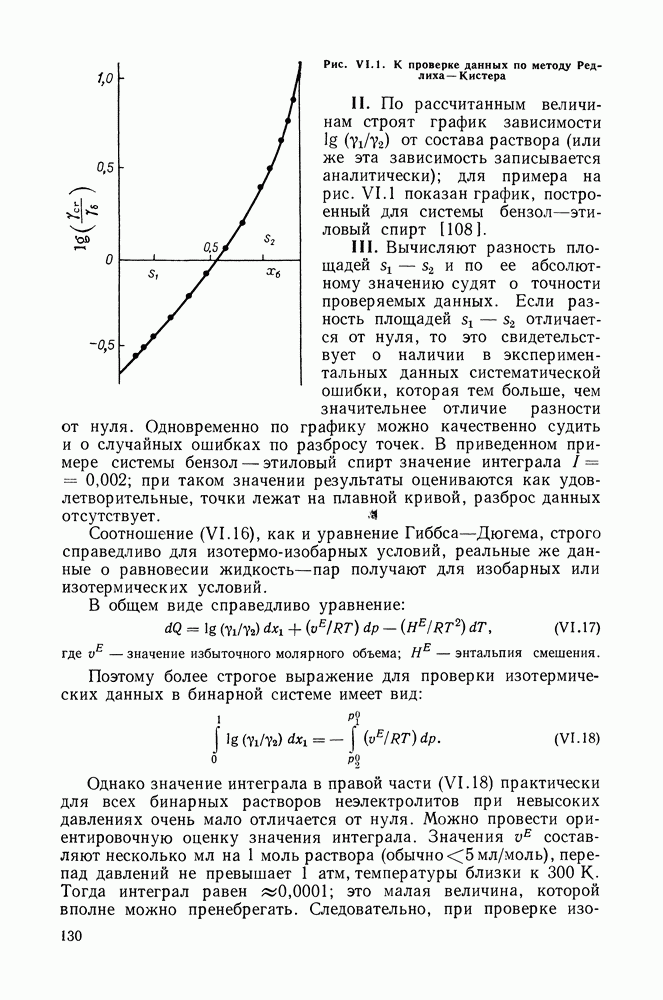
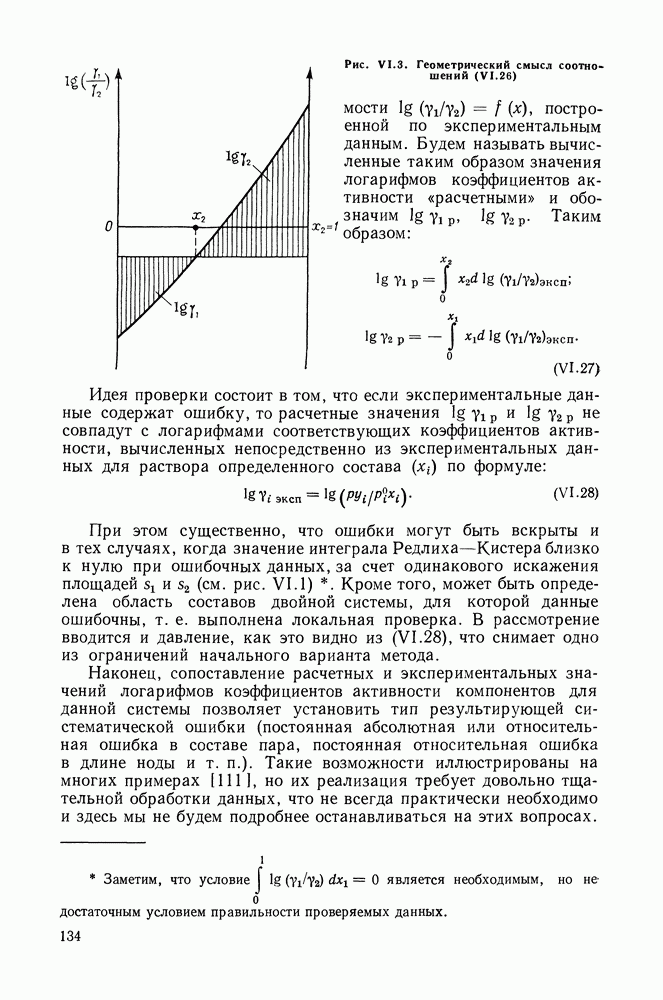
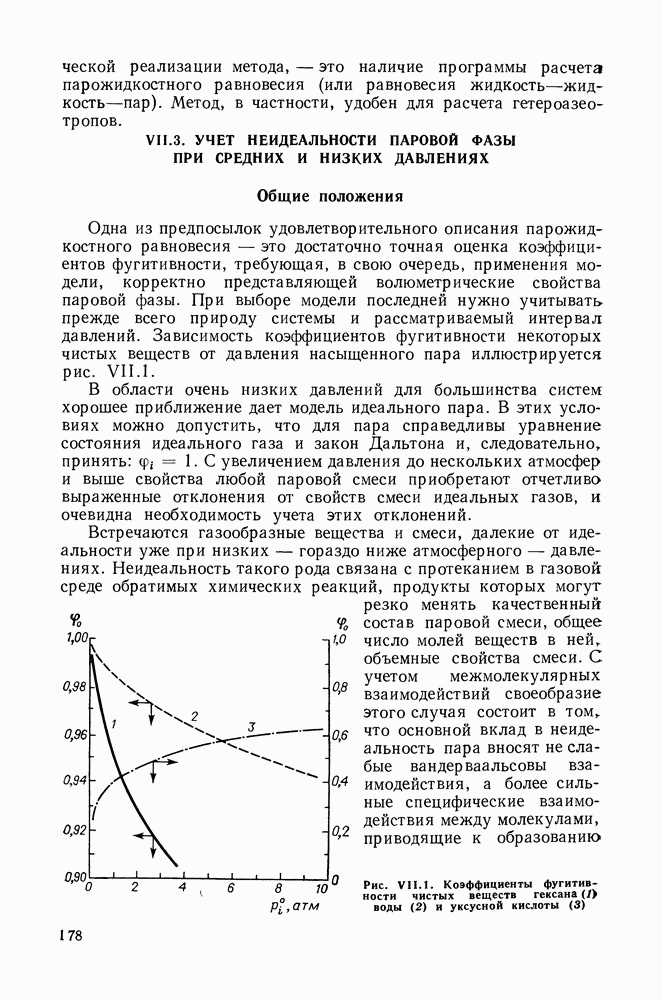
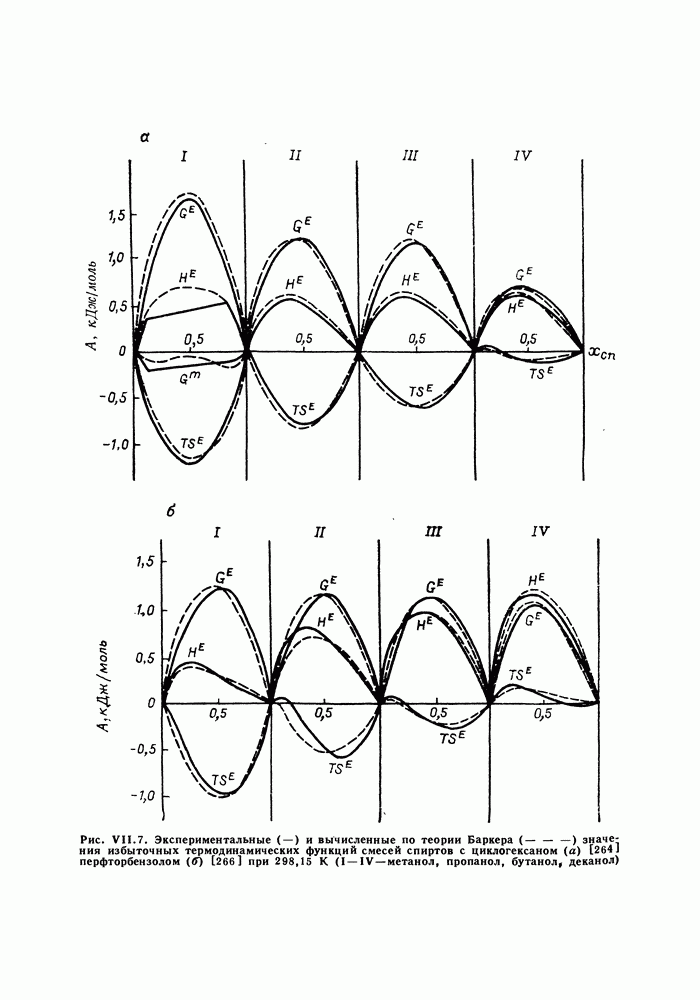
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
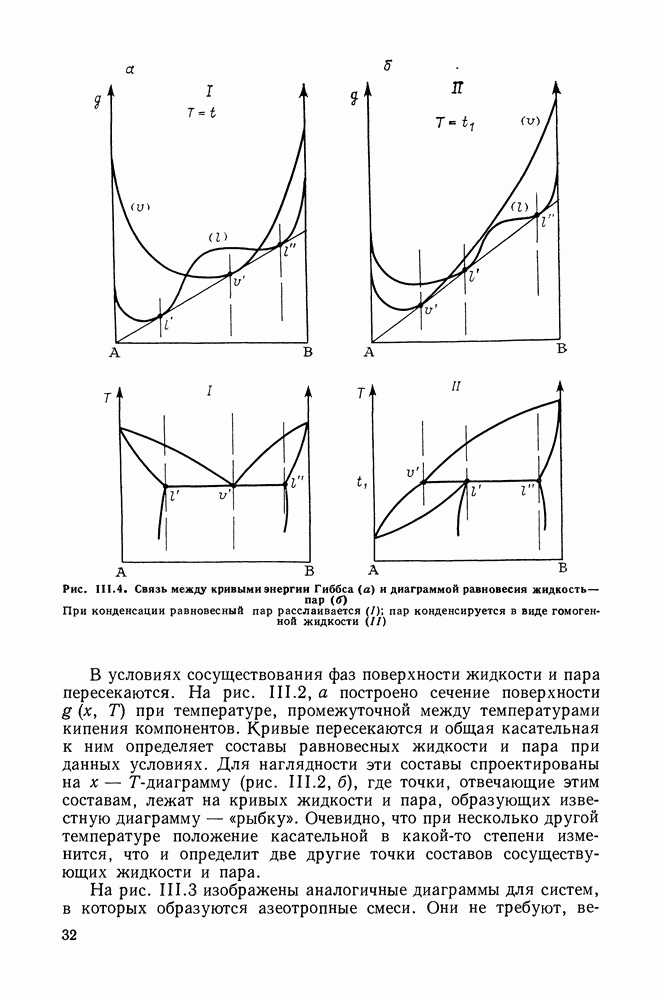
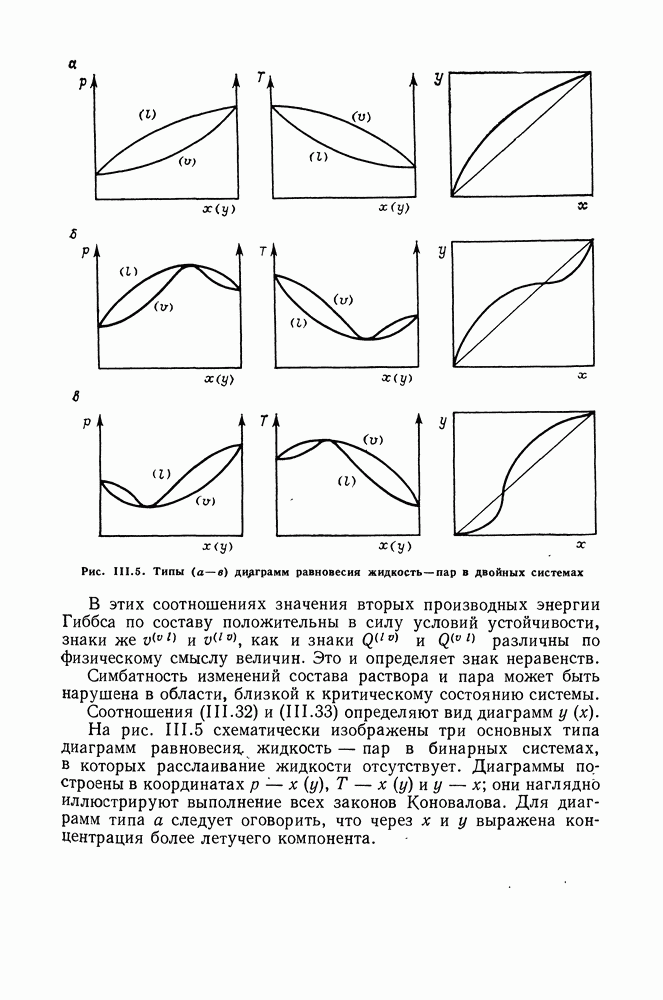
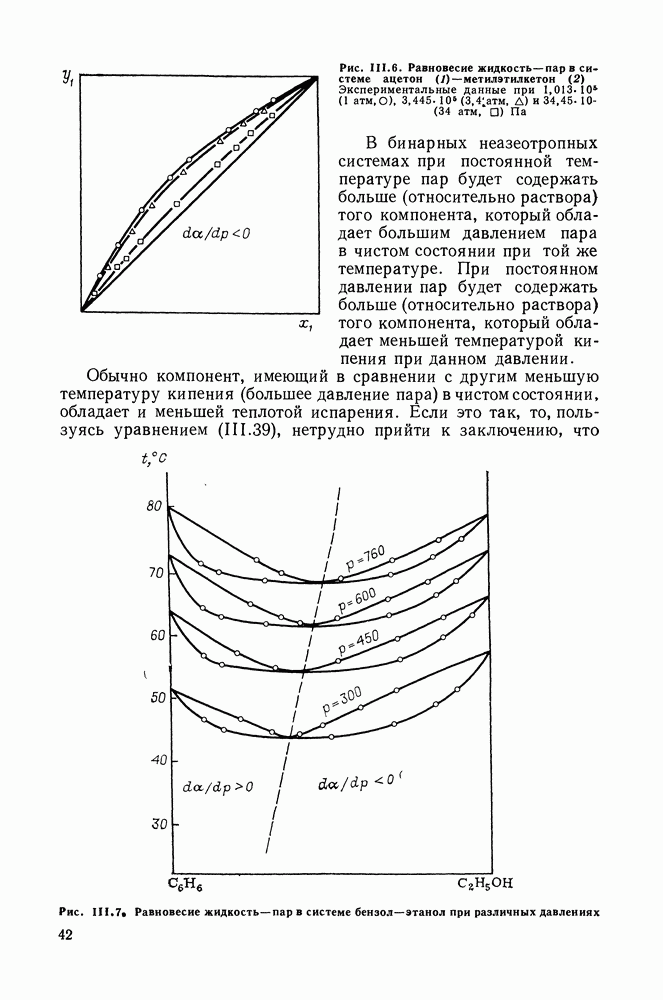
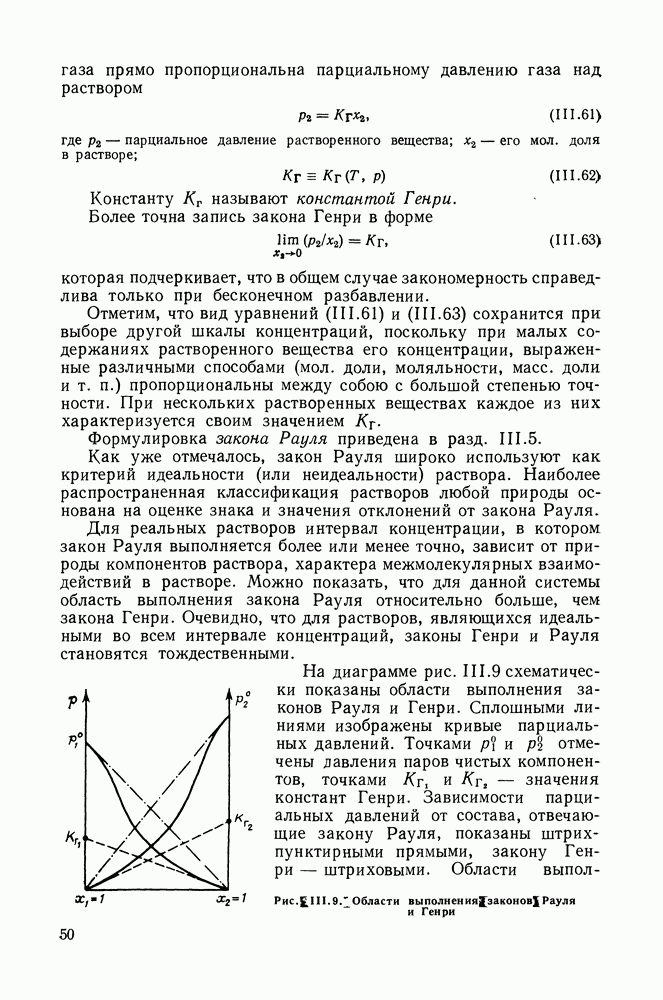
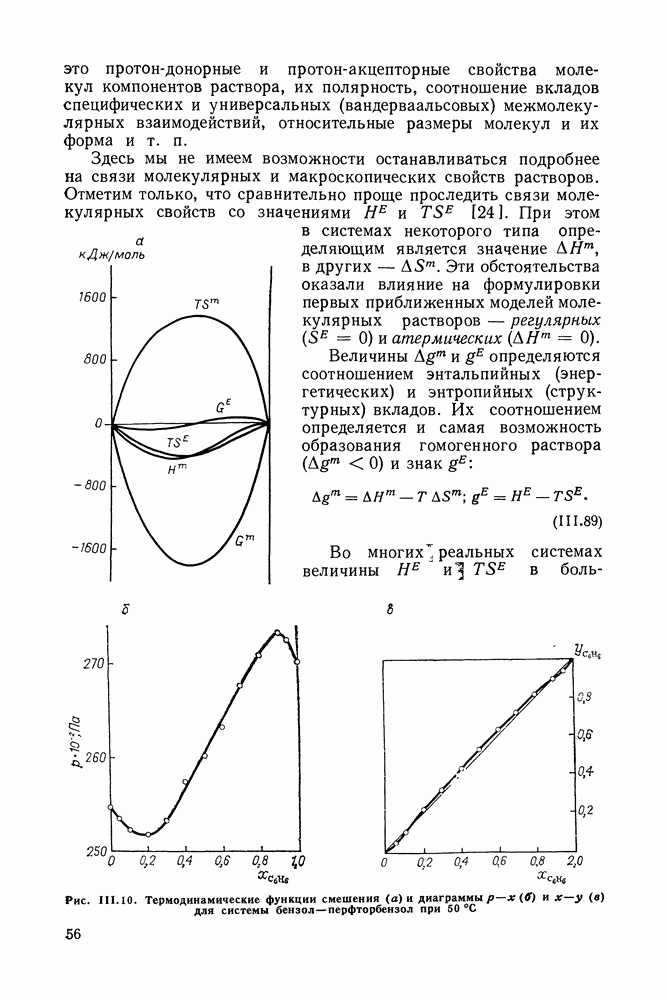
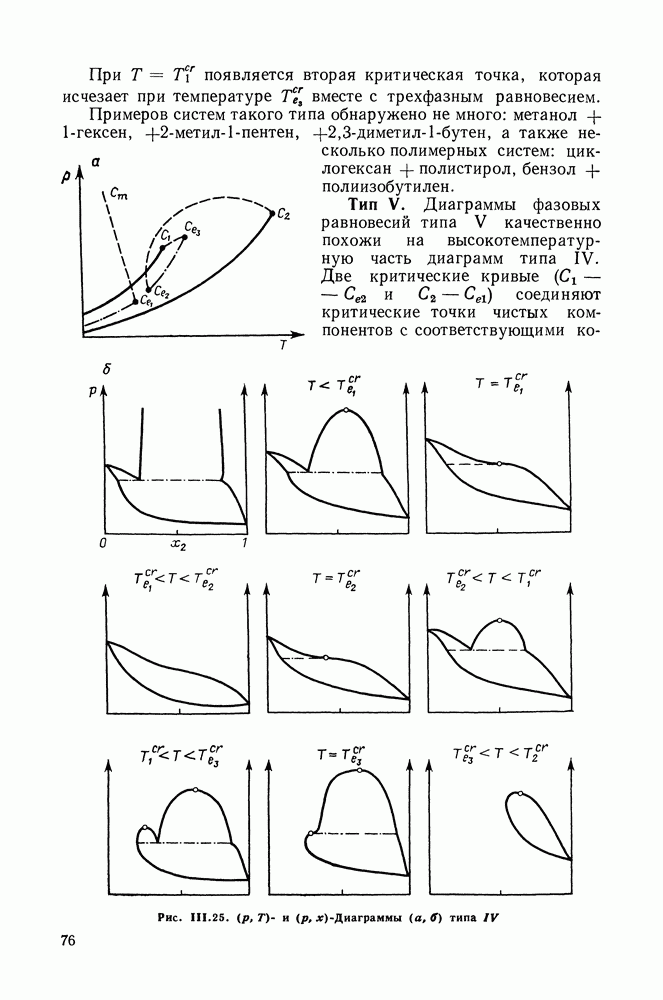
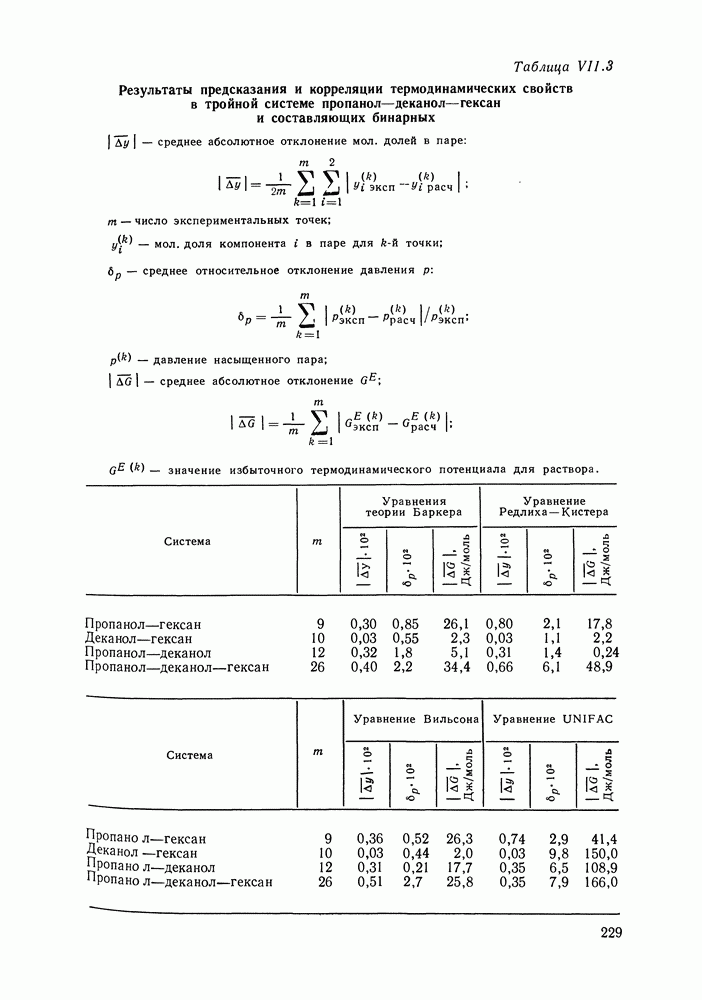
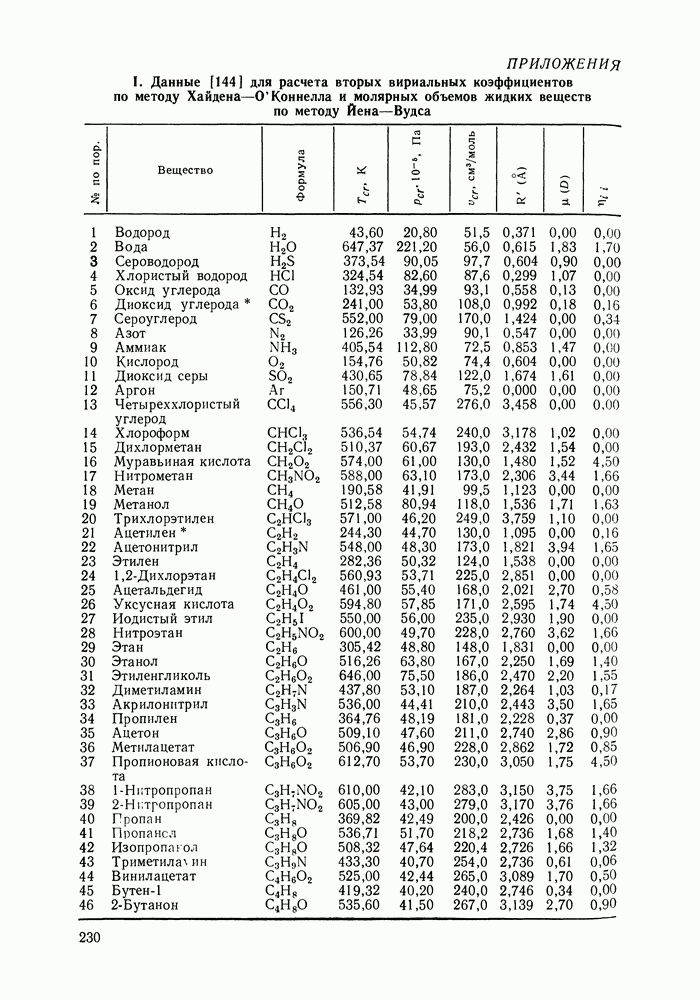
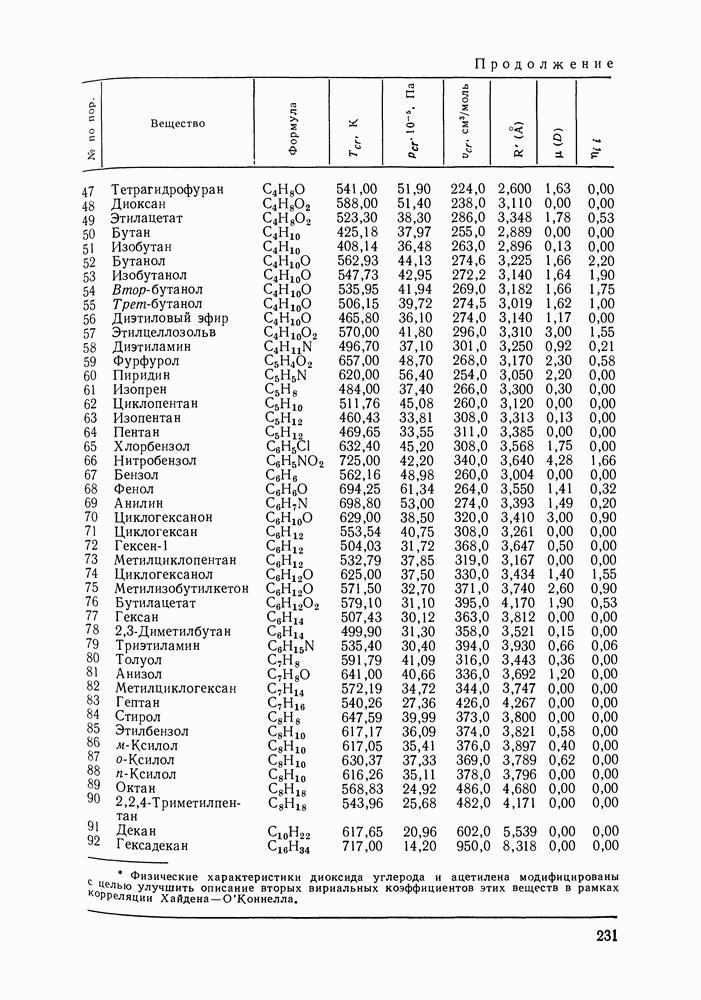
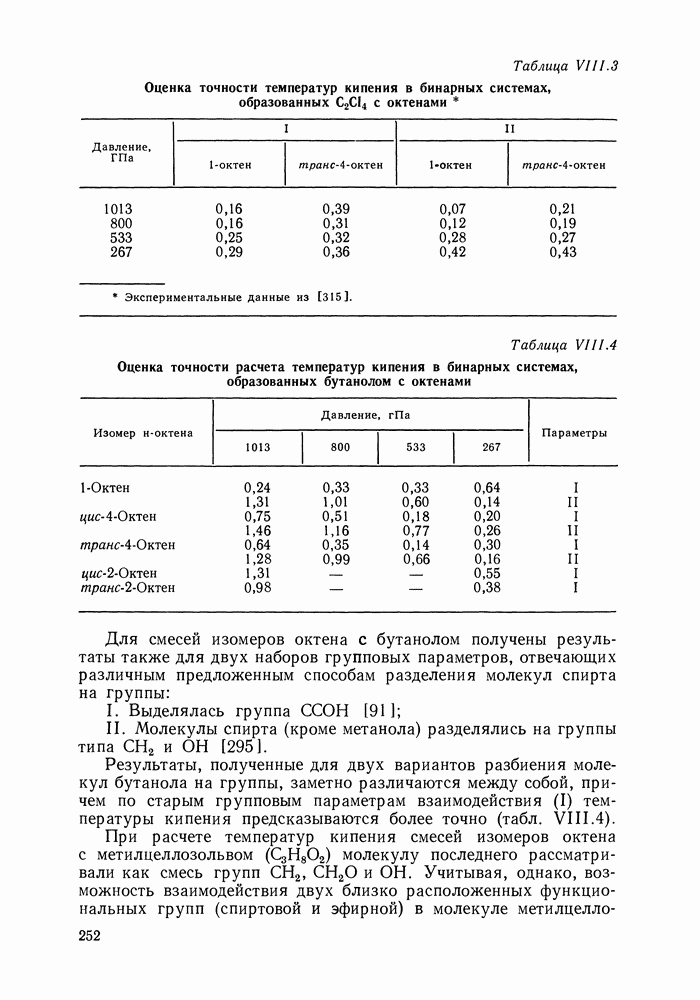
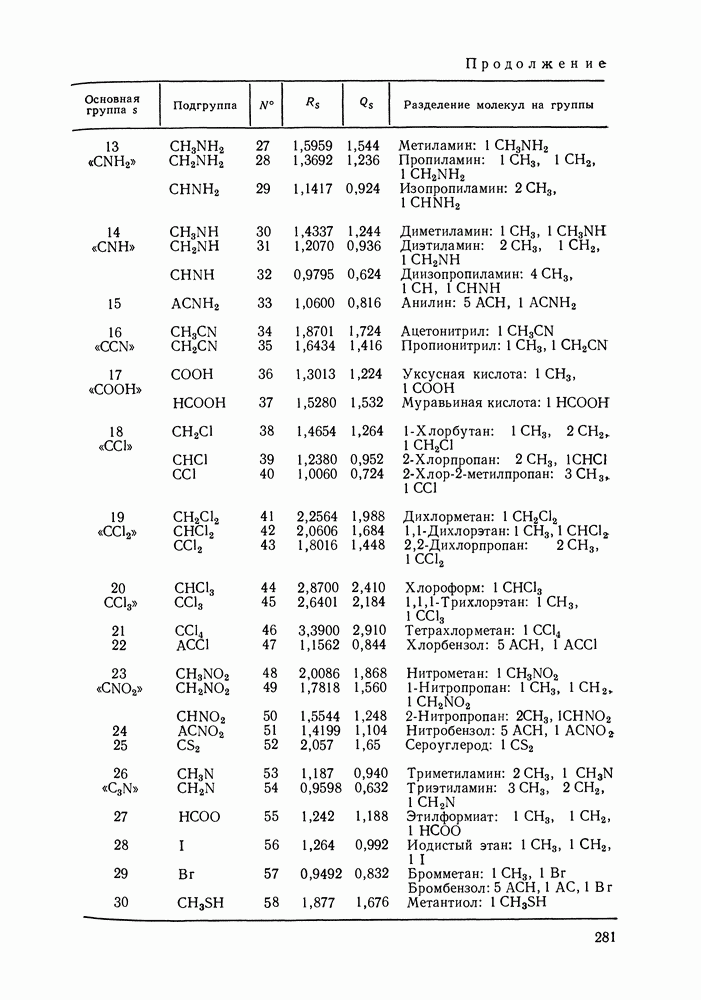
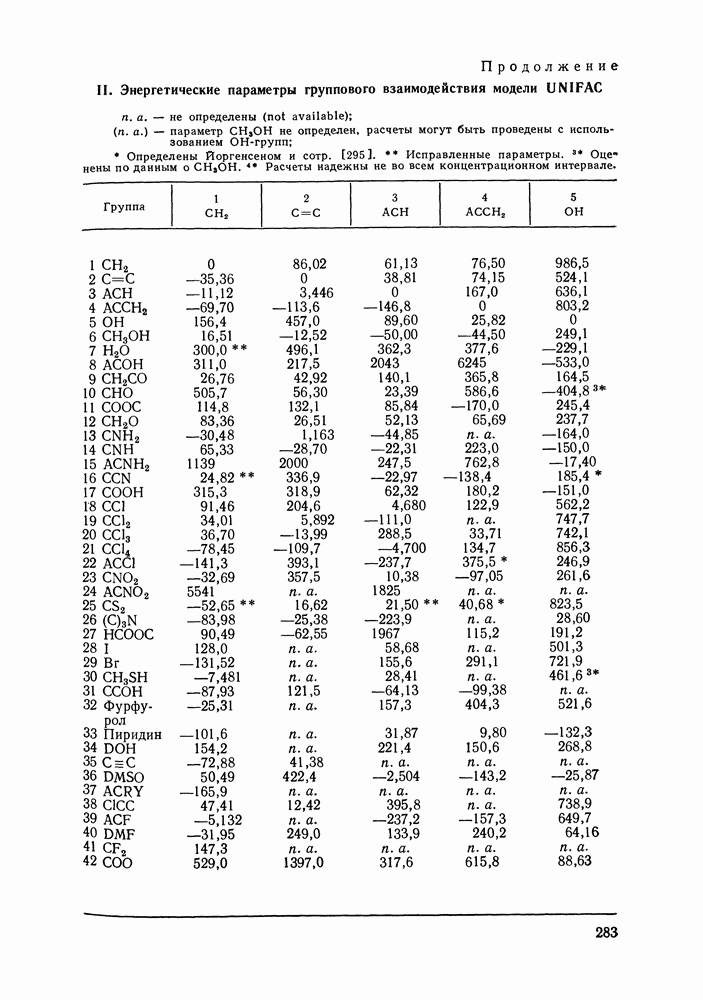
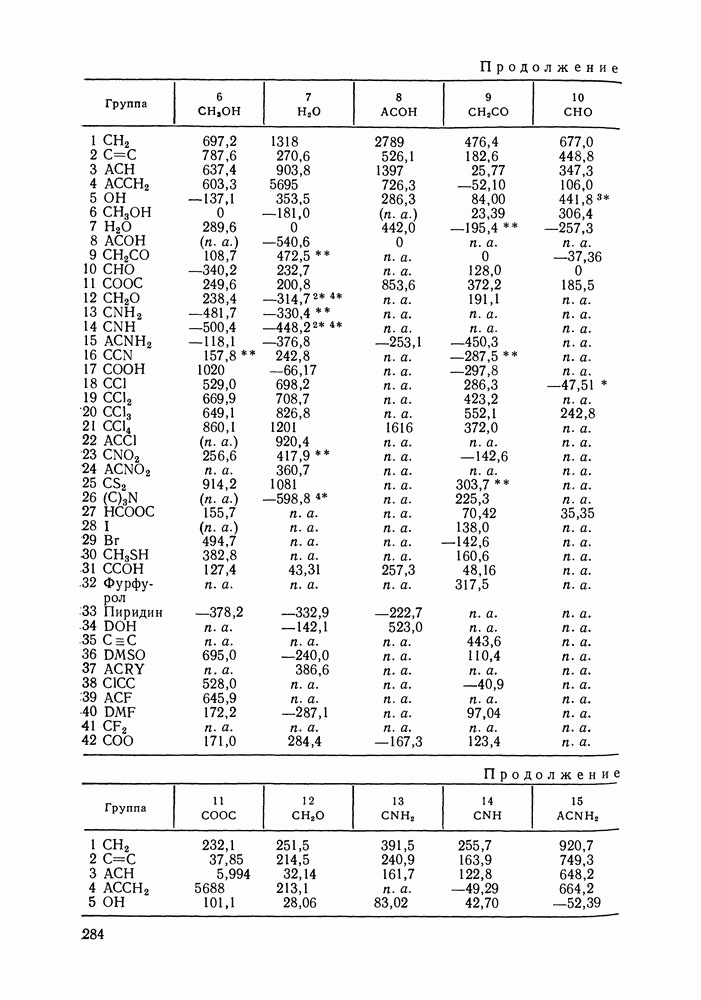
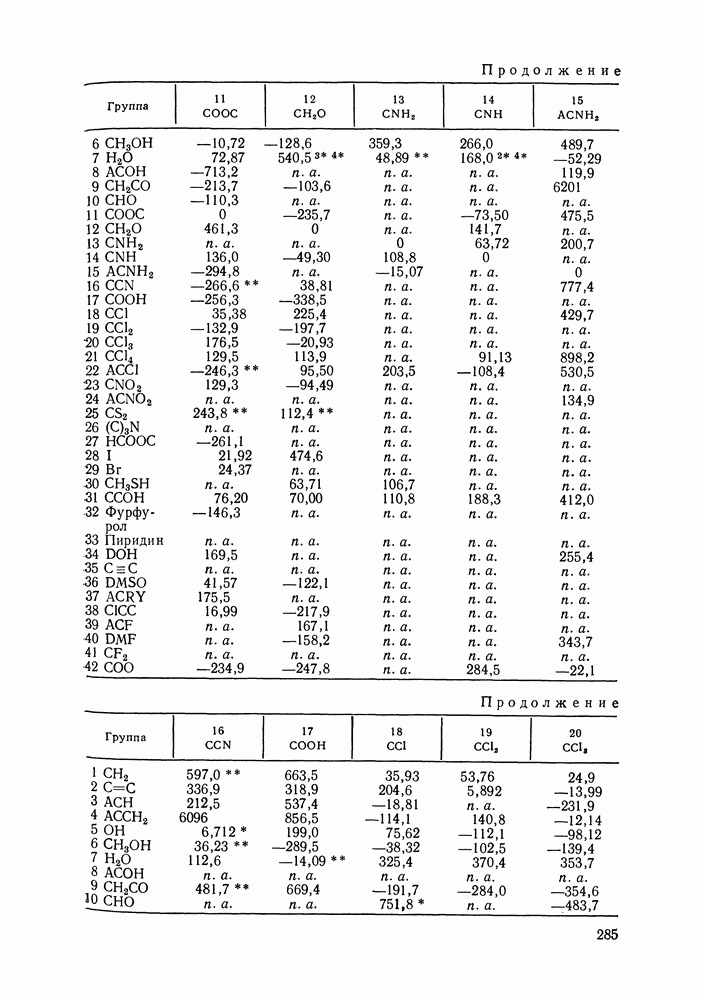
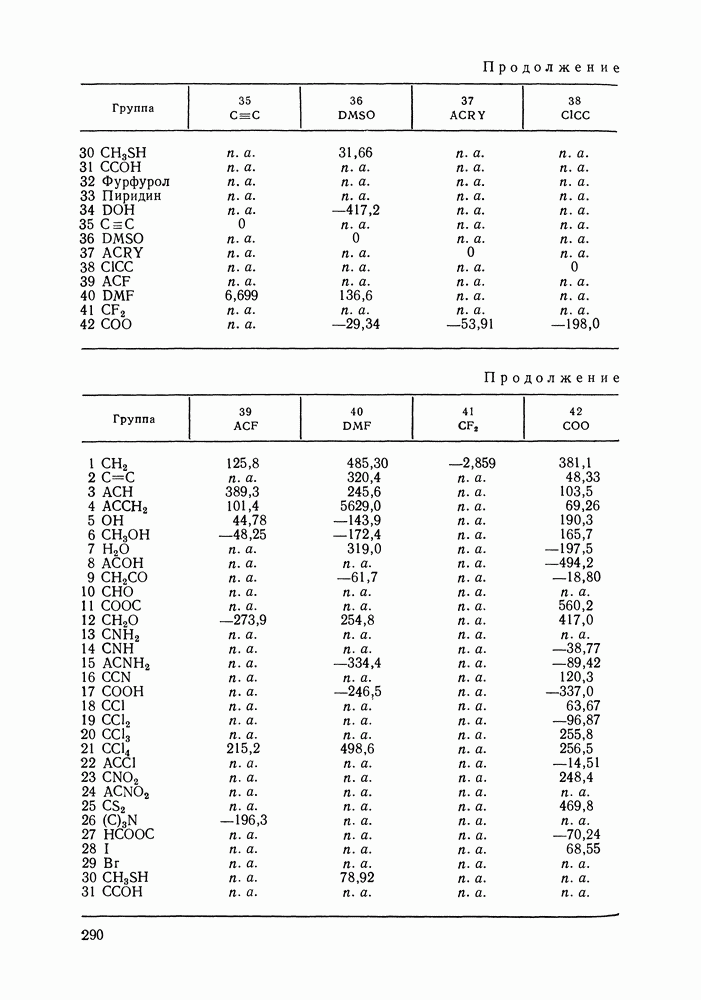
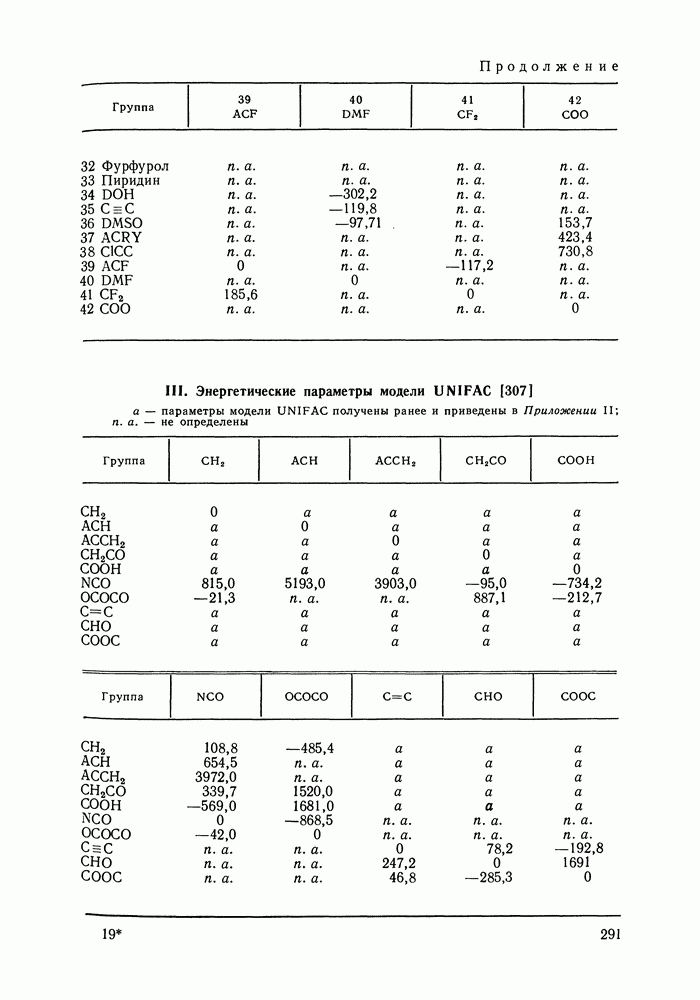
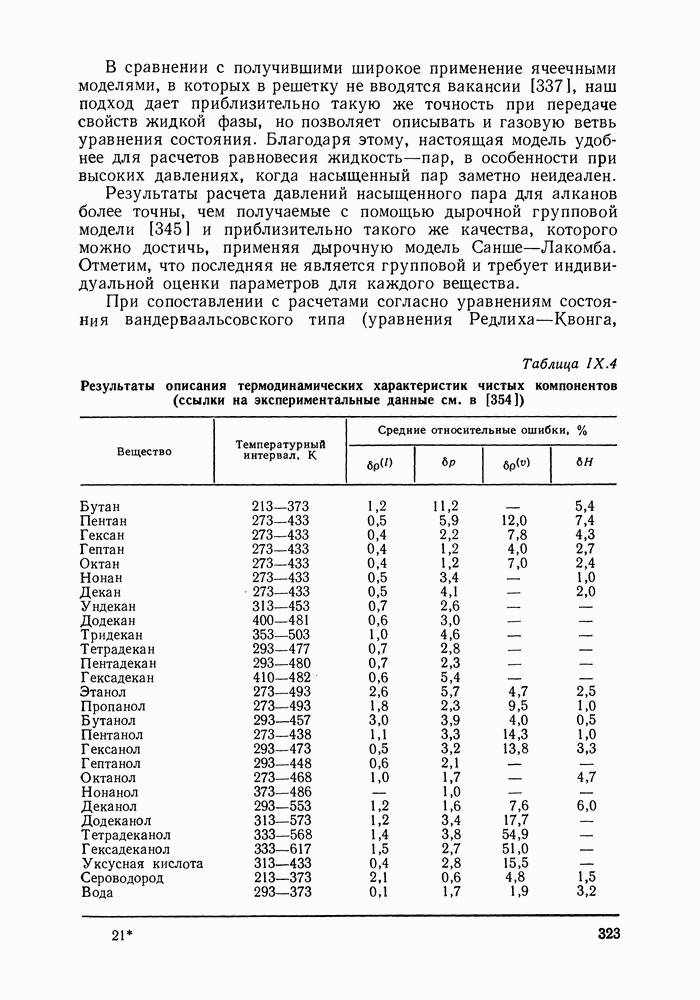
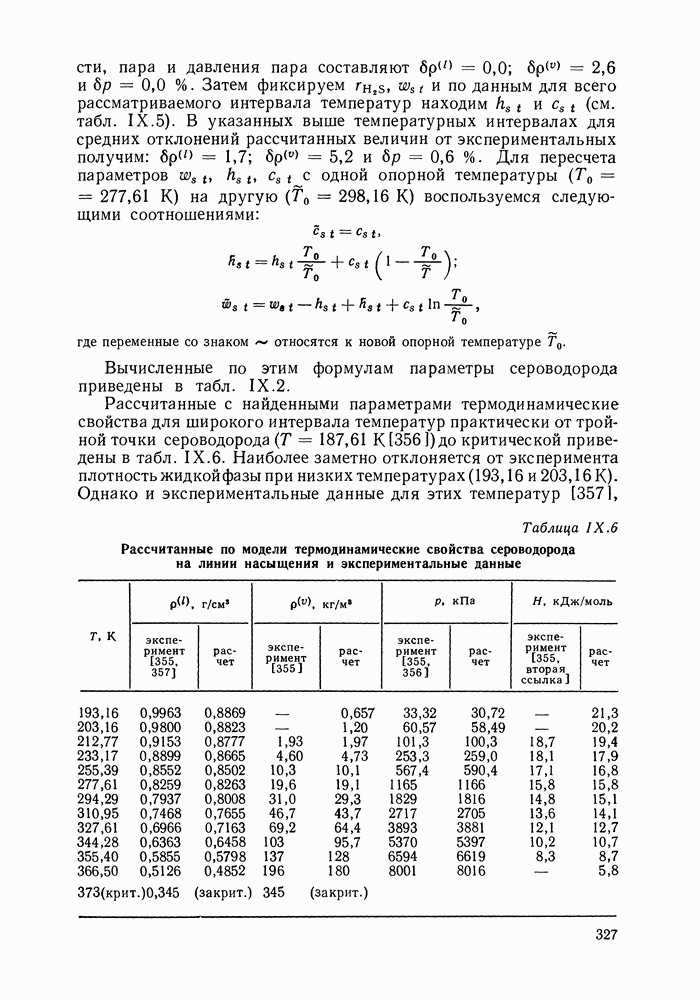
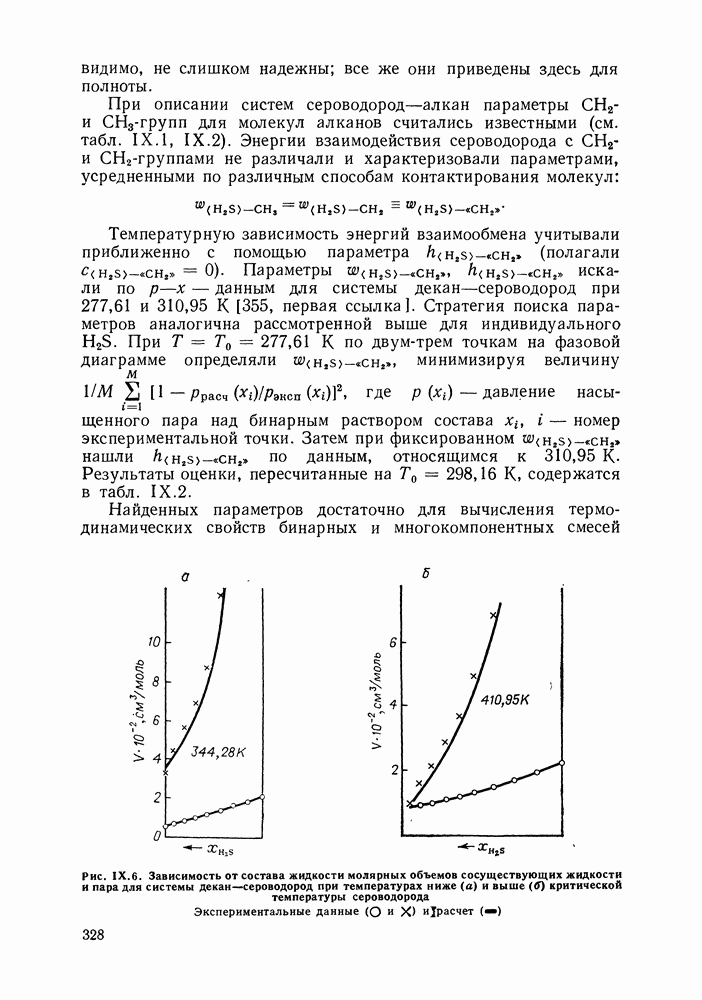
VIII.3. КВАЗИХИМИЧЕСКИЕ ГРУППОВЫЕ МОДЕЛИСохраняя общие черты рассмотренных выше групповых моделей (предположение об аддитивности вкладов групп, разделение остаточного и комбинаторного вкладов в избыточные функции), квазихимические модели обладают той особенностью, что оценка чисел пар разного рода, образуемых соседними группами (чисел контактов), проводится в квазихимическом приближении. Все эти модели являются по существу развитием модели Баркера— Гуггенгейма (см. разд. VII.8). В большинстве работ рассматриваются модели, основанные на представлении о системе без вакансий и предназначенные, подобно самой модели Баркера, исключительно для описания функций смешения [328]. Возможность одновременного описания и жидкой, и паровой фазы дает дырочная квазихимическая групповая модель (см. гл. IX). Впервые групповая квазихимическая модель растворов была сформулирована Кехиаяном и сотр. [325]. Отличие этой модели от модели Баркера состоит прежде всего в том, что вместо контактных участков молекулы рассматриваются группы в целом, и параметры взаимодействия предполагаются для данной пары групп постоянными при заданной температуре, независимо от того, какой конкретно молекуле принадлежат группы. По-иному интерпретируются величины и Величина При использовании модели учитывается температурная зависимость энергии взаимообмена, при этом в общем случае для каждой пары разноименных групп вводится три энергетических параметра. Расчеты по модели проводили для многих систем, включающих алканы, бензол, алкилбензолы, алканоны, фенил-алканоны, амины, алифатические и ароматические эфиры, альдегиды, хлоралканы. В числе исследуемых свойств были Показано, что модель хорошо описывает смеси неполярных и слабо полярных жидкостей, но результаты для растворов сильно ассоциированных жидкостей, например, спиртов неудовлетворительны. Несколько лучшее описание дает модификация модели, названная DISQUAC (DISpersive QUAsi Chemical model), где помимо квазихимического, вводится «дисперсионный» вклад, оцениваемый в приближении беспорядочной смеси [329]. Однако и эта модификация не обеспечивает хороших результатов для сильно ассоциированных смесей. Несколько иные и близкие друг другу варианты групповой квазихимической модели рассмотрены в работах Специфика подхода [326] состоит в том, что на сложных группах выделяются различные контактные участки (допустим, на гидроксильной группе — контактные участки Применение модели к некоторым ассоциированным системамОстановимся подробнее на модели [326] и ее применении для расчета термодинамических свойств и фазовых равновесий в бинарных и трехкомпонентных растворах неэлектролитов. Комбинаторная составляющая Их молярные значения следующие:
т. е. при
Величина остаточного вклада
где
Здесь Удобство переменных Избыточная энтальпия раствора определяется следующим: выражением:
где Температурная зависимость энергий взаимообмена
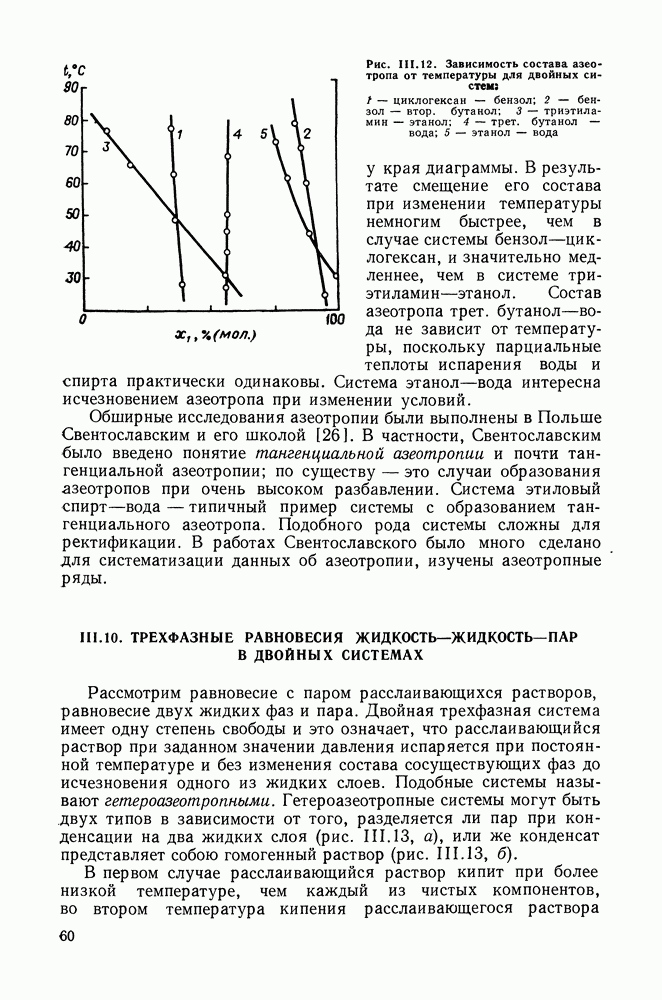
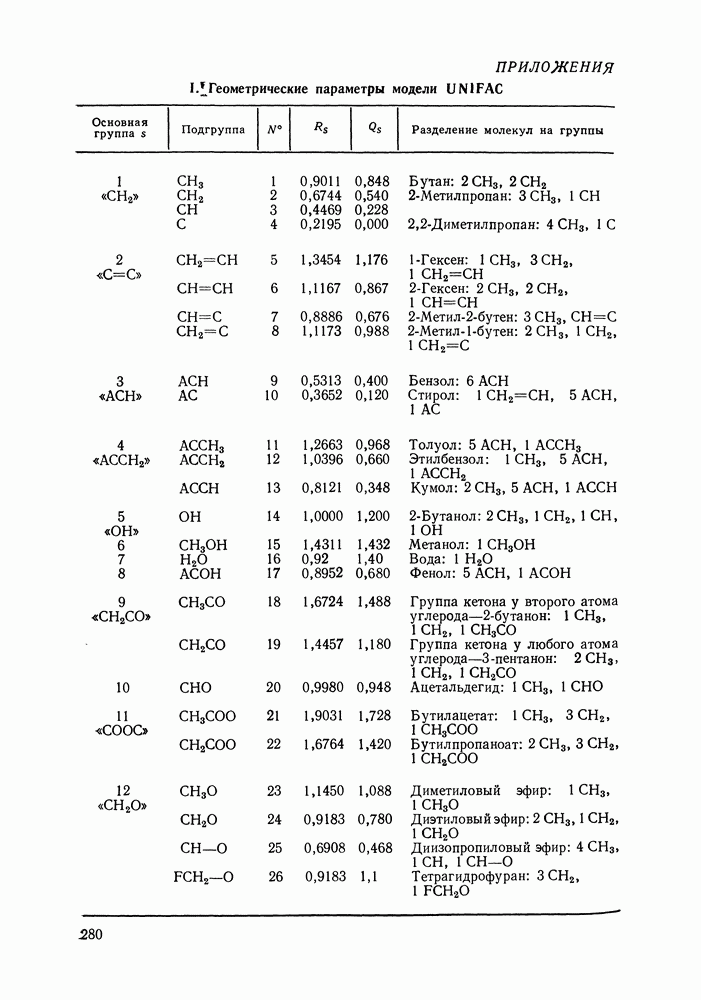
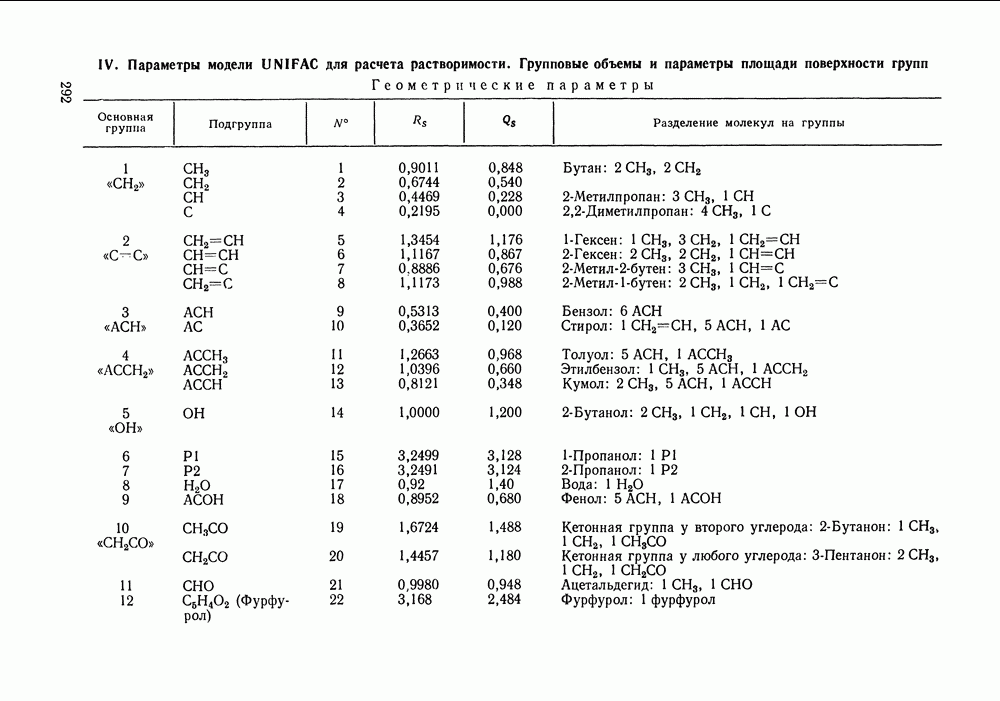
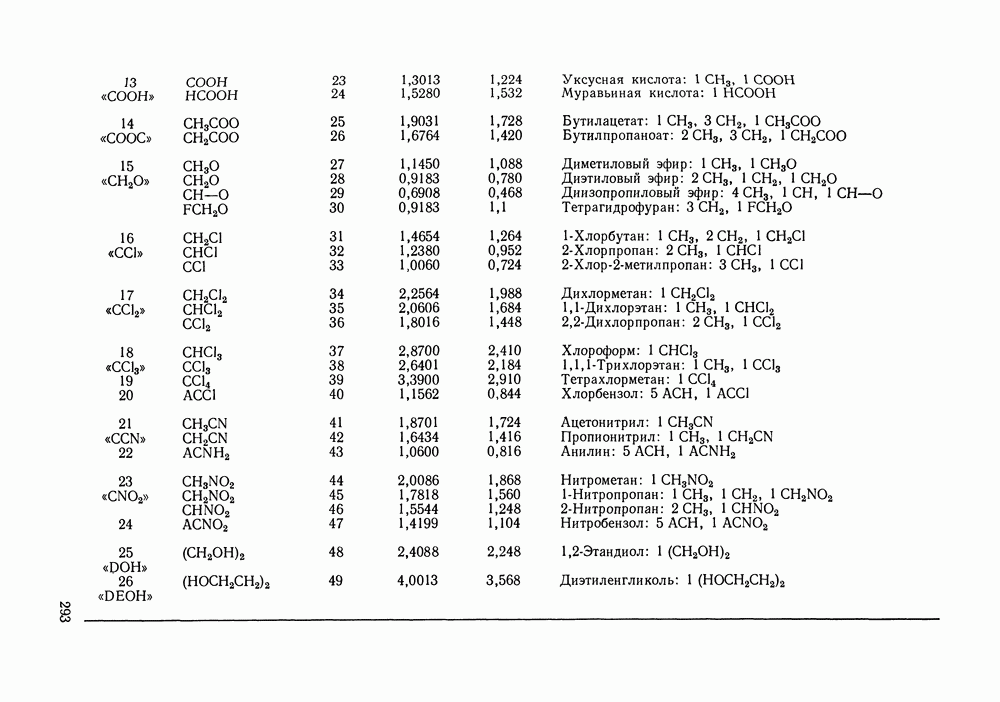
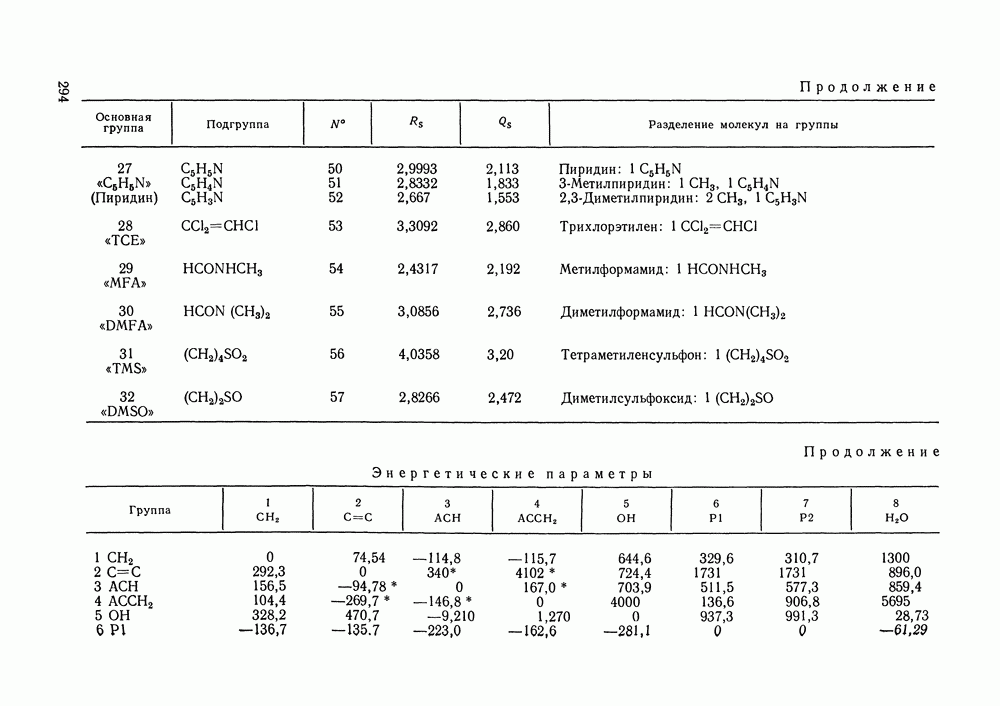
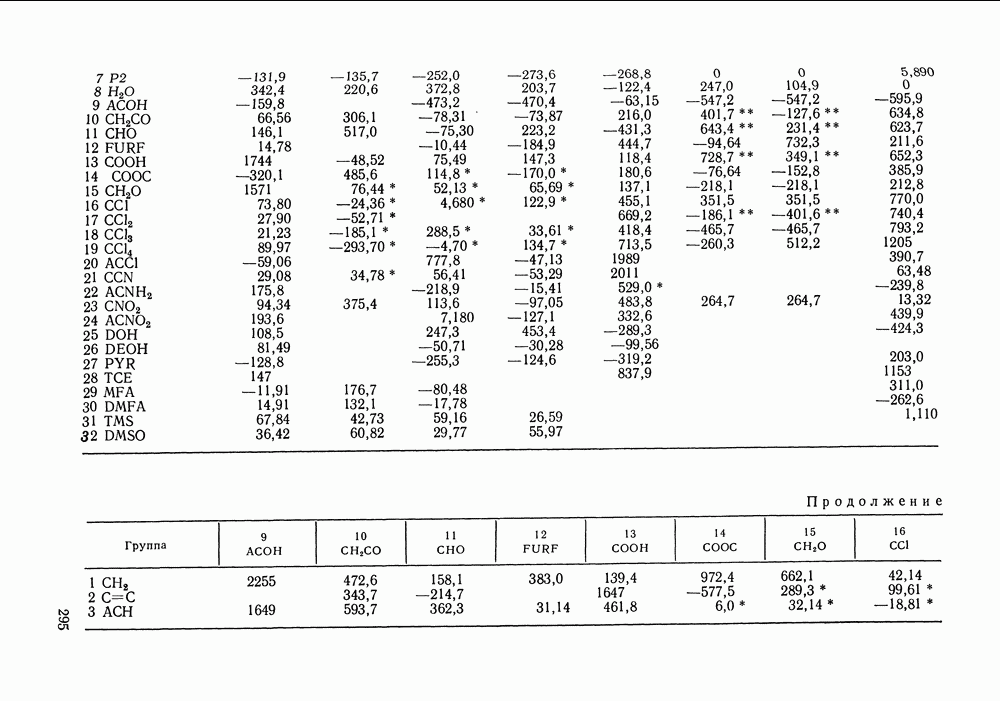
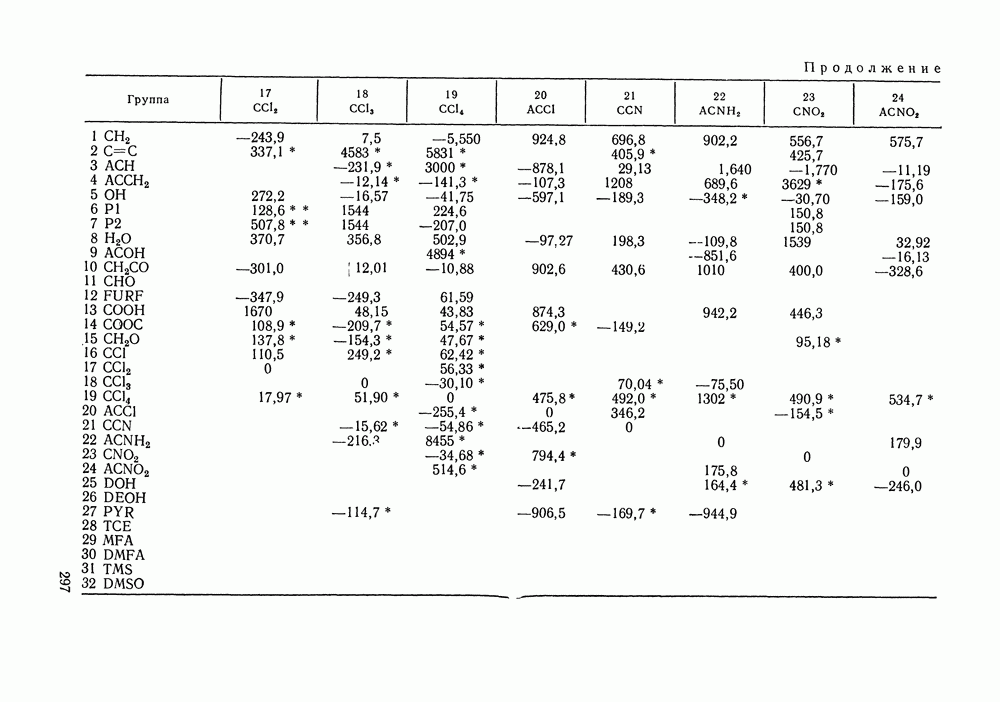
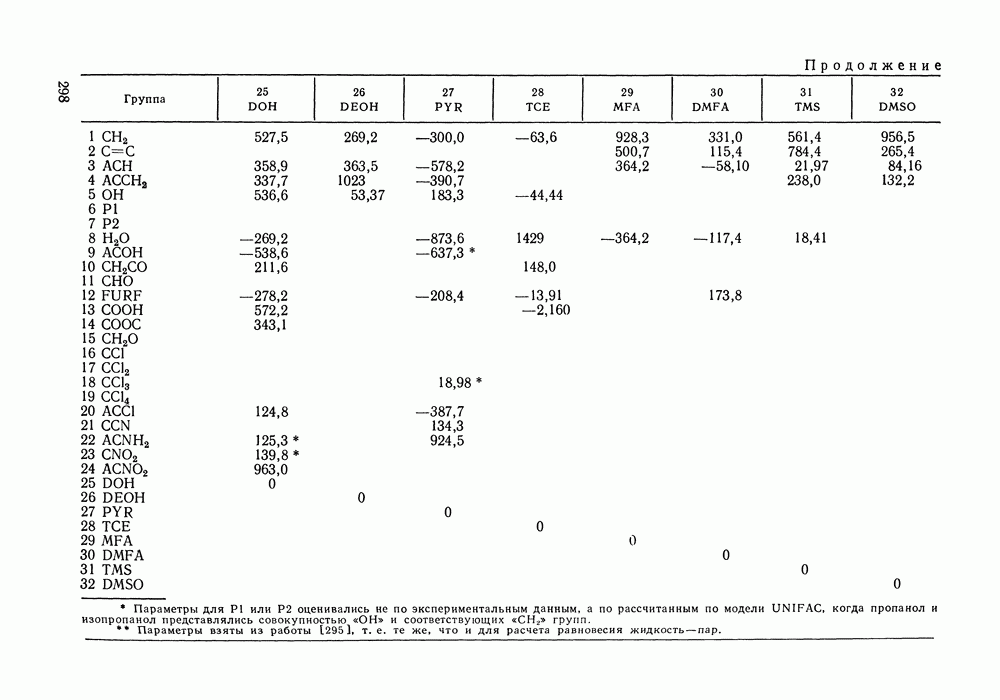
Температурная зависимость Модель применяли для расчетов Деление молекул на группы было следующим: I. Молекулы алканов образованы группами II. Молекулы алкенов и алкинов содержат группы III. Молекулы перфторалканов — группы Таблица VIII.8 (см. скан) Геометрические параметры квазихимической групповой модели. Условно принято: IV. Молекулы алканолов — группы V. Молекулы целлозольвов включают группы При определении геометрических параметров групп Однако, имея смысл некоторых эффективных характеристик, эти параметры могут в модели принимать значения, несколько отличные от единицы для Для выявления чувствительности модели к значению координационного числа практически неизменными. В связи с этим все дальнейшие расчеты по этой модели проводились при Заметим, что при расчетах, где группы рассматривались в целом и отдельные контактные участки не выделялись, для ряда систем с ориентационными эффектами было обнаружено заметное влияние координационного числа Энергетические параметры модели оценивали по экспериментальным данным о коэффициентах активности и энтальпиях смешения в бинарных системах. В качестве целевой функции при этом использовали следующую
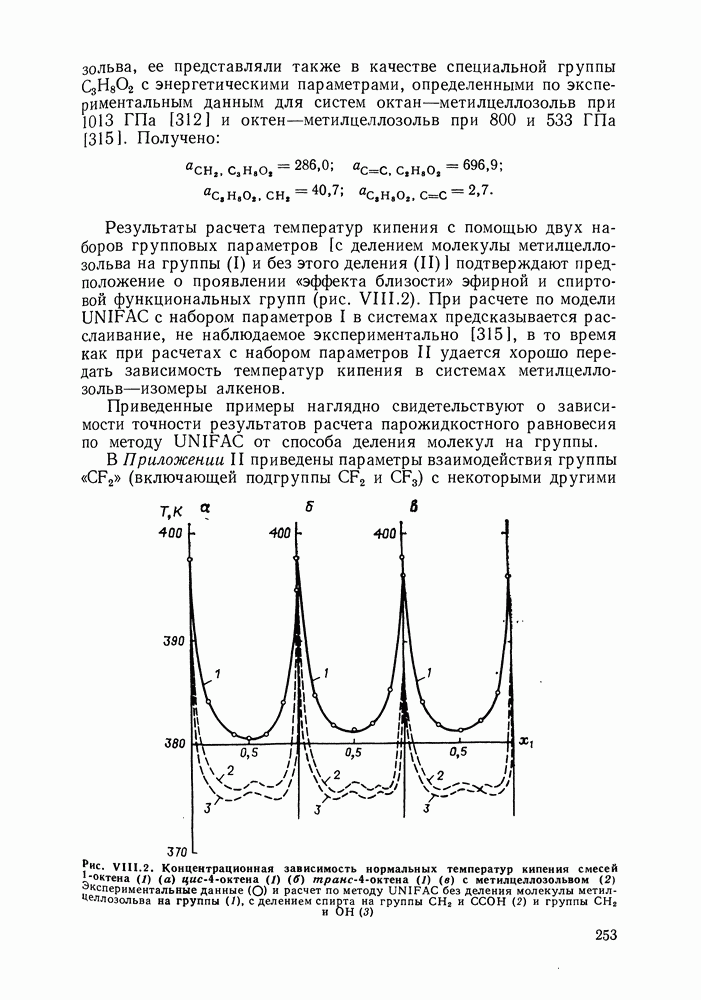
где Полученные энергетические параметры модели представлены в табл. VIII.9. Групповая квазихимическая модель позволила с удовлетворительной точностью описывать данные о равновесиях жидкость-пар и жидкость—жидкость в бинарных и тройных системах (рис. VIII.5; VIII.6; табл. Хорошо согласуются с экспериментом результаты расчета равновесия жидкость—жидкость и гетероазеотропов для смесей алканол—перфторалкан, характеризующихся малой взаимной растворимостью компонентов (см. табл. VIII. 10). Результаты предсказания равновесия жидкость— жидкость в тройных системах пропанол — гексан — перфторгексан и
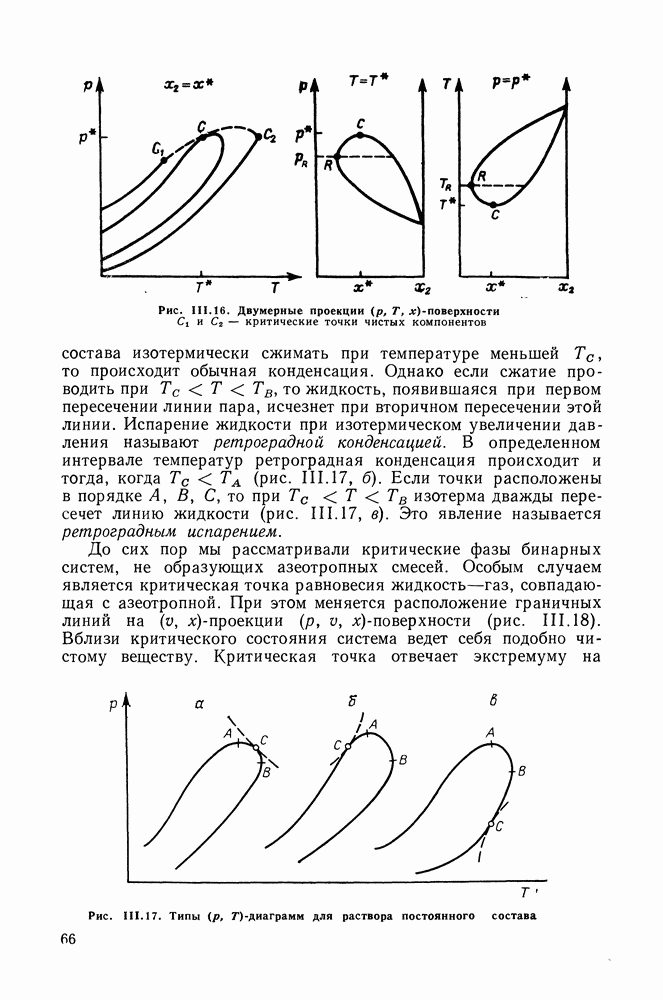
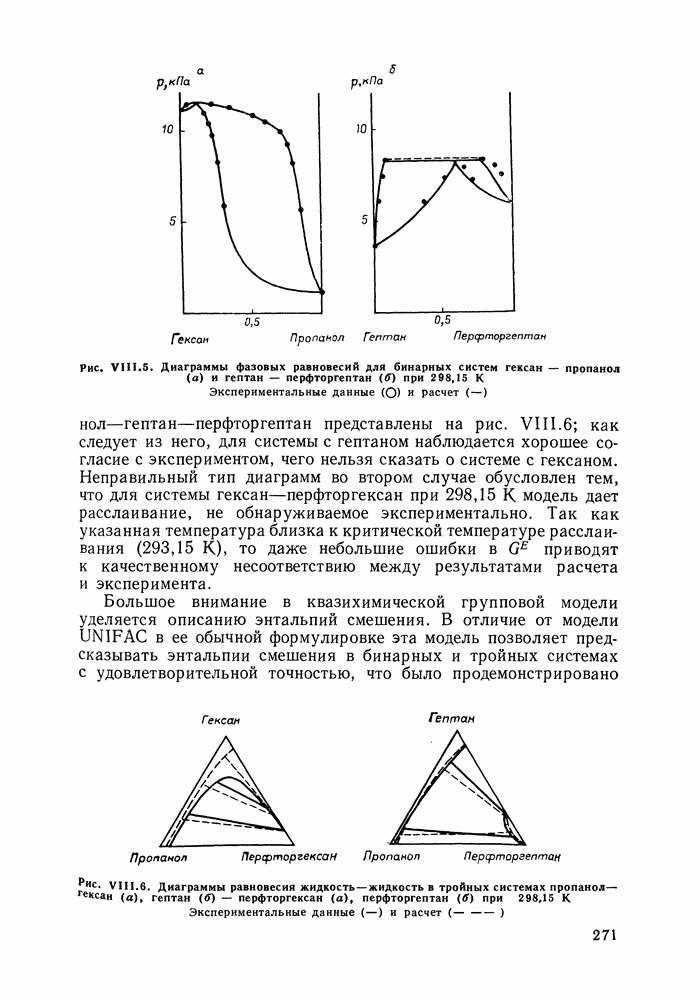
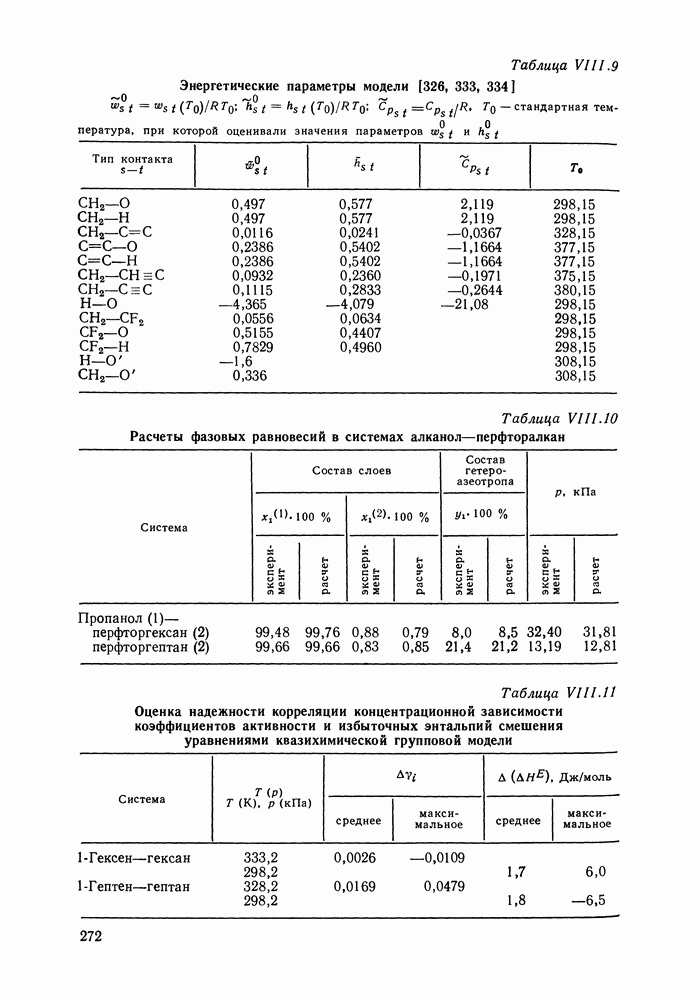
Рис. VIII.5. Диаграммы фазовых равновесий для бинарных систем гексан — пропанол (а) и гептан — перфторгептан (б) при Экспериментальные данные пропанол—гептан—перфторгептан представлены на рис. VIII.6; как следует из него, для системы с гептаном наблюдается хорошее согласие с экспериментом, чего нельзя сказать о системе с гексаном. Неправильный тип диаграмм во втором случае обусловлен тем, что для системы гексан—перфторгексан при 298,15 К модель дает расслаивание, не обнаруживаемое экспериментально. Так как указанная температура близка к критической температуре расслаивания (293,15 К), то даже небольшие ошибки в Большое внимание в квазихимической групповой модели уделяется описанию энтальпий смешения. В отличие от модели UNIFAC в ее обычной формулировке эта модель позволяет предсказывать энтальпии смешения в бинарных и тройных системах с удовлетворительной точностью, что было продемонстрировано
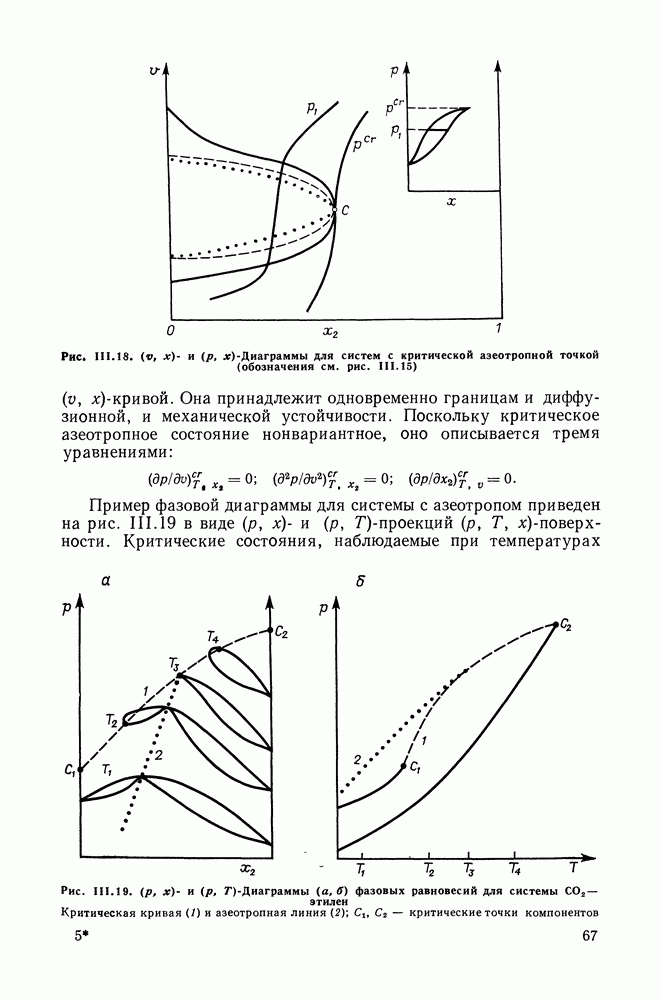
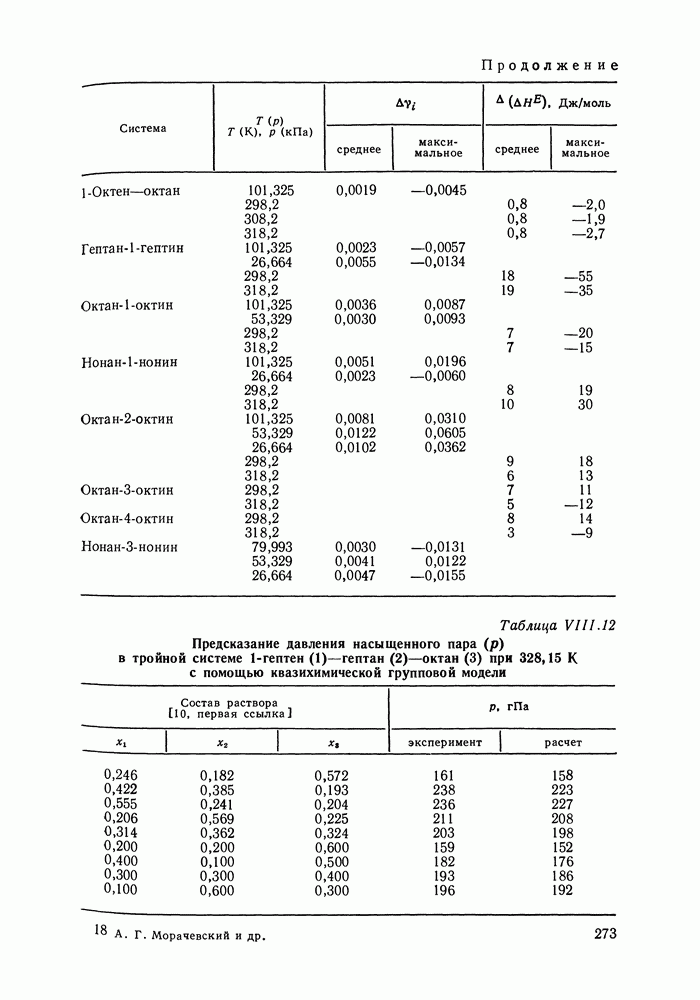
Рис. VIII.6. Диаграммы равновесия жидкость—жидкость в тройных системах пропанол—гексан (см. скан) (см. скан) Рис. VIII.7. (см. скан) Энтальпии смешения на примере систем алкан—алканол (рис. VIII.7, VIII.8) [326] алкан—алкен и алкан—алкин [см. табл. VIII.11, VIII.12» рис. VIII.9) [334]. Результаты предсказания энтальпий смешения значительно улучшаются при учете температурной зависимости энтальпий взаимообмена, т. е. при использовании формулы (VIII.31). С помощью групповой квазихимической модели предсказывались предельные коэффициенты активности компонентов в бинарных и трехкомпонентных системах, содержащих алканы, алканолы и перфторалканы (см. табл. VIII.5 и VIII.6). Если учесть значительность интервала изменения величин
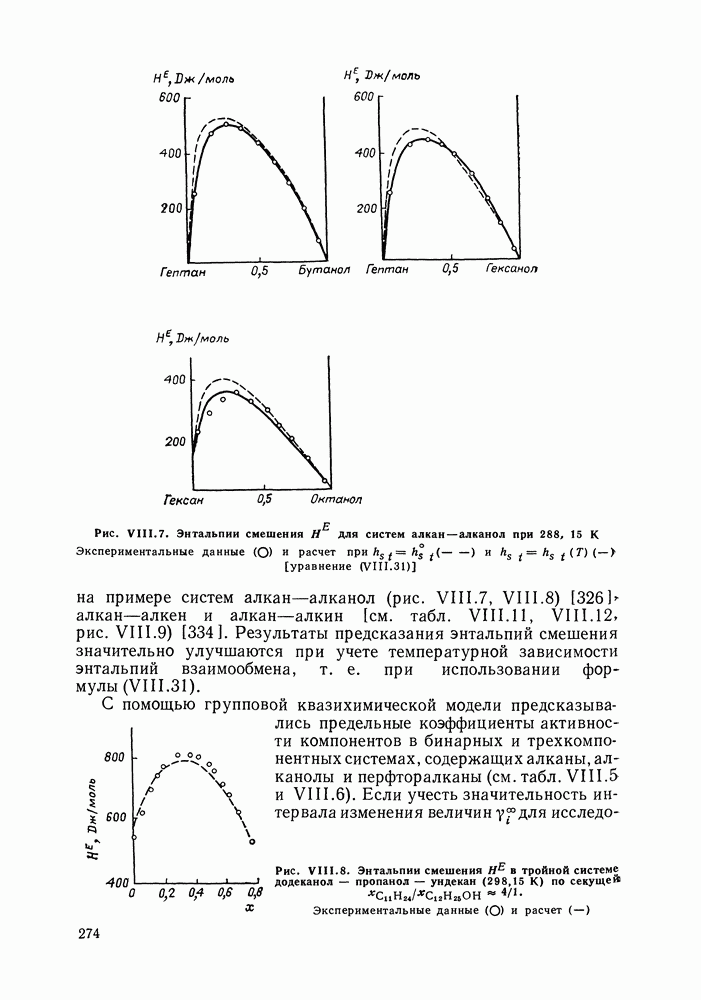
Рис. VIII.8. Энтальпии смешения
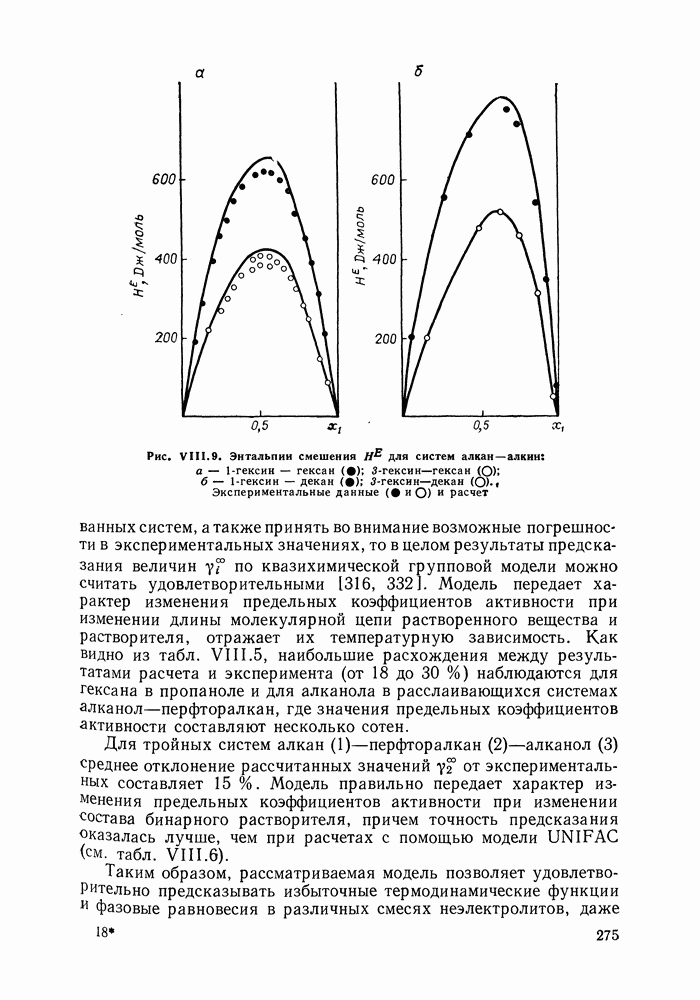
Рис. VIII.9. Энтальпии смешения исследованных систем, а также принять во внимание возможные погрешности в экспериментальных значениях, то в целом результаты предсказания величин Для тройных систем алкан Таким образом, рассматриваемая модель позволяет удовлетворительно предсказывать избыточные термодинамические функции и фазовые равновесия в различных смесях неэлектролитов, даже с такими сильно ассоциированными компонентами, как спирты. Представляется, что выделение контактных участков Модель растворов с образованием внутримолекулярных водородных связейОсобый случай — это растворы, в которых возможно образование не только можно и внутримолекулярных связей. К таким: системам относятся, в частности, растворы целлозольвов (моноэфиры этиленгликоля), где внутримолекулярная водородная связь возникает между водородом гидроксила и кислородом эфирной группы. Компонент, способный к образованию внутримолекулярных связей, обозначим через А. Модель Баркера и рассмотренные выше квазихимические групповые модели не различают внутримолекулярных и межмолекулярных связей. В соответствии с этими моделями в бесконечно разбавленном растворе какие-либо контакты между группами (контактными участками) молекул растворенного вещества А отсутствуют, имеются контакты только с молекулами растворителя Модификация квазихимической групповой модели для описания растворов, в которых один из компонентов образует и межмолекулярные, и внутримолекулярные водородные связи, предложена в работе [333]. Эта модель сочетает обычный вариант квазихимической групповой модели с теорией ассоциативных равновесий. Считаем, что бинарный раствор
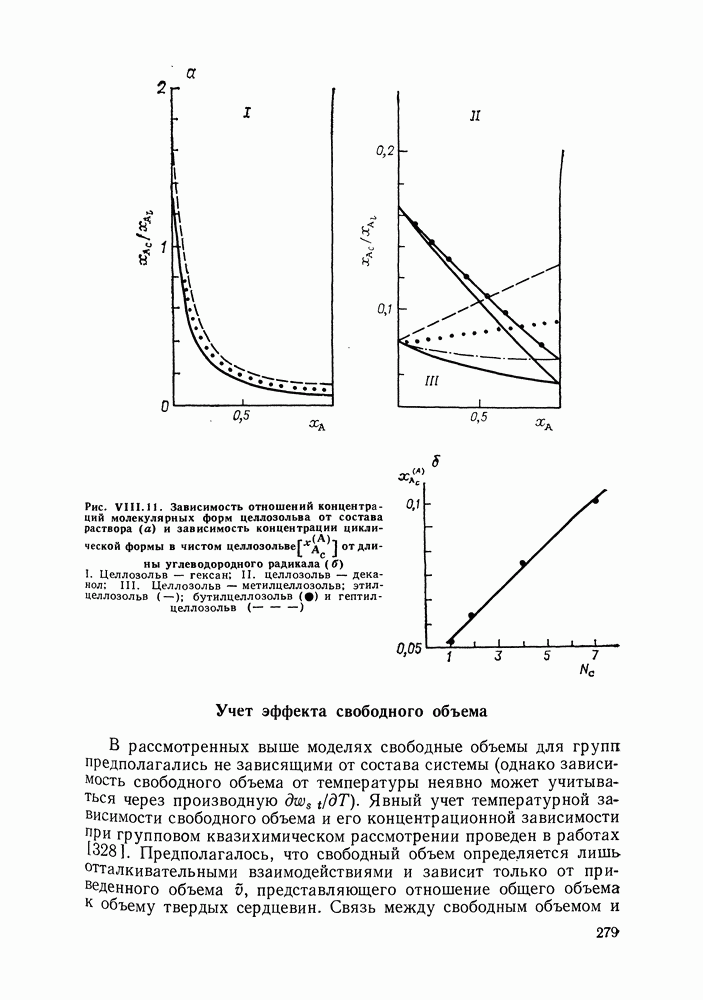
Применяя формулы квазихимической групповой модели [326] для оценки избыточных химических потенциалов молекулярных форм
где
величины Для расчета коэффициента активности по формулам
где величина
Здесь По спектральным данным для метилцеллозольва, растворенного в гексане, Параметры модели, определенные для растворов целлозольв— гексан (в частности, значение
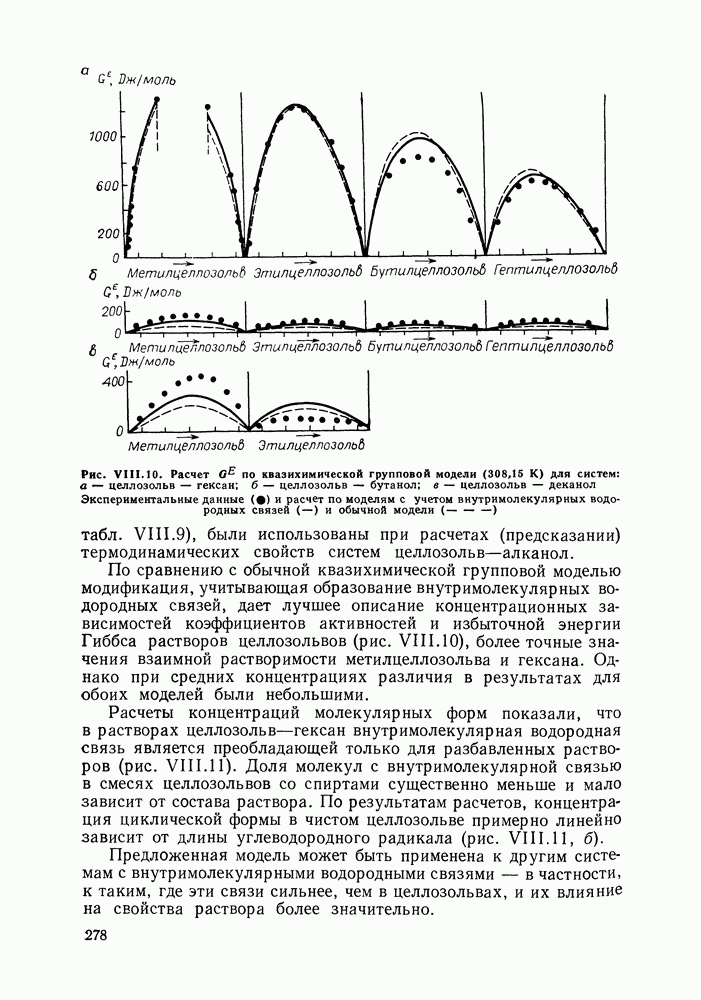
Рис. VIII. 10. Расчет табл. VIII.9), были использованы при расчетах (предсказании) термодинамических свойств систем целлозольв—алканол. По сравнению с обычной квазихимической групповой моделью модификация, учитывающая образование внутримолекулярных водородных связей, дает лучшее описание концентрационных зависимостей коэффициентов активностей и избыточной энергии Гиббса растворов целлозольвов (рис. VIII.10), более точные значения взаимной растворимости метилцеллозольва и гексана. Однако при средних концентрациях различия в результатах для обоих моделей были небольшими. Расчеты концентраций молекулярных форм показали, что в растворах целлозольв—гексан внутримолекулярная водородная связь является преобладающей только для разбавленных растворов (рис. VIII. 11). Доля молекул с внутримолекулярной связью в смесях целлозольвов со спиртами существенно меньше и мало зависит от состава раствора. По результатам расчетов, концентрация циклической формы в чистом целлозольве примерно линейно зависит от длины углеводородного радикала (рис. VI 11.11, б). Предложенная модель может быть применена к другим системам с внутримолекулярными водородными связями — в частности, к таким, где эти связи сильнее, чем в целлозольвах, и их влияние на свойства раствора более значительно. Рис. VIII. 11. (см. скан) Зависимость отношений концентраций молекулярных форм целлозольва от состава раствора (а) и зависимость концентрации циклической формы в чистом целлозольвед Учет эффекта свободного объемаВ рассмотренных выше моделях свободные объемы для групп: предполагались не зависящими от состава системы (однако зависимость свободного объема от температуры неявно может учитываться через производную (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) (см. скан) приведенным устанавливалась с помощью уравнения состояния флюида твердых сфер в приближении Карнахана-Старлинга. От приведенного объема зависела и энергия контактов
где Выражения для энергии взаимодействия пар и ячеечные статистические суммы вводили в статистическую сумму системы; наиболее вероятные числа пар и число конфигураций оценивали согласно квазихимическому методу Баркера-Гуггенгейма. Полученные выражения для термодинамических функций помимо членов, фигурирующих в решеточных теориях и рассмотренных выше их групповых модификациях, содержат также члены, связанные с эффектом свободного объема. Модель дала возможность совместно рассматривать многие свойства: молекулярные объемы жидкостей, энергии испарения, коэффициенты активности компонентов раствора, энтальпии смешения и др. С хорошей точностью в широком интервале температур были описаны свойства чистых алканов, алканолов, алканонов и их смесей; успешно были предсказаны свойства ряда систем, не включенных в расчеты при оценке параметров. Модель была применена для описания термодинамических свойств воды и водных растворов алканов, алканолов, алканонов. Учет зависимости свободного объема и энергий взаимообмена от приведенного объема, изменяющегося с изменением состава системы, безусловно, делает модель раствора более адекватной по сравнению с обсужденными ранее. В то же время расчеты становятся более трудоемкими, так как включают большее число параметров и требуют совместного рассмотрения нескольких свойств (в том числе, обязательно объемных). Трудно сказать, насколько учет эффекта свободного объема улучшает корреляцию и предсказание коэффициентов активности и избыточных энтальпий. Результаты расчетов этих свойств для систем алканон—алкан [325, первая ссылка], где эффект свободного объема не учитывался, были не хуже, чем полученные по модели [328, первая ссылка]. Однако учет свободного объема увеличивает число свойств, которые могут быть описаны моделью при одном и том же наборе параметров, расширяет температурную область приложения модели и, по-видимому, круг охватываемых моделью систем. Действительно, учет связи свободного объема с параметрами взаимодействия (хотя бы через приведенный объем) приближает уровень рассмотрения к молекулярному, а на этом уровне в большей степени оправдано допущение об аддитивности вкладов групп в результирующие свойства. Явный учет теорией эффекта свободного объема должен уменьшить зависимость параметров взаимодействия от положения группы в молекуле, от наличия других групп. Заметим, что в работе [328] удалось с одними и теми же энергетическими параметрами удовлетворительно описать свойства и индивидуальных спиртов от метанола до деканола и свойства их растворов, тогда как при расчетах без учета эффекта свободного объема для метанола требуются особые параметры. Как уже отмечалось, необходимым является учет эффекта свободного объема для растворов полимеров. Дальнейшее развитие квазихимических групповых моделей связано с введением в решетку вакансий, что позволит описывать одновременно как жидкую, так и газовую фазу. Применению таких моделей посвящена гл. IX.
|
 |
Оглавление
|















































































































































































































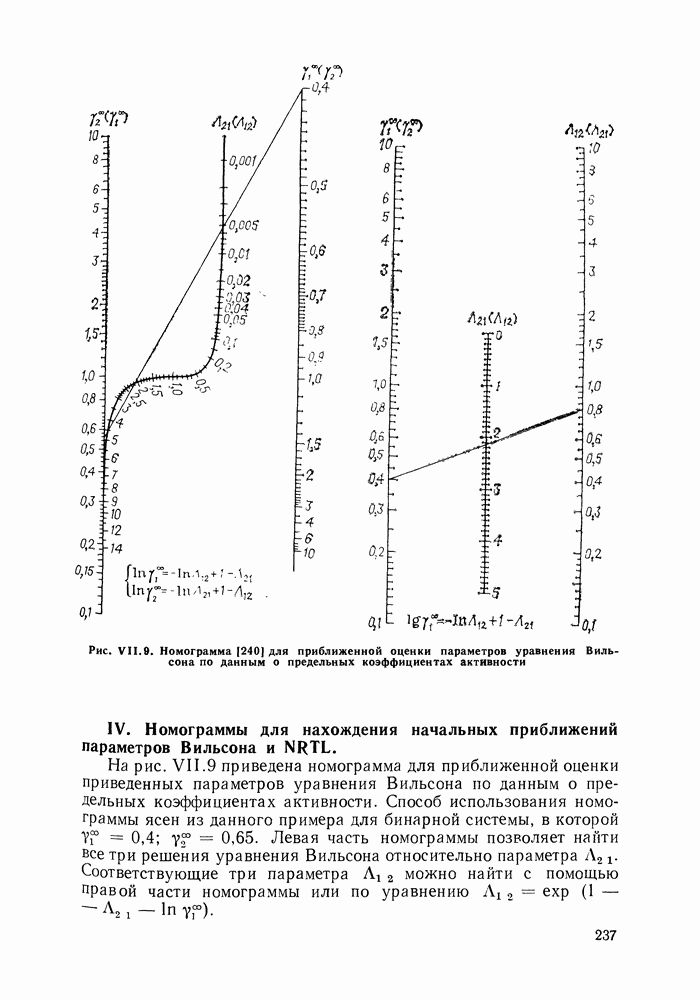
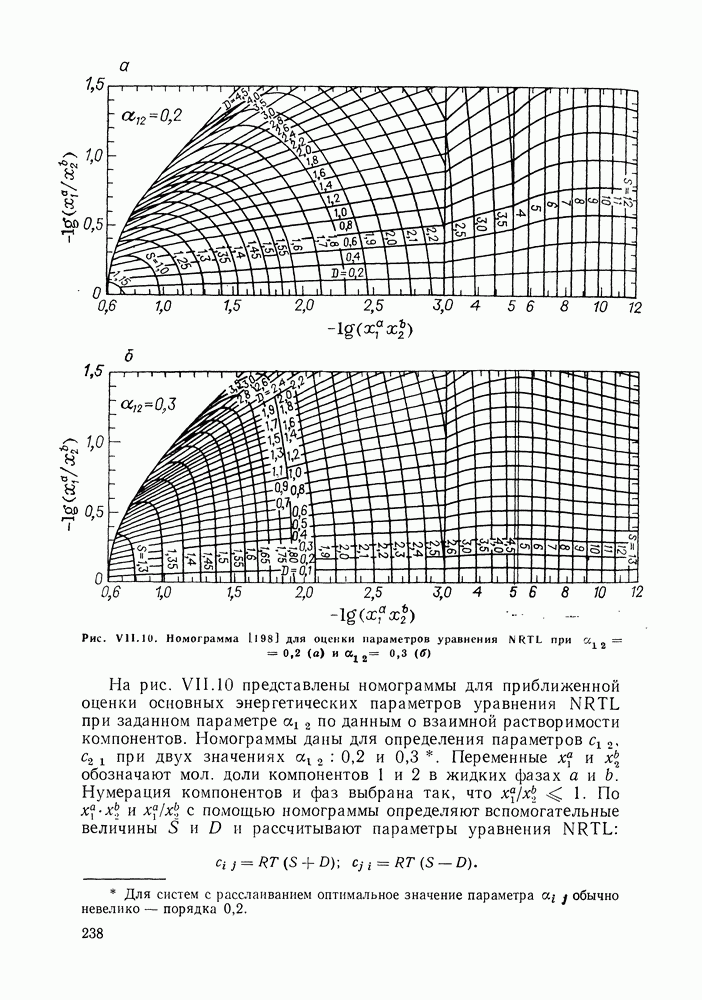





























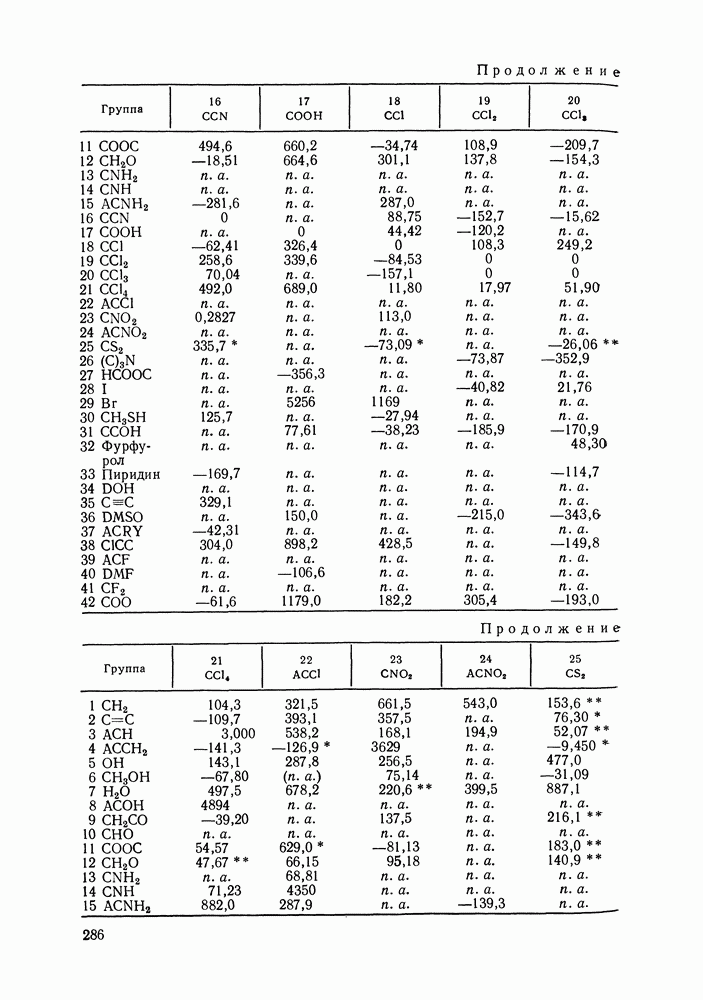
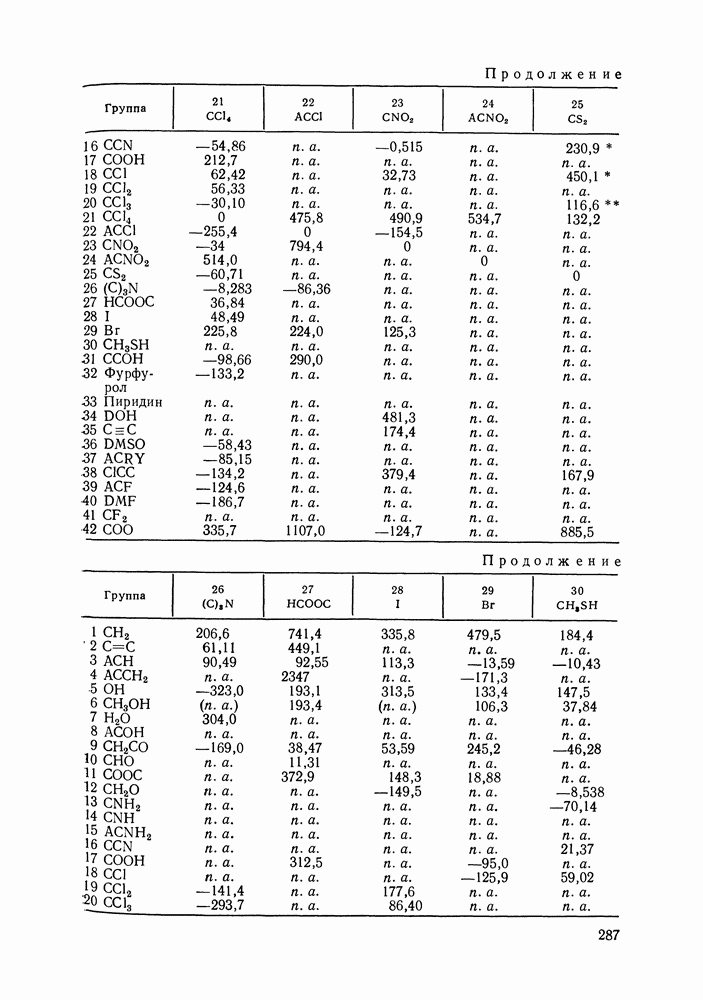
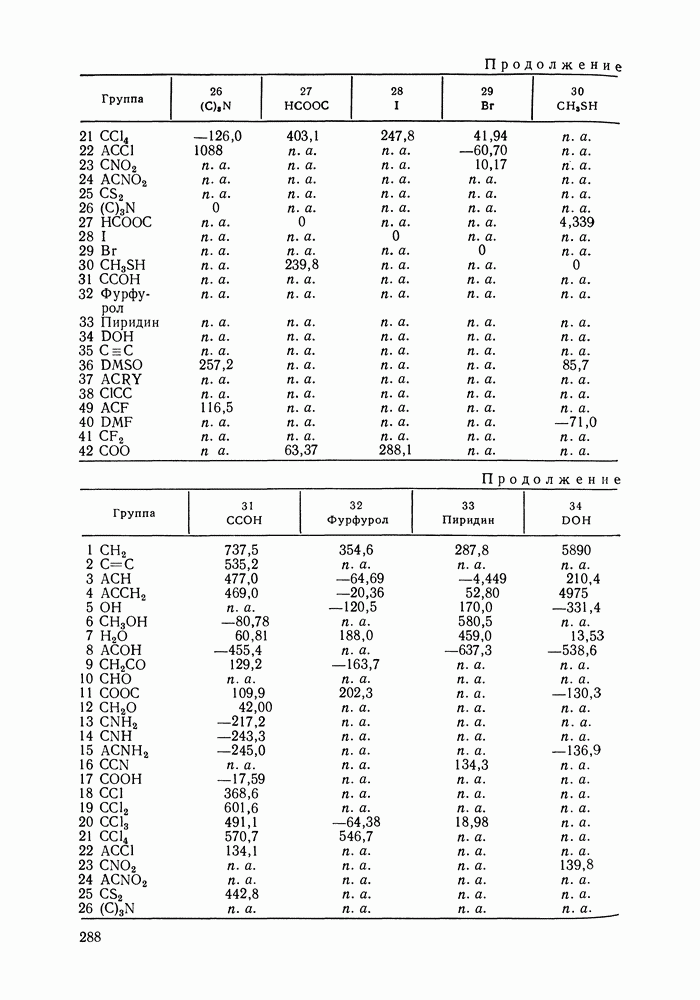
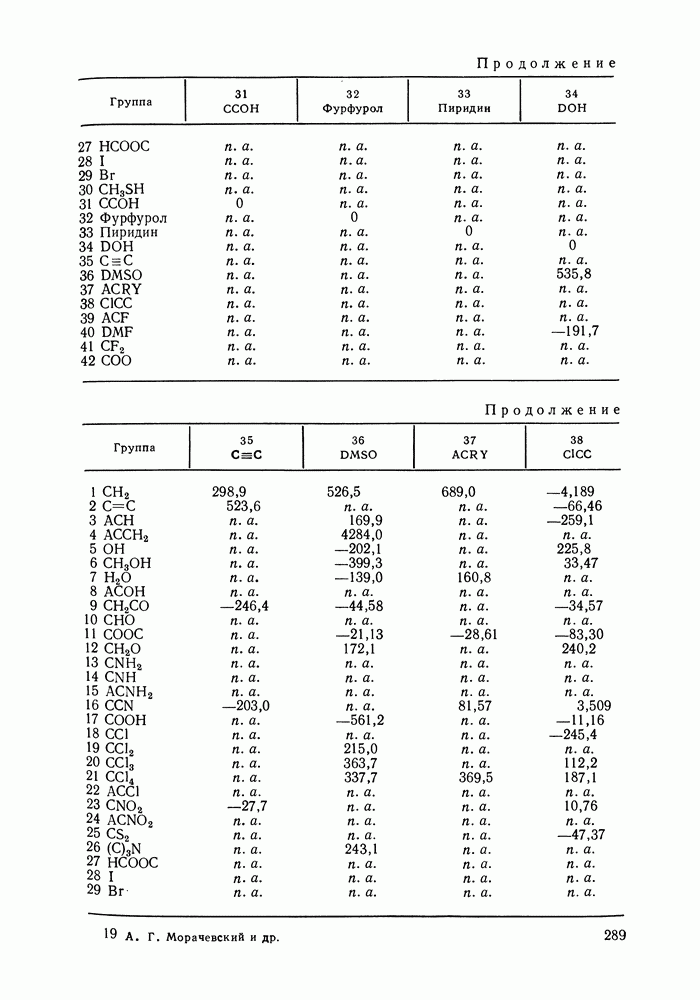




















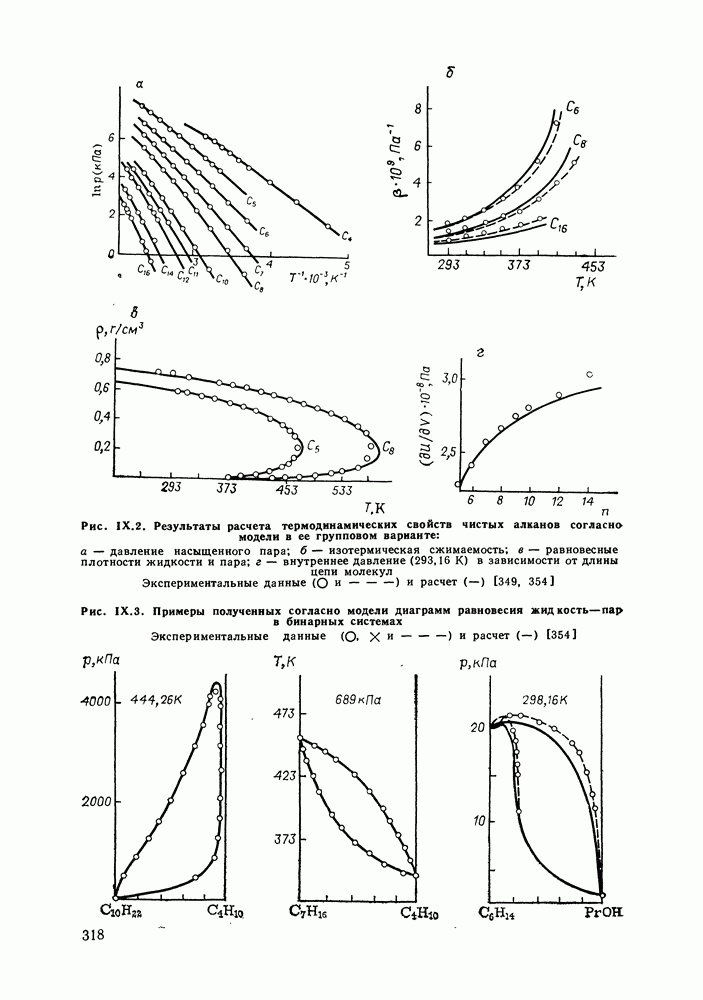
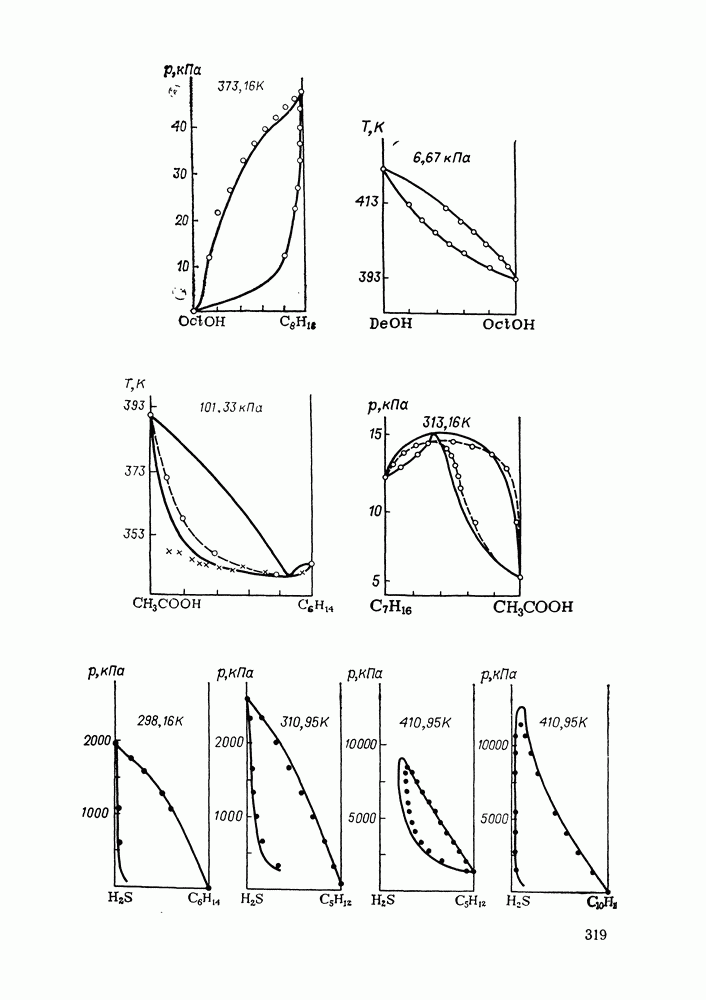

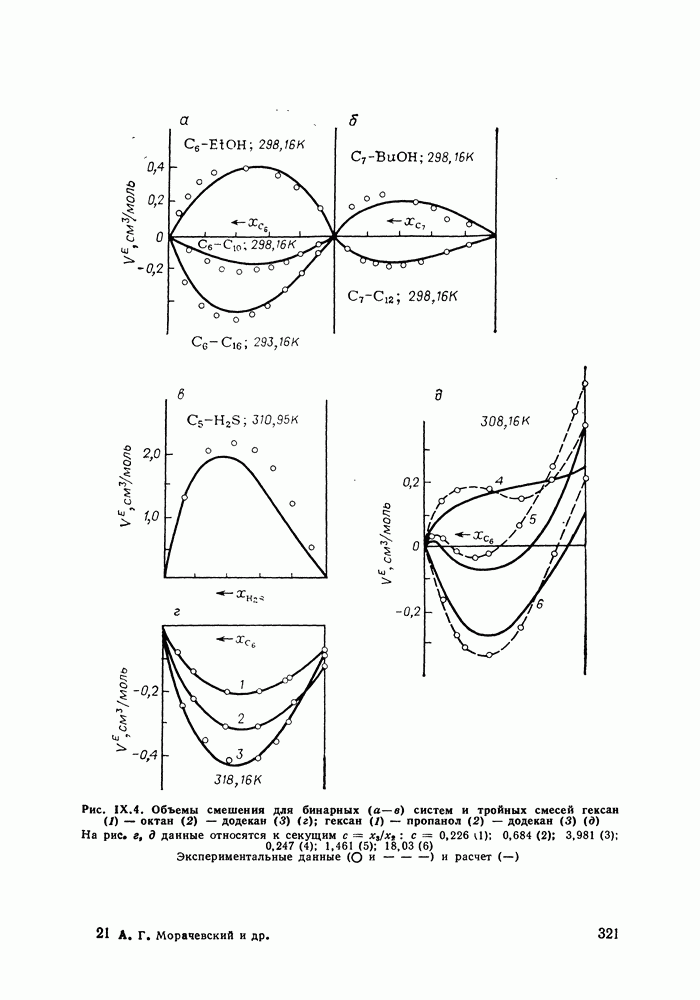
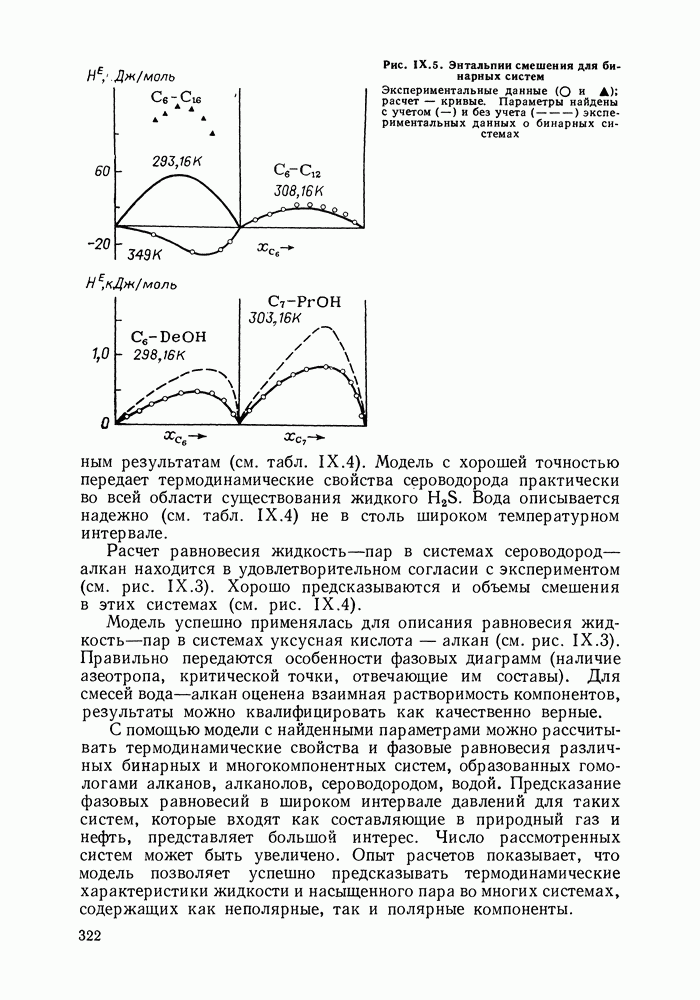
















 жидких растворов [279, с. 87; 325—327]. Предложен Вариант модели, учитывающий вклад свободного объема и позволяющий включить в рассмотрение также объемные эффекты
жидких растворов [279, с. 87; 325—327]. Предложен Вариант модели, учитывающий вклад свободного объема и позволяющий включить в рассмотрение также объемные эффекты
 которые уже не связаны с определенной геометрией решетки, а имеют смысл общего объема и поверхности молекулы и находятся по таблицам Бонди [220].
которые уже не связаны с определенной геометрией решетки, а имеют смысл общего объема и поверхности молекулы и находятся по таблицам Бонди [220].
 выступает скорее не как координационное число, а как параметр, позволяющий учесть ориентационную упорядоченность (предельный случай
выступает скорее не как координационное число, а как параметр, позволяющий учесть ориентационную упорядоченность (предельный случай  отвечает отсутствию упорядоченности). Авторы нашли, что в случае систем, молекулы которых неоднородны по структуре, т. е. содержат различные группы, изменение числа
отвечает отсутствию упорядоченности). Авторы нашли, что в случае систем, молекулы которых неоднородны по структуре, т. е. содержат различные группы, изменение числа  для групп влияет на асимметрию рассчитанных кривых избыточных функций (в наибольшей степени это влияние проявляется для растворов с одним полярным компонентом). Координационное число рассматривают как варьируемый параметр и допускают, что различным группам можно приписывать разные значения
для групп влияет на асимметрию рассчитанных кривых избыточных функций (в наибольшей степени это влияние проявляется для растворов с одним полярным компонентом). Координационное число рассматривают как варьируемый параметр и допускают, что различным группам можно приписывать разные значения  Хотя, вообще говоря, такой подход возражения не вызывает, возникают трудности в применении его к многокомпонентным системам.
Хотя, вообще говоря, такой подход возражения не вызывает, возникают трудности в применении его к многокомпонентным системам.
 взаимная растворимость жидкостей.
взаимная растворимость жидкостей.

 как и в модели Баркера, разд. VII.8). Такое сочетание группового описания с моделью контактных участков позволяет лучше передать ориентационные эффекты и ассоциацию, чем модели, рассматривающие группу в целом. Детализация модели контактных участков позволяет отказаться от варьирования координационных чисел для групп, которое требуется для модели [325], и в то же время хорошо описывать, в частности, сильно ассоциированные системы, что будет показано далее на примере растворов спиртов.
как и в модели Баркера, разд. VII.8). Такое сочетание группового описания с моделью контактных участков позволяет лучше передать ориентационные эффекты и ассоциацию, чем модели, рассматривающие группу в целом. Детализация модели контактных участков позволяет отказаться от варьирования координационных чисел для групп, которое требуется для модели [325], и в то же время хорошо описывать, в частности, сильно ассоциированные системы, что будет показано далее на примере растворов спиртов. 
 согласно модели определена формулой (VIII.11), т. е. так же, как и в модели UNIFAC. Оценка объема и поверхности групп также проводится по таблицам Бонди [220], но объем у и поверхность а стандартного сегмента выбирается в соответствии с рекомендациями работы [330].
согласно модели определена формулой (VIII.11), т. е. так же, как и в модели UNIFAC. Оценка объема и поверхности групп также проводится по таблицам Бонди [220], но объем у и поверхность а стандартного сегмента выбирается в соответствии с рекомендациями работы [330].



 модели UNIFAC соответственно
модели UNIFAC соответственно 
 получается, как уже отмечалось, при переходе в общих формулах Баркера к суммированию только по типам групп. Для рассматриваемого члена в [326, первая ссылка] предложена следующая модифицированная форма
получается, как уже отмечалось, при переходе в общих формулах Баркера к суммированию только по типам групп. Для рассматриваемого члена в [326, первая ссылка] предложена следующая модифицированная форма

 номер групп) — решение системы уравнений
номер групп) — решение системы уравнений

 молярная энергия взаимообмена для контактов
молярная энергия взаимообмена для контактов  поверхностная доля группы
поверхностная доля группы  в чистом компоненте
в чистом компоненте  поверхностная доля группы
поверхностная доля группы  в растворе;
в растворе;  площадь поверхности группы (контактного участка)
площадь поверхности группы (контактного участка)  в молекуле
в молекуле  площадь поверхности молекулы
площадь поверхности молекулы  (все в относительных единицах);
(все в относительных единицах);  решение системы уравнений (VIII.29) для чистого компонента
решение системы уравнений (VIII.29) для чистого компонента 
 по сравнению с обычно используемыми
по сравнению с обычно используемыми  состоит в том, что с изменением концентрации раствора они изменяются меньше, чем переменные X В предельно разбавленных растворах переменные
состоит в том, что с изменением концентрации раствора они изменяются меньше, чем переменные X В предельно разбавленных растворах переменные  остаются конечными и уравнения модели могут служить, в частности, для расчетов коэффициентов активности при предельном разбавлении. Для атермического раствора, т. е. в случае, когда
остаются конечными и уравнения модели могут служить, в частности, для расчетов коэффициентов активности при предельном разбавлении. Для атермического раствора, т. е. в случае, когда  при любых значениях
при любых значениях  для всех концентраций раствора.
для всех концентраций раствора.

 - энтальпия взаимообмена для контактов
- энтальпия взаимообмена для контактов 
 в общем случае представляется в виде:
в общем случае представляется в виде:

 учитывается с помощью параметра
учитывается с помощью параметра  называемого теплоемкостью взаимообмена. В этом случае взаимодействие пары
называемого теплоемкостью взаимообмена. В этом случае взаимодействие пары  характеризуется тремя параметрами:
характеризуется тремя параметрами:  Однако в ряде систем, где для оценки
Однако в ряде систем, где для оценки  недостаточно данных, этот параметр приравнивается нулю и для пары разноименных групп учитываются два параметра:
недостаточно данных, этот параметр приравнивается нулю и для пары разноименных групп учитываются два параметра:  Это приближение обычно оказывается удовлетворительным, если расчеты охватывают небольшой температурный интервал.
Это приближение обычно оказывается удовлетворительным, если расчеты охватывают небольшой температурный интервал.
 предельных коэффициентов активности, равновесия жидкость—пар и взаимной растворимости жидкостей в бинарных и тройных системах, образованных алканами, алканолами, перфторалканами [316, 326, 331, 332]. Исследовались также смеси алканов и алканолов с целлозольвами [331, 333], алкенами и алкинами [334]. Рассмотрим некоторые результаты приложения модели.
предельных коэффициентов активности, равновесия жидкость—пар и взаимной растворимости жидкостей в бинарных и тройных системах, образованных алканами, алканолами, перфторалканами [316, 326, 331, 332]. Исследовались также смеси алканов и алканолов с целлозольвами [331, 333], алкенами и алкинами [334]. Рассмотрим некоторые результаты приложения модели.
 включающими подгруппы
включающими подгруппы  которые считали эквивалентными по энергетическим характеристикам;
которые считали эквивалентными по энергетическим характеристикам;
 и соответственно группы
и соответственно группы  или
или  ;
;
 состоящие из энергетически эквивалентных подгрупп
состоящие из энергетически эквивалентных подгрупп 
 поскольку конкретные значения гц и
поскольку конкретные значения гц и  не существенны
не существенны
 и гидроксильную группу, имеющую энергетически неэквивалентные контактные участки
и гидроксильную группу, имеющую энергетически неэквивалентные контактные участки  которые условно будем называть группами
которые условно будем называть группами 
 эфирную группу
эфирную группу  и гидроксильную группу с контактными участками
и гидроксильную группу с контактными участками 
 в модели учитывалось, что комбинаторная составляющая термодинамических функций зависит лишь от суммарных значений гон
в модели учитывалось, что комбинаторная составляющая термодинамических функций зависит лишь от суммарных значений гон  но на остаточный вклад влияет значение каждой из величин
но на остаточный вклад влияет значение каждой из величин  При этом величины
При этом величины  (в модели Баркера имеющие смысл числа контактных участков) должны отразить способность гидроксильной группы образовывать одну водородную связь через водород и две связи через кислород.
(в модели Баркера имеющие смысл числа контактных участков) должны отразить способность гидроксильной группы образовывать одну водородную связь через водород и две связи через кислород.
 и от двух для
и от двух для  (таблицы Бонди при
(таблицы Бонди при  дают
дают  Расчеты с различными значениями
Расчеты с различными значениями  показали, что результаты чувствительны к способу разбиения поверхности на группы и целесообразно выбрать
показали, что результаты чувствительны к способу разбиения поверхности на группы и целесообразно выбрать  так как в этом случае наблюдается наилучшее согласие с экспериментом. Геометрические параметры модели приведены в табл. VIII.8.
так как в этом случае наблюдается наилучшее согласие с экспериментом. Геометрические параметры модели приведены в табл. VIII.8.
 для систем алканол—алкан были вычислены термодинамические функции при
для систем алканол—алкан были вычислены термодинамические функции при  [326], первая ссылка]. Результаты в обоих случаях оказались близкими, значения энергетических параметров модели при различных
[326], первая ссылка]. Результаты в обоих случаях оказались близкими, значения энергетических параметров модели при различных  были
были

 на асимметрию рассчитанных концентрационных кривых избыточных термодинамических функций [335]. В этом случае, по-видимому, координационное число
на асимметрию рассчитанных концентрационных кривых избыточных термодинамических функций [335]. В этом случае, по-видимому, координационное число  выступает в роли дополнительного параметра, отражающего в некоторой степени ориентационные эффекты. При более детальном описании специфических взаимодействий необходимость в варьировании
выступает в роли дополнительного параметра, отражающего в некоторой степени ориентационные эффекты. При более детальном описании специфических взаимодействий необходимость в варьировании  отпадает.
отпадает.

 коэффициент активности
коэффициент активности  компонента
компонента  энтальпия смешения для
энтальпия смешения для  состава
состава  системы;
системы;  число систем;
число систем;  общее число исследованных составов для
общее число исследованных составов для  системы.
системы.
 Как видно из рис. VIII.5, для системы гексан—пропанол (рис. VIII.5, а) получено хорошее согласие с экспериментом давления и состава пара, равновесного с раствором. Для системы гептан—перфторгептан (рис. VIII.5, б) расчет с большой точностью передает наблюдающуюся в эксперименте область расслаивания. Хорошо предсказываются фазовые равновесия в системах алкан—алкен и алкан— алкин (см. табл.
Как видно из рис. VIII.5, для системы гексан—пропанол (рис. VIII.5, а) получено хорошее согласие с экспериментом давления и состава пара, равновесного с раствором. Для системы гептан—перфторгептан (рис. VIII.5, б) расчет с большой точностью передает наблюдающуюся в эксперименте область расслаивания. Хорошо предсказываются фазовые равновесия в системах алкан—алкен и алкан— алкин (см. табл. 


 и расчет
и расчет 
 приводят к качественному несоответствию между результатами расчета и эксперимента.
приводят к качественному несоответствию между результатами расчета и эксперимента.

 гептан
гептан  перфторгексан
перфторгексан  перфторгептан (б) при
перфторгептан (б) при  Экспериментальные данные
Экспериментальные данные  и расчет
и расчет 
 для систем алкан — алканол при 288, 15 К Экспериментальные данные
для систем алкан — алканол при 288, 15 К Экспериментальные данные  и расчет при
и расчет при  [уравнение (VIII.31)]
[уравнение (VIII.31)]
 для
для

 в тройной системе додеканол — пропанол — ундекан (298,15 К) по секущей
в тройной системе додеканол — пропанол — ундекан (298,15 К) по секущей  Экспериментальные данные
Экспериментальные данные 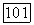 и расчет
и расчет 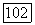

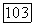 для систем алкан—алкин:
для систем алкан—алкин: 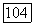 .
.
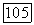 по квазихимическои групповой модели можно считать удовлетворительными [316, 332]. Модель передает характер изменения предельных коэффициентов активности при изменении длины молекулярной цепи растворенного вещества и растворителя, отражает их температурную зависимость. Как видно из табл. VIII.5, наибольшие расхождения между результатами расчета и эксперимента (от 18 до 30 %) наблюдаются для гексана в пропаноле и для алканола в расслаивающихся системах алканол—перфторалкан, где значения предельных коэффициентов активности составляют несколько сотен.
по квазихимическои групповой модели можно считать удовлетворительными [316, 332]. Модель передает характер изменения предельных коэффициентов активности при изменении длины молекулярной цепи растворенного вещества и растворителя, отражает их температурную зависимость. Как видно из табл. VIII.5, наибольшие расхождения между результатами расчета и эксперимента (от 18 до 30 %) наблюдаются для гексана в пропаноле и для алканола в расслаивающихся системах алканол—перфторалкан, где значения предельных коэффициентов активности составляют несколько сотен.
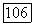 -перфторалкан
-перфторалкан 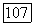 -алканол (3) среднее отклонение рассчитанных значений
-алканол (3) среднее отклонение рассчитанных значений 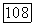 от экспериментальных составляет
от экспериментальных составляет 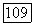 Модель правильно передает характер изменения предельных коэффициентов активности при изменении постава бинарного растворителя, причем точность предсказания оказалась лучше, чем при расчетах с помощью модели UNIFAC (см. табл. VIII.6).
Модель правильно передает характер изменения предельных коэффициентов активности при изменении постава бинарного растворителя, причем точность предсказания оказалась лучше, чем при расчетах с помощью модели UNIFAC (см. табл. VIII.6).
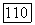 на гидроксильной группе явилось важным моментом описания. Отметим, что модель [327], где такого выделения не проводилось дала для растворов алканол—алкан менее точные результаты, чем модель [326].
на гидроксильной группе явилось важным моментом описания. Отметим, что модель [327], где такого выделения не проводилось дала для растворов алканол—алкан менее точные результаты, чем модель [326].
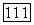 В действительности же внутримолекулярные связи образуются и при
В действительности же внутримолекулярные связи образуются и при 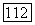 что должно заметно влиять на парциальные избыточные функции компонента А при его малых концентрациях.
что должно заметно влиять на парциальные избыточные функции компонента А при его малых концентрациях.
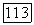 состоит из трех форм:
состоит из трех форм: 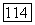 молекула А, в которой имеется внутримолекулярная связь (циклическая форма),
молекула А, в которой имеется внутримолекулярная связь (циклическая форма), 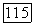 молекула А без внутримолекулярной связи (линейная форма) и молекула В. Согласно условиям химического равновесия
молекула А без внутримолекулярной связи (линейная форма) и молекула В. Согласно условиям химического равновесия

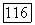 из равенства (VIII.33) получим:
из равенства (VIII.33) получим:


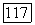 являются решениями системы уравнений
являются решениями системы уравнений 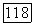 соответствующие величины для чистого вещества
соответствующие величины для чистого вещества 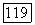 переменные с верхним индексом
переменные с верхним индексом 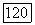 в выражении (VIII.34) относятся к циклической форме в чистой жидкости А.
в выражении (VIII.34) относятся к циклической форме в чистой жидкости А.
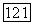 (VIII.36) помимо той информации, которая требуется в обычном варианте квазихимической групповой модели (геометрические параметры групп рассматриваемых молекулярных форм, энергетические параметры взаимодействия), дополнительно нужно знать концентрацию циклической формы в растворе заданного состава
(VIII.36) помимо той информации, которая требуется в обычном варианте квазихимической групповой модели (геометрические параметры групп рассматриваемых молекулярных форм, энергетические параметры взаимодействия), дополнительно нужно знать концентрацию циклической формы в растворе заданного состава 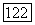 В соответствии с моделью:
В соответствии с моделью:

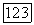 определяется формулой (
определяется формулой (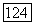 ; величина
; величина 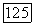 аналогичным выражением;
аналогичным выражением; 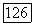 постоянная для вещества при данной температуре; этот параметр, не зависящий от природы и концентрации компонента В, можно найти, если имеются данные о значениях
постоянная для вещества при данной температуре; этот параметр, не зависящий от природы и концентрации компонента В, можно найти, если имеются данные о значениях 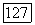 для любого раствора, в частности, бесконечно разбавленного:
для любого раствора, в частности, бесконечно разбавленного:

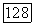 величины
величины 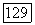 относятся к бесконечному разбавлению (именно через них проявляется влияние растворителя В на значение
относятся к бесконечному разбавлению (именно через них проявляется влияние растворителя В на значение 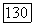
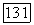 при
при 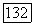 К [336]. Так как в других целлозольвах гидроксил и эфирный кислород разделены тем же мостиком
К [336]. Так как в других целлозольвах гидроксил и эфирный кислород разделены тем же мостиком 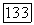 разумно принять, что и для гомологов метилцеллозольва в гексане значение
разумно принять, что и для гомологов метилцеллозольва в гексане значение 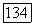 приблизительно такое же. Подстановка
приблизительно такое же. Подстановка 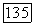 в формулу (VIII.38) позволяет найти
в формулу (VIII.38) позволяет найти  для рассматриваемого целлозольва. Далее, концентрации форм
для рассматриваемого целлозольва. Далее, концентрации форм 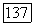 для произвольного состава раствора могут быть рассчитаны по уравнению (VIII.37), которое рассматривается совместно
для произвольного состава раствора могут быть рассчитаны по уравнению (VIII.37), которое рассматривается совместно 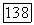 и с аналогичным выражением для
и с аналогичным выражением для 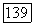 (задача может быть решена итерационно). Подстановка найденных значений
(задача может быть решена итерационно). Подстановка найденных значений 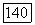 в формулу (VIII.34) дает
в формулу (VIII.34) дает 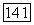 при заданном составе раствора А.
при заданном составе раствора А.
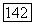 для разных целлозольвов) (см.
для разных целлозольвов) (см.

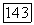 по квазихимической групповой модели (308,15 К) для систем: а — целлозольв — гексан;
по квазихимической групповой модели (308,15 К) для систем: а — целлозольв — гексан; 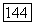 целлозольв — бутанол; в — целлозольв — деканол Экспериментальные данные
целлозольв — бутанол; в — целлозольв — деканол Экспериментальные данные 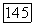 и расчет по моделям с учетом внутримолекулярных водородных связей
и расчет по моделям с учетом внутримолекулярных водородных связей 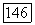 и обычной модели
и обычной модели 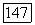
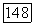 от длины углеводородного радикала
от длины углеводородного радикала 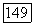 Целлозольв — гексан; II. целлозольв — дека-нол; III. Целлозольв — метилцеллозольв; этил-целлозольв (—); бутилцеллозольв
Целлозольв — гексан; II. целлозольв — дека-нол; III. Целлозольв — метилцеллозольв; этил-целлозольв (—); бутилцеллозольв  и гептил-целлозольв
и гептил-целлозольв 
 Явный учет температурной зависимости свободного объема и его концентрационной зависимости при групповом квазихимическом рассмотрении проведен в работах 1328]. Предполагалось, что свободный объем определяется лишь отталкивательными взаимодействиями и зависит только от приведенного объема 0, представляющего отношение общего объема к объему твердых сердцевин. Связь между свободным объемом и
Явный учет температурной зависимости свободного объема и его концентрационной зависимости при групповом квазихимическом рассмотрении проведен в работах 1328]. Предполагалось, что свободный объем определяется лишь отталкивательными взаимодействиями и зависит только от приведенного объема 0, представляющего отношение общего объема к объему твердых сердцевин. Связь между свободным объемом и

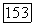 полная энергия контакта
полная энергия контакта 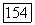 параметр неспецифического взаимодействия; объем твердых сердцевин и величина
параметр неспецифического взаимодействия; объем твердых сердцевин и величина 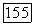 считались зависящими от температуры;
считались зависящими от температуры; 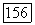 вклад специфического взаимодействия.
вклад специфического взаимодействия.