Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
7.7. Влияние ионной силы растворителяРассмотрим теперь изменения скорости реакций, которые могут быть вызваны добавлением солей. Возникающие при этом эффекты обычно подразделяют на две категории. К первой относятся изменения активности ионов или полярных молекул. Этот эффект называют первичным, чтобы отличить его от вторичного солевого эффекта, обусловленного тем, что ионная сила раствора влияет на истинную концентрацию ионов, образующихся при диссоциации слабых электролитов. Хорошо известным примером является замедление реакции, катализируемой кислотой или основанием, под действием соли, что связано с подавлением ионизации каталитической кислоты или основания. Оба солевых эффекта описываются в рамках электростатических представлений с помощью уравнений, выведенных Бренстедом, Бьеррумом и Кристиансеном. Все эти уравнения основаны на теории Дебая — Хюккеля, но каждое из них опирается на несколько различающиеся допущения, которые сопоставлены в известной книге Амиса [3]. Все три уравнения можно свести к простому общему виду
который считается справедливым для разбавленных растворов с концентрацией не выше Неоднократные неудачи, связанные с применением, электростатических теорий для количественного описания солевых эффектов (и даже для качественного описания, если вспомнить, к примеру, правило Олсона — Симонсона, можно объяснить в предположении, что активность иона определяется не только его зарядом и радиусом, как утверждает теория Дебая — Хюккеля, но еще и его донорными и акцепторными свойствами. По общему признанию эти свойства зависят не только от общего заряда. Если исходить из простого утверждения, что катионы — это акцепторы, а анионы — доноры, то солевые эффекты очень хорошо согласуются с представлениями о химических координационных эффектах растворителя. Отсюда вытекают два основных следствия. а) Ионы электролита проявляют донорные и акцепторные свойства в большей (или сравнимой) степени, чем истинные реагенты. Следовательно, компоненты электролита будут конкурировать с реагентом при сольватации. Удаление молекул растворителя из внешней сольватной оболочки реагентов может привести к изменению их активности. Простым примером является выделение диоксида углерода при подсаливании напитка кока-кола (это не что иное, как «высаливание»). То, что причина заключена не в общем заряде как таковом, видно при добавлении сахара, которое оказывает тот же самый эффект. Эти наблюдения несложно объяснить тем, что диоксид углерода очень слабо гидратирован, и, для того чтобы удержать его в растворе, требуется множество молекул воды. (Слабогидратированные частицы стабилизируются только большим числом молекул воды, включенных во внешнюю гидратную оболочку, и это усиливает внутреннюю гидратную оболочку.) Сахар и соль гидратируются сильнее, принимают растворитель на себя, поэтому б) Иоцы электролита проявляют донорные и акцепторные свойства в большей (или сравнимой) степени, чем молекулы растворителя. Поэтому и те и другие могут конкурировать за присоединение к истинным реагентам («координация во внешней оболочке» в отличие от «сольватации во внешней оболочке»). Образование ионных ассоциатов и комплексов во внешней оболочке может привести к резкому изменению активности. Последовательное вытеснение молекул растворителя конкурирующими молекулами, действующими как лиганды, характерно для координационных сфер катионов переходных металлов. Теперь мы имеем грубое представление об очень сложных взаимодействиях растворенное вещество — растворенное вещество — растворитель, которые должны зависеть от относительной донорной и акцепторной способности каждого из компонентов: реагентов, растворителя, электролита. С этой точки зрения уместно сравнить систему растворитель — соль с системой растворитель — растворитель, так как общее описание солевых эффектов по сложности сравнимо с описанием смесей растворителей. Однако рассмотрение упрощается, если ограничиться электролитами, которые являются слабыми донорами и акцепторами (например, большие одновалентные ионы) в явно амфотерных растворителях, таких, как вода. Обычно для одновалентных электролитов наблюдается линейная зависимость метод расчета коэффициентов активности по Дебаю и Хюккелю был успешно распространен на апротонные растворители при использовании в качестве электролита тетрабутиламмонийперхлората [128]. Это позволяет определить константы скорости при нулевой ионной силе, что необходимо для сравнения констант скоростей реакций в неводных растворителях, получаемых в различных условиях. Следует подчеркнуть, что пределы пригодности уравнения (7-16) задаются не только концентрациями, что не имеет исключений, но также и природой электролитов, т.е. их химическими координирующими свойствами. Широко распространенные попытки не учитывать пределы применения электростатических теорий породили множество запутанных и запутывающих работ по солевым эффектам. Некоторые противоречия отмечены Гамметом в работе [49]. Нужно еще раз напомнить, что для обсуждения изменений параметров пригодны только константы скорости определенных отдельных стадий реакции. Однако зачастую этот важный момент не принимается во внимание, особенно в более ранних, «классических» сообщениях, где, к примеру, обычно рассматриваются константы скорости псевдопервого порядка (см. [97]). Учитывая сложность химии растворов электролитов, мы считаем, что изучение солевых эффектов не может найти широкого применения как средство для исследования механизмов реакции. Тем не менее наличие солевых эффектов вызывает необходимость проверять ионную силу при кинетических исследованиях. Если ионная сила не мала настолько, что солевые эффекты незначительны, то нужно провести серию измерений при разных значениях ионной силы и экстраполировать данные к нулю. Только тогда получаемые константы скорости можно сравнивать друг с другом.
|
 |
Оглавление
|















































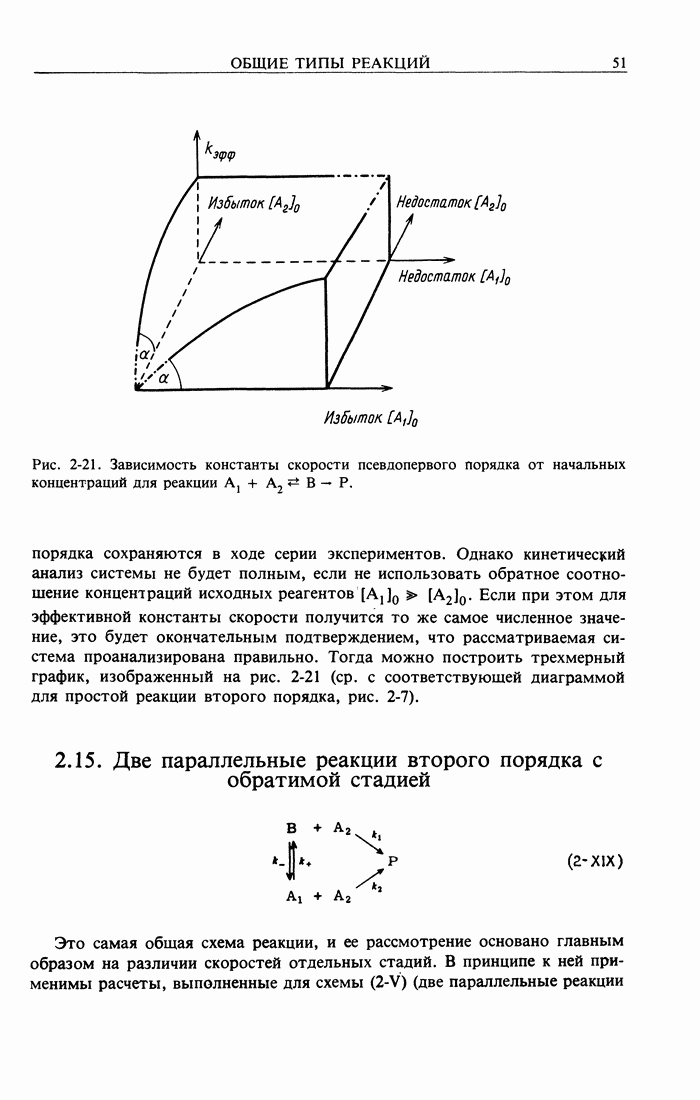



























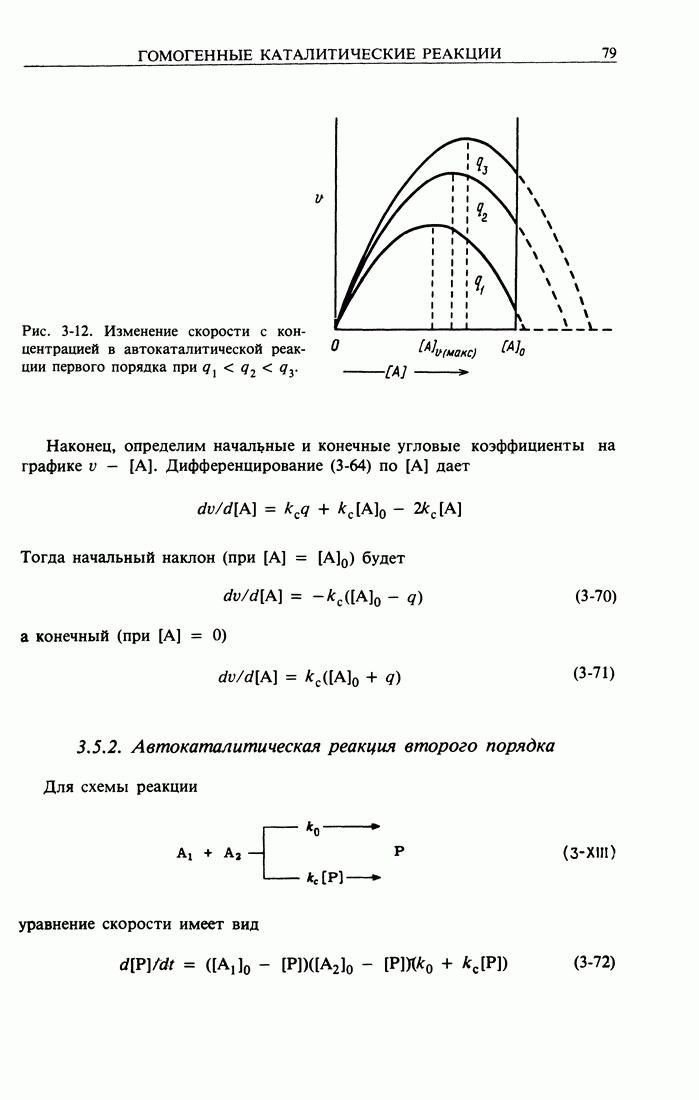




















































































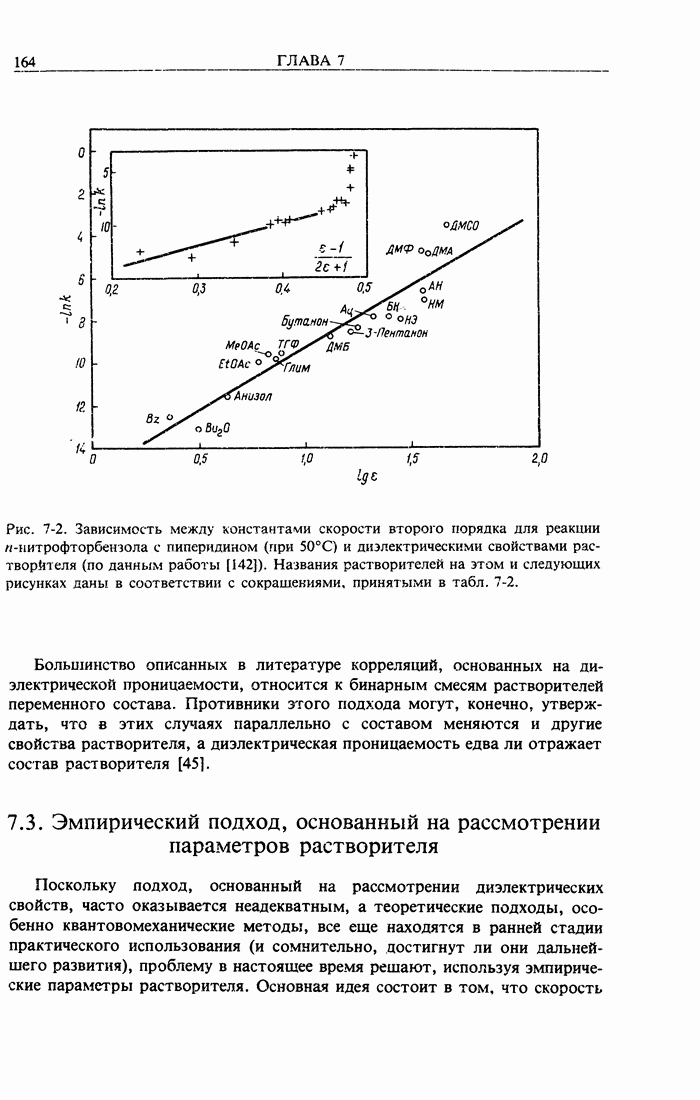



































































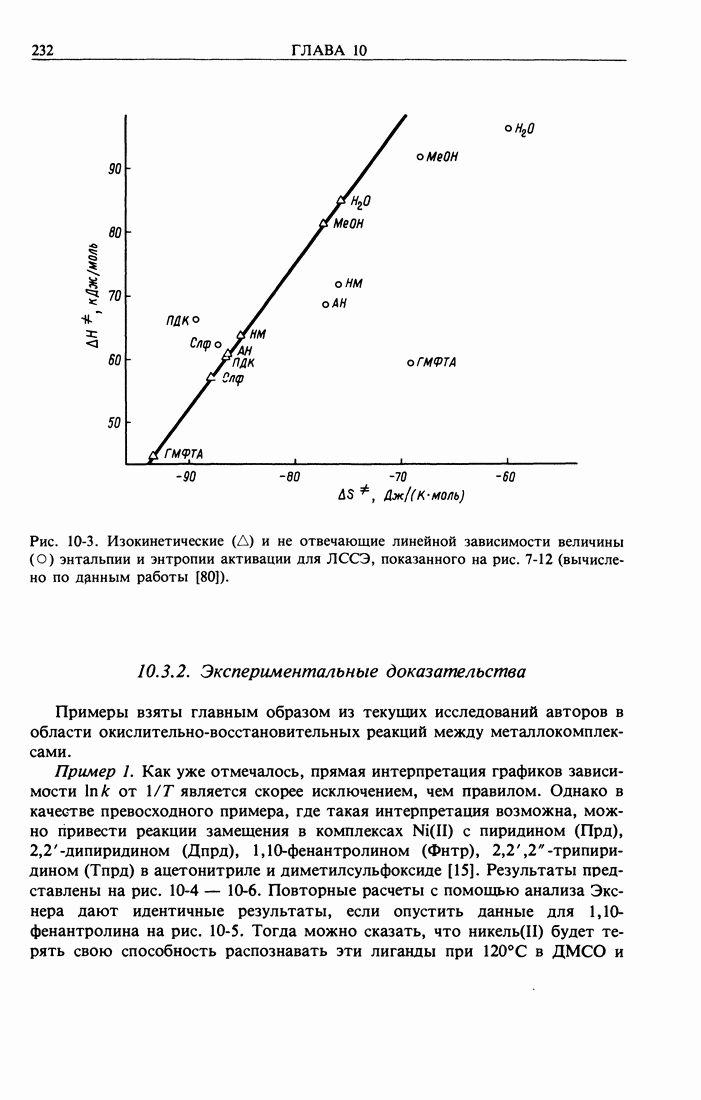






















 Здесь
Здесь  означает ионную силу системы, а
означает ионную силу системы, а  константу скорости при нулевой ионной силе. Поэтому оказывается возможной линейная зависимость
константу скорости при нулевой ионной силе. Поэтому оказывается возможной линейная зависимость  к от
к от  Однако практически часто наблюдаются криволинейные графики, иногда они могут быть выражены переменным степенным рядом по
Однако практически часто наблюдаются криволинейные графики, иногда они могут быть выражены переменным степенным рядом по  [49].
[49].
 освобождается из раствора. Иногда может меняться приток молекул растворителя при почти неизменной концентрации самого реагента. В случае структурированных растворителей добавление электролита может изменить структуру раствора и, следовательно, его координирующие свойства. Тогда можно говорить о новой интерпретации коэффициента активности в терминах химической координации или структуры; эта весьма реалистическая точка зрения была развита Гутманом [42].
освобождается из раствора. Иногда может меняться приток молекул растворителя при почти неизменной концентрации самого реагента. В случае структурированных растворителей добавление электролита может изменить структуру раствора и, следовательно, его координирующие свойства. Тогда можно говорить о новой интерпретации коэффициента активности в терминах химической координации или структуры; эта весьма реалистическая точка зрения была развита Гутманом [42].
 к от квадратного корня из ионной силы. Заслуживает внимания тот факт, что
к от квадратного корня из ионной силы. Заслуживает внимания тот факт, что