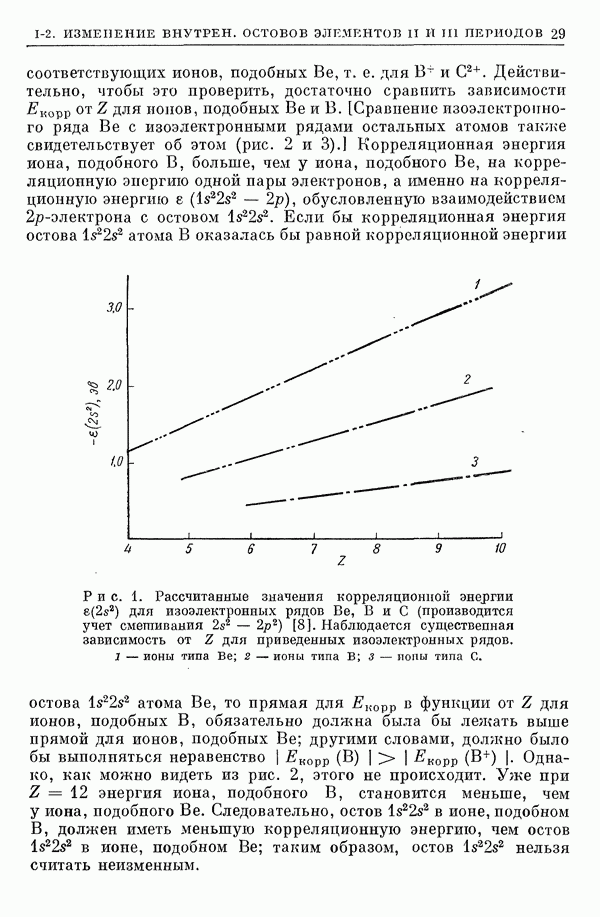
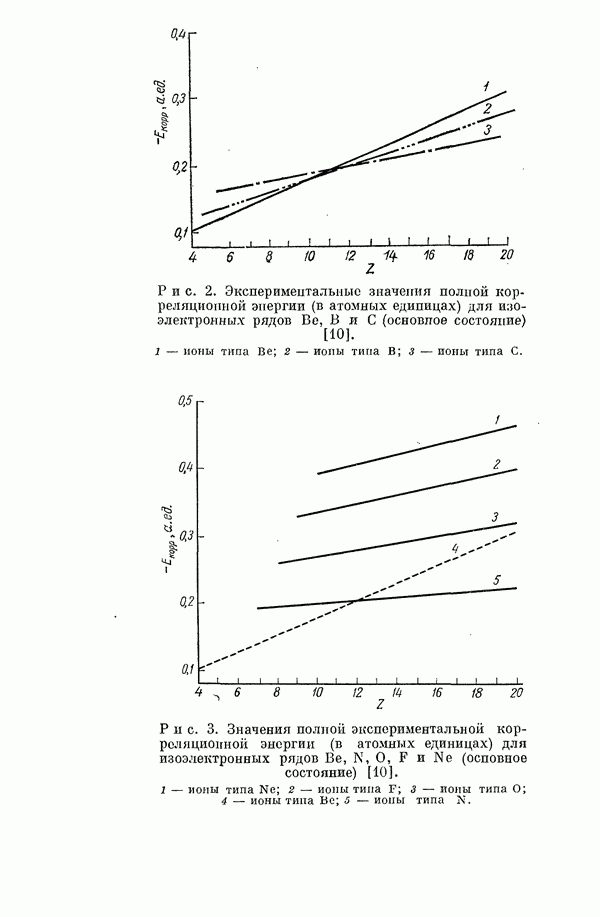
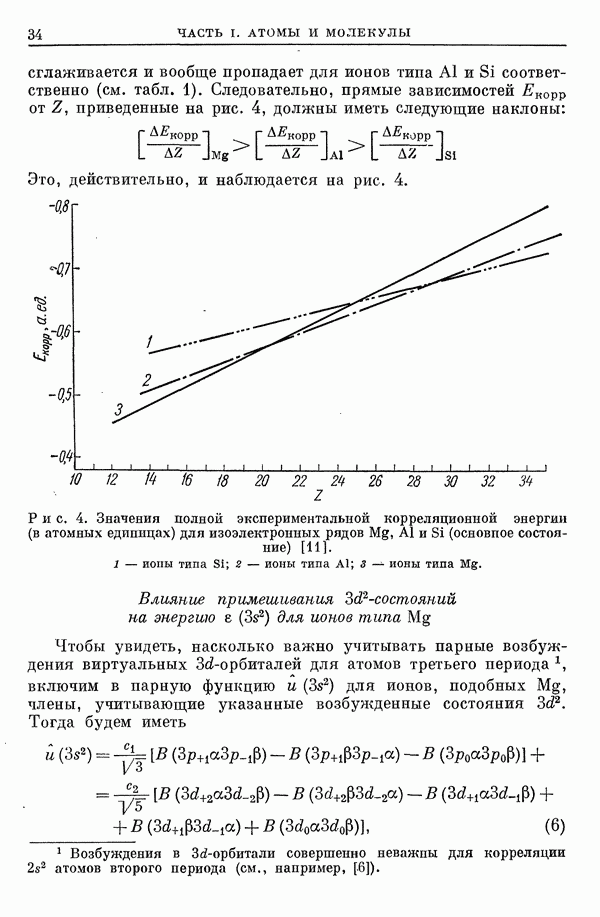
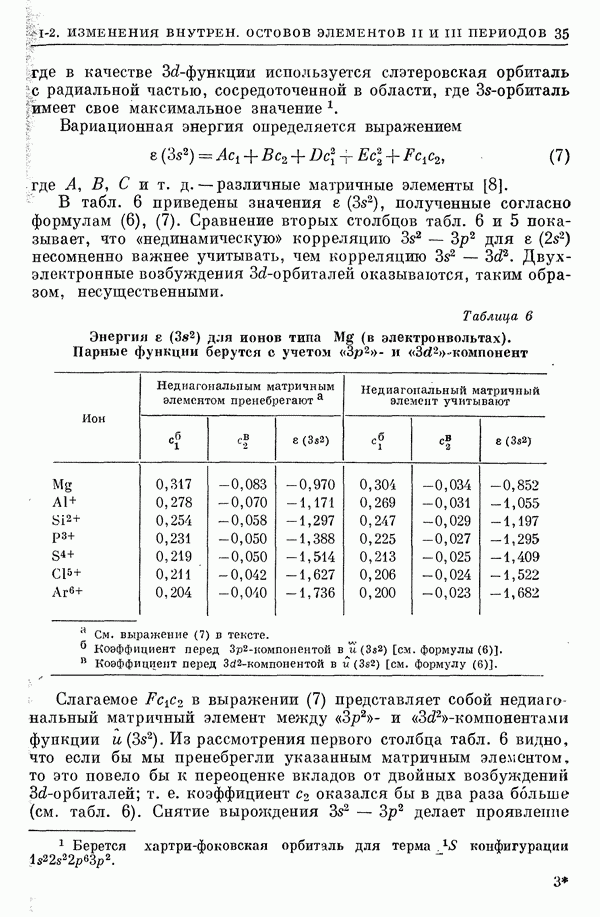
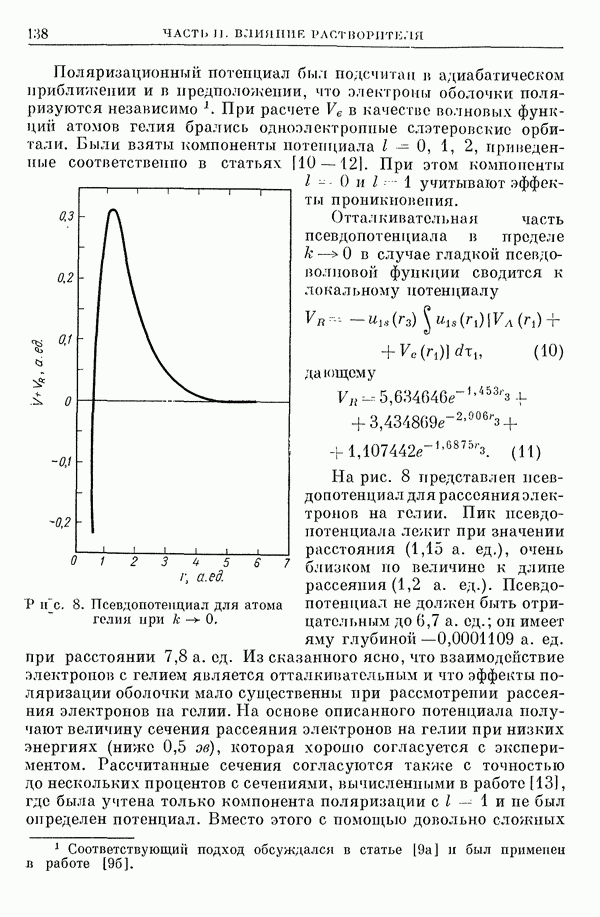
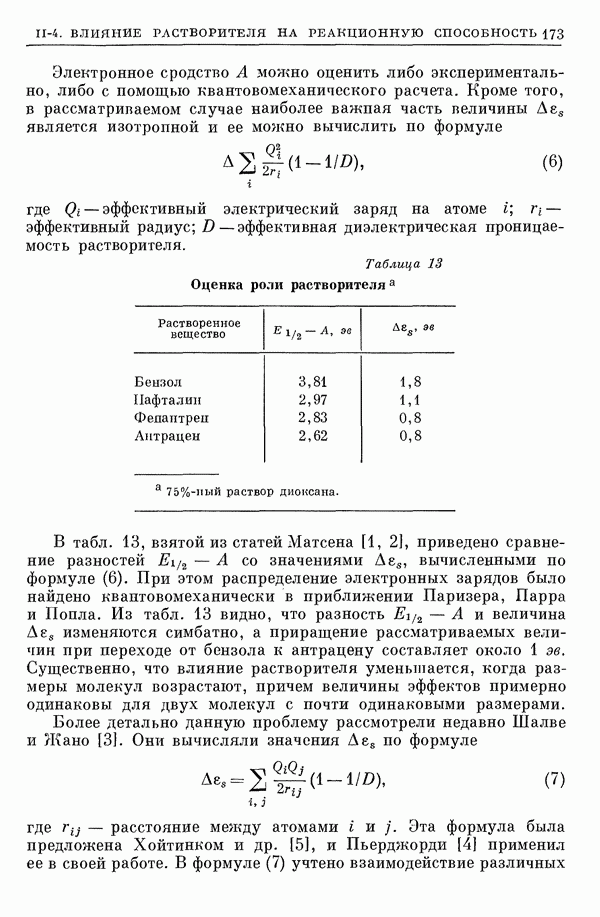
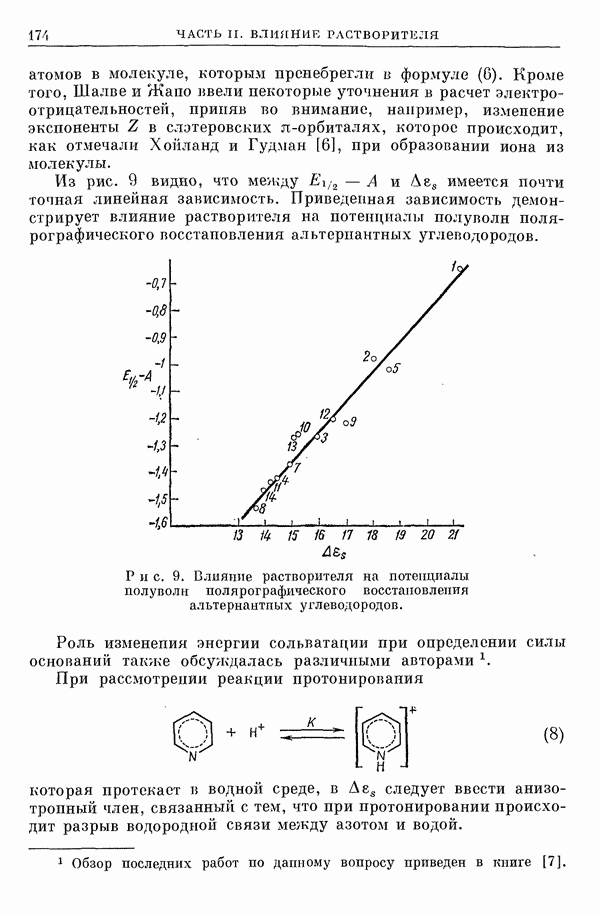
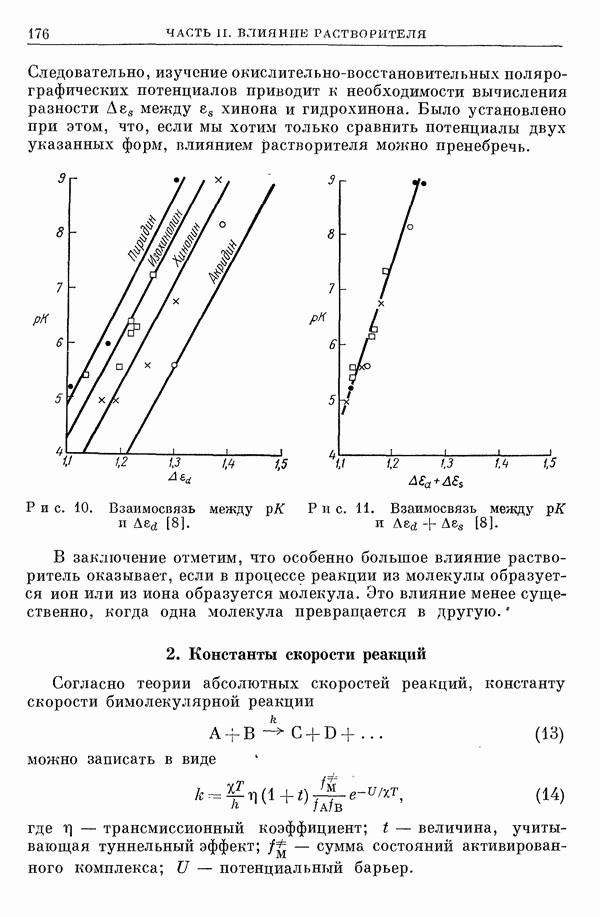
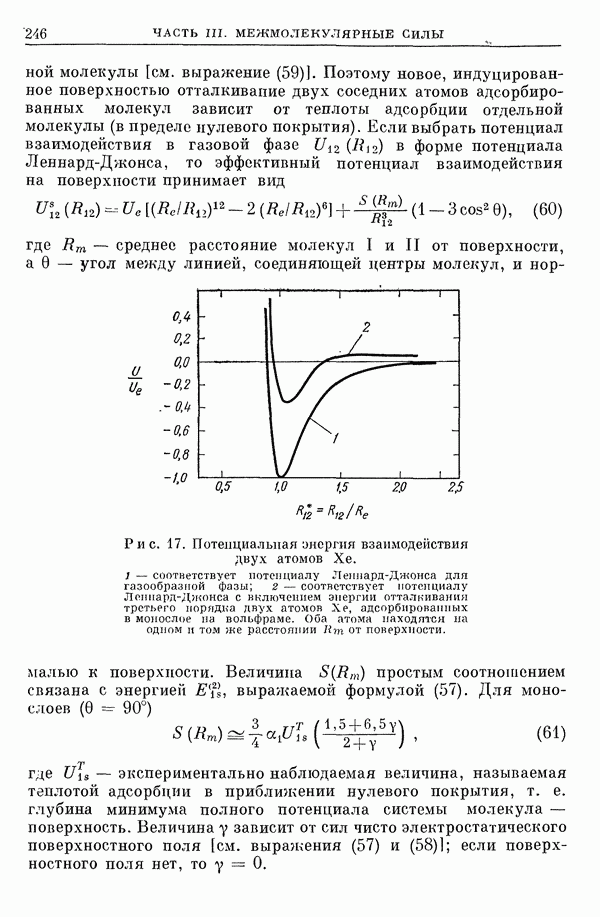
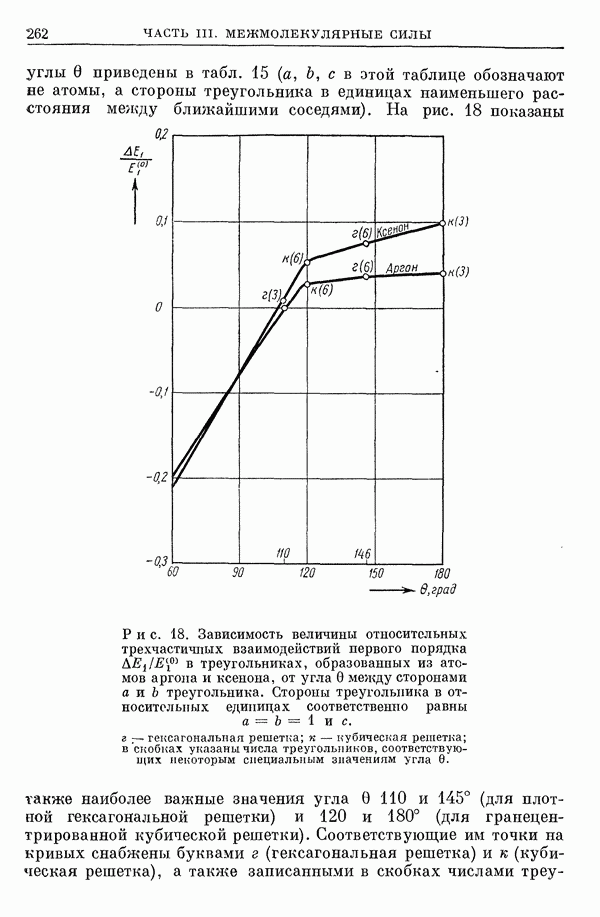
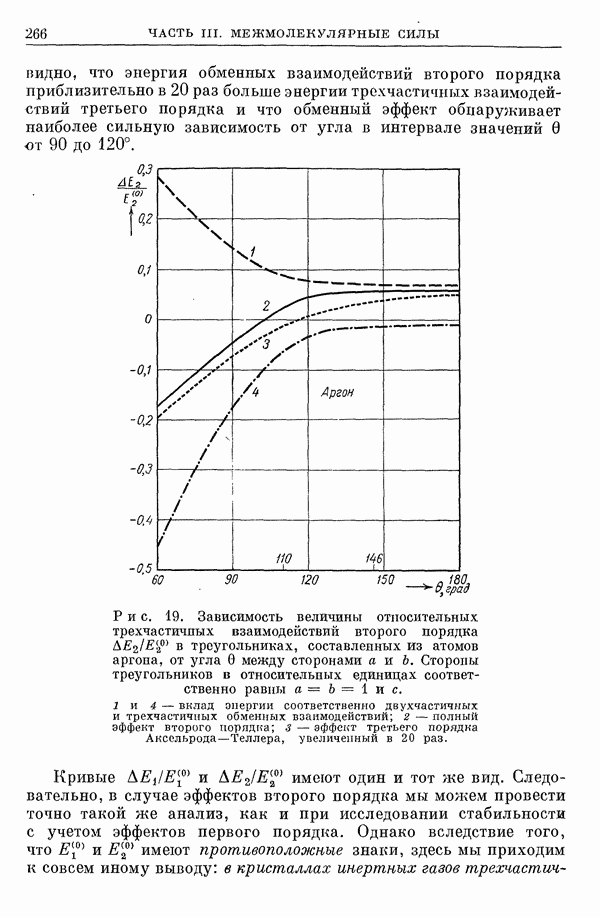
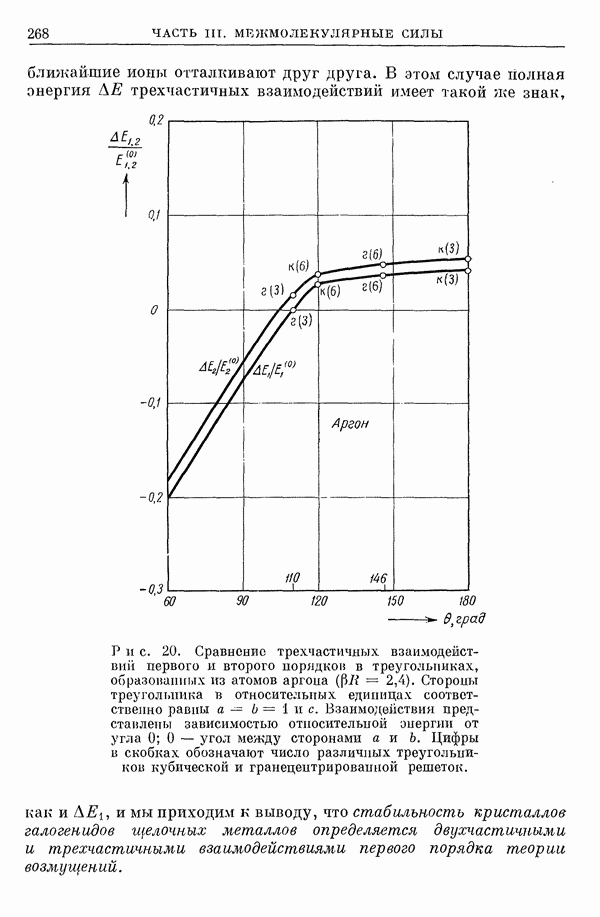
I-6. Энергия диссоциации
С. Холистер и О. Синаноглу
1. Введение
Корреляционная энергия — важная характеристика молекул. Во многих случаях большая часть энергии химической связи молекулы определяется корреляционной энергией; расчеты по методу Хартри — Фока часто оказываются не в состоянии правильно предсказать энергию связи. Так, например, для молекулы  рассчитанная хартри-фоковская энергия химической связи равна —0,060 а. ед. (1 а. ед. = 27,2 эв) [1]; экспериментальное же значение этой энергии
рассчитанная хартри-фоковская энергия химической связи равна —0,060 а. ед. (1 а. ед. = 27,2 эв) [1]; экспериментальное же значение этой энергии  а. ед. Таким образом, ясно, что для молекулярных систем с неизвестпой «экспериментальной» энергией связи очень важно уметь определять корреляционную энергию независимо от хартри-фоковских расчетов. Для молекулярных систем с известной энергией связи знание корреляционной энергии можно использовать для предсказания значений хартри-фоковской энергии; такие значения могут оказаться полезными для контроля хартри-фоковских расчетов.
а. ед. Таким образом, ясно, что для молекулярных систем с неизвестпой «экспериментальной» энергией связи очень важно уметь определять корреляционную энергию независимо от хартри-фоковских расчетов. Для молекулярных систем с известной энергией связи знание корреляционной энергии можно использовать для предсказания значений хартри-фоковской энергии; такие значения могут оказаться полезными для контроля хартри-фоковских расчетов.
В работах [2а, б] были подробно изучены вопросы о построении общего выражения для корреляционной энергии многоэлектронной системы, о связи корреляционной энергии определенной группы электронов с системой остальных электронов и, наконец, о различных корреляционных эффектах и их относительной важности. Теперь возникает задача, как, рассматривая отдельные группы электронов молекулы и сравнивая их с соответствующими группами электронов атомов (для которых корреляциопная энергия обычно известна), можно получить корреляционную энергию молекулы. Мы изложим два нолуэмпирических метода, с помощью которых можно решить указанную задачу [3]. Эти методы должны давать оценки корреляционной энергии сверху и снизу, поскольку в них молекула рассматривается с двух различных предельных точек зрения. Ниже среди прочих рассматриваются двухатомные молекулы из элементов второго периода, для которых имеются данные хартри-фоковских расчетов. Применяя к этим молекулам оба полуэмпирических метода расчета корреляционной энергии, мы получаем возможность проверить наши результаты по результатам хартри-фоковских расчетов и убедиться в эффективности излагаемых методов.
2. Эффекты «исключения»
О влиянии окружения электронов на корреляцию данной пары электронов [2а, б] уже подробно говорилось в разд. 1-2. Эта корреляция может резко изменяться при переходе от системы к системе, если только мы имеем дело с корреляцией «нединамического» типа. Такие изменения главным образом связаны с эффектами «исключения» для занятых орбиталей.
Ранее в работах [4, 2] было показано, что величина  становится несущественной с ростом заселенности
становится несущественной с ростом заселенности  -оболочки для атомов и ионов (см. также разд. 1-2). Так, атом
-оболочки для атомов и ионов (см. также разд. 1-2). Так, атом  характеризуется конфигурацией
характеризуется конфигурацией  и в приближении, когда парная функция
и в приближении, когда парная функция  выражается функцией по методу взаимодействия конфигураций, мы получаем, что
выражается функцией по методу взаимодействия конфигураций, мы получаем, что  Будет ли корреляция оставаться малой при переходе к молекуле
Будет ли корреляция оставаться малой при переходе к молекуле  Конфигурация молекулы следующая:
Конфигурация молекулы следующая:  причем от указанных занятых орбиталей близко расположены незанятые орбитали и Зстц, подобные незанятым
причем от указанных занятых орбиталей близко расположены незанятые орбитали и Зстц, подобные незанятым  -орбиталям атома азота К. Так что опять мы имеем дело со случаем «почти вырождения», и энергия
-орбиталям атома азота К. Так что опять мы имеем дело со случаем «почти вырождения», и энергия  должна быть малой, однако следует ожидать, что энергия
должна быть малой, однако следует ожидать, что энергия  несколько увеличится в молекуле
несколько увеличится в молекуле  по сравнению с атомом
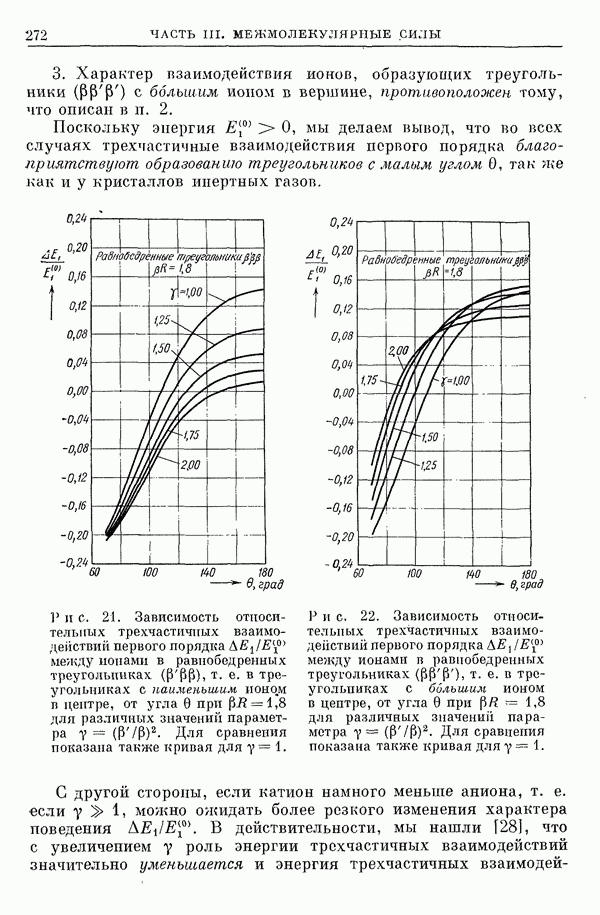
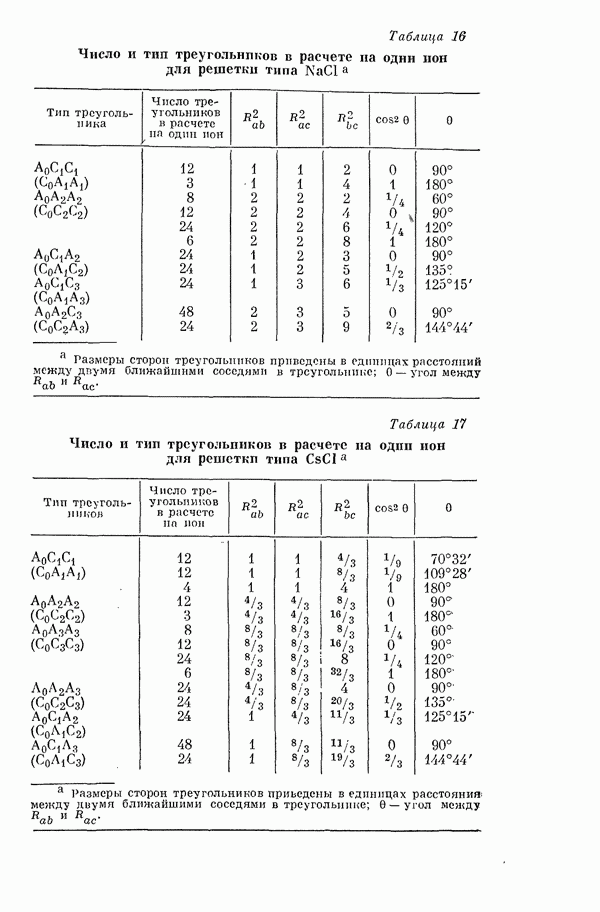
по сравнению с атомом  ибо некоторая доля
ибо некоторая доля  -электронов «выталкивается» из области ядер в область связи.
-электронов «выталкивается» из области ядер в область связи.
Чтобы проверить справедливость этого утверждения, были проведены расчеты молекулы  с учетом четырех
с учетом четырех  взаимодействующих конфигураций, при этом в качестве базисных использовались орбитали Рэнсила [5]. Взятая парная функция имела вид
взаимодействующих конфигураций, при этом в качестве базисных использовались орбитали Рэнсила [5]. Взятая парная функция имела вид

где

Коэффициенты в формуле (1) определялись с помощью вариационной процедуры, подробно описанной в работах [2а, б], т. е. с помощью минимизации выражения

Было проведено три расчета: в качестве  брали орбитали
брали орбитали  со спинами
со спинами  Далее было использовано унитарное преобразование
Далее было использовано унитарное преобразование  которое переводит молекулярные орбитали
которое переводит молекулярные орбитали
в локализованные «основные» орбитали (которые заняты  -элек-тронами каждого атома
-элек-тронами каждого атома  Такое преобразование было предложено Петерсом [6]. Если отвлечься от примеси орбиталей
Такое преобразование было предложено Петерсом [6]. Если отвлечься от примеси орбиталей  преобразование
преобразование  в точности дает атомные
в точности дает атомные  -орбитали, ибо оно преобразует
-орбитали, ибо оно преобразует  Если же в унитарном преобразовании
Если же в унитарном преобразовании  затрагиваются орбитали
затрагиваются орбитали  то преобразование
то преобразование  только приближенно совпадает с преобразованием к эквивалентным орбиталям, хотя примесь орбитали
только приближенно совпадает с преобразованием к эквивалентным орбиталям, хотя примесь орбитали  в локализованной орбитали и будет незначительной; так что локализованная орбиталь
в локализованной орбитали и будет незначительной; так что локализованная орбиталь  будеть очень близкой к атомной
будеть очень близкой к атомной  -орбитали. Энергия (2) преобразованием
-орбитали. Энергия (2) преобразованием  сводится к виду
сводится к виду 

где  — индексы
— индексы  — орбитали
— орбитали  . Рассчитанная энергия
. Рассчитанная энергия  для каждого атома
для каждого атома  равна
равна  таким образом, энергия остова
таким образом, энергия остова  атома
атома  в молекуле
в молекуле  примерно на
примерно на  мепьше, чем энергия соответствующего остова свободного атома
мепьше, чем энергия соответствующего остова свободного атома  Поэтому для молекулы
Поэтому для молекулы  мы должны включить
мы должны включить  в энергию химической связи!
в энергию химической связи!
Изложенные расчеты грубые. Они показывают только большую величину эффекта «исключения». Чтобы получить более надежные данные, необходимо обратиться к методу конфигурационного взаимодействия с учетом большого числа хартри-фоковских молекулярных орбиталей.
Тем не менео для оценок мы будем пользоваться значительно более простым методом, в котором не нужно знать молекулярные хартри-фоковские орбитали и энергии. Так, мы можем построить графики  для атомов с данным значением
для атомов с данным значением  в зависимости от числа
в зависимости от числа  электронов с параллельными спинами, используя значения
электронов с параллельными спинами, используя значения  рассчитанные в работе [4]. Этот график позволяет провести интерполяцию и определить
рассчитанные в работе [4]. Этот график позволяет провести интерполяцию и определить  в случае дробных чисел заполнения
в случае дробных чисел заполнения  электронов; такие дробные числа заполнения как раз возникают в молекулах вследствие имеющего место смещения электронов в область химической связи (в силу чего числа заполнения
электронов; такие дробные числа заполнения как раз возникают в молекулах вследствие имеющего место смещения электронов в область химической связи (в силу чего числа заполнения  для электронов оказываются, вообще говоря, несколько меньшими, чем для свободных атомов). Такого рода интерполяция для молекулы
для электронов оказываются, вообще говоря, несколько меньшими, чем для свободных атомов). Такого рода интерполяция для молекулы  дает в качестве значения
дает в качестве значения  величину
величину  на отдельный атом
на отдельный атом  т. е. мы получаем примерно то же число, которое было рассчитано выше по методу взаимодействия конфигураций. Ниже всюду для энергии
т. е. мы получаем примерно то же число, которое было рассчитано выше по методу взаимодействия конфигураций. Ниже всюду для энергии  мы будем использовать указанные интерполяционные значения.
мы будем использовать указанные интерполяционные значения.
3. Метод I
Модель объединенного атома для молекулы, очевидно, дает верхнюю оценку для величины корреляционной энергии, поскольку, во-первых, все нужные пары электронов присутствуют и в объединенном атоме и, во-вторых, все они занимают в объединенном атоме меньшую область пространства, чем в молекуле. Вместе с тем такая модель молекулы весьма нереалистична, потому что: 1) в ней ошибочно учитывается сильное отталкивание атомных остовов, возникающее при  и 2) внутренние электроны, оказываются более прочно связанными с ядрами ввиду увеличившегося заряда ядра. Рассматривая в методе ЛKAO-MO коэффициенты перед
и 2) внутренние электроны, оказываются более прочно связанными с ядрами ввиду увеличившегося заряда ядра. Рассматривая в методе ЛKAO-MO коэффициенты перед  убеждаемся, что низкие МО довольно точно могут быть аппроксимированы орбиталями электронов атомных остовов молекулы; эти электропы сосредоточены в непосредственной близости от ядер. Напротив, в модели объединенного атома энергии соответствующих низких орбиталей очень далеки от энергий истинных орбиталей. Модель объединенного атома, исторически возникшая из рассмотрения простейшего иона
убеждаемся, что низкие МО довольно точно могут быть аппроксимированы орбиталями электронов атомных остовов молекулы; эти электропы сосредоточены в непосредственной близости от ядер. Напротив, в модели объединенного атома энергии соответствующих низких орбиталей очень далеки от энергий истинных орбиталей. Модель объединенного атома, исторически возникшая из рассмотрения простейшего иона  оказывается, таким образом, полезной только для гидридов, но не для более сложных молекул, содержащих по нескольку атомных остовов.
оказывается, таким образом, полезной только для гидридов, но не для более сложных молекул, содержащих по нескольку атомных остовов.
Для большинства молекул более близкой является модель, которую мы здесь назовем моделью атома с сокращенным остовом. В этой модели остов результирующего атома действует на внешние МО так же, как настоящие атомные остовы молекулы. Атомные орбитали атома с сокращенным остовом и их энергии ближе соответствуют МО и их энергиям. То же нужно сказать в отношении корреляционных энергий отдельных пар электронов и о влиянии на них эффектов «исключения».
Поясним на примере, что представляет собой модель атома с сокращенным остовом. Возьмем молекулу  имеющую 12 электронов и находящуюся в состоянии, описываемом термом
имеющую 12 электронов и находящуюся в состоянии, описываемом термом  Простой расчет по методу ЛKAO-MO [5] показывает, что атомные остовы молекулы образованы четырьмя
Простой расчет по методу ЛKAO-MO [5] показывает, что атомные остовы молекулы образованы четырьмя  -электронами, движущимися в окрестностях своих ядер. Таким образом, молекулярный остов имеет вид
-электронами, движущимися в окрестностях своих ядер. Таким образом, молекулярный остов имеет вид  Оставшиеся 8 внешних электронов (молекулярных орбиталей) испытывают влияние остова «тина
Оставшиеся 8 внешних электронов (молекулярных орбиталей) испытывают влияние остова «тина  по крайней мере при удалении в радиальном направлении от межмолекулярной оси; причем положительный заряд остова оказывается примерно суммой зарядов двух
по крайней мере при удалении в радиальном направлении от межмолекулярной оси; причем положительный заряд остова оказывается примерно суммой зарядов двух  В модели атома с сокращенным остовом указанные 8 электронов, занимающие МО, довольно размазанные по молекуле, заменяются внешними орбиталями атома с остовом
В модели атома с сокращенным остовом указанные 8 электронов, занимающие МО, довольно размазанные по молекуле, заменяются внешними орбиталями атома с остовом  и зарядом ядра, равным полному заряду ядер. Таким атомом является неон.
и зарядом ядра, равным полному заряду ядер. Таким атомом является неон.
Следовательно, состояние молекулы представляется в виде  Соответственно корреляционная энергия дается выражением
Соответственно корреляционная энергия дается выражением

Хотя описанным методом очень легко пользоваться, следует проявлять известную осторожность. Во-первых, необходимо заботиться о сохранении соответствующего подобия, а именно в модели атома с сокращенным остовом должно содержаться такое же количество электронов, что и в исходной молекуле. Например, для  соответствующим атомом с сокращенным остовом будет атом С. В этом случае к тому же нельзя использовать основное состояние атома С, поскольку оно характеризуется термом
соответствующим атомом с сокращенным остовом будет атом С. В этом случае к тому же нельзя использовать основное состояние атома С, поскольку оно характеризуется термом  и имеет два неспарепных электрона, в то время как
и имеет два неспарепных электрона, в то время как  находится в состоянии и вовсе не имеет неспаренных электронов. Нужно
находится в состоянии и вовсе не имеет неспаренных электронов. Нужно
Таблица 7а (см. скан) Значения корреляционной энергии, рассчитанные для некоторых молекул
всегда заботиться о правильном выборе состояния атома; иначе нам не удастся правильно учесть с помощью него эффекты «исключения». Во-вторых, и это также связано с правильным учетом эффектов «исключения», можно по-разному разбивать электроны молекулы на электроны остова и внешние электроны. Так, например, молекулу  можно рассматривать либо с помощью модели
можно рассматривать либо с помощью модели  как это мы делали выше, либо с помощью модели
как это мы делали выше, либо с помощью модели  Какая из этих возможностей лучше — неясно. Это зависит от величины перекрывания
Какая из этих возможностей лучше — неясно. Это зависит от величины перекрывания  -орбиталей на разных атомах. Если возможна первая модель, значение
-орбиталей на разных атомах. Если возможна первая модель, значение  надо брать непосредственно для свободных ионов
надо брать непосредственно для свободных ионов  так как вряд ли имеется какое-либо влияние «исключения» на «динамическую» корреляцию
так как вряд ли имеется какое-либо влияние «исключения» на «динамическую» корреляцию  При второй возможности, напротив, остовы
При второй возможности, напротив, остовы  имеют «нединамическую»
имеют «нединамическую»  -корреляцию с энергией
-корреляцию с энергией  на которую существенно влияет «исключение» (и потому ее нельзя считать неизменной при переходе от одной молекулы к другой). Таким образом, энергия
на которую существенно влияет «исключение» (и потому ее нельзя считать неизменной при переходе от одной молекулы к другой). Таким образом, энергия  не равна
не равна  а приблизительно та же, что
а приблизительно та же, что  или
или 
Модель атома с сокращенным остовом применима, разумеется, только к двухатомным молекулам, а также к простейшим многоатомным молекулам — гидридам  Следует ожидать, что там, где эта модель оказывается эффективной, она всегда дает оценку сверху для величины
Следует ожидать, что там, где эта модель оказывается эффективной, она всегда дает оценку сверху для величины 
Результаты применения такой модели к расчетам корреляционной энергии двухатомных молекул, составленных из элементов второго периода, и сравнепие их с результатами расчетов по изложенному ниже методу II приведены в табл. 7а.
4. Метод II
Как показано в разд. 1-2 и 1-7, корреляционная энергия  для основного состояния молекулы с заполненными оболочками или основного состояния с незаполненными оболочками, представляемого однодетерминантной функцией, приближенно равна
для основного состояния молекулы с заполненными оболочками или основного состояния с незаполненными оболочками, представляемого однодетерминантной функцией, приближенно равна  т. е. Екорр представляется суммой корреляций соответствующих пар
т. е. Екорр представляется суммой корреляций соответствующих пар  обозначают хартри-фоковские спин-орбитали). В рамках метода ЛКАО-МО в этой сумме легко выделить межатомные и внутриатомные пары.
обозначают хартри-фоковские спин-орбитали). В рамках метода ЛКАО-МО в этой сумме легко выделить межатомные и внутриатомные пары.
Хартри-фоковская волновая функция уже учитывает основные эффекты дальнодействующего кулоновского взаимодействия, которые и обусловливают большие деформации атомов в молекуле. Остающаяся часть корреляции (в основном «динамического»
характера) является результатом действия близкодействующих флуктуирующих потенциалов [2]. Так что в молекулу переносится не полная энергия атомов, а корреляционная энергия, будучи более «локальным» свойством (после учета эффектов «исключения» и «почти вырождения»). В более ранних методах «атомов в молекулах» [8] имели дело главным образом с полными энергиями атомов, основываясь на представлениях о валентных связях.
Вообще говоря, для определения молекулярной корреляции по данным для отдельных атомов, а также для правильного описания областей перекрывапия зарядов существует несколько возможных способов, но здесь мы обсудим лишь один из этих способов.
Исследуем волновую функцию с ограниченным базисом с помощью анализа заселенностей в методе  В этом методе вместо АО возникают члены
В этом методе вместо АО возникают члены  и
и  Члены первого типа связаны с атомными заселенностями и характеризуют время, которое электронная пара проводит вблизи данного ядра. Члены второго типа включают два центра
Члены первого типа связаны с атомными заселенностями и характеризуют время, которое электронная пара проводит вблизи данного ядра. Члены второго типа включают два центра  и
и  они являются мерой заселенности областей перекрывания и характеризуют время, которое электроны проводят в области химической связи. Сумма атомных заселенностей и заселенностей областей перекрывания (последнюю условно распределяют между атомами по Малликену [9]) для данной АО дает эффективную атомную заселенность; эта величина показывает, как электроны распределяются между атомами, и учитывает заселенности областей перекрывания и взаимодействия электронов, находящихся на различных центрах. Таким образом, для вычисления молекулярной корреляционной энергии мы получаем возможность использовать значения атомных парных корреляционных энергий
они являются мерой заселенности областей перекрывания и характеризуют время, которое электроны проводят в области химической связи. Сумма атомных заселенностей и заселенностей областей перекрывания (последнюю условно распределяют между атомами по Малликену [9]) для данной АО дает эффективную атомную заселенность; эта величина показывает, как электроны распределяются между атомами, и учитывает заселенности областей перекрывания и взаимодействия электронов, находящихся на различных центрах. Таким образом, для вычисления молекулярной корреляционной энергии мы получаем возможность использовать значения атомных парных корреляционных энергий 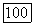 вводя их как коэффициенты перед заселенностями.
вводя их как коэффициенты перед заселенностями.
Рассмотрим молекулу 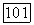 Анализ заселенностей [10] для волновой функции но методу ЛКАО-МО с использованием ограниченного базиса [5] дает следующую картину заселенностей для каждого атома:
Анализ заселенностей [10] для волновой функции но методу ЛКАО-МО с использованием ограниченного базиса [5] дает следующую картину заселенностей для каждого атома: 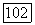 Мы видим, что некоторая доля
Мы видим, что некоторая доля 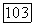 -электронов оказалась промотированной в
-электронов оказалась промотированной в 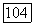 -электроны. Кроме того, в молекуле
-электроны. Кроме того, в молекуле 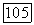 -электроны оказываются спаренными, чего не было в атоме азота
-электроны оказываются спаренными, чего не было в атоме азота 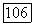 теперь имеется 1,602 пары
теперь имеется 1,602 пары 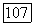 -электронов. Кроме того, согласно изложенному выше, энергия
-электронов. Кроме того, согласно изложенному выше, энергия 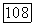 должна несколько увеличиться, а дробная доля заселенности для
должна несколько увеличиться, а дробная доля заселенности для 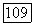 -электронов немного уменьшиться из-за соседства незанятых орбиталей. Соответствующие значения
-электронов немного уменьшиться из-за соседства незанятых орбиталей. Соответствующие значения 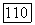 следует определять
следует определять
по атомным данным с помощью графического интерполяционного метода. Неизменяемые же «динамические» парные корреляции, такие, как 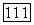 можно взять прямо такими, как для атомов [4].
можно взять прямо такими, как для атомов [4].
Описанный выше метод будем называть «методом заселенностей пар»; его удобно использовать, если нам известны волновые функции в методе ЛKAO-MO с ограниченной системой базисных орбиталей ССП. Заселенности изменяются, вероятно, не очень сильно при использовании расширенной базисной системы молекулярных орбиталей ССП (обзор недавних расчетов и соответствующую терминологию см. в статье [11]). Следует отметить, что часто расчеты оказываются нечувствительными к точной структуре используемых орбиталей; поэтому мы здесь и использовали заселенности, полученные при расчетах с ограниченным базисом.
Результаты применения методов I и II к расчету корреляционной энергии молекул обсуждаются ниже (см. также табл. 7а).
5. Результаты
Типичные результаты применения методов I и II к некоторым двухатомным и малым многоатомным молекулам, составленным из атомов второго периода, приведены в табл. 7а.
Во всех случаях при использовании метода I (модель атома с сокращенным остовом) в качестве остовов 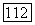 принимали остов
принимали остов 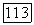 Поэтому эффекты «исключения» для
Поэтому эффекты «исключения» для 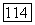 учитывались автоматически при рассмотрении атома с сокращенным остовом, заменяющим молекулу. Таким образом, при применении метода I нужно было знать только хартри-фоковские и полные энергии для рассматриваемых атомов и для ионов типа Не; использованные здесь хартри-фоковские результаты для атомов были взяты из работы Клементи [12].
учитывались автоматически при рассмотрении атома с сокращенным остовом, заменяющим молекулу. Таким образом, при применении метода I нужно было знать только хартри-фоковские и полные энергии для рассматриваемых атомов и для ионов типа Не; использованные здесь хартри-фоковские результаты для атомов были взяты из работы Клементи [12].
В методе II (метод заселенностей пар) корреляцию 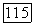 также принимали равной энергии
также принимали равной энергии 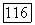 для ионов типа Не [12]. При определении
для ионов типа Не [12]. При определении 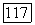 была использована графическая интерполяция по данным Мак-Коя и Синаноглу [4]. Значения величин
была использована графическая интерполяция по данным Мак-Коя и Синаноглу [4]. Значения величин 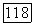 а также
а также 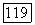 он
он 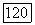 были взяты из работы [41.
были взяты из работы [41.
Анализ заселенностей, проведенный для волновой функции 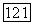 построенной по методу ЛKAO-MO с использованием ограниченного базиса, дает для каждого атома С
построенной по методу ЛKAO-MO с использованием ограниченного базиса, дает для каждого атома С 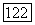 Так как электроны могут иметь любые спины, то на каждую атомную орбиталь приходится половина от каждой пары электронов. Проводя анализ заселенностей для свободного атома и сравнивая его результаты с анализом заселенностей для атомов
Так как электроны могут иметь любые спины, то на каждую атомную орбиталь приходится половина от каждой пары электронов. Проводя анализ заселенностей для свободного атома и сравнивая его результаты с анализом заселенностей для атомов
в молекуле, мы сразу получаем следующее выражение для

В приведенной формуле значения корреляционных эпергий свободных атомов для занятых пар орбиталей, таких, как 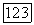 или
или 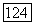 просто умножались на разницу молекулярных и атомных парных заселенностей при условии, что эти корреляции являются неизменяемыми при переходе от одного окружения к другому. В случае корреляции
просто умножались на разницу молекулярных и атомных парных заселенностей при условии, что эти корреляции являются неизменяемыми при переходе от одного окружения к другому. В случае корреляции 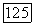 использовались различные значения для свободного атома и атома в молекуле. Межорбитальные корреляции определялись следующим образом: сначала находили их величину на одну орбиталь без учета спина, затем полученную величину умножали на дробное число электронов каждого тина, определяемое в анализе заселенностей. Так, например, атомная корреляция
использовались различные значения для свободного атома и атома в молекуле. Межорбитальные корреляции определялись следующим образом: сначала находили их величину на одну орбиталь без учета спина, затем полученную величину умножали на дробное число электронов каждого тина, определяемое в анализе заселенностей. Так, например, атомная корреляция 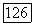 определяется непосредственно по энергии
определяется непосредственно по энергии 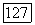 для атома С. Допускалось, что
для атома С. Допускалось, что 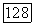 и для значения корреляции спин-орбиталей бралась величина
и для значения корреляции спин-орбиталей бралась величина 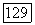 Для атома (Г в молекуле мы имеем 2,33
Для атома (Г в молекуле мы имеем 2,33 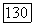 -спин-орбитали и
-спин-орбитали и 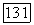 -орбитали. Так что молекулярная корреляция
-орбитали. Так что молекулярная корреляция 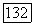 в расчете на один атом
в расчете на один атом 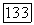 равна
равна 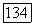 Подставляя в формулу (5) значения парных корреляций из работы [4], получаем
Подставляя в формулу (5) значения парных корреляций из работы [4], получаем

Эта формула приведена здесь так подробно, чтобы было ясно, какую долю вносит каждый корреляционный эффект в энергию связи. Метод II очень легко применим к молекулам любого размера, если только, конечно, нам известны достаточно надежные коэффициенты перед АО в методе 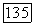 (см. табл. 7а). Из табл. 7а видно, что, несмотря на большое различие методов I и
(см. табл. 7а). Из табл. 7а видно, что, несмотря на большое различие методов I и
II, эти методы, тем не менее, дают согласующиеся результаты. Наиболее важно учитывать корреляционную энергию для связей с небольшим ионным характером; это заключение подтверждается
также результатами имеющихся хартри-фоковских расчетов 
Предсказываемые значения хартри-фоковских энергий в табл. 7а определялись на основе результатов метода II по формуле 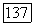 (мол) —
(мол) — 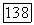 (метод II); при этом предполагалось, что релятивистской поправкой
(метод II); при этом предполагалось, что релятивистской поправкой 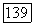 можно пренебречь. Как видно из табл. 7а, получалось очень хорошее согласие с рассчитанными хартри-фоковскими молекулярными энергиями, причем предсказываемое нами значение лежит всегда ниже вычисленного, как это и должно быть.
можно пренебречь. Как видно из табл. 7а, получалось очень хорошее согласие с рассчитанными хартри-фоковскими молекулярными энергиями, причем предсказываемое нами значение лежит всегда ниже вычисленного, как это и должно быть.
ЛИТЕРАТУРА
(см. скан)