21.14. Генетические дефекты липидного обмена
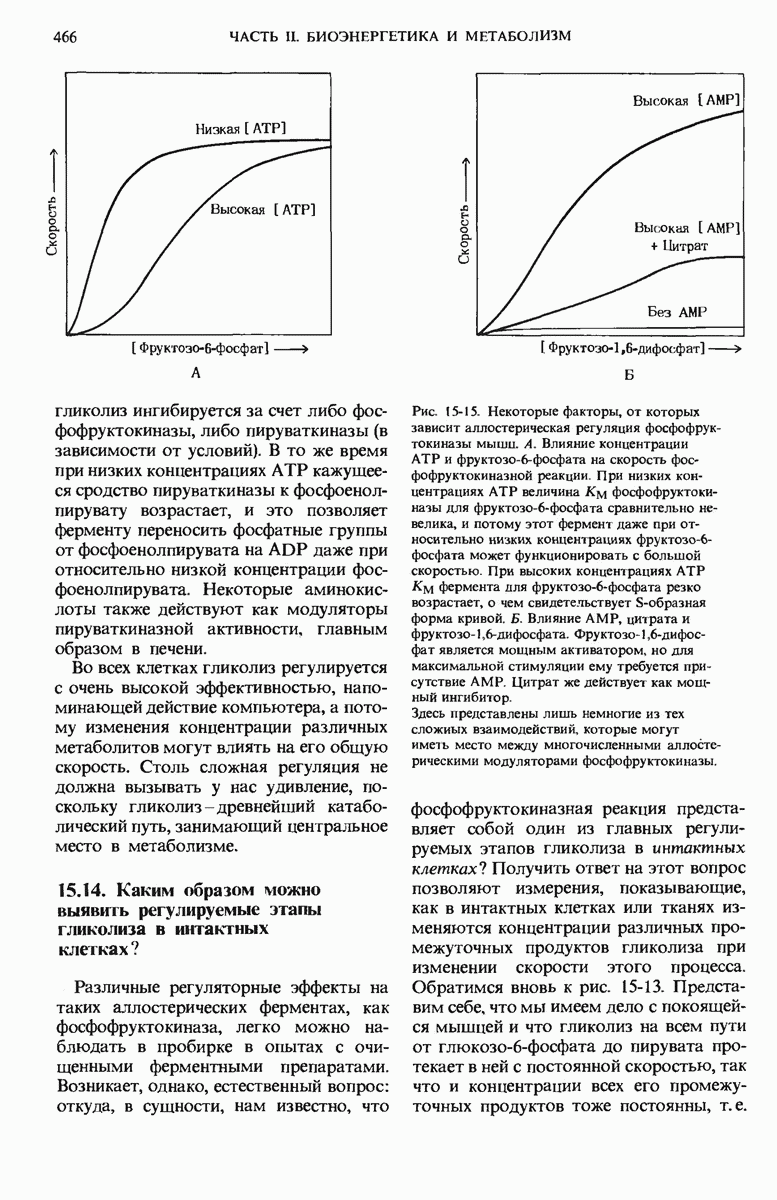
Все полярные липиды в мембранах постоянно обновляются в процессе метаболизма; при нормальных условиях в клетке устанавливается динамическое стационарное состояние, при котором скорость синтеза липидов равна скорости их распада. Расщепление липидов катализируется гидролитическими ферментами, способными расщеплять строго определенные ковалентные связи. Например, расщепление фосфатидилхолина, главного мембранного липида, происходит при помощи нескольких разных фосфолипаз. Способ их действия показан на рис. 21-20.
Метаболизм мембранных сфинголипидов, к которым относятся сфингомиелин, цереброзиды и ганглиозиды (разд. 12.6), особенно подвержен нарушениям, обусловленным генетическими дефектами ферментов, участвующих в их деградации.

Рис. 21-20. Места действия фосфолипаз на фосфатидилхолин. и  - длинноцепочечные остатки жирных кислот.
- длинноцепочечные остатки жирных кислот.
При этом сфинголипиды или продукты их частичного распада накапливаются в тканях в больших количествах, поскольку синтез сфинголипидов протекает нормально, а их расщепление нарушено. Например, при такой редкой генетической аномалии, как болезнь Нима-на-Пика, сфингомиелин накапливается в мозгу, селезенке и печени. Болезнь проявляется у детей уже вскоре после рождения и приводит к задержке умственного развития и смерти в раннем возрасте. Причиной болезни Нимана-Пика является генетически обусловленное нарушение процесса расщепления сфингомиелина ферментом сфингомиелиназой, которая отщепляет фосфохолин от сфингомиелина (строение этих соединений изображено на рис. 12-11).
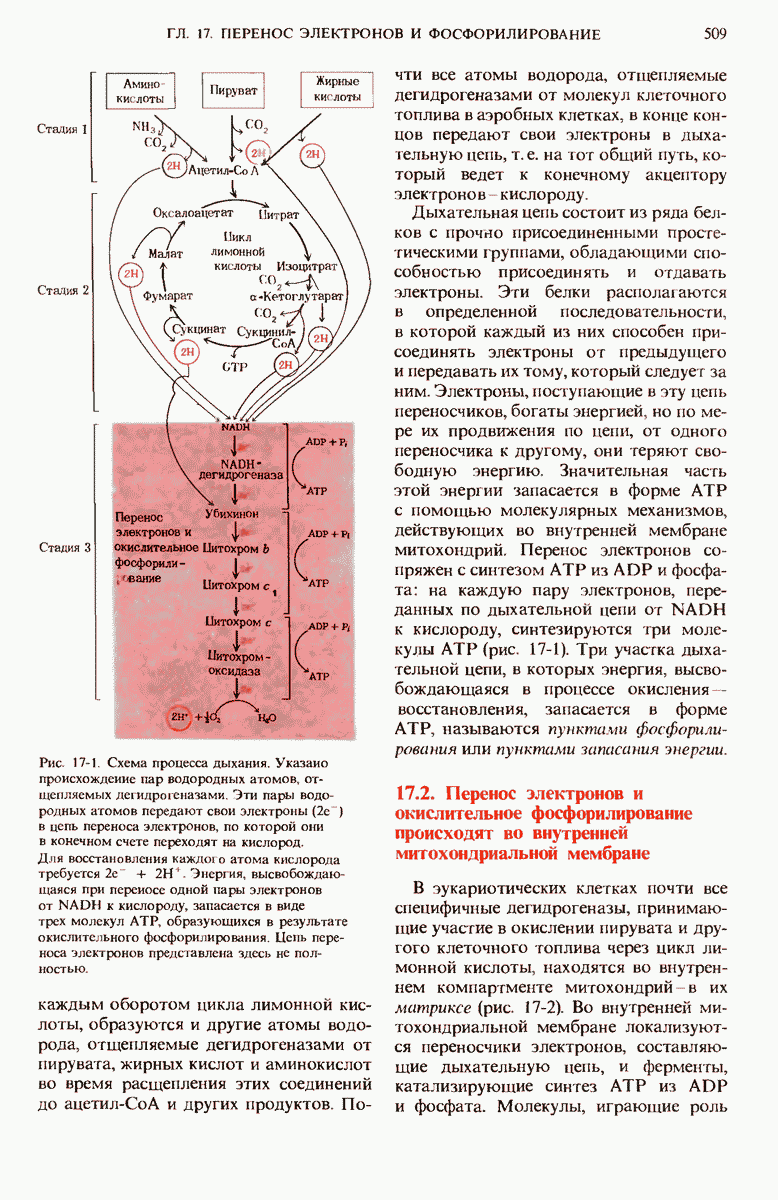
Значительно чаще встречается болезнь Тея - Сакса, при которой из-за отсутствия лизосомного фермента N-ацетилгексоз-аминидазы, осуществляющего гидролиз специфической связи между остатками ТЧ-ацетил-О-галактозамина и D-галактозы в полярной голове молекулы ган-глиозида (рис. 21-21), в мозгу и селезенке накапливаются ганглиозиды определенного типа. Вследствие того что расщепление ганглиозидов прекращается на одном из промежуточных этапов, происходит накопление больших количеств частично расщепленных ганглиозидов, что приводит в результате дегенеративных процессов в нервной системе к задержке умственного развития, слепоте и смерти в раннем возрасте (рис. 21-22).
Если считать по отношению ко всему населению в целом, то болезнь Тея-Сакса-явление редкое (1 случай на 300 000 рождений), однако среди евреев ашкенази (выходцев из Центральной Европы), которые составляют 90% всего еврейского населения Америки, ее частота очень высока (1 случай на 3600 новорожденных); каждый 28-й еврей ашкенази является носителем рецессивного дефектного гена.

Рис. 21-21. Генетический дефект при болезни Тея - Сакса. В результате нормального расщепления ганглиозида  под действием лизосомного фермента N-ацетилгексозаминидазы образуется N-ацетил-D-галактозамин и ганглиозид
под действием лизосомного фермента N-ацетилгексозаминидазы образуется N-ацетил-D-галактозамин и ганглиозид  . При болезни Тея-Сакса этот фермент поврежден и поэтому в лизосомах, главным образом в клетках мозга, накапливается ганглиозид
. При болезни Тея-Сакса этот фермент поврежден и поэтому в лизосомах, главным образом в клетках мозга, накапливается ганглиозид  .
.

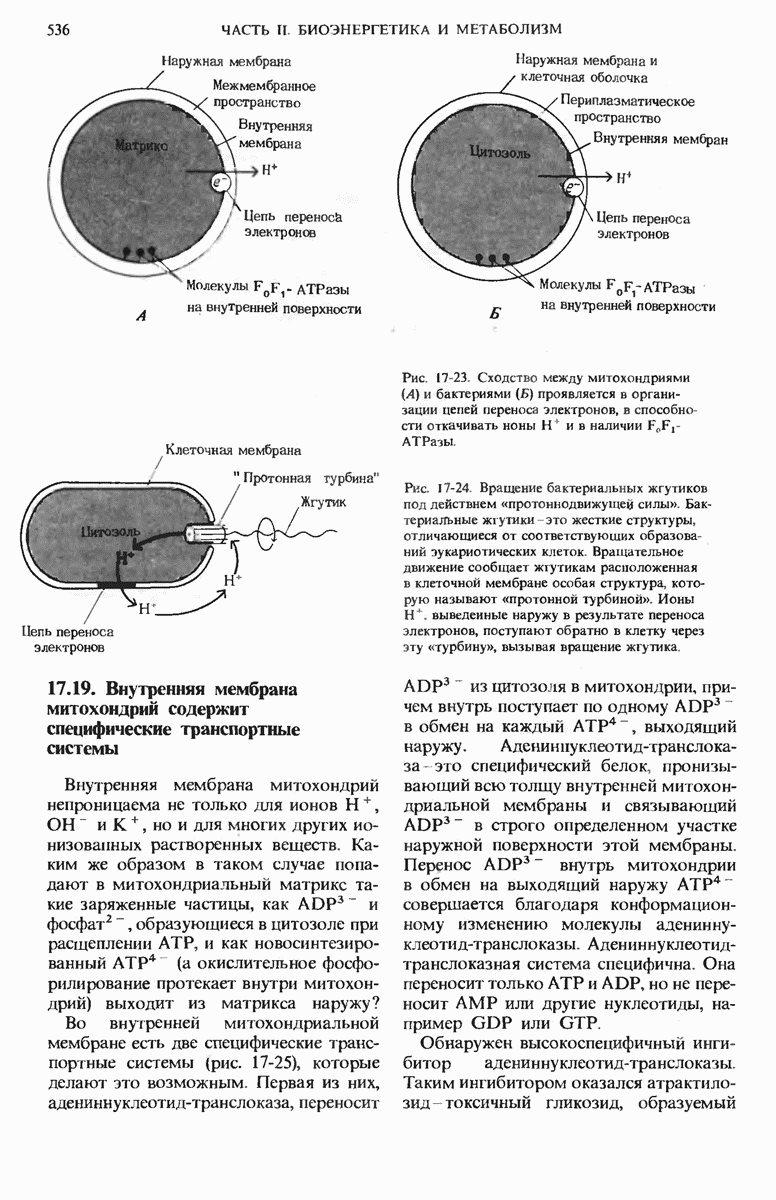
Рис. 21-22. А. Годовалый ребенок с болезнью Тея-Сакса. Уже началось поражение мозга, вслед за которым наступит слепота. Лишь немногие дети с этим заболеванием доживают до пяти лет. Б. Электронная микрофотография части пораженной клетки мозга, на которой видны аномальные скопления ганглнозидов в лизосомах.
Если дефектный ген имеется у обоих родителей, вероятность проявления болезни Тея-Сакса у детей сильно возрастает. Из-за необратимого характера заболевания, приводящего к тяжелой инвалидности, важное значение приобретает генетическое консультирование супругов. Разработаны тесты, позволяющие выявлять наличие рецессивного гена у будущих родителей. Носителей этого гена можно выявить, определив активность гексозаминидазы А в культурах фибробластов, полученных путем биопсии кожи. Генетический статус плода можно также установить, определив активность фермента в клетках, полученных из амниотической жидкости. Метод исследования образцов амниотической жидкости называется амниоцентезом.