Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
2. Молекулярные «сигма»-орбиталиДля интерпретации свойств больших органических молекул в терминах квантовой механики мы вынуждены в общем случае использовать довольно смелые приближения. В связи с этим часто используют подход, в котором ищут и выделяют «инвариантные» составляющие системы. Известное рассмотрение сопряженных молекул в Как хорошо известно, проекция углового момента на ось молекулы имеет определенное значение, т. е. является хорошим квантовым числом. В нелинейных многоатомных молекулах можно использовать понятие Для обсуждения электронной структуры молекул в рамках приближения Борна — Оппенгеймера мы выберем многоэлектронный гамильтониан (в атомных единицах) для органической молекулы в виде
где
Мы не знаем, каков аналитический вид функции называемым «корреляционным» ошибкам, которые в основном обусловлены пренебрежением столкновениями электронов. Такие ошибки мы не будем пока учитывать; о корреляционных эффектах см. в т. 2 настоящей книги. В одноэлектронном приближении функция
причем правая часть представляет собой детерминант
где
где
Во всем предыдущем рассмотрении мы не задавались определенной формой каждой молекулярной орбитали определенного числа известных функций
Для минимизации энергии коэффициенты линейных комбинаций должны удовлетворять уравнениям Фока, которые в этом случае сводятся к виду
где
Выражение для энергии принимает вид
Далее можно принять три вида линейных комбинаций. 1. Метод ЛKAO: 2. Метод ЛКВО: 3. Метод ЛКСО: При необходимости можно использовать при дальнейшем решении также метод конфигурационного взаимодействия, как это обычно делается в ЛКАО-ССП, ЛКВО-ССП и ЛКСО-ССП соответственно. Применению метода ЛКАО-ССП для простых молекул посвящено большое число работ, которые отражены в обзоре Аллена и Каро [2]. Метод ЛКВО-ССП применялся только для специфических простых случаев 13, 4]. Кроме того, Имамура и др. [5] выполнили расчет по методу ЛКВО-АСМО для метана. Леннард-Джонс и др. В некоторых из этих методов
где
Наилучшие функции, даваемые линейной комбинацией вида (5), можно определить путем решения системы линейных уравнений относительно коэффициентов
где
Интегралы
Это упрощение можно применять не только в методе ЛКСО-ССП, но также и в методах ЛКАО-ССП и ЛКВО-ССП. Упрощенные методы, упомянутые выше, мы предлагаем называть ЛКАО-ХМО (молекулярная орбиталь Хюккеля), ЛКВО-ХМО и ЛКСО-ХМО соответственно, поскольку сходный метод, в котором в качестве Во всех методах ЛКСО-ХМО [7а-з] в качестве базисных функций в линейной комбинации применяются только связывающие орбитали для каждой связи и не принимаются во внимание разрыхляющие орбитали. В рамках этой схемы мы не можем рассматривать высшие одноэлектронные уровни, поскольку линейная комбинация орбиталей связи дает только общее число орбиталей, заполненных электронами. Это обстоятельство оказывается серьезным недостатком такого метода при обсуждении, ианример, проблем химической реакционной способности. Для того чтобы избежать этого недостатка, Метод ЛКАО-ХМО был нредложен для практического использования Гофманом [8а, б] при рассмотрении углеводородов с большим молекулярным весом. Для вычислений подобного типа первоначально Малликеп и др. резонансного интеграла
Если необходимо рассмотрение, выходящее за рамки приближения Хюккеля, то мы можем вернуться к антисимметризованному выражению (3) и с помощью метода Хартри — Фока получить более точные сведения о занятых одноэлектронных уровнях. Такая процедура чрезвычайно эффективна, поскольку она позволяет получить определенную информацию об изменении молекулярной энергии при любом изменении конформации молекулярной системы. Таким образом, в принципе мы можем определить наиболее устойчивую конфигурацию молекулы. Это особенно важно также для теории химических реакций в связи с выбором конфигурации «активированного комплекса» и «промежуточных продуктов реакции». Однако указанный подход все же не совершенен, поскольку понятие «связи», которое восстанавливается в методе ЛКАО, благодаря идее об эквивалентной орбитали становится еще более неясным. (При использовании представления о «заселенности связи» [14а] указанное понятие несколько уточняется. Но однозначное вычисление заселенности связи вызывает затруднения.) Нам не нужно строго сохранять консервативное классическое понятие о связи; однако при сопоставлении теории с химической практикой мы еще часто вынуждены к нему обращаться. При этом лучше воспользоваться методом ЛКВО, в котором валентные орбитали вытянуты в направлении В этом направлении выполнено несколько работ [15а-г]. По ряду причин здесь остановимся на методе, предложенном в работе Фукуи и др. [15г], в которой главное внимание уделено рассмотрению углеводородов. Базисные функции в линейной комбинации, т. е.
где Оценка различных интегралов проводится полуэмпирически. Кулоновские интегралы
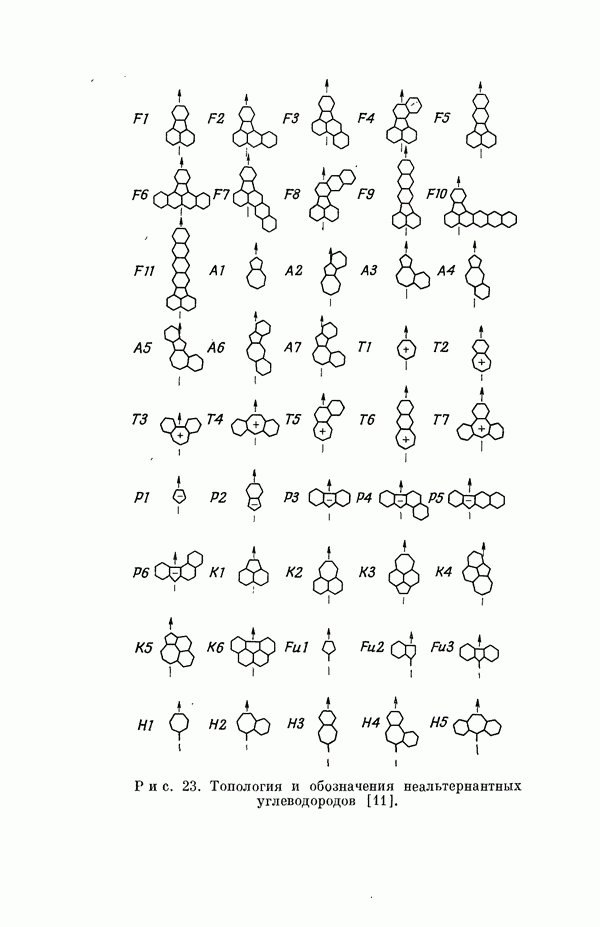
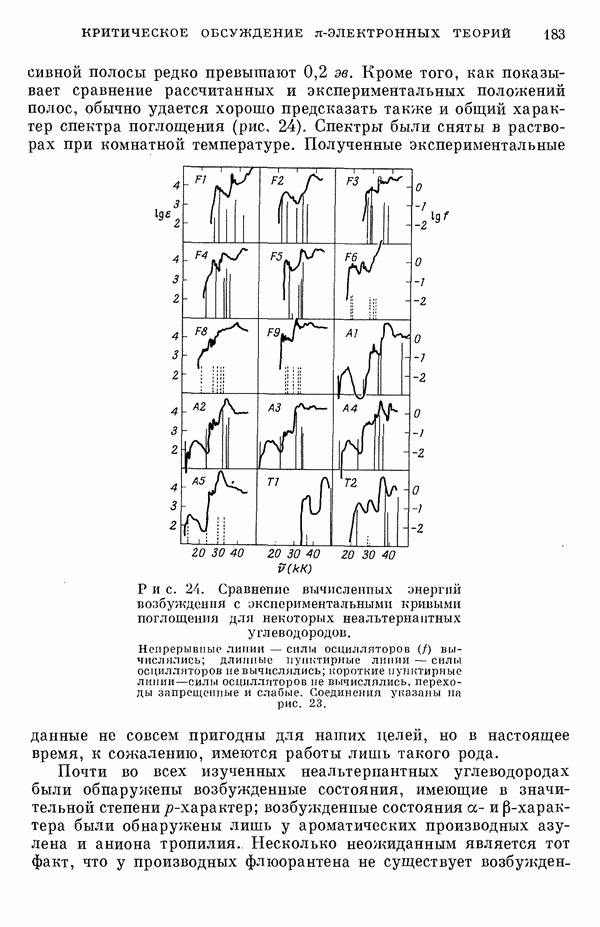
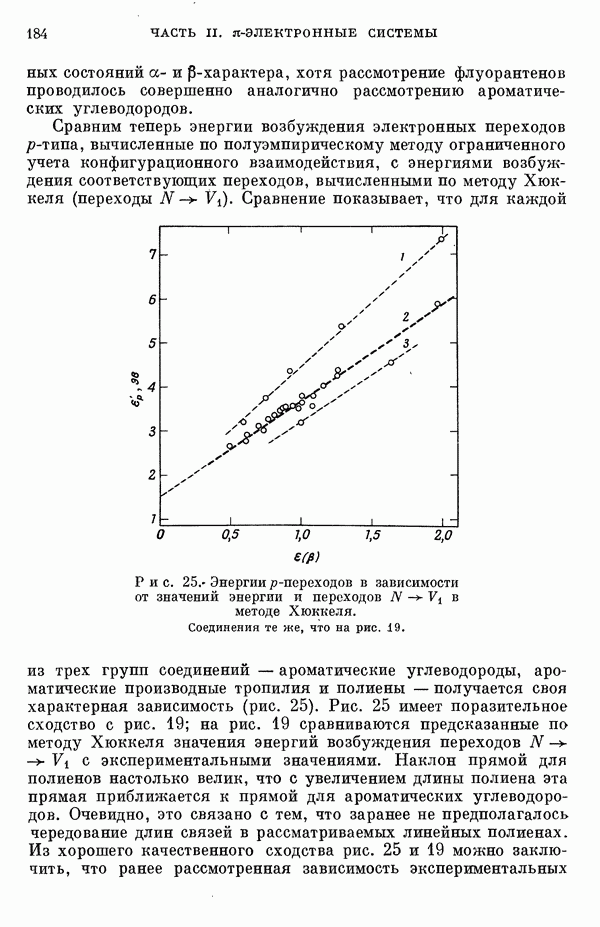
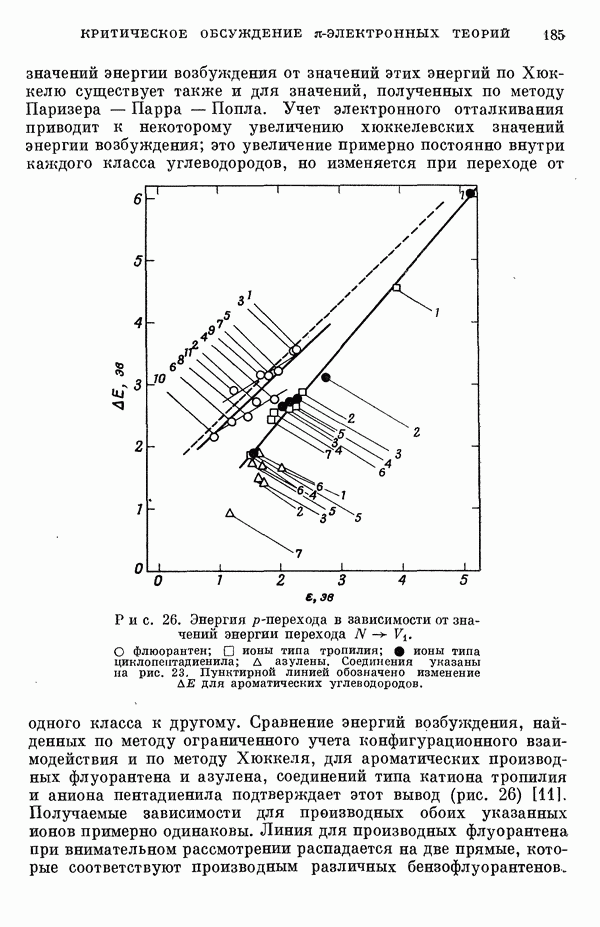
Резонансные интегралы
Все другие, пренебрежимо малые резонапсные интегралы не учитывались. Интегралы перекрыванияСандорфи [15а] принял во внимание интегралы перекрывания, но Фукуи и др. [15г] не стали учитывать их, из-за чего получили картину со смещенными уровнями энергии, на распределении Для Таблица 5
оценке интегралов перекрывания, Фукуи и др. [15а] приняли следующие численные значения, согласующиеся с различными эмпирическими результатами: (см. скан) Интегралы для других типов атомов также определялись аналогичным путем Примеры: Полная плотность а-электронов на каждой валентной атомной орбитали
(кликните для просмотра скана) (см. скан)
|
 |
Оглавление
|






























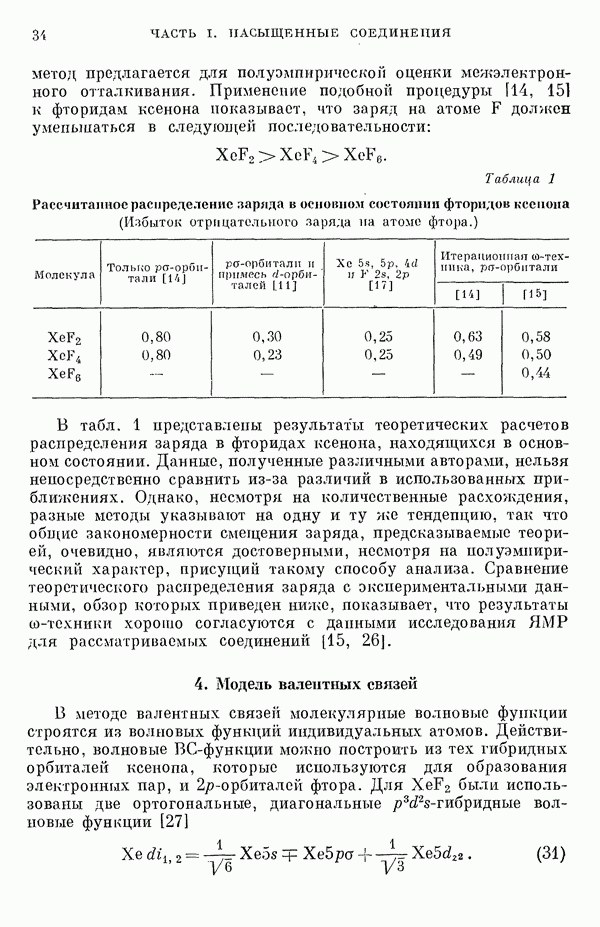




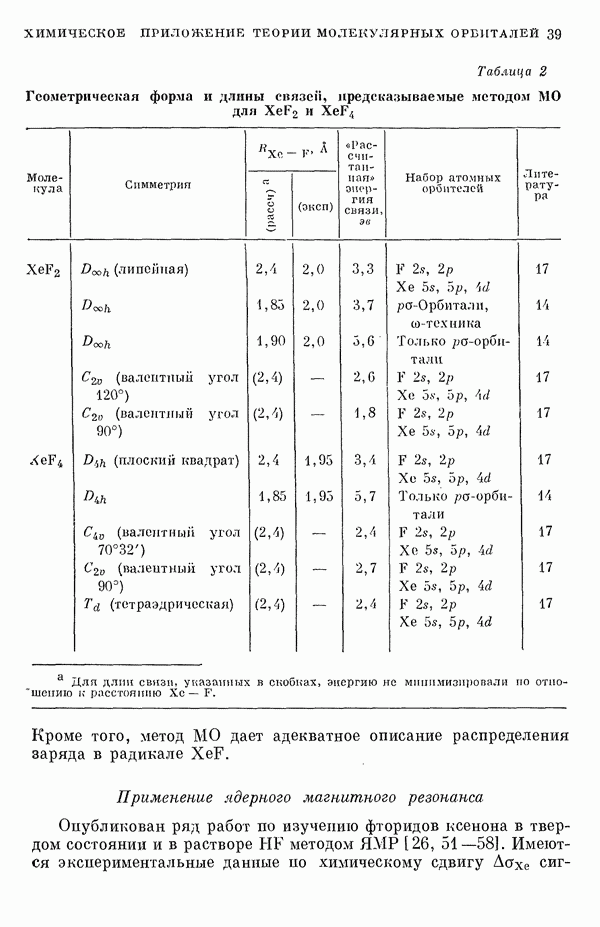
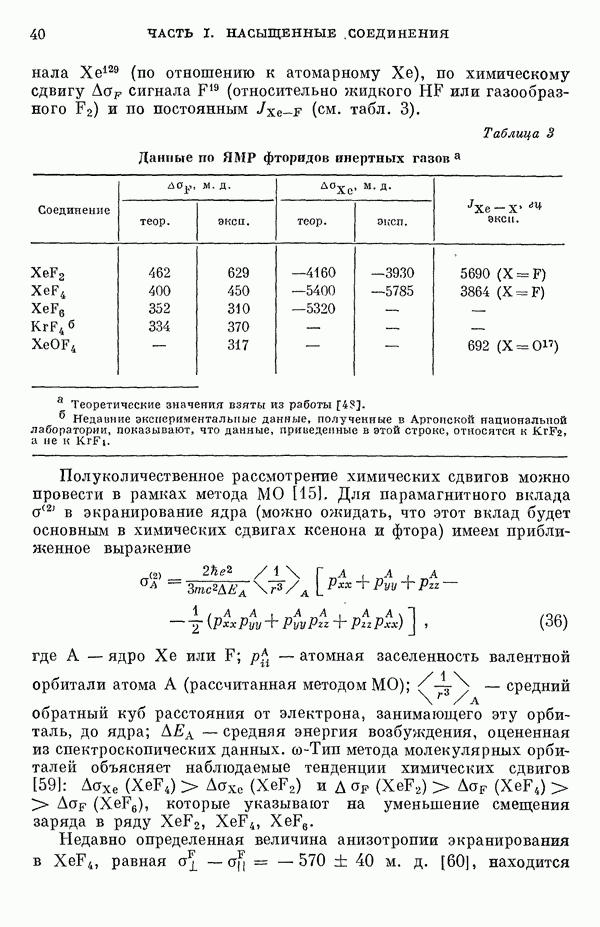



























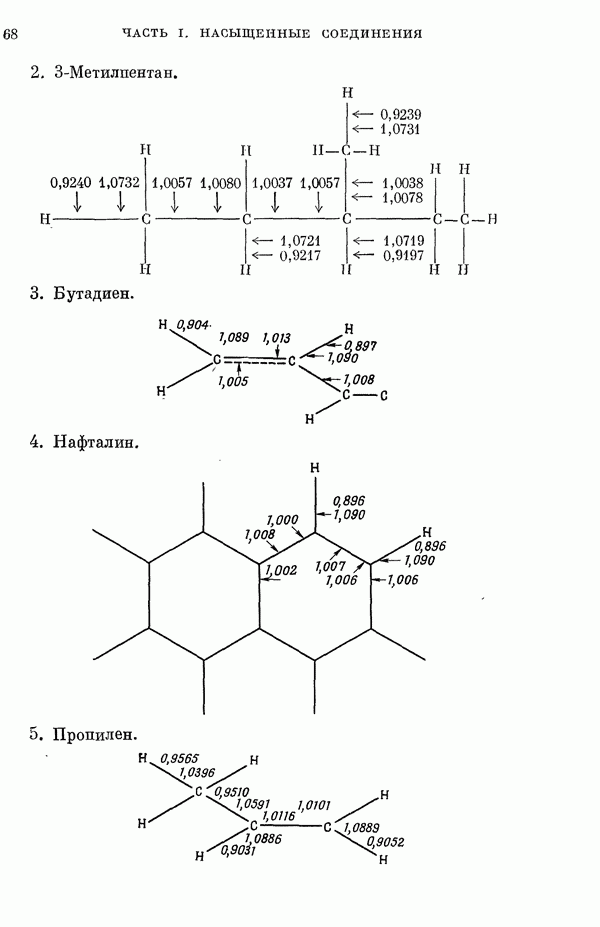









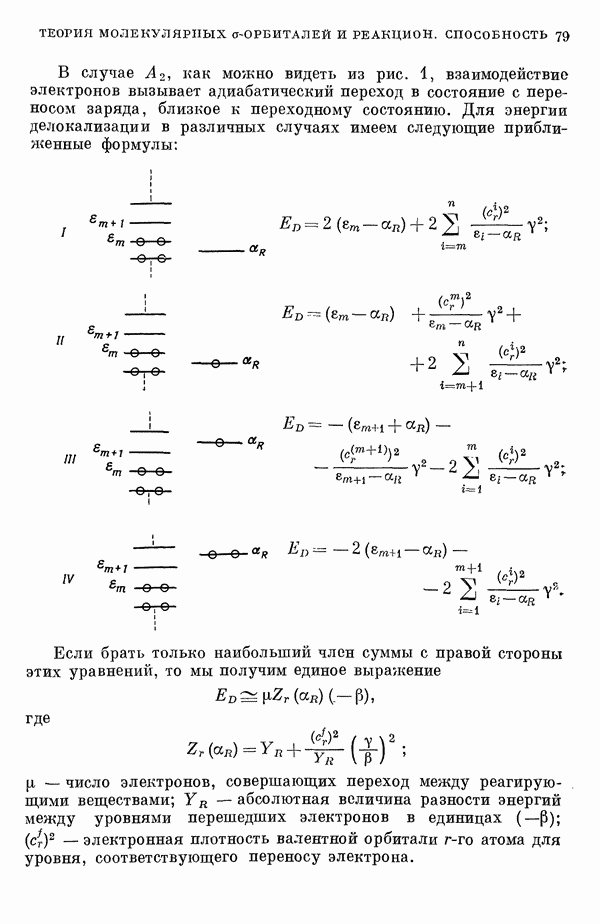


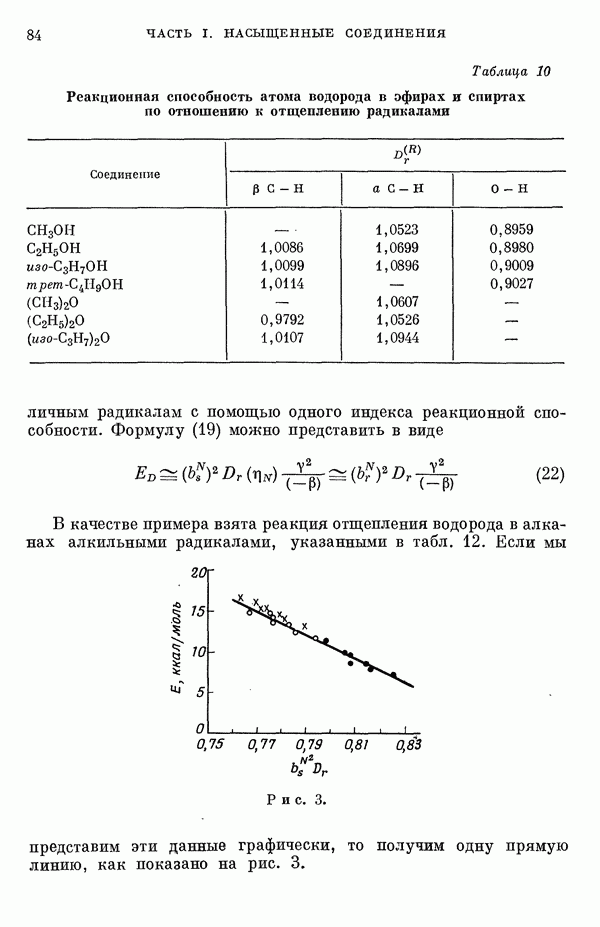
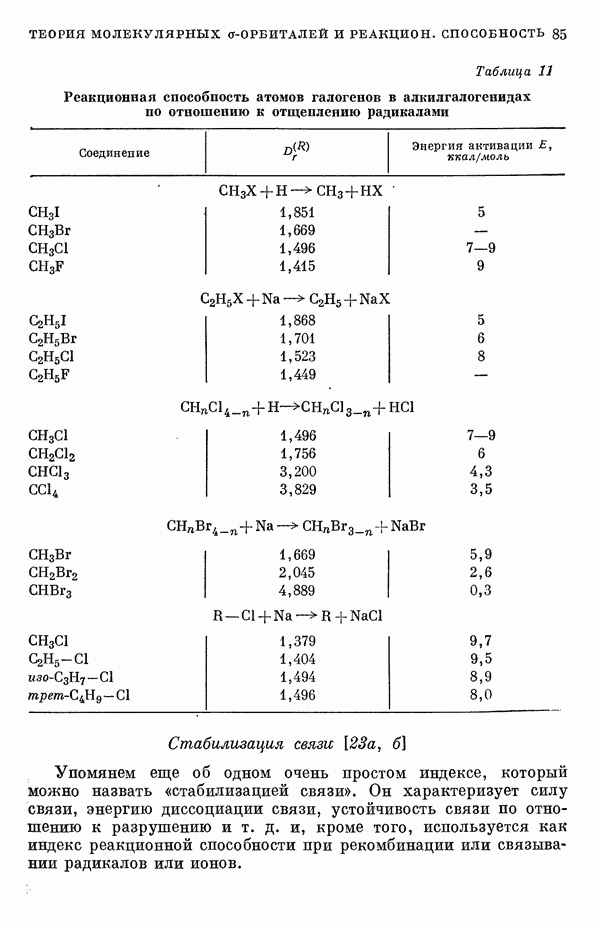
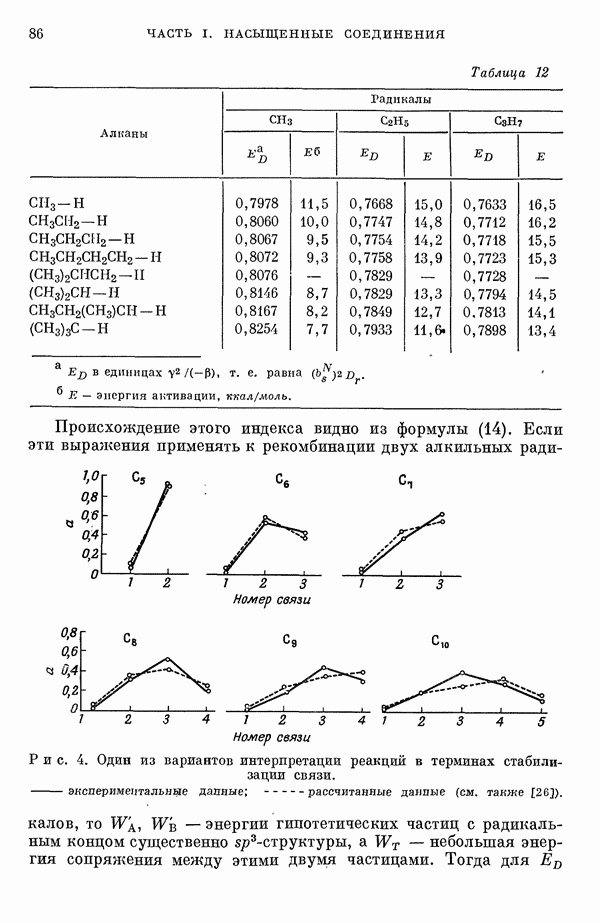
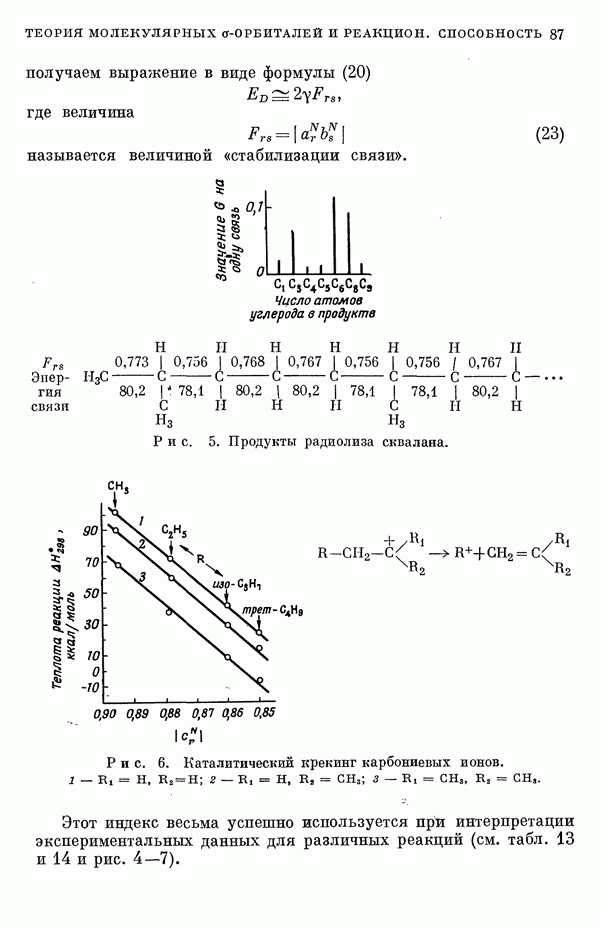


































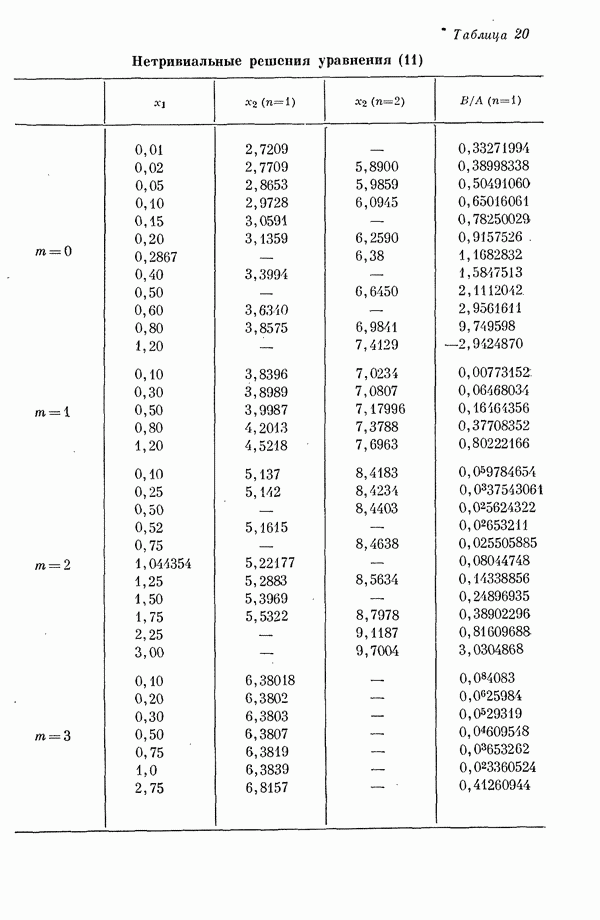






















































































































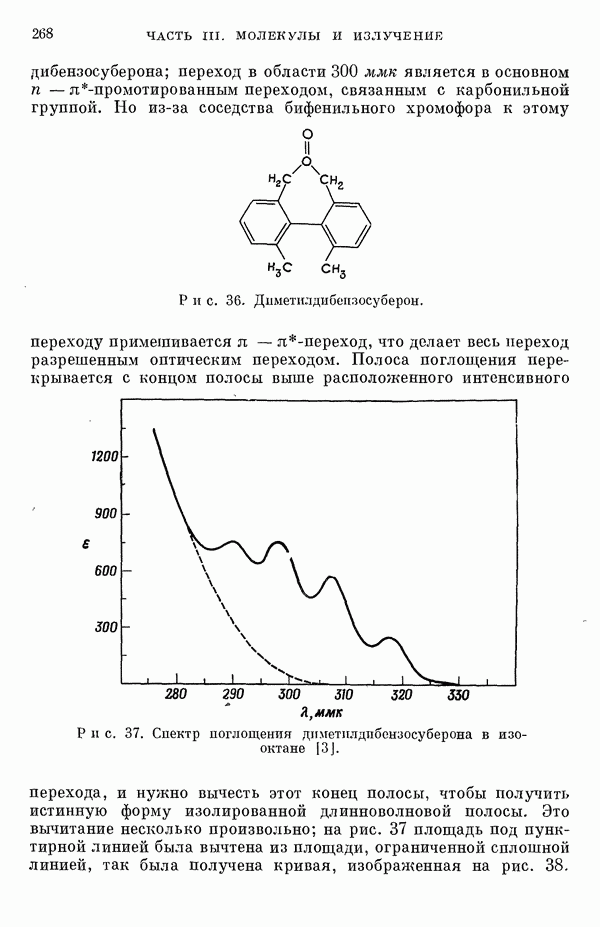
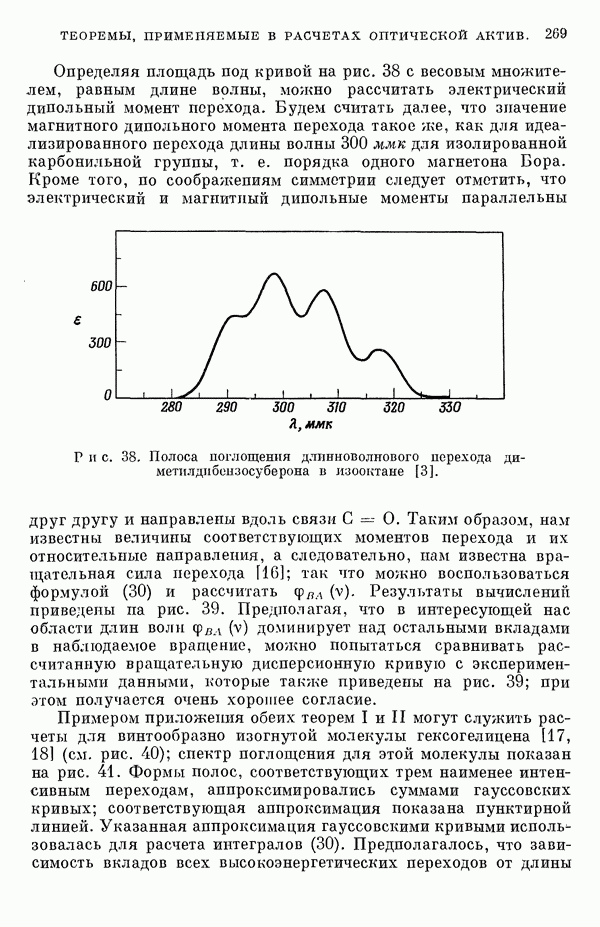
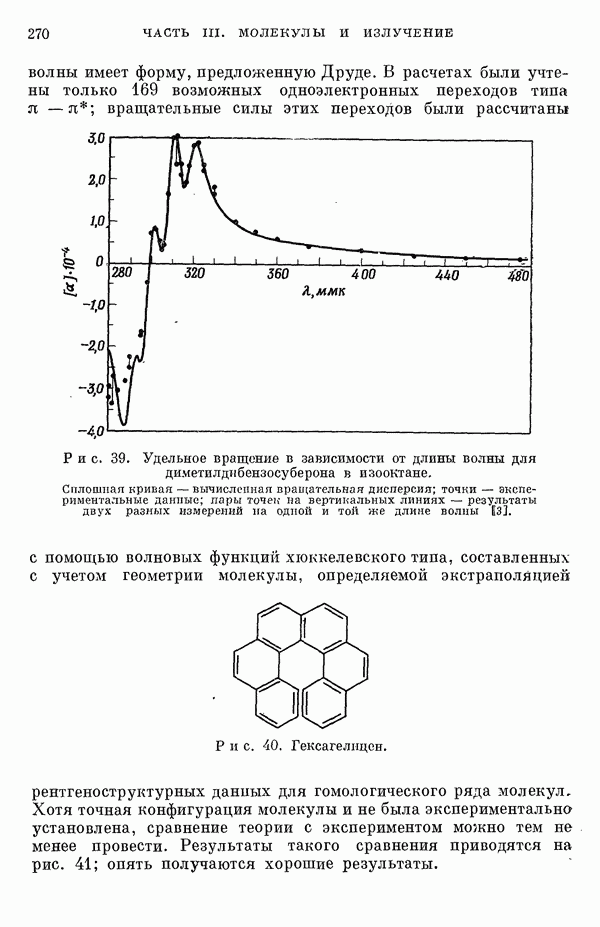
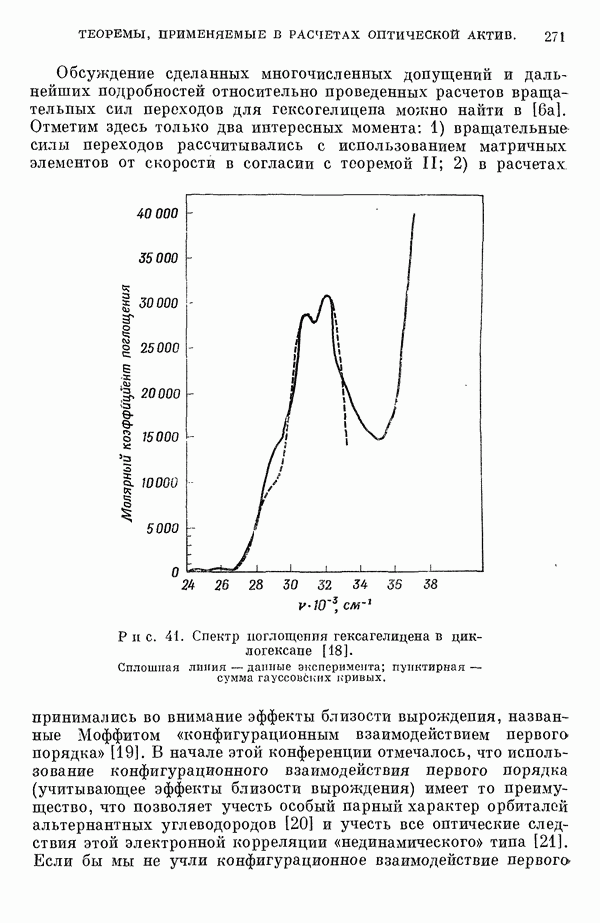

























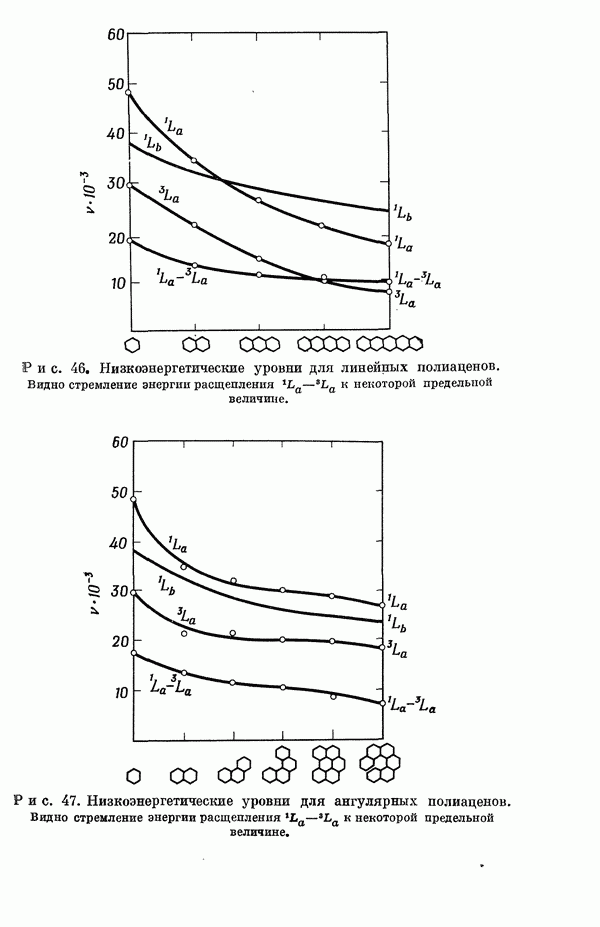
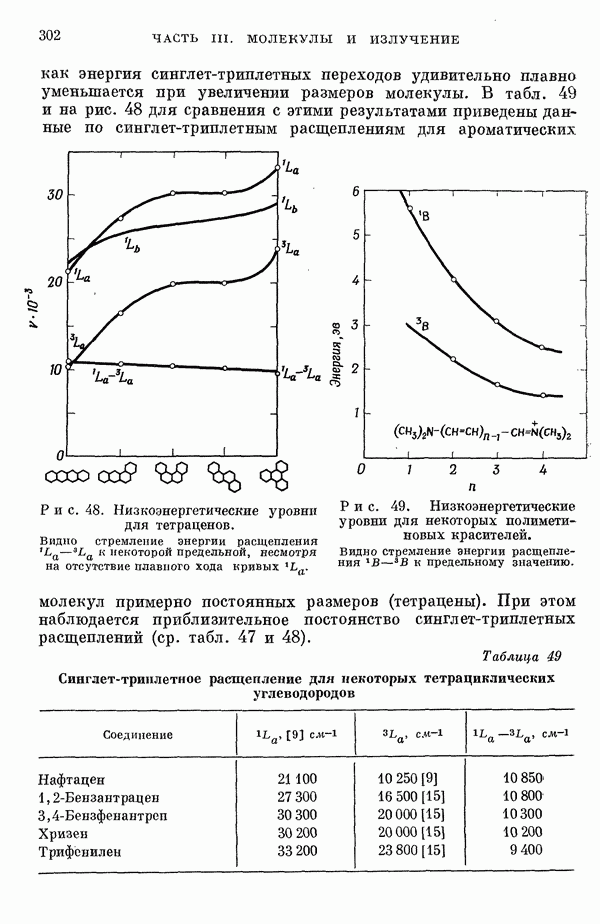

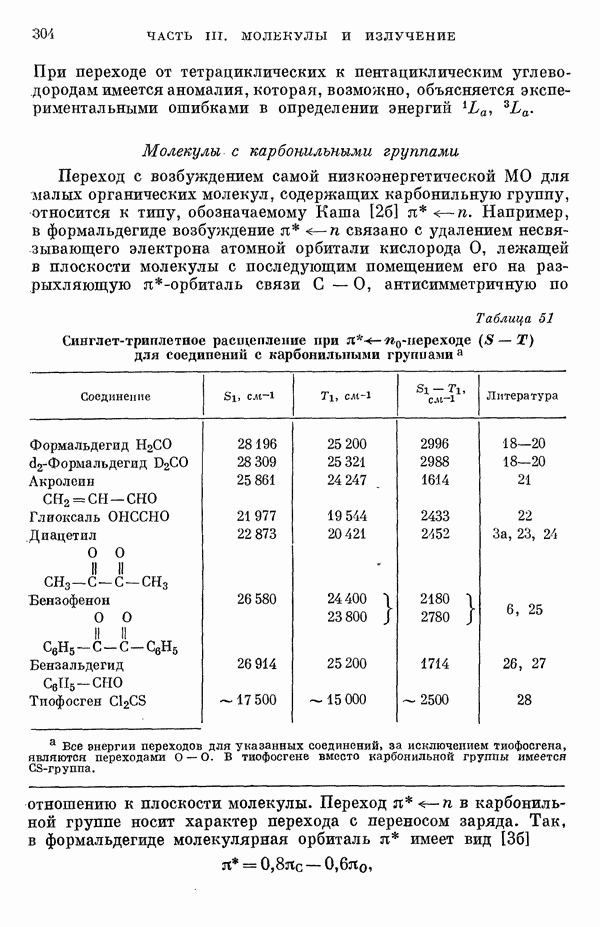

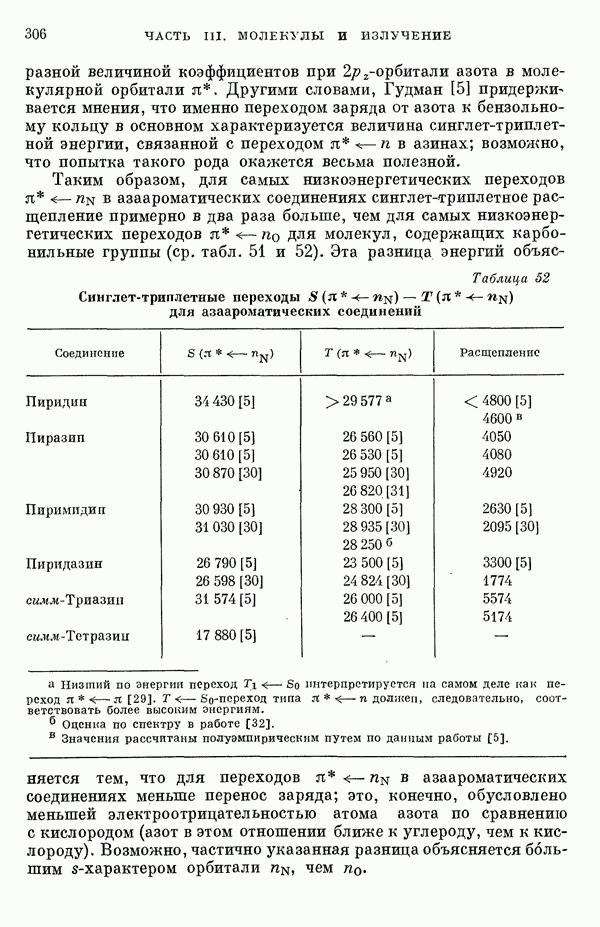




























 -электронном приближении представляет собой один из таких примеров. В общем случае для насыщенных органических молекул «методы инвариантного разделения» не дают заметного упрощения, если не считать обычного исключения электронов внутренних оболочек из рассмотрения, что принято также и в этом разделе.
-электронном приближении представляет собой один из таких примеров. В общем случае для насыщенных органических молекул «методы инвариантного разделения» не дают заметного упрощения, если не считать обычного исключения электронов внутренних оболочек из рассмотрения, что принято также и в этом разделе.
 -схема была предложена в связи с обсуждением двухатомных или линейных молекул, у которых
-схема была предложена в связи с обсуждением двухатомных или линейных молекул, у которых
 -электроных в приближенном смысле. В сопряженных органических молекулах понятие
-электроных в приближенном смысле. В сопряженных органических молекулах понятие  -электроны» имеет особое значение. В плоской молекуле, если мы используем подходящее приближение,
-электроны» имеет особое значение. В плоской молекуле, если мы используем подходящее приближение,  и
и  -части волновой функции отделены друг от друга. Физические причины применимости
-части волновой функции отделены друг от друга. Физические причины применимости  -электронного приближения в плоских сопряженных молекулах детально обсуждаются Ликосом и Парром
-электронного приближения в плоских сопряженных молекулах детально обсуждаются Ликосом и Парром  Кроме того, по аналогии со случаем двухатомных молекул в насыщенных органических молекулах одинарная связь, в которой распределение связывающих электронов обладает аксиальной симметрией относительно направления связи, обозначается обычно как
Кроме того, по аналогии со случаем двухатомных молекул в насыщенных органических молекулах одинарная связь, в которой распределение связывающих электронов обладает аксиальной симметрией относительно направления связи, обозначается обычно как  -связь.
-связь.


 — заряд остова, связанный с атомом а, у которого удалены все валентные электроны;
— заряд остова, связанный с атомом а, у которого удалены все валентные электроны;  — расстояние между ядром а и электроном
— расстояние между ядром а и электроном  — расстояние между электронами
— расстояние между электронами  и
и  Суммирование в формуле (1) проводится по всем валентным электронам. Волновая функция основного состояния
Суммирование в формуле (1) проводится по всем валентным электронам. Волновая функция основного состояния  определяется как функция, которая минимизирует следующее выражение:
определяется как функция, которая минимизирует следующее выражение:

 за исключением того, что она во всяком случае является функцией
за исключением того, что она во всяком случае является функцией  координат, где
координат, где  — число валентных электронов. В качестве приближенного многоэлектронного рассмотрения мы используем метод Хартри—Фока, в котором для образования полной волновой функции
— число валентных электронов. В качестве приближенного многоэлектронного рассмотрения мы используем метод Хартри—Фока, в котором для образования полной волновой функции  вводится понятие об одноэлектронной волновой функции (орбитали). В этом приближении принято, что электроны движутся независимо, если не считать эффекта взаимного влияния, обусловленного усредненным полем. Даже «наилучшая» хартри-фоковская волновая функция для молекулы приводит к так
вводится понятие об одноэлектронной волновой функции (орбитали). В этом приближении принято, что электроны движутся независимо, если не считать эффекта взаимного влияния, обусловленного усредненным полем. Даже «наилучшая» хартри-фоковская волновая функция для молекулы приводит к так
 аппроксимируется линейной комбинацией слэтеровских детерминантов
аппроксимируется линейной комбинацией слэтеровских детерминантов

 характеризующийся своими диагональными элементами; образуют некоторый подходящий набор ортонормированных одноэлектронных орбиталей, состоящих из координатной части
характеризующийся своими диагональными элементами; образуют некоторый подходящий набор ортонормированных одноэлектронных орбиталей, состоящих из координатной части  и спиновой части
и спиновой части  . В качестве первого приближения мы рассмотрим случай, когда
. В качестве первого приближения мы рассмотрим случай, когда  аппроксимируется одним детерминантом. Устойчивые органические молекулы, вообще говоря, обладают электронной структурой, в которой каждая пара электронов занимает две орбитали
аппроксимируется одним детерминантом. Устойчивые органические молекулы, вообще говоря, обладают электронной структурой, в которой каждая пара электронов занимает две орбитали  с одинаковыми координатными и противоположными спиновыми функциями. Такая «замкнутая оболочка» описывается волновой функцией вида
с одинаковыми координатными и противоположными спиновыми функциями. Такая «замкнутая оболочка» описывается волновой функцией вида

 Для энергии
Для энергии  имеем выражение
имеем выражение


 Так же, как и при расчетах атомов, можно найти наилучшие
Так же, как и при расчетах атомов, можно найти наилучшие  решив уравнения Фока. Соответствующая процедура решения системы нелинейных дифференциальных уравнений всегда является трудной задачей, поэтому воспользуемся приближенными выражениями для
решив уравнения Фока. Соответствующая процедура решения системы нелинейных дифференциальных уравнений всегда является трудной задачей, поэтому воспользуемся приближенными выражениями для  . В качестве приближения мы используем «липейиый вариационный метод», в котором
. В качестве приближения мы используем «липейиый вариационный метод», в котором  представляют в виде линейной комбинации
представляют в виде линейной комбинации



 число взятых орбиталей
число взятых орбиталей  — число занятых орбиталей
— число занятых орбиталей  как было отмечено выше, и, кроме того,
как было отмечено выше, и, кроме того,


 — все атомные орбитали, скажем орбитали всех атомов, слэтеровского типа
— все атомные орбитали, скажем орбитали всех атомов, слэтеровского типа  которые связаны с химической валентностью.
которые связаны с химической валентностью.
 — содержит валентные орбитали, ассоциируемые со связывающими электронами, включая обособленные электронные пары; например, орбиталь
— содержит валентные орбитали, ассоциируемые со связывающими электронами, включая обособленные электронные пары; например, орбиталь  и гибридные орбитали атома углерода и орбиталь
и гибридные орбитали атома углерода и орбиталь  атома водорода в углеводородах; гибридными орбиталями углерода могут быть
атома водорода в углеводородах; гибридными орбиталями углерода могут быть  орбитали.
орбитали.
 — орбитали связи, выбранные соответствующим образом и локализованные на каждой связи.
— орбитали связи, выбранные соответствующим образом и локализованные на каждой связи.
 -электронном приближении. Для краткости мы предлагаем называть три метода, упомянутые выше, как
-электронном приближении. Для краткости мы предлагаем называть три метода, упомянутые выше, как
 показали, что для симметричной молекулы набор
показали, что для симметричной молекулы набор  полученных по методу ЛКАО, можно при помощи унитарного преобразования перевести в другой набор эквивалентных орбиталей
полученных по методу ЛКАО, можно при помощи унитарного преобразования перевести в другой набор эквивалентных орбиталей  в основном локализованных на каждой связи или на каждой орбитали обособленной пары. Орбитали, полученные таким образом, называются «эквивалентными орбиталями», и разумно ожидать, что они будут сходны с базисными функциями, входящими в линейные комбинации в методе ЛКСО. Другими словами, неопределенность понятия связи в методе ЛКАО исключается, согласно концепции Леннард-Джонса, при введении понятия об эквивалентных орбиталях. Метод Леннард-Джонса позволил дать физическое обоснование для понятия «орбитали связи» при обсуждении свойств насыщенных молекул и вызвал появление ряда более упрощенных методов
в основном локализованных на каждой связи или на каждой орбитали обособленной пары. Орбитали, полученные таким образом, называются «эквивалентными орбиталями», и разумно ожидать, что они будут сходны с базисными функциями, входящими в линейные комбинации в методе ЛКСО. Другими словами, неопределенность понятия связи в методе ЛКАО исключается, согласно концепции Леннард-Джонса, при введении понятия об эквивалентных орбиталях. Метод Леннард-Джонса позволил дать физическое обоснование для понятия «орбитали связи» при обсуждении свойств насыщенных молекул и вызвал появление ряда более упрощенных методов  .
.
 представляют в виде линейной комбинации орбиталей связи, так же как в методе ЛKCO. При этом, однако, используется гамильтониан не вида (1), а следующий:
представляют в виде линейной комбинации орбиталей связи, так же как в методе ЛKCO. При этом, однако, используется гамильтониан не вида (1), а следующий:

 — определенный оператор, включающий члены отталкивания, содержащиеся в выражении (1) в усредненной форме. В этой схеме для энергии замкнутой оболочки выражение (4) сводится квиду
— определенный оператор, включающий члены отталкивания, содержащиеся в выражении (1) в усредненной форме. В этой схеме для энергии замкнутой оболочки выражение (4) сводится квиду



 — одноэлектронная энергия
— одноэлектронная энергия  орбитали, определяемая из хорошо известного секулярного уравнения
орбитали, определяемая из хорошо известного секулярного уравнения

 соответственно кулоновский, резонансный интеграл и интеграл перекрывания
соответственно кулоновский, резонансный интеграл и интеграл перекрывания

 оцениваются полуэмпирически. Уравнение для энергии принимает вид
оцениваются полуэмпирически. Уравнение для энергии принимает вид

 берут
берут  -атомные орбитали углерода, был введен ранее в
-атомные орбитали углерода, был введен ранее в  -электронном приближении Хюккелем. Первые два из указанных методов при условии соответствующей параметризации в основном эквивалентны.
-электронном приближении Хюккелем. Первые два из указанных методов при условии соответствующей параметризации в основном эквивалентны.
 должны использовать методы ЛКАО или ЛКВО, в которых обычно число
должны использовать методы ЛКАО или ЛКВО, в которых обычно число  равно числу валентных электронов. Это число орбиталей является необходимым и достаточным для построения почти такого числа незанятых молекулярных орбиталей (помимо занятых
равно числу валентных электронов. Это число орбиталей является необходимым и достаточным для построения почти такого числа незанятых молекулярных орбиталей (помимо занятых  чтобы мы могли рассматривать более высокие одноэлектронные уровни так же, как в методе Хюккеля для
чтобы мы могли рассматривать более высокие одноэлектронные уровни так же, как в методе Хюккеля для  -электронных систем.
-электронных систем.
 а затем еще несколько авторов
а затем еще несколько авторов  предложили различные способы приближенной оценки
предложили различные способы приближенной оценки


 -связей. Таким образом, мы получим метод, в котором все еще применяется понятие связи и который в то же время удобен для обсуждения химического взаимодействия насыщенных молекул.
-связей. Таким образом, мы получим метод, в котором все еще применяется понятие связи и который в то же время удобен для обсуждения химического взаимодействия насыщенных молекул.
 в формуле (5), выбраны в следующем виде:
в формуле (5), выбраны в следующем виде:

 — соответственно
— соответственно  и
и  -слэтеровские орбитали атома углерода, расположенные в направлении а-связи.
-слэтеровские орбитали атома углерода, расположенные в направлении а-связи.


 электронов это почти не отразилось. При обсуждении химической реакционной способности можно ввести это упрощающее предложение.
электронов это почти не отразилось. При обсуждении химической реакционной способности можно ввести это упрощающее предложение.
 -параметров, относящихся к аналитической форме орбиталей Слэтера и теоретической
-параметров, относящихся к аналитической форме орбиталей Слэтера и теоретической
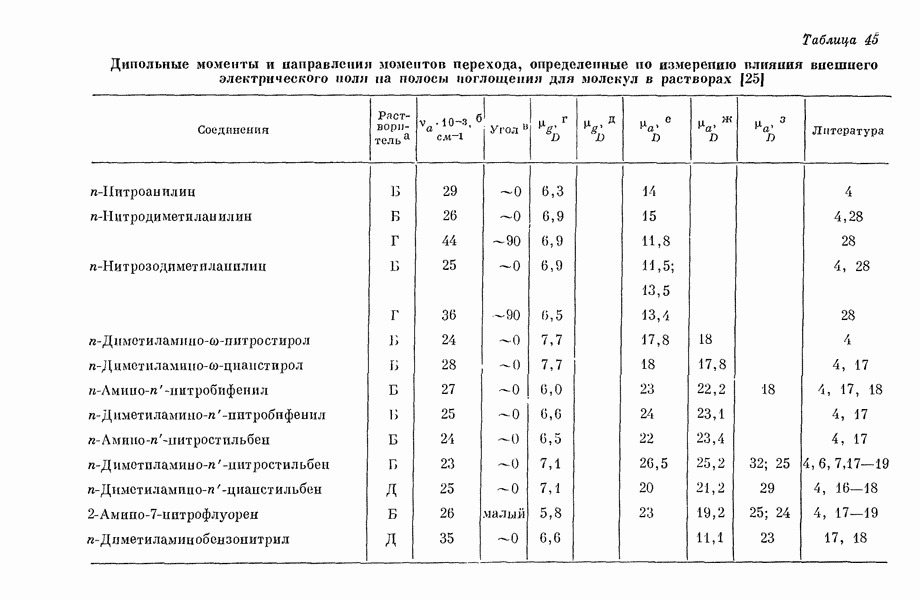
 -Дипольные моменты,
-Дипольные моменты, 

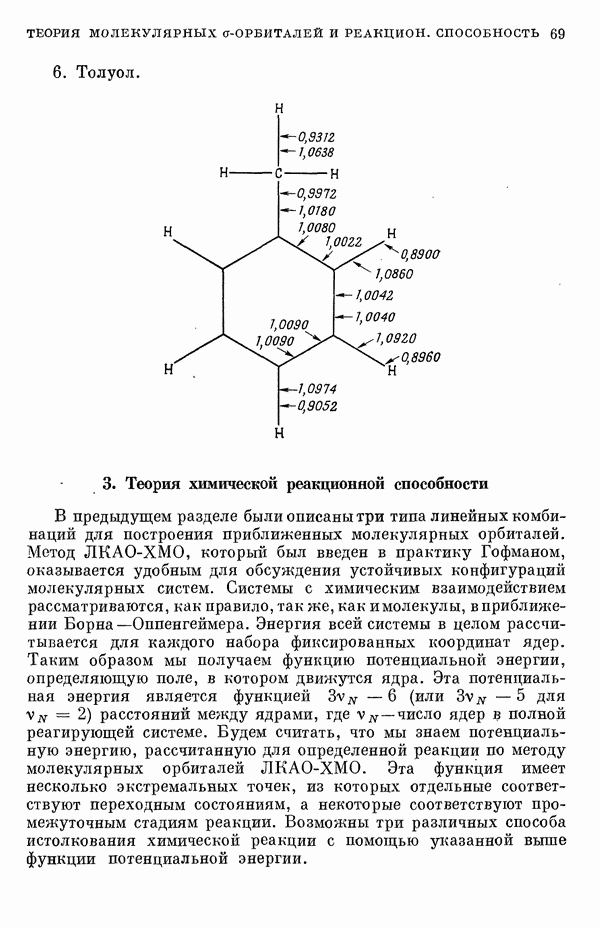
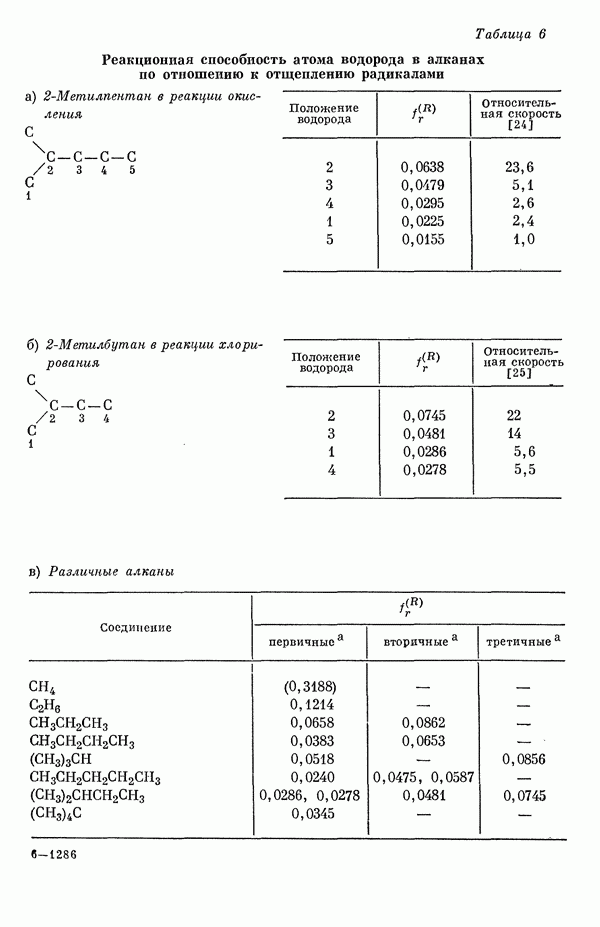
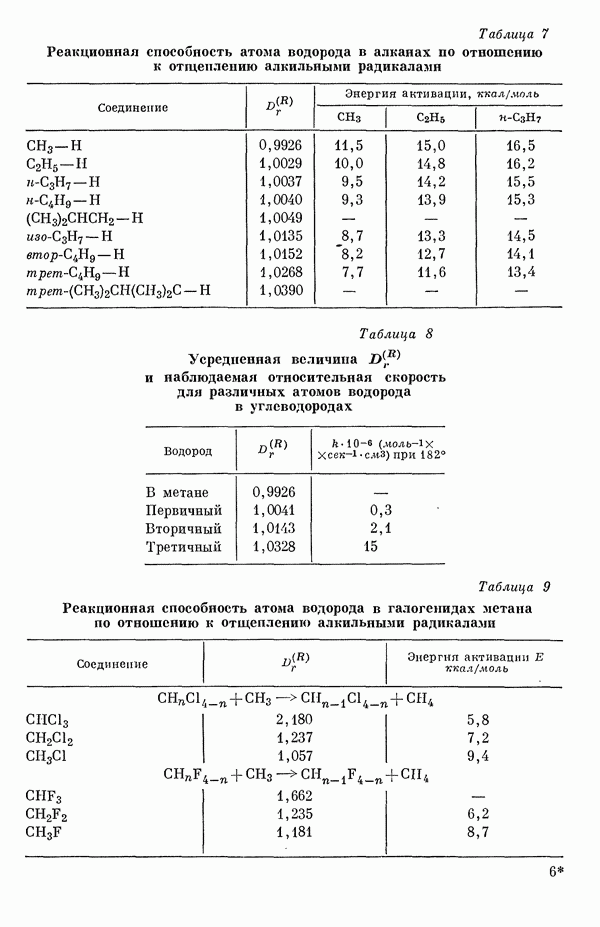
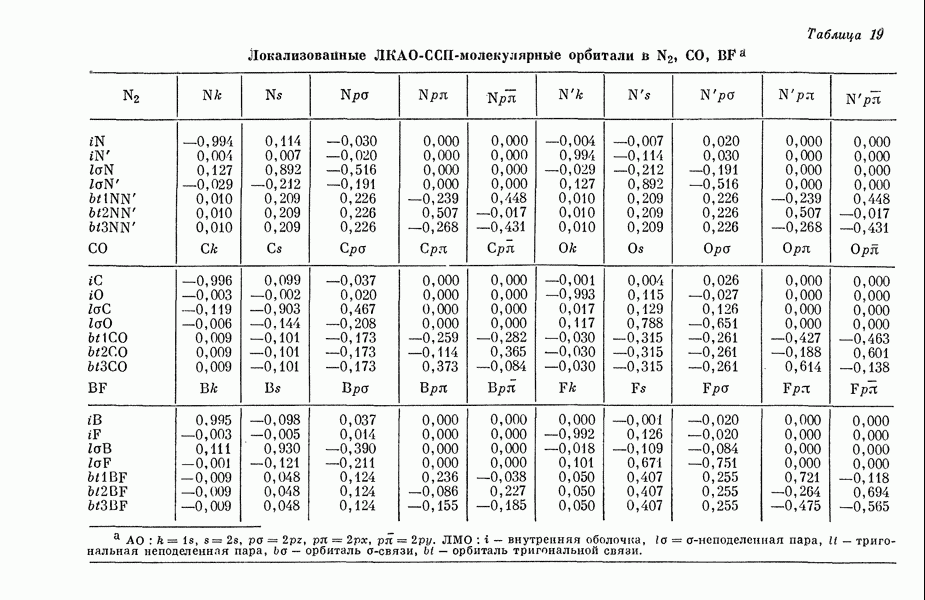
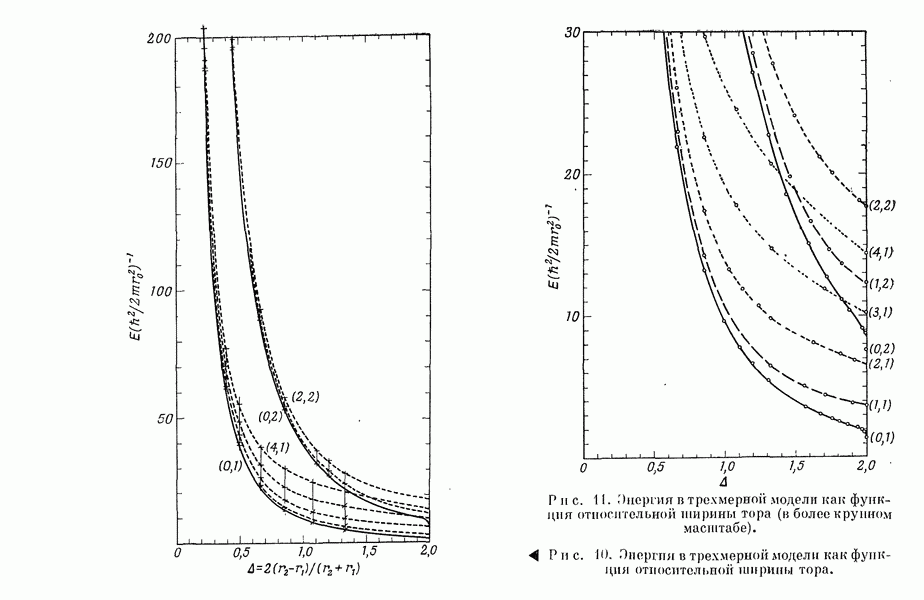
 Этим методом исследовались сг-электронные структуры и а-дипольные моменты (табл. 5) сопряженных молекул [15д].
Этим методом исследовались сг-электронные структуры и а-дипольные моменты (табл. 5) сопряженных молекул [15д].

