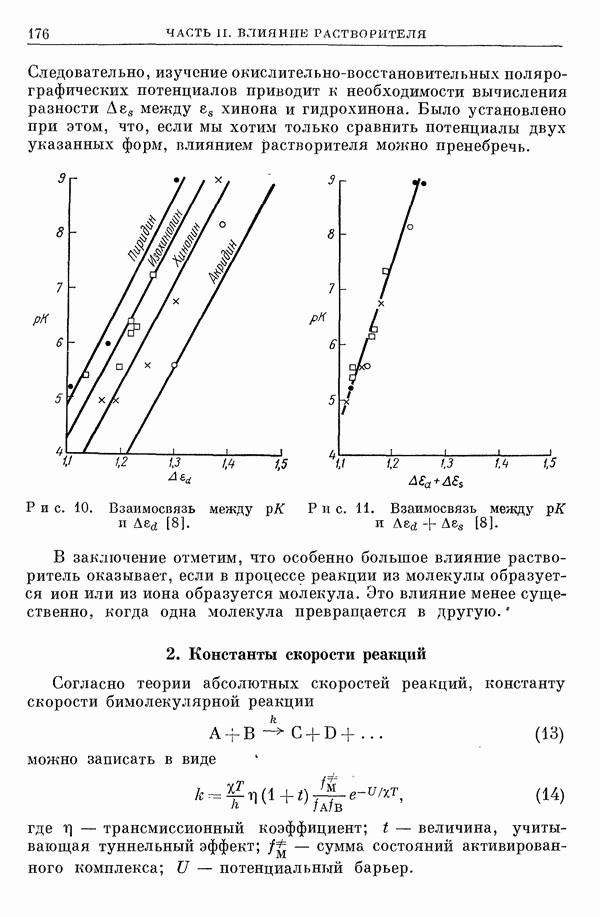
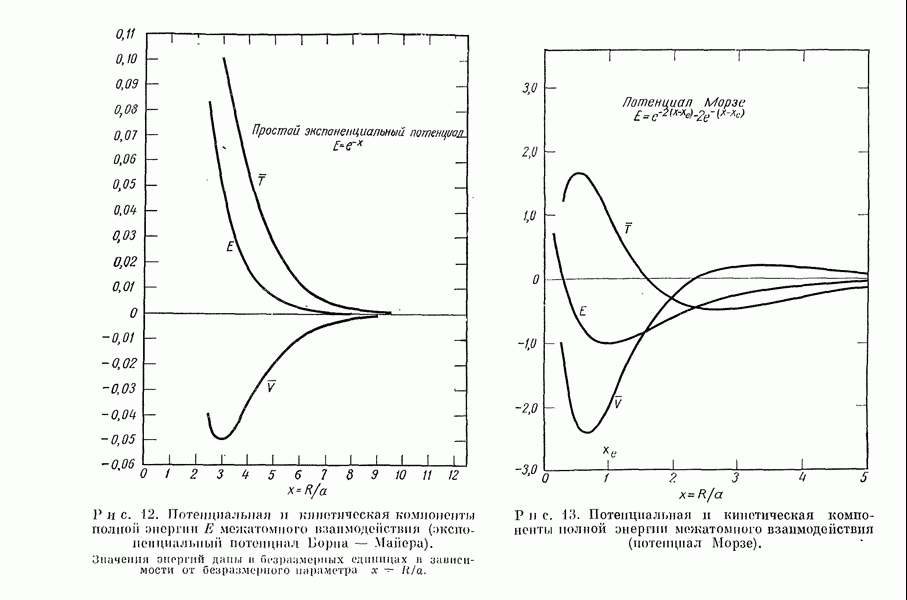
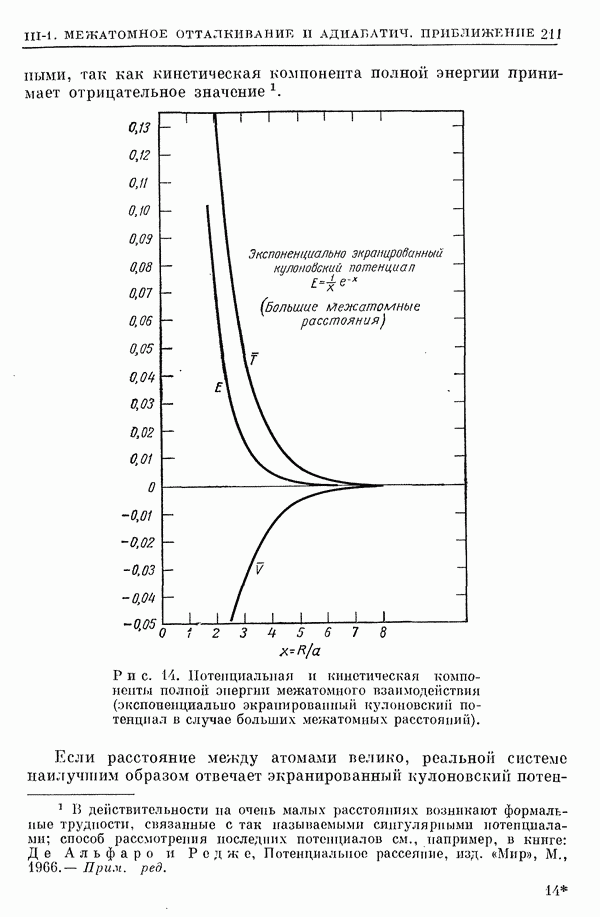
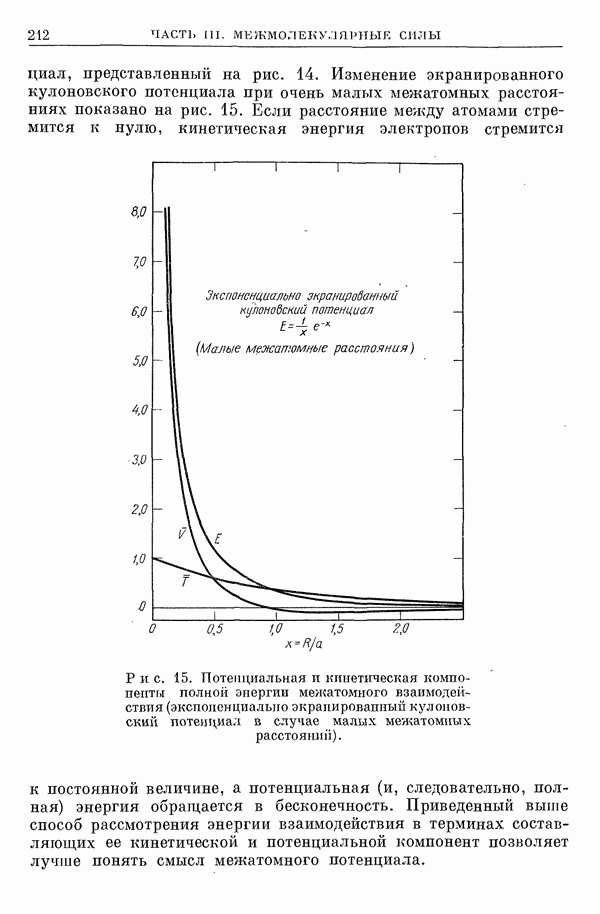
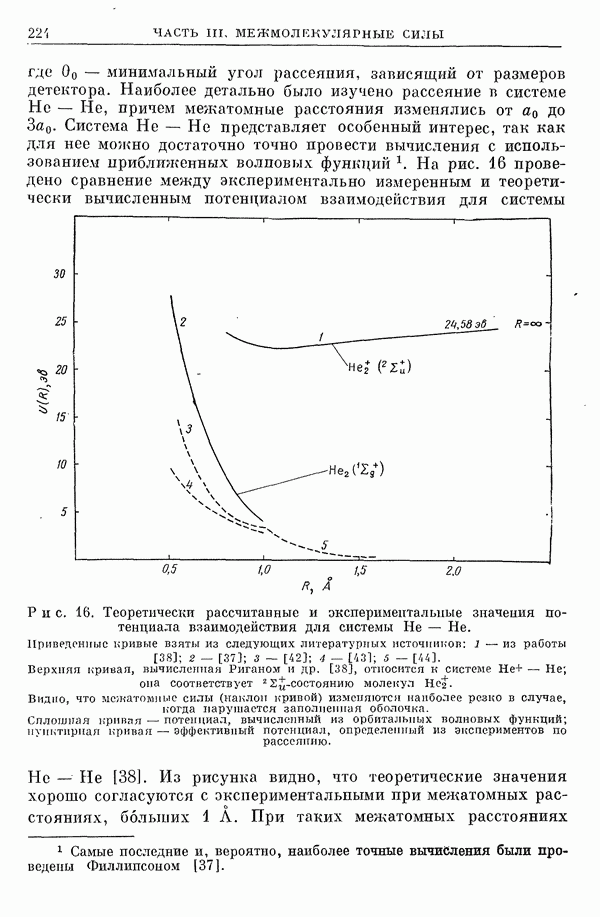
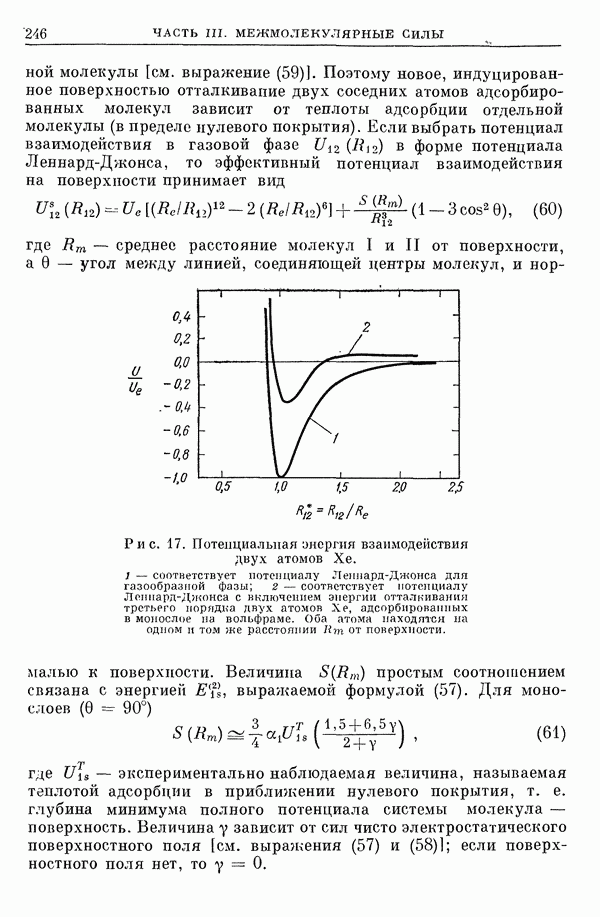
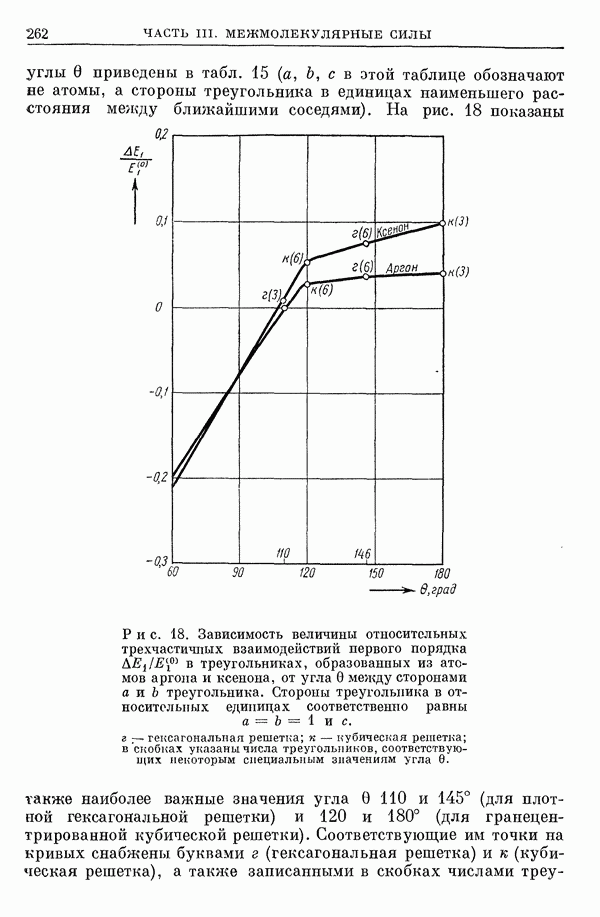
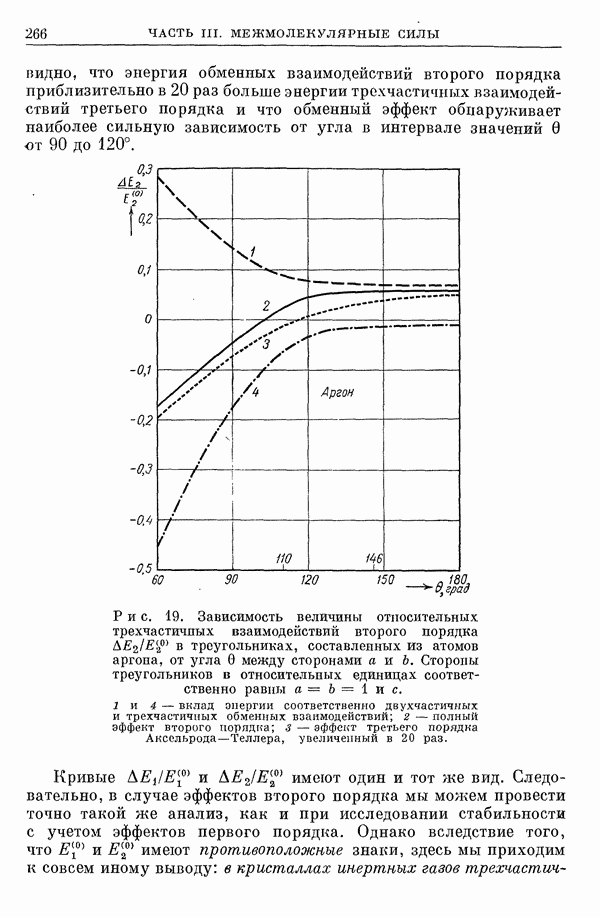
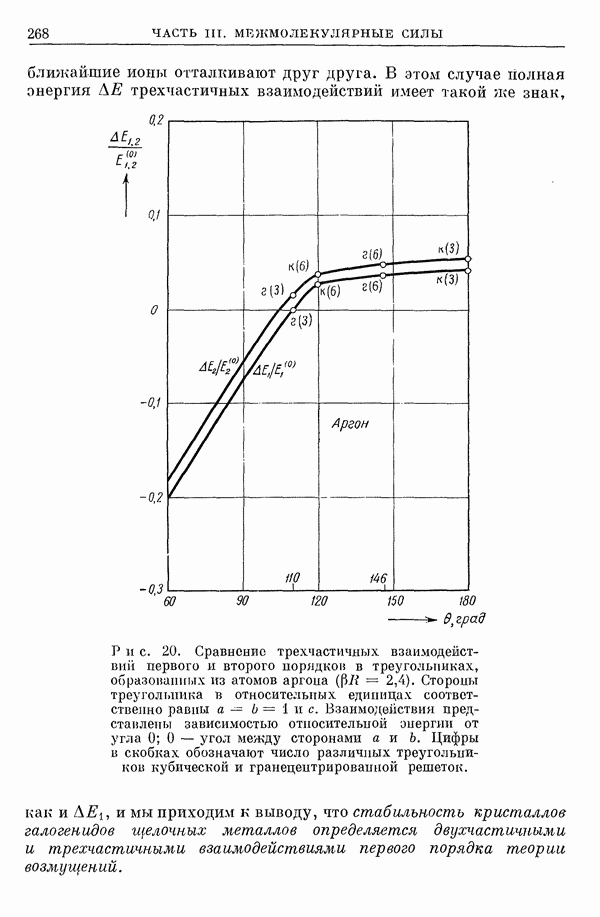
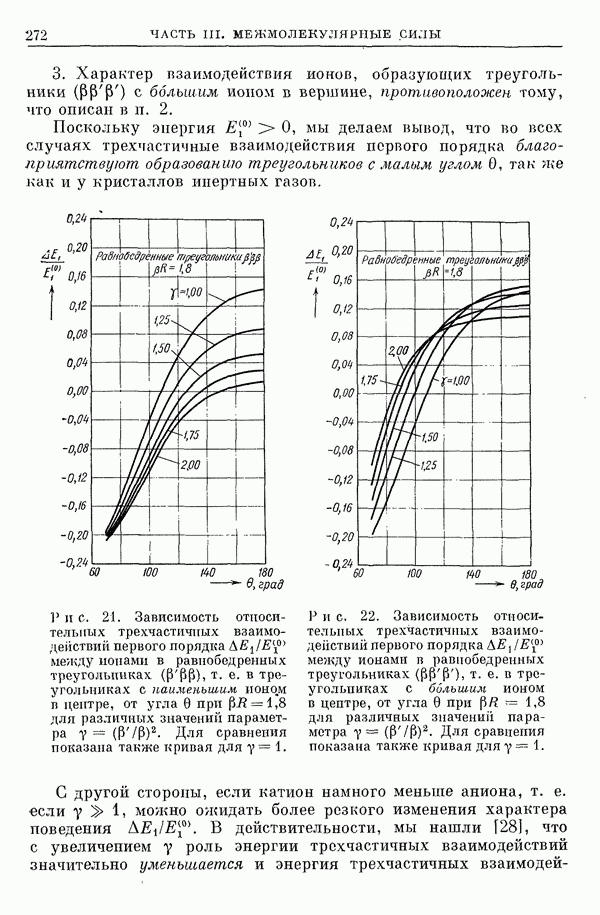
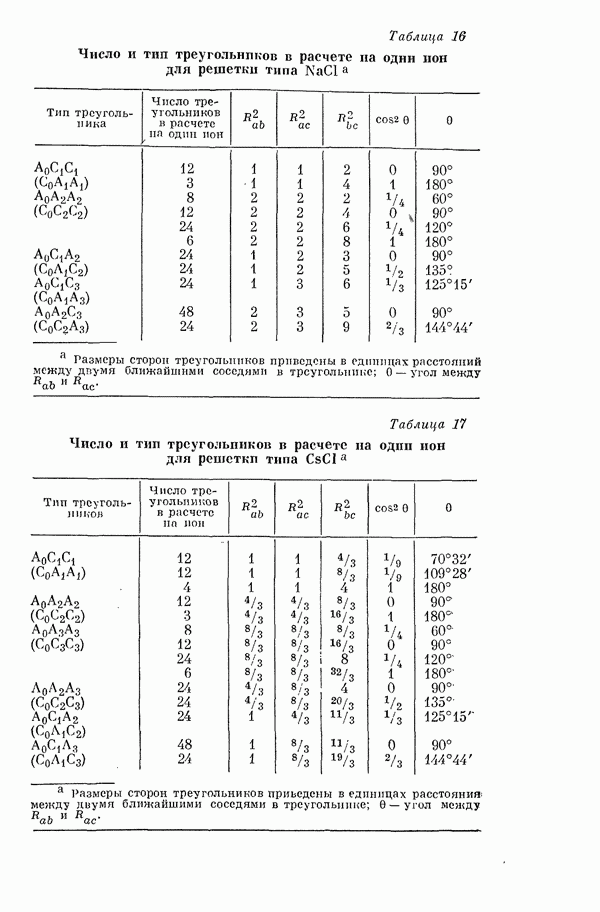
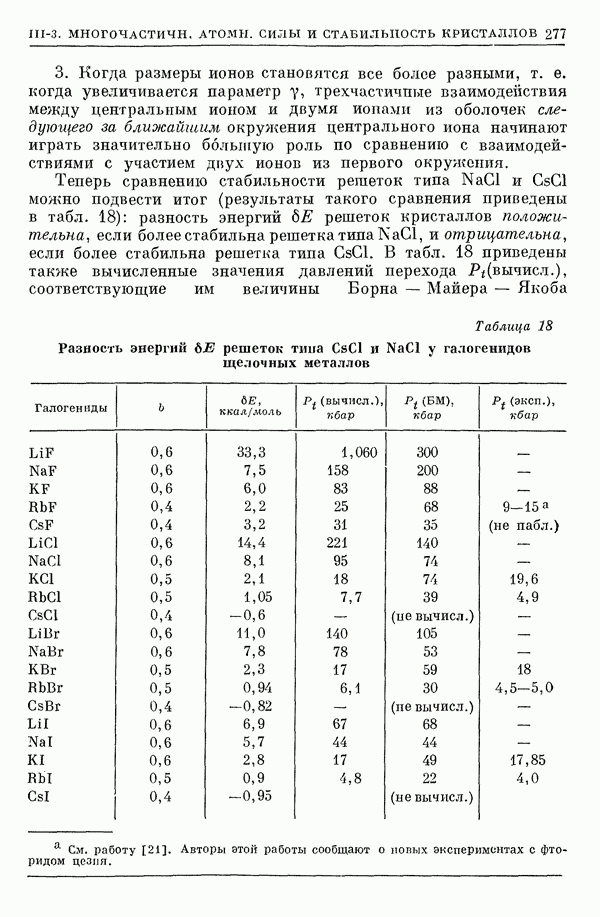
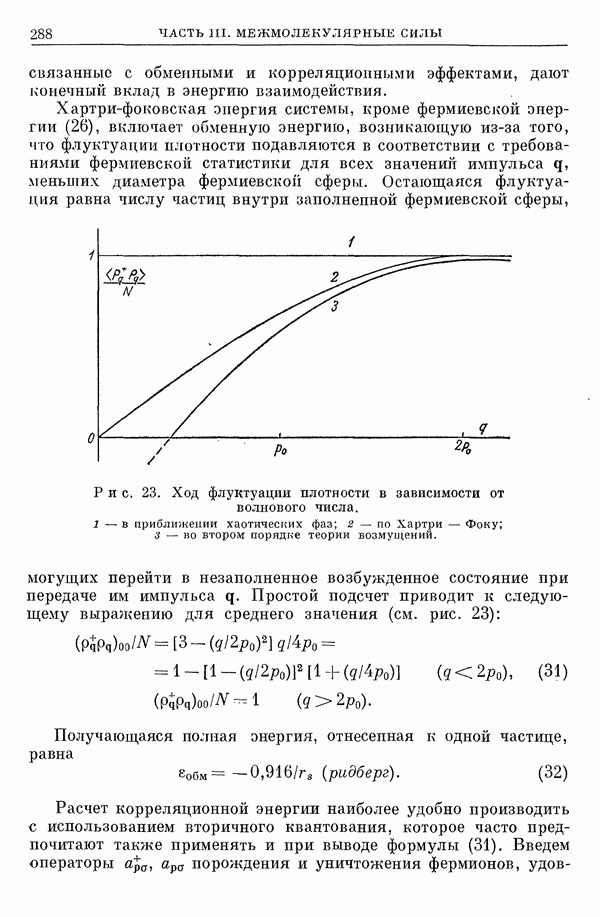
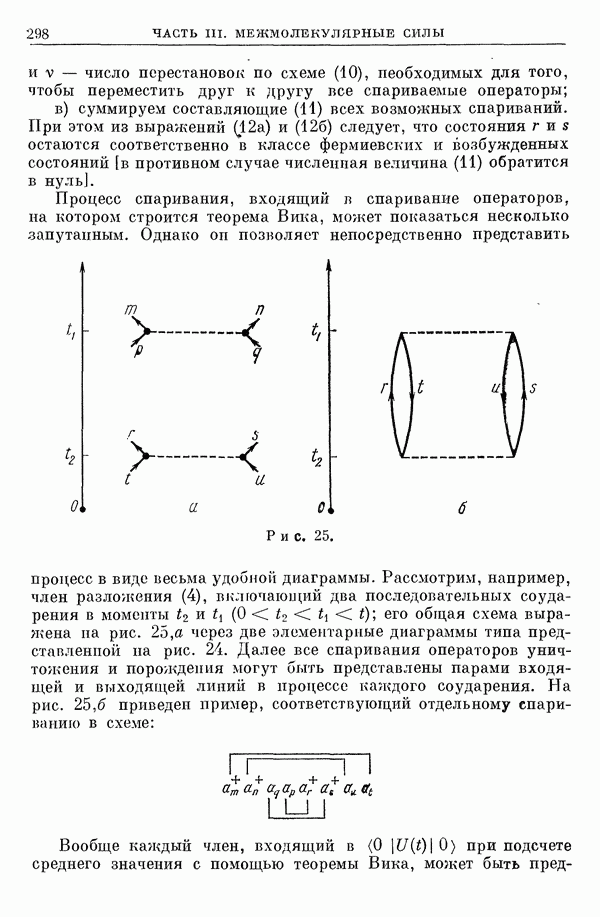
2. Потенциал взаимодействия двух молекул
При интерпретации термодинамических данных или данных, полученных при изучении явлений переноса [1], кривые потенциальной энергии взаимодействия двух молекул приближенно представляют полуэмпирическими функциями. При этом наиболее часто используют зависящий от двух параметров потенциал Леннард-Джонса

Реже употребляют зависящий от трех параметров потенциал Ридберга [2, 3]

где

Потенциал Ридберга довольно хорошо [4] передает зависимость (вида  ) энергии взаимодействия от расстояния в области притяжения при больших
) энергии взаимодействия от расстояния в области притяжения при больших  и сочетает в себе простоту формы и универсальность.
и сочетает в себе простоту формы и универсальность.
В выражении (1) глубина потенциальной ямы  и положение
и положение  минимума на потенциальной кривой полностью определяют форму кривой, в частности коэффициент
минимума на потенциальной кривой полностью определяют форму кривой, в частности коэффициент  при члене
при члене  Выражение (2) содержит, кроме того, константу
Выражение (2) содержит, кроме того, константу  которая характеризует ширину потенциальной ямы.
которая характеризует ширину потенциальной ямы.
Непосредственный способ определения потенциала между двумя молекулами состоит в изучении рассеяния молекулярных пучков. При этом рассеяние частиц с высокой энергией позволяет детально исследовать область сил отталкивания при малых  Экспериментальные методы, используемые при нахождении вида потенциала в данной области, описаны Эрджинсоем в предыдущем разделе. Рассеяние частиц с малой энергией позволяет определить параметры
Экспериментальные методы, используемые при нахождении вида потенциала в данной области, описаны Эрджинсоем в предыдущем разделе. Рассеяние частиц с малой энергией позволяет определить параметры  и константу 1 С при члене
и константу 1 С при члене  а также найти число связанных состояний в потенциальной яме и анизотропию нецентральных потенциалов. По этому поводу мы рекомендуем читателю прекрасный обзор Берпстейна [61.
а также найти число связанных состояний в потенциальной яме и анизотропию нецентральных потенциалов. По этому поводу мы рекомендуем читателю прекрасный обзор Берпстейна [61.
Параметры  можно найти не только из опытов с молекулярными пучками, но и из макроскопических данных по второму вириальному коэффициенту
можно найти не только из опытов с молекулярными пучками, но и из макроскопических данных по второму вириальному коэффициенту  вязкости газа и т. д. [1], взятых в виде функции температуры; при этом, однако, значения параметров часто зависят от того, по какому физическому свойству эти параметры определяются. Обычно считают, что вириальный коэффициент
вязкости газа и т. д. [1], взятых в виде функции температуры; при этом, однако, значения параметров часто зависят от того, по какому физическому свойству эти параметры определяются. Обычно считают, что вириальный коэффициент  не чувствителен к виду потенциала и что по нему нельзя найти форму кривой
не чувствителен к виду потенциала и что по нему нельзя найти форму кривой  Однако Кингстон [7] заметил, что на основании последних данных, относящихся к широкому температурному интервалу, можно отличить одну форму потенциала
Однако Кингстон [7] заметил, что на основании последних данных, относящихся к широкому температурному интервалу, можно отличить одну форму потенциала  от другой.
от другой.
Теоретическое квантовомеханическое вычисление потенциала — задача сложная. Так, например, для системы Не — Не величина минимума на потенциальной кривой  рассчитанная теоретически, составляет лишь около
рассчитанная теоретически, составляет лишь около  что очень мало даже в сравнении с корреляционной энергией атома Не, составляющей
что очень мало даже в сравнении с корреляционной энергией атома Не, составляющей  не говоря уже о полной его энергии.
не говоря уже о полной его энергии.
Как было отмечено выше, в квантовой теории межмолекулярных взаимодействий обычно независимо рассматривают два предельных случая больших и малых расстояний. Если расстояние  между молекулами небольшое, то используют представление
между молекулами небольшое, то используют представление
о «составной молекуле» и применяют для описания такой системы метод молекулярных орбиталей (МО). Вычисления по методу молекулярных орбиталей для небольших межатомных расстояний, когда существенными являются только силы отталкивания, в особенности вычисления для системы Не — Не, рассмотрены в предыдущем разд. III-1. С увеличением расстояния  перекрывание между двумя молекулами уменьшается, и в конце концов при некотором
перекрывание между двумя молекулами уменьшается, и в конце концов при некотором  таким перекрыванием можно пренебречь. Тогда, раскладывая потенциал взаимодействия обеих молекул но мультиполям и ограничиваясь в разложении только дипольным членом, приходим к выражению
таким перекрыванием можно пренебречь. Тогда, раскладывая потенциал взаимодействия обеих молекул но мультиполям и ограничиваясь в разложении только дипольным членом, приходим к выражению  для потенциала дальнодействующих лондоиовских дисперсионных сил притяжения. Следует отметить, однако, что неясно, при каких расстояниях между атомами указанное разложение справедливо. Выражение, полученное с точностью до членов второго порядка теории возмущений и описывающее взаимодействия лондоновского типа, содержит сумму по всем возбужденным виртуальным состояниям обоих атомов. При этом, конечно, предполагается, что отсутствует перекрывание между волновыми функциями не только в основном, но и в возбужденных состояниях. Однако боровский радиус пропорционален
для потенциала дальнодействующих лондоиовских дисперсионных сил притяжения. Следует отметить, однако, что неясно, при каких расстояниях между атомами указанное разложение справедливо. Выражение, полученное с точностью до членов второго порядка теории возмущений и описывающее взаимодействия лондоновского типа, содержит сумму по всем возбужденным виртуальным состояниям обоих атомов. При этом, конечно, предполагается, что отсутствует перекрывание между волновыми функциями не только в основном, но и в возбужденных состояниях. Однако боровский радиус пропорционален  и поэтому разложение по мультиполям должно быть справедливым при значительно больших расстояниях, чем это обычно предполагается [8].
и поэтому разложение по мультиполям должно быть справедливым при значительно больших расстояниях, чем это обычно предполагается [8].
Мы не будем здесь подробно обсуждать способы вычисления коэффициента С, который характеризует лондоновское взаимодействие между атомами; это является отдельной задачей. Отметим только, что теоретические значения С, определенные по формуле Лондона

для ряда атомов и молекул, таких, как  почти в 2,2 раза меньше экспериментальных [9]. Такое расхождение теории с экспериментом, по-видимому, объясняется наличием многоэлектронных эффектов [8, 9], хотя и имеются сомнения в правильности интерпретации экспериментальных данных в этих работах [7].
почти в 2,2 раза меньше экспериментальных [9]. Такое расхождение теории с экспериментом, по-видимому, объясняется наличием многоэлектронных эффектов [8, 9], хотя и имеются сомнения в правильности интерпретации экспериментальных данных в этих работах [7].
Коэффициент С можно также определить по экспериментальным значениям сил осцилляторов с помощью правил сумм [10а, б; 11а, б]. Кроме того, имеются способы определения и оценок С, предложенные Питцером [9], Салемом [12], Матером [13] (см. также разд. 1-3) и Кестнером [14].
При очень больших расстояниях между молекулами нужно учитывать эффекты запаздывания (при этом большими считаются расстояния, удовлетворяющие неравенству  например,
например,
при  имеем 11 1000 Л). С учетом эффектов запаздывания, которые несколько уменьшают силы притяжения между частицами, выражение (3) принимает вид [15]- [17]
имеем 11 1000 Л). С учетом эффектов запаздывания, которые несколько уменьшают силы притяжения между частицами, выражение (3) принимает вид [15]- [17]

Для средних межатомных расстояний  энергия взаимодействия была вычислена Мак-Лахланом [16].
энергия взаимодействия была вычислена Мак-Лахланом [16].
Рассмотрим теперь теорию межмолекулярного взаимодействия, которая позволяет получить кривую потенциальной энергии взаимодействия при всех расстояниях в рамках единых представлений, не накладывая никаких ограничений на расстояпия между частицами, на перекрывание их волновых функций и т. д. Заметим, что в силу своего общего характера эта теория применима также и для изучения взаимодействий между различными локализованными и непосредственно несвязанными друг с другом группами электронов внутри одной и той же молекулы.
Итак, допустим, что взаимодействуют две молекулы А и В, имеющие заполненные электронные оболочки. Состояние сложной молекулярной системы  будем описывать хартри-фоковской однодетермипантной волновой функцией
будем описывать хартри-фоковской однодетермипантной волновой функцией

где  — молекулярные сиин-орбитали (МО), распространяющиеся на обе молекулы А и В. Представление о двух отдельных молекулах все еще сохраняется, несмотря на формулу (5). Введем унитарную матрицу
— молекулярные сиин-орбитали (МО), распространяющиеся на обе молекулы А и В. Представление о двух отдельных молекулах все еще сохраняется, несмотря на формулу (5). Введем унитарную матрицу  которая преобразует молекулярные спин-орбитали к молекулярной системы
которая преобразует молекулярные спин-орбитали к молекулярной системы  в локализованные спин-орбитали
в локализованные спин-орбитали  молекул А и В:
молекул А и В:

где  — транспонированная комплексно-сопряженная матрица к матрице
— транспонированная комплексно-сопряженная матрица к матрице  Допустим, что пА локализованных спин-орбиталей принадлежат молекуле А и
Допустим, что пА локализованных спин-орбиталей принадлежат молекуле А и  локализованных спин-орбиталей принадлежат молекуле В. В силу инвариантности детерминанта при унитарном преобразовании имеем
локализованных спин-орбиталей принадлежат молекуле В. В силу инвариантности детерминанта при унитарном преобразовании имеем

Если молекулы А и В идентичны, матрицу преобразования можно полностью определить только из соображений симметрии. Если же
молекулы А и В не идентичны, матрицу унитарного преобразования следует определять, например, из условия минимума обменных отталкиваний, как это было предложено Рюденбергом (т. 1, разд. 1-4).
Точная энергия сложной молекулярной системы  с заполненными электронными оболочками равна
с заполненными электронными оболочками равна

где  — хартри-фоковская энергия
— хартри-фоковская энергия  вычисленная методом
вычисленная методом  на основе волновой функции (8) при фиксированном значении межъядерного расстояпия
на основе волновой функции (8) при фиксированном значении межъядерного расстояпия  — оставшаяся часть точной энергии, обусловленная корреляцией электронов системы
— оставшаяся часть точной энергии, обусловленная корреляцией электронов системы 
Точную энергию каждой из молекул А и В, имеющих заполненные электронные оболочки, можно представить аналогичными выражениями вида (9). Тогда потенциальная энергия взаимодействия  равна
равна

где, например,  Из выражения (10) видно, что полная потенциальная энергия состоит из хартри-фоковской компоненты
Из выражения (10) видно, что полная потенциальная энергия состоит из хартри-фоковской компоненты  и корреляционной компоненты
и корреляционной компоненты  При этом компонента
При этом компонента  описывает главным образом межэлектронное отталкивание, происходящее от электростатических и обменных эффектов. Большая часть сил притяжения между молекулами А и В, большая часть энергии минимума
описывает главным образом межэлектронное отталкивание, происходящее от электростатических и обменных эффектов. Большая часть сил притяжения между молекулами А и В, большая часть энергии минимума  на кривой потенциальной энергии и энергия в области дисперсионных сил
на кривой потенциальной энергии и энергия в области дисперсионных сил  обусловлены компонентой
обусловлены компонентой  Кестнер и др. [18], проведя недавно методом
Кестнер и др. [18], проведя недавно методом  вычисления для системы Не — Не, вообще не обнаружили никакого минимума для
вычисления для системы Не — Не, вообще не обнаружили никакого минимума для 
Перепишем теперь выражение (9) для точной энергии системы  вводя в рассмотрение корреляционную функцию
вводя в рассмотрение корреляционную функцию

где  — хартри-фоковские молекулярные спин-орбитали сложной системы
— хартри-фоковские молекулярные спин-орбитали сложной системы  В — двухэлектронный оператор антисимметризации
В — двухэлектронный оператор антисимметризации  — точная корреляционная функция спин-орбиталей
— точная корреляционная функция спин-орбиталей  и
и  Из точной
Из точной  -электронной волновой функции
-электронной волновой функции
(см. работу [8]) следует условие

которое мы называем эффектом «исключения».
Хартри-фоковская энергия  системы
системы  вычисленная при помощи однодетерминантной волновой функции
вычисленная при помощи однодетерминантной волновой функции  образованной из молекулярных орбиталей, равна
образованной из молекулярных орбиталей, равна

где  — энергия электрона на
— энергия электрона на  молекулярной спин-орбитали в поле экранированных остовов молекул А и В, т. е.
молекулярной спин-орбитали в поле экранированных остовов молекул А и В, т. е.

 — обычные кулоновский и обменный интегралы, составленные для спин-орбиталей. Если теперь перейти к локализованным спин-орбиталям, то вид выражений (13) не изменится, но интегралы
— обычные кулоновский и обменный интегралы, составленные для спин-орбиталей. Если теперь перейти к локализованным спин-орбиталям, то вид выражений (13) не изменится, но интегралы  и К надо будет вычислять в этом случае для локализованных спин-орбиталей
и К надо будет вычислять в этом случае для локализованных спин-орбиталей  молекул А и В. При переходе к локализованным спин-орбиталям легко получить картину взаимодействия двух возмущенных молекул А и В, если сгруппировать определенным образом члерш и представить энергию в виде
молекул А и В. При переходе к локализованным спин-орбиталям легко получить картину взаимодействия двух возмущенных молекул А и В, если сгруппировать определенным образом члерш и представить энергию в виде

где

и

причем величины, входящие в выражения (14б) — (14г), имеют следующий смысл:

Заметим, что индексы  относятся к локализованным спин-орбиталям молекулы А, а индексы
относятся к локализованным спин-орбиталям молекулы А, а индексы  и
и  — к локализованным спин-орбиталям молекулы В.
— к локализованным спин-орбиталям молекулы В.
Выражения (146) и (14в) определяют записанные через локализованные орбитали хартри-фоковские энергии возмущенных молекул А и В в функции от расстояния  между ними, а выражение (14г) — записанную через локализованные орбитали энергию электростатического и обменного взаимодействий между А и В. Если
между ними, а выражение (14г) — записанную через локализованные орбитали энергию электростатического и обменного взаимодействий между А и В. Если  то
то

и

где  — энергии невозмущенных молекул А и В.
— энергии невозмущенных молекул А и В.
Точные выражения корреляционной энергии в формулах (9) и  также можно записать через локализованные орбитали. Прежде всего отметим, что точные корреляционные функции между локализованными спин-орбиталями, скажем между орбиталями
также можно записать через локализованные орбитали. Прежде всего отметим, что точные корреляционные функции между локализованными спин-орбиталями, скажем между орбиталями  и
и  связаны с корреляционными функциями между молекулярными спин-орбиталями, например между орбиталями
связаны с корреляционными функциями между молекулярными спин-орбиталями, например между орбиталями  следующим преобразованием [8]:
следующим преобразованием [8]:

причем  , где
, где  Заметим, что унитарная матрица
Заметим, что унитарная матрица  в формуле (15) та же, что и в формуле (7). С учетом формулы (15) выражение
в формуле (15) та же, что и в формуле (7). С учетом формулы (15) выражение  для полной энергии взаимодействия молекул А и В, записанное через локализованные спин-орбитали, принимает вид
для полной энергии взаимодействия молекул А и В, записанное через локализованные спин-орбитали, принимает вид

где  любые две локализованные спиц-орбитали. Комбинируя выражения (16) и (14а), получаем
любые две локализованные спиц-орбитали. Комбинируя выражения (16) и (14а), получаем

Здесь  — полные энергии возмущенных молекул А и В с учетом их корреляционной энергии, т. е.
— полные энергии возмущенных молекул А и В с учетом их корреляционной энергии, т. е.

а  корреляционная энергия, включающая по одной локализованной орбитали на каждую молекулу:
корреляционная энергия, включающая по одной локализованной орбитали на каждую молекулу:

Из выражений (10) и (17) непосредственно можно получить выражение для потенциальпой энергии при любом фиксированном расстоянии между молекулами А и В

где, например,  характеризует изменение полной энергии молекулы А вследствие возмущения ее молекулой В, находящейся от нее на расстоянии
характеризует изменение полной энергии молекулы А вследствие возмущения ее молекулой В, находящейся от нее на расстоянии 
Отметим, что выражения  (16), (18) и (19) для корреляционной эпергии
(16), (18) и (19) для корреляционной эпергии  формальны и могут оказаться полезными только в том случае, если известны точные волновые функции и энергии. Сама форма приведенных выражений не позволяет обнаружить многие соответствующие корреляционные эффекты [8], которые, хотя и невелики по сравнению с полной корреляционной энергией
формальны и могут оказаться полезными только в том случае, если известны точные волновые функции и энергии. Сама форма приведенных выражений не позволяет обнаружить многие соответствующие корреляционные эффекты [8], которые, хотя и невелики по сравнению с полной корреляционной энергией  вносят существенный вклад в потенциальную энергию
вносят существенный вклад в потенциальную энергию  Для вычислений или просто для того, чтобы выявить указанные эффекты в явной форме, следует взять точную волновую функцию
Для вычислений или просто для того, чтобы выявить указанные эффекты в явной форме, следует взять точную волновую функцию  подставить ее в формулу
подставить ее в формулу  и вариационным методом найти энергию системы. Тогда члены с корреляционной энергией Екорр в только что указанных выражениях будут непосредственно составляться из сумм независимых парных электронных корреляций, а также из тройных корреляций и корреляций с большим числом электронов.
и вариационным методом найти энергию системы. Тогда члены с корреляционной энергией Екорр в только что указанных выражениях будут непосредственно составляться из сумм независимых парных электронных корреляций, а также из тройных корреляций и корреляций с большим числом электронов.
Мы не останавливаемся здесь на деталях вычислений, которые ложно найти в работе [81. Отметим только, что члены, включающие трехэлектрошгае корреляции, теперь для межмолекулярных взаимодействий могут быть такого же порядка величины, как и парные корреляционные эффекты. Поэтому, вероятно, обычное пренебрежение указанными корреляционными эффектами и приводит к тому, что теоретически вычисленная эпергия дисперсионных взаимодействий двух атомов с заполненными  -электронпыми оболочками при
-электронпыми оболочками при  оказывается почти в 2 раза меньше экспериментально найденной [8, 9].
оказывается почти в 2 раза меньше экспериментально найденной [8, 9].
Соотношение (10) для  справедливо при всех расстояниях
справедливо при всех расстояниях  независимо от того, вычислена ли полная энергия с помощью молекулярных орбиталей
независимо от того, вычислена ли полная энергия с помощью молекулярных орбиталей  или локализованных орбиталей
или локализованных орбиталей  Рассматривая соответствующие вариационные
Рассматривая соответствующие вариационные
выражения [8, 14], находим, что описание системы  в терминах МО более удобно, когда
в терминах МО более удобно, когда  а описание в терминах
а описание в терминах 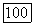 когда
когда 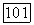 Предварительные вычисления, выполненные для системы Не — Не [14], позволяют оценить вклад различных эффектов при
Предварительные вычисления, выполненные для системы Не — Не [14], позволяют оценить вклад различных эффектов при 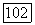 си 3 А), хотя получить точное значение
си 3 А), хотя получить точное значение 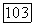 даже в этом простом случае довольно трудно. Чтобы сделать вышеприведенные формулы более конкретными и выяснить пределы применимости выражений (11) и (20) при
даже в этом простом случае довольно трудно. Чтобы сделать вышеприведенные формулы более конкретными и выяснить пределы применимости выражений (11) и (20) при 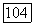 рассмотрим два примера.
рассмотрим два примера.
1. Молекула 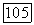 с заполненной электронной оболочкой находится в состоянии
с заполненной электронной оболочкой находится в состоянии 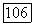 описываемом однодетерминантной волновой функцией
описываемом однодетерминантной волновой функцией 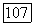

Точную волновую функцию молекулы 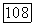 запишем в виде
запишем в виде

где

Обозначим локализованные орбитали каждого из атомов через 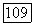 Тогда
Тогда

Подставляя выражение (23) в (21), найдем, что

так как, например, 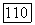 Кроме того,
Кроме того,

При 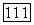 локализованные орбитали
локализованные орбитали 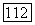 возмущенных атомов Н переходят соответственно в орбитали
возмущенных атомов Н переходят соответственно в орбитали 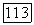
Таким образом, для потенциальной энергии 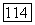 молекулы
молекулы 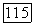 получим следующее выражение:
получим следующее выражение:

В рассматриваемом случае нет никакой внутриатомной корреляции. Второе слагаемое в выражении (26а) приближенно равно

Компонента потенциальной энергии 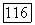 характеризует отталкивание, а компонента
характеризует отталкивание, а компонента 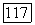 притяжение.
притяжение.
При достаточно больших 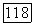 можно приближенно принять
можно приближенно принять

выражение (26а) сводится тогда к выражению для лондоновской дисперсионной энергии.
Действительно, второе слагаемое в уравнении (28) описывает мгновенное диполь-дипольпое взаимодействие, причем тензор 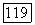 равен
равен

где I — единичный тензор, 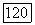 — единичный вектор в направлении
— единичный вектор в направлении 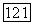 Величина с в выражении (29) — вариационная константа. Комбинируя формулы (266) и (29), находим, что
Величина с в выражении (29) — вариационная константа. Комбинируя формулы (266) и (29), находим, что

Подставляя сюда

и

(где а — поляризуемость, 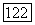 — средпяя энергия возбуждения), убеждаемся, что формула (31) не отличается от формулы Лондона (3); причем заметим, что Лондон полагал
— средпяя энергия возбуждения), убеждаемся, что формула (31) не отличается от формулы Лондона (3); причем заметим, что Лондон полагал 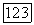 , где
, где 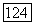 — потенциал ионизации (см., однако, [9]).
— потенциал ионизации (см., однако, [9]).
Приближения 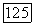 вообще говоря, не обязательны. Величину
вообще говоря, не обязательны. Величину 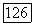 определяемую выражением (266), можно вычислить, выбрав подходящую пробную функцию для
определяемую выражением (266), можно вычислить, выбрав подходящую пробную функцию для 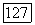 или для
или для 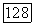 и минимизируя выражения (266) или (26а) при любом фиксированном межатомном расстоянии
и минимизируя выражения (266) или (26а) при любом фиксированном межатомном расстоянии 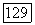
2. Система Не — Не. Для описания системы Не — Не можно использовать как молекулярные, так и локализованные орбитали при любых межатомных расстояниях. Так как система Не — 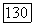 является системой с заполненными оболочками, то в силу принципа Паули орбитали МО могут быть строго представлены как линейные комбинации атомных орбиталей. Хартри-фоковская волновая функция системы Не — Не, составленная из орбиталей
является системой с заполненными оболочками, то в силу принципа Паули орбитали МО могут быть строго представлены как линейные комбинации атомных орбиталей. Хартри-фоковская волновая функция системы Не — Не, составленная из орбиталей 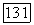 имеет вид
имеет вид

Локализованные спин-орбитали 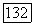 определяются из соображений симметрии. Таким образом, для рассматриваемой системы преобразование (7) имеет вид
определяются из соображений симметрии. Таким образом, для рассматриваемой системы преобразование (7) имеет вид

где 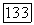 — локализованные спин-орбитали атома А;
— локализованные спин-орбитали атома А; 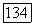 — локализованные спин-орбитали атома В. При
— локализованные спин-орбитали атома В. При 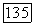

Если расстояние  между атомами Не не очень мало, то хартри-фоковские молекулярные орбитали, из которых составлена полная однодетерминантная волновая функция системы, можно приближенно заменить простыми линейными комбинациями (ЛКАО МО), составленными из хартри-фоковских орбиталей
между атомами Не не очень мало, то хартри-фоковские молекулярные орбитали, из которых составлена полная однодетерминантная волновая функция системы, можно приближенно заменить простыми линейными комбинациями (ЛКАО МО), составленными из хартри-фоковских орбиталей 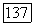 изолированных атомов Не. В этом случае для локализованных орбиталей
изолированных атомов Не. В этом случае для локализованных орбиталей 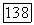 получим следующие выражения:
получим следующие выражения:

где

при этом интеграл перекрывания равен

В принятом приближении орбитали 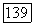 совпадают с ортогонализованными атомными орбиталями ОАО [19, 20].
совпадают с ортогонализованными атомными орбиталями ОАО [19, 20].
Хартри-фоковская компонента 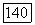 [см. формулу (10)] потенциальной эпергии системы Не — Не была вычислена в приближении линейных комбинаций хартри-фоковских атомных орбиталей ЛKAO (ХФ) МО [14]. Численные результаты для межатомных расстояний
[см. формулу (10)] потенциальной эпергии системы Не — Не была вычислена в приближении линейных комбинаций хартри-фоковских атомных орбиталей ЛKAO (ХФ) МО [14]. Численные результаты для межатомных расстояний 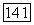 А можно представить в виде формулы
А можно представить в виде формулы

Имеются также и более точные вычисления величины 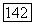 полным методом самосогласованного поля (МО ССП) [18].
полным методом самосогласованного поля (МО ССП) [18].
Корреляционная компонента 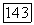 потенциальной энергии
потенциальной энергии 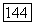 как это следует из формул (18) — (20), равна
как это следует из формул (18) — (20), равна

указанная энергии составляет основную часть энергии минимума потенциальной энергии системы.
Формулы (39), (18) и (19) являются точными. Они содержат в неявном виде трехэлектронные корреляции, которые вносят заметный вклад в энергию в вариационных вычислениях [8, 9, 14]. Выражение для энергии второго порядка 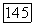 имеет ту же форму (39), (18), (19). Указанная энергия
имеет ту же форму (39), (18), (19). Указанная энергия 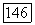 характеризует лондоновское взаимодействие, которое при
характеризует лондоновское взаимодействие, которое при 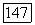 описывается выражением (3). Однако выражение для энергии
описывается выражением (3). Однако выражение для энергии 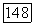 недостаточно точно, так как не учитывает трехэлектронных корреляционных эффектов, впервые появляющихся в
недостаточно точно, так как не учитывает трехэлектронных корреляционных эффектов, впервые появляющихся в 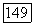
Вычисленная при помощи молекулярных орбиталей корреляционная энергия Не — Не равна

где

Отметим, что выражения (26а, б), минимизация которых дает верхний предел для энергии 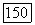 учитывают трехэлектронные корреляции между молекулярными орбиталями [8].
учитывают трехэлектронные корреляции между молекулярными орбиталями [8].
Выясним теперь, как отдельные энергии 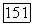 выраженные через молекулярные орбитали, связаны с энергиями внутри-
выраженные через молекулярные орбитали, связаны с энергиями внутри- 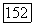 и межатомных корреляций
и межатомных корреляций 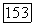 выраженными через локализованные орбитали. Полная корреляционная энергия системы Не — Не конечно инвариантна при переходе от МО и
выраженными через локализованные орбитали. Полная корреляционная энергия системы Не — Не конечно инвариантна при переходе от МО и 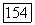 так что
так что

Отдельные энергии парных корреляций (в описании МО) выражаются через 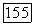 с помощью формул (6), (11) и (15). После соответствующих подстановок получаем
с помощью формул (6), (11) и (15). После соответствующих подстановок получаем

где звездочкой обозначены комплексно-сопряженные величины.
Первый член в правой части формулы (42) является суммой по всем парам локализованных орбиталей

Второй член — сумма не диагональных (перекрестных) парных корреляций

Недиагоналыше парные корреляции аналогичны недиагональным хартри-фоковским орбитальным энергиям, которые возникают, если записать уравнения Хартри — Фока через локализованные орбитали (см., например, т. 1, разд. 1-4). Локализованные орбитали не принадлежат к неприводимым представлениям группы перестановок полного (а также хартри-фоковского) гамильтониана. Поэтому у систем с незаполненными электронными оболочками сохраняются недиагональные орбитальные и корреляционные энергии; у систем с заполненными электронными оболочками указанные величины исчезают при суммировании по всем орбиталям.
Таким образом, члены, входящие в выражение (41) для энергии системы Не — Не, принимают вид

Заметим, что корреляционные энергии 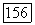 которые описывают корреляции
которые описывают корреляции 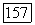 непосредственно преобразуют в выражения, которые в свою очередь описывают корреляции пар
непосредственно преобразуют в выражения, которые в свою очередь описывают корреляции пар 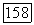 Эти последние пары аналогичны парам молекулярных орбиталей, описывающим состояние 32? молекулы
Эти последние пары аналогичны парам молекулярных орбиталей, описывающим состояние 32? молекулы 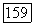 в вариационных вычислениях может быть использована аналогичная пробная функция
в вариационных вычислениях может быть использована аналогичная пробная функция 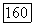 Недиагональные парные корреляции для локализованных орбиталей в формулах
Недиагональные парные корреляции для локализованных орбиталей в формулах 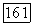 исчезают при
исчезают при 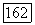 В этом пределе получаем
В этом пределе получаем

Таким образом, корреляционные энергии для пар орбиталей 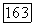 и
и 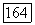 каждая в отдельности составляют почти половину полной корреляционной энергии атома Не. Это обусловлено тем, что молекулярные орбитали сильно делокализованы, и энергии орбиталей
каждая в отдельности составляют почти половину полной корреляционной энергии атома Не. Это обусловлено тем, что молекулярные орбитали сильно делокализованы, и энергии орбиталей 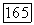 близки между собой; кроме того, надо учитывать соображения симметрии. Небольшое различие в энергиях молекулярных орбиталей
близки между собой; кроме того, надо учитывать соображения симметрии. Небольшое различие в энергиях молекулярных орбиталей 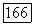 возникающее при расщеплении энергий атомных орбиталей Не, приводит к появлению межатомного отталкивания, которое описывается хартри-фоковской компонентой энергии
возникающее при расщеплении энергий атомных орбиталей Не, приводит к появлению межатомного отталкивания, которое описывается хартри-фоковской компонентой энергии 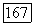 Подобным образом в выражениях
Подобным образом в выражениях 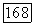 небольшое различие между энергиями для пар электронов с противоположными спинами, а также между энергиями
небольшое различие между энергиями для пар электронов с противоположными спинами, а также между энергиями 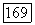 для пар электронов с одинаковыми спинами непосредственно приводит к возникновению вандерваальсовых сил притяжения.
для пар электронов с одинаковыми спинами непосредственно приводит к возникновению вандерваальсовых сил притяжения.
Как отмечалось выше, систему Не — Не для всех значений 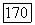 можно описывать и при помощи молекулярных орбиталей и при помощи локализованных орбиталей. Выражения (49) показывают, почему вычисления с использованием МО при учете конфигурационного взаимодействия менее удобны в случае больших
можно описывать и при помощи молекулярных орбиталей и при помощи локализованных орбиталей. Выражения (49) показывают, почему вычисления с использованием МО при учете конфигурационного взаимодействия менее удобны в случае больших 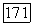 и почему многие члены отвечают только небольшому притяжению [22], в то время как даже более простые вычисления с использованием локализованных орбиталей (что эквивалентно методу Гайтлера — Лондона с учетом конфигурационного взаимодействия) приводят к значительно большему притяжению [14, 23, 24]. При использовании для вычислений локализованных орбиталей большинство атомных энергий
и почему многие члены отвечают только небольшому притяжению [22], в то время как даже более простые вычисления с использованием локализованных орбиталей (что эквивалентно методу Гайтлера — Лондона с учетом конфигурационного взаимодействия) приводят к значительно большему притяжению [14, 23, 24]. При использовании для вычислений локализованных орбиталей большинство атомных энергий 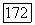 взаимно компенсируются. Однако остающаяся часть энергии, которая входит в выражение (39), вносит все еще заметный вклад в потенциальную энергию
взаимно компенсируются. Однако остающаяся часть энергии, которая входит в выражение (39), вносит все еще заметный вклад в потенциальную энергию 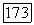 и она должна быть аккуратно вычислена. Указанные эффекты возмущения становятся еще большими при
и она должна быть аккуратно вычислена. Указанные эффекты возмущения становятся еще большими при 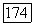 Поэтому метод МО оказывается наиболее удобным только для межатомных расстояний
Поэтому метод МО оказывается наиболее удобным только для межатомных расстояний