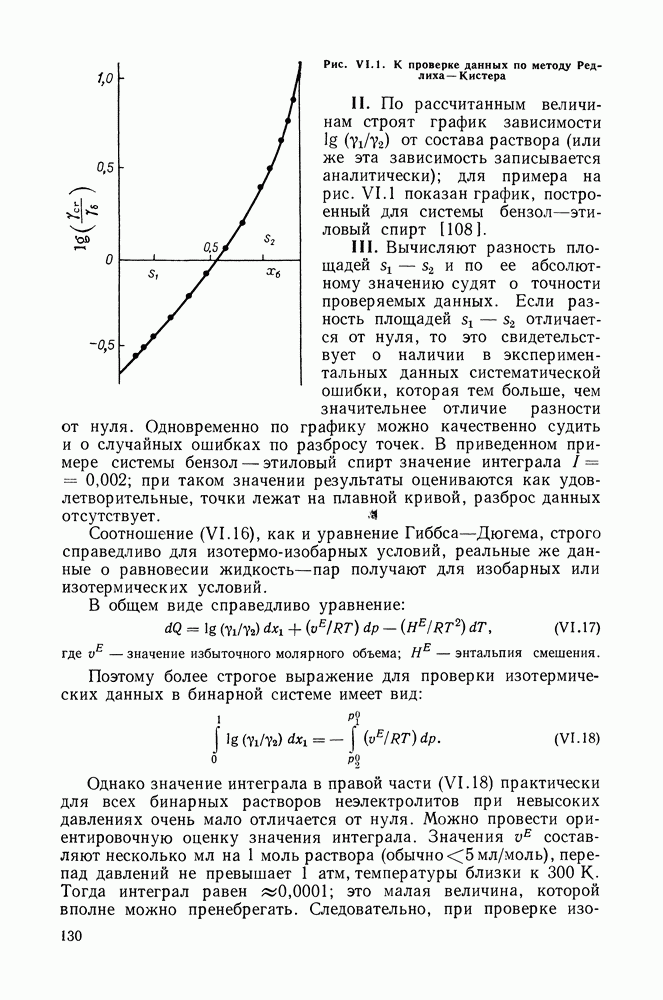
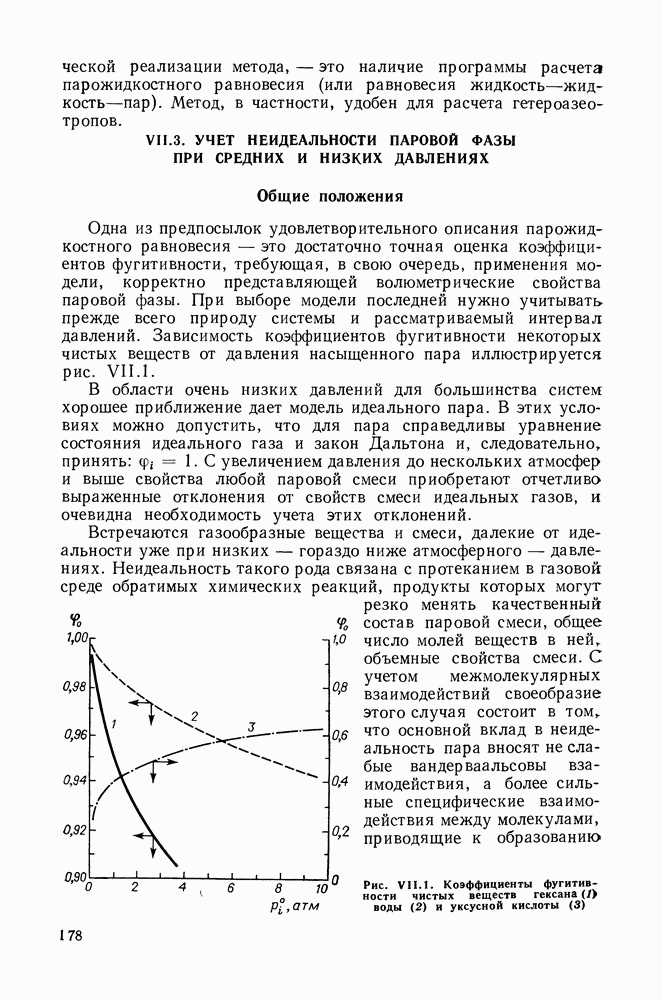
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
VII.8. РЕШЕТОЧНЫЕ МОДЕЛИ. МОДЕЛЬ БАРКЕРАБольшую роль в развитии методов расчета фазовых равновесий сыграли решеточные модели. На их основе удалось вскрыть многие закономерности концентрационной и температурной зависимости термодинамических функций, хотя очевидно, что предположение о расположении молекул в узлах правильной решетки, не отражает действительную структуру жидких систем. Простейшая модель, оказавшая заметное влияние на развитие решеточного подхода — модель строго регулярного раствора. Она относится к системам из небольших молекул примерно одинакового размера и сферической формы, занимающих по одному узлу в решетке, и взаимодействующих посредством центральных сил. Сама модель в настоящее время интереса для жидких растворов не представляет, но на базе ее были развиты модели, позволяющие описывать весьма сложные системы, которые пока трудно поддаются строгому описанию. В решеточных моделях развиты способы учета влияния размера и формы молекул, различий в энергиях их взаимодействий на термодинамические функции. В рамках этих моделей оказывается возможным учесть зависимость энергии взаимодействия молекул от их взаимной ориентации, что оказывается особенно важным при описании ассоциированных растворов [175, с. 3]. Обычно в выражениях для избыточных термодинамических Функций выделяют комбинаторный вклад, обусловленный различием в размерах молекул, и остаточный, обусловленный различием в энергиях взаимодействия. Если последним можно пренебречь, получаем модель атермического раствора. Для учета комбинаторного вклада предложены различные приближения. К числу наиболее распространенных относятся: приближение Флори [212] —
приближение Гуггенгейма для открытых цепей
Здесь
число контактных участков молекулы Более общим является приближение Гуггенгейма — Ставермана [218], позволяющее рассматривать не только линейные, но и «объемные» молекулы. Выражение для
где
так называемый фактор объемности; для открытой цепи и цикла равно соответственно Потенциальная энергия межмолекулярных взаимодействий обычно представляется как сумма энергий взаимодействия пар ближайших соседей.
где В выражения для избыточных термодинамических функций входит параметр
называемый энергией взаимообмена и учитывающий различие в энергиях взаимодействия смешанных пар Для оценки наиболее вероятных чисел пар
Если в системе отсутствуют сильные ориентационные эффекты, то при отрицательной энергии взаимообмена и при положительной энергии взаимообмена вдали от критической точки расслаивания, оба приближения достаточно хорошо согласуются между собой и с результатами более строгих подходов. Однако приближение Брэгга — Вильямса не позволяет описать ориентационную упорядоченность, и для моделирования систем с полярными и ассоциированными компонентами необходимо обратиться к квазихимическому или другим приближениям. Остановимся на модели Баркера (Баркера — Гуггенгейма), рассматривающей ориентационные эффекты в квазихимическом приближении и одновременно позволяющей включать в рассмотрение факторы размера молекул [255]. Модель предназначена для оценки коэффициентов активности Как и в теории строгорегулярного раствора, в теории Баркера принимается квазикристаллическая модель. Молекулярные размеры учитываются в приближении Гуггенгейма для линейных
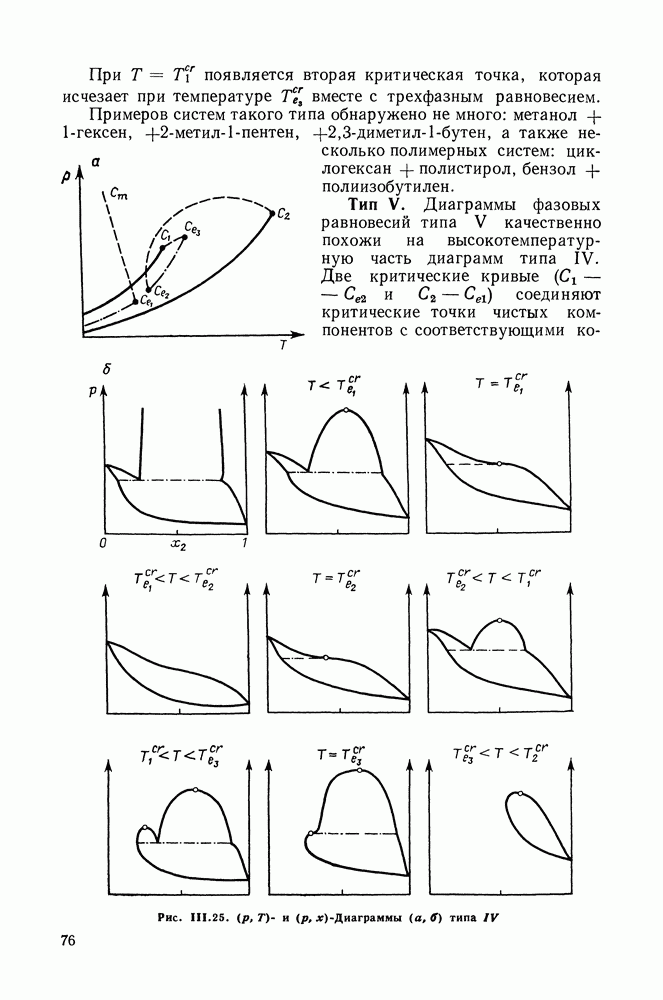
Рис. VII.6. Модель молекул алканола по Баркеру Контактные участки: Одинаковые контактные участки относят к одному типу. Можно предположить, что у таких молекул, как алканы, перфторалканы, четыреххлористый углерод все контактные участки однотипны. У молекулы спирта выделяют три типа контактных участков: Для молекул спирта, модель которых изображена на рис. VII.6, водородной связи соответствуют контакты Термодинамические функции
Комбинаторный вклад оценивается по формулам Гуггенгейма для атермической смеси
где Величины
где
Для избыточной энтальпии, которая определяется только остаточным вкладом
где В случае неспецифических взаимодействий в самом грубом приближении величину Следовательно, модель Баркера включает как геометрические При выборе модели контактных участков для конкретной системы (выделении различных типов контактных участков, чисел участков определенного типа) следует учитывать химическую природу и размеры молекул. Энергетические параметры при расчетах по теории Баркера обычно выступают как подгоночные и определяются с учетом экспериментальных данных об избыточных функциях. Иногда оценивают все параметры или некоторые из них для каждой исследуемой системы, иногда рассматривают согласованные наборы параметров для серий систем, аналогичных по химической природе. Для специфических взаимодействий, например, водородной связи, значения параметров часто фиксируют заранее с учетом независимых данных, например, спектральных [256, 257]. Модель Баркера нашла широкое применение особенно в работах 60—70-х гг. В последнее десятилетие интерес сместился к групповым модификациям этой модели (см. разд. VIII.3). Избыточные термодинамические функции по теории Баркера рассчитывали как для сильно ассоциированных растворов, так и для систем без ассоциации. Многочисленны работы, в которых с помощью этой теории исследованы термодинамические свойства растворов спиртов в различных растворителях: в четыреххлористом углероде [255, 256, 258], алканах и циклоалканах [175, с. 43; 259—264], ароматических углеводородах [255, 259, 263, 265], перфторбензоле [266]. Изучены системы с другими полярными компонентами, такими как амины [259, 261, 262], эфиры [257, 259, 261, 267—270], кетоны [259, 261, 262, 267, 271]. Примеры приложения модели к системам с неполярными и слабо полярными компонентами — это расчеты для смесей углеводородов одной или разных гомологических серий [259, 261, 262, 267, 271—275] растворов углеводород — галоидзамещенный углеводород [268, 272, 276, 277]. В большинстве работ модель применяли к бинарным растворам при средних концентрациях. Однако ее использовали и для описания свойств систем при предельных разбавлениях [278; 279, с. 179; 280, 281 ]. Выполнены расчеты (хотя и немногочисленные) свойств тройных систем [278; 279, с. 179; 282], обсуждены возможности модели при описании диаграмм растворимости не только с верхней, но и с нижней критической точкой [283]. Вычисленные значения термодинамических функций, как правило, сопоставляли с экспериментальными, относящимися к процессам смешения при Остановимся подробнее на результатах применения модели Баркера к двум сериям растворов спиртов (от метилового до децилового) в циклогексане [264] и перфторбензоле [266]. Эти системы с интенсивными, приводящими к ассоциации, взаимодействиями (водородная связь). Расчеты для указанных серий позволяют наглядно выявить возможности модели при описании избыточных функций и равновесия жидкость — пар в смесях со сложным характером межмолекулярных взаимодействий. Совместное рассмотрение родственных систем представляет особый интерес, поскольку дает основу для более надежных оценок энергетических параметров модели, уяснения самого характера взаимодействий в растворах спиртов с циклогексаном и перфторбензолом. Была принята следующая модель раствора. Координационное число квазирешетки Типы контактных участков и их числа были следующими: циклогексан — В системах спирт — циклогексан единственное сильное взаимодействие — это образование водородной связи между Таблица VII.1 (см. скан) Значения параметров молекулами спирта. Модель взаимодействий включала два параметра:
В системах с перфторбензолом характер взаимодействия более сложен, поскольку наряду с образованием водородной связи между молекулами спирта возможно также слабое специфическое взаимодействие между молекулами спирта и кольцом молекулы перфторбензола, по аналогии с тем, что имеется в системах с бензолом. Для учета этого взаимодействия в системах с перфторбензолом вводили третий параметр
Обсуждались и другие варианты, в частности, Учитывалась температурная зависимость параметра — вводился параметр Найдено [175, с. 3; 255], что избыточная энтропия
(кликните для просмотра скана)
Рис. VII.8. Экспериментальные Отрицательные значения параметра Значения Модель позволяет выявить наличие расслаивания (рис. Теория Баркера позволяет не только рассчитывать избыточные термодинамические функции, но и оценивать числа специфических и неспецифических контактов в системе. Так, доли несвязанных гидроксильных групп для спиртов в их растворах с циклогексаном и перфторбензолом, рассчитанные по модели Баркера, удовлетворительно согласовывались со спектральными данными [264, 266]. Как показывают расчеты, хорошие результаты теория Баркера Дает при нахождении характеристик предельно разведенных растворов. Модель способна передать сильную концентрационную зависимость коэффициента активности ассоциированного компонента при больших разбавлениях. Значения предельных коэффициентов активности лучше согласуются с экспериментом, чем при Таблица VII.2 (см. скан) Значения предельных коэффициентов активности в растворах спирт использовании ряда известных корреляционных уравнений (уравнение Вильсона, NRTL, UNIFAC [280] (табл. VII.2). Модель Баркера позволяет успешно предсказывать свойства тройных систем различного типа по составляющим бинарным [278; 279, с. 179; 282]. Такие расчеты проводились, в частности, для систем типа спирт — два неполярных компонента [278] и неполярный компонент — два спирта [279, с. 179]. Точность предсказания Возможности модели Баркера, однако, не следует переоценивать. Она способна отразить основные особенности ассоциированных систем, определяемые образованием сильных связей, но слишком груба для описания сравнительно тонких эффектов, связанных с неспецифическими взаимодействиями. Как и все решеточные модели, основанные на представлении о жесткой решетке без вакансий, она не позволяет учесть объемные эффекты смешения. Недостаток модели — известный произвол в выборе геометрических параметров, использование подгоночных значений энергетических параметров, которые выступают не как молекулярные, а как полуфеноменологические, что затрудняет применение модели в практических расчетах фазовых равновесий. Однако при некоторой унификации схемы взаимодействий, согласованном рассмотрении серий систем, полезность модели для целей предсказания свойств возрастает. Наиболее полное развитие такой подход получил в групповых модификациях модели Баркера, о которых речь пойдет в гл. VIII. (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) III. Алгоритм расчета параметров моделей жидкой фазы по методу Ньютона-Гаусса. Метод состоит в минимизации суммы квадратов отклонений расчетных значений от заданных; применяют его тогда когда расчетные уравнения нелинейны относительно искомых параметров. При изложении метода использованы определения и условные обозначения разд. VII.6. Необходимое условие минимальности функции
которая с учетом (VII. 148) и (VII. 149) преобразуется к виду:
Систему В соответствии с методом Ньютона — Гаусса, функцию
где Формула (3), точная для линейной зависимости Все
где
Величины В процессе минимизации функции
Вследствие значительной нелинейности функций 1. При нарушении ограничений на значения параметров. Указанные ограничения могут быть обусловлены физическим смыслом параметров или математической формой уравнений для коэффициентов активности. Так, для параметра 2. При возрастании значения минимизируемой функции. Если первая релаксация не была успешной, релаксацию повторяют до тех пор, пока значения После того, как успешный шаг по параметрам сделан, полагают Сходимость метода Ньютона — Гаусса в среднем высокая, причем в большинстве случаев потребность в применении релаксационной методики не возникает. Обычное число итераций при оценке двух энергетических параметров моделей локального состава по данным для бинарной системы, при аналитическом расчете производных Рис. VII.9. (см. скан) Номограмма IV. Номограммы для нахождения начальных приближений параметров Вильсона и NRTL. На рис. VII.9 приведена номограмма для приближенной оценки приведенных параметров уравнения Вильсона по данным о предельных коэффициентах активности. Способ использования номограммы ясен из данного примера для бинарной системы, в которой Рис. VII.10. (см. скан) Номограмма На рис. VII.10 представлены номограммы для приближенной оценки основных энергетических параметров уравнения NRTL при заданном параметре
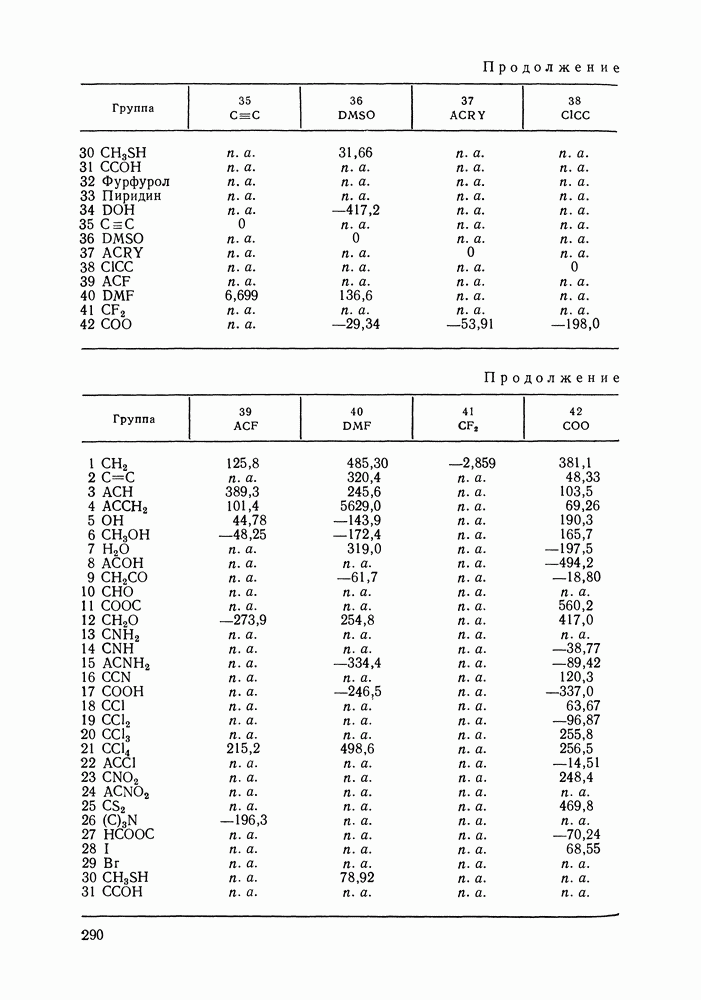
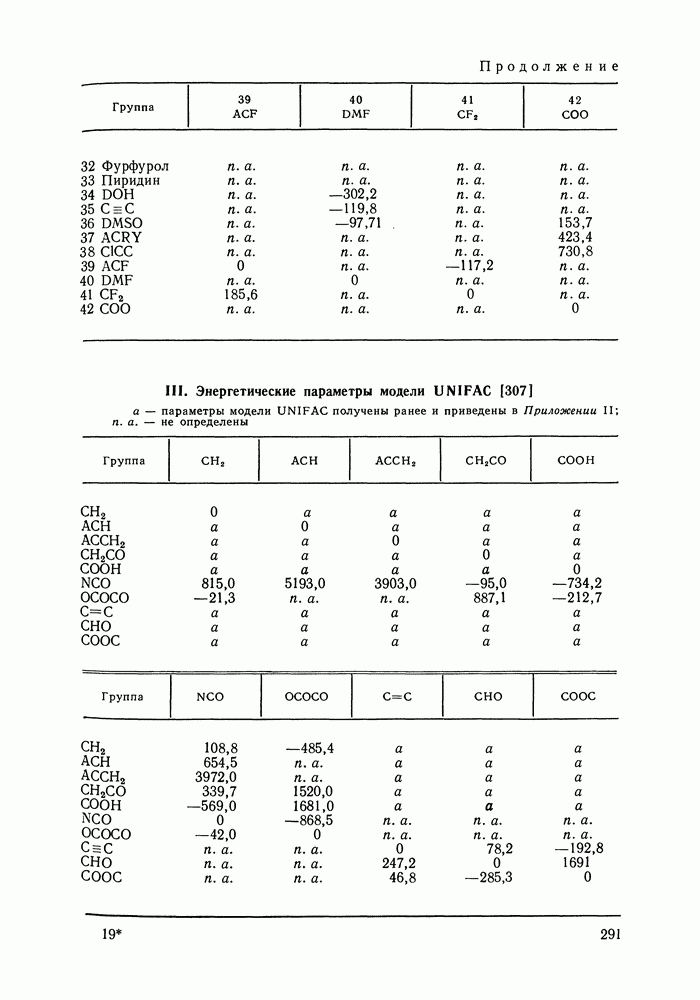
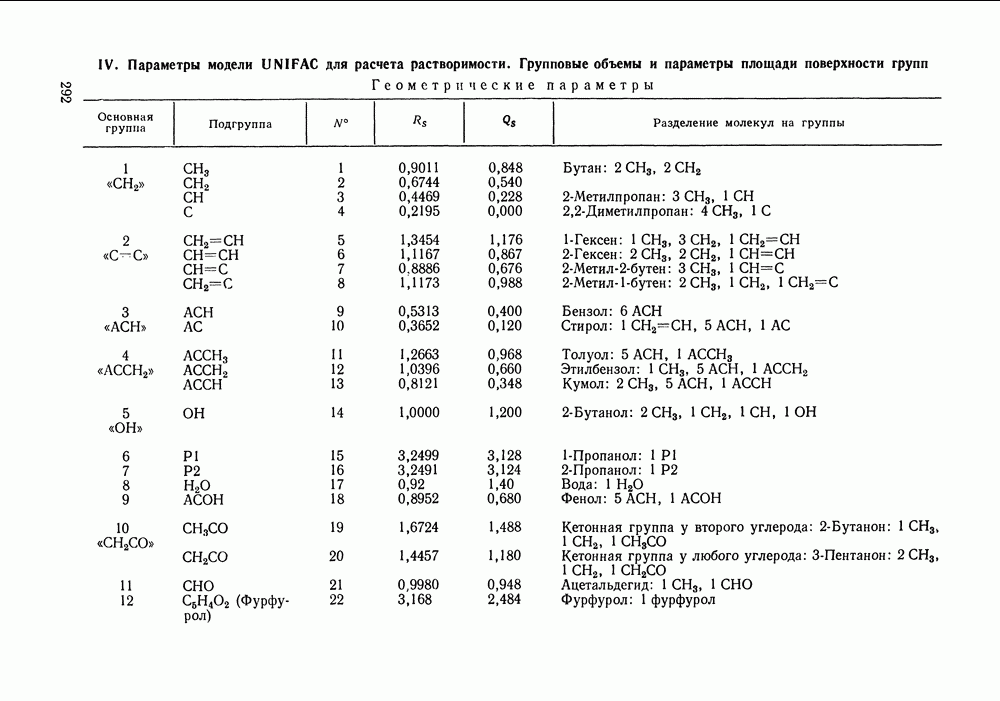
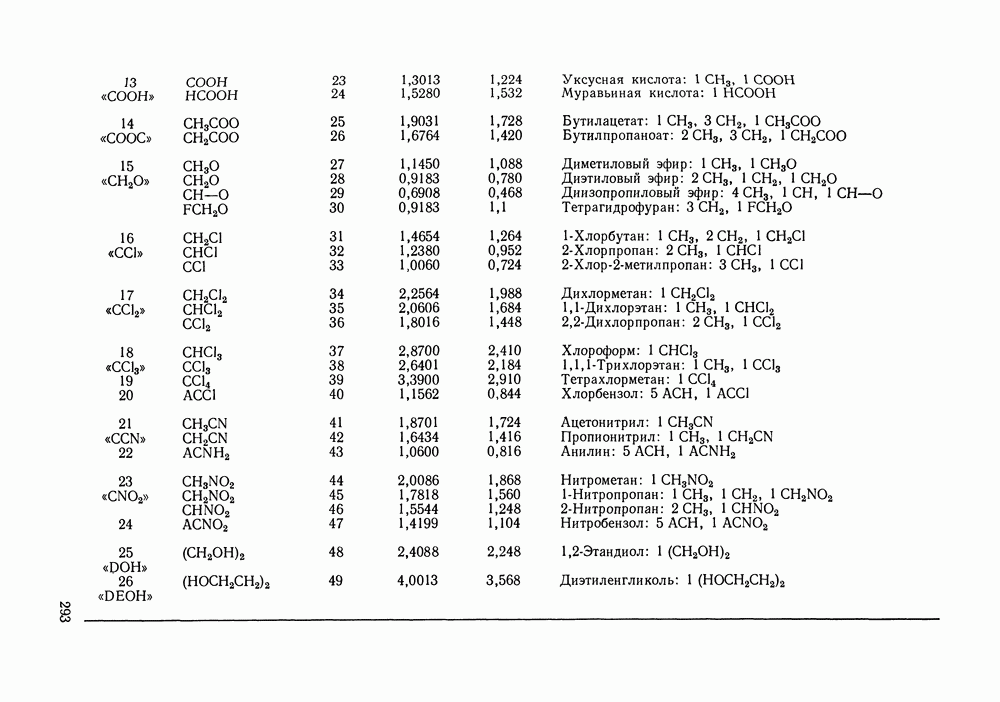
|
 |
Оглавление
|














































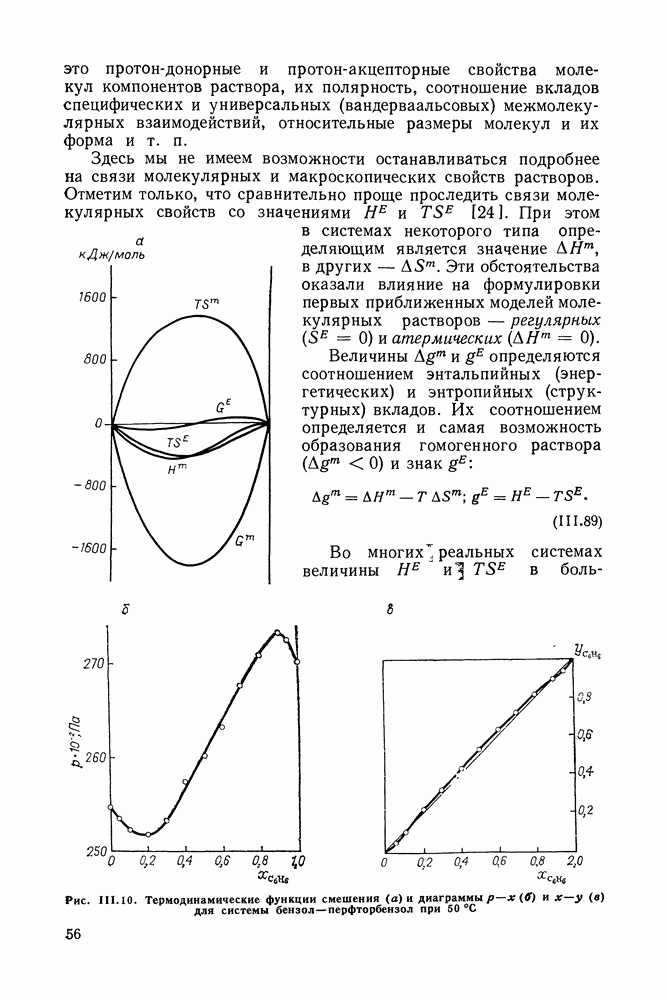



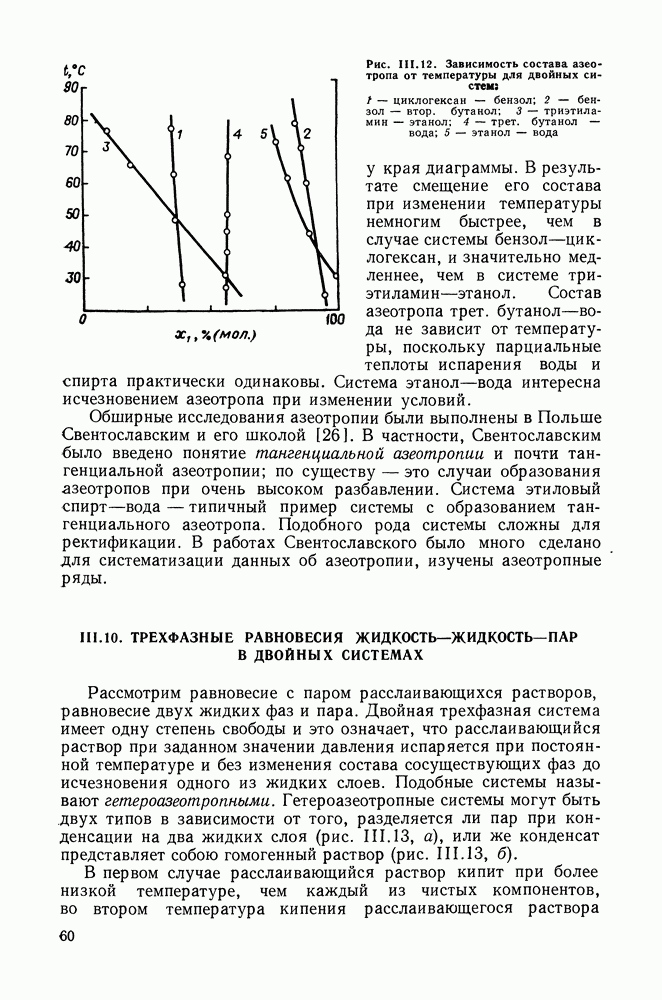





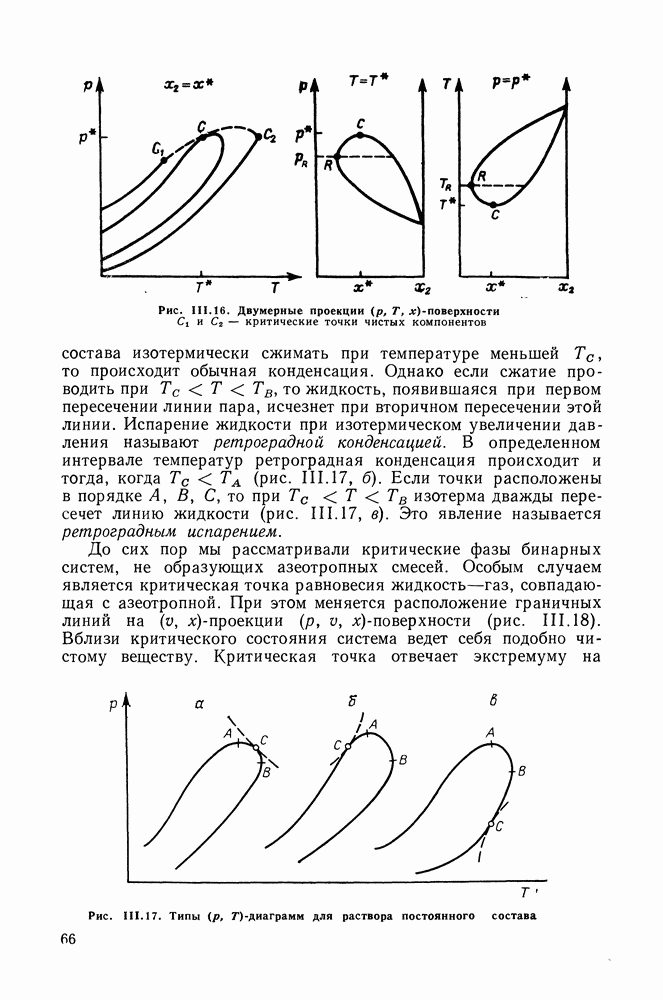
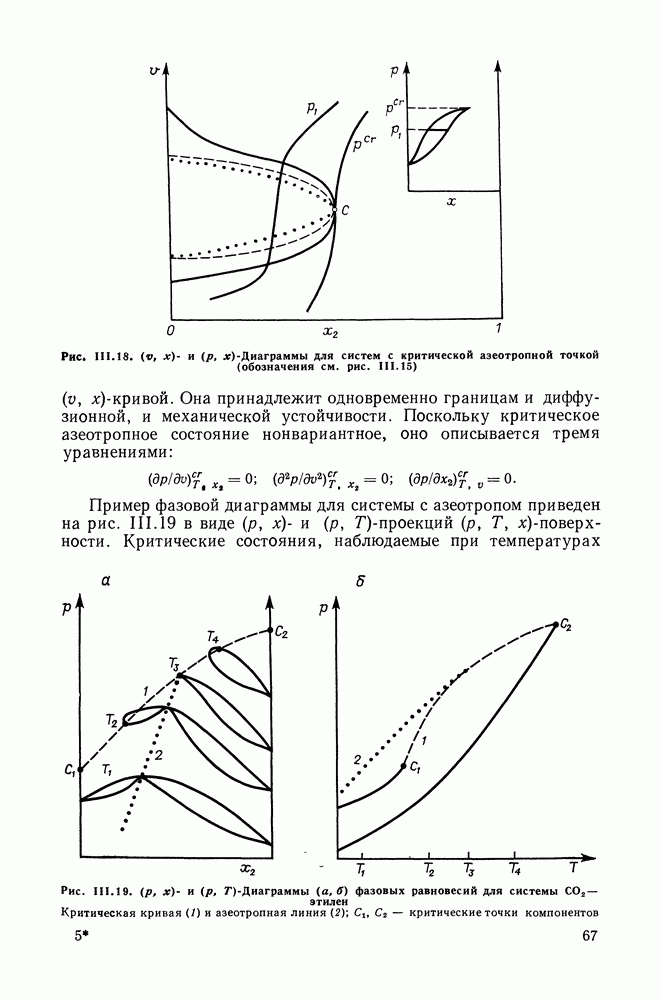




































































































































































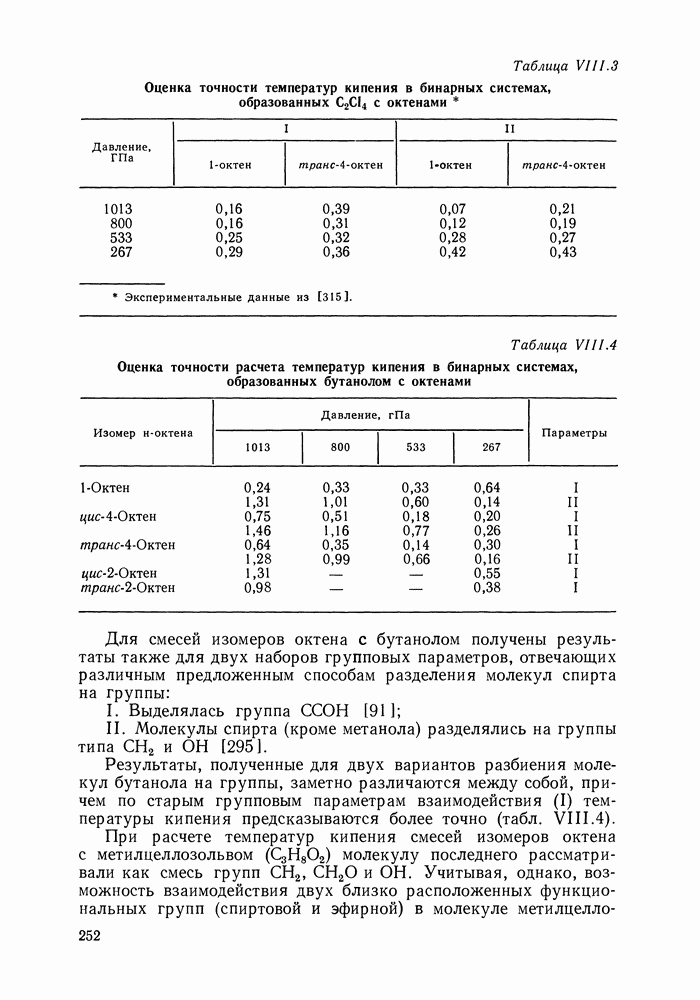















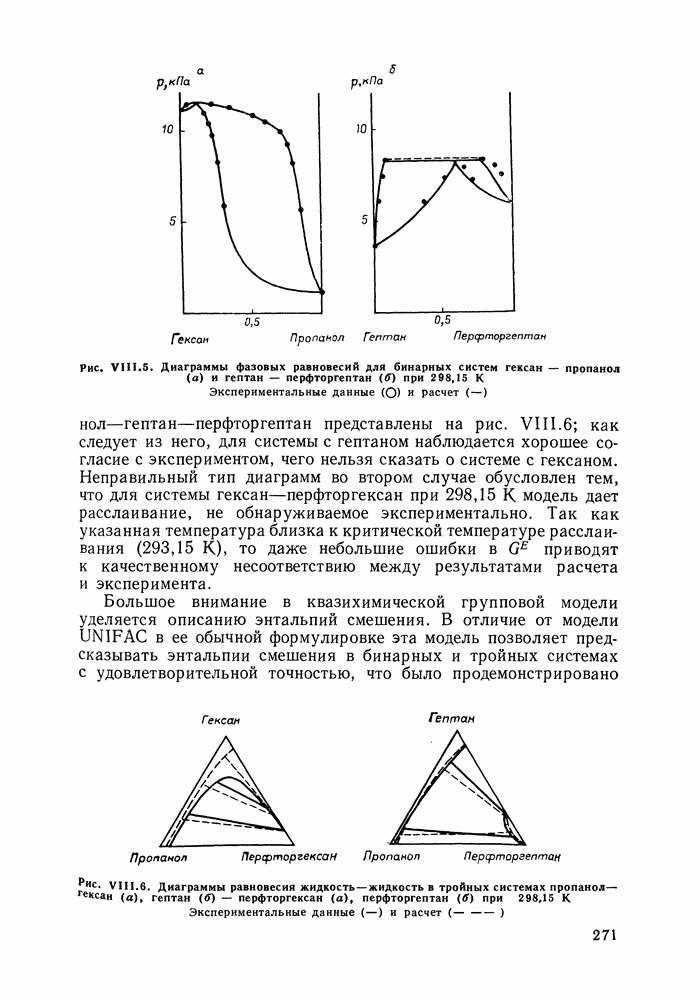
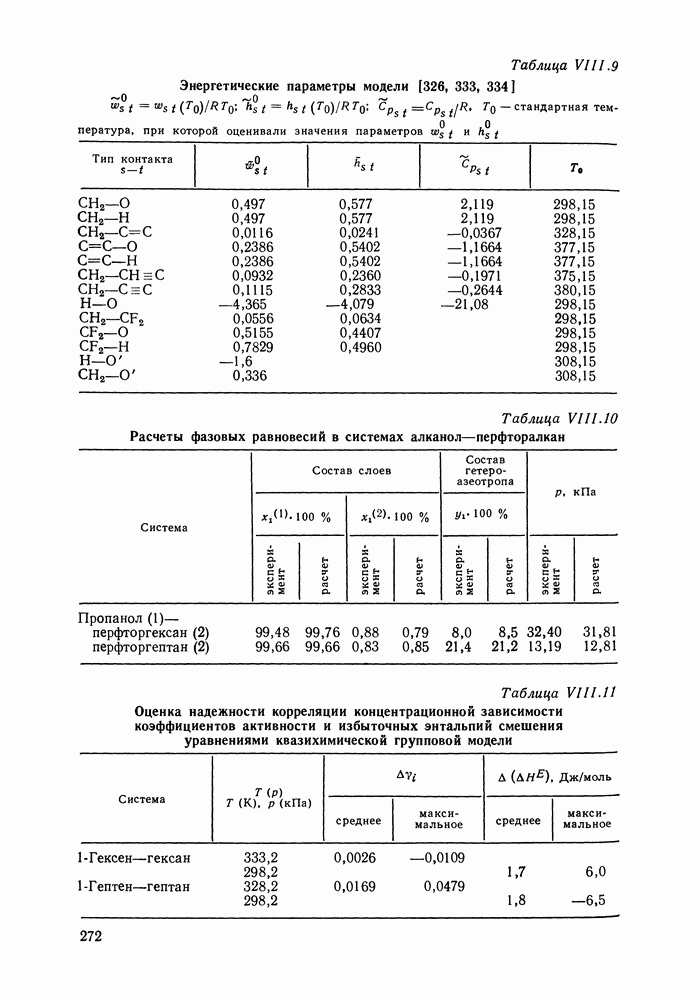
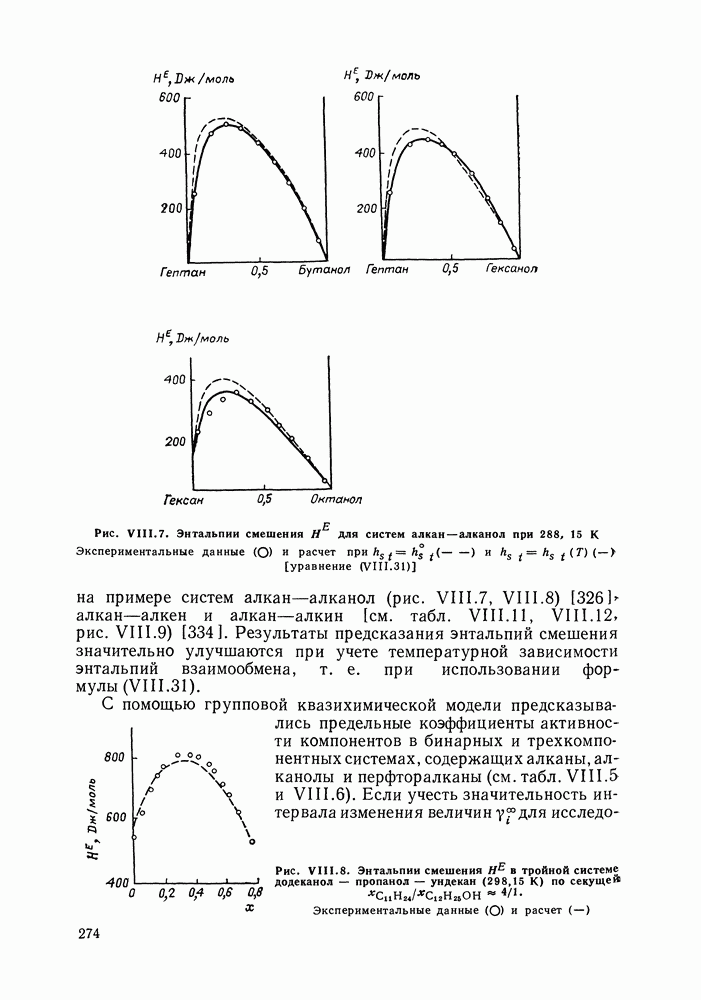


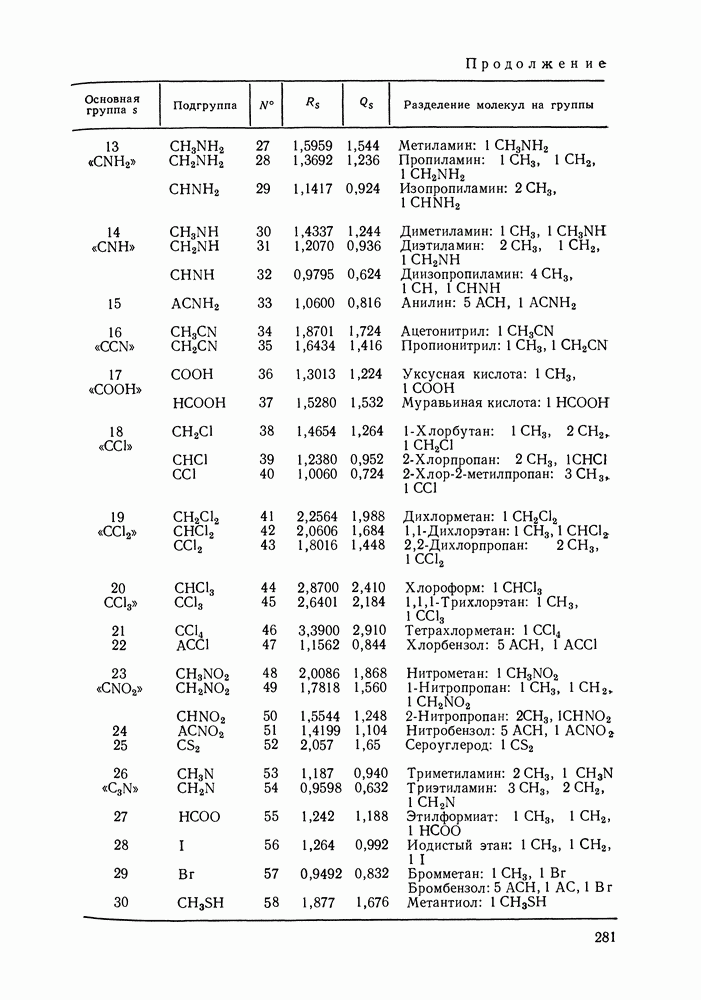

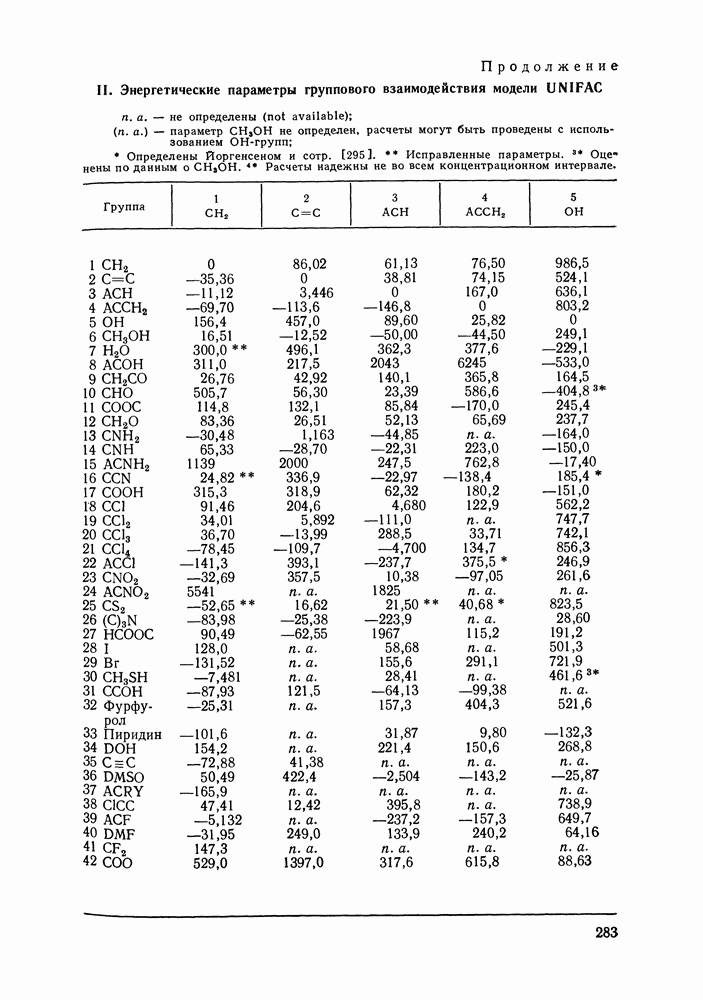
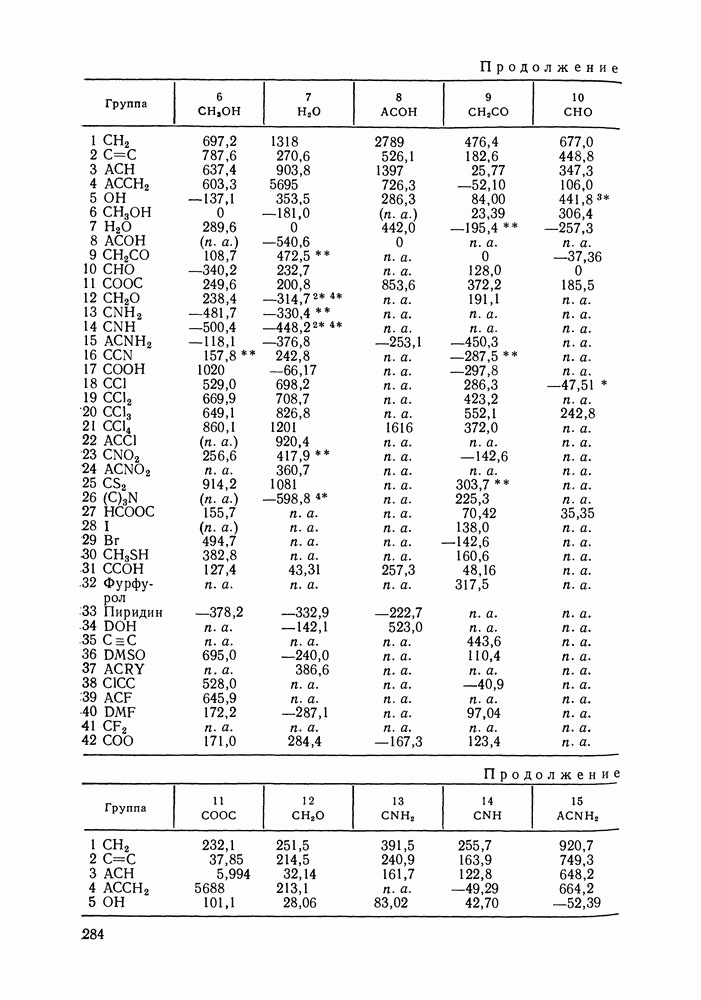
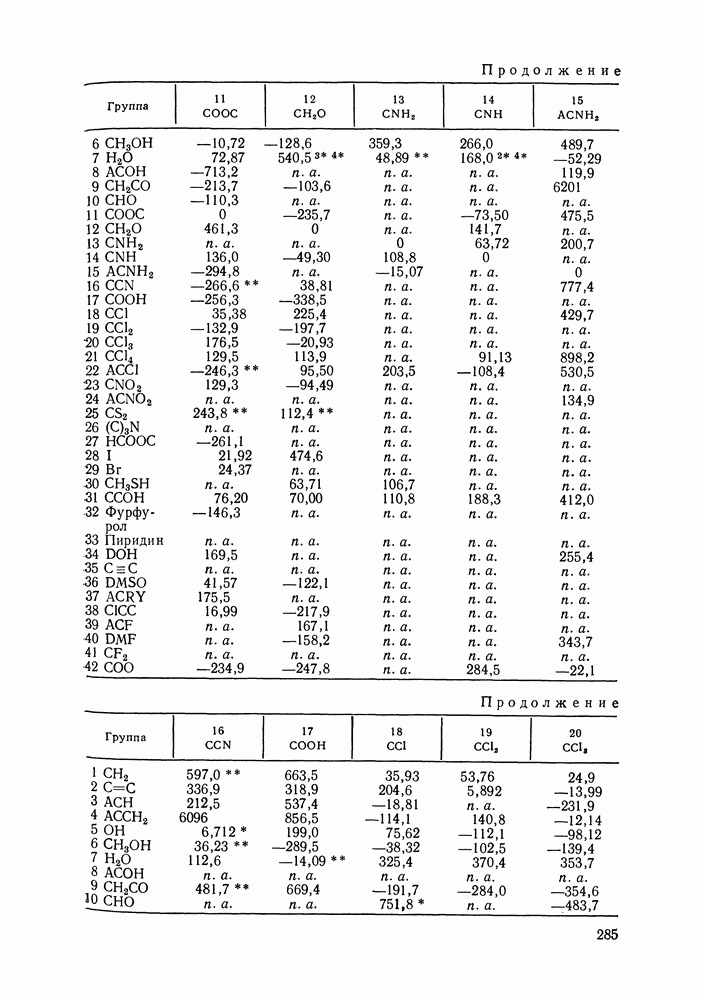
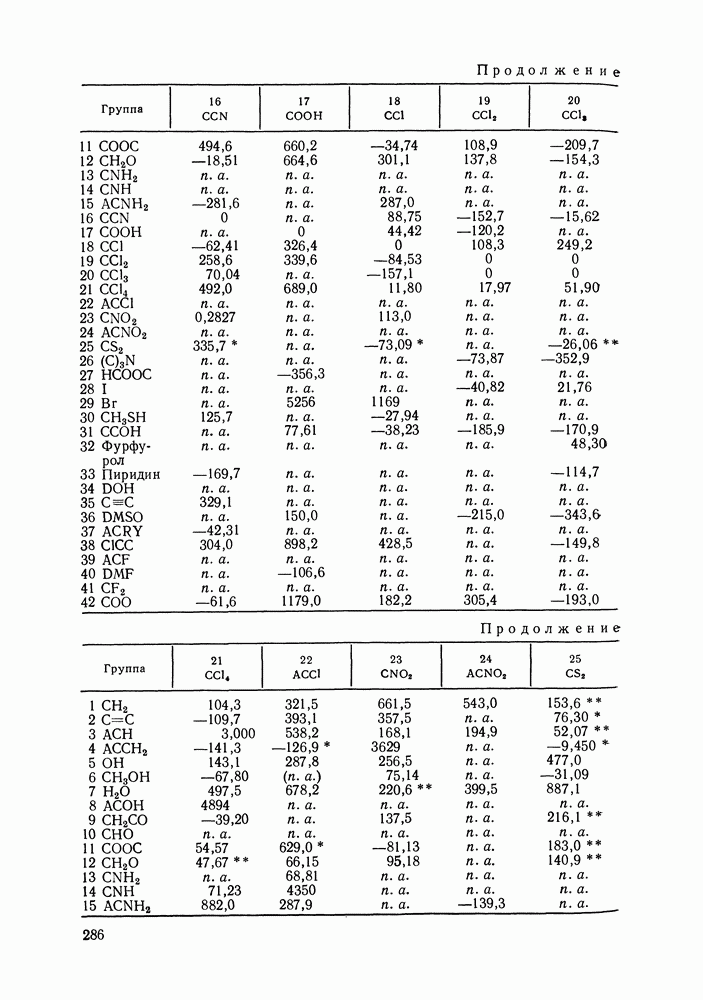
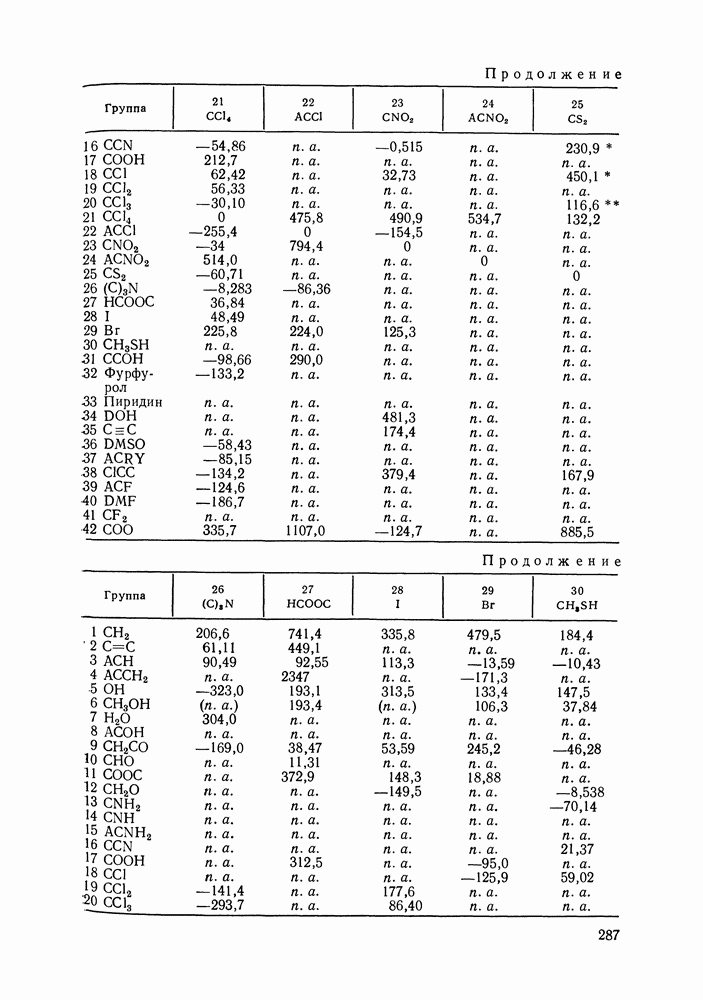
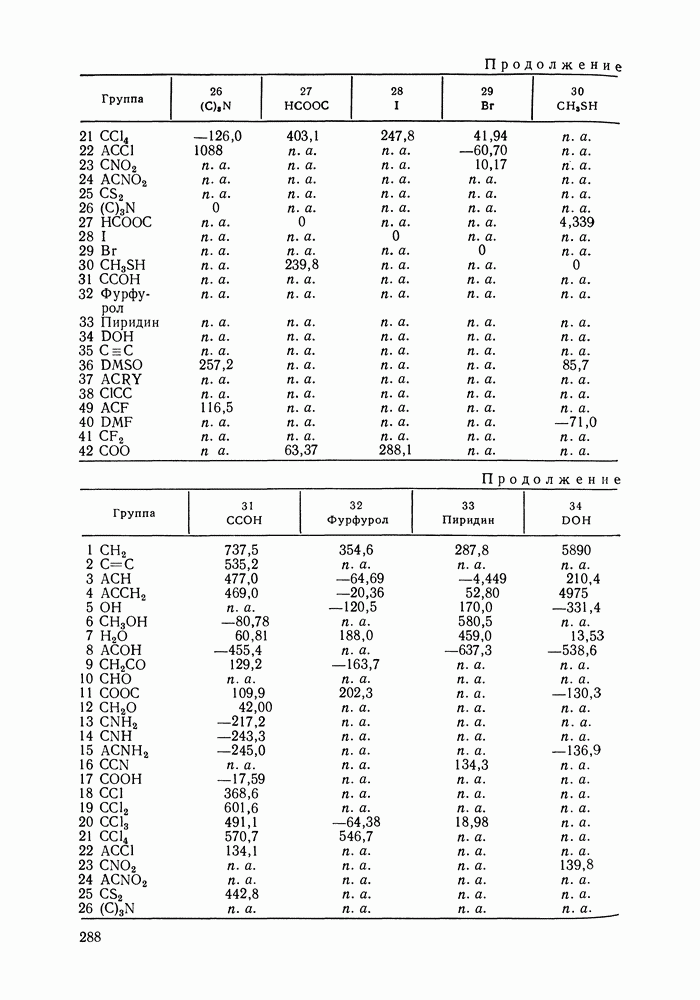
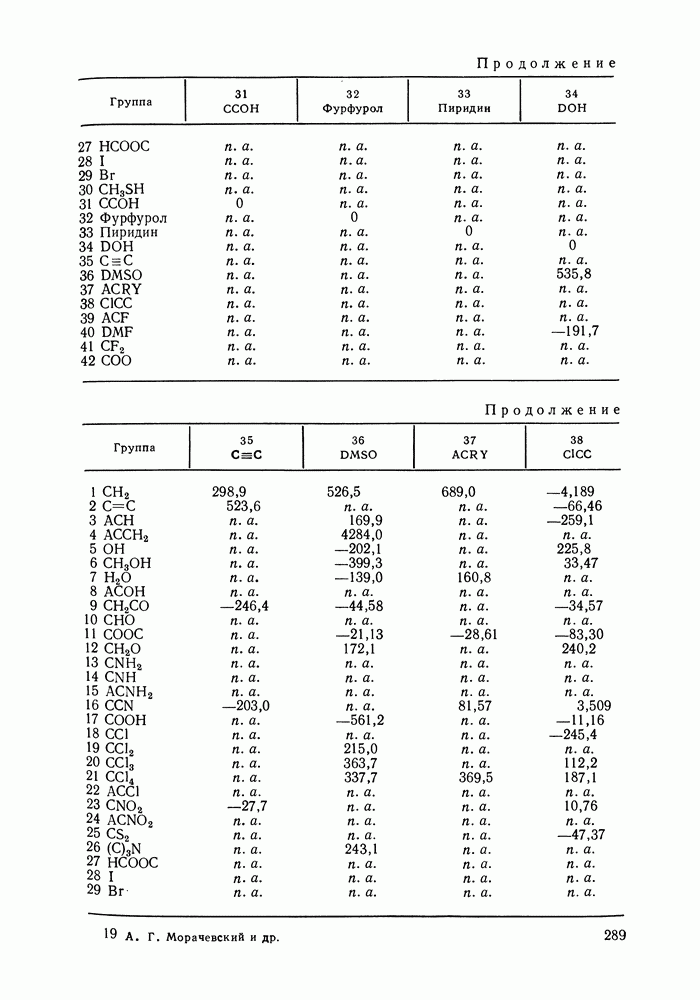




















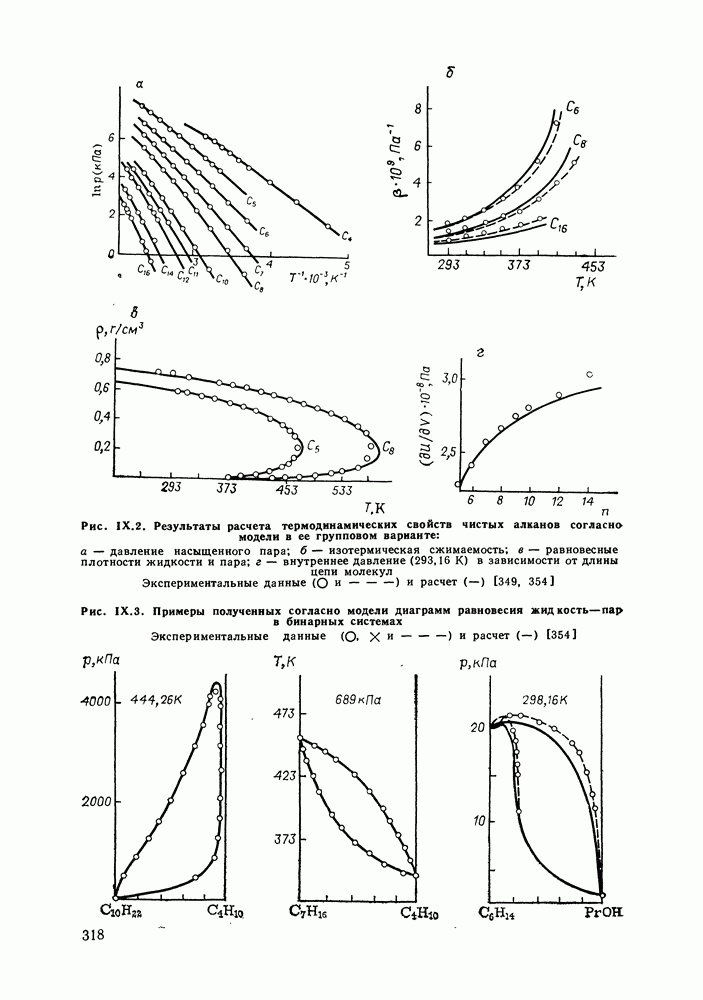
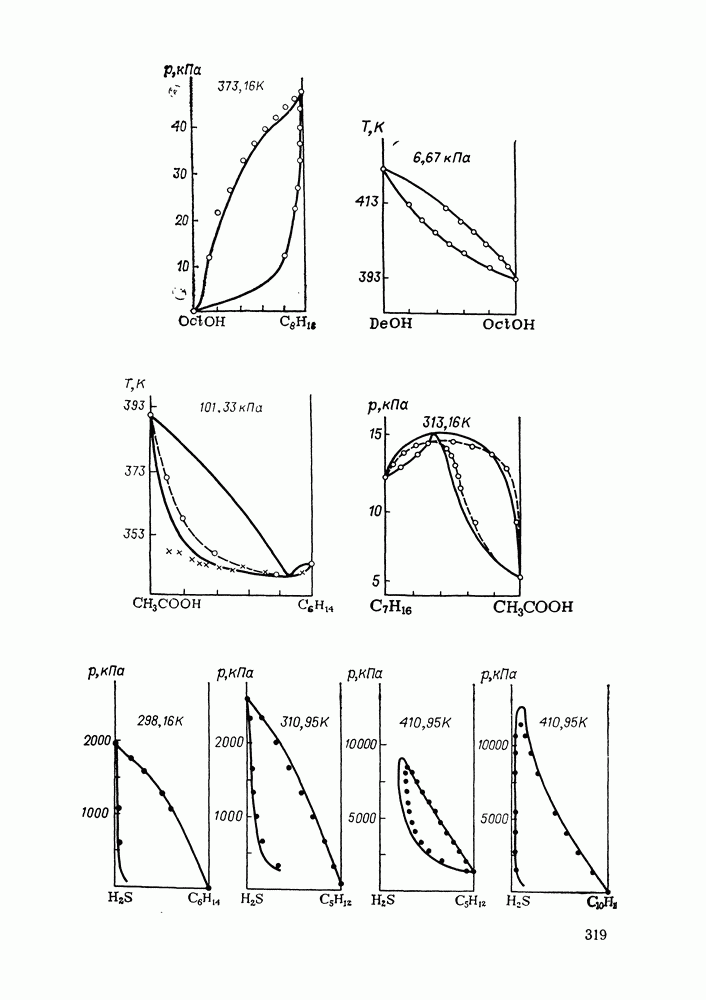

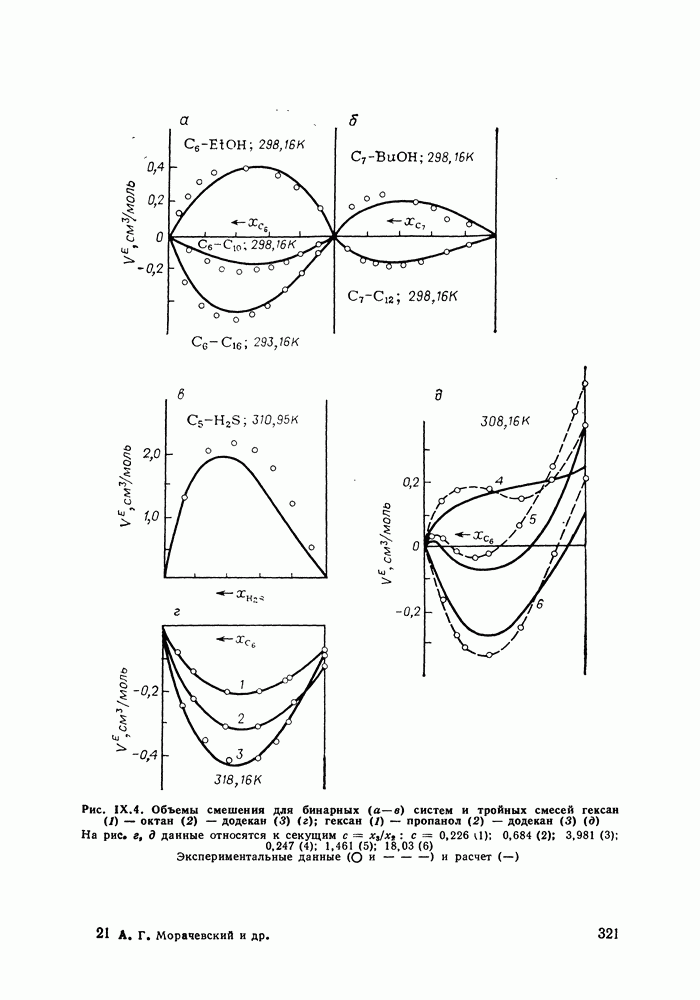
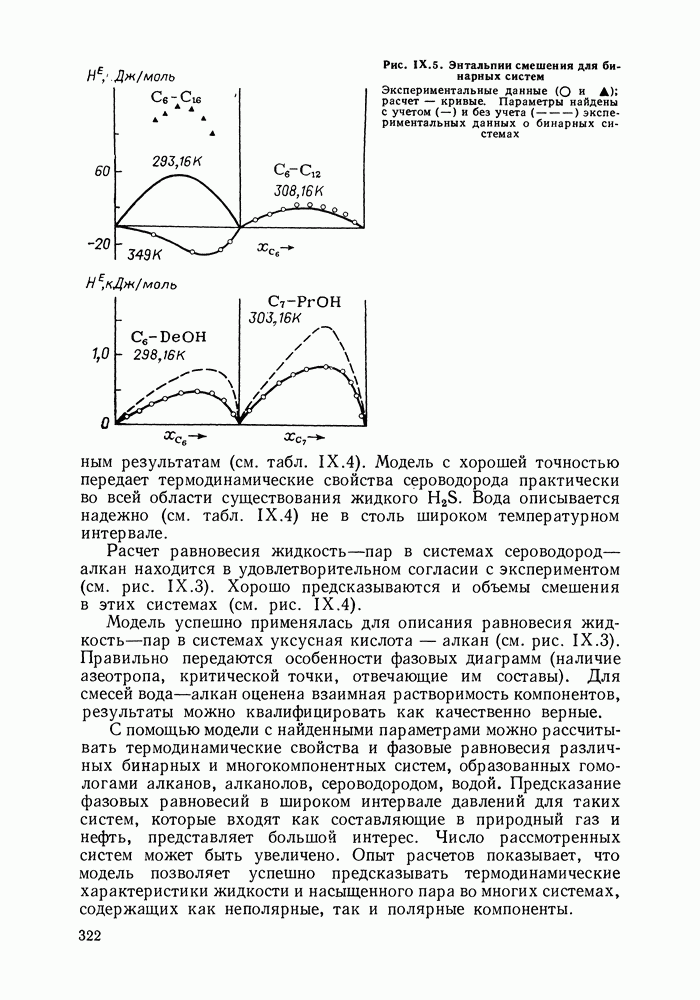

















 меров) [215] —
меров) [215] —

 объемная доля компонента
объемная доля компонента  поверхностная доля компонента
поверхностная доля компонента  число мест квазирешетки, занятых молекулой
число мест квазирешетки, занятых молекулой 


 в этом приближении совпадает с (VII. 155), избыточный химический потенциал определяется формулой:
в этом приближении совпадает с (VII. 155), избыточный химический потенциал определяется формулой:


 и 1.
и 1.

 число пар типа
число пар типа  энергия взаимодействия пар
энергия взаимодействия пар 

 и однотипных
и однотипных  Для неполярных и слабополярных молекул вводится параметр взаимодействия, усредненный по всей «поверхности» молекул. Уравнения для избыточных термодинамических функций бинарной системы 1—2 в таком случае включают один параметр — энергию взаимообмена
Для неполярных и слабополярных молекул вводится параметр взаимодействия, усредненный по всей «поверхности» молекул. Уравнения для избыточных термодинамических функций бинарной системы 1—2 в таком случае включают один параметр — энергию взаимообмена  Энергетика взаимодействий в бинарных системах с полярными компонентами характеризуется двумя или более энергиями взаимообмена.
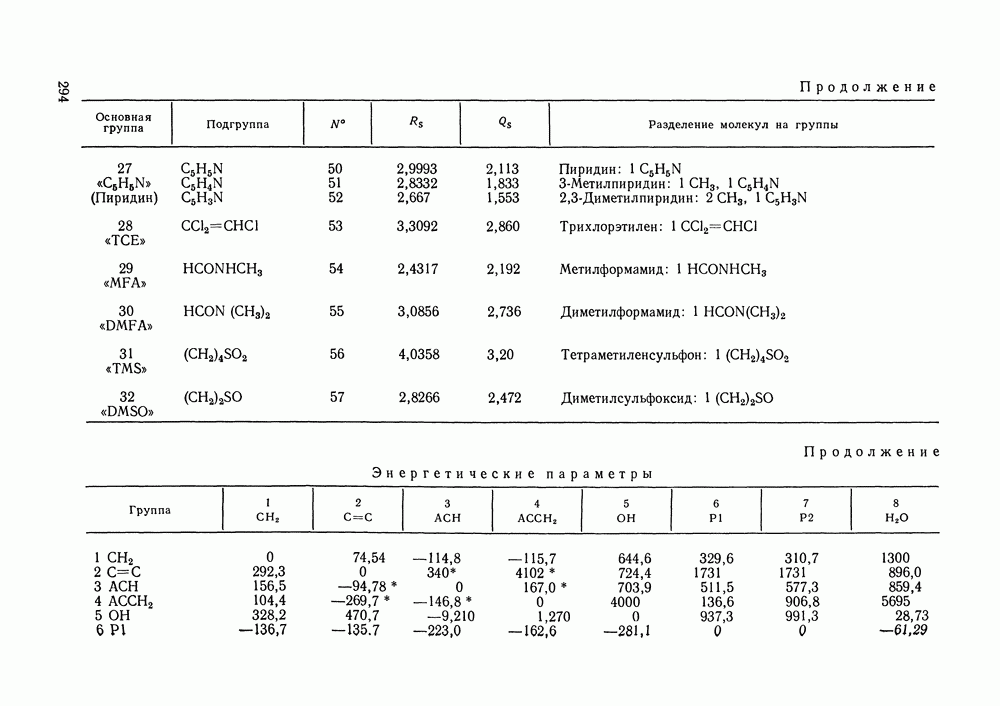
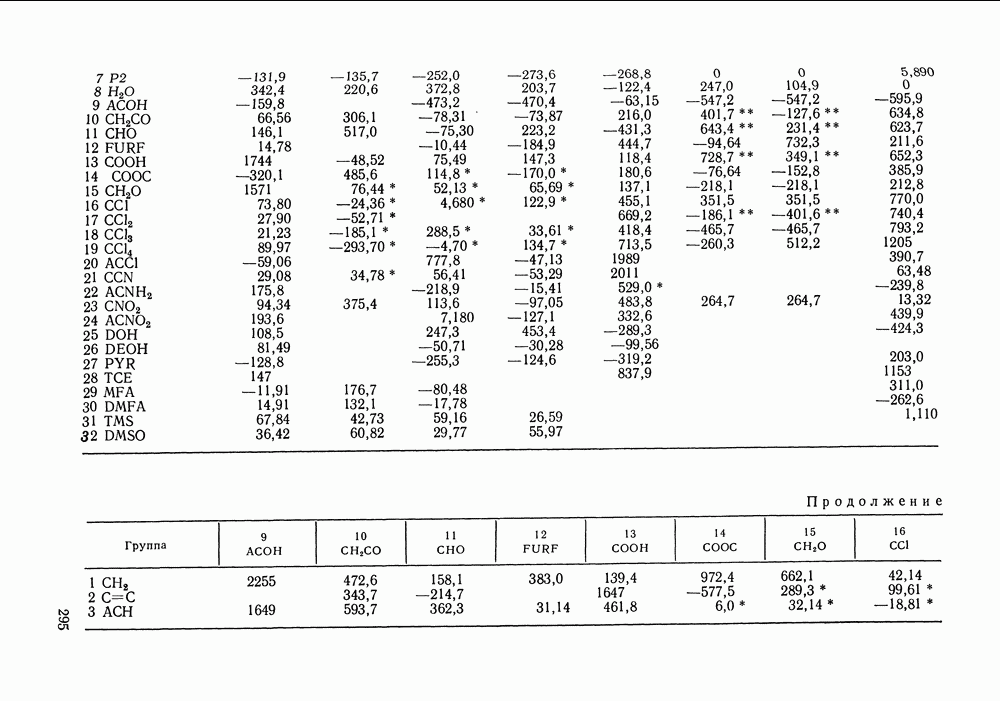
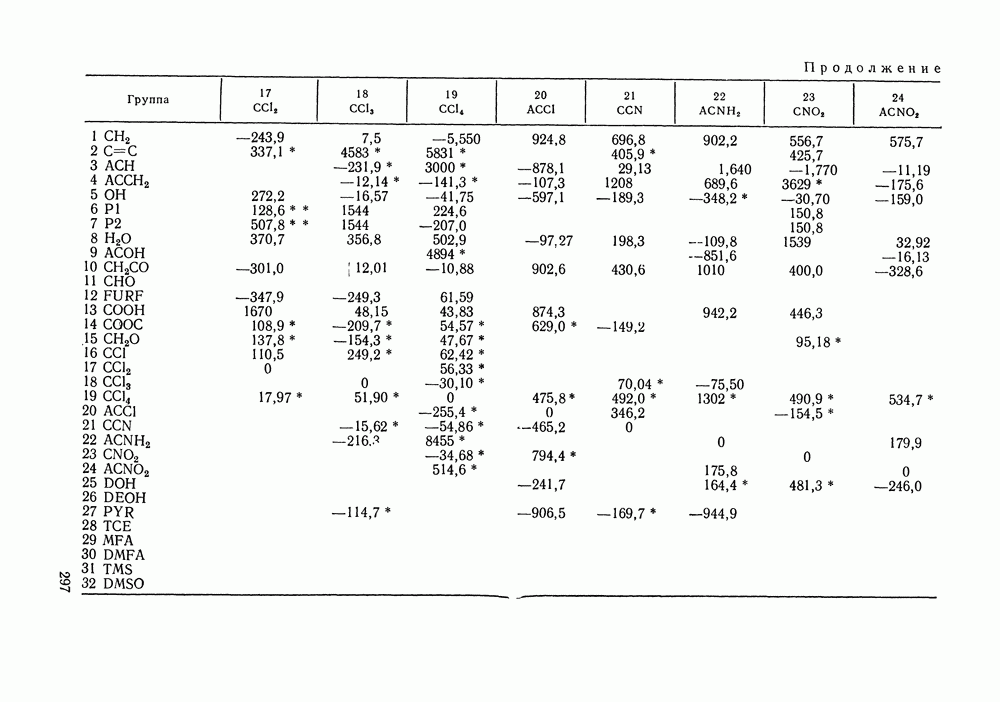
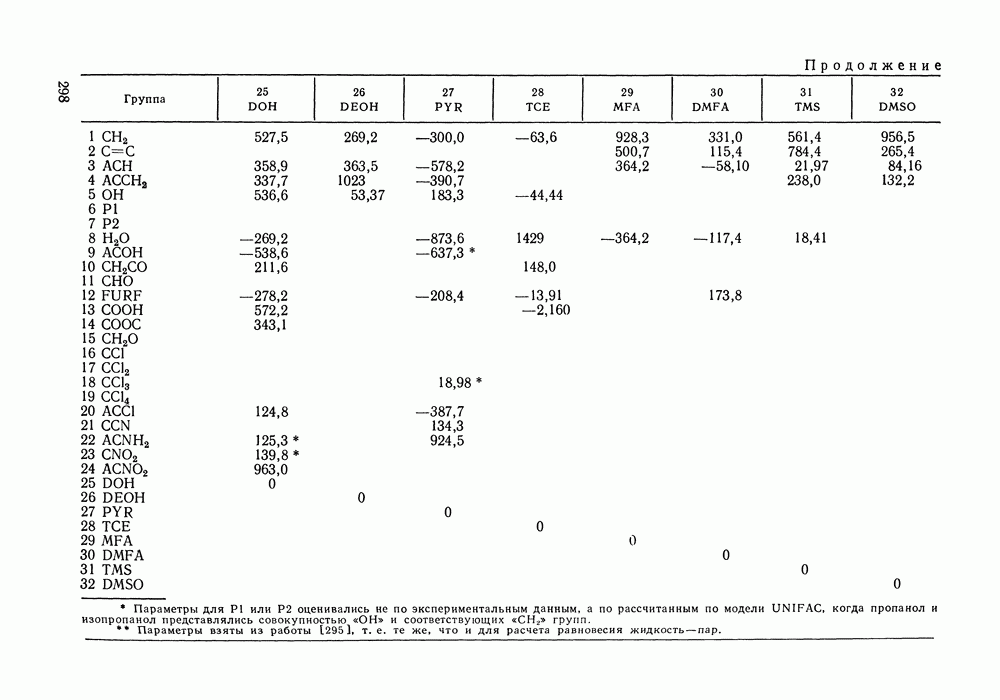
Энергетика взаимодействий в бинарных системах с полярными компонентами характеризуется двумя или более энергиями взаимообмена.
 [см. (VII.160)] чаще всего используют приближения Брэгга — Вильямса или квазихимическое [215]. Первое соответствует предположению о беспорядочном распределении частиц по узлам решетки. Более последовательное квазихимическое приближение учитывает корреляцию ближайших соседей. Наиболее вероятные числа пар связаны соотношением:
[см. (VII.160)] чаще всего используют приближения Брэгга — Вильямса или квазихимическое [215]. Первое соответствует предположению о беспорядочном распределении частиц по узлам решетки. Более последовательное квазихимическое приближение учитывает корреляцию ближайших соседей. Наиболее вероятные числа пар связаны соотношением:

 избыточных функций
избыточных функций  Объемные эффекты смешения моделью не учитываются
Объемные эффекты смешения моделью не учитываются 
 -меров. Предполагается, что все узлы решетки заняты, вакансии отсутствуют. Молекуле сорта
-меров. Предполагается, что все узлы решетки заняты, вакансии отсутствуют. Молекуле сорта  приписывается
приписывается  контактных Участков по числу ближайших соседних узлов. Величина
контактных Участков по числу ближайших соседних узлов. Величина  определяется формулой (VII. 157). Контактные участки могут отличаться по энергетическим характеристикам взаимодействия.
определяется формулой (VII. 157). Контактные участки могут отличаться по энергетическим характеристикам взаимодействия.

 кислород и водород гидроксила; I — углеводородный радикал;
кислород и водород гидроксила; I — углеводородный радикал;  число гтомов углерода в углеродной цепи
число гтомов углерода в углеродной цепи
 водород и кислород гидроксила, I — контактные участки углеводородного радикала (рис. VII.6). Введение нескольких энергий взаимообмена позволяет выделить способы контактирования, отвечающие как неспецифическим взаимодействиям ван-дерваальсова типа, так и интенсивным специфическим взаимодействиям, например, водородной связи.
водород и кислород гидроксила, I — контактные участки углеводородного радикала (рис. VII.6). Введение нескольких энергий взаимообмена позволяет выделить способы контактирования, отвечающие как неспецифическим взаимодействиям ван-дерваальсова типа, так и интенсивным специфическим взаимодействиям, например, водородной связи.

 как обычно в решеточных моделях, представляются суммой комбинаторного вклада и остаточного:
как обычно в решеточных моделях, представляются суммой комбинаторного вклада и остаточного:

 -меров [см. (VII. 156) и (VII.158)]. Выражение для остаточного вклада в избыточный химический потенциал имеет вид:
-меров [см. (VII. 156) и (VII.158)]. Выражение для остаточного вклада в избыточный химический потенциал имеет вид:

 доля контактных участков типа
доля контактных участков типа  от общего числа
от общего числа  участков молекулы
участков молекулы  число контактных участков типа
число контактных участков типа  молекулы
молекулы  доля контактных участков типа
доля контактных участков типа  в растворе заданного состава;
в растворе заданного состава;  общее число типов контактных участков в системе.
общее число типов контактных участков в системе.
 решения системы уравнений типа:
решения системы уравнений типа:

 решения аналогичной системы уравнений для чистого компонента
решения аналогичной системы уравнений для чистого компонента 


 можно записать:
можно записать:

 энтальпия взаимообмена.
энтальпия взаимообмена.
 приравнивают энергии взаимообмена
приравнивают энергии взаимообмена  Однако в системах с ассоциацией требуется учет температурной зависимости параметров
Однако в системах с ассоциацией требуется учет температурной зависимости параметров 
 так и энергетические
так и энергетические  параметры.
параметры.
 Некоторые авторы, однако, более обоснованным считают сопоставление с экспериментальными данными о функциях смешения при
Некоторые авторы, однако, более обоснованным считают сопоставление с экспериментальными данными о функциях смешения при  [256, 257, 272]. Единого мнения на этот счет нет.
[256, 257, 272]. Единого мнения на этот счет нет.
 предполагали равным 4. Допускали, что молекула спирта занимает в решетке
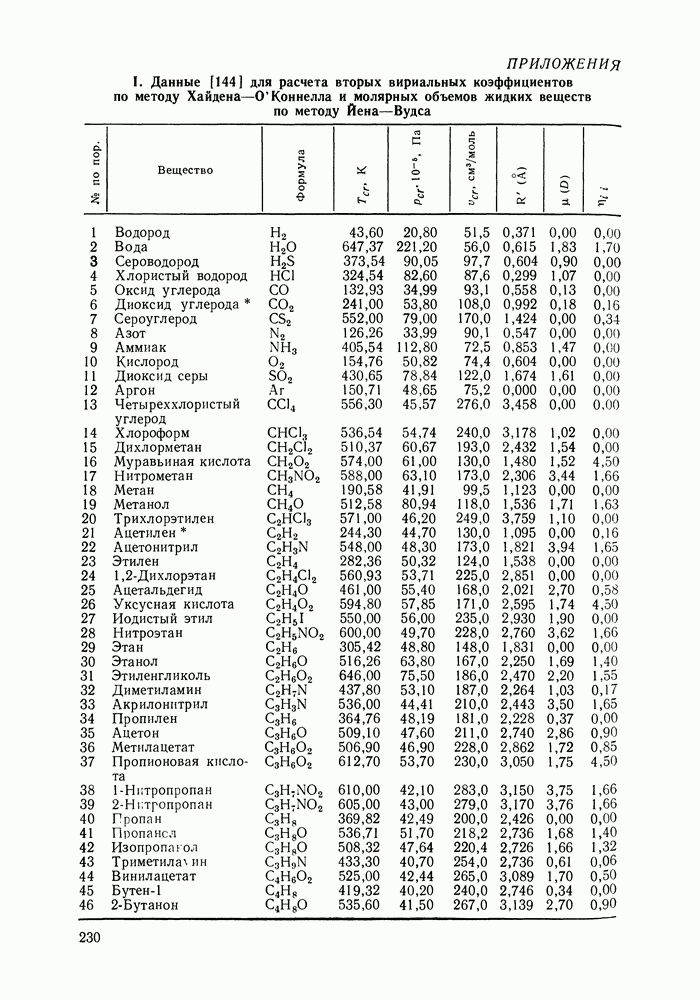
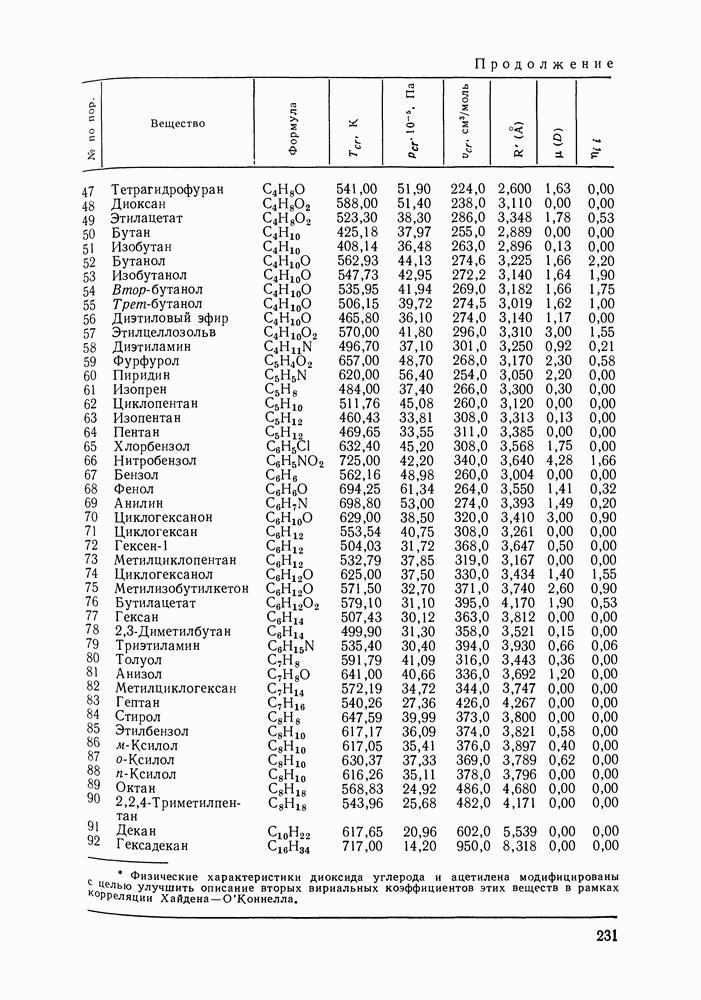
предполагали равным 4. Допускали, что молекула спирта занимает в решетке  мест (см. рис. VII.6). Исходя из соотношений молекулярных объемов метилового спирта и циклогексана, молекуле последнего отводилось 5 мест в решетке. Заметим, что для линейных алканов число мест обычно приравнивается числу атомов углерода в цепи
мест (см. рис. VII.6). Исходя из соотношений молекулярных объемов метилового спирта и циклогексана, молекуле последнего отводилось 5 мест в решетке. Заметим, что для линейных алканов число мест обычно приравнивается числу атомов углерода в цепи  Значение
Значение  для циклогексана можно рассматривать как эмпирический учет цикличности молекул. Число мест, занимаемых молекулой пер-фторбензола, по аналогии с циклогексаном и бензолом также приравнивалось 5.
для циклогексана можно рассматривать как эмпирический учет цикличности молекул. Число мест, занимаемых молекулой пер-фторбензола, по аналогии с циклогексаном и бензолом также приравнивалось 5.
 перфторбензол —
перфторбензол —  спирт —
спирт —  Полагали, что все контактные участки молекул циклогексана (перфтор-бензола) одного типа,
Полагали, что все контактные участки молекул циклогексана (перфтор-бензола) одного типа,  Поскольку кислород гидроксильной группы способен участвовать в двух водородных связях, а водород — в одной, для чисел контактных участков спирта были приняты значения
Поскольку кислород гидроксильной группы способен участвовать в двух водородных связях, а водород — в одной, для чисел контактных участков спирта были приняты значения  (см. рис. VII.6).
(см. рис. VII.6).
 для систем спирт—циклогексан и спирт—перфторбензол при
для систем спирт—циклогексан и спирт—перфторбензол при 
 энергия (свободная энергия) взаимообмена при образовании водородной связи между молекулами спирта;
энергия (свободная энергия) взаимообмена при образовании водородной связи между молекулами спирта;
 параметр, характеризующий усредненную энергию взаимообмена для неспецифических взаимодействий вандерваальсова типа.
параметр, характеризующий усредненную энергию взаимообмена для неспецифических взаимодействий вандерваальсова типа.


 [266]. Параметр
[266]. Параметр  считали одинаковым для обеих серий систем, т. е. допускали постоянство энергии образования водородной связи в различных растворителях.
считали одинаковым для обеих серий систем, т. е. допускали постоянство энергии образования водородной связи в различных растворителях.
 энтальпия взаимообмена. Значение
энтальпия взаимообмена. Значение  полагали равным —
полагали равным —  (по результатам спектральных измерений). Остальные энергии взаимообмена определяли варьированием по экспериментальным данным об избыточных функциях.
(по результатам спектральных измерений). Остальные энергии взаимообмена определяли варьированием по экспериментальным данным об избыточных функциях.
 чувствительна, главным образом, к параметру, характеризующему направленные взаимодействия в системе, и мало зависит от энергии взаимообмена для вандерваальсовых взаимодействий. Поэтому параметр оценивали по экспериментальным данным
чувствительна, главным образом, к параметру, характеризующему направленные взаимодействия в системе, и мало зависит от энергии взаимообмена для вандерваальсовых взаимодействий. Поэтому параметр оценивали по экспериментальным данным  для систем с циклогексаном, параметр
для систем с циклогексаном, параметр  по данным
по данным  для систем с перфторбензолом. Для каждой из исследуемых систем параметр оценивали по данным об энтальпии смешения
для систем с перфторбензолом. Для каждой из исследуемых систем параметр оценивали по данным об энтальпии смешения  (табл. VII. 1).
(табл. VII. 1).


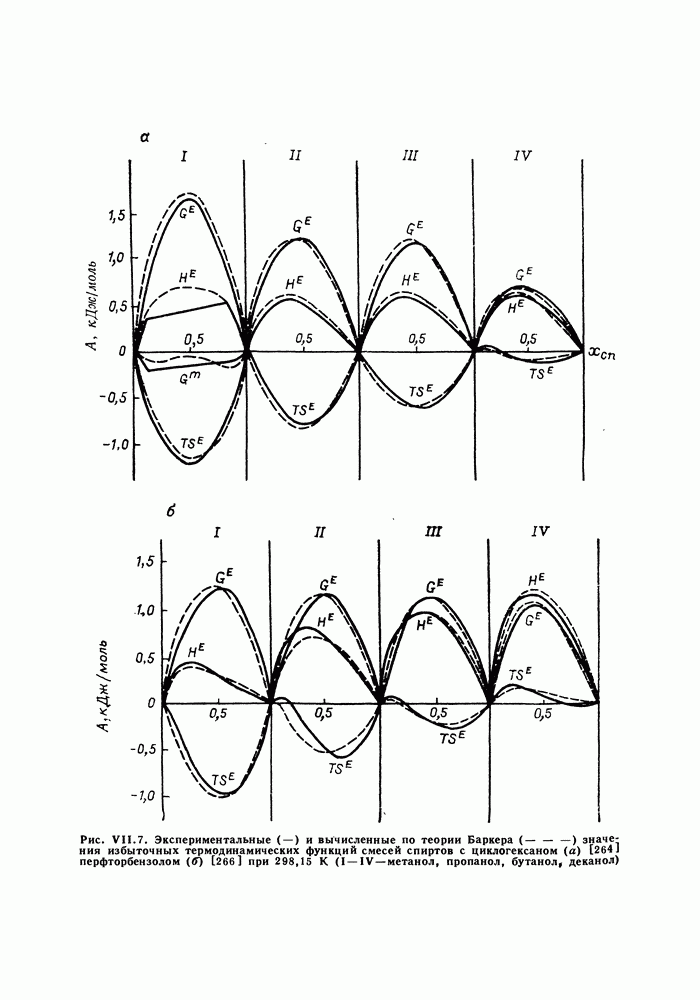
 и вычисленные
и вычисленные  по теории Баркера значения концентрационной и температурной зависимостей избыточной энтальпии
по теории Баркера значения концентрационной и температурной зависимостей избыточной энтальпии  для системы метанол — перфторбензол при различных температурах
для системы метанол — перфторбензол при различных температурах
 действительно свидетельствуют об энергетически выгодном взаимодействии между спиртом и перфторбензолом
действительно свидетельствуют об энергетически выгодном взаимодействии между спиртом и перфторбензолом  Найдено, что значение избыточных термодинамических функций чувствительно только к разности
Найдено, что значение избыточных термодинамических функций чувствительно только к разности  [258, 266], и вывод о наличии специфического взаимодействия спирт — перфторбензол, таким образом, может быть сделан только при учете данных для систем спирт — циклогексан.
[258, 266], и вывод о наличии специфического взаимодействия спирт — перфторбензол, таким образом, может быть сделан только при учете данных для систем спирт — циклогексан.
 и
и  рассчитанные по модели Баркера со значениями параметров из табл. VII. 1, и найденные экспериментально, приведены на рис. VII.7; они хорошо согласуются. Модель в состоянии описать различную форму кривых, в частности, отразить сложный
рассчитанные по модели Баркера со значениями параметров из табл. VII. 1, и найденные экспериментально, приведены на рис. VII.7; они хорошо согласуются. Модель в состоянии описать различную форму кривых, в частности, отразить сложный  -образный ход кривой
-образный ход кривой 
 передать температурную зависимость избыточных термодинамических функций, в частности,
передать температурную зависимость избыточных термодинамических функций, в частности,  (рис. VII.8). Описание термодинамических функций при одной температуре не требует введения параметров энтальпии взаимообмена, т. е. достаточным оказывается приближение
(рис. VII.8). Описание термодинамических функций при одной температуре не требует введения параметров энтальпии взаимообмена, т. е. достаточным оказывается приближение  (см. рис. VII.7).
(см. рис. VII.7).
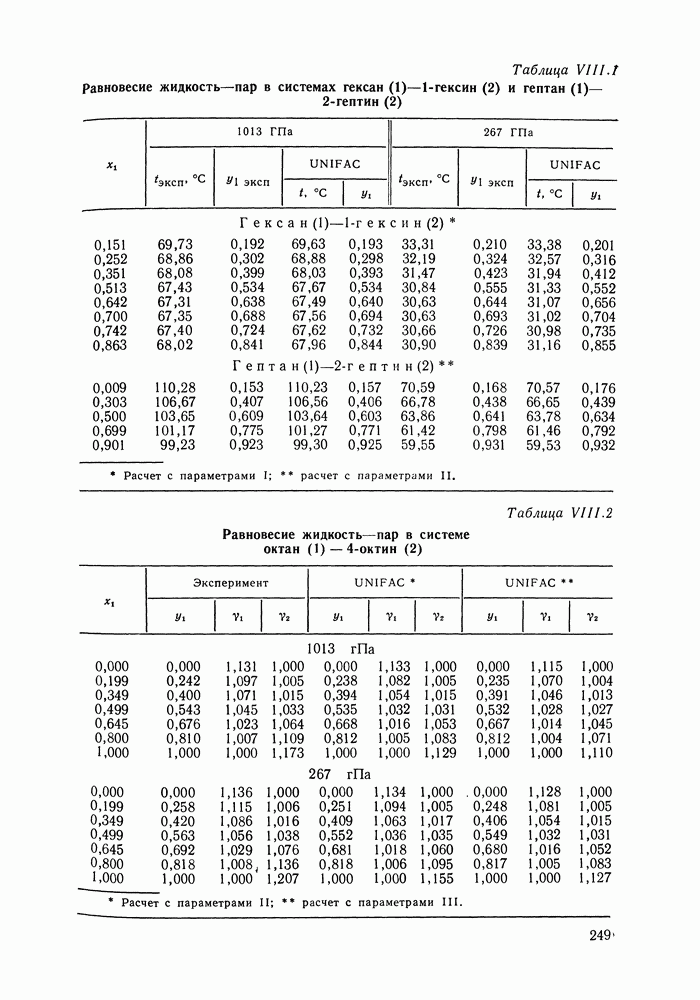
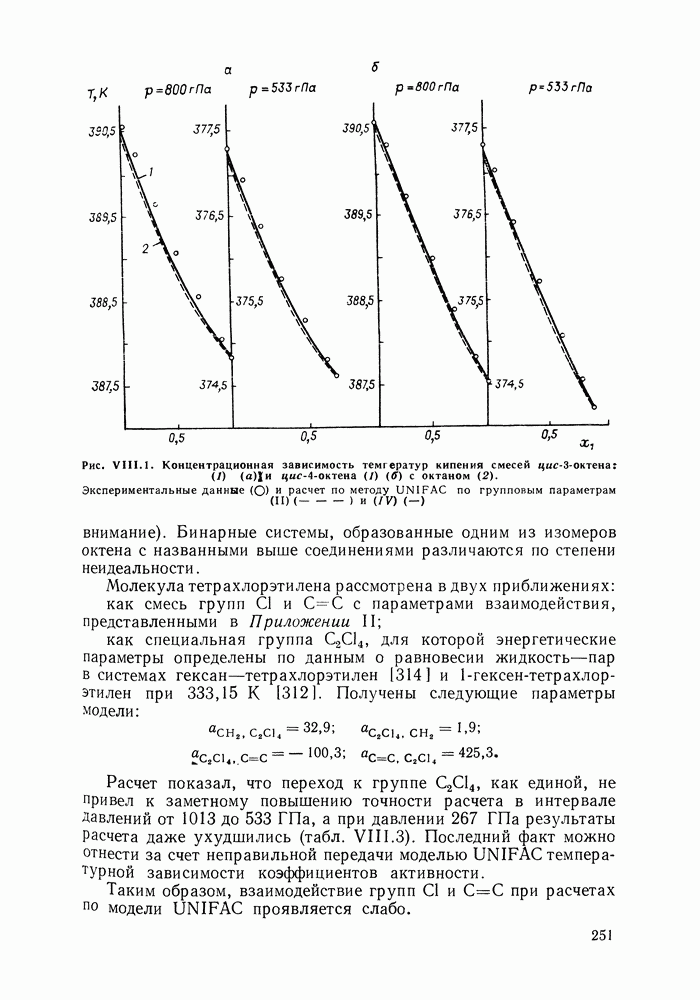
 -гексан (2), рассчитанные различными методами
-гексан (2), рассчитанные различными методами
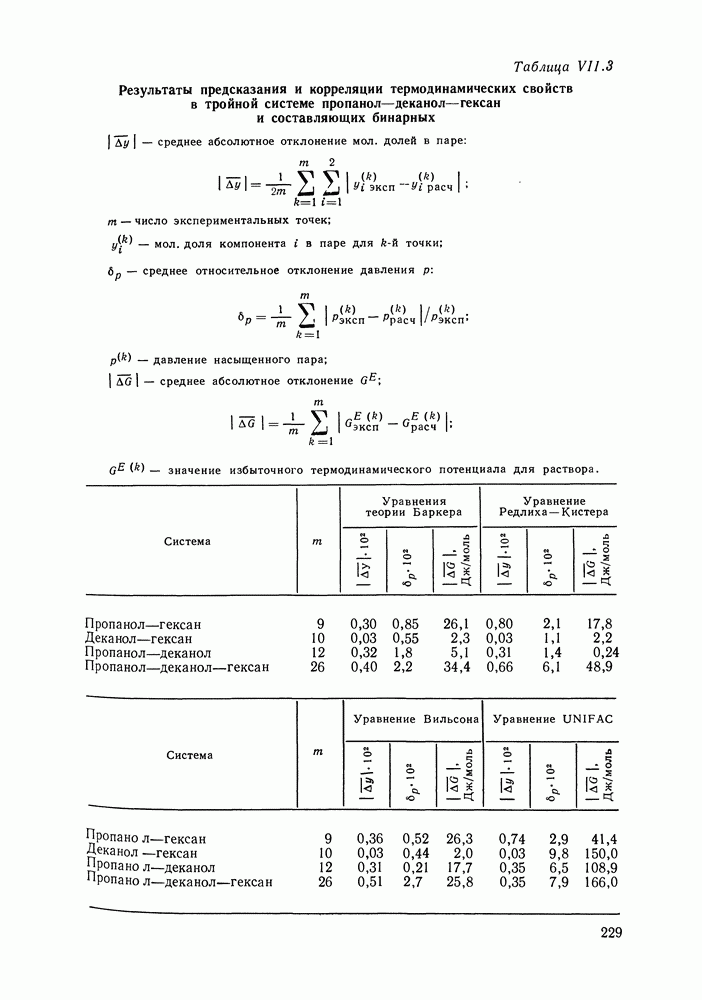
 была примерно такой же, а в некоторых случаях и лучшей, чем при использовании уравнений Редлиха — Кистера, Вильсона, UNIFAC. Иллюстрация для тройной системы пропанол — деканол — гексан приведена в табл. VII.3. Наряду с успехами в расчете фазовых равновесий к достоинствам модели Баркера следует отнести и то, что она позволяет уяснить роль таких молекулярных факторов, как размер, форма молекул на термодинамические свойства раствора, способствует уяснению представлений о структуре раствора.
была примерно такой же, а в некоторых случаях и лучшей, чем при использовании уравнений Редлиха — Кистера, Вильсона, UNIFAC. Иллюстрация для тройной системы пропанол — деканол — гексан приведена в табл. VII.3. Наряду с успехами в расчете фазовых равновесий к достоинствам модели Баркера следует отнести и то, что она позволяет уяснить роль таких молекулярных факторов, как размер, форма молекул на термодинамические свойства раствора, способствует уяснению представлений о структуре раствора.
 выразим в виде следующей системы уравнений в частных производных:
выразим в виде следующей системы уравнений в частных производных:


 уравнений с
уравнений с  неизвестными, в принципе разрешимую, нельзя решить алгебраически, так как и
неизвестными, в принципе разрешимую, нельзя решить алгебраически, так как и  нелинейны относительно искомых переменных
нелинейны относительно искомых переменных 
 разлагают в окрестности точки, соответствующей начальным или текущим значениям параметров
разлагают в окрестности точки, соответствующей начальным или текущим значениям параметров  в ряд Тейлора, ограниченный членами, содержащими производные первого порядка:
в ряд Тейлора, ограниченный членами, содержащими производные первого порядка:

 текущие приближенные значения параметров;
текущие приближенные значения параметров;  поправки к ним.
поправки к ним.
 в общем случае дает лишь приближенные оценки
в общем случае дает лишь приближенные оценки  точность оценок возрастает в окрестности оптимума.
точность оценок возрастает в окрестности оптимума.
 в точке
в точке  принимают конкретные числовые значения. Следовательно, подстановка (3) в (2) с учетом (VII. 149) дает систему
принимают конкретные числовые значения. Следовательно, подстановка (3) в (2) с учетом (VII. 149) дает систему 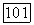 уравнений с
уравнений с 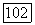 неизвестными
неизвестными 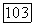 линейную относительно поправок и легко разрешимую. После необходимых преобразований эту систему можно представить в простом виде:
линейную относительно поправок и легко разрешимую. После необходимых преобразований эту систему можно представить в простом виде:


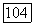 рассчитывают при значениях параметров
рассчитывают при значениях параметров 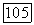
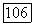 на каждом итерационном шаге решением системы (4) при текущих значениях с рассчитывают поправки
на каждом итерационном шаге решением системы (4) при текущих значениях с рассчитывают поправки 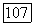 их помощью находят уточненные значения параметров:
их помощью находят уточненные значения параметров:

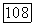 на отдельных этапах расчета приращения
на отдельных этапах расчета приращения 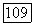 могут оказаться неоправданно большими. В этом случае производят релаксацию значений параметров: все
могут оказаться неоправданно большими. В этом случае производят релаксацию значений параметров: все 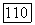 домножают на один и тот же множитель, меньший единицы (например, 0,5), и шаг (7) повторяют из исходной точки в том же направлении — но, соответственно, меньшей длины. Релаксационную методику применяют в двух случаях:
домножают на один и тот же множитель, меньший единицы (например, 0,5), и шаг (7) повторяют из исходной точки в том же направлении — но, соответственно, меньшей длины. Релаксационную методику применяют в двух случаях:
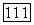 уравнения NRTL разумно положить
уравнения NRTL разумно положить 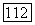 Для приведенных параметров уравнения Вильсона, определяемых уравнением (VII.119), должно выполняться
Для приведенных параметров уравнения Вильсона, определяемых уравнением (VII.119), должно выполняться 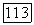 Релаксацию расчетных
Релаксацию расчетных 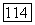 при фиксированных
при фиксированных 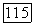 при необходимости, производят несколько раз — до возвращения траектории поиска в область допустимых значений параметров. Лишь после этого можно приступить к расчету значения
при необходимости, производят несколько раз — до возвращения траектории поиска в область допустимых значений параметров. Лишь после этого можно приступить к расчету значения 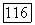 при новых значениях
при новых значениях 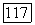
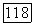 не станут меньше, чем на предыдущем итерационном шаге. При приближении к точке
не станут меньше, чем на предыдущем итерационном шаге. При приближении к точке 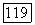 такое рано или поздно случится, так как локально, в окрестности этой точки, направление поиска оценивается верно.
такое рано или поздно случится, так как локально, в окрестности этой точки, направление поиска оценивается верно.
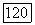 и приступают к дальнейшему уточнению параметров. Расчет заканчивают тогда, когда на данном шаге приращения параметров и изменение минимизируемой функции окажутся меньше заданных погрешностей величин
и приступают к дальнейшему уточнению параметров. Расчет заканчивают тогда, когда на данном шаге приращения параметров и изменение минимизируемой функции окажутся меньше заданных погрешностей величин 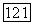
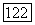 составляет от 5 до 15. При численном расчете производных число итераций выше. Скорость сходи мости падает с уменьшением степени неидеальности системы.
составляет от 5 до 15. При численном расчете производных число итераций выше. Скорость сходи мости падает с уменьшением степени неидеальности системы.
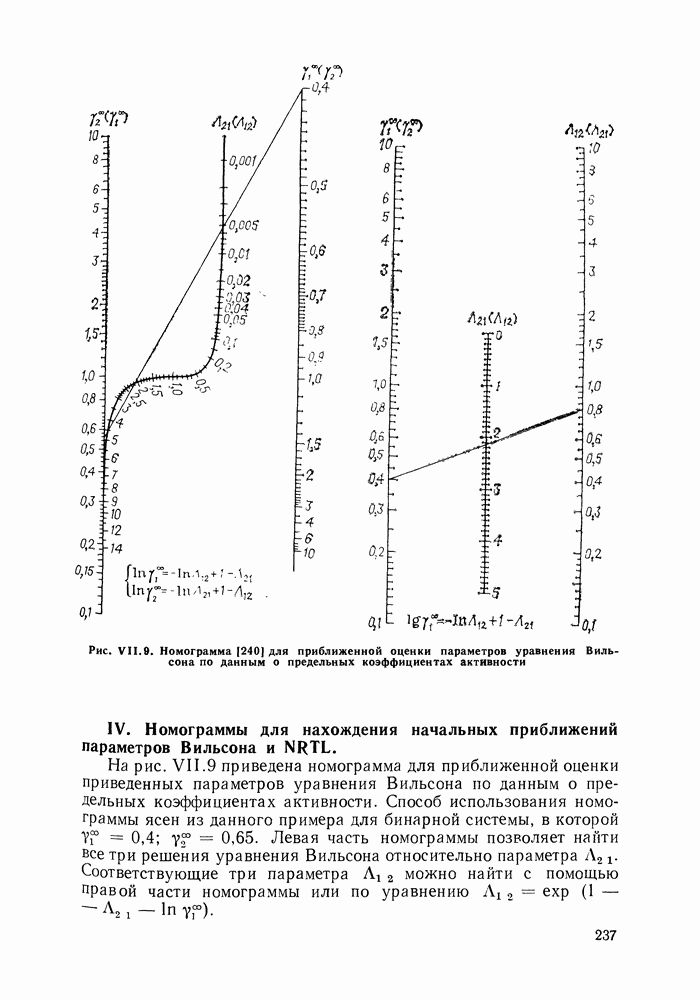
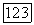 для приближенной оценки параметров уравнения Вильсона по данным о предельных коэффициентах активности
для приближенной оценки параметров уравнения Вильсона по данным о предельных коэффициентах активности
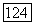 Левая часть номограммы позволяет найти все три решения уравнения Вильсона относительно параметра
Левая часть номограммы позволяет найти все три решения уравнения Вильсона относительно параметра 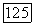 Соответствующие три параметра
Соответствующие три параметра 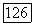 можно найти с помощью правой части номограммы или по уравнению
можно найти с помощью правой части номограммы или по уравнению 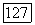
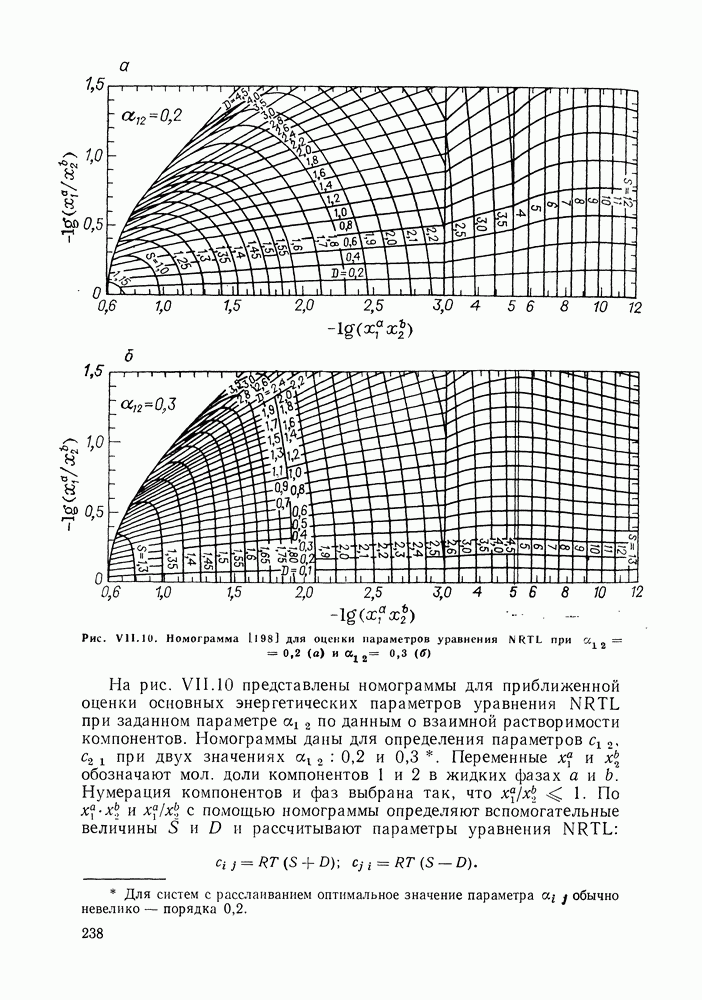
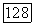 для оценки параметров уравнения NRTL при
для оценки параметров уравнения NRTL при 
 по данным о взаимной растворимости компонентов. Номограммы даны для определения параметров
по данным о взаимной растворимости компонентов. Номограммы даны для определения параметров  при двух значениях
при двух значениях  и
и 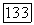 Переменные
Переменные 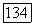 обозначают мол. доли компонентов 1 и 2 в жидких фазах
обозначают мол. доли компонентов 1 и 2 в жидких фазах 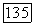 Нумерация компонентов и фаз выбрана так, что
Нумерация компонентов и фаз выбрана так, что  По
По 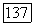 с помощью номограммы определяют вспомогательные величины
с помощью номограммы определяют вспомогательные величины  и рассчитывают параметры уравнения NRTL:
и рассчитывают параметры уравнения NRTL:
