Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
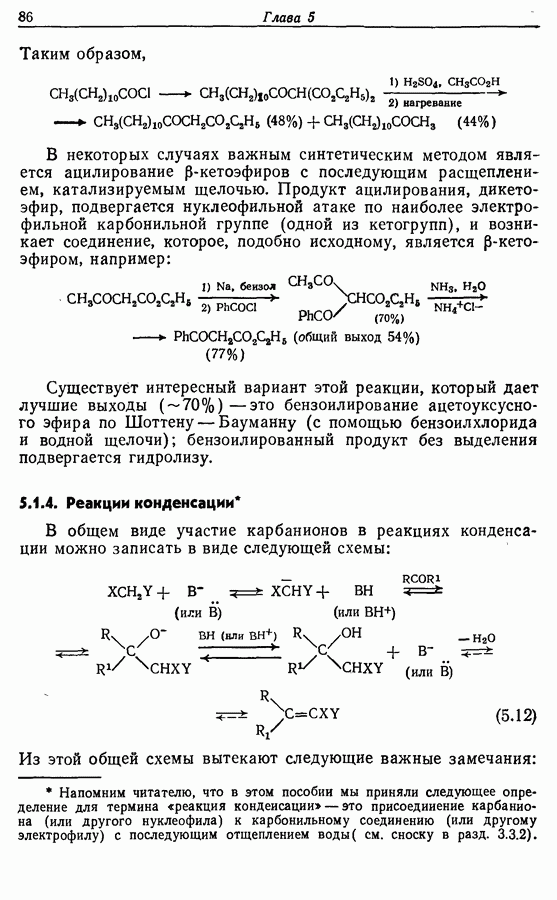
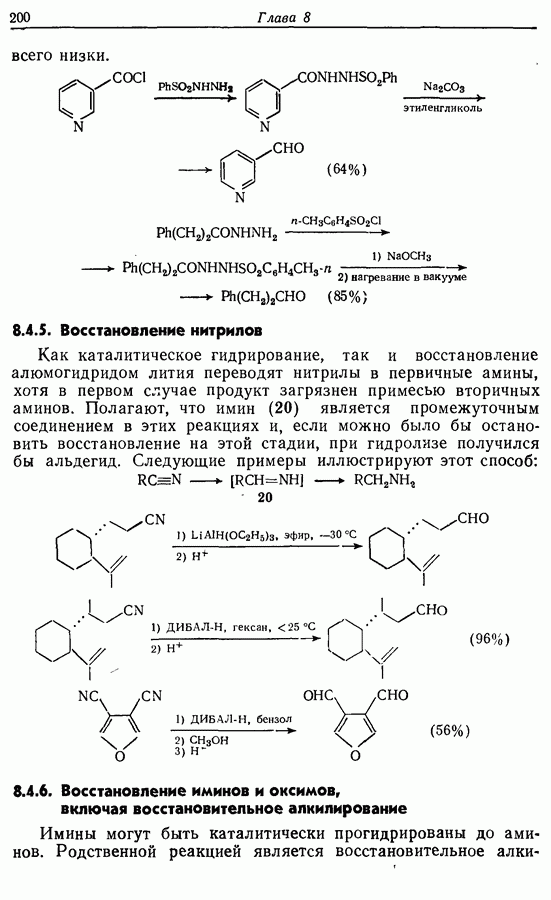
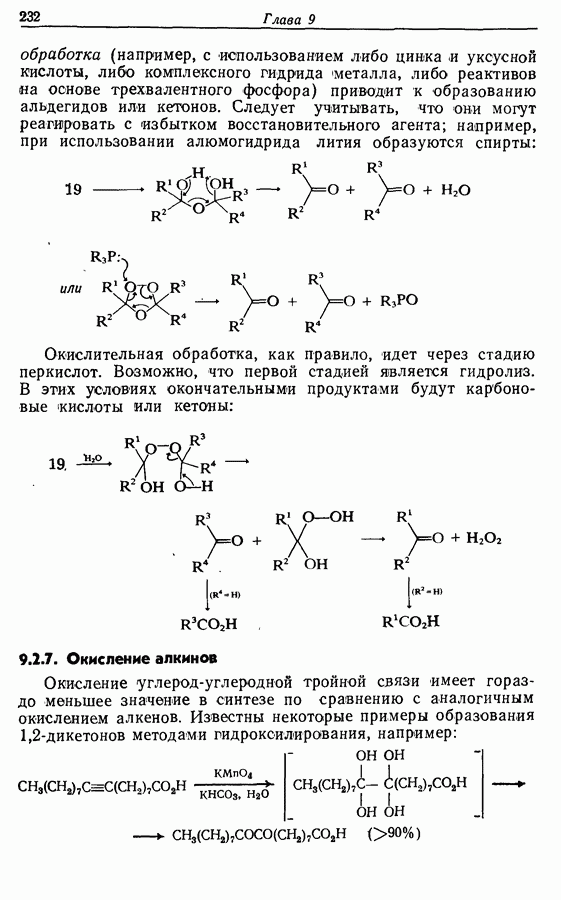
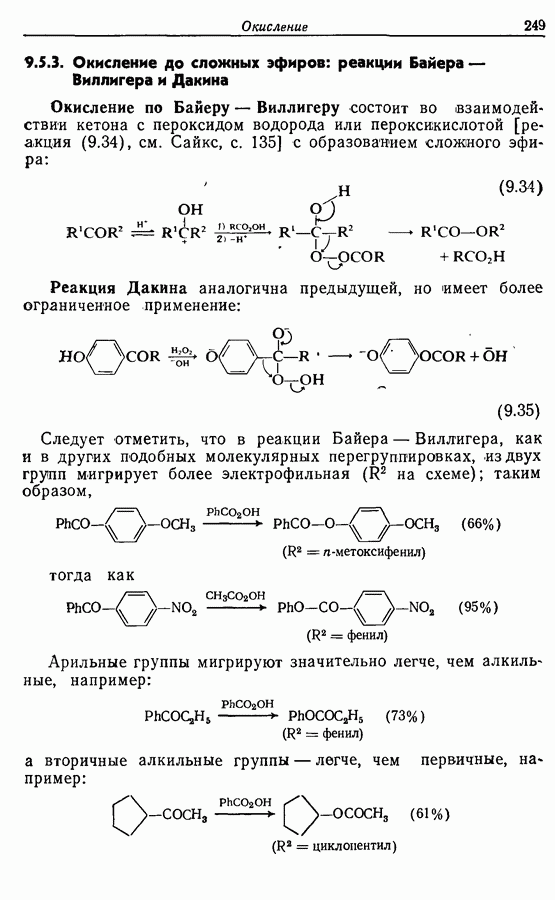
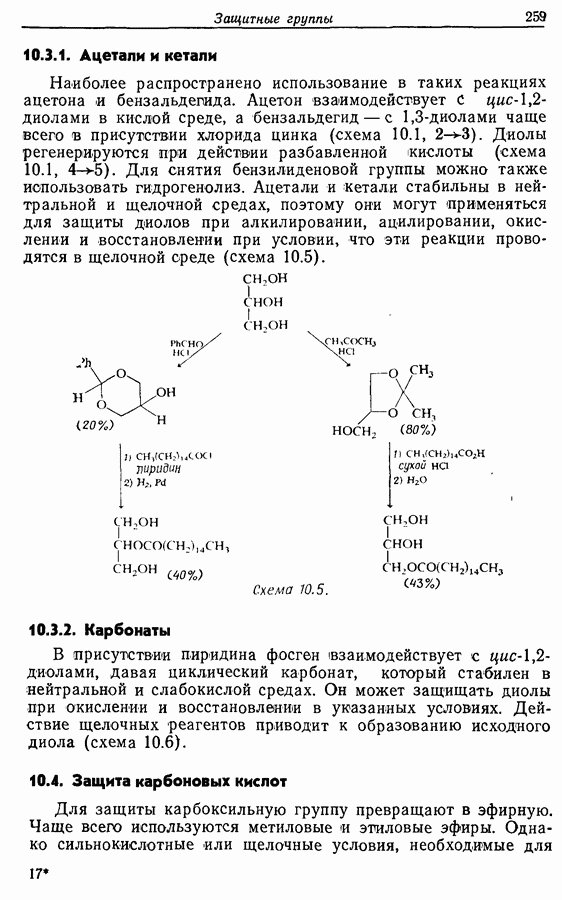
5.4. Алкены, арены и гетероарены как нуклеофилыДо сих пор в данной главе рассматривались реакции с образованием углерод-углеродной связи, в которых нуклеофильный компонент формально нес на себе отрицательный заряд, т. е. был карбанионом. Теперь же мы рассмотрим группу реакций, в которых нуклеофил — нейтральная молекула; простые алкены, арены и гетероарены входят в эту группу. Однако, как уже отмечалось в разд. 2.2, 2.4-2.6 и 3.4.3, существует лишь немного подобных синтетически полезных лабораторных реакций (в отличие от промышленных процессов) этого типа, в которых происходит образование углерод-углеродной связи. Конечно, исключение составляет реакция Фриделя — Крафтса. В разд. 3.4.3 также уже отмечалось, что электронодонорный обогащенными электронами гетероароматическими циклическими системами. (Конечно, енолы являются таутомерными формами альдегидов и кетонов, и реакции енолов, описанные далее, более точно следует рассматривать как реакции карбонильных соединений с енолизацией на первой стадии.) 5.4.1. АлкилированиеАлкилирование — наименее важная среди реакций образования связи
Аналогично
Если несимметричный кетон может давать два енамина, в смеси продуктов преобладает наиболее стабильный. Из
альдегидов через енамины имеет ограниченное применение из-за возможности побочных реакций
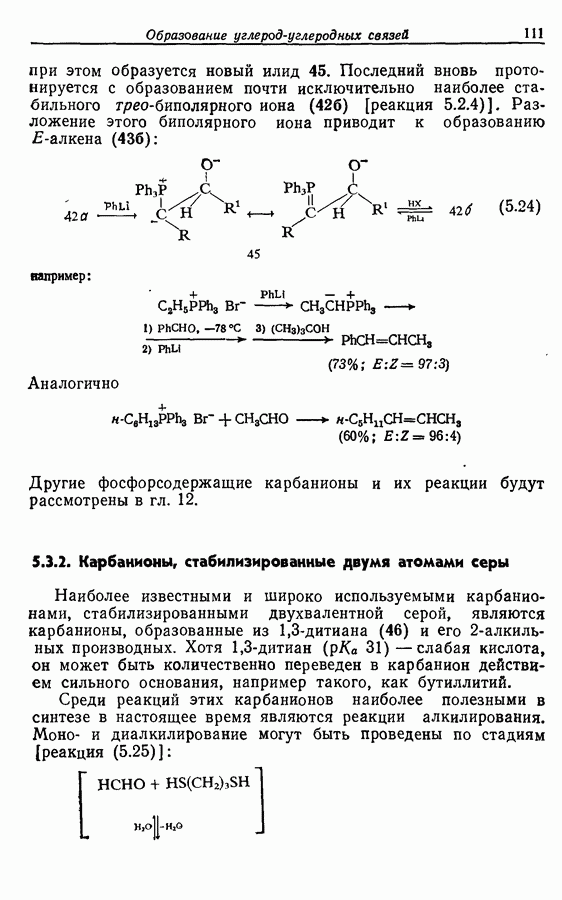
Для получения альдегидов через дигидро-1,3-оксазины также удобно использовать енаминный интермедиат 51: (см. скан) 5.4.2. АцилированиеВ отличие от алкилирования, применение которого в синтезе ограничено, ацилирование активированных алкенов, аренов и гетероаренов имеет очень большое синтетическое значение. Многие из этих реакций являются вариантами реакции Фриделя—Крафтса, например:
Ацилирование енаминов, как и алкилирование, может идти и по углероду, и по азоту, но поскольку в последнем случае реакция легко обратима и в результате ее получается N-ациламмониевая соль, которая представляет собой ацилирующий агент, обычно можно достичь высоких выходов С-ацилирован-ных продуктов. Реакции ацилирования часто проводят в присутствии основания, такого, как триэтиламин, поскольку первоначальный продукт ацилирования (52) является кислотой средней силы и может в отсутствие более сильного основания лротонировать непрореагировавший енамин:
Однако если ацилирующий агент содержит а-водород, он может реагировать с добавленным основанием, давая кетен, который в свою очередь вступает в реакцию циклоприсоединения к енамину. В подобных случаях может быть получено лишь небольшое количество продукта ацилирования. Ниже приведены примеры ацилирования енаминов:
Последняя реакция, хотя и не является формально ацилированием, имеет родственный механизм. Среди методов формилирования наиболее распространен метод Вильсмейера — Хаака — Арнольда, который заключается в использовании
Аналогично
В первоначальном варианте (реакция Вильсмейера — Хаака) метод использовался для формилирования активированных аренов и гетероаренов, например:
Аналогично
Активированные алкены также достаточно легко реагируют с иминиевыми солями, например:
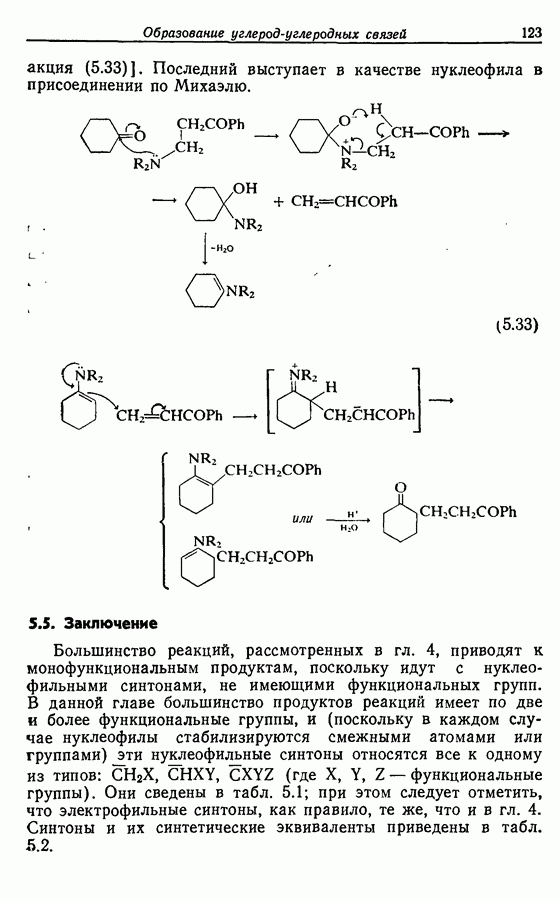
Однако в большинстве случаев простые продукты формилирования получить не удается. Две другие реакции, которые формально можно считать ацилированием, заслуживают краткого упоминания. Обе они в принципе связаны с ацилированием фенолов: реакция Реймера-Тимана, в которой электрофилом служит дихлоркарбен, и реакция Кольбе — Шмитта, в которой электрофилом является диоксид углерода (см. Сайке, с. 269):
Оба типа реакций применяли не только к феноксид-ионам, но и к иным нуклеофилам, например:
Однако в реакции Реймера — Тимана выходы обычно низки, и часто получаются побочные продукты. Ни один из этих методов необщеприменим, хотя каждый в отдельности имеет определенное значение в специфических случаях.
|
 |
Оглавление
|








































































































































































































































































































































 -заместитель значительно повышает нуклеофильность алкенов и аренов. Такая «активация» позволяет указанным соединениям взаимодействовать с углеродными электрофилами, обладающими меньшей электрофильностью, чем используемые в реакции Фриделя — Крафтса (относительно последней см. Сайке, с. 147—150). В настоящей главе мы рассмотрим реакции таких электрофилов с енолами и енаминами, их ароматическими аналогами, фенолами и ариламинами. В тех случаях, когда это возможно, будет проведено сравнение их с
-заместитель значительно повышает нуклеофильность алкенов и аренов. Такая «активация» позволяет указанным соединениям взаимодействовать с углеродными электрофилами, обладающими меньшей электрофильностью, чем используемые в реакции Фриделя — Крафтса (относительно последней см. Сайке, с. 147—150). В настоящей главе мы рассмотрим реакции таких электрофилов с енолами и енаминами, их ароматическими аналогами, фенолами и ариламинами. В тех случаях, когда это возможно, будет проведено сравнение их с
 для этой группы нуклеофилов. Алкилирование альдегидов и кетонов, как правило, проводят чаще через карбанионы (еноляты), а не через енолы (разд. 5.2.1 и 5.2.3), а алкилирование фенолов, ариламинов и гетероаренов идет чаще по гетероатому, а не по углероду. Алкилирование енаминов имеет некоторое препаративное значение, поскольку на нем основан косвенный метод
для этой группы нуклеофилов. Алкилирование альдегидов и кетонов, как правило, проводят чаще через карбанионы (еноляты), а не через енолы (разд. 5.2.1 и 5.2.3), а алкилирование фенолов, ариламинов и гетероаренов идет чаще по гетероатому, а не по углероду. Алкилирование енаминов имеет некоторое препаративное значение, поскольку на нем основан косвенный метод  -алкилирования альдегидов и кетонов в отсутствие сильных оснований (разд. 5.2.3). Наиболее полезным является С-алкилирование енаминов с использованием высокоэлектрофильного алкилгалогенида (см. приведенные ниже примеры); в других же случаях главной реакцией будет
-алкилирования альдегидов и кетонов в отсутствие сильных оснований (разд. 5.2.3). Наиболее полезным является С-алкилирование енаминов с использованием высокоэлектрофильного алкилгалогенида (см. приведенные ниже примеры); в других же случаях главной реакцией будет  -алкилирование:
-алкилирование:


 -тетралона образуется почти исключительно сопряженный енамин 49а, а не несопряженный изомер 496. В случае
-тетралона образуется почти исключительно сопряженный енамин 49а, а не несопряженный изомер 496. В случае  -метилцикло-гексанона основным продуктом взаимодействия с пирролидином является 50а, так как во втором изомере (506) имеется дестабилизирующее стерическое отталкивание между метальной группой и
-метилцикло-гексанона основным продуктом взаимодействия с пирролидином является 50а, так как во втором изомере (506) имеется дестабилизирующее стерическое отталкивание между метальной группой и  -водородом гетероциклического кольца:
-водородом гетероциклического кольца:

 -алкилирования енаминов и самоконденсации альдегидов). Однако в некоторых случаях такое алкилирование можно провести успешно, например:
-алкилирования енаминов и самоконденсации альдегидов). Однако в некоторых случаях такое алкилирование можно провести успешно, например:






 -дизамещенного формамида и хлороксида фосфора
-дизамещенного формамида и хлороксида фосфора  , Действительным электрофилом в таких реакциях является хлорометилениминиевый ион (53) [реакция (5.28)]:
, Действительным электрофилом в таких реакциях является хлорометилениминиевый ион (53) [реакция (5.28)]:






