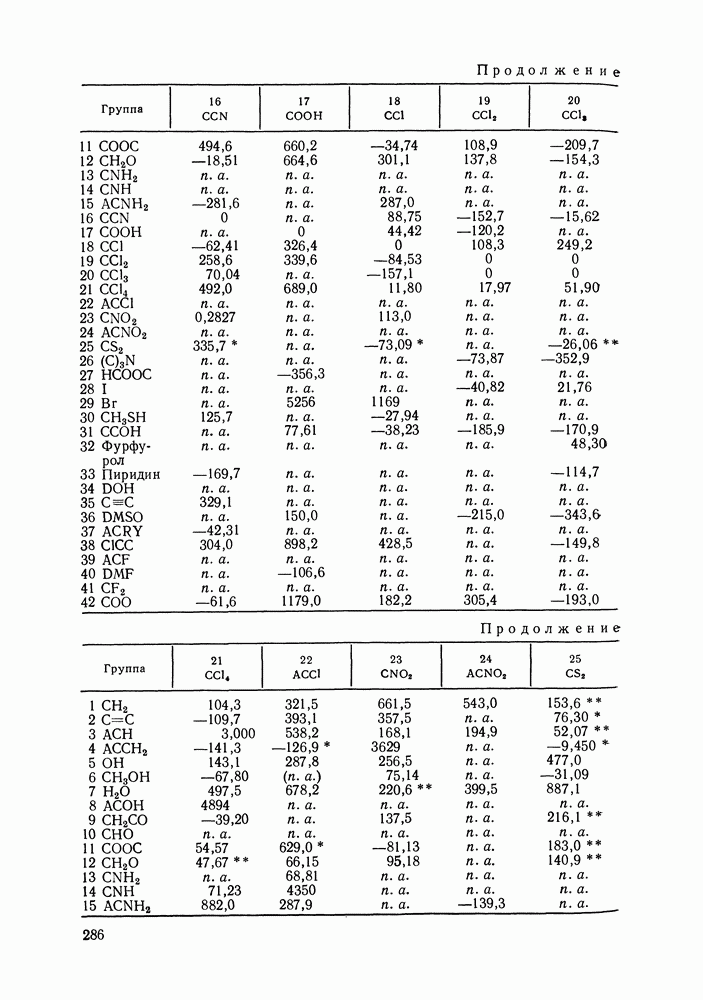
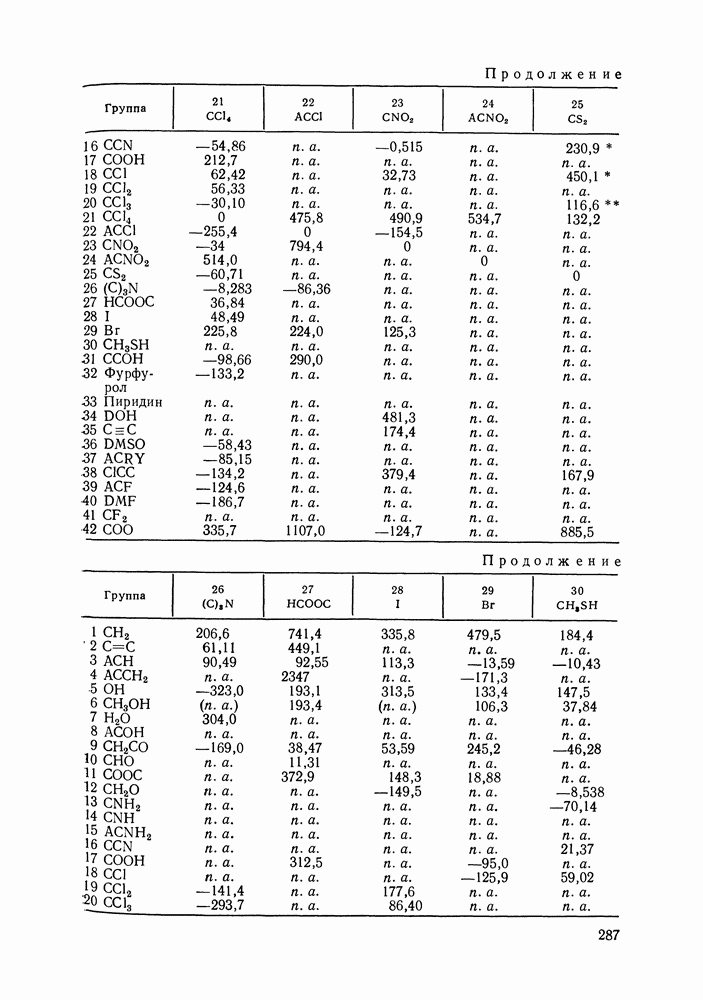
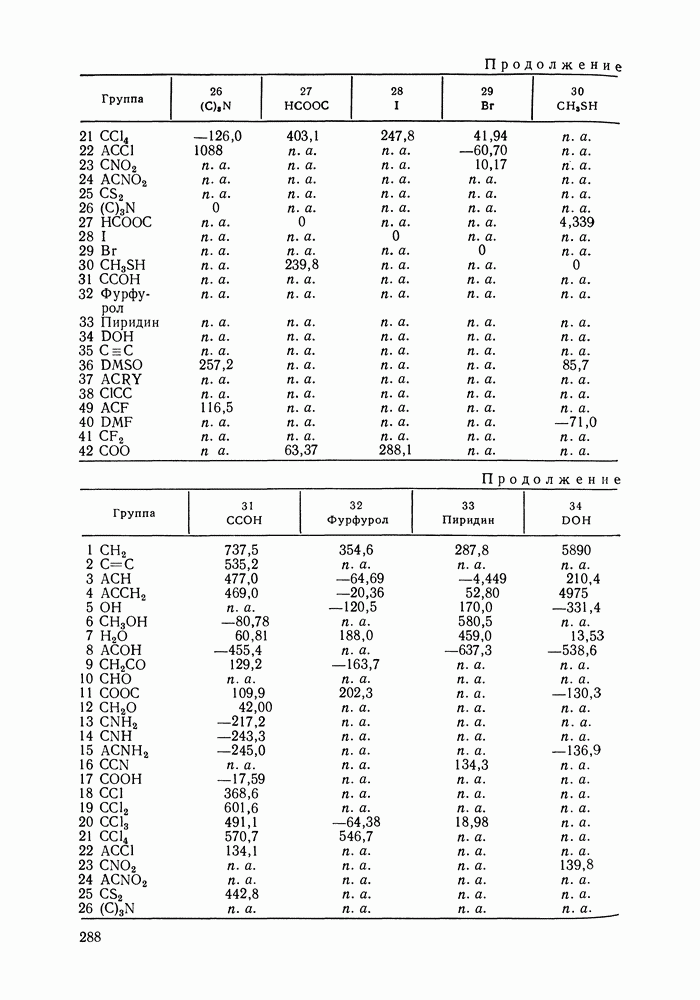
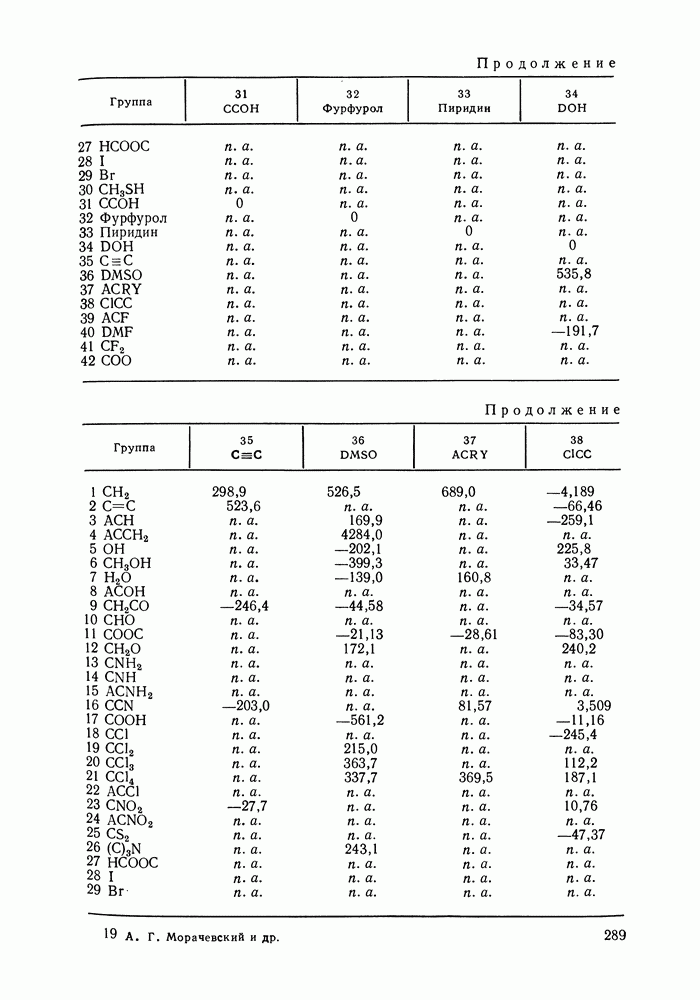
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
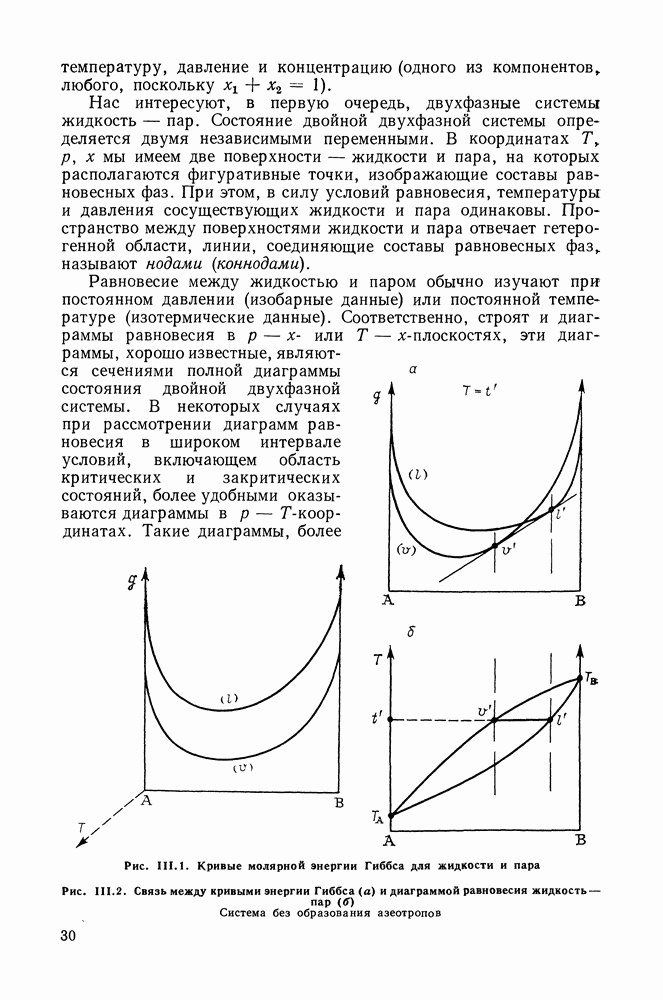
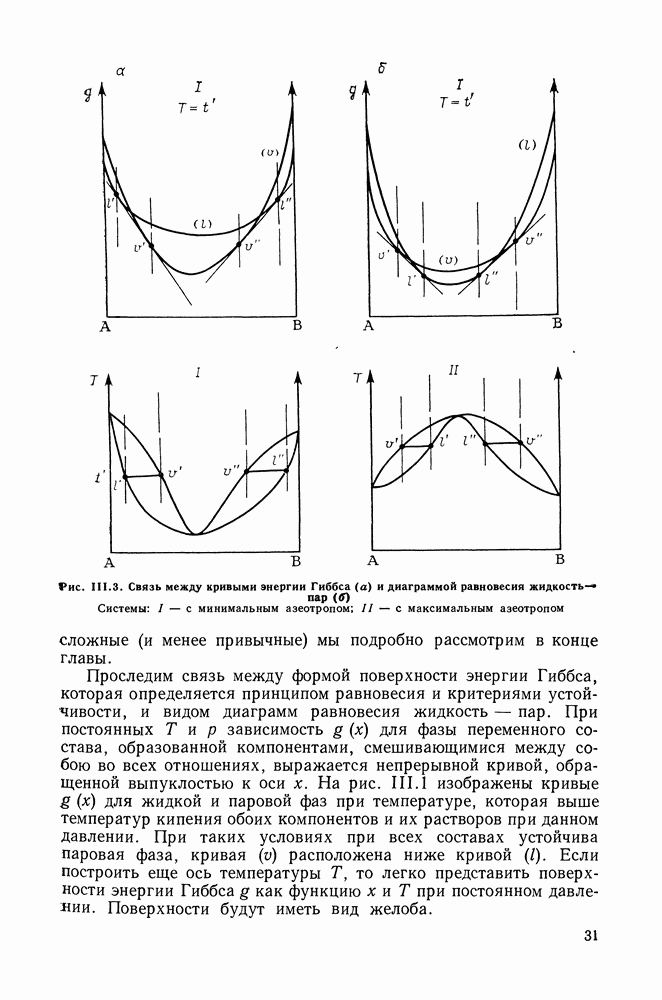
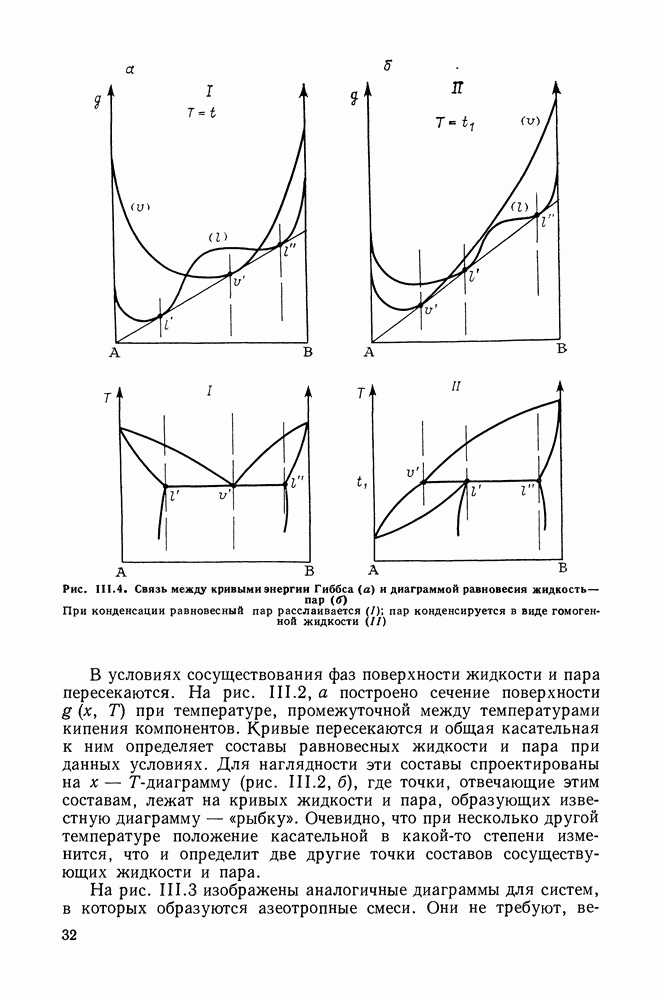
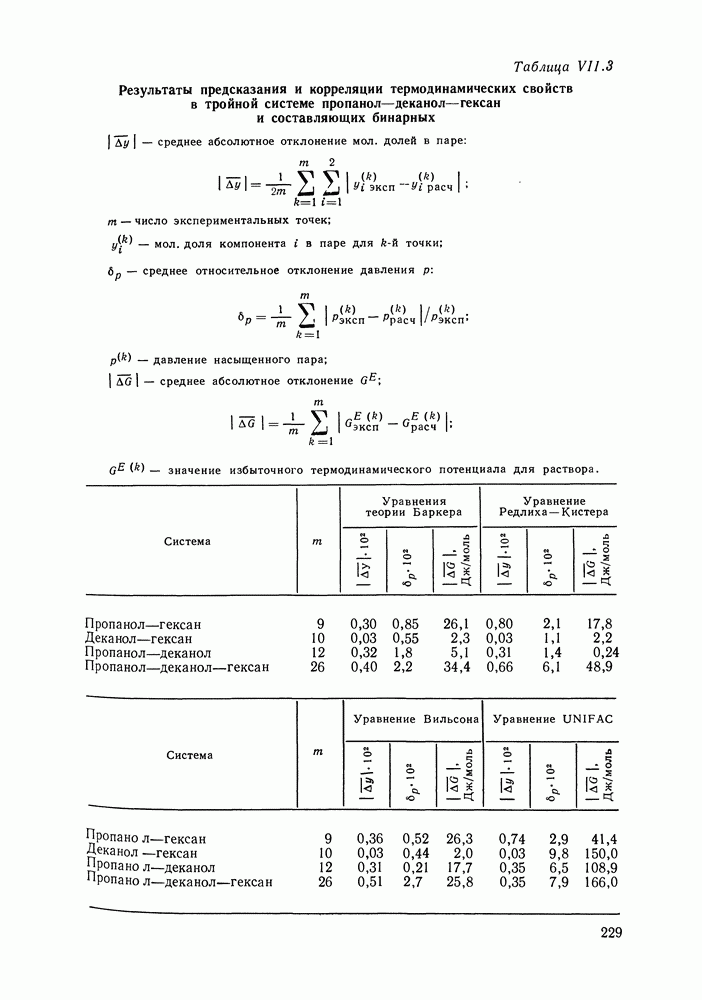
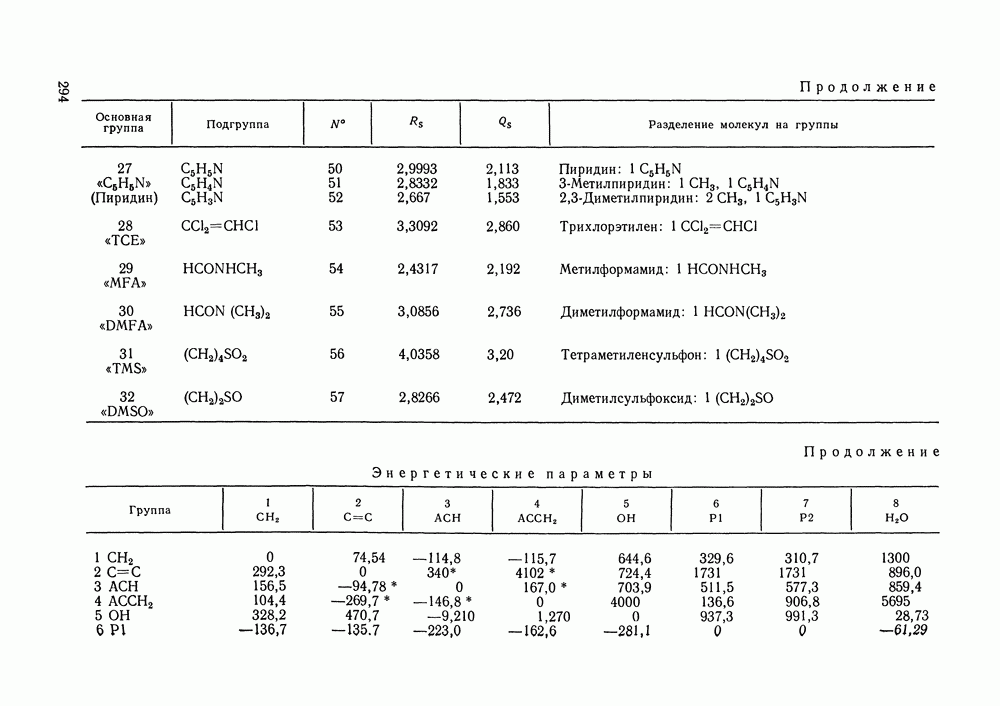
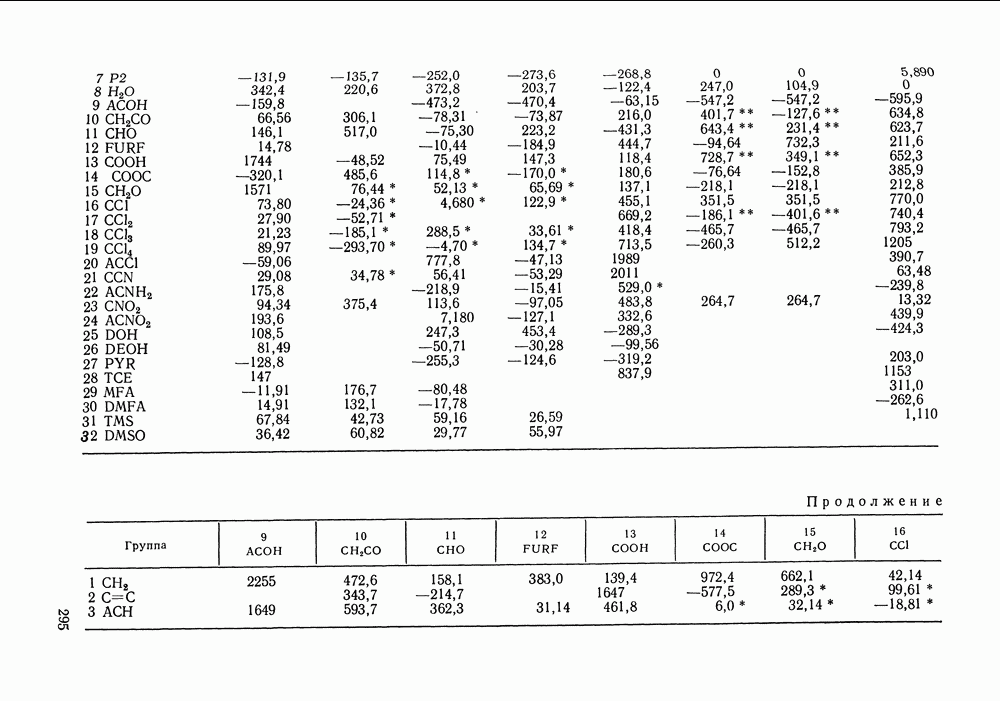
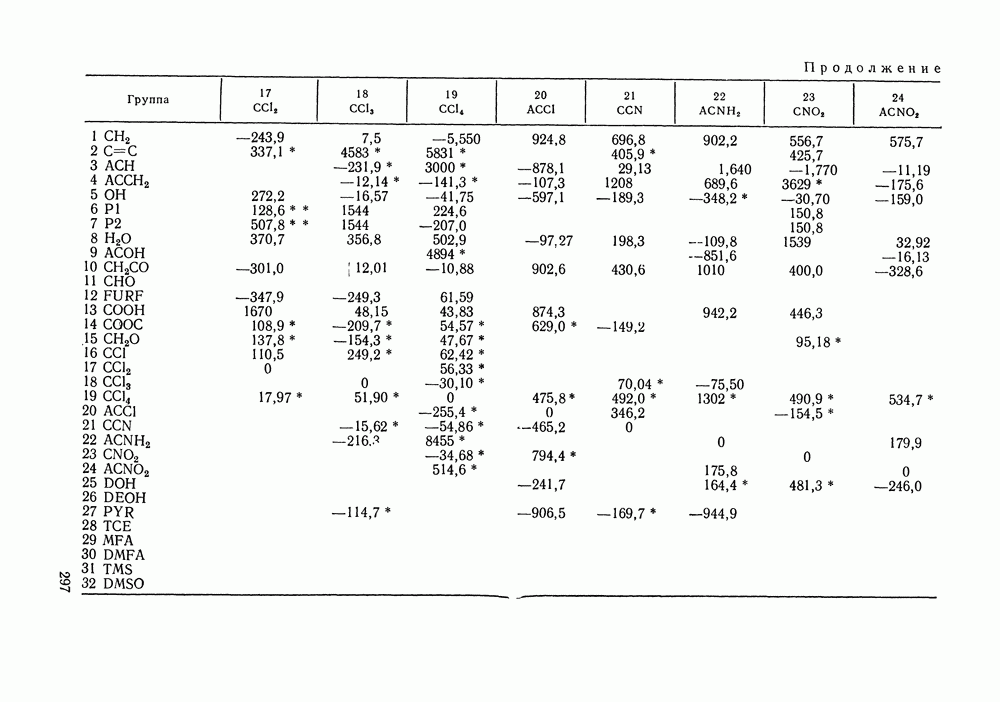
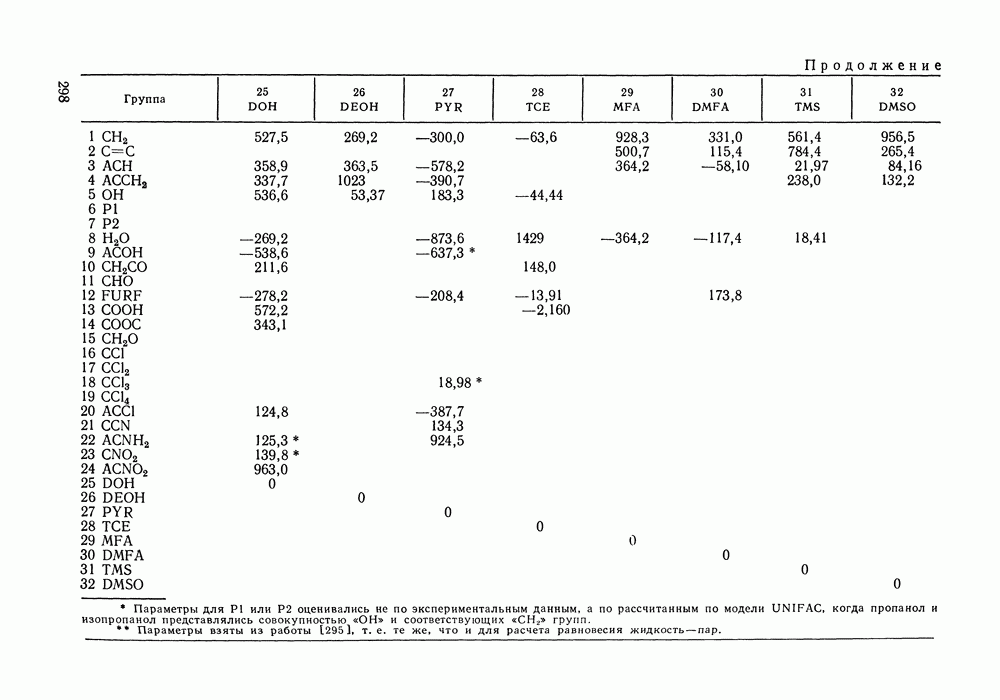
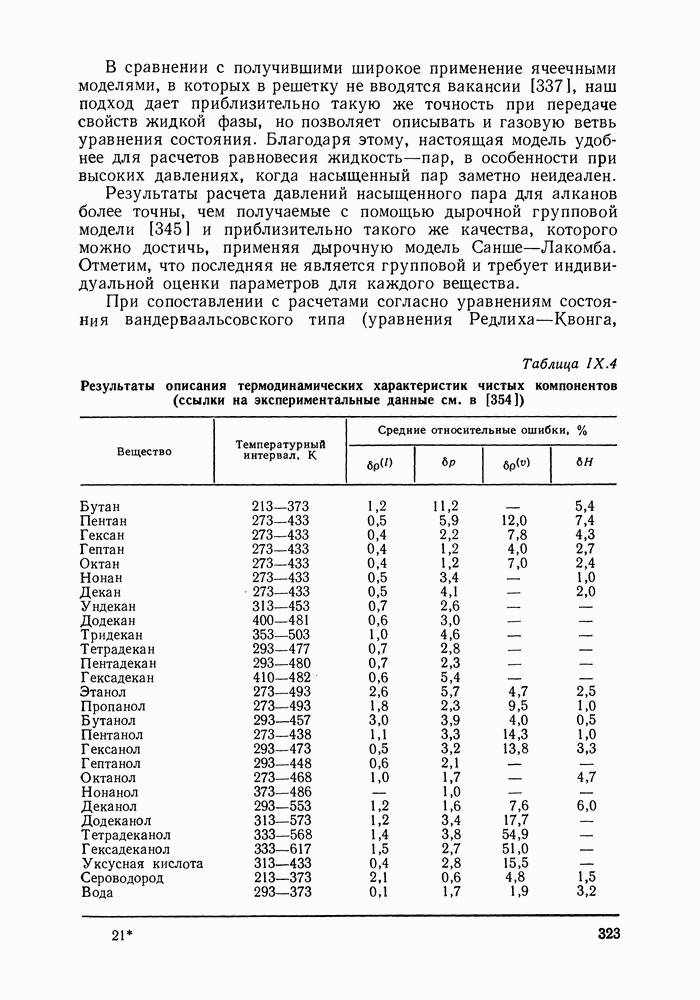
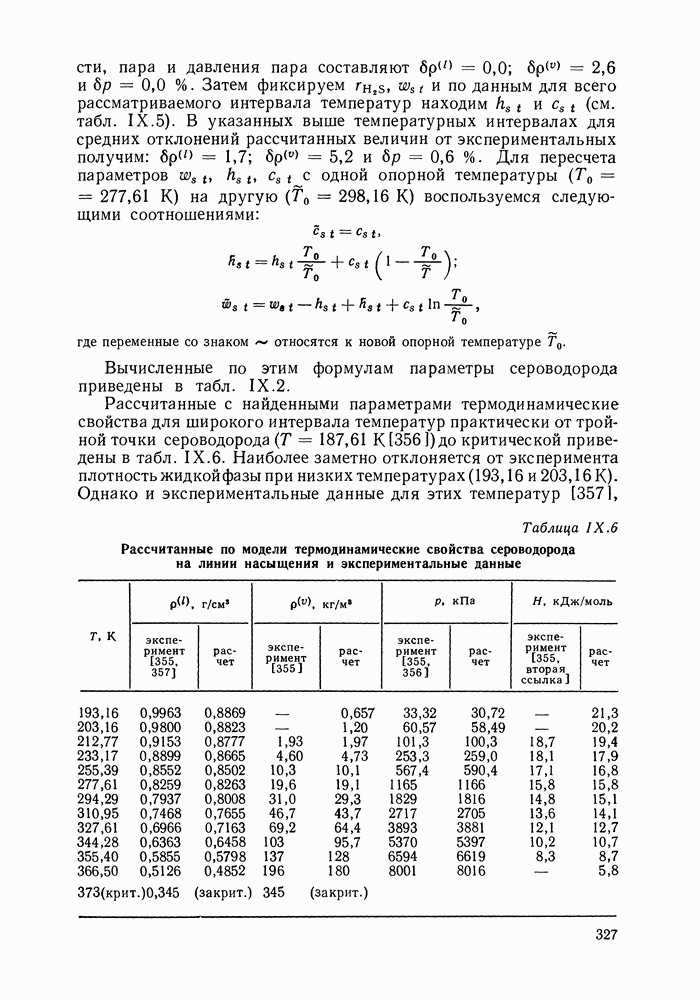
III.8. ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ СМЕШЕНИЯ И РАВНОВЕСИЕ ЖИДКОСТЬ—ПАРТермодинамические свойства неидеального раствора принято характеризовать избыточными термодинамическими функциями, которые представляют собою разность между функциями смешения данного раствора и идеального. Избыточные функции обозначают символом
Для индивидуальных веществ (чистых компонентов) все функции смешения равны нулю и, соответственно,
Для изучения равновесий жидкость—пар особое значение имеют избыточная энергия Гиббса и избыточный химический потенциал, которые прямо связаны с активностями и коэффициентами активности:
Подчеркнем, что при вычислении функций смешения пользуются симметричной системой нормировки. Значение и знак Для идеального раствора, когда
При отклонениях от закона Рауля:
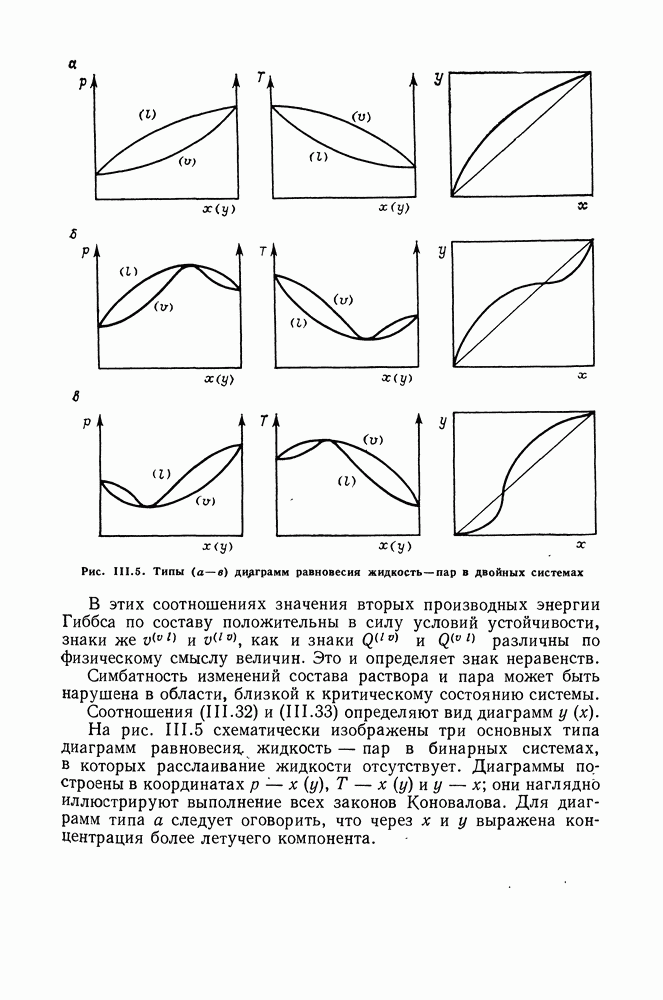
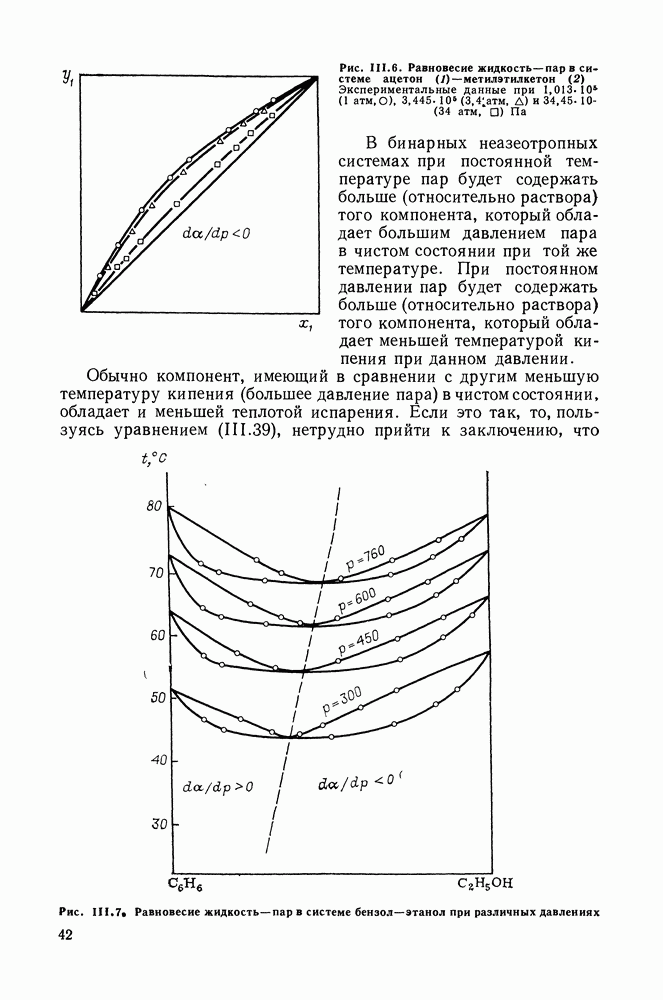
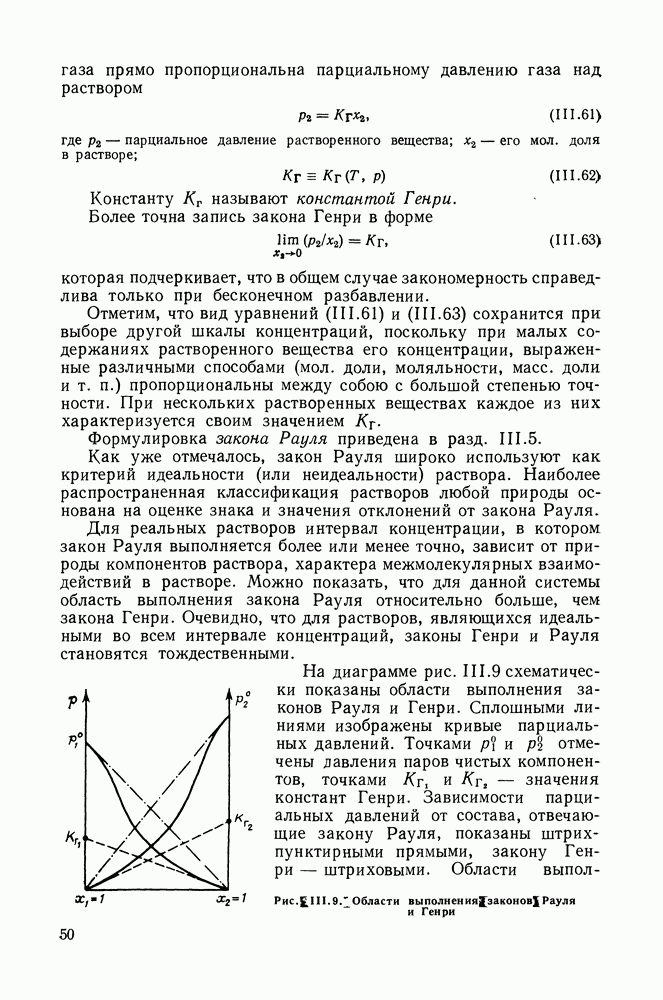
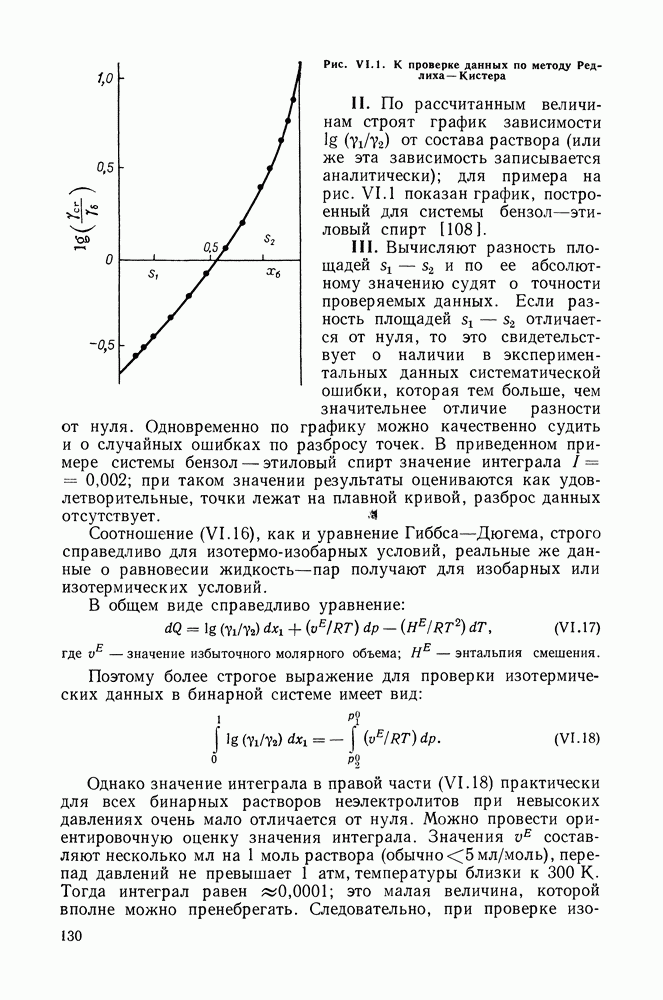
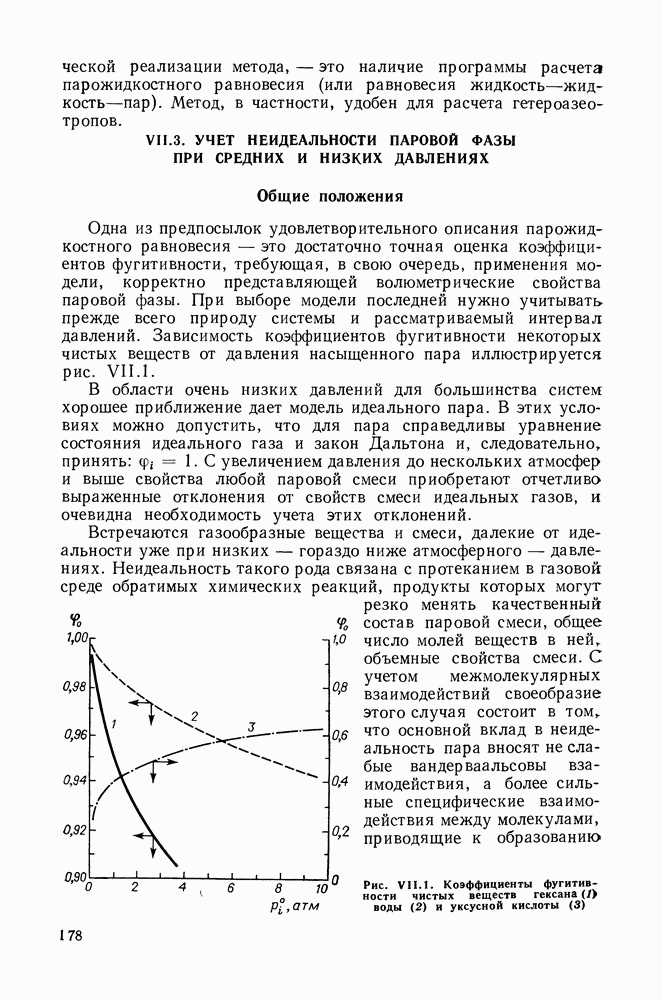
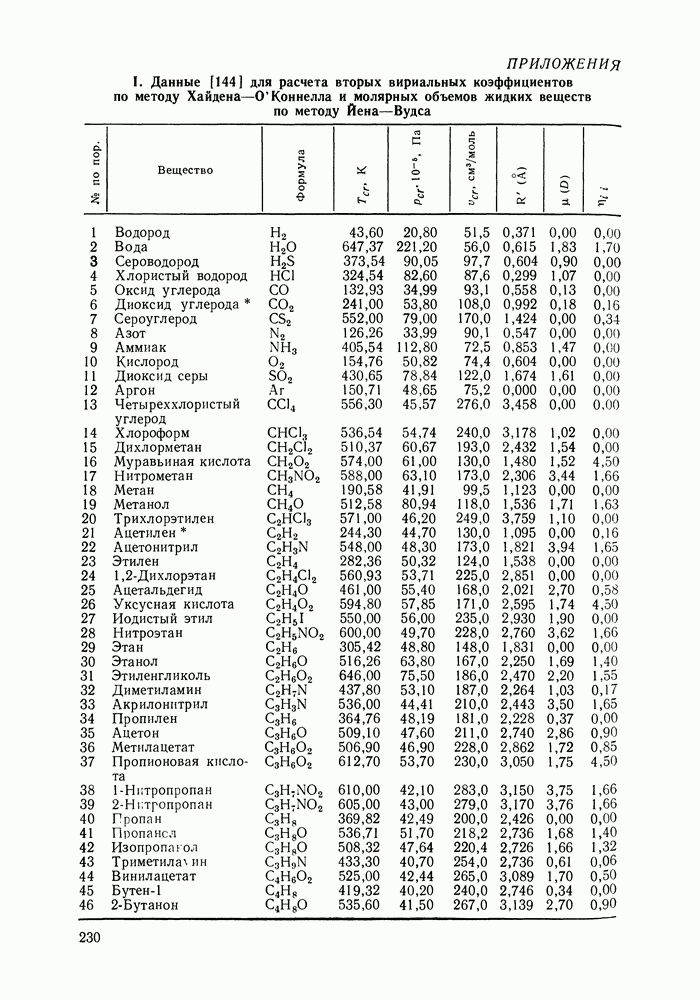
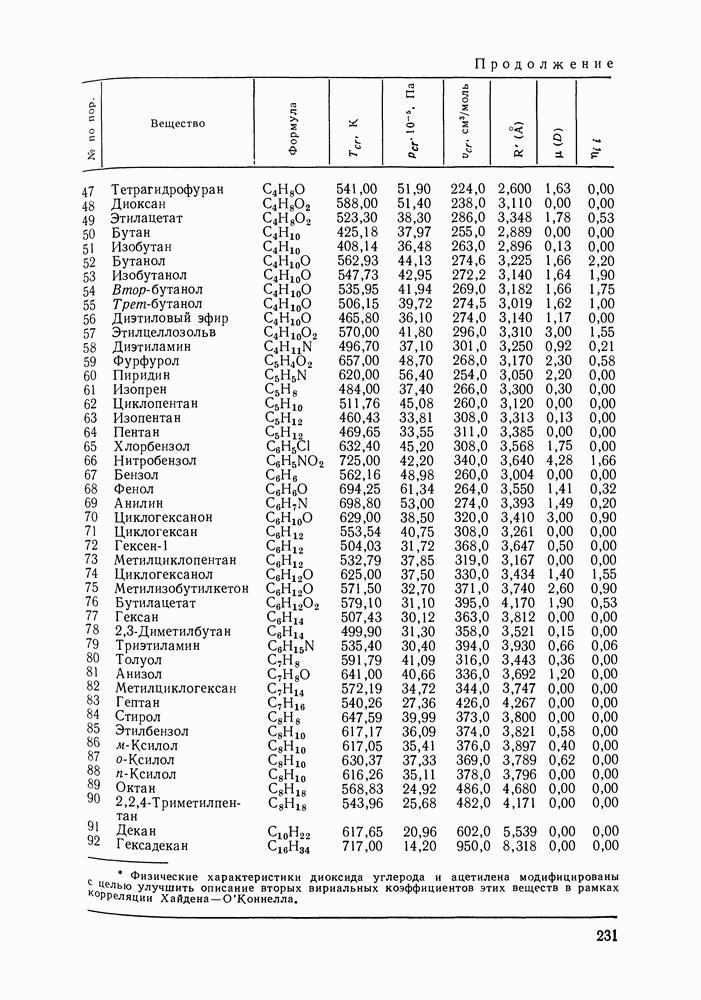
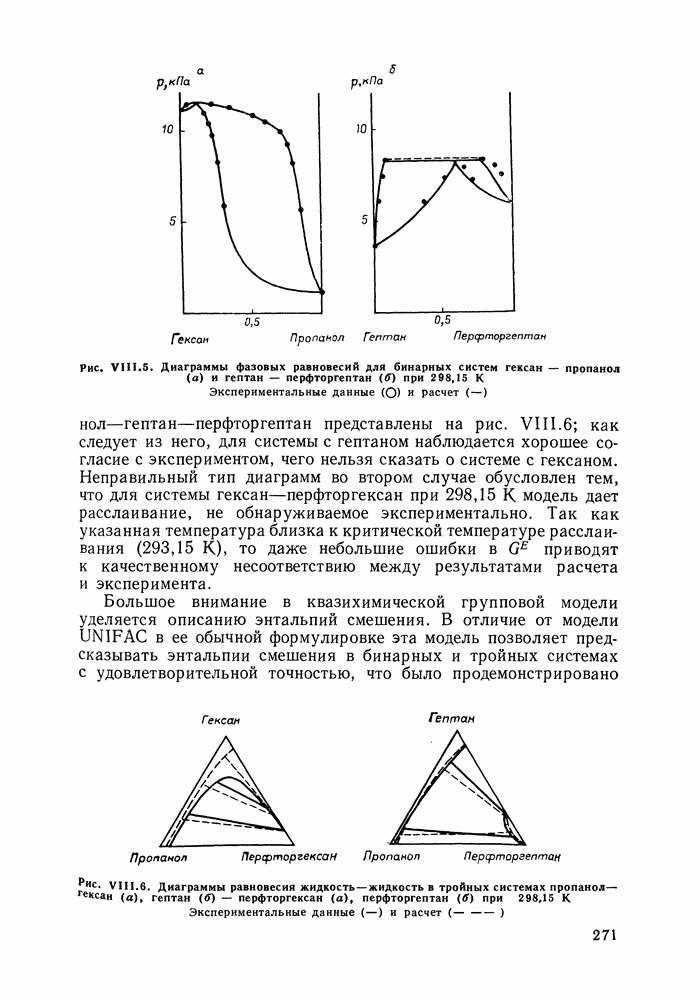
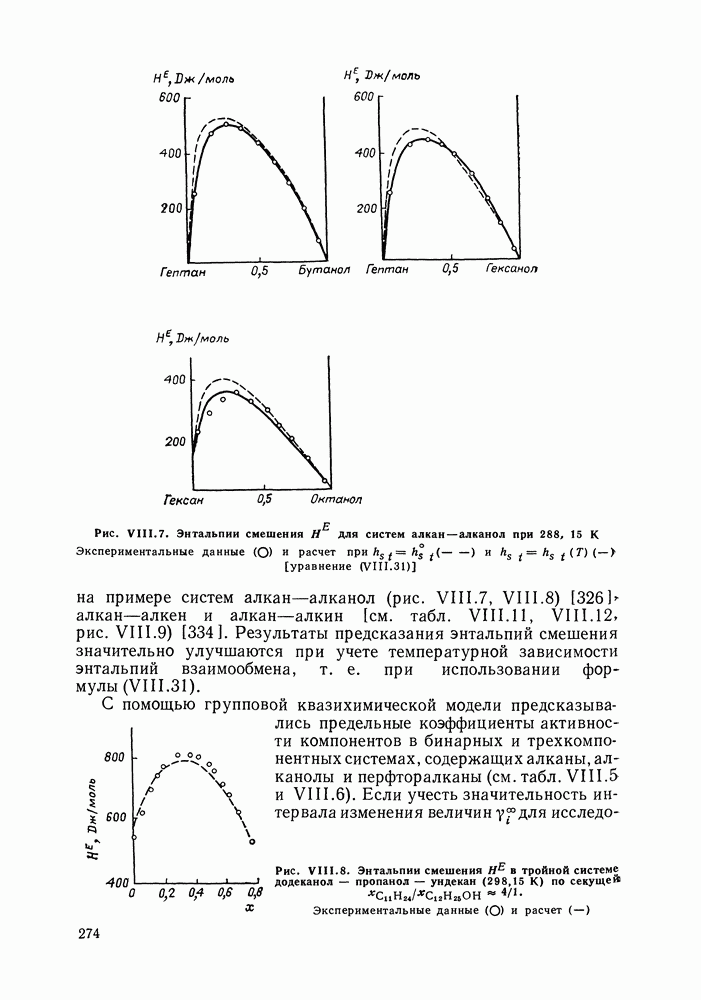
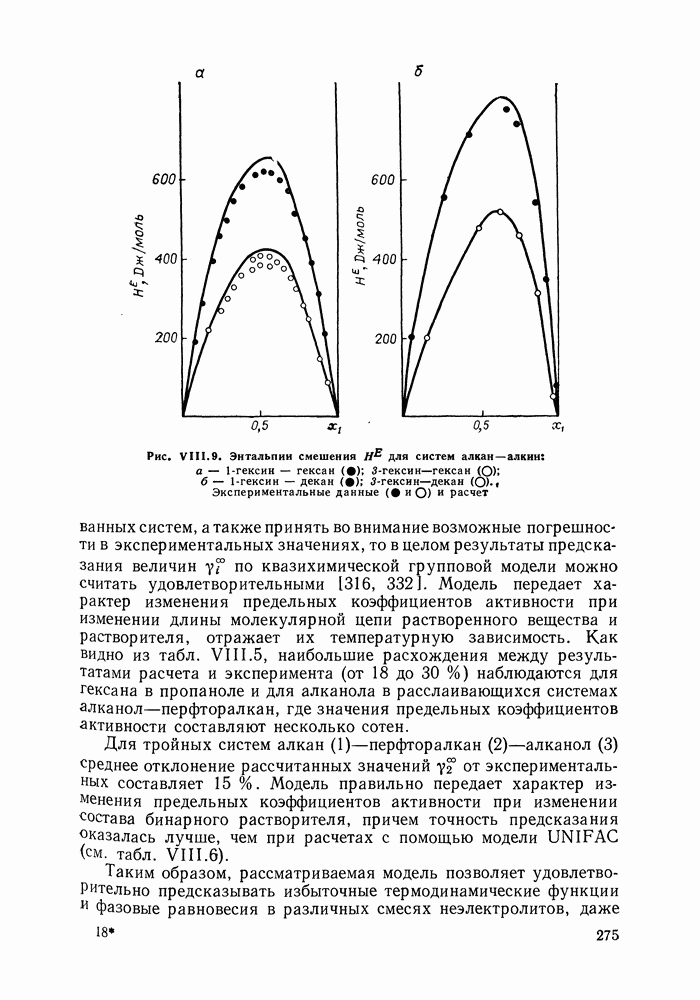
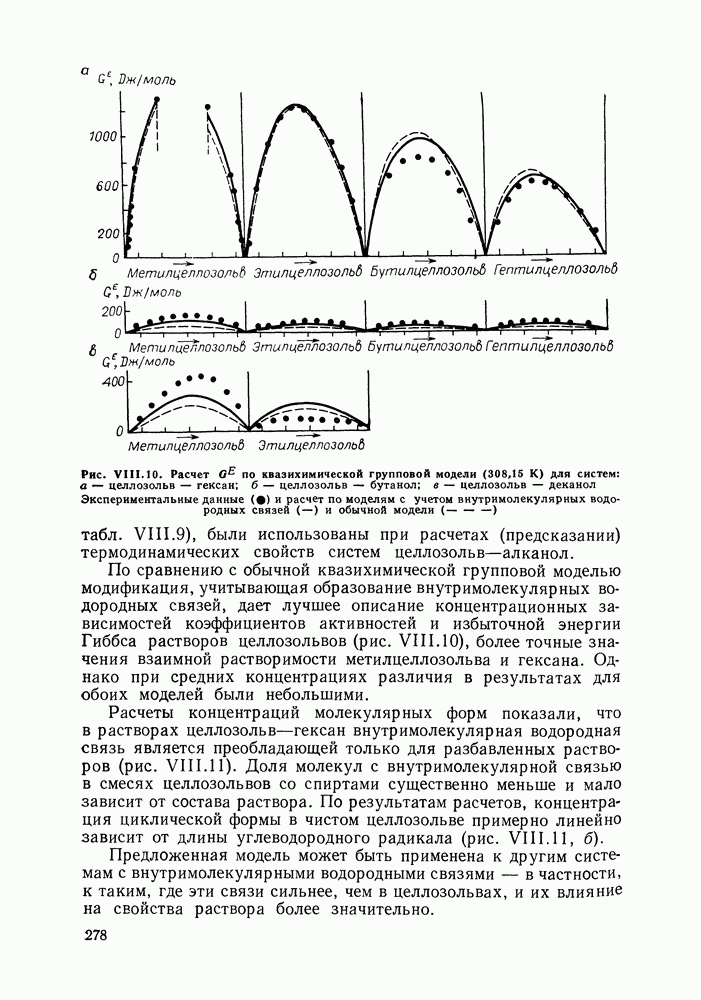
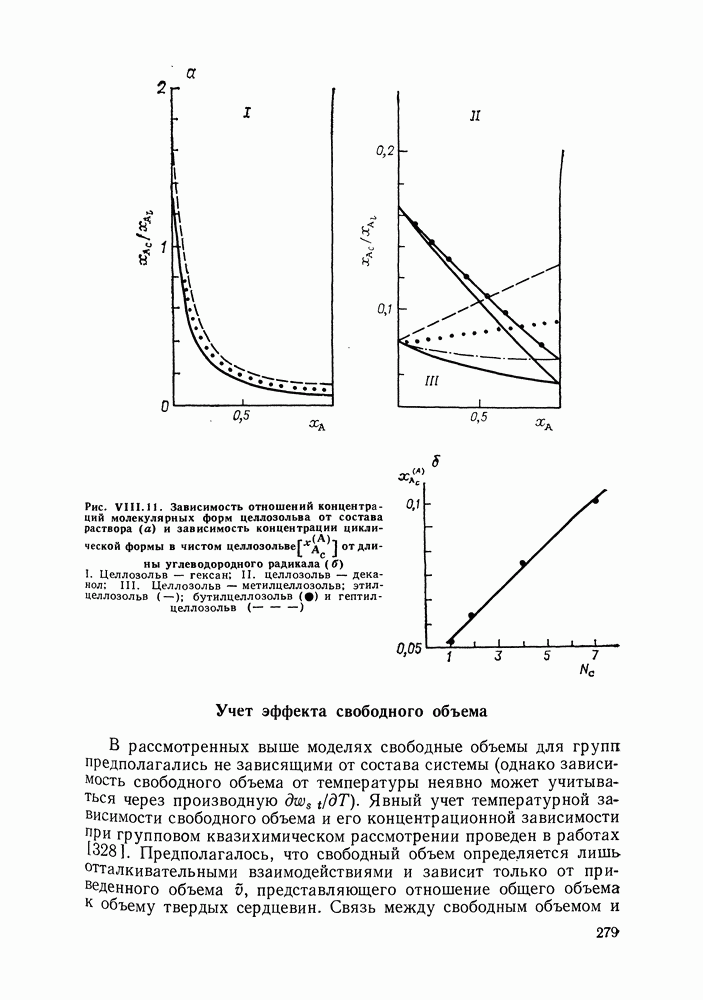
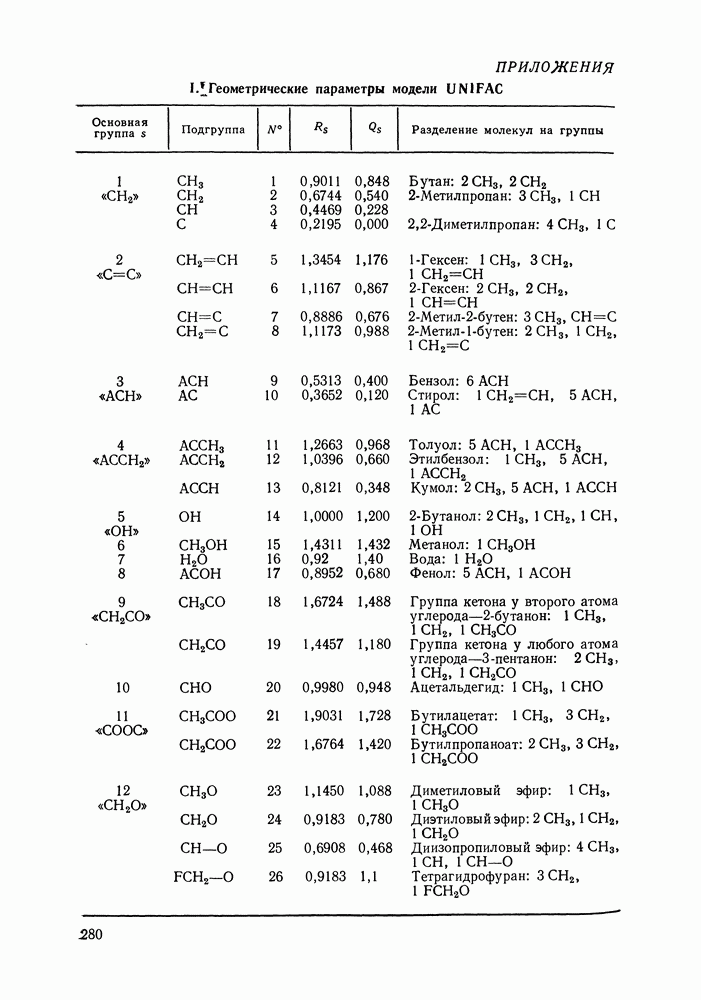
Очевидно, что если для двойной или многокомпонентной систем при небольших давлениях каким-либо путем, из эксперимента или расчетом, вычислены значения С другой стороны, и это важно, для растворов неэлектролитов изотермические данные о равновесии жидкость — пар являются основным, а для очень многих систем и единственным источником получения сведений о значениях коэффициента активности, К настоящему времени основной объем данных о термодинамических функциях смешения для растворов неэлектролитов накоплен или сочетанием исследования равновесия жидкость—пар в изотермических условиях с калориметрическими измерениями тепловых эффектов смешения, или же на основании политермических исследований равновесий жидкость—пар и соответствующей обработки полученных данных. Сопоставим характер диаграмм равновесия жидкость—пар со значениями функций смешения. Первые работы в этой области были выполнены Долежалеком, который связал положительные отклонения от закона Рауля с диссоциацией ассоциированного компонента, а отрицательные — с образованием соединения. В общем, положения Долежалека остаются справедливыми. Но следует иметь в виду, что значения функций смешения определяются совокупным влиянием многих факторов, в первую очередь это протон-донорные и протон-акцепторные свойства молекул компонентов раствора, их полярность, соотношение вкладов специфических и универсальных (вандерваальсовых) межмолеку-лярных взаимодействий, относительные размеры молекул и их форма и т. п. Здесь мы не имеем возможности останавливаться подробнее на связи молекулярных и макроскопических свойств растворов. Отметим только, что сравнительно проще проследить связи молекулярных свойств со значениями Величины
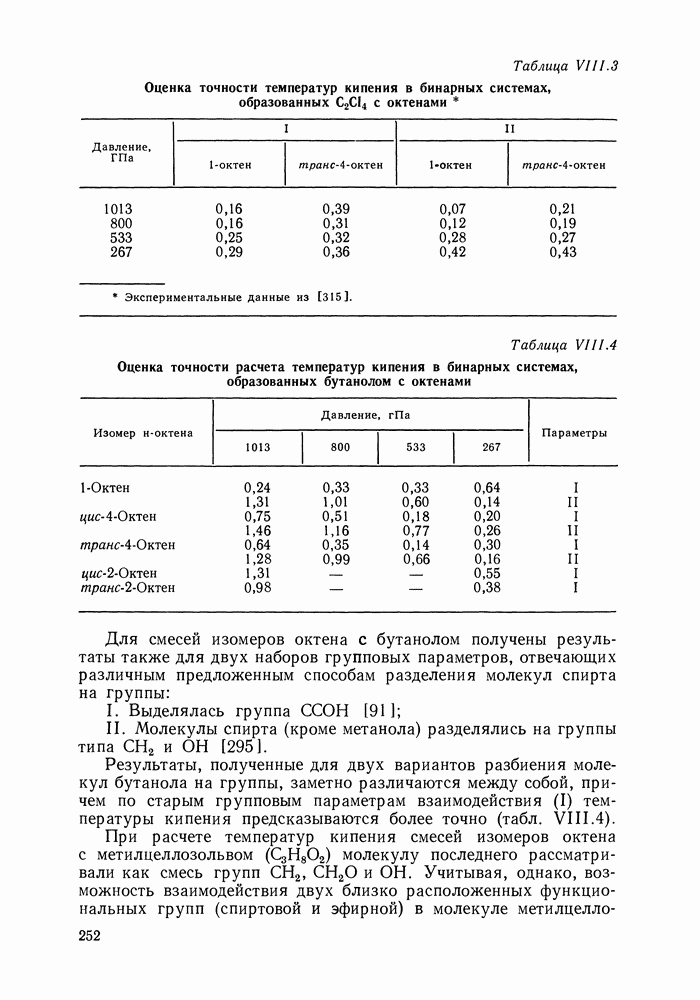
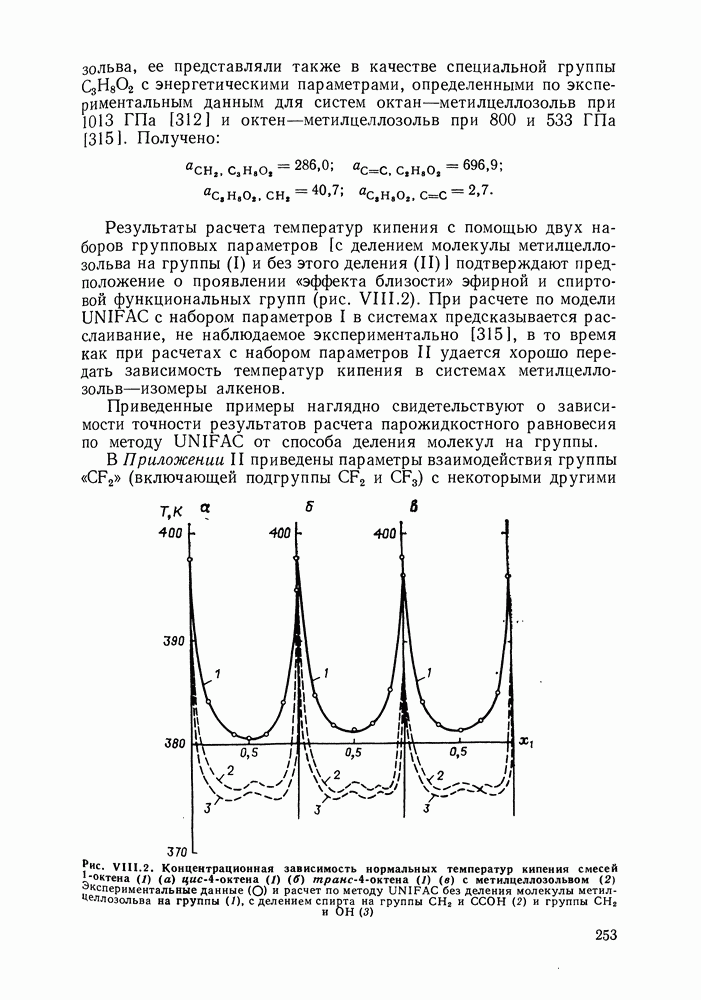
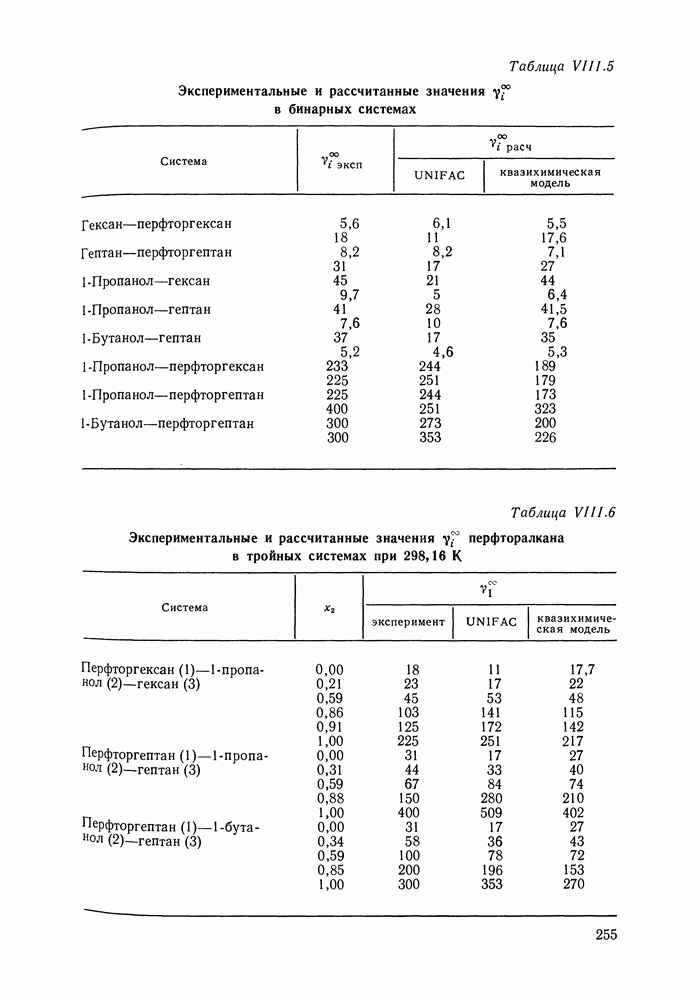
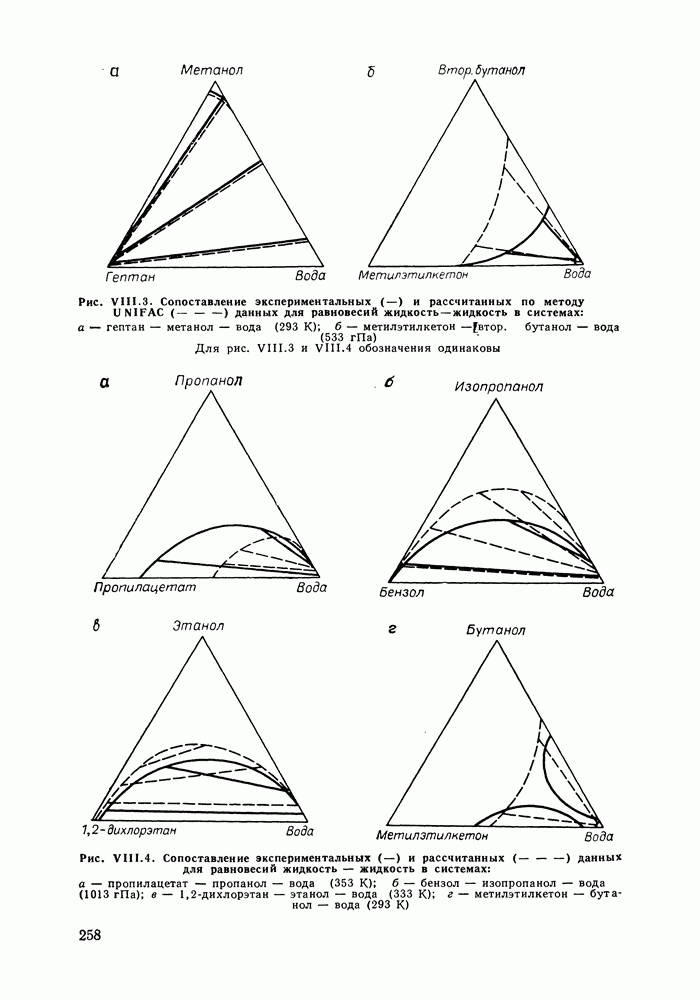
Во многих реальных системах величины Рис. III.10 (см. скан) Термодинамические функции смешения (а) и диаграммы большей или меньшей степени компенсируют друг друга, уменьшая абсолютные значения Для выполнения практических расчетов по равновесиям жидкость — пар особое значение имеют сведения об энтальпиях смешения. В этой главе ранее приводились уравнения, которые определяют влияние изменений температуры и давления на состав пара, на относительную летучесть смесей, на смещение состава азеотропных смесей и т. п. В эти уравнения входили значения парциальных молярных теплот испарения компонентов Прямых калориметрических измерений теплот испарения растворов очень мало. В гл. II были указаны источники сведений о теплотах испарения индивидуальных веществ. Данных о теплотах смешения неэлектролитов в литературе много, они собраны и обобщены в справочных изданиях [24, 25]. В [24], кроме справочных таблиц, обсуждены величины Данных о теплотах смешения много, но все же меньше, чем о теплотах испарения чистых веществ (последние значительно легче и определить при необходимости), поэтому для целей практических расчетов привлекательна возможность замены Соотношение слагаемых зависит не только от природы системы, но и от состава раствора, для которого выполняется расчет. Энтальпии средних концентраций, вблизи от эквимолярного состава. Значения
|
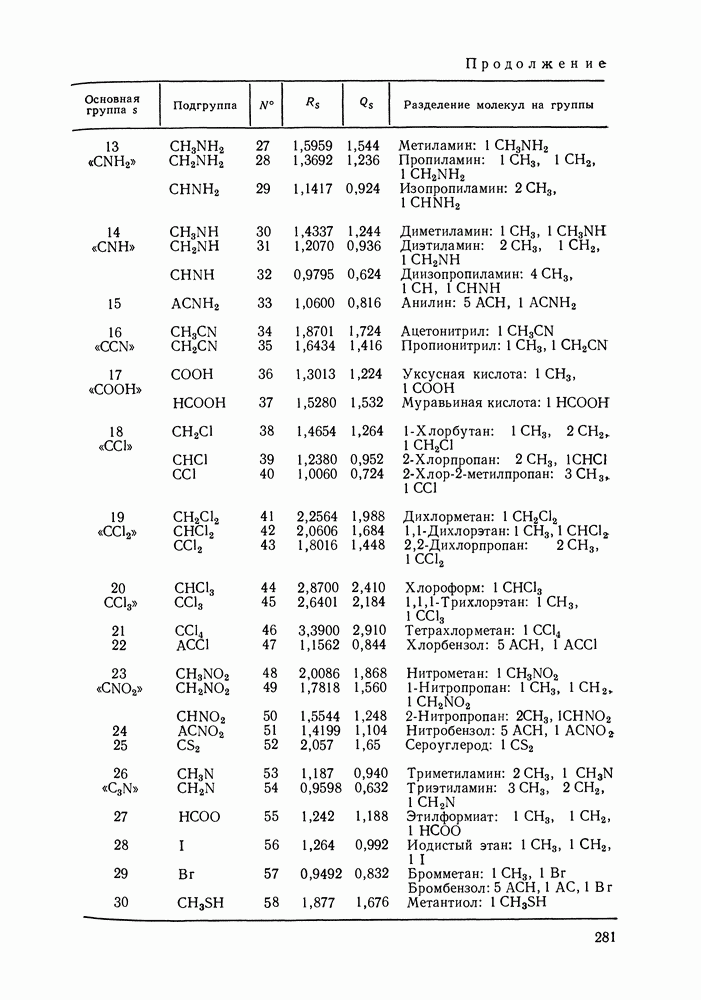
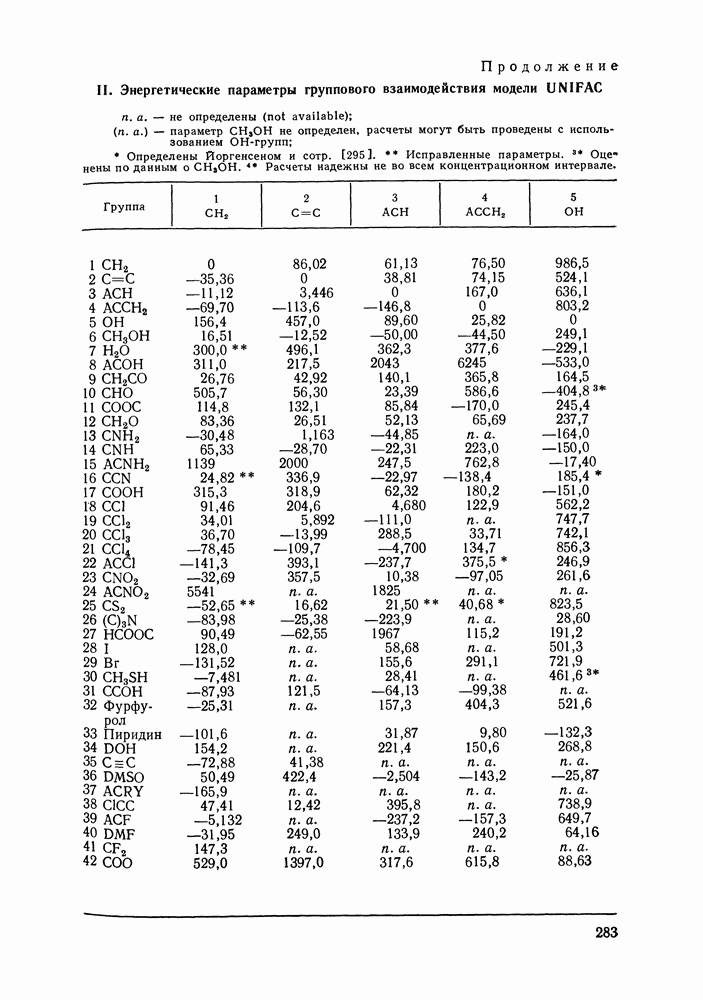
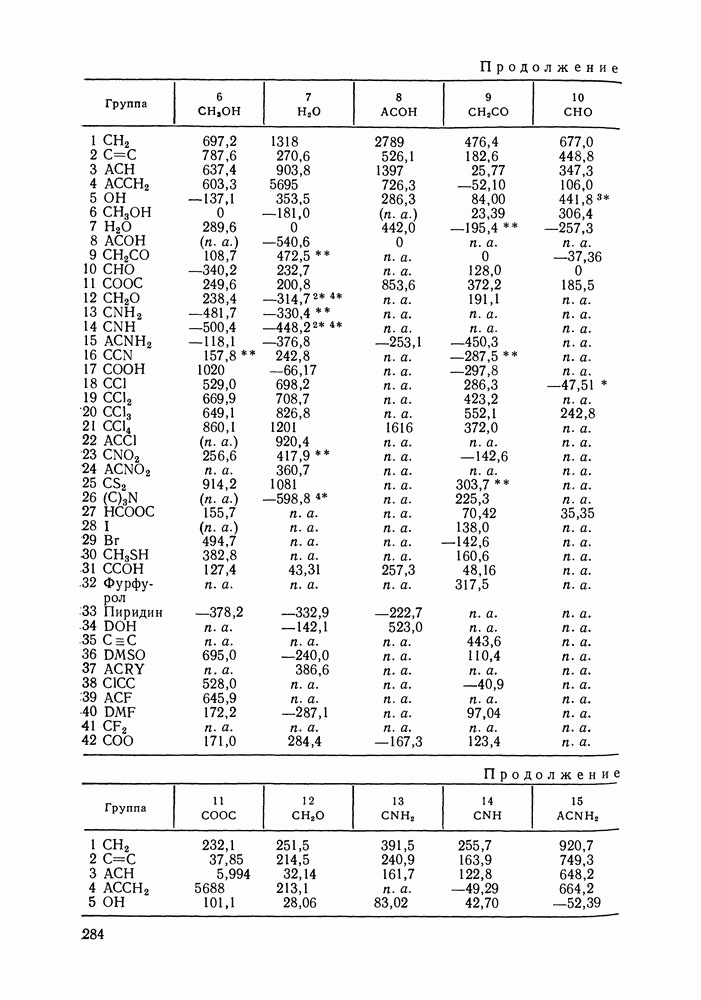
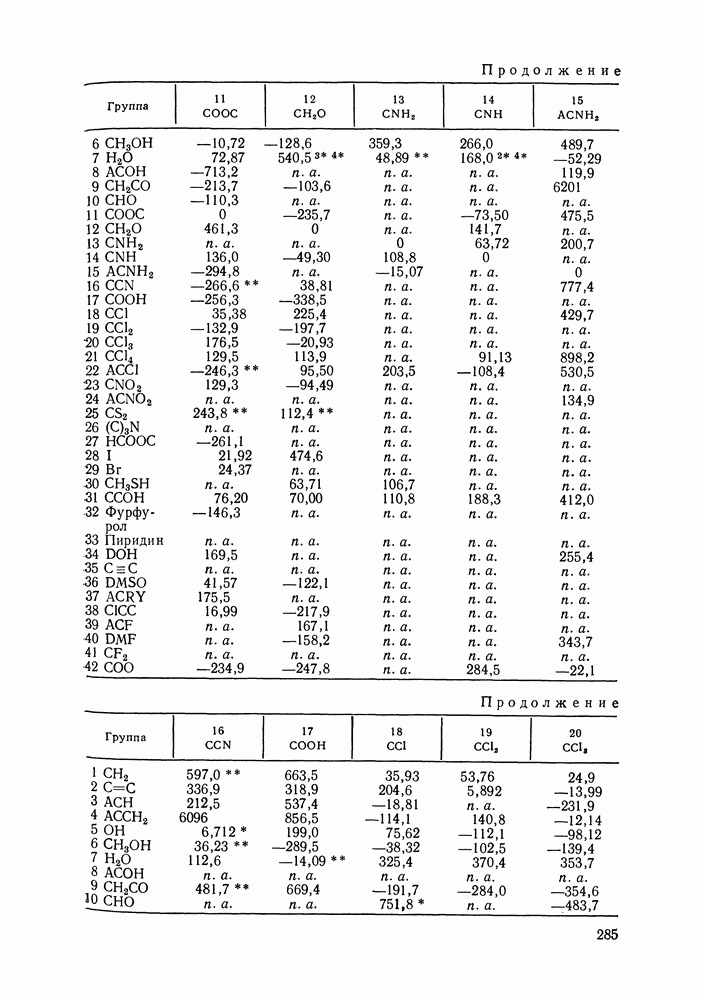
 |
Оглавление
|

















































































































































































































































































 По определению:
По определению:

 Значения функций смешения идеального раствора были определены ранее [см. (111.50)-(111.54)]. Для реального раствора любой природы имеем:
Значения функций смешения идеального раствора были определены ранее [см. (111.50)-(111.54)]. Для реального раствора любой природы имеем:


 определяют характер и степень отклонения раствора от идеального поведения.
определяют характер и степень отклонения раствора от идеального поведения.
 выполняется закон Рауля:
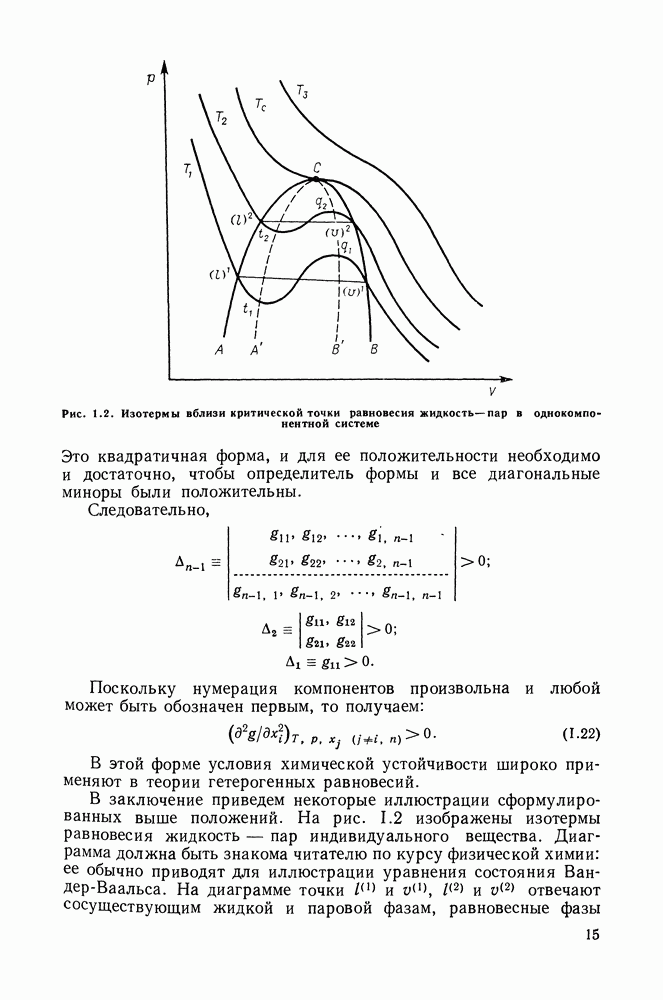
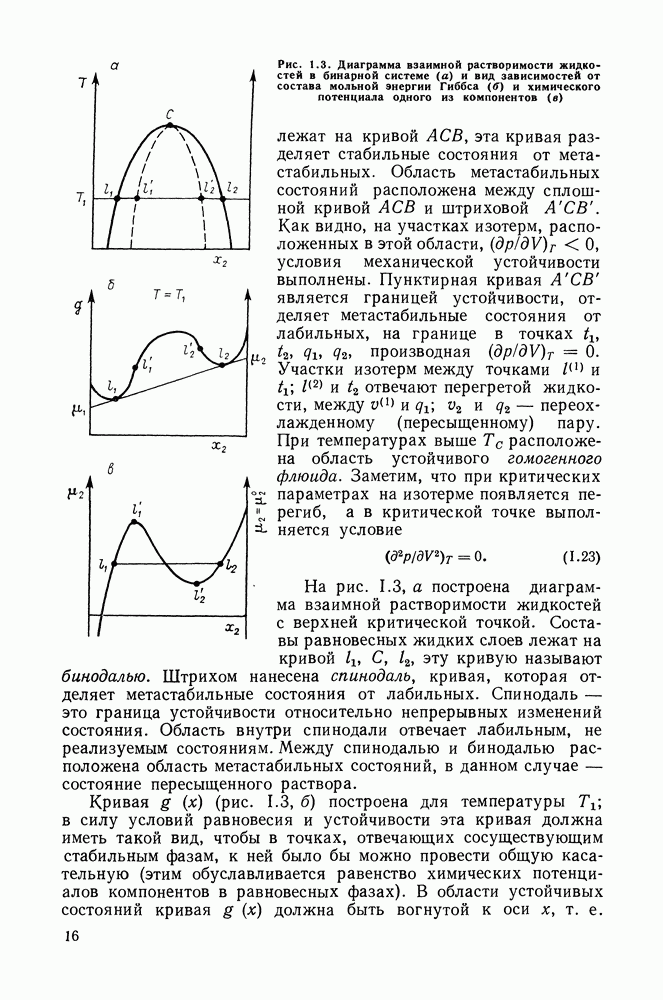
выполняется закон Рауля:  Для неидеального раствора (но при условии применимости к пару законов идеальных газов):
Для неидеального раствора (но при условии применимости к пару законов идеальных газов):


 то это означает, что получены полные данные о равновесии жидкость—пар.
то это означает, что получены полные данные о равновесии жидкость—пар.
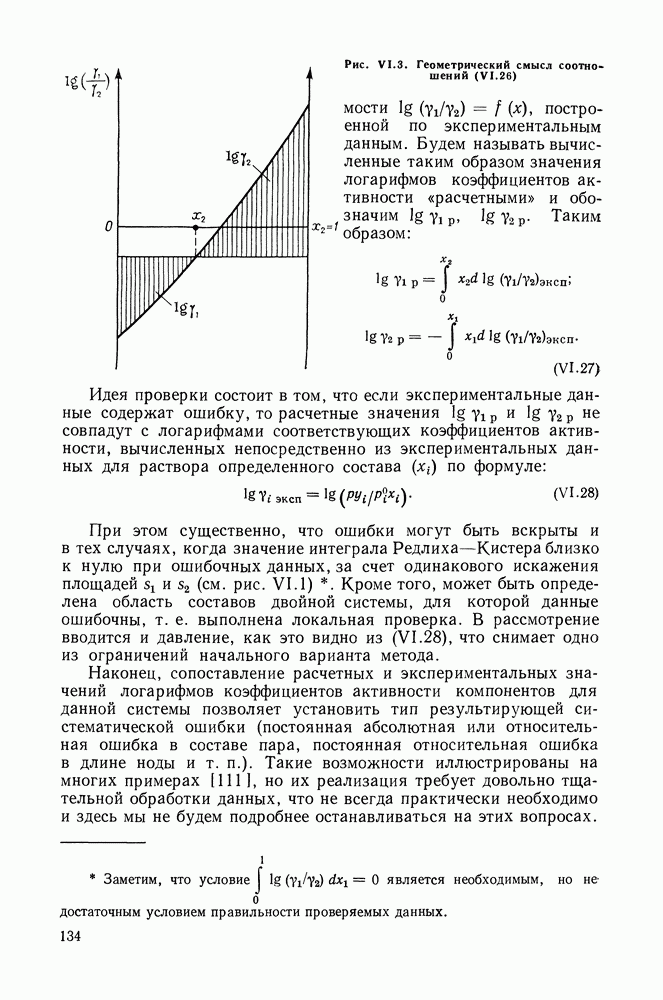
 Если же получены политермические данные о равновесии жидкость—пар, то по ним, пользуясь формулами разд. III.7, можно вычислить и другие избыточные функции смешения —
Если же получены политермические данные о равновесии жидкость—пар, то по ним, пользуясь формулами разд. III.7, можно вычислить и другие избыточные функции смешения — 
 и
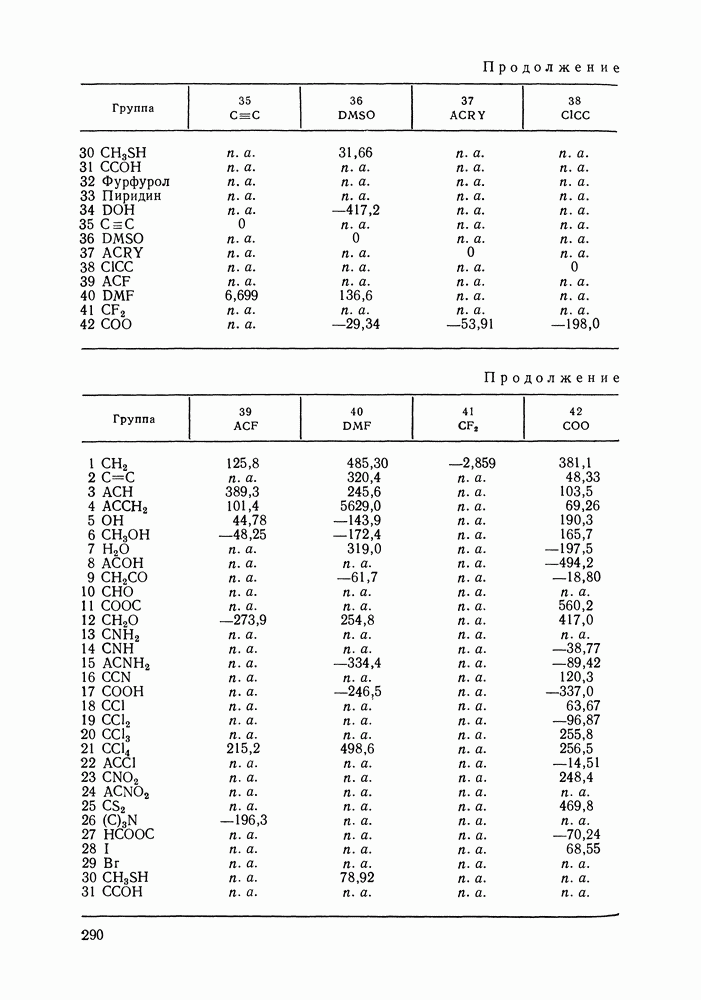
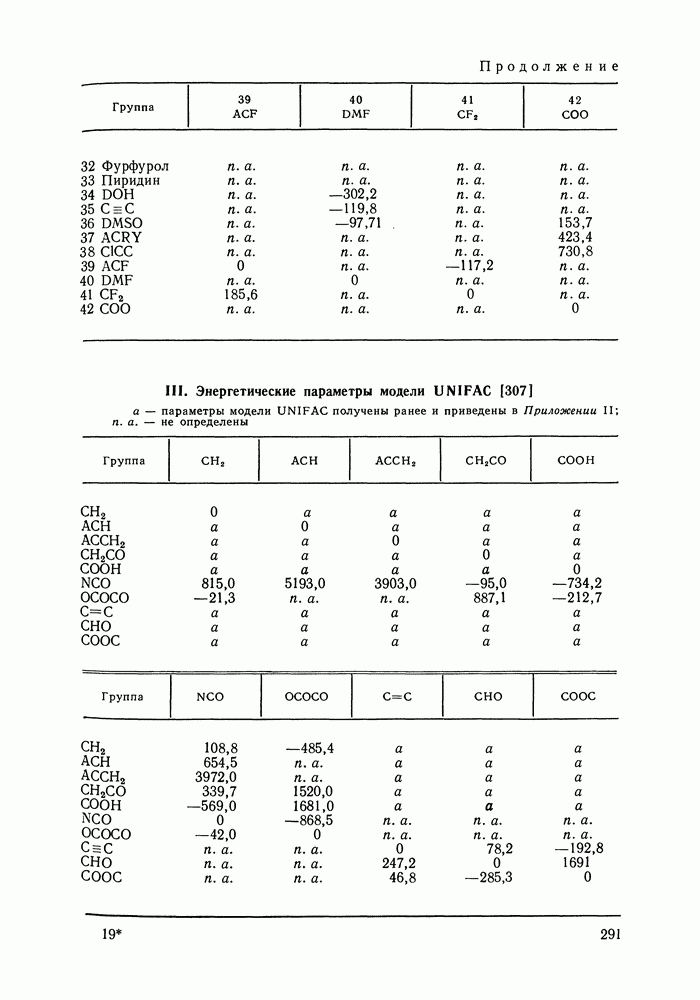
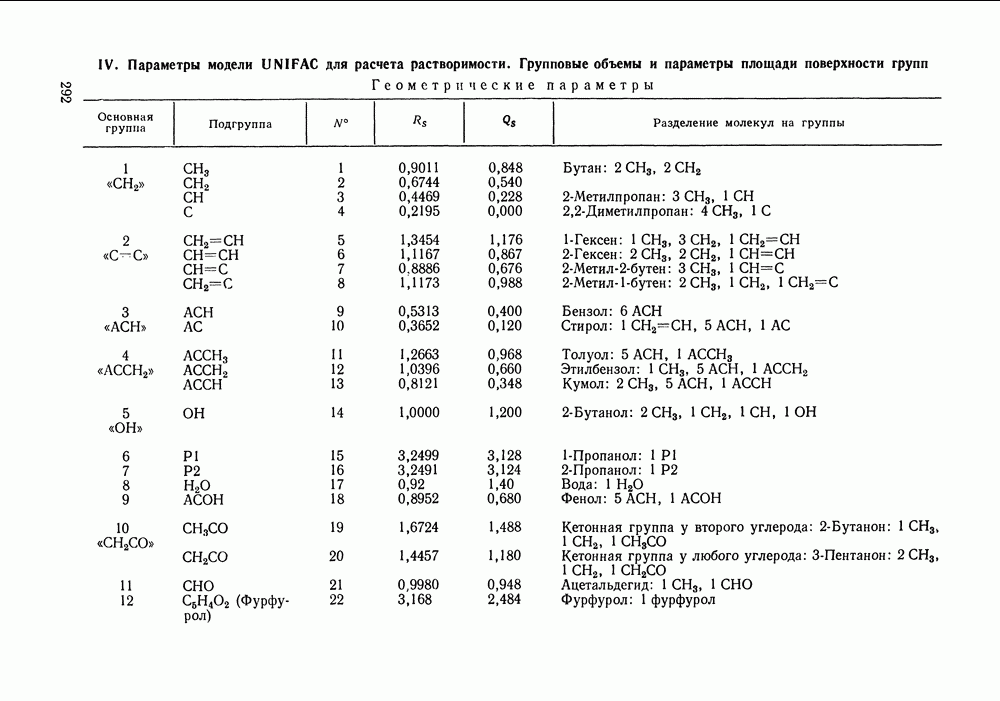
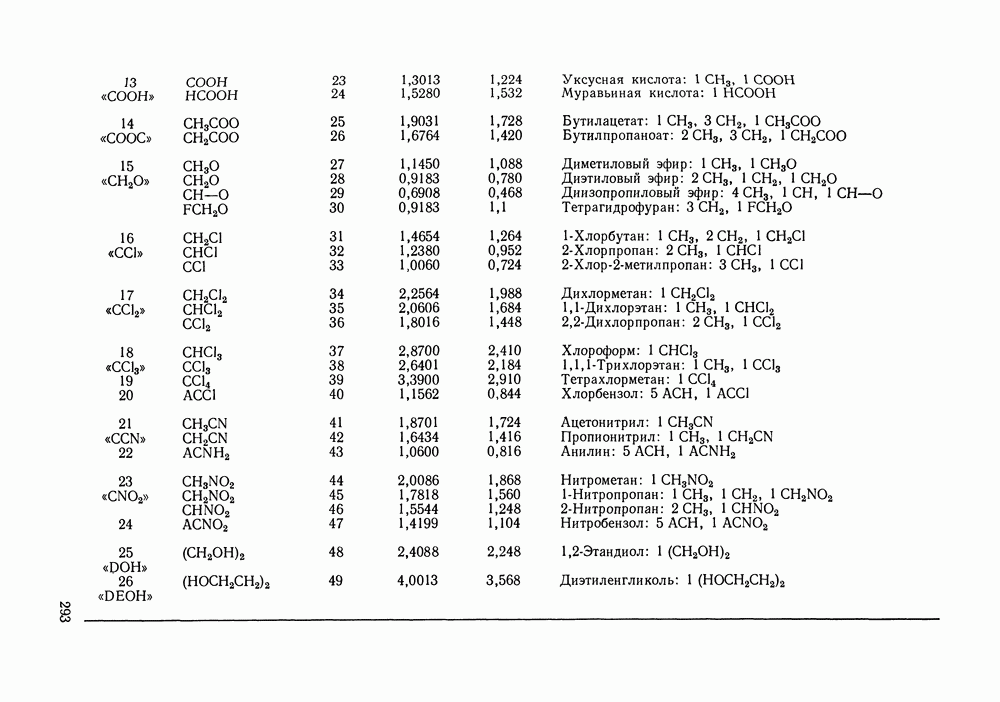
и  [24]. При этом в системах некоторого типа определяющим является значение
[24]. При этом в системах некоторого типа определяющим является значение  в других —
в других —  Эти обстоятельства оказали влияние на формулировки первых приближенных моделей молекулярных растворов — регулярных
Эти обстоятельства оказали влияние на формулировки первых приближенных моделей молекулярных растворов — регулярных  и атермических
и атермических 
 определяются соотношением энтальпийных (энергетических) и энтропийных (структурных) вкладов. Их соотношением определяется и самая возможность образования гомогенного раствора
определяются соотношением энтальпийных (энергетических) и энтропийных (структурных) вкладов. Их соотношением определяется и самая возможность образования гомогенного раствора  и знак
и знак 

 и
и  в
в
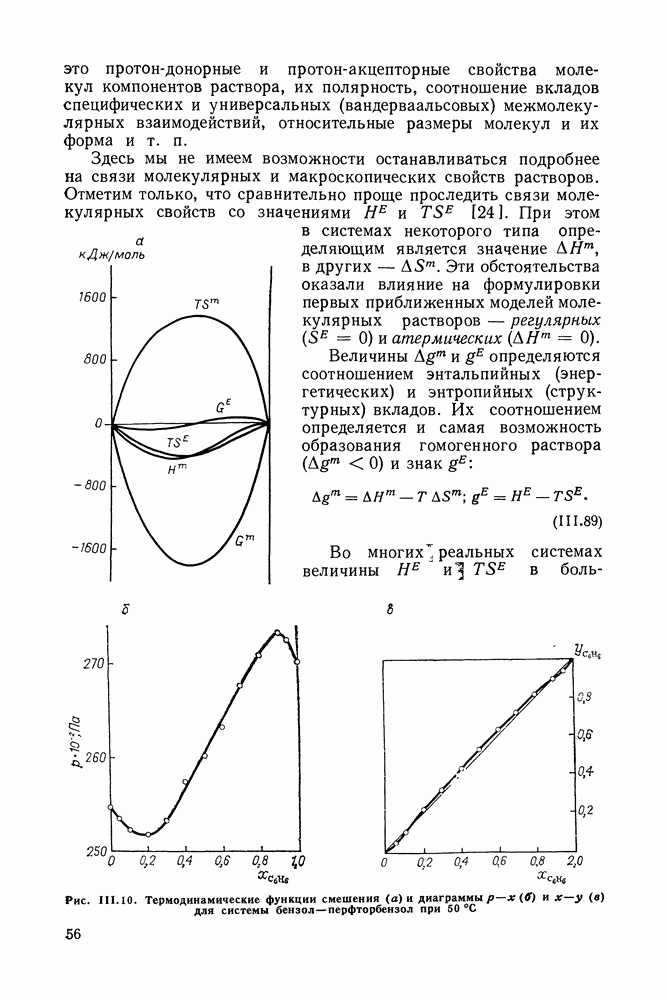
 для системы бензол—перфторбензол при
для системы бензол—перфторбензол при 
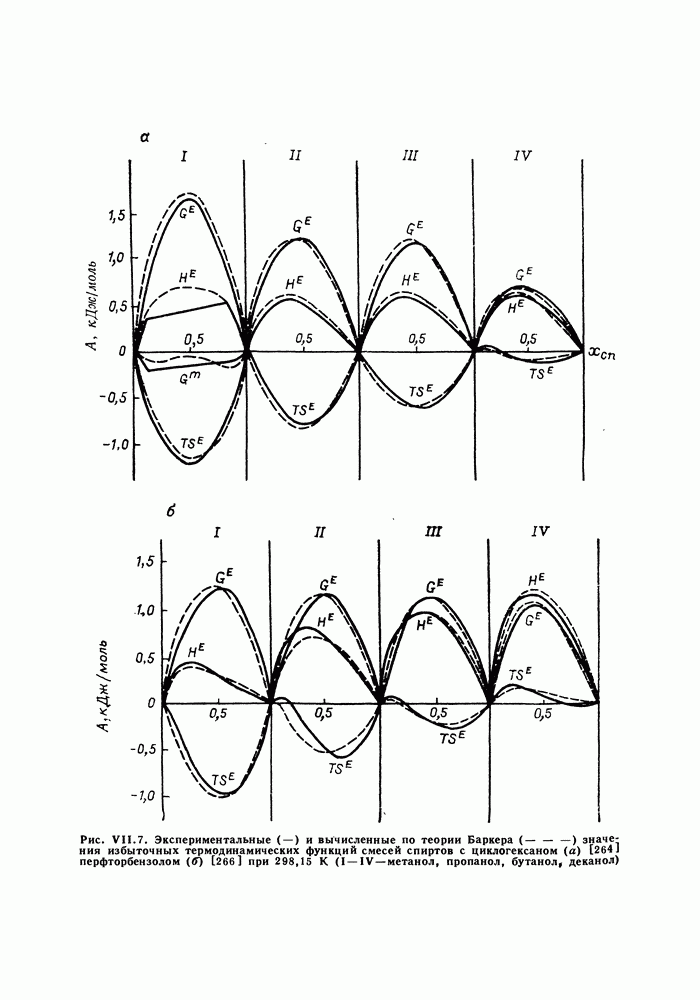
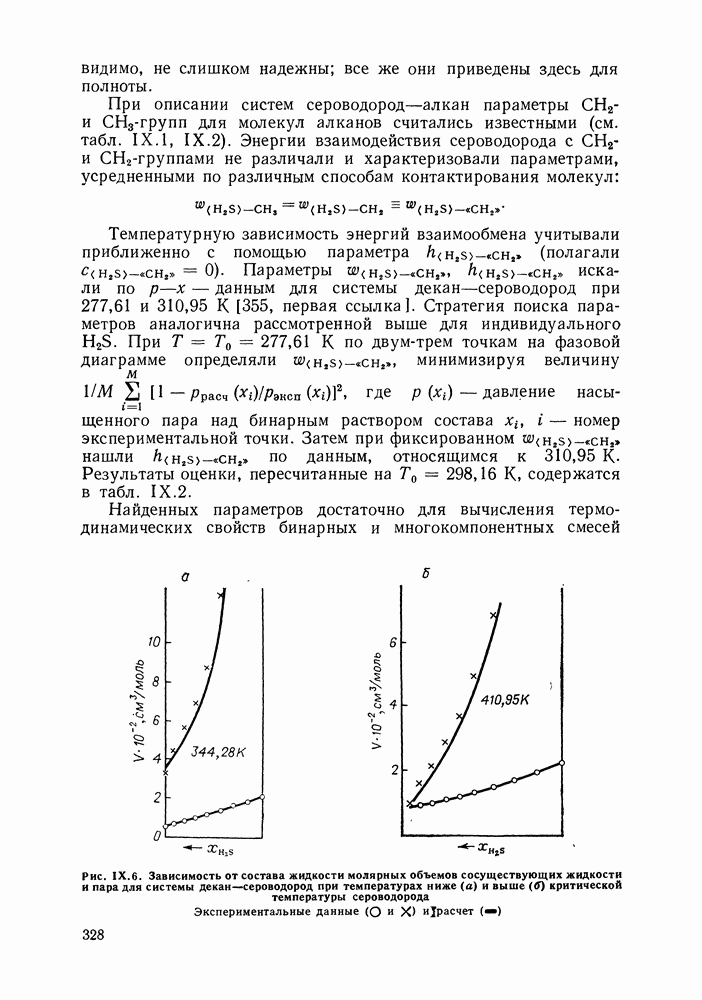
 Показателен пример системы бензол-перфторбензол [23], которая отличается сложным характером межмолекулярных взаимодействий. Но для этой системы значения
Показателен пример системы бензол-перфторбензол [23], которая отличается сложным характером межмолекулярных взаимодействий. Но для этой системы значения  и
и  очень близки, значение
очень близки, значение  очень мало и система близка к идеальной. Но по характеру равновесия система необычна — в ней образуются два азеотропа и это, насколько нам известно, единственный пример бинарной системы из неэлектролитов, для которой установлен факт образования двух азеотропов (рис. III.10). На рис. 111.10, а построены функции смешения для этой системы, а на рис. III.10, б, в — диаграммы равновесия жидкость—пар.
очень мало и система близка к идеальной. Но по характеру равновесия система необычна — в ней образуются два азеотропа и это, насколько нам известно, единственный пример бинарной системы из неэлектролитов, для которой установлен факт образования двух азеотропов (рис. III.10). На рис. 111.10, а построены функции смешения для этой системы, а на рис. III.10, б, в — диаграммы равновесия жидкость—пар.
 которые, как правило, находят по теплотам испарения чистых веществ
которые, как правило, находят по теплотам испарения чистых веществ  и парциальным теплотам смешения
и парциальным теплотам смешения 
 в системах различной химической природы, закономерности изменения
в системах различной химической природы, закономерности изменения  в рядах родственных систем, точность расчета тепловых эффектов из политермических данных о равновесии жидкость—пар.
в рядах родственных систем, точность расчета тепловых эффектов из политермических данных о равновесии жидкость—пар.
 на
на  Это можно сделать достаточно часто, поскольку в уравнения обычно входит разность
Это можно сделать достаточно часто, поскольку в уравнения обычно входит разность  и замена вполне допустима, когда
и замена вполне допустима, когда 
 это парциальные молярные величины, и они равны между собою в экстремуме
это парциальные молярные величины, и они равны между собою в экстремуме  их разность мала в окрестности точки экстремума, которая обычно лежит в области
их разность мала в окрестности точки экстремума, которая обычно лежит в области
 и
и  для растворенных веществ и их разность почти всегда будут велики для разбавленных растворов. Подробнее об этих вопросах см. в [24].
для растворенных веществ и их разность почти всегда будут велики для разбавленных растворов. Подробнее об этих вопросах см. в [24].