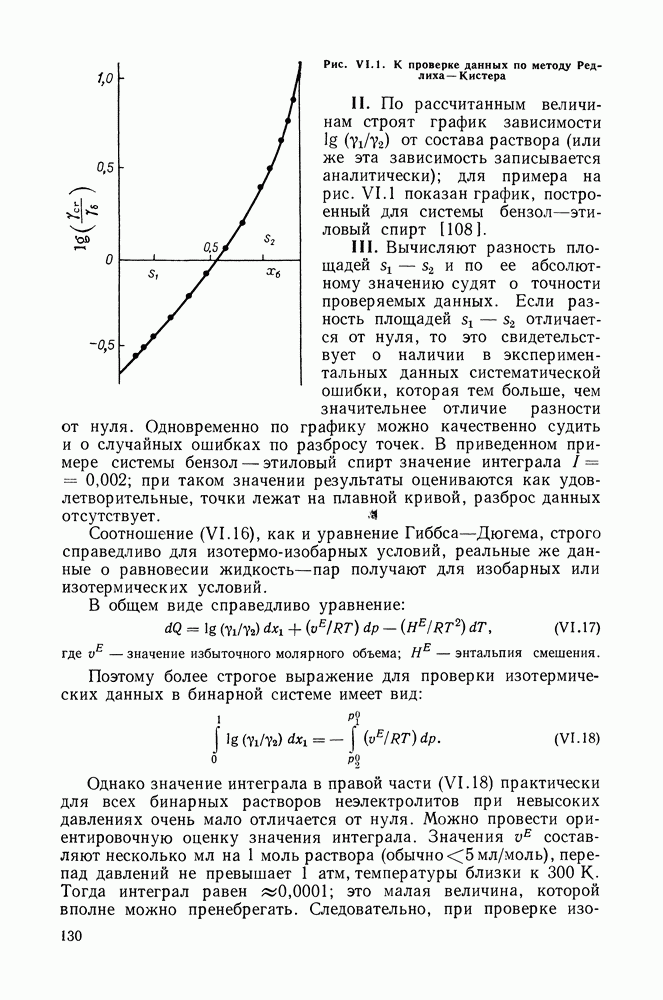
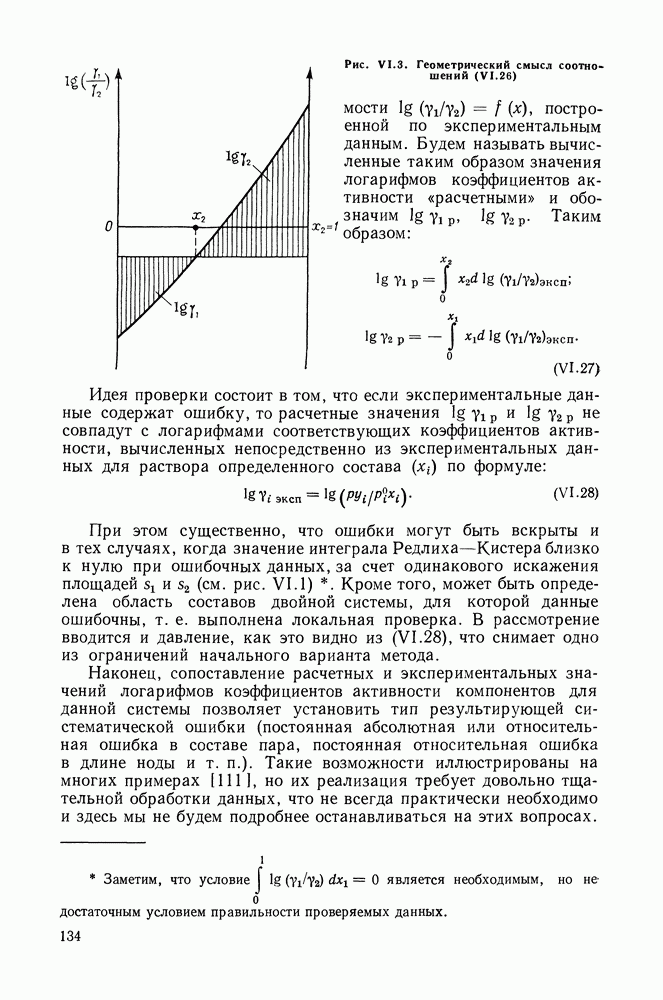
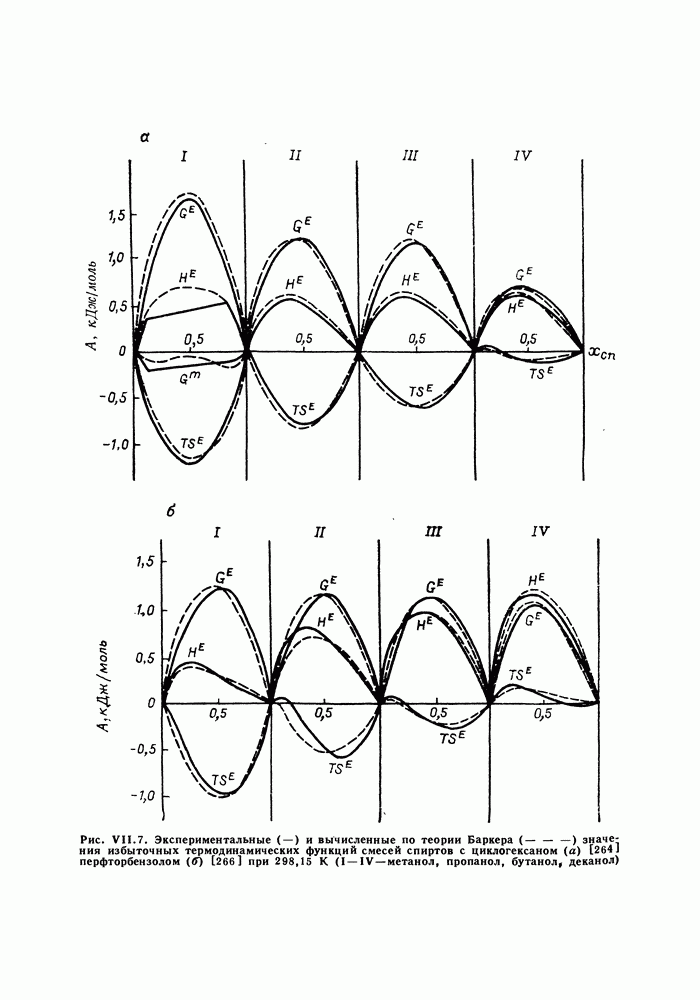
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
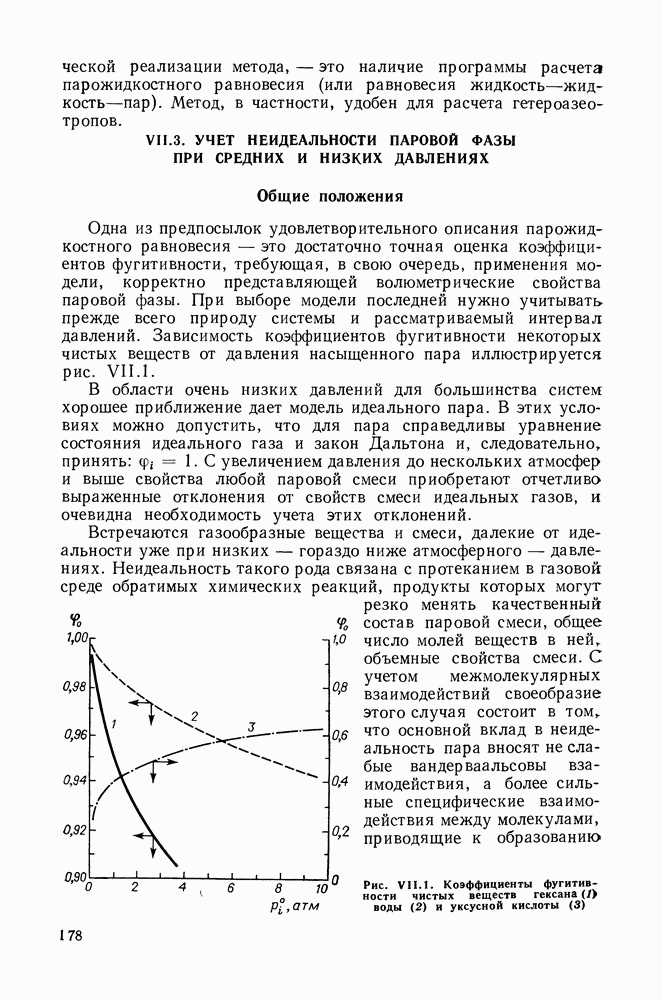
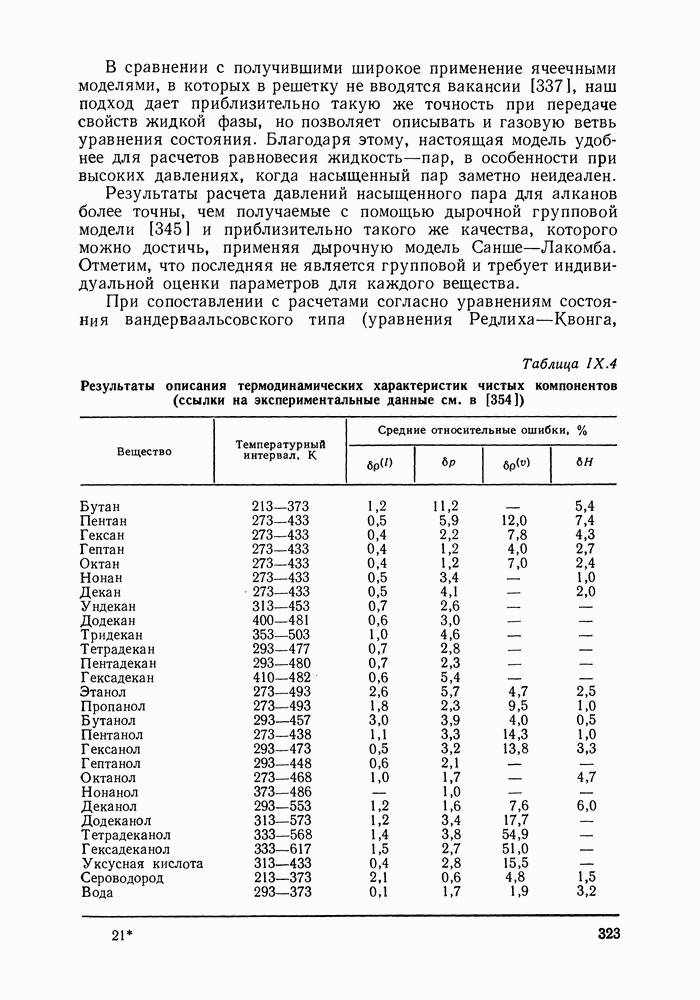
VII.3. УЧЕТ НЕИДЕАЛЬНОСТИ ПАРОВОЙ ФАЗЫ ПРИ СРЕДНИХ И НИЗКИХ ДАВЛЕНИЯХОбщие положения Одна из предпосылок удовлетворительного описания парожидкостного равновесия — это достаточно точная оценка коэффициентов фугитивности, требующая, в свою очередь, применения модели, корректно представляющей волюметрические свойства паровой фазы. При выборе модели последней нужно учитывать прежде всего природу системы и рассматриваемый интервал давлений. Зависимость коэффициентов фугитивности некоторых чистых веществ от давления насыщенного пара иллюстрируется рис. VII.1. В области очень низких давлений для большинства систем хорошее приближение дает модель идеального пара. В этих условиях можно допустить, что для пара справедливы уравнение состояния идеального газа и закон Дальтона и, следовательно, принять: Встречаются газообразные вещества и смеси, далекие от идеальности уже при низких — гораздо ниже атмосферного — давлениях. Неидеальность такого рода связана с протеканием в газовой среде обратимых химических реакций, продукты которых могут резко менять качественный состав паровой смеси, общее число молей веществ в нейг объемные свойства смеси. С учетом межмолекулярных взаимодействий своеобразие этого случая состоит в том что основной вклад в неидеальность пара вносят не слабые вандерваальсовы взаимодействия, а более сильные специфические взаимодействия между молекулами, приводящие к образованию
Рис. VII.1. Коэффициенты фугитивности чистых веществ гексана (1) воды (2) и уксусной кислоты (3) относительно устойчивых соединений — ассоциатов. Известный пример соединений, сильно ассоциирующих в парах и важных в практическом отношении — карбоновые кислоты, которые в парообразном состоянии образуют связанные водородной связью относительно устойчивые димеры. Соотношение вкладов в неидеальность паровой фазы вандер-ваальсовых взаимодействий и ассоциации приходится учитывать при выборе модели пара. Явление ассоциации, типичное для жидкости, в паровой фазе обычно или отсутствует, или выражено слабо. Свойства такого пара адекватно описываются вириальным уравнением состояния. Учет неидеальности сильно ассоциированного пара требует специального подхода, учитывающего химическую сущность ассоциативных равновесий, что, к сожалению, приводит к разнообразию моделей паровой фазы. При давлениях порядка атмосферного и пониженных в сильно ассоциированных парах основной вклад в их свойства вносит ассоциация, и во внимание можно принять только ее. Последнее приводит к модели так называемой идеальной ассоциированной смеси — идеальной смеси всех присутствующих в паре мономеров и ассоциатов. При повышенных давлениях вкладом неспецифических взаимодействий, действующих между всеми присутствующими в паре молекулярными образованиями, уже нельзя пренебречь. Его можно оценить методами, развитыми для неассоциированных смесей. Рассмотрим методы расчета коэффициентов фугитивности при использовании различных моделей паровой фазы. Расчет коэффициентов фугитивности на основе вириального уравнения состоянияВириальное уравнение состояния [170] выражает зависимость коэффициента сжимаемости
Оба бесконечных ряда эквивалентны, а их коэффициенты однозначно связаны:
Коэффициенты Вириальное разложение, предложенное первоначально как эмпирическое, поддается строгой молекулярно-статистической трактовке [171], в рамках которой второй вириальный коэффициент обусловлен парными взаимодействиями молекул, третий — тройными взаимодействиями — и так далее, а концентрационная зависимость вириальных коэффициентов смеси имеет вид:
Здесь В теоретическом обосновании вириального уравнения состоит его главнее преимущество перед другими уравнениями состояния. Так, большое практическое значение имеет возможность связать через вириальные коэффициенты макро- и микросвойства фазы: экспериментальные данные о вириальных коэффициентах позволяют получить сведения о потенциалах межмолекулярного взаимодействия, так же как молекулярные характеристики Ееществ могут служить для оценки величин вириальных коэффициентов, а с ними и объемных свойств газов. Последнее реализуется, в частности, в некоторых корреляциях для второго вириального коэффициента, нашедших применение при расчетах парожидкостного равновесия. Выгодно отличает вириальное уравнение наличие строгого соотношения для смесей (VII.58) и (VII.59), а также имеющаяся определенность относительно области применимости уравнения, его экстраполяционных возможностей. Вириальное уравнение состояния имеет и свои недостатки, которые достаточно полно изучены и сформулированы. Область его применимости — математически — ограничена радиусом сходимости степенного ряда. Сходимость в соответствии с формой уравнения ухудшается с увеличением плотности. С учетом молекулярной теории применимость уравнения ограничена областью состояний, в которой справедливо представление о молекулах, как об относительно свободных, сохраняющих свою индивидуальность частицах. Вириальное уравнение неприменимо для конденсированных фаз, в области сверхвысоких давлений, приводящих к деформации электронных оболочек молекул и атомов, в области очень высоких температур, при которых происходит ионизация газа, и в случае протекания в газе химических реакций. Для расчета парожидкостного равновесия в области средних и низких давлений существенно последнее ограничение: вириальное уравнение не годится для описания сильно ассоциированного пара. Соотношения (VII.57) — (VII.59) являются строгими для бесконечного вириального ряда, на практике же приходится использовать его усеченную форму, применимость которой зависит от того, насколько значимы в рассматриваемой области состояний опущенные слагаемые. Вопрос о числе слагаемых вириального ряда, необходимых для описания газа с заданной точностью при различных внешних условиях, исследован в работах [172]. При расчете парожидкостного равновесия свойства пара обычно аппроксимируются вириальным уравнением только со вторым вириальным коэффициентом, так как экспериментальные данные о третьем, и тем более высших, вириальных коэффициентах, труднодоступны, а имеющиеся корреляции [1731 для третьего вириального коэффициента описывают лишь неполярные вещества. Выбрав в качестве независимой переменной давление, а не объем, используем в дальнейшем вириальный ряд по степеням давления. Ограничившись двумя первыми его слагаемыми и приняв во внимание (VII.57), вириальное уравнение состояния со вторым вириальным коэффициентом можем записать так:
Это уравнение обеспечивает вполне удовлетворительную точность расчета коэффициентов фугитивности при плотностях пара менее половины критического значения. Праусницем [174] предложен способ ориентировочной оценки области давлений, в которой применимо уравнение (VII.60), на основе критических свойств чистых компонентов:
Уравнения (VI1.58) и (VI1.60) вместе с общим определением парциальных молярных величин, приводят к следующему выражению для парциального молярного объема:
Из (VII. 11) и (VII.60) следует рабочее соотношение для расчета коэффициентов фугитивности чистых веществ
а из (VII.10) и (VI1.62) — соотношения для коэффициентов фуги тивности компонентов паровой смеси
В случае бинарной системы
где
С учетом
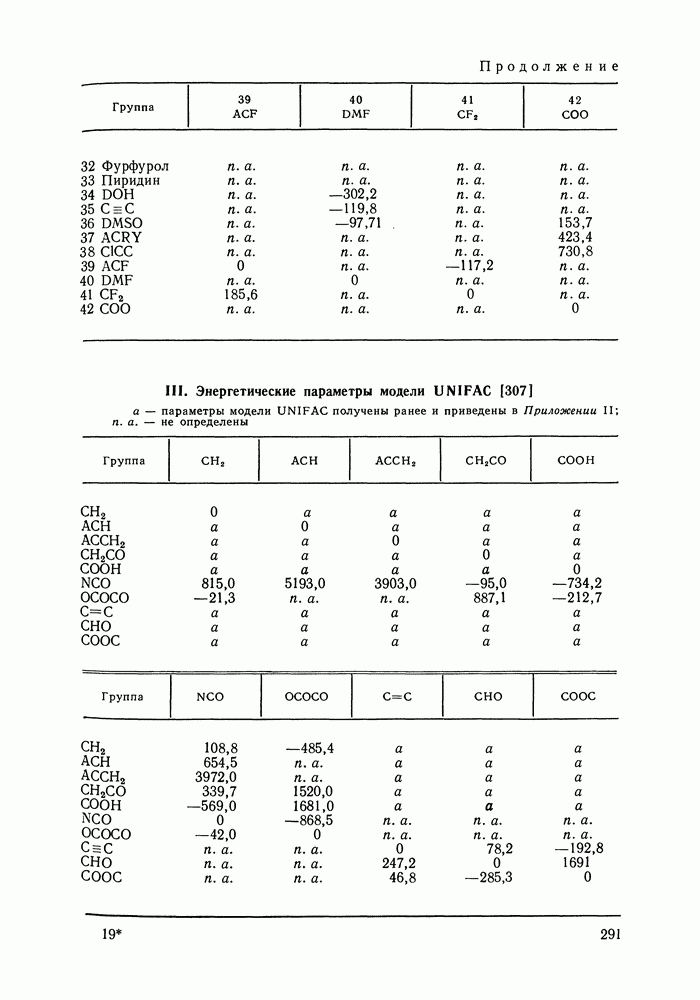
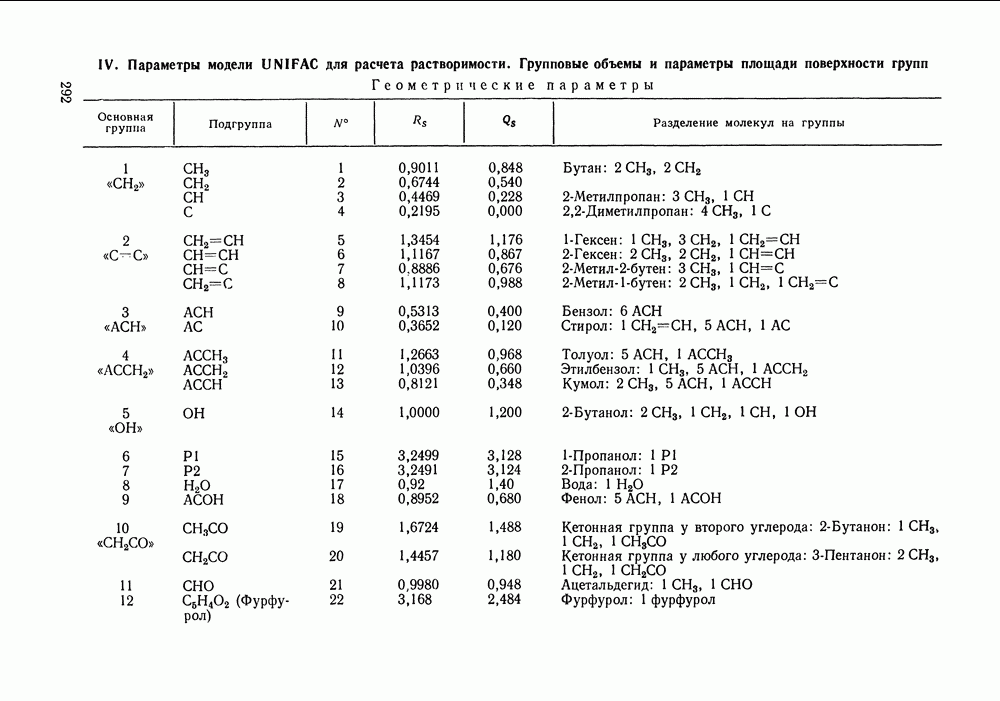
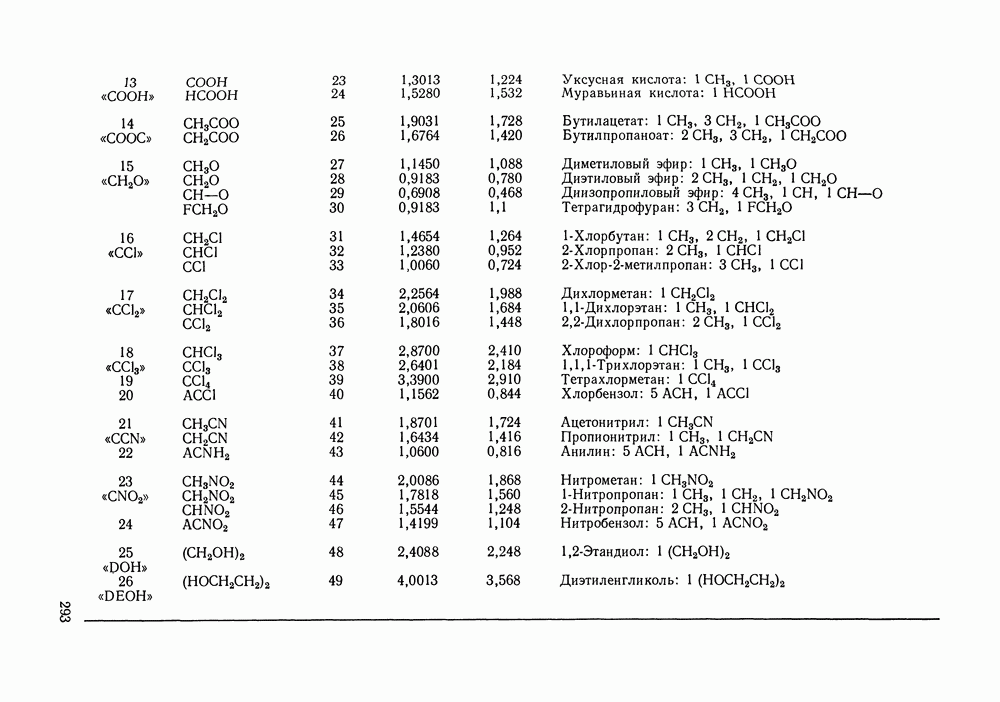
которое в отличие от (VII.24) не разделяет вклады При расчете по вышеприведенным формулам необходимо знать вторые вириальные коэффициенты. Имеющиеся сводки экспериментальных значений этих величин [16, 17, 175, с. 212] и их публикации в отдельных работах, к сожалению, не могут удовлетворить всех потребностей, возникающих при описании разнообразных технологических смесей. Особенно ограничены данные о смешанных вириальных коэффициентах Для смесей некоторых неполярных веществ коэффициент При описании парожидкостного равновесия в системах разнообразной природы значения коэффициентов сочетании с эмпирически подобранными комбинационными правилами для смесей, позволяет оценивать не только Последние две корреляции сравнивались в работе [167, стр. 347]. Результаты сравнения показывают, что относительные погрешности предсказания второго вириального коэффициента при использовании обеих корреляций в большинстве случаев сопоставимы и обычно составляют от долей до нескольких процентов. Однако для отдельных веществ корреляция Цонопулоса дает грубую ошибку в расчетном Применение описанного метода учета неидеальности пара дает несомненное улучшение качества корреляции и предсказания парожидкостного равновесия в случае повышенных давлений. При атмосферном или пониженном давлении ответ на вопрос о необходимости такого учета не является однозначным. Уже при атмосферном давлении при отсутствии в паре сильной ассоциации коэффициенты В различных системах при одном и том же давлении коэффициенты к тому, что дает модель идеального пара, и будет чисто формальным. Учет неидеальности пара при атмосферном давлении вряд ли принесет ощутимую пользу, если высокая точность описания просто не требуется (например, при моделировании ректификации в легко разделяемой системе), или, если не точны исходные данные о чистых веществах и смесях. Влияние учета неидеальности пара на его вычисленный состав, когда расчет не осложнен экспериментальными ошибками, исследовано в работе [183] на примере почти полутора сотен наборов «псевдоэкспериментальных» данных о равновесии жидкость—пар—термодинамически согласованных данных, аппроксимирующих реальные экспериментальные зависимости в системах различного типа. Показано, что погрешность вычисления состава паровой фазы, вызванная допущением Такой компенсации не происходит, когда задача состоит в нахождении по данным о равновесии жидкость—пар значений термодинамических функций. При решении этой задачи поправка на неидеальность пара должна вноситься по возможности во всех случаях. Вопрос о влиянии модели пара на точность расчета термодинамических функций систем подробно рассматривался Белоусовым и Морачевским [105]. Авторы, в частности, показали, что при атмосферном давлении погрешность, вносимая допущением Корреляция Хайдена-О’Коннелла для вторых вириальных коэффициентовСреди методов расчета второго вириального коэффициента, пригодных для применения в технологической практике, метод Хайдена-О'Коннелла [180] является, по-видимому, наиболее удовлетворительным как по точности, так и по универсальности, применимости ко многим веществам и смесям. По точности для неполярных и слабополярных веществ этот метод сопоставим с другими [176—179], но превосходит их в случае применения к веществам с отчетливо выраженными полярными свойствами. Исходными данными для расчета вириального коэффициента Параметры ассоциации и сольватации, отражающие стремление молекул газообразных веществ к образованию однородных и, соответственно, смешанных ассоциатов, носят эмпирический характер, и в общем случае должны быть оценены по имеющимся экспериментальным данным о вторых вириальных коэффициентах. Существенно, однако, то, что в корреляции эти параметры обычно близки для всего класса веществ или смесей, объединенных наличием тех или иных полярных групп. Последнее говорит о предсказательных возможностях корреляции: параметры ассоциации (сольватации), найденные по свойствам отдельных представителей данного класса веществ (смесей), могут быть использованы для приближенного расчета свойств других представителей того же класса. При выводе корреляции использованы трехпараметрическая теория соответственных состояний и ряд результатов молекулярностатистической теории газов, в частности, идеи работ [184], а также накопленный экспериментальный материал о вторых вириальных коэффициентах. Исходя из того, что различные виды межмолекулярных сил по-разному вносят свой вклад в свойства газа, авторы корреляции представили второй вириальный коэффициент как сумму следующих составляющих:
где Величина
а величину
Первых два слагаемых отражают эффект слабой ассоциации (сольватации) веществ, последнее слагаемое необходимо только для учета сильной ассоциации в газовой фазе карбоновых кислот. Константа димеризации карбоновой кислоты (определение константы смотри в следующем разделе) связана с величиной В соотношением:
которое может служить для оценки Опуская подробности вывода корреляционных соотношений, приведем ниже их полный перечень — в последовательности, соответствующей последовательности расчета. Ряд числовых коэффициентов, фигурирующих в формулах оригинальной работы [180], изменен в соответствии с более поздним изложением корреляции [144]. Из переменных, являющихся промежуточными при расчете, укажем только: Вычисления, которые необходимо проделать, достаточно громоздки и требуют применения ЭВМ. В случае I. Расчет переменных
II. Расчет переменных
Значение В обоих случаях
Для всех остальных сочетаний значений
III. Дальнейшие расчетные соотношения для чистых веществ
Все найденные к настоящему моменту величины зависят от природы смеси, но не зависят от параметров ее состояния. Следовательно, при расчетах в пределах одной системы их достаточно вычислить один раз. IV. Расчет величин, зависящих от температуры смеси:
Результирующие выражения для вкладов в вириальный коэффициент имеют вид:
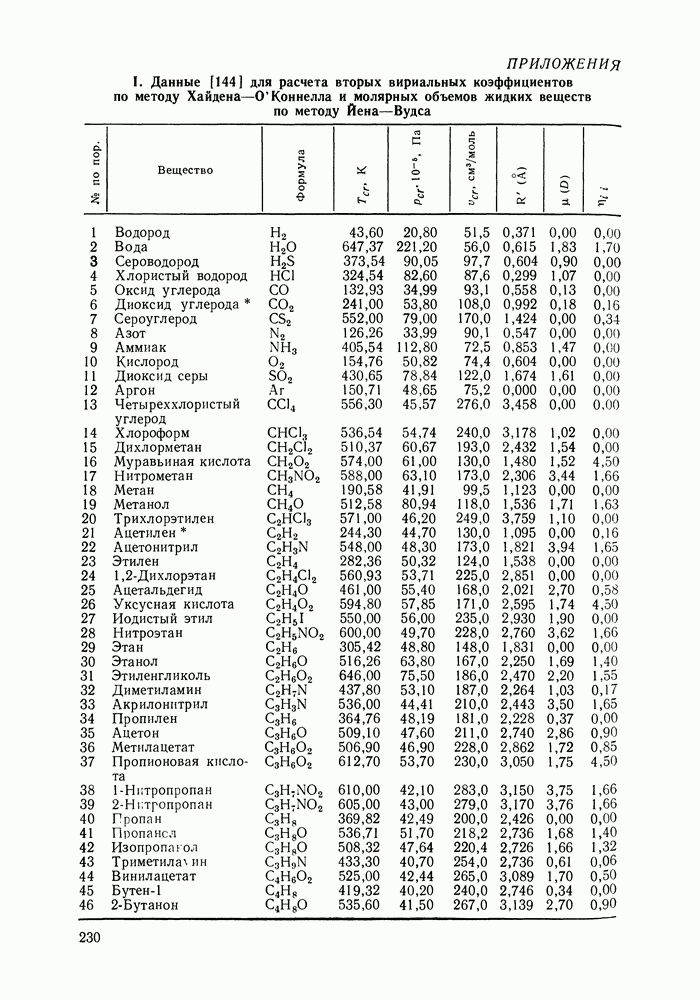
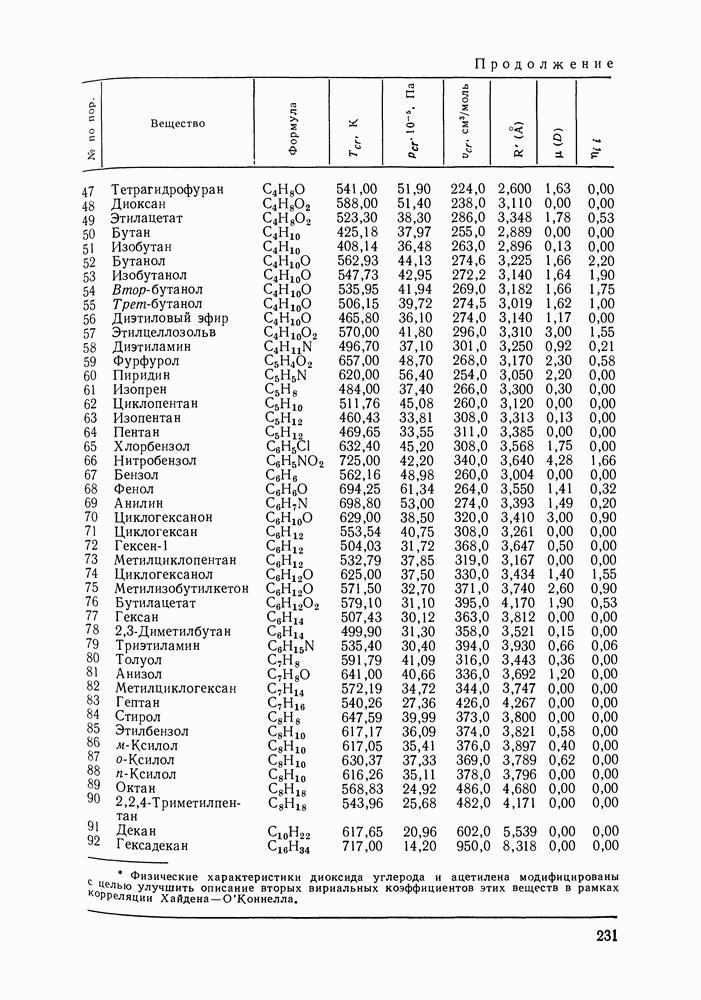
Подстановка (VI1.74) - (VII.77) в (VI1.70) - (VI1.72) дает вклады Данные для расчета вторых вириальных коэффициентов по методу Хайдена-О’Коннелла для 92 распространенных веществ приведены в Приложениях I и II; там же помещены значения критических объемов веществ, необходимые для оценки их жидкофазных молярных объемов методом Йена и Вудса (см. разд. VII. 1). Для веществ, не представленных в Приложении I, необходимую информацию часто можно найти в различной справочной литературе. Так, сведения об экспериментальных значениях критических параметров и дипольных моментах веществ, а также методах их расчета по групповым вкладам содержатся, например, в [148, 185—187]. Средний радиус вращения [188], определяемый главными моментами инерции
Максимальная ошибка в оценке углеводородов с полярными соединениями В системах с выраженными специфическими взаимодействиями компонентов параметры ассоциации и сольватации обычно принимают значения от нескольких десятых до 1—2 единиц. При отсутствии сведений Учет неидеальности сильно ассоциированного параИз распространенных органических растворителей сильно ассоциируют в паровой фазе карбоновые кислоты и их замещенные, причем значительная ассоциация молекул кислот происходит в широком интервале температур и давлений, включая область весьма низких давлений. Метод учета неидеальности сильно ассоциированного пара принципиально отличается от описанного выше метода, использующего вириальное уравнение состояния. Сильная ассоциация молекул приводит к тому, что истинное число молей веществ в смеси существенно меньше, чем определяемое обычным анализом. Аналитический состав отражает суммарное число свободных и связанных мономерных частиц во всех присутствующих в паре молекулярных образованиях. Расчеты на его основе без учета эффекта ассоциации приводят к грубым ошибкам в значениях термодинамических функций системы. Теоретически обоснованную и физически ясную интерпретацию свойств ассоциированной смеси можно дать, исходя из ее истинного состава. Последний играет в свойствах равновесной ассоциированной смеси такую же роль, что и обычный состав — в свойствах неассоциированной смеси. Истинный состав можно однозначно вывести из аналитического, если известны протекающие в системе химические реакции и константы их равновесия. При изменении внешних условий ассоциативные равновесия смещаются, в соответствии со значениями констант равновесия. Это приводит к изменению истинного состава смеси и, как следствие, своеобразному характеру изменения ее макроскопических свойств. Специфика данного случая отражена в теории ассоциативных равновесий. Приложению теории ассоциативных равновесий к практическим расчетам свойств равновесных фаз уделено достаточно много внимания (укажем, например, работы [86, 113; 175, с. 166; 193, 194]). Наиболее полные результаты получены для случая идеальной ассоциированной смеси, модель которой хорошо описывает свойства ассоциированных систем при атмосферном и пониженном давлении. Покажем, как применить эту модель к расчету равновесий между жидкостью и ассоциированным паром. Рассмотрим Поскольку при умеренных давлениях ассоциация сводится преимущественно к образованию димерных молекулярных форм, ограничимся случаем димеризации. Таким образом, в n-компонентном паре в общем случае протекает
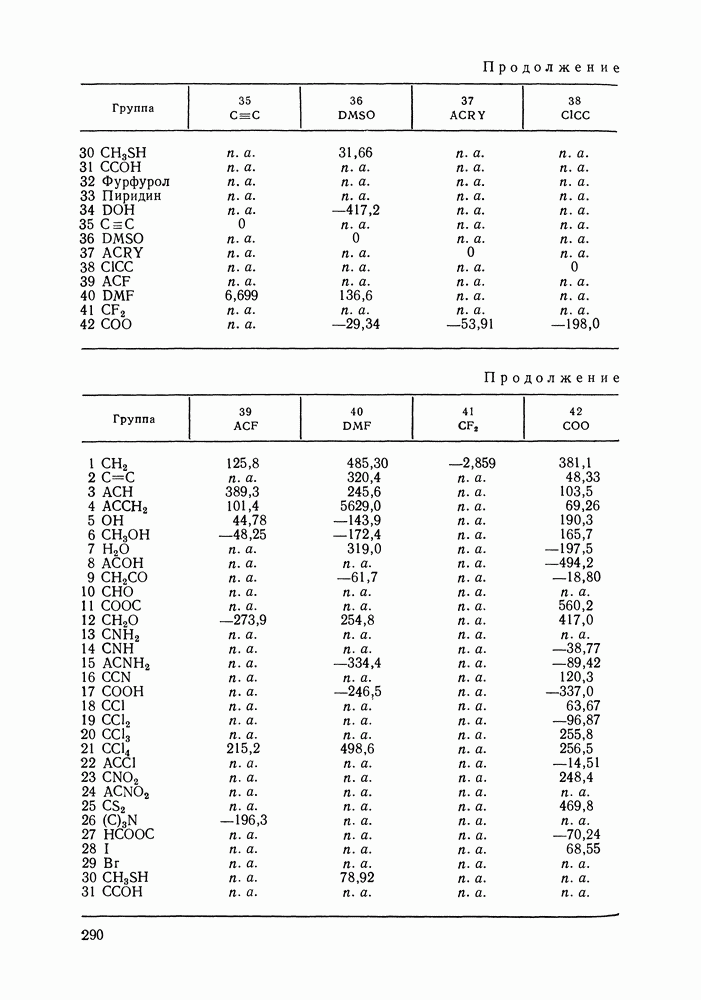
где В димере вид молекул Парциальные давления Если значения констант димеризации известны, то по парциальным давлениям мономеров легко рассчитать парциальные давления димерных форм:
затем найти общее давление пара
и истинные мол. доли
Истинные мол. доли подчиняются следующему уравнению материального баланса:
Чтобы связать найденные параметры с практически используемыми аналитическими концентрациями, подсчитаем общее число
Уравнение связи
Аналитический состав можно выразить непосредственно через парциальные давления молекулярных форм. Из (VII.81), (VII.83) и (VII.84) следует:
В то время как аналитический состав смеси однозначно определяется истинным составом, последний зависит не только от аналитического, но также от температуры и давления смеси. Уравнения (VII.79)-(VII.85) позволяют найти истинные мол. доли или парциальные давления молекулярных форм по При расчете парожидкостного равновесия необходимо определить фугитивности компонентов пара и стандартные фугитивности компонентов жидкости. Связь фугитивности компонента с параметрами молекулярных форм можно установить, если принять во внимание строго установленный в термодинамике ассоциативных равновесий [5] факт равенства химического потенциала ассоциирующего компонента химическому потенциалу его мономера
С учетом выражения, связывающего фугитивность с химическим потенциалом, и совпадения стандартных состояний для компонента в целом и мономера (идеально-разреженный газ полностью диссоциирован до мономерной формы) получаем аналогичное важное выражение для фугитивности:
В случае идеального ассоциированного пара и в пренебрежении поправкой Пойнтинга, при симметричной нормировке коэффициентов активности строгие выражения (VII.87) приводятся к виду:
где Условия фазового равновесия (VII.2) с учетом (VII.9) и (VII.88) в случае идеального ассоциированного пара можно представить в следующей форме:
Откуда:
Значение
Для неассоциирующего в чистом состоянии компонента
При расчете давления и состава пара по составу и температуре жидкости в первую очередь вычисляют коэффициенты активности (по модели жидкой фазы) и давления паров чистых компонентов при заданной температуре. Далее расчет ведут последовательным применением формул (VII.91) или (VII.92), (VII.89), (VII.79), (VII.80), (VII.85). Температуру кипения жидкости и состав пара по составу жидкости и общему давлению можно рассчитать итерационным методом, описанным в разд. VII.2, выполняя на каждом итерационном шаге по температуре приведенную выше последовательность операций. Расчет коэффициентов активности по уравнению (VII.90) требует решения системы уравнений (VII.80), (VII.85) относительно Приведенный выше способ описания равновесия между жидкостью и идеальным ассоциированным паром не связан с применением коэффициентов фугитивности. В некоторых случаях, однако, введение этих величин целесообразно. Например, при желании проводить расчет в рамках стандартных алгоритмов, использующих формулы (VII.20)-(VII.24). Из определения (VII.8) и соотношений (VII.15) и (VII.88) следует:
Нетрудно заметить, что для всех неассоциирующих компонентов
где При решении однотипных задач и особенно при расчетах «вручную» изложенный обобщенный способ описания можно конкретизировать применительно к типу системы. Это позволяет устранить лишние операции с нулевыми константами Проведем такую конкретизацию модели для п-компонентной системы с одним ассоциирующим компонентом, который обозначим как компонент а. Единственный вид ассоциативного равновесия в системе — реакция самоассоциации
Введем вспомогательную величину
Когда расчет идет в направлении от параметров жидкости к параметрам пара, необходимые значения
Коэффициенты фугитивности определены уравнениями (VII.93). Коэффициенты При описании равновесий между жидкостью и ассоциированным паром необходимы значения констант ассоциации. Данные о константах димеризации карбоновых кислот собраны в [195, 196]. Их зависимость от температуры чаще всего представляют уравнением вида
где Значения Константы ассоциации для смешанных димеров, как правило, неизвестны. Часто их оценивают по константам чистых веществ на основе приближенной формулы [197, 198]:
которая для карбоновых кислот дает удовлетворительные результаты. Более точное выражение для
Для некоторых типов соединений соотношение (VII.95) может приводить к далеким от действительности результатам. Например, для смесей галогензамещенных органических кислот экспериментально определенные значения В завершение остановимся на методе описания реального ассоциированного пара, т. е. пара, в свойства которого значимый вклад вносят, наряду с ассоциацией, вандерваальсовы взаимодействия между имеющимися молекулярными образованиями. Эффектом взаимодействий этого вида нельзя пренебречь, когда ассоциированный пар находится при повышенных давлениях. Модель реального ассоциированного пара, так же как ее только что рассмотренный частный случай, опирается на представление об истинном молекулярном составе смеси, однако, вместо парциальных давлений молекулярных форм, использует их фугитивности. Так, выражение для константы димеризации имеет вид:
Константа Связь фугитивности молекулярной формы а с общим давлением и истинным составом пара формально выражают с помощью коэффициента фугитивности
Отклонения 0а от единицы обусловлены вандерваальсовыми взаимодействиями частиц ассоциированной смеси. Коэффициенты фугитивности компонентов связаны с коэффициентами фугитивности молекулярных форм и составом пара в соответствии с уравнениями (VII.8), (VII.87), (VII.98) следующим образом:
где Пусть
которая, согласно (VII.97) и (VII.98), связана с
Для чистого неассоциирующего компонента
При заданных Задача нахождения смеси с единственным ассоциирующим компонентом а решение имеет вид:
Как следует из соотношения (VII. 102), величины
Стадия, помеченная звездочкой, описана в предыдущем абзаце; она также может содержать свой итерационный цикл при фиксированных Определенные трудности при применении обсуждаемой модели связаны с оценкой величин Метод, используемый в работе [144], связывает константы Константы
Коэффициенты 0? чистых молекулярных форм (гипотетических состояний) определяются, по аналогии с коэффициентами фугитивности чистых неассоциирующих веществ, соотношением При произвольных концентрациях молекулярных форм коэффициенты
Это значительно упрощает расчет, так как коэффициенты
Существуют и принципиально иные подходы к описанию реального ассоциированного пара, применимые к системам в широком интервале давлений и температур. Так, в работе
|
 |
Оглавление
|













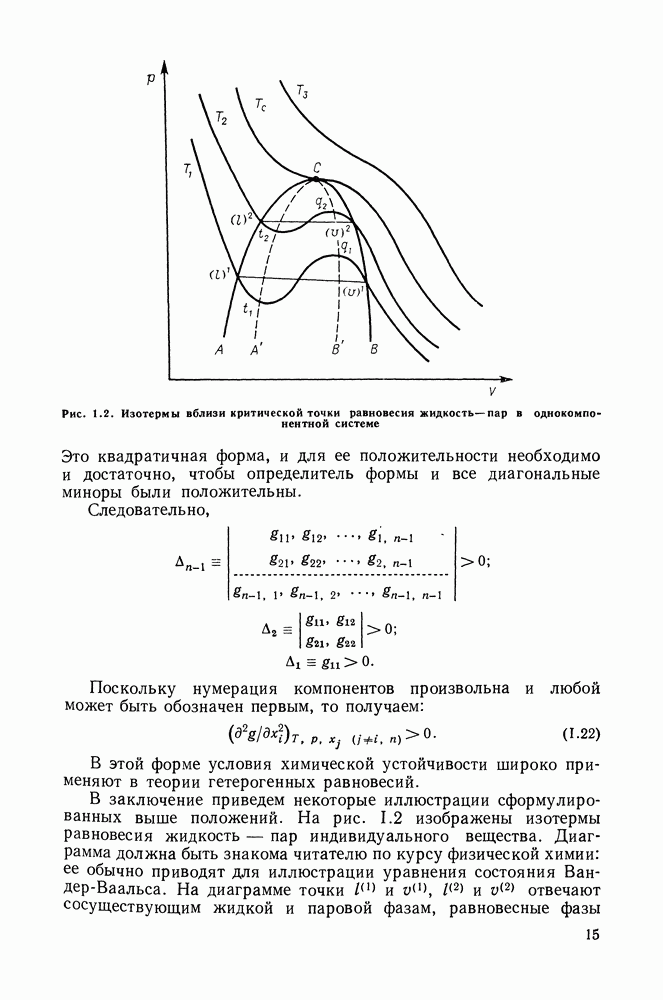






























































































































































































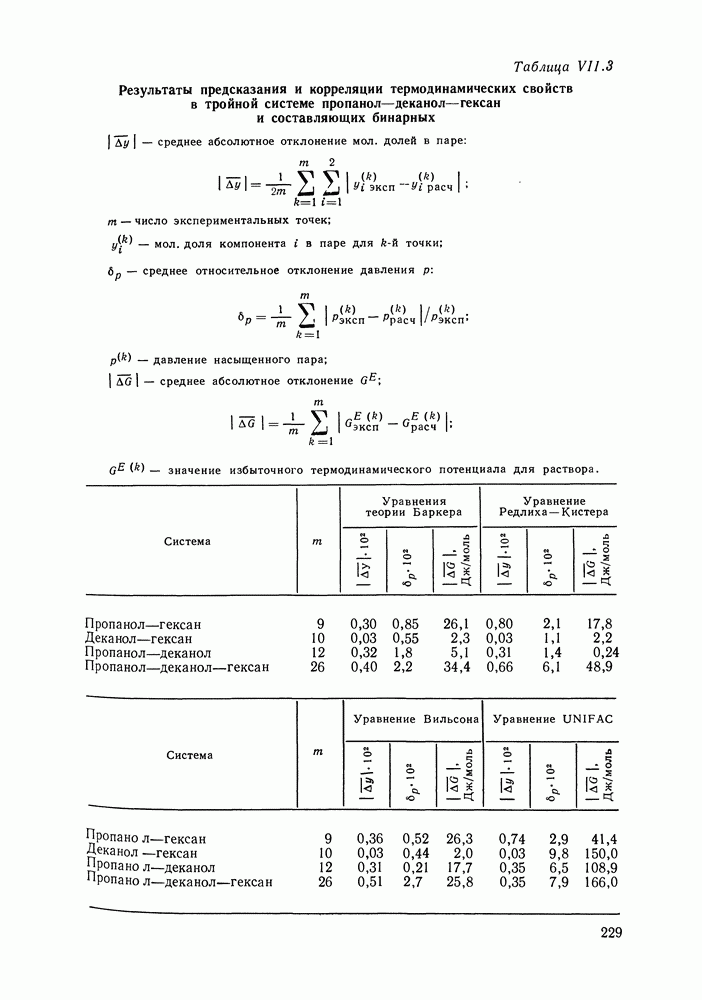















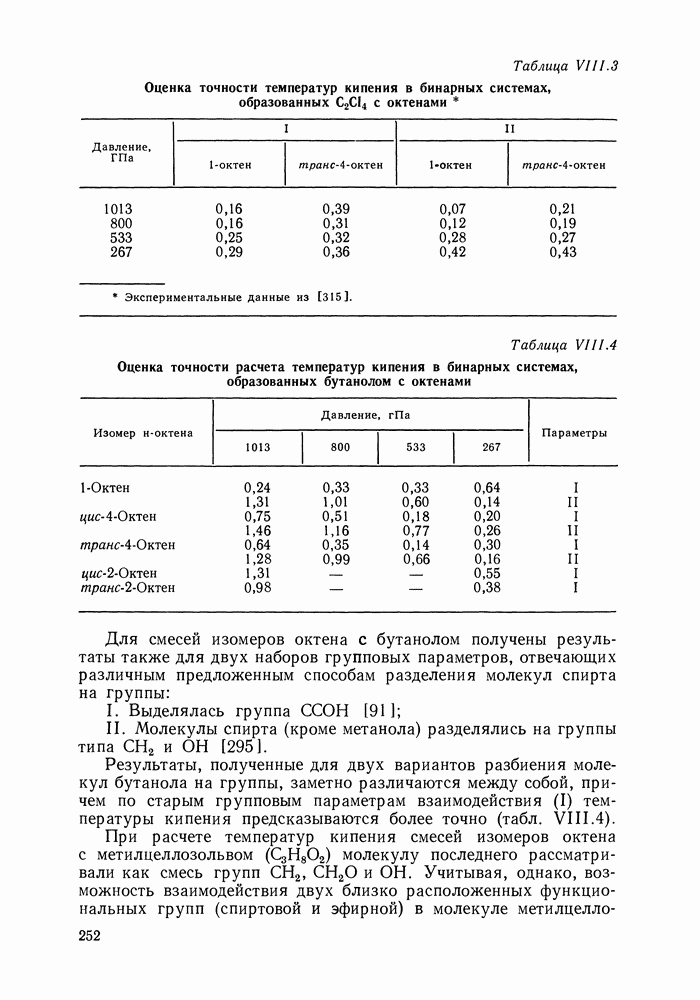
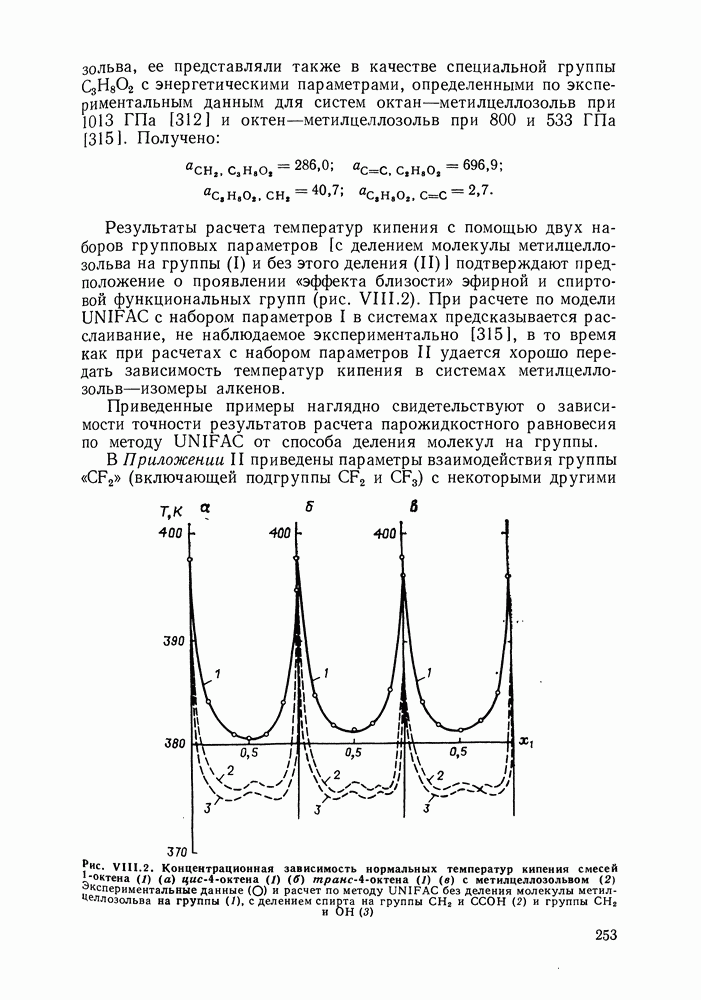

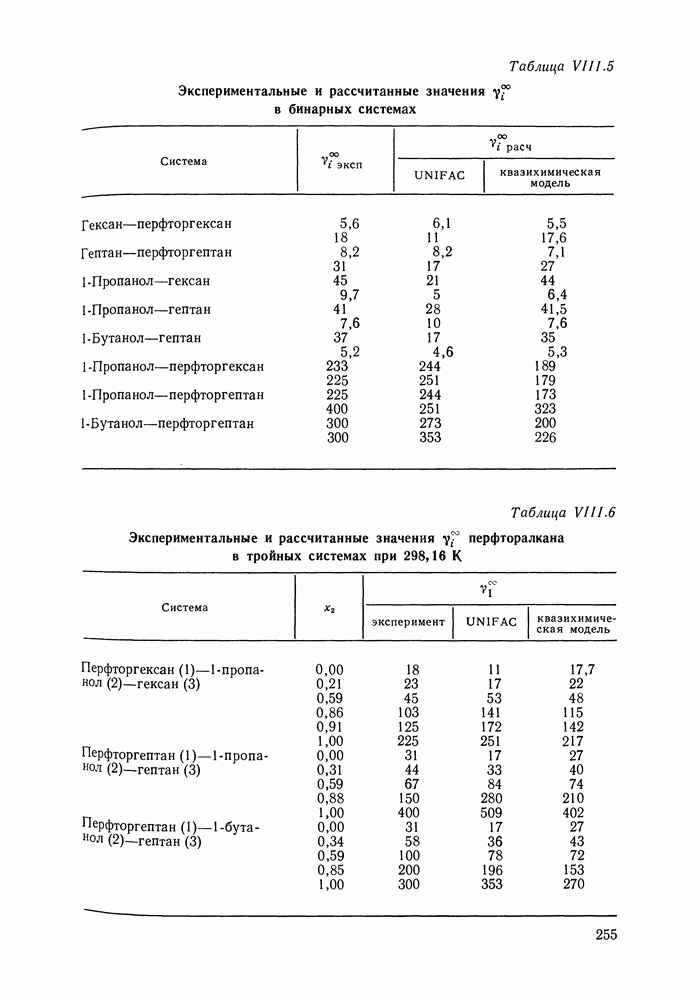


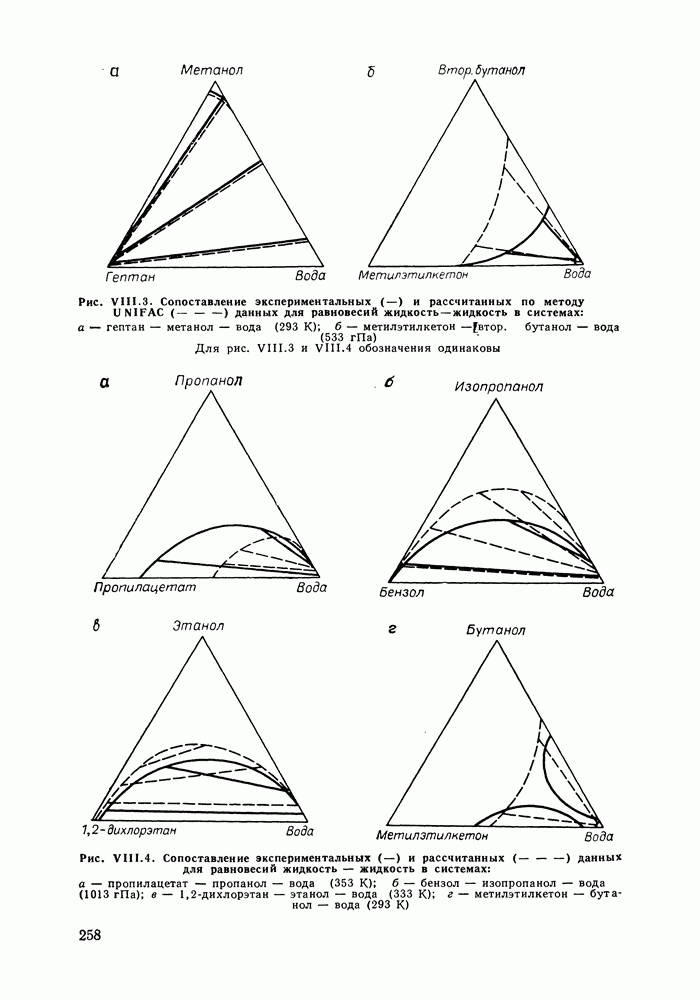












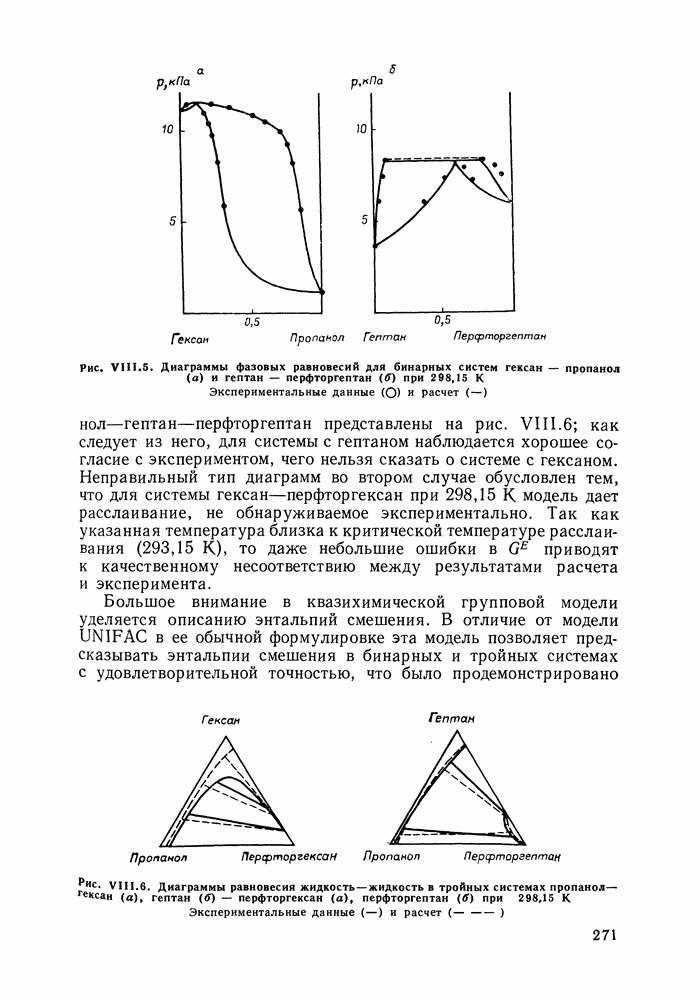
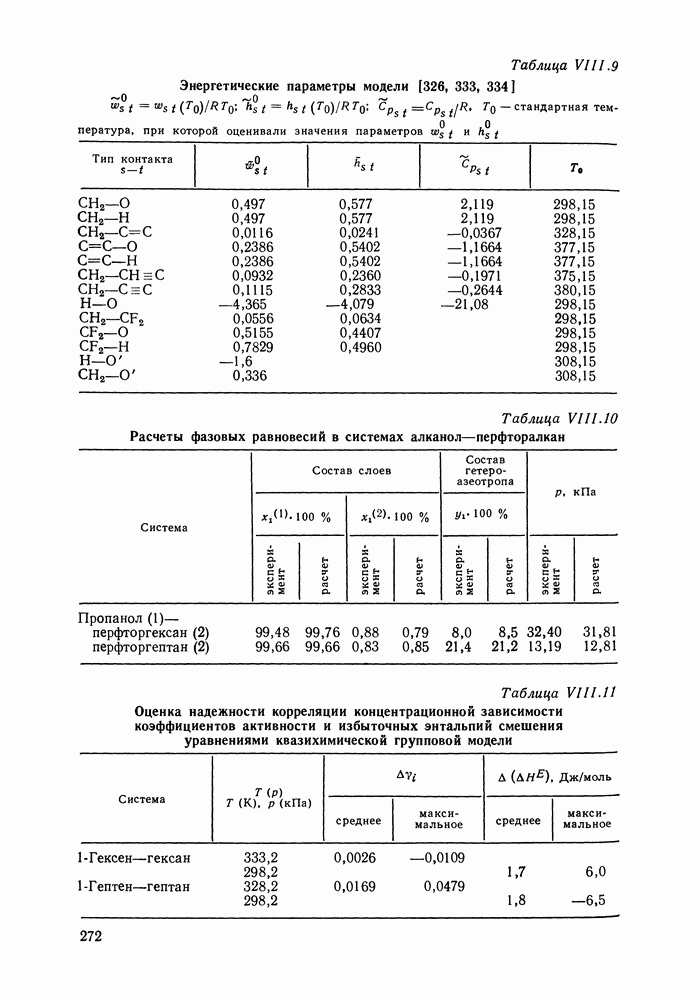
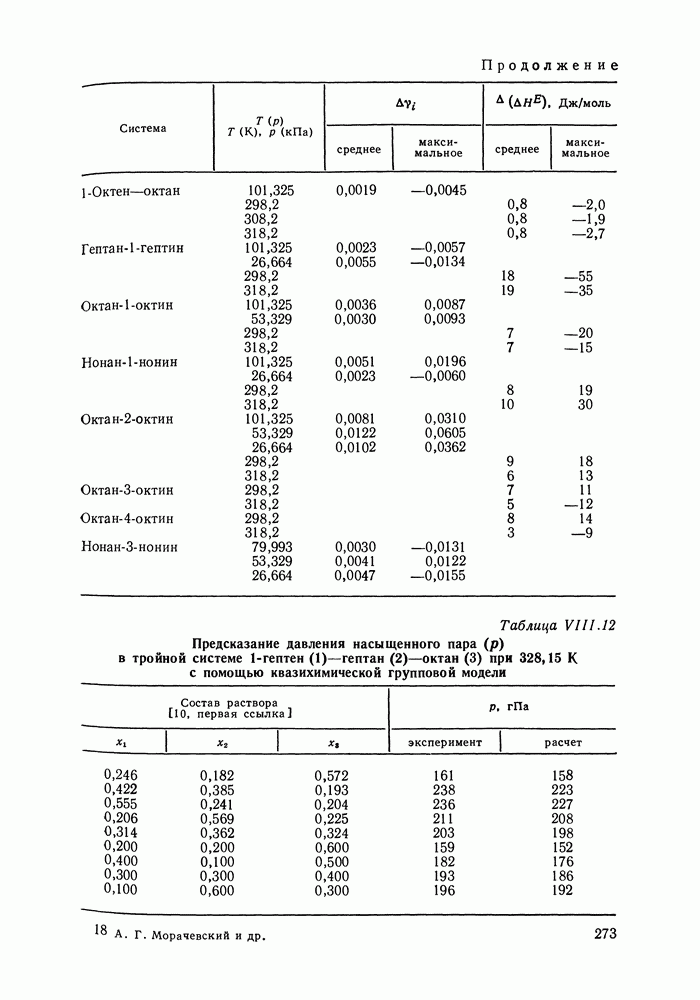
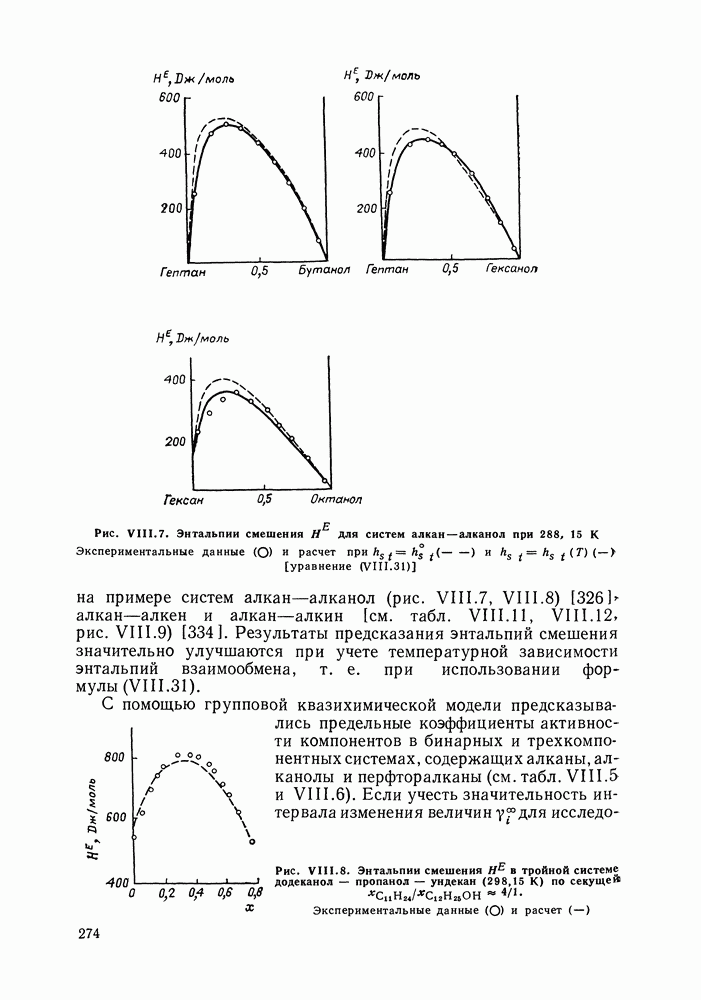























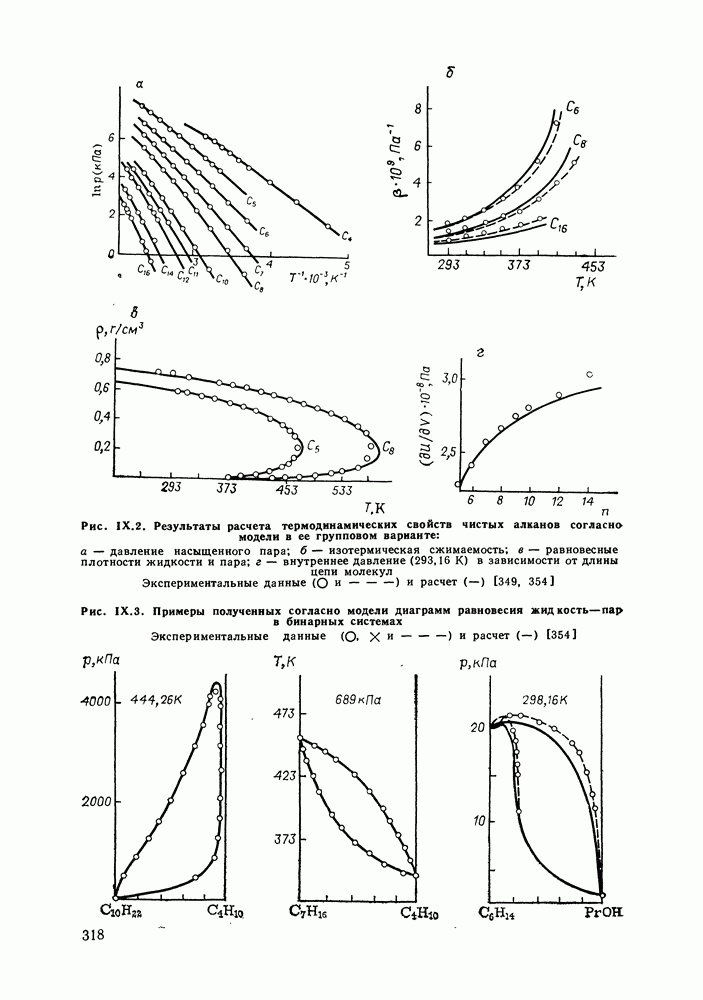
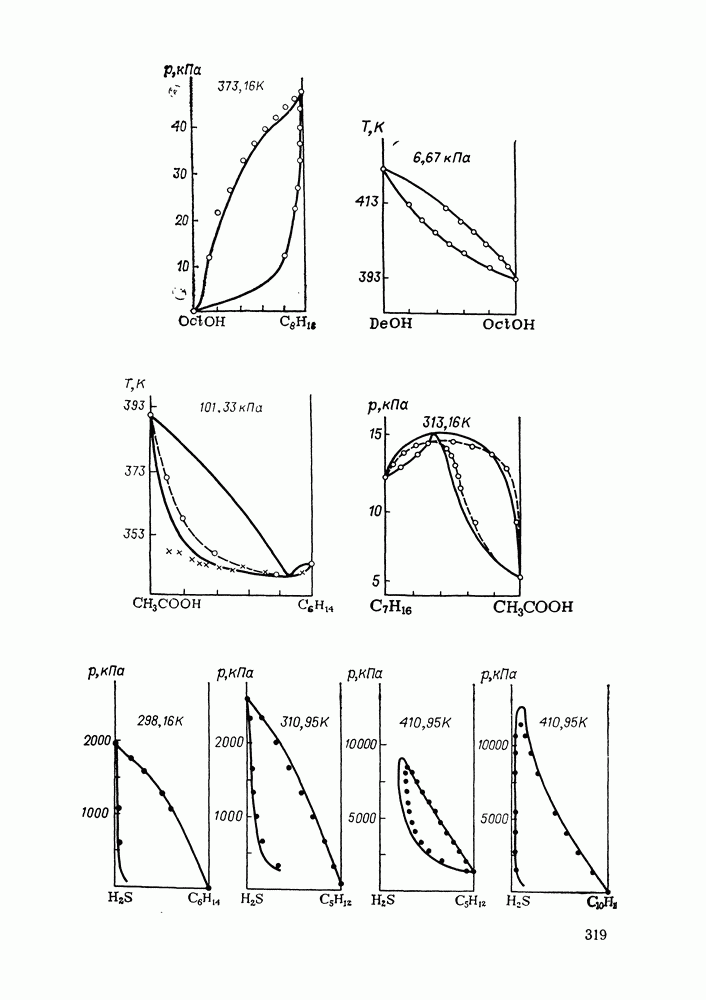

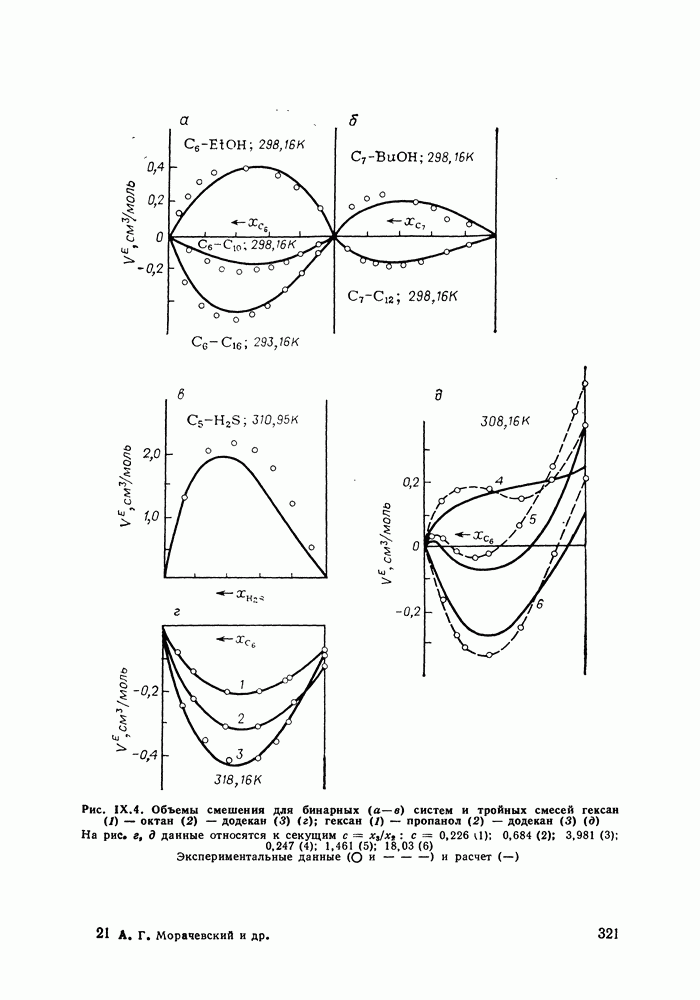
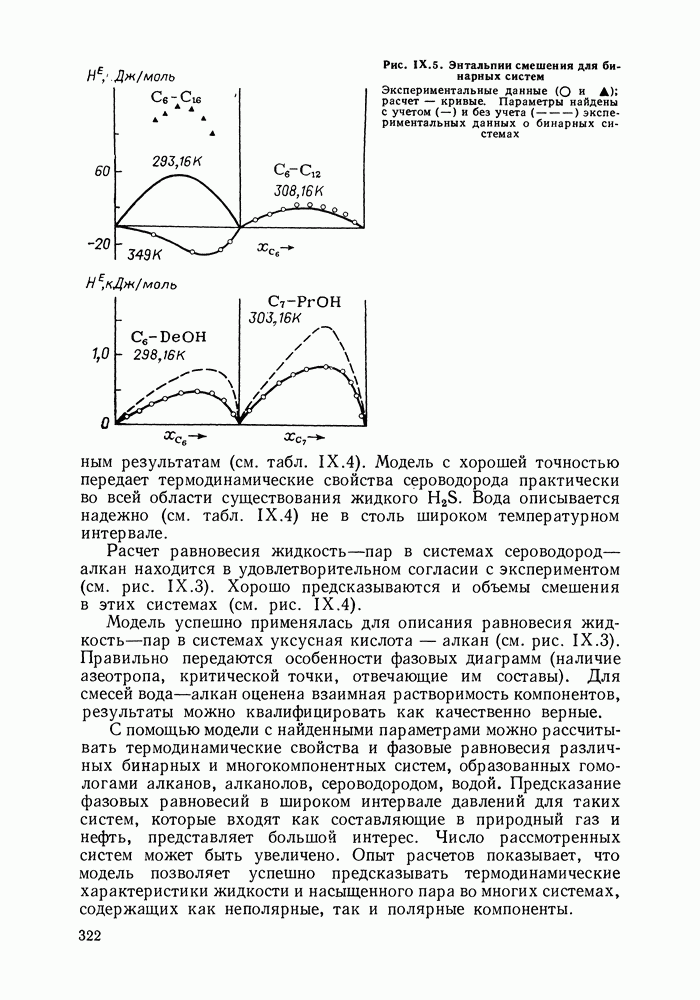
















 С увеличением давления до нескольких атмосфер и выше свойства любой паровой смеси приобретают отчетлива выраженные отклонения от свойств смеси идеальных газов, и очевидна необходимость учета этих отклонений.
С увеличением давления до нескольких атмосфер и выше свойства любой паровой смеси приобретают отчетлива выраженные отклонения от свойств смеси идеальных газов, и очевидна необходимость учета этих отклонений.

 от параметров состояния фазы в виде бесконечного степенного ряда по молярной плотности или давлению:
от параметров состояния фазы в виде бесконечного степенного ряда по молярной плотности или давлению:


 называют вторым, третьим — и так далее — вириальными коэффициентами, они зависят от природы, состава и температуры фазы, но не зависят от ее плотности или давления.
называют вторым, третьим — и так далее — вириальными коэффициентами, они зависят от природы, состава и температуры фазы, но не зависят от ее плотности или давления.

 вириальные коэффициенты, отражающие вклад взаимодействий молекул веществ
вириальные коэффициенты, отражающие вклад взаимодействий молекул веществ  и зависящие только от природы веществ и температуры;
и зависящие только от природы веществ и температуры;  вириальные коэффициенты чистого
вириальные коэффициенты чистого  -го вещества.
-го вещества.





 соотношения для
соотношения для  можно представить в такой форме:
можно представить в такой форме:


 и
и  выражение для коэффициентов
выражение для коэффициентов  суммарно учитывающих вклад неидеальности пара и поправки Пойнтинга в рассчитываемые величины, в случае бинарной системы переходит в следующее:
суммарно учитывающих вклад неидеальности пара и поправки Пойнтинга в рассчитываемые величины, в случае бинарной системы переходит в следующее:

 и поправки Пойнтинга, но часто используется, как несколько более экономное при расчетах.
и поправки Пойнтинга, но часто используется, как несколько более экономное при расчетах.
 которые являются величинами того же порядка, что и вириальные коэффициенты чистых веществ, и в той же мере влияют на результат расчета.
которые являются величинами того же порядка, что и вириальные коэффициенты чистых веществ, и в той же мере влияют на результат расчета.
 близок к среднеарифметическому вириальных коэффициентов чистых
близок к среднеарифметическому вириальных коэффициентов чистых  В этом случае
В этом случае  и расчетные соотношения для
и расчетные соотношения для  -компонентной смеси упрощаются:
-компонентной смеси упрощаются:  Заметим, что
Заметим, что  при этом описывается в соответствии с приближенным законом Амага,
при этом описывается в соответствии с приближенным законом Амага,  в соответстви с приближенным правилом Льюиса. Следует предостеречь от произвольного применения этих соотношений — вне области справедливости допущения о среднеарифметическом
в соответстви с приближенным правилом Льюиса. Следует предостеречь от произвольного применения этих соотношений — вне области справедливости допущения о среднеарифметическом  Рассчитанные значения равновесных свойств системы могут при этом оказаться даже дальше от истинных значений, чем рассчитанные в предположении идеальности пара [105].
Рассчитанные значения равновесных свойств системы могут при этом оказаться даже дальше от истинных значений, чем рассчитанные в предположении идеальности пара [105].
 чаще всего оценивают с помощью полуэмпирических корреляционных соотношений [176—181] — ряд из них приведен в монографии [9]. В этих корреляциях полезные результаты дало применение расширенного принципа соответственных состояний, который, в
чаще всего оценивают с помощью полуэмпирических корреляционных соотношений [176—181] — ряд из них приведен в монографии [9]. В этих корреляциях полезные результаты дало применение расширенного принципа соответственных состояний, который, в
 но и
но и  на основе преимущественно свойств чистых компонентов. Заметным этапом в развитии методов расчета вторых вириальных коэффициентов явилась простая и весьма точная корреляция Питцера-Керла [176] для неполярных веществ. Для предсказания вторых вириальных коэффициентов разнообразных по при роде и степени полярности веществ и смесей широко используют корреляции Цонопулоса [179] и Хайдена-О'Коннелла [180].
на основе преимущественно свойств чистых компонентов. Заметным этапом в развитии методов расчета вторых вириальных коэффициентов явилась простая и весьма точная корреляция Питцера-Керла [176] для неполярных веществ. Для предсказания вторых вириальных коэффициентов разнообразных по при роде и степени полярности веществ и смесей широко используют корреляции Цонопулоса [179] и Хайдена-О'Коннелла [180].
 от 13 до
от 13 до  (например, для воды, метанола, метилпропилкетона, пиридина, нитрометана). Корреляция Хайдена-О'Коннелла обеспечивает гораздо более стабильные результаты, с максимальными ошибками ниже
(например, для воды, метанола, метилпропилкетона, пиридина, нитрометана). Корреляция Хайдена-О'Коннелла обеспечивает гораздо более стабильные результаты, с максимальными ошибками ниже  уровня (максимальные погрешности в 8-9 % получены для воды, ацетальдегида и метиламина). Расчетные формулы метода Хайдена-О'Коннелла приведены ниже (стр. 184).
уровня (максимальные погрешности в 8-9 % получены для воды, ацетальдегида и метиламина). Расчетные формулы метода Хайдена-О'Коннелла приведены ниже (стр. 184).
 достаточно близки к единице, в таком случае парожидкостное равновесие часто описывают, используя модель идеального пара (говоря об идеальном паре, здесь и ниже предполагаем также, что поправка Пойнтинга пренебрежимо мала). Пользу учета парофазной неидеальности при атмосферном давлении иногда ставят под сомнение [182]. Окончательный ответ на вопрос, обязательно ли принимать во внимание неидеальность пара при низких давлениях, зависит от целей расчета, качества исходных данных и особенностей конкретной системы.
достаточно близки к единице, в таком случае парожидкостное равновесие часто описывают, используя модель идеального пара (говоря об идеальном паре, здесь и ниже предполагаем также, что поправка Пойнтинга пренебрежимо мала). Пользу учета парофазной неидеальности при атмосферном давлении иногда ставят под сомнение [182]. Окончательный ответ на вопрос, обязательно ли принимать во внимание неидеальность пара при низких давлениях, зависит от целей расчета, качества исходных данных и особенностей конкретной системы.
 могут отличаться от единицы в разной степени. Например, в бинарных системах, в соответствии с (VII.69), отличие
могут отличаться от единицы в разной степени. Например, в бинарных системах, в соответствии с (VII.69), отличие  от единицы зависит от подэкспоненциальных слагаемых
от единицы зависит от подэкспоненциальных слагаемых  каждый из которых, независимо от другого, может быть и положительным, и отрицательным. В результате абсолютные значения слагаемых могут как складываться, так и частично взаимно компенсировать друг друга [105]. Ясно, что во втором случае допущение
каждый из которых, независимо от другого, может быть и положительным, и отрицательным. В результате абсолютные значения слагаемых могут как складываться, так и частично взаимно компенсировать друг друга [105]. Ясно, что во втором случае допущение  внесет меньшую погрешность в расчет, чем в первом, хотя приближение второго случая
внесет меньшую погрешность в расчет, чем в первом, хотя приближение второго случая
 при атмосферном давлении, составляет, в среднем, от 0,3 до
при атмосферном давлении, составляет, в среднем, от 0,3 до  (мол.) и не зависит от степени неидеальности жидкой фазы. Следует добавить, что указанная погрешность на самом деле может оказаться меньше, если при расчете параметров модели жидкой фазы пар также считался идеальным: погрешности решения обратной и прямой задач фазового равновесия в какой-то мере могут компенсироваться.
(мол.) и не зависит от степени неидеальности жидкой фазы. Следует добавить, что указанная погрешность на самом деле может оказаться меньше, если при расчете параметров модели жидкой фазы пар также считался идеальным: погрешности решения обратной и прямой задач фазового равновесия в какой-то мере могут компенсироваться.
 в рассчитанные избыточные энергии Гиббса, в среднем составляет
в рассчитанные избыточные энергии Гиббса, в среднем составляет  а в отдельных системах (например, ацетон—хлороформ) может доходить до
а в отдельных системах (например, ацетон—хлороформ) может доходить до  Особенно важен корректный учет неидеальности пара при расчете термодинамических функций, являющихся производными от избыточной энергии Гиббса. Так, использование модели идеального пара или неточных значений вторых вириальных коэффициентов приводит к большим — до
Особенно важен корректный учет неидеальности пара при расчете термодинамических функций, являющихся производными от избыточной энергии Гиббса. Так, использование модели идеального пара или неточных значений вторых вириальных коэффициентов приводит к большим — до  погрешностям расчета избыточной энтальпии растворов.
погрешностям расчета избыточной энтальпии растворов.
 в корреляции Хайдена-О'Коннелла, кроме температуры системы, являются критические температура и давление
в корреляции Хайдена-О'Коннелла, кроме температуры системы, являются критические температура и давление  вещества —
вещества —  его дипольный момент —
его дипольный момент —  средний радиус вращения молекулы —
средний радиус вращения молекулы —  параметр ассоциации
параметр ассоциации  Для расчета коэффициента
Для расчета коэффициента  кроме уже перечисленных свойств чистых веществ
кроме уже перечисленных свойств чистых веществ  необходим параметр сольватации
необходим параметр сольватации  Все исходные данные, за исключением параметров ассоциации и сольватации, являются относительно доступными физико-химическими характеристиками чистых веществ или могут быть рассчитаны по таким характеристикам.
Все исходные данные, за исключением параметров ассоциации и сольватации, являются относительно доступными физико-химическими характеристиками чистых веществ или могут быть рассчитаны по таким характеристикам.

 вклад в вириальный коэффициент, вносимый вандерваальсовыми взаимодействиями между свободными частицами газа;
вклад в вириальный коэффициент, вносимый вандерваальсовыми взаимодействиями между свободными частицами газа;  вклад, обусловленный образованием связанных молекулярных пар (димеров).
вклад, обусловленный образованием связанных молекулярных пар (димеров).
 в свою очередь, разбивается на неполярную и полярную составляющие:
в свою очередь, разбивается на неполярную и полярную составляющие:

 рассматривают как сумму вкладов, приписываемых метастабильно связанным, связанным и химически связанным молекулярным парам:
рассматривают как сумму вкладов, приписываемых метастабильно связанным, связанным и химически связанным молекулярным парам:


 карбоновой кислоты, когда экспериментальные значения ее неизвестны.
карбоновой кислоты, когда экспериментальные значения ее неизвестны.
 -характеристическую энергию взаимодействия молекул
-характеристическую энергию взаимодействия молекул  параметр молекулярного размера,
параметр молекулярного размера,  неполярный ацентрический фактор.
неполярный ацентрический фактор.
 -компонентной системы задача состоит в расчете
-компонентной системы задача состоит в расчете  величин
величин  величин
величин  Нахождение всей матрицы значений
Нахождение всей матрицы значений  излишне, ввиду равенства
излишне, ввиду равенства  Соответственно, расчет всех промежуточных вспомогательных величин достаточно провести, например, для всех
Соответственно, расчет всех промежуточных вспомогательных величин достаточно провести, например, для всех  от 1 до
от 1 до  , и при каждом данном
, и при каждом данном  для всех значений
для всех значений  от
от  до
до  Вычисления удобно разбить на следующие этапы.
Вычисления удобно разбить на следующие этапы.
 характеризующих чистые вещества:
характеризующих чистые вещества:

 для сочетаний веществ
для сочетаний веществ 

 для смеси сильно полярного
для смеси сильно полярного  и неполярного
и неполярного  веществ рассчитывают так: при
веществ рассчитывают так: при  полагаем
полагаем  а при
а при  полагаем
полагаем 



 и их сочетаний
и их сочетаний  совпадают:
совпадают:



 и искомые вторые вириальные коэффициенты
и искомые вторые вириальные коэффициенты 
 и массой
и массой  молекулы, может быть строго рассчитан из картезианских координат и масс атомов. Исходные для вычисления межъядерные расстояния и углы между связями для различных веществ приведены в [185, 189]. Для линейных молекул
молекулы, может быть строго рассчитан из картезианских координат и масс атомов. Исходные для вычисления межъядерные расстояния и углы между связями для различных веществ приведены в [185, 189]. Для линейных молекул  для нелинейных
для нелинейных  Удовлетворительный альтернативный метод определения
Удовлетворительный альтернативный метод определения  дает его корреляция с парахором
дает его корреляция с парахором  [190]:
[190]:

 по формуле (VII.78) составляет ±0,3 А, что соответствует максимальной ошибке, вносимой в значение второго вириального коэффициента,
по формуле (VII.78) составляет ±0,3 А, что соответствует максимальной ошибке, вносимой в значение второго вириального коэффициента,  Значения парахора и методы его расчета по вкладам групп можно найти в [185, 187]. Наиболее трудно оценить эмпирические параметры ассоциации
Значения парахора и методы его расчета по вкладам групп можно найти в [185, 187]. Наиболее трудно оценить эмпирические параметры ассоциации  и сольватации
и сольватации  Для неполярных газов, углеводородов, их галогенпроизводных, простых эфиров и сульфидов
Для неполярных газов, углеводородов, их галогенпроизводных, простых эфиров и сульфидов  Сольватационный параметр во многих случаях также нулевой, например, для смесей неполярных газов, углеводородов различного класса, а также смесей алканов или циклоалканов с любыми полярными соединениями. Для многих смесей непредельных и ароматических
Сольватационный параметр во многих случаях также нулевой, например, для смесей неполярных газов, углеводородов различного класса, а также смесей алканов или циклоалканов с любыми полярными соединениями. Для многих смесей непредельных и ароматических
 также нулевой — например, для смесей с водой, спиртами — или значение его невелико в пределах
также нулевой — например, для смесей с водой, спиртами — или значение его невелико в пределах 
 для рассматриваемой системы эти параметры приходится оценивать априорно, по аналогии с другими близкими по природе веществами и системами. Оценка упрощается, когда компоненты
для рассматриваемой системы эти параметры приходится оценивать априорно, по аналогии с другими близкими по природе веществами и системами. Оценка упрощается, когда компоненты  принадлежат к одному классу соединений. В этом случае параметры
принадлежат к одному классу соединений. В этом случае параметры  обычно или совпадают, или близки.
обычно или совпадают, или близки.
 -компонентную идеальную ассоциированную паровую смесь, в которой произвольное число компонентов участвует в реакциях ассоциации. В соответствии с моделью, считаем, что вандерваальсовы взаимодействия между частицами смеси отсутствуют и неидеальность последней связана только с протеканием обратимых реакций. Условно примем все компоненты способными вступать в реакции, что позволит при записи соотношений не различать ассоциирующие и неассоциирующие компоненты. При расчетах различие между первыми и вторыми проявится только через соответствующие значения констант ассоциации.
-компонентную идеальную ассоциированную паровую смесь, в которой произвольное число компонентов участвует в реакциях ассоциации. В соответствии с моделью, считаем, что вандерваальсовы взаимодействия между частицами смеси отсутствуют и неидеальность последней связана только с протеканием обратимых реакций. Условно примем все компоненты способными вступать в реакции, что позволит при записи соотношений не различать ассоциирующие и неассоциирующие компоненты. При расчетах различие между первыми и вторыми проявится только через соответствующие значения констант ассоциации.
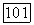 реакций вида;
реакций вида;

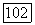 мономерные формы компонентов
мономерные формы компонентов 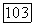 и
и 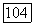 их димерная форма.
их димерная форма.
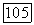 может совпадать (самоассоциация вещества) и быть различным (сольватация).
может совпадать (самоассоциация вещества) и быть различным (сольватация).
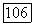 участников реакции связаны константой химического равновесия
участников реакции связаны константой химического равновесия 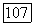 которая в области применимости модели идеальной ассоциированной смеси зависит только от природы
которая в области применимости модели идеальной ассоциированной смеси зависит только от природы 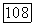 и температуры. Константы
и температуры. Константы 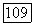 отвечающие условным, не существующим в действительности димерам, при расчете полагаются равными нулю. (Это автоматически приводит к нулевым расчетным парциальным давлениям условных димеров).
отвечающие условным, не существующим в действительности димерам, при расчете полагаются равными нулю. (Это автоматически приводит к нулевым расчетным парциальным давлениям условных димеров).


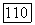 молекулярных форм в паровой смеси:
молекулярных форм в паровой смеси:


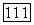 моль свободных и связанных мономерных частиц каждого
моль свободных и связанных мономерных частиц каждого 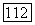 компонента в одном истинном моле смеси:
компонента в одном истинном моле смеси:

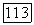 получается подстановкой соотношения (VII.83) в определение аналитической мол. доли:
получается подстановкой соотношения (VII.83) в определение аналитической мол. доли:


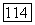 -данным, но расчет, за исключением некоторых частных случаев, требует использования метода последовательных приближений. Например, расчет
-данным, но расчет, за исключением некоторых частных случаев, требует использования метода последовательных приближений. Например, расчет 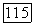 необходимых величин парциальных давлений мономеров может быть осуществлен решением системы, состоящей из
необходимых величин парциальных давлений мономеров может быть осуществлен решением системы, состоящей из 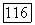 независимых уравнений вида (VII.85) и уравнения
независимых уравнений вида (VII.85) и уравнения 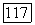 с учетом (VII.79).
с учетом (VII.79).



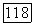 парциальное давление мономера I в чистом веществе
парциальное давление мономера I в чистом веществе 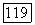 при температуре раствора.
при температуре раствора.


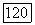 в зависимости от типа задачи рассчитывают по свойствам жидкой фазы в соответствии с уравнением (VII.89) или по данным о равновесном паре на основе уравнений (VII.79), (VII.80) и (VII.85). Значение
в зависимости от типа задачи рассчитывают по свойствам жидкой фазы в соответствии с уравнением (VII.89) или по данным о равновесном паре на основе уравнений (VII.79), (VII.80) и (VII.85). Значение 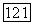 дается решением квадратного уравнения
дается решением квадратного уравнения 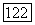 Выбирая корень уравнения, отвечающий физическому смыслу величины
Выбирая корень уравнения, отвечающий физическому смыслу величины 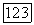 для чистого ассоциирующего компонента
для чистого ассоциирующего компонента 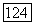 получаем:
получаем:


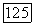 по полным данным о равновесии жидкость—пар, в общем случае
по полным данным о равновесии жидкость—пар, в общем случае
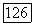 методом последовательных приближений.
методом последовательных приближений.

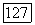 Поделив числитель и знаменатель в (VII.93) на общее давление (для чистого вещества
Поделив числитель и знаменатель в (VII.93) на общее давление (для чистого вещества 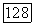 можем также записать:
можем также записать:

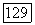 истинная мол. доля мономера в чистом компоненте
истинная мол. доля мономера в чистом компоненте 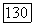
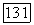 неассоциирующих компонентов.
неассоциирующих компонентов.
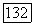 и единственная ненулевая константа димеризации —
и единственная ненулевая константа димеризации — 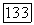 Присвоив всем остальным (условным) константам
Присвоив всем остальным (условным) константам 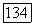 нулевые значения и подставив имеющийся набор констант в уравнения модели, после необходимых преобразований получаем следующие расчетные формулы:
нулевые значения и подставив имеющийся набор констант в уравнения модели, после необходимых преобразований получаем следующие расчетные формулы:

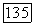 тогда:
тогда:

 находят по уравнению (VII.89) с учетом (VII.91) и (VII.92). Если заданы параметры паровой фазы, для расчета величин
находят по уравнению (VII.89) с учетом (VII.91) и (VII.92). Если заданы параметры паровой фазы, для расчета величин 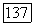 использовать метод последовательных приближений не требуется, так как квадратное Уравнение, связывающее величины
использовать метод последовательных приближений не требуется, так как квадратное Уравнение, связывающее величины 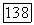 в данном случае легко решить:
в данном случае легко решить:

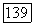 неассоциирующих компонентов равны между собой, выражение для них приводится к виду:
неассоциирующих компонентов равны между собой, выражение для них приводится к виду: 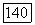

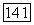 параметры, характеризующие вещество
параметры, характеризующие вещество 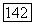
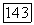 для карбоновых кислот дает также корреляция Хайдена-О’Коннелла [см. (VII.73)].
для карбоновых кислот дает также корреляция Хайдена-О’Коннелла [см. (VII.73)].

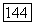 карбоновых кислот имеет вид [199]:
карбоновых кислот имеет вид [199]:

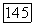 в несколько раз превышают значения, предсказываемые формулой (VII.95) [200]. Следовательно, если в системах рассматриваемого типа значения
в несколько раз превышают значения, предсказываемые формулой (VII.95) [200]. Следовательно, если в системах рассматриваемого типа значения 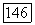 неизвестны, они должны быть оценены по имеющимся данным для соответствующих бинарных систем.
неизвестны, они должны быть оценены по имеющимся данным для соответствующих бинарных систем.

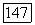 зависит от природы веществ
зависит от природы веществ 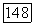 и температуры; при низком давлении она совпадает с
и температуры; при низком давлении она совпадает с 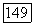
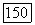 молекулярной формы:
молекулярной формы:


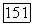 коэффициент фугитивности молекулярной формы I в парах [чистого компонента
коэффициент фугитивности молекулярной формы I в парах [чистого компонента 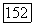 при температуре системы и давлении
при температуре системы и давлении 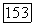
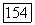 известны; рассмотрим, как оцениваются
известны; рассмотрим, как оцениваются 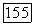 (а отсюда и
(а отсюда и 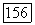 при расчете фазовых равновесий или коэффициентов активности по данным о равновесии. В качестве вспомогательной переменной используем концентрационную константу димеризации
при расчете фазовых равновесий или коэффициентов активности по данным о равновесии. В качестве вспомогательной переменной используем концентрационную константу димеризации

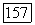 соотношением
соотношением

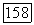 следовательно,
следовательно, 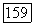 Для чистого ассоциирующего компонента решение уравнения материального баланса
Для чистого ассоциирующего компонента решение уравнения материального баланса 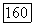 дает:
дает:

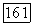 соотношения (VII.100) и (VII.103) дают величины
соотношения (VII.100) и (VII.103) дают величины 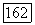
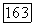 по У, или
по У, или 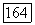 по
по 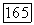 далее,
далее, 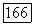 при фиксированных
при фиксированных 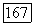 по сути, аналогична той, которая решалась Для идеальной ассоциированной смеси. Взаимосвязь
по сути, аналогична той, которая решалась Для идеальной ассоциированной смеси. Взаимосвязь 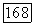 определяется строгими соотношениями материального баланса. При заданном истинном составе пара величины
определяется строгими соотношениями материального баланса. При заданном истинном составе пара величины 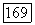 вычисляют непосредственно по уравнениям (VII.83) и (VII.84). При заданном аналитическом составе пара величины
вычисляют непосредственно по уравнениям (VII.83) и (VII.84). При заданном аналитическом составе пара величины 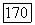 в общем случае находят решением системы уравнений
в общем случае находят решением системы уравнений 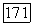 методом последовательных приближений (см. [144, с. 135]). В частном случае
методом последовательных приближений (см. [144, с. 135]). В частном случае

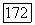 зависят от коэффициентов фугитивности
зависят от коэффициентов фугитивности 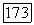 следовательно, изменяются с составом смеси. Это приводит к необходимости итерационного расчета по схеме:
следовательно, изменяются с составом смеси. Это приводит к необходимости итерационного расчета по схеме:

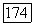 Начальные приближения
Начальные приближения 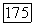 определяют, используя модель идеальной ассоциированной смеси. Итерации заканчиваются, когда рассчитываемые величины перестают изменяться. После этого можно рассчитать величины
определяют, используя модель идеальной ассоциированной смеси. Итерации заканчиваются, когда рассчитываемые величины перестают изменяться. После этого можно рассчитать величины 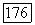
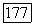 . В работах [144, 178] предложены приближенные способы оценки
. В работах [144, 178] предложены приближенные способы оценки 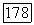 для области давлений, не превышающих несколько атмосфер.
для области давлений, не превышающих несколько атмосфер.
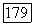 и коэффициенты
и коэффициенты 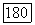 со вкладами во второй вириальный коэффициент, обусловленными явлением парофазной димеризации и, соответственно, неспецифическими взаимодействиями свободных молекул. В данной работе величины вкладов рассчитывают по корреляционным соотношениям Хайдена — О’Коннелла (смотри стр. 184), где эти вклады обозначены как
со вкладами во второй вириальный коэффициент, обусловленными явлением парофазной димеризации и, соответственно, неспецифическими взаимодействиями свободных молекул. В данной работе величины вкладов рассчитывают по корреляционным соотношениям Хайдена — О’Коннелла (смотри стр. 184), где эти вклады обозначены как 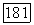 и
и 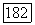 Независимо от числа сильно ассоциирующих компонентов, паровая фаза рассматривается как совокупность продуктов всех реакций димеризации, возможных в системе с данным числом компонентов.
Независимо от числа сильно ассоциирующих компонентов, паровая фаза рассматривается как совокупность продуктов всех реакций димеризации, возможных в системе с данным числом компонентов.
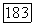 для однородных димеров рассчитывают по уравнению (VII.73), а гетеродимеров — по уравнению
для однородных димеров рассчитывают по уравнению (VII.73), а гетеродимеров — по уравнению

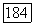 При этом для димерных форм
При этом для димерных форм 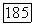 полагают
полагают 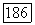
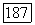 определяются в соответствии с правилом Льюиса:
определяются в соответствии с правилом Льюиса:

 оказываются независимыми от состава смеси. С учетом принятых допущений расчетные формулы имеют вид:
оказываются независимыми от состава смеси. С учетом принятых допущений расчетные формулы имеют вид:

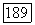 использован подход, сочетающий теорию ассоциативных равновесий с уравнением состояния флюидных фаз. При расчете свойств обратимо диссоциирующих газов нашел применение метод [202], основанный на групповых разложениях термодинамических функций (по степеням активности мономерных частиц). К реальному ассоциированному пару приложима дырочная модель, описанная в гл. IX.
использован подход, сочетающий теорию ассоциативных равновесий с уравнением состояния флюидных фаз. При расчете свойств обратимо диссоциирующих газов нашел применение метод [202], основанный на групповых разложениях термодинамических функций (по степеням активности мономерных частиц). К реальному ассоциированному пару приложима дырочная модель, описанная в гл. IX.