Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
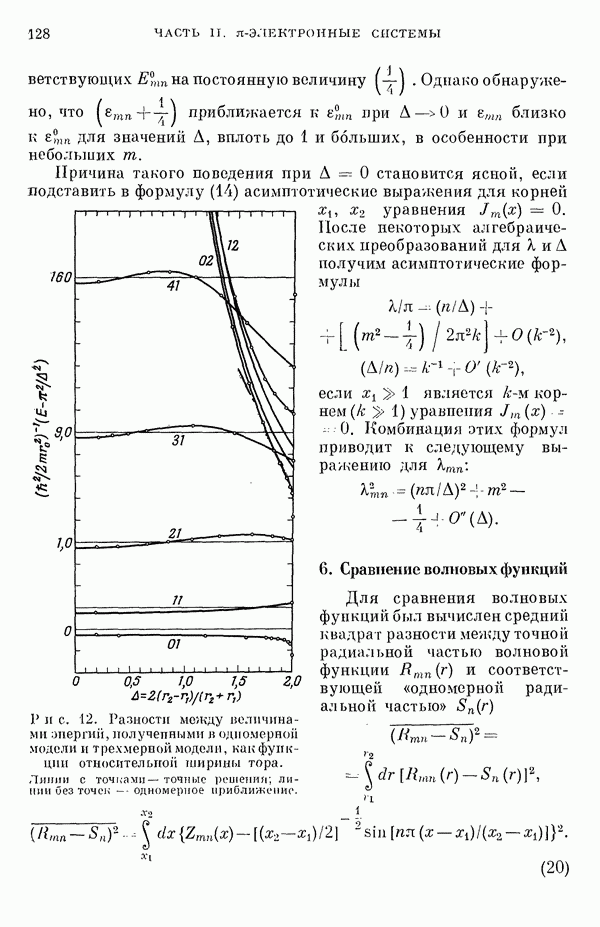
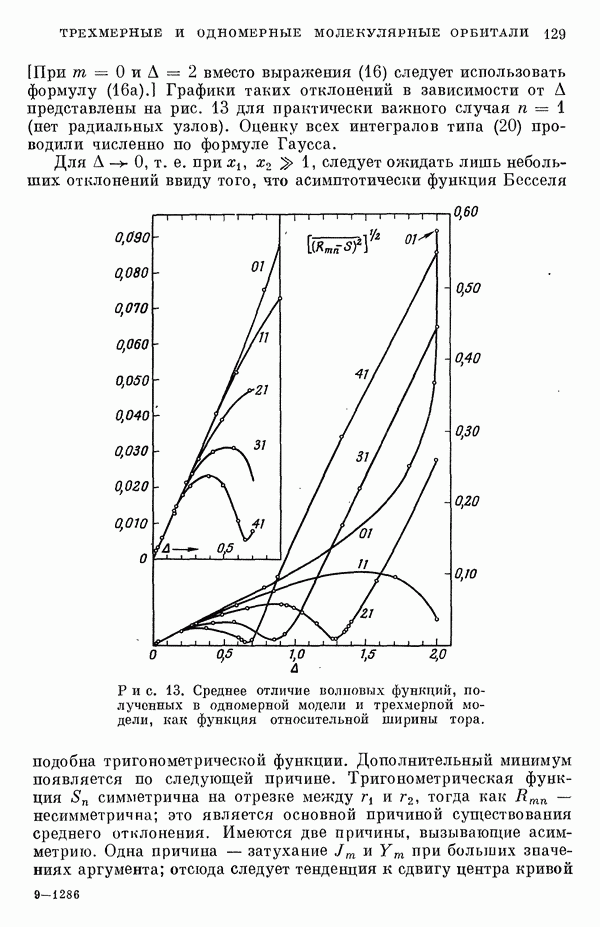
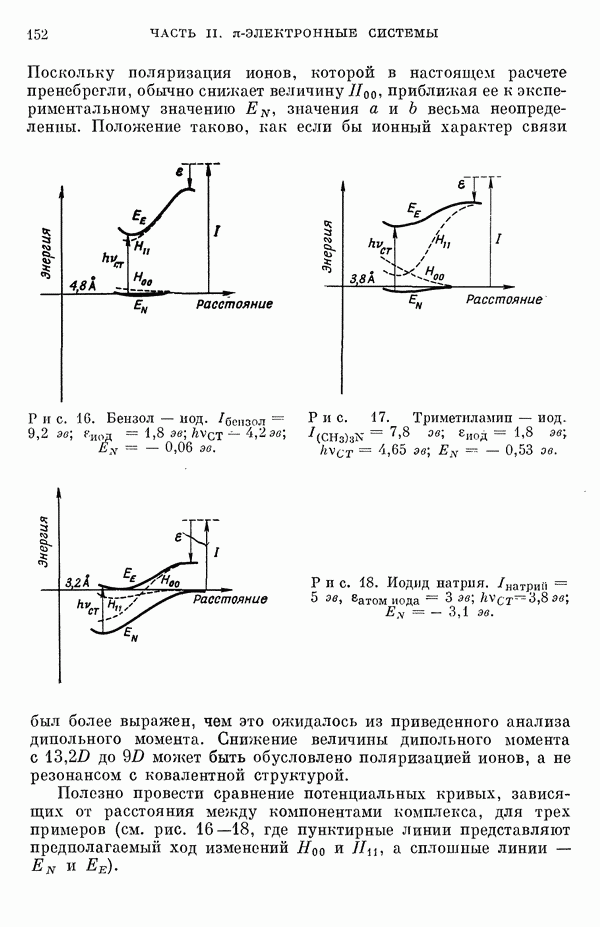
3. Модель молекулярных орбиталейДля описания фторидов ксенона используют метод молекулярных орбиталей (МО) в его обычном виде [7, 9—20]. Связь в соединениях ксенона и фтора описывают в терминах долокализованных молекулярных орбиталей, образованных в основном из орбиталей Полуэмпирическое рассмотрение сводится к простому методу ЛКАО, эквивалентному тому, который применяется для гетероатомных
где
Матричные элементы можно выразить через эффективный одноэлектронный гамильтониан
Тогда полная энергия в одноэлектронном приближении выражается следующим образом:
где
где
Рассмотрим приближения, использованные при выводе формулы (10). В методе Хартри — Фока сумма энергий, взятая но всем орбиталям, не равна полной энергии атомной или молекулярной системы, потому что должным образом не учтено кулоновское отталкивание между парами электронов и обменное взаимодействие между электронами с параллельными спинами. Поскольку одноэлектронный эффективный гамильтониан С другой стороны, в выражении для энергии молекулы опущен член, учитывающий взаимное отталкивание ядер. Если Припять во внимание эти эффекты (а обменными эффектами пренебречь), то уравнение (10) примет вид
При выводе формулы (10) предполагается, что два последних лена выражения (11) взаимно уничтожаются [21, 22]. Однако полного сокращения этих членов не происходит даже для простых молекулярных систем [22], и абсолютные величины энергий связи, полученные из выражения (10), обычно завышены в 2—3 раза. Нельзя ожидать, чтобы полуэмпирический метод ЛКАО дал падежвые величины энергий связи, которые не получаются даже при тораздо более тщательных вычислениях по методу самосогласованного поля (ССП). С другой стороны, можно ожидать, что лолуэмпирическое рассмотрение будет чрезвычайно полезным для вычисления разностей между энергиями ядерных или электронных конфигураций. Из собственных векторов секулярной матрицы можно извлечь сведения о распределении заряда в молекуле. При проведении такого расчета нужно учесть неортогональность атомных орбиталей. Условие нормировки для
и интегралы перекрывания можно рассматривать как компоненты метрического тензора в пространстве, заданном коэффициентами молекулярных орбиталей. Рассмотрим совокупность коэффициентов
Тогда заряд
Вернемся теперь к подробному полуэмпирическому рассмотрению фторидов ксенопа. Пренебрежем влиянием
Орбитали
и
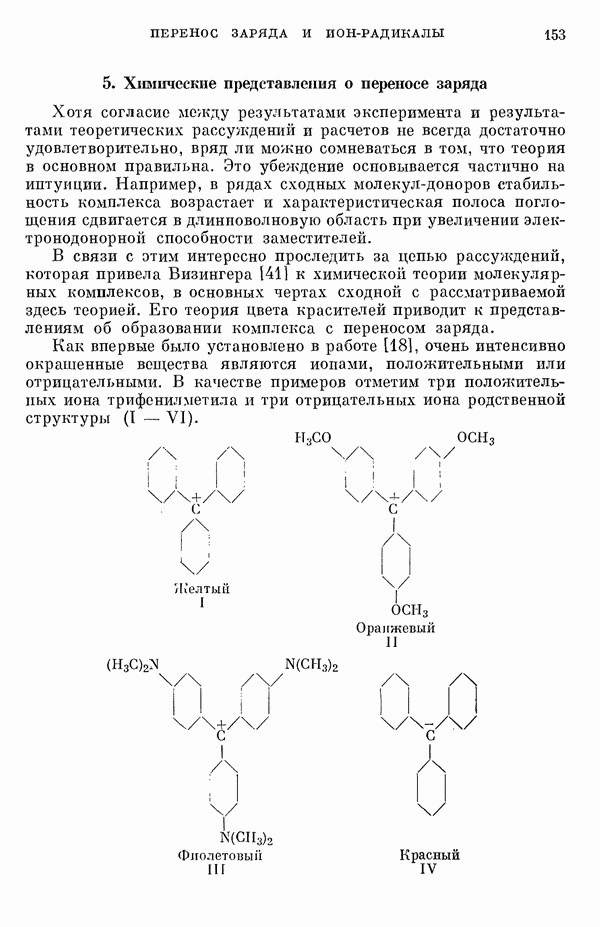
Из секулярного уравнения легко получаем выражения для энергии уровней:
где
и
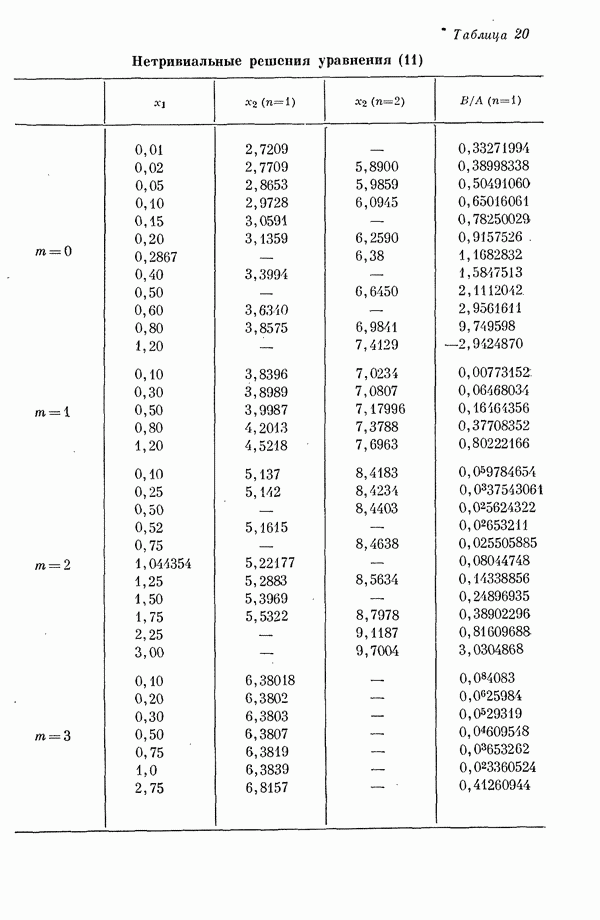
Вначале рассмотрим Величины энергий Для энергетических уровней
Поскольку в основном состоянии молекулы же, как если бы электроны оставались у своих разделенных ядер, что также свидетельствует в пользу
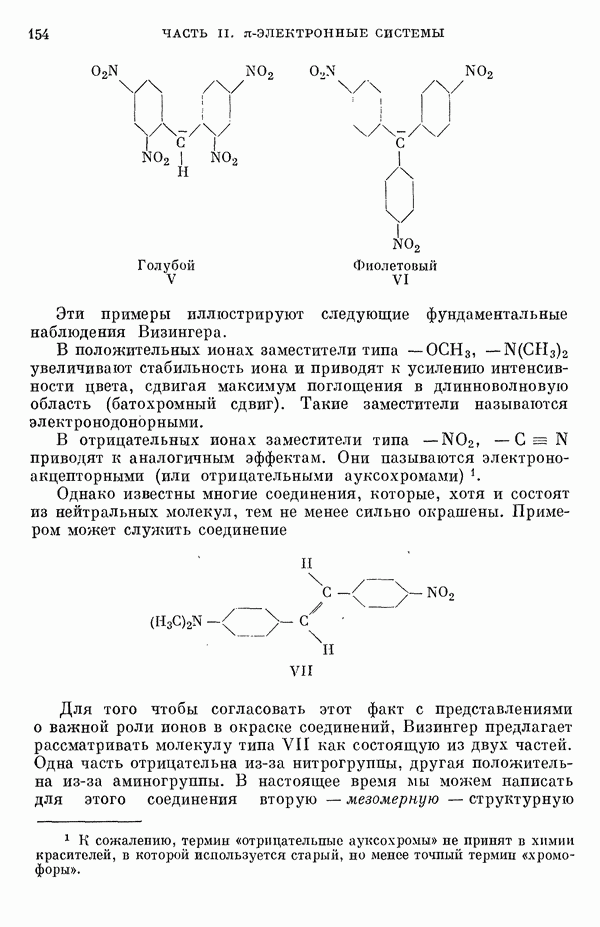
Таким образом, термин «делокализация» не следует понимать буквально. Преобразованные орбитали описывают локализованные Рассмотрим далее структуру плоской квадратной молекулы
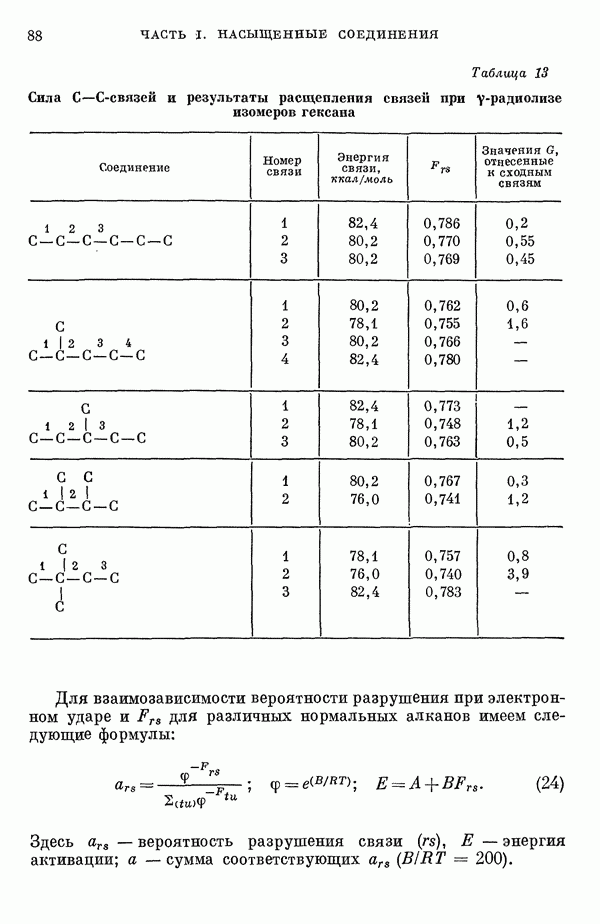
где Другие
Следует отметить, что теперь нельзя пренебрегать взаимодействием смежных атомов фтора (расстояние между которыми составляет 2,82 А). Энергии соответствующих орбиталей равны
Таким образом, взаимодействие между соседними
В основном состоянии Были проведены детальные численные расчеты, основанные на указанной выше полуэмпирической схеме [9, 11, 14—17, 24]. Следует иметь в виду, что численные результаты, полученные таким образом, довольно чувствительны к использованным приближениям. Для того чтобы учесть взаимодействие на расстояниях порядка равновесных межатомных расстояний, для атомов
Кроме того, коэффициенты пропорциональности кулоновских интегралов, определяли следующей формулой:
или
Для постоянной Результаты детальных расчетов показывают, что связывающие взаимодействия включают главным образом Эти расчеты указывают, что имеется существенное смещение отрицательного заряда от ксенона к фтору. Как отмечалось ранее, связывающие и разрыхляющие В рамках метода молекулярных орбиталей влияние перераспределения заряда на величину кулоновских интегралов может быть учтено с помощью итерационной
где метод предлагается для полуэмпирической оценки межэлектронного отталкивания. Применение подобной процедуры [14, 15] к фторидам ксенона показывает, что заряд на атоме
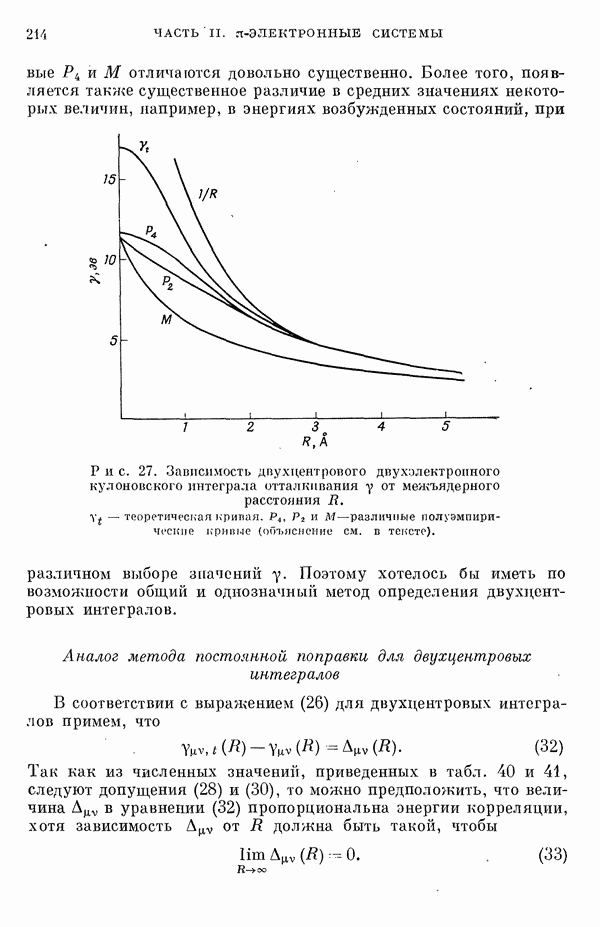
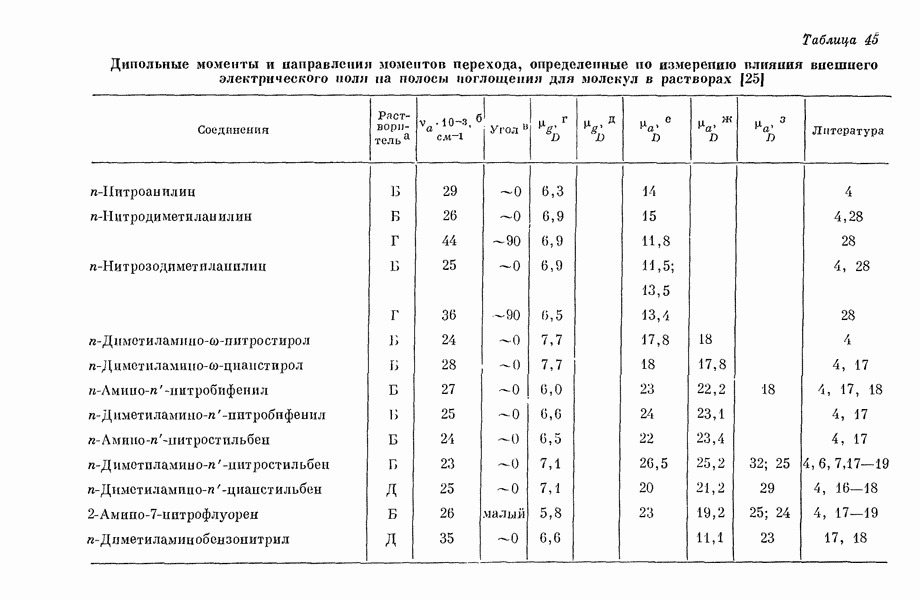
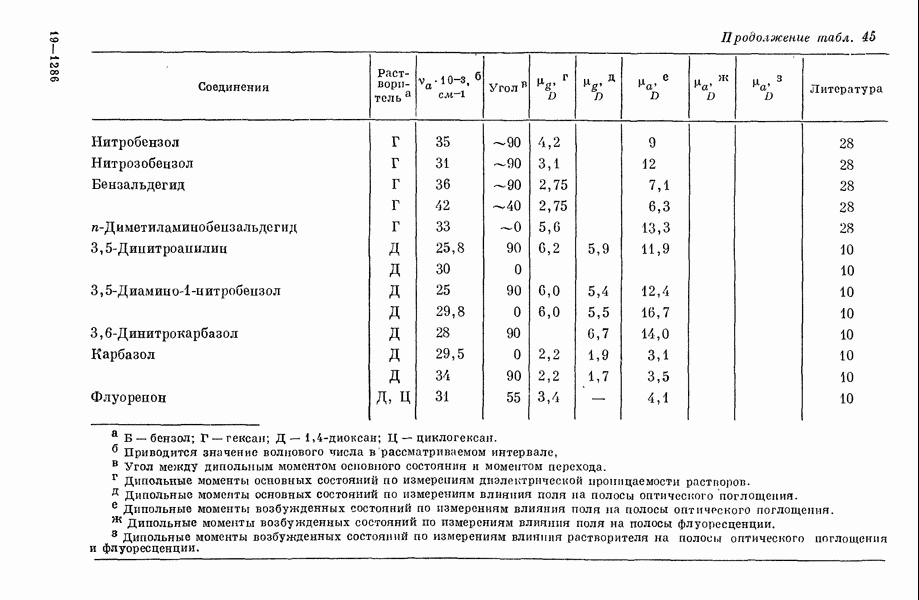
Таблица 1. Рассчитанное распределение заряда в основном состоянии фторидов ксенона
(Избыток отрицательного заряда на атоме фтора.) В табл. 1 представлены результаты теоретических расчетов распределения заряда в фторидах ксенона, находящихся в основном состоянии. Данные, полученные различными авторами, нельзя непосредственно сравнить из-за различий в использованных приближениях. Однако, несмотря на количественные расхождения, разные методы указывают на одну и ту же тендепцию, так что общие закономерности смещения заряда, предсказываемые теорией, очевидно, являются достоверными, несмотря на полуэмнирический характер, присущий такому способу анализа. Сравнение теоретического распределения заряда с экспериментальными данными, обзор которых приведен ниже, показывает, что результаты
|
 |
Оглавление
|























































































































































































































































































 -типа атомов ксенона и фтора. Таким образом, в
-типа атомов ксенона и фтора. Таким образом, в  учитывают одну заполненную атомную
учитывают одну заполненную атомную  -орбиталь
-орбиталь  и две атомные
и две атомные  -орбитали
-орбитали  каждая из которых занята одпим электроном. Аналогичное рассмотрение применимо к
каждая из которых занята одпим электроном. Аналогичное рассмотрение применимо к  для чего надо начинать с четырех
для чего надо начинать с четырех  -орбиталей
-орбиталей  и двух
и двух  -орбиталей Хе. В нулевом приближении
-орбиталей Хе. В нулевом приближении  и
и  -орбитали
-орбитали  и
и  и
и  -орбитали
-орбитали  можно рассматривать как несвязывающие. В более тщательных вычислениях учитывают влияние
можно рассматривать как несвязывающие. В более тщательных вычислениях учитывают влияние  -связи и используют в методе
-связи и используют в методе  -орбитали
-орбитали  и
и  -орбитали
-орбитали 
 -электронных систем с тем исключением, что используются орбитали различной симметрии. В обычном методе ЛКАО молекулярные орбитали
-электронных систем с тем исключением, что используются орбитали различной симметрии. В обычном методе ЛКАО молекулярные орбитали  записывают следующим образом:
записывают следующим образом:

 — атомные орбитали. Секулярные уравнения имеют вид
— атомные орбитали. Секулярные уравнения имеют вид

 (который не совсем точно определен в этом методе), так что имеем
(который не совсем точно определен в этом методе), так что имеем


 — число заполнения
— число заполнения  молекулярной орбитали. Энергия атомов при разведении их на бесконечность определяется выражением
молекулярной орбитали. Энергия атомов при разведении их на бесконечность определяется выражением

 — число заполнения
— число заполнения  атомной орбитали для атомов, находящихся в основном состоянии. Тогда энергия, приходящаяся на одну связь (считаем, что в молекулярной системе имеется
атомной орбитали для атомов, находящихся в основном состоянии. Тогда энергия, приходящаяся на одну связь (считаем, что в молекулярной системе имеется  связей), аппроксимируется следующей формулой [14, 16, 17]:
связей), аппроксимируется следующей формулой [14, 16, 17]:

 представляет гамильтониан самосогласованного поля, формула (8) дважды включает энергию кулоповского межэлектронного отталкивания
представляет гамильтониан самосогласованного поля, формула (8) дважды включает энергию кулоповского межэлектронного отталкивания 

 имеет вид
имеет вид

 определяемых
определяемых

 на атоме
на атоме  можно представить в виде
можно представить в виде

 -орбиталей
-орбиталей  -орбиталей
-орбиталей  Используя линейную комбинацию трех атомных орбиталей и выбрав те же фазы, что и в работах [23, 14], можно построить три молекулярные орбитали, обладающие
Используя линейную комбинацию трех атомных орбиталей и выбрав те же фазы, что и в работах [23, 14], можно построить три молекулярные орбитали, обладающие  -симметрией. Три соответствующие молекулярные орбитали
-симметрией. Три соответствующие молекулярные орбитали  -типа для линейной молекулы
-типа для линейной молекулы  имеют вид
имеют вид

 -типа записываются в виде
-типа записываются в виде





 -орбитали, поскольку они обусловливают связь в этих соединениях.
-орбитали, поскольку они обусловливают связь в этих соединениях.
 -орбиталей образуют следующую последовательность:
-орбиталей образуют следующую последовательность:  так что в основном состоянии
так что в основном состоянии  связывающая
связывающая  и несвязывающая
и несвязывающая  -opбитали каждая заняты двумя электронами, тогда как разрыхляющая орбиталь
-opбитали каждая заняты двумя электронами, тогда как разрыхляющая орбиталь  остается свободной.
остается свободной.
 -орбиталей имеем аналогичные выражения. Два значения энергии орбиталей
-орбиталей имеем аналогичные выражения. Два значения энергии орбиталей  получаются из формулы (18) при подстановке
получаются из формулы (18) при подстановке  соответственно вместо
соответственно вместо  Таким образом, получаем три дважды вырожденные занятые
Таким образом, получаем три дважды вырожденные занятые  -орбитали
-орбитали  Далее, орбитали
Далее, орбитали  очень близки по энергии к атомным орбиталям фтора
очень близки по энергии к атомным орбиталям фтора  потому что в этих случаях перекрывание очень мало. В более высоком приближении, т. е. при учете
потому что в этих случаях перекрывание очень мало. В более высоком приближении, т. е. при учете  и
и  -орбиталей
-орбиталей  энергия как
энергия как  так и
так и  -орбиталей уменьшится, орбиталь
-орбиталей уменьшится, орбиталь  будет, вероятно, лежать ниже, чем орбиталь
будет, вероятно, лежать ниже, чем орбиталь  потому что
потому что  -перекрывание обычно больше, чем
-перекрывание обычно больше, чем  -перекрывание. Кроме того, вследствие большего перекрывания размазанность (т. е. неопределенность в положениии) энергий
-перекрывание. Кроме того, вследствие большего перекрывания размазанность (т. е. неопределенность в положениии) энергий  -орбиталей будет превосходить размазанность энергий
-орбиталей будет превосходить размазанность энергий  -орбиталей. Таким образом, приходим к выводу, что в отсутствие спин-орбитального взаимодействия энергии орбиталей будут располагаться в следующем порядке:
-орбиталей. Таким образом, приходим к выводу, что в отсутствие спин-орбитального взаимодействия энергии орбиталей будут располагаться в следующем порядке:

 -орбитали полностью заняты, распределение
-орбитали полностью заняты, распределение  -электронов является таким
-электронов является таким
 -связи. В работе [20] было принято, что
-связи. В работе [20] было принято, что  -орбитали
-орбитали  образуют трехцентровую четырехэлектронную связь. Однако такое качественное описание совершенно ненадежно. Поэтому интересно другое представление волновой функции основного состояния
образуют трехцентровую четырехэлектронную связь. Однако такое качественное описание совершенно ненадежно. Поэтому интересно другое представление волновой функции основного состояния  Легко показать, что волновая функция основного состояния системы, представленная с помощью одного слэтеровского детерминанта, сводится к виду, характерному для двух локализованных
Легко показать, что волновая функция основного состояния системы, представленная с помощью одного слэтеровского детерминанта, сводится к виду, характерному для двух локализованных  связей. Поэтому мы можем рассматривать только орбитали с одной спиновой функцией. Поскольку волновая функция, представленная в виде детерминанта, инвариантна по отношению к унитарному преобразованию, очевидно, что
связей. Поэтому мы можем рассматривать только орбитали с одной спиновой функцией. Поскольку волновая функция, представленная в виде детерминанта, инвариантна по отношению к унитарному преобразованию, очевидно, что

 связи, но эти новые орбитали не ортогональны, и они малоэффективны при оценке распределения заряда.
связи, но эти новые орбитали не ортогональны, и они малоэффективны при оценке распределения заряда.
 (группа симметрии
(группа симметрии  ). Из орбиталей соответствующей симметрии легко построить молекулярные орбитали. Следует отметить, что дважды вырожденная
). Из орбиталей соответствующей симметрии легко построить молекулярные орбитали. Следует отметить, что дважды вырожденная  -орбиталь содержит вклад
-орбиталь содержит вклад  -орбиталей фтора. Запишем эту молекулярную орбиталь в следующем виде:
-орбиталей фтора. Запишем эту молекулярную орбиталь в следующем виде:

 — коэффициенты молекулярных орбиталей. Если пренебречь взаимодействием между парами атомов фтора, находящихся на расстоянии 3,9 А, то получим три уровня энергии
— коэффициенты молекулярных орбиталей. Если пренебречь взаимодействием между парами атомов фтора, находящихся на расстоянии 3,9 А, то получим три уровня энергии  -типа (т. е. два дважды вырожденных
-типа (т. е. два дважды вырожденных  -уровня и один дважды вырожденный
-уровня и один дважды вырожденный  -уровень).
-уровень).
 -орбитали в
-орбитали в  можно представить в виде
можно представить в виде


 -орбита-лями
-орбита-лями  которые перпендикулярны друг к другу, приводит к расщеплению энергий орбиталей. Разность энергий определяется выражением
которые перпендикулярны друг к другу, приводит к расщеплению энергий орбиталей. Разность энергий определяется выражением

 орбиталь
орбиталь  не занята, и основное состояние молекулы описывается как
не занята, и основное состояние молекулы описывается как 
 и
и  использовали атомные орбитали слэтеровского типа. Параметры орбиталей (т. е. величины а в
использовали атомные орбитали слэтеровского типа. Параметры орбиталей (т. е. величины а в  ) подбирали либо с помощью правил Слэтера, либо путем использования экспериментальных данных по диамагнитной восприимчивости атомов. Для расчета взаимодействия смежных атомов
) подбирали либо с помощью правил Слэтера, либо путем использования экспериментальных данных по диамагнитной восприимчивости атомов. Для расчета взаимодействия смежных атомов  в
в  нельзя использовать орбитали слэтеровского типа, потому что здесь существенны большие расстояния (2,8 А); соответственно были использованы атомные волновые ССП-функции. Кулоновские интегралы были приняты равными потенциалам ионизации атомов (или полуэмпирическим потенциалам ионизации валентных состояний), а обменные интегралы — пропорциональпыми интегралам перекрывания:
нельзя использовать орбитали слэтеровского типа, потому что здесь существенны большие расстояния (2,8 А); соответственно были использованы атомные волновые ССП-функции. Кулоновские интегралы были приняты равными потенциалам ионизации атомов (или полуэмпирическим потенциалам ионизации валентных состояний), а обменные интегралы — пропорциональпыми интегралам перекрывания:

 принятые равными арифметическому или геометрическому среднему
принятые равными арифметическому или геометрическому среднему


 были использованы различные значения в интервале
были использованы различные значения в интервале  Такая процедура является обычной для полуэмпирического рассмотрения ароматических молекул и неорганических соединений переходных металлов. Такое представление обменных интегралов следует из приближения Малликена для вычисления много центровых интегралов. Таким образом в рамках описанной полуэмпирической схемы расчеты методом МО сводятся к вычислению интегралов перекрывания.
Такая процедура является обычной для полуэмпирического рассмотрения ароматических молекул и неорганических соединений переходных металлов. Такое представление обменных интегралов следует из приближения Малликена для вычисления много центровых интегралов. Таким образом в рамках описанной полуэмпирической схемы расчеты методом МО сводятся к вычислению интегралов перекрывания.
 -орбитали. Было найдено очень небольшое смешивание орбиталей
-орбитали. Было найдено очень небольшое смешивание орбиталей 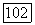 Несколько больший вклад вносит орбиталь
Несколько больший вклад вносит орбиталь 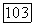 но результаты сильно зависят от выбранных параметров. В терминах классической химии все полученные результаты указывают на то, что гибридизация
но результаты сильно зависят от выбранных параметров. В терминах классической химии все полученные результаты указывают на то, что гибридизация 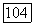 или
или 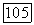 для атома ксенона пренебрёжимо мала; степень
для атома ксенона пренебрёжимо мала; степень 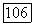 -гибридизации для
-гибридизации для 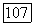 -орбиталей фтора, возможно, несколько большая [17], но она все еще мала по сравнению с обычным смешиванием, в котором различные орбитали участвуют с близким по величине весом.
-орбиталей фтора, возможно, несколько большая [17], но она все еще мала по сравнению с обычным смешиванием, в котором различные орбитали участвуют с близким по величине весом.
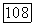 -орбитали заняты, и, следовательно, они не вносят вклада в смещение заряда. Очевидно, что заряд будет перемещаться от
-орбитали заняты, и, следовательно, они не вносят вклада в смещение заряда. Очевидно, что заряд будет перемещаться от 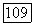 к
к 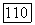 поскольку первые потенциалы ионизации равны 12,12 и 17,42 эв соответственно. Несомненно также, что низкий потенциал ионизации центрального атома и электроотрицательный характер лигандов имеют решающее значение.
поскольку первые потенциалы ионизации равны 12,12 и 17,42 эв соответственно. Несомненно также, что низкий потенциал ионизации центрального атома и электроотрицательный характер лигандов имеют решающее значение.
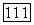 -техники [25]. При этом принимается, что кулоновские интегралы зависят от полного заряда на атоме
-техники [25]. При этом принимается, что кулоновские интегралы зависят от полного заряда на атоме 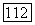

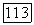 — соответственно кулоновский интеграл и отрицательный заряд на атоме
— соответственно кулоновский интеграл и отрицательный заряд на атоме 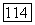 полученные в
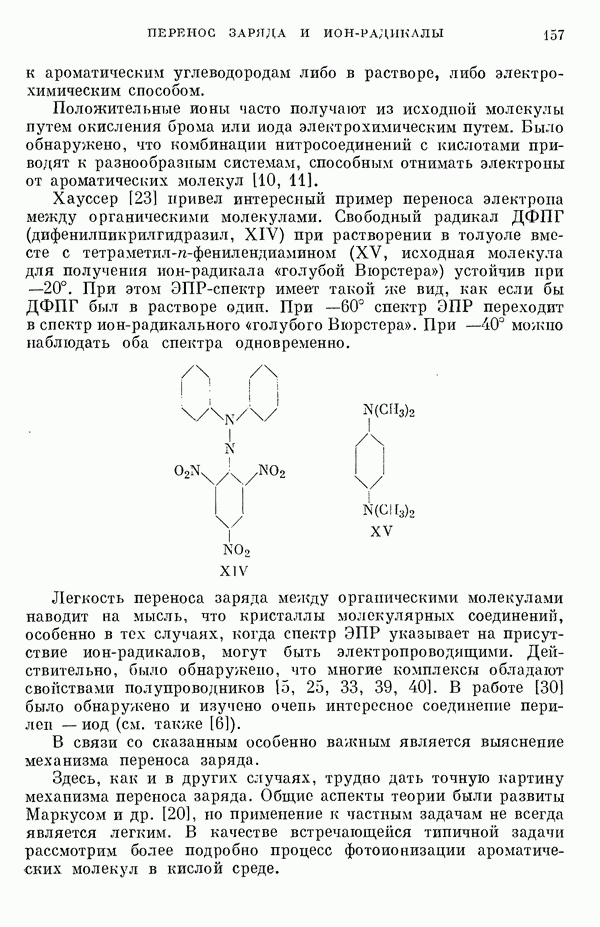
полученные в 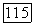 итерации. Этот
итерации. Этот
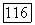 должен уменьшаться в следующей последовательности:
должен уменьшаться в следующей последовательности:


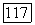 -техники хорошо согласуются с данными исследования ЯМР для рассматриваемых соединений [15, 26].
-техники хорошо согласуются с данными исследования ЯМР для рассматриваемых соединений [15, 26].