Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
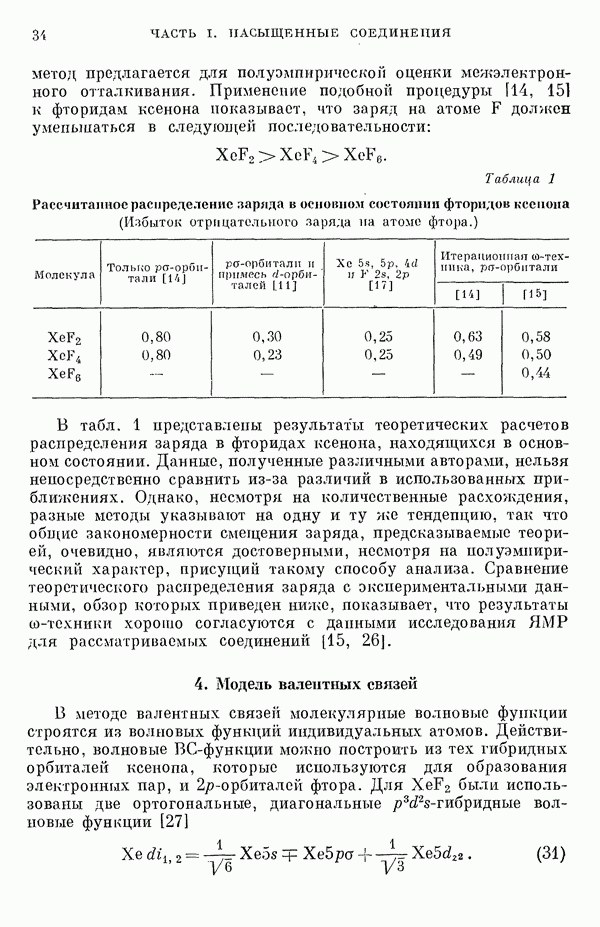
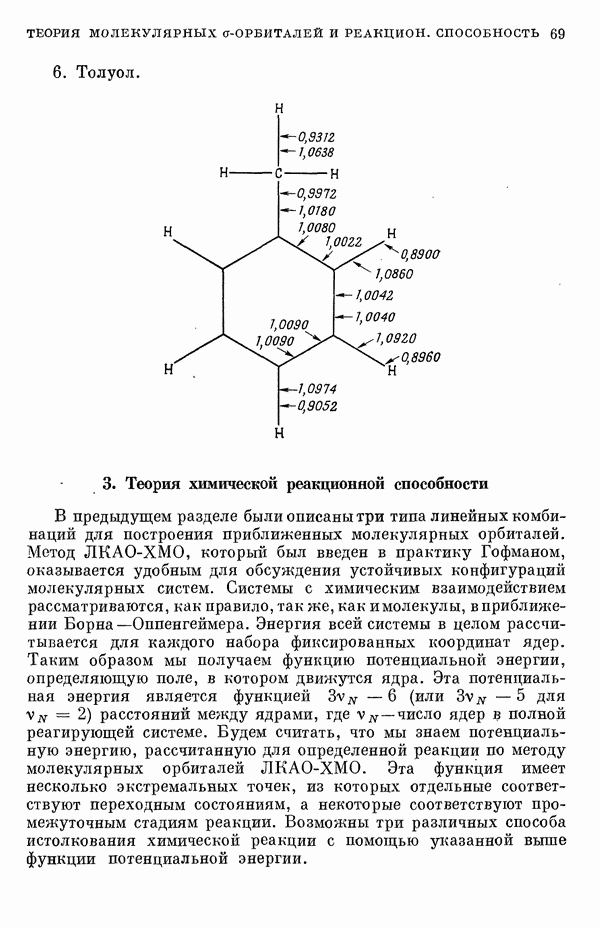
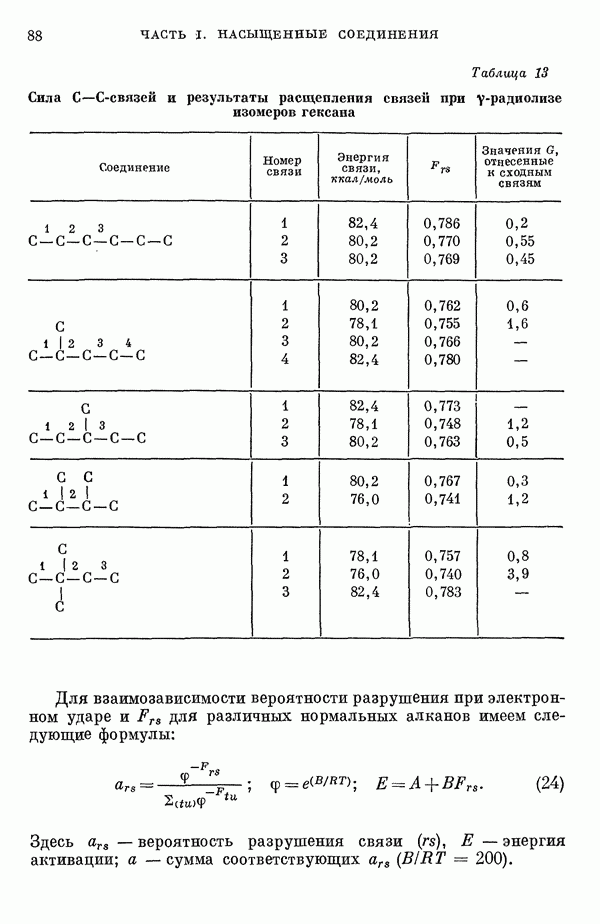
3. Теория химической реакционной способностиВ предыдущем разделе были описаны три типа линейных комбинаций для построения приближенных молекулярных орбиталей. Метод ЛKAO-XMO, который был введен в практику Гофманом, оказывается удобным для обсуждения устойчивых конфигураций молекулярных систем. Системы с химическим взаимодействием рассматриваются, как правило, так же, как имолекулы, вприближении Борна—Оппенгеймера. Энергия всей системы в целом рассчитывается для каждого набора фиксированных координат ядер. Таким образом мы получаем функцию потенциальной энергии, определяющую поле, в котором движутся ядра. Эта потенциальная энергия является функцией Теория соударенийРешают классические уравнения Гамильтона для движения Квантовомеханическая теория абсолютных скоростей реакцийС помощью выражения для потенциальной энергии и пользуясь приемами квантовой механики, рассчитывают коэффициент прохождения ядер при движении по координате реакции; из значения этого коэффициента при соответствующем усреднении получается скорость реакции [17]. Квазиклассическая теория абсолютных скоростей реакцийНаиболее удобным является использование функции потенциальной энергии лишь при определении конфигурации переходного состояния для расчета статистической суммы состояния, необходимой для статистического нахождения скорости при использовании специально подобранной величины коэффициента прохождения. В этом состоит известное приближение Эйринга [18]. Значение указанных неэмпирических методов возрастает с развитием техники вычислительных машин. Однако по крайней мере в настоящий момент их трудно практически применять при рассмотрении сложных органических реакций. Так, например, весьма привлекательный метод, основанный на теории соударений, никогда не применялся в задаче более чем для трех тел. Если не применять сложные методы ортодоксальной теории абсолютных скоростей реакций, то надо сказать, что полученные сведения о конфигурации активированного комплекса чрезвычайно полезны, поскольку они имеют большое значение для теории химической реакционной способности органических молекул. Метод Гофмана в его настоящем виде является несовершенным для расчета расстояний между ядрами активированного комплекса, поскольку он не вполне приспособлен для рассмотрения заряженных частиц. Предпринимается модификация метода [14], чтобы улучшить параметризацию и достичь приближенной самосогласо-ванности для величин кулоновских интегралов Гофман [8] показал, что для следующих конфигураций в некоторых углеводородах энергия экстремальна:
Полагают, что они являются прототипом активированного комплекса или промежуточного продукта соответственно реакций
(Общее число атомных орбиталей равно 61, расчет для каждой конфигурации занимает 5,04 мин на машине IBM 7090.)
Отрицательный заряд
Такой подход позволяет нам грубо оценить энергию активации в духе представлений классической кинетической теории реакций и приводит к ожидаемому количественному выражению для так называемого стерического эффекта. Независимо от развития теории столкновений и теории абсолютных скоростей химических реакций, отмеченных выше, с начала XX века продолжаются попытки собрать в единой системе огромное количество разнообразных экспериментальных результатов. Результатом этих усилий является «электронная теория органических соединений». Приблизительно с 1930 г. делаются попытки обобщить и дать количественное выражение этой теории на основе квантовой механики. Данная теория была применена также к проблеме химических реакций, и на этой основе была построена так называемая квантовомеханическая теория реакционной способности. По некоторым причинам эта теория впервые возникла в химии ароматических молекул и затем была распространена на другие типы молекул, включая насыщенные соединения. Цель квантовомеханической теории реакционной способности состоит просто в предсказании или объяснении относительной вероятности некоторой реакции для любой данной, в общем случае сложной молекулы. В этой теории используется простая и ясная квантовомеханическая характеристика, которая имеет тот же порядок величины, что и энергия активации, и называется индексом реакционной способности; она различна для разных типов реакций. Наиболее общим индексом реакционной способности является разность между энергиями переходного состояния и начального состояния, определяемая из функции потенциальной энергии всей реагирующей системы, т. е. квантовомеханическое выражение энергии активации. Для того, чтобы рассчитать энергию активации, следует найти энергию
и есть квантовомеханическое выражение энергии активации, которую следует сравнивать с экспериментальной величиной. Помимо того, в некоторых случаях учитывают влияние взаимодействия с молекулами растворителя или катализатора на потенциальную энергию. Такой подход часто является очень эффективным для объяснения «эффектов сольватации» или «рабочего механизма» катализатора. Однако, по крайней мере в существующей ситуации, такие методы ни в коей мере не удобны для практического использования, и рассчитанная таким образом величина В предельно упрощенной форме теории реакционной способности мы используем «приближение изолированной молекулы». При таком подходе мы используем индексы реакционной способности, которые связаны только со свойствами изолированной молекулы реагента. При рассмотрении реакций в ароматических соединениях в качестве примера таких величин можно взять полную плотность При обсуждении квантовомеханической теории реакционной способности мы выделяли понятие энергии активации. Для получения теоретической информации о скорости реакции необходимо наряду с энергией активации знать величину так называемого «энтропийного члена». Из изложенного мы уже знаем, что теория столкновений и теория абсолютных скоростей позволяют рассчитать энтропийный член при помощи квантовой механики. Но если нас интересует ряд сходных реакций с близким видом функции потенциальной энергии для переходного состояния, можно обойтись и без подобных сложных методов, поскольку в этом случае имеет место своеобразная компенсация между изменениями энтропии и энтальпии активации. В таких случаях можно ограничиваться обсуждением относительной скорости реакции только в терминах энергии активации.
|
 |
Оглавление
|








































































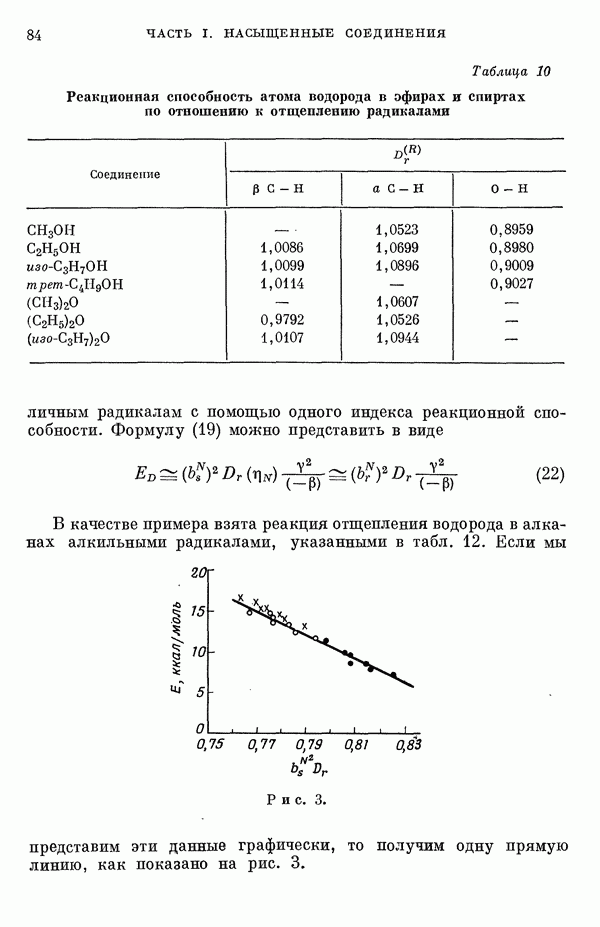
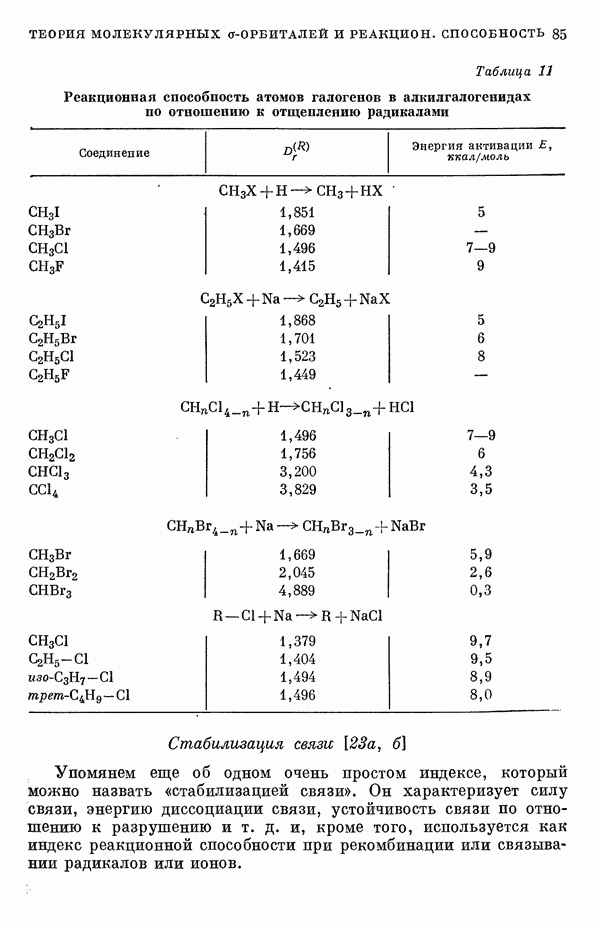
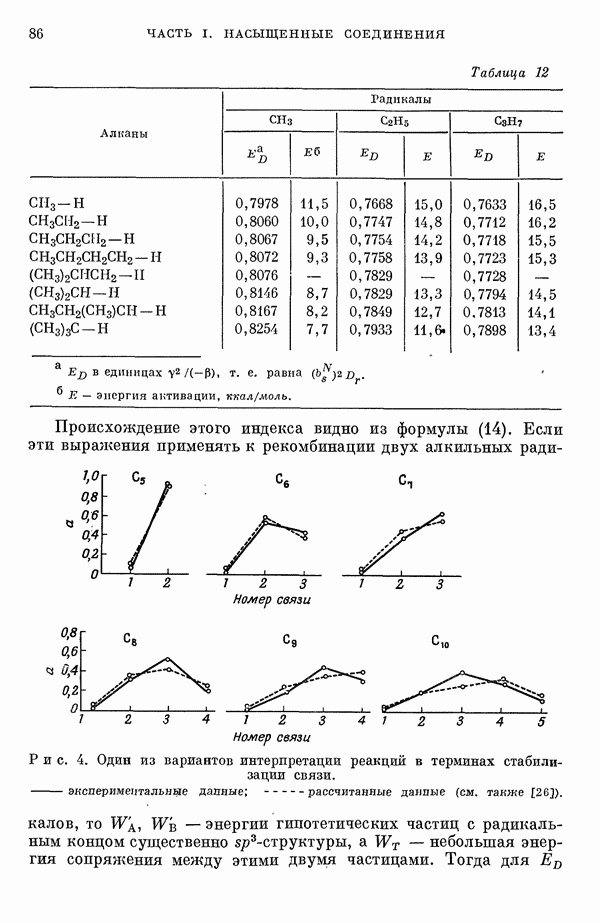
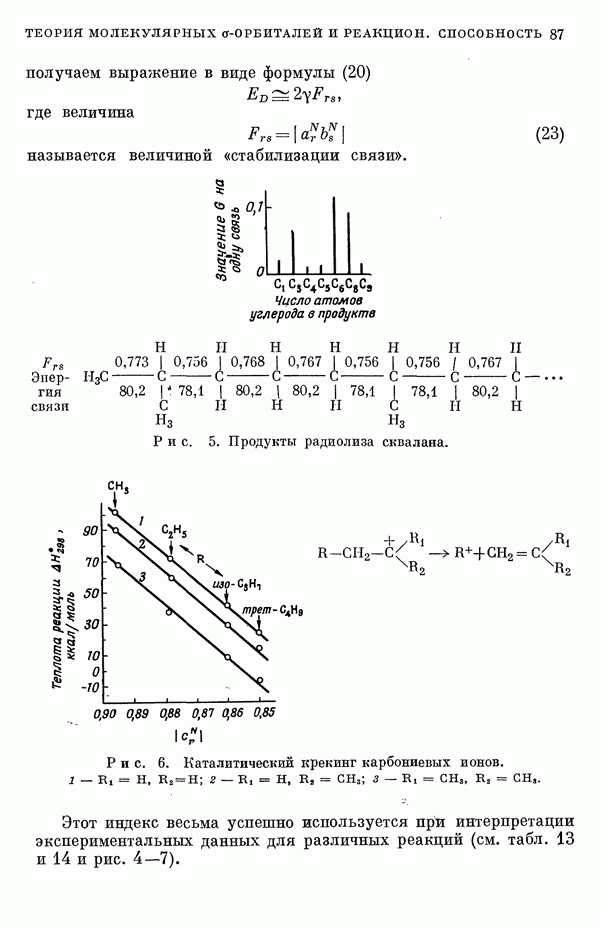


































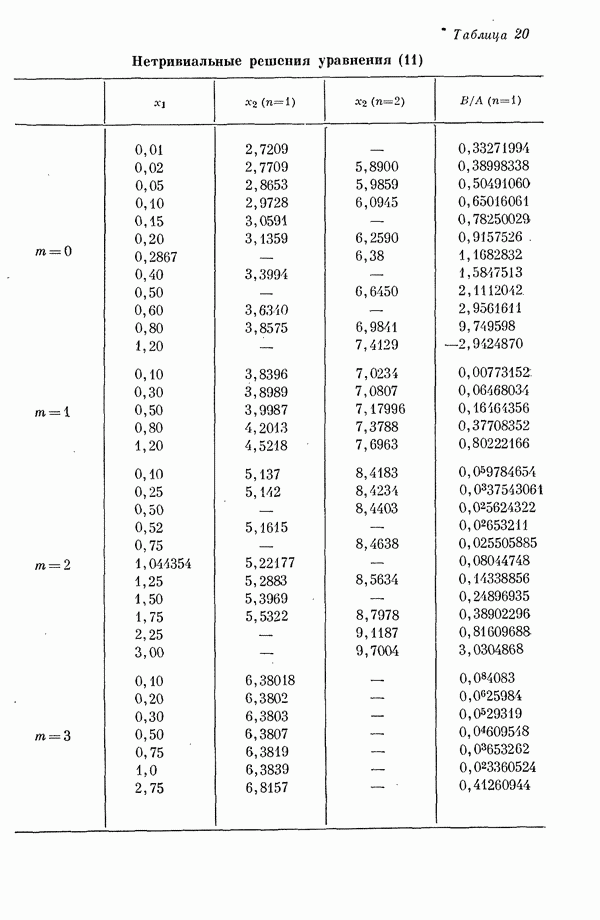

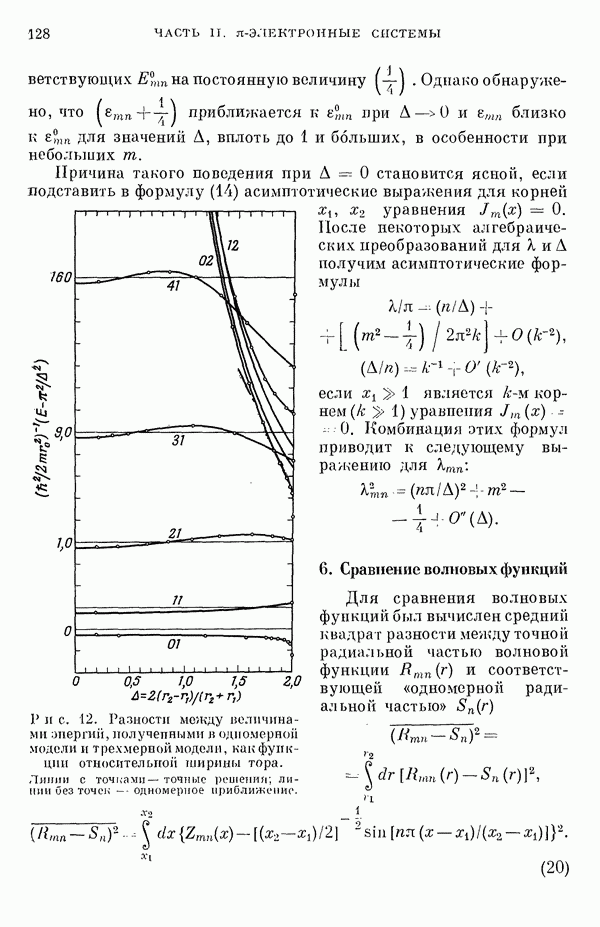
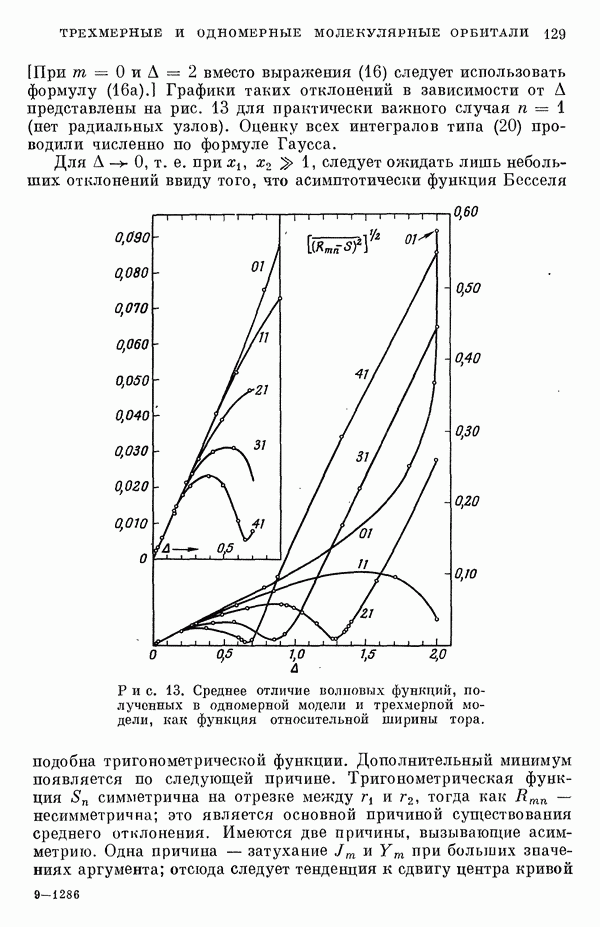






















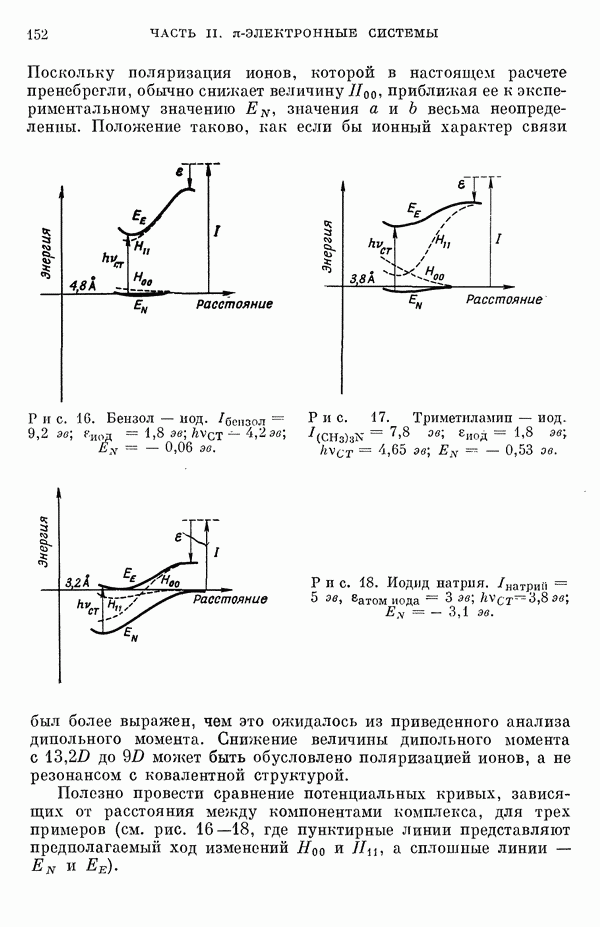
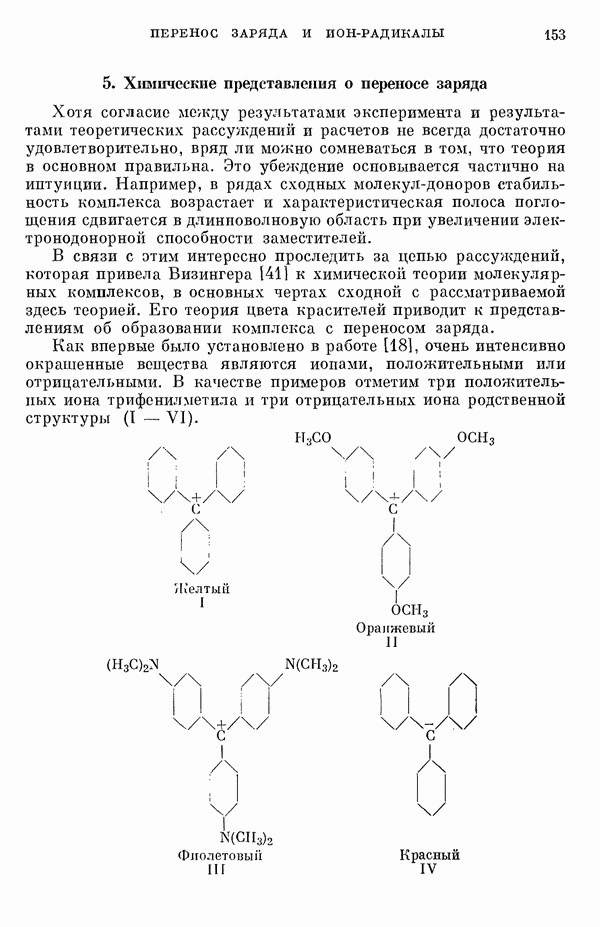
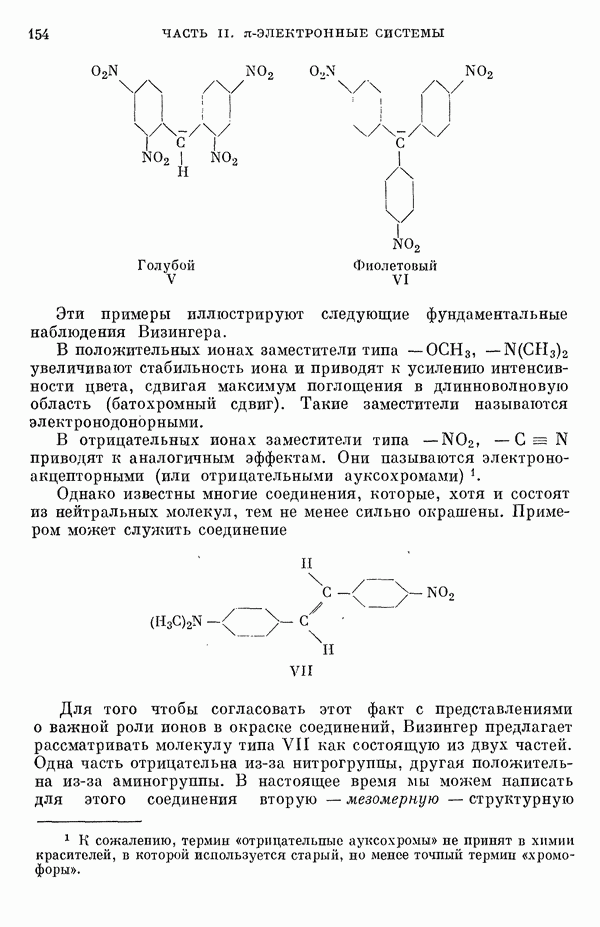
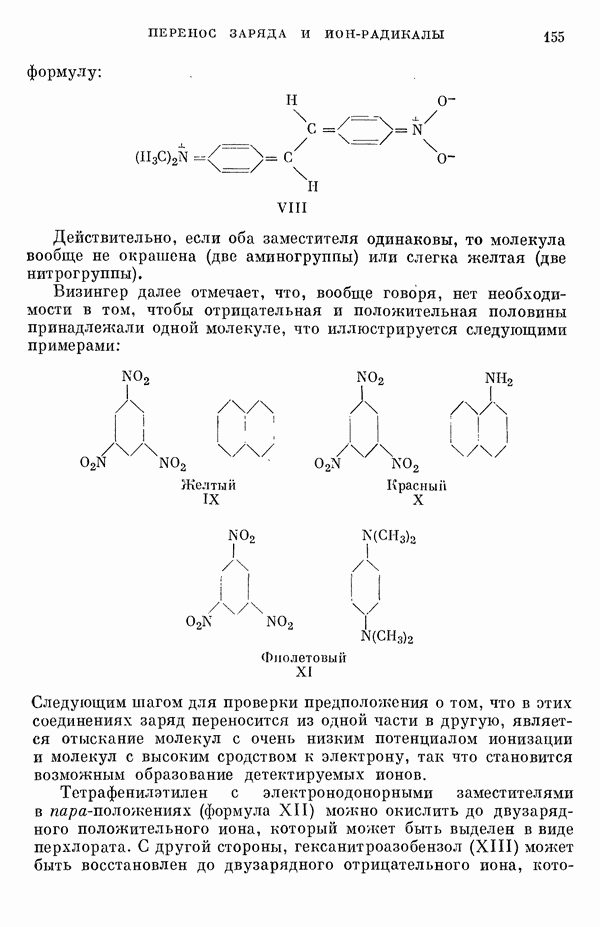
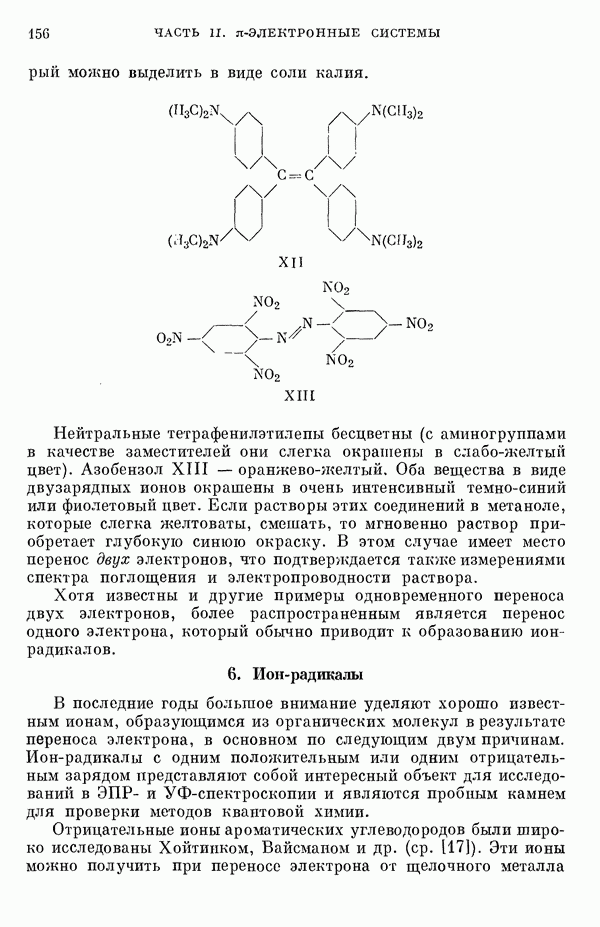





















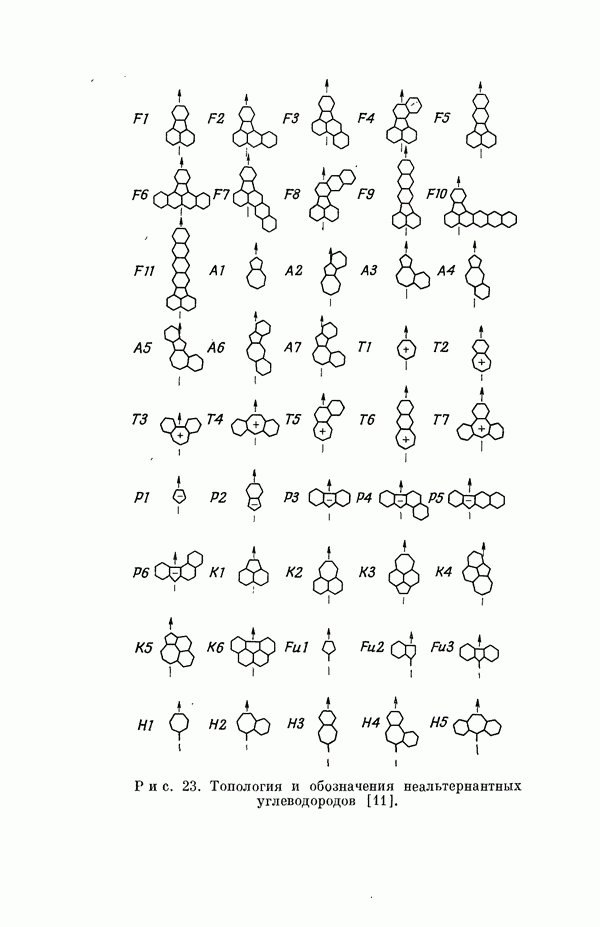










































































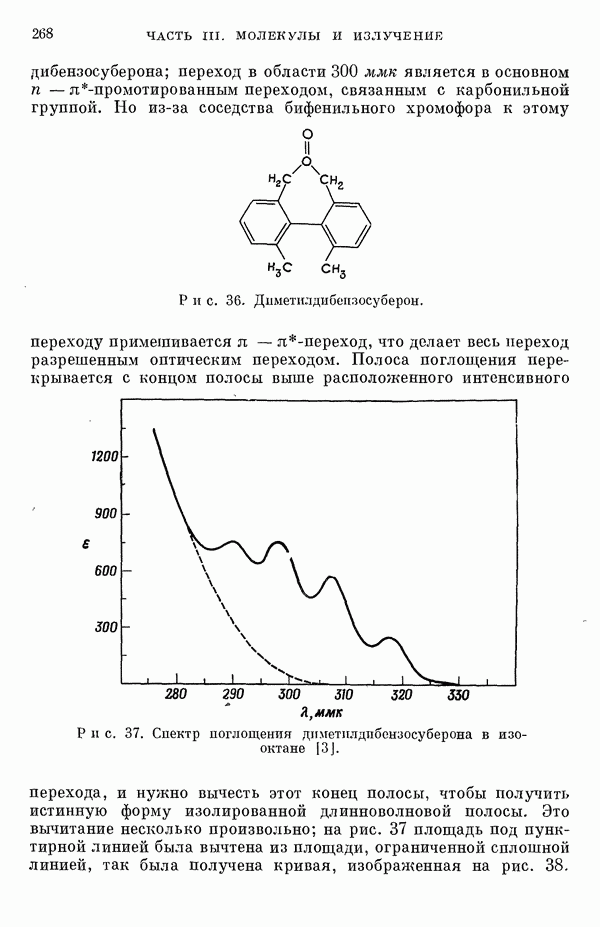
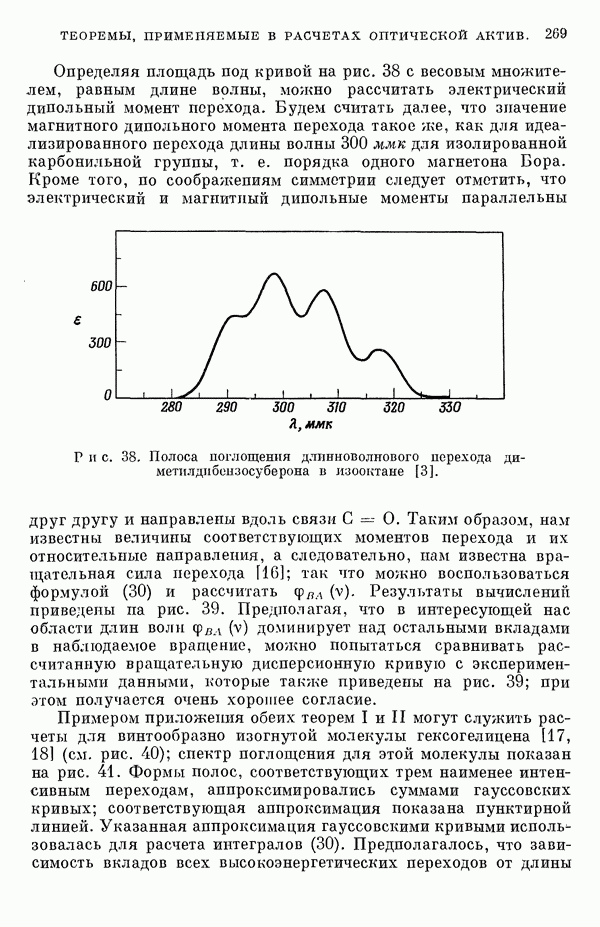
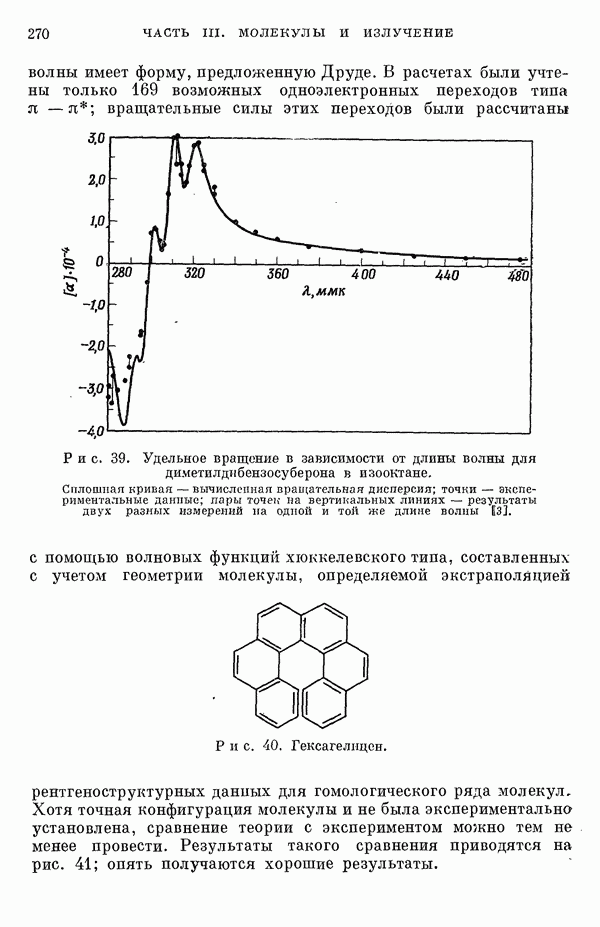
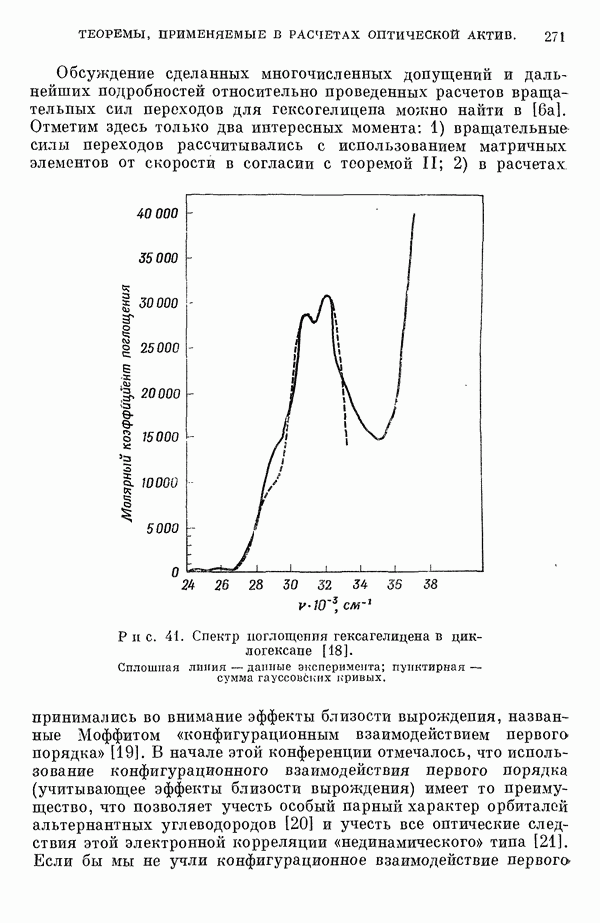























































 (или
(или  для
для  ) расстояний между ядрами, где
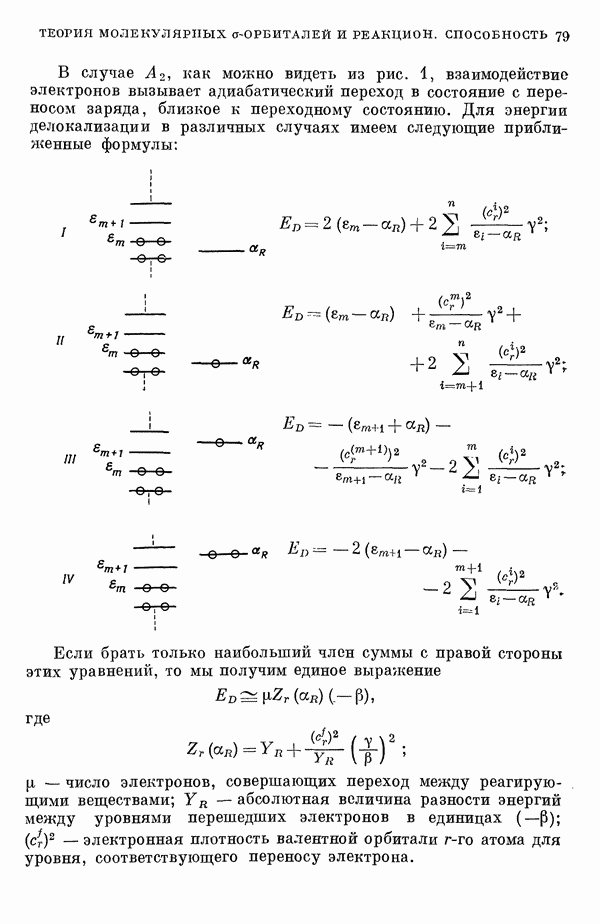
) расстояний между ядрами, где  — число ядер в полной реагирующей системе. Будем считать, что мы знаем потенциальную энергию, рассчитанную для определенной реакции по методу молекулярных орбиталей ЛKAO-XMO. Эта функция имеет несколько экстремальных точек, из которых отдельные соответствуют переходным состояниям, а некоторые соответствуют промежуточным стадиям реакции. Возможны три различных способа истолкования химической реакции с помощью указанной выше функции потенциальной энергии.
— число ядер в полной реагирующей системе. Будем считать, что мы знаем потенциальную энергию, рассчитанную для определенной реакции по методу молекулярных орбиталей ЛKAO-XMO. Эта функция имеет несколько экстремальных точек, из которых отдельные соответствуют переходным состояниям, а некоторые соответствуют промежуточным стадиям реакции. Возможны три различных способа истолкования химической реакции с помощью указанной выше функции потенциальной энергии.
 тел в заданном потенциальном поле. Постоянная скорости реакции получается при усреднении поперечного сечения по начальным состояниям
тел в заданном потенциальном поле. Постоянная скорости реакции получается при усреднении поперечного сечения по начальным состояниям 


 -миграции водорода и замещения в ароматическом кольце. Фукуи и др., кроме того, рассмотрели с помощью метода Гофмана переходное состояние следующих реакций [19]:
-миграции водорода и замещения в ароматическом кольце. Фукуи и др., кроме того, рассмотрели с помощью метода Гофмана переходное состояние следующих реакций [19]:


 непрерывно передается другому атому
непрерывно передается другому атому  по мере того как увеличивается угол
по мере того как увеличивается угол  ; в переходном состоянии заселенность связи в
; в переходном состоянии заселенность связи в  положительна и велика, что соответствует классическому описанию связи
положительна и велика, что соответствует классическому описанию связи  в виде
в виде

 [формула (7)] для каждого изолированного реагента
[формула (7)] для каждого изолированного реагента  , кроме того, для полной реагирующей системы в переходном состоянии; эти величины обозначаются как
, кроме того, для полной реагирующей системы в переходном состоянии; эти величины обозначаются как  соответственно. Разность энергий
соответственно. Разность энергий

 не соответствует строго по смыслу индексу реакционной способности. В следующем разделе будет описано возможное упрощение такого подхода.
не соответствует строго по смыслу индексу реакционной способности. В следующем разделе будет описано возможное упрощение такого подхода.
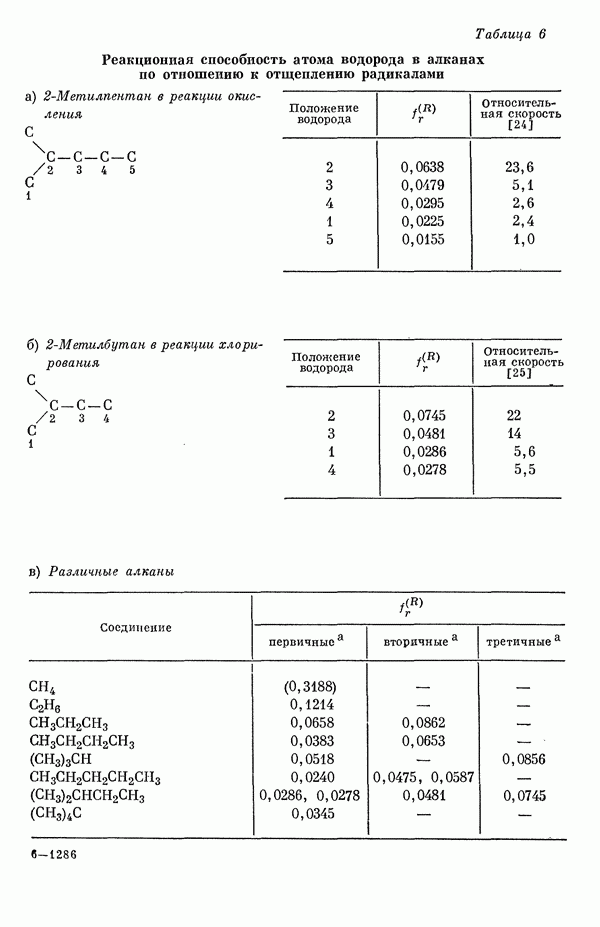
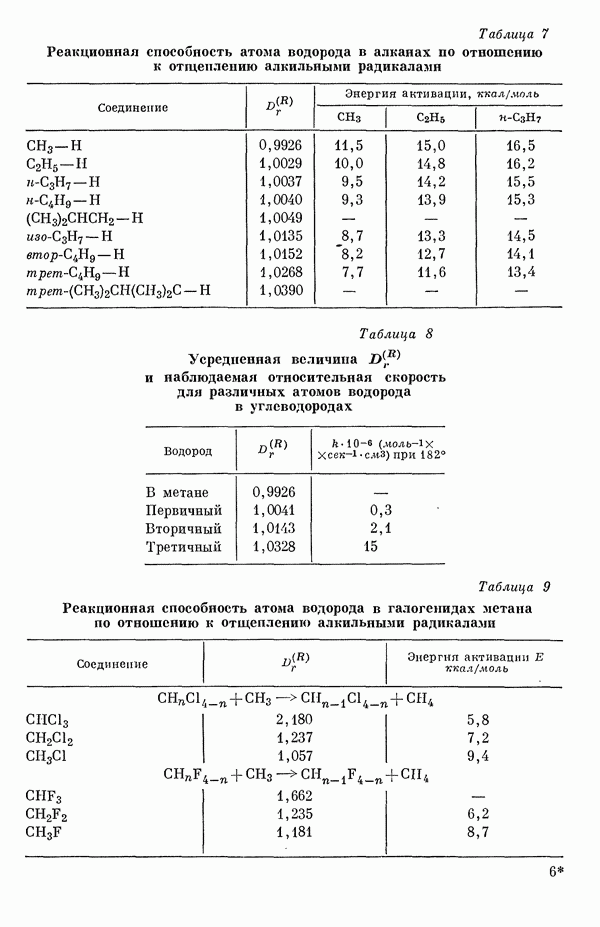
 -электронов, свободную валентность, собственную поляризуемость атома, энергию делокализации, граничную плотность электронов и т. д. [20а-ж]. Было показано, что для каждой из этих величин до некоторой степени наблюдается соответствие с экспериментальными результатами.
-электронов, свободную валентность, собственную поляризуемость атома, энергию делокализации, граничную плотность электронов и т. д. [20а-ж]. Было показано, что для каждой из этих величин до некоторой степени наблюдается соответствие с экспериментальными результатами.