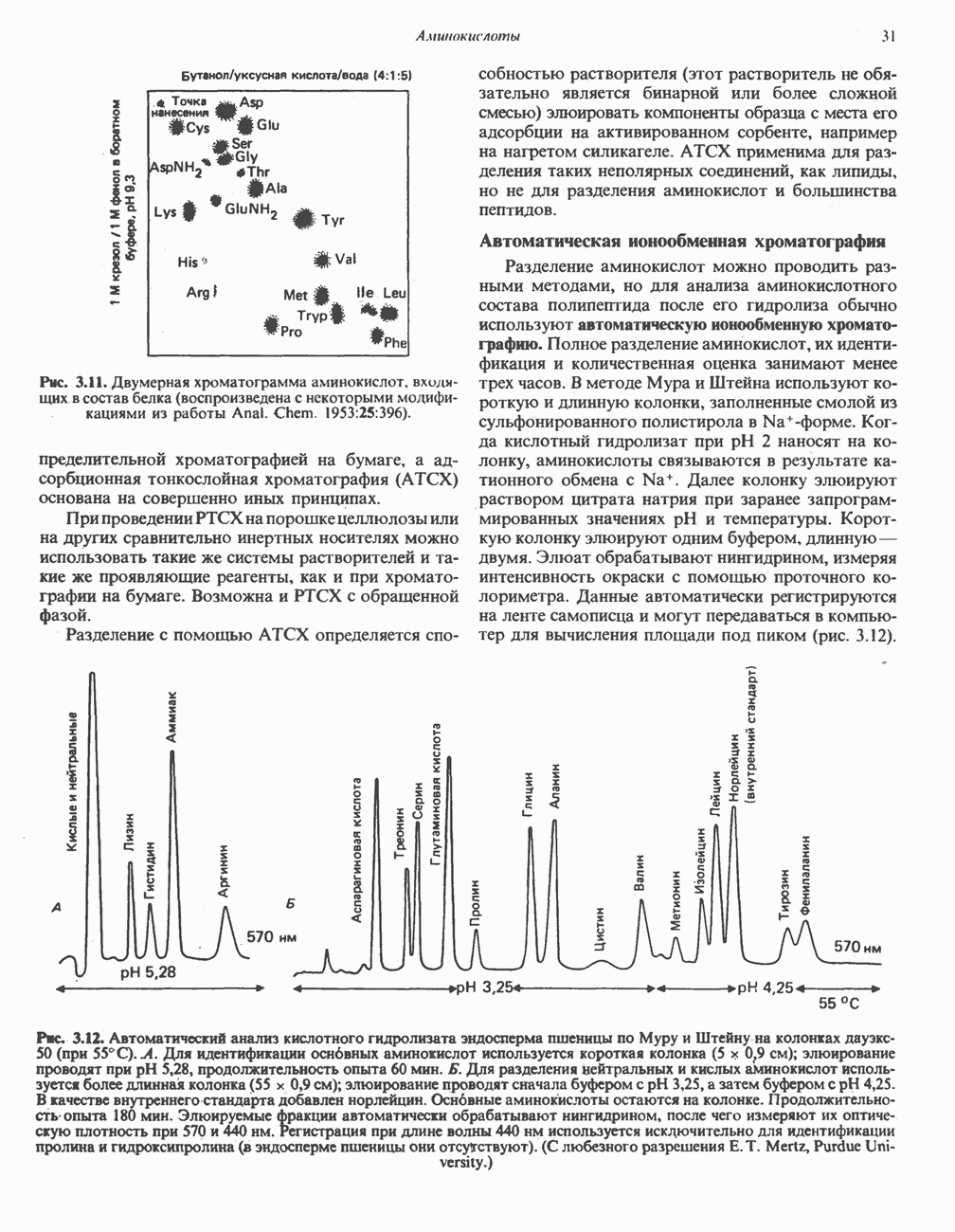
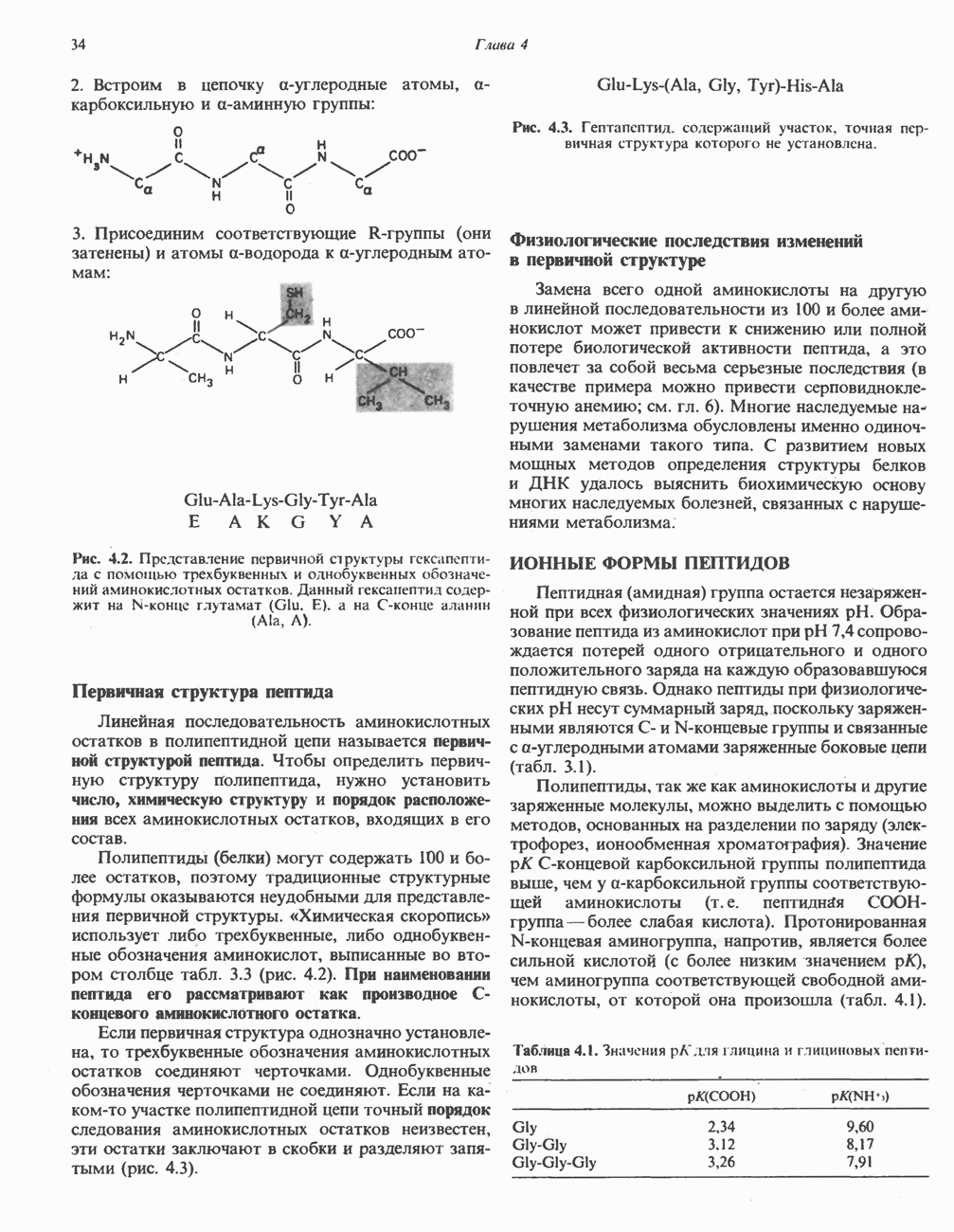
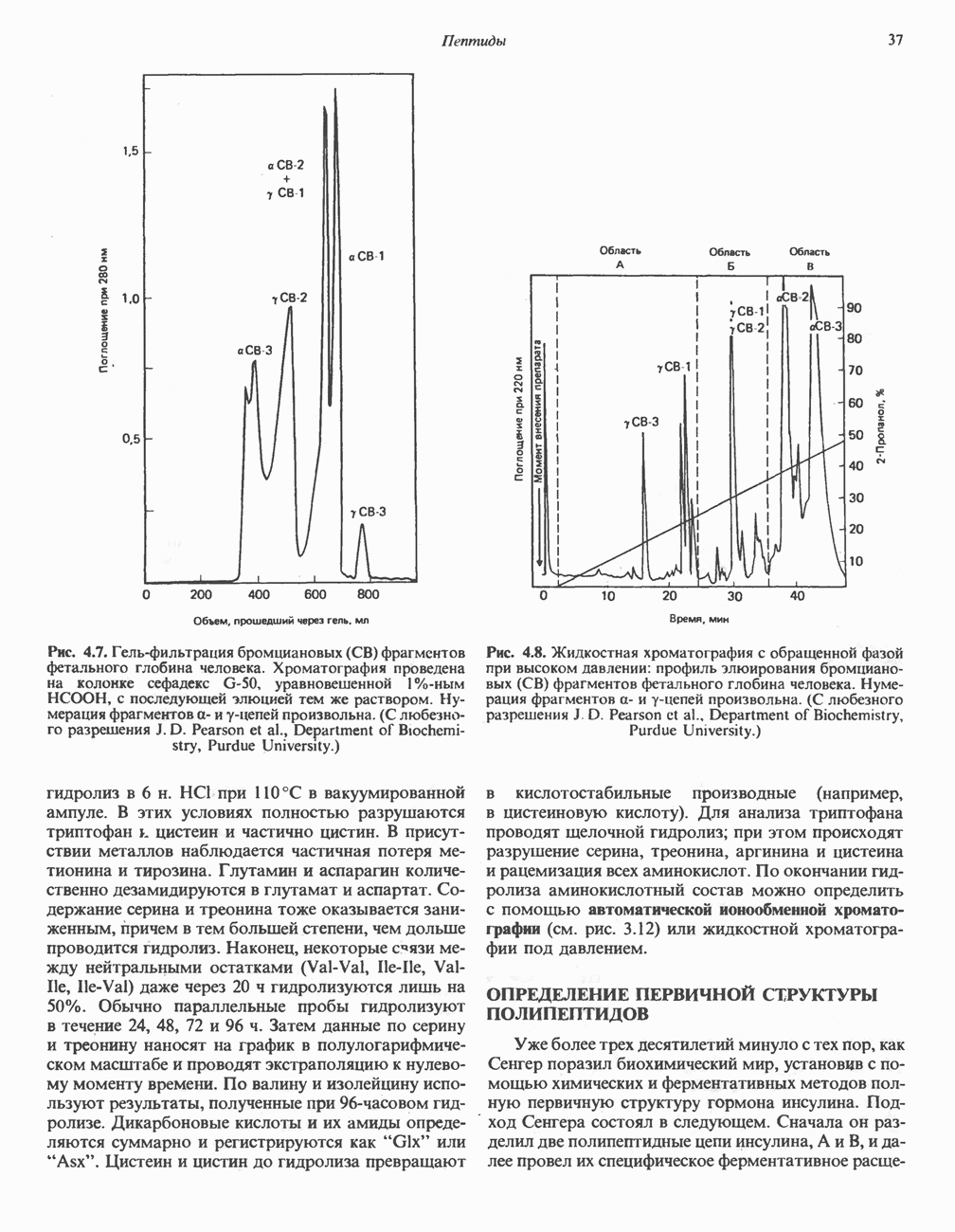
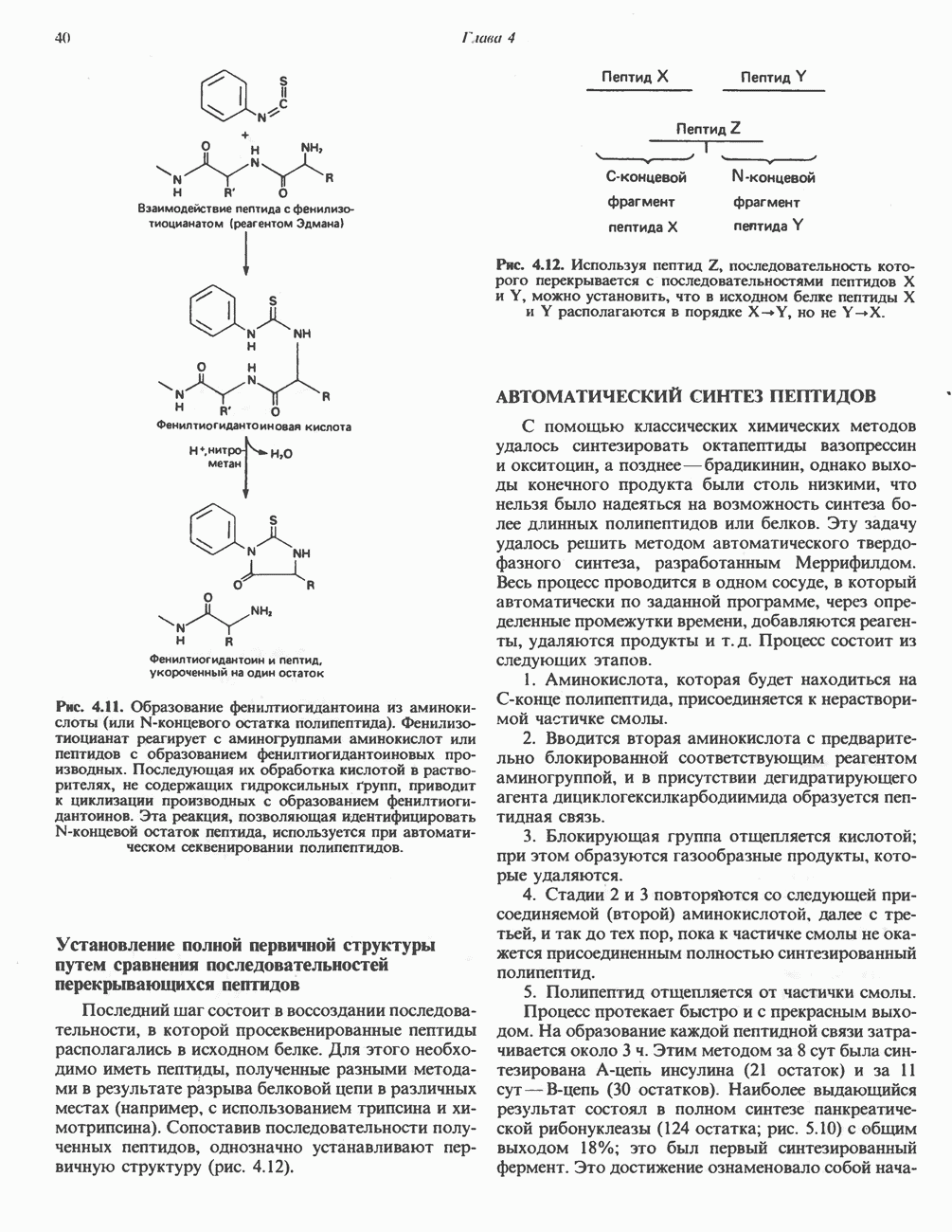
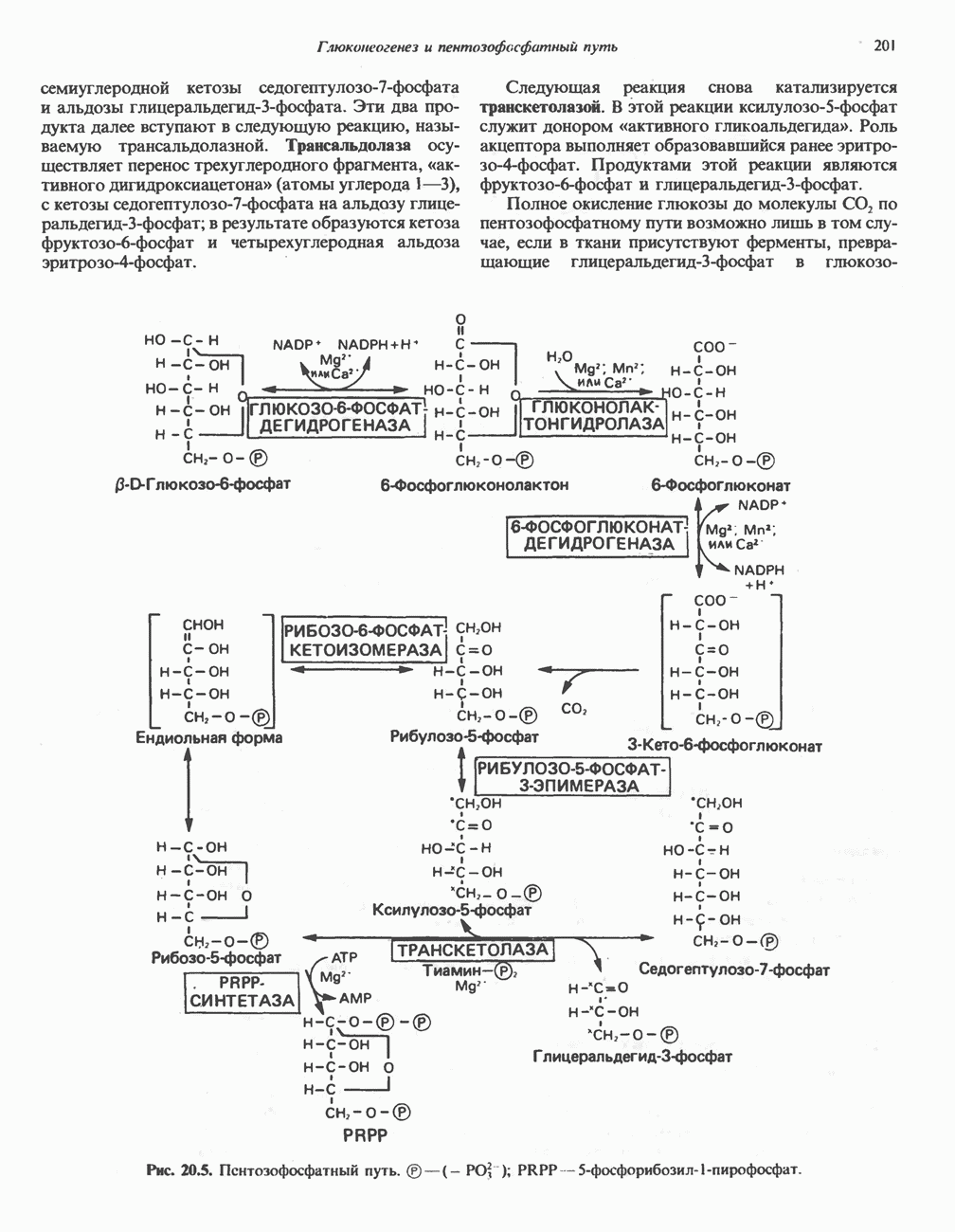
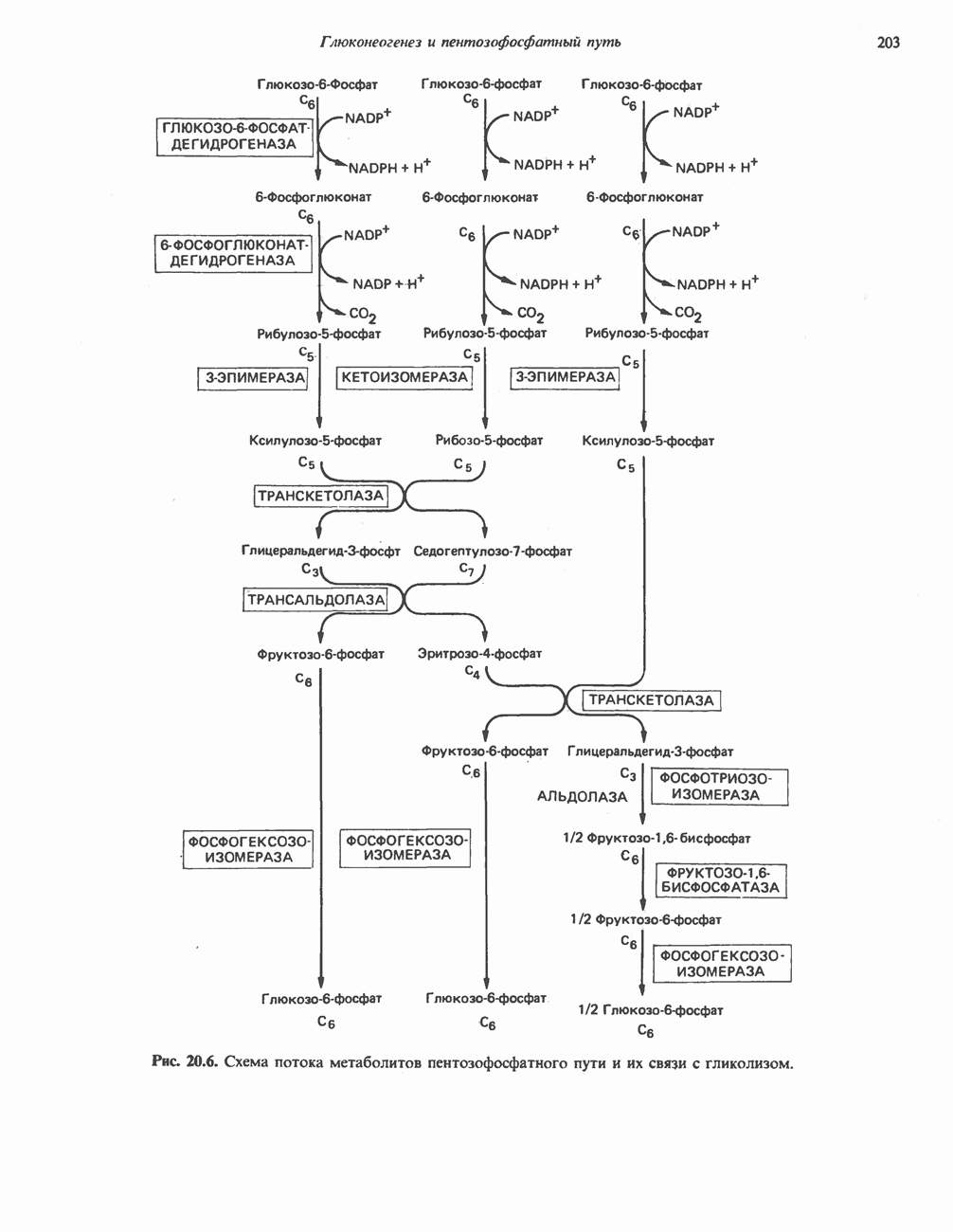
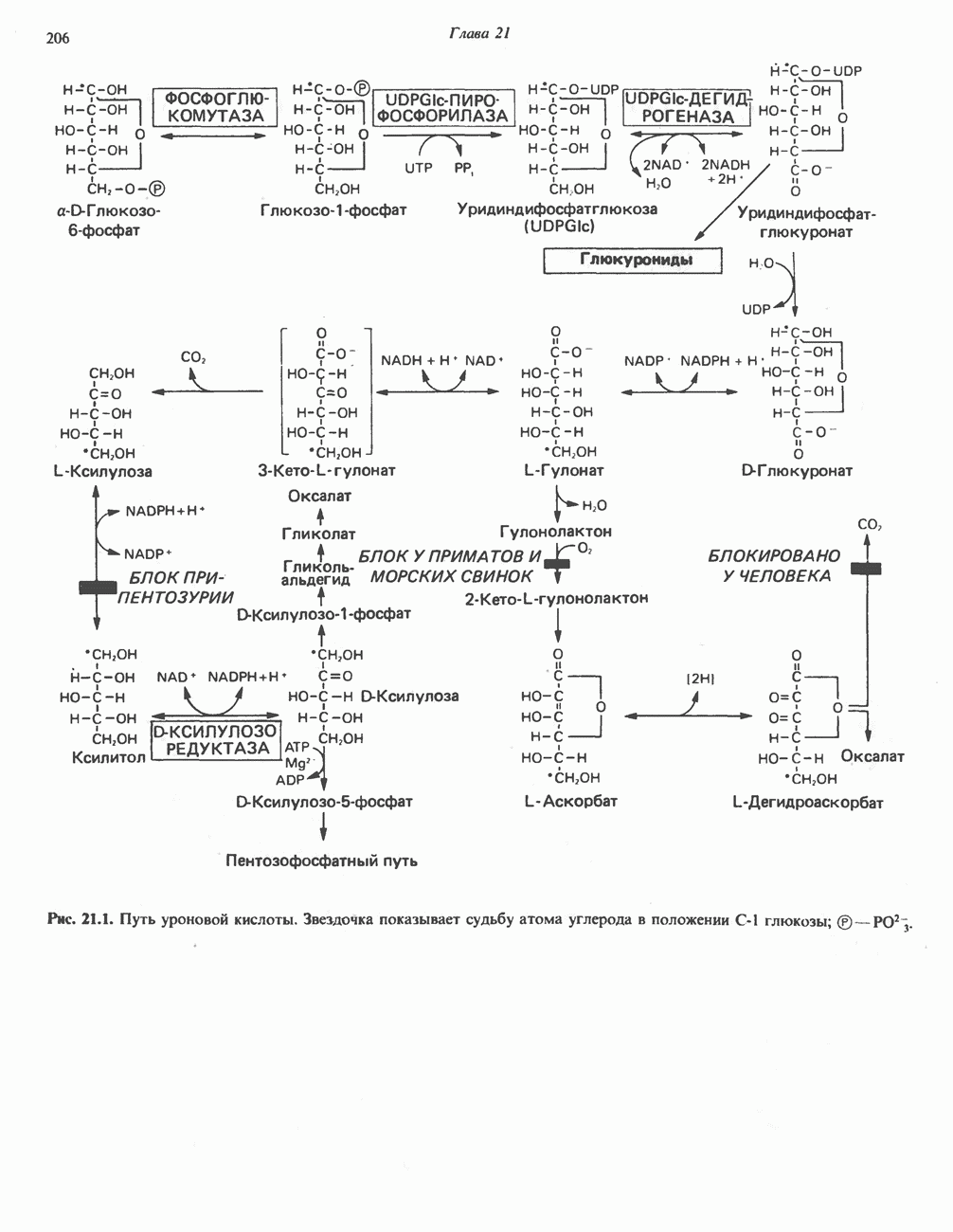
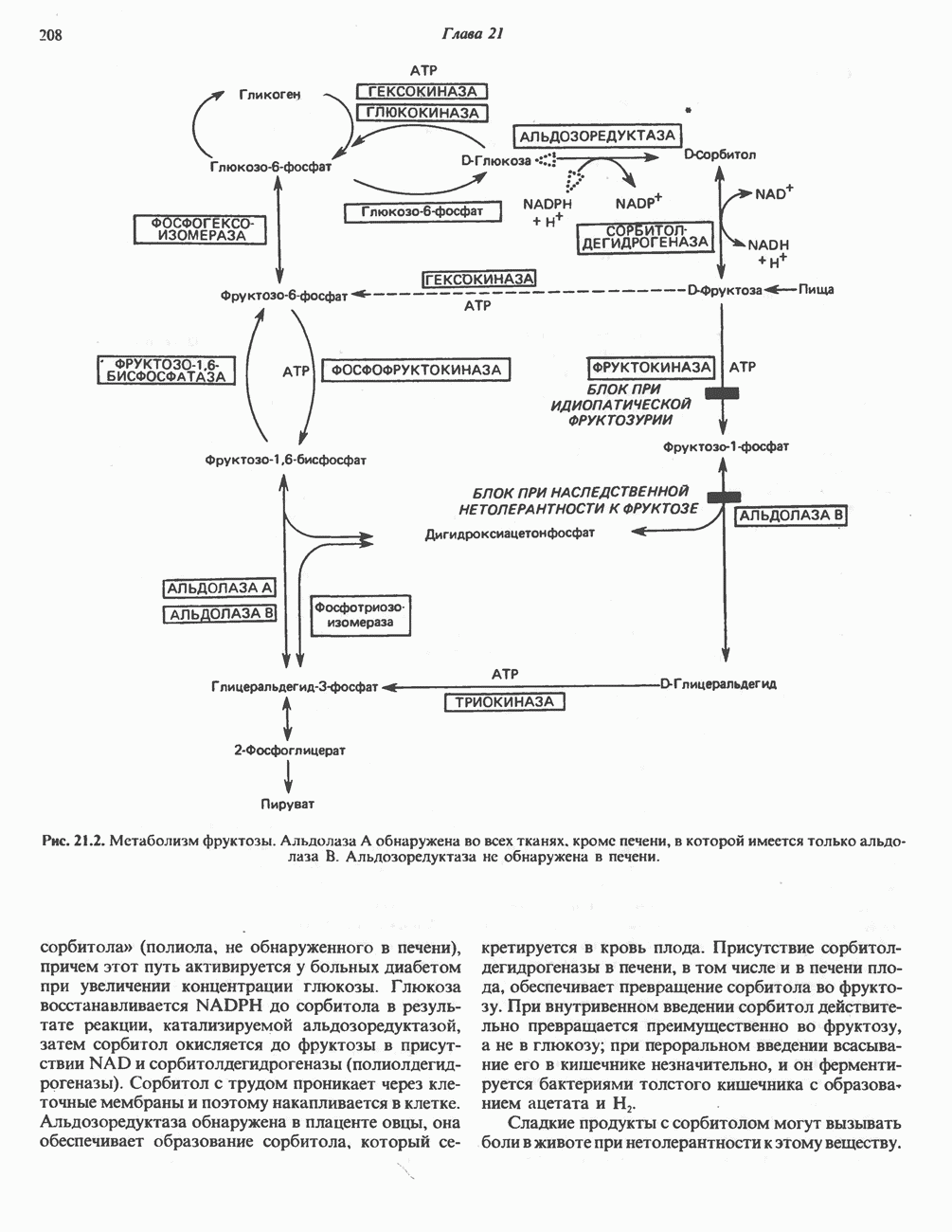
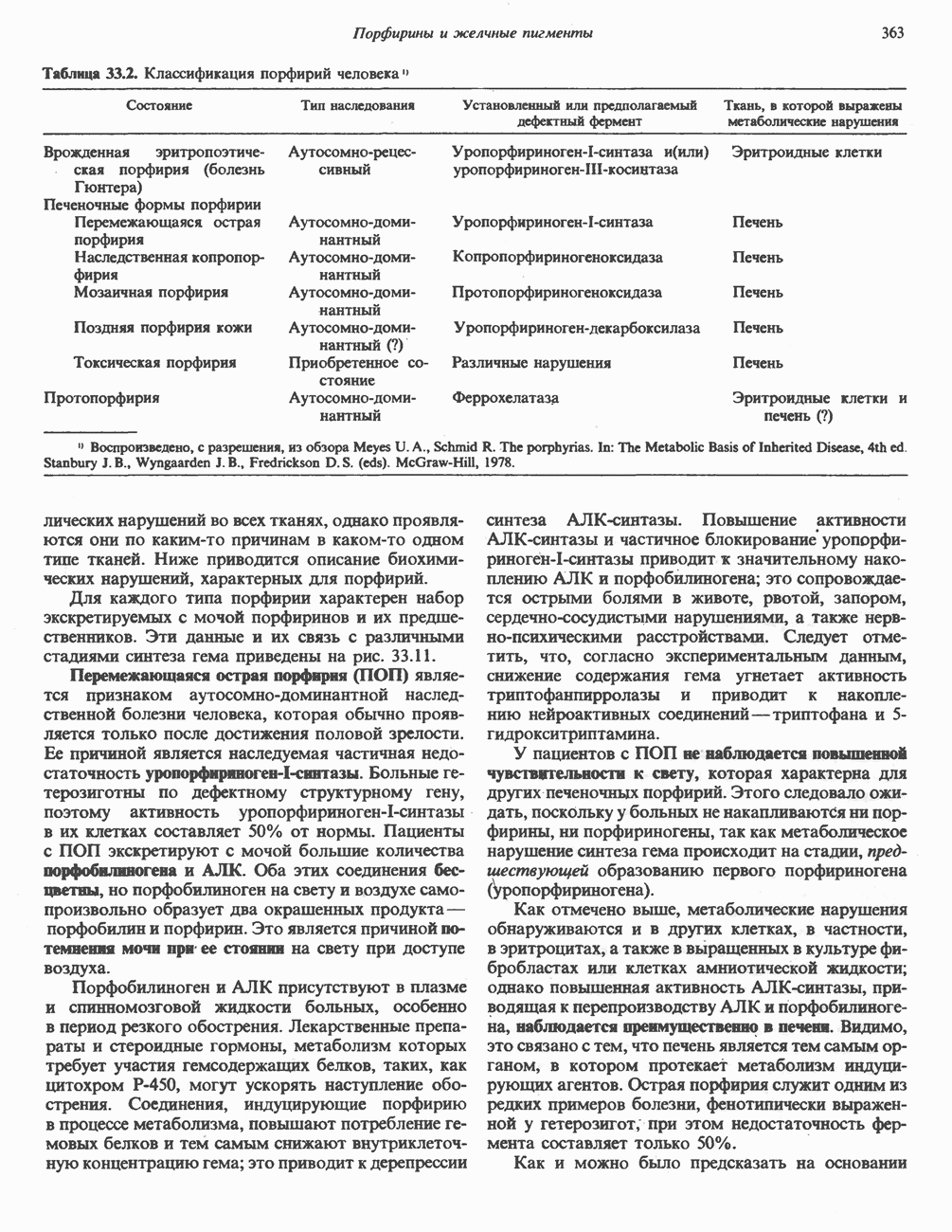
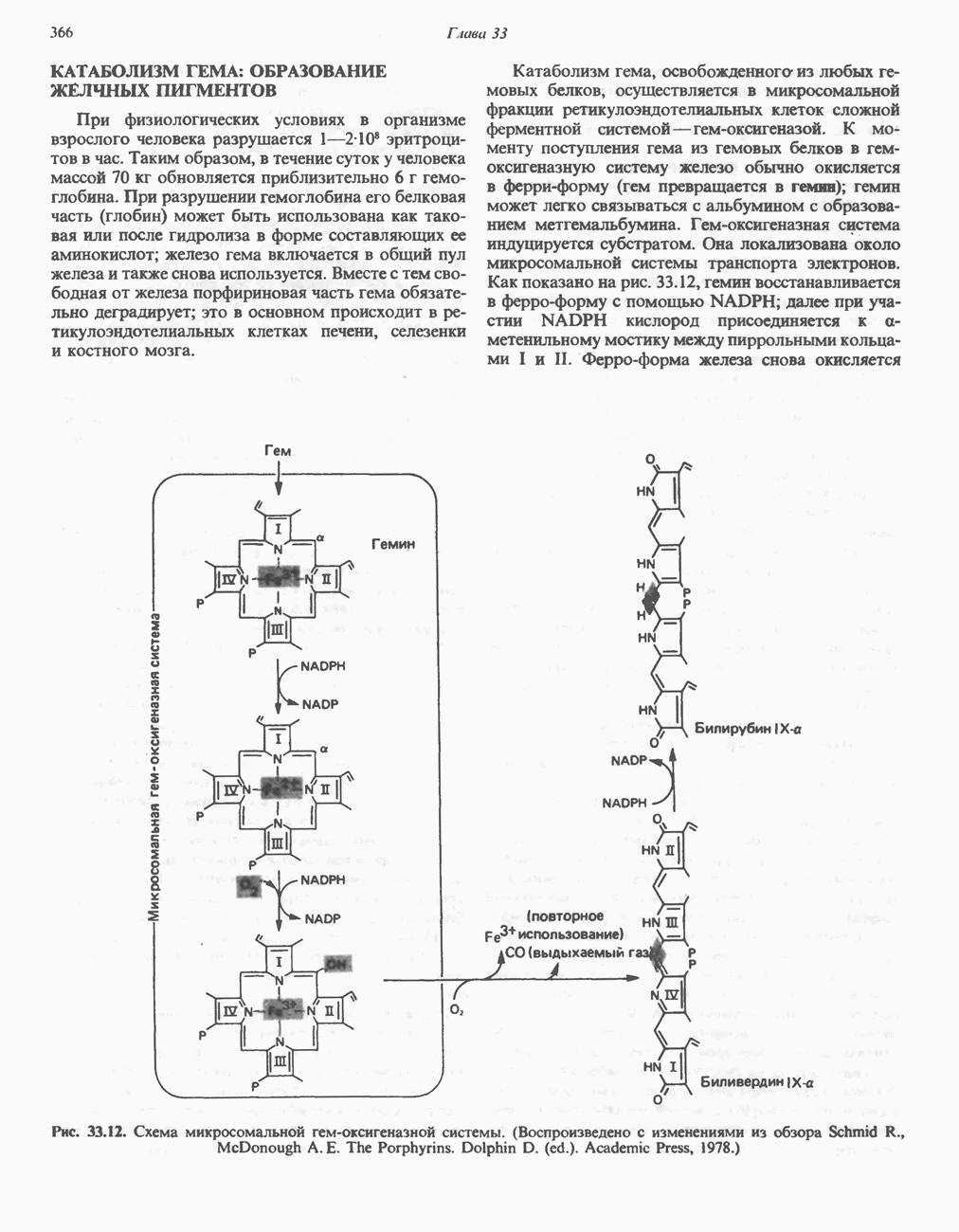
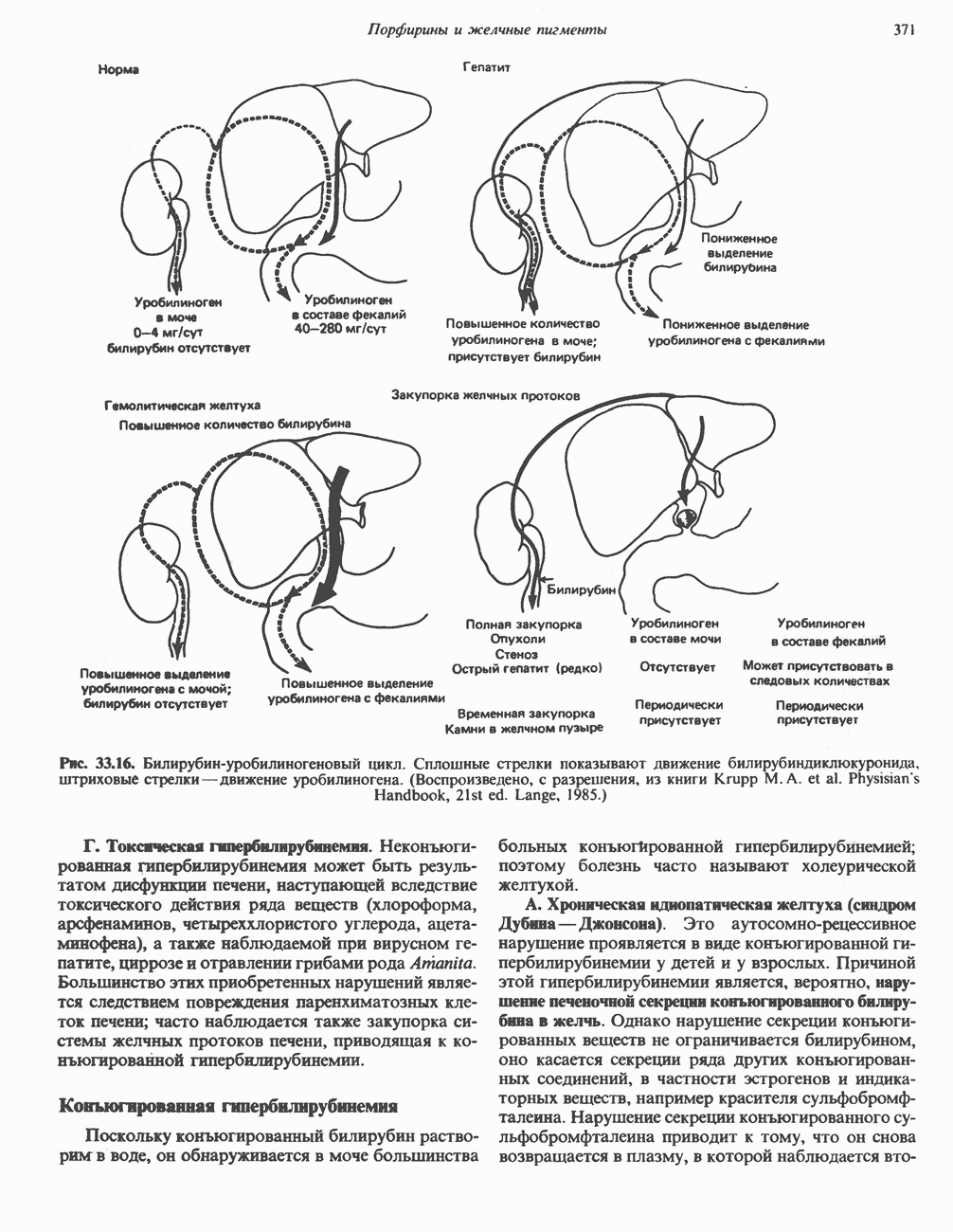
Пред.
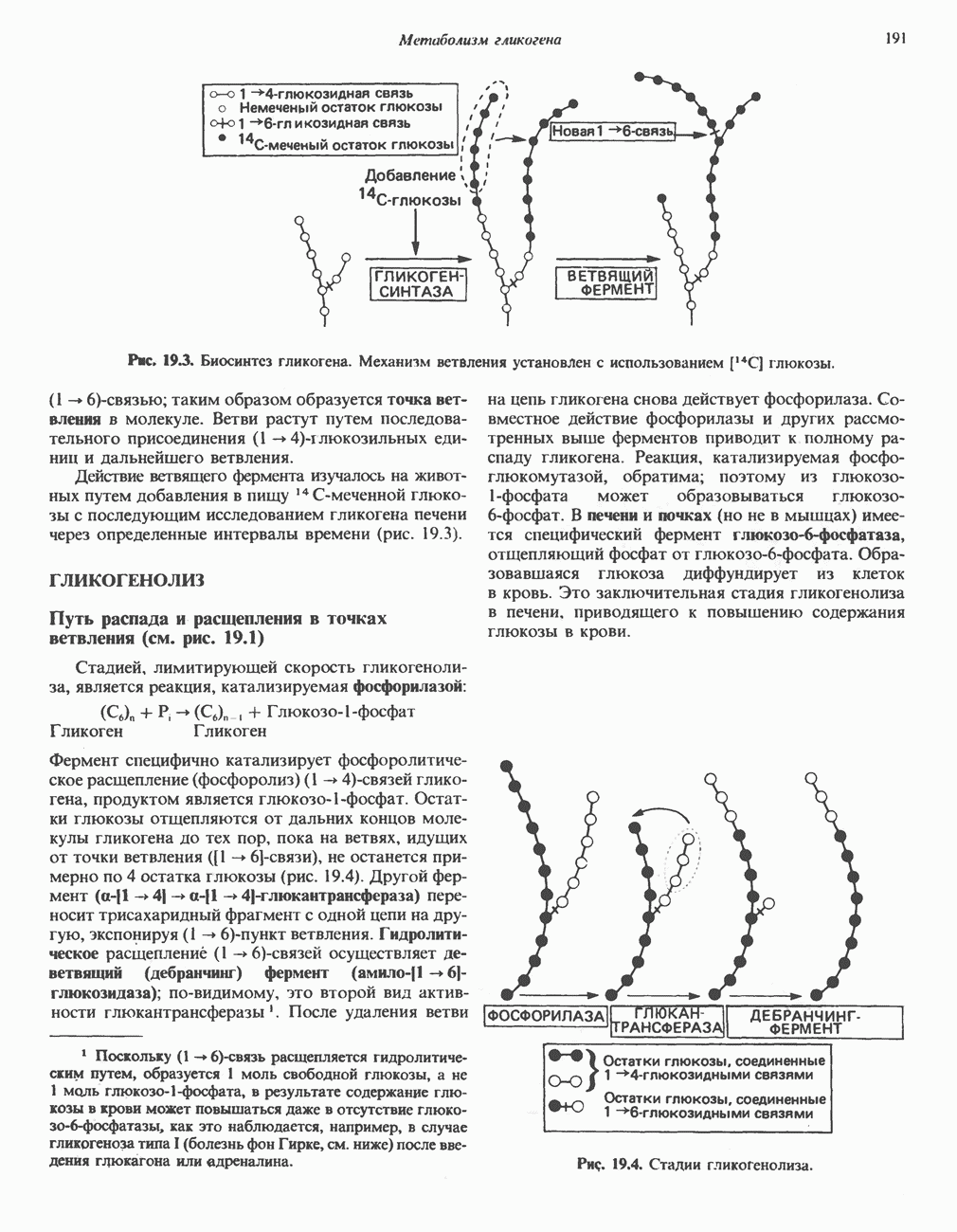
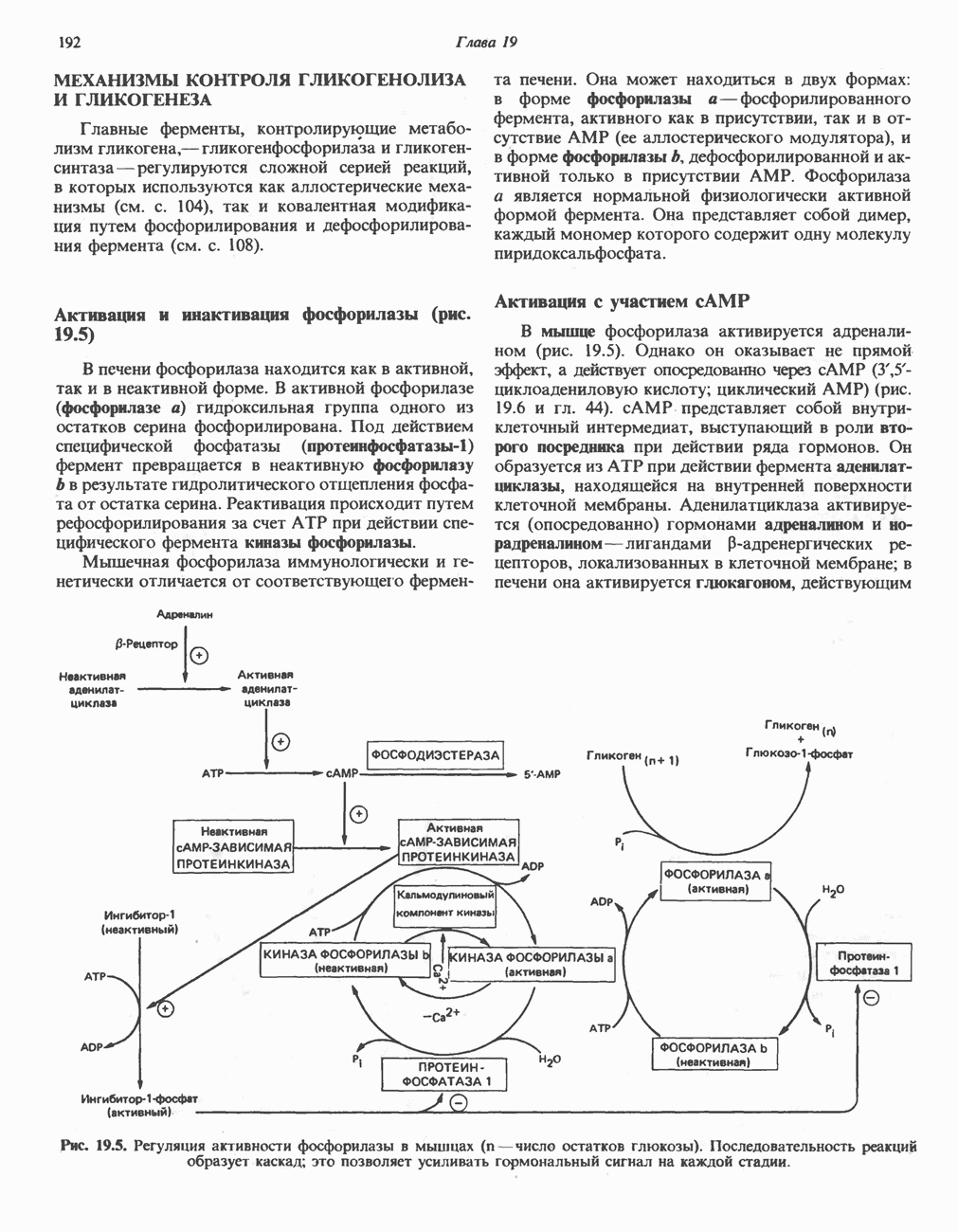
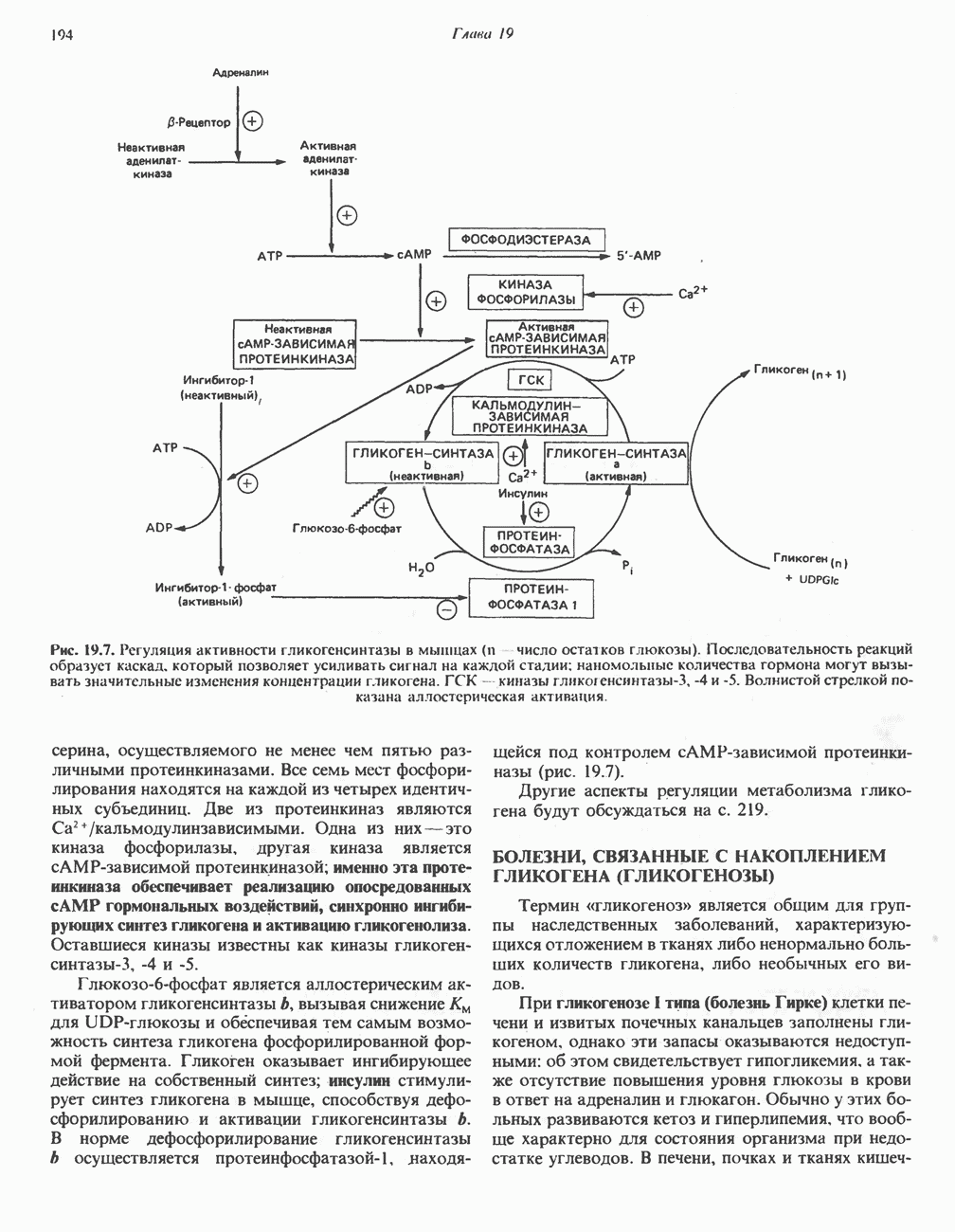
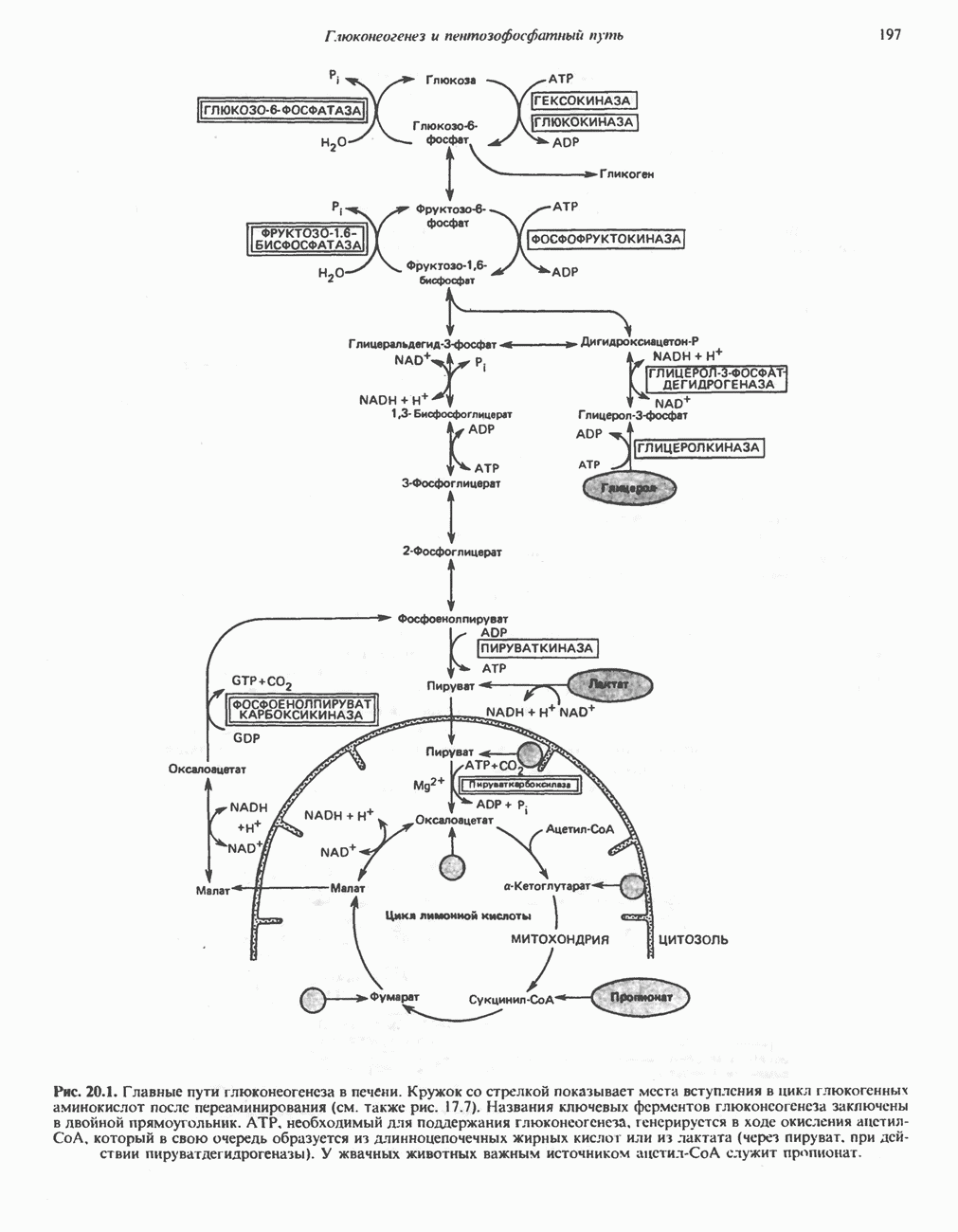
След.
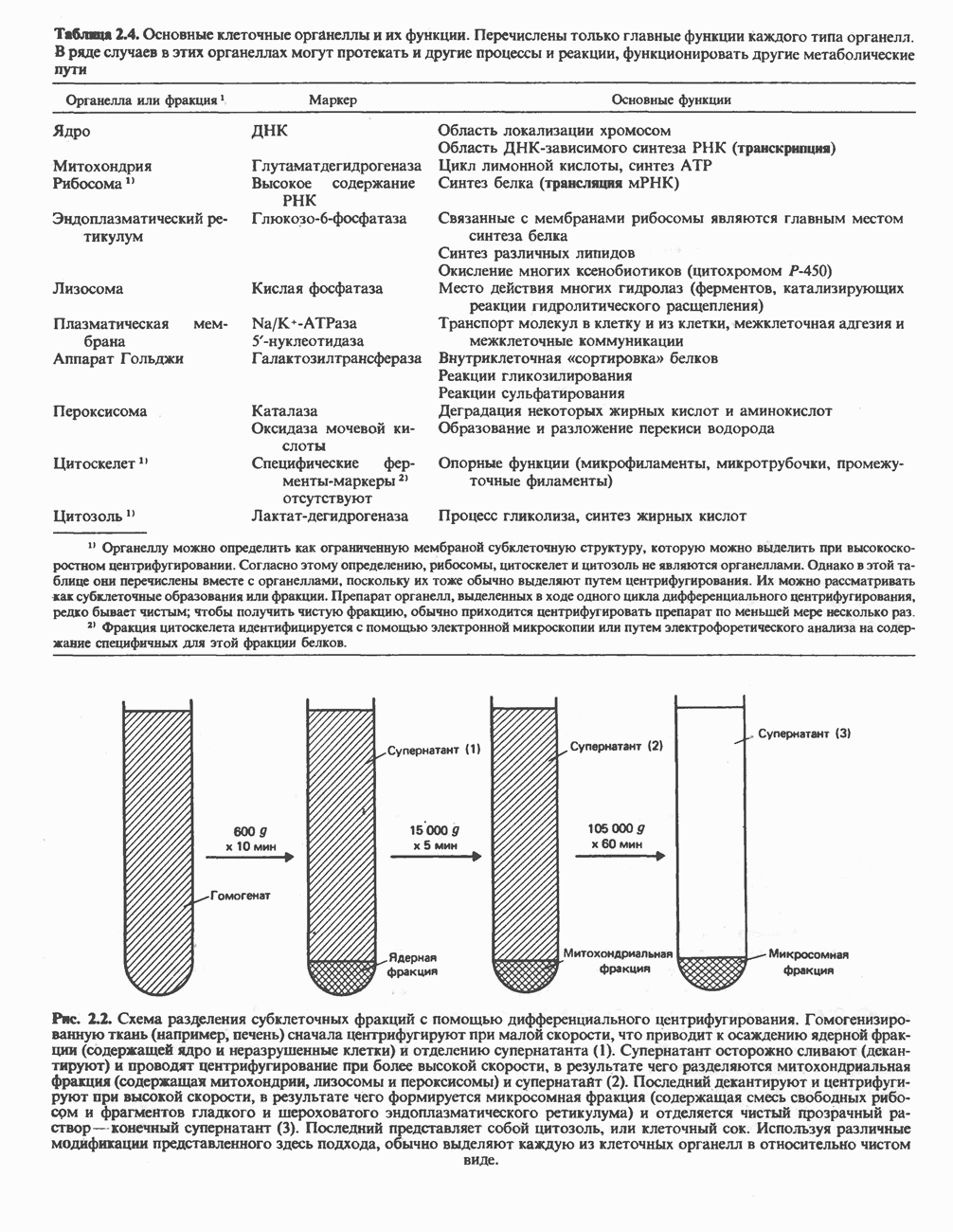
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
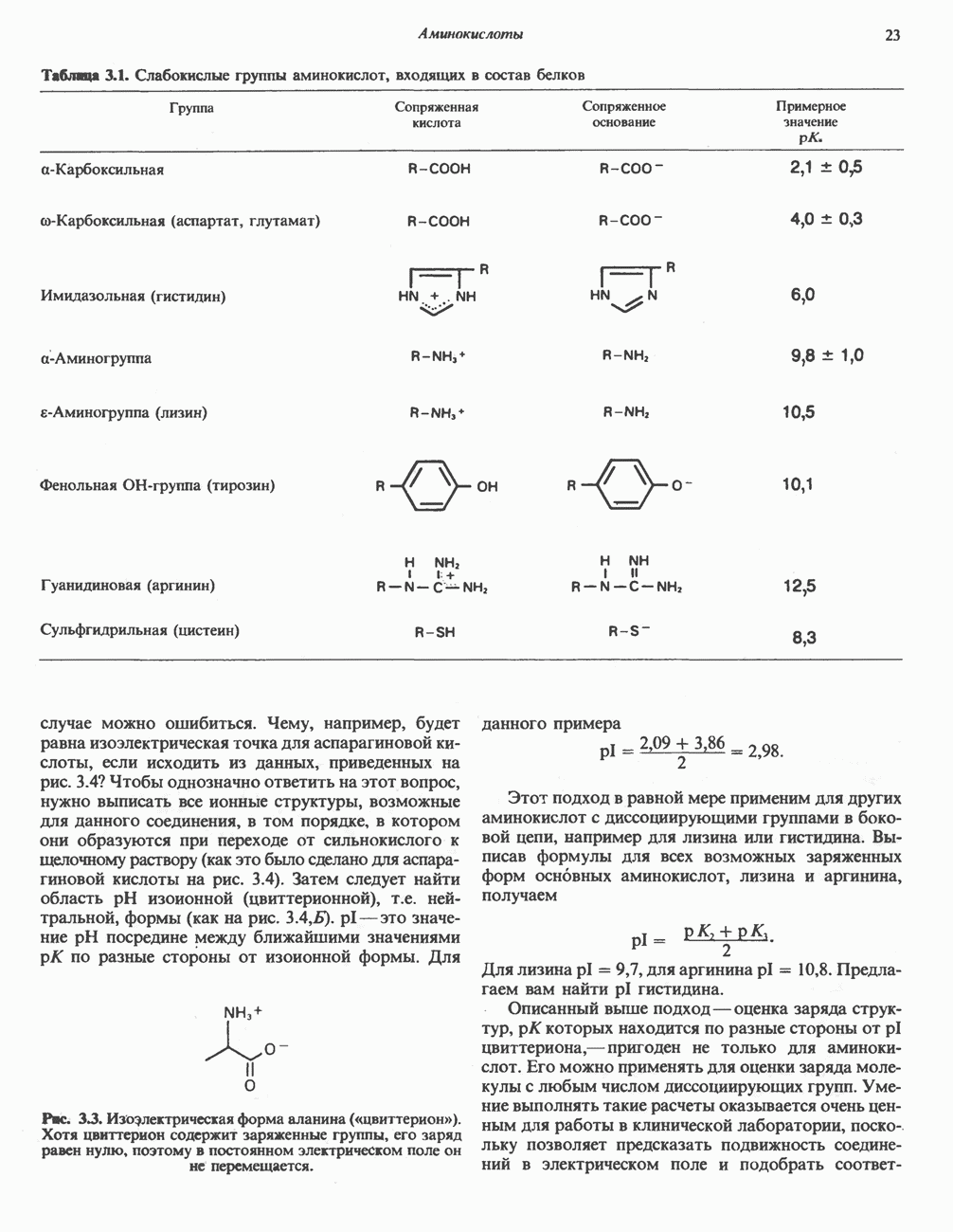
АМИНОКИСЛОТЫ, ОБРАЗУЮЩИЕ ПИРУВАТНиже приведена общая схема превращения углеродного скелета аланина, цистеина, глицина, треонина и серина в пирува
ГлицинВ число амфиболических интермедиатов, образующихся из глицина, входят пируват,
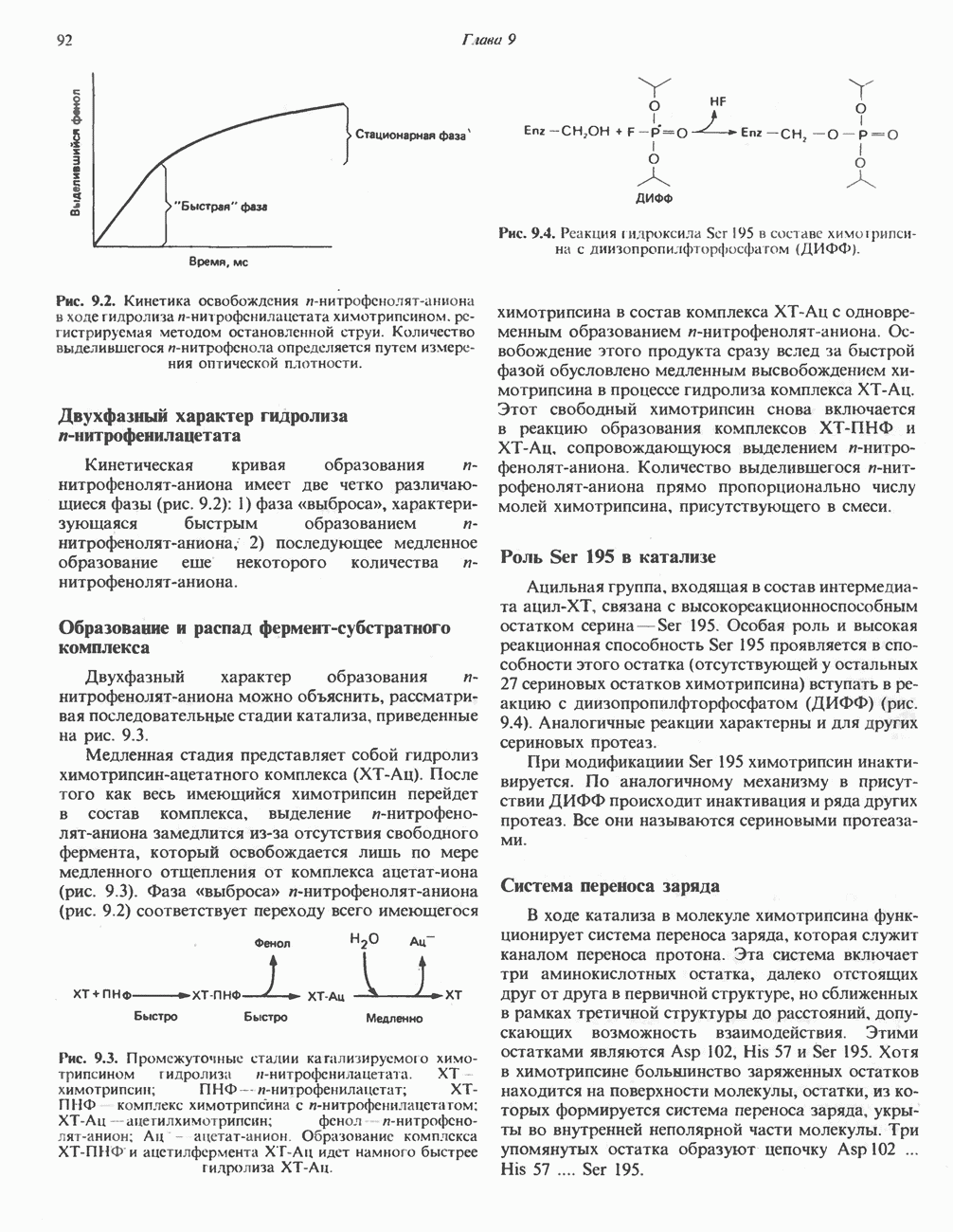
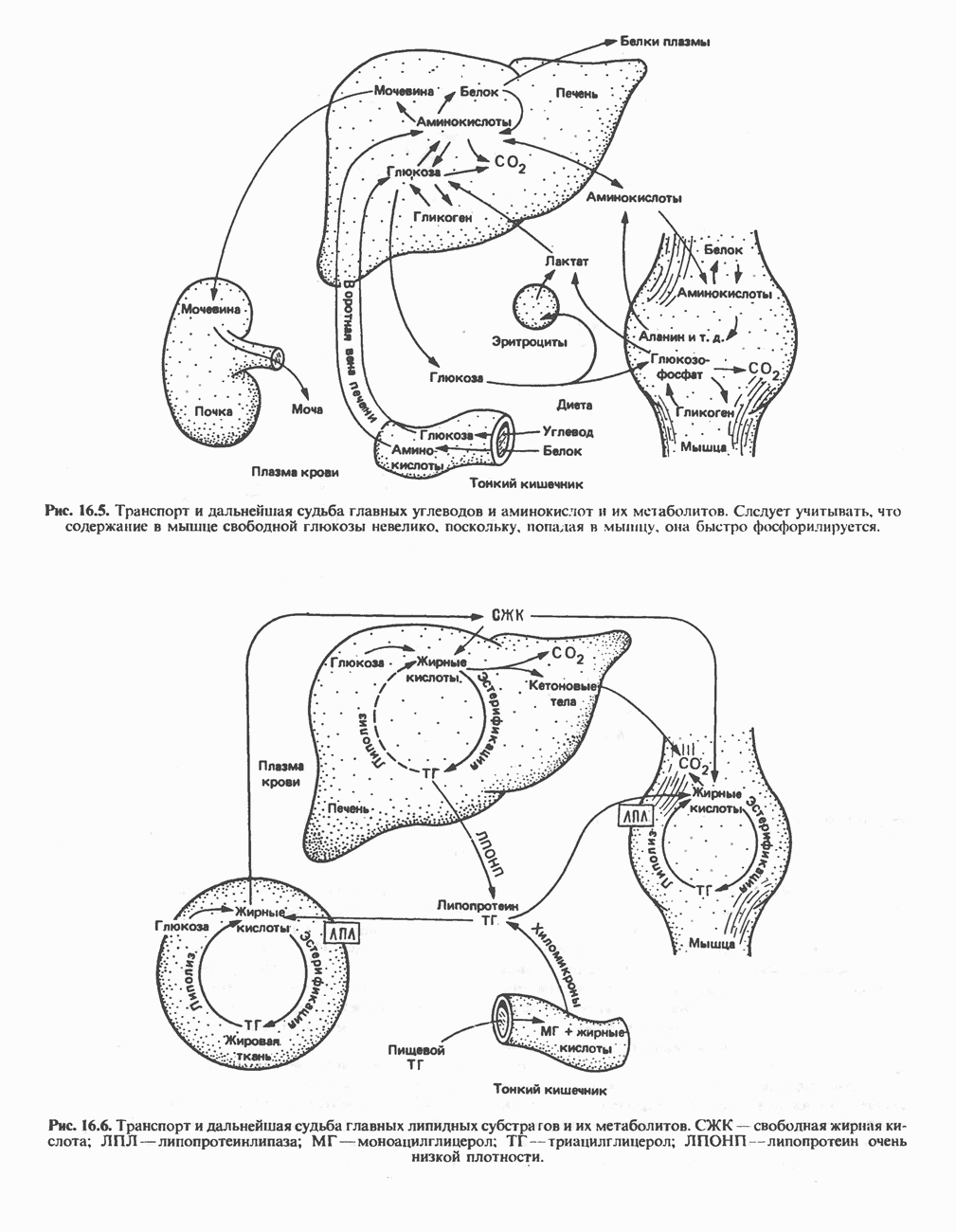
Рис. 31.5. Легко обратимая реакция, катализируемая се-рингидроксиметилтрансферазой. Главный путь катаболизма глицина у позвоночных — это катализируемое глицинсинтазным комплексом превращение, в результате которого образуются По-видимому, у человека и многих других позвоночных эта реакция является основным путем катаболизма не только глицина, но и серина (см. ниже раздел «Серин»). Метаболические нарушения катаболизма глицина. Ниже рассматриваются два вида нарушений метаболизма глицина. А. Глицннурия. Глицинурия наблюдалась лишь в одной семье. Она характеризуется повышенной экскрецией глицина с мочой и ассоциируется с тенденцией к образованию оксалатных камней в почках, при этом содержание оксалата в моче остается в пределах нормы. Глицинурия, по всей видимости наследуется как доминантный признак, сцепленный, вероятно, с Х-хромосомой. Уровень содержания глицина в плазме остается нормальным, тогда как количество глицина, экскретируемого с мочой, достигает 600—1000 Б. Первичная гипероксалурия. Первичная гипероксалурия характеризуется постоянно высокой экскрецией оксалата с мочой, независимо от поступления оксалата с пищей. При развитии болезни наблюдается прогрессирующее двустороннее образование оксалатных камней в мочевыводящих путях;
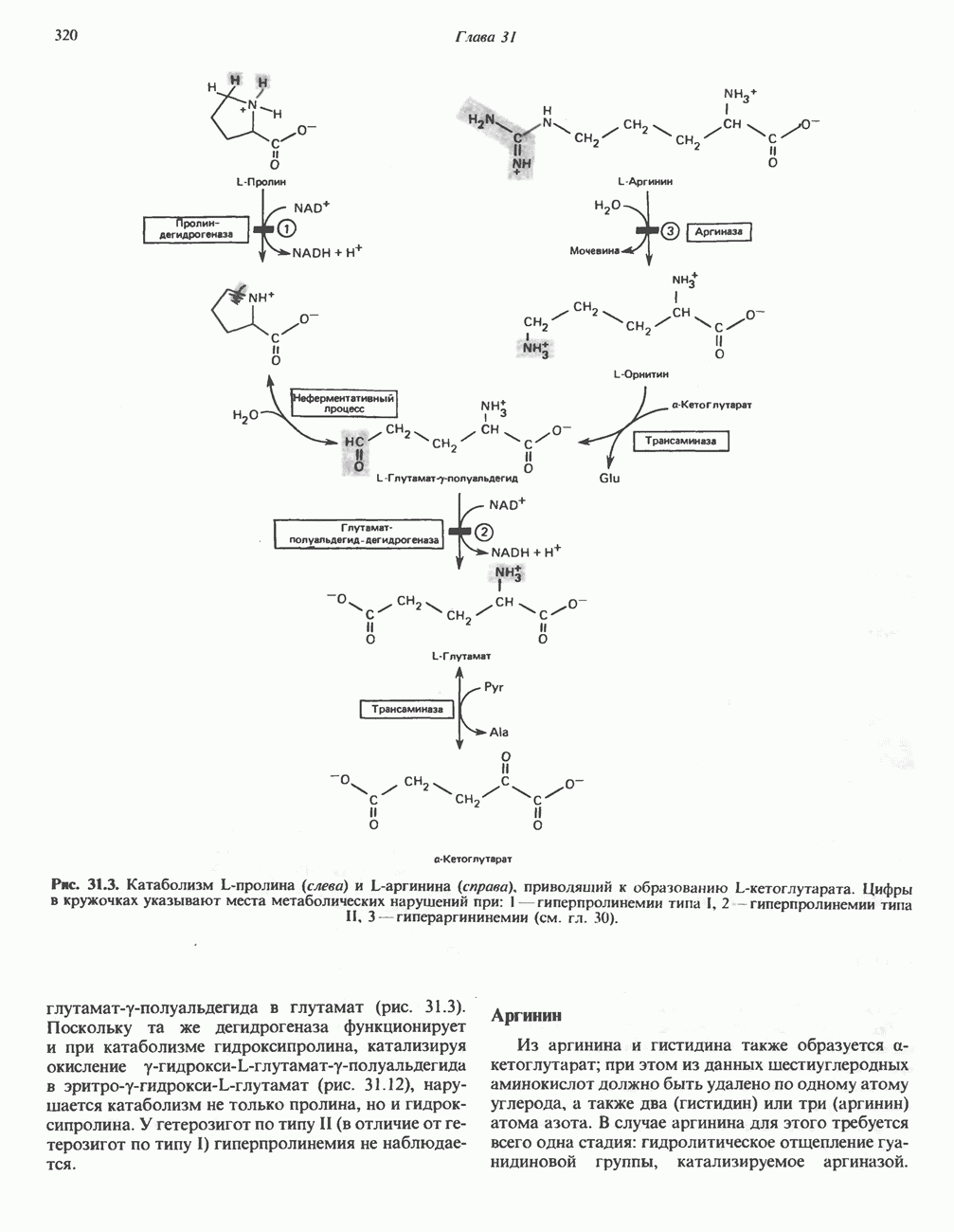
Рис. 31.6. Обратимое расщепление глицина митохондриальным глицинсинтазным комплексом. PLP пиридоксальфосфат. далее развивается нефрокальциноз и рецидивирующая инфекция мочевыводящих путей. Летальный исход наступает в детском или молодом возрасте от почечной недостаточности или гипертонии. Избыток оксалата, очевидно, имеет эндогенное происхождение; вероятно, он образуется из глицина (при дезаминировании последнего образуется глиоксилат — предшественник оксалата). Метаболический дефект, как полагают, состоит в нарушении метаболизма глиоксилата, точнее его превращения в формиат или (путем пераминирования) в глицин. В результате избыток глиоксилата окисляется до оксалата. Вероятно, наследственное нарушение метаболизма — первичную гипероксалурию — можно объяснить сочетанием недостаточности глицин-трансаминазы и нарушения окисления глиоксилата в формиат. АланинВ результате переаминировання L-аланина (рис. 31.7) образуется пируват, который далее может декарбоксилироваться с образованием ацетил-СоА. Вероятно, по тем же причинам, которые были рассмотрены при обсуждении катаболизма глутамата и аспартата, метаболических нарушений катаболизма а-аланина не обнаружено. СеринПревращение серина в пируват, катализируемое сериндегидратазой (пиридоксальфосфатсодержа-щим белком), включает элиминирование воды и гидролитическое удаление аммонийной группы из образующегося интермедиата (рис. 31.7). Печень крысы и морской свинки богата сериндегидратазой; у этих видов превращение серина в пируват при участии данного фермента имеет существенное физиологическое значение, в то время как у человека и многих других позвоночных серин деградирует преимущественно с образованием глицина и N5, N10-метилентетрагидрофолата. Эта реакция катализируется серин-гидроксиметилтрансферазой (рис. 31.5). Дальнейший катаболизм серина идет по пути катаболизма глицина (рис. 31.6). ЦистинПодобно азоту и углероду, сера совершает в биосфере непрерывный кругооборот, который осуществляется за счет метаболической активности прокариотических и эукариотических организмов. Млекопитающие не имеют систем перевода серы в оргарическую форму они участвуют в кругообороте, осуществляя катаболическое превращение органических соединений серы в неорганические. Например, человек экскретирует приблизительно 20—30 ммоль серы в сутки, из них по меньшей мере 80% приходится на неорганический сульфат. Главный метаболический путь цистина у млекопитающих — превращение в цистеин в результате реакции, катализируемой цистинредуктазой (рис. 31.8). Далее катаболизм цистина совпадает с катаболизмом цистеина (см. ниже).
Рис. 31.7. Превращение аланина и серина в пируват. Аланин-трансаминаза и сериндегидратаза в качестве кофактора используют пиридоксальфосфат. В результате реакции, катализируемой сериндегидратазои, происходит элиминирование
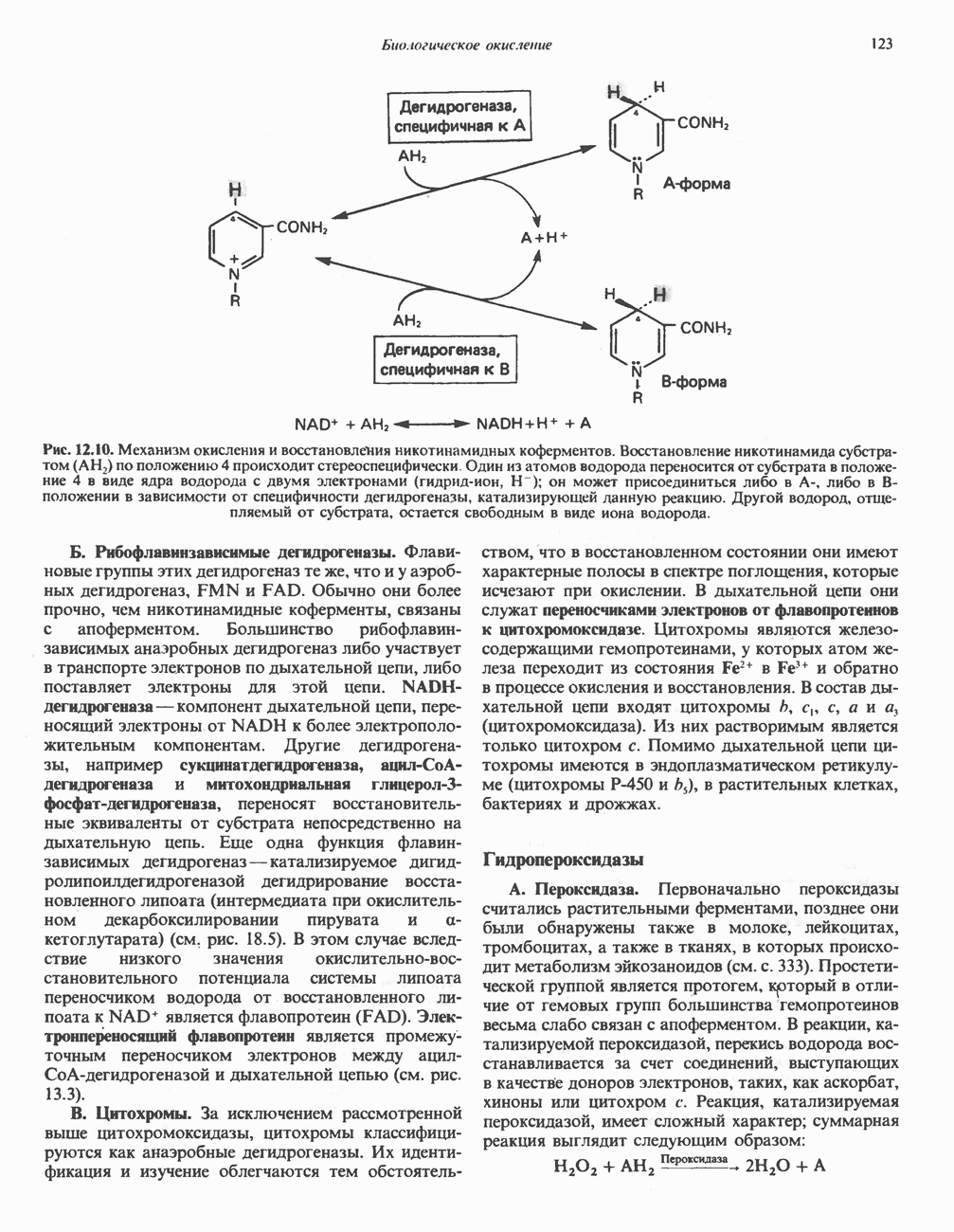
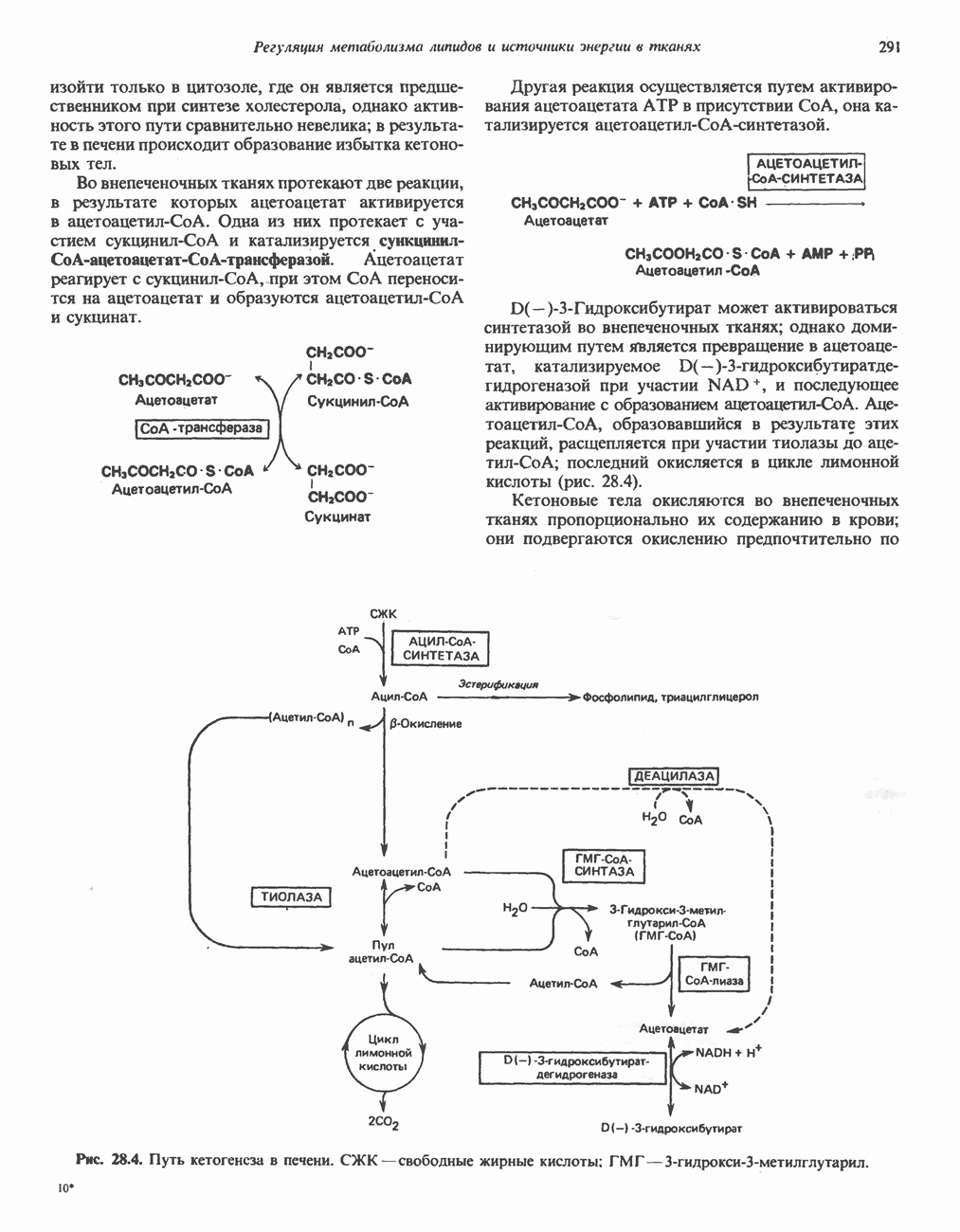
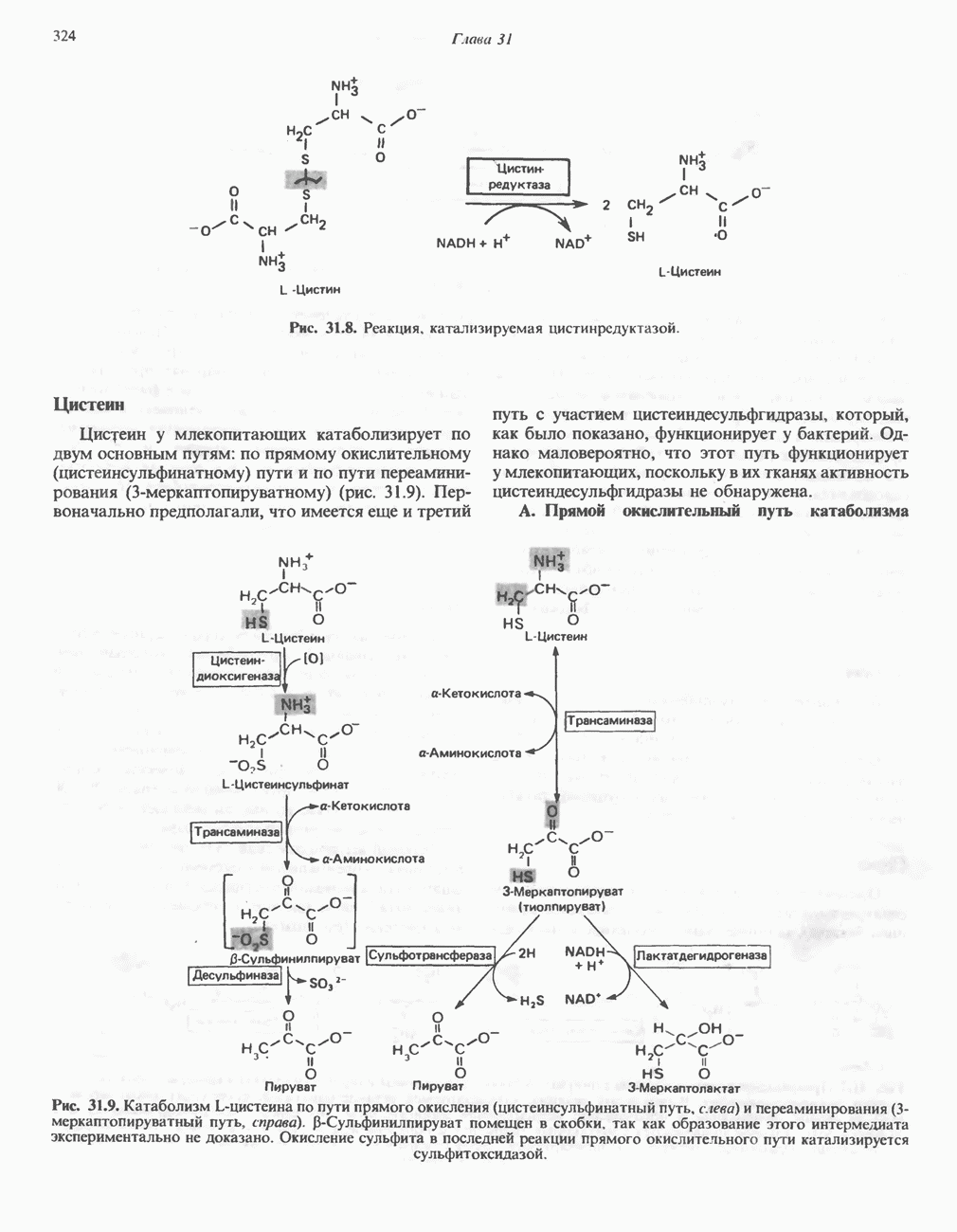
Рис. 31.8. Реакция, катализируемая цистинрсдуктазой. ЦистеинЦистеин у млекопитающих катаболизирует по двум основным путям: по прямому окислительному (цистеинсульфинатному) пути и по пути переаминирования ( А. Прямой окислительный путь катаболизма
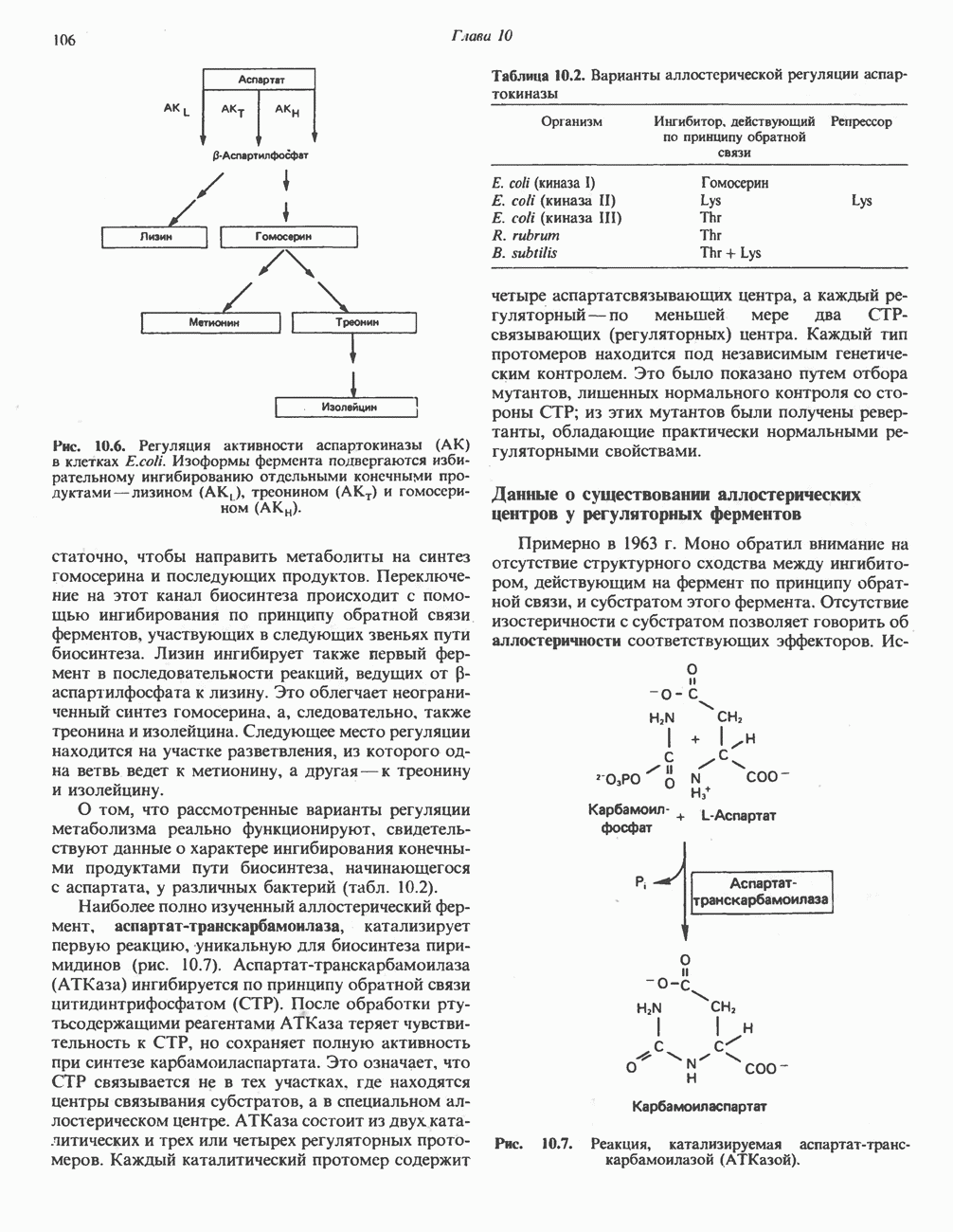
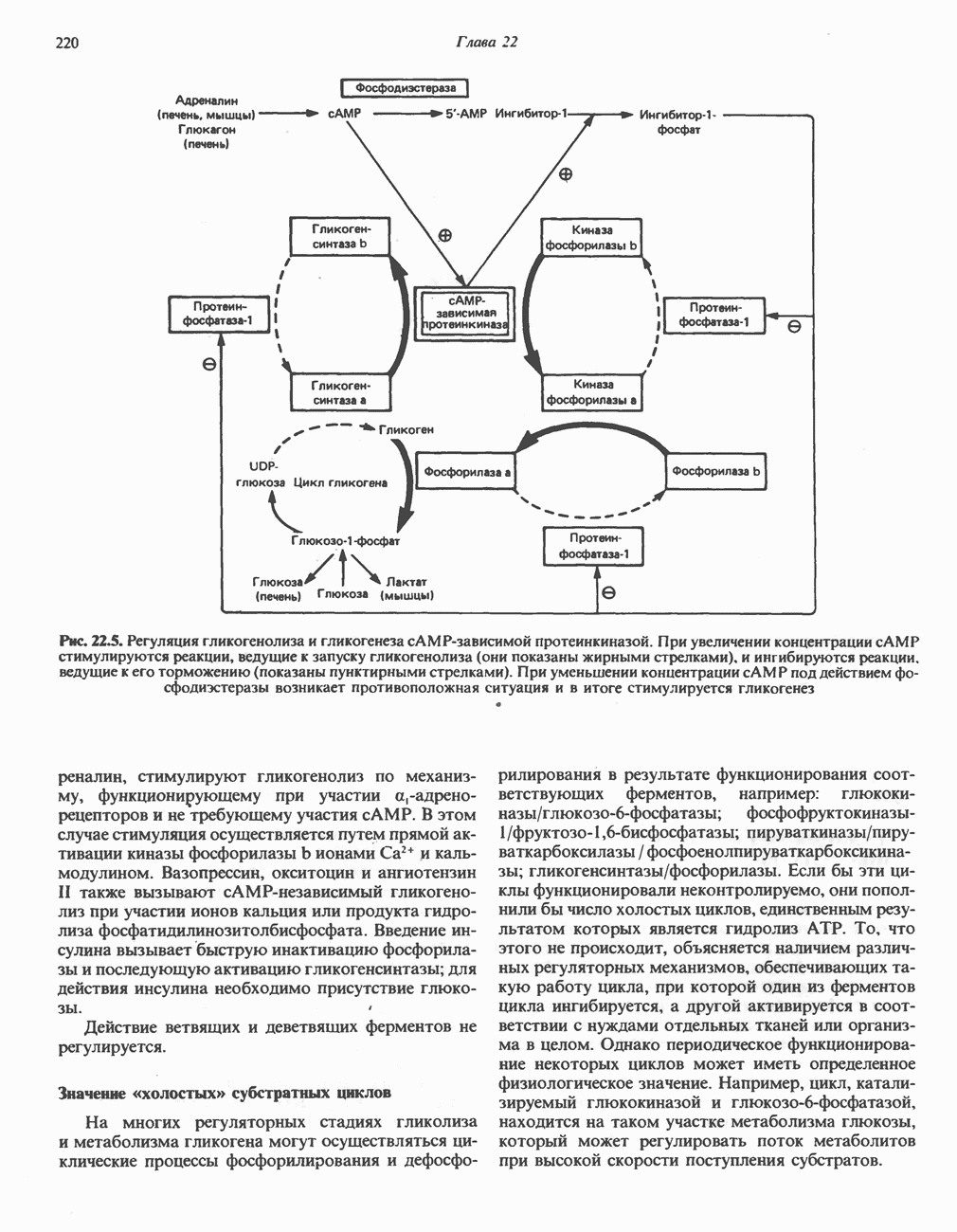
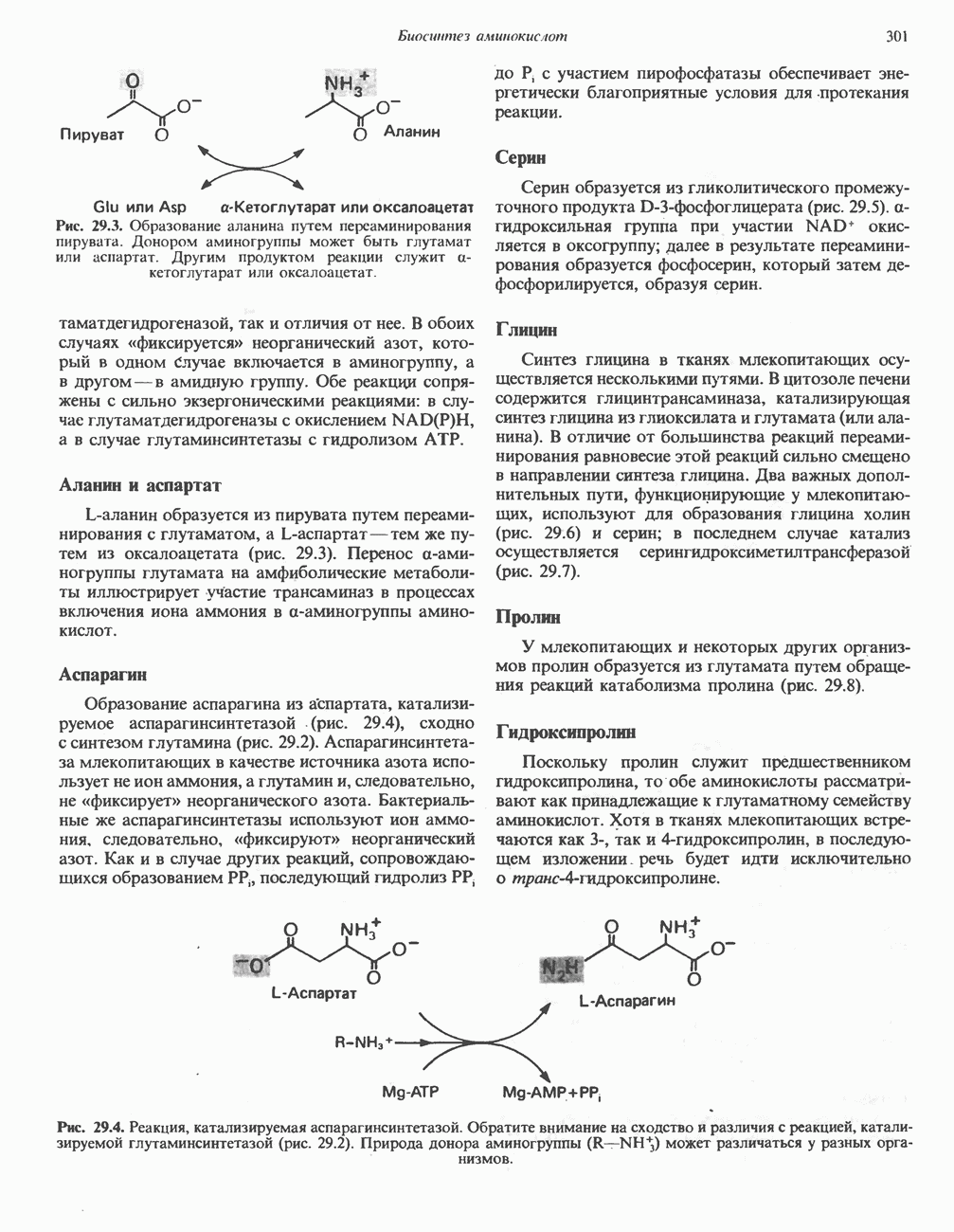
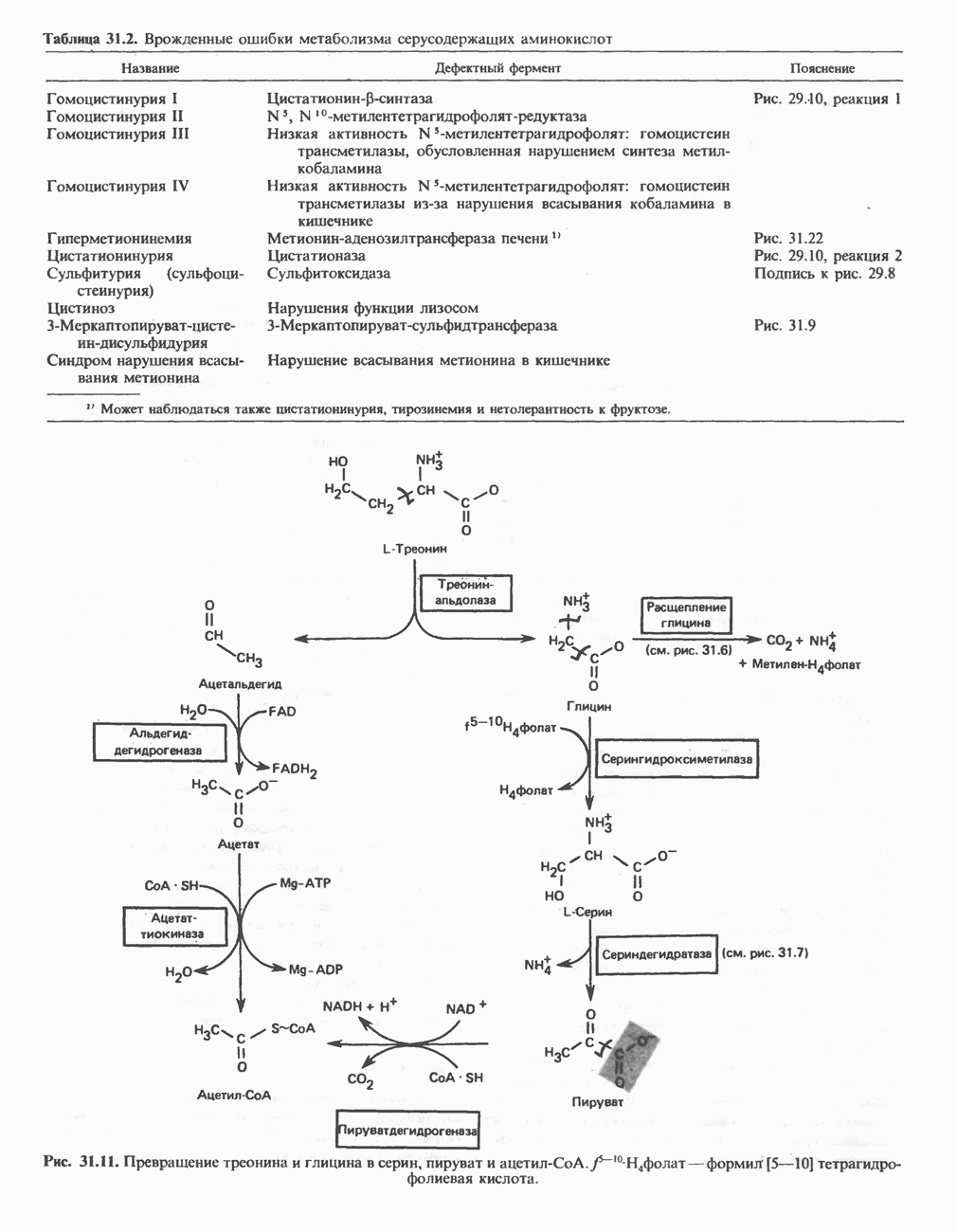
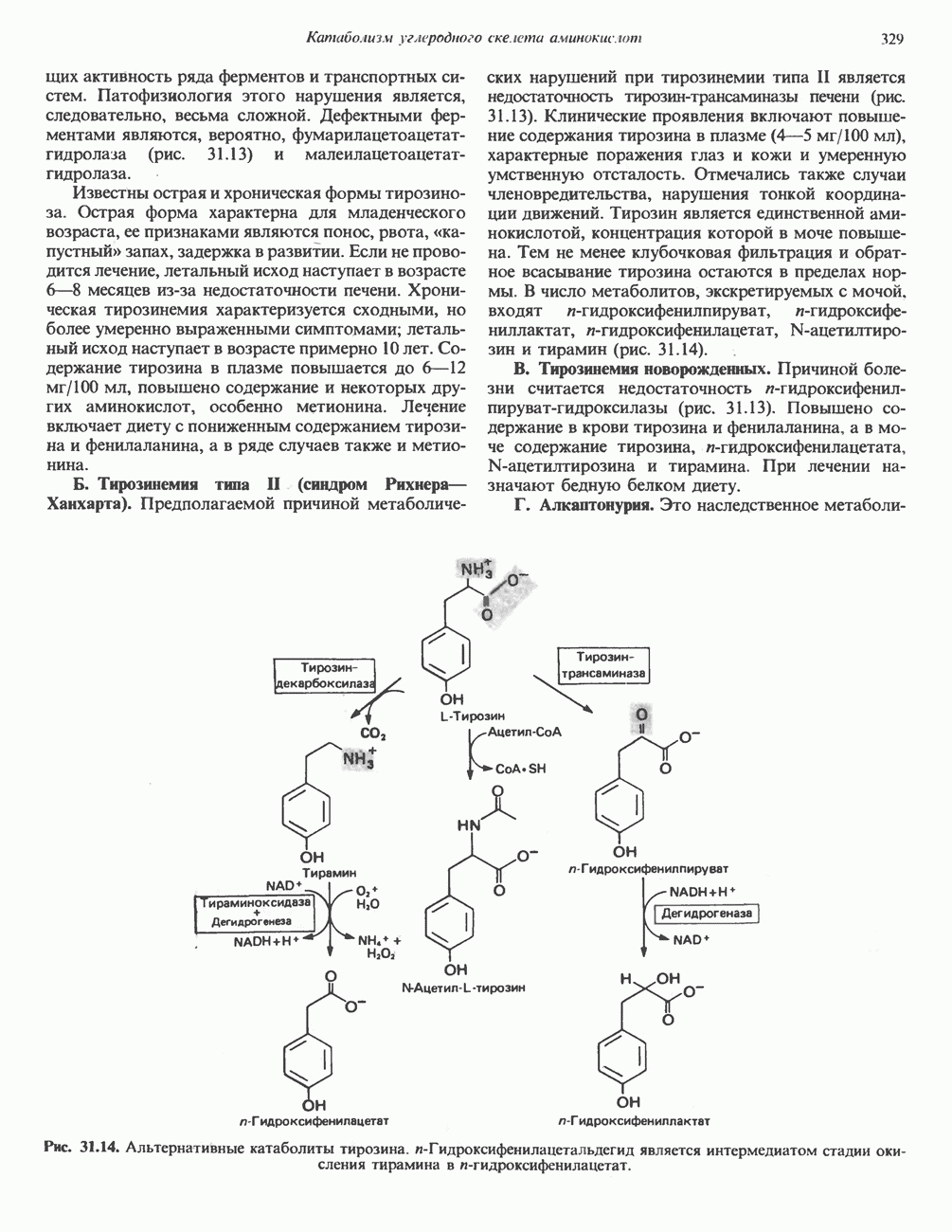
Рис. 31.9. Катаболизм L-цистеина по пути прямого окисления (цистеинсулъфинатный путь, слева) и переаминировання ( цистеина. Превращение цистеина в цистеинсульфинат (рис. 31.9) катализируется цистеин-диоксигеназой — ферментом, функционирующим при участии Б. Трансаминазный (З-меркаптопируватный) путь катаболизма цистеина. Обратимое пераминирование цистеина в 3-меркаптопируват (гиолпируват) катализируется специфическими цистеиновыми транс-аминазами или аспарагиновой и глутаматной транс-аминазами печени и почек млекопитающих (рис. 31.9). З-Меркаптопируват может далее восстанавливаться в ходе реакции, катализируемой L-лактатдегидрогеназой. Образующийся продукт 3-меркап-голактат является нормальным компонентом мочи человека в форме смешанного дисульфида с цистеином; содержание последнего в моче пациентов с меркаптолактат-цистеин-дисульфидурией возрастает. Альтернативное превращение 3-меркапто-пирувата идет по пути отщепления Нарушения метаболизма серусодержащих аминокислот. В табл. 31.2 приведена сводка данных о нарушениях метаболизма серусодержащих аминокислот. Некоторые из них рассмотрены ниже. А. Цистинурия (цистин-лизинурия). При этом наследуемом метаболическом заболевании экскреция цистина с мочой в 20 - 30 раз превышает норму. Значительно повышается также экскреция лизина, аргинина и орнитина. Цистинурию рассматривают как следствие нарушения процессов транспорта в почках. Значительное увеличение у пациентов с цистинурией экскреции с мочой наряду с цистином лизина, аргинина и орнитина позволяет предполагать, что нарушается процесс обратного всасывания всех этих четырех аминокислот, который, возможно, осуществляется в общем для них «участке» реабсорбции; поэтому вместо термина «цистинурия» в настоящее время предпочитают термин цистинлизинурия. Поскольку цистин слаборастворим, у больных цистинурией может происходить образование цистиновых камней в почечных канальцах. Если такого осложнения не возникает, цистинурия протекает сравнительно доброкачественно и во многих случаях остается недиагностируемой.
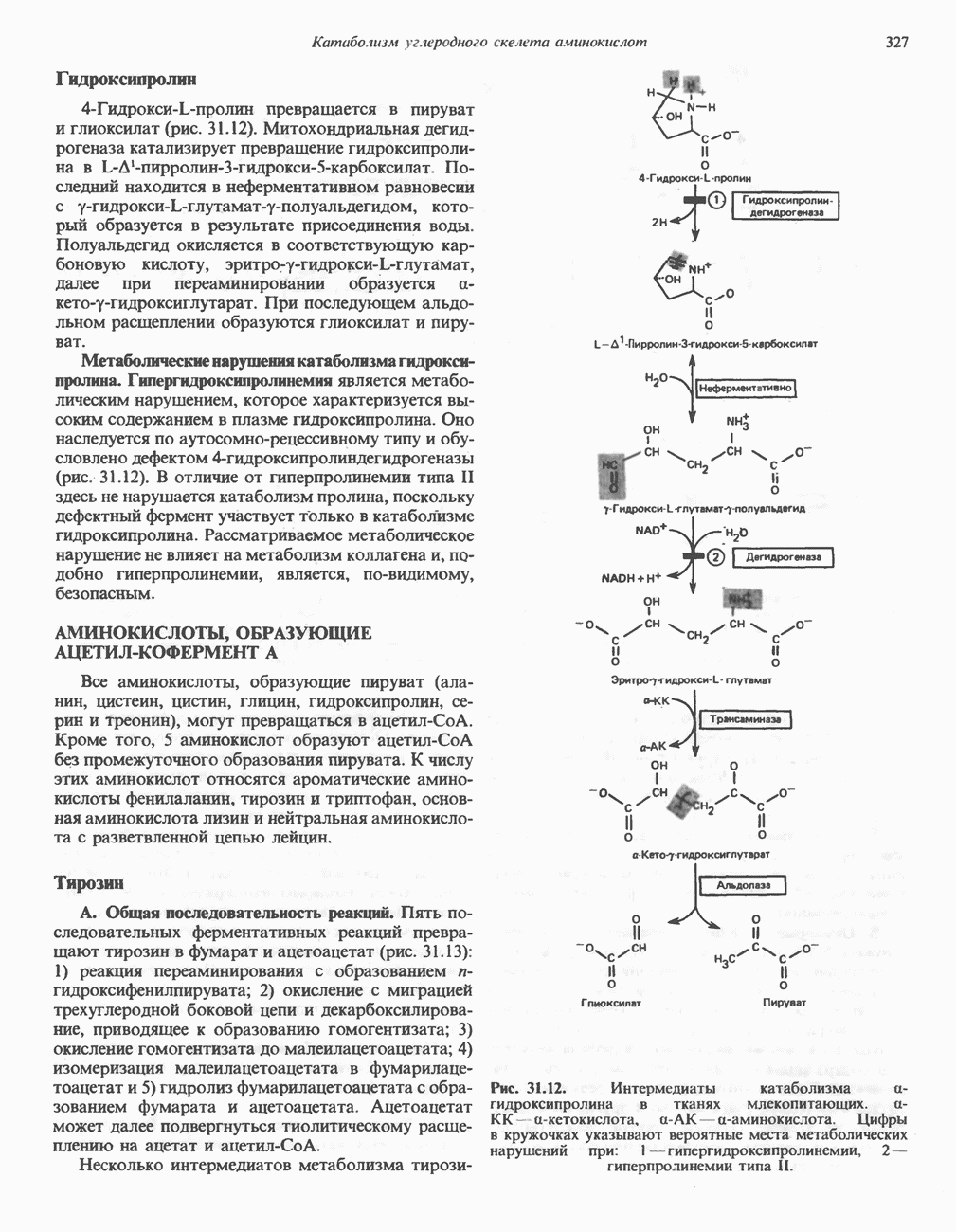
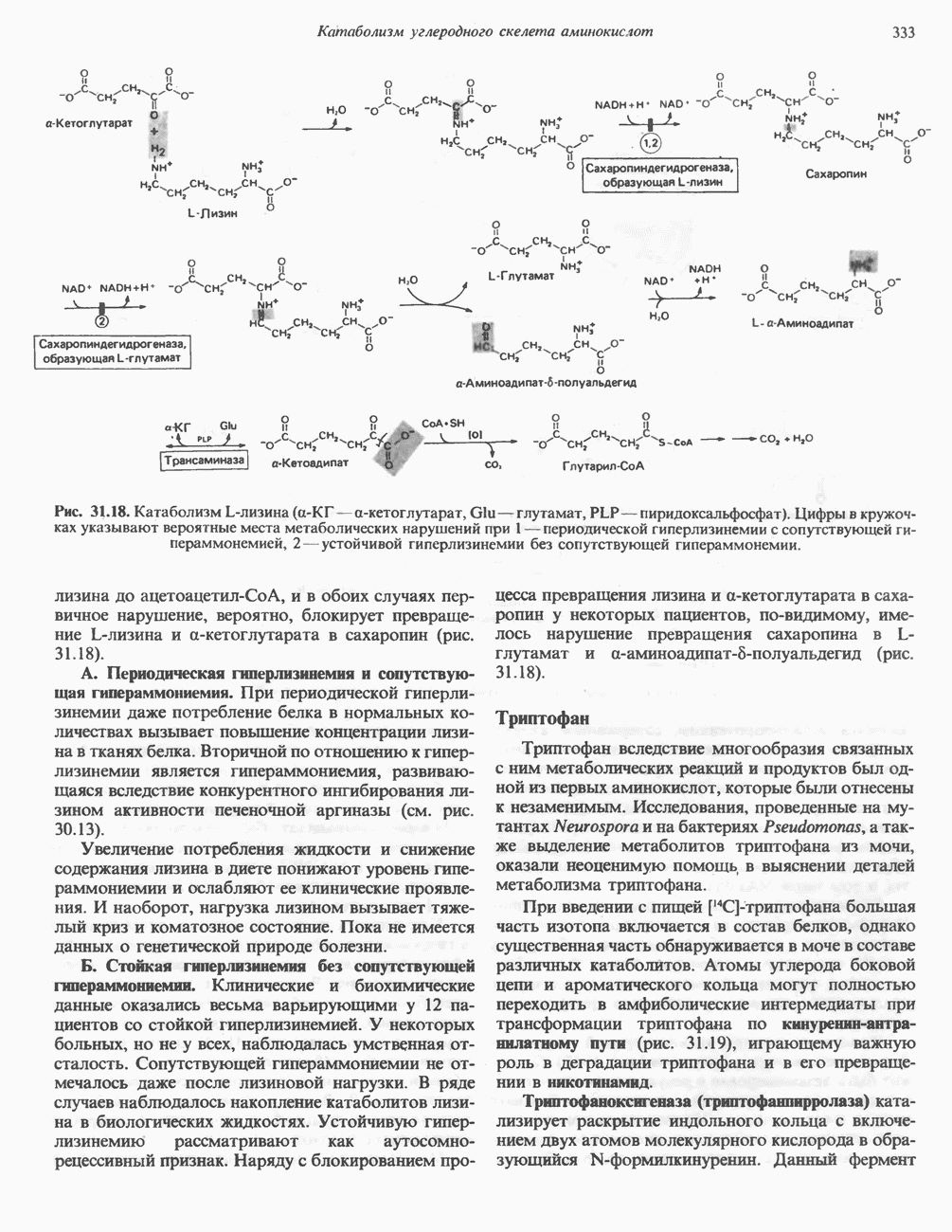
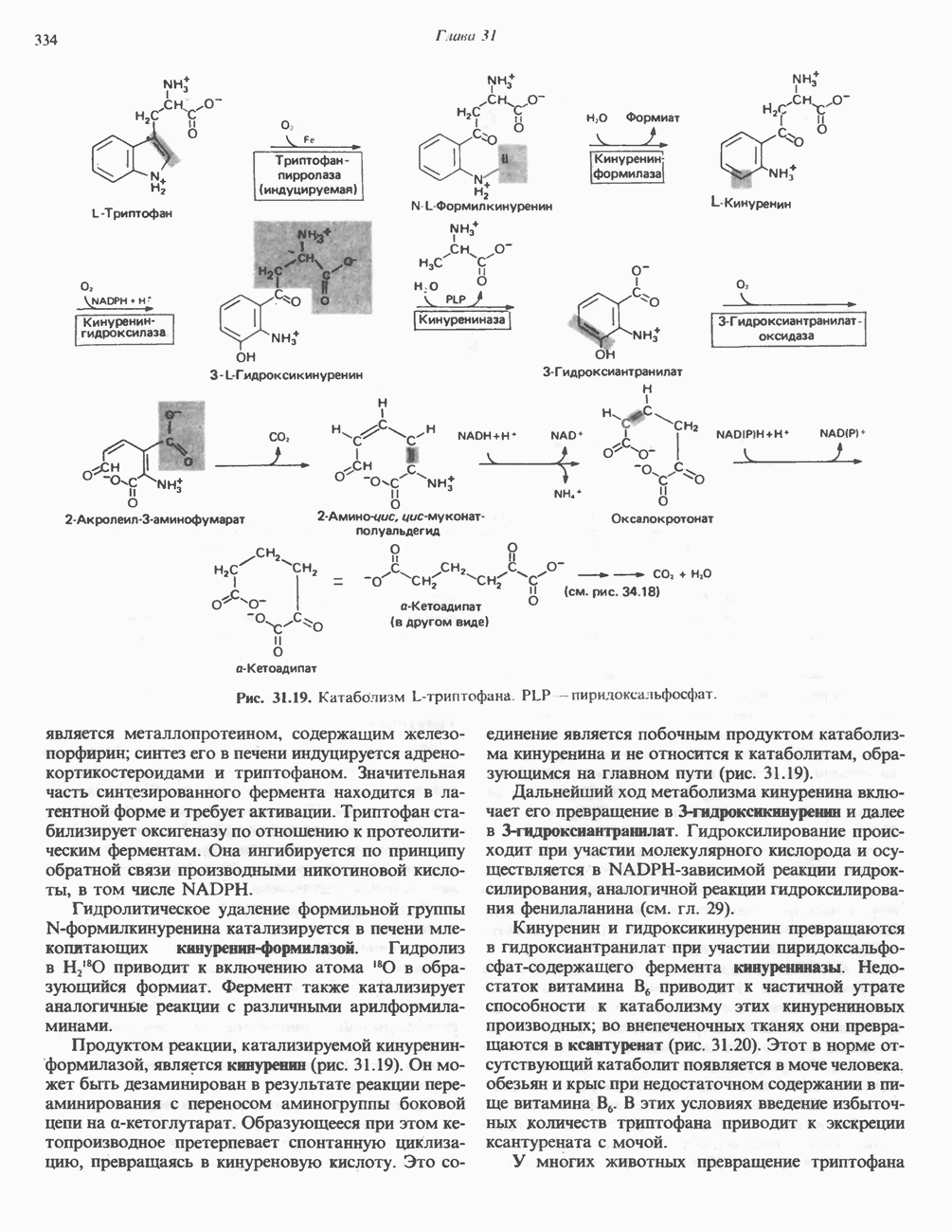
Рис. 31.10. Смешанный дисульфид цистеина и гомоцистеина. В моче пациентов с цистинурией был также обнаружен смешанный дисульфид L-цистеина и L-гомоцистеина (рис. 31.10). Это соединение имеет несколько большую растворимость, чем цистин; в той мере, в какой оно образуется вместо цистина, уменьшается тенденция к образованию цистиновых камней. Б. Цистиноз (болезнь накопления цистина). При цистинозе, который также является наследственным заболеванием, происходит формирование кристаллов цистина во многих тканях и органах (особенно в ретикулоэндотелиальной системе). Обычно при этом заболевании наблюдается общая аминоацидурия, т. е. повышение содержания в моче всех аминокислот. Серьезно нарушается и ряд других функций почек; летальный исход обычно наступает в раннем возрасте при явлениях острой почечной недостаточности. Согласно полученным в последнее время данным, В. Гомоцистинурии. Частота наследственных нарушений катаболизма метионина оценивается как 1 на 160000 новорожденных. Гомоцистин экскретируется с мочой (до 300 мг в сутки), в ряде случаев вместе с Повышается содержание метионина в плазме. Причиной гомоцистинурии могут быть по крайней мере четыре метаболических нарушения (табл. 31.2). При гомоцистинурии типа 1 клиническими симптомами являются тромбоз, остеопороз, смещение хрусталика глаза и, часто, умственная отсталость. Известны две формы заболевания: витамин В6 - чувствительная и витамин В6 - нечувствительная. Диета с низким содержанием метионина и высоким содержанием цистина может предотвратить патологические изменения, если она соблюдается с раннего возраста. Другие типы цистинурии связаны с нарушениями в цикле реметилирования (табл. 31.2). ТреонинТреонин расщепляется треонинальдолазой на ацетальдегид и глицин, из ацетальдегида затем образуется ацетил-СоА (рис. 31.11). Катаболизм глицина обсуждался выше. Таблица 31.2. Врожденные ошибки метаболизма серусодержащих аминокислот (см. скан) (см. скан) Рис. 31.11. Превращение треонина и глицина в серин, пируват и ацетил-СоА. Гидроксипролин4-Гидрокси L-пролин превращается в пируват и глиоксилат (рис. 31.12). Митохондриальная дегидрогеназа катализирует превращение гидроксипролина в Метаболические нарушения катаболизма гидроксипролин а. Гипергидроксипролинемия является метаболическим нарушением, которое характеризуется высоким содержанием в плазме гидроксипролина. Оно наследуется по аутосомно-рецессивному типу и обусловлено дефектом 4-гидроксипролиндегидрогеназы (рис. 31.12). В отличие от гиперпролинемии типа II здесь не нарушается катаболизм пролина, поскольку дефектный фермент участвует только в катаболизме гидроксипролина. Рассматриваемое метаболическое нарушение не влияет на метаболизм коллагена и, подобно гиперпролинемии, является, по-видимому, безопасным.
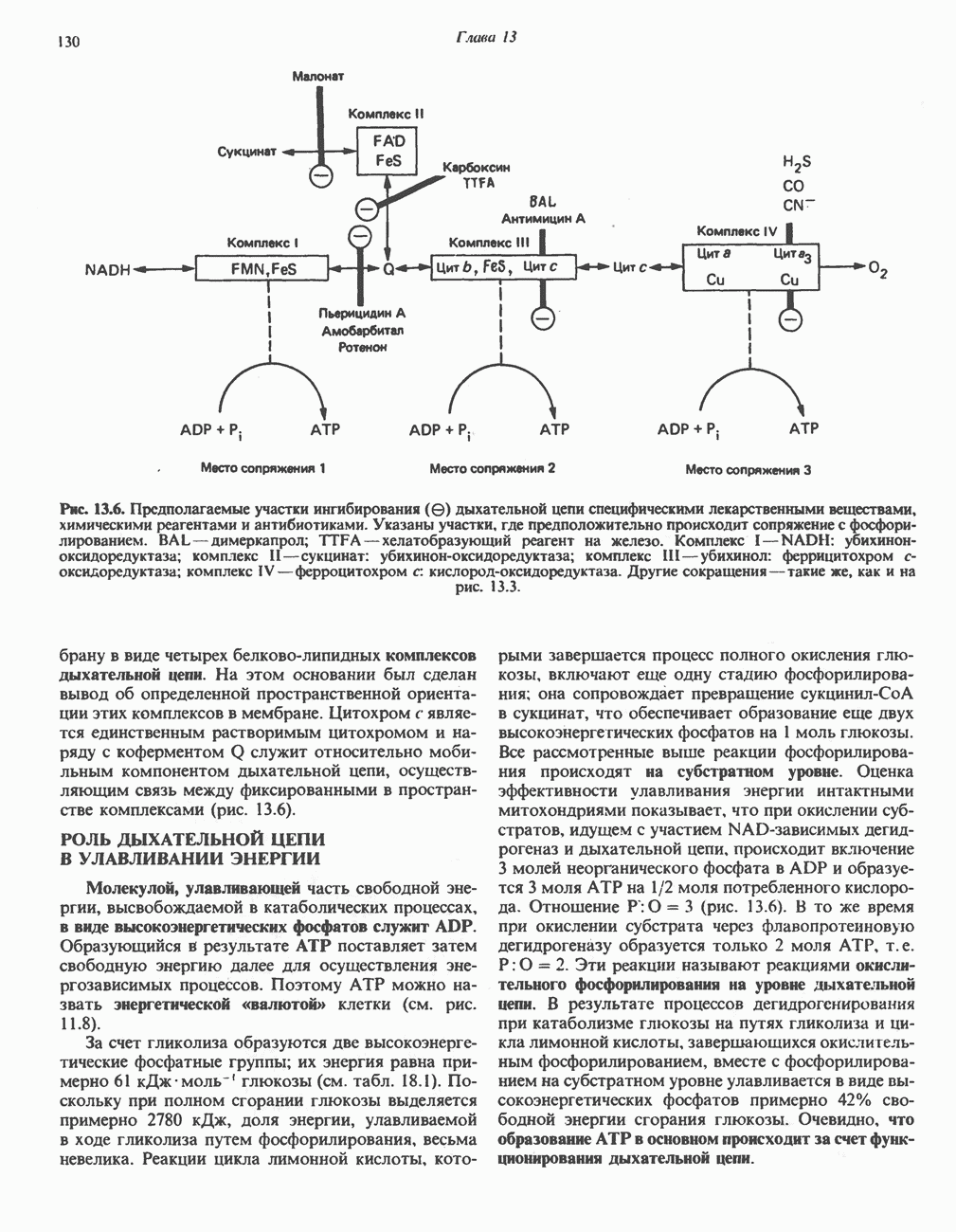
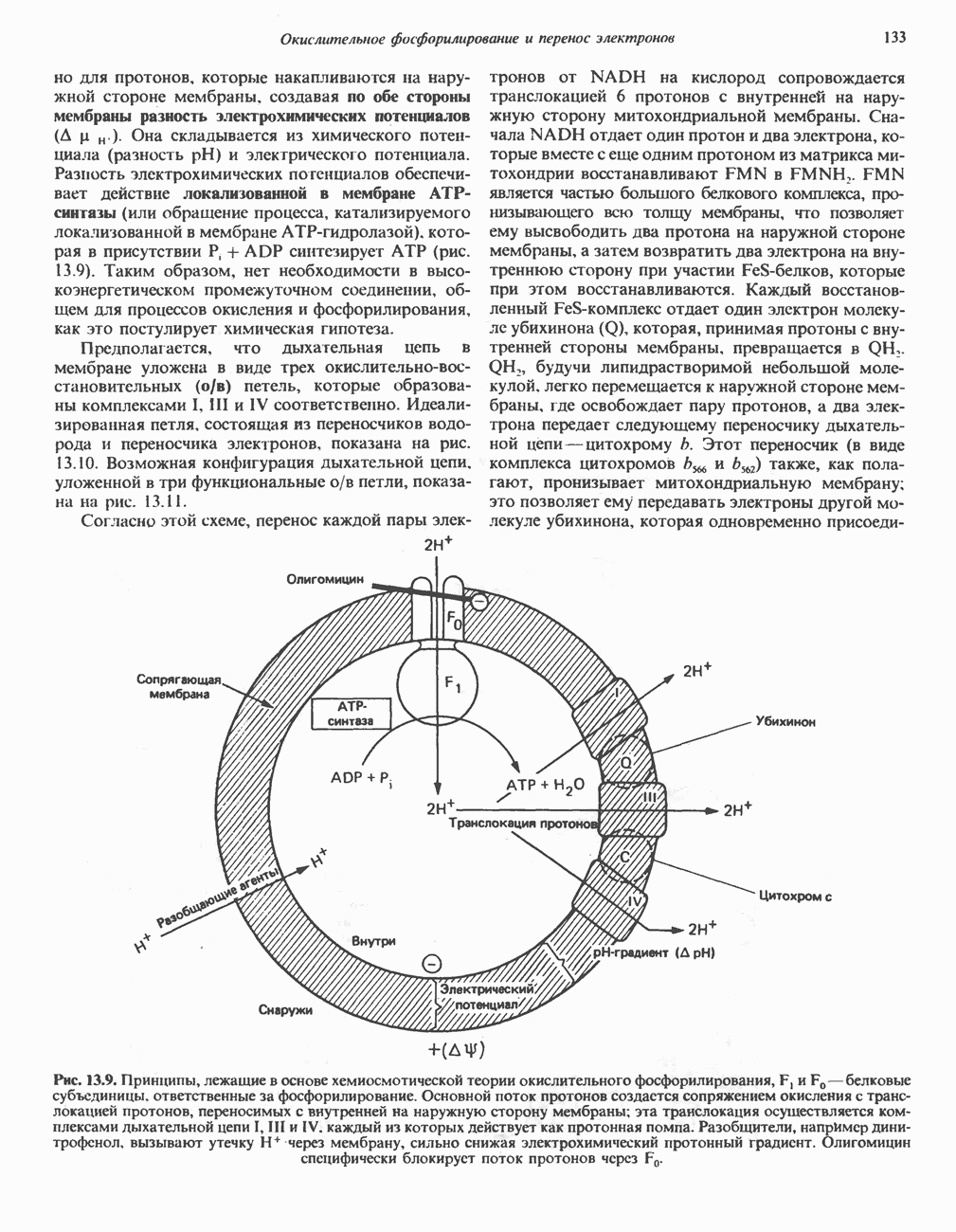
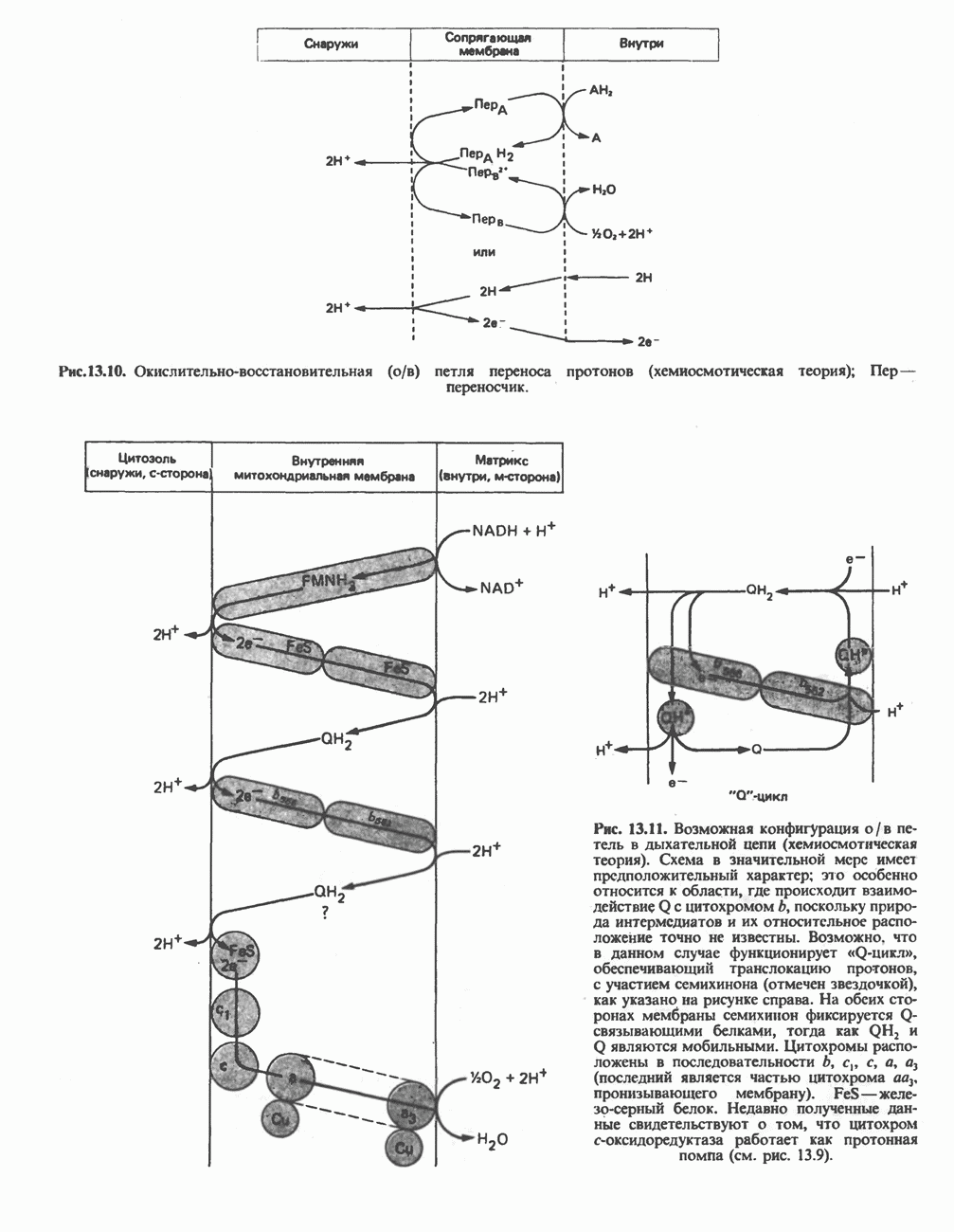
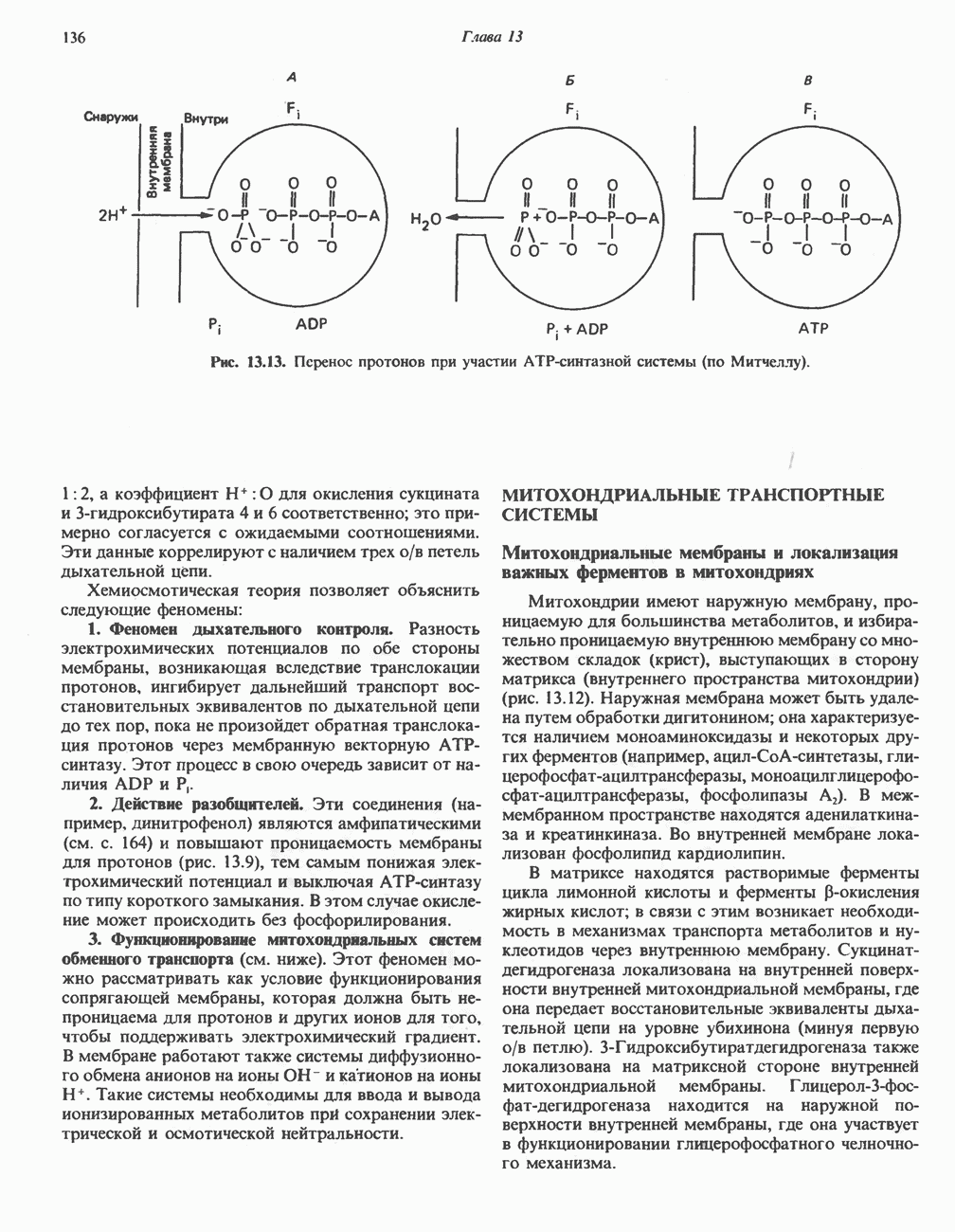
|
 |
Оглавление
|


































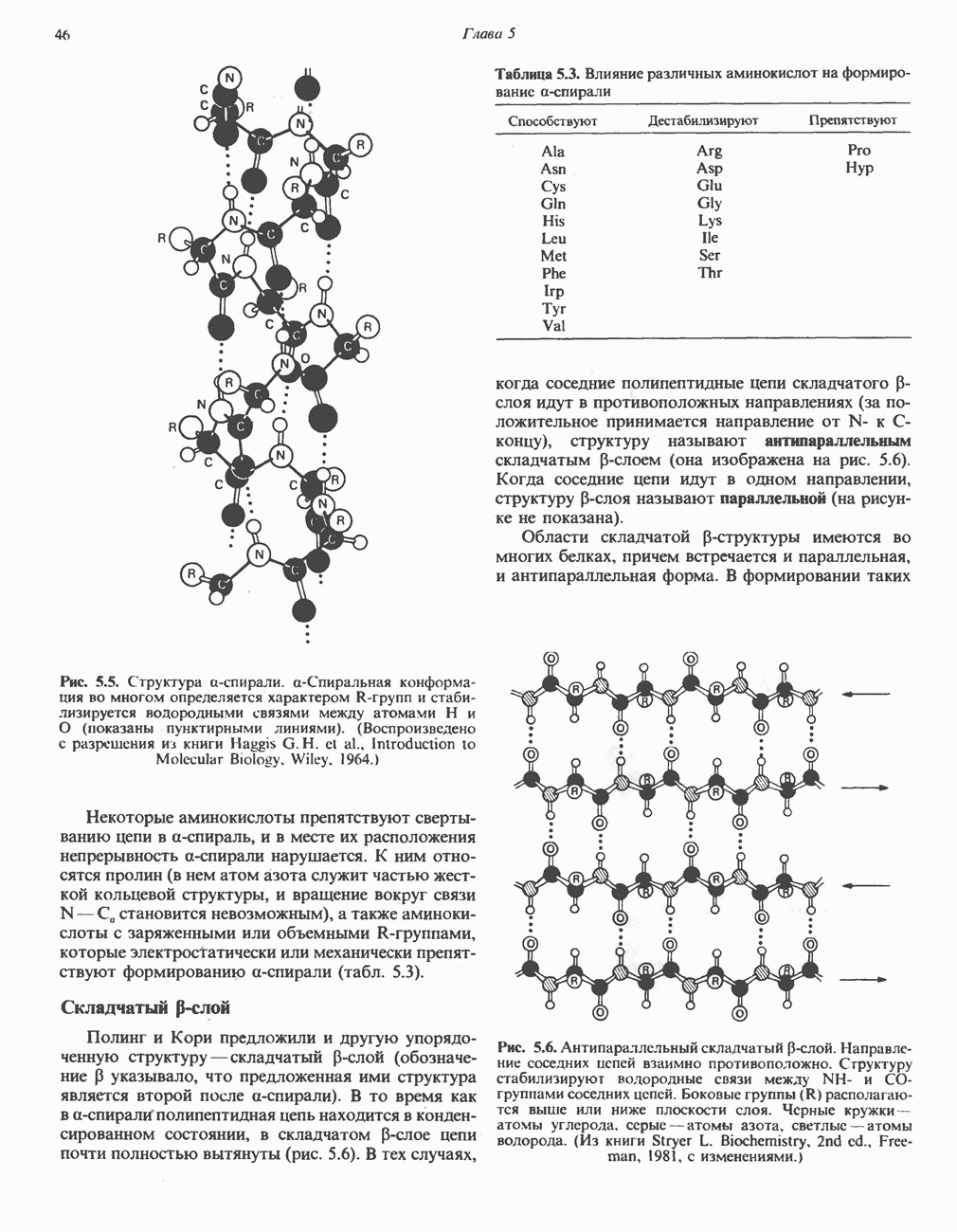









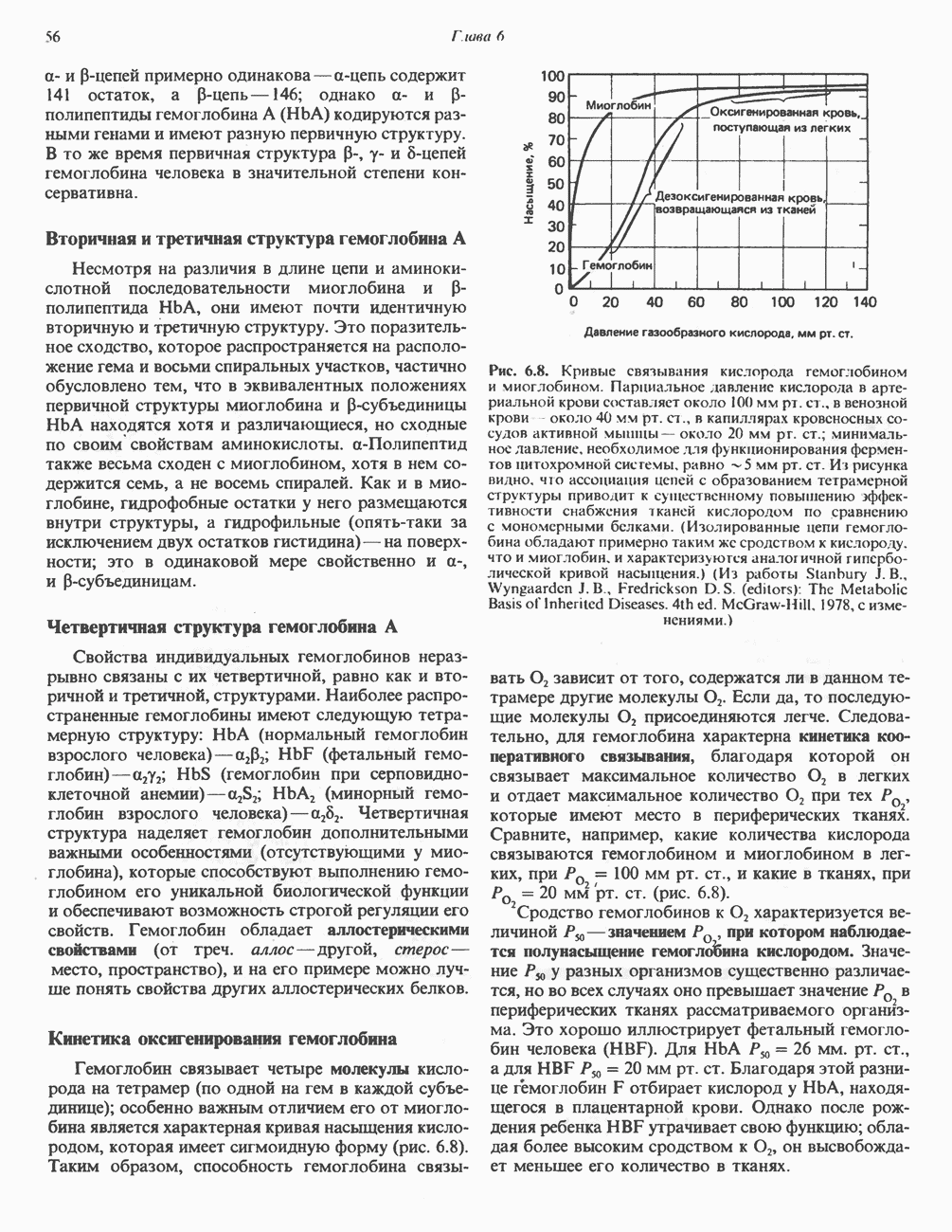

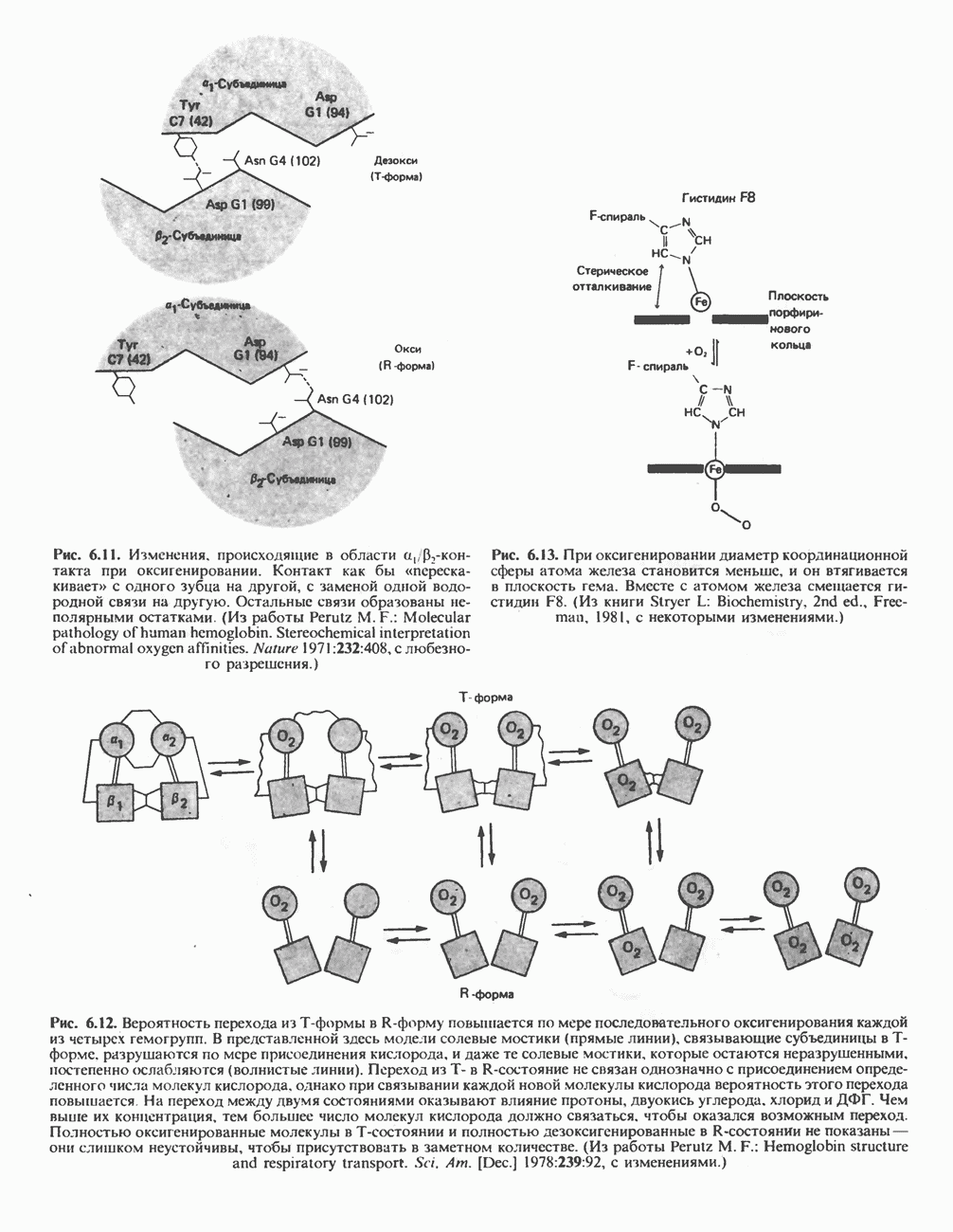
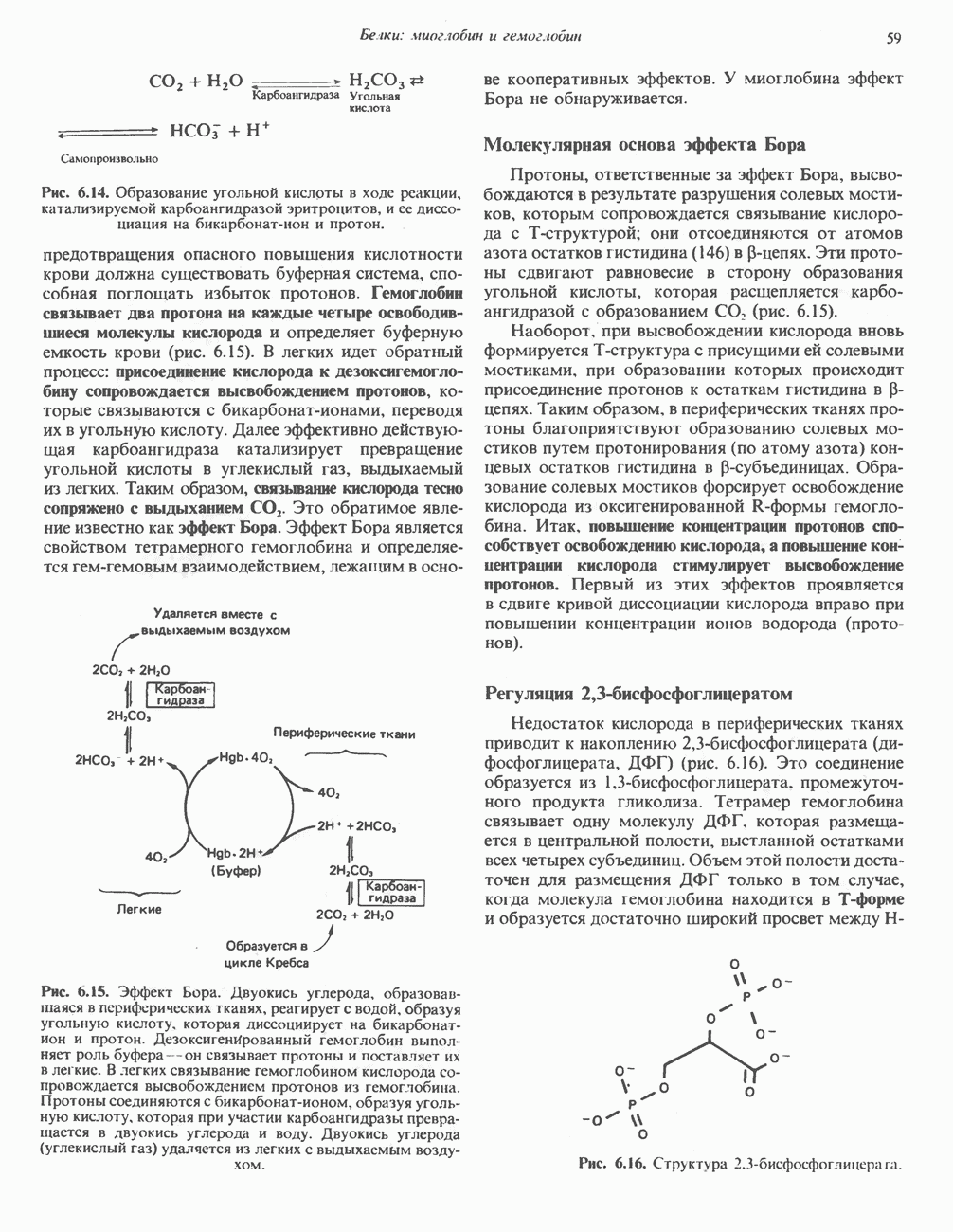









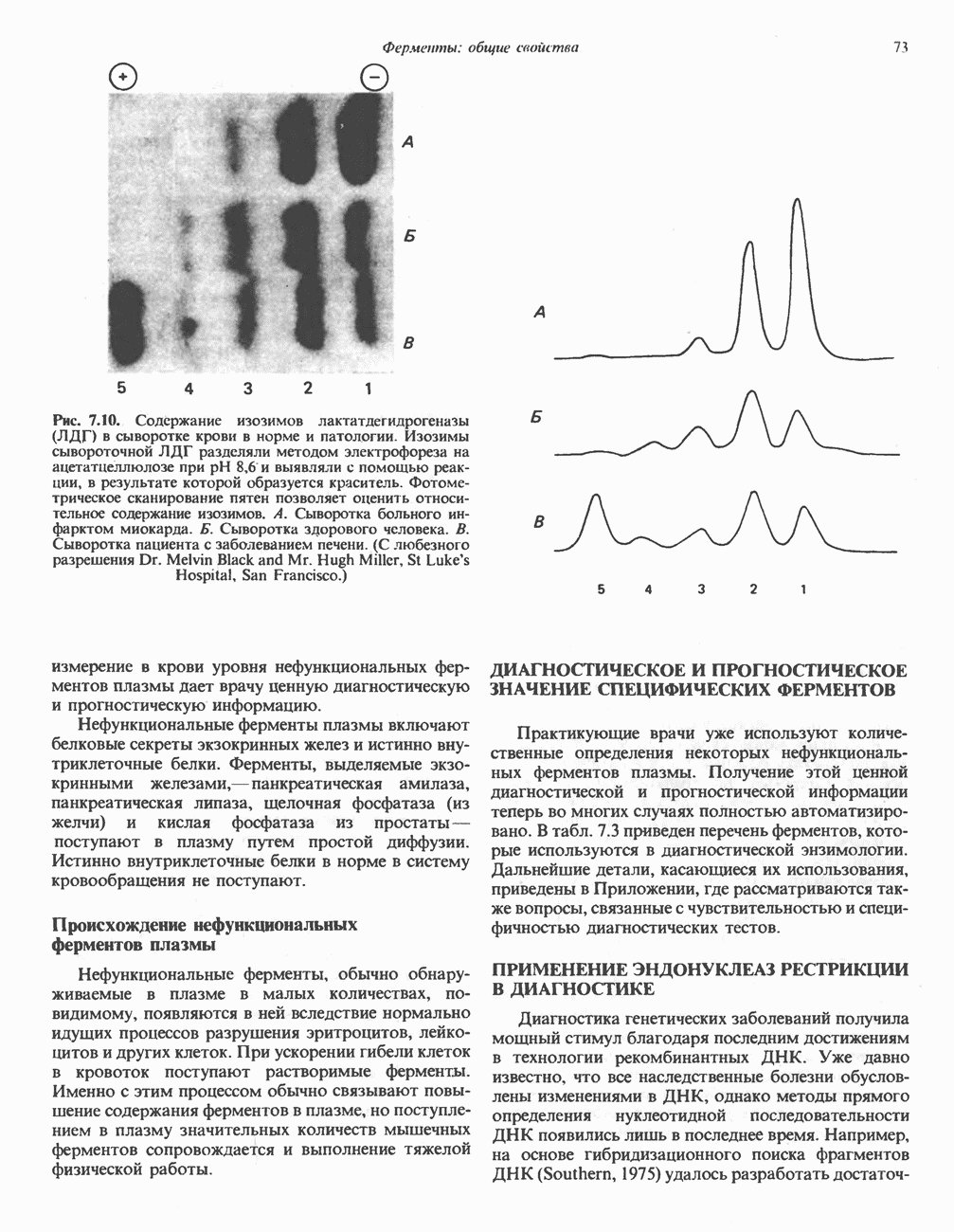



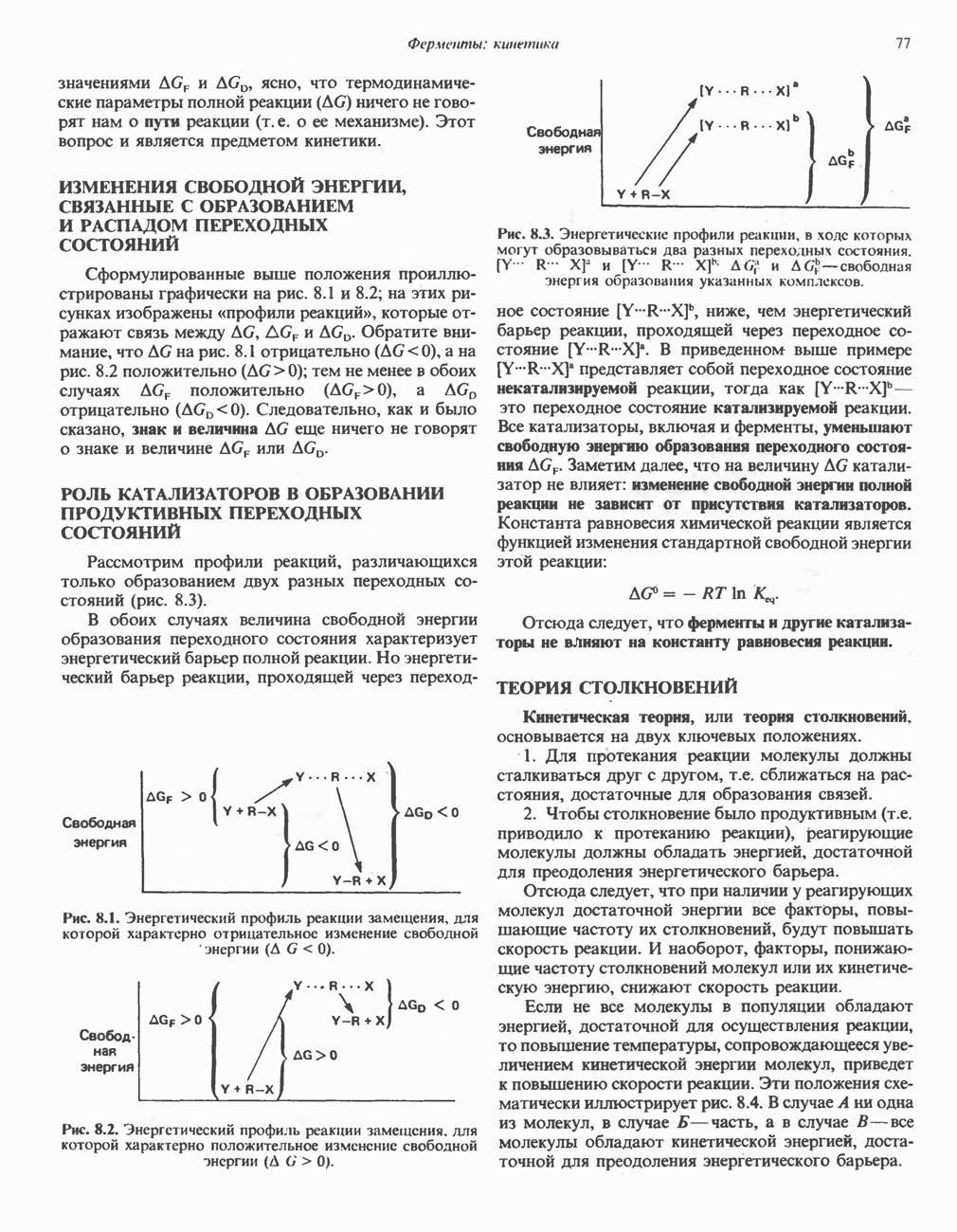


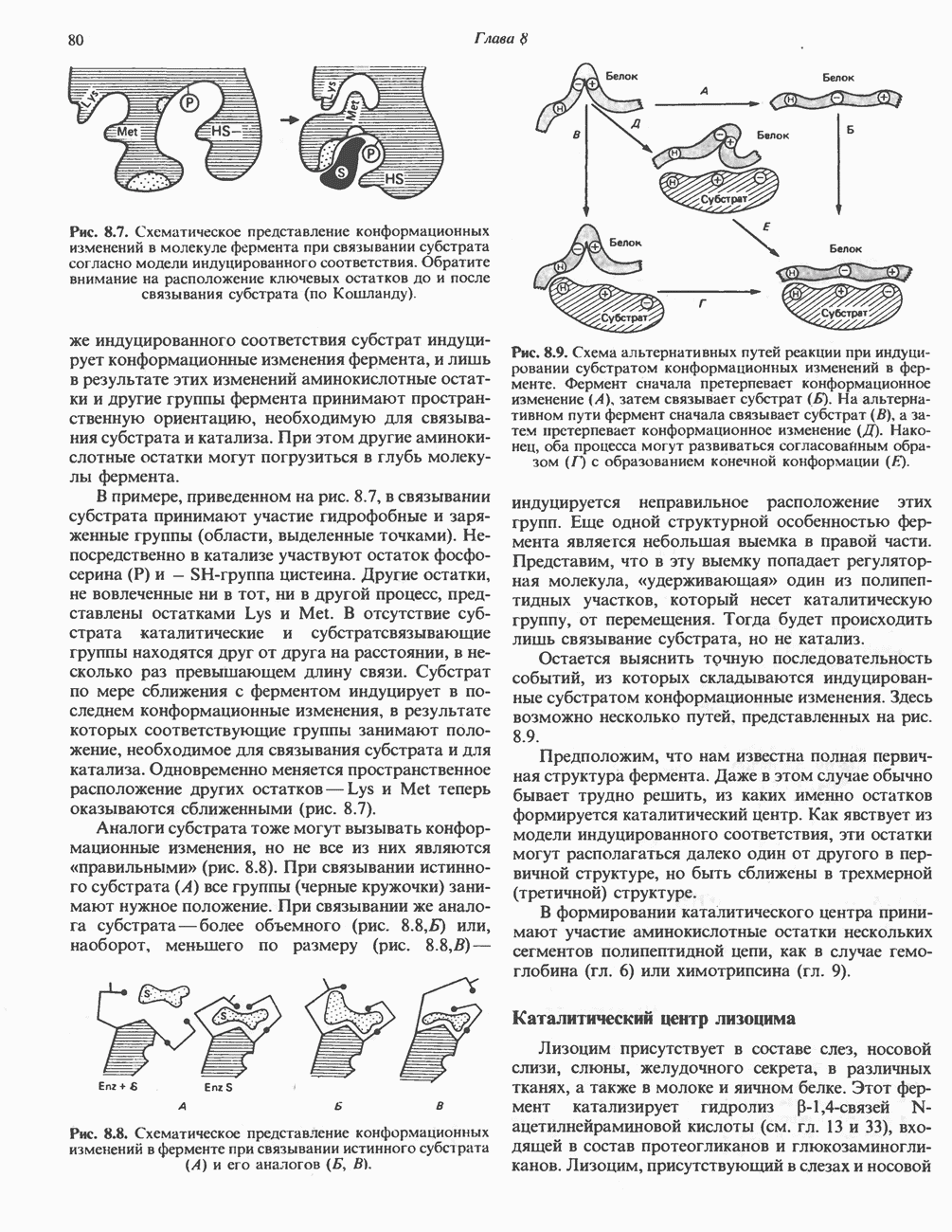

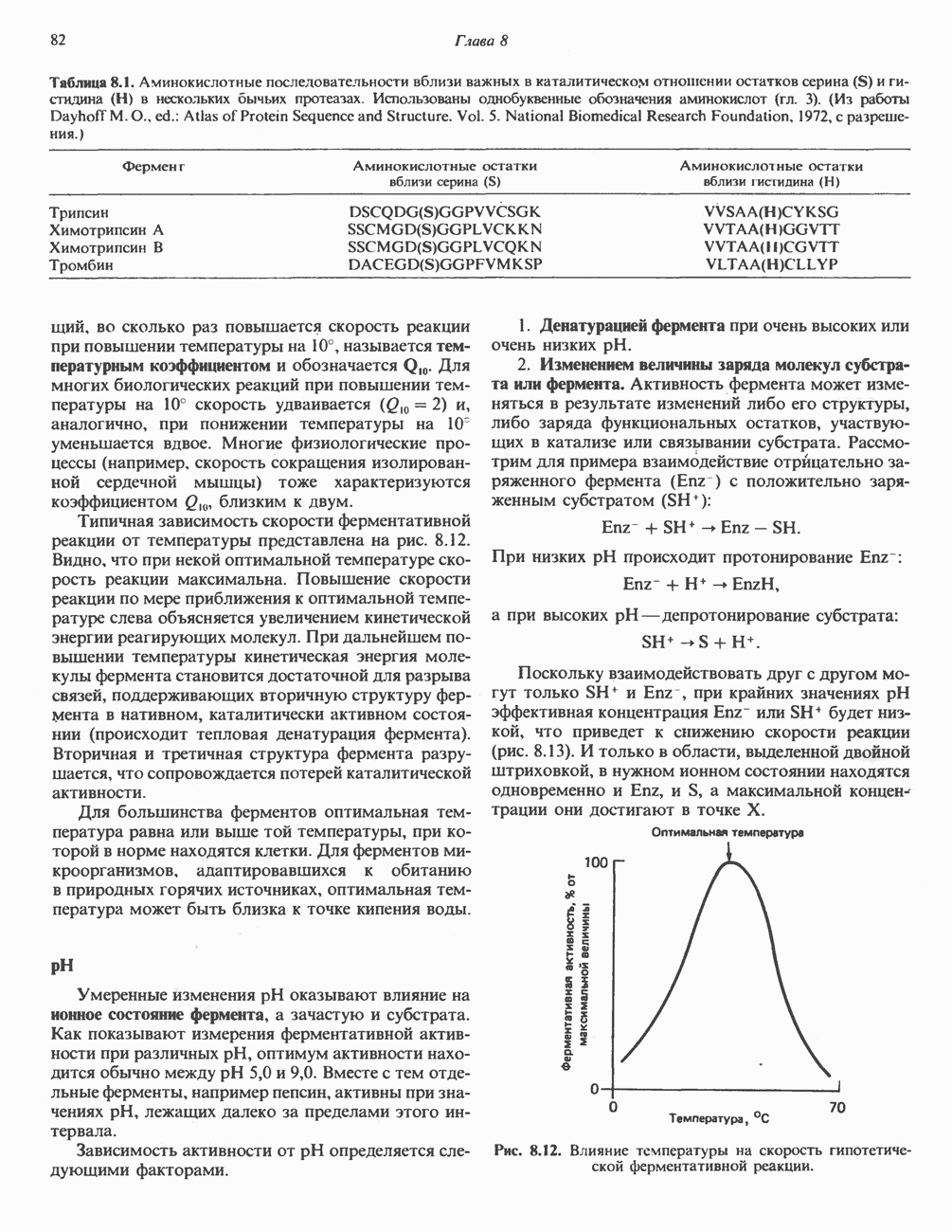































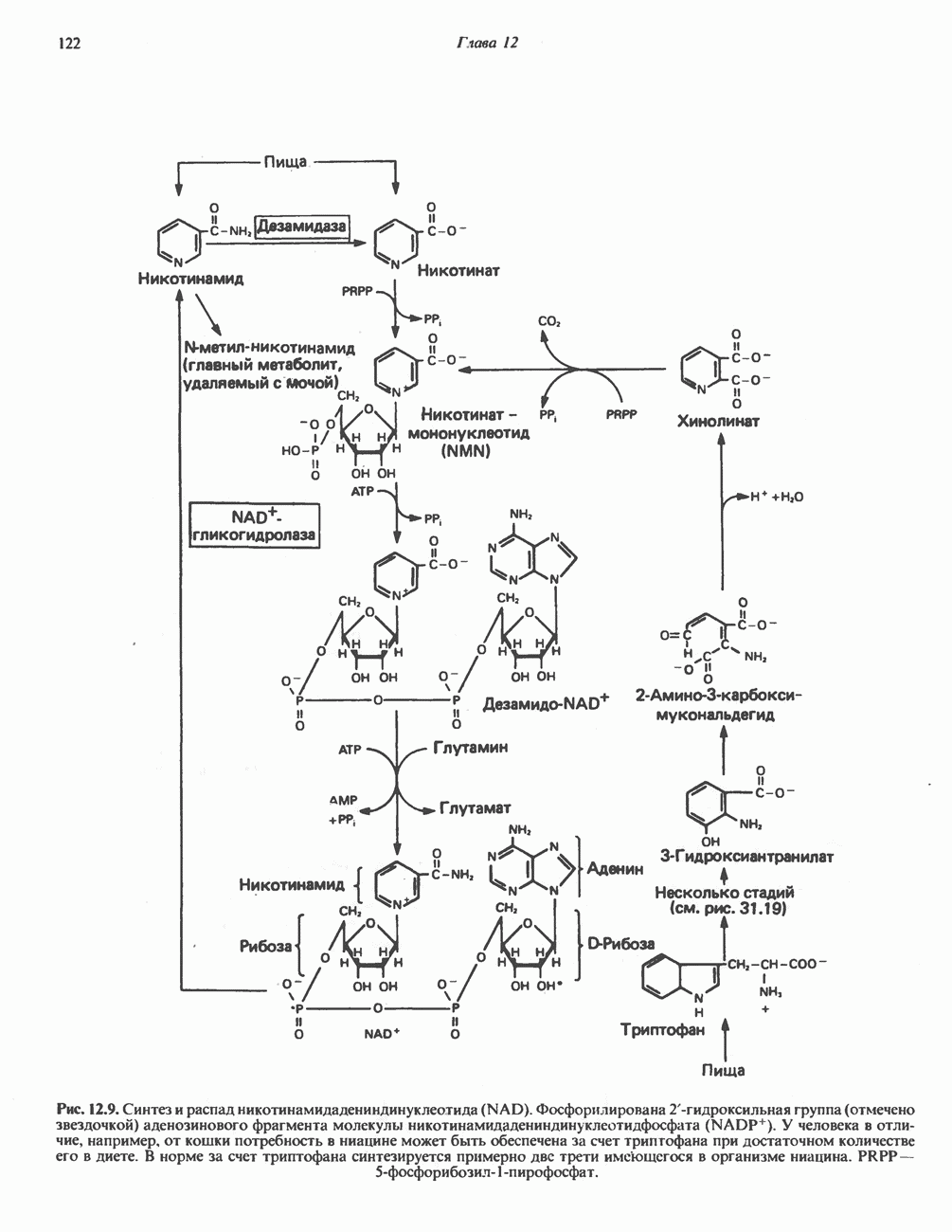







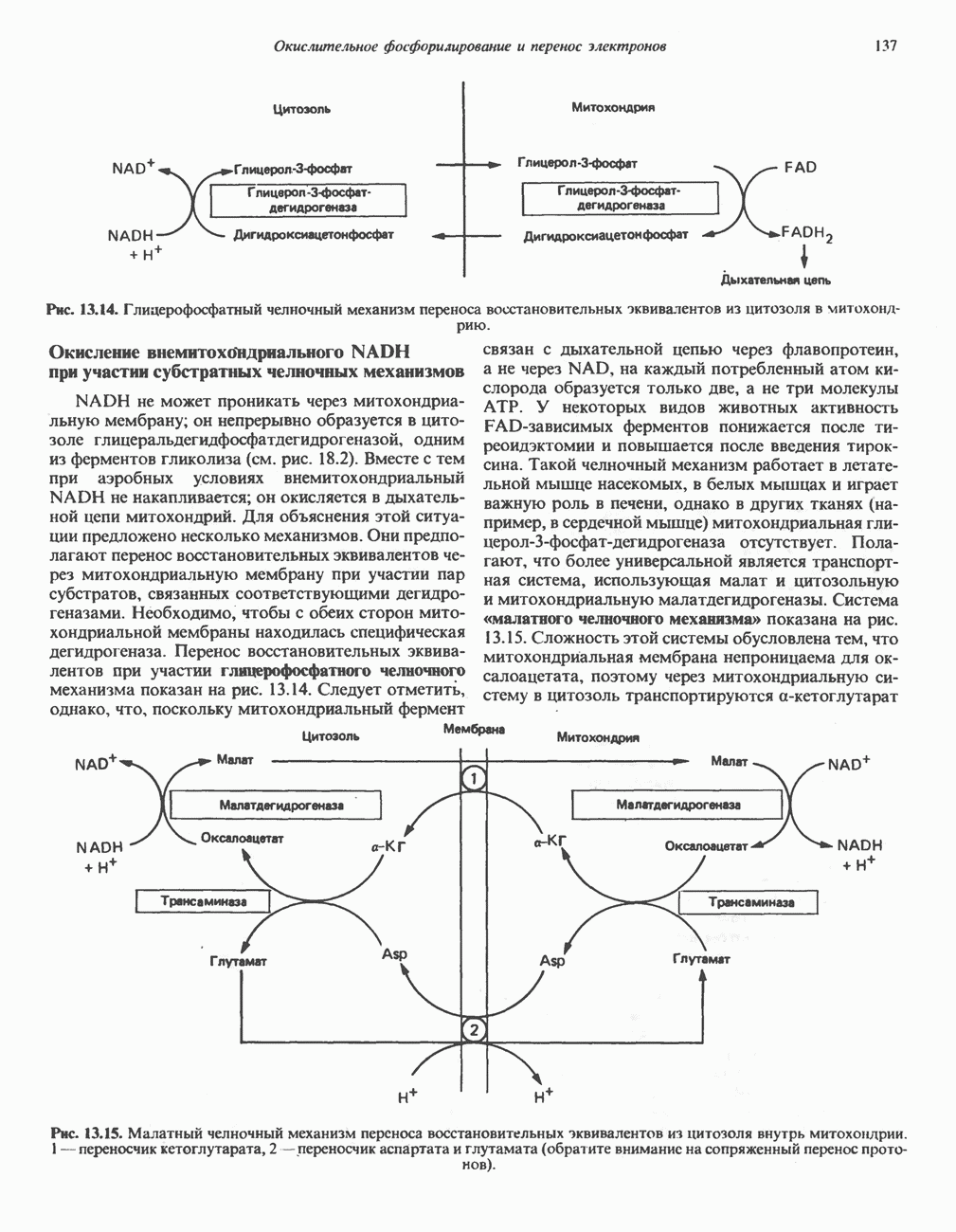



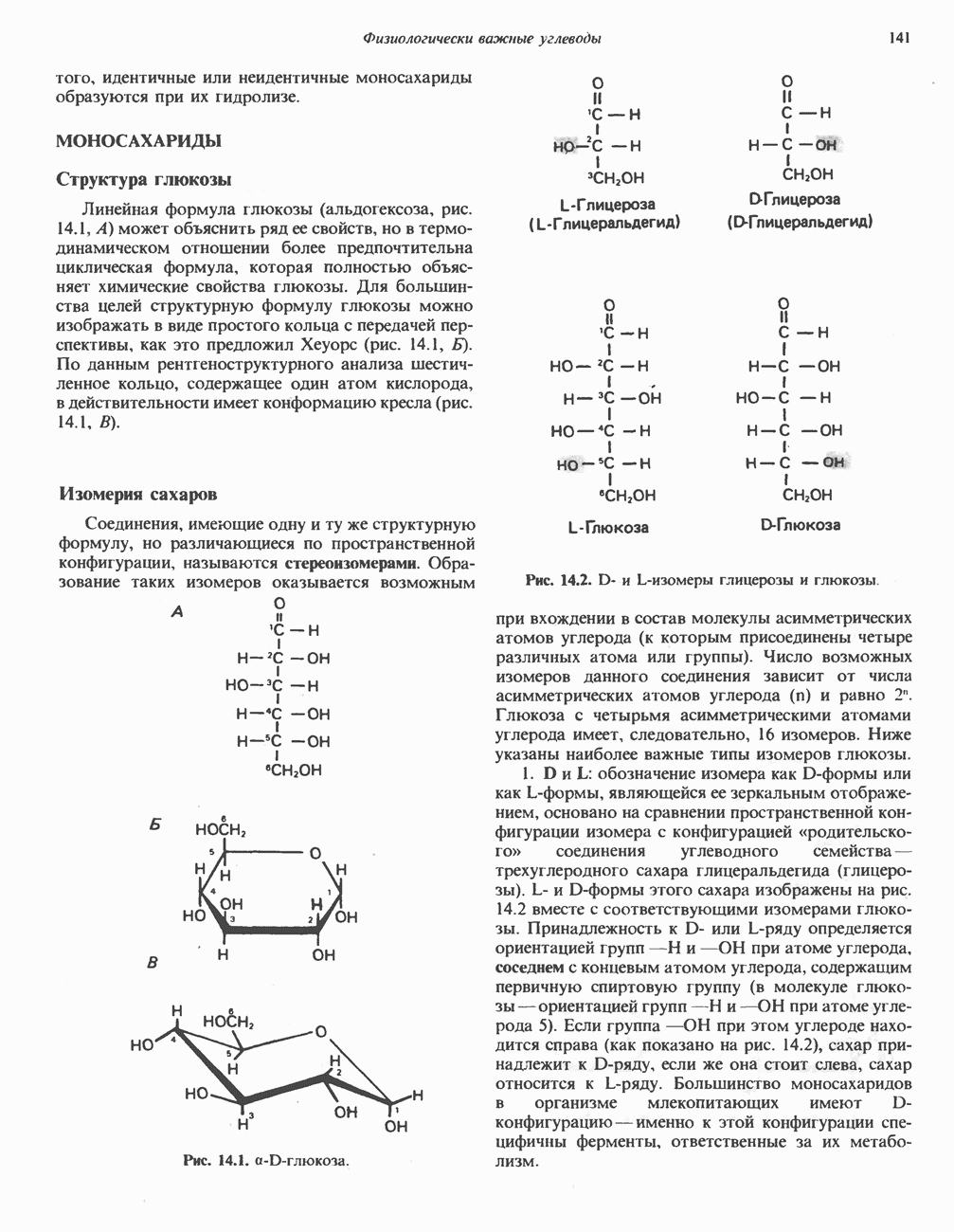
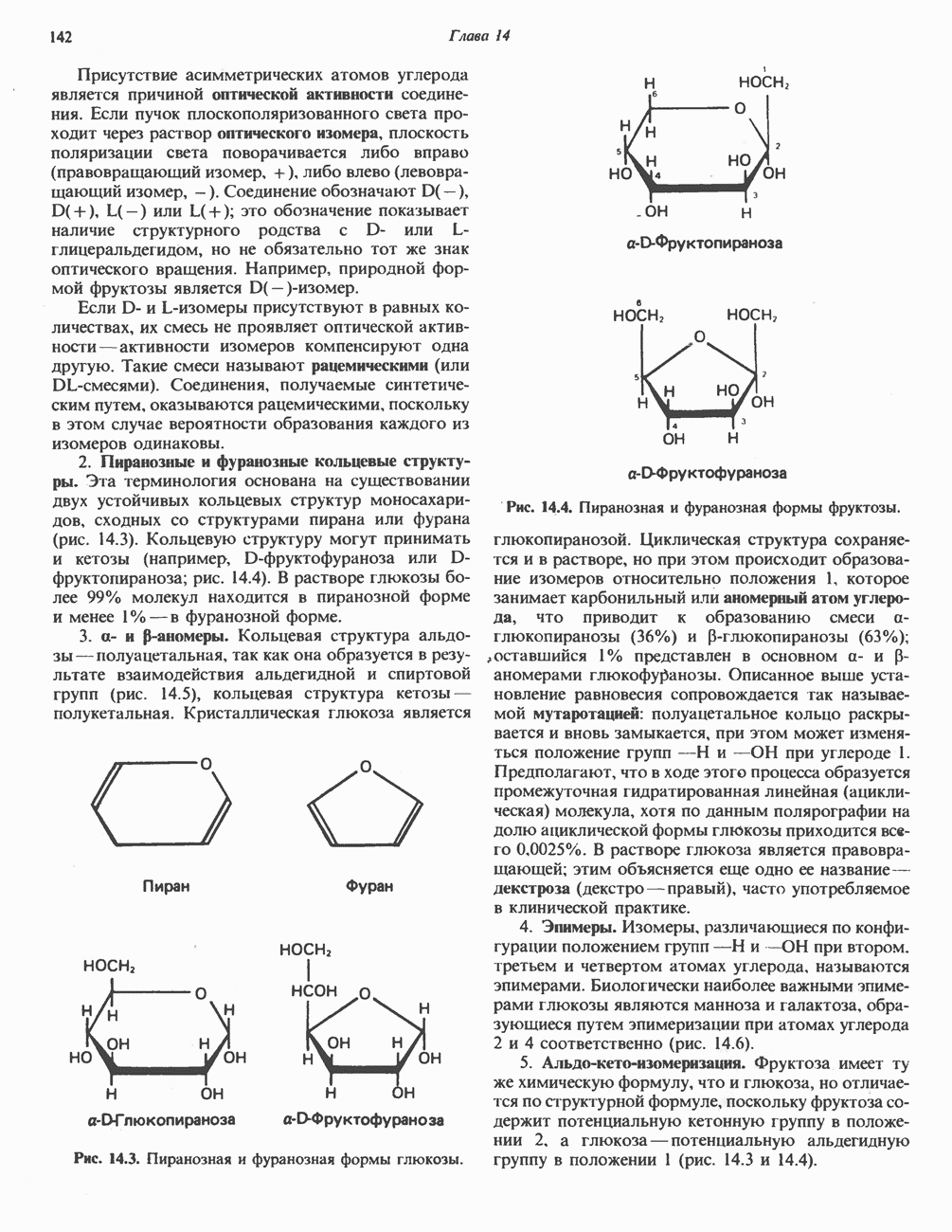
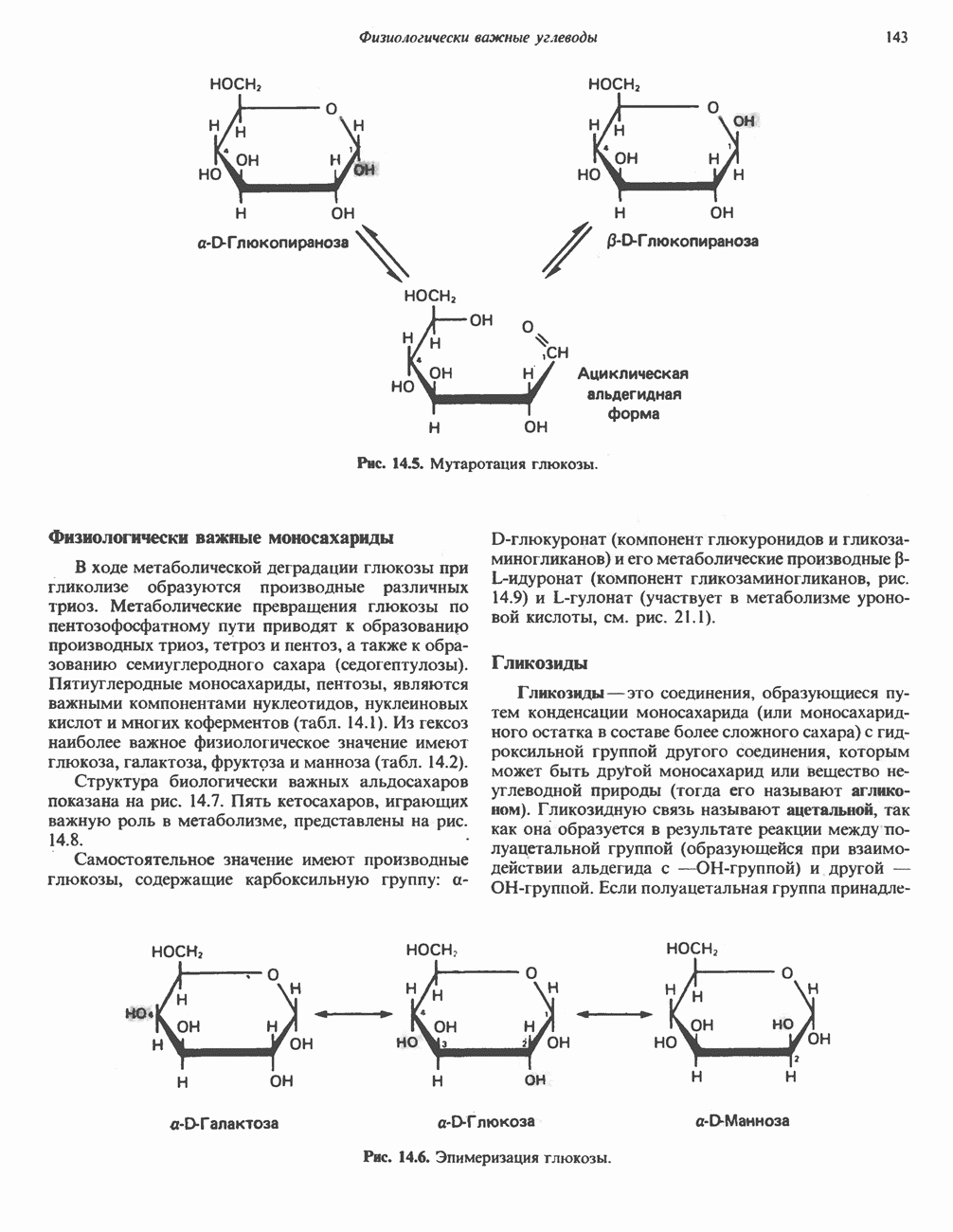
















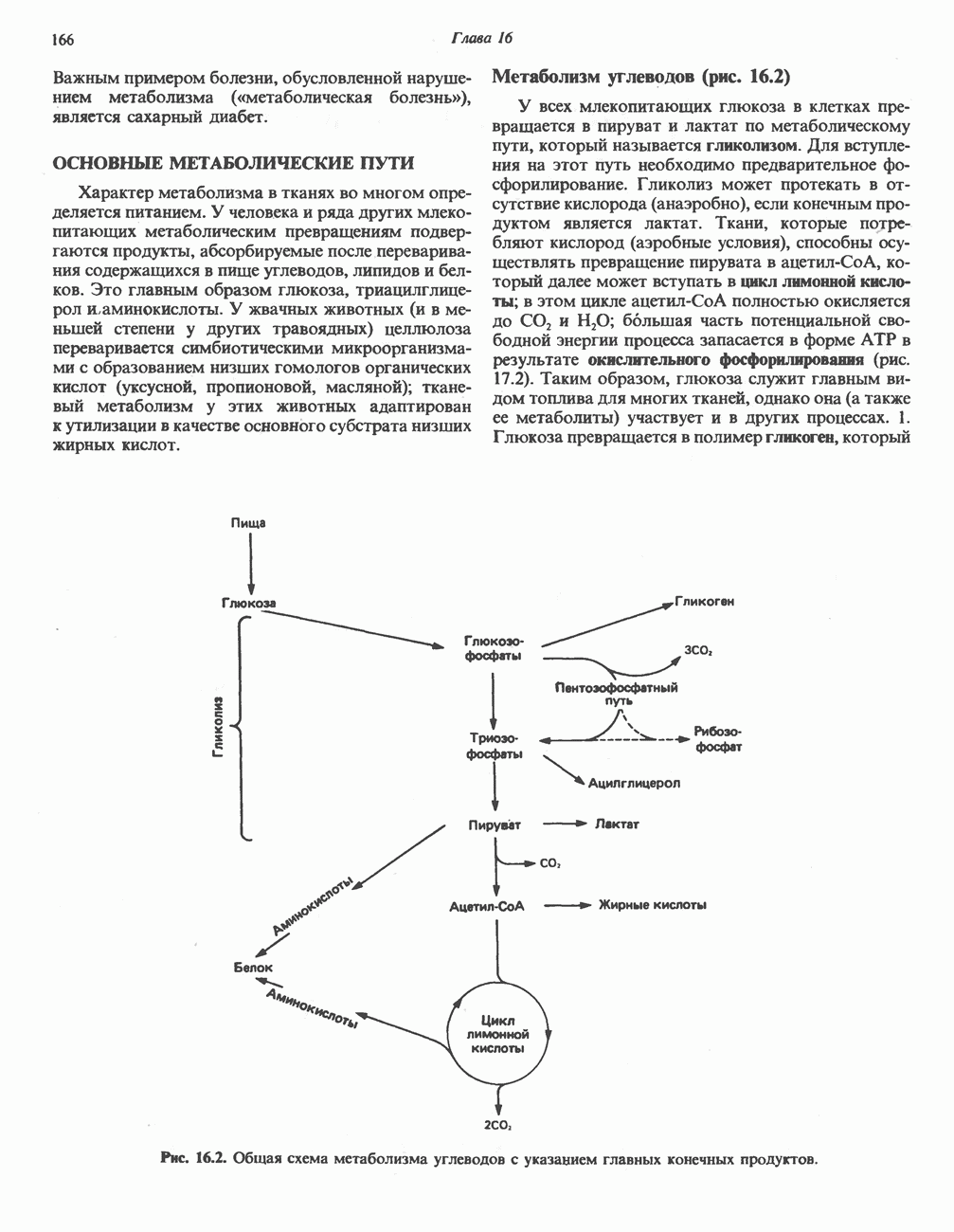


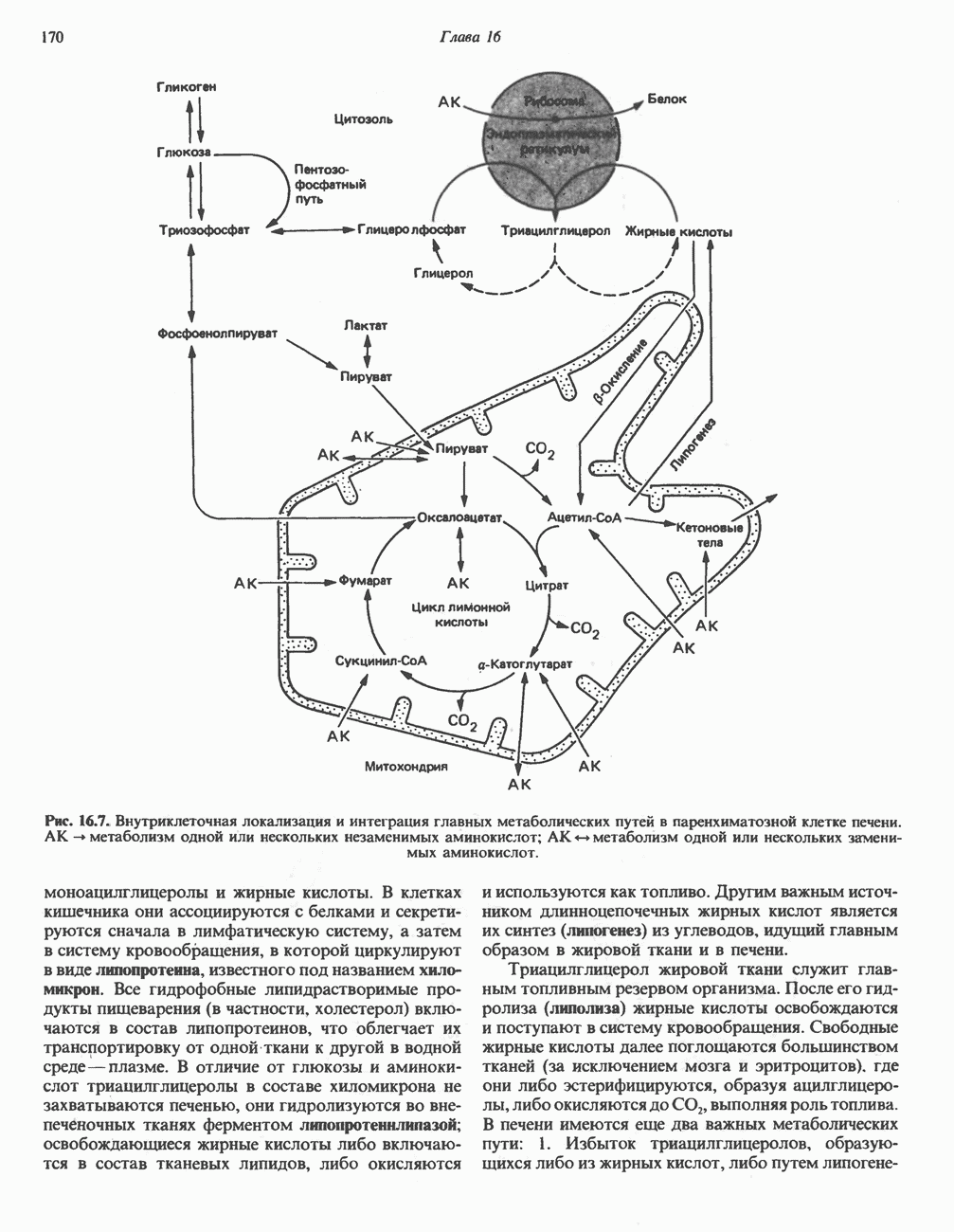


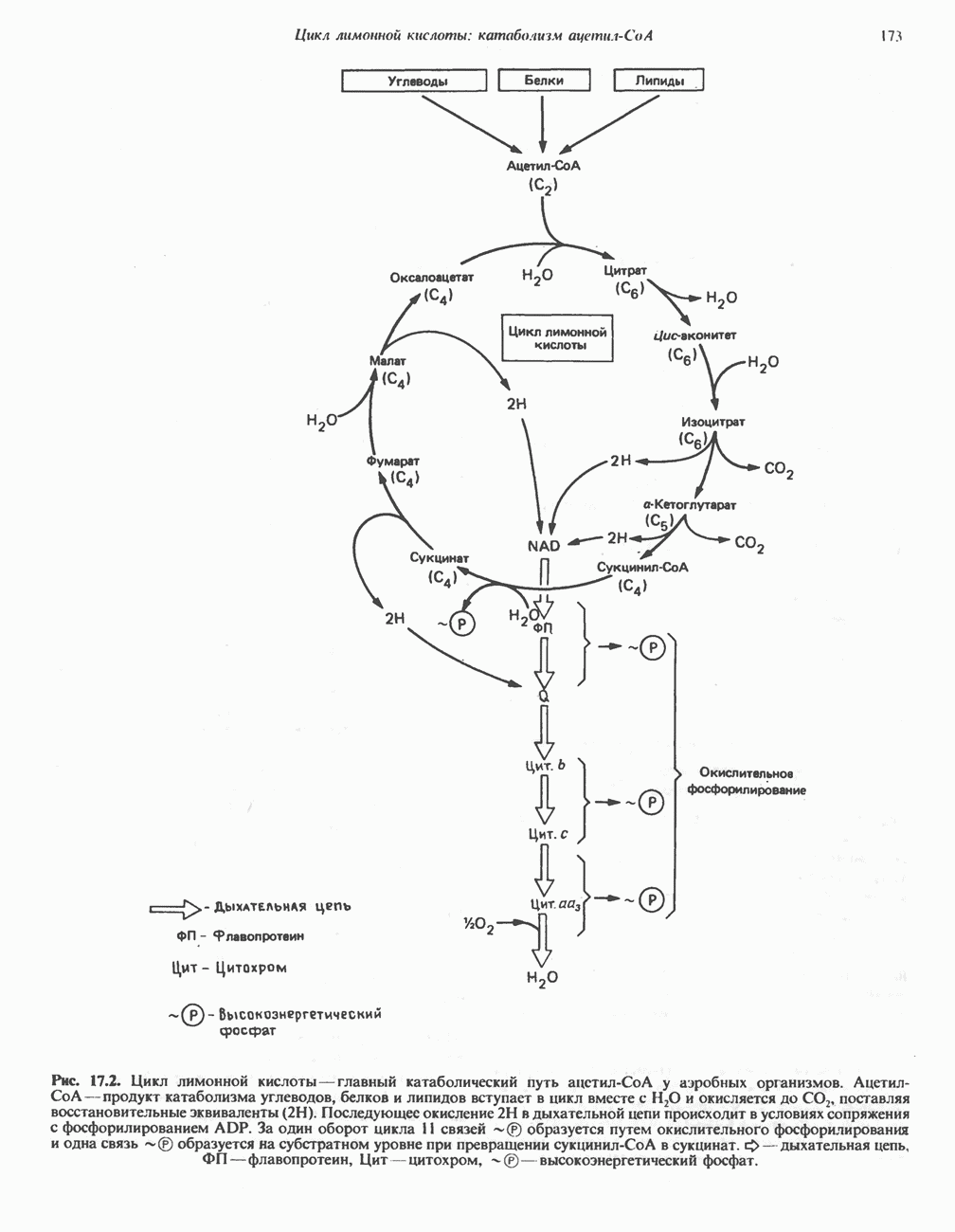

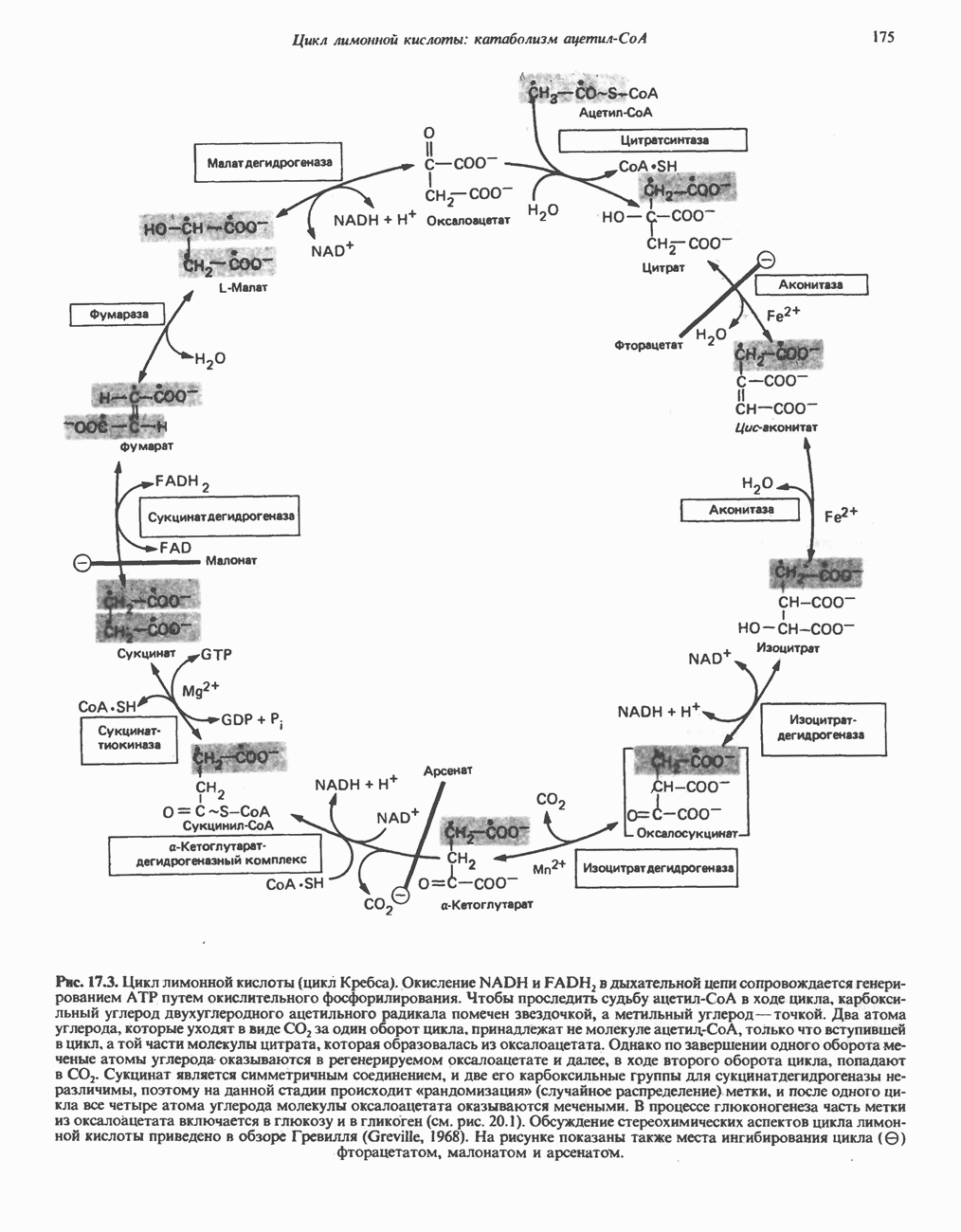


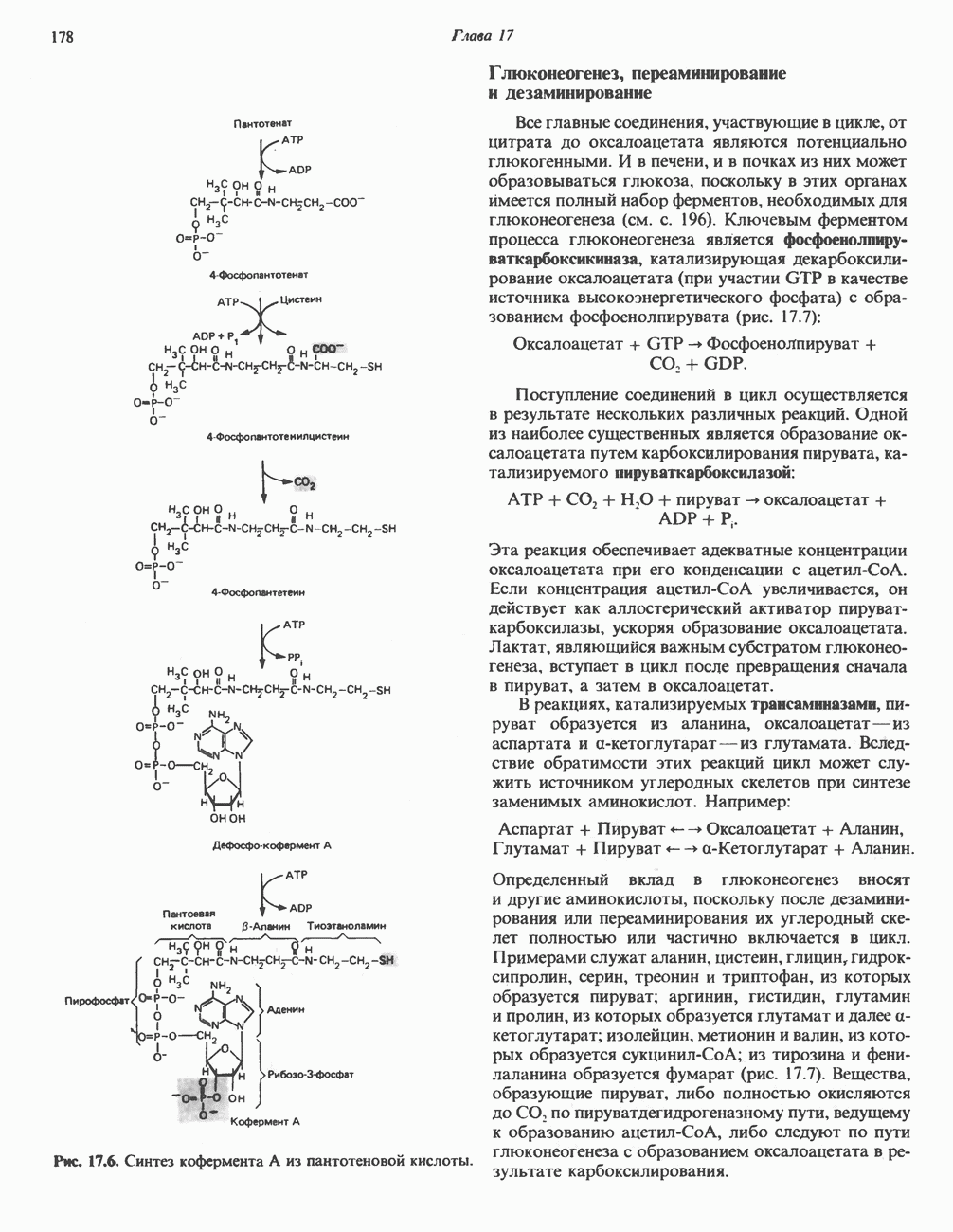
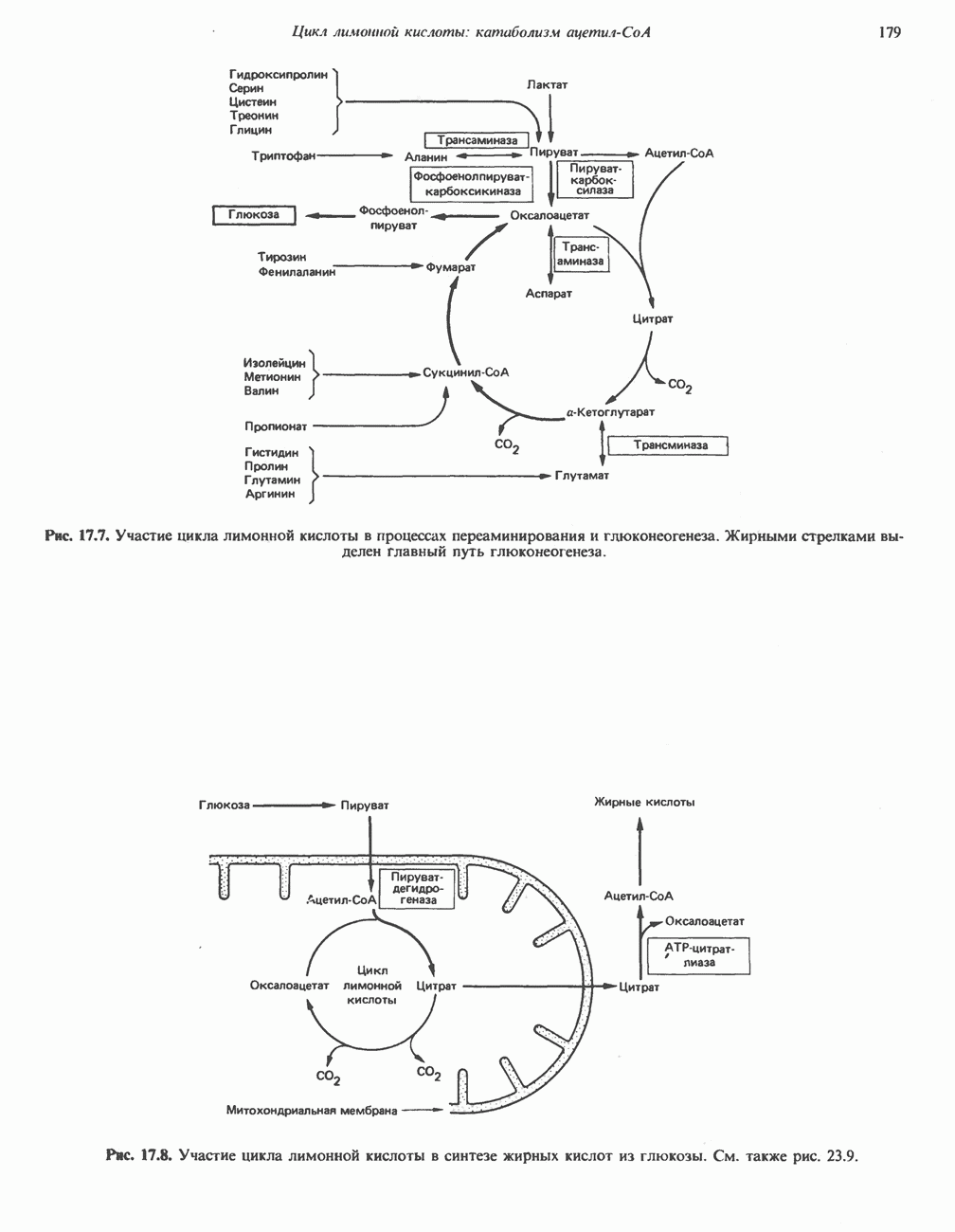



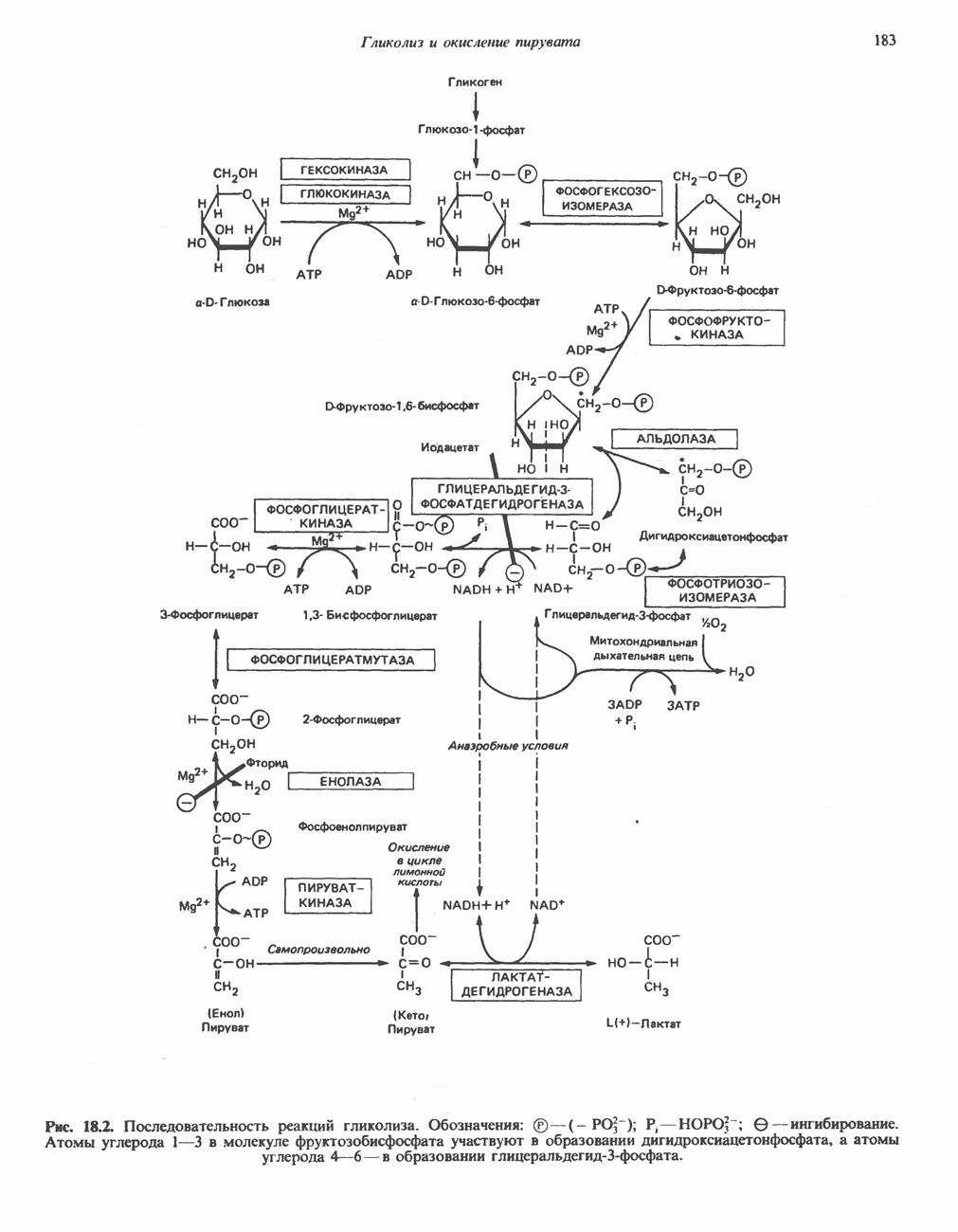



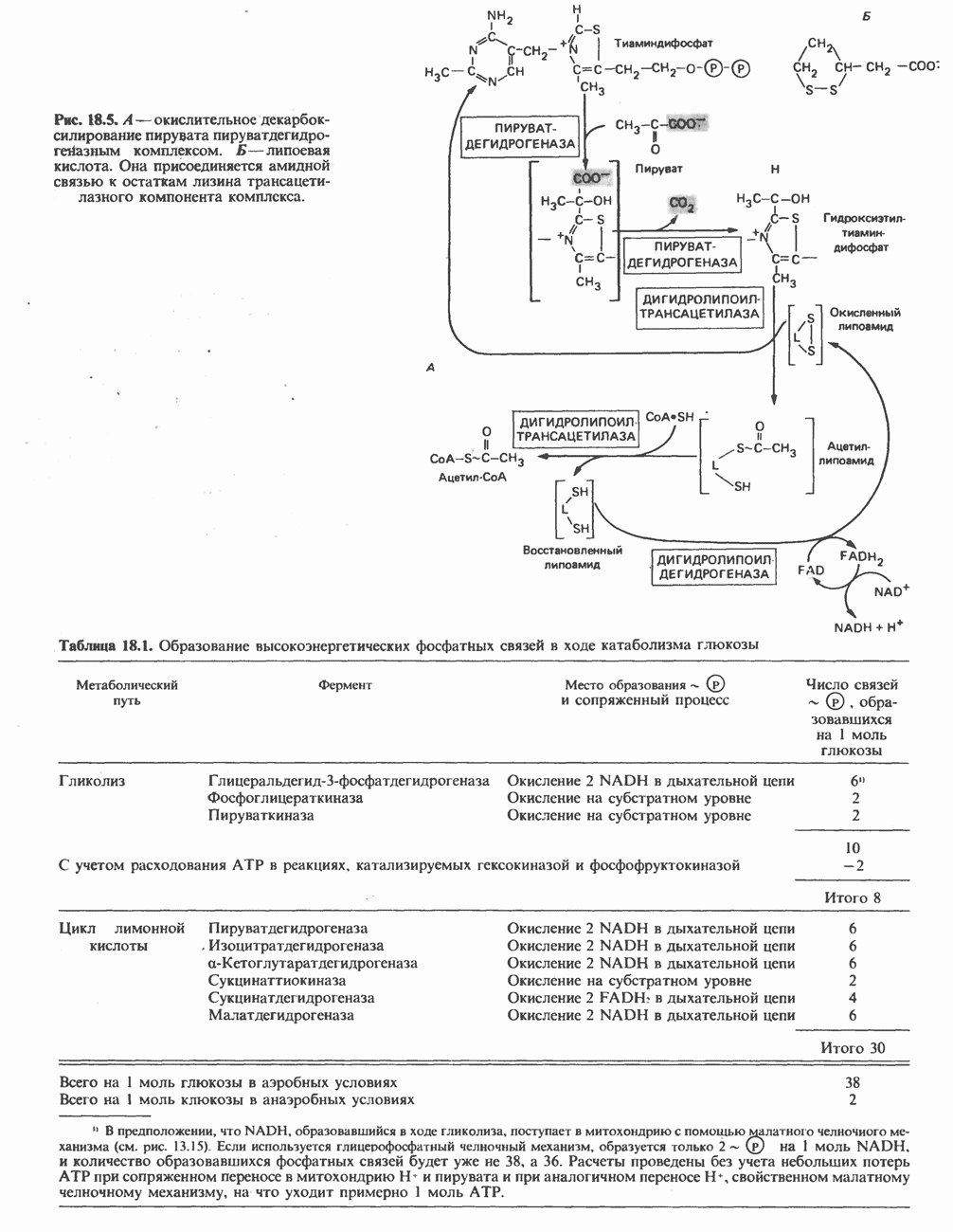


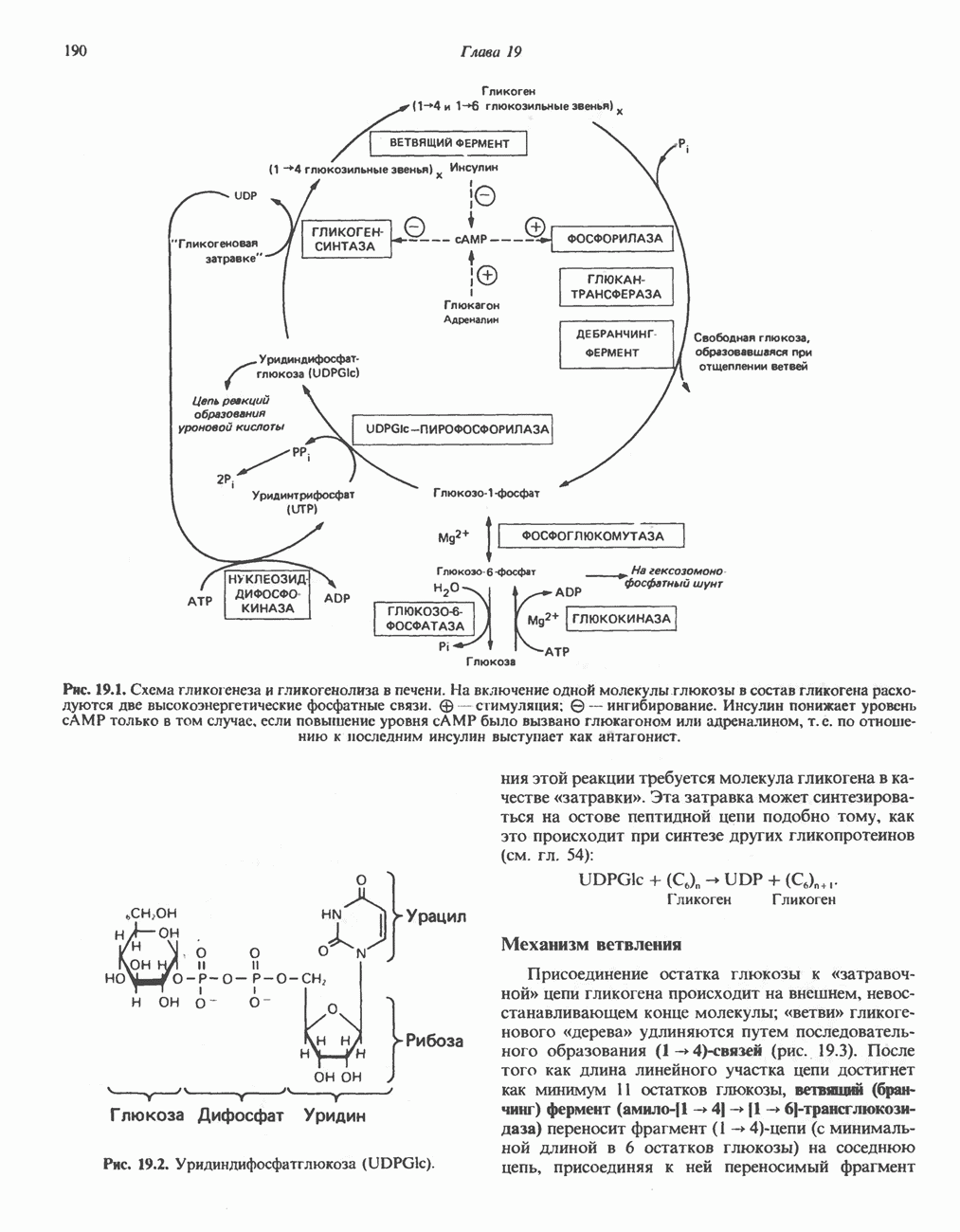










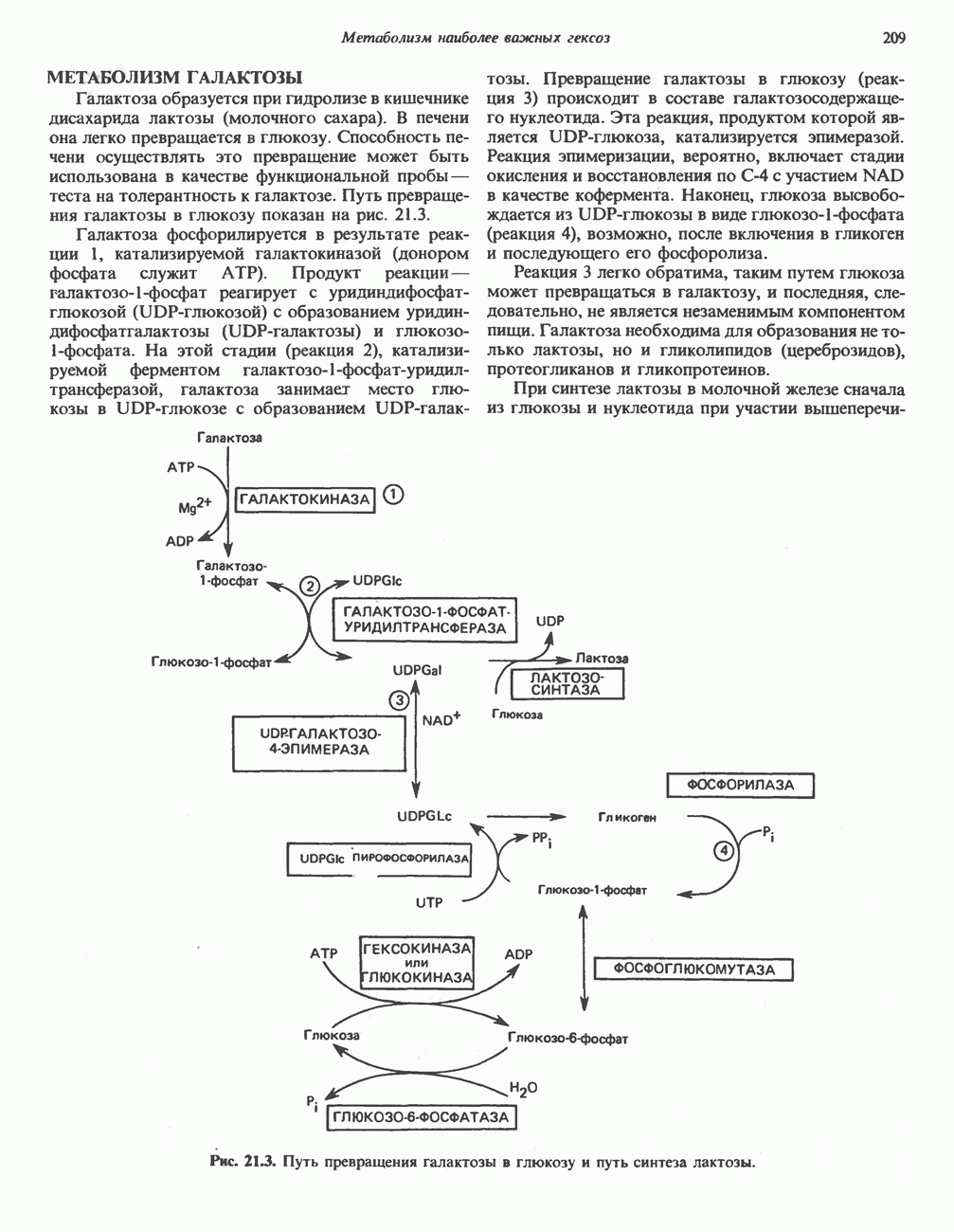
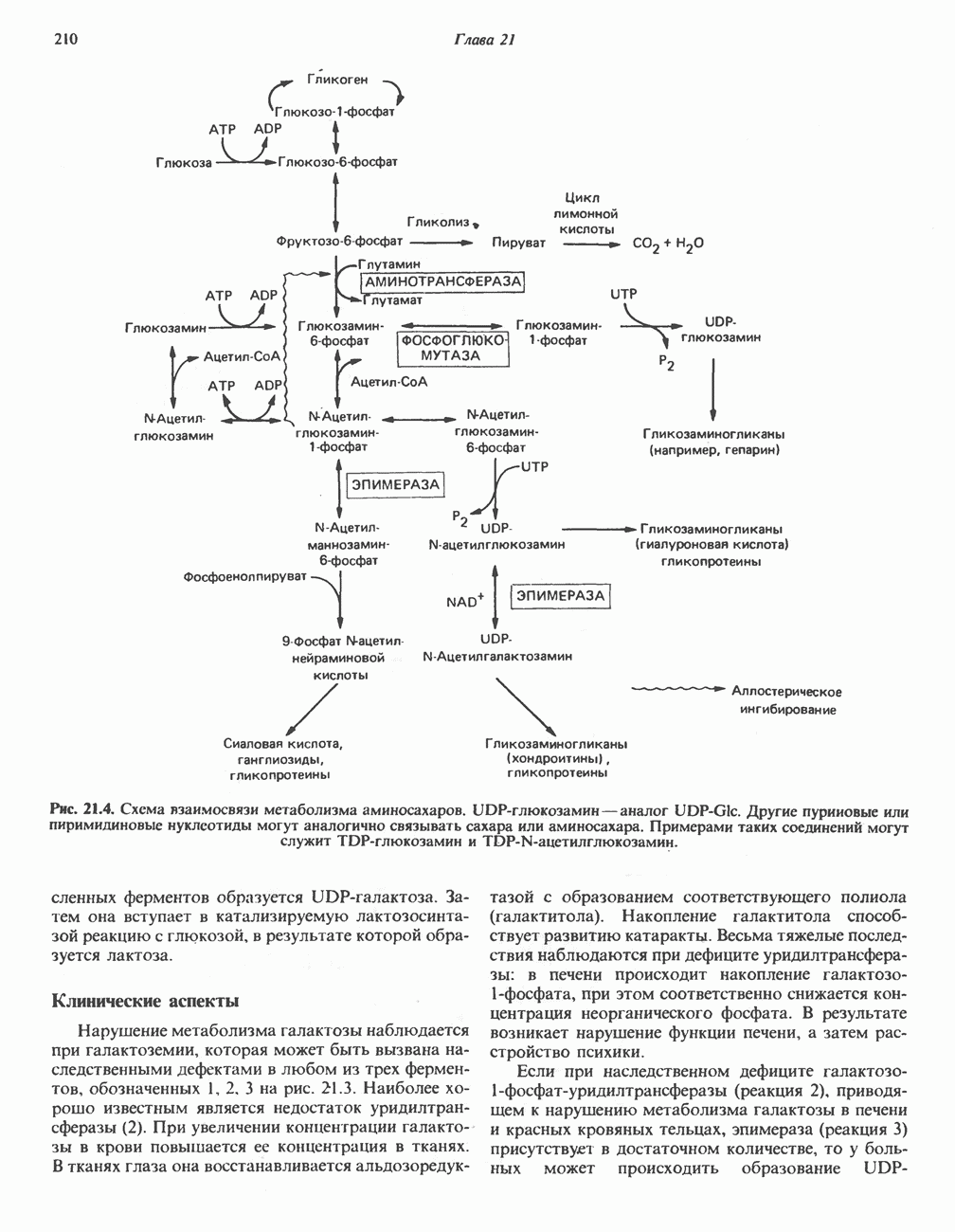


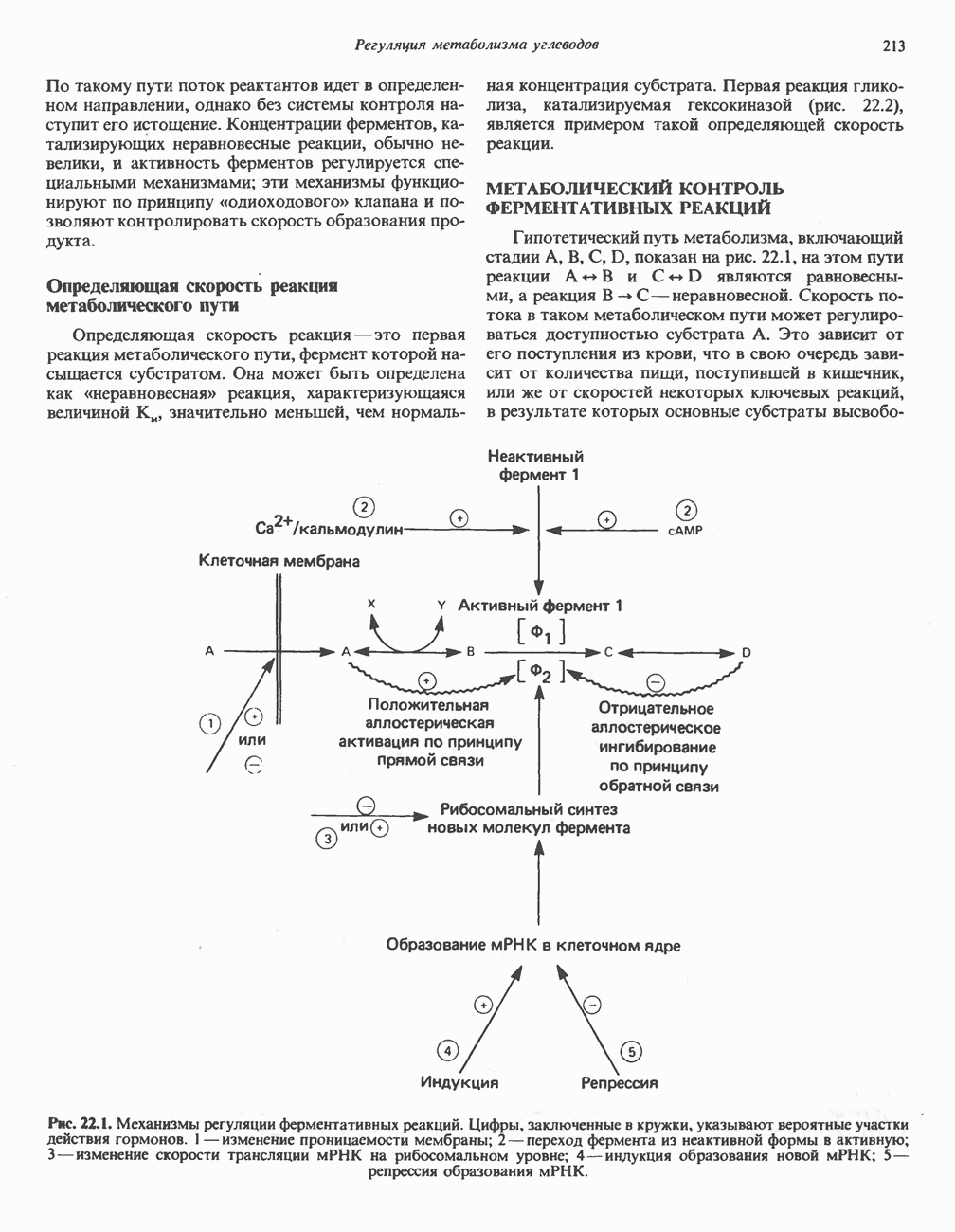

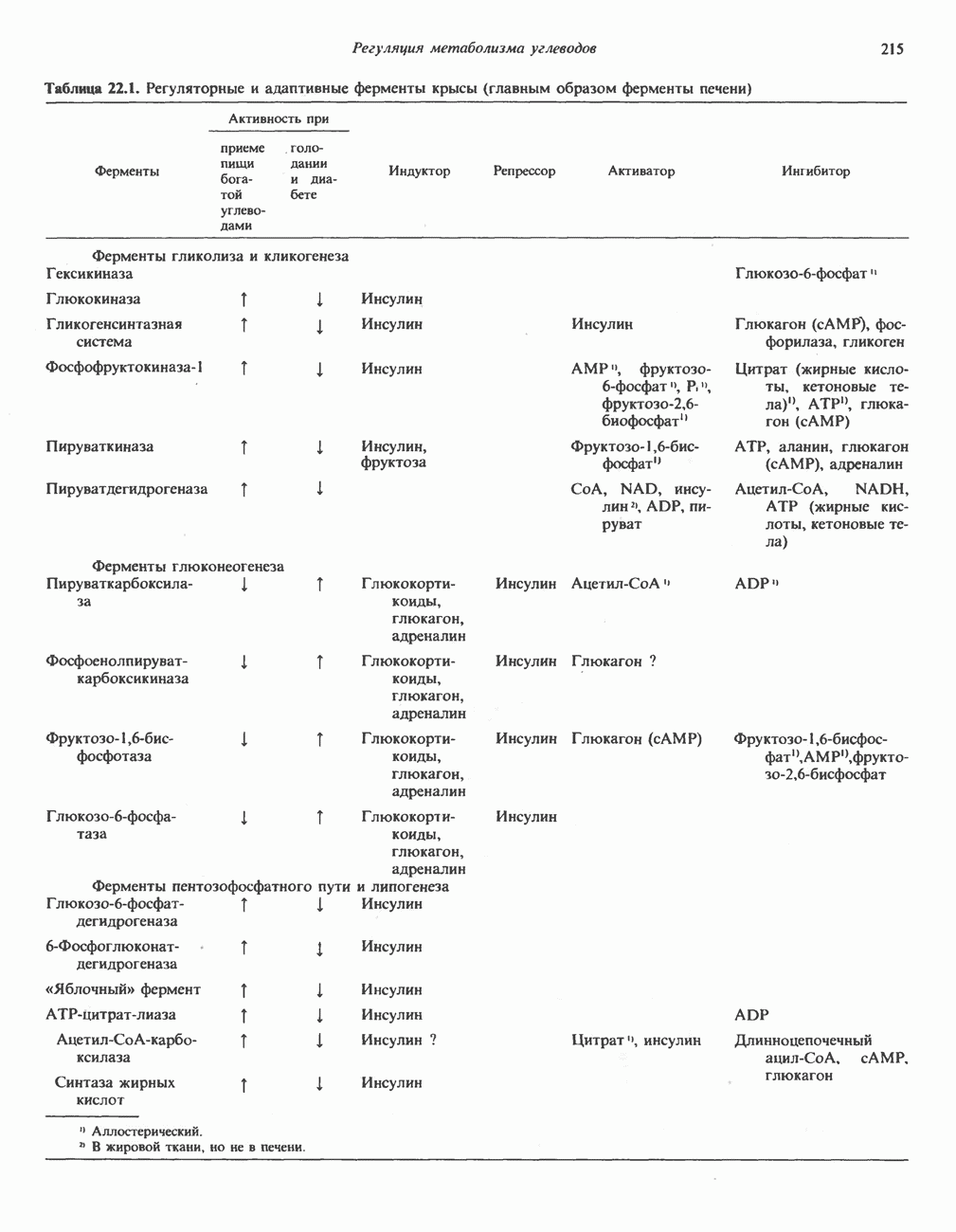
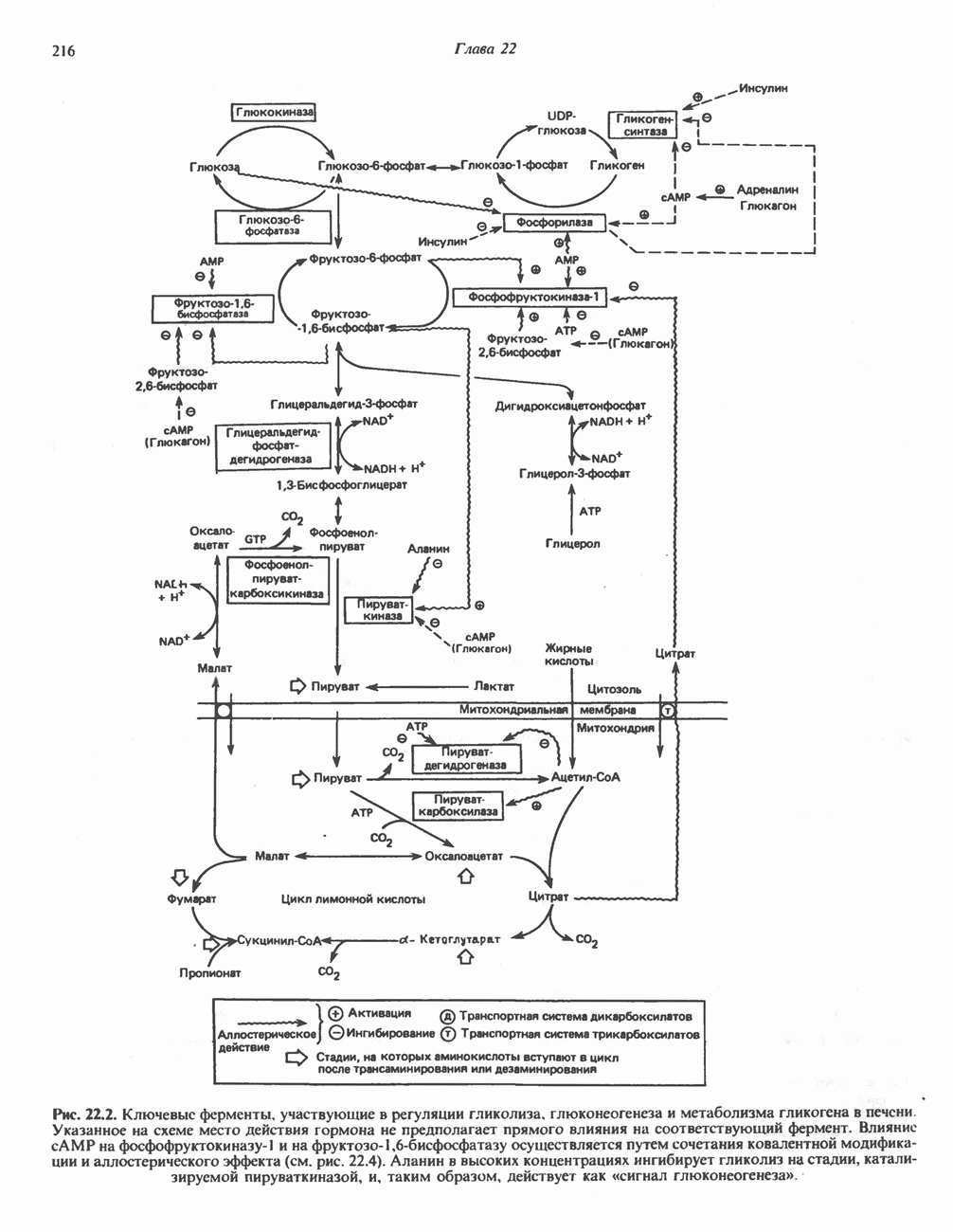

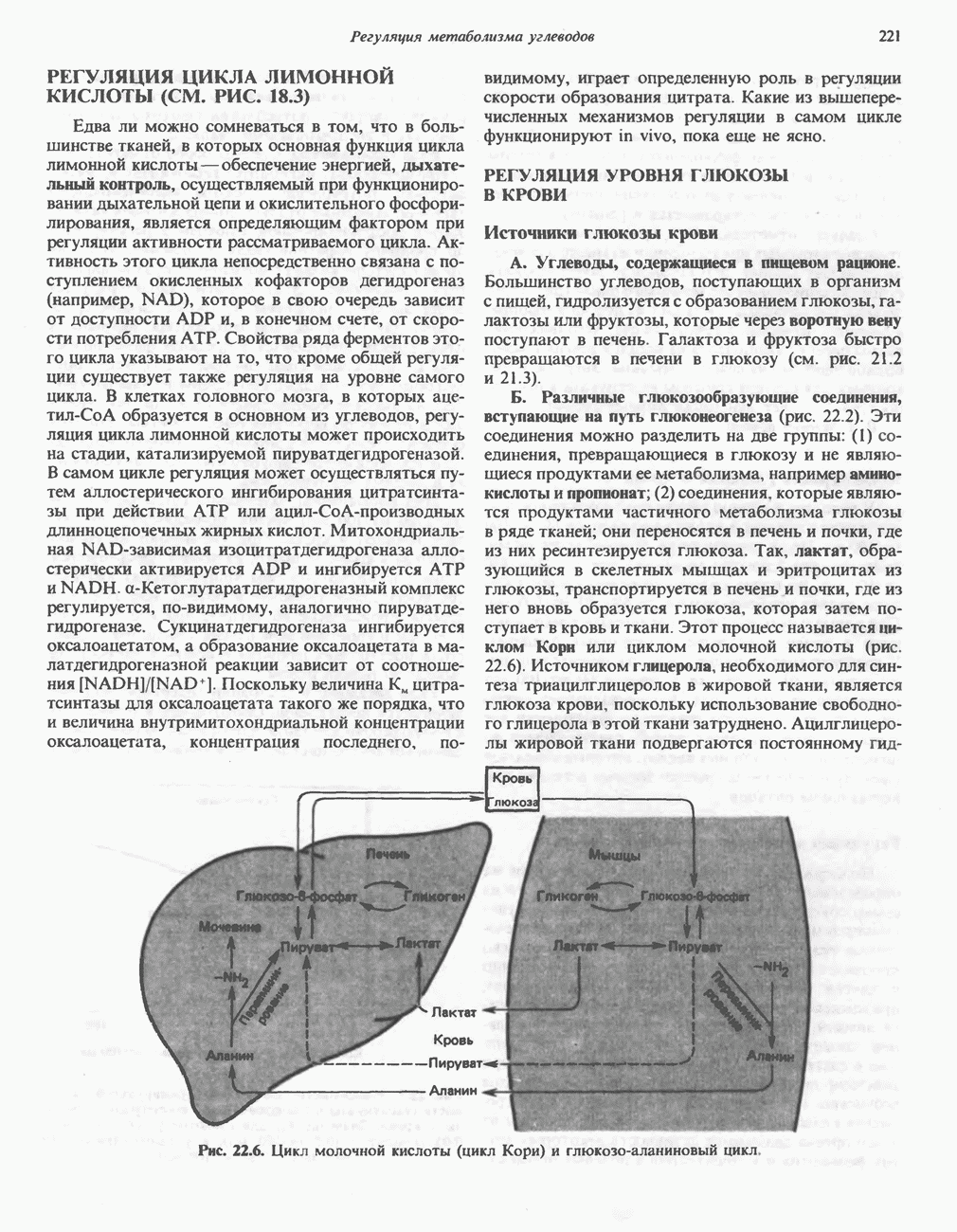










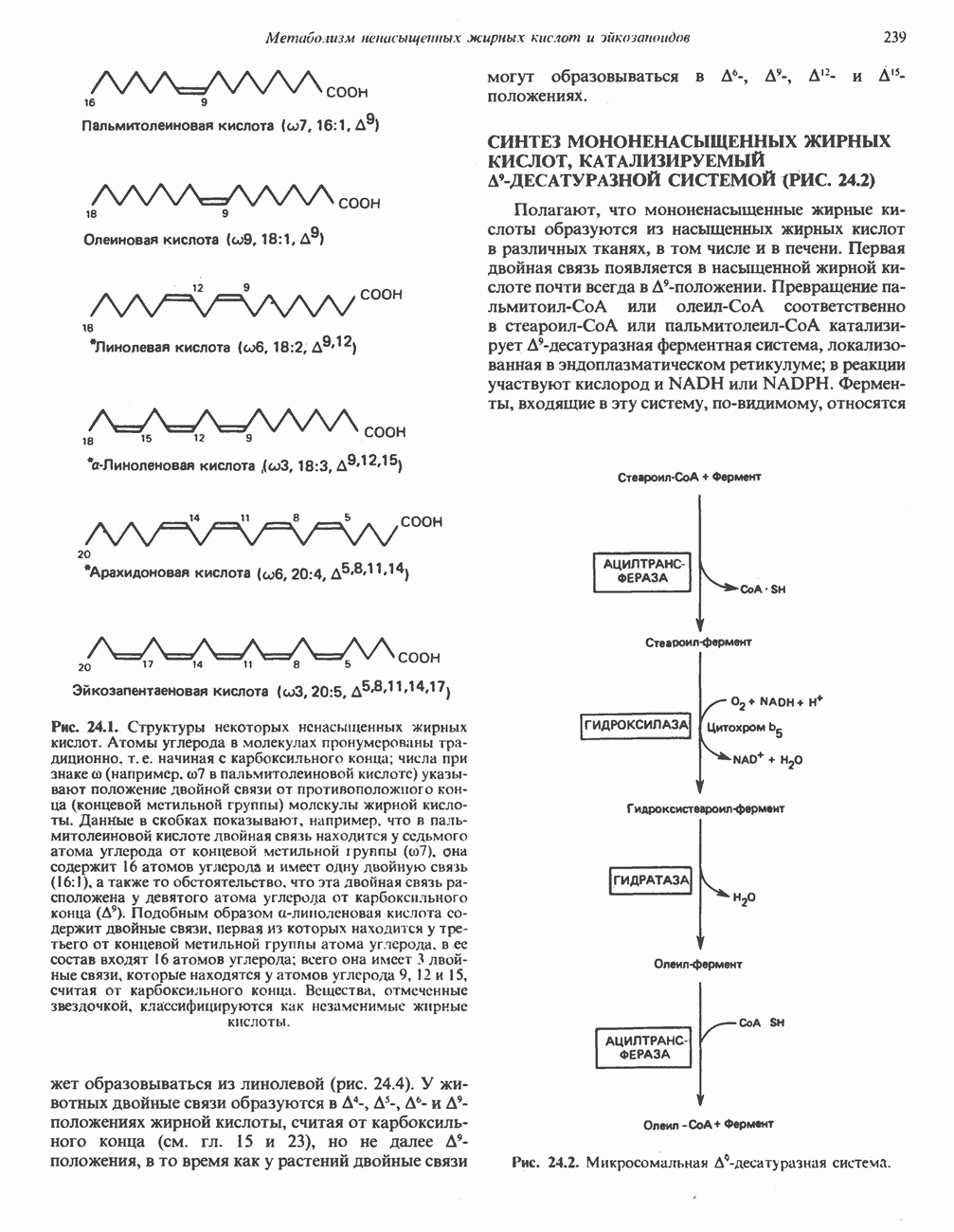
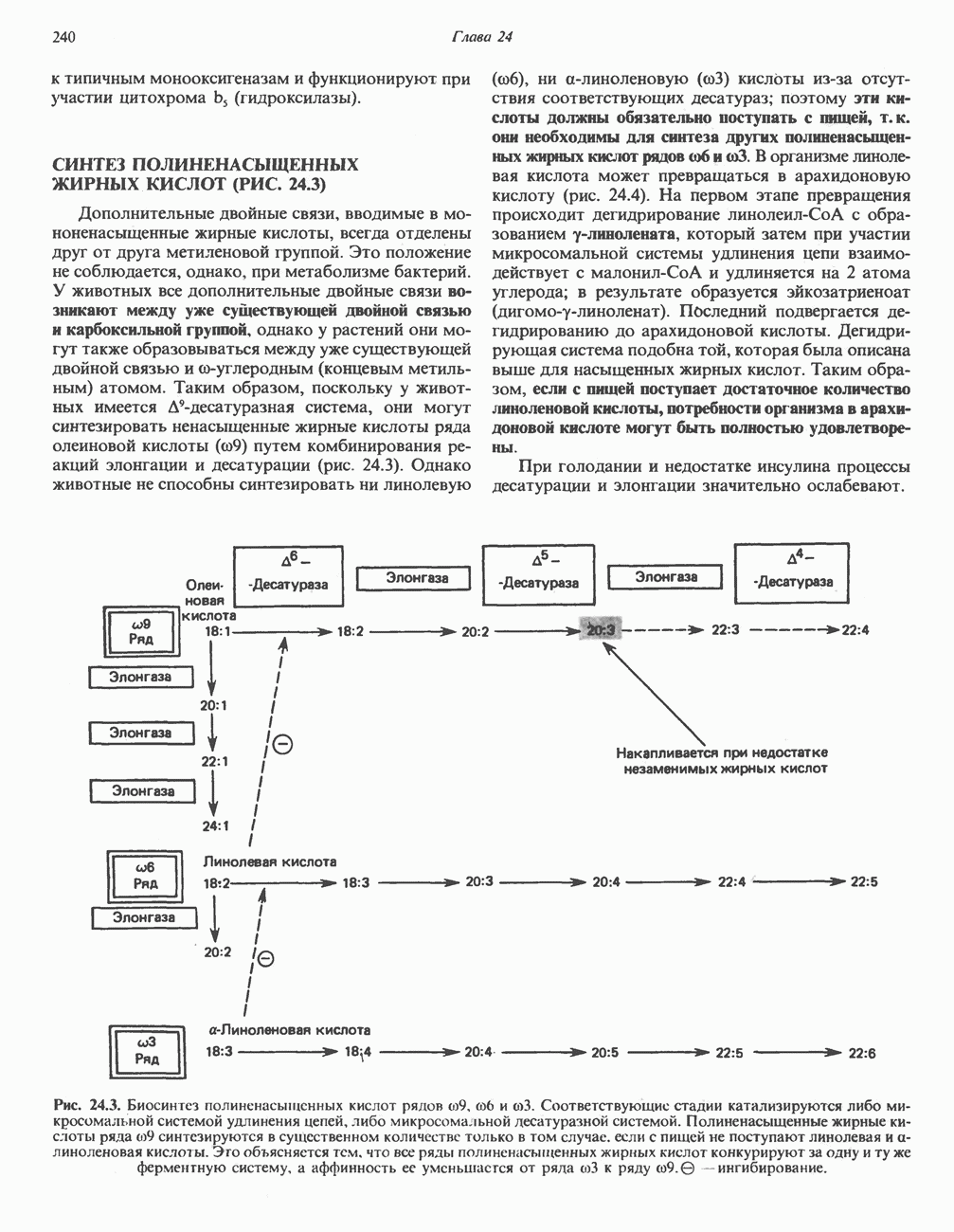
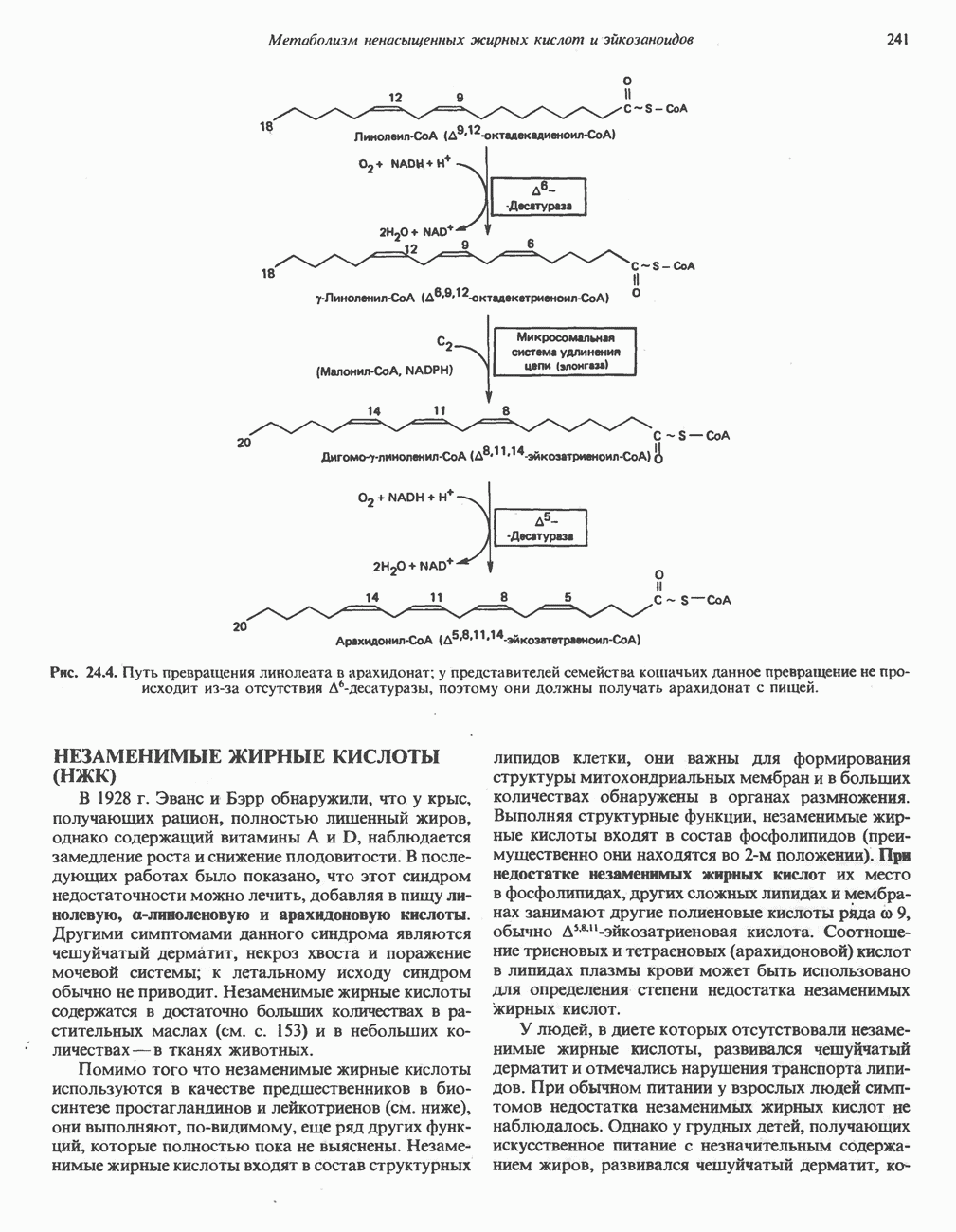

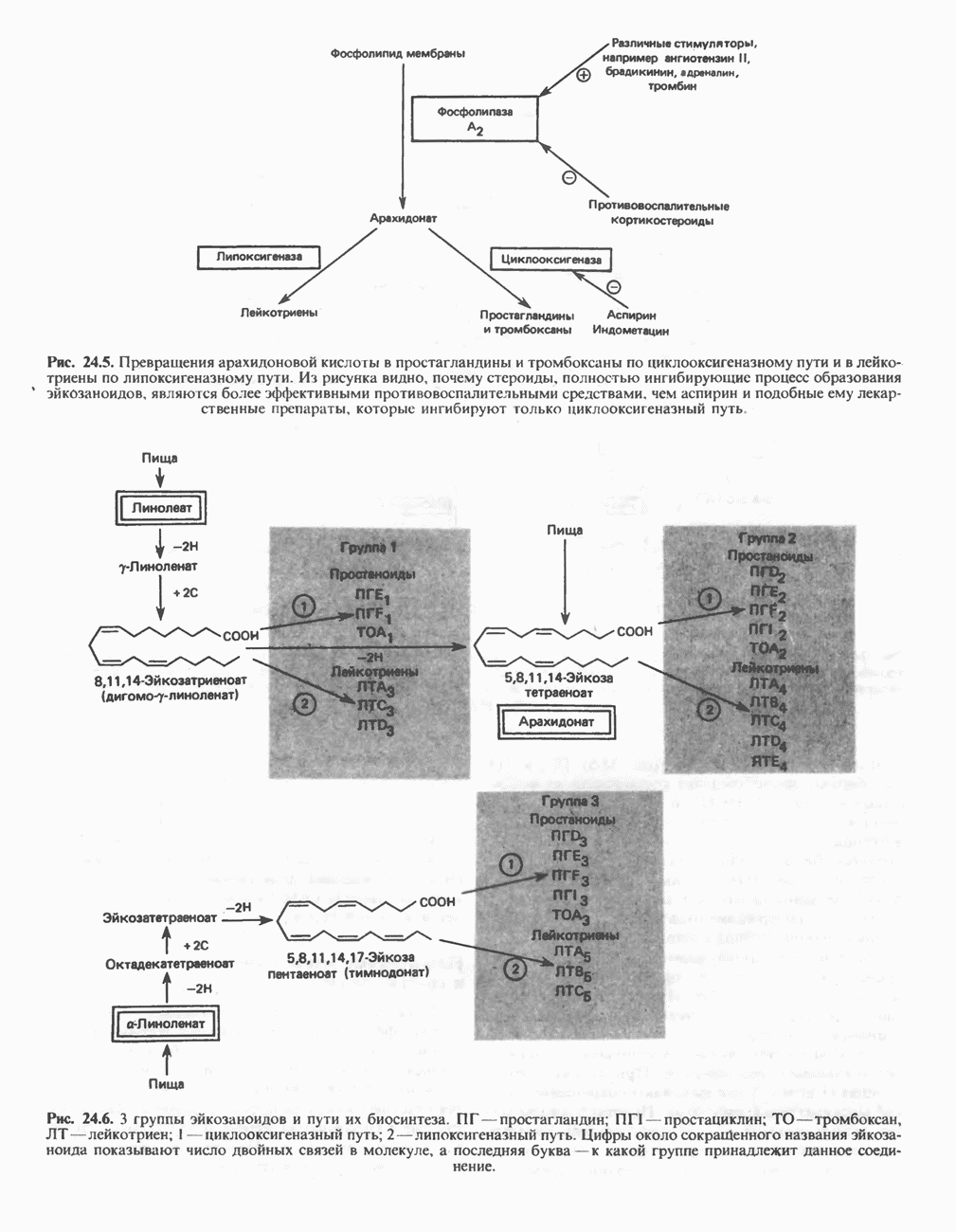





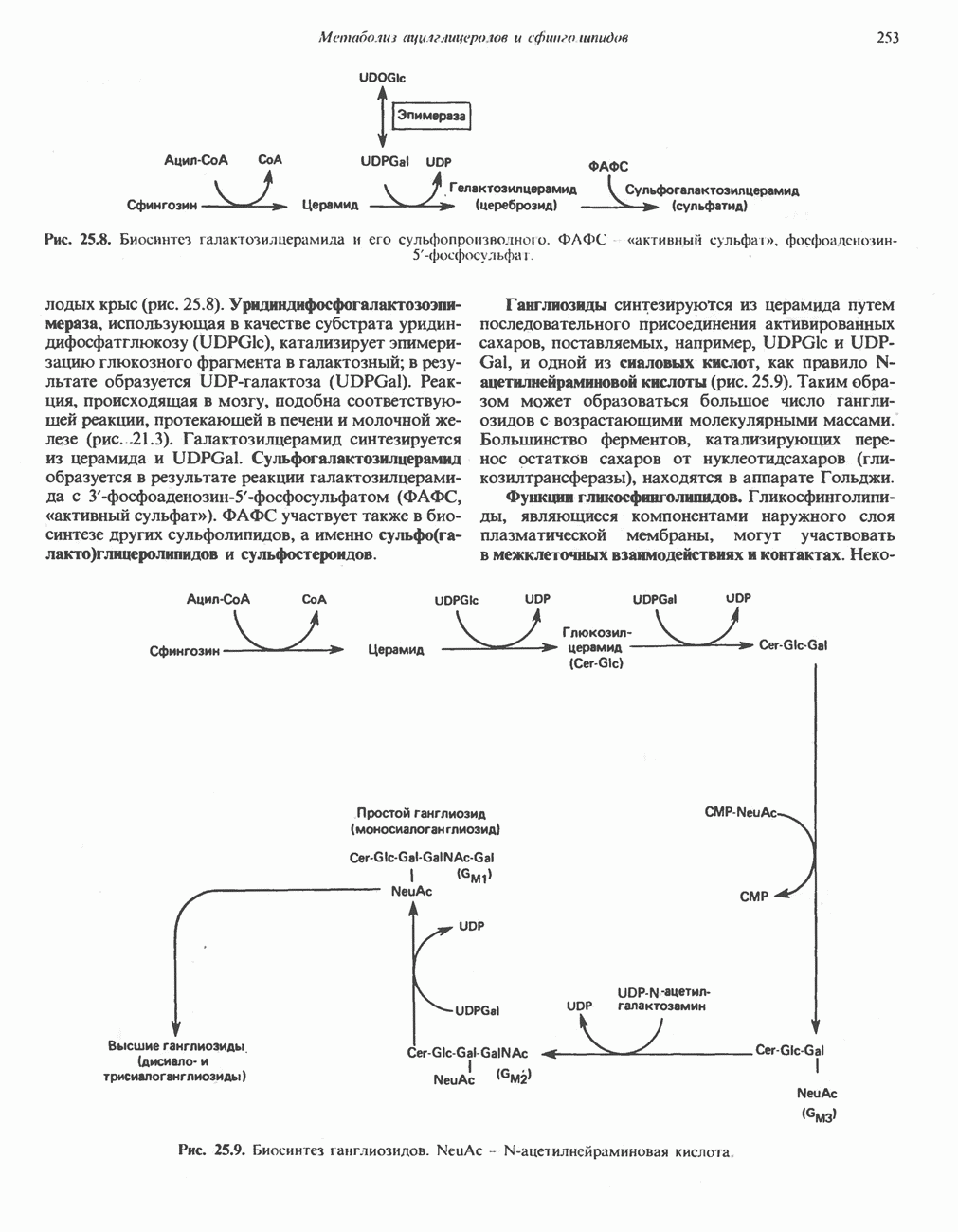
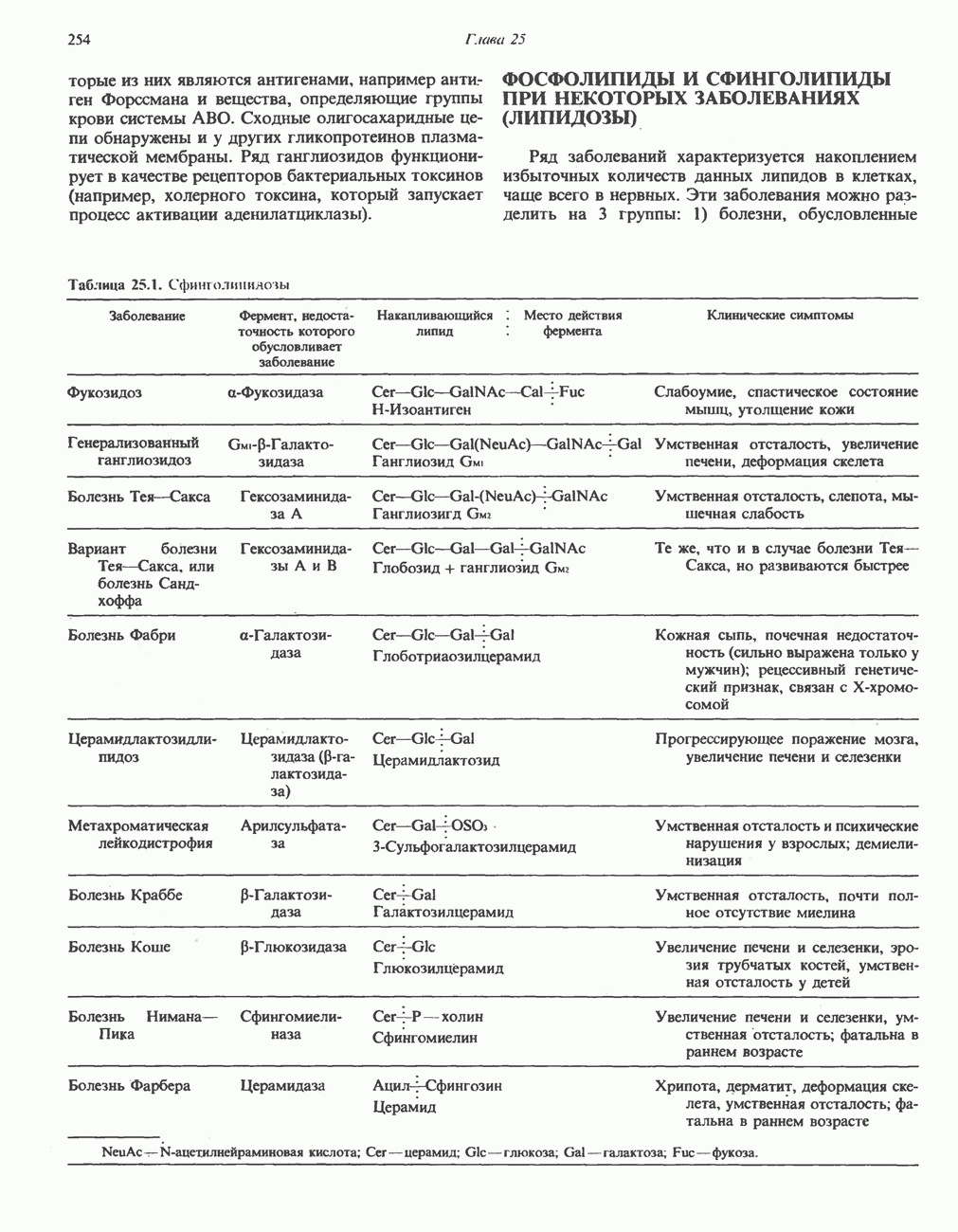


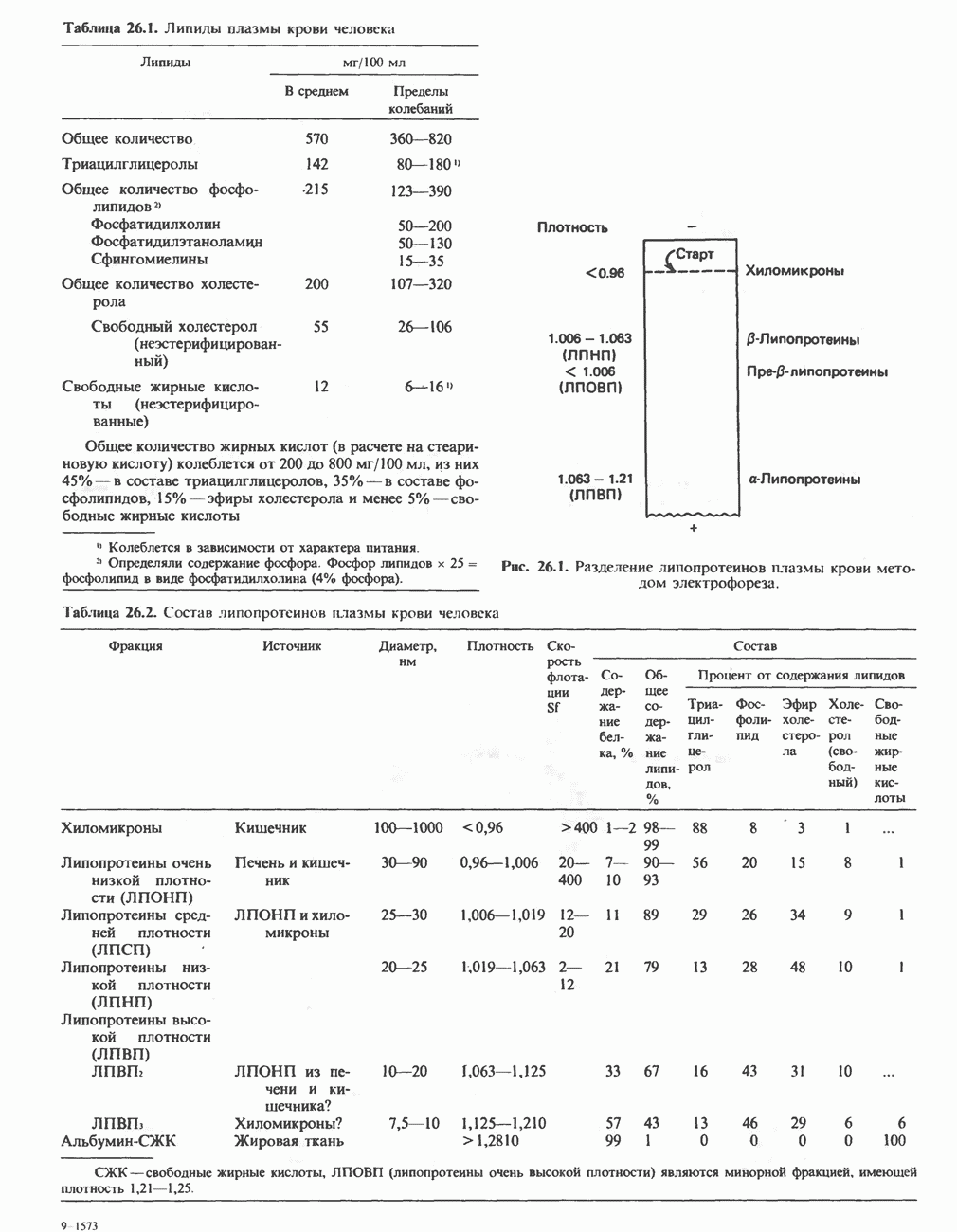
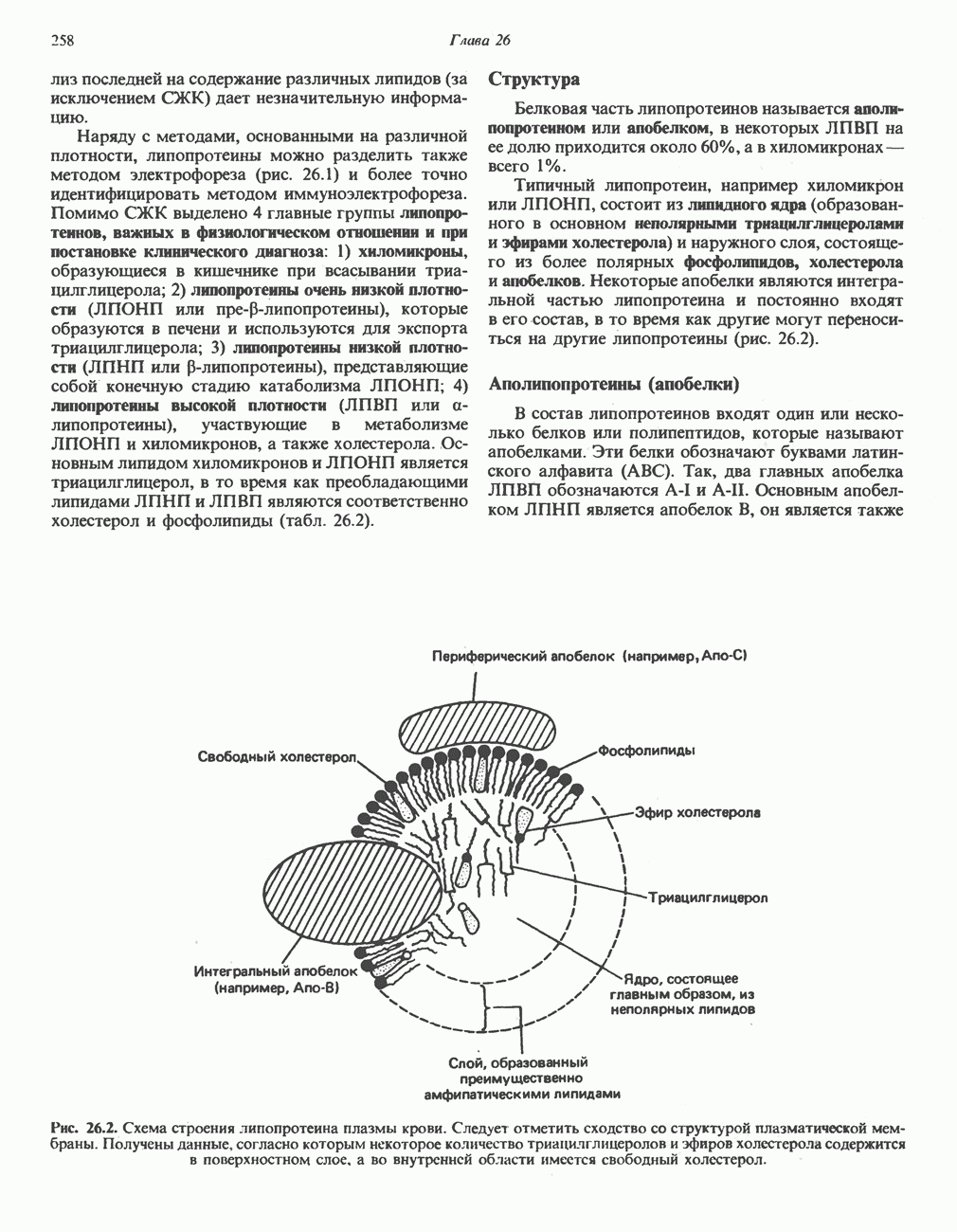






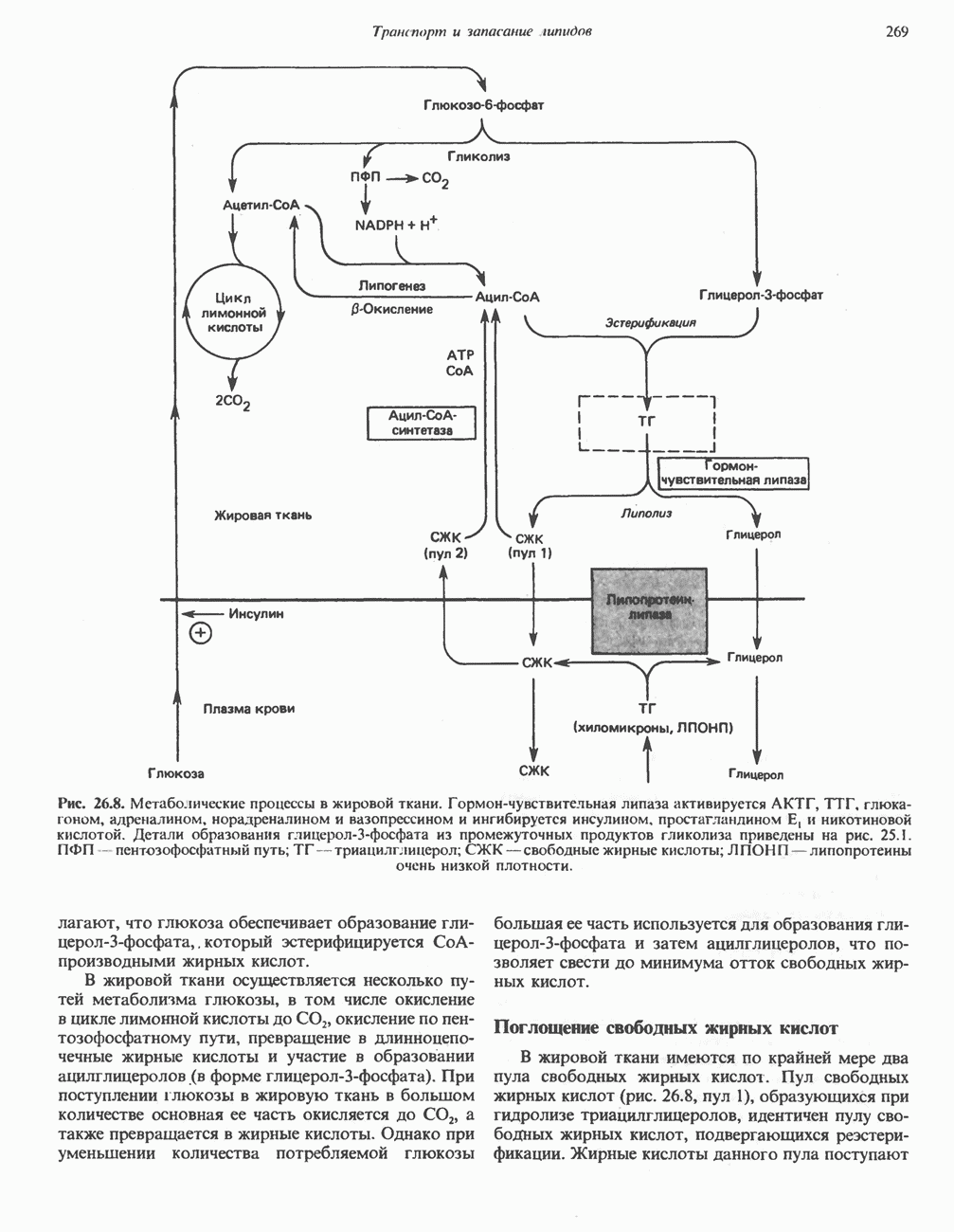

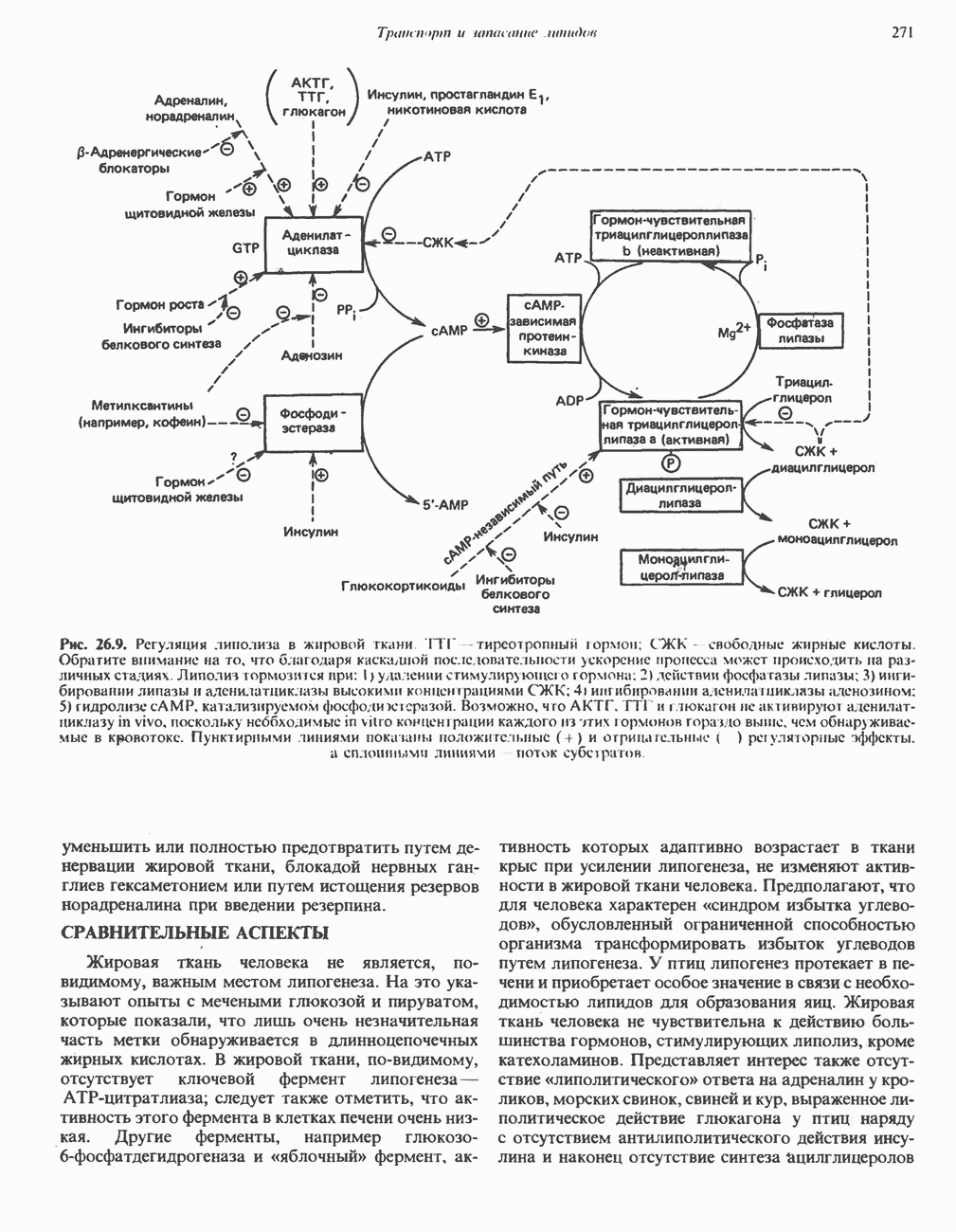




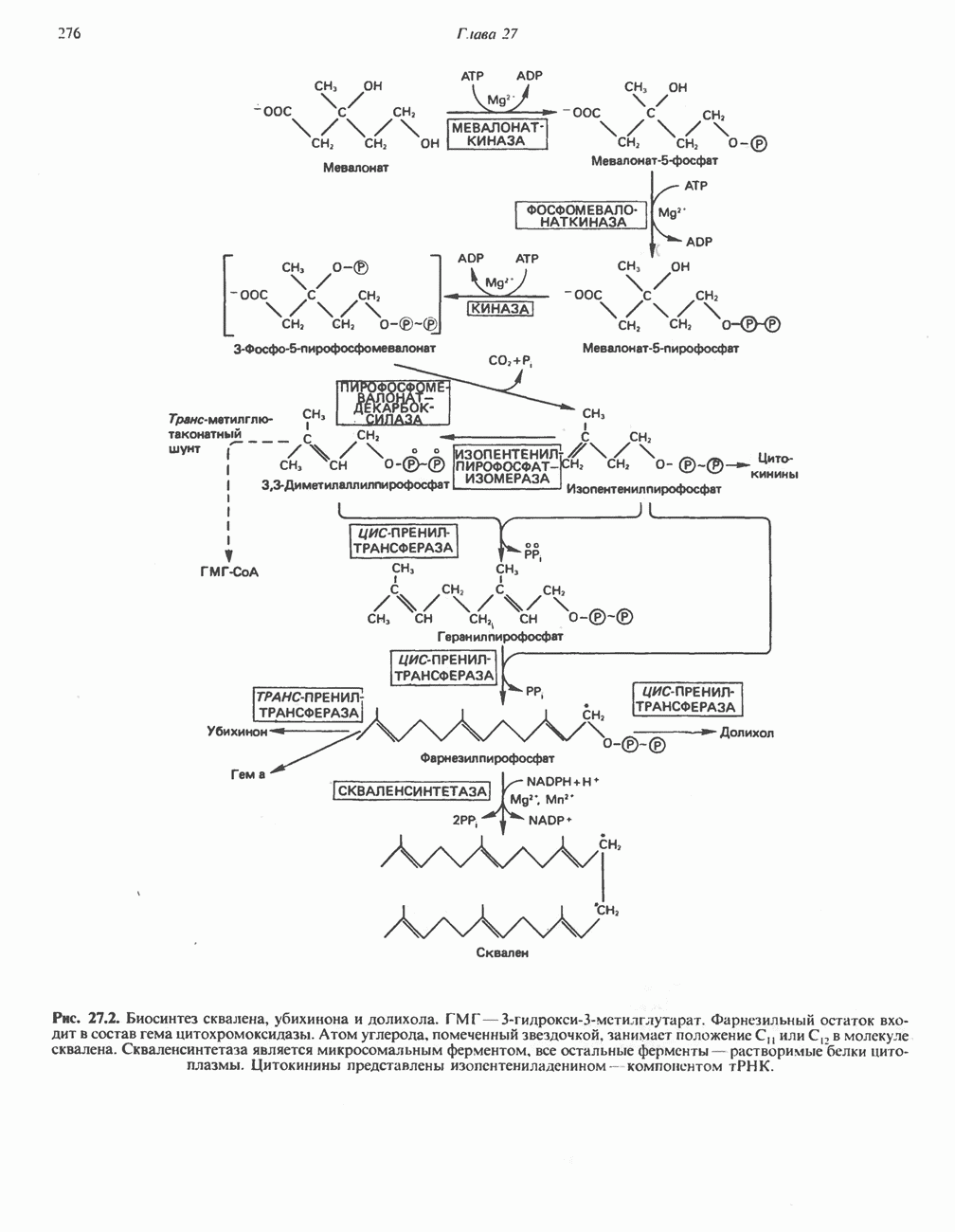
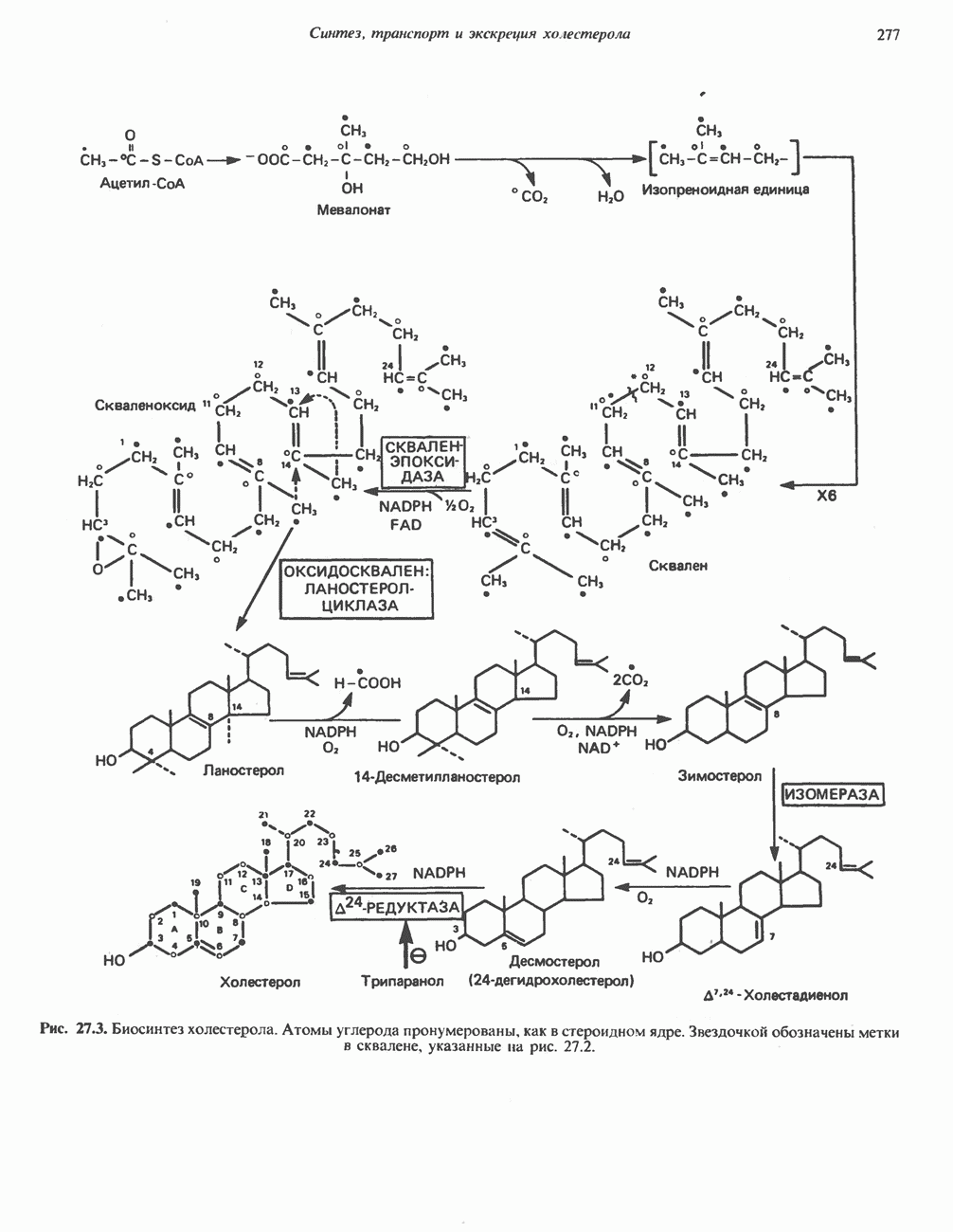
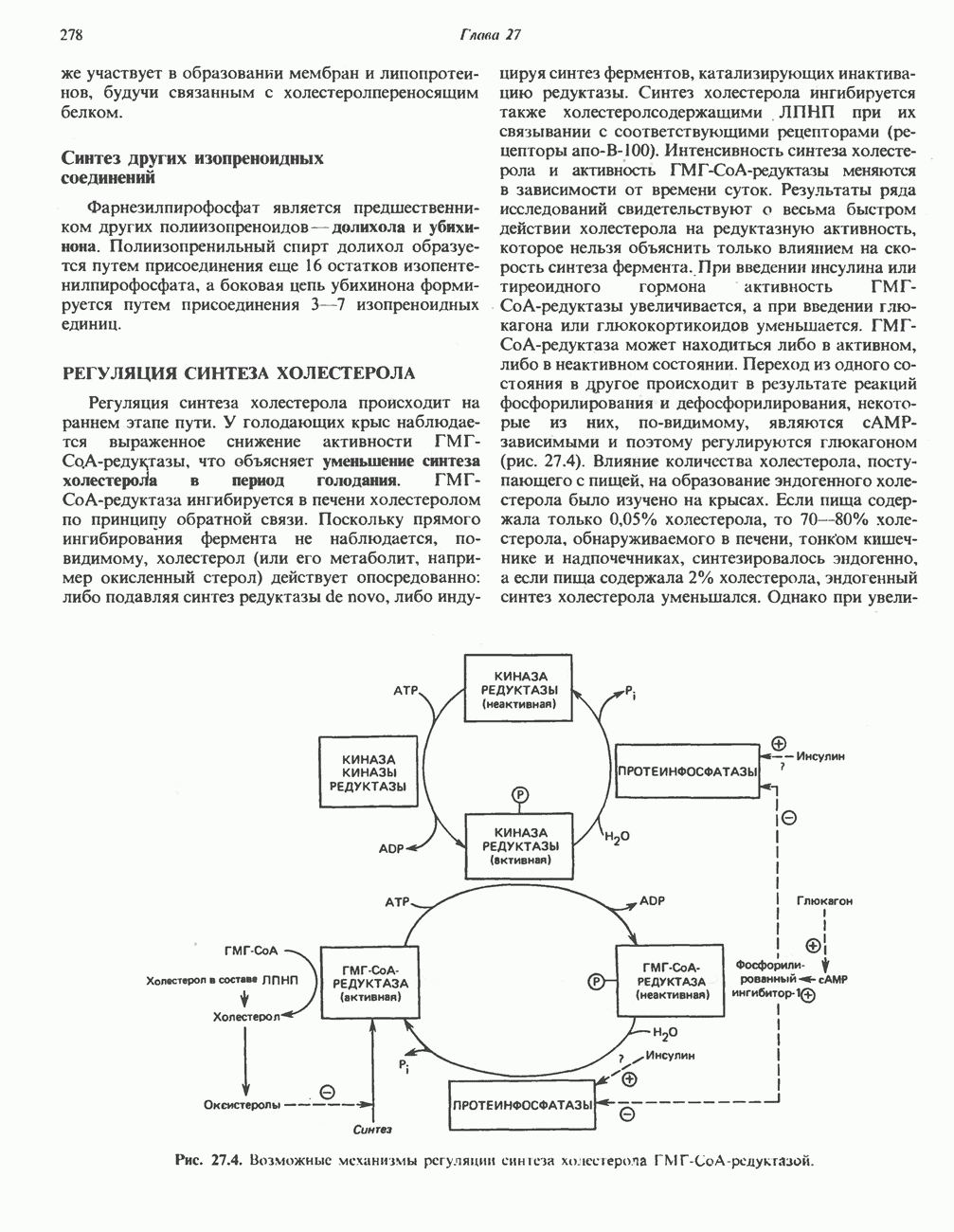
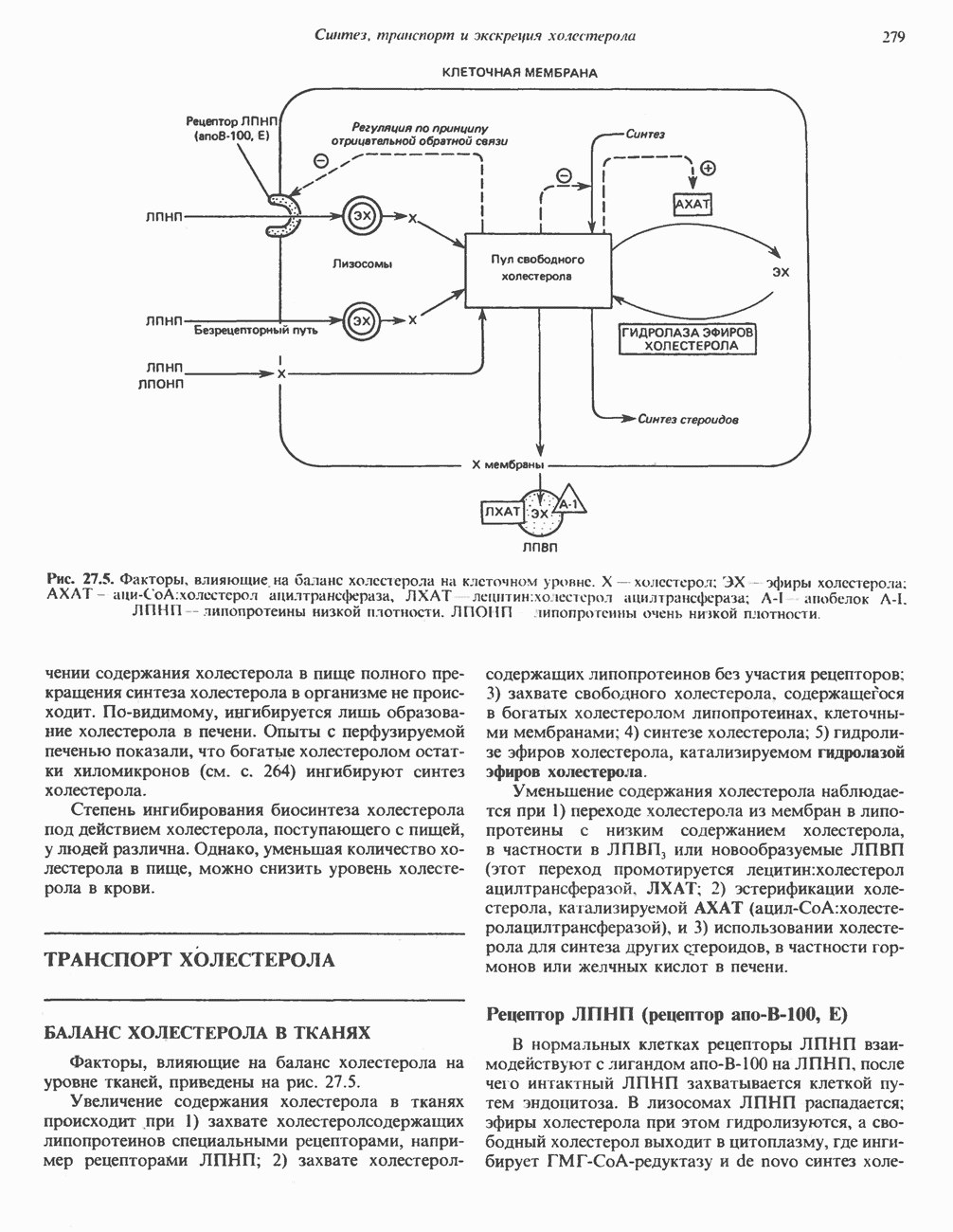
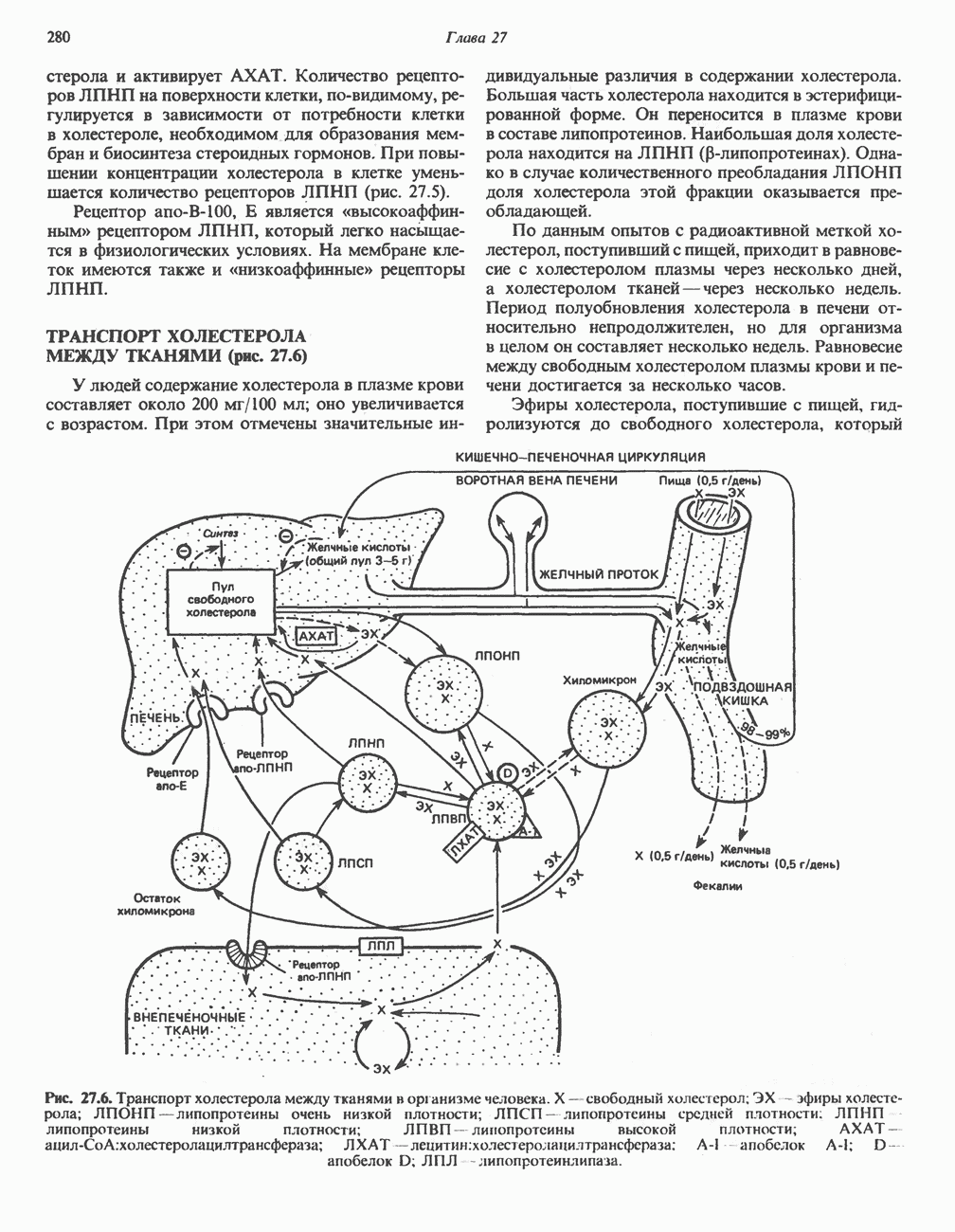

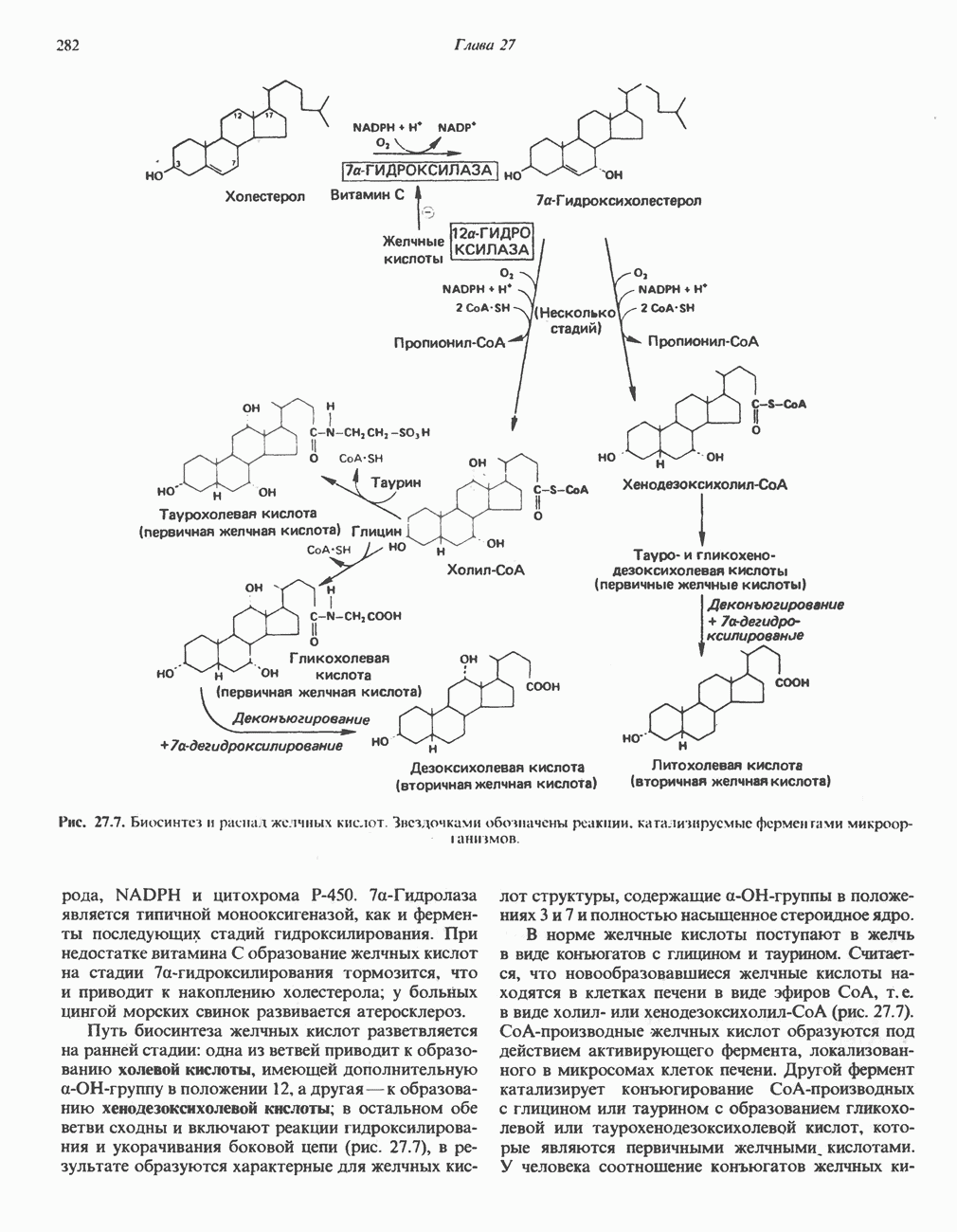









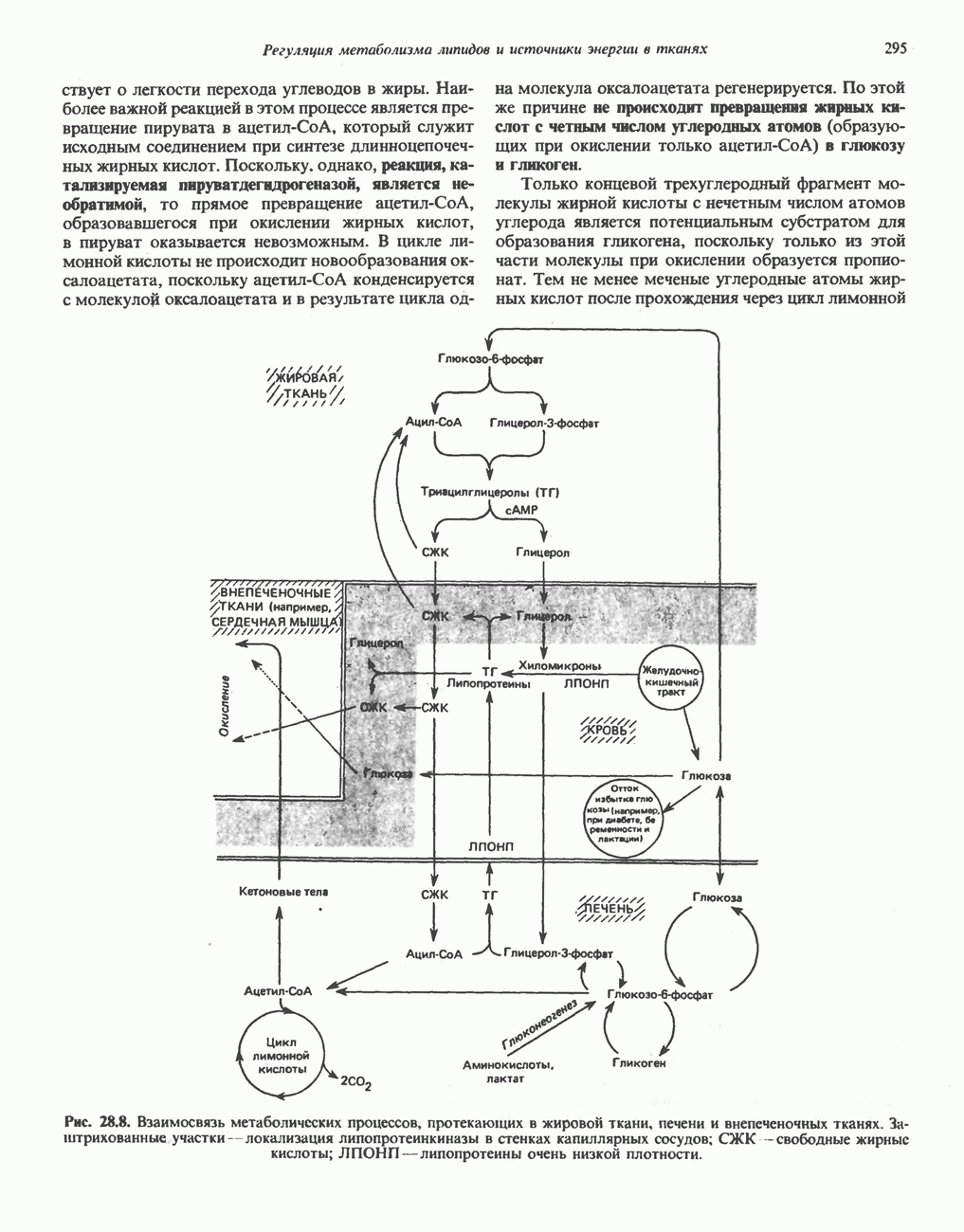
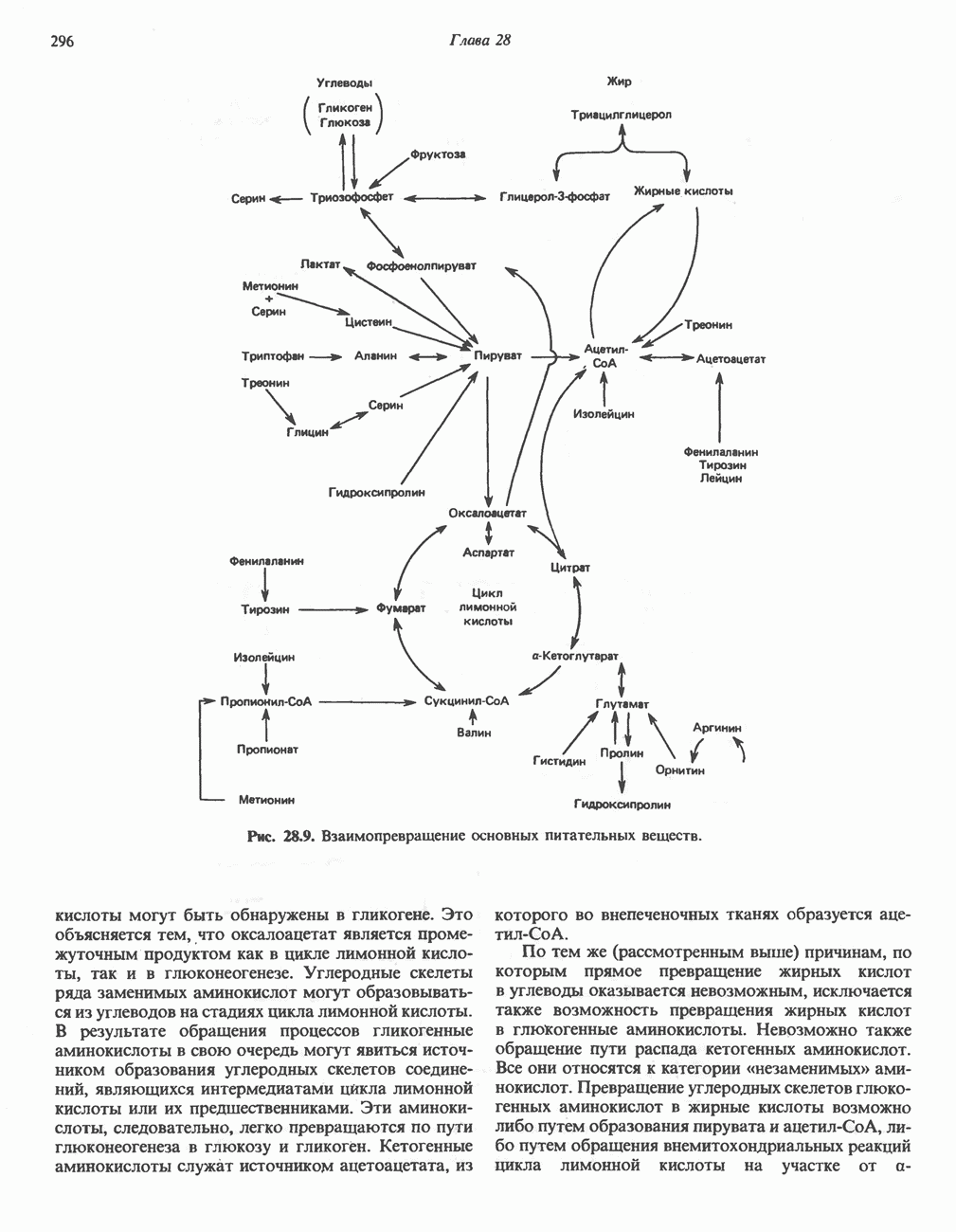



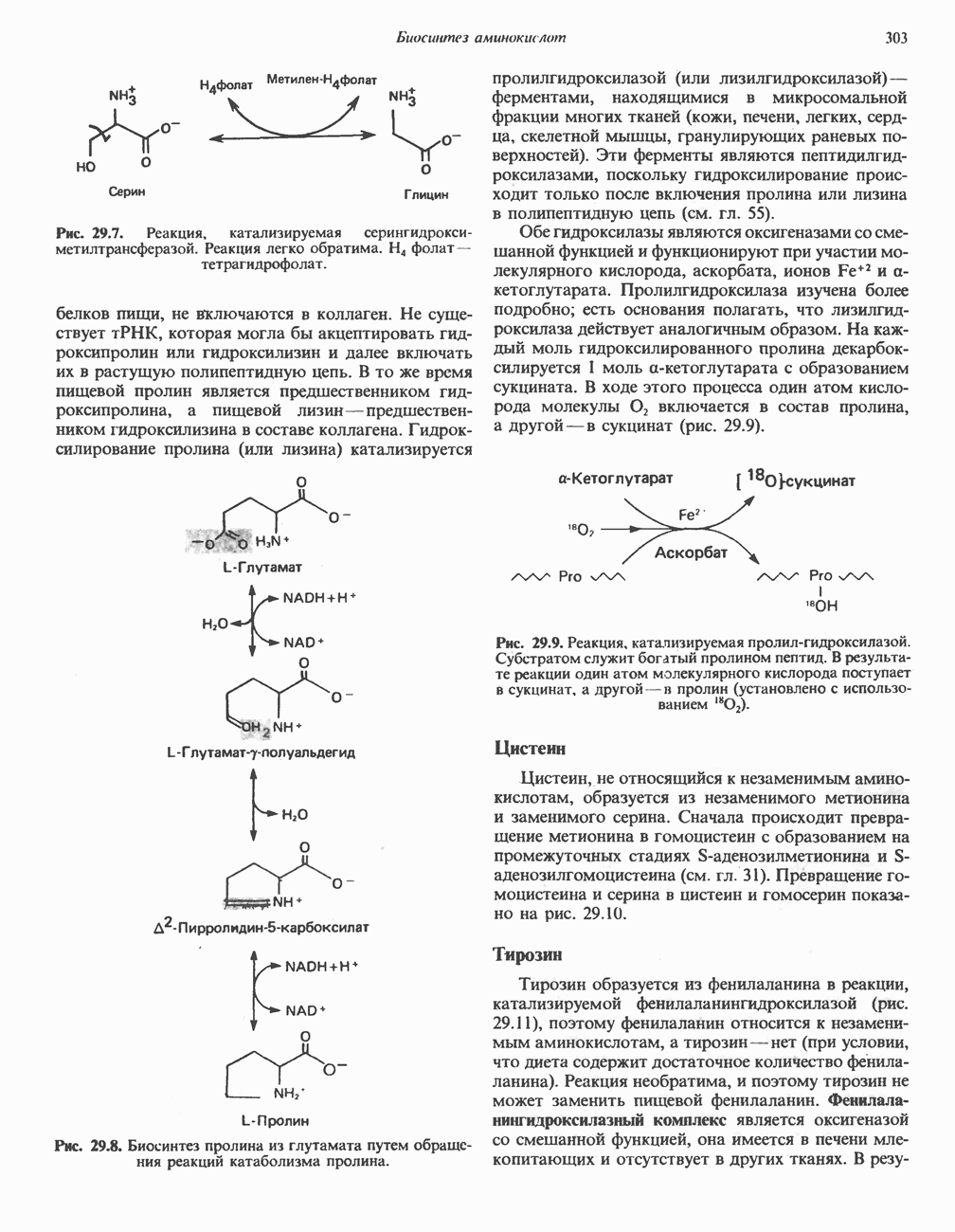
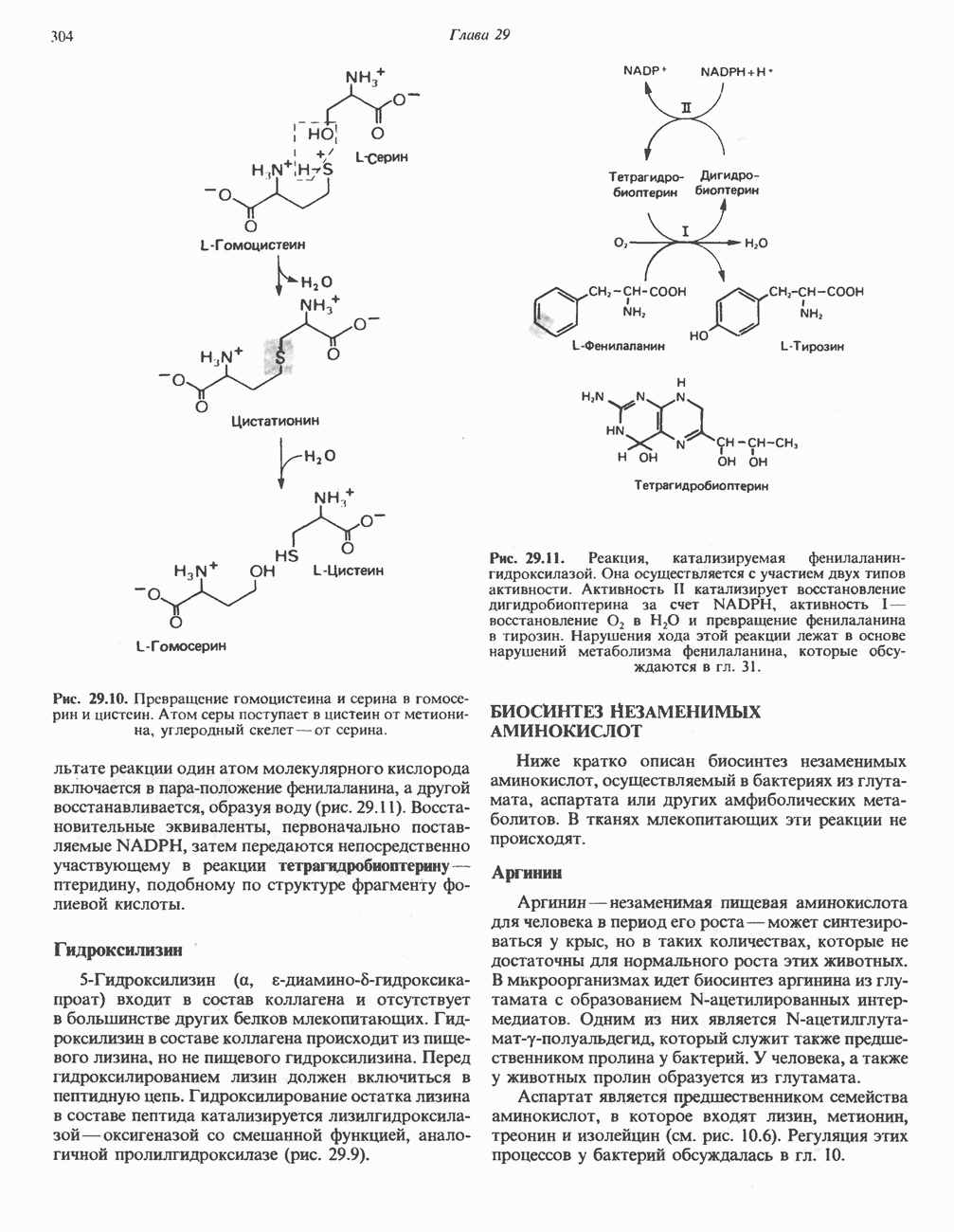





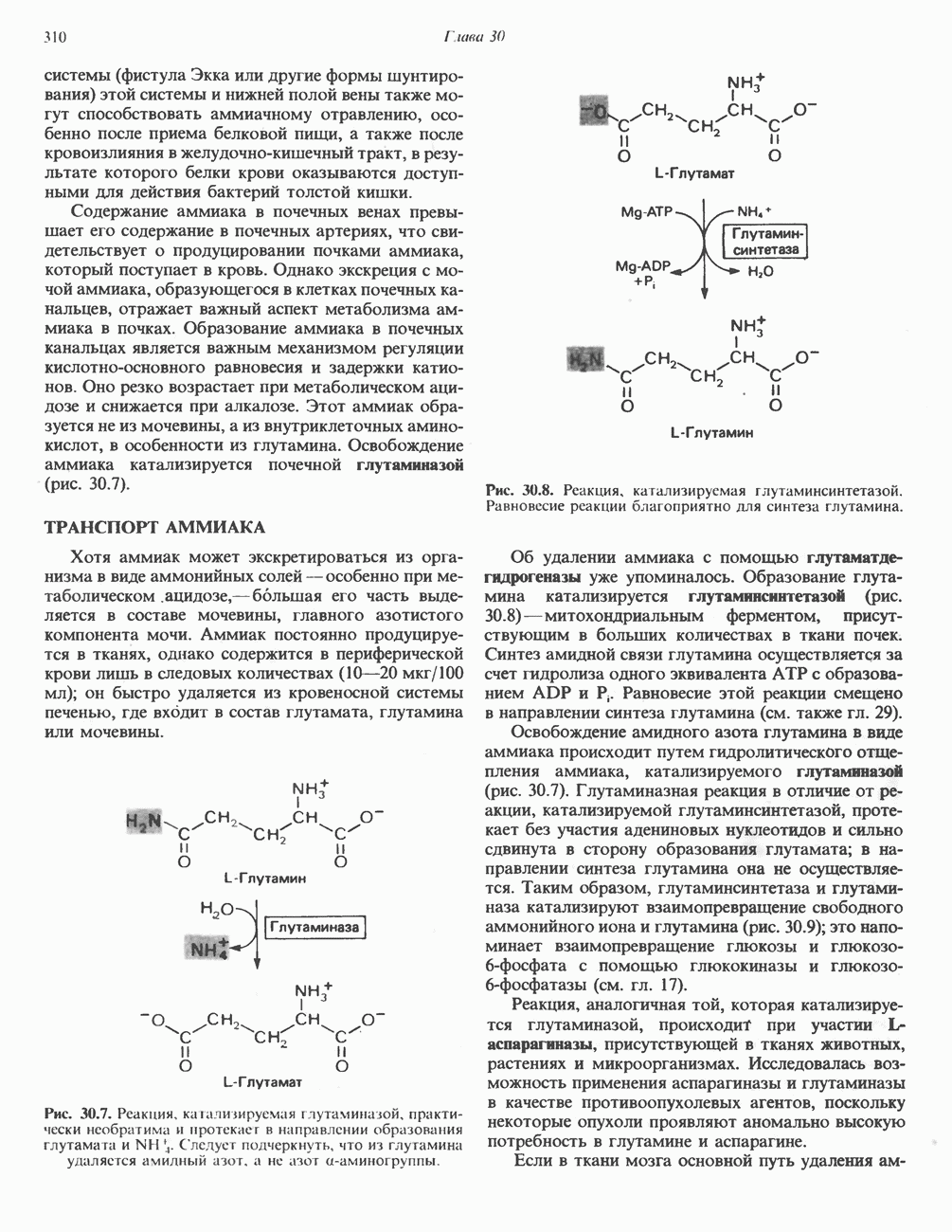



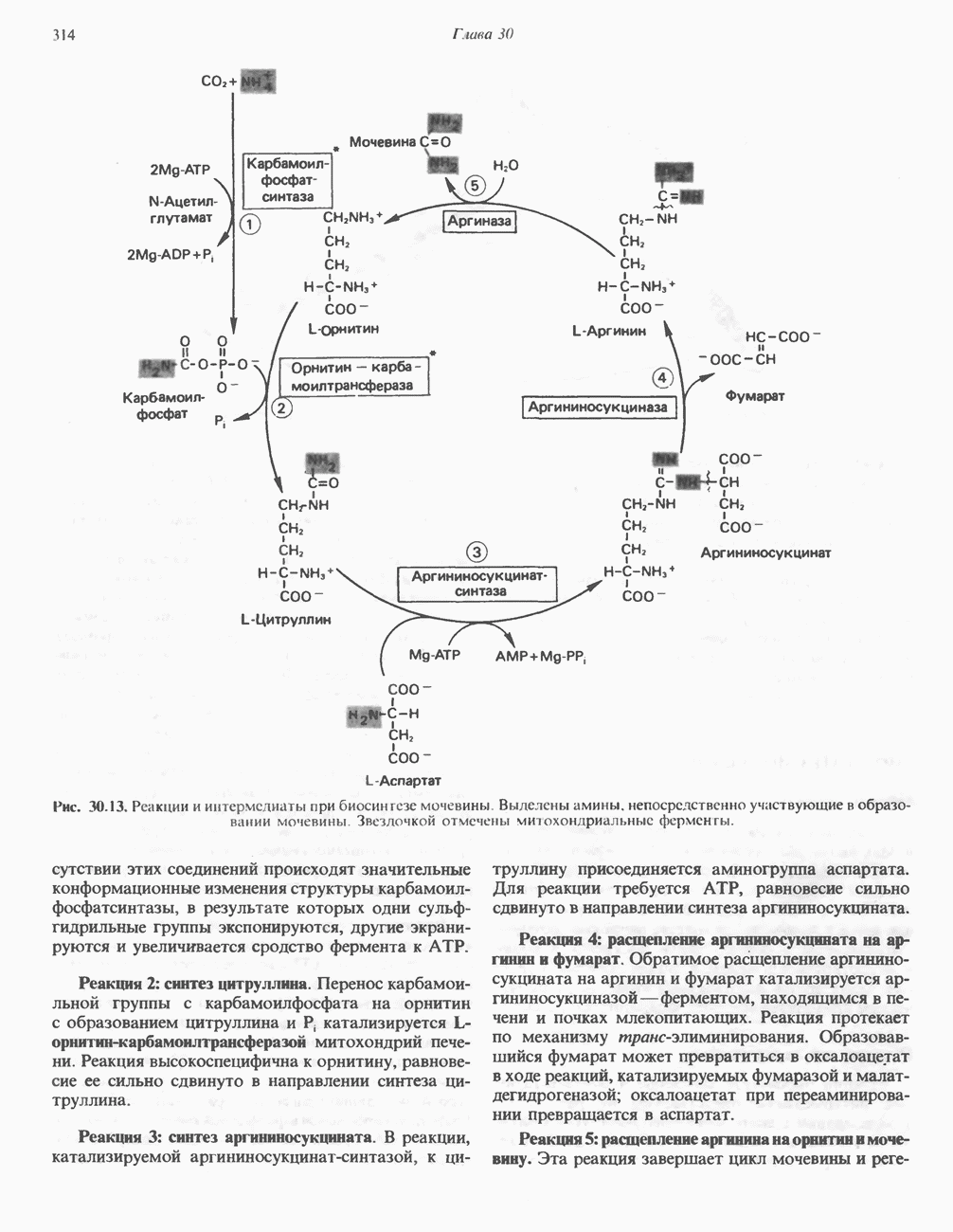



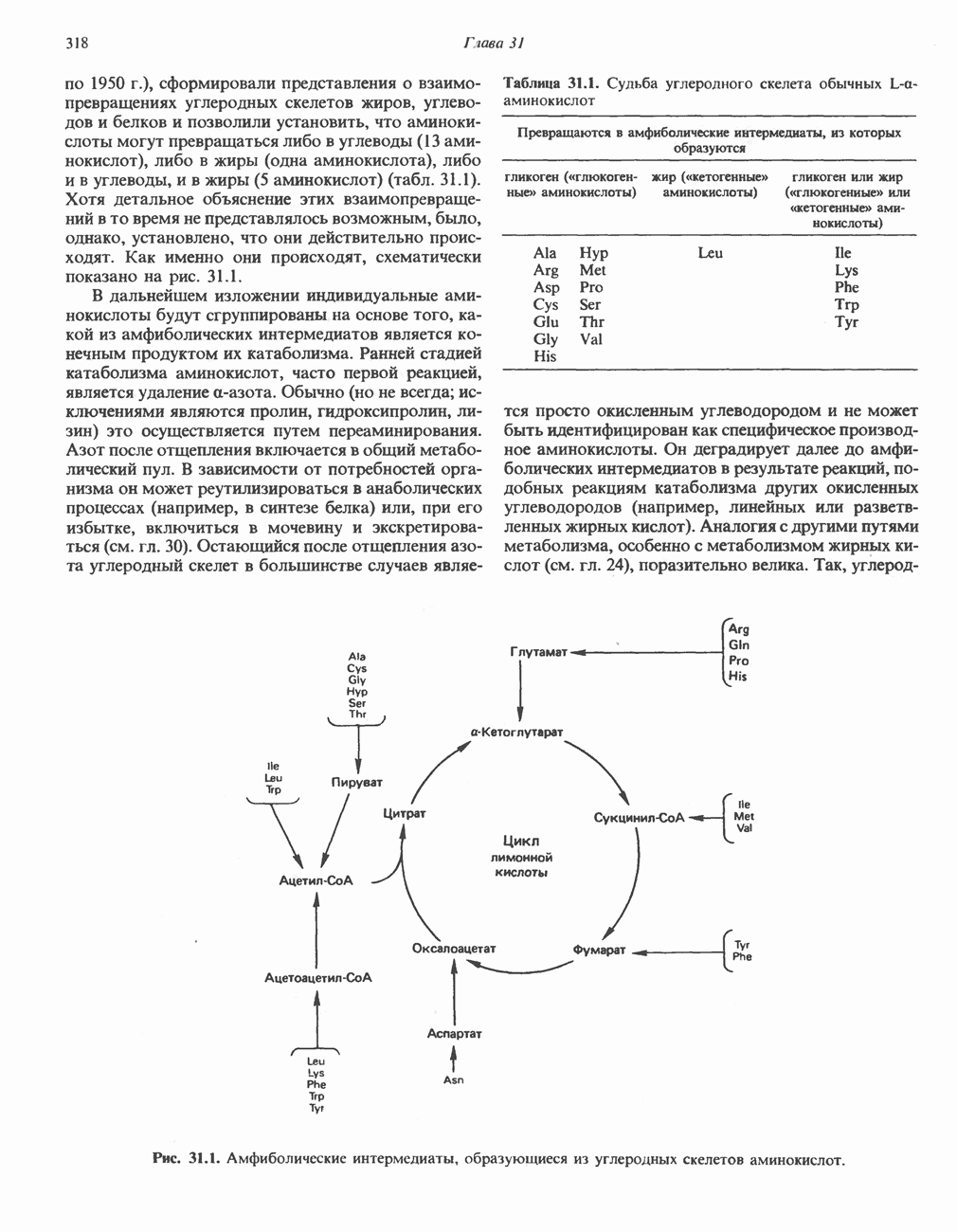
































 В состав пирувата включаются все атомы углерода глицина, аланина, цистеина и серина и только два атома углерода треонина. Далее пируват может превращаться в ацетил-СоА.
В состав пирувата включаются все атомы углерода глицина, аланина, цистеина и серина и только два атома углерода треонина. Далее пируват может превращаться в ацетил-СоА.

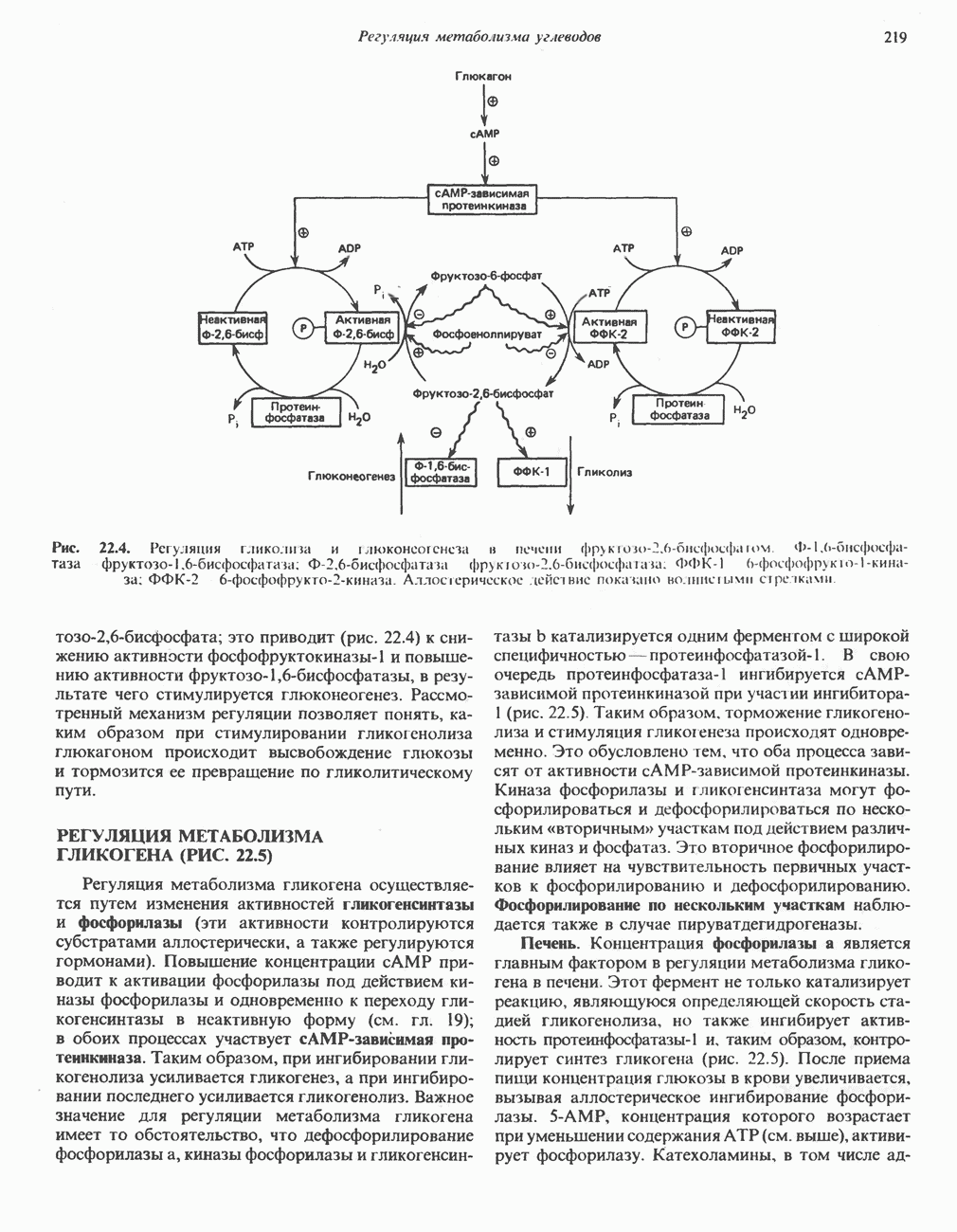
 -метилентетрагидрофолат. При образовании пирувата из глицина последний сначала превращается в серин в результате реакции, катализируемой серин-гидроксиметилтрансферазой (рис. 31.5), а затем из серина образуется пируват (реакция катализируется сериндегидратазой) (рис. 31.7; см также следующий ниже раздел «Серин»).
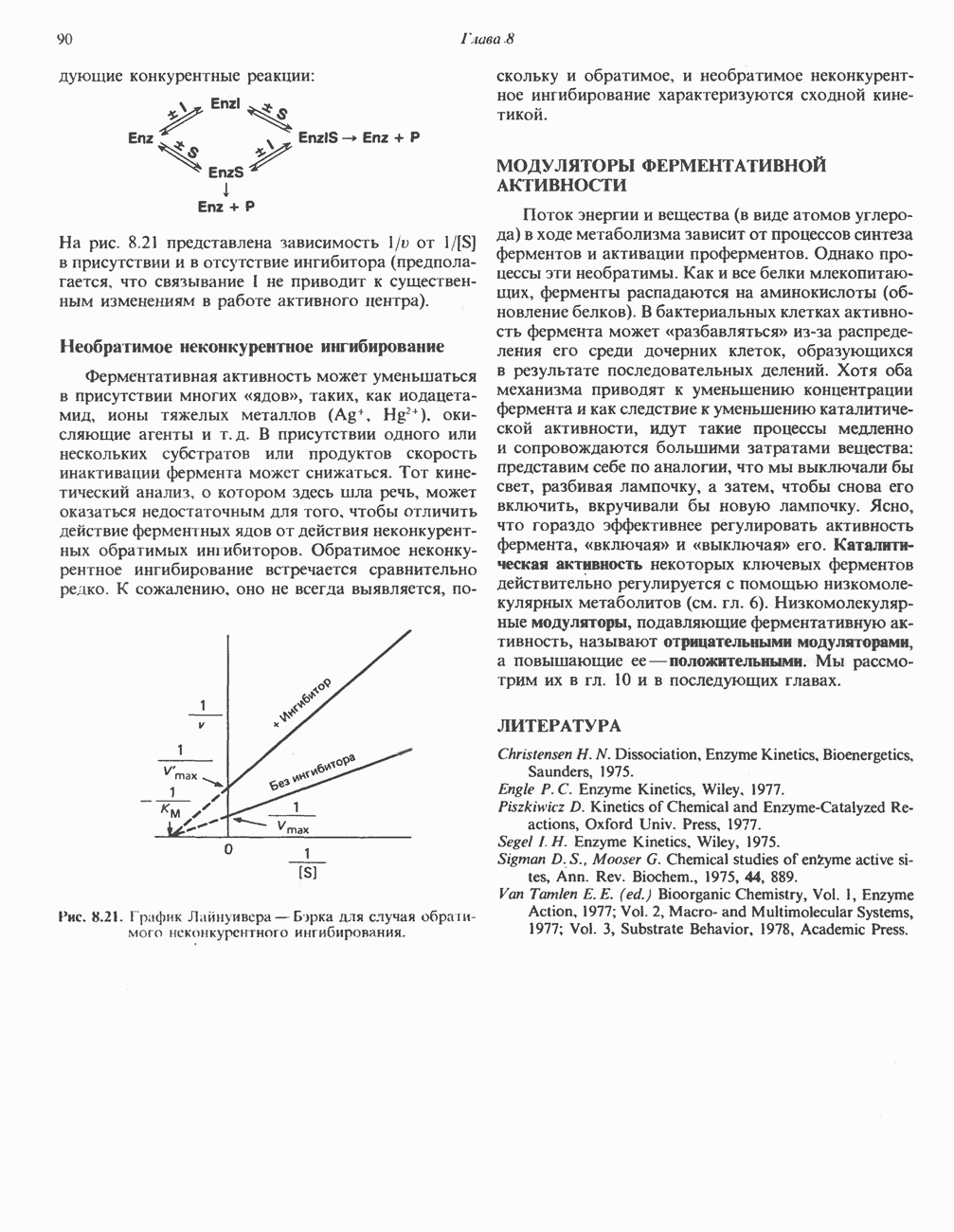
-метилентетрагидрофолат. При образовании пирувата из глицина последний сначала превращается в серин в результате реакции, катализируемой серин-гидроксиметилтрансферазой (рис. 31.5), а затем из серина образуется пируват (реакция катализируется сериндегидратазой) (рис. 31.7; см также следующий ниже раздел «Серин»).

 тетрагидрофолат.
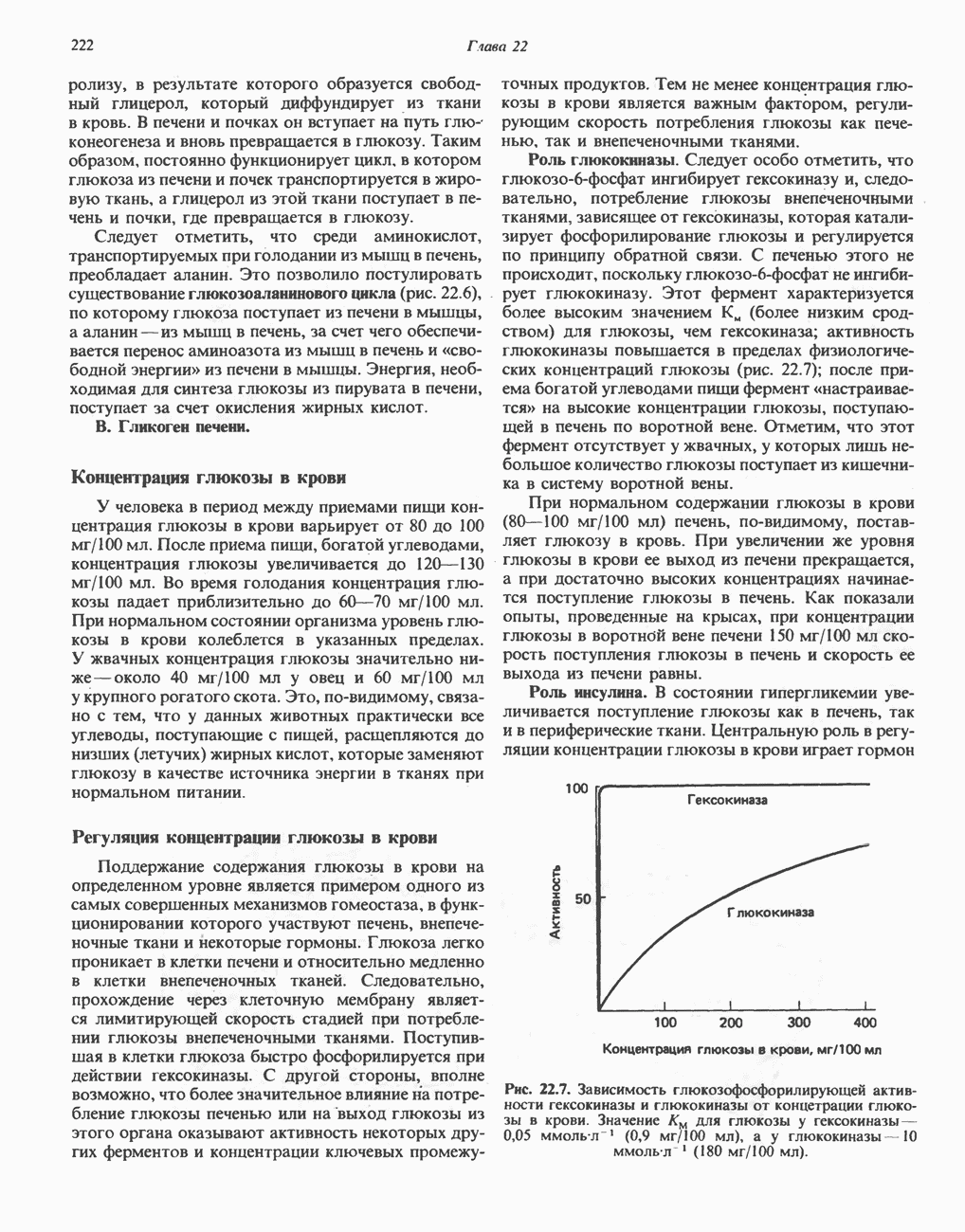
тетрагидрофолат.
 и NH, а метиленовая группа переносится на тетрагидрофолат с образованием
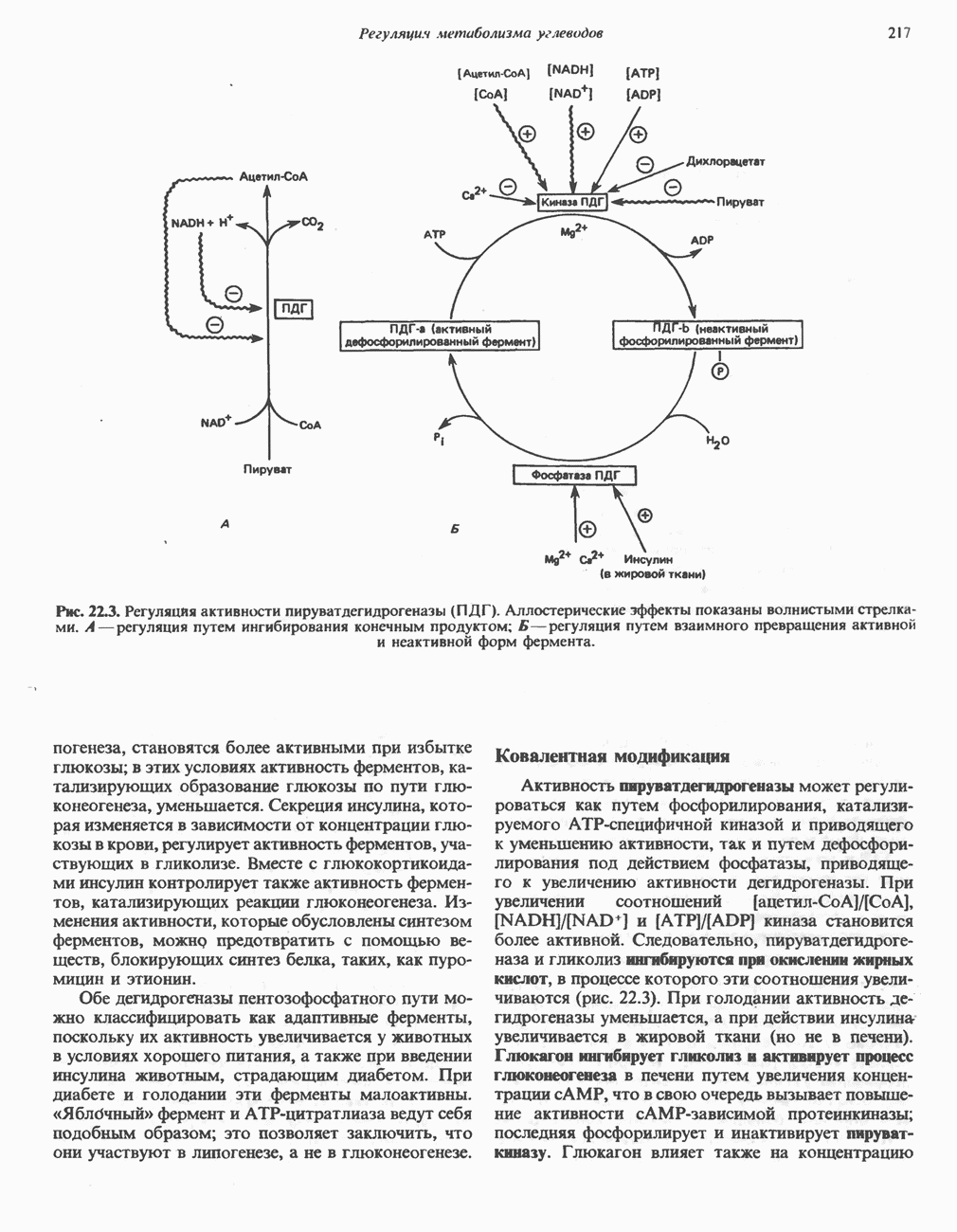
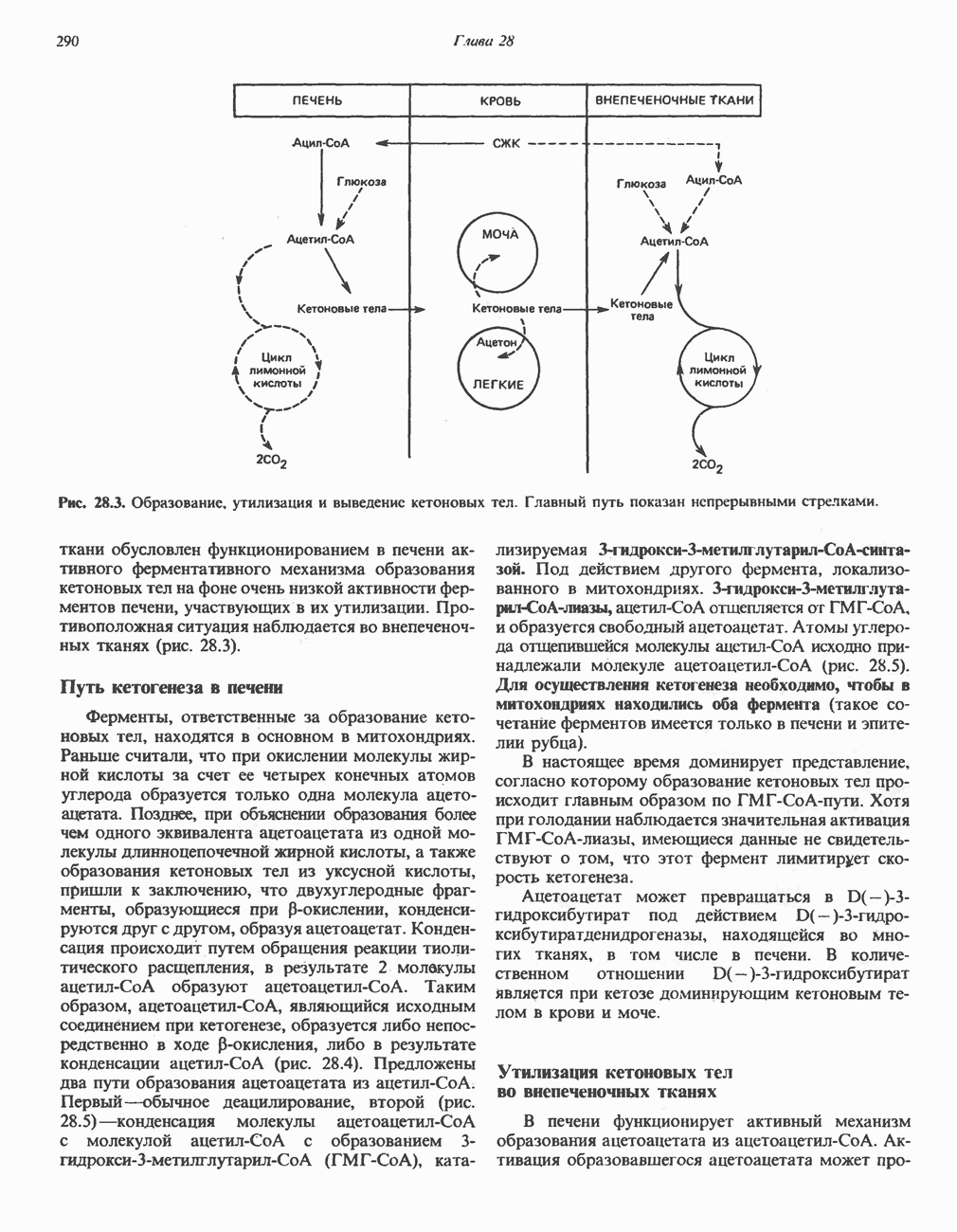
и NH, а метиленовая группа переносится на тетрагидрофолат с образованием  -метилентетрагидрофолата. Эта обратимая реакция (рис. 31.6) напоминает превращение пирувата в ацетил-СоА ферментами пируватде идрогеназного комплекса. Оба комплекса находятся в митохондриях печени и представляют собой макромолекулярные агрегаты. Реакция расщепления глицина протекает в печени большинства позвоночных включая человека и других млекопитающих, а также птиц и рептилии
-метилентетрагидрофолата. Эта обратимая реакция (рис. 31.6) напоминает превращение пирувата в ацетил-СоА ферментами пируватде идрогеназного комплекса. Оба комплекса находятся в митохондриях печени и представляют собой макромолекулярные агрегаты. Реакция расщепления глицина протекает в печени большинства позвоночных включая человека и других млекопитающих, а также птиц и рептилии
 . Это позволяет сделать заключение, что глицинурия связана с нарушением реабсорбции глицина в почечных канальцах.
. Это позволяет сделать заключение, что глицинурия связана с нарушением реабсорбции глицина в почечных канальцах.


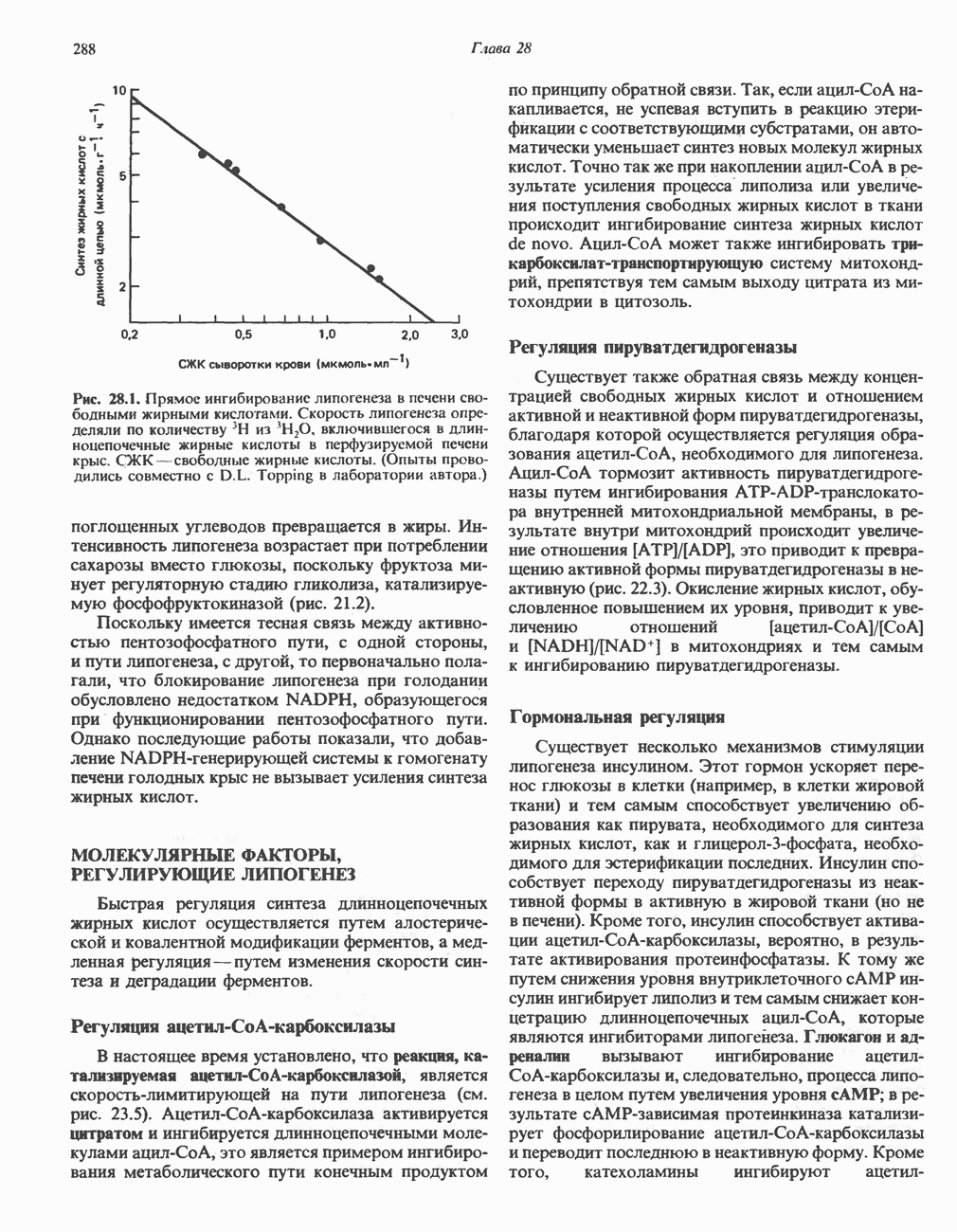
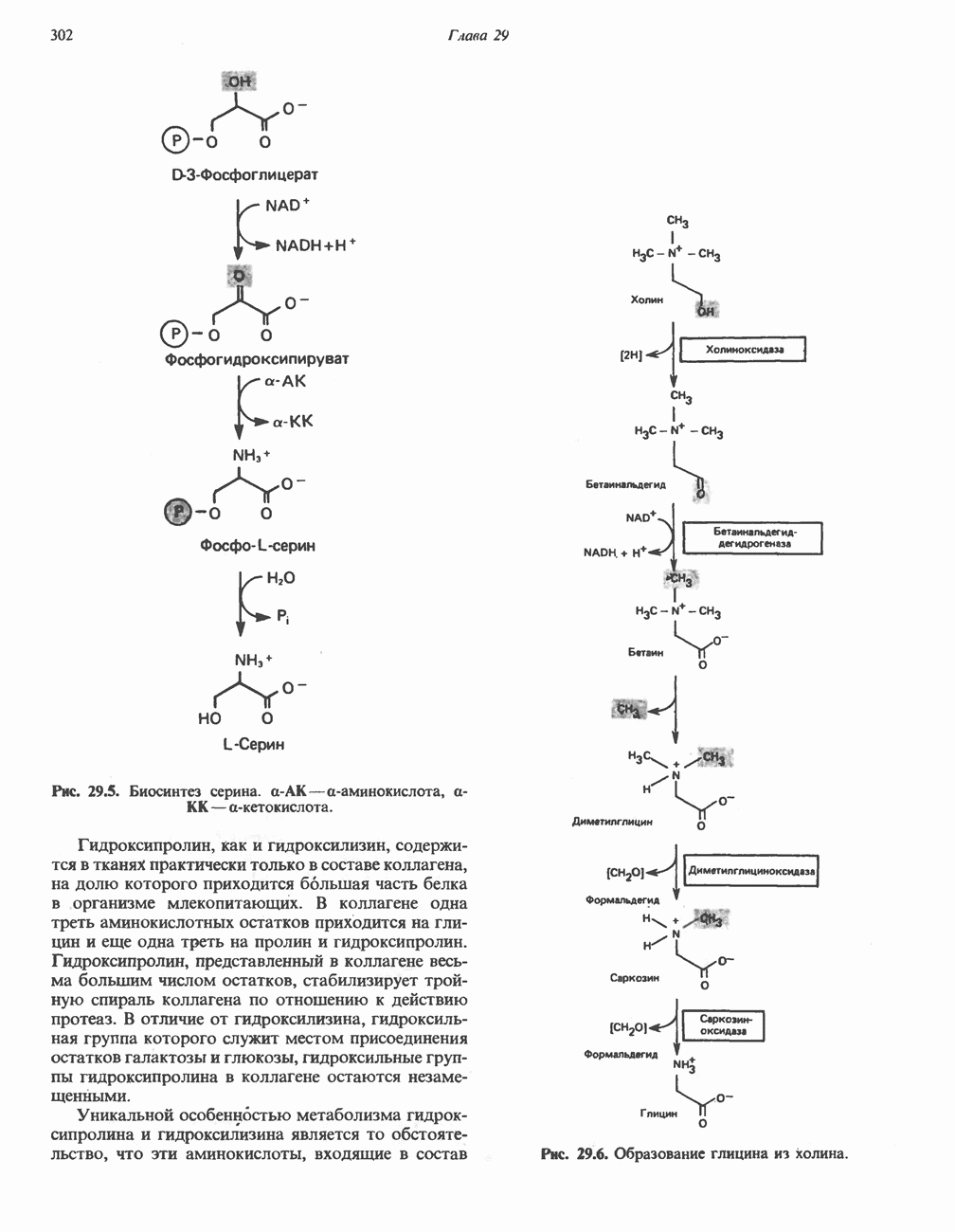
 из серина, приводящее к образованию ненасыщенной аминокислоты. Последняя перегруппировывается в а-иминокислоту, которая подвергается спонтанному гидролизу с образованием пирувата и аммиака. Таким образом, в уравнение суммарной реакции, катализируемой сериндегидратазой, вода не входит.
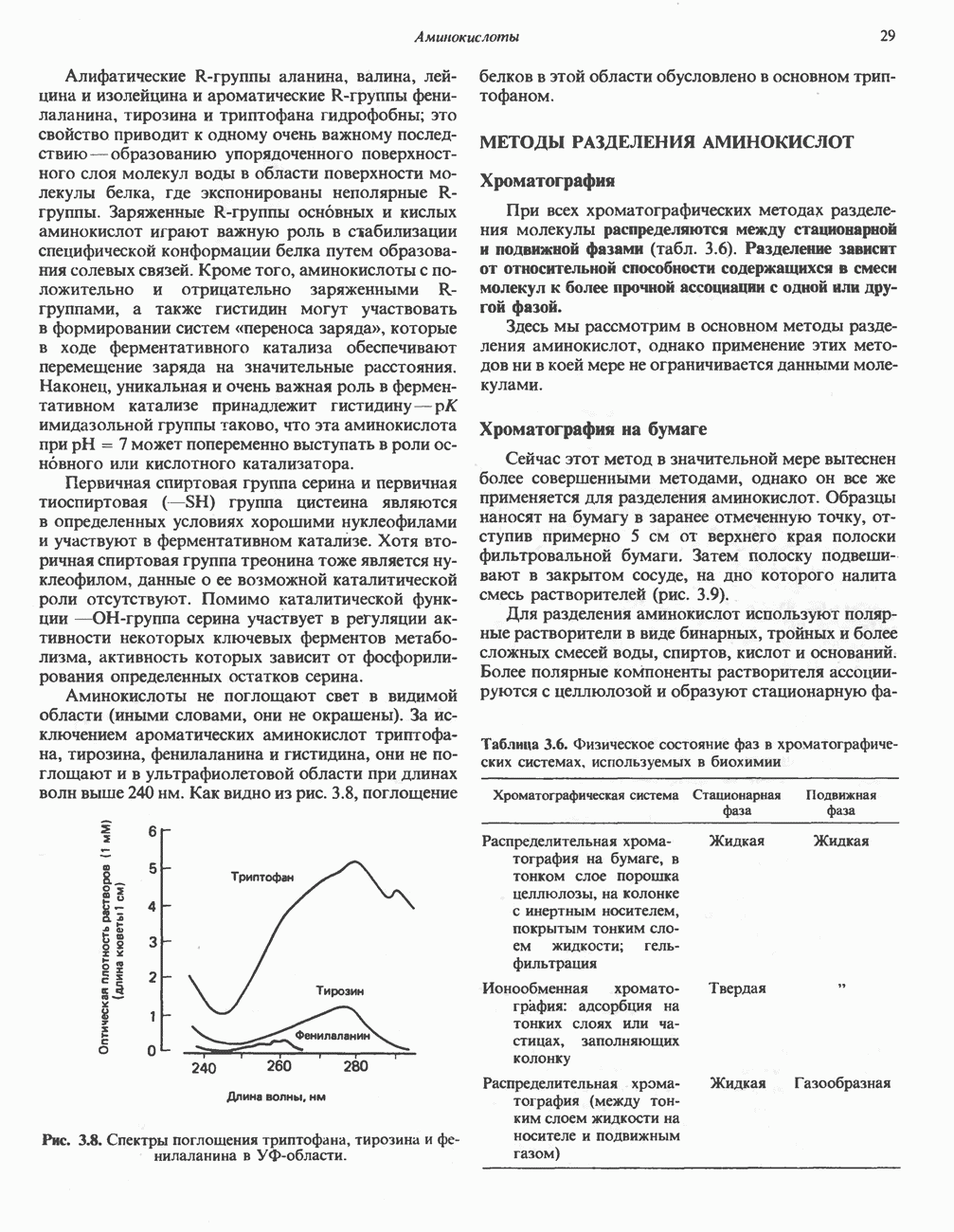
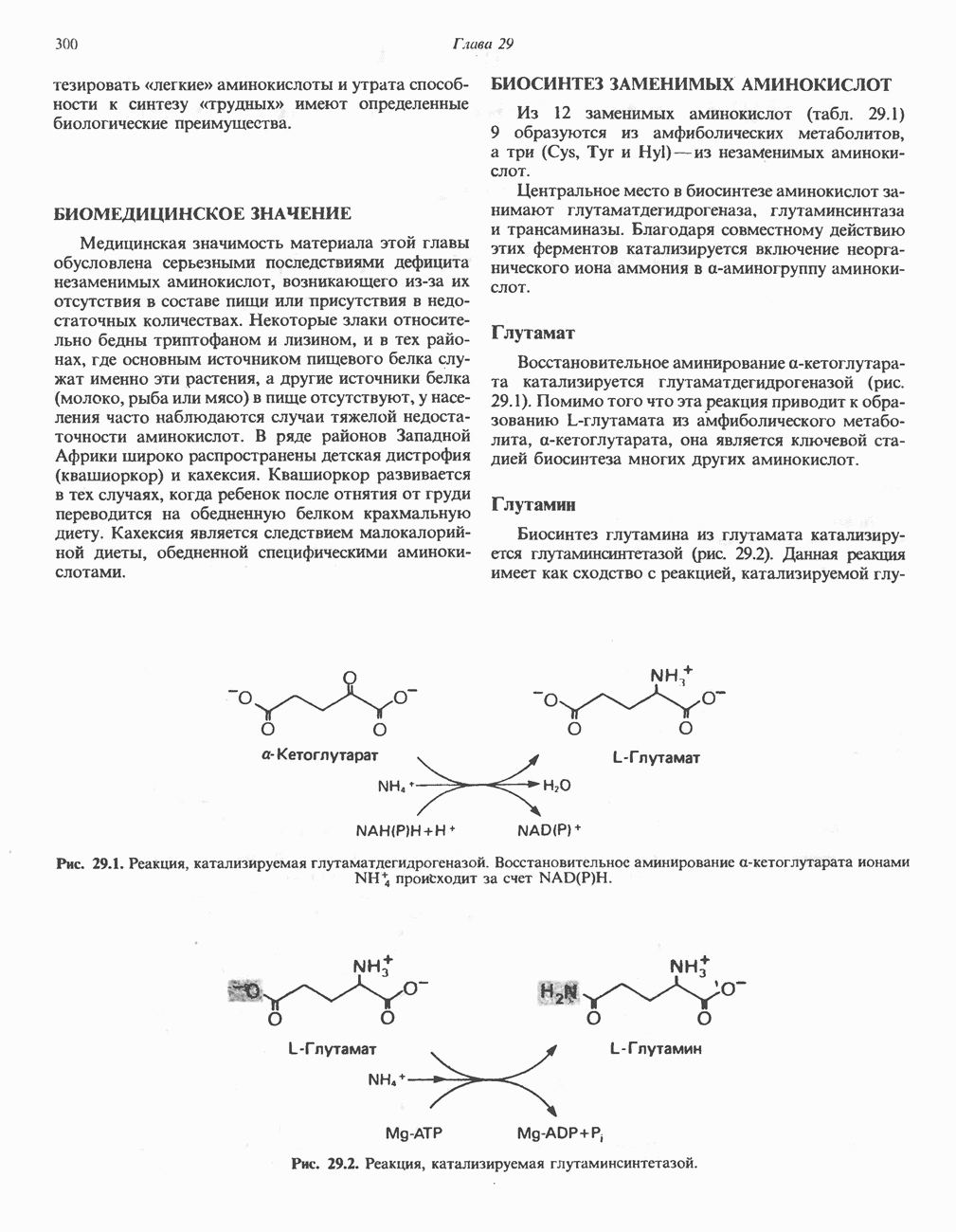
из серина, приводящее к образованию ненасыщенной аминокислоты. Последняя перегруппировывается в а-иминокислоту, которая подвергается спонтанному гидролизу с образованием пирувата и аммиака. Таким образом, в уравнение суммарной реакции, катализируемой сериндегидратазой, вода не входит.  глутамат, а КГ - а-кетоглутарат
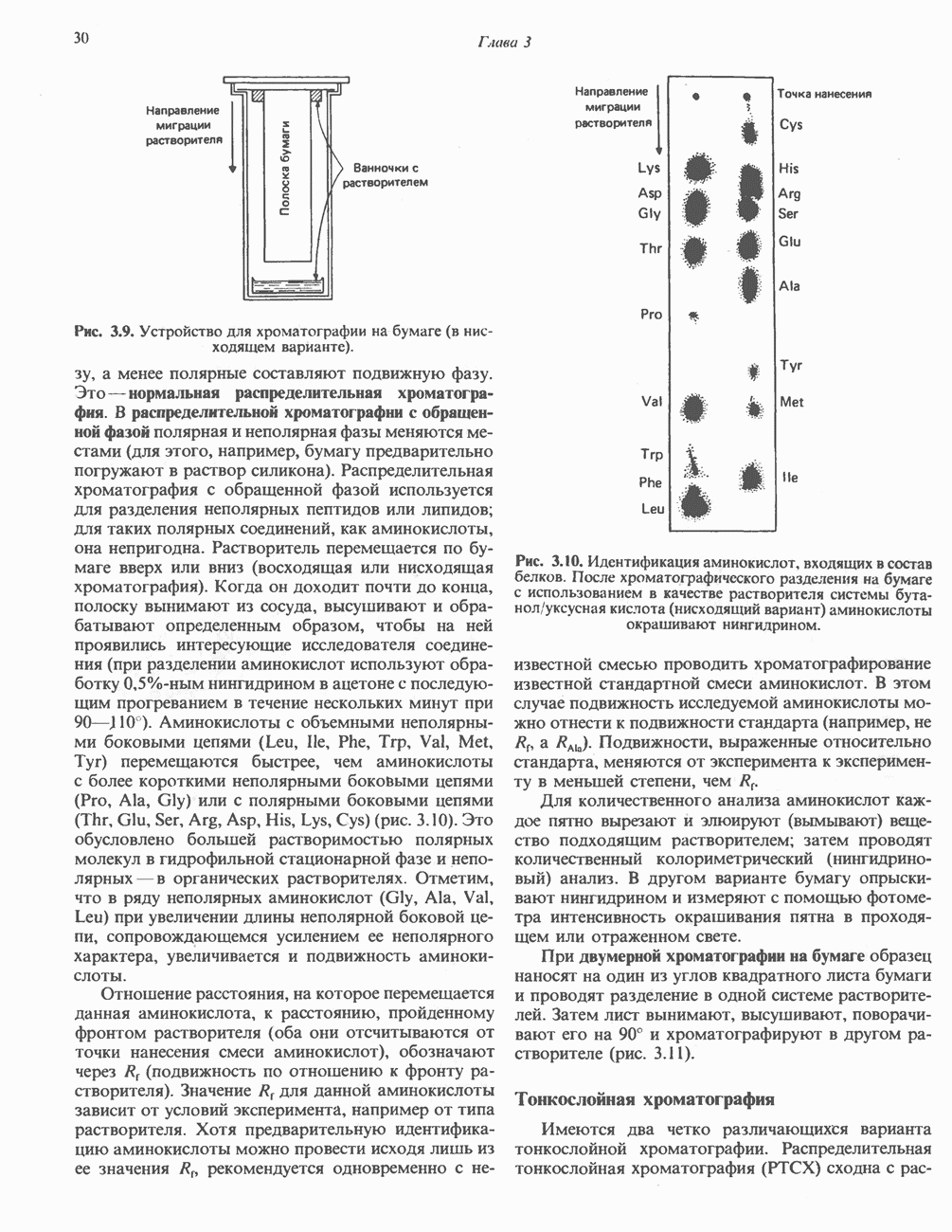
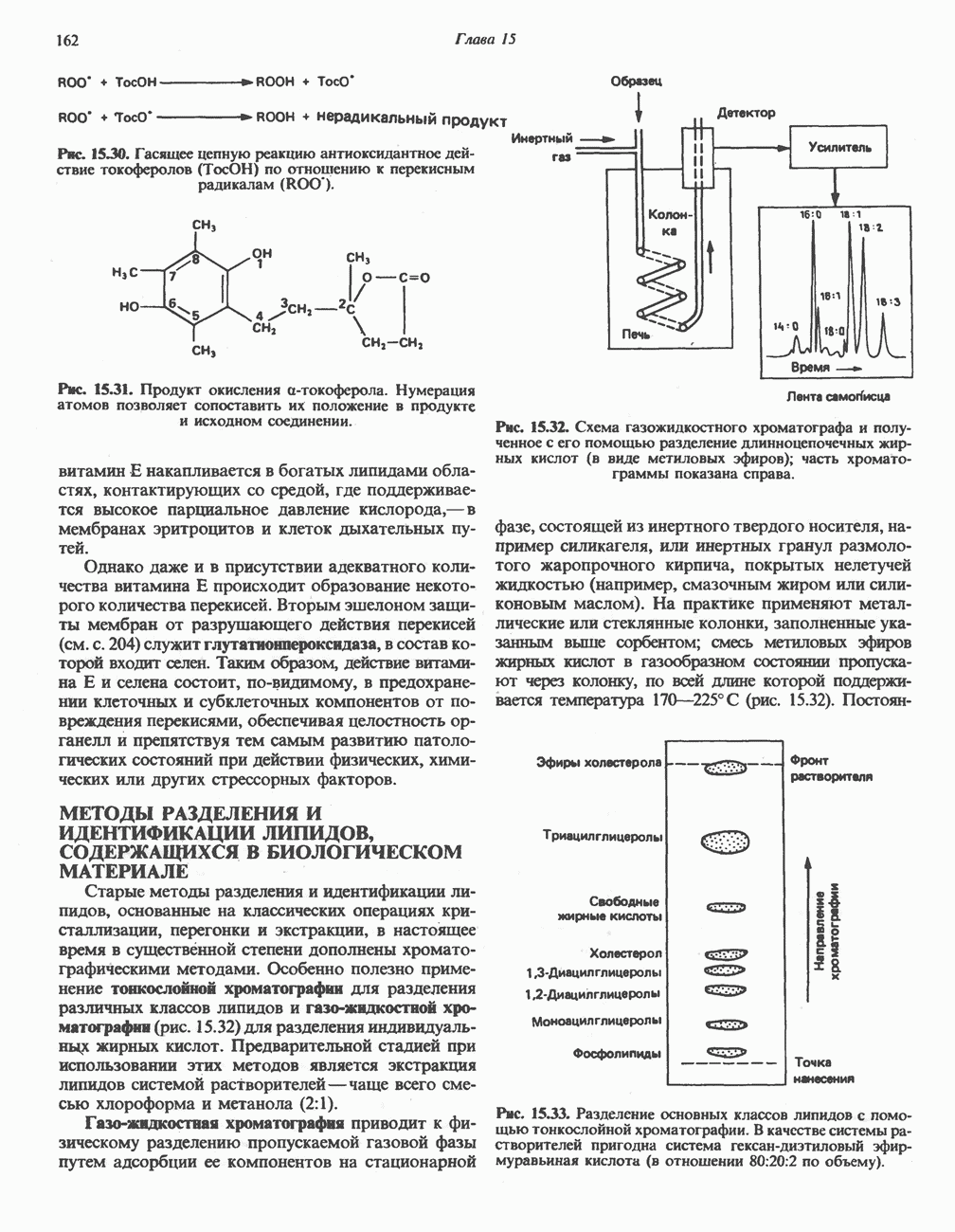
глутамат, а КГ - а-кетоглутарат

 -меркаптопируватному) (рис. 31.9). Первоначально предполагали, что имеется еще и третий путь с участием цистеиндесульфгидразы, который, как было показано, функционирует у бактерий. Однако маловероятно, что этот путь функционирует у млекопитающих, поскольку в их тканях активность цистеиндесульфгидразы не обнаружена.
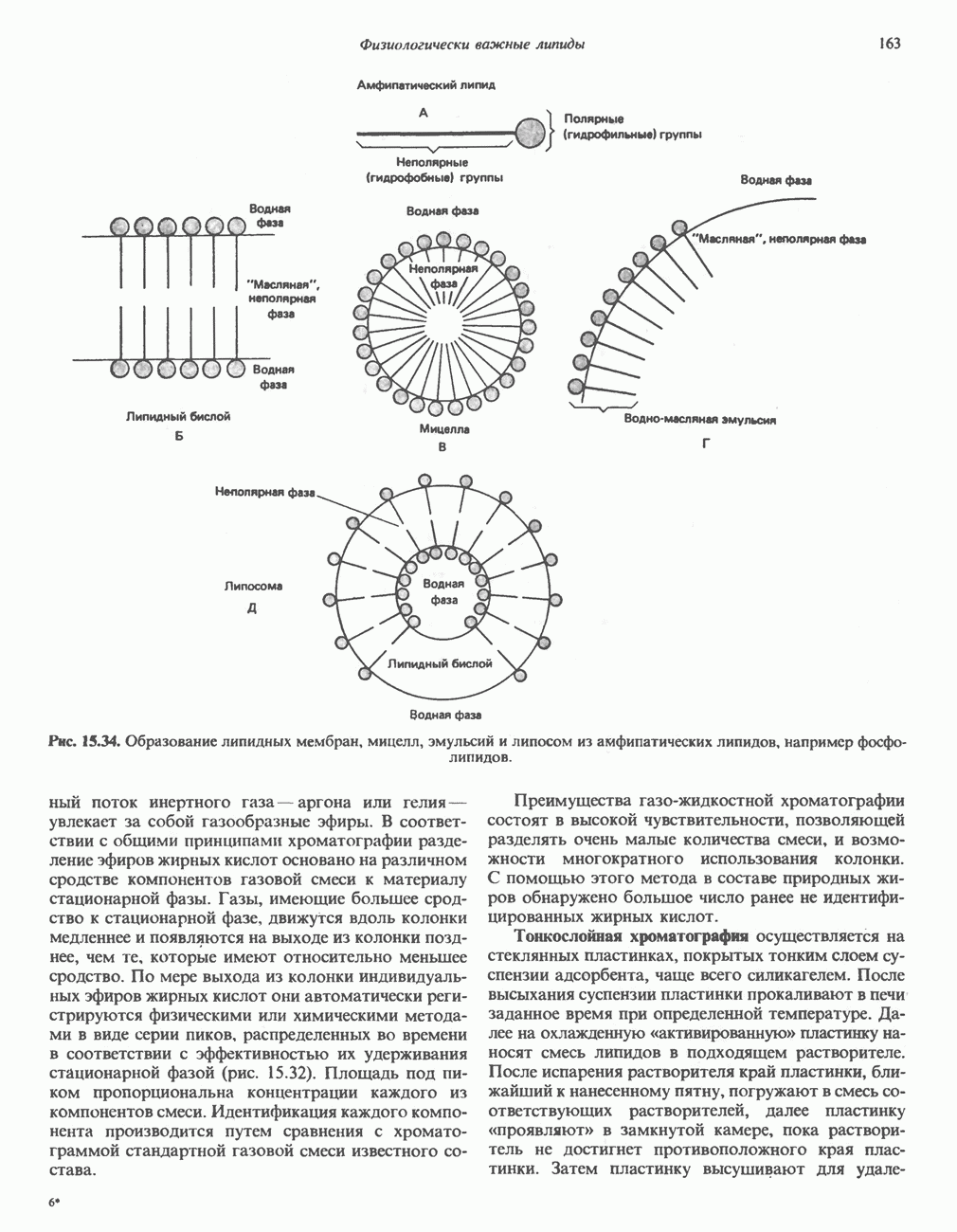
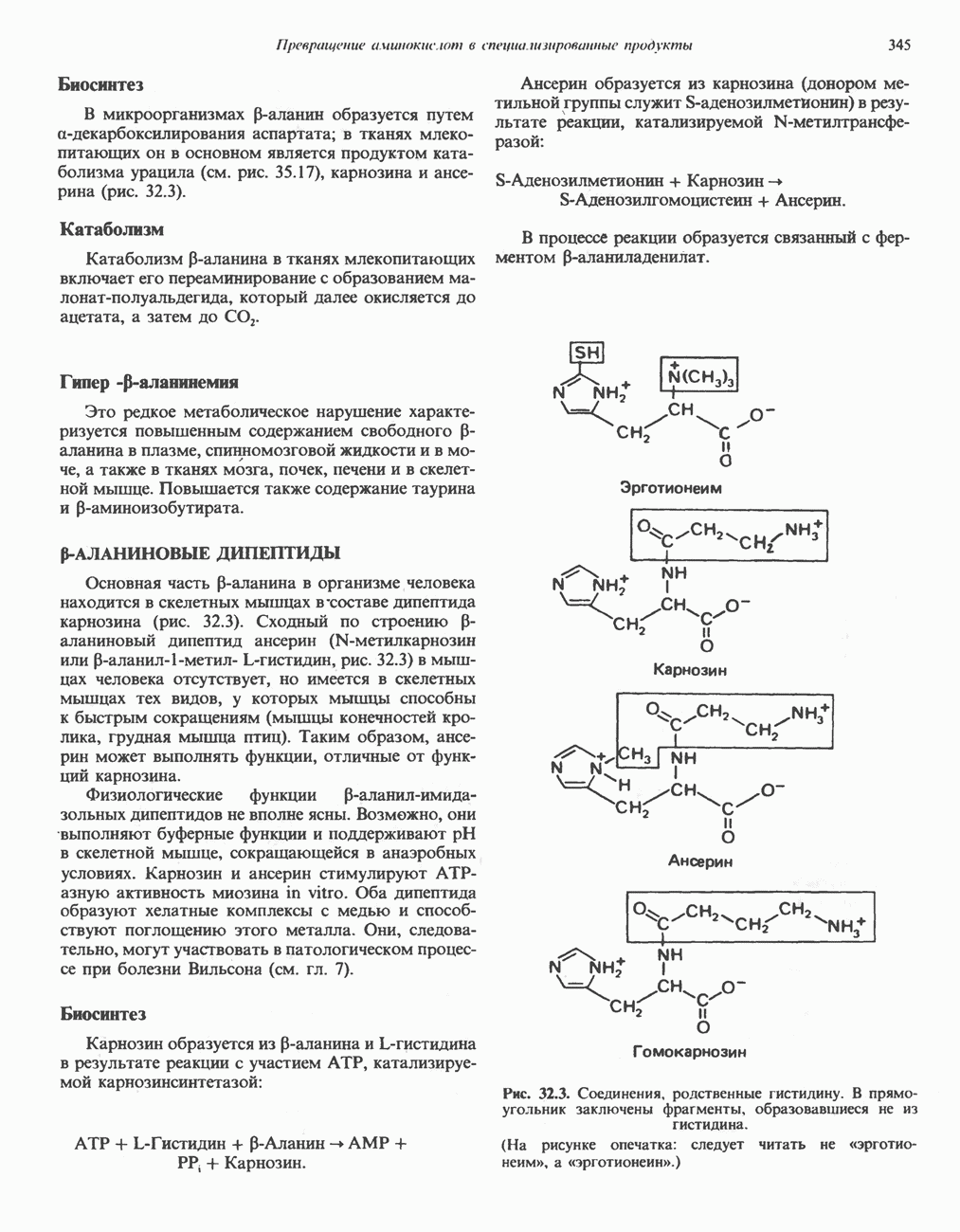
-меркаптопируватному) (рис. 31.9). Первоначально предполагали, что имеется еще и третий путь с участием цистеиндесульфгидразы, который, как было показано, функционирует у бактерий. Однако маловероятно, что этот путь функционирует у млекопитающих, поскольку в их тканях активность цистеиндесульфгидразы не обнаружена.

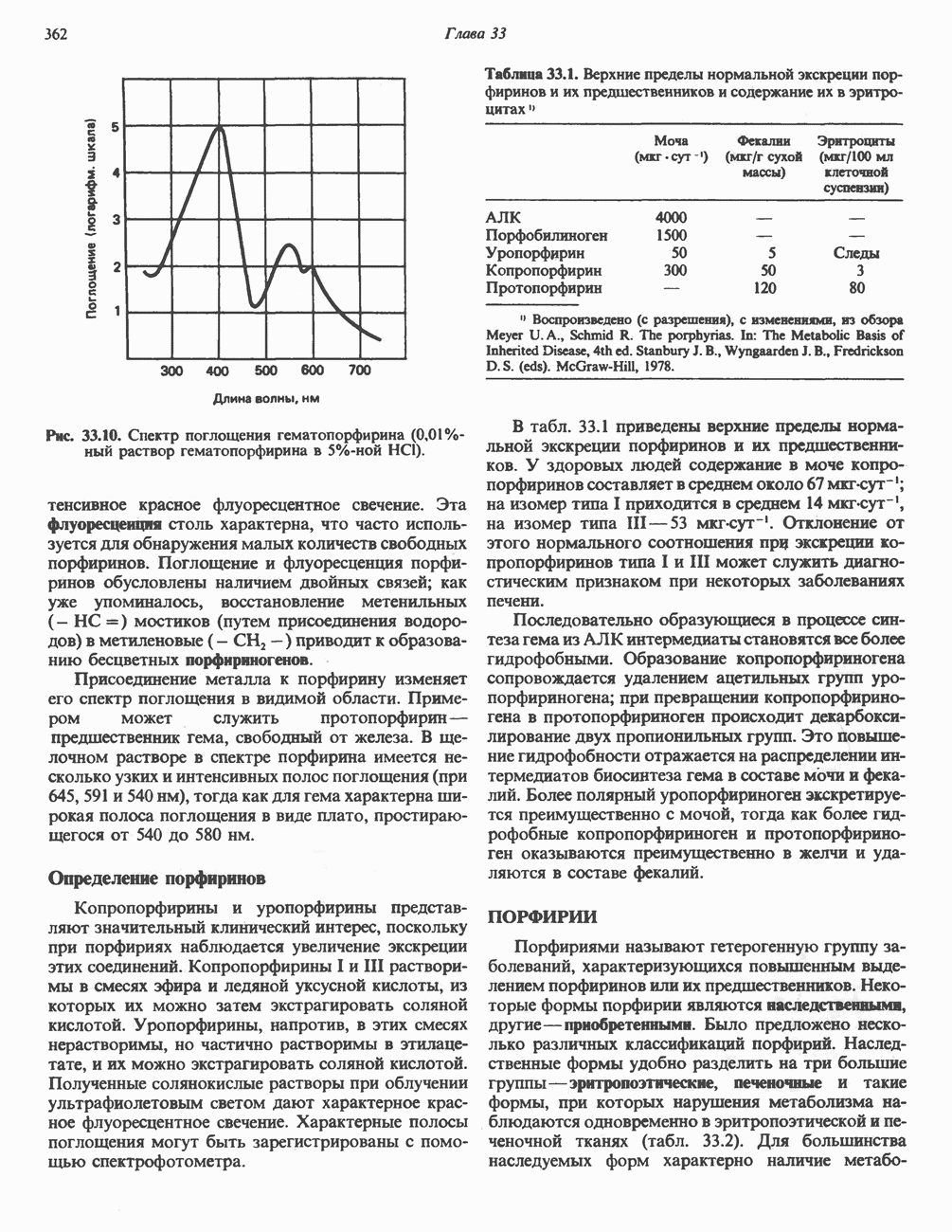
 -меркаптопируватный путь, справа). Р-Сульфинилпируват помещен в скобки, так как образование этого интермедиата экспериментально не доказано. Окисление сульфита в последней реакции прямого окислительного пути катализируется сульфитоксидазой.
-меркаптопируватный путь, справа). Р-Сульфинилпируват помещен в скобки, так как образование этого интермедиата экспериментально не доказано. Окисление сульфита в последней реакции прямого окислительного пути катализируется сульфитоксидазой.
 . Дальнейший катаболизм цистеинсульфината включает, вероятно, переаминирование с образованием Р-сульфинилпи-рувата. Возможно, что трансаминаза тканей млекопитающих, использующая цистеинсульфинат как донор аминогруппы, идентична классической глутамат:аспартат трансаминазе. Следует, однако, отметить, что предполагаемый продукт трансаминирования Р-сульфинилпирува
. Дальнейший катаболизм цистеинсульфината включает, вероятно, переаминирование с образованием Р-сульфинилпи-рувата. Возможно, что трансаминаза тканей млекопитающих, использующая цистеинсульфинат как донор аминогруппы, идентична классической глутамат:аспартат трансаминазе. Следует, однако, отметить, что предполагаемый продукт трансаминирования Р-сульфинилпирува  пока еще не был идентифицирован в системе катаболизма цистеинсульфината. Превращение постулируемого интермедиата Р-сульфинилпирувата в пируват осуществляется, вероятно, неферментативным путем.
пока еще не был идентифицирован в системе катаболизма цистеинсульфината. Превращение постулируемого интермедиата Р-сульфинилпирувата в пируват осуществляется, вероятно, неферментативным путем.
 с образованием пирувата (рис. 31.9).
с образованием пирувата (рис. 31.9).

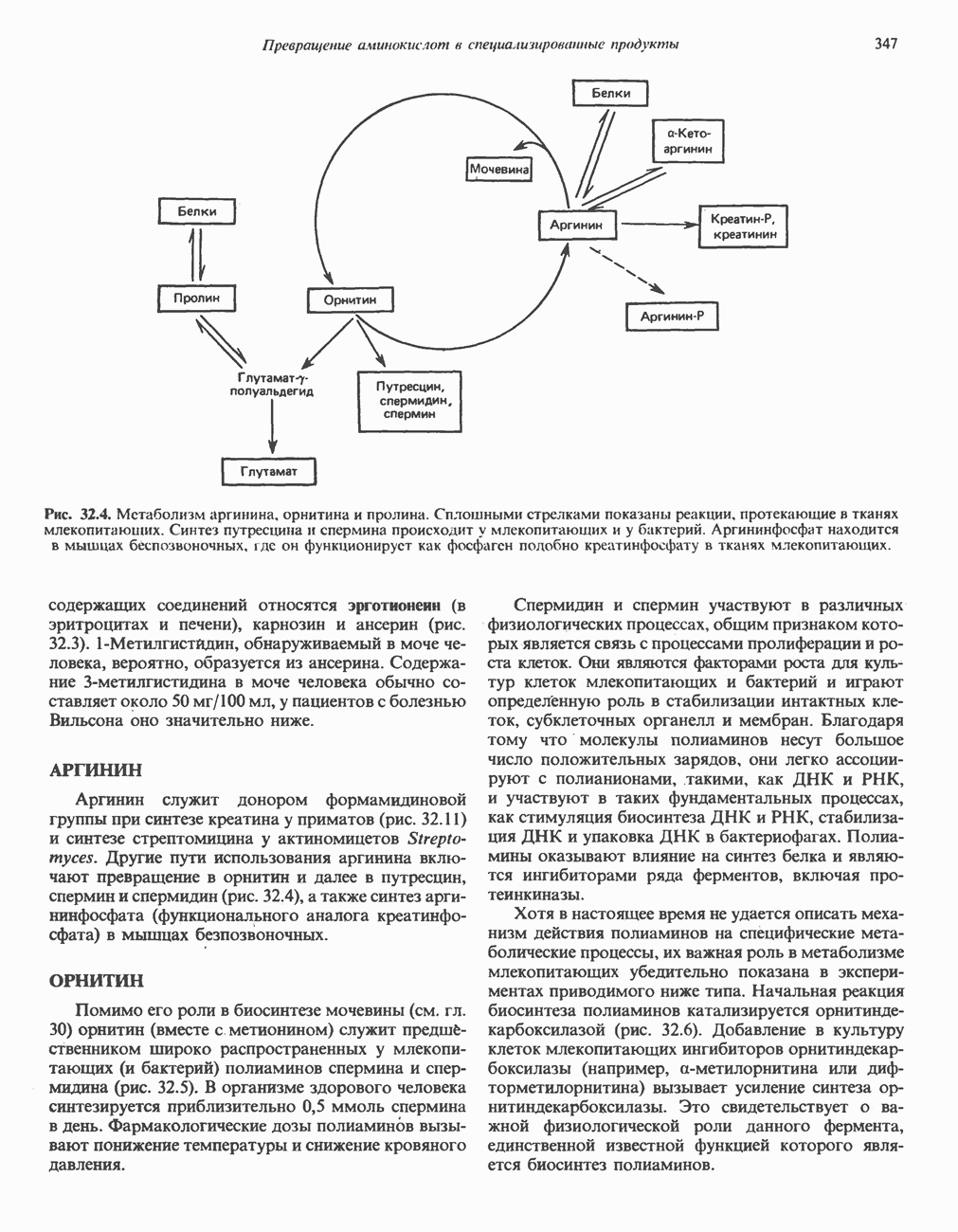
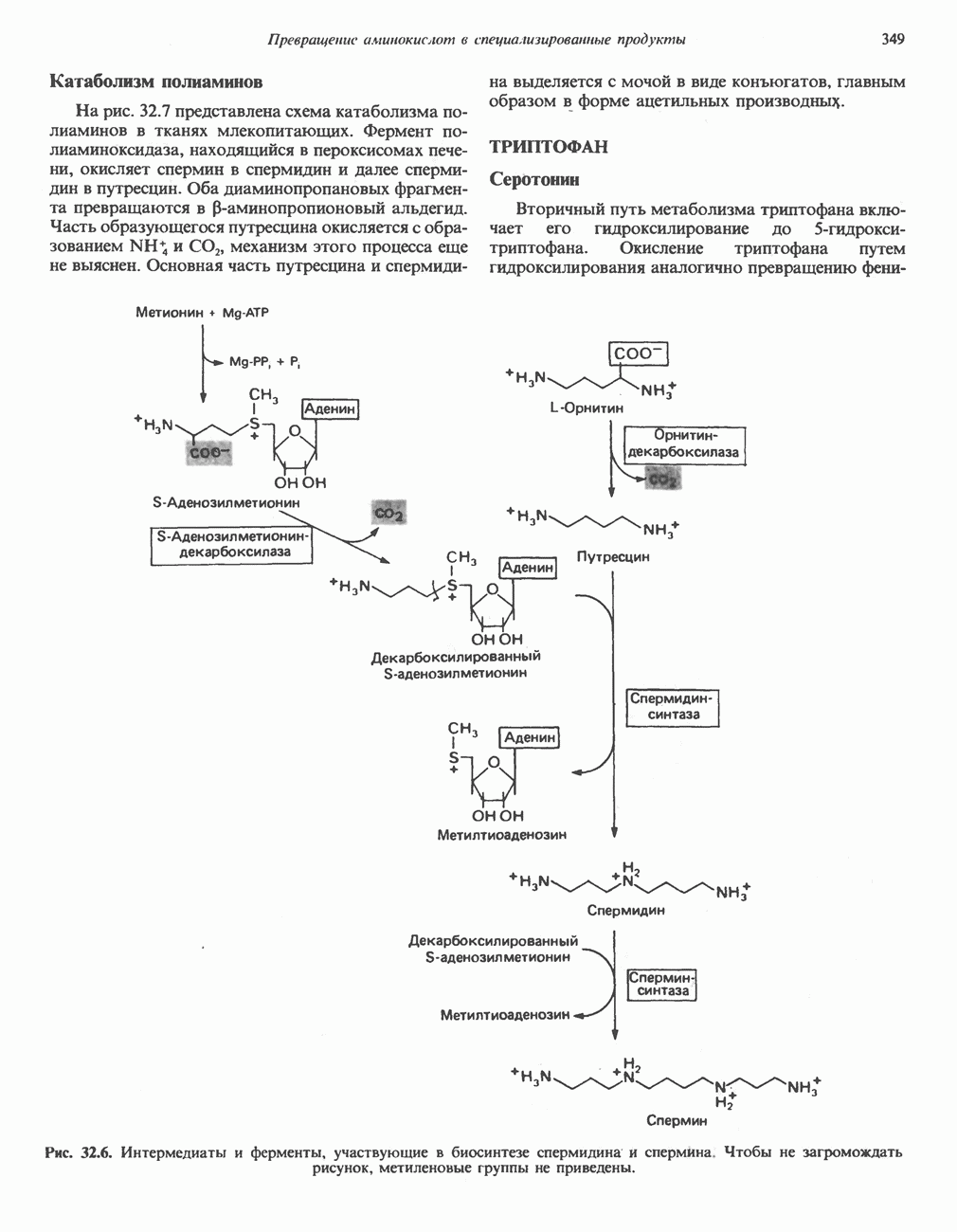
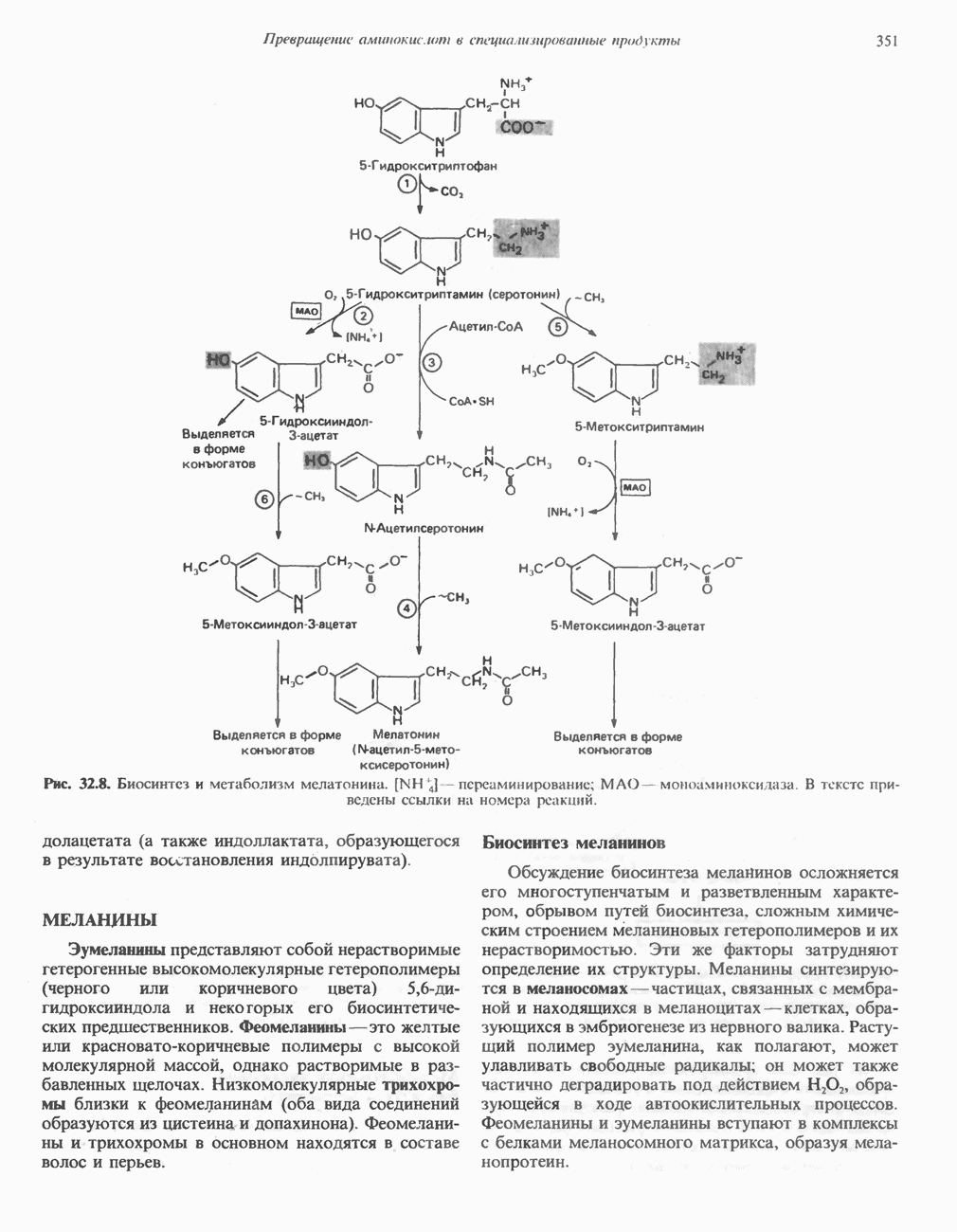
 причиной болезни является нарушение функции лизосом.
причиной болезни является нарушение функции лизосом.
 -аденозилметионином.
-аденозилметионином.
 фолат-формил [5—10] тетрагидрофолиевая кислота.
фолат-формил [5—10] тетрагидрофолиевая кислота.
 Последний находится в неферментативном равновесии с у-гидрокси
Последний находится в неферментативном равновесии с у-гидрокси  -глутамат-у-полуальдегидом, который образуется в результате присоединения воды. Полуальдегид окисляется в соответствующую карбоновую кислоту,
-глутамат-у-полуальдегидом, который образуется в результате присоединения воды. Полуальдегид окисляется в соответствующую карбоновую кислоту,  далее при переаминировании образуется а-кето-у-гидроксиглутарат. При последующем альдольном расщеплении образуются глиоксилат и пируват.
далее при переаминировании образуется а-кето-у-гидроксиглутарат. При последующем альдольном расщеплении образуются глиоксилат и пируват.