Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
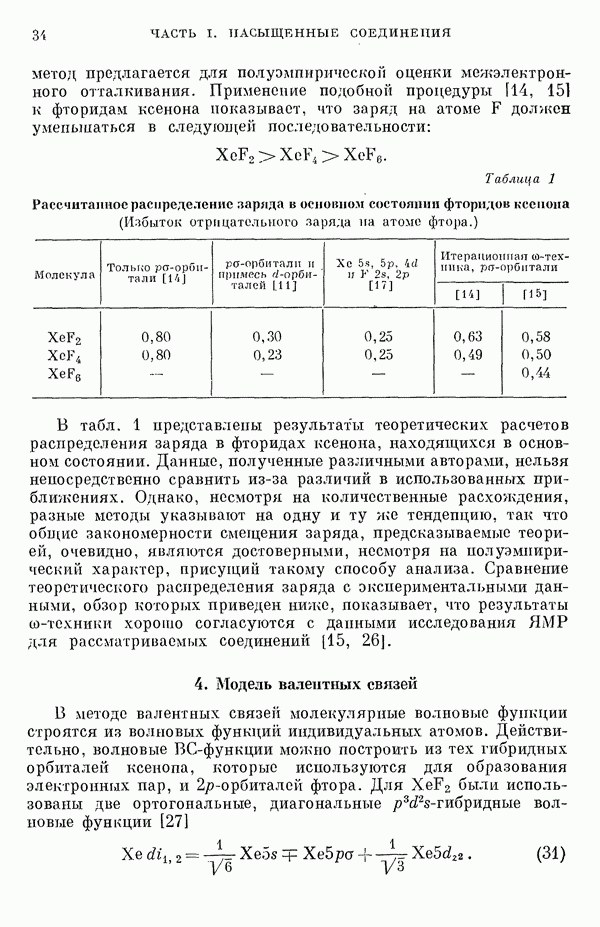
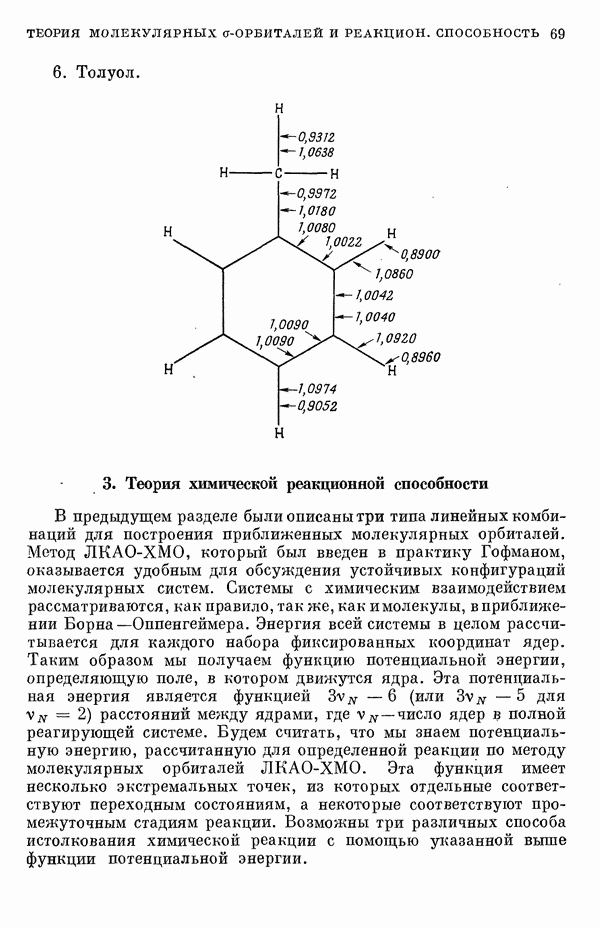
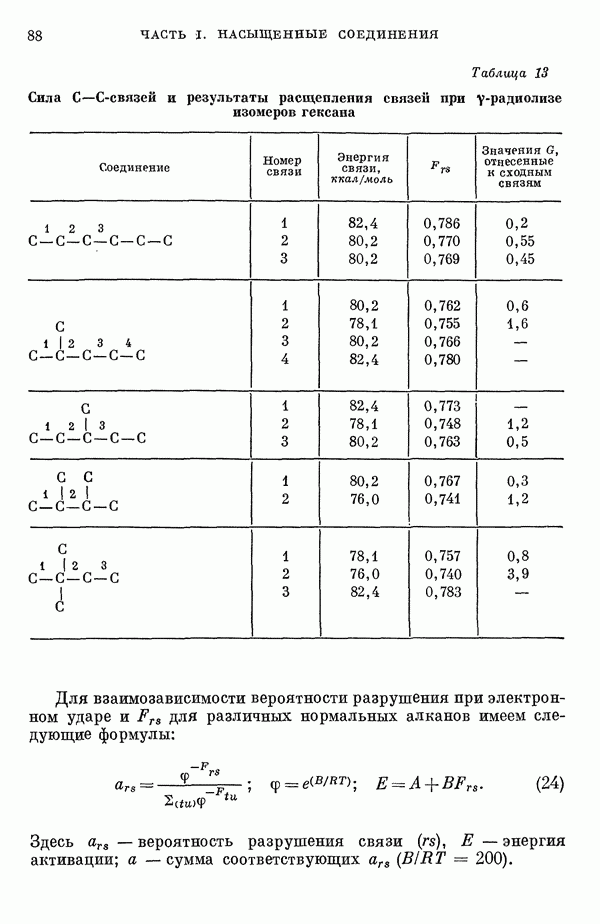
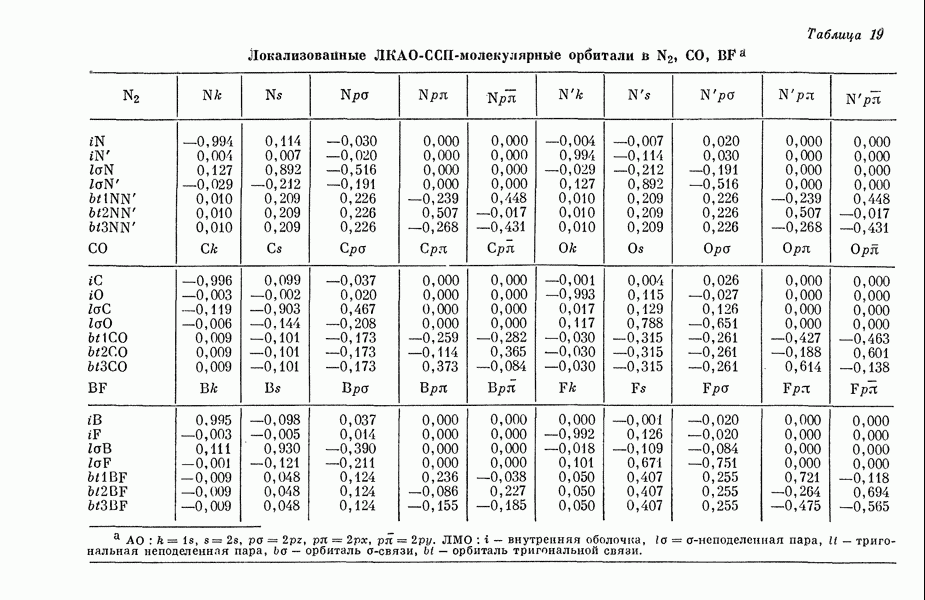
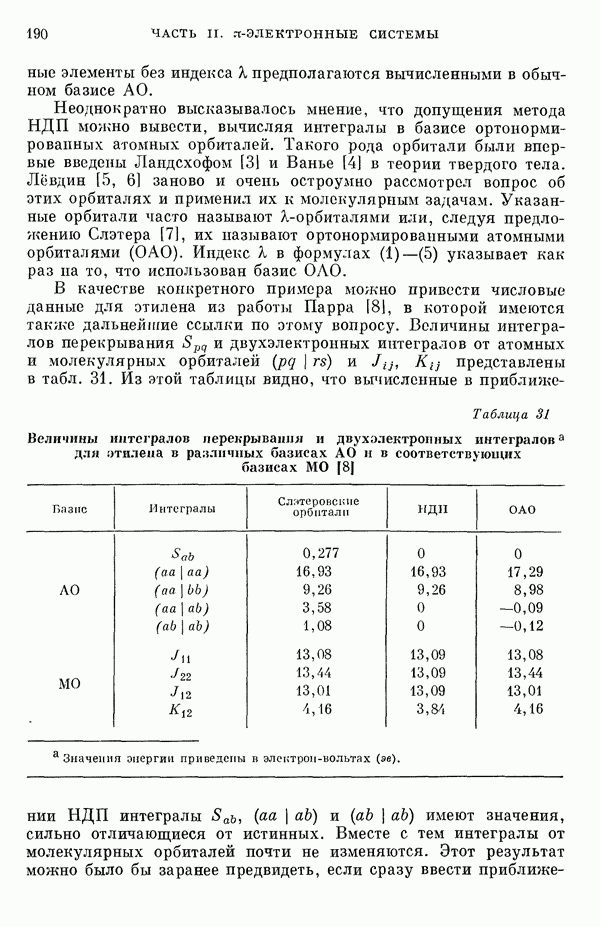
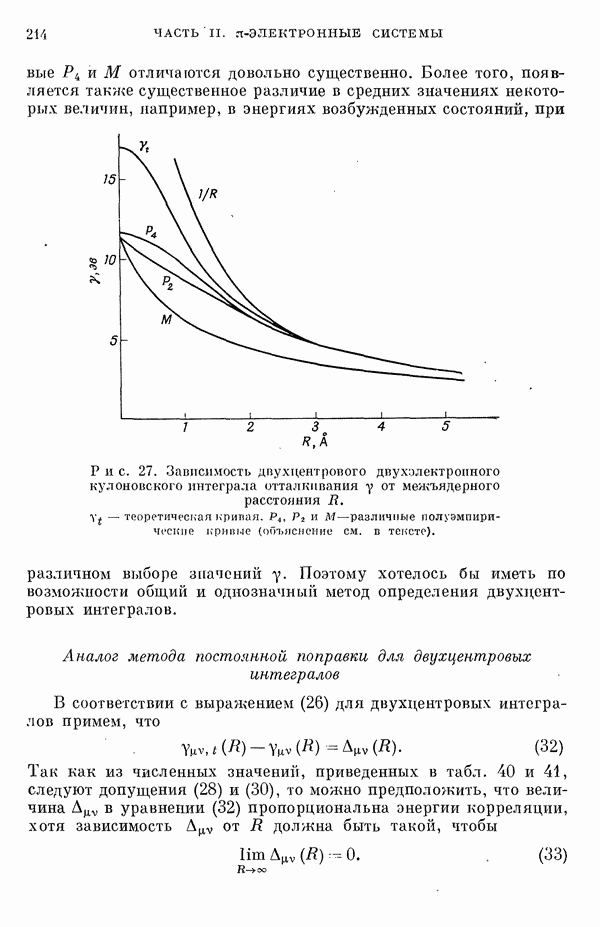
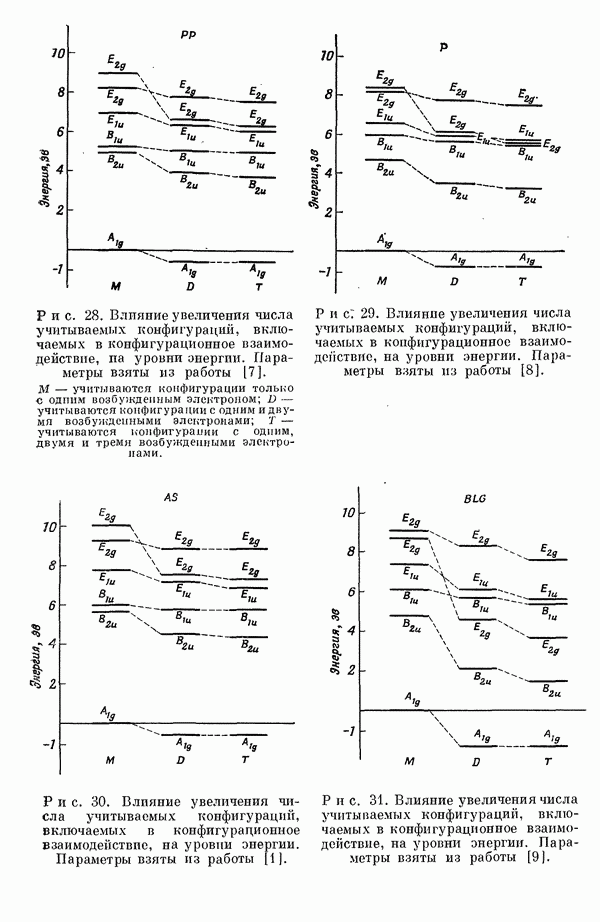
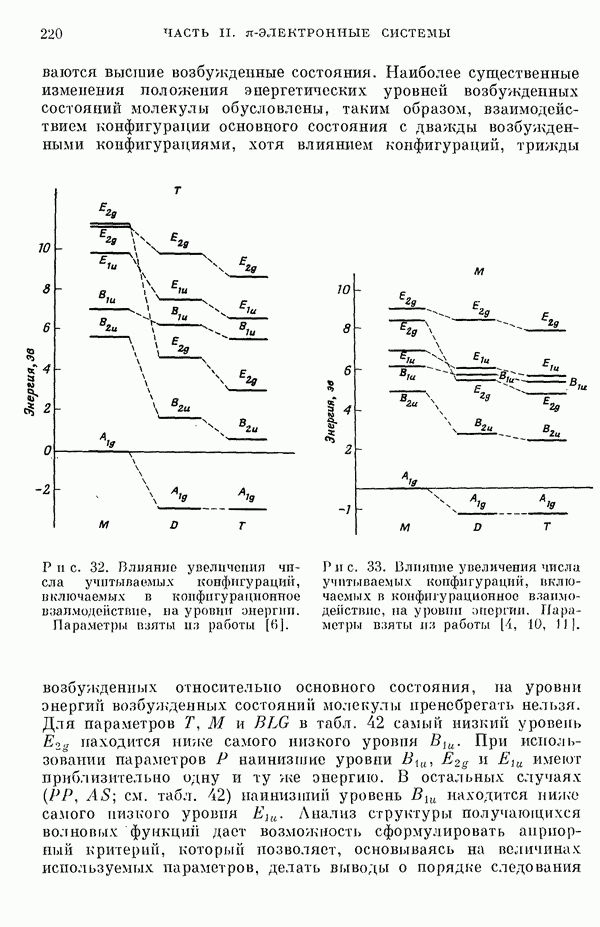
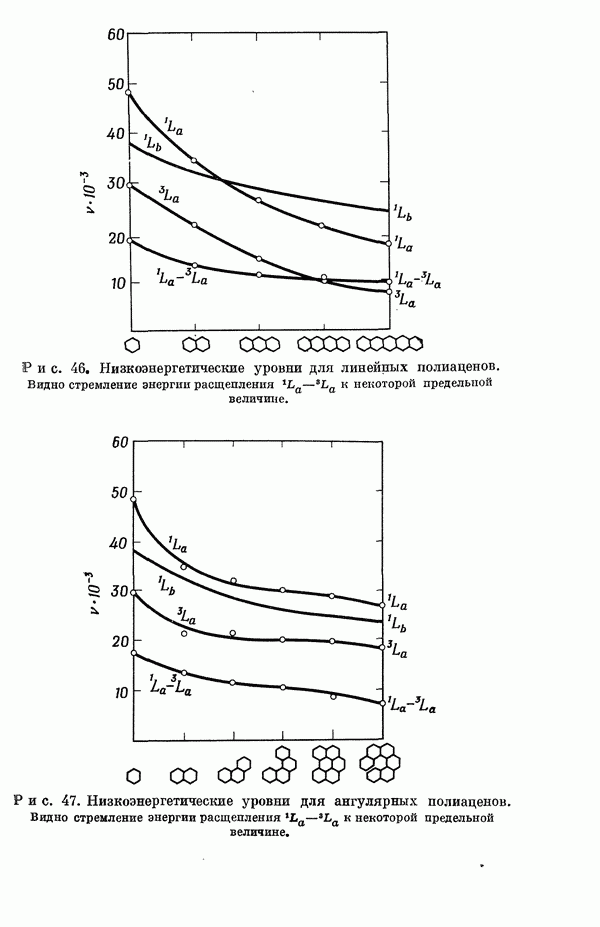
II-6. Критическое обсуждение «пи»-электронных теорийII-6A. ПРЕДЕЛЫ ПРИМЕНИМОСТИ МЕТОДОВ ТИПА МЕТОДОВ ХЮККЕЛЯ И ПАРИЗЕРА - ПАРРАЯ. Коутецкий Для предсказания физических и химических свойств больших молекул с сопряженными двойными связями наиболее часто используются два квантовохимических метода. В органической химии применяется метод Хюккеля ввиду его крайней математической простоты. В молекулярной спектроскопии, кроме метода Хюккеля, используется также метод Паризера — Парра — Поп-ла [1], основанный на определенных полуэмпирических предположениях о величинах атомных интегралов. В последнем методе учитываются лишь наиболее простые одноэлектронные возбужденные конфигурации. Большое распространение метода Паризера — Парра — Попла связано не только с его простотой и возможностями использования вычислительных машин, но и тем обстоятельством, что этот метод оказался успешным в ряде важных случаев. Однако подробный анализ исходных предположений метода показывает, что его успехи удивительны и непонятны. В этой работе мы проведем подробное сравнение электронных спектров, предсказанных по методам Хюккеля и Паризера — Парра, с экспериментальными данными, а также сравнение методов Хюккеля и Паризера — Парра между собой. Может показаться, что метод Хюккеля, как более простой, должен всегда давать менее точные результаты. Это, однако, неверно, что видно, например, из сравнения энергии возбуждения самой длинноволновой полосы интенсйвного синглет-синглетного перехода в УФ-спектрах ненасыщенных молекул и соответствующей теоретической величины, вычисленной по методу Хюккеля [2, 3]. Экспериментальные величины энергий возбуждения определялись по положению так называемой
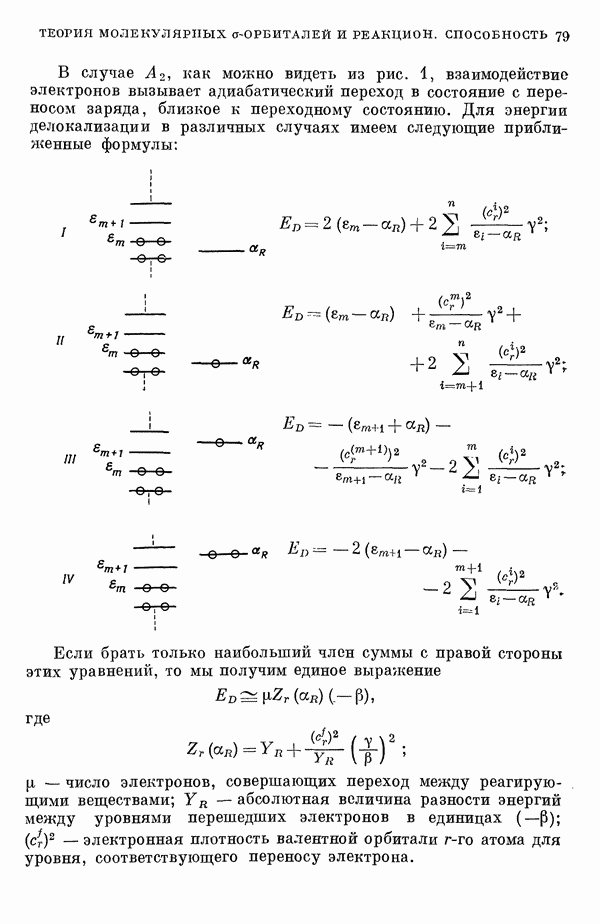
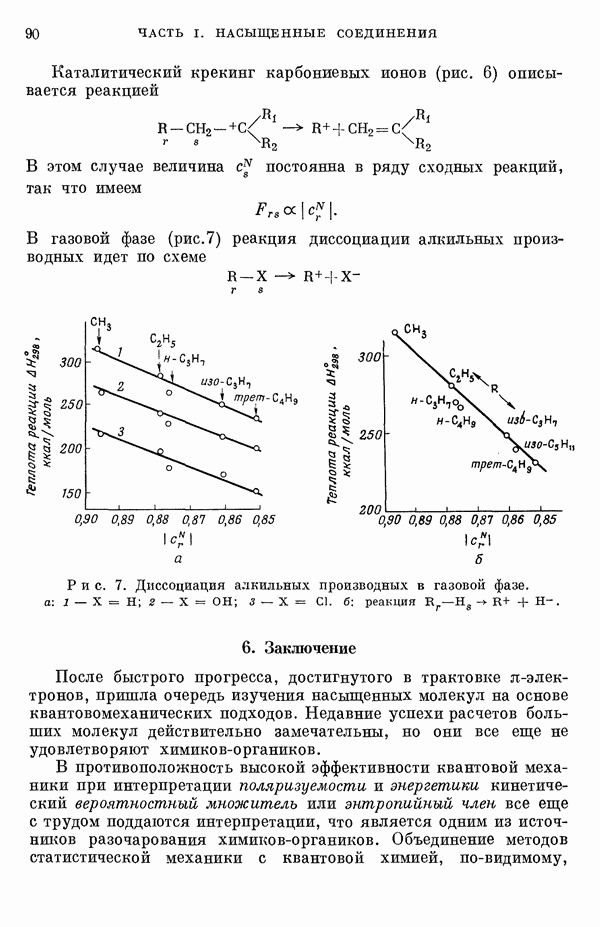
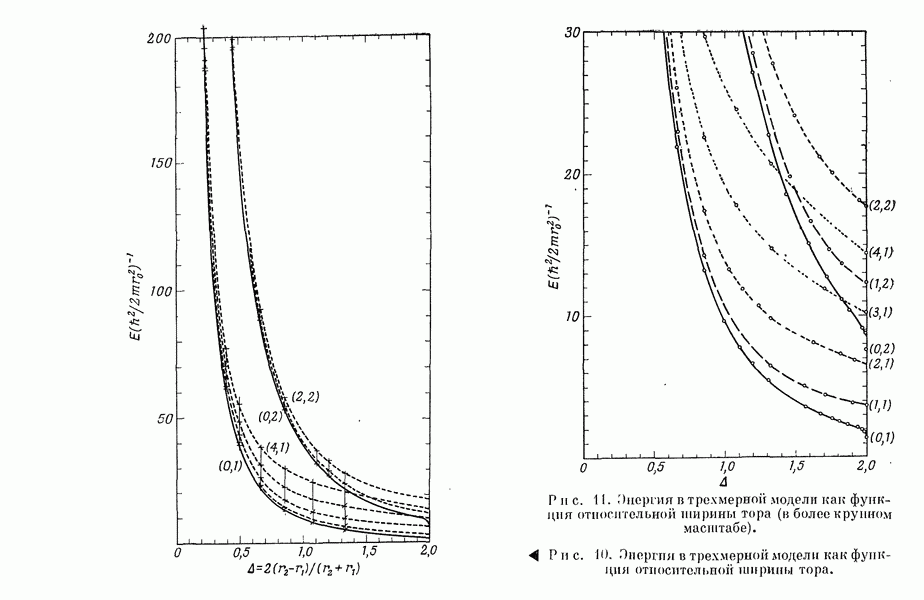
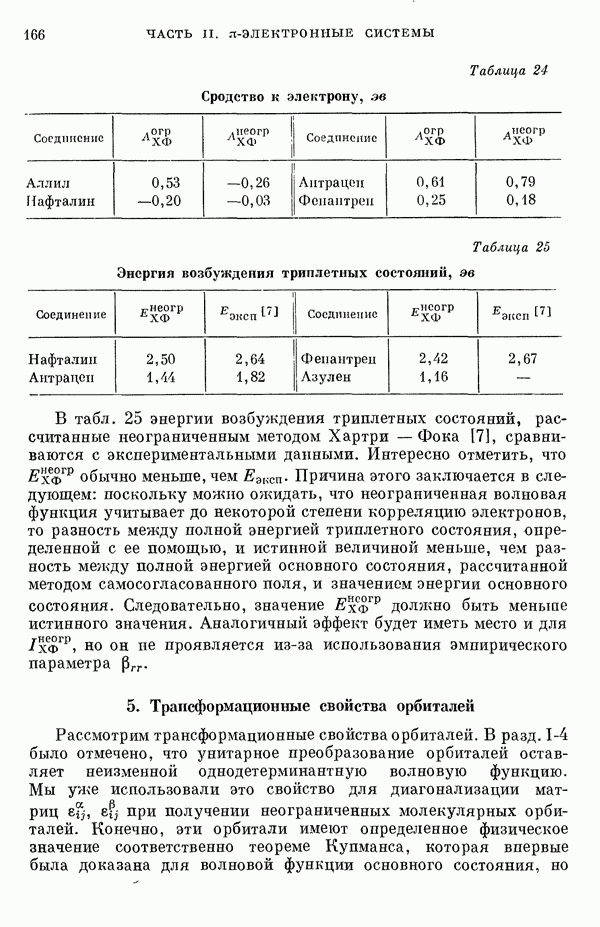
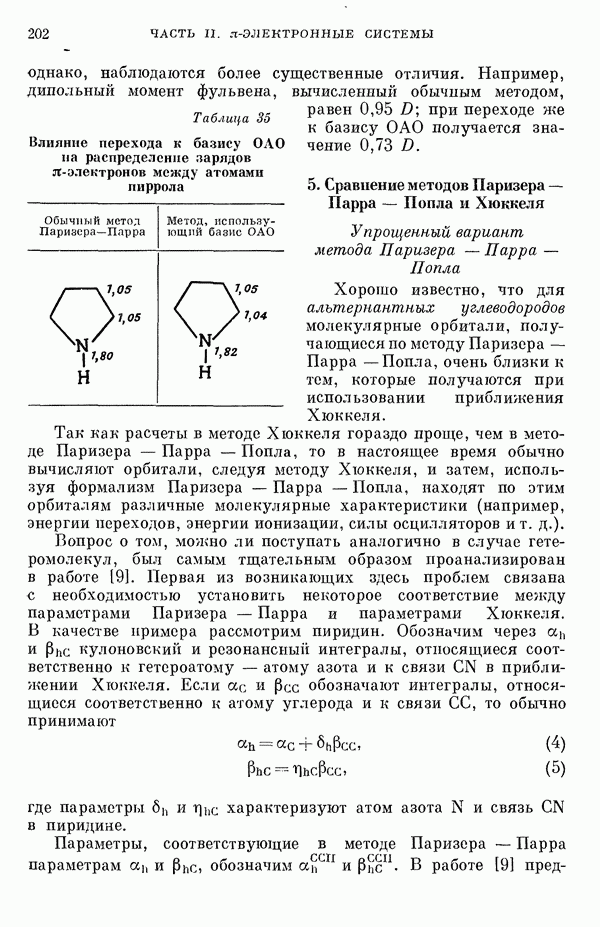
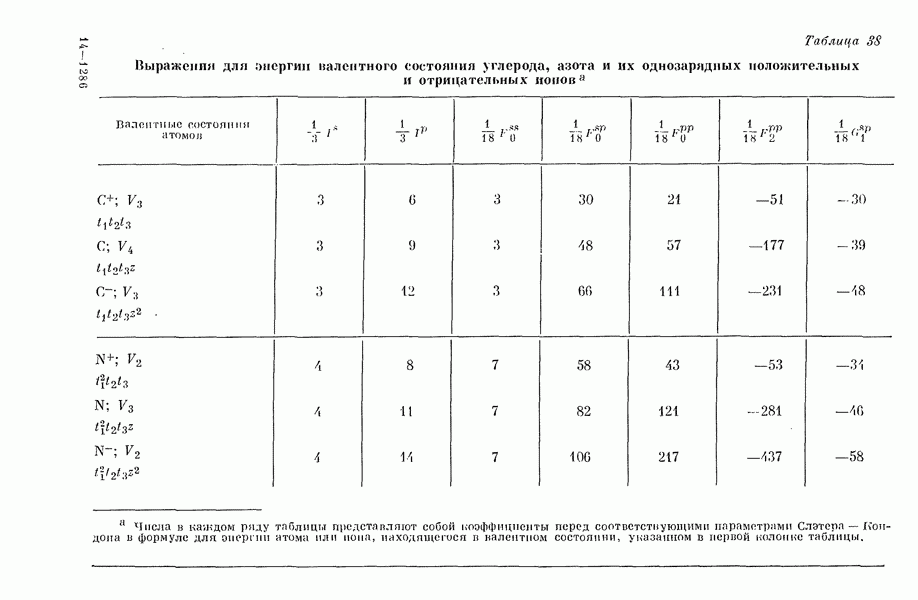
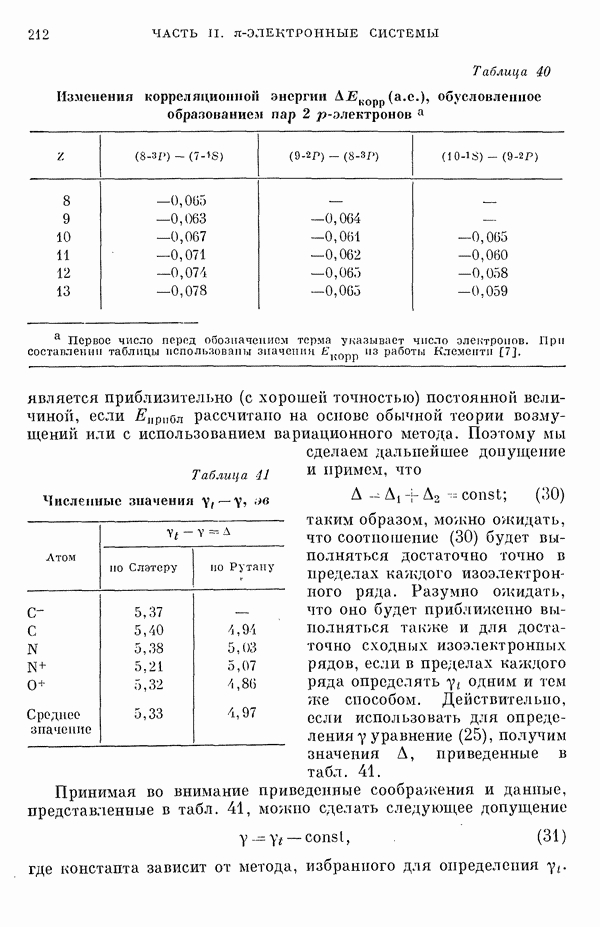
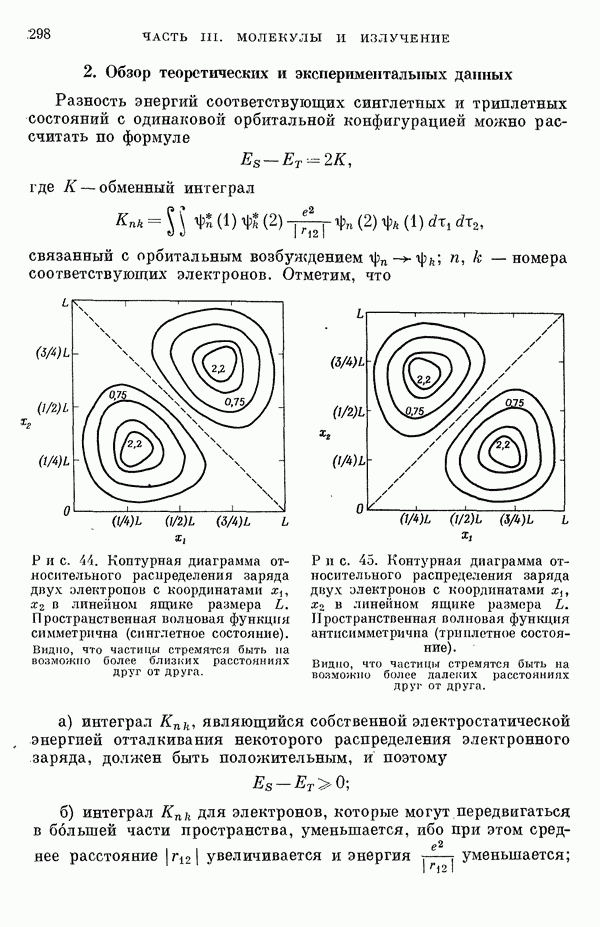
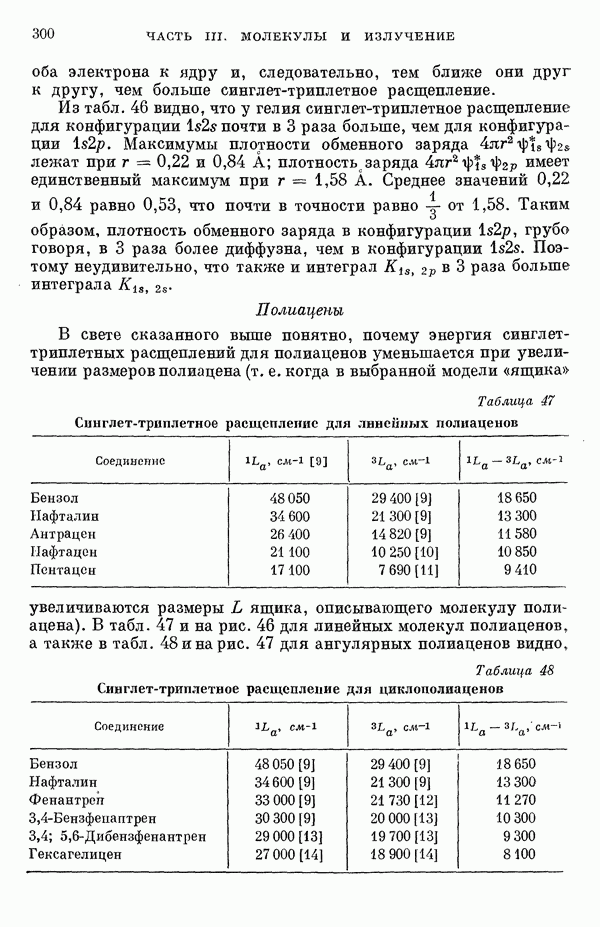
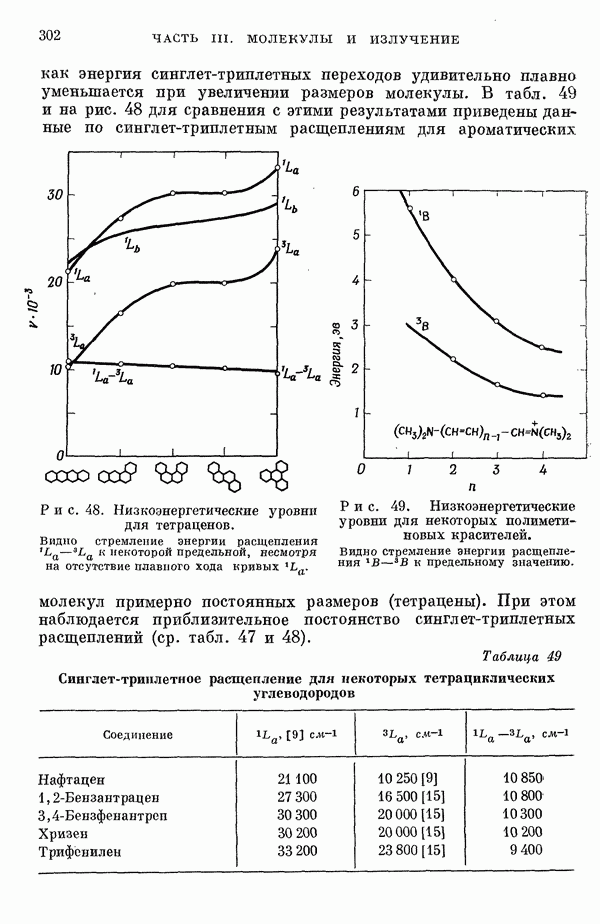
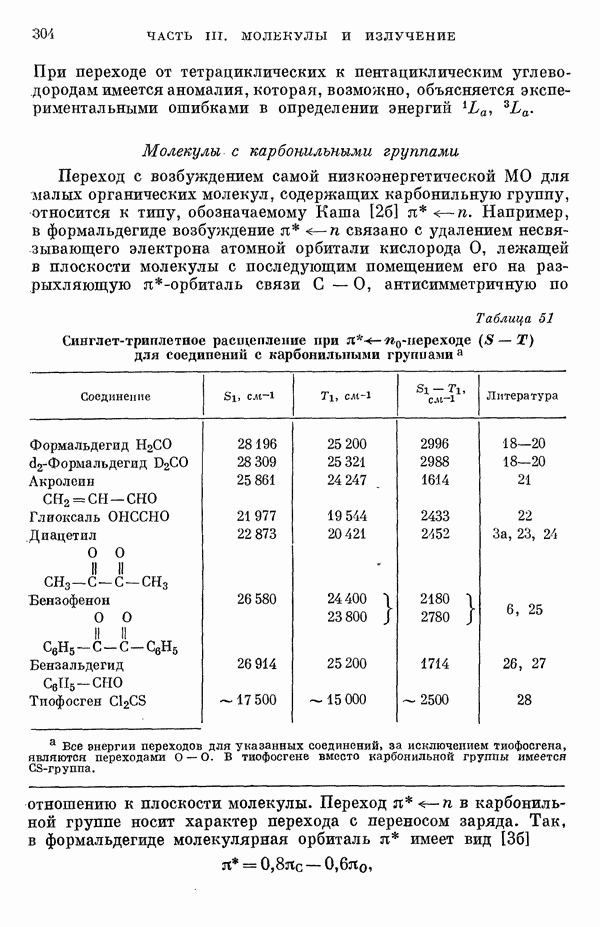
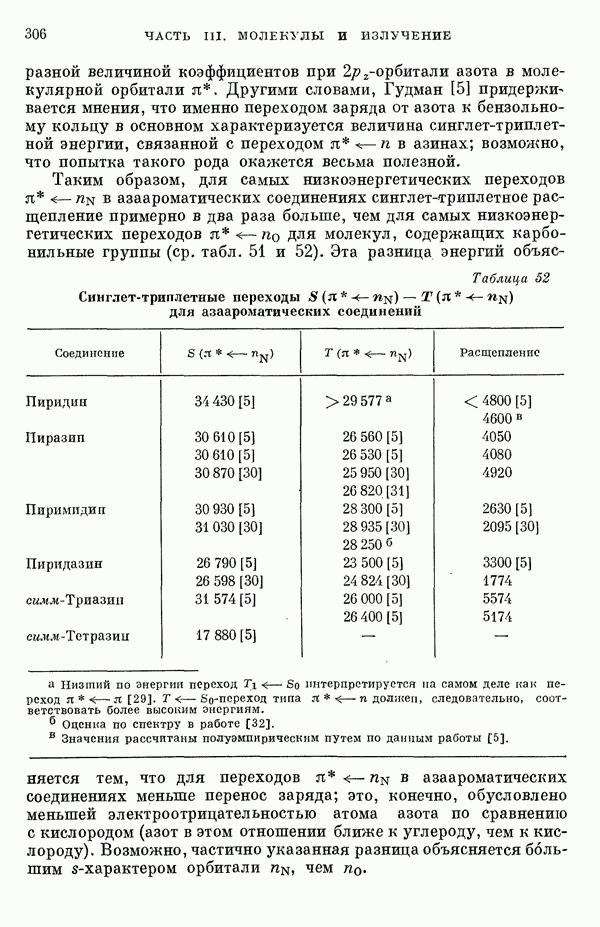
Рис. 19. Зависимость энергии тропилия [5], имеется очевидная корреляция между теоретическими и экспериментальными величинами (рис. 19) [3]. Соответствующие зависимости примерно укладываются на прямые, не проходящие через начало координат. Постоянные члены в линейных зависимостях, соответствующие отрезкам, отсекаемым на оси ординат, различны и убывают в ряду — линейные полнены, ароматические углеводороды, тропилий. Для Это изменение характера соединений при удлинении цепи отчетливо видно на рис. 19; данные для По теории Хюккеля для всех углеводородов следовало бы ожидать единой линейной зависимости с нулевым постоянным членом, т. е. единственной прямой линии, проходящей через начало координат. Поэтому наличие постоянных членов в зависимостях, представленных на рис. 19, непонятно и нуждается в объяснении. В то же время из рис. 19 сразу следует, что в пределах каждого класса углеводородов простой метод Хюккеля позволяет правильно оценивать относительные положения рассматриваемой самой длинноволновой синглетной полосы. Это несколько неожиданный результат, так как кажется бйлее естественным сравнивать теоретические энергии возбуждений, вычисленные по методу Хюккеля, только с положениями центров тяжести полос возбуждения как в синглетное, так и в триплетное состояния. Указанная корреляция между экспериментальными и теоретическими значениями энергий возбуждения отражает влияние аннелирования бензольных колец в определенной основной группе или влияние удлинения полиеновой цепи в молекуле. В случае ароматических углеводородов основной группой молекулы является само бензольное кольцо, в случае бензотропилиевых углеводородов — семичленное кольцо тронилия и т. д. Поэтому полученный результат можно сформулировать следующим образом: метод Хюккеля качественно правильно и достаточно хорошо количественно описывает изменения положений полос в спектрах поглощения соединепий в процессе их аннелирования. В то же время метод никак не объясняет природы этих полос и причины изменений положений этих полос при переходе от одного класса углеводородов к другому. Сформулированное правило, верное почти во всех случаях, имеет все же несколько исключений, наиболее важное из которых, по-видимому, связано с поведением ароматических производных азулена [6]. Следует подчеркнуть, что величина изменения положений полос очень чувствительна как к топологии аннелирования (т. е. к взаимному расположению аннелированных бензольных колец), так и к характеру основной структурной группы, с которой аннелированы бензольные кольца. Успех метода Хюккеля в описании положений полос в спектрах поглощения нельзя объяснить тем, что этот метод по своей природе всегда предсказывает батохромные изменения 3 при увеличении сопряженной системы а в эксперименте по большей части как раз такие и встречались. Во-первых, хорошо известны случаи, когда увеличение сопряженной системы приводит к гипсохромным изменениям. Примерами такого рода являются ароматические производные фульвена и гептафульвена, причем аномальные, на первый взгляд, изменения положений полос поглощения для них были предсказаны успешно с помощью метода Хюккеля. Во-вторых, сами батохромные сдвиги сильно различны при линейном и ангулярном аннелировании (ср. пентацен и пентафен. Приступим теперь к изложению результатов, полученных с помощью одного из вариантов метода ограниченного учета конфигурационного взаимодействия в приближении Паризера — Парра, и проведем сравнение этих результатов как с результатами соответствующих расчетов по методу Хюккеля, так и с экспериментальными данными по ультрафиолетовым спектрам поглощения. В методе Паризера — Парра используется много приближений, и из- этого вытекают два нежелательных следствия. Первое из них заключается в том, что в литературе трудно найти достаточное число соединений, для которых вычисления проводились бы на основе одинаковых предположений. Второе следствие связано с тем, что почти всегда неизвестно, насколько полученные результаты чувствительны к выбору параметров и другим исходным предположениям метода, а насколько они связаны с самой сущностью метода. Ввиду указанных трудностей мы выбрали в качестве предлагаемого стандартного метода следующий вариант метода Паризера — Парра. Геометрия молекул предполагается идеализированной, а именно все длины связей считаются одинаковыми. При составлении слэтеровских детерминантов используются простые молекулярные орбитали Хюккеля. Значения интегралов для атомных орбиталей берутся по Паризеру — Парру — Поплу [1]. Численные значения интегралов электронного отталкивания оцениваются на основе работы [7]. Величина резонансного интеграла выбирается на основе экспериментальных данных по положению Сравнение вычисленных энергий возбуждения для синглетных Результаты расчетов по полуэмпирическому методу ограниченного учета конфигурационного взаимодействия в случае неальтернантных углеводородов оказались очень хорошими [11]. Единственное требование при этом — чтобы полиеновый характер соединения не был бы доминирующим, т. е. чтобы было возможным написать для данного соединения несколько структур Кекуле. В то же время использовались те же самые предположения, как и в случае альтернантных углеводородов, включая предположение ние об упрощенной геометрии молекулы. Систематически были исследованы ароматические производные флуорантена, азулена, катиона тропилия, аниона циклопентадиенила, фульвена и гептафульвена (рис. 23).
Рис. 20. Зависимость экспериментальных значений энергии еэксп возбуждения
Рис. 21. Зависимость экспериментальных значений энергии возбуждения a-полосы от теоретических значений энергии возбуждения (метод ограниченного учета конфигурационного взаимодействия). Те же соединения, что и на рис. 20.
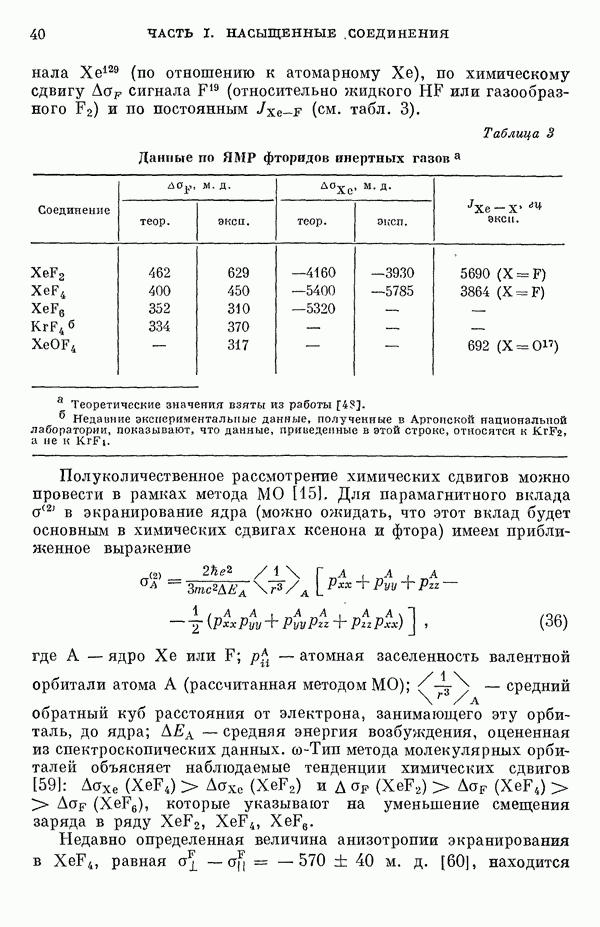
Рис. 22. Зависимость экспериментальных значений энергии возбуждения Те же соединения, что и на рис. 20. Хотя согласие с экспериментом в данном случае не столь хорошее как в случае ароматических углеводородов, ошибки рассчитанных положений самой длинноволновой интенсивной
(кликните для просмотра скана) полосы редко превышают
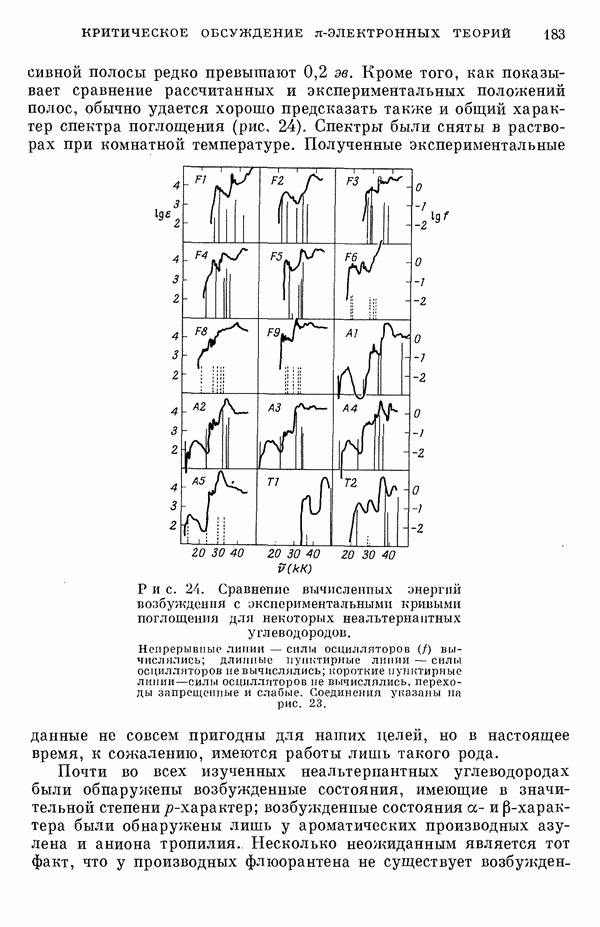
Рис. 24. Сравнение вычисленных энергий возбуждения с экспериментальными кривыми поглощения для некоторых неалътернантных углеводородов. Непрерывные линии — силы осцилляторов Почти во всех изученных неальтерпантных углеводородах были обпаружепы возбужденные состояния, имеющие в значительной степени состояний а- и Сравним теперь энергии возбуждения электронных переходов р-типа, вычисленные по полуэмпирическому методу ограниченного учета конфигурационного взаимодействия, с энергиями возбуждения соответствующих переходов, вычисленными по методу Хюккеля (переходы
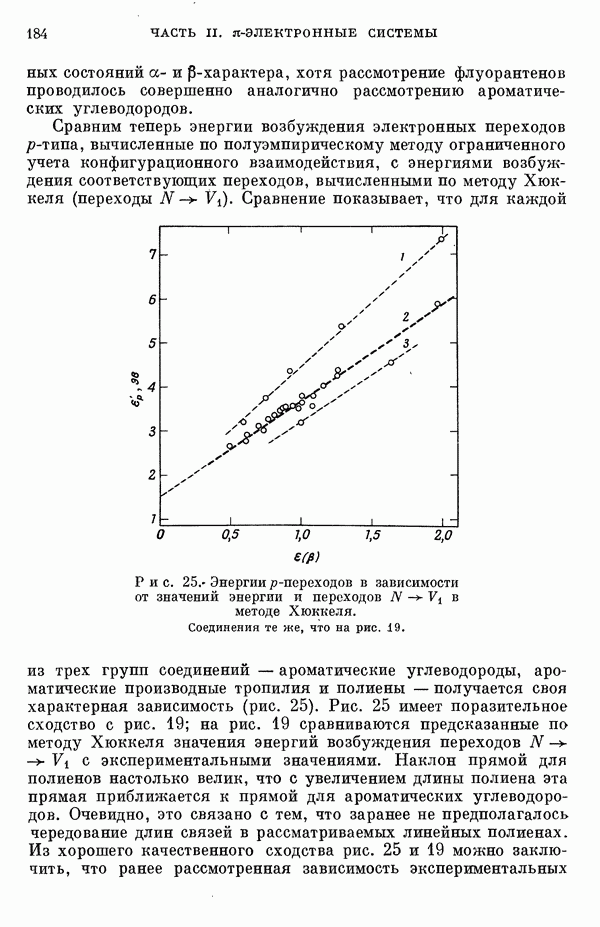
Рис. 25. Энергии Рис. 25 имеет поразительное сходство с рис. 19; на рис. 19 сравниваются предсказанные по методу Хюккеля значения энергий возбуждения переходов значений энергии возбуждения от значений этих энергий по Хюккелю существует также и для значений, полученных по методу Паризера — Парра — Попла. Учет электронного отталкивания приводит к некоторому увеличению хюккелевских значений энергии возбуждения; это увеличение примерно постоянно внутри каждого класса углеводородов, но изменяется при переходе от одного класса к другому.
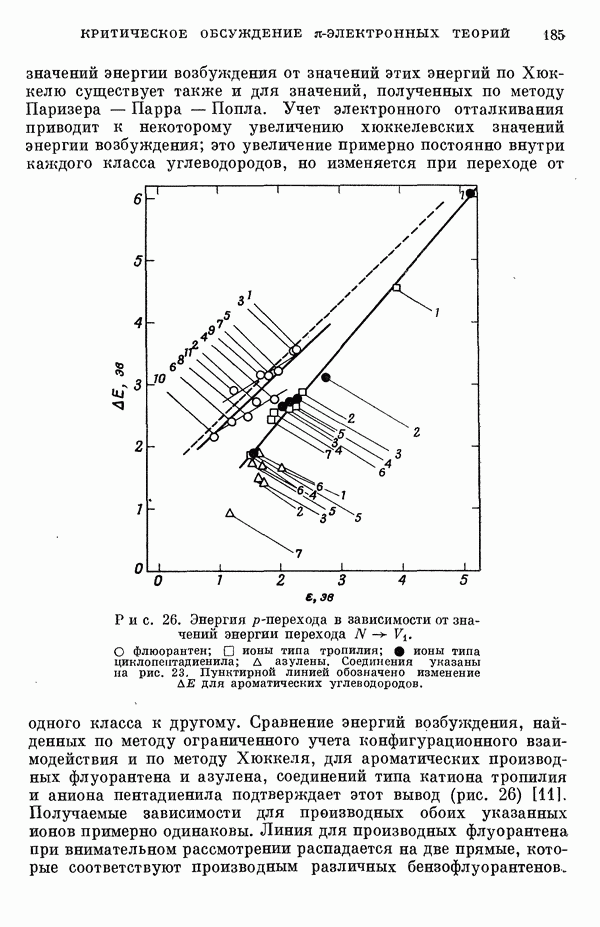
Рис. 26. Энергия Сравнение энергий возбуждения, найденных по методу ограниченного учета конфигурационного взаимодействия и по методу Хюккеля, для ароматических производных флуорантена и азулена, соединений типа катиона тропилия и аниона пентадиенила подтверждает этот вывод (рис. 26) [11]. Получаемые зависимости для производных обоих указанных ионов примерно одинаковы. Линия для производных флуорантена при внимательном рассмотрении распадается на две прямые, которые соответствуют производным различных бензофлуорантенов. Результаты для производных азулена плохо коррелируют между собой, что находится в согласии с тем известным фактом, что для этой группы веществ по теории Хюккеля не удается правильно предсказать влияние аннелирования. Только с помощью более точных методов для этих веществ удается получить согласие теоретических предсказаний с экспериментальными данными. Ясно, что основной причиной расщепления линии, дающей зависимость экспериментальных значений энергии возбуждения от энергий переходов Остается невыясненным следующий вопрос: в какой степени установленная корреляция значений энергии возбуждения Причины столь тесной корреляции результатов этих методов можно объяснять ссылками на определяющее значение топологии молекулы, хотя, с другой стороны, до сих пор не удавалось просто объяснить эту корреляцию, исходя из точных выражений для энергий возбуждения в обоих методах. Использованный нами подход можно обобщить различными способами: например, расширить базис атомных орбиталей (скажем, произвести добавление конфигураций, то эти два метода, строго говоря, не могут быть эквивалентными. С другой стороны, трудности, упомянутые в связи с веществами типа полиенов, могут быть частично преодолены, если при составлении слэтеровских детерминантов воспользоваться молекулярными орбиталями ССП. Метод ССП объясняет реально существующее ослабление простых связей в структурах Кекуле для этих молекул. Правда, следует сказать, что всегда имеется возможность априорного предположения об ослаблении этих связей; для этого надо только использовать уменьшенные значения резонансных интегралов в простом методе Хюккеля. Таким путем мы сразу, хотя и довольпо искусственно, приходим к результирующим молекулярным орбиталям, которые учитывают полиеновый характер молекулы. Наконец, существует третья возможность — считать резонансные интегралы функциями длин или порядков связей. При этом имеется даже два варианта. В первом налагается условие самосогласования, при котором получаемые для молекулярных орбиталей функции исправляют до тех пор, пока разница между получаемыми порядками связей на электронов. Отметим, что даже для ароматических углеводородов различия в длинах связей могут быть довольно значительными. Мы полагаем поэтому, что непосредственное влияние длин связей не столь существенно, как учет влияния полиенового характера соединения. Другие важные проблемы касаются вопросов, связанных с оценками интегралов, содержащих атомные орбитали, и с приближением нулевого дифференциального перекрывания. Поскольку эти проблемы обсуждаются в разд. II-1 и II-6Б, мы не считаем необходимым рассматривать их здесь. Одним из важных предположений, которые делаются в обычных приближенных методах, является пренебрежение высшими возбужденными конфигурациями. Недавно стало известно, однако, что учет этих конфигураций ведет к значительным изменениям вычисляемых энергетических уровней для углеводородов. Теоретические вычисления показали, что может существенно измениться даже порядок расположения основных энергетических уровней важнейших возбужденных состояний. Поэтому остается актуальным вопрос об учете двукратно возбужденных и высших возбужденных конфигураций (см. разд.II-6Д). В этом разделе проведено сравнение результатов наиболее часто используемых полуэмпирических методов. Казалось бы, более целесообразно было рассмотреть совсем другие методы, которые позволяют преодолевать связанные с корреляционными эффектами трудности совершенно иными путями. Однако если рассматривать имеющуюся статистическую корреляцию между результатами простого метода Хюккеля и экспериментальными данными просто как эмпирическую закономерность, то представляется, что именно на ней следует строить будущую теорию. Можно считать, что обнаруженные соотношения приближенно отражают некоторые новые закономерности, знание которых позволит на основе объединения эмпирических закономерностей разработать новые методы квантовохимических расчетов. Ясно, что упрощения такого рода важны при практических применениях квантовой химии, несмотря на успехи, достигнутые в современной вычислительной технике. ЛИТЕРАТУРА(см. скан) (см. скан)
|
 |
Оглавление
|








































































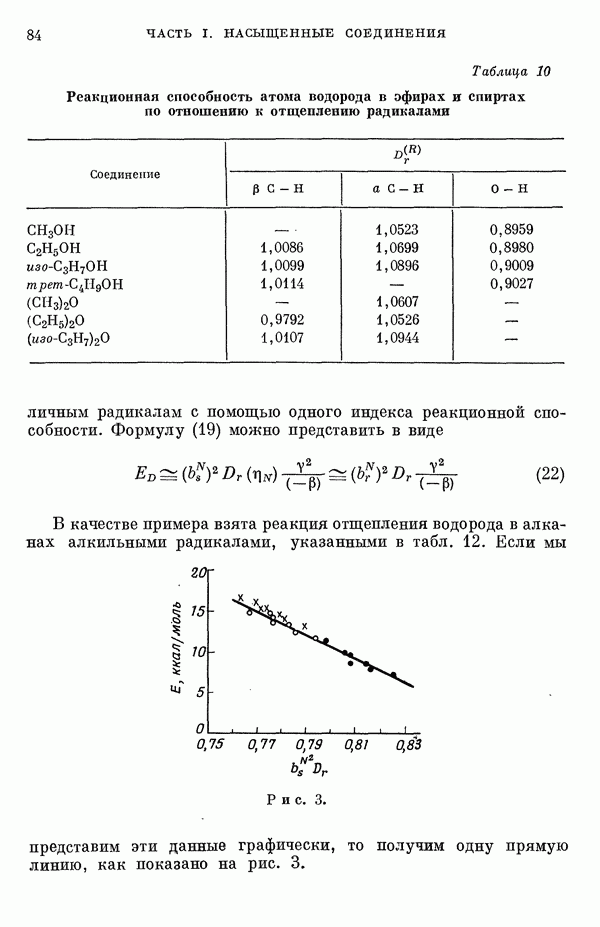
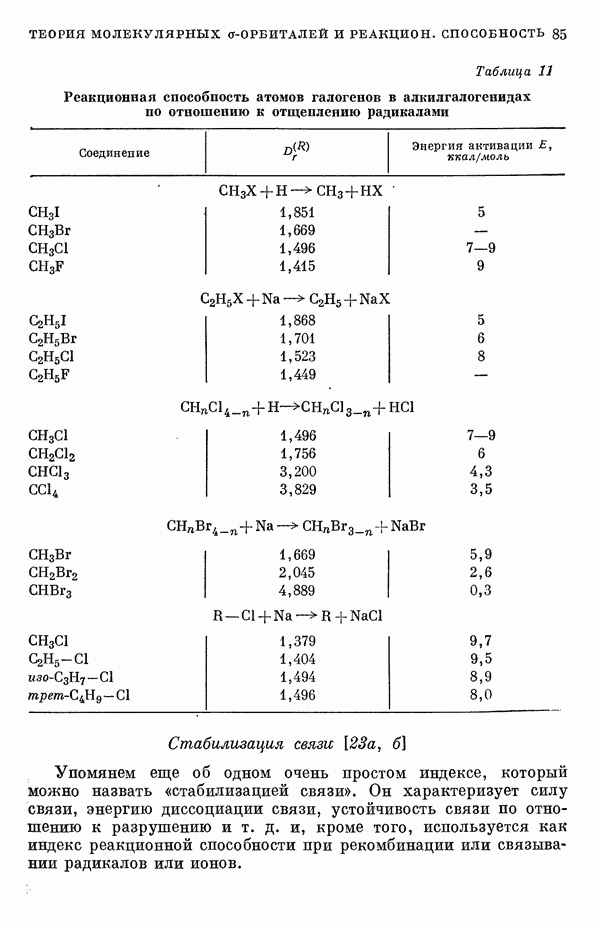
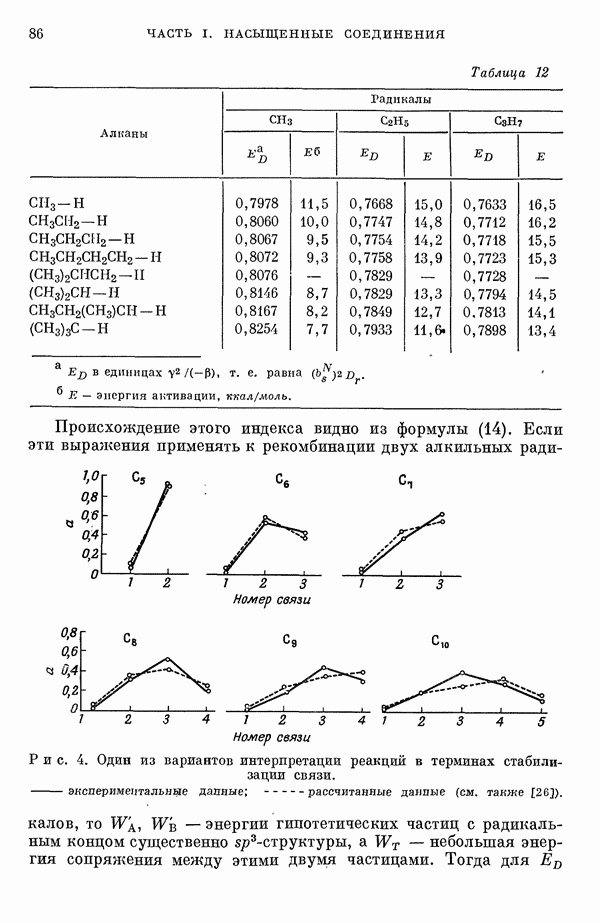
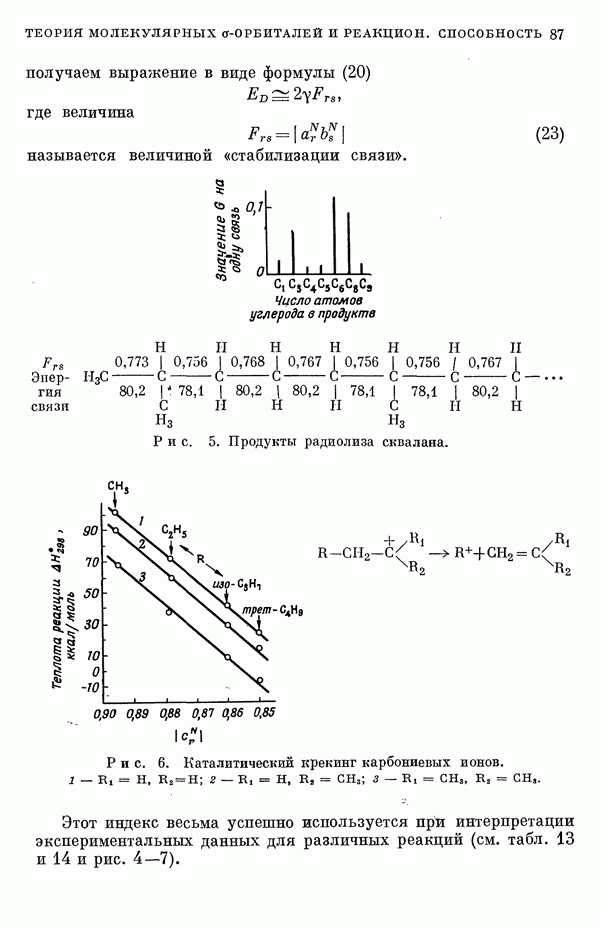

























































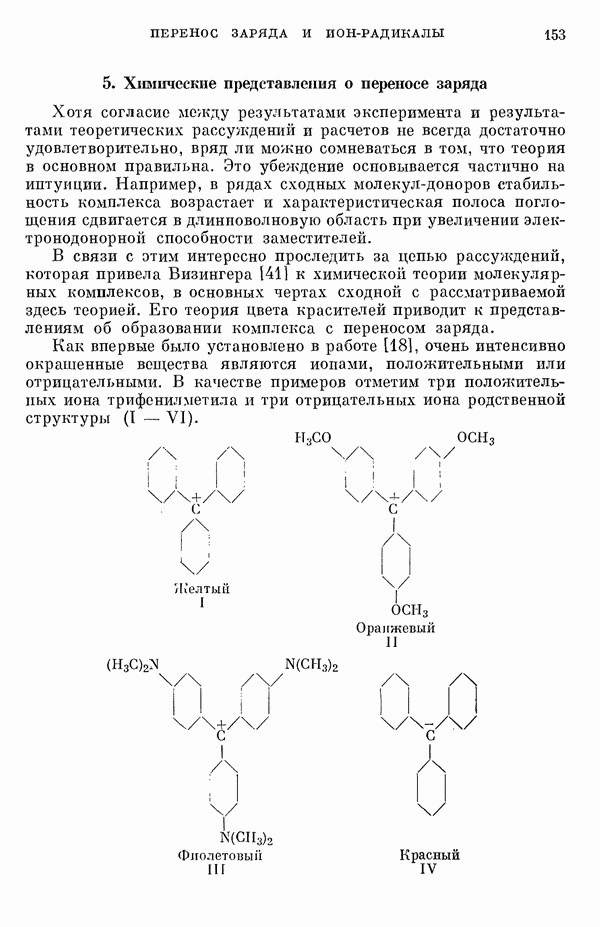
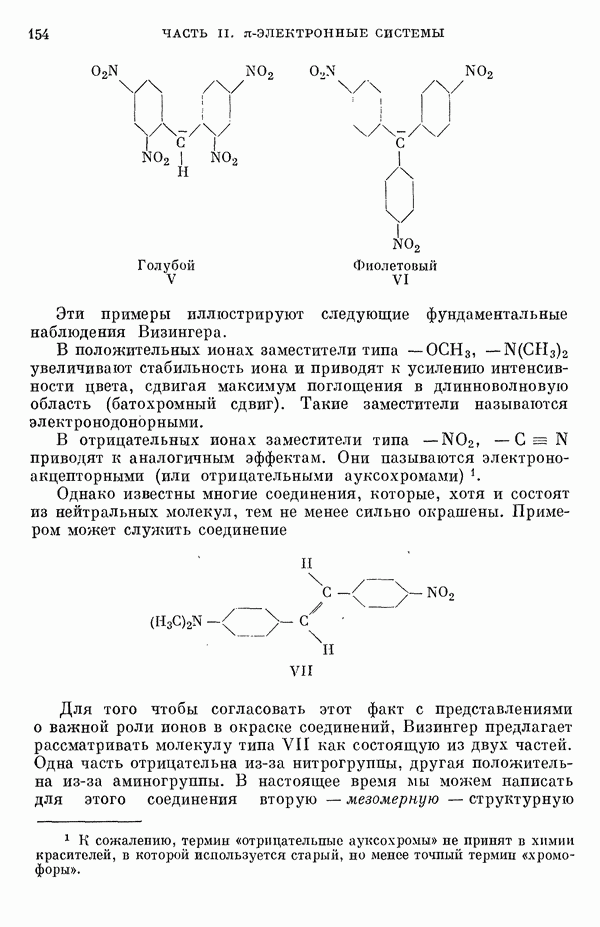
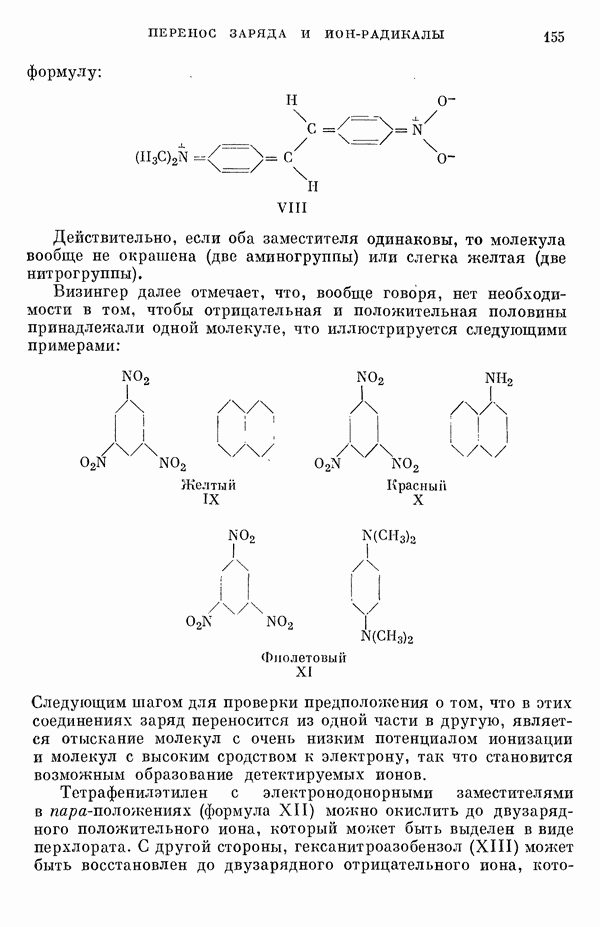
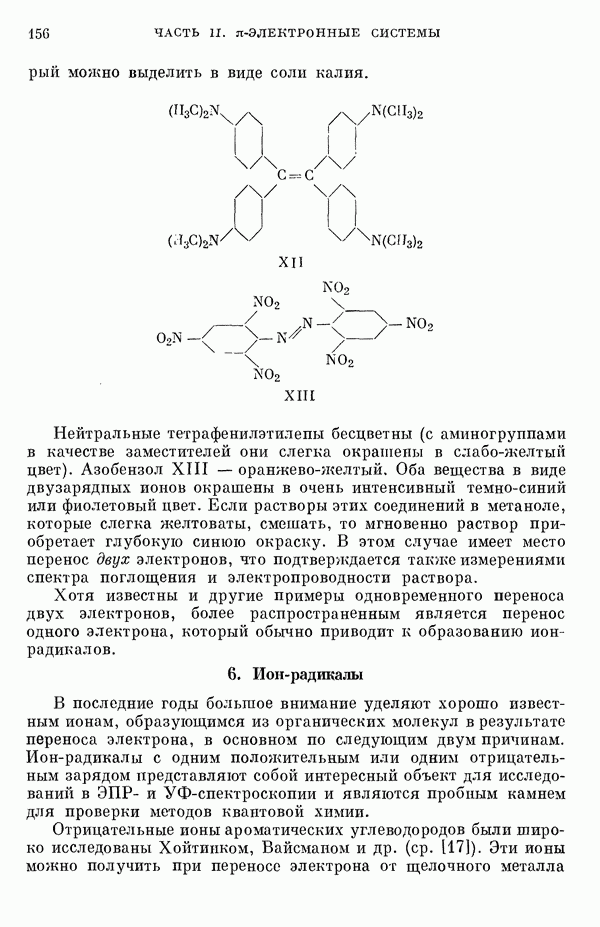















































































































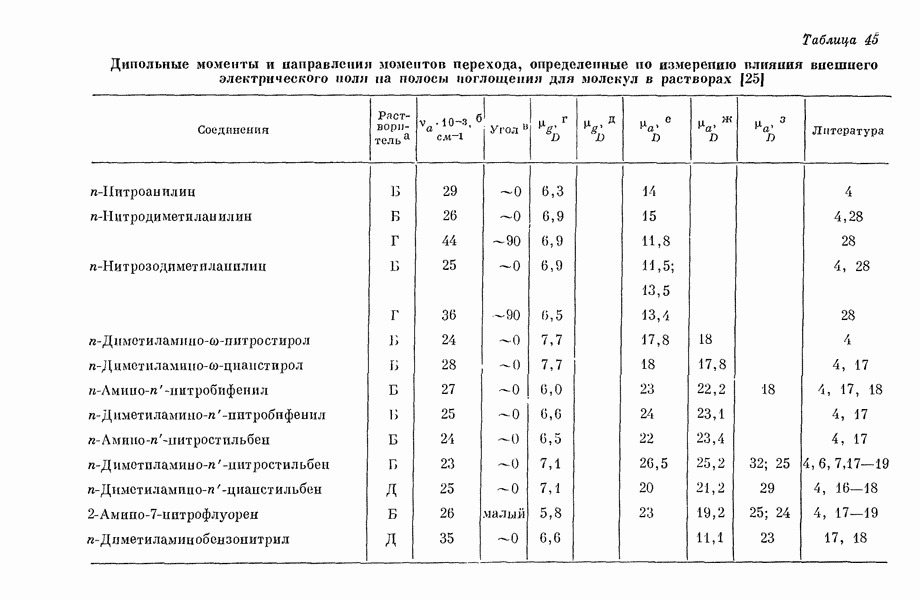
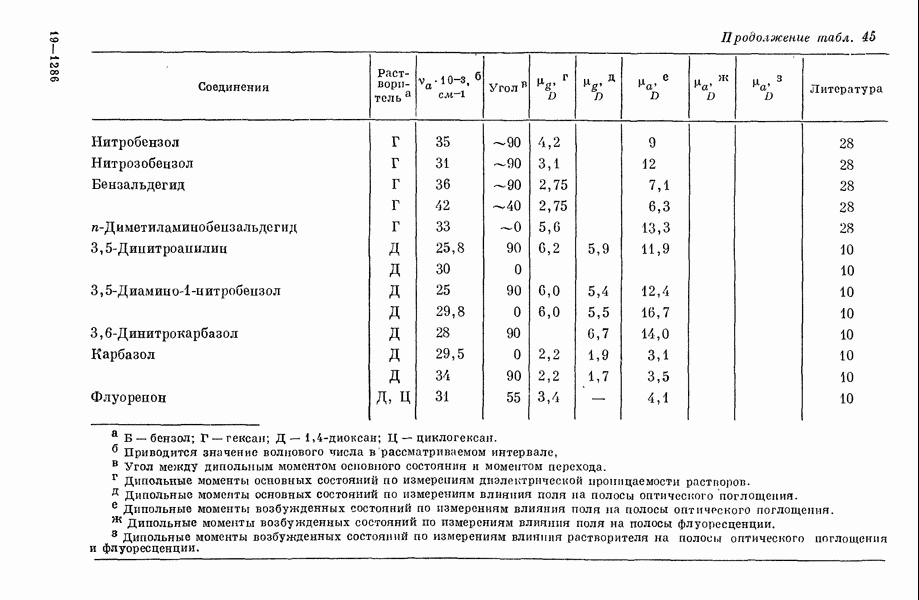







































 -полосы по классификации Клара [4]. Идентификация этой полосы сама по себе является сложной задачей и требует должного внимания. Теоретические величины энергий возбуждения вычислялись по методу Хюккеля как разности энергий между высшим из занятых и низшим из незанятых одноэлектронных уровней. Геометрию молекулы при этом рассматривали упрощенно, принимая все длины связей одинаковыми. Внутри каждого класса углеводородов, например ароматических углеводородов, линейных полиенов и ароматических производных
-полосы по классификации Клара [4]. Идентификация этой полосы сама по себе является сложной задачей и требует должного внимания. Теоретические величины энергий возбуждения вычислялись по методу Хюккеля как разности энергий между высшим из занятых и низшим из незанятых одноэлектронных уровней. Геометрию молекулы при этом рассматривали упрощенно, принимая все длины связей одинаковыми. Внутри каждого класса углеводородов, например ароматических углеводородов, линейных полиенов и ароматических производных

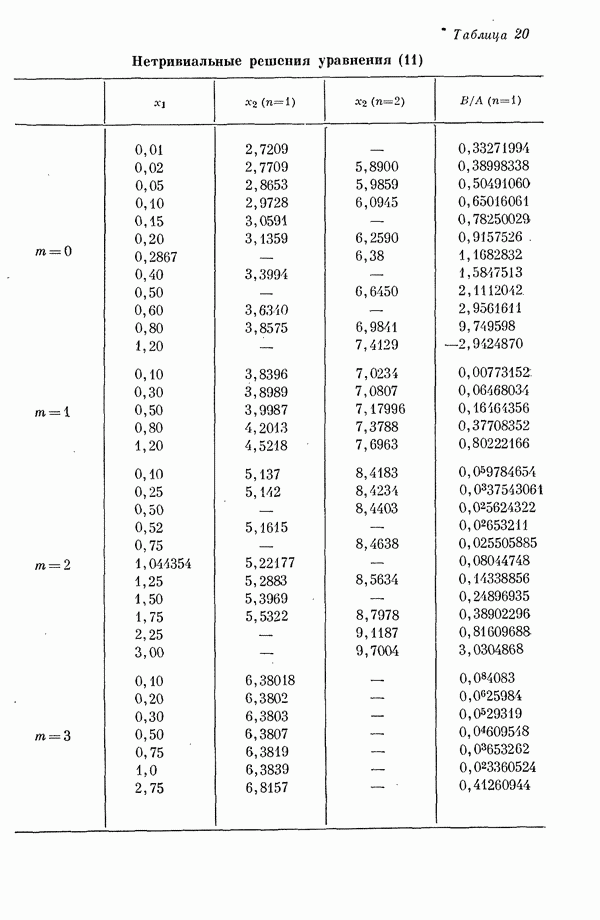
 первого самого низкочастотного интенсивного максимума поглощения от энергии
первого самого низкочастотного интенсивного максимума поглощения от энергии  перехода
перехода  линейные полнены; 2 — ароматические углеводороды; 3 — ионы типа тропилия; 4 — а,
линейные полнены; 2 — ароматические углеводороды; 3 — ионы типа тропилия; 4 — а,  -дифенилполиены.
-дифенилполиены.
 -дифенил-полиенов при удлинении линейной цепи наблюдается непрерывный переход от имеющего почти ароматический характер дифенила к углеводородам типа полиенов.
-дифенил-полиенов при удлинении линейной цепи наблюдается непрерывный переход от имеющего почти ароматический характер дифенила к углеводородам типа полиенов.
 -дифенилпо-лиенов располагаются на
-дифенилпо-лиенов располагаются на  -образной кривой, соединяющей прямую линию, соответствующую ароматическим углеводородам, с прямой линией, соответствующей линейным полиенам.
-образной кривой, соединяющей прямую линию, соответствующую ароматическим углеводородам, с прямой линией, соответствующей линейным полиенам.
 -полосы в молекуле бензола. Данный выбор параметров ближе к тому, который был предложен в работе Паризера 18], чем в работе Паризера — Парра [9].
-полосы в молекуле бензола. Данный выбор параметров ближе к тому, который был предложен в работе Паризера 18], чем в работе Паризера — Парра [9].
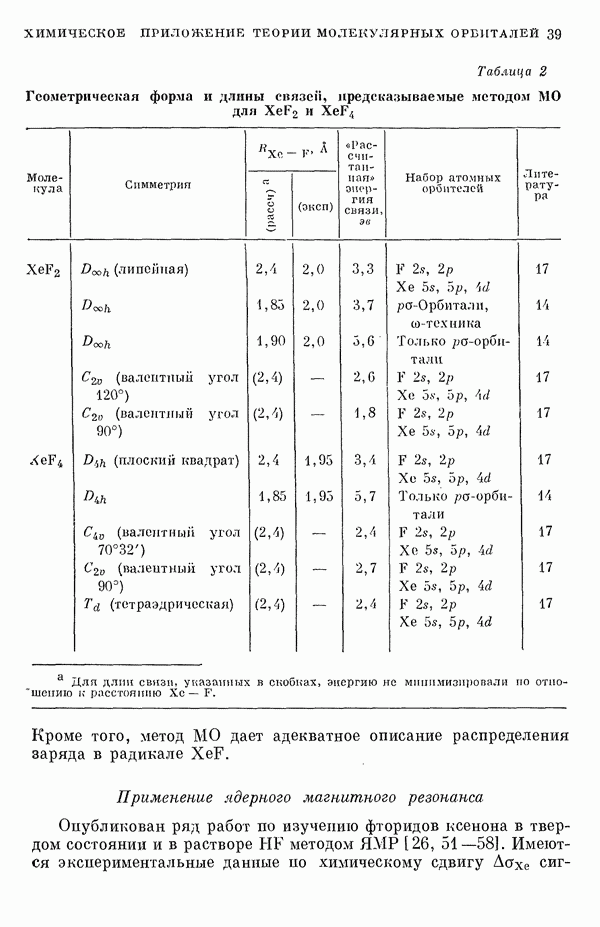
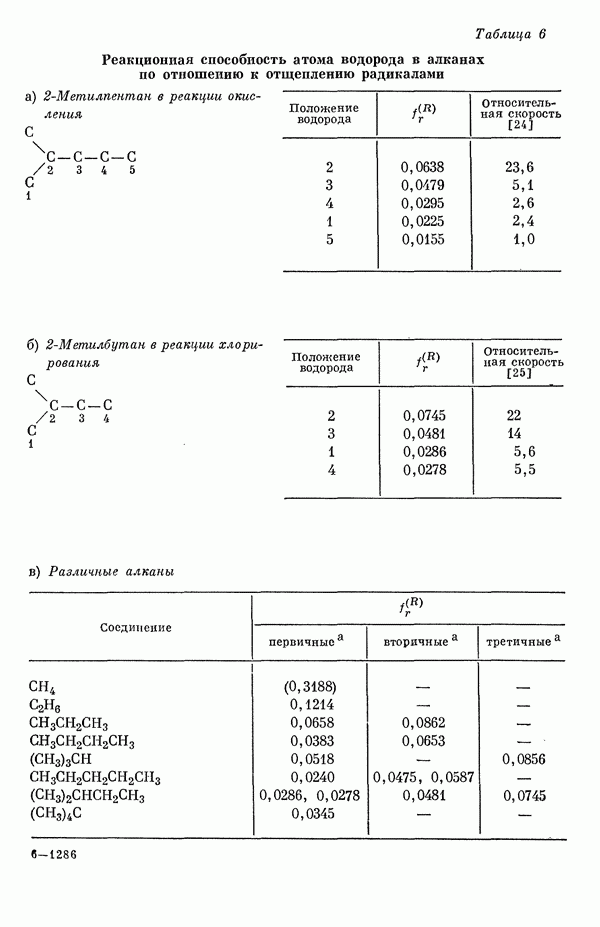
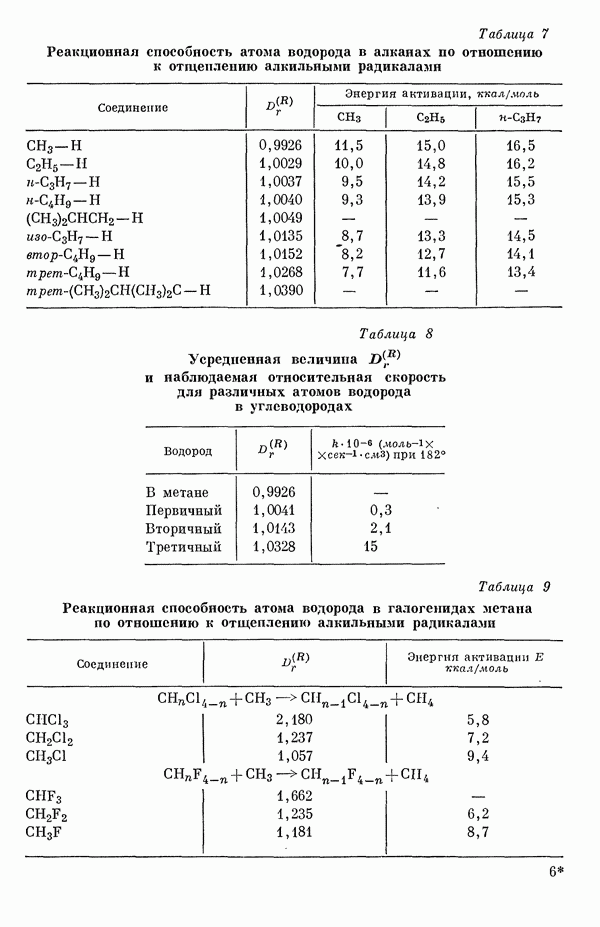
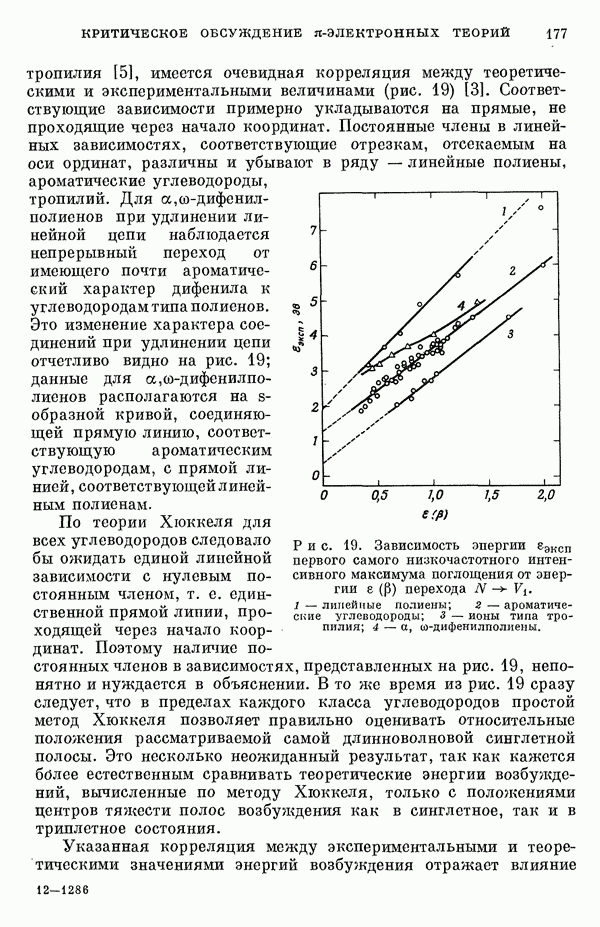
 -полос с экспериментальными данными, проведенное для 20 ароматических углеводородов, показывает необычайно хорошее согласие теоретических и экспериментальных результатов (рис. 20) [3]. Учет конфигурационных взаимодействий с включением только одновозбужденных конфигураций приводит к несколько заниженным значениям энергии возбуждения (в среднем примерно на 0,1 эв). Это очень незначительное понижение объясняется слабостью конфигурационного взаимодействия. С другой стороны, только благодаря учету конфигурационного взаимодействия удается получить очень хорошее согласие с экспериментом при сделанном выборе параметров. Рассчитанные положения а- и
-полос с экспериментальными данными, проведенное для 20 ароматических углеводородов, показывает необычайно хорошее согласие теоретических и экспериментальных результатов (рис. 20) [3]. Учет конфигурационных взаимодействий с включением только одновозбужденных конфигураций приводит к несколько заниженным значениям энергии возбуждения (в среднем примерно на 0,1 эв). Это очень незначительное понижение объясняется слабостью конфигурационного взаимодействия. С другой стороны, только благодаря учету конфигурационного взаимодействия удается получить очень хорошее согласие с экспериментом при сделанном выборе параметров. Рассчитанные положения а- и  -полос согласуются с экспериментом менее удовлетворительно (рис. 21 и 22) [10]. Появляются систематические отклонения, поскольку теория предсказывает положения
-полос согласуются с экспериментом менее удовлетворительно (рис. 21 и 22) [10]. Появляются систематические отклонения, поскольку теория предсказывает положения  -полосы в области более длинных волн. Однако изменив соответствующим образом параметры, удается получить также для а- и
-полосы в области более длинных волн. Однако изменив соответствующим образом параметры, удается получить также для а- и  -полос ароматических углеводородов хорошее согласие с экспериментом, как и для
-полос ароматических углеводородов хорошее согласие с экспериментом, как и для  -полос. Обозначим посредством
-полос. Обозначим посредством  конфигурации, в которых
конфигурации, в которых  молекулярная орбиталь заменяется на некоторую до того не занятую
молекулярная орбиталь заменяется на некоторую до того не занятую  орбиталь, причем молекулярные орбитали, занятые в основном состоянии системы, нумеруются в порядке убывания энергии, а незанятые молекулярные орбитали — в порядке возрастания энергии. Когда в линейной комбинации слэтеровских детерминантов, описывающих некоторое возбужденное состояние, доминирует конфигурация
орбиталь, причем молекулярные орбитали, занятые в основном состоянии системы, нумеруются в порядке убывания энергии, а незанятые молекулярные орбитали — в порядке возрастания энергии. Когда в линейной комбинации слэтеровских детерминантов, описывающих некоторое возбужденное состояние, доминирует конфигурация  , то говорят, что это состояние имеет выраженный
, то говорят, что это состояние имеет выраженный  -характер. Когда в линейной комбинации конфигураций доминируют две конфигурации
-характер. Когда в линейной комбинации конфигураций доминируют две конфигурации  , то говорят, что такие состояния имеют а- или Р-харак-тер в зависимости от того, мала или велика интенсивность соответствующего электронного перехода. В ароматических углеводородах
, то говорят, что такие состояния имеют а- или Р-харак-тер в зависимости от того, мала или велика интенсивность соответствующего электронного перехода. В ароматических углеводородах  а- или
а- или  -характер соответствующих полос весьма четко определен.
-характер соответствующих полос весьма четко определен.

 -полосы от вычисленных по методу ограниченного конфигурационного взаимодействия энергий возбуждения е. 1 — бензол; 2 — нафталин; 3 — фенантрен; 4 — антрацен; 5 — 3,4-бензфенан-трен; в — пирен; 7 — хризен; 8 -
-полосы от вычисленных по методу ограниченного конфигурационного взаимодействия энергий возбуждения е. 1 — бензол; 2 — нафталин; 3 — фенантрен; 4 — антрацен; 5 — 3,4-бензфенан-трен; в — пирен; 7 — хризен; 8 - -беизантрацен; 9 — тетрадей; 10 — пицен; 11 — перилен; 12 — пентафен;
-беизантрацен; 9 — тетрадей; 10 — пицен; 11 — перилен; 12 — пентафен;  — 3.4-бензпирен; 14 - 1,2-; 3,4-дибензан-трацен; 16-1,2-; 5,6-дибензаитрацен; 16 — пентацещ 17 — 1,2-бензиирен; 18 — 3.4-; 9,10-дибензпирен; 19 - 3,4-; 8,9-дибензпирен; 20 — коронен.
— 3.4-бензпирен; 14 - 1,2-; 3,4-дибензан-трацен; 16-1,2-; 5,6-дибензаитрацен; 16 — пентацещ 17 — 1,2-бензиирен; 18 — 3.4-; 9,10-дибензпирен; 19 - 3,4-; 8,9-дибензпирен; 20 — коронен.


 -полосы от теоретических значений энергии возбуждения (метод ограниченного учета конфигурационного взаимодействия).
-полосы от теоретических значений энергии возбуждения (метод ограниченного учета конфигурационного взаимодействия).

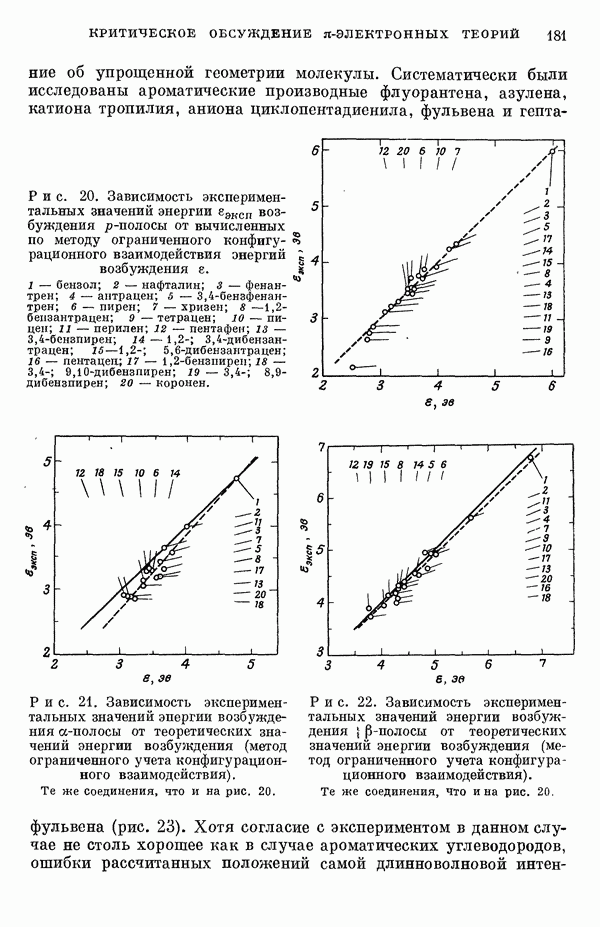
 Кроме того, как показывает сравнение рассчитанных и экспериментальных положений полос, обычно удается хорошо предсказать также и общий характер спектра поглощения (рис. 24). Спектры были сняты в растворах при комнатной температуре. Полученные экспериментальные данные не совсем пригодны для наших целей, но в настоящее время, к сожалению, имеются работы лишь такого рода.
Кроме того, как показывает сравнение рассчитанных и экспериментальных положений полос, обычно удается хорошо предсказать также и общий характер спектра поглощения (рис. 24). Спектры были сняты в растворах при комнатной температуре. Полученные экспериментальные данные не совсем пригодны для наших целей, но в настоящее время, к сожалению, имеются работы лишь такого рода.

 вычислялись; длинные пунктирные линии — силы осцилляторов не вычислялись; короткие пунктирные линии—силы осцилляторов не вычислялись, переходы запрещенные и слабые. Соединения указаны на рис. 23.
вычислялись; длинные пунктирные линии — силы осцилляторов не вычислялись; короткие пунктирные линии—силы осцилляторов не вычислялись, переходы запрещенные и слабые. Соединения указаны на рис. 23.
 -характер; возбужденные состояния
-характер; возбужденные состояния  -характера были обнаружены лишь у ароматических производных азулена и аниона тропилия. Несколько неожиданным является тот факт, что у производных флюорантена не существует возбужденных
-характера были обнаружены лишь у ароматических производных азулена и аниона тропилия. Несколько неожиданным является тот факт, что у производных флюорантена не существует возбужденных
 -характера, хотя рассмотрение флуорантенов проводилось совершенно аналогично рассмотрению ароматических углеводородов.
-характера, хотя рассмотрение флуорантенов проводилось совершенно аналогично рассмотрению ароматических углеводородов.
 Сравнение показывает, что для каждой из трех групп соединений — ароматические углеводороды, ароматические производные тропилия и полнены — получается своя характерная зависимость (рис. 25).
Сравнение показывает, что для каждой из трех групп соединений — ароматические углеводороды, ароматические производные тропилия и полнены — получается своя характерная зависимость (рис. 25).

 -переходов в зависимости от значений энергии и переходов
-переходов в зависимости от значений энергии и переходов  в методе Хюккеля. Соединения те же, что на рис. 19.
в методе Хюккеля. Соединения те же, что на рис. 19.
 с экспериментальными значениями. Наклон прямой для полиенов настолько велик, что с увеличением длины полиена эта прямая приближается к прямой для ароматических углеводородов. Очевидно, это связано с тем, что заранее не предполагалось чередование длин связей в рассматриваемых линейных полиенах. Из хорошего качественного сходства рис. 25 и 19 можно заключить, что ранее рассмотренная зависимость экспериментальных
с экспериментальными значениями. Наклон прямой для полиенов настолько велик, что с увеличением длины полиена эта прямая приближается к прямой для ароматических углеводородов. Очевидно, это связано с тем, что заранее не предполагалось чередование длин связей в рассматриваемых линейных полиенах. Из хорошего качественного сходства рис. 25 и 19 можно заключить, что ранее рассмотренная зависимость экспериментальных

 -перехода в зависимости от значений энергии перехода
-перехода в зависимости от значений энергии перехода  флюорантен;
флюорантен;  ионы типа тропилия;
ионы типа тропилия;  ионы типа циклопептадиенила;
ионы типа циклопептадиенила;  азулены. Соединения указаны на рис. 23. Пунктирной линией обозначено изменение
азулены. Соединения указаны на рис. 23. Пунктирной линией обозначено изменение  для ароматических углеводородов.
для ароматических углеводородов.
 рассчитанных по Хюккелю, не может являться неучет различия в длинах связей в классическом приближении Хюккеля. Очевидно производные катиона тропилия и аниона пентадиенила имеют очень сходные свойства, несмотря на различие длин связей у этих веществ, хотя при количественных оценках энергии возбуждения и необходимо учитывать различия в длинах связей. Для производных фульвена и гентафульвена теоретические предсказания по методу ограниченного учета конфигурационного взаимодействия в пренебрежении различиями в длинах связей не согласуются с экспериментальными результатами. Линия зависимости энергий возбуждения
рассчитанных по Хюккелю, не может являться неучет различия в длинах связей в классическом приближении Хюккеля. Очевидно производные катиона тропилия и аниона пентадиенила имеют очень сходные свойства, несмотря на различие длин связей у этих веществ, хотя при количественных оценках энергии возбуждения и необходимо учитывать различия в длинах связей. Для производных фульвена и гентафульвена теоретические предсказания по методу ограниченного учета конфигурационного взаимодействия в пренебрежении различиями в длинах связей не согласуются с экспериментальными результатами. Линия зависимости энергий возбуждения  -переходов по Паризеру — Парру от энергий возбуждения переходов
-переходов по Паризеру — Парру от энергий возбуждения переходов  по Хюккелю идет для производных фульвена и гептафульвена очень круто, как и в случае линейных пояиенов.
по Хюккелю идет для производных фульвена и гептафульвена очень круто, как и в случае линейных пояиенов.
 -полосы, которые рассчитываются в используемой здесь модификации метода Паризера — Парра и по методу Хюккеля, зависит от выбора параметров (ср. расчеты 18, 12-15])?
-полосы, которые рассчитываются в используемой здесь модификации метода Паризера — Парра и по методу Хюккеля, зависит от выбора параметров (ср. расчеты 18, 12-15])?
 или
или  -атомных орбиталей, см. 116, 171), без учета которых можно сомневаться в пригодности используемых молекулярных орбиталей. Кроме того, при расчете спектральных свойств, возможно, следует отдать предпочтение теоретически более обоснованным молекулярным орбиталям, получаемым методом ССП. Опираясь на проведенные до сих пор исследования, к сожалению, нельзя дать на этот вопрос достаточно уверенного ответа. Ясно, что метод ограниченного учета конфигурационного взаимодействия приводит к тем же результатам, что и метод ССП, по крайней мере для основного состояния. Но если ограничиться учетом только однократно возбужденных
-атомных орбиталей, см. 116, 171), без учета которых можно сомневаться в пригодности используемых молекулярных орбиталей. Кроме того, при расчете спектральных свойств, возможно, следует отдать предпочтение теоретически более обоснованным молекулярным орбиталям, получаемым методом ССП. Опираясь на проведенные до сих пор исследования, к сожалению, нельзя дать на этот вопрос достаточно уверенного ответа. Ясно, что метод ограниченного учета конфигурационного взаимодействия приводит к тем же результатам, что и метод ССП, по крайней мере для основного состояния. Но если ограничиться учетом только однократно возбужденных
 шагах процедуры не станет меньше, чем некоторая наперед заданная величина [181. По исправленным таким образом функциям молекулярных орбиталей следует строить далее слэтеровские детерминанты и т. д. Второй вариант состоит в использовании исправленных значений резонансных интегралов при расчетах матричных элементов в методе конфигурационного взаимодействия, основанном на обычных хюккелевских орбиталях. Этот вариант, конечно, менее удовлетворителен, чем первый, хотя и было показано, что если взять для резонансных интегралов подходящую зависимость от порядка связей, то удается получить хорошее согласие с экспериментальными данными даже для углеводородов, принадлежащих разным классам [19]. Однако зависимость резонансного интеграла от порядка связи будет получаться очень сильной, если взаимодействие между электронами не учитывается в явном виде. Вероятно, это объясняется тем, что различия во вкладах от кулоновского отталкивания между электронами для различных классов углеводородов не принимаются во внимание. Зависимость от порядка связей, по-видимому, отражает эти последние вклады. В выражениях для энергии возбуждения
шагах процедуры не станет меньше, чем некоторая наперед заданная величина [181. По исправленным таким образом функциям молекулярных орбиталей следует строить далее слэтеровские детерминанты и т. д. Второй вариант состоит в использовании исправленных значений резонансных интегралов при расчетах матричных элементов в методе конфигурационного взаимодействия, основанном на обычных хюккелевских орбиталях. Этот вариант, конечно, менее удовлетворителен, чем первый, хотя и было показано, что если взять для резонансных интегралов подходящую зависимость от порядка связей, то удается получить хорошее согласие с экспериментальными данными даже для углеводородов, принадлежащих разным классам [19]. Однако зависимость резонансного интеграла от порядка связи будет получаться очень сильной, если взаимодействие между электронами не учитывается в явном виде. Вероятно, это объясняется тем, что различия во вкладах от кулоновского отталкивания между электронами для различных классов углеводородов не принимаются во внимание. Зависимость от порядка связей, по-видимому, отражает эти последние вклады. В выражениях для энергии возбуждения  -полосы по обоим методам получаются подобные члены; из-за этого результаты обоих методов и оказываются сходными. Поэтому до некоторой степени можно компенсировать пренебрежение одним эффектом, учитывая увеличенный вклад от другого эффекта. По-видимому, этим и объясняются хорошие результаты, полученные для молекул, для которых было выписано по нескольку структур Кекуле, даже без учета различий в длинах связей, но с учетом в явном виде отталкивания
-полосы по обоим методам получаются подобные члены; из-за этого результаты обоих методов и оказываются сходными. Поэтому до некоторой степени можно компенсировать пренебрежение одним эффектом, учитывая увеличенный вклад от другого эффекта. По-видимому, этим и объясняются хорошие результаты, полученные для молекул, для которых было выписано по нескольку структур Кекуле, даже без учета различий в длинах связей, но с учетом в явном виде отталкивания