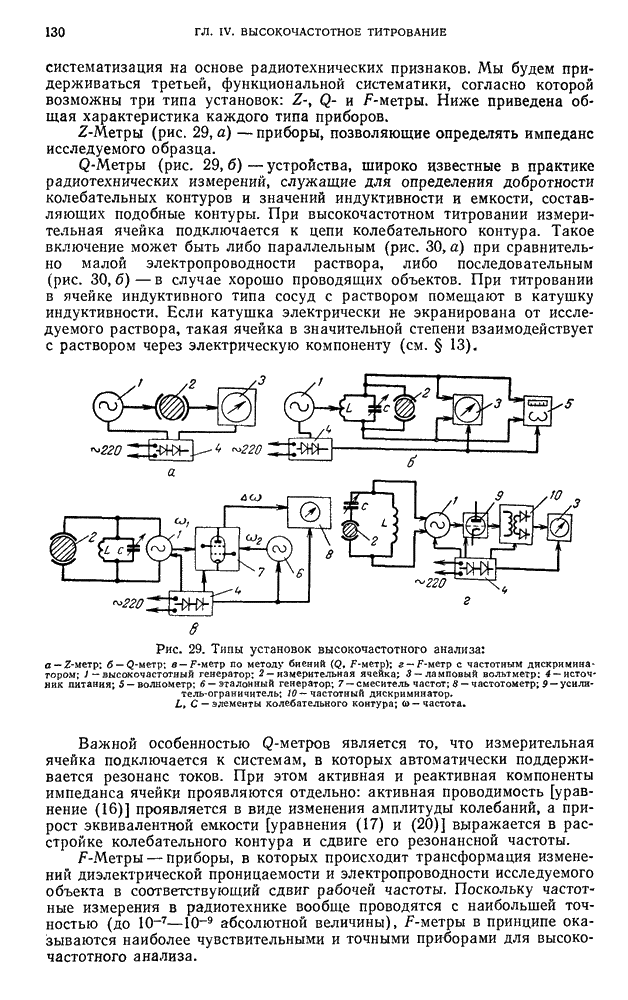
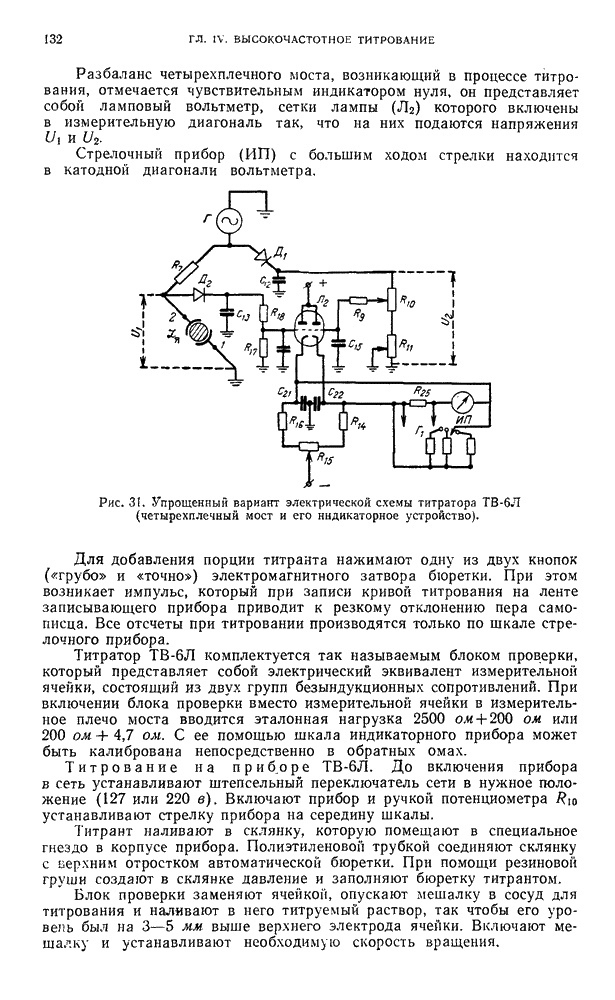
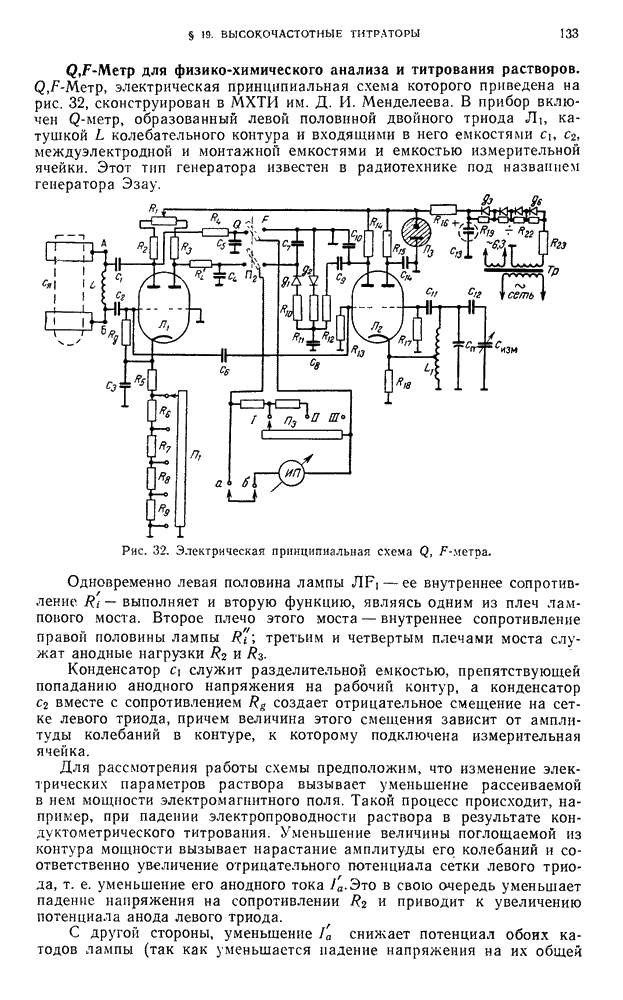
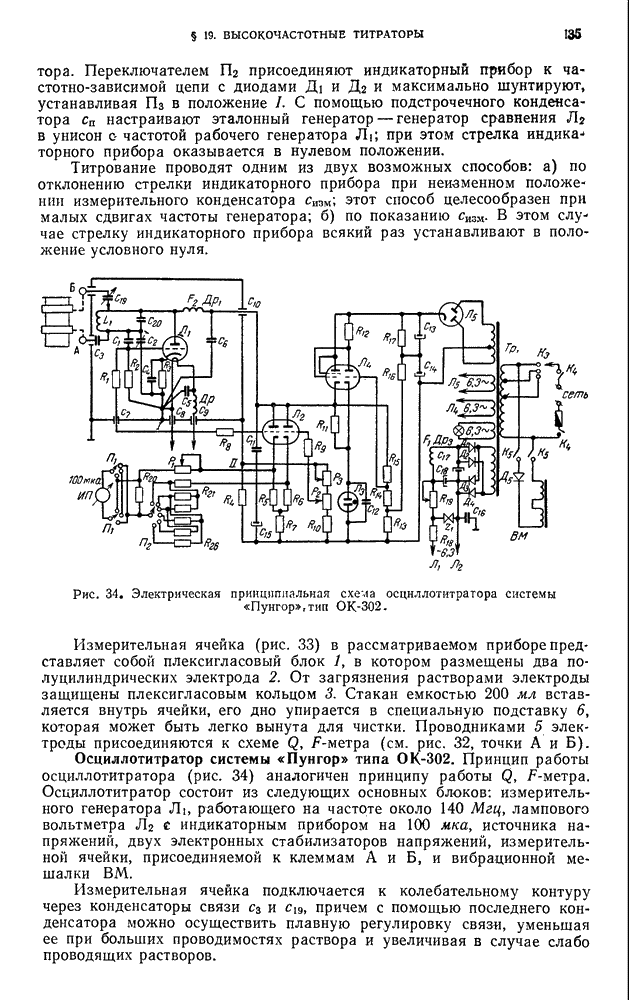
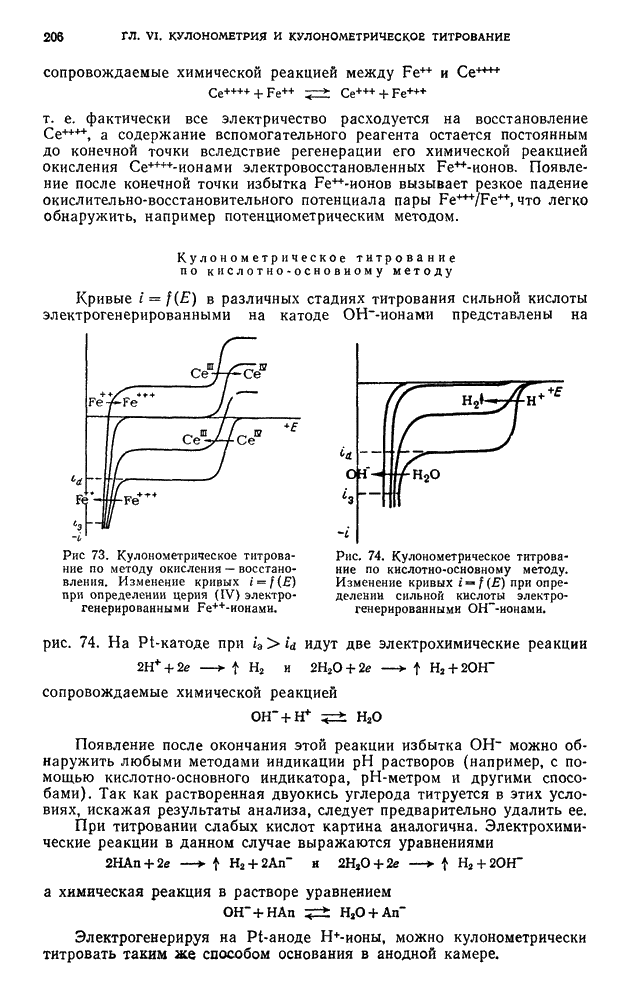
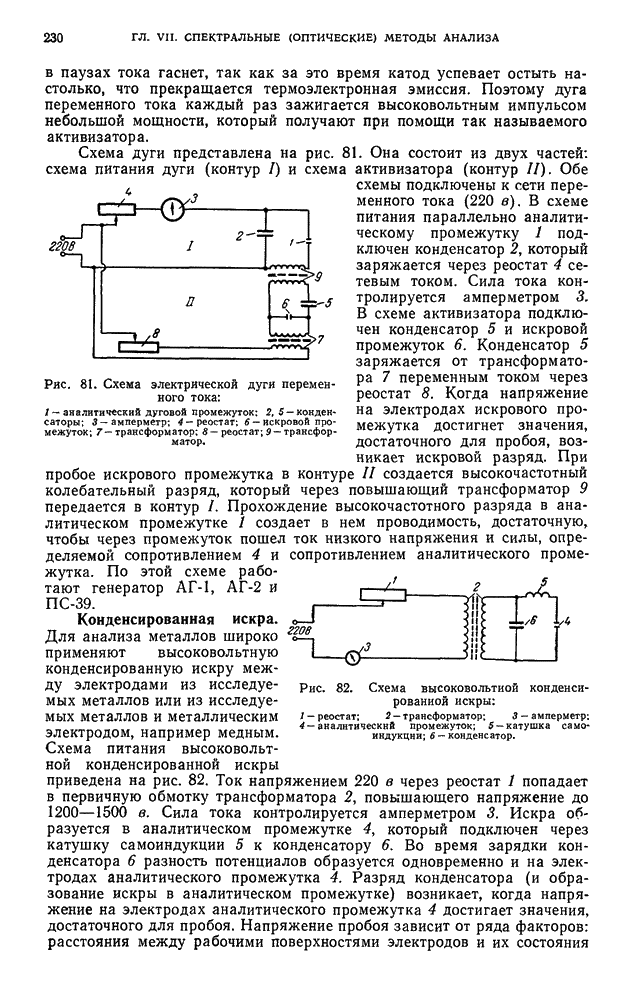
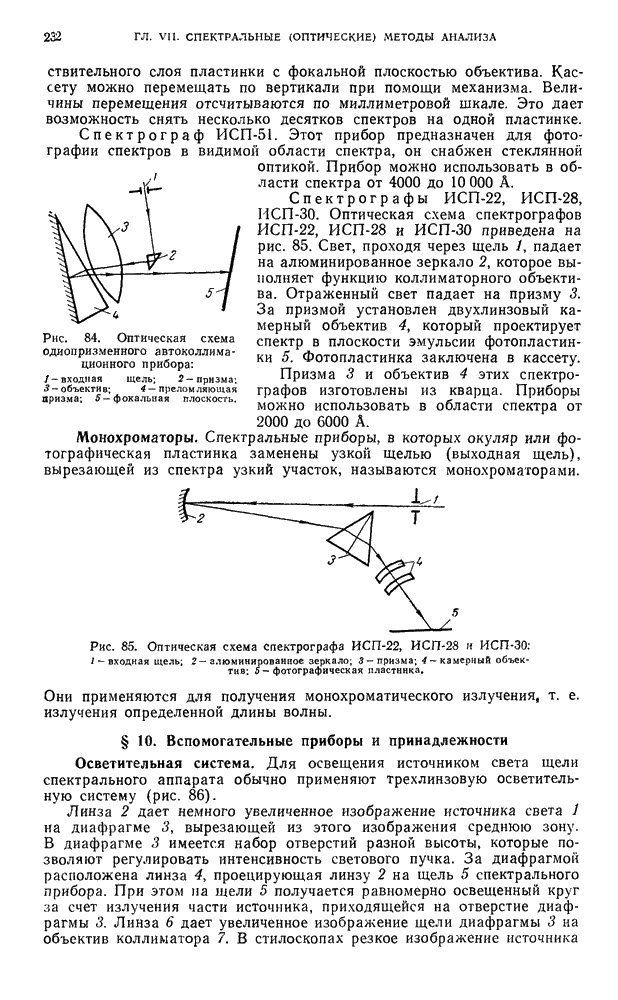
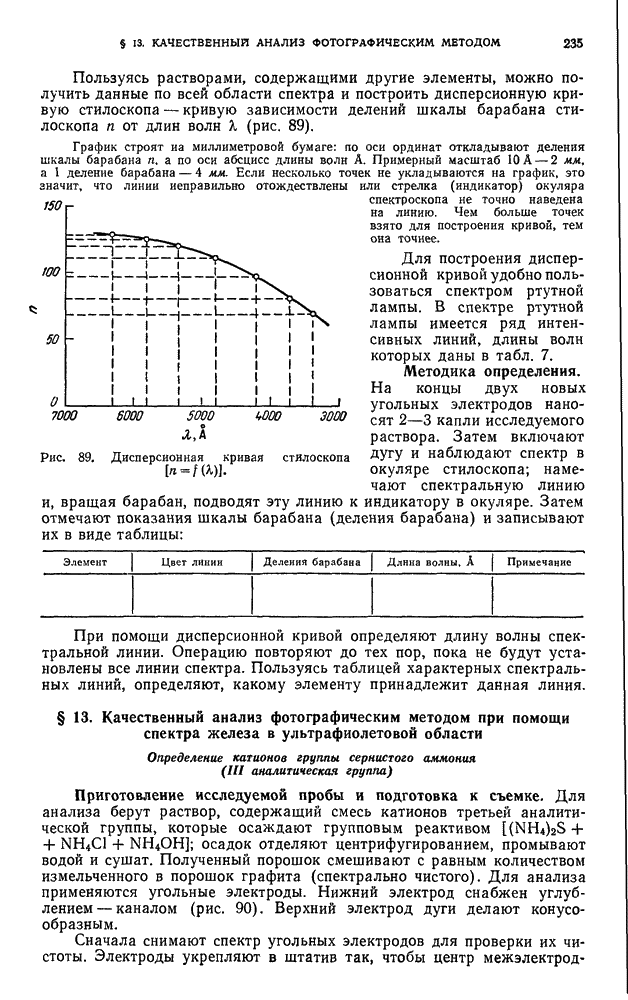
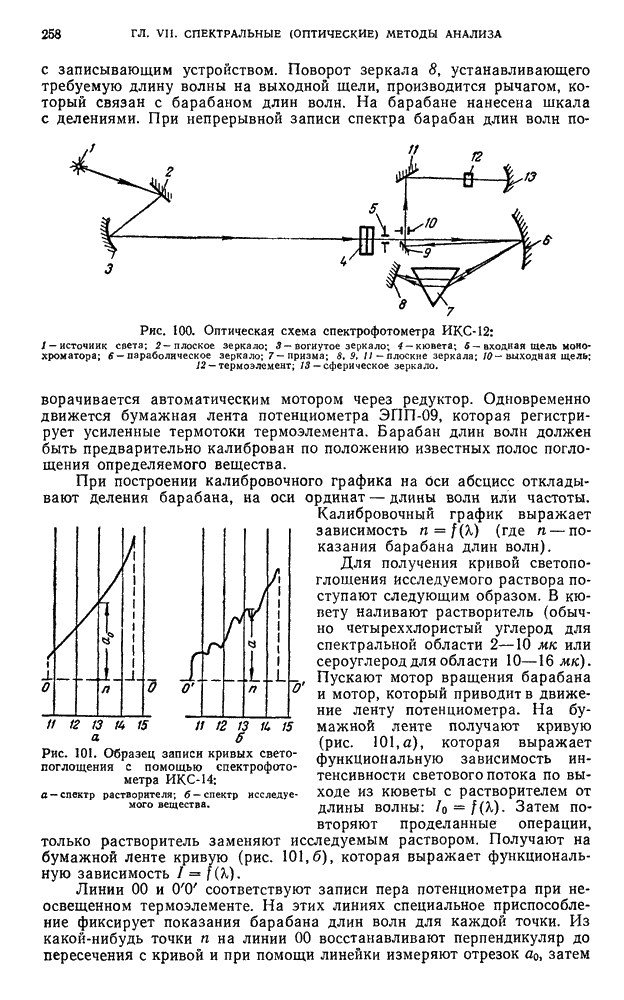
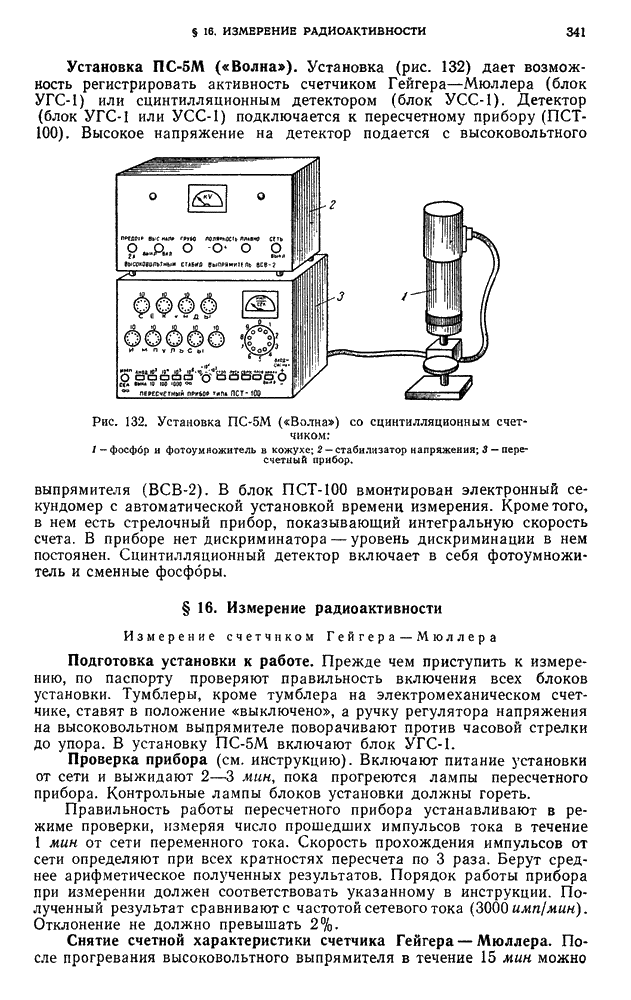
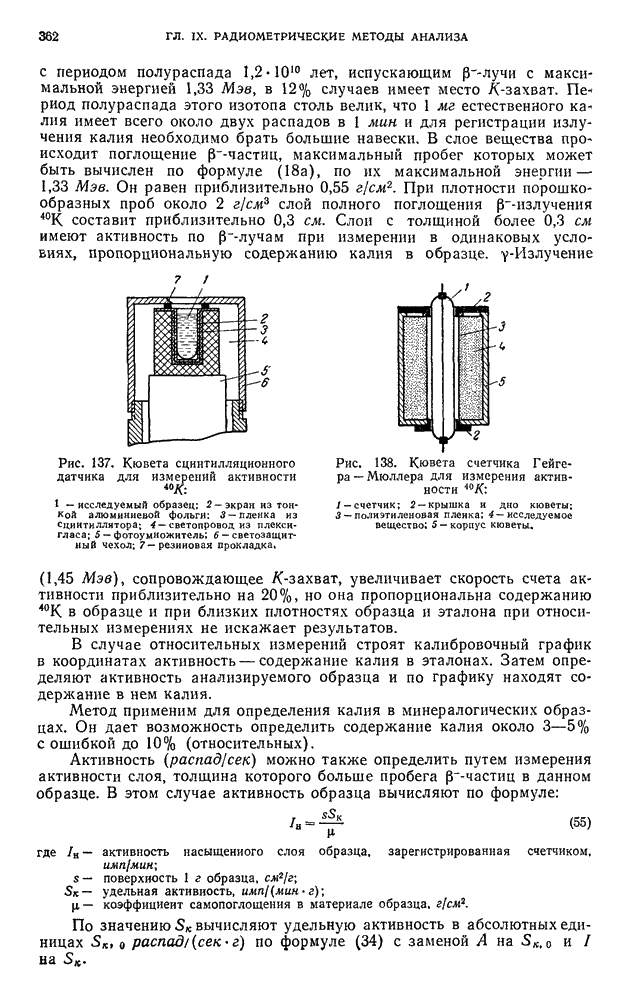
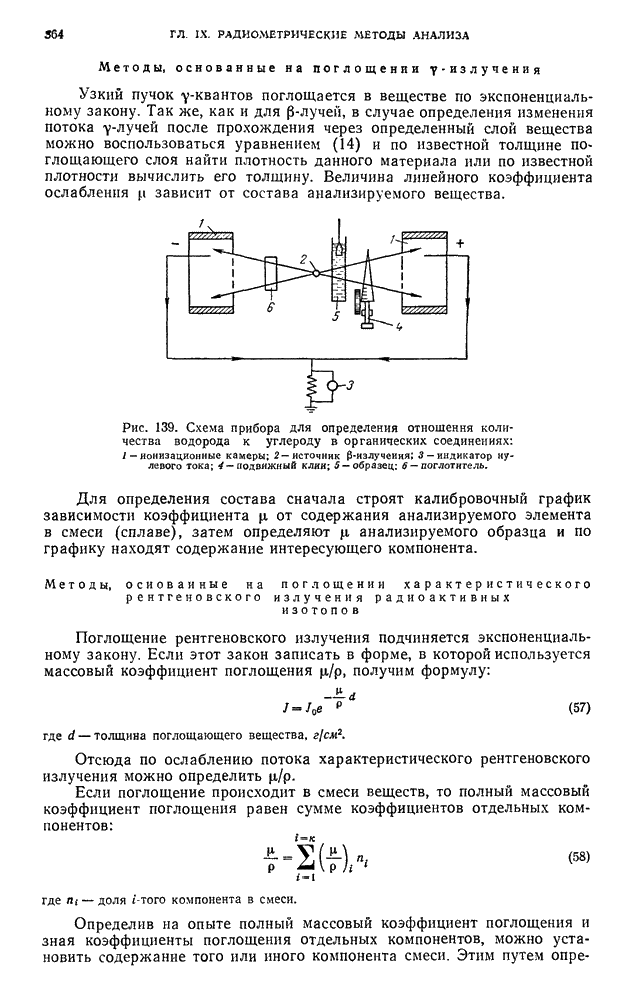
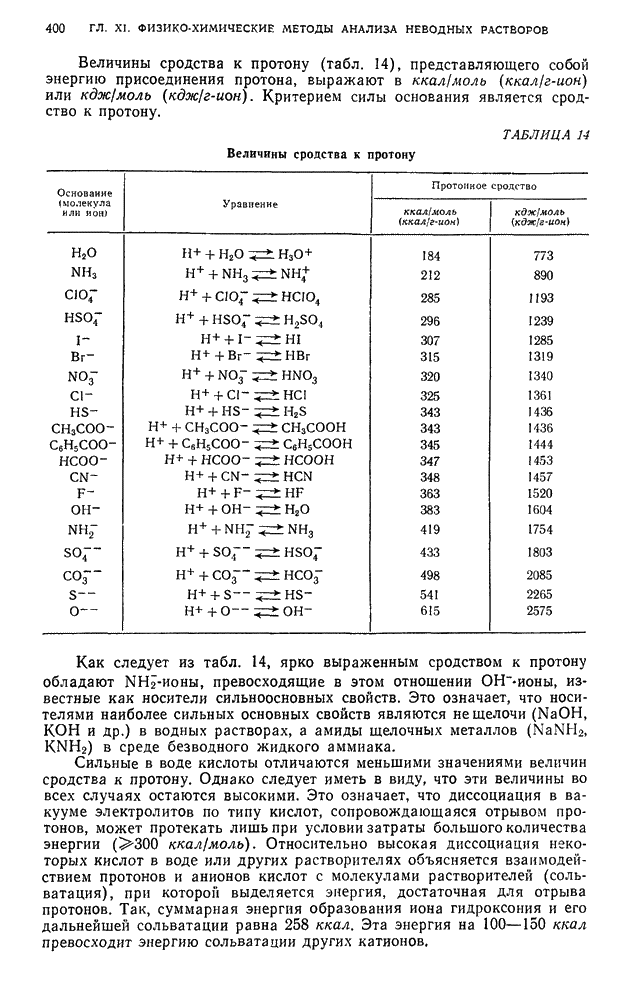
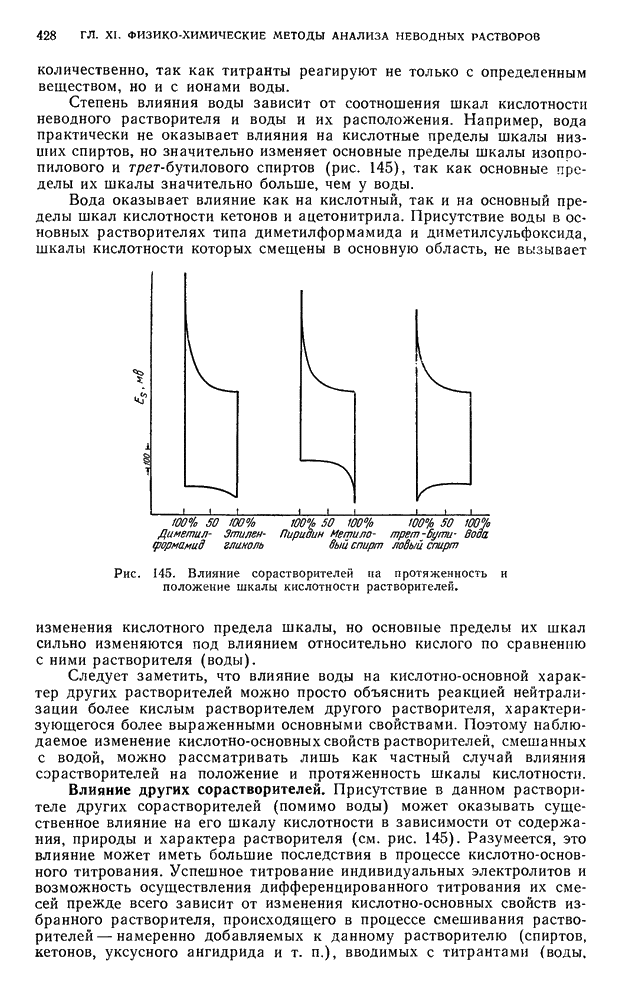
В. ПРАКТИЧЕСКИЕ РАБОТЫ
§ 15. Работы по методу адсорбционной (жидкостной и газовой) хроматографии
Разделение пигментов зеленых листьев растений методом адсорбционно-жидкостной хроматографии
Хлорофилл — зеленый пигмент растений — состоит из смеси нескольких пигментов. При пропускании раствора хлорофилла через колонку происходит сорбция и разделение пигментов в соответствии с их избирательной сорбируемостью.
Хроматографическое разделение пигментов зеленых листьев растений состоит из следующих этапов: 1) экстрагирование пигментов из растительного материала; 2) образование хроматограмм; 3) получение растворов хлорофилла- ,
,  и каротиноидов.
и каротиноидов.
При подготовке раствора для хроматографирования пигменты извлекают из растительного материала полярным растворителем, а затем растворяют в смеси неполярных растворителей. Полученный раствор хроматографируют. Первичную хроматограмму промывают полярным растворителем и в вытекающем растворе собирают отдельные фракции пигментов.
Методика определения. Для анализа берут навеску растительного материала из свежих зеленых (3—5 г) или сухих листьев (1 г). Свежий растительный материал помещают в фарфоровую ступку и тщательно растирают. Для лучшего растирания прибавляют мелкое стекло или кварцевый песок. Если для опыта взят сухой материал, то его измельчают в ступке.
В полученную растертую массу добавляют  смеси бензина и бензола
смеси бензина и бензола  ацетона, снова растирают и перемешивают. Смесь переносят на стеклянный фильтр № 2. Ступку очищают, ополаскивая ее чистым ацетоном. Промывную жидкость также переносят на фильтр № 2. Экстракт пигментов отфильтровывают. Для более полного отделения пигментов оставшуюся на фильтре массу промывают несколькими порциями ацетона (по
ацетона, снова растирают и перемешивают. Смесь переносят на стеклянный фильтр № 2. Ступку очищают, ополаскивая ее чистым ацетоном. Промывную жидкость также переносят на фильтр № 2. Экстракт пигментов отфильтровывают. Для более полного отделения пигментов оставшуюся на фильтре массу промывают несколькими порциями ацетона (по  ) до тех пор, пока вытекающий фильтрат не станет бесцветным, т. е. не будет содержать пигментов.
) до тех пор, пока вытекающий фильтрат не станет бесцветным, т. е. не будет содержать пигментов.
Полученный после фильтрования экстракт пигментов переносят из приемника в делительную воронку, отделяют ацетон путем осторожного введения в воронку  дистиллированной воды. Жидкость в делительной воронке слегка взбалтывают. Следует избегать сильного взбалтывания, так как может образоваться стойкая эмульсия.
дистиллированной воды. Жидкость в делительной воронке слегка взбалтывают. Следует избегать сильного взбалтывания, так как может образоваться стойкая эмульсия.
После того как произойдет расслаивание двух несмешивающихся жидких фаз, нижнюю фазу (вода — ацетон) сливают. В делительную воронку осторожно вводят новую порцию воды ( ). После расслаивания жидкостей водную фазу сливают. Операцию отмывания ацетона повторяют 10 раз. Бензино-бензольный экстракт просушивают после удаления всех водорастворимых веществ. Для этого экстракт переносят в колбу емкостью
). После расслаивания жидкостей водную фазу сливают. Операцию отмывания ацетона повторяют 10 раз. Бензино-бензольный экстракт просушивают после удаления всех водорастворимых веществ. Для этого экстракт переносят в колбу емкостью  , в которую добавляют 2—3 г прокаленного сульфата натрия. Экстракт отфильтровывают через стеклянный или бумажный фильтр.
, в которую добавляют 2—3 г прокаленного сульфата натрия. Экстракт отфильтровывают через стеклянный или бумажный фильтр.
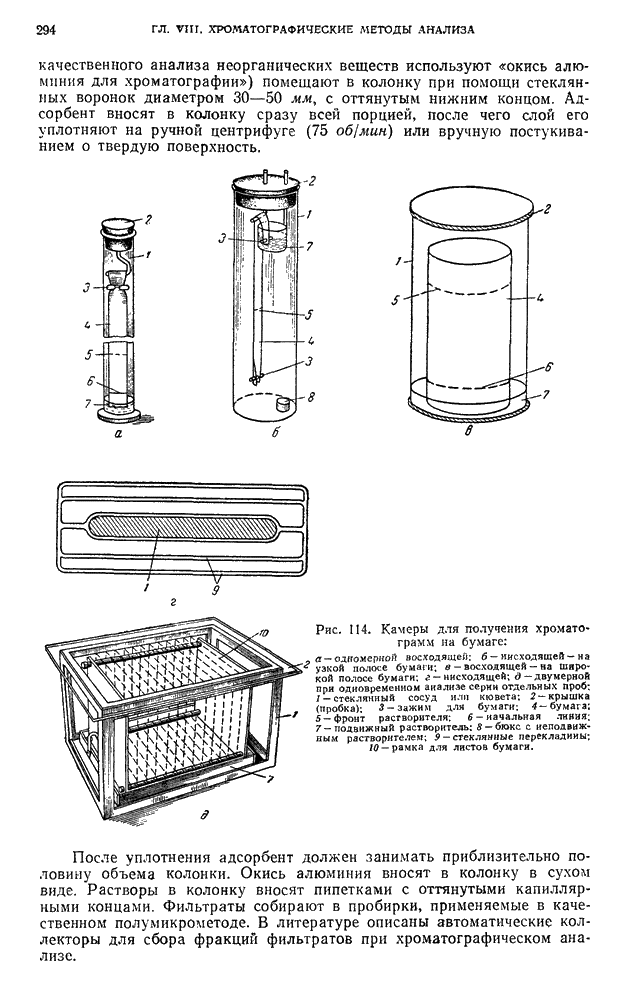
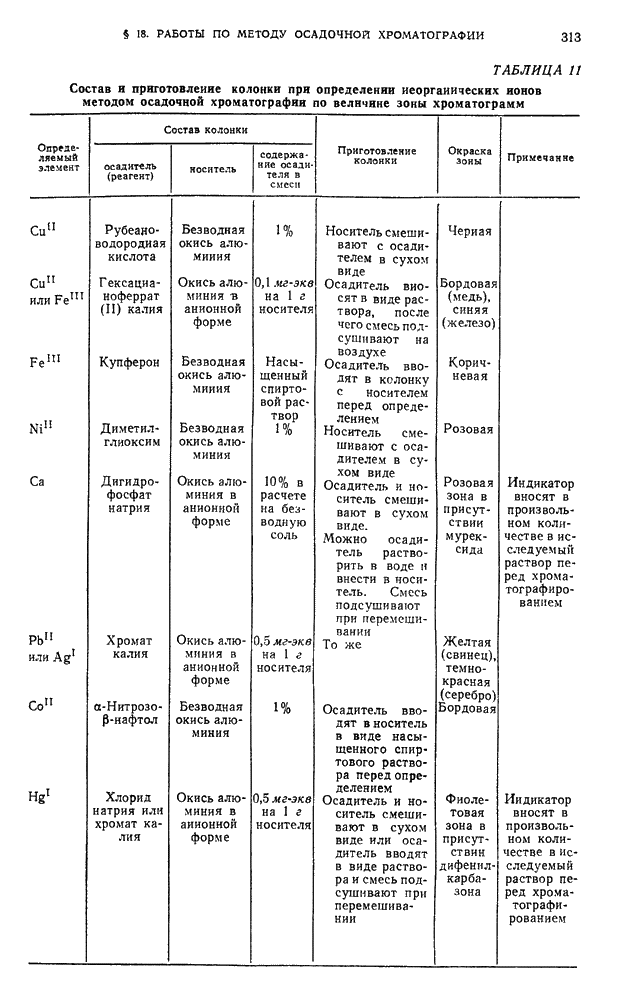
Хроматографирование проводят в стеклянной колонке размером 20 X 1 см (см. рис. 111). Внизу колонки помещают ватный тампон и вносят небольшими порциями сорбент  или сахарный порошок. Уплотнение сорбента проводят постукиванием трубки о твердую поверхность.
или сахарный порошок. Уплотнение сорбента проводят постукиванием трубки о твердую поверхность.
В подготовленную колонку вводят порцию ( ) экстракта. Полученную первичную хроматограмму промывают смесью бензина и бензола
) экстракта. Полученную первичную хроматограмму промывают смесью бензина и бензола  . При промывании колонки происходит разделение зон, вверху колонки образуется зеленая зона
. При промывании колонки происходит разделение зон, вверху колонки образуется зеленая зона  -хлорофилла, затем сине-зеленая
-хлорофилла, затем сине-зеленая  -хлорофилла, несколько ниже располагается зона желтого цвета (каротиноиды).
-хлорофилла, несколько ниже располагается зона желтого цвета (каротиноиды).
При дальнейшем промывании колонки растворителем в фильтрат переводятся сначала желтые пигменты (каротиноиды), затем  -хлорофилл и
-хлорофилл и  -хлорофилл. Фракции собирают в отдельные приемники.
-хлорофилл. Фракции собирают в отдельные приемники.
Анализ многокомпонентной смеси газов методом газо-адсорбционной хроматографии
Разделение многокомпонентной смеси газов, состоящей из кислорода, азота, двуокиси азота, окиси углерода, закиси азота и двуокиси углерода, достигается при десорбции хроматографируемых компонентов смеси путем изменения температуры.
Адсорбируют компоненты смеси газов на силикагеле марки МСМ при низкой температуре. При этой же температуре проводят десорбцию кислорода, азота, двуокиси азота и окиси углерода. Закись азота и двуокись углерода десорбируют при комнатной температуре. Результаты исследований регистрируются самописцем. По хроматограмме рассчитывают содержание каждого компонента газовой смеси в объемных процентах.
Методика определения. Стеклянную колонку  -образной формы, длиной 180 см, с внутренним диаметром 6 мм заполняют предварительно высушенным в токе воздуха при 400° С измельченным силикагелем марки МСМ (фракция
-образной формы, длиной 180 см, с внутренним диаметром 6 мм заполняют предварительно высушенным в токе воздуха при 400° С измельченным силикагелем марки МСМ (фракция  мм). Колонку помещают в криостат с холодильной смесью, состоящей из сухого льда и ацетона. В качестве криостата используют цилиндрический стеклянный сосуд Дьюара. Устанавливают температуру в криостате не выше —70° С, продувают всю систему гелием, подаваемым из баллона, со скоростью
мм). Колонку помещают в криостат с холодильной смесью, состоящей из сухого льда и ацетона. В качестве криостата используют цилиндрический стеклянный сосуд Дьюара. Устанавливают температуру в криостате не выше —70° С, продувают всю систему гелием, подаваемым из баллона, со скоростью  и устанавливают на самописце нулевую линию. Затем вводят сухую анализируемую смесь газов, состоящую из шести компонентов (хранить смесь газов над водой нельзя), и подсоединяют колонку к хроматографу типа
и устанавливают на самописце нулевую линию. Затем вводят сухую анализируемую смесь газов, состоящую из шести компонентов (хранить смесь газов над водой нельзя), и подсоединяют колонку к хроматографу типа  или ГСТЛ-3.
или ГСТЛ-3.
Наблюдают запись результатов анализа на самописце. При температуре ниже —70° С происходит десорбция сначала кислорода, который выходит из колонки через 3 мин, затем азота (через 4 мин), двуокиси аэота (через 10 мин) и окиси углерода (через 13 мин).
После десорбции указанных газов колонку вынимают из криостата, нагревают до комнатной температуры и снова продувают гелием. При этом первым на графике появляется пик, соответствующий закиси азота, и вторым — двуокиси углерода.
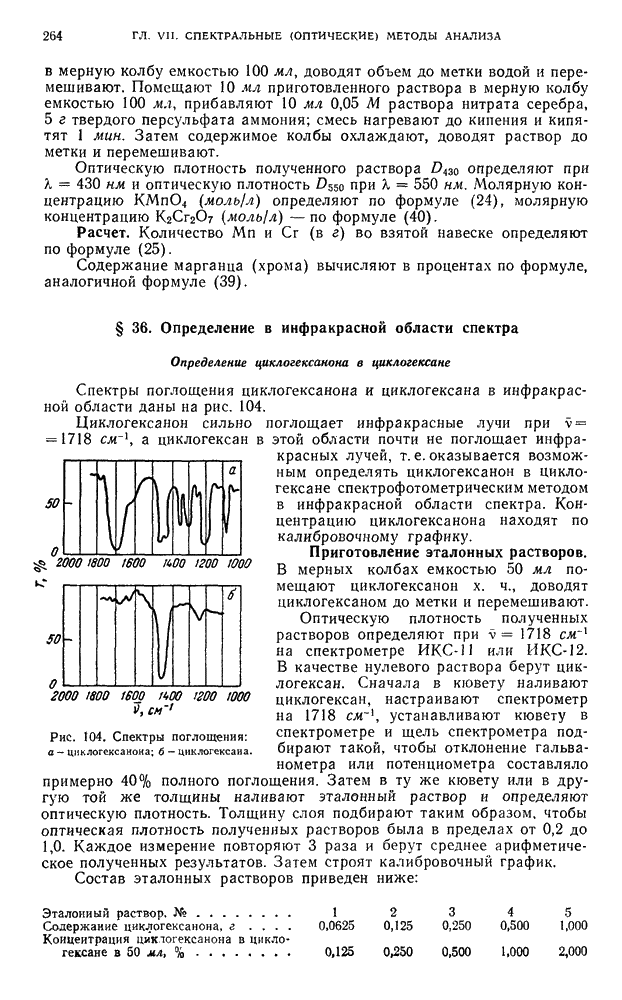
Расчет. Для количественного определения состава анализируемой смеси по хроматограммам измеряют высоту максимумов пиков или площадь пиков.
Концентрация компонента в максимуме пропорциональна высоте или площади пика и количеству вещества, взятого для исследования.
Высоту пика определяют, опуская перпендикуляр из максимума пика на нулевую линию.
Площадь пика определяют с помощью планиметра или вычисляют, умножая высоту (АВ) на ширину (CD) (рис. 116).
Расчет можно проводить по методу «нормировки», при котором сумма высот всех пиков компонентов смеси принимается равной 100%. Высота пика одного из компонентов, выраженная в процентах, соответствует процентному содержанию компонента в смеси.

Рис. 116. Выходная кривая хроматографического разделения.
Анализ смеси газообразных углеводородов C1-C5 методом газо-жидкостной хроматографии
Газообразные углеводороды разделяются на колонке, в которой жидкая фаза нанесена на носитель. При пропускании газа-носителя через колонку, содержащую анализируемую смесь, компоненты ее последовательно вытесняются и выносятся из колонки, после чего газ проходит через детектор; возникающий в детекторе импульс фиксируется на ленте в виде пика.
По высоте или площади пика определяют количественное содержание каждого компонента анализируемой газовой смеси.
Методика определения. В колонку длиной  , диаметром 6 мм вносят носитель-диатомит, измельченный до величины зерна
, диаметром 6 мм вносят носитель-диатомит, измельченный до величины зерна  мм, и уплотняют. Диатомит пропитывают жидким диизоамилфталатом в соотношении 15% к твердой фазе.
мм, и уплотняют. Диатомит пропитывают жидким диизоамилфталатом в соотношении 15% к твердой фазе.
Колонку устанавливают в хроматограф  или
или  , проверяют его герметичность и включают ток газа-носителя, в качестве которого служит водород, подаваемый из баллона (осторожно!). Прибор включают в электрическую сеть и подготавливают его к анализу, т. е. устанавливают температуру термостата колонки
, проверяют его герметичность и включают ток газа-носителя, в качестве которого служит водород, подаваемый из баллона (осторожно!). Прибор включают в электрическую сеть и подготавливают его к анализу, т. е. устанавливают температуру термостата колонки  , давление газа-носителя на входе в колонку (1,5 атм) и скорость прохождения газа-носителя
, давление газа-носителя на входе в колонку (1,5 атм) и скорость прохождения газа-носителя  . После того как на самописце будет вычерчиваться нулевая линия в виде прямой, параллельной оси объема, прибор считают подготовленным к анализу.
. После того как на самописце будет вычерчиваться нулевая линия в виде прямой, параллельной оси объема, прибор считают подготовленным к анализу.
Анализируемую газообразную смесь углеводородов  любого состава в количестве
любого состава в количестве  вводят шприцем в колонку, закрытую самоуплотняющимся резиновым колпачком. Момент впуска смеси отмечается автоматически на нулевой линии самописца. Пропускают через колонку газ-носитель. Разделение смеси углеводородов продолжается 20—25 мин.
вводят шприцем в колонку, закрытую самоуплотняющимся резиновым колпачком. Момент впуска смеси отмечается автоматически на нулевой линии самописца. Пропускают через колонку газ-носитель. Разделение смеси углеводородов продолжается 20—25 мин.
Порядок выхода компонентов при данных условиях опыта будет следующий: метан, этан, этилен, пропан, пропилен, изобутан,  -бутан, изобутилен вместе с бутиленом-1, транс-бутен-2, цис-бутен-2, изопентан,
-бутан, изобутилен вместе с бутиленом-1, транс-бутен-2, цис-бутен-2, изопентан,  -метилбутен-1,
-метилбутен-1,  -пентан, пентен-1,
-пентан, пентен-1,  .
.
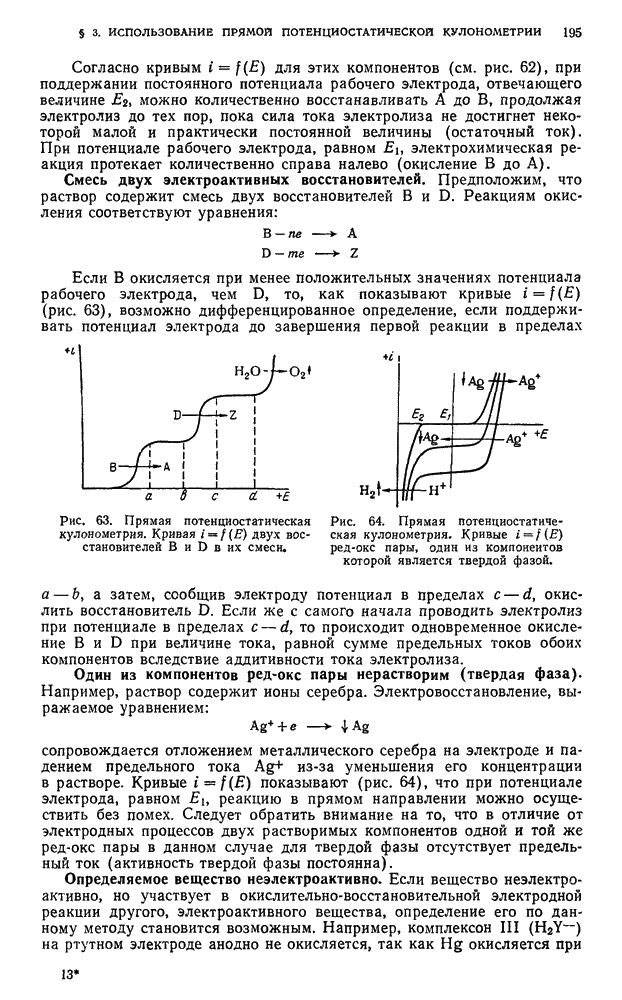
По окончании опыта отключают ток газа-носителя, выключают прибор из электросети, вынимают ленту из самописца, расшифровывают хроматограмму и определяют количественный состав смеси.