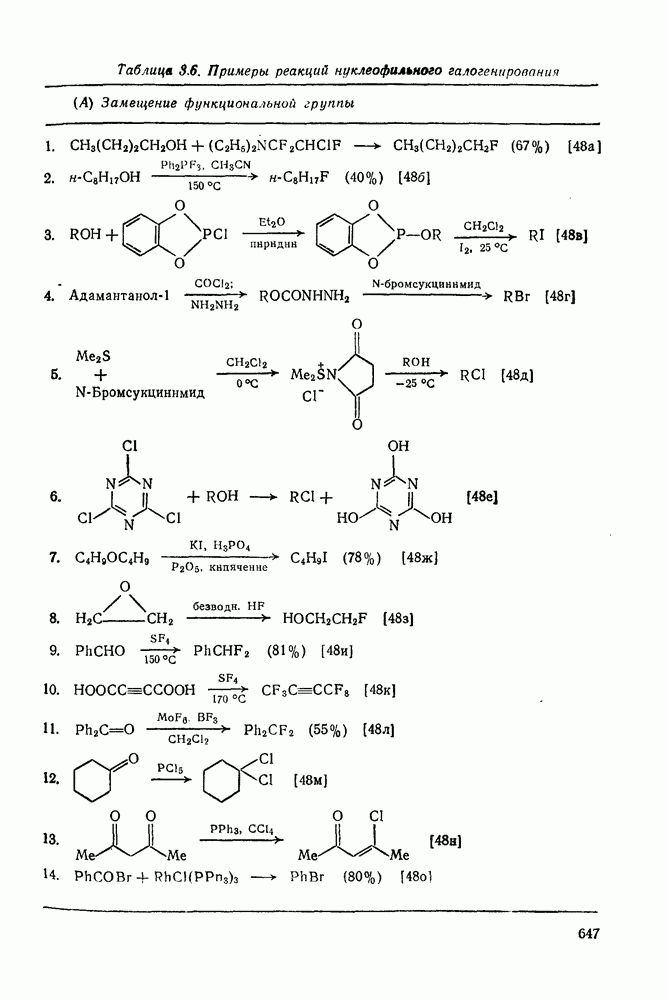


































 к отрыву водорода фенильными радикалами [23]
к отрыву водорода фенильными радикалами [23]

 в переходном состоянии, например (16), поэтому сила связи начинает играть большую роль и энергия переходного состояния снижается за счет любых факторов, стабилизующих первоначальный радикал. Это хорошо иллюстрируется относительной реакционной способностью в реакциях отрыва водорода, выделенного в структурах (17) и (18), радикалами
в переходном состоянии, например (16), поэтому сила связи начинает играть большую роль и энергия переходного состояния снижается за счет любых факторов, стабилизующих первоначальный радикал. Это хорошо иллюстрируется относительной реакционной способностью в реакциях отрыва водорода, выделенного в структурах (17) и (18), радикалами  и
и  . Оба соединения обладают сходной реакционной способностью по отношению к
. Оба соединения обладают сходной реакционной способностью по отношению к  , однако в случае отрыва водорода радикалом
, однако в случае отрыва водорода радикалом  , когда переходное состояние по строению больше напоминает первоначальный радикал, соединение (17) в 800 раз реакционноспособнее (18) в силу большей способности фенильной группы к стабилизации радикала. При отрыве водорода фенильным радикалом реакционная способность соединения (17) примерно в 5 раз выше. Влияние некоторых других
, когда переходное состояние по строению больше напоминает первоначальный радикал, соединение (17) в 800 раз реакционноспособнее (18) в силу большей способности фенильной группы к стабилизации радикала. При отрыве водорода фенильным радикалом реакционная способность соединения (17) примерно в 5 раз выше. Влияние некоторых других  -заместителей на скорость отрыва водорода фенильными радикалами видно из табл. 2.8.2 [23]. Следует отметить большое увеличение скорости отрыва винильной группой, что родственно легкости аллильного замещения в подобных соединениях [24]. Относительная реакционная способность к отрыву водорода метильными радикалами от связей
-заместителей на скорость отрыва водорода фенильными радикалами видно из табл. 2.8.2 [23]. Следует отметить большое увеличение скорости отрыва винильной группой, что родственно легкости аллильного замещения в подобных соединениях [24]. Относительная реакционная способность к отрыву водорода метильными радикалами от связей  и
и  представлена рядом (19) [1а].
представлена рядом (19) [1а].

 ], является лимитирующей стадией; для фенильного радикала эта стадия экзотермична
], является лимитирующей стадией; для фенильного радикала эта стадия экзотермична  и при обычных условиях, по-видимому, необратима. Образующийся резонансно стабилизованный циклогексадиенильный радикал (32) не реагирует с субстратом и не отщепляет спонтанно атом водорода с образованием продукта замещения (33), а подвергается быстрым радикал-радикальным реакциям [уравнения
и при обычных условиях, по-видимому, необратима. Образующийся резонансно стабилизованный циклогексадиенильный радикал (32) не реагирует с субстратом и не отщепляет спонтанно атом водорода с образованием продукта замещения (33), а подвергается быстрым радикал-радикальным реакциям [уравнения  ]. Для реакции дибензоилпероксида с бензолом при
]. Для реакции дибензоилпероксида с бензолом при  были определены константы скорости:
были определены константы скорости:  . В этой реакции дибен-зоилпероксид разлагается также за счет взаимодействия с фенилциклогексадиенильными радикалами.
. В этой реакции дибен-зоилпероксид разлагается также за счет взаимодействия с фенилциклогексадиенильными радикалами.
