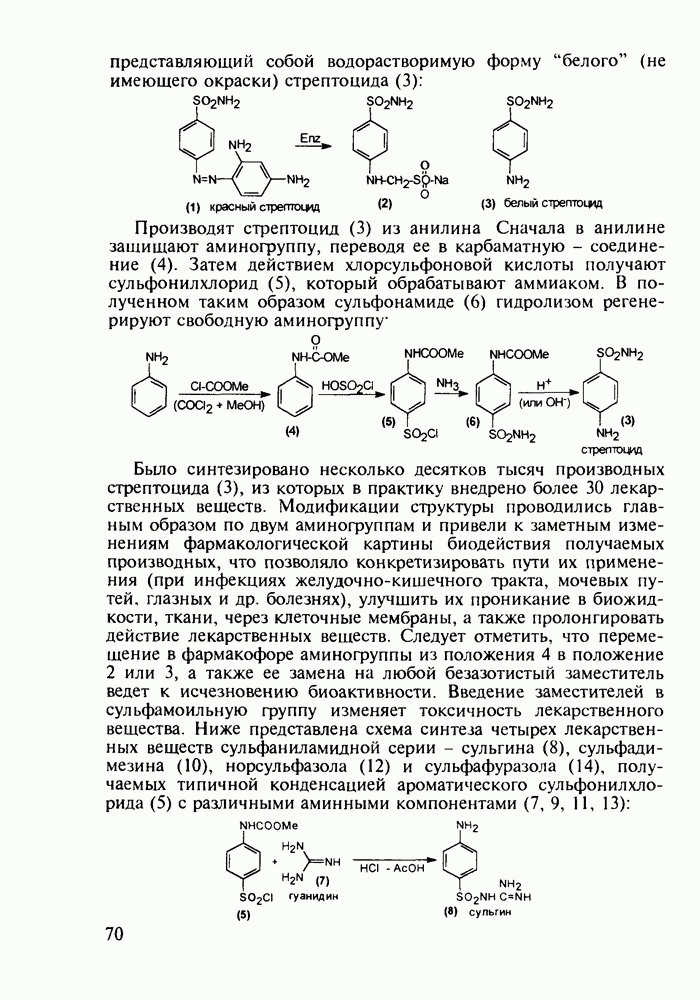
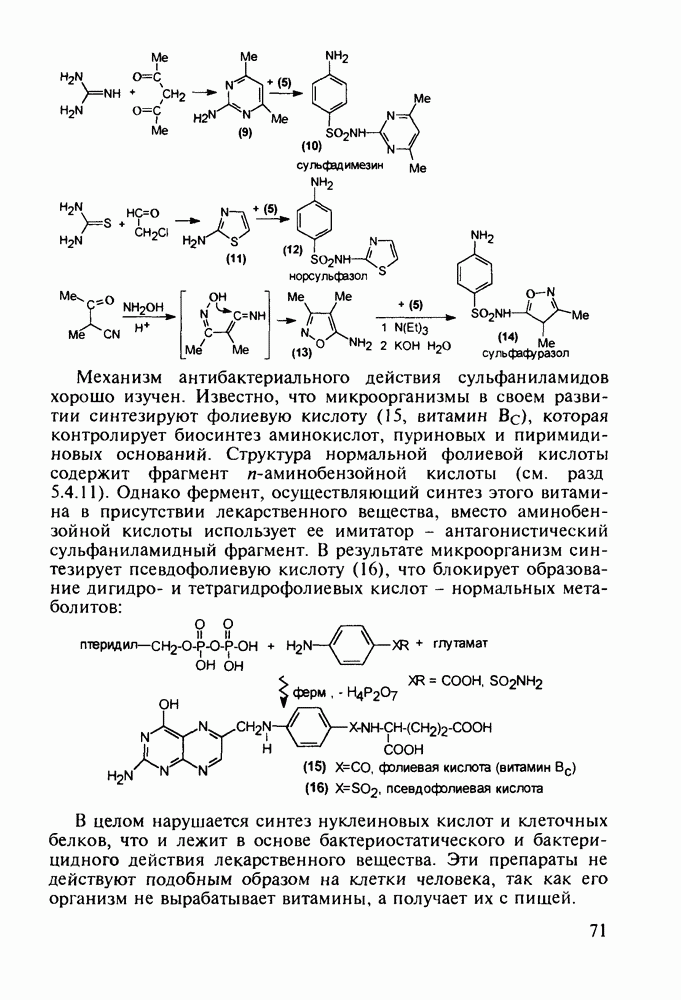
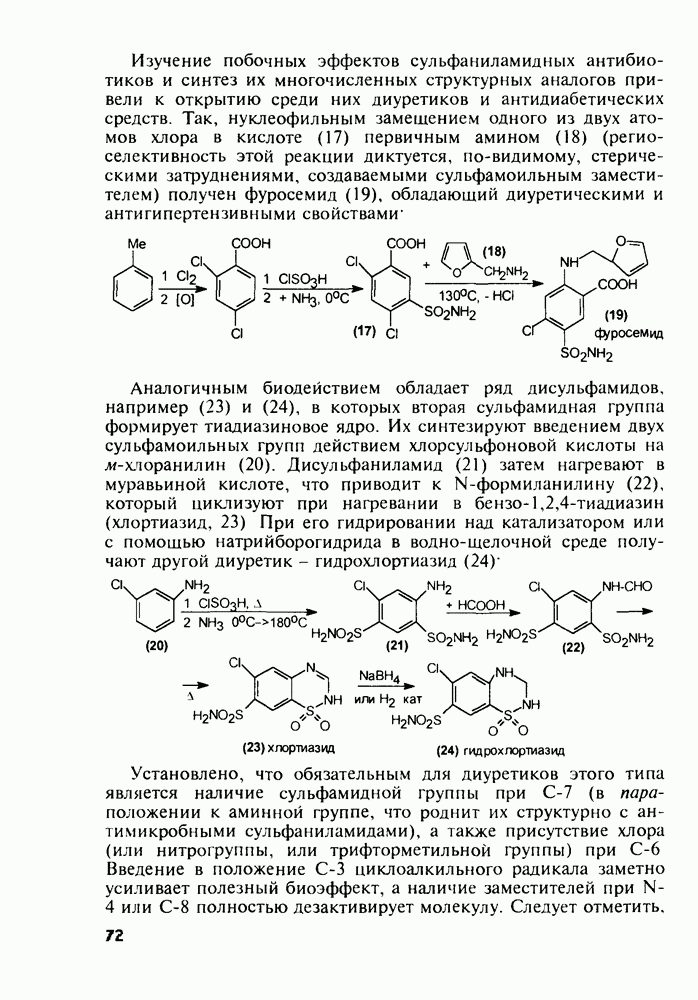
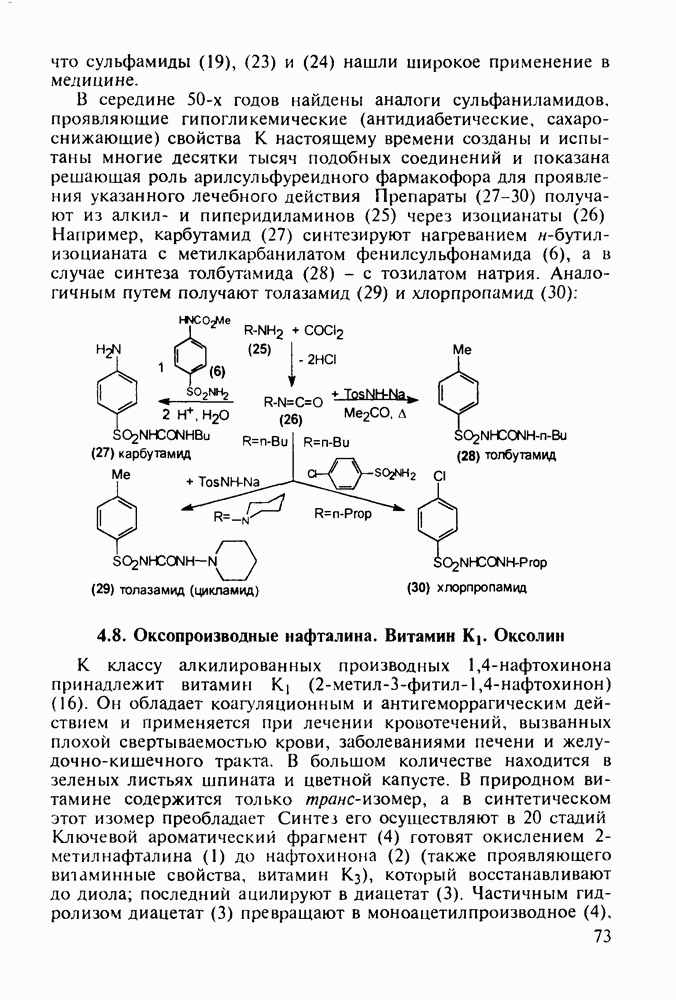
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
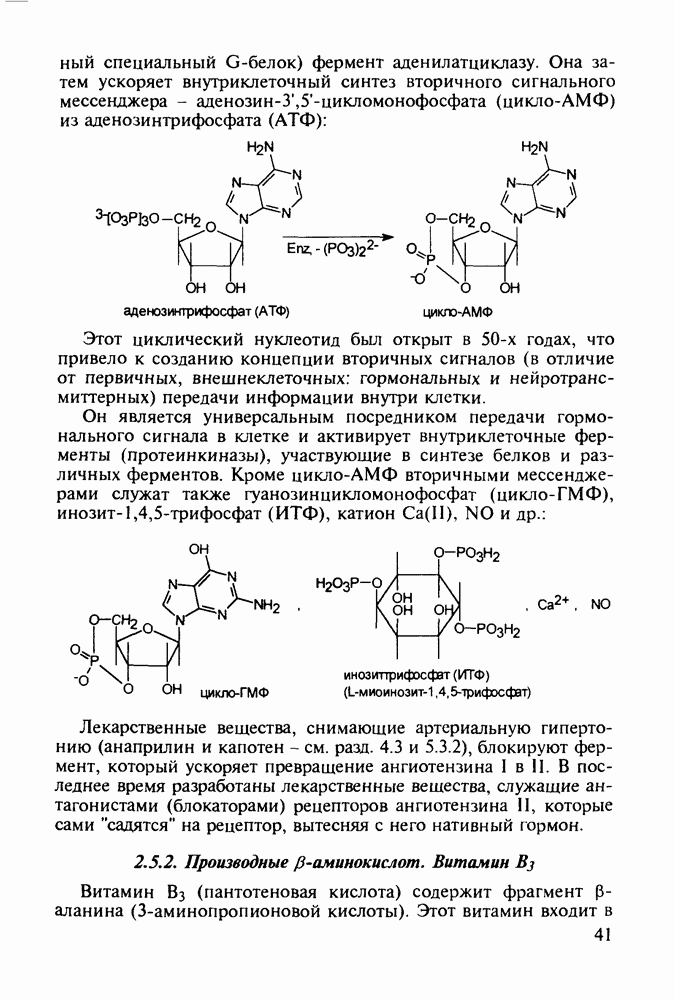
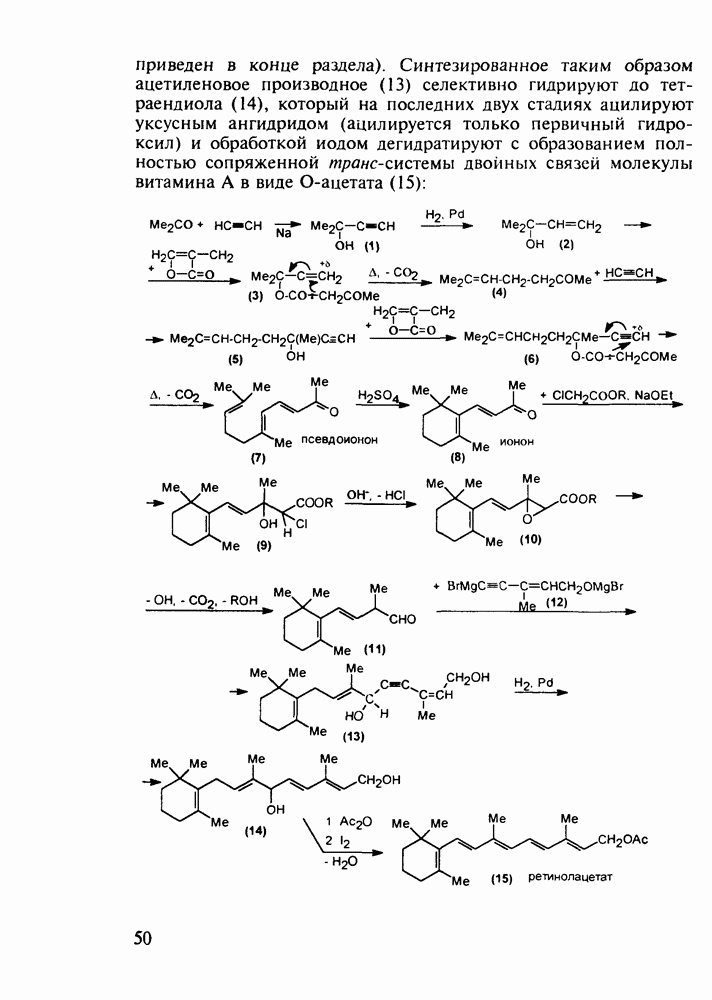
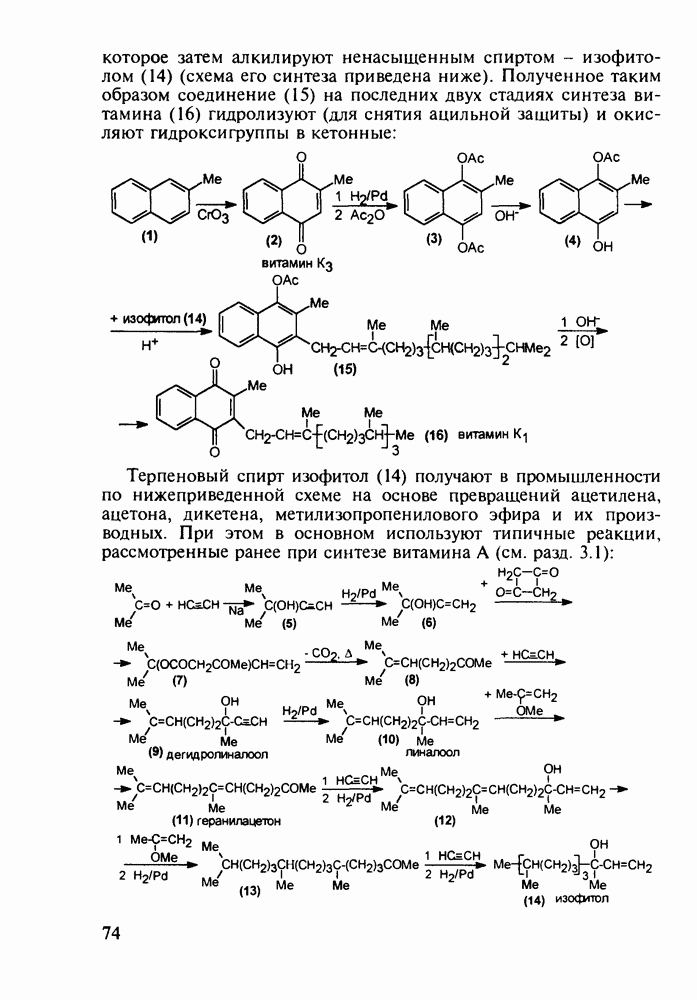
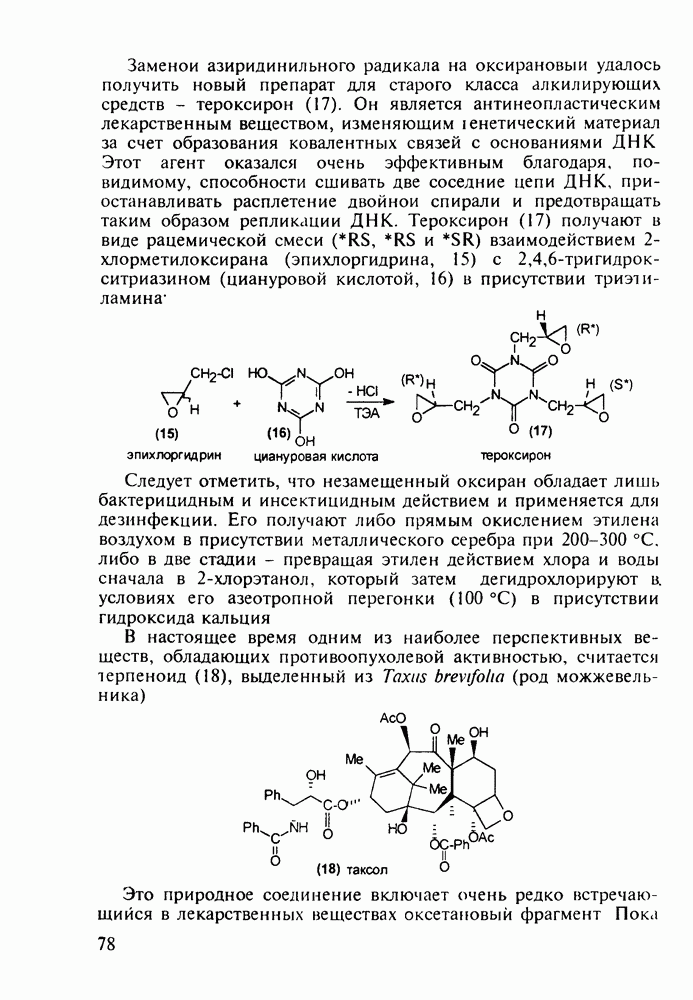
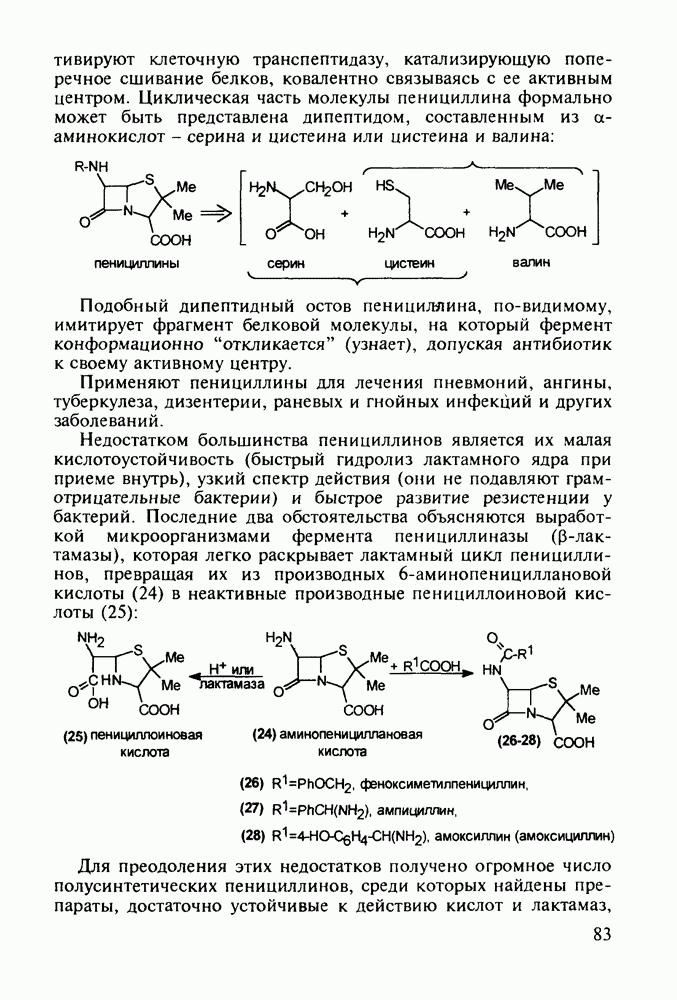
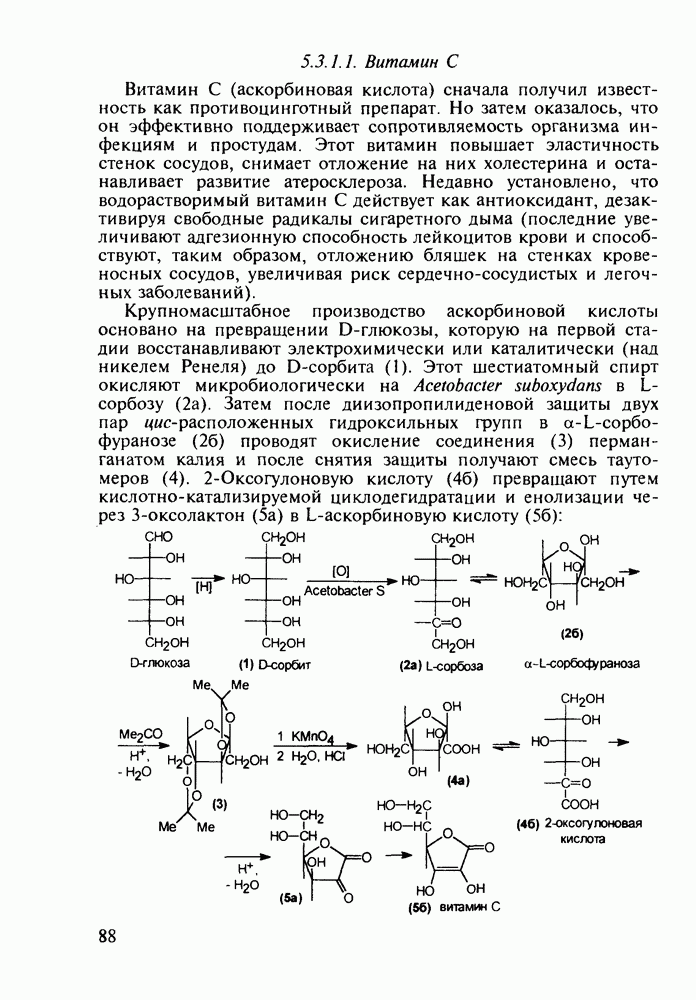
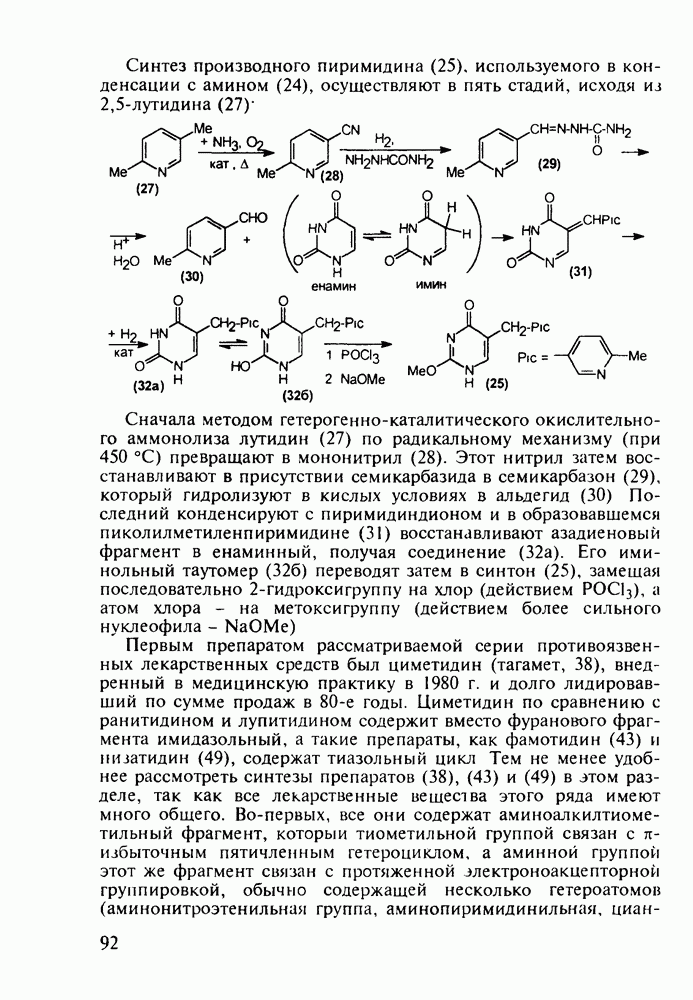
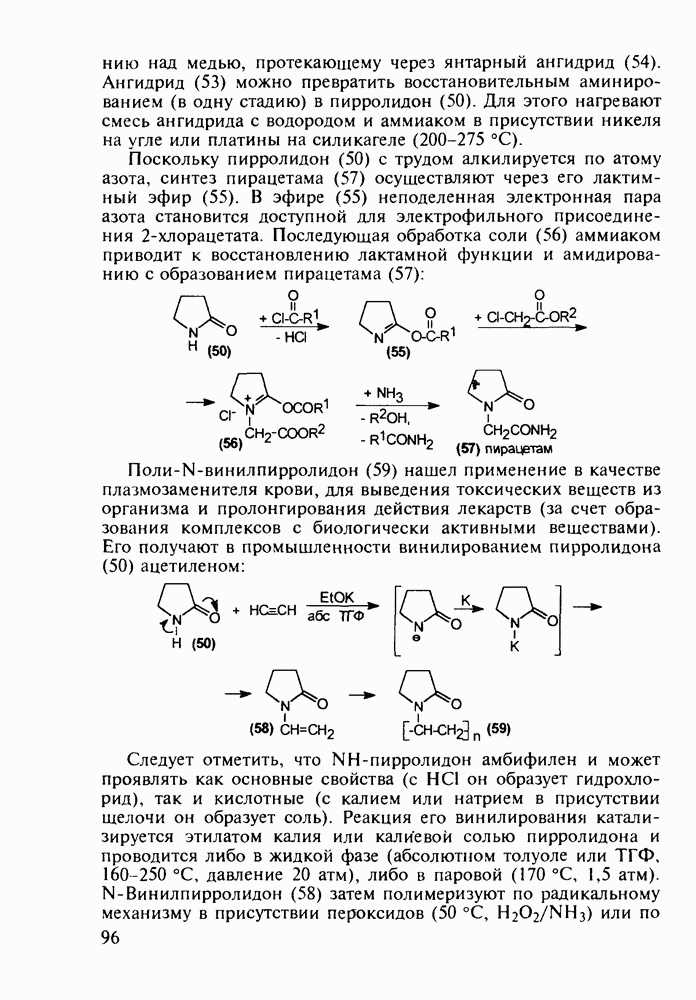
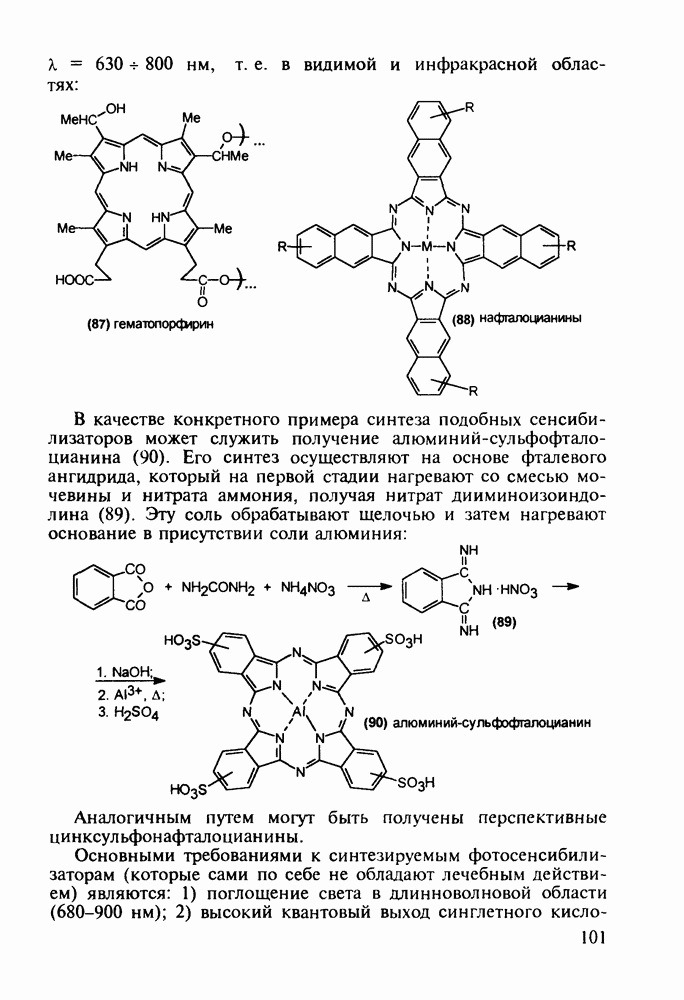
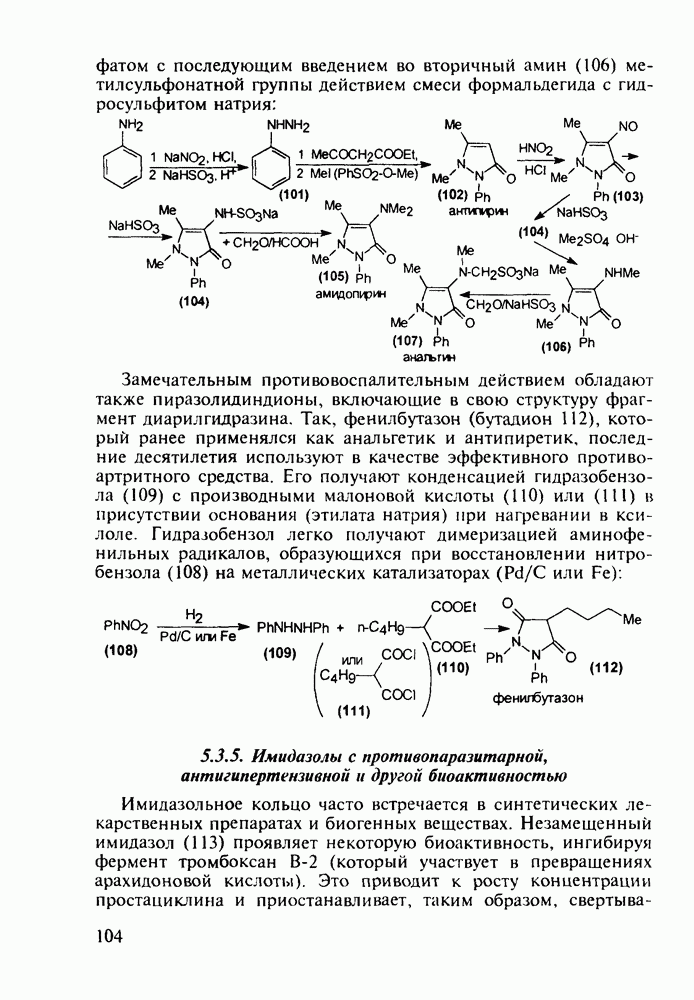
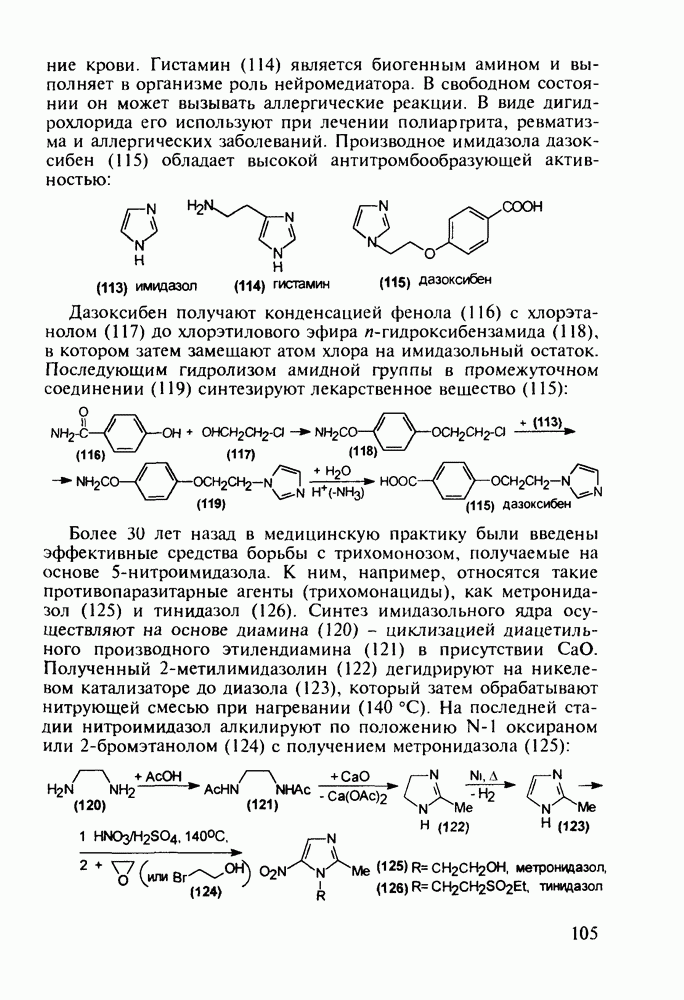
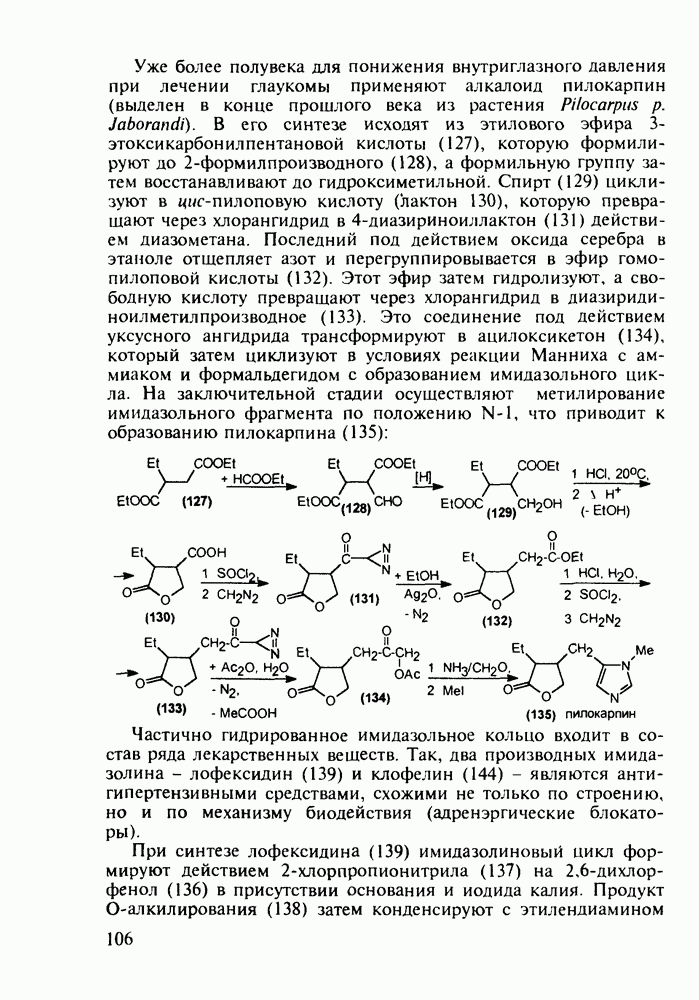
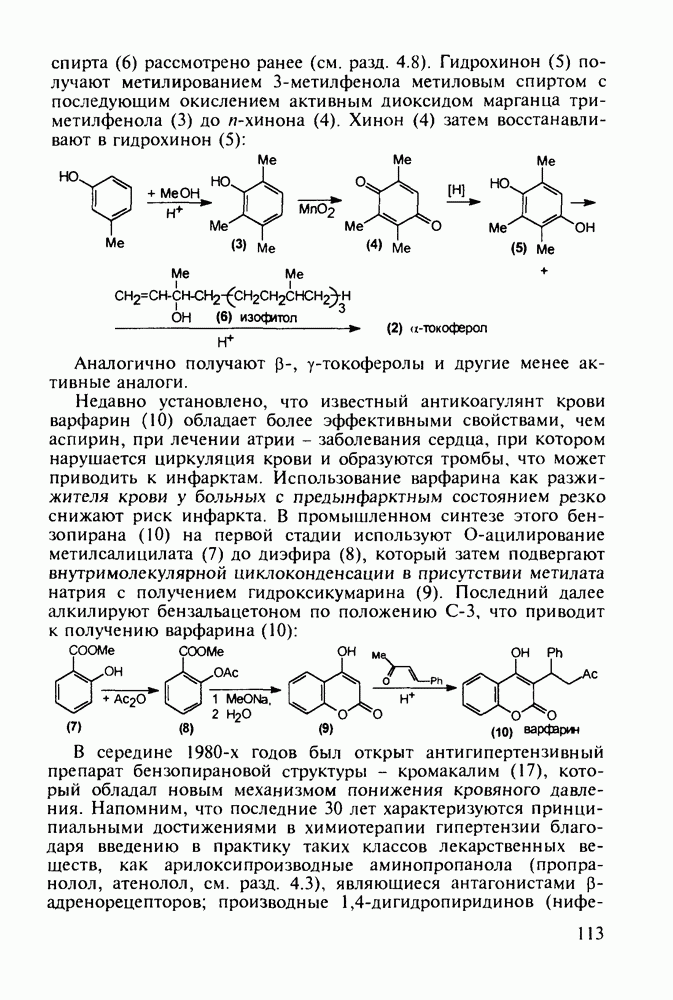
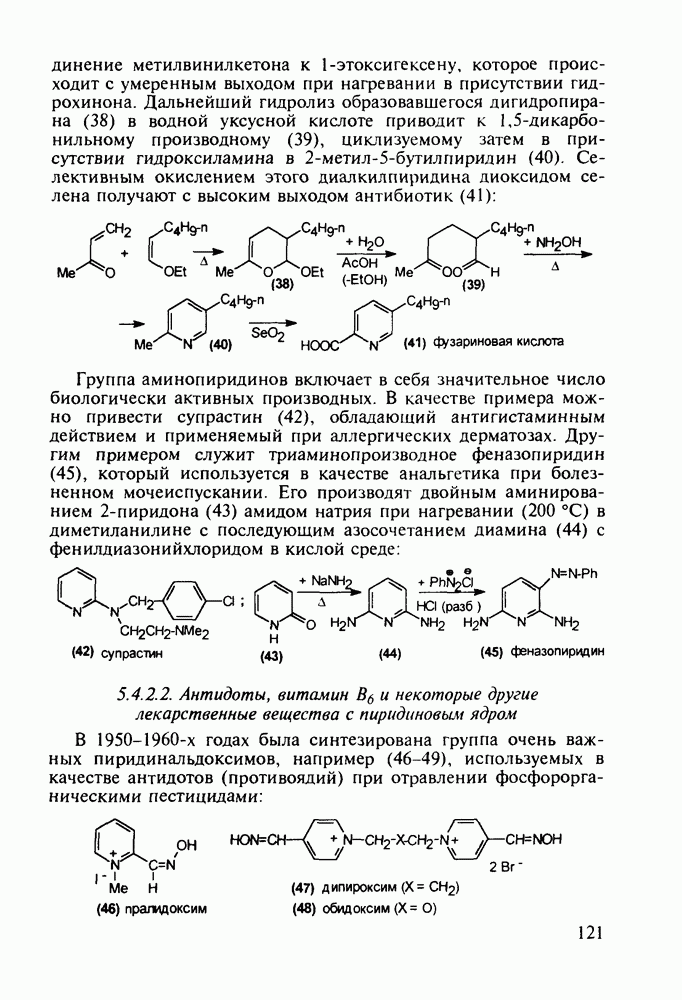
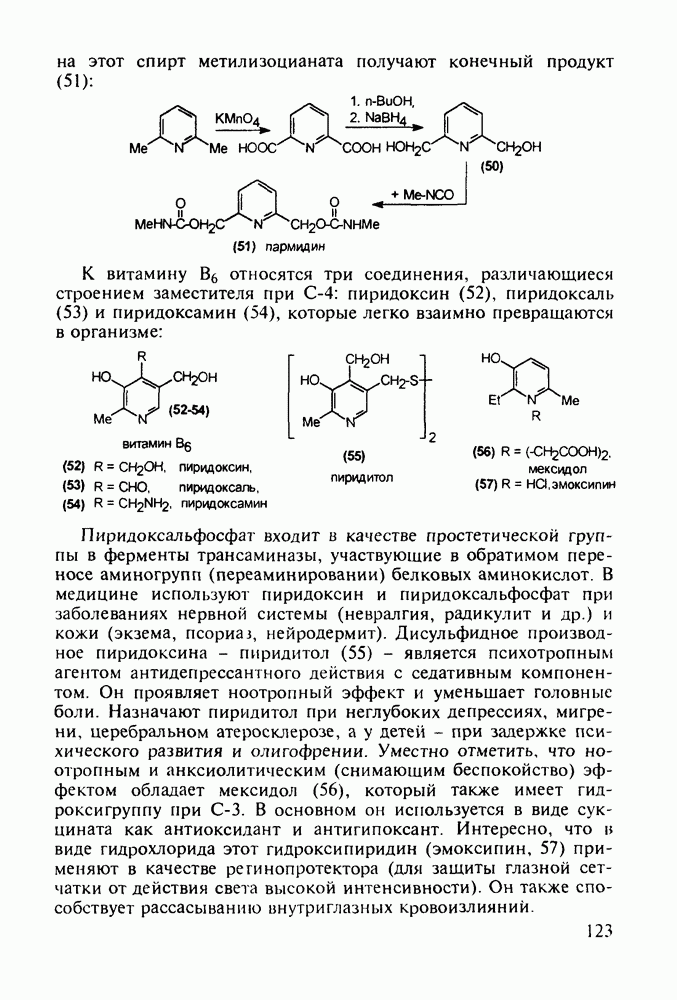
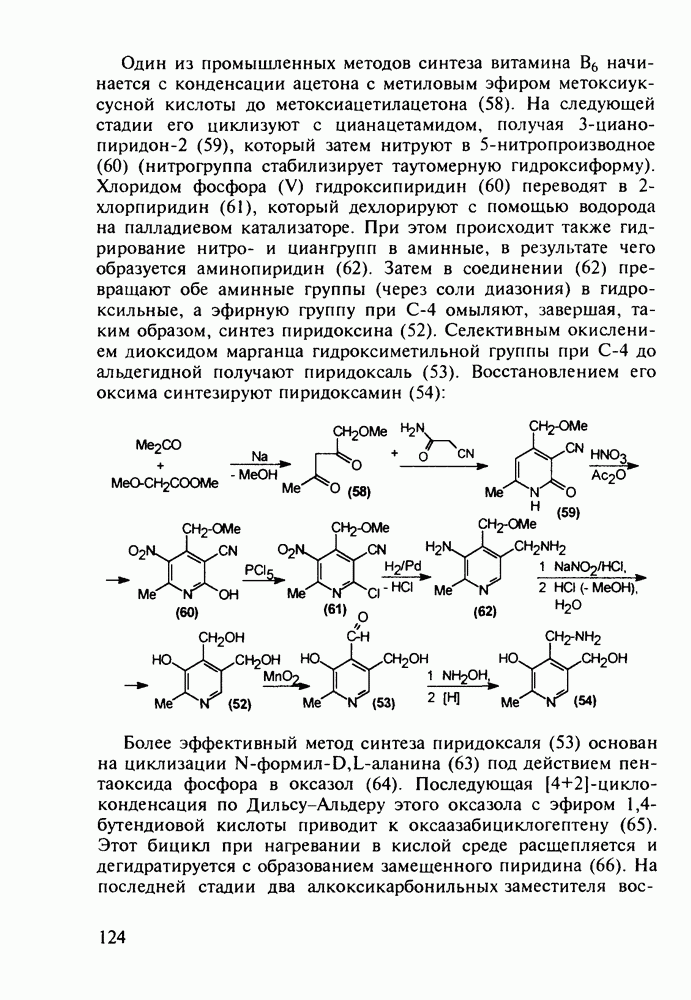
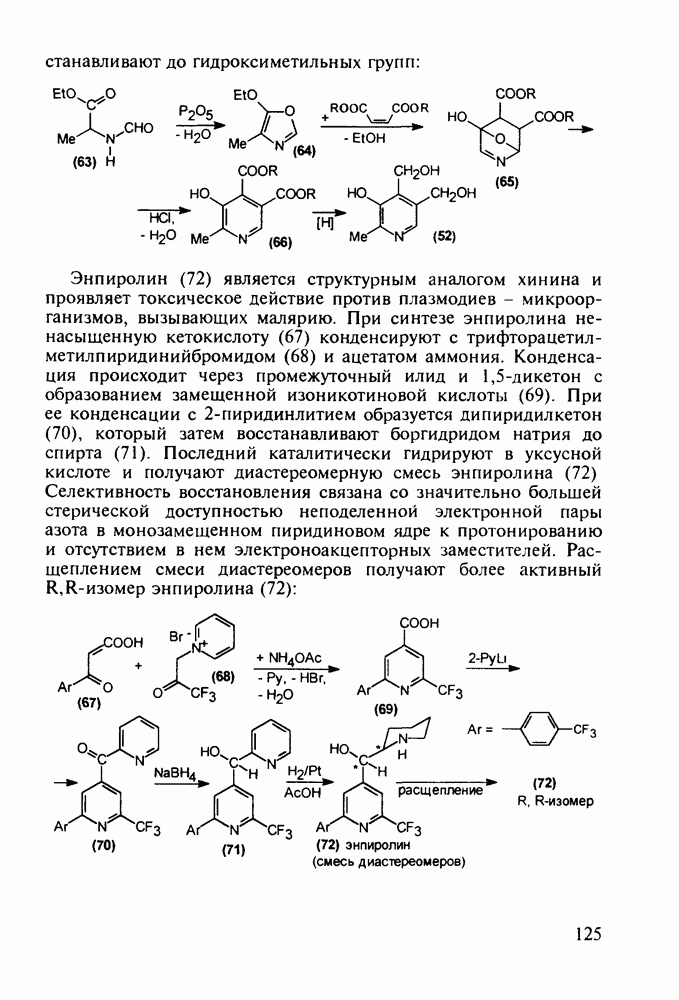
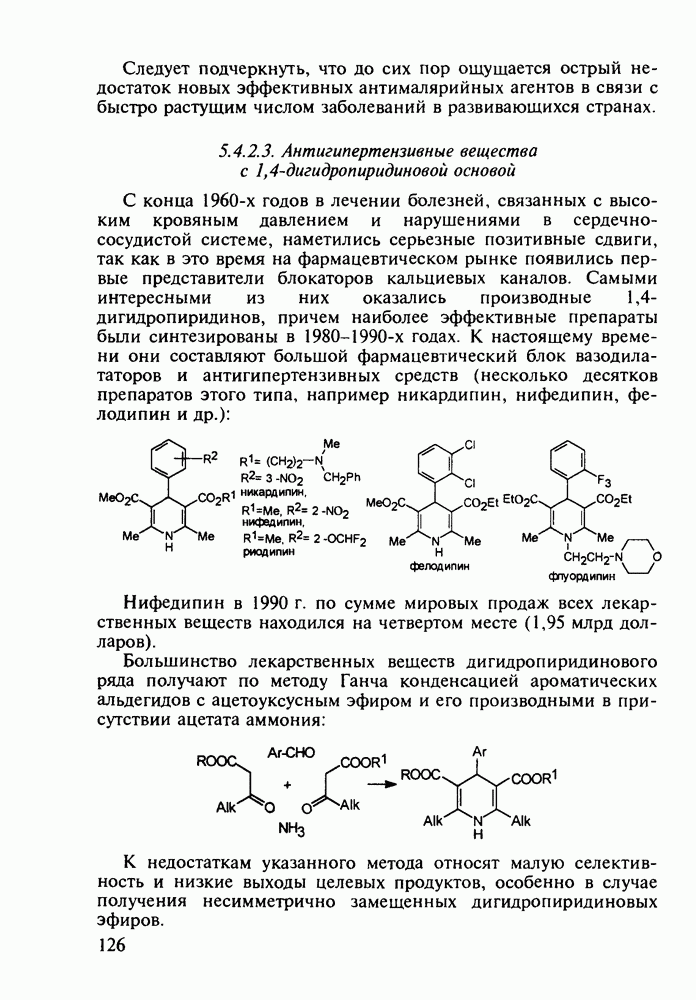
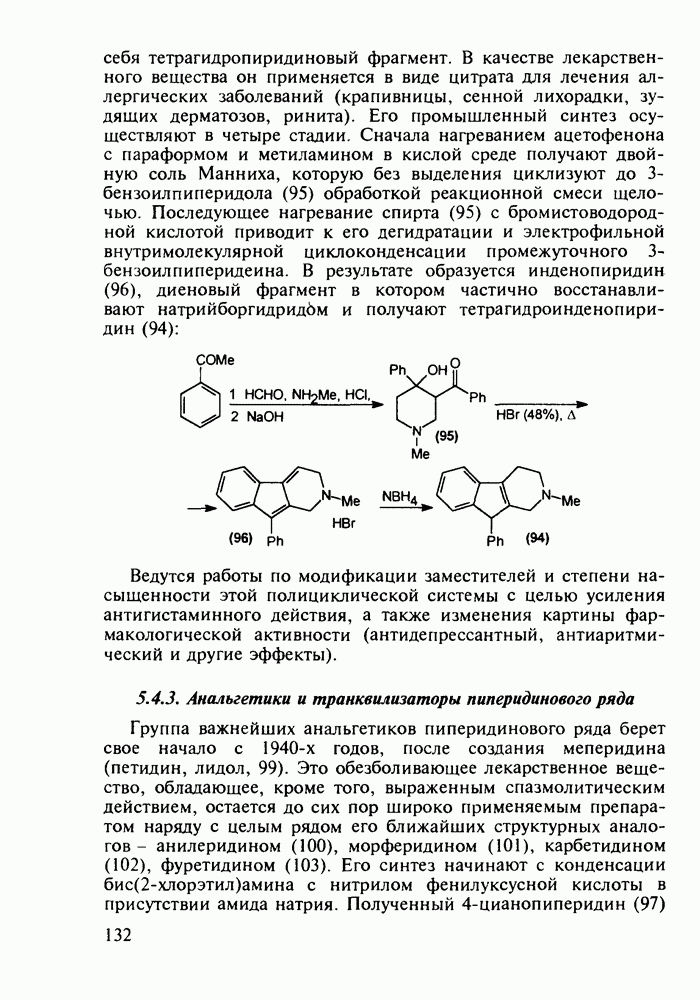
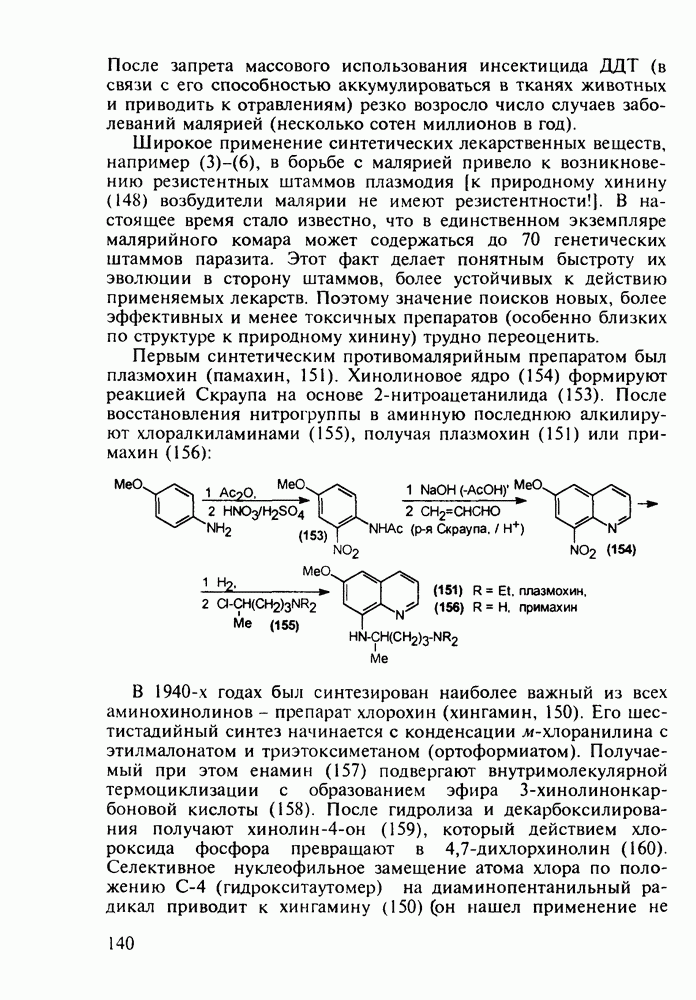
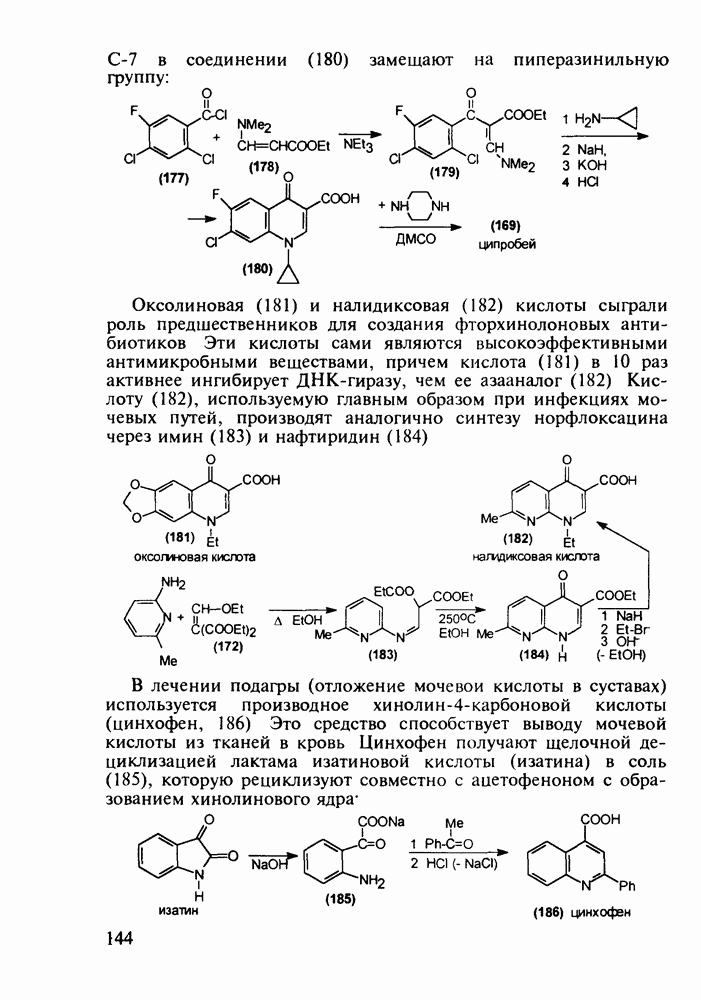
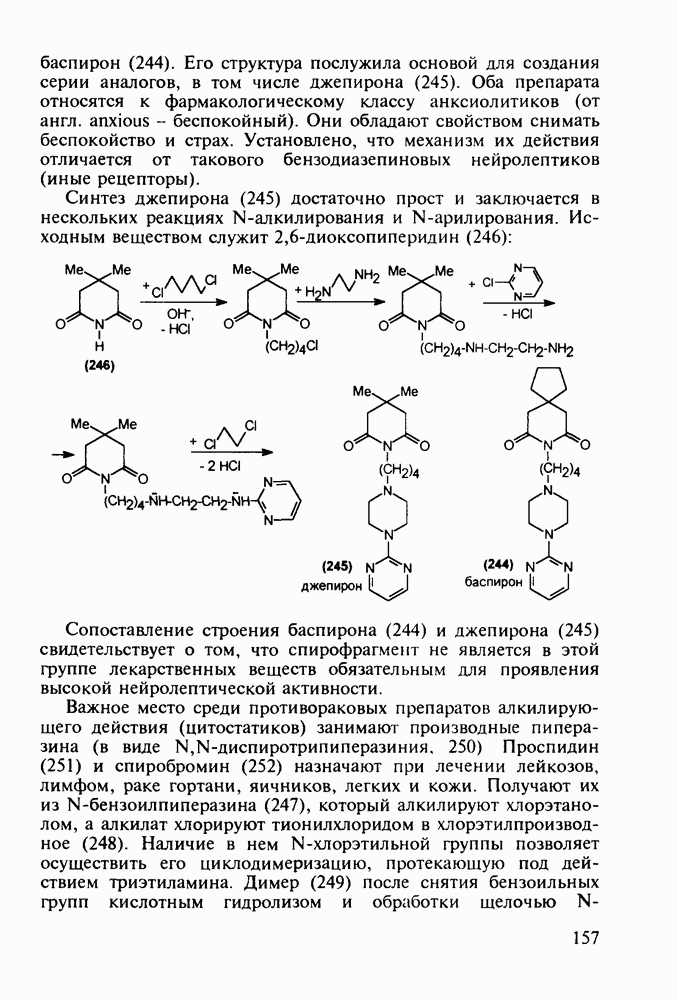
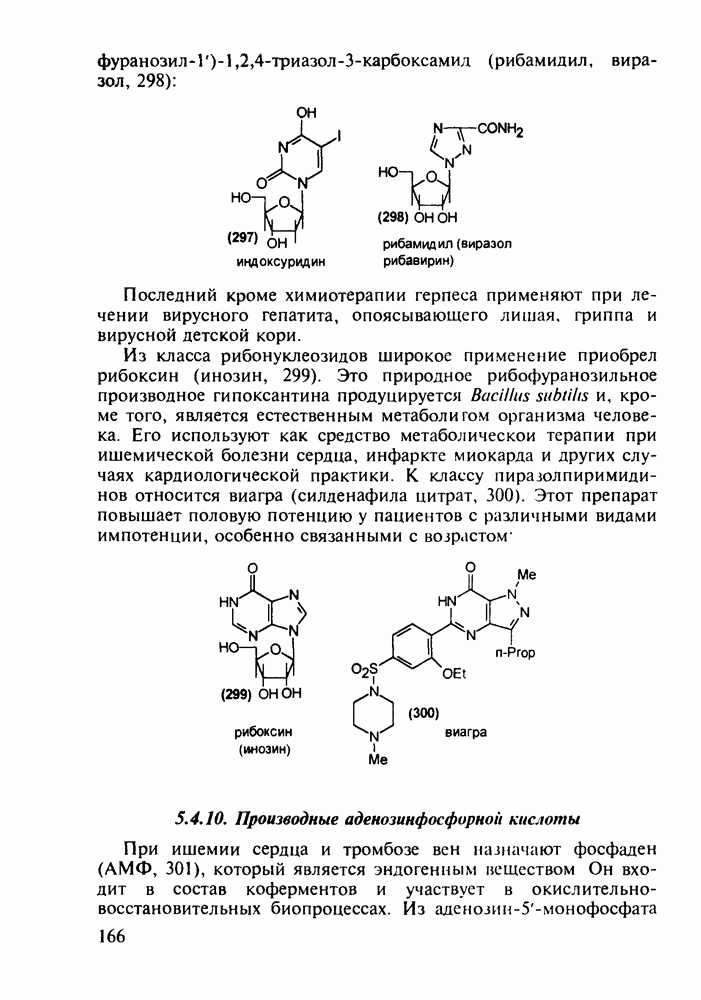
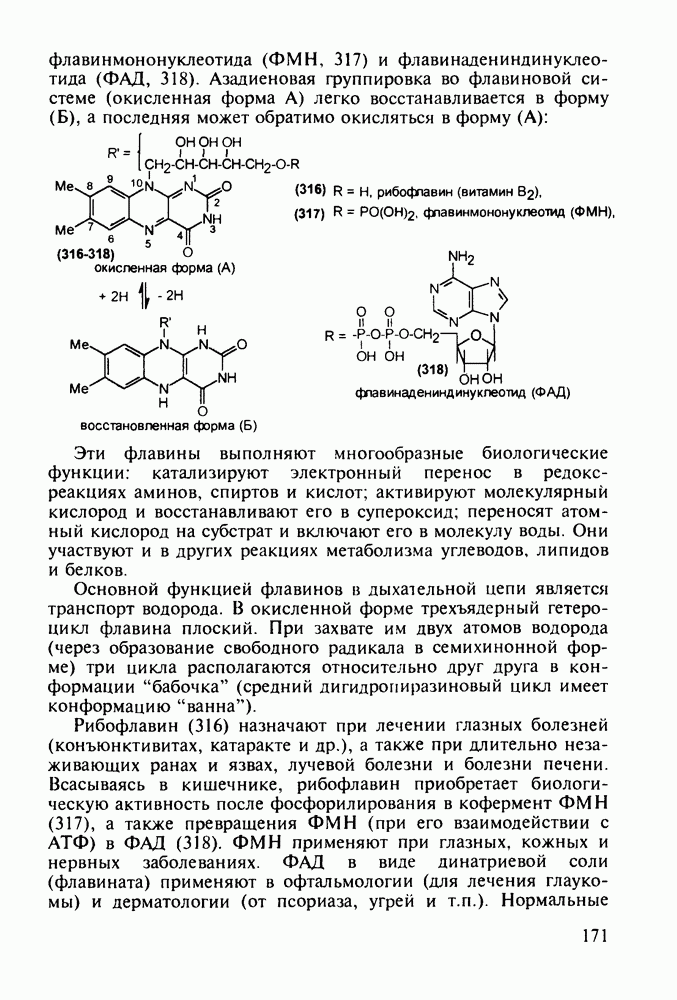
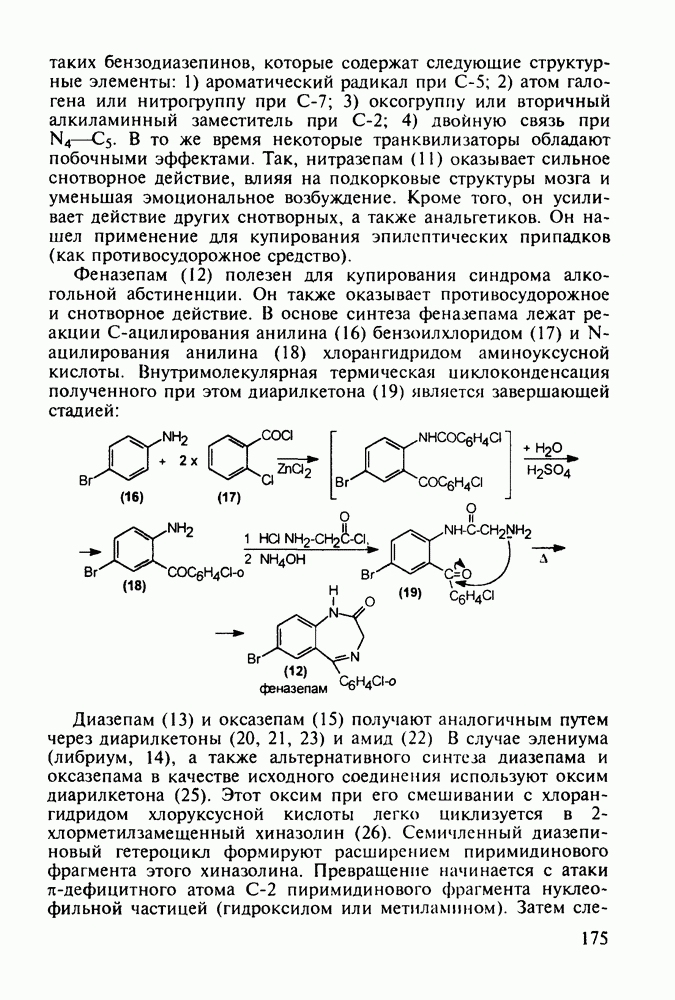
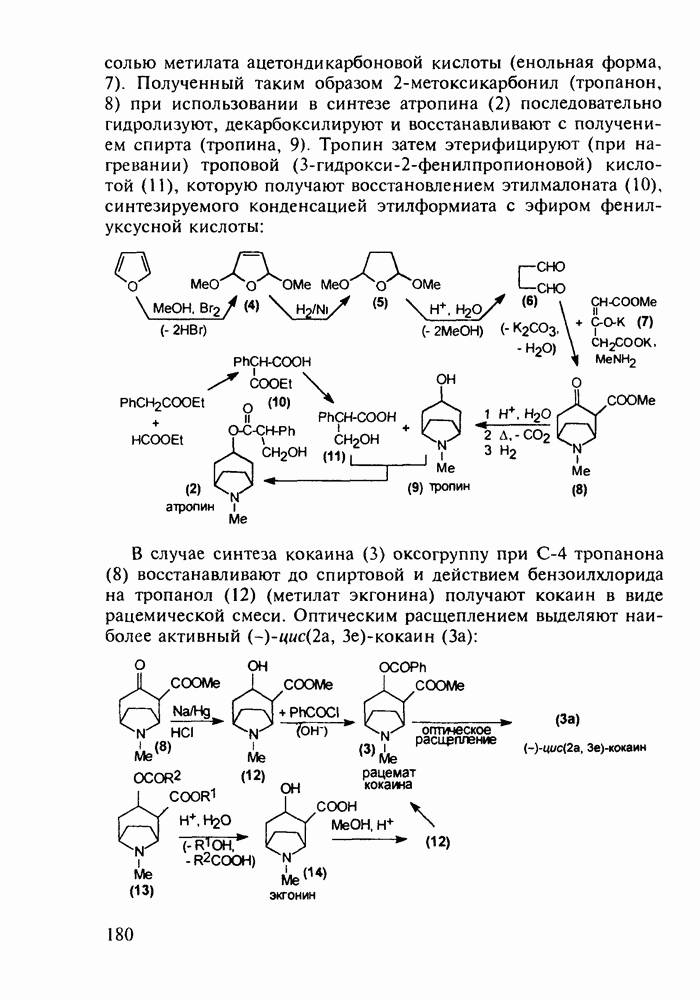
5.4.6. Производные пиримидинов5.4.6.1. Снотворные на основе триоксопиримидиновБарбитуровая кислота (202) лежит в основе большого класса широко используемых снотворных веществ, например барбитуратов (202-209):
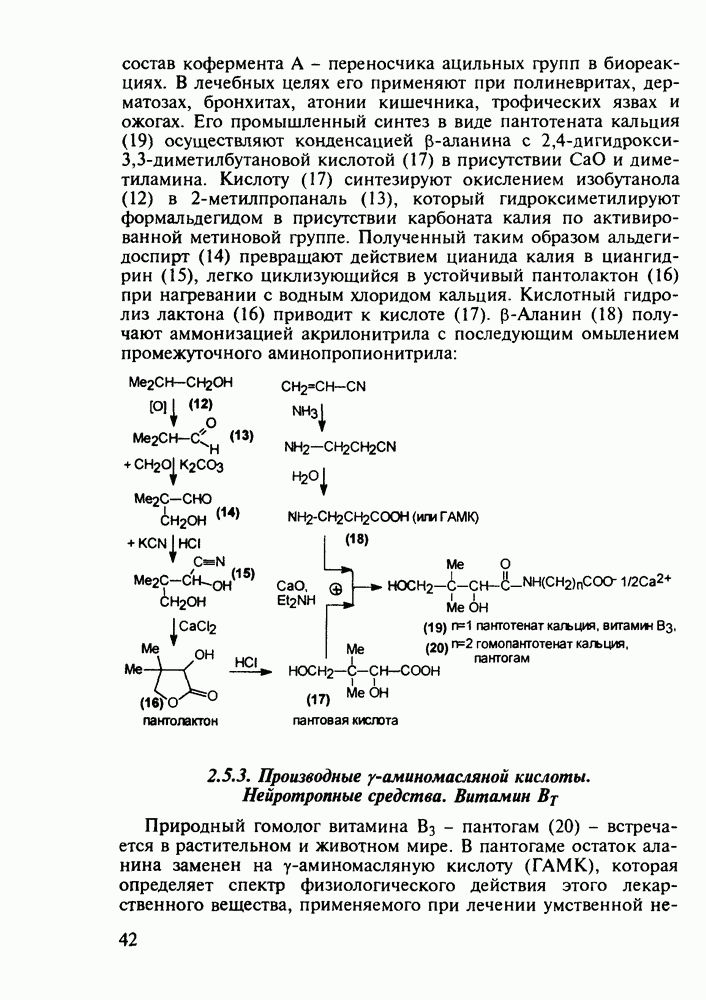
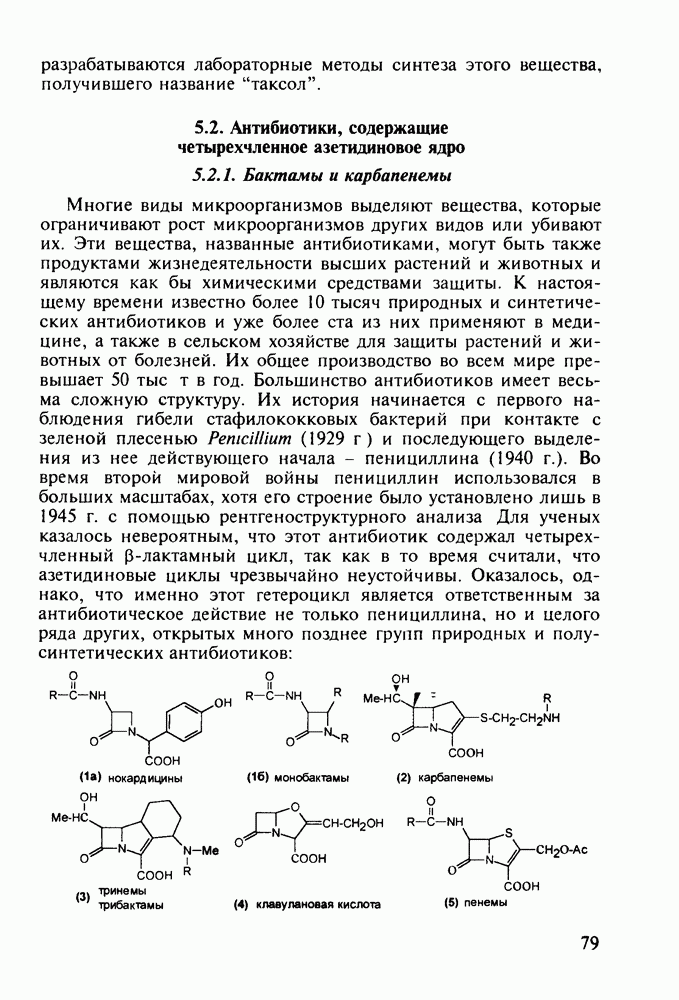
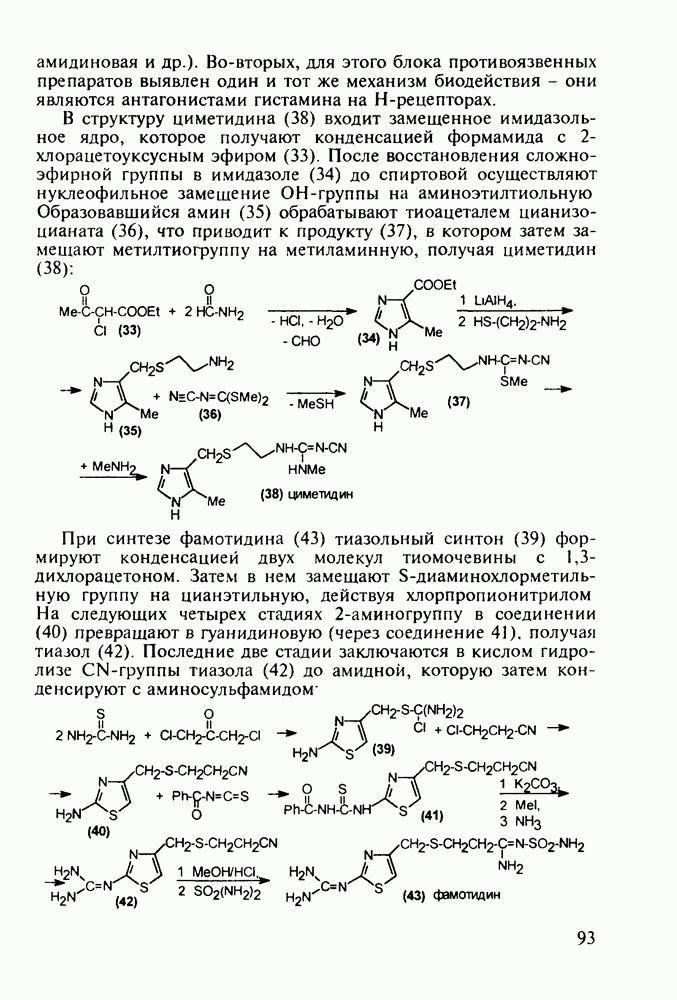
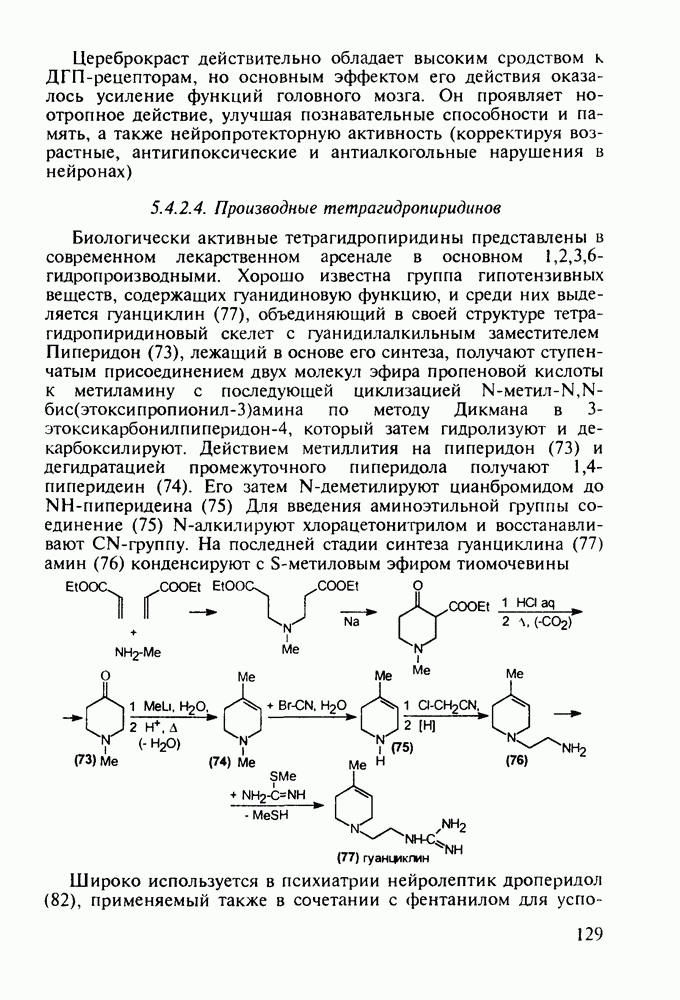
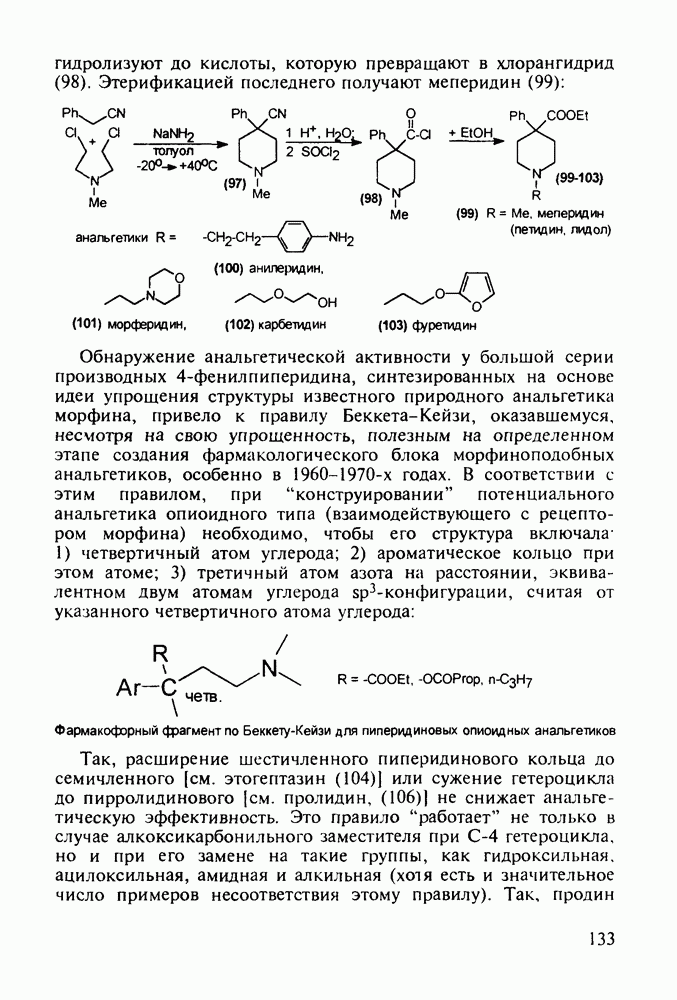
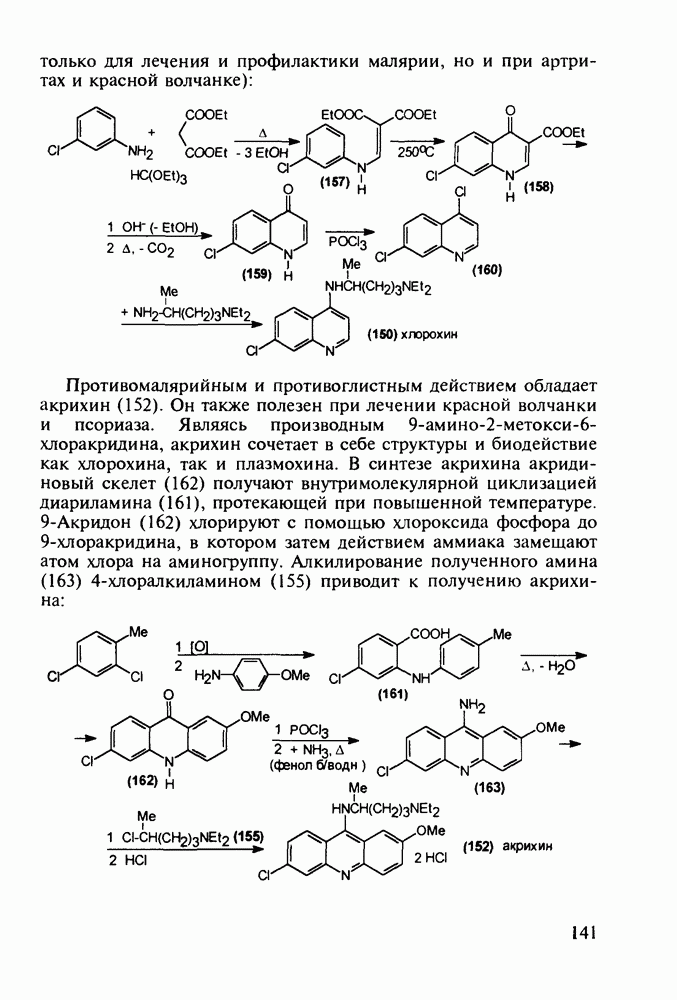
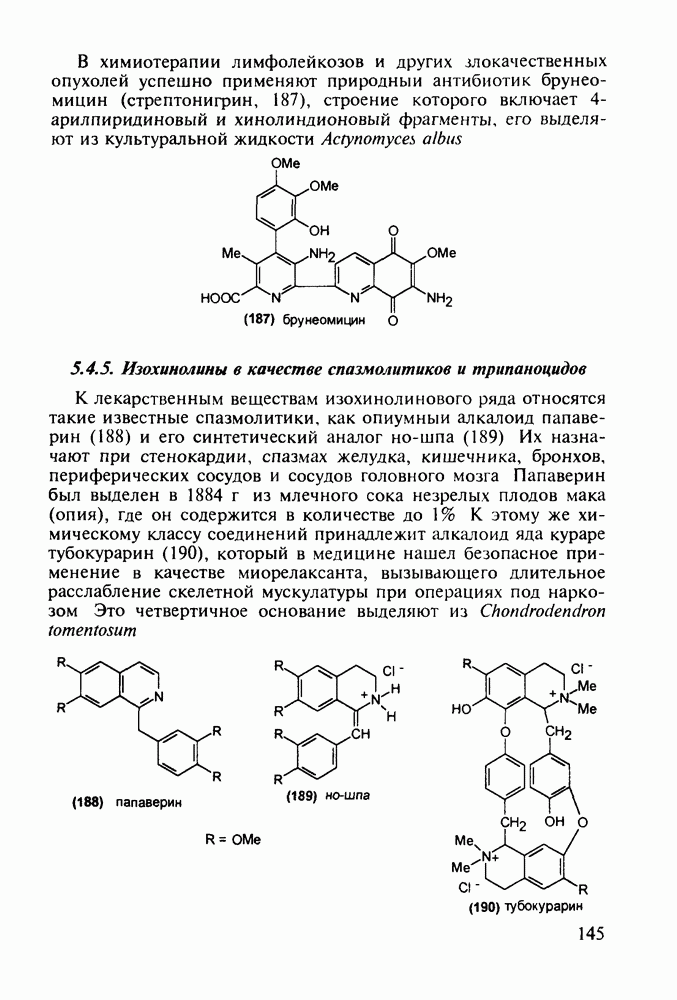
Механизм их действия пока не раскрыт в деталях. Предположительно они активируют функцию у-аминомасляной кислоты - природного нейромедиатора торможения передачи нервных импульсов в ЦНС (см. разд. 2.5.3). Некоторые из барбитуратов оказывают антиконвульсивное действие (фенобарбитал, 206), другие - анестезирующее [гексенал (203) и тиопентал-натрий (204) используют в неингаляционном наркозе для общего обезболивания организма]. Ранее их применяли также как седативные средства, но к настоящему времени они вытеснены в этом плане бензодиазепинами - более эффективными успокаивающими средствами. Отмечены случаи физического привыкания к некоторым снотворным этой группы циклических уреидов. Первый представитель снотворных рассматриваемого типа - веронал (205) - начал использоваться с 1903 г. С тех пор получено множество 5,5-дизамещенных производных триоксопиримидинов, в которых протоны NH-группы обладают кислотностью (в иминольной форме) благодаря акцепторному действию трех С=0-фупп. Это обстоятельство позволяет применять большую часть барбитуратов в виде их натриевых солей. Особенно успешно подобные барбитураты стали применять совместно с ингаляционными анестетиками в хирургии (при инъекциях срок их действия - до трех часов, а прием в виде пилюль обеспечивает сон до шести и более часов) Установлено, что сила и длительность снотворного действия барбитуратов возрастает с удлинением алкильной цепи при Общая схема получения барбитуратов (211) основана на циклоконденсации замещенных эфиров малоновой кислоты с мочевиной или гуанидином в присутствии этоксида натрия В первом случае образуется промежуточная натриевая соль (210), а во втором - промежуточный имин (212), которые затем переводят в уреиды (211)
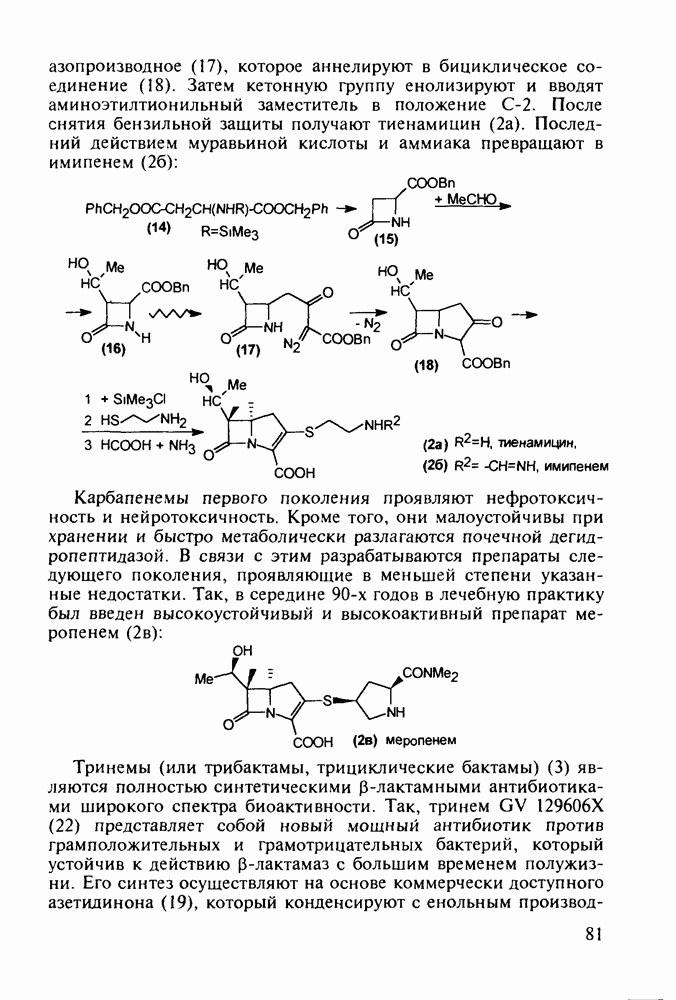
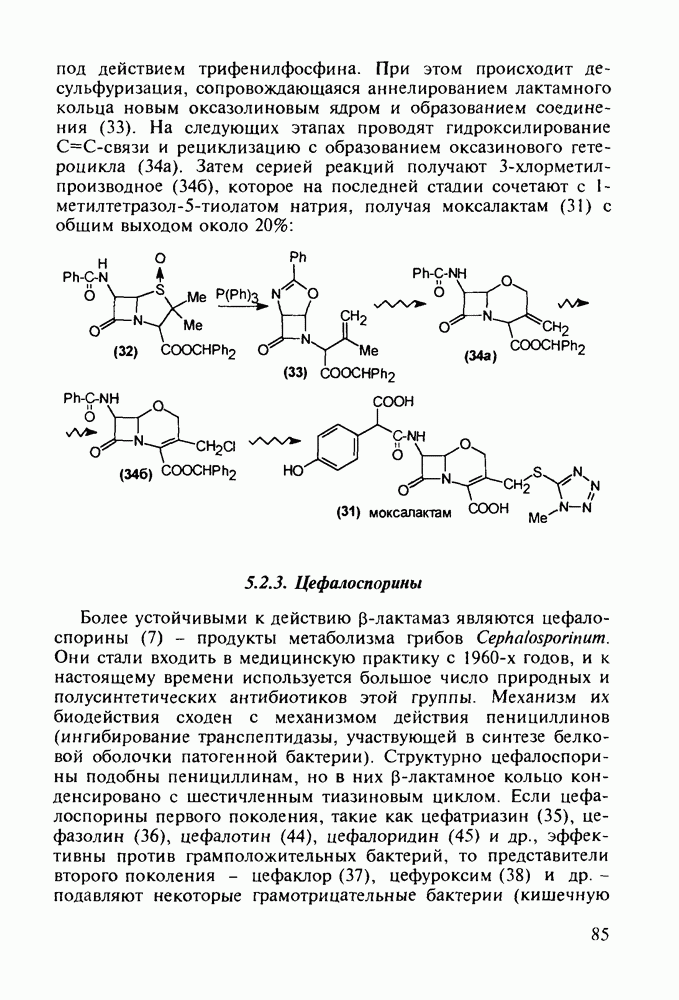
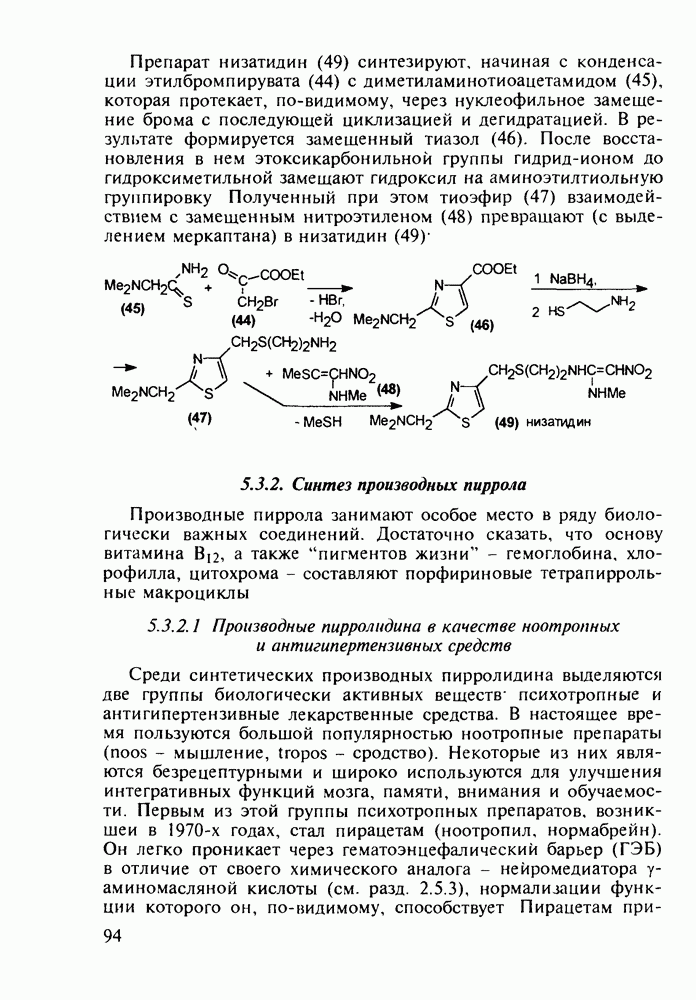
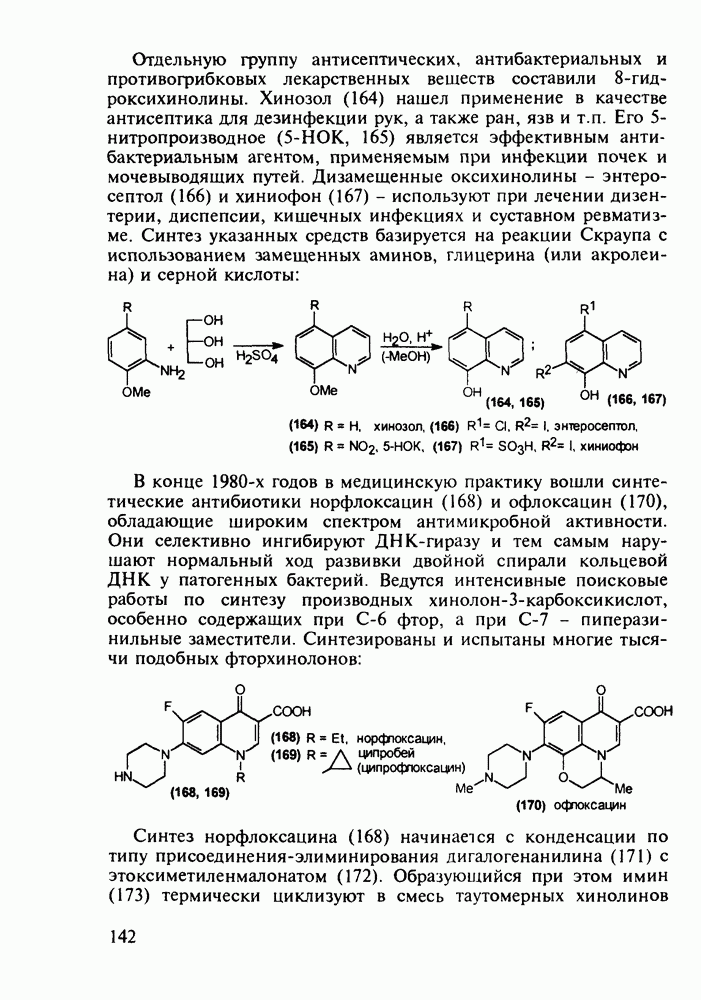
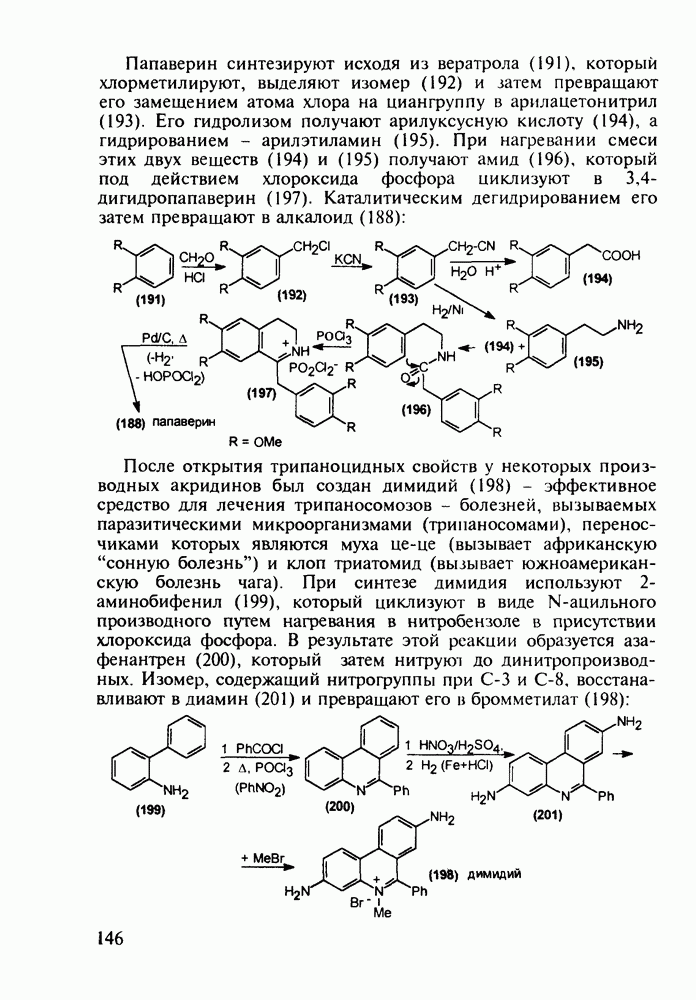
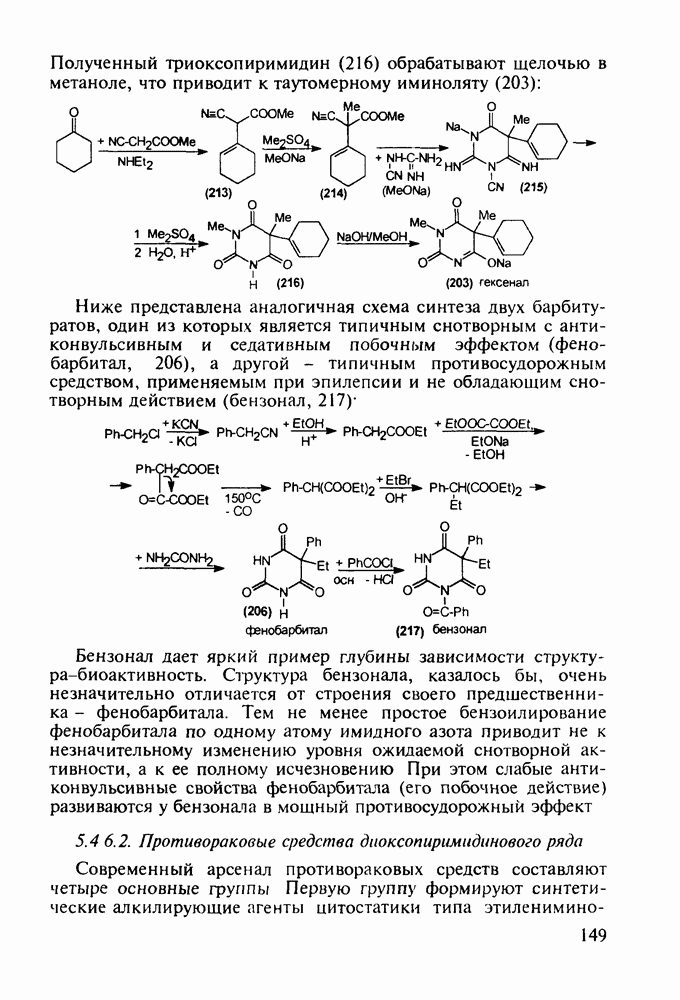
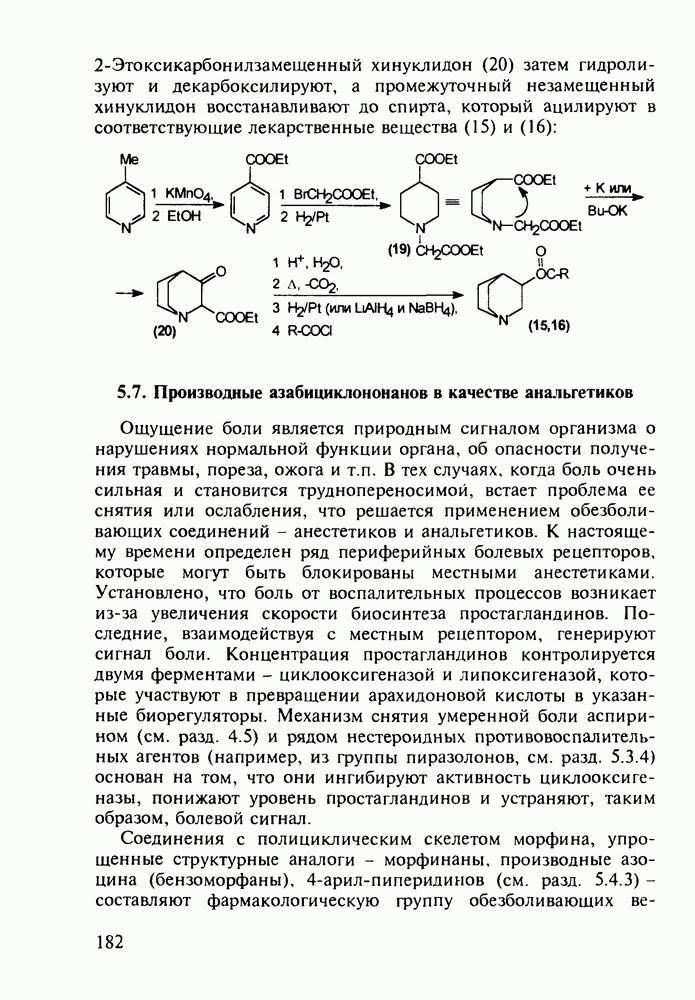
Отметим, что в этих процессах конденсации неосновные атомы азота в карбамиде и гуанидине сохраняют нуклеофильность, достаточную для успешной атаки карбонильных атомов углерода в исходном малонате Обычно получение 5,5-дизамещенных барбитуратов не вызывает больших проблем, но необходимость ввести циклоалифатические или ароматические заместители усложняет синтез. При получении гексенала (203) сначала синтезируют ключевое производное мононитрилмалоната (214) Для этого конденсируют циклогексанон с цианацетатом до производного циклогексена (213), которое затем метилируют диметилсульфатом. Обе однотипные реакции катализируются основаниями Дизамещенный малонат (214) далее циклизуют с циангуанидином Образовавшееся при этом натриевое производное диазина Полученный триоксопиримидин (216) обрабатывают щелочью в метаноле, что приводит к таутомерному иминоляту (203):
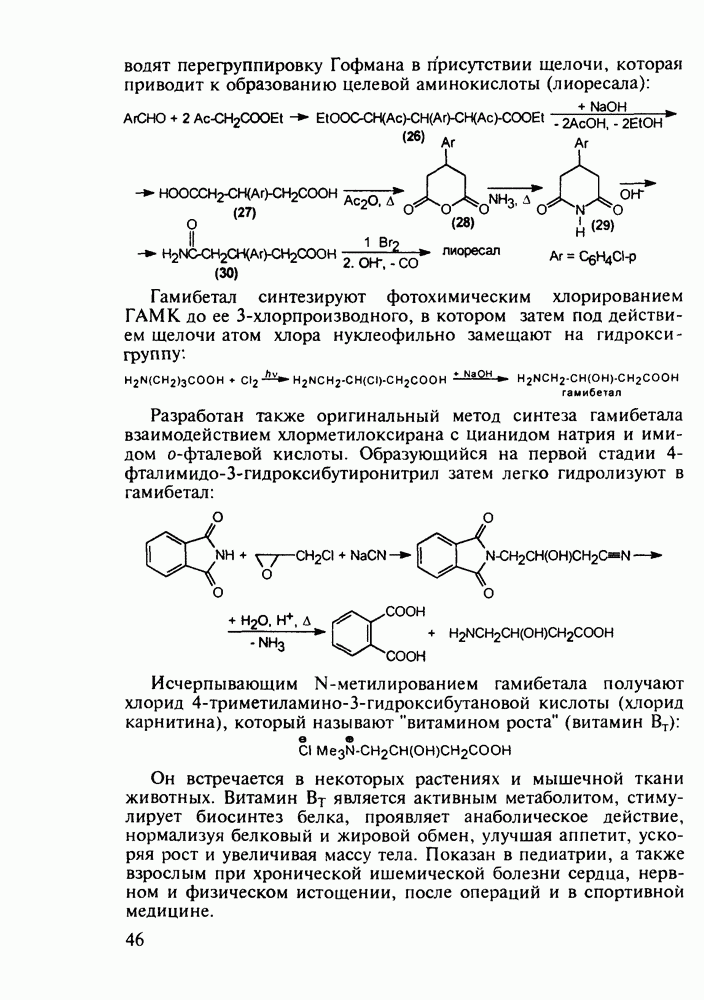
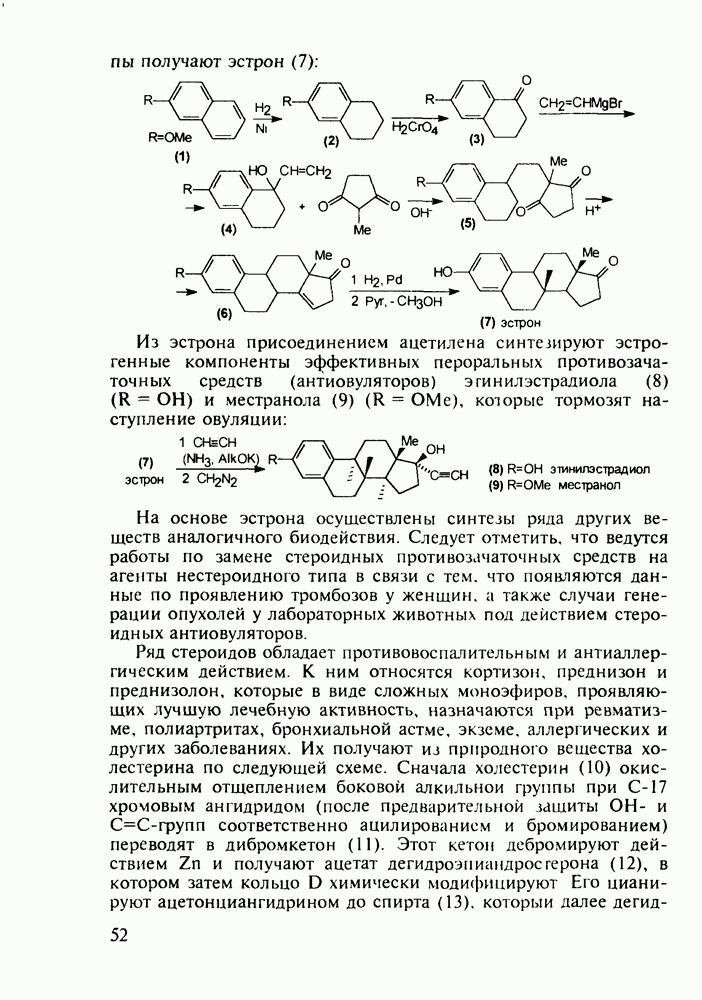
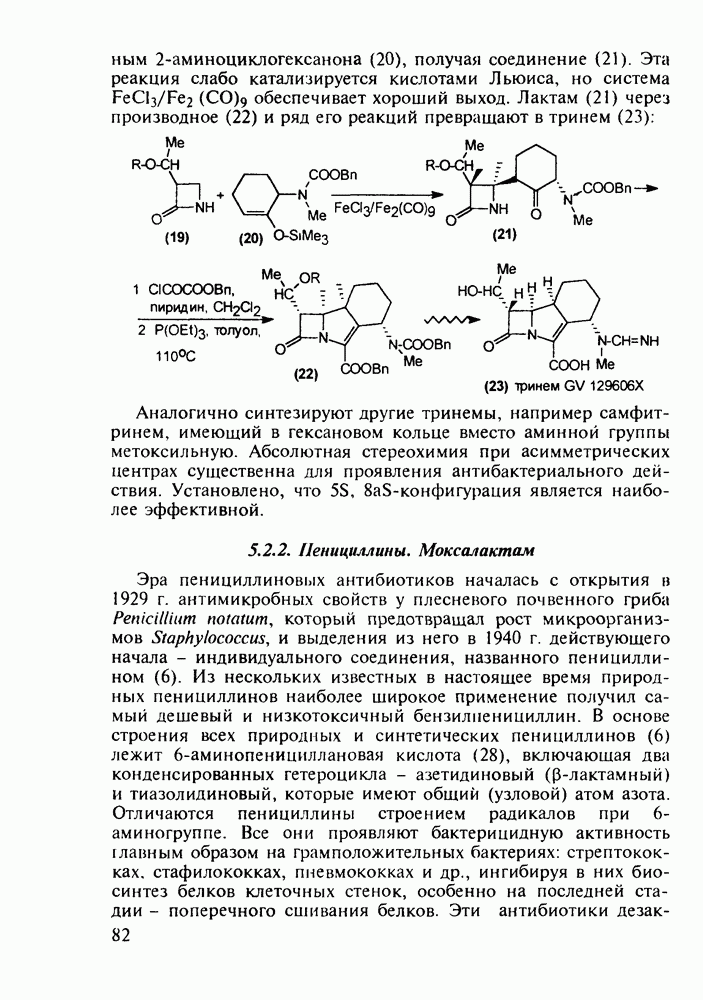
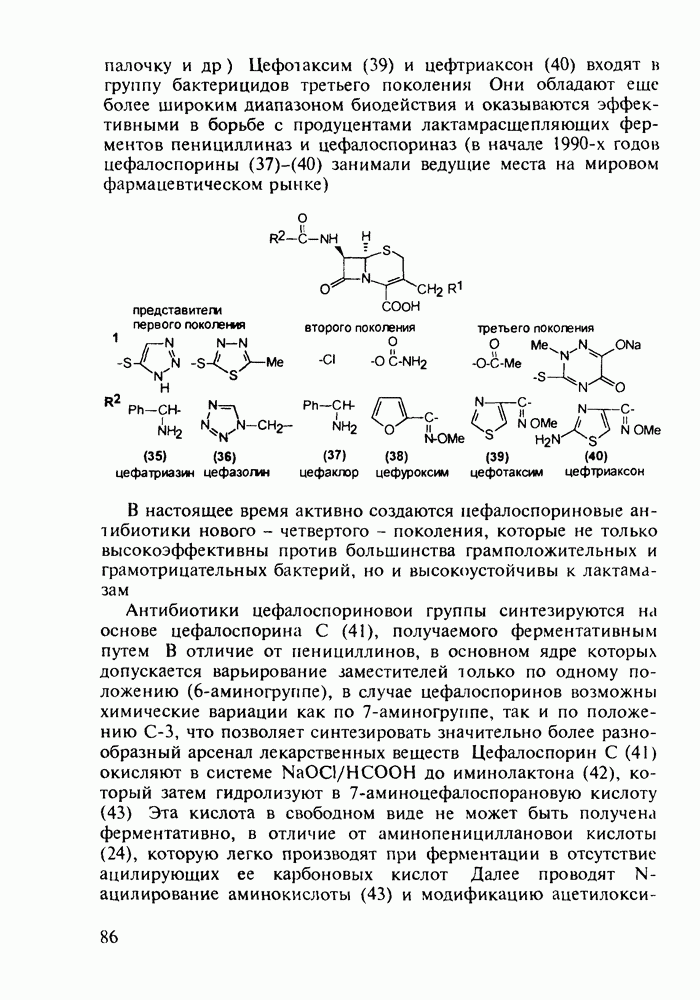
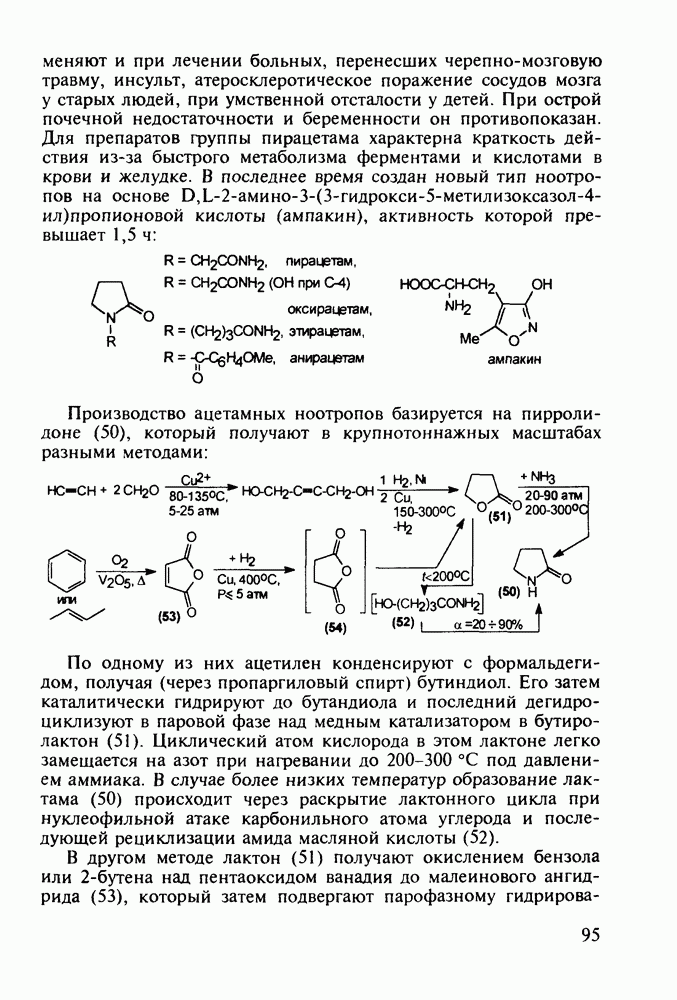
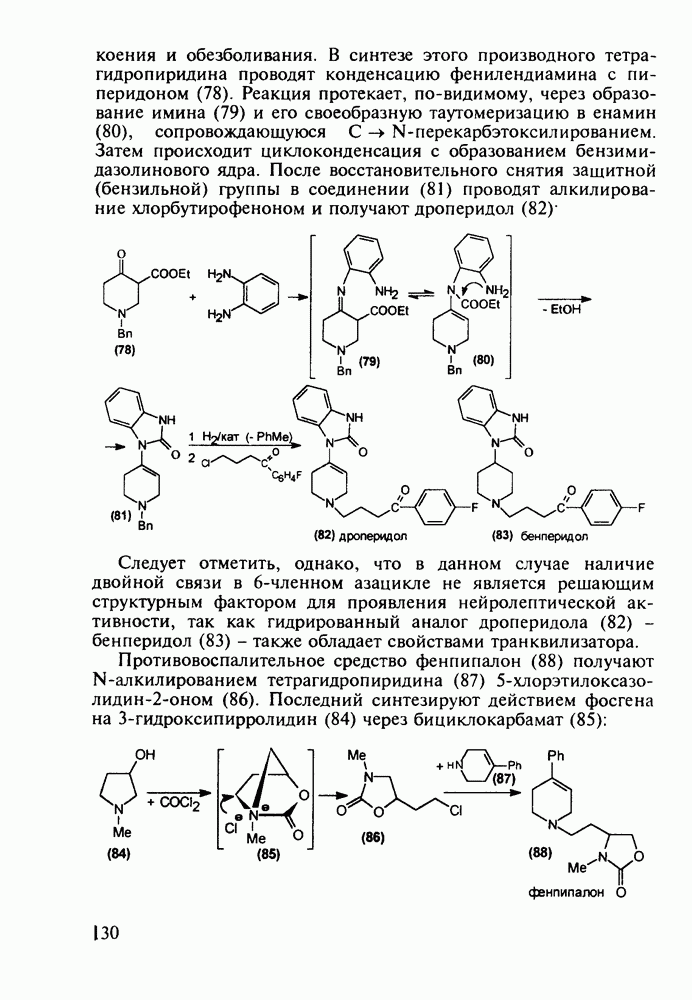
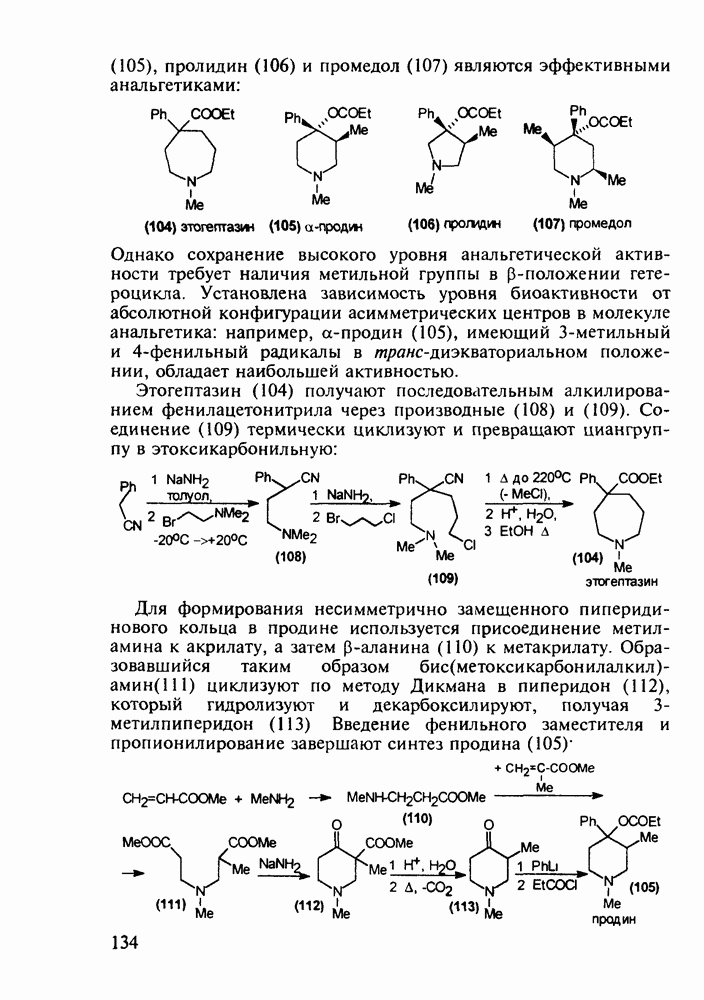
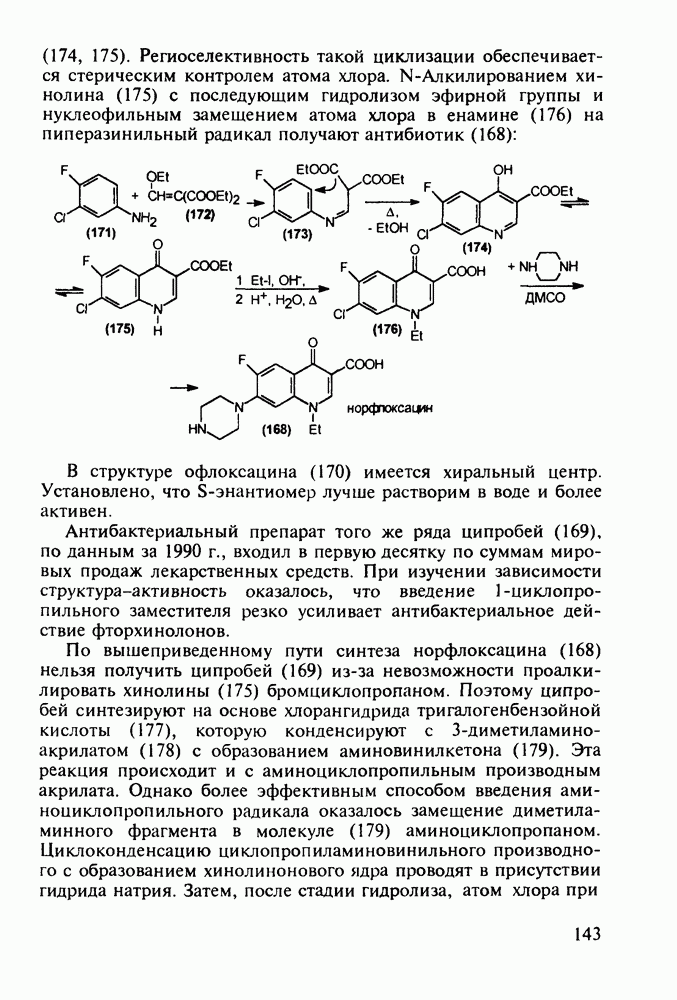
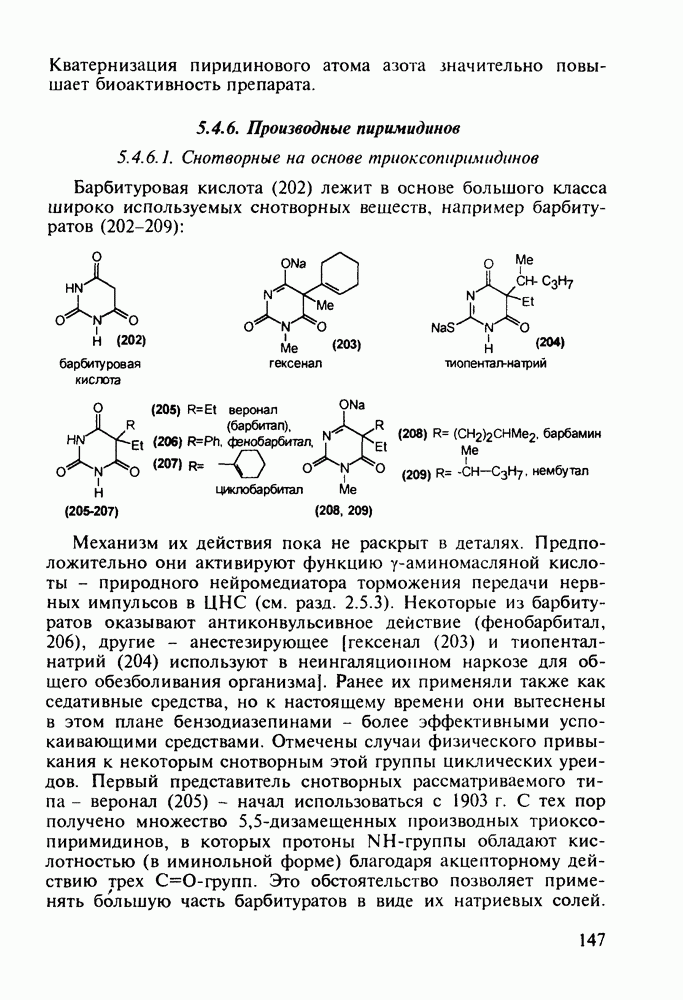
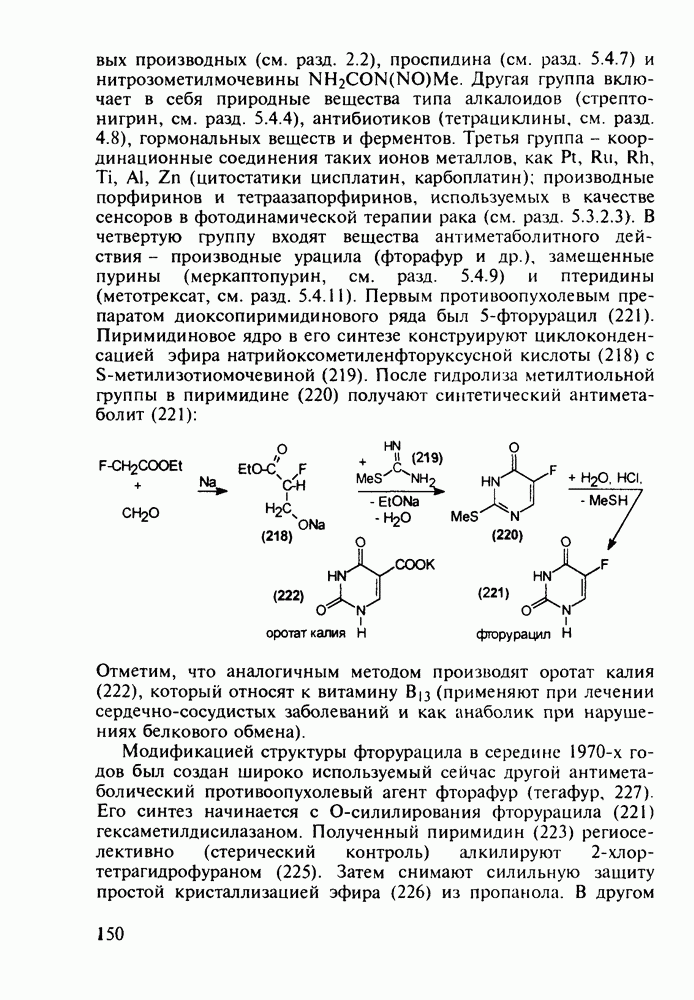
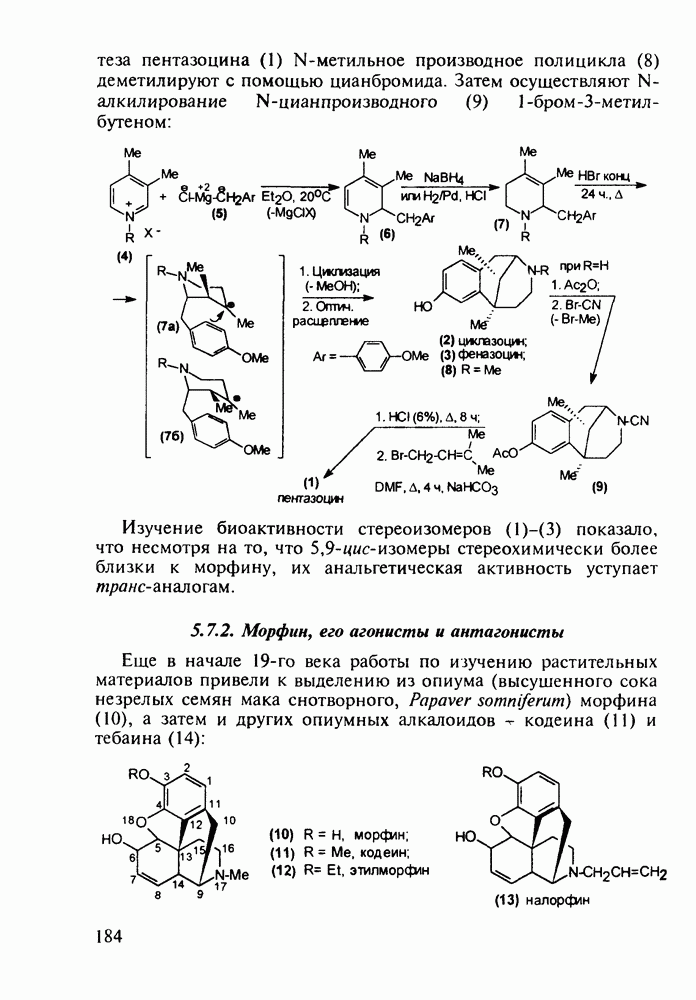
Ниже представлена аналогичная схема синтеза двух барбитуратов, один из которых является типичным снотворным с антиконвульсивным и седативным побочным эффектом (фенобарбитал, 206), а другой - типичным противосудорожным средством, применяемым при эпилепсии и не обладающим снотворным действием (бензонал, 217)
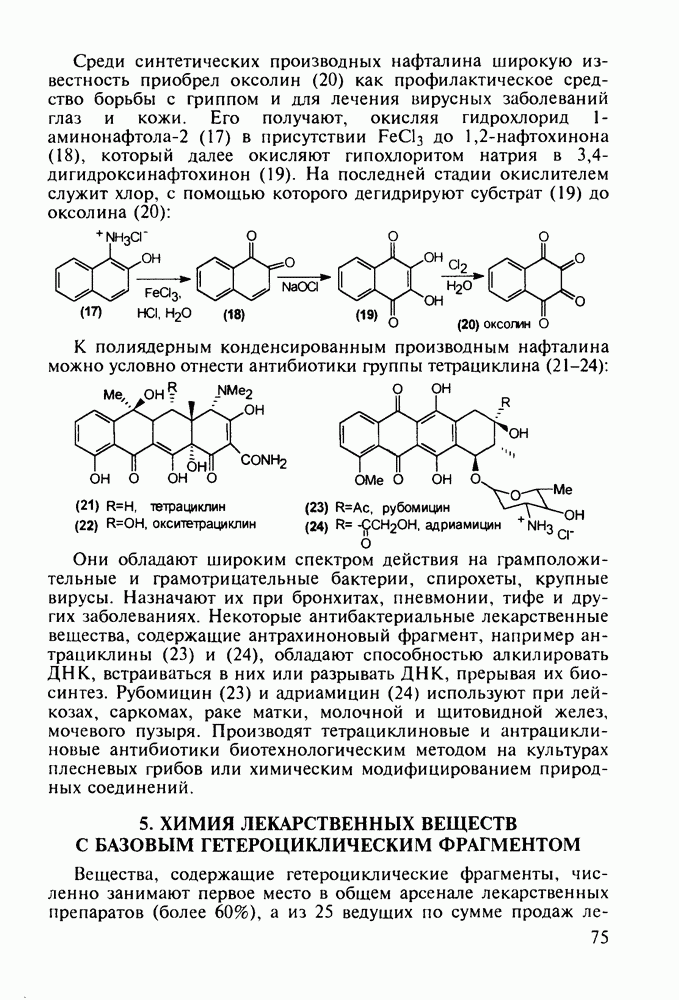
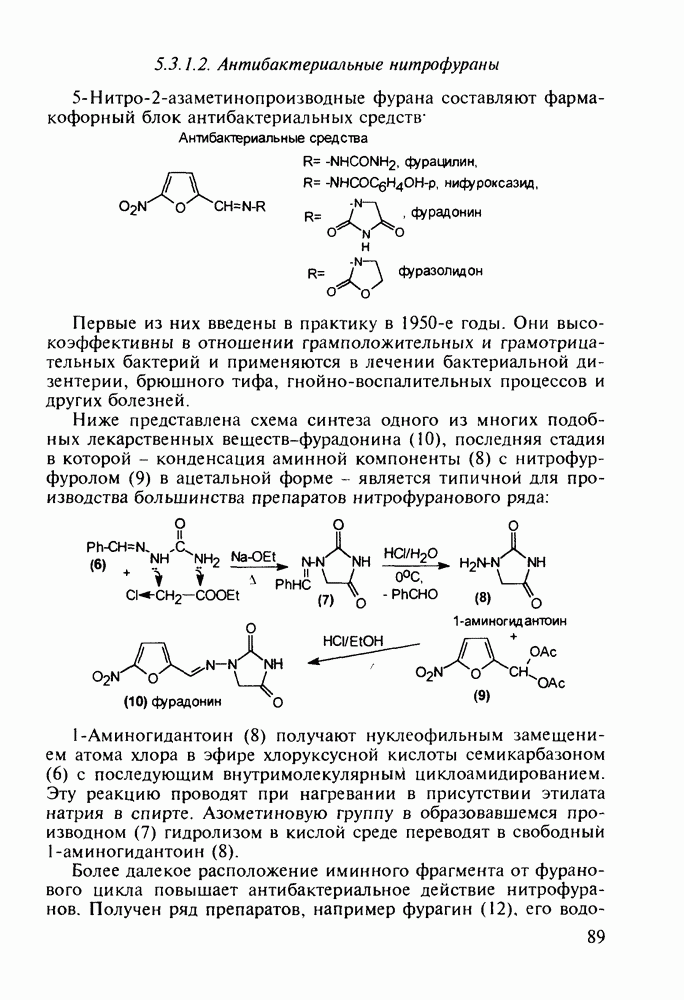
Бензонал дает яркий пример глубины зависимости структура-биоактивность. Структура бензонала, казалось бы, очень незначительно отличается от строения своего предшественника - фенобарбитала. Тем не менее простое бензоилирование фенобарбитала по одному атому имидного азота приводит не к незначительному изменению уровня ожидаемой снотворной активности, а к ее полному исчезновению При этом слабые антиконвульсивные свойства фенобарбитала (его побочное действие) развиваются у бензонааа в мощный противосудорожный эффект 5.4 6.2. Противораковые средства даоксопиримидинового рядаСовременный арсенал противораковых средств составляют четыре основные группы Первую группу формируют синтетические алкилирующие агенты цитостатики типа этилениминовых производных (см. разд. 2.2), проспидина (см. разд. 5.4.7) и нитрозометилмочевины
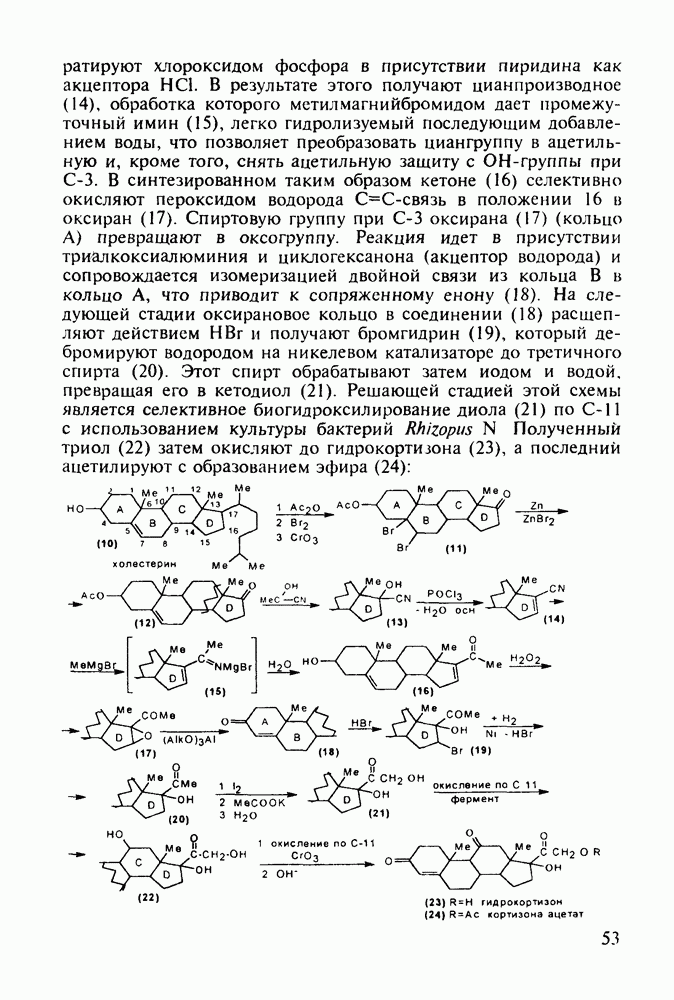
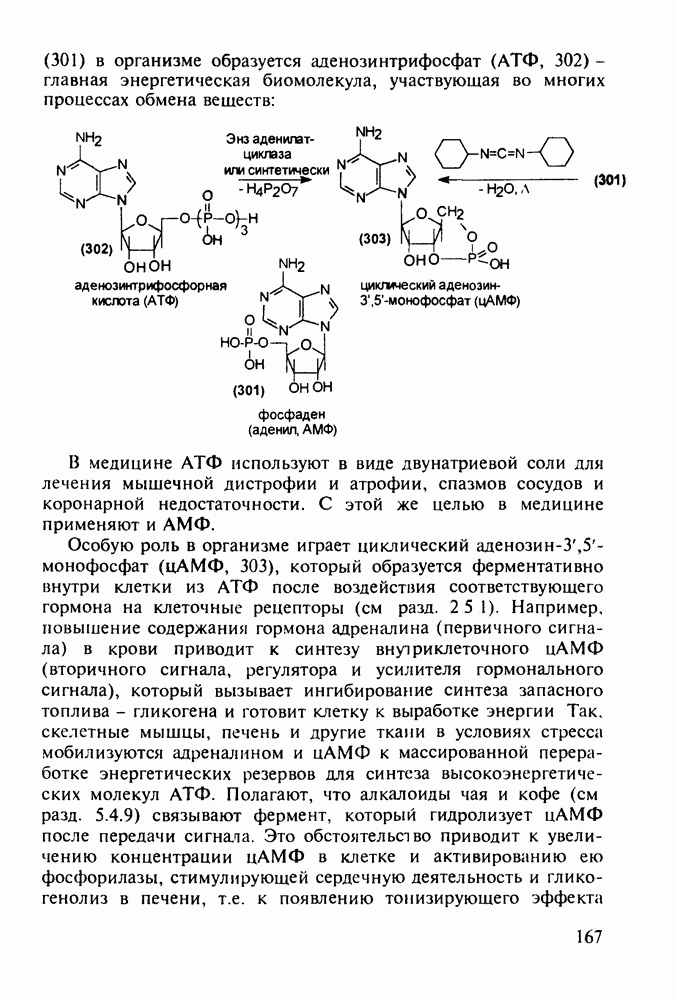
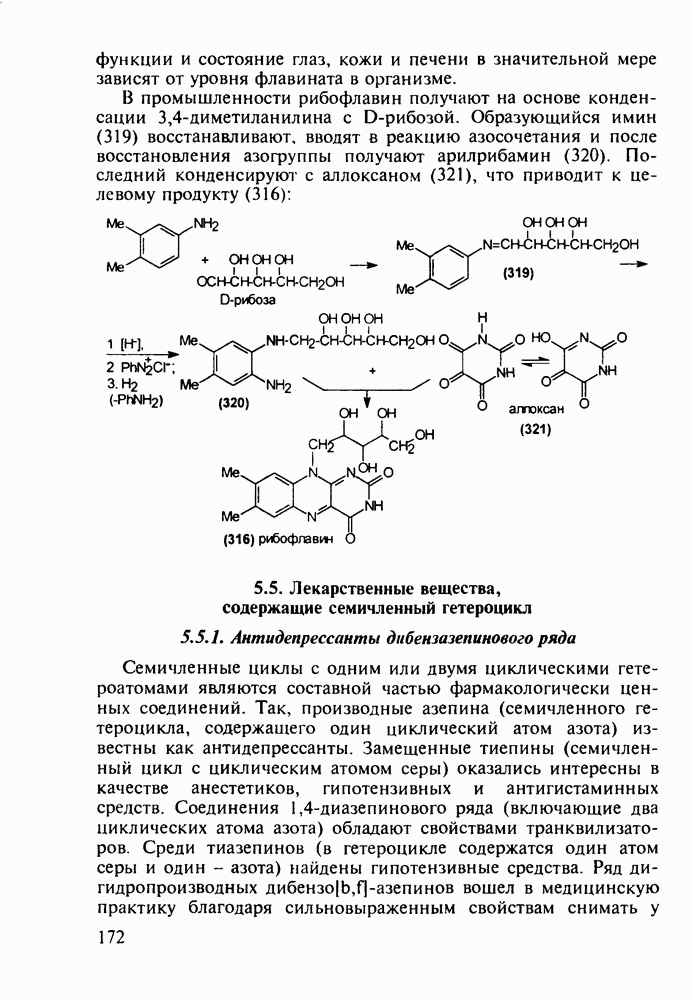
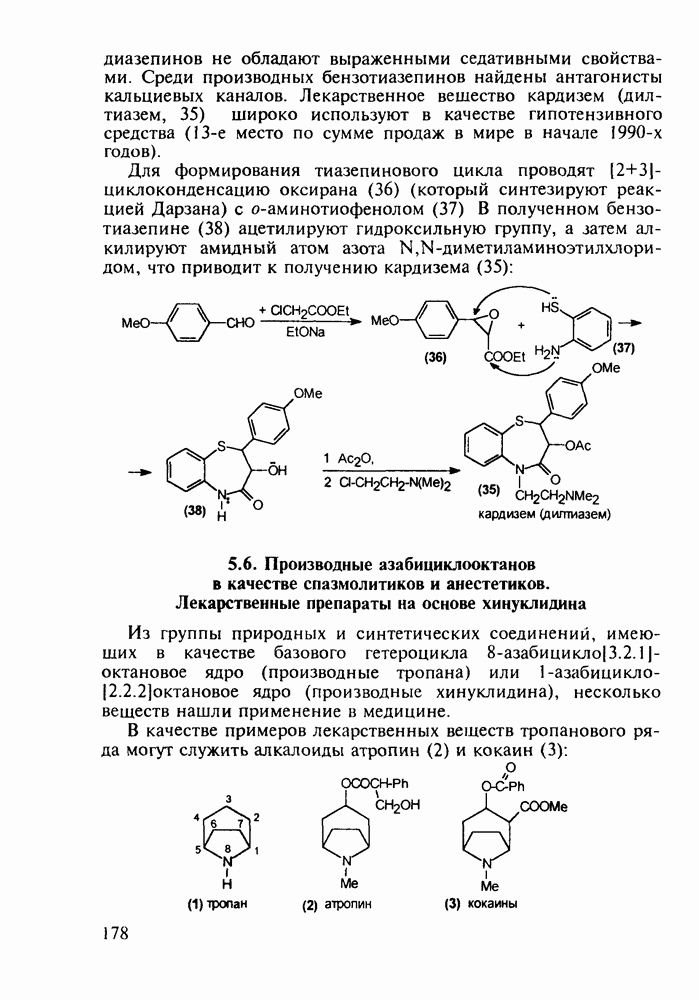
Отметим, что аналогичным методом производят оротат калия (222), который относят к витамину Модификацией структуры фторурацила в середине 1970-х годов был создан широко используемый сейчас другой антиметаболический противоопухолевый агент фторафур (тегафур, 227). Его синтез начинается с О-силилирования фторурацила (221) гексаметилдисилазаном. Полученный пиримидин (223) региоселективно (стерический контроль) алкилируют 2-хлор-тетрагидрофураном (225). Затем снимают силильную защиту простой кристаллизацией эфира (226) из пропанола. В другом методе пиримидин (223) алкилируют дигидрофураном (224) в присутствии тетрахлорида олова:
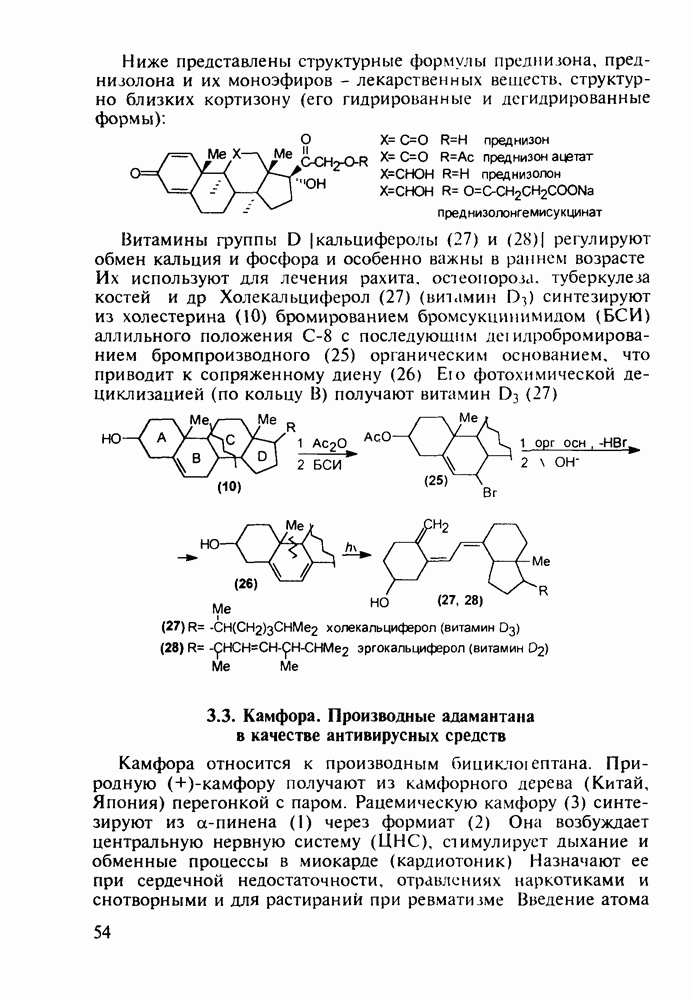
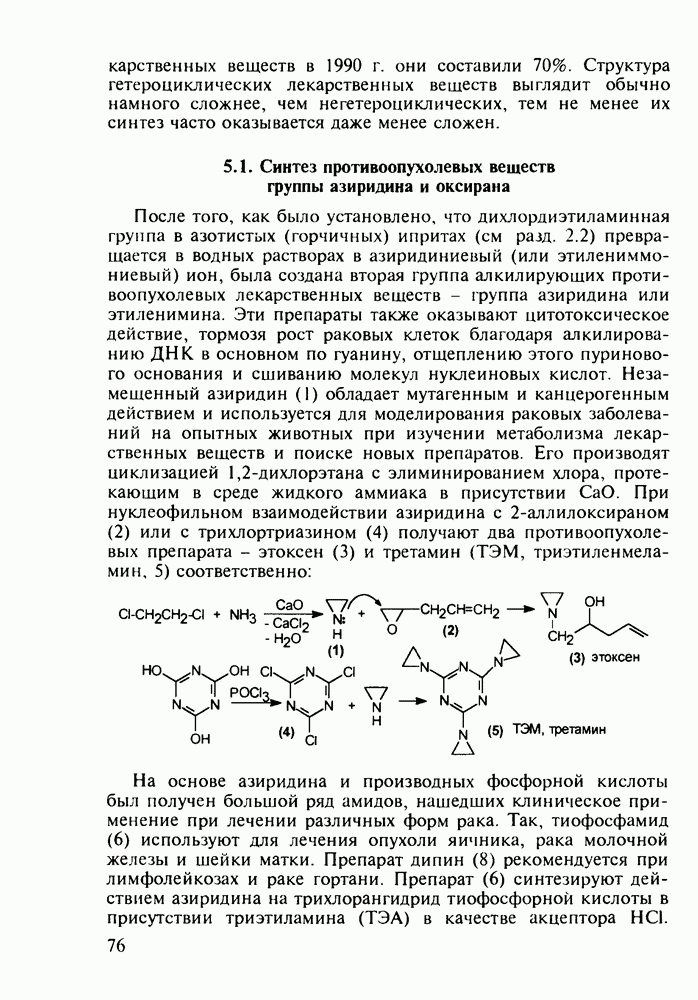
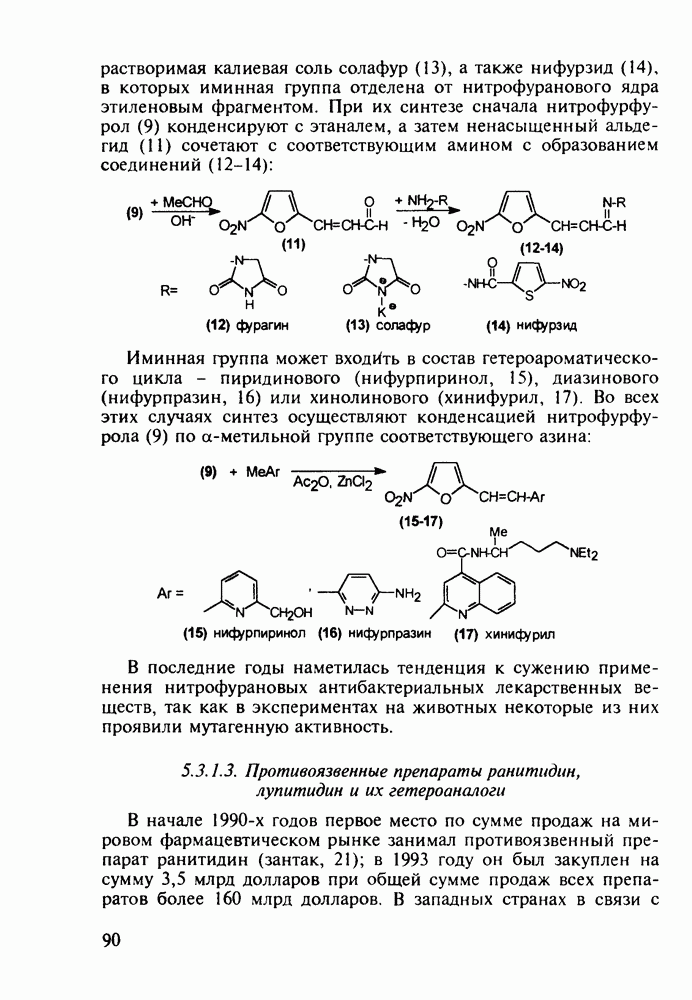
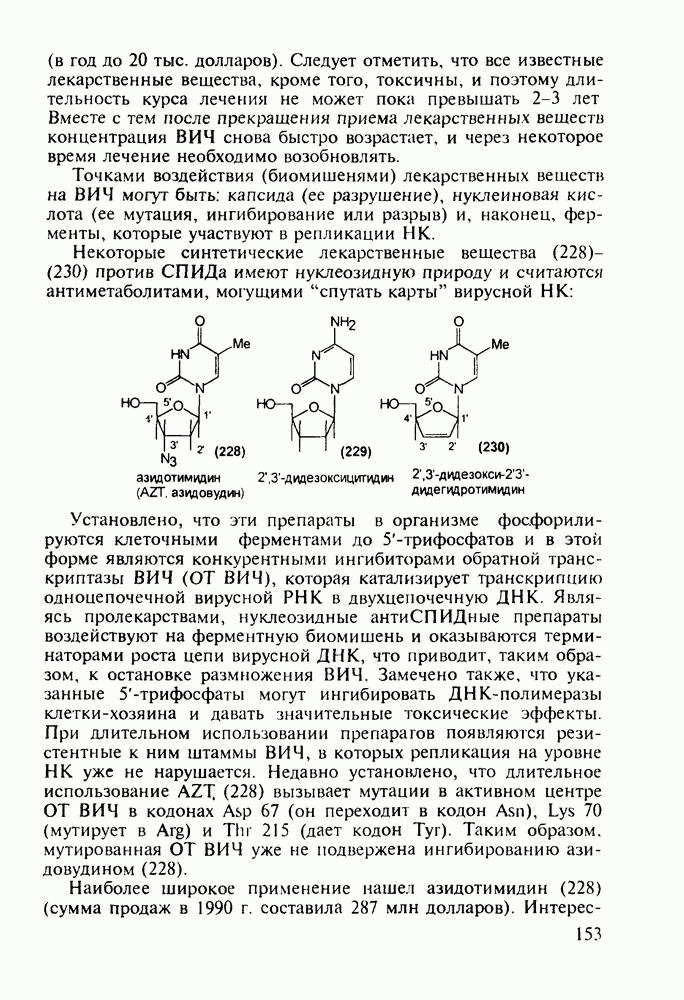
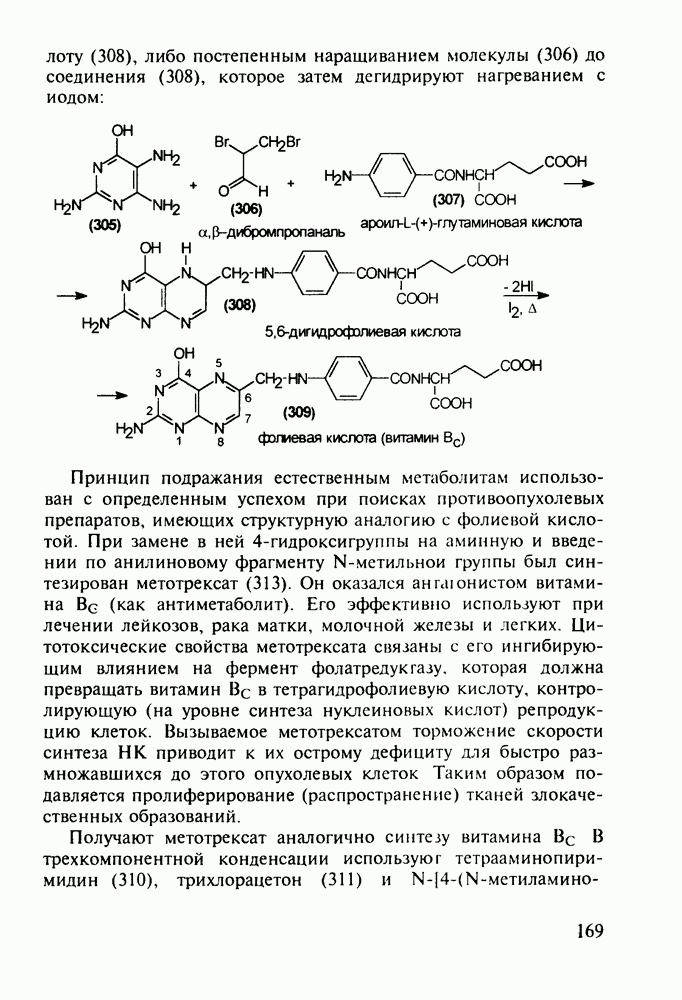
Оба лекарственных вещества (221) и (227) применяют при злокачественных опухолях желудка и других отделов желудочно-кишечного тракта. Первый вводят внутривенно, ибо он плохо всасывается при пероральном приеме. Второй же легко всасывается в случае приема в виде таблеток. Попадая в раковые клетки, эти соединения превращаются в 5-фтордезокси-уридинмонофосфаты и в качестве таковых ингибируют тимидилатсинтетазу. При этом тормозится биосинтез одного из оснований ДНК - тимидина - и тем самым подавляется синтез ДНК раковой клетки. Фторурацил и фторафур имеют определенную токсичность и для здоровых клеток, что диктует необходимость создания новых лекарственных веществ на основе плодотворной идеи антиметаболитов. 5.4.6.3. Производные пиримидинов с антивирусной (антыСПИДноы) и антимикробной активностьюВ 20-м веке наука добилась больших успехов в борьбе со многими, в том числе и с когда-то считавшимися неизлечимыми инфекционными болезнями, например чумой, оспой, проказой и т.д. Но эра передаваемых (заразных) болезней, к сожалению, далека от завершения, несмотря на бурное развитие в последнюю четверть столетия науки и практики органического и фармацевтического синтеза, генетики и биотехнологии, молекулярной биологии и медицины в целом. Некоторые старые, казалось бы, побежденные болезни, такие как туберкулез и малярия, начали снова выходить из-под контроля. В то же время продолжают возникать новые инфекционные заболевания, например СПИД, болезнь легионеров, гантавирусный легочный синдром. Поэтому химики, биологи, медики и другие специалисты всегда должны быть готовы к борьбе с появляющимися неизвестными ранее патогенными микроорганизмами и вызываемыми ими инфекционными болезнями. Одной из самых серьезных является открытая в США в начале 1980-х годов болезнь, названная синдромом приобретенного иммунодефицита (СПИД; по-английски AIDS - acquired immunodeficiency syndrom). Ее опасность заключается не только в том, что она приводит к летальному исходу, но и в том, что это - инфекционное (вирусное) заболевание. К настоящему времени СПИД приобрел пандемический характер и охватывает почти все страны мира. На 1998 г. число инфицированных вирусом иммунодефицита человека (ВИЧ) во всем мире достигло 30 млн человек (в США - от 600 до 900 тыс., в России - от 30 до 100 тыс. человек). Передается этот вирус от ВИЧ-инфицированного через кровь или половые контакты. Любой вирус (варион) состоит из нуклеиновой кислоты (НК), защищаемой капсидой (цилиндрической или сферической оболочкой белкового типа, иногда с включением липидов и сахаров). Капсида выполняет также функцию взаимодействия с клетками чужого организма, способствуя проникновению вирусной НК внутрь клетки-хозяина и запуску там синтеза новых вирусных молекул. В случае ВИЧ сложность заключается в том, что в чужом организме он встраивается в клетки самой иммунной системы (в лейкоциты, фагоциты, лимфоциты), призванной бороться с патогенными микроорганизмами. И как только зараженный организм включает в действие защитную иммунную систему, вместе с размножением собственных иммунных клеток начинается бурный рост числа ВИЧ, и клетка-хозяин теряет генетический контроль над биопроцессами. Иммунные силы (сопротивляемость) организма, таким образом, слабеют, и у больных СПИДом возрастает вероятность заражения другими инфекциями - туберкулезом, пневмонией, лейкозами и т.д. К настоящему времени для лечения от СПИДа, причисляемого к “болезням XXI века”, применяют уже дюжину синтетических лекарственных веществ, однако все они только понижают концентрацию вируса. Более того, этот вирус очень быстро эволюционирует, давая уже за один репродуктивный цикл одну мутацию. Возникающая у разновидностей ВИЧ резистентность приводит к необходимости использовать композиции (“коктейли”) из двух-трех лекарств. Это удорожает курс лечения (в год до 20 тыс. долларов). Следует отметить, что все известные лекарственные вещества, кроме того, токсичны, и поэтому длительность курса лечения не может пока превышать 2-3 лет Вместе с тем после прекращения приема лекарственных веществ концентрация ВИЧ снова быстро возрастает, и через некоторое время лечение необходимо возобновлять. Точками воздействия (биомишенями) лекарственных веществ на ВИЧ могут быть: капсида (ее разрушение), нуклеиновая кислота (ее мутация, ингибирование или разрыв) и, наконец, ферменты, которые участвуют в репликации НК. Некоторые синтетические лекарственные вещества (228)- (230) против СПИДа имеют нуклеозидную природу и считаются антиметаболитами, могущими “спутать карты” вирусной НК:
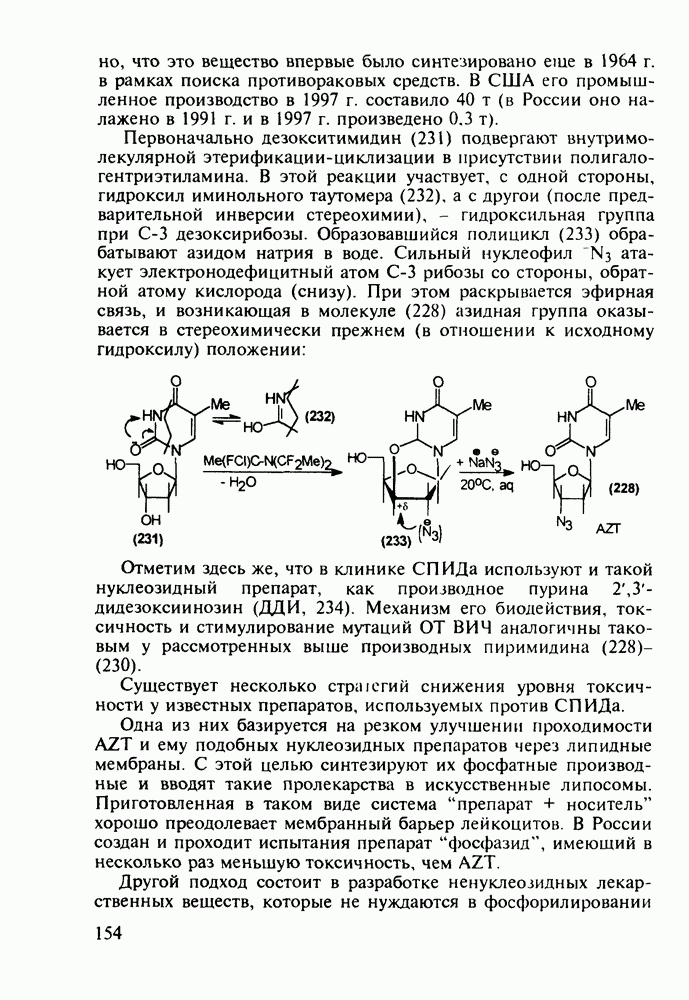
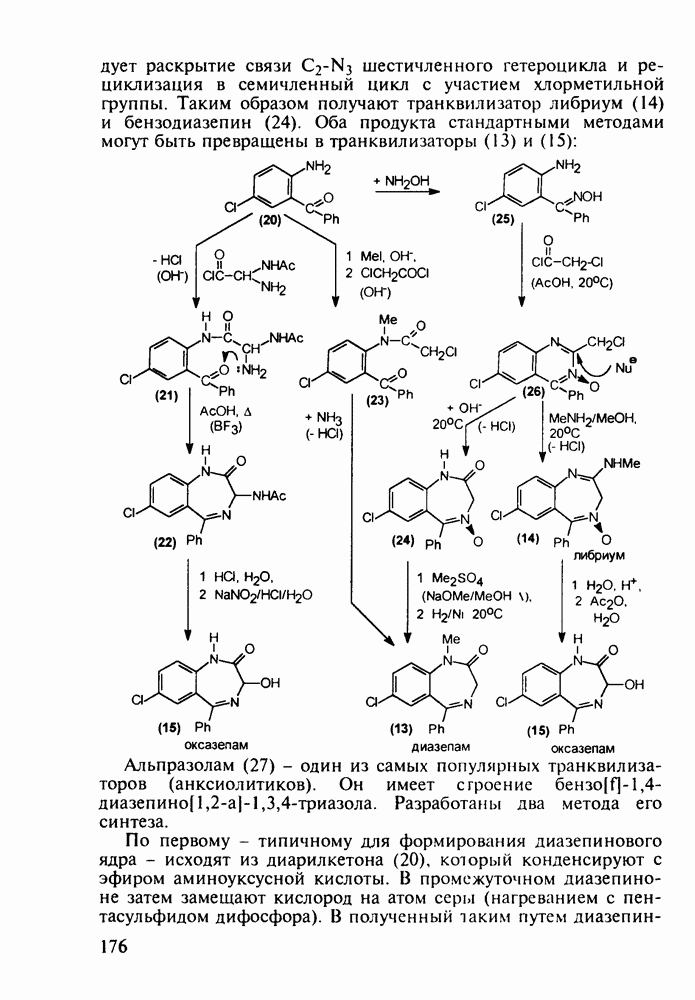
Установлено, что эти препараты в организме фосфорилируются клеточными ферментами до Наиболее широкое применение нашел азидотимидин (228) (сумма продаж в 1990 г. составила 287 млн долларов). Интересно, что это вещество впервые было синтезировано еще в 1964 г. в рамках поиска противораковых средств. В США его промышленное производство в 1997 г. составило 40 т (в России оно налажено в 1991 г. и в 1997 г. произведено 0.3 т). Первоначально дезокситимидин (231) подвергают внутримолекулярной этерификации-циклизации в присутствии полигало-гентриэтиламина. В этой реакции участвует, с одной стороны, гидроксил иминольного таутомера (232), а с другой (после предварительной инверсии стереохимии), - гидроксильная группа при
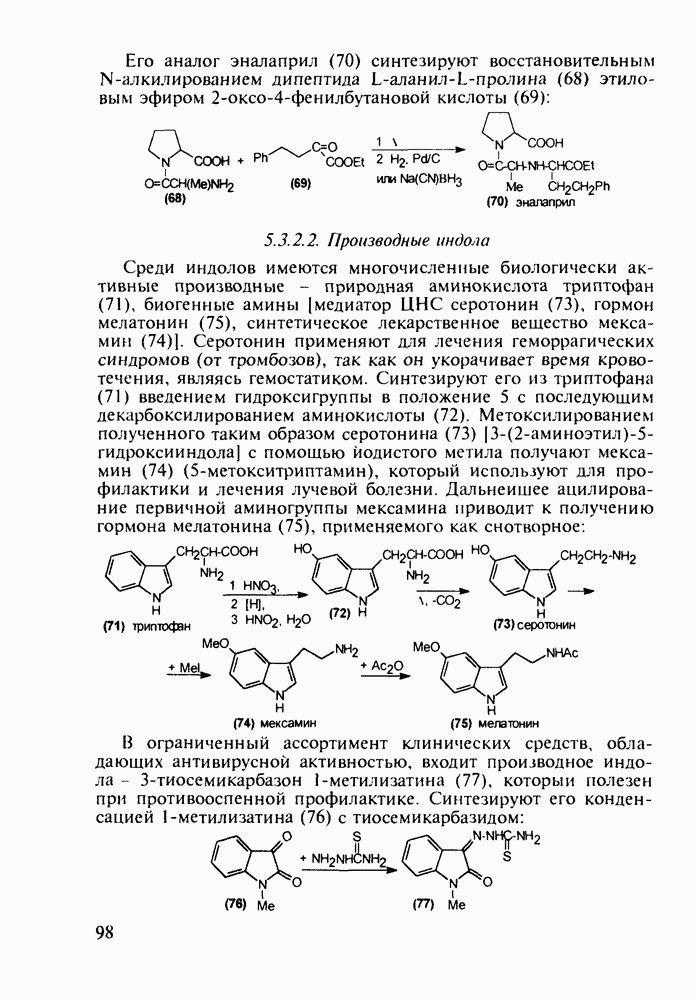
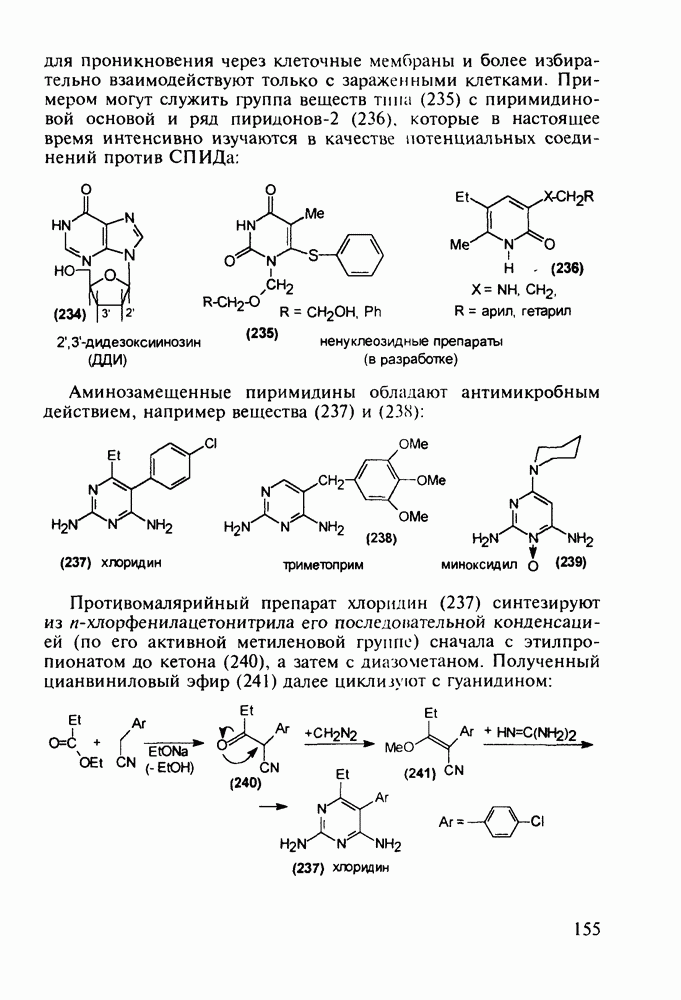
Отметим здесь же, что в клинике СПИДа используют и такой нуклеозидный препарат, как производное пурина Существует несколько стратегий снижения уровня токсичности у известных препаратов, используемых против СПИДа. Одна из них базируется на резком улучшении проходимости AZT и ему подобных нуклеозидных препаратов через липидные мембраны. С этой целью синтезируют их фосфатные производные и вводят такие пролекарства в искусственные липосомы. Приготовленная в таком виде система “препарат + носитель” хорошо преодолевает мембранный барьер лейкоцитов. В России создан и проходит испытания препарат “фосфазид", имеющий в несколько раз меньшую токсичность, чем AZT. Другой подход состоит в разработке ненуклеозидных лекарственных веществ, которые не нуждаются в фосфорилировании для проникновения через клеточные мембраны и более избирательно взаимодействуют только с зараженными клетками. Примером могут служить группа веществ типа (235) с пиримидиновой основой и ряд пиридонов-2 (236), которые в настоящее время интенсивно изучаются в качестве потенциальных соединений против СПИДа:
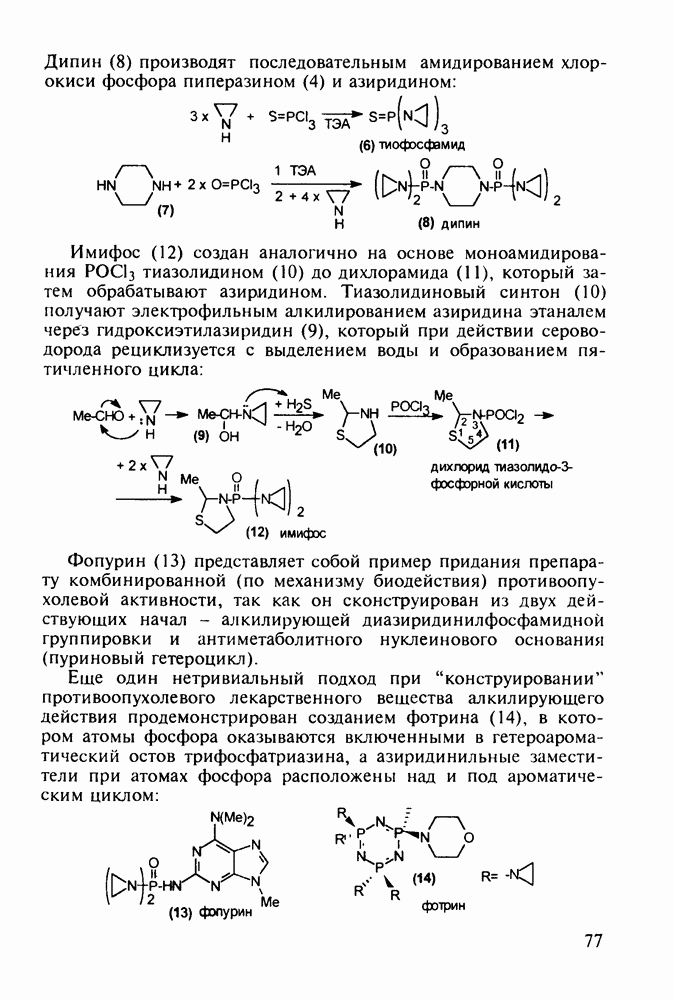
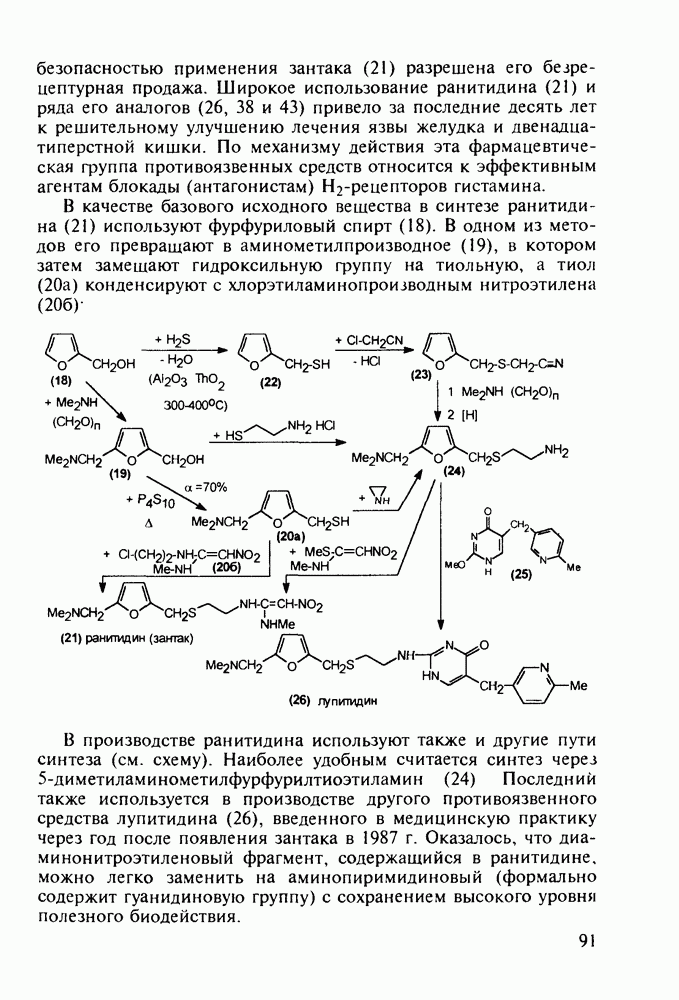
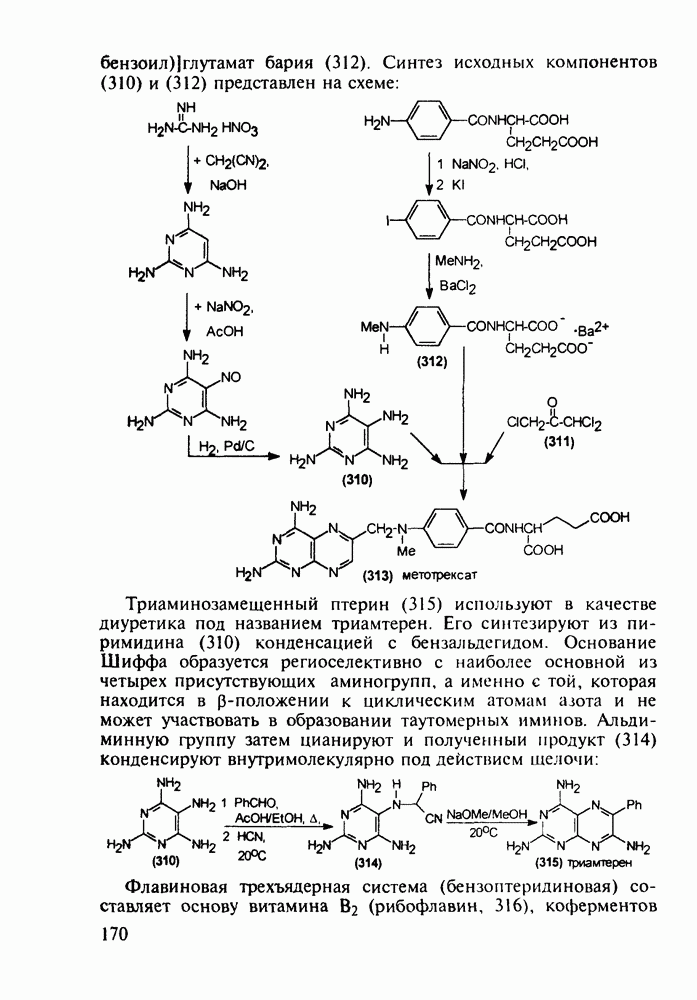
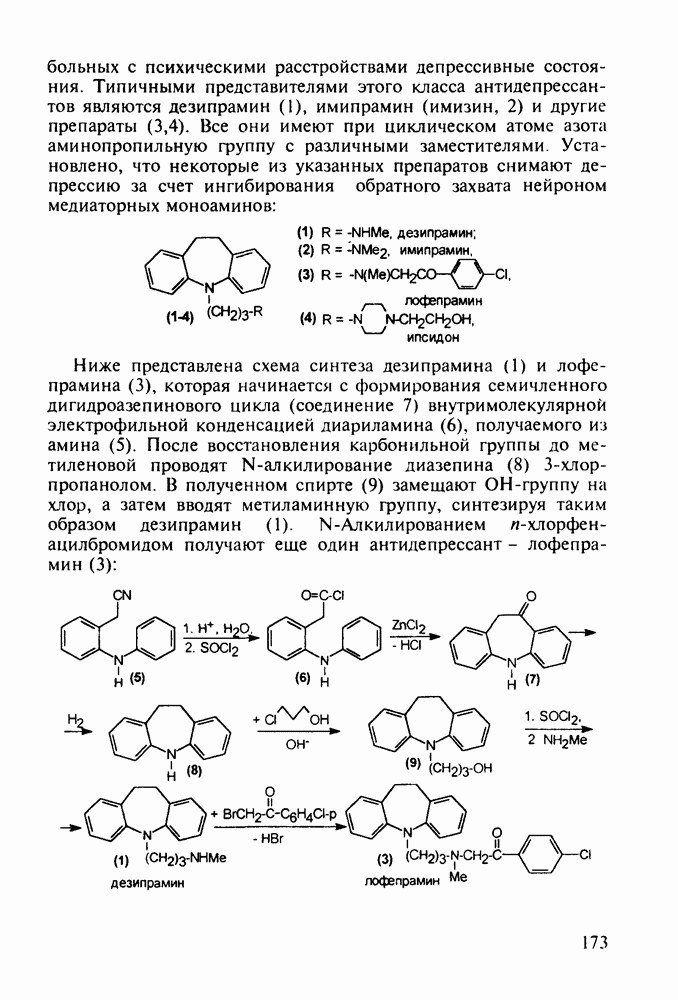
Аминозамещенные пиримидины обладают антимикробным действием, например вещества (237) и (238):
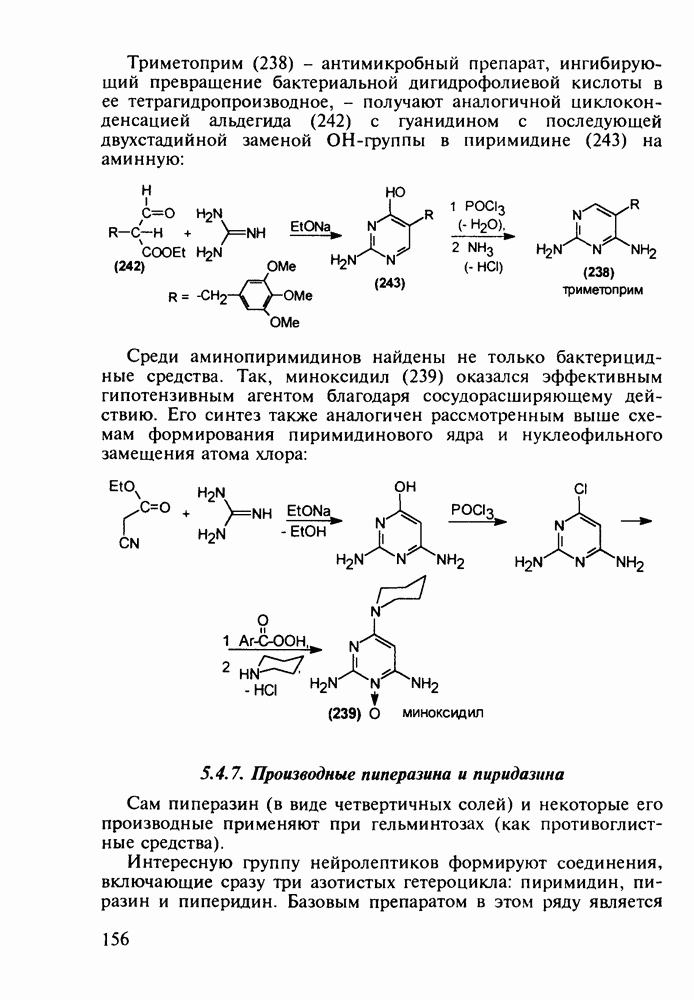
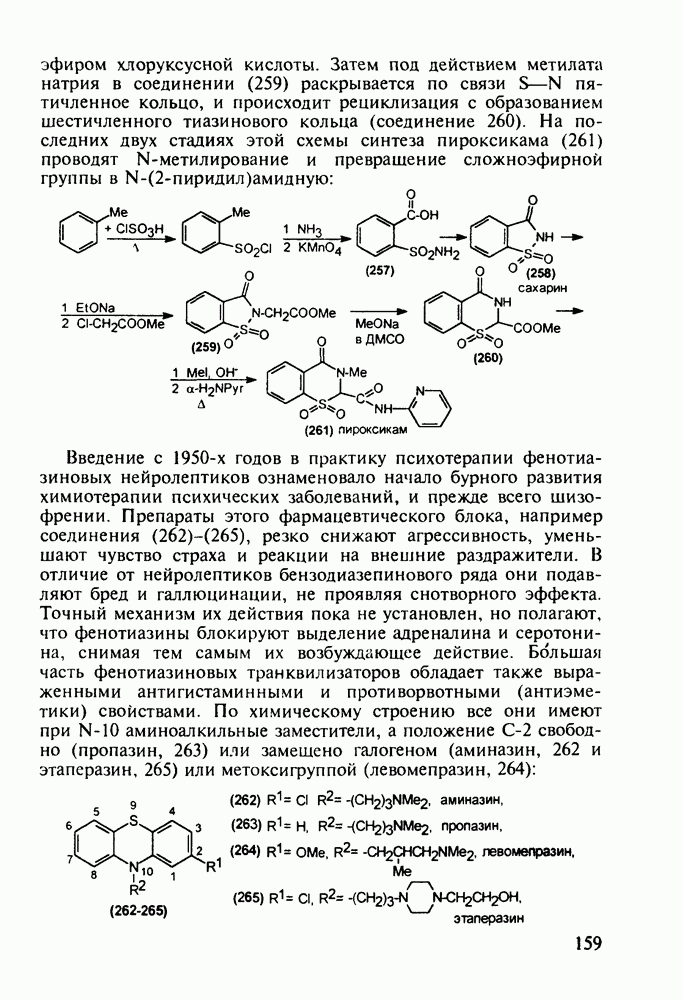
Противомалярийный препарат хлорндин (237) синтезируют из
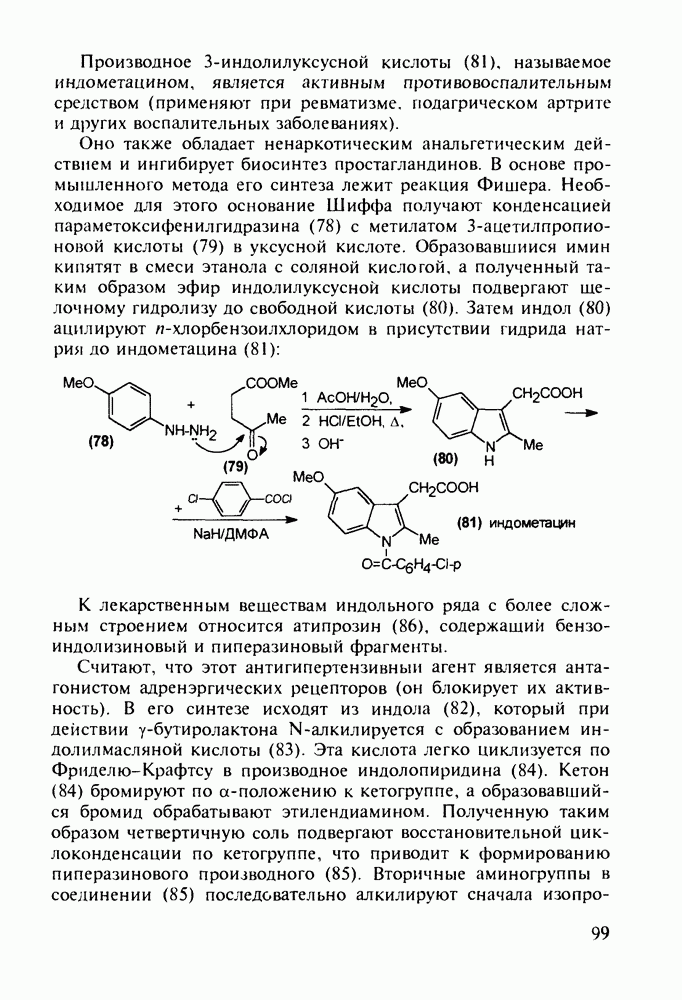
Триметоприм (238) - антимикробный препарат, ингибирующий превращение бактериальной дигидрофолиевой кислоты в ее тетрагидропроизводное, - получают аналогичной циклоконденсацией альдегида (242) с гуанидином с последующей двухстадийной заменой ОН-группы в пиримидине (243) на аминную:
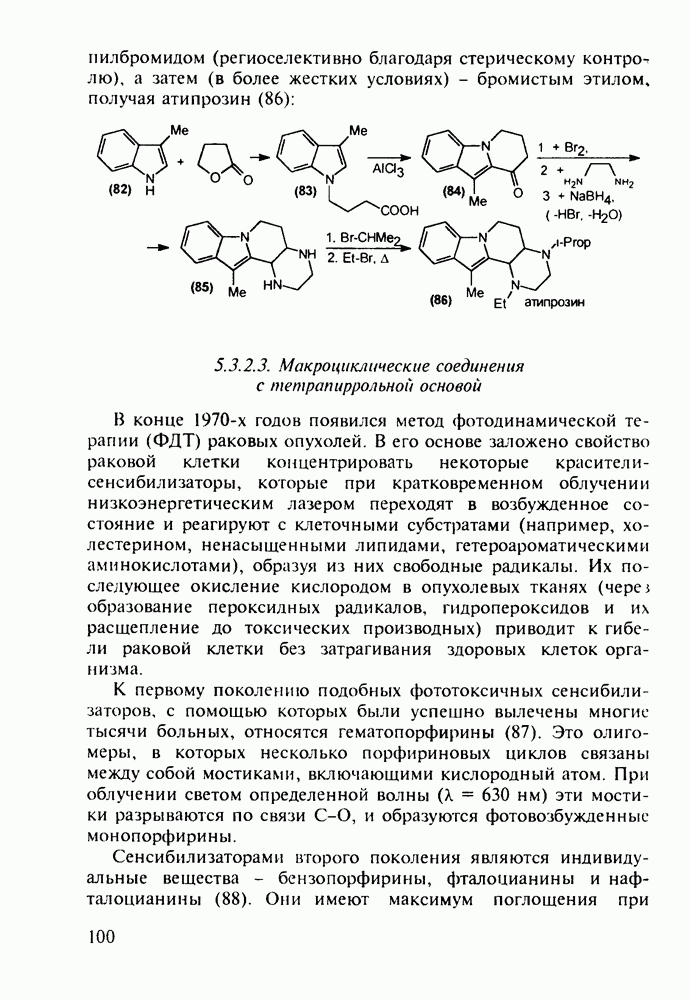
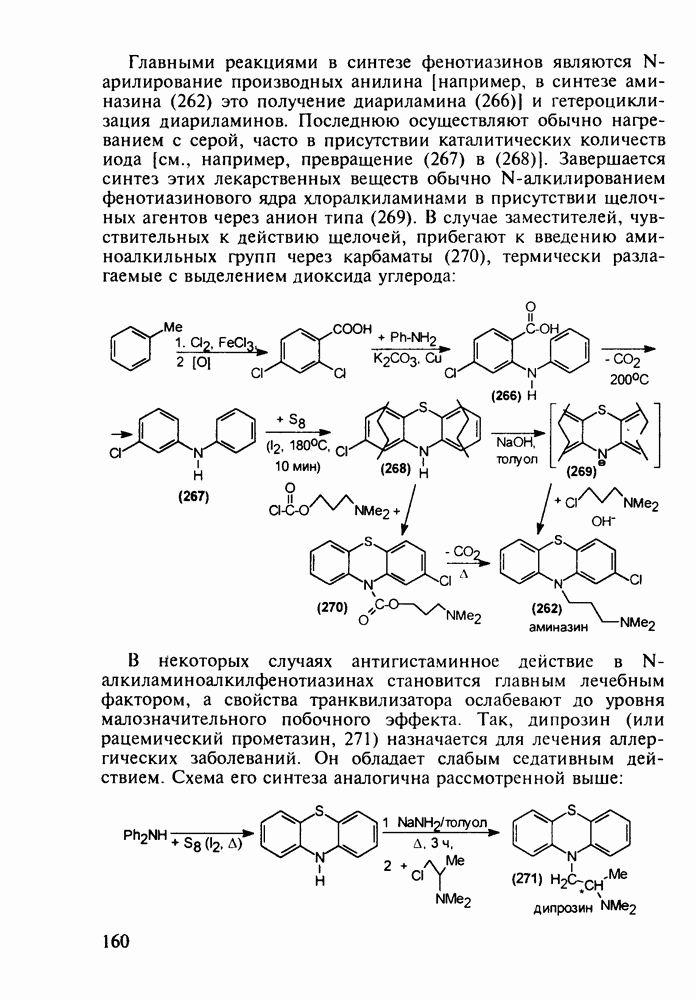
Среди аминопиримидинов найдены не только бактерицидные средства. Так, миноксидил (239) оказался эффективным гипотензивным агентом благодаря сосудорасширяющему действию. Его синтез также аналогичен рассмотренным выше схемам формирования пиримидинового ядра и нуклеофильного замещения атома хлора:
|
 |
Оглавление
|






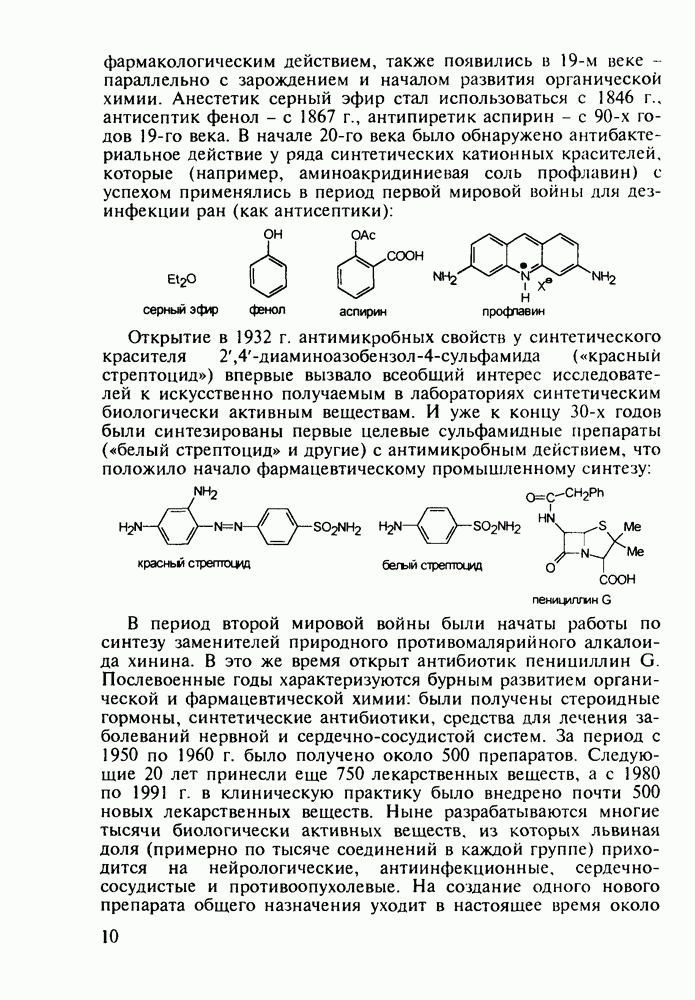





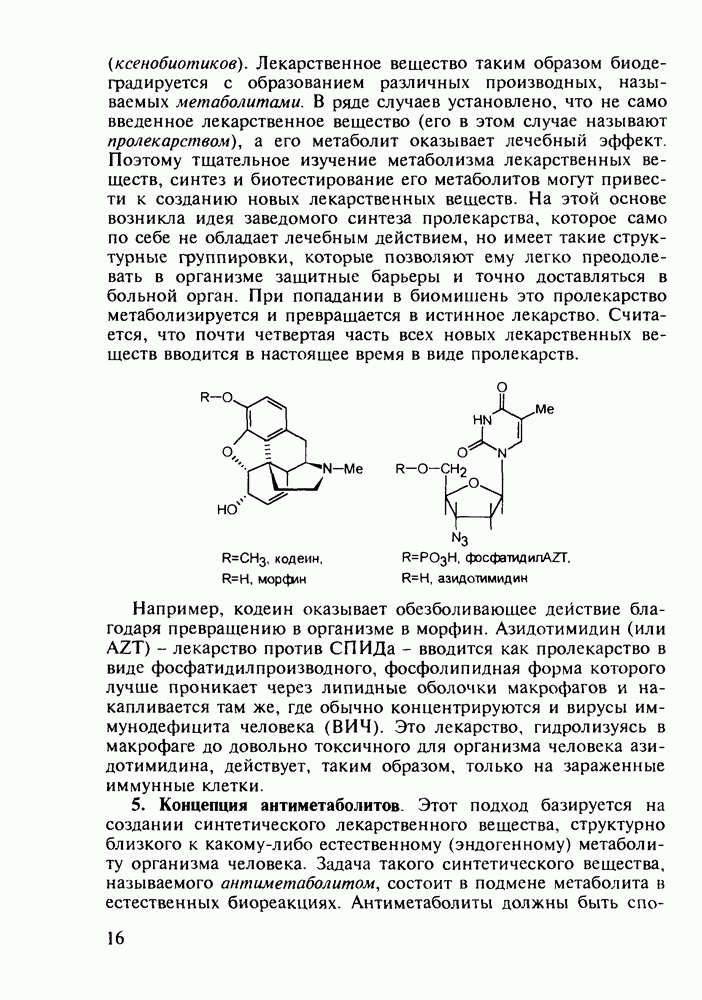















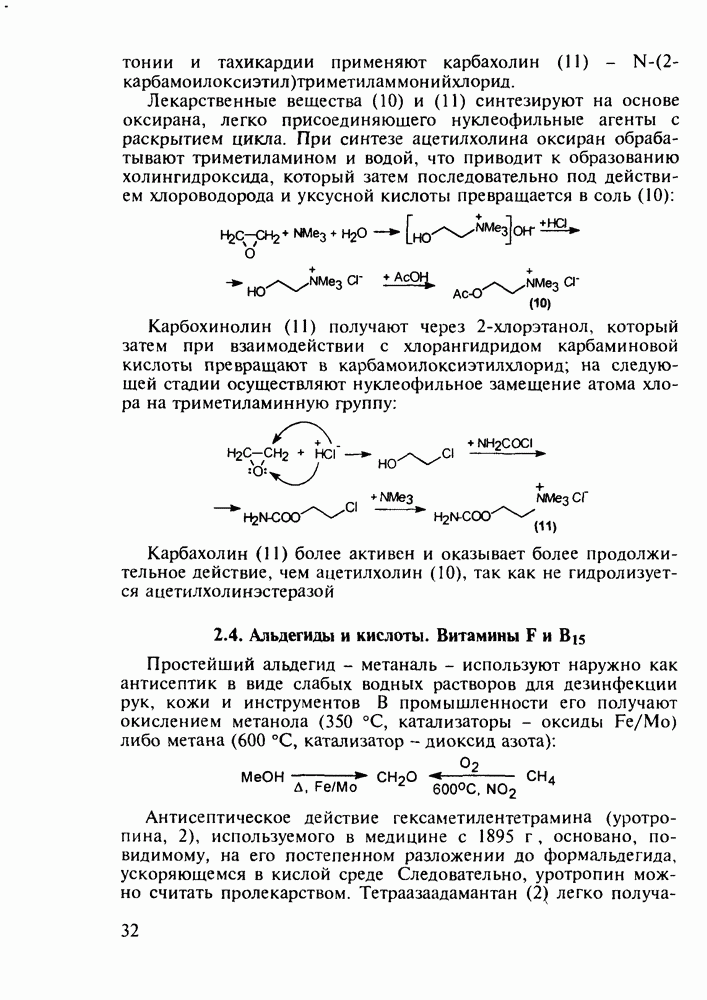




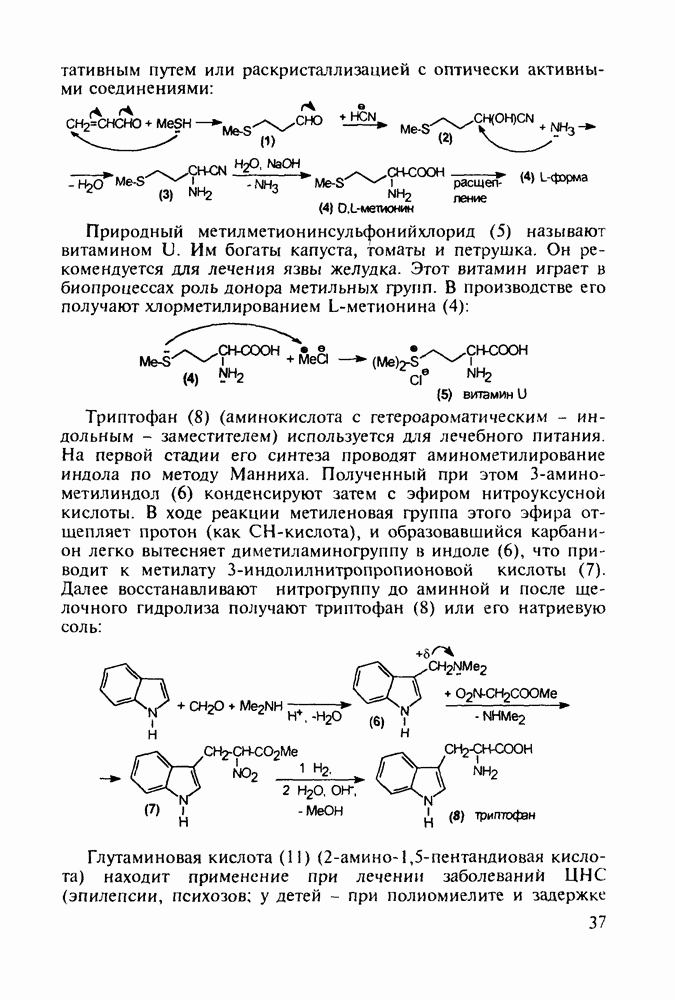









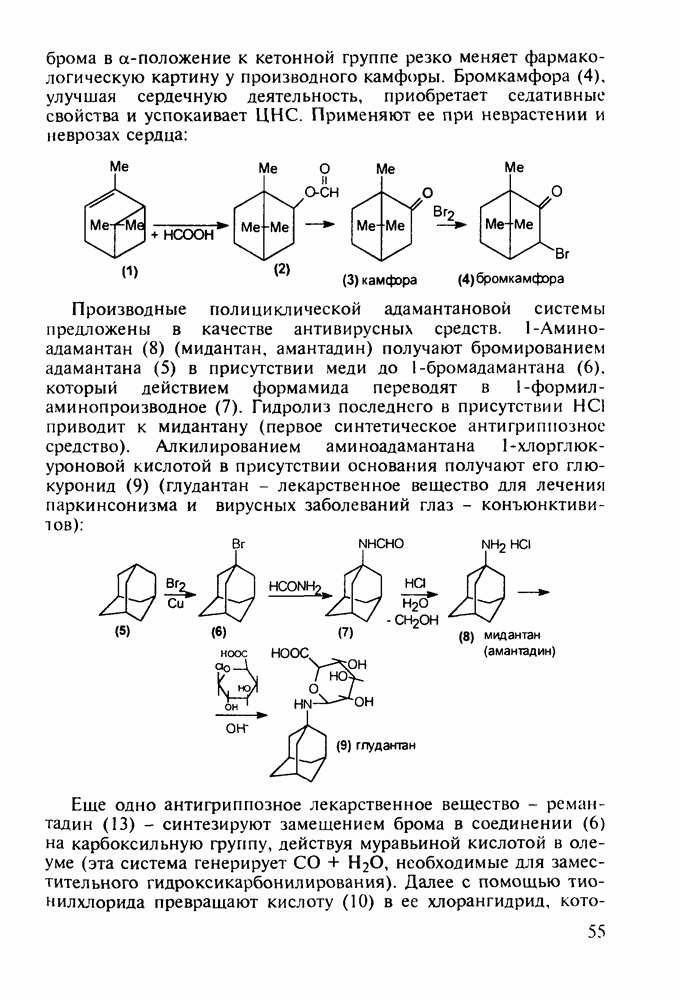
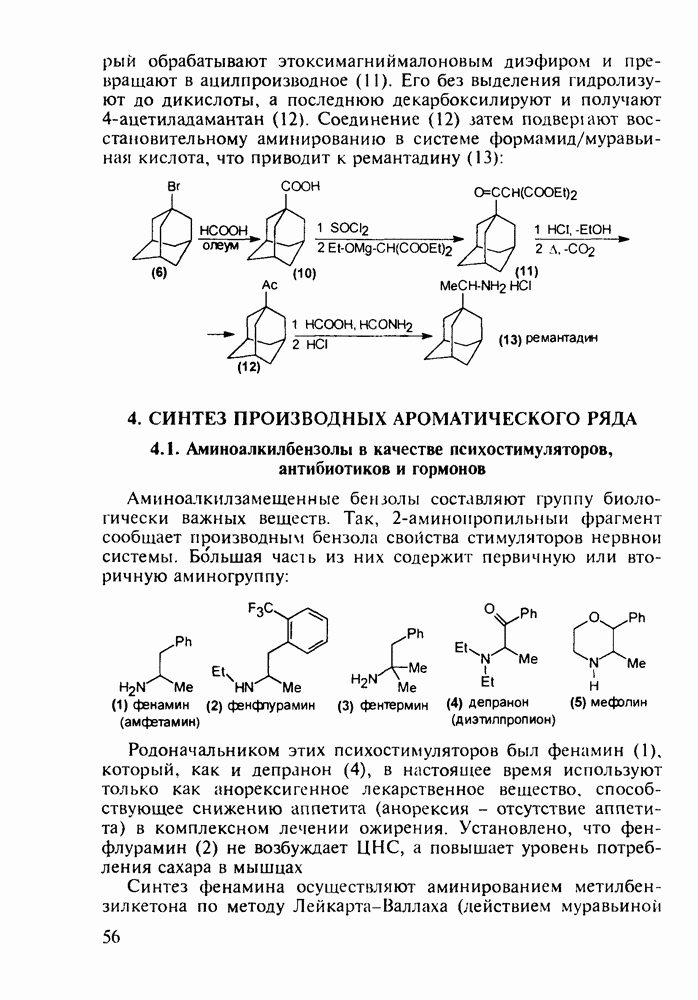


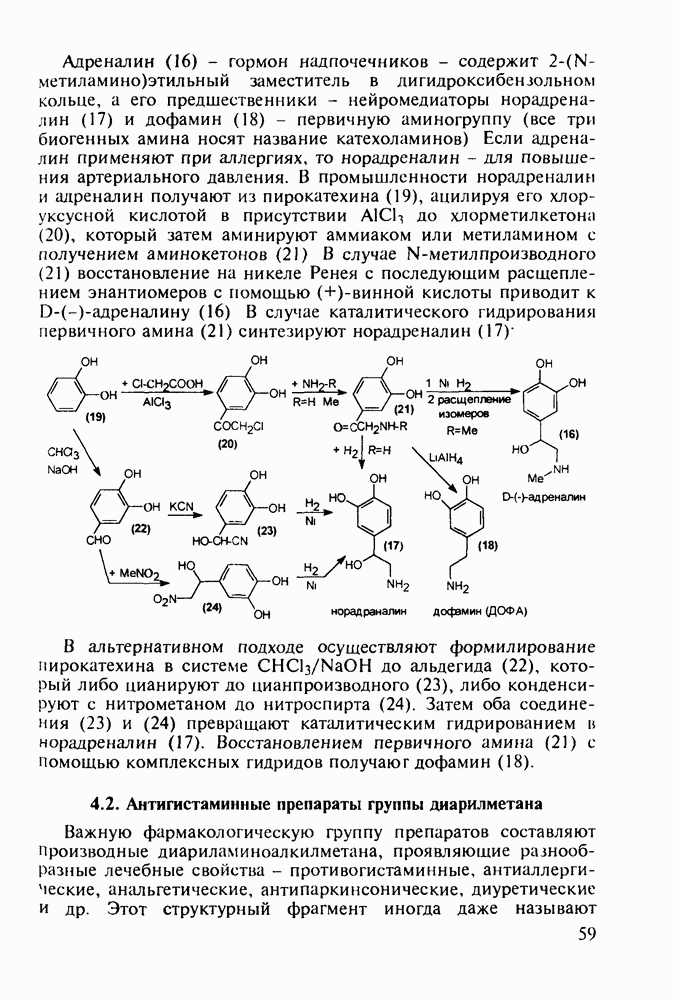
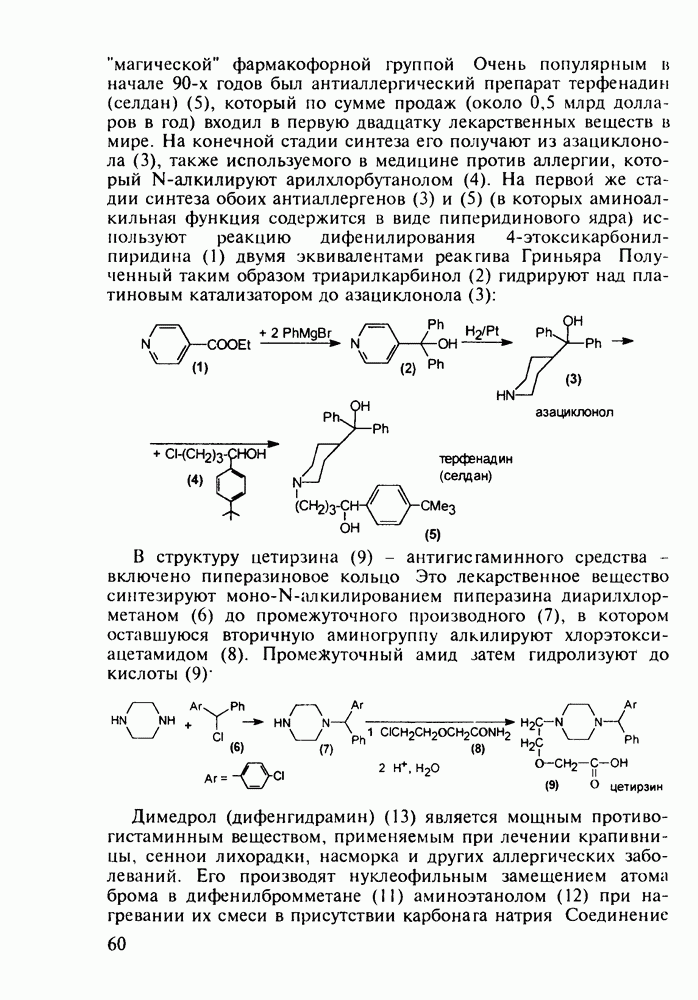

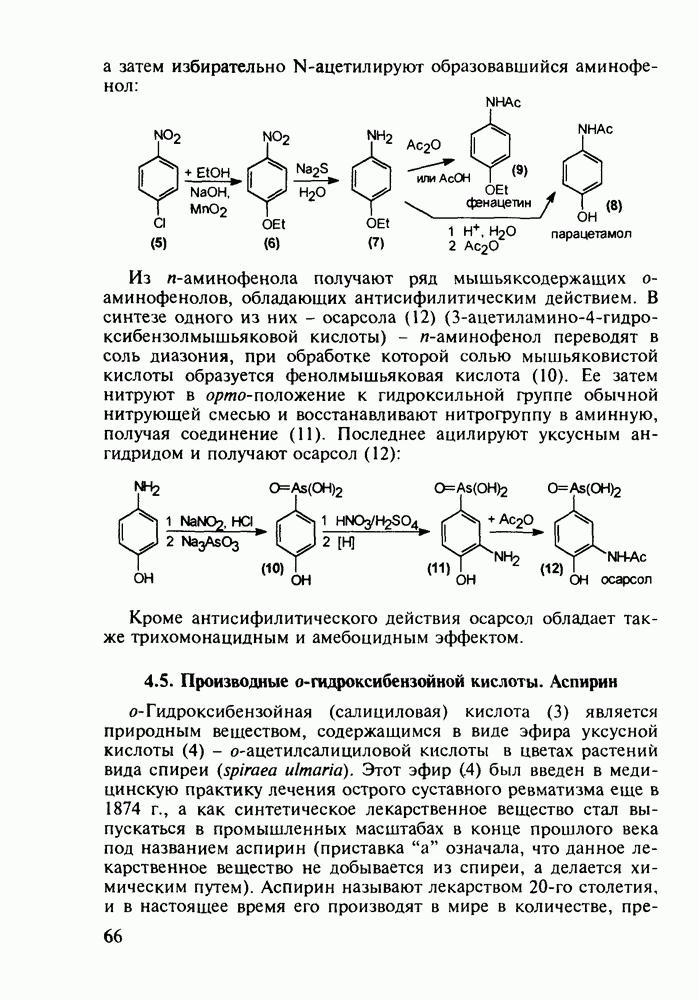
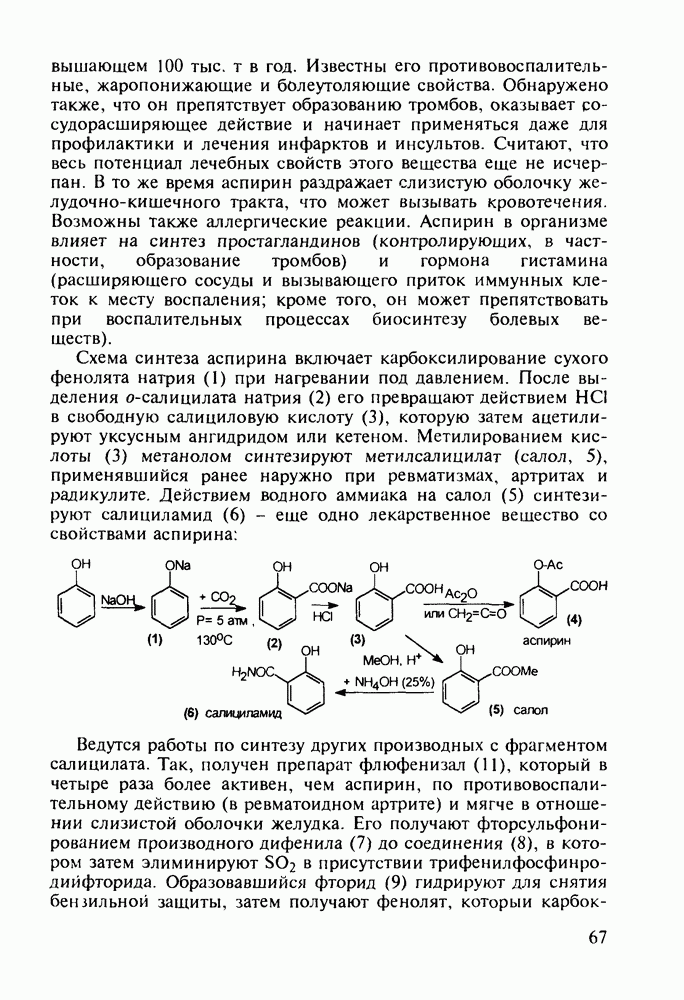
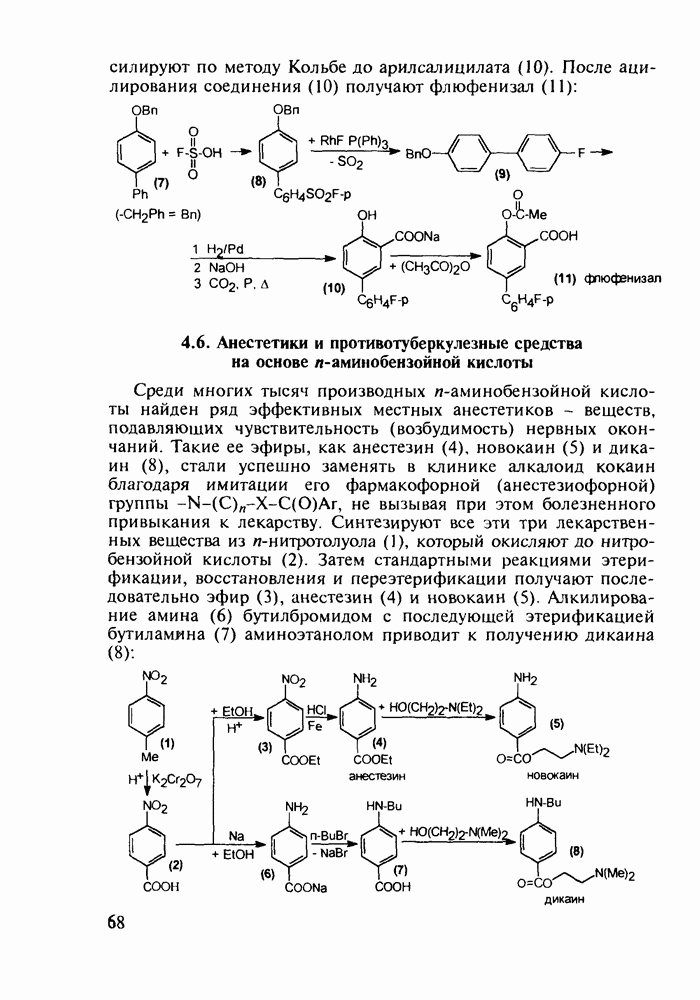
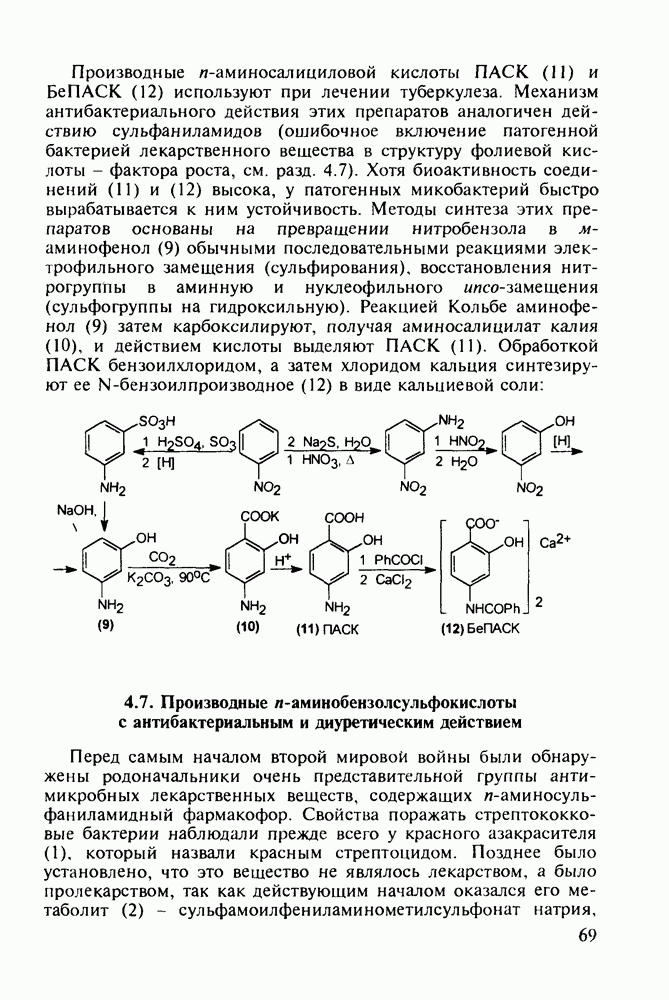








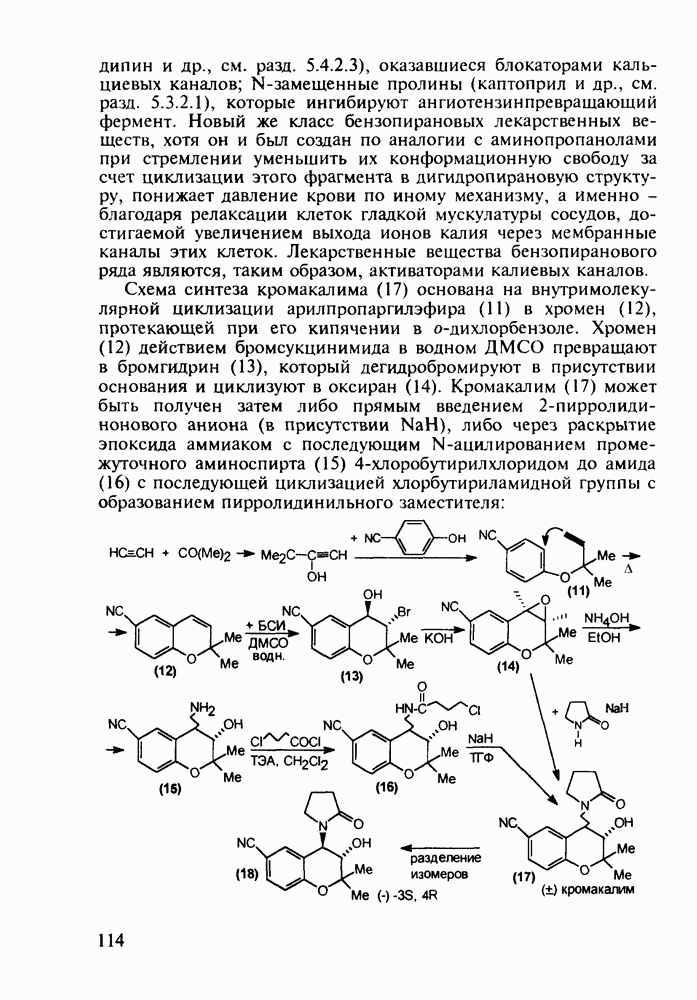



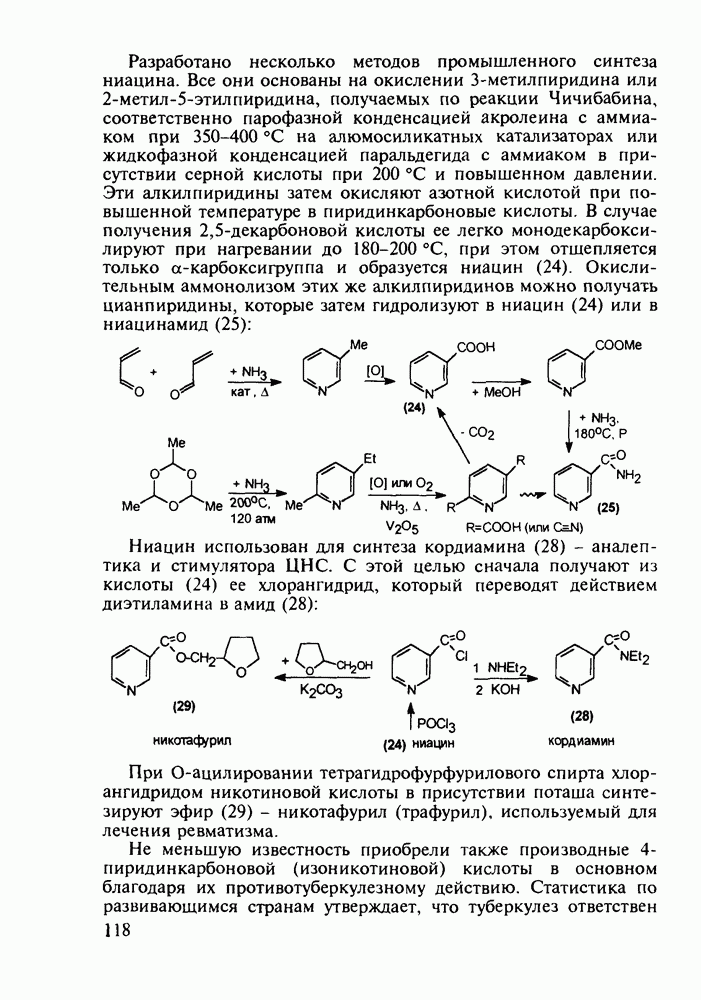
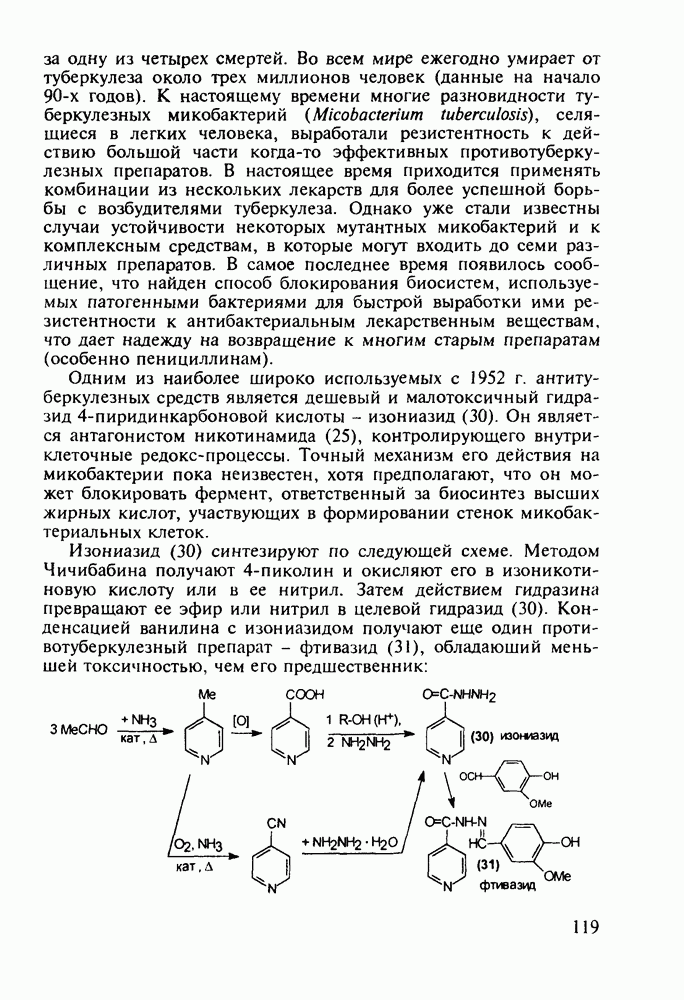
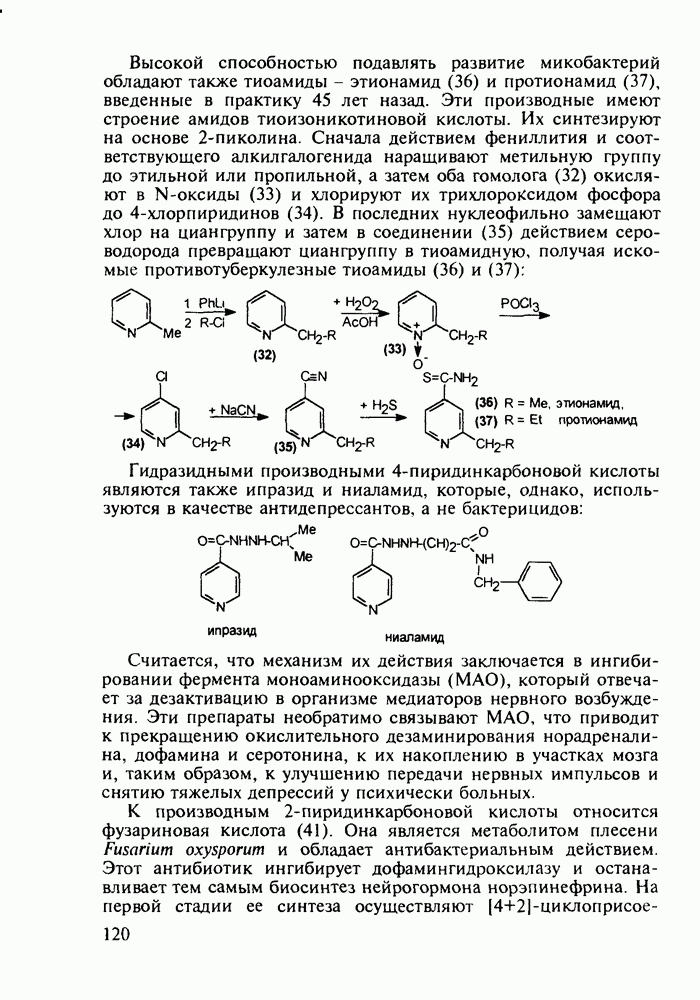



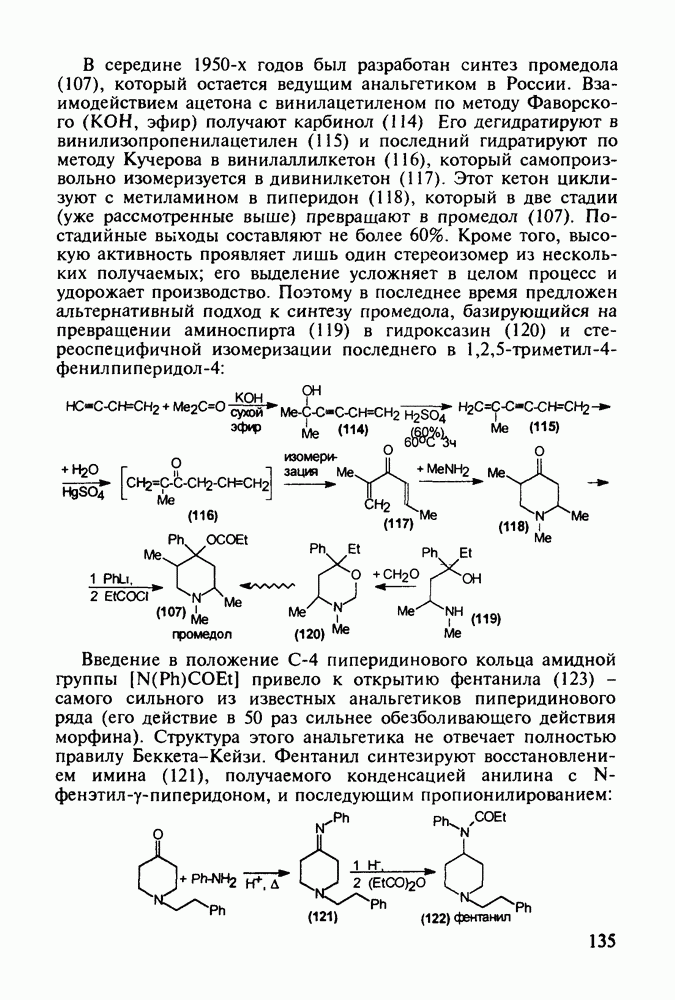
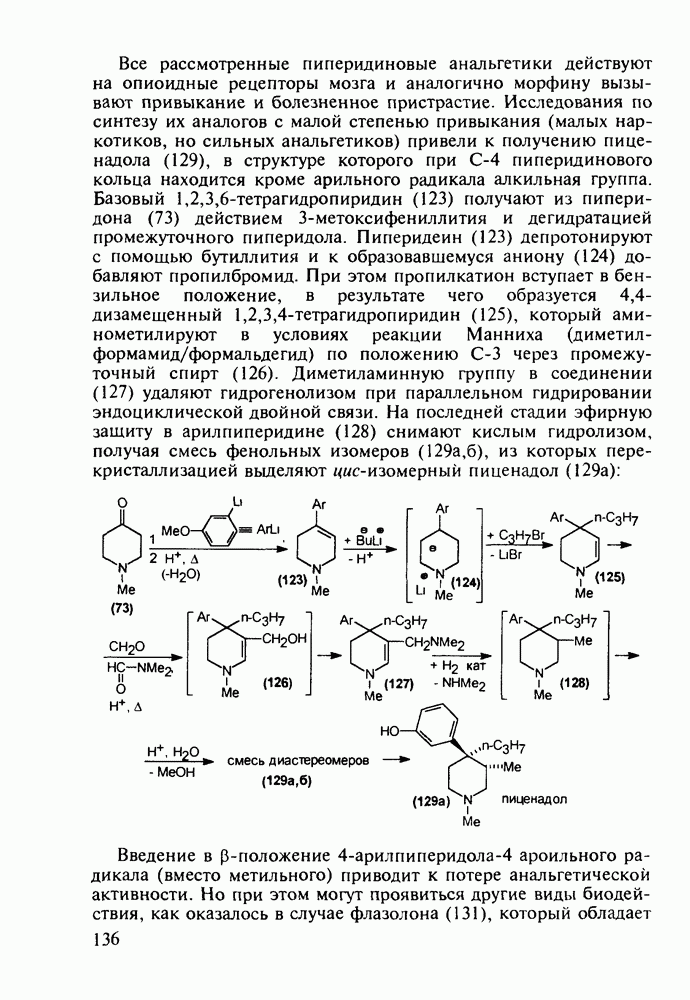
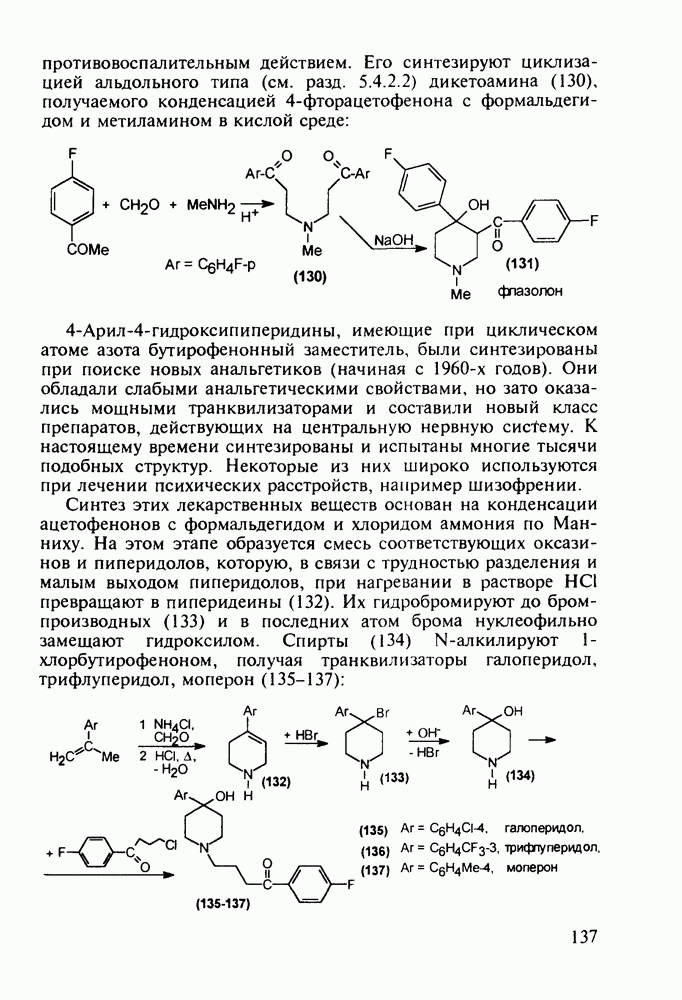
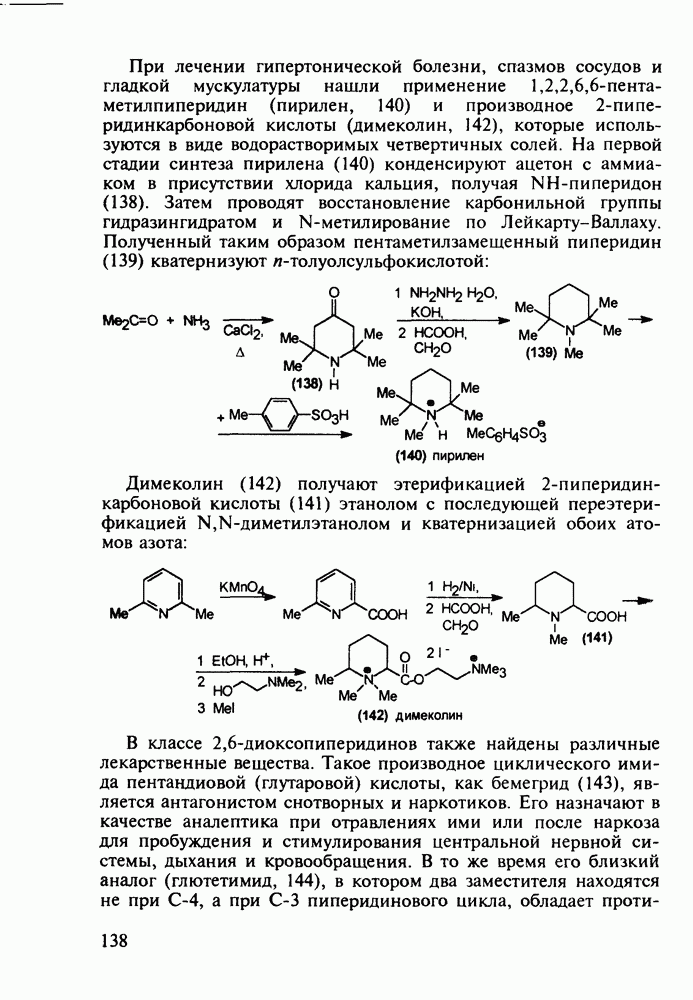
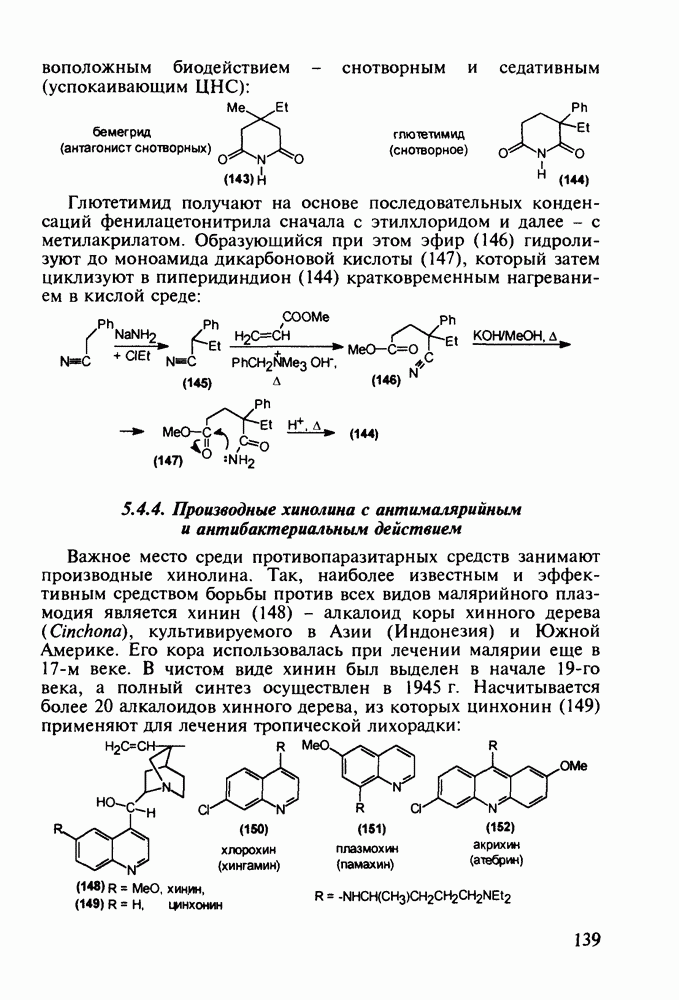
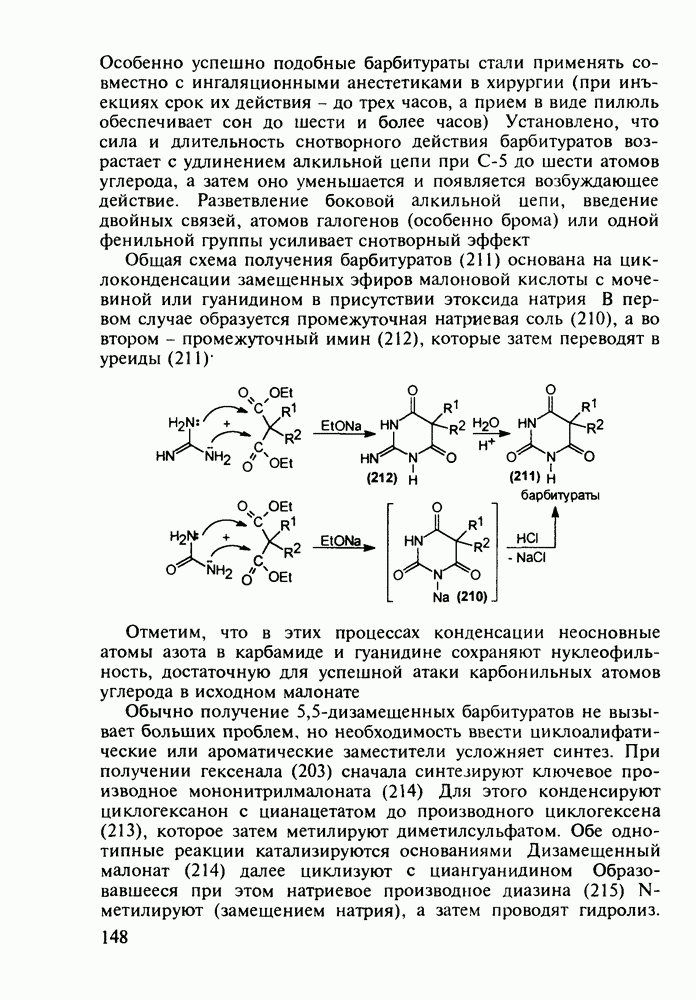










 до шести атомов углерода, а затем оно уменьшается и появляется возбуждающее действие. Разветвление боковой алкильной цепи, введение двойных связей, атомов галогенов (особенно брома) или одной фенильной группы усиливает снотворный эффект
до шести атомов углерода, а затем оно уменьшается и появляется возбуждающее действие. Разветвление боковой алкильной цепи, введение двойных связей, атомов галогенов (особенно брома) или одной фенильной группы усиливает снотворный эффект

 (замещением натрия), а затем проводят гидролиз.
(замещением натрия), а затем проводят гидролиз.


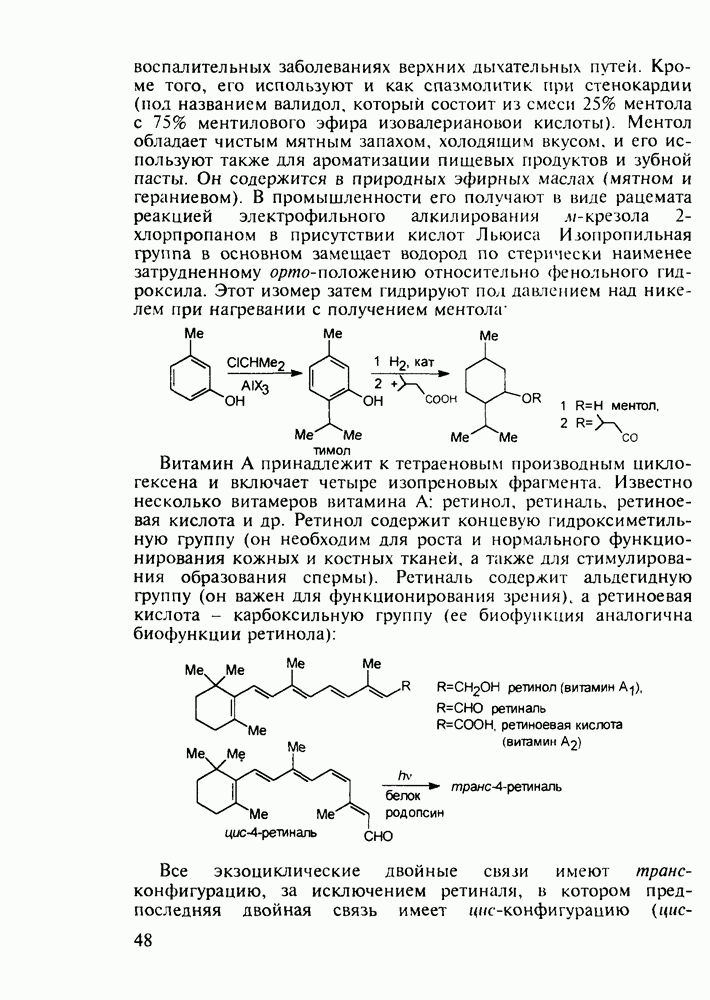
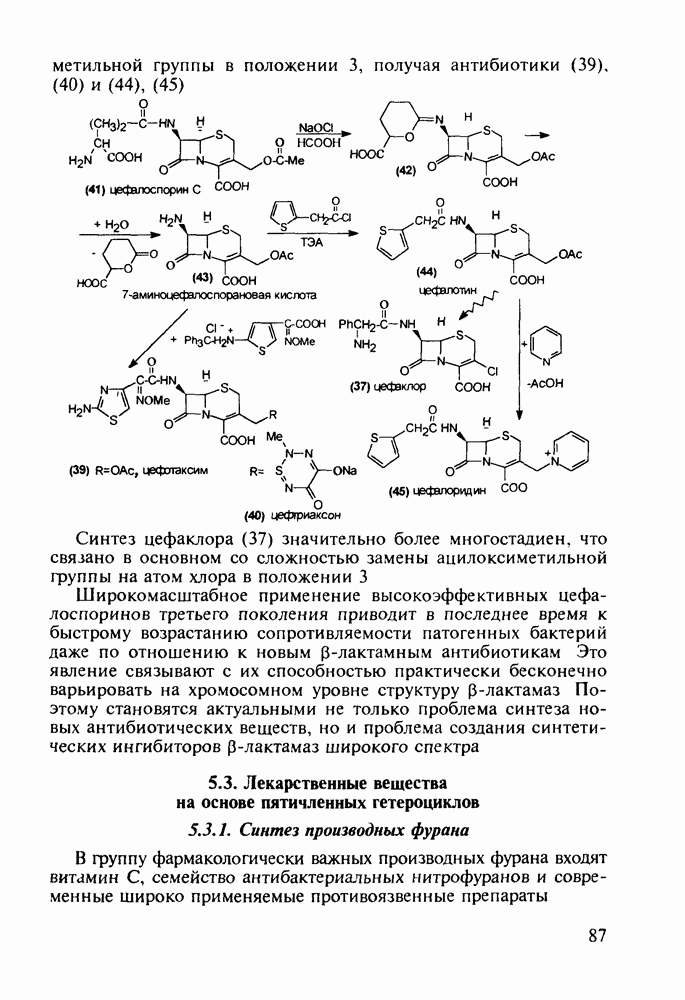
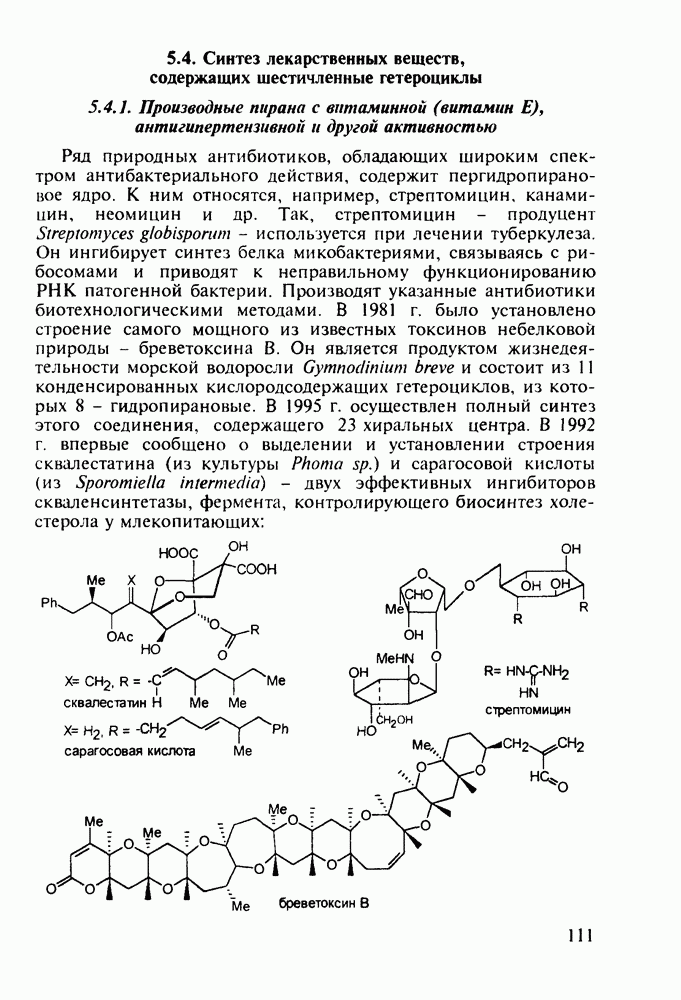
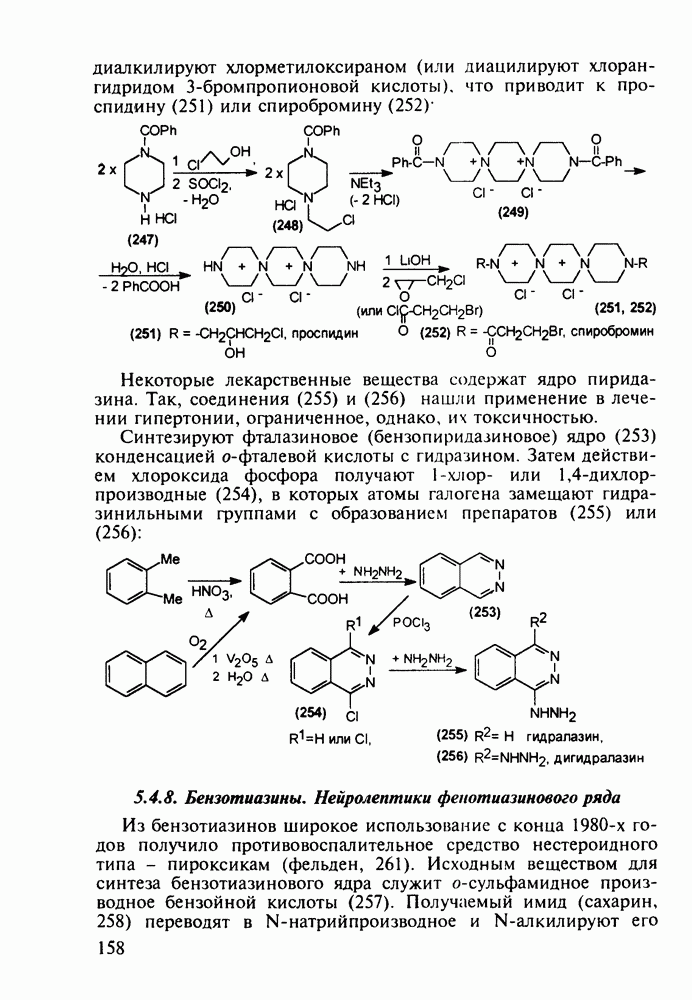
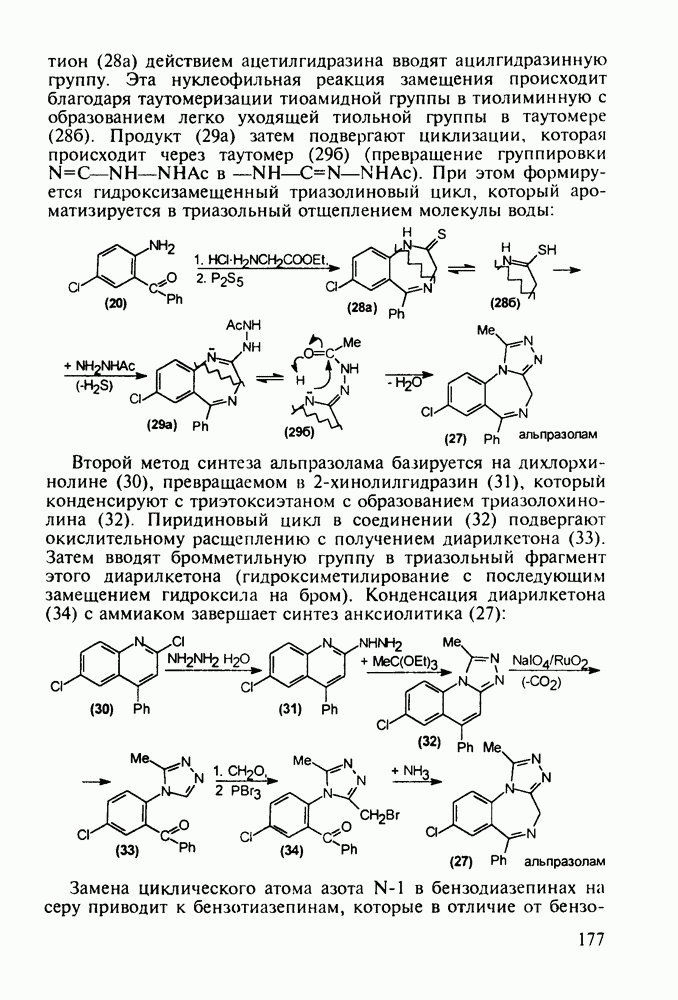
 Другая группа включает в себя природные вещества типа алкалоидов (стрепто-нигрин, см. разд. 5.4.4), антибиотиков (тетрациклины, см. разд. 4.8), гормональных веществ и ферментов. Третья группа - координационные соединения таких ионов металлов, как
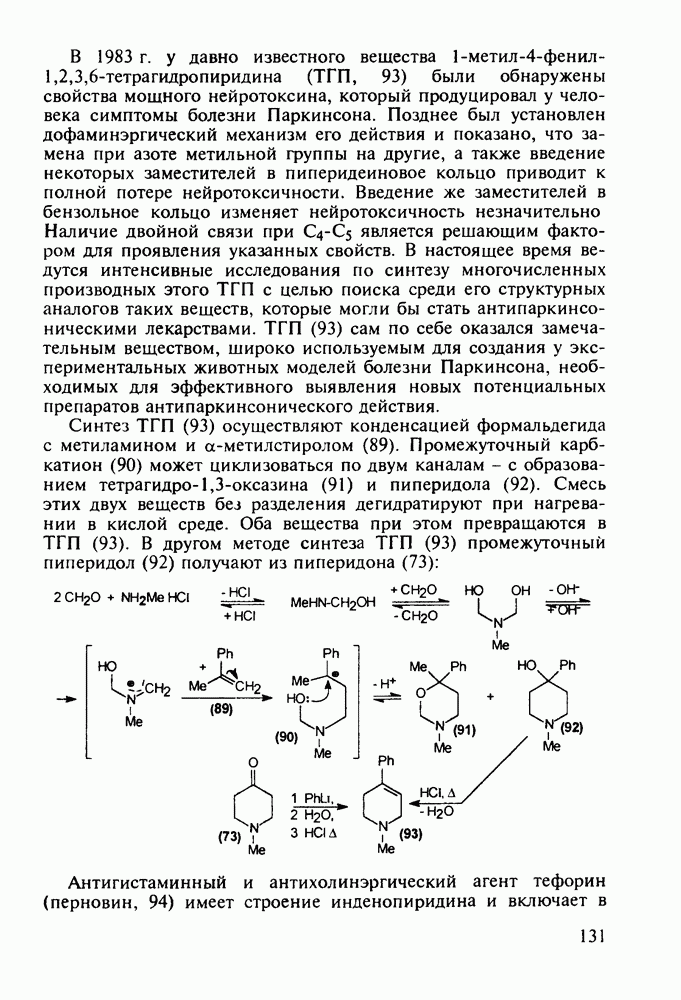
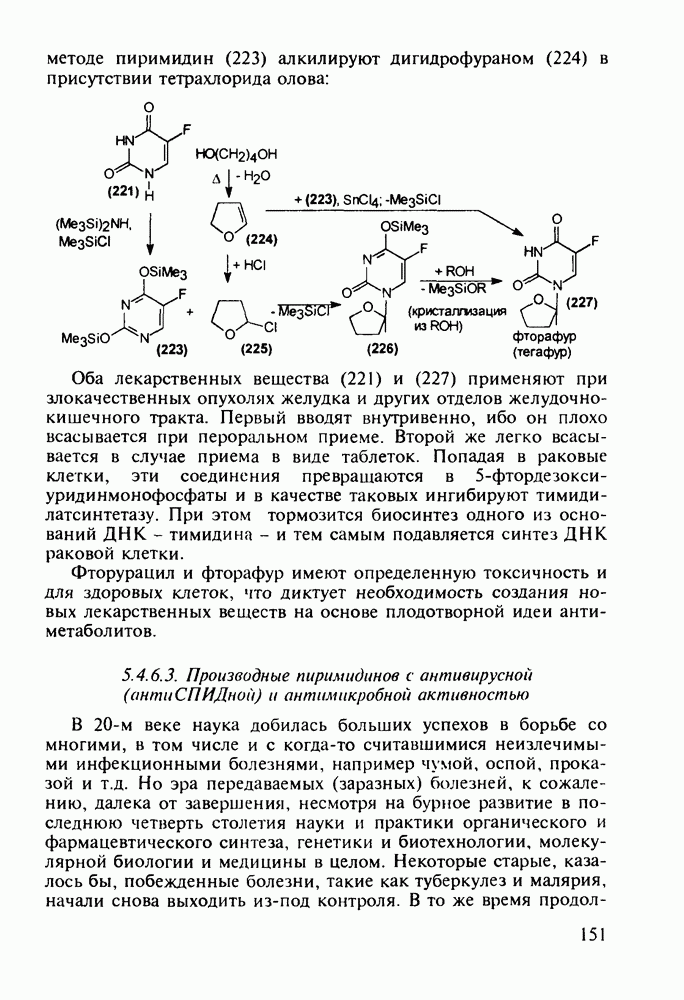
Другая группа включает в себя природные вещества типа алкалоидов (стрепто-нигрин, см. разд. 5.4.4), антибиотиков (тетрациклины, см. разд. 4.8), гормональных веществ и ферментов. Третья группа - координационные соединения таких ионов металлов, как  цисплатин, карбоплатин); производные порфиринов и тетраазапорфиринов, используемых в качестве сенсоров в фотодинамической терапии рака (см. разд. 5.3.2.3). В четвертую группу входят вещества антиметаболитного действия - производные урацила (фторафур и др.), замещенные пурины (меркаптопурин, см. разд. 5.4.9) и птеридины (метотрексат, см. разд. 5.4.11). Первым противоопухолевым препаратом диоксопиримидинового ряда был 5-фторурацил (221). Пиримидиновое ядро в его синтезе конструируют циклоконденсацией эфира натрийоксометиленфторуксусной кислоты (218) с S-метилизотиомочевиной (219). После гидролиза метилтиольной группы в пиримидине (220) получают синтетический антиметаболит (221):
цисплатин, карбоплатин); производные порфиринов и тетраазапорфиринов, используемых в качестве сенсоров в фотодинамической терапии рака (см. разд. 5.3.2.3). В четвертую группу входят вещества антиметаболитного действия - производные урацила (фторафур и др.), замещенные пурины (меркаптопурин, см. разд. 5.4.9) и птеридины (метотрексат, см. разд. 5.4.11). Первым противоопухолевым препаратом диоксопиримидинового ряда был 5-фторурацил (221). Пиримидиновое ядро в его синтезе конструируют циклоконденсацией эфира натрийоксометиленфторуксусной кислоты (218) с S-метилизотиомочевиной (219). После гидролиза метилтиольной группы в пиримидине (220) получают синтетический антиметаболит (221):

 (применяют при лечении сердечно-сосудистых заболеваний и как анаболик при нарушениях белкового обмена).
(применяют при лечении сердечно-сосудистых заболеваний и как анаболик при нарушениях белкового обмена).


 -трифосфатов и в этой форме являются конкурентными ингибиторами обратной транс-криптазы ВИЧ (ОТ ВИЧ), которая катализирует транскрипцию одноцепочечной вирусной РНК в двухцепочечную ДНК. Являясь пролекарствами, нуклеозидные антиСПИДные препараты воздействуют на ферментную биомишень и оказываются терминаторами роста цепи вирусной ДНК, что приводит, таким образом, к остановке размножения ВИЧ. Замечено также, что указанные
-трифосфатов и в этой форме являются конкурентными ингибиторами обратной транс-криптазы ВИЧ (ОТ ВИЧ), которая катализирует транскрипцию одноцепочечной вирусной РНК в двухцепочечную ДНК. Являясь пролекарствами, нуклеозидные антиСПИДные препараты воздействуют на ферментную биомишень и оказываются терминаторами роста цепи вирусной ДНК, что приводит, таким образом, к остановке размножения ВИЧ. Замечено также, что указанные  -трифосфаты могут ингибировать ДНК-полимеразы клетки-хозяина и давать значительные токсические эффекты. При длительном использовании препаратов появляются резистентные к ним штаммы ВИЧ, в которых репликация на уровне НК уже не нарушается. Недавно установлено, что длительное использование AZX (228) вызывает мутации в активном центре ОТ ВИЧ в кодонах
-трифосфаты могут ингибировать ДНК-полимеразы клетки-хозяина и давать значительные токсические эффекты. При длительном использовании препаратов появляются резистентные к ним штаммы ВИЧ, в которых репликация на уровне НК уже не нарушается. Недавно установлено, что длительное использование AZX (228) вызывает мутации в активном центре ОТ ВИЧ в кодонах  (он переходит в кодон
(он переходит в кодон  ), Lys 70 (мутирует в
), Lys 70 (мутирует в  )
)  (дает кодон
(дает кодон  ). Таким образом, мутированная ОТ ВИЧ уже не подвержена ингибированию азидовудином (228).
). Таким образом, мутированная ОТ ВИЧ уже не подвержена ингибированию азидовудином (228).
 дезоксирибозы. Образовавшийся полицикл (233) обрабатывают азидом натрия в воде. Сильный нуклеофил
дезоксирибозы. Образовавшийся полицикл (233) обрабатывают азидом натрия в воде. Сильный нуклеофил  атакует электронодефицитный атом
атакует электронодефицитный атом  рибозы со стороны, обратной атому кислорода (снизу). При этом раскрывается эфирная связь, и возникающая в молекуле (228) азидная группа оказывается в стереохимически прежнем (в отношении к исходному гидроксилу) положении:
рибозы со стороны, обратной атому кислорода (снизу). При этом раскрывается эфирная связь, и возникающая в молекуле (228) азидная группа оказывается в стереохимически прежнем (в отношении к исходному гидроксилу) положении:

 -дидезоксиинозин (ДДИ, 234). Механизм его биодействия, токсичность и стимулирование мутаций ОТ ВИЧ аналогичны таковым у рассмотренных выше производных пиримидина (228)-(230).
-дидезоксиинозин (ДДИ, 234). Механизм его биодействия, токсичность и стимулирование мутаций ОТ ВИЧ аналогичны таковым у рассмотренных выше производных пиримидина (228)-(230).


 -хлорфенилацетонитрила его последонательной конденсацией (по его активной метиленовой группе) сначала с этилпропионатом до кетона (240), а затем с диазометаном. Полученный цианвиниловый эфир (241) далее циклизуют с гуанидином:
-хлорфенилацетонитрила его последонательной конденсацией (по его активной метиленовой группе) сначала с этилпропионатом до кетона (240), а затем с диазометаном. Полученный цианвиниловый эфир (241) далее циклизуют с гуанидином:


