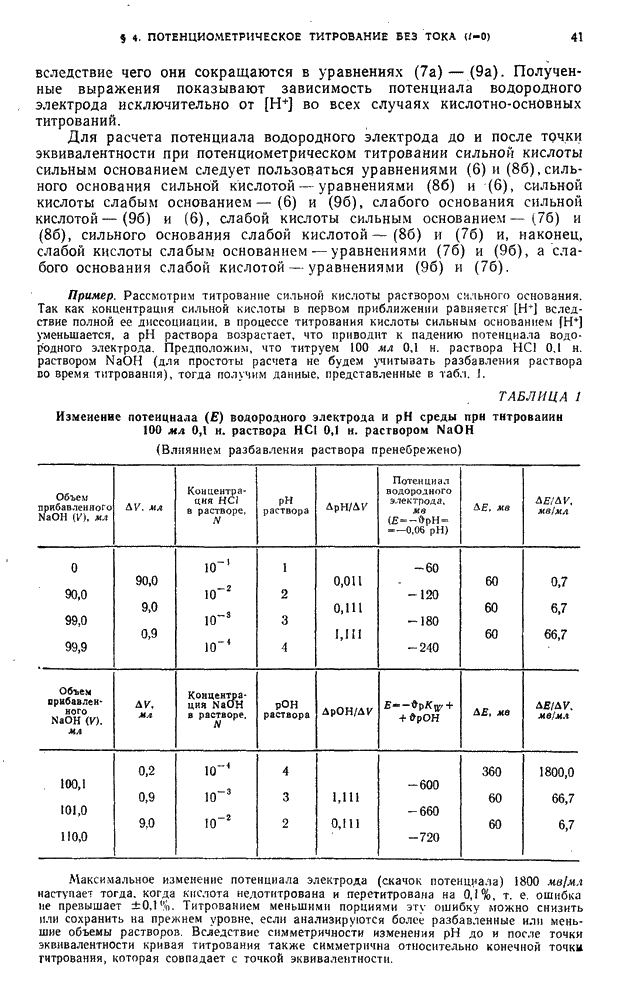
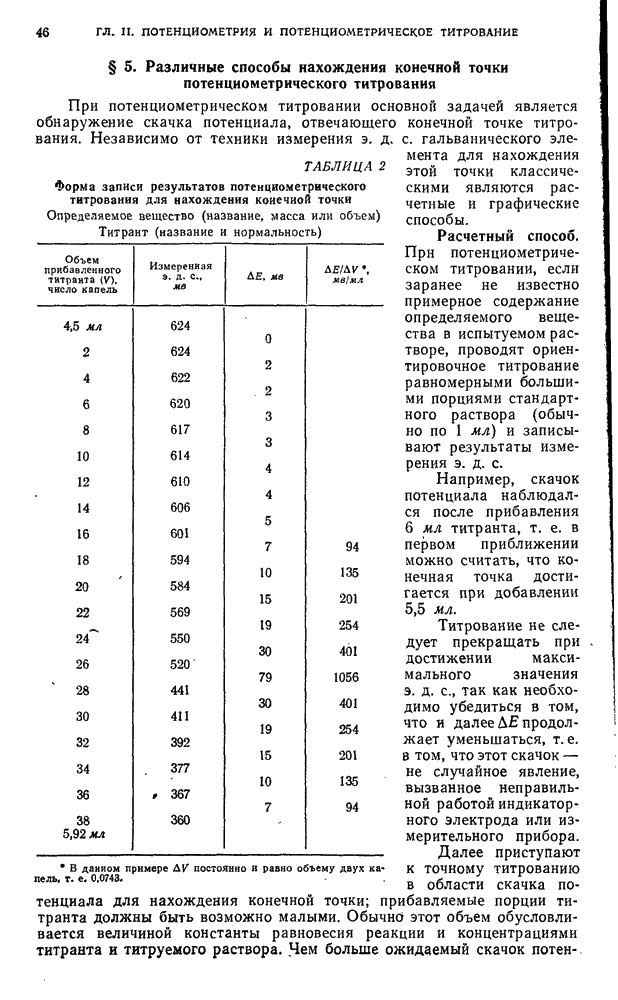
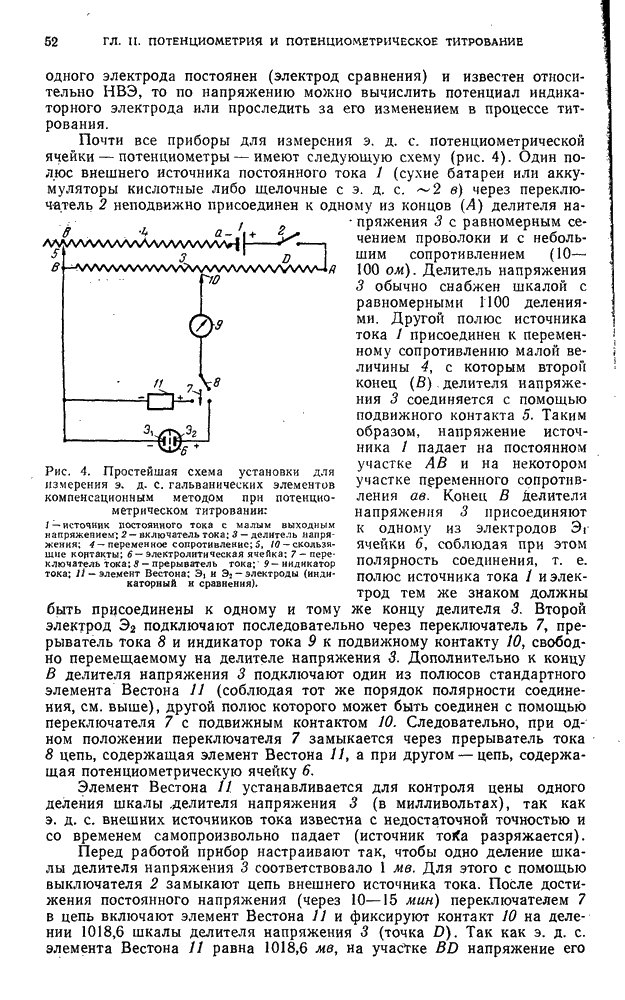
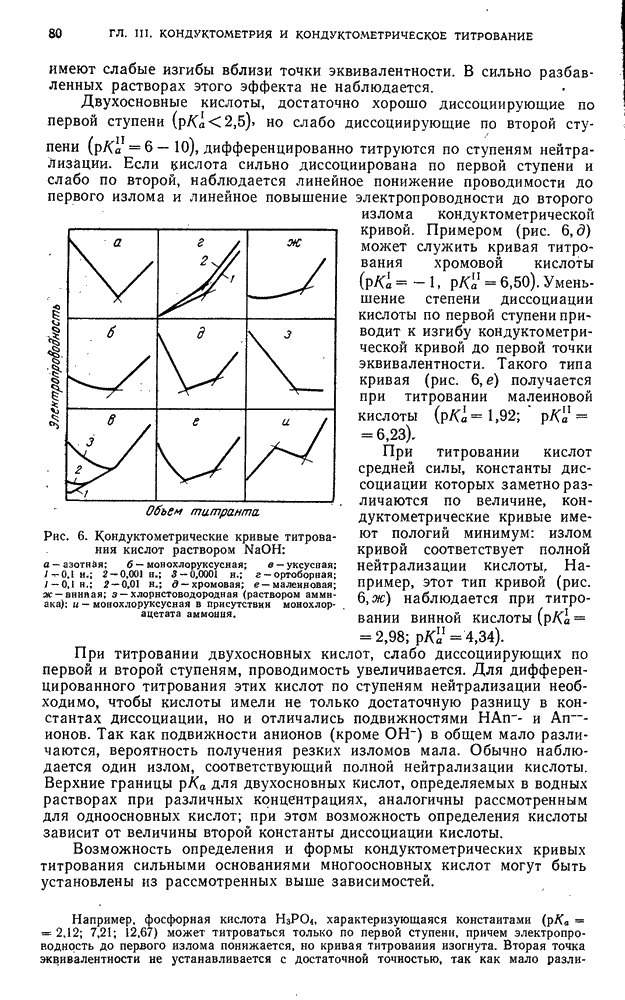
§ 4. Работы, выполняемые экстракционно-фотометрическим методом
Определение микроколичеств тория
Для определения очень малых количеств тория окрашенное соединение тория с арсеназо III экстрагируют изоамиловым спиртом в присутствии хлорида дифенилгуанидиния и монохлоруксусной кислоты. Метод сочетает собственно определение тория с его экстракционным концентрированием и дает возможность определять торий при разбавлении  .
.
Калибровочный график. Для построения калибровочного графика в делительные воронки емкостью  вводят
вводят  стандартного раствора
стандартного раствора  с содержанием
с содержанием  , по
, по  -ного раствора арсеназо III,
-ного раствора арсеназо III,  н. раствора азотной кислоты, по 1 г монохлоруксусной кислоты, по
н. раствора азотной кислоты, по 1 г монохлоруксусной кислоты, по  -ного раствора хлорида дифенилгуанидиния и по
-ного раствора хлорида дифенилгуанидиния и по  изоамилового спирта. Смесь встряхивают 2 мин, дают разделиться фазам, органический слой фильтруют через бумажный фильтр в сухую кювету и, измеряют оптическую плотность по отношению к раствору сравнения, полученному аналогично и не содержащему торий.
изоамилового спирта. Смесь встряхивают 2 мин, дают разделиться фазам, органический слой фильтруют через бумажный фильтр в сухую кювету и, измеряют оптическую плотность по отношению к раствору сравнения, полученному аналогично и не содержащему торий.
Методика определения. Анализируемый раствор проводят через все описанные выше операции. Содержание тория находят по калибровочному графику.
Определение урана
Арсеназо III образует с уранил-ионом комплексное соединение зеленого цвета с максимумом светопоглощения 655 нм. Чувствительность определения  урана, коэффициент молярного поглощения
урана, коэффициент молярного поглощения  равен 75 500. Оптимальная область
равен 75 500. Оптимальная область  . Определению не мешают сульфаты, фториды, оксалаты, фосфаты. Из катионов мешают только торий, цирконий, алюминий, хром (III) и редкоземельные элементы, однако их можно замаскировать введением подходящих веществ (сульфосалициловая кислота в 0,05 н. хлористоводородной кислоте для алюминия, щавелевая кислота для циркония и гафния и т. д.).
. Определению не мешают сульфаты, фториды, оксалаты, фосфаты. Из катионов мешают только торий, цирконий, алюминий, хром (III) и редкоземельные элементы, однако их можно замаскировать введением подходящих веществ (сульфосалициловая кислота в 0,05 н. хлористоводородной кислоте для алюминия, щавелевая кислота для циркония и гафния и т. д.).
Восстанавливая уран (VI) до урана (IV), можно резко повысить селективность определения, проводя его в 5—6 н. хлористоводородной кислоте в присутствии щавелевой кислоты. В качестве восстановителя лучше всего применять гранулированный цинк в присутствии аскорбиновой кислоты, предотвращающей частичное окисление урана (IV) кислородом воздуха.
Экстракционно-фотометрический вариант определения урана (VI) основан на способности соединения урана с арсеназо III экстрагироваться бутиловым спиртом из растворов, содержащих комплексон III и хлорид дифенилгуанидиния.
Комплексен III маскирует катионы, образующие устойчивые комплексонаты. Определению не мешают хлориды, нитраты, фториды (при содержании до 1 мг); сульфат и фосфат осаждаются хлоридом дифенилгуанидиния и после насыщения ими растворителя флотируются. Осадок отделяют фильтрованием экстрактов; влияние тория устраняют прибавлением фторида (5 мг фторида калия на  раствора).
раствора).
Калибровочный график. Для приготовления серий стандартов в ряд пробирок вводят  урана с интервалом
урана с интервалом  , в виде раствора нитрата уранила с содержанием
, в виде раствора нитрата уранила с содержанием  и погружают их в кипящую водяную баню, раствор выпаривают досуха, пропуская через него воздух, сухой остаток растворяют в
и погружают их в кипящую водяную баню, раствор выпаривают досуха, пропуская через него воздух, сухой остаток растворяют в  н. хлористоводородной кислоты. Вводят
н. хлористоводородной кислоты. Вводят  -ного раствора комплексона III,
-ного раствора комплексона III,  -ного раствора арсеназо III,
-ного раствора арсеназо III,  -ного раствора хлорида дифенилгуанидиния и
-ного раствора хлорида дифенилгуанидиния и  бутилового спирта. Экстрагируют, энергично встряхивают пробирки около 30 сек и после разделения фаз измеряют оптическую плотность органического слоя на фотометре или фотоэлектроколориметре по отношению к воде.
бутилового спирта. Экстрагируют, энергично встряхивают пробирки около 30 сек и после разделения фаз измеряют оптическую плотность органического слоя на фотометре или фотоэлектроколориметре по отношению к воде.
Методика определения. В образце определяют уран аналогично после разложения пробы любым подходящим способом.
Примечание. Можно предварительно экстрагировать уран  -ным раствором трибутилфосфата в хлороформе с применением нитрата аммония в качестве высаливателя и комплексона III для удержания мешающих элементов в водной фазе, а затем реэкстрагировать ураи раствором арсеназо III и определять уран измерением оптической плотности водной фазы.
-ным раствором трибутилфосфата в хлороформе с применением нитрата аммония в качестве высаливателя и комплексона III для удержания мешающих элементов в водной фазе, а затем реэкстрагировать ураи раствором арсеназо III и определять уран измерением оптической плотности водной фазы.
Определение молибдена в сталях
Молибден (V) образует с роданидами окрашенные соединения, состав которых зависит от кислотности среды и концентрации роданида. Соединения молибдена (VI) восстанавливают до пятивалентного состояния хлоридом олова (II), иодидом калия, аскорбиновой кислотой, тиомочевиной в присутствии солей меди (И) и другими восстановителями. Наиболее надежные результаты получаются при использовании последних трех восстановителей. На процесс восстановления молибдена (VI) сильно влияет кислотность раствора.
В зависимости от концентрации роданида могут образовываться соединения с мольным отношением  до
до  . Наиболее интенсивно окрашены соединения с мольным отношением
. Наиболее интенсивно окрашены соединения с мольным отношением  (коэффициент молярного поглощения
(коэффициент молярного поглощения  равен 15000) и
равен 15000) и  (коэффициент молярного поглощения
(коэффициент молярного поглощения  равен 12600).
равен 12600).
Определению молибдена роданидным методом не мешают ионы алюминия, кобальта, урана, тантала, натрия, калия, кремния, кальция, магния, титана, ванадия, хрома, марганца, никеля, цинка, мышьяка, серебра, олова, сурьмы и ртути. Соединения железа (III) и меди усиливают интенсивность окраски, вероятно, вследствие образования многоядерных комплексов, содержащих молибден, железо (или медь) и роданид. Мешающее влияние вольфрама устраняют введением винной кислоты, препятствующей образованию роданидных комплексов вольфрама.
Известно большое число различных вариантов фотометрического роданидного метода определения молибдена, разработанных с целью достижения высокой чувствительности и получения надежных и воспроизводимых результатов. Одним из них является экстракционно-фотометрический вариант определения молибдена в сталях.
Калибровочный график. В пять делительных воронок емкостью  вводят по
вводят по  -ного раствора сульфата железа (III), от 0,5 до
-ного раствора сульфата железа (III), от 0,5 до  раствора молибдата натрия (аммония) с содержанием
раствора молибдата натрия (аммония) с содержанием  серной кислоты
серной кислоты  :
:  ),
),  -ного раствора роданида калия и энергично встряхивают 2—3 мин. Затем прибавляют
-ного раствора роданида калия и энергично встряхивают 2—3 мин. Затем прибавляют  -ного раствора хлорида олова (II) и вновь энергично встряхивают 1—2 мин. При этом растворы постепенно окрашиваются в янтарный или красновато-бурый цвет.
-ного раствора хлорида олова (II) и вновь энергично встряхивают 1—2 мин. При этом растворы постепенно окрашиваются в янтарный или красновато-бурый цвет.
К растворам прибавляют по  диэтилового эфира, экстрагируют соединение молибдена 2—3 мин, эфирный слой сливают в сухие мерные колбы емкостью
диэтилового эфира, экстрагируют соединение молибдена 2—3 мин, эфирный слой сливают в сухие мерные колбы емкостью  и повторяют экстрагирование. Эфирные экстракты разбавляют диэтиловым эфиром до
и повторяют экстрагирование. Эфирные экстракты разбавляют диэтиловым эфиром до  и измеряют их оптическую плотность на фотоэлектроколориметре в кюветах, закрытых крышками для предотвращения испарения эфира. По полученным данным строят калибровочный график.
и измеряют их оптическую плотность на фотоэлектроколориметре в кюветах, закрытых крышками для предотвращения испарения эфира. По полученным данным строят калибровочный график.
Методика определения. Навеску  г стали с содержанием
г стали с содержанием  молибдена помещают в стакан емкостью
молибдена помещают в стакан емкостью  , прибавляют
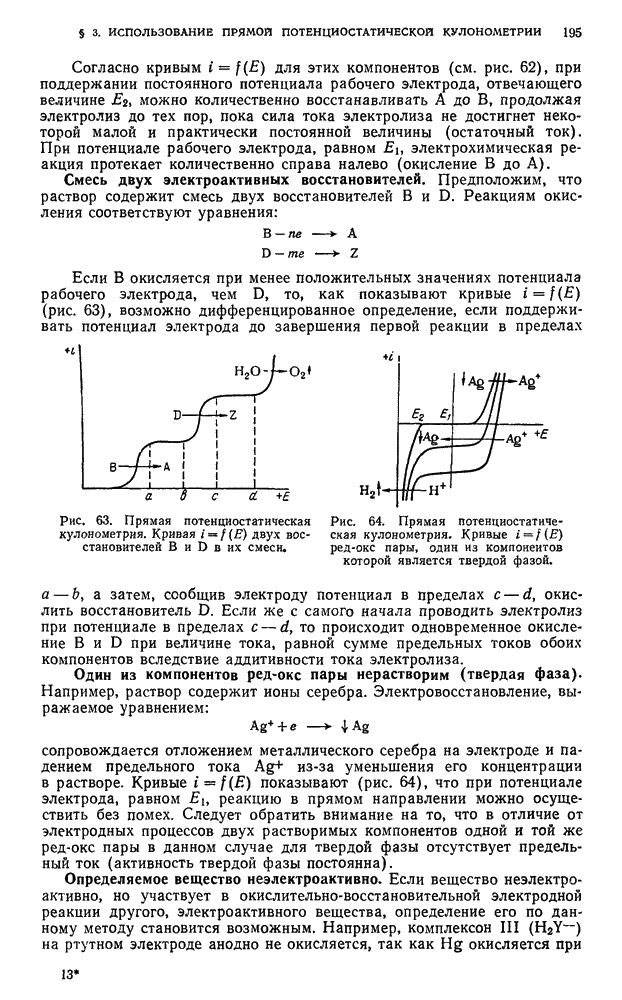
, прибавляют  серной кислоты
серной кислоты  и нагревают до 60—70° С. По окончании растворения прибавляют
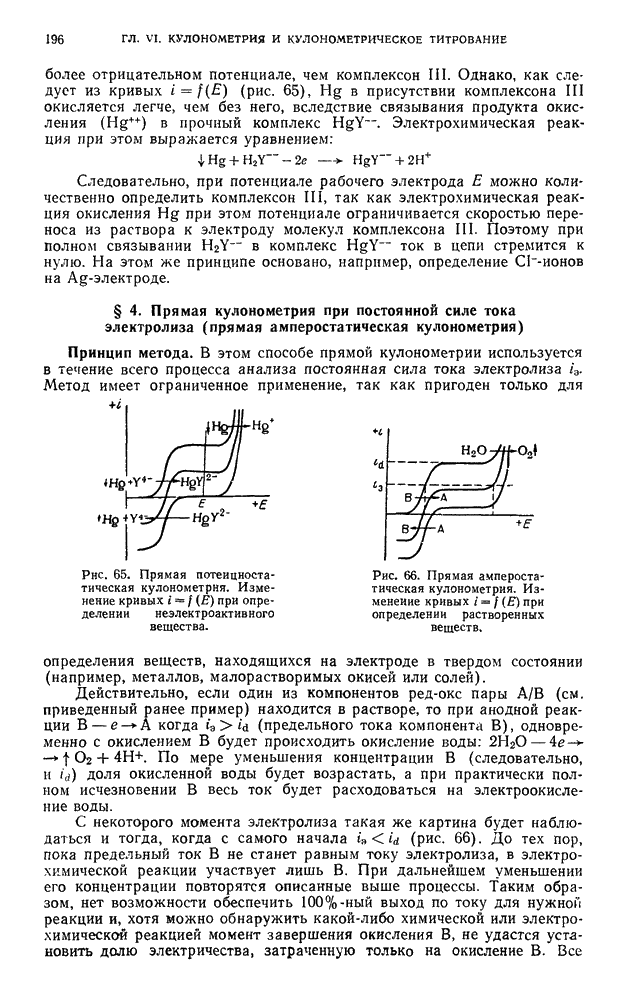
и нагревают до 60—70° С. По окончании растворения прибавляют  -ной перекиси водорода, раствор кипятят несколько минут для окисления карбидов, затем фильтруют через бумажный фильтр для удаления углерода, нерастворимый остаток промывают водой и отбрасывают.
-ной перекиси водорода, раствор кипятят несколько минут для окисления карбидов, затем фильтруют через бумажный фильтр для удаления углерода, нерастворимый остаток промывают водой и отбрасывают.
Фильтрат и промывные воды объединяют, упаривают в стакане до объема  . При анализе простых углеродистых и легированных сталей или сталей, содержащих вольфрам, остаток не отфильтровывают. Прибавляют 0,5 г винной (лимонной) кислоты, если сталь содержит вольфрам, затем нейтрализуют
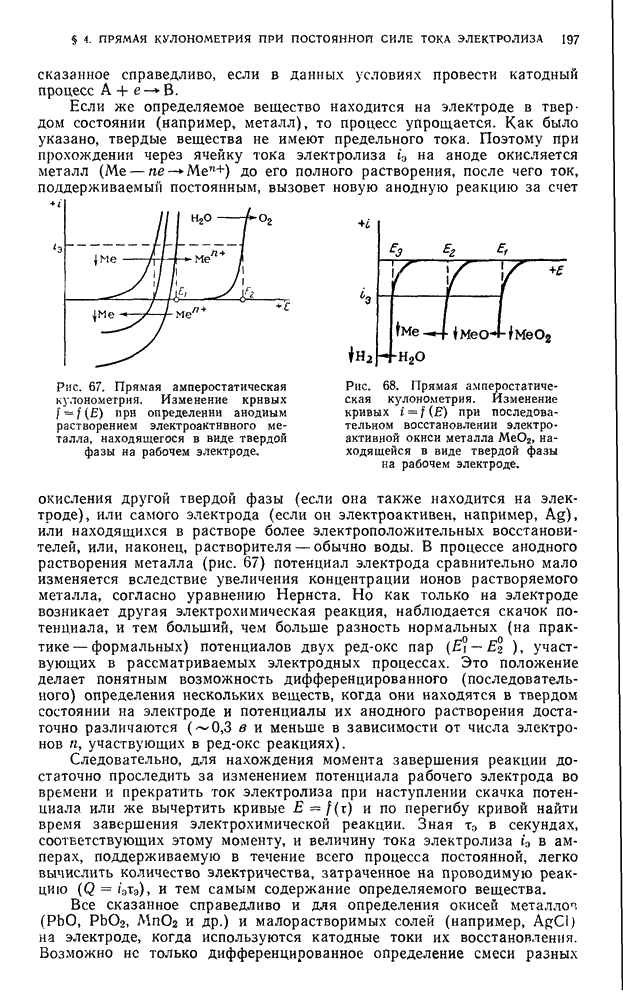
. При анализе простых углеродистых и легированных сталей или сталей, содержащих вольфрам, остаток не отфильтровывают. Прибавляют 0,5 г винной (лимонной) кислоты, если сталь содержит вольфрам, затем нейтрализуют  -ным раствором едкого натра до
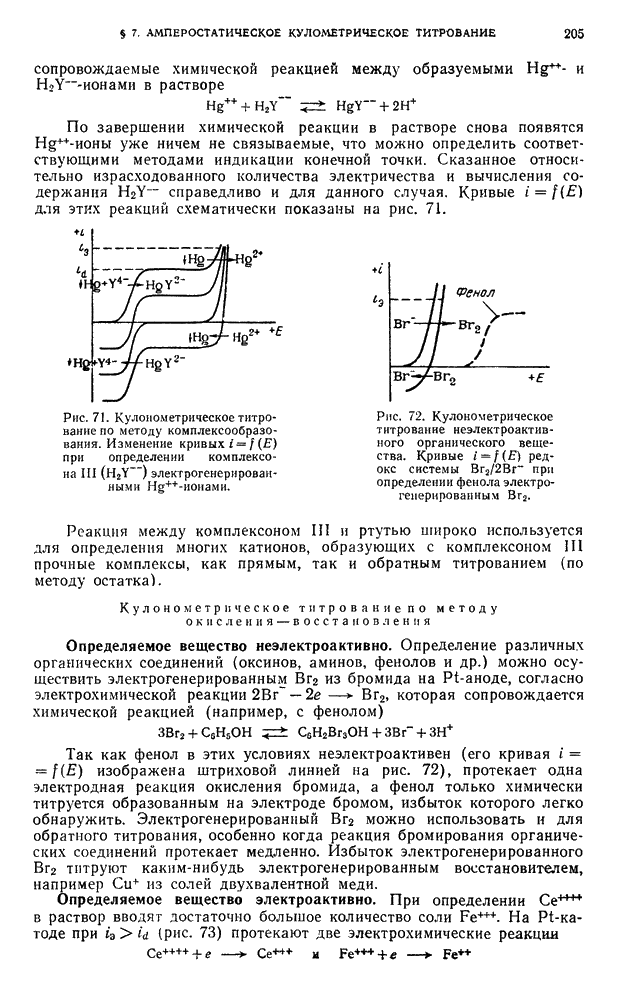
-ным раствором едкого натра до  по универсальной индикаторной бумаге, прибавляют
по универсальной индикаторной бумаге, прибавляют  серной кислоты
серной кислоты  и переносят в делительную воронку емкостью
и переносят в делительную воронку емкостью  . Прибавляют
. Прибавляют  -ного раствора роданида калия и далее поступают, как описано при построении калибровочного графика. Содержание молибдена в навеске находят по калибровочному графику.
-ного раствора роданида калия и далее поступают, как описано при построении калибровочного графика. Содержание молибдена в навеске находят по калибровочному графику.
Определение галлия
Принцип метода. Метод основан на взаимодействии ионов галлия с родамином С в среде 6 н. хлористоводородной кислоты, сопровождающемся последующей экстракцией хлоргаллата родамина подходящим органическим растворителем. В качестве экстрагентов могут быть использованы: бензол, смесь его с диэтиловым эфиром  , с бутилацетатом
, с бутилацетатом  также смесь хлорбензола с четыреххлористым углеродом. Молярный коэффициент поглощения е бензольного экстракта при 565 нм составляет 60 000, а при экстракции смесью хлорбензола и четыреххлористого углерода
также смесь хлорбензола с четыреххлористым углеродом. Молярный коэффициент поглощения е бензольного экстракта при 565 нм составляет 60 000, а при экстракции смесью хлорбензола и четыреххлористого углерода  ) молярный коэффициент поглощения е экстракта при 562 нм равен 78 900.
) молярный коэффициент поглощения е экстракта при 562 нм равен 78 900.
Определению мешают ионы железа (III), сурьмы (V), мышьяка (V), молибдена (VI), таллия (III), теллура (IV), селена (IV), алюминия  мг), меди
мг), меди  мг). Соединения алюминия и меди отделяются при экстракции галлия бутилацетатом из 6 н. раствора
мг). Соединения алюминия и меди отделяются при экстракции галлия бутилацетатом из 6 н. раствора  . Влияние
. Влияние  , Те,
, Те,  и TI устраняют введением
и TI устраняют введением  .
.
Калибровочный график. В делительные воронки емкостью  помещают растворы, содержащие
помещают растворы, содержащие  галлия, с интервалом
галлия, с интервалом  , добавляют до объема
, добавляют до объема  н. раствора
н. раствора  , затем вводят
, затем вводят  -ного раствора треххлористого титана,
-ного раствора треххлористого титана,  смеси бензола и бутилацетата и
смеси бензола и бутилацетата и  -ного раствора родамина С. Взбалтывают 2 мин.
-ного раствора родамина С. Взбалтывают 2 мин.
После окончания разделения фаз нижний слой отбрасывают, а окрашенный экстракт фильтруют через слой стеклянной ваты. Измеряют оптическую плотность экстрактов на спектрофотоколориметре ФЭК с зеленым светофильтром в кюветах с толщиной слоя 1 см относительно того же органического растворителя и строят калибровочный график.
Методика определения. В анализируемом растворе определяют галлий, как описано выше, и содержание его находят по калибровочному графику.
Определение галлия в алюминии
Наилучшим методом отделения галлия от сопутствующих элементов для последующего фотометрического определения является экстракция его из 6 н. раствора  диэтиловым эфиром или менее летучим бутилацетатом при соотношении фаз
диэтиловым эфиром или менее летучим бутилацетатом при соотношении фаз  . Коэффициент распределения в последнем случае приблизительно равен 400. Реэкстрагируют галлий водой. Определение галлия проводят родаминовым методом.
. Коэффициент распределения в последнем случае приблизительно равен 400. Реэкстрагируют галлий водой. Определение галлия проводят родаминовым методом.
Методика определения. Навеску 0,25 г образца металлического алюминия растворяют при умеренном нагревании в  н. растворе
н. растворе  . К полученному раствору прибавляют по каплям
. К полученному раствору прибавляют по каплям  -ный раствор
-ный раствор  до фиолетовой окраски раствора. Выдерживают 2—3 мин и переводят в делительную воронку емкостью
до фиолетовой окраски раствора. Выдерживают 2—3 мин и переводят в делительную воронку емкостью  , ополаскивая стакан
, ополаскивая стакан  п. хлористоводородной кислотой. К раствору прибавляют равный объем бутилацетата и энергично встряхивают 1 мин. После разделения фаз нижний слой отбрасывают, экстракт промывают 2 раза 6 н. хлористоводородной кислотой (по
п. хлористоводородной кислотой. К раствору прибавляют равный объем бутилацетата и энергично встряхивают 1 мин. После разделения фаз нижний слой отбрасывают, экстракт промывают 2 раза 6 н. хлористоводородной кислотой (по  . Реэкстрагируют галлий
. Реэкстрагируют галлий  воды, взбалтывая в течение 1 мин. После расслаивания нижнюю фазу переводят в фарфоровую чашку. Повторяют реэкстракцию галлия водой, сливая реэкстракт в ту же чашку, прибавляют 0,1 г
воды, взбалтывая в течение 1 мин. После расслаивания нижнюю фазу переводят в фарфоровую чашку. Повторяют реэкстракцию галлия водой, сливая реэкстракт в ту же чашку, прибавляют 0,1 г  , упаривают на водяной бане досуха. Сухой остаток растворяют в 6 н. хлористоводородной кислоте, раствор переводят в мерную колбу емкостью
, упаривают на водяной бане досуха. Сухой остаток растворяют в 6 н. хлористоводородной кислоте, раствор переводят в мерную колбу емкостью  и объем доводят до метки этой же кислотой. Отбирают
и объем доводят до метки этой же кислотой. Отбирают  раствора, переносят в делительную воронку емкостью
раствора, переносят в делительную воронку емкостью  и поступают далее, как указано в предыдущей методике.
и поступают далее, как указано в предыдущей методике.
Определение германия
Метод основан на взаимодействии германия (IV) с фенилфлуороном в кислом растворе с образованием красного осадка. При малом количестве германия появляется суспензия, которую можно стабилизовать добавлением защитного коллоида. Соединение германия с фенилфлуороном не экстрагируется органическими растворителями, но флотируется. Изменение концентрации кислоты в сравнительно широких пределах  н.) не влияет на образование этого соединения. При меньшей кислотности выпадает осадок реактива. Максимум поглощения окрашенных растворов фенилфлуората германия находится при 500 нм, однако при измерениях лучше пользоваться светофильтром с максимумом светопропускания 530 нм. Молярный коэффициент поглощения (е) комплекса
н.) не влияет на образование этого соединения. При меньшей кислотности выпадает осадок реактива. Максимум поглощения окрашенных растворов фенилфлуората германия находится при 500 нм, однако при измерениях лучше пользоваться светофильтром с максимумом светопропускания 530 нм. Молярный коэффициент поглощения (е) комплекса  .
.
С фенилфлуороном реагируют также титан, цирконий, гафний, олово (IV), ниобий, тантал, сурьма (III), теллур, молибден, вольфрам. Окислители ванадий  (VI), марганец (VII) и церий (IV) окисляют реагент. Ионы галлия и мышьяка в кислых растворах не реагируют с фенилфлуороном. Не мешают определению фторид
(VI), марганец (VII) и церий (IV) окисляют реагент. Ионы галлия и мышьяка в кислых растворах не реагируют с фенилфлуороном. Не мешают определению фторид  мг в
мг в  и железо (III) (
и железо (III) ( ).
).
Калибровочный график. В ряд мерных колб емкостью  вводят
вводят  германия с интервалом
германия с интервалом  (в виде раствора с содержанием
(в виде раствора с содержанием  ), оставляя одну колбу пустой. Прибавляют во все колбы по
), оставляя одну колбу пустой. Прибавляют во все колбы по  н. раствора
н. раствора  , воду до
, воду до  -ной желатины и перемешивают. Затем вводят по
-ной желатины и перемешивают. Затем вводят по  -ного раствора фенилфлуорона, перемешивают и доводят водой до метки. Оставляют стоять 30 мин. Измеряют оптическую плотность на фотоэлектроколориметре или фотометре, пользуясь светофильтром с максимумом пропускания при
-ного раствора фенилфлуорона, перемешивают и доводят водой до метки. Оставляют стоять 30 мин. Измеряют оптическую плотность на фотоэлектроколориметре или фотометре, пользуясь светофильтром с максимумом пропускания при  , относительно раствора сравнения, полученного в холостом опыте.
, относительно раствора сравнения, полученного в холостом опыте.
Методика определения. При анализе различных объектов германий отделяют от мешающих ионов экстракцией четыреххлористым углеродом из 9 М раствора  или дистилляцией
или дистилляцией  (в присутствии окислителя).
(в присутствии окислителя).
Для определения германия в угле 1 г пробы помещают в платиновую чашку, прибавляют 0,5 г  , перемешивают, приливают
, перемешивают, приливают  насыщенного раствора
насыщенного раствора  мг в
мг в  общего объема
общего объема  выпаривают сначала на водяной бане, а потом на песочной. После этого озоляют в муфельной печи, медленно нагревая до 400—450° С для сгорания основной массы углистых частиц, затем повышают температуру печи до 700—800° С. Слишком резкое нагревание в начале сжигания приводит к вспышке и распылению сжигаемой массы. После сгорания основной массы угля смесь 2—3 раза перемешивают шпателем для ускорения выгорания углистых частиц. Сжигание продолжают до получения белого или буроватого порошка. Охлаждают, приливают по каплям при перемешивании
выпаривают сначала на водяной бане, а потом на песочной. После этого озоляют в муфельной печи, медленно нагревая до 400—450° С для сгорания основной массы углистых частиц, затем повышают температуру печи до 700—800° С. Слишком резкое нагревание в начале сжигания приводит к вспышке и распылению сжигаемой массы. После сгорания основной массы угля смесь 2—3 раза перемешивают шпателем для ускорения выгорания углистых частиц. Сжигание продолжают до получения белого или буроватого порошка. Охлаждают, приливают по каплям при перемешивании  азотной кислоты и выпаривают на водяной бане досуха. К остатку прибавляют
азотной кислоты и выпаривают на водяной бане досуха. К остатку прибавляют  фтористоводородной кислоты и снова упаривают досуха. Прибавляют
фтористоводородной кислоты и снова упаривают досуха. Прибавляют  фтористоводородной и
фтористоводородной и  фосфорной кислоты и выпаривают сначала на водяной бане, а затем на песочной до удаления HF и получения сиропообразного остатка, который смывают
фосфорной кислоты и выпаривают сначала на водяной бане, а затем на песочной до удаления HF и получения сиропообразного остатка, который смывают  воды в стакан и нагревают до распадения комков.
воды в стакан и нагревают до распадения комков.
При анализе железных окисных руд германий может быть извлечен разложением навески фтористоводородной, азотной и фосфорной кислотами. Навеску  г помещают в платиновую чашку, прибавляют
г помещают в платиновую чашку, прибавляют  азотной и
азотной и  фтористоводородной кислоты и выпаривают на водяной бане досуха, затем добавляют
фтористоводородной кислоты и выпаривают на водяной бане досуха, затем добавляют  фтористоводородной и
фтористоводородной и  фосфорной кислоты и выпаривают до удаления HF. Сиропообразный остаток смывают
фосфорной кислоты и выпаривают до удаления HF. Сиропообразный остаток смывают  воды в стакан и нагревают до растворения остатка.
воды в стакан и нагревают до растворения остатка.
При анализе силикатов их разлагают смесью фтористоводородной, серной и азотной кислот (или смесью  ). Навеску 0,5 г помещают в платиновую чашку, прибавляют
). Навеску 0,5 г помещают в платиновую чашку, прибавляют  серной кислоты
серной кислоты  концентрированной азотной и
концентрированной азотной и  фтористоводородной кислоты. Выпаривают до появления паров
фтористоводородной кислоты. Выпаривают до появления паров  . Добавляют
. Добавляют  воды и нагревают до растворения осадка.
воды и нагревают до растворения осадка.
Экстракция германия. Полученный тем или иным способом раствор (объем  ) охлаждают до комнатной температуры, прибавляют
) охлаждают до комнатной температуры, прибавляют  концентрированной хлористоводородной кислоты (на возможное помутнение не обращают внимания), охлаждают, переносят в делительную воронку емкостью
концентрированной хлористоводородной кислоты (на возможное помутнение не обращают внимания), охлаждают, переносят в делительную воронку емкостью  , вводят
, вводят  четыреххлористого углерода и экстрагируют 2 раза по 2 мин. Экстракт промывают 3 раза
четыреххлористого углерода и экстрагируют 2 раза по 2 мин. Экстракт промывают 3 раза  -ным раствором солянокислого гидроксиламина в 9 н. хлористоводородной кислоте (каждый раз по
-ным раствором солянокислого гидроксиламина в 9 н. хлористоводородной кислоте (каждый раз по  ), взбалтывая в течение 1 мин. Реэкстрагируют германий
), взбалтывая в течение 1 мин. Реэкстрагируют германий  воды, повторяя операцию 2 раза, взбалтывая каждый раз в течение 1 мин.
воды, повторяя операцию 2 раза, взбалтывая каждый раз в течение 1 мин.
Водные вытяжки переносят в мерную колбу емкостью  и далее поступают так же, как при построении калибровочного графика.
и далее поступают так же, как при построении калибровочного графика.
Определение селена
Принцип метода. Селен (IV) образует с  -диаминобензидином в нейтральной, щелочной или кислой среде окрашенное в желтый цвет соединение, плохо растворимое в воде.
-диаминобензидином в нейтральной, щелочной или кислой среде окрашенное в желтый цвет соединение, плохо растворимое в воде.
Образующийся дифенилдипиазоселенол хорошо экстрагируется толуолом при  это используется при экстракционно-фотометрическом определении. В разбавленных водных растворах пиазоселенол образует окрашенные растворы, оптические плотности которых пропорциональны концентрациям селена в интервале
это используется при экстракционно-фотометрическом определении. В разбавленных водных растворах пиазоселенол образует окрашенные растворы, оптические плотности которых пропорциональны концентрациям селена в интервале  . Молярный коэффициент поглощения
. Молярный коэффициент поглощения  составляет 10 200 при 347—349° С и 0,1 н. концентрации кислоты. Окраска растворов развивается в этих условиях через 50 мин и сохраняется в течение
составляет 10 200 при 347—349° С и 0,1 н. концентрации кислоты. Окраска растворов развивается в этих условиях через 50 мин и сохраняется в течение  .
.
Определению не мешают алюминий, барий, кальций, кадмий, кобальт, калий, магний, марганец, молибден (VI), никель, теллур (IV), натрий, цинк, аммоний, бромид, хлорид, нитрат, фосфат, сульфат, цитрат, оксалат и тартрат.
Железо (III) можно маскировать фторидом, оксалатом.
 -Диаминобензидин применяется для фотометрического определения селена в чистом индии, мышьяке и их полупроводниковых соединениях.
-Диаминобензидин применяется для фотометрического определения селена в чистом индии, мышьяке и их полупроводниковых соединениях.
Калибровочный график. В ряд стаканов емкостью  вводят стандартный раствор, содержащий
вводят стандартный раствор, содержащий  селена, с интервалом
селена, с интервалом  , в один стакан стандартный раствор не вводят. Добавляют по
, в один стакан стандартный раствор не вводят. Добавляют по  воды,
воды,  -ного раствора комплексона III,
-ного раствора комплексона III,  муравьиной кислоты
муравьиной кислоты  и нейтрализуют аммиаком
и нейтрализуют аммиаком  по крезоловому красному до
по крезоловому красному до  (желтая окраска индикатора). Затем вводят
(желтая окраска индикатора). Затем вводят  -ного свежеприготовленного раствора
-ного свежеприготовленного раствора  -диаминобензидина в хлористоводородной кислоте и оставляют на 30 мин. Прибавляют аммиак
-диаминобензидина в хлористоводородной кислоте и оставляют на 30 мин. Прибавляют аммиак  до
до  (фиолетовая окраска раствора индикатора), переносят в делительные воронки емкостью
(фиолетовая окраска раствора индикатора), переносят в делительные воронки емкостью  , добавляют
, добавляют  толуола или бензола и экстрагируют окрашенное соединение 1 мин. Экстракты фильтруют через сухой бумажный фильтр в кювету с толщиной слоя 3 см и измеряют оптическую плотность на фотоэлектроколориметре ФЭК-Н-54 при 415—420 нм и строят калибровочный график.
толуола или бензола и экстрагируют окрашенное соединение 1 мин. Экстракты фильтруют через сухой бумажный фильтр в кювету с толщиной слоя 3 см и измеряют оптическую плотность на фотоэлектроколориметре ФЭК-Н-54 при 415—420 нм и строят калибровочный график.
Методика определения селена в мышьяке. Навеску 1,0 г мышьяка растворяют при умеренном нагревании в  смеси равных объемов азотной и хлористоводородной кислот, избегая бурной реакции. Упаривают почти досуха, прибавляют
смеси равных объемов азотной и хлористоводородной кислот, избегая бурной реакции. Упаривают почти досуха, прибавляют  воды, нагревают до получения прозрачного раствора, который после охлаждения разбавляют водой до
воды, нагревают до получения прозрачного раствора, который после охлаждения разбавляют водой до  , вводят комплексон III и муравьиную кислоту и далее, как описано выше (см. Калибровочный график).
, вводят комплексон III и муравьиную кислоту и далее, как описано выше (см. Калибровочный график).
Методика определения селена в индии. Навеску 0,5 г индия растворяют при умеренном нагревании в  азотной кислоты, раствор упаривают почти досуха. Остаток растворяют в
азотной кислоты, раствор упаривают почти досуха. Остаток растворяют в  соляной кислоты (
соляной кислоты ( ), прибавляют раствор, содержащий 50 мг мышьяка,
), прибавляют раствор, содержащий 50 мг мышьяка,  раствора хлорида олова (II),
раствора хлорида олова (II),  -ного раствора гипофосфита натрия в хлористоводородной кислоте
-ного раствора гипофосфита натрия в хлористоводородной кислоте  и немного мацерированной бумаги. Раствор нагревают до кипения, кипятят 5—7 мин до появления осадка, разбавляют хлористоводородной кислотой
и немного мацерированной бумаги. Раствор нагревают до кипения, кипятят 5—7 мин до появления осадка, разбавляют хлористоводородной кислотой  до
до  и оставляют на ночь.
и оставляют на ночь.
На следующий день осадок отфильтровывают через тампон из мацерированной бумаги и промывают 8—10 раз хлористоводородной кислотой  .
.
Промытый осадок вместе с тампоном переносят в стакан, в котором проводили осаждение, прибавляют  хлористоводородной кислоты
хлористоводородной кислоты  и 3—5 капель азотной кислоты
и 3—5 капель азотной кислоты  . Стакан погружают в водяную баню и выдерживают при 70—80° С до растворения осадка. Прибавляют
. Стакан погружают в водяную баню и выдерживают при 70—80° С до растворения осадка. Прибавляют  горячей воды,
горячей воды,  -ного раствора мочевины для разрушения избытка окислителя, хорошо перемешивают и отфильтровывают на фильтре «белая лента», фильтр промывают 2—3 раза теплой водой. К фильтрату добавляют
-ного раствора мочевины для разрушения избытка окислителя, хорошо перемешивают и отфильтровывают на фильтре «белая лента», фильтр промывают 2—3 раза теплой водой. К фильтрату добавляют  -ного раствора лимонной кислоты, охлаждают и далее, как описано выше (см. Калибровочный график).
-ного раствора лимонной кислоты, охлаждают и далее, как описано выше (см. Калибровочный график).
Методика определения селена в арсениде и антимониде индия. Навеску 0,5 г арсенида или антимонида индия растворяют при умеренном нагревании в  смеси равных объемов концентрированных хлористоводородной и азотной кислот и воды и упаривают почти досуха.
смеси равных объемов концентрированных хлористоводородной и азотной кислот и воды и упаривают почти досуха.
При анализе арсенида индия остаток растворяют при нагревании в  хлористоводородной кислоты
хлористоводородной кислоты  , прибавляют
, прибавляют  раствора хлорида олова (II),
раствора хлорида олова (II),  раствора гипофосфита натрия и немного мацерированной бумаги, в холостой опыт вводят дополнительно
раствора гипофосфита натрия и немного мацерированной бумаги, в холостой опыт вводят дополнительно  раствора арсената натрия (50 мг мышьяка).
раствора арсената натрия (50 мг мышьяка).
При анализе антимонида индия остаток растворяют при нагревании в  -ного раствора лимонной кислоты, затем вводят
-ного раствора лимонной кислоты, затем вводят  хлористоводородной кислоты
хлористоводородной кислоты  раствора арсената натрия,
раствора арсената натрия,
 раствора хлорида олова (II),
раствора хлорида олова (II),  раствора гипофосфита натрия и немного мацерированной бумаги.
раствора гипофосфита натрия и немного мацерированной бумаги.
Далее в обоих случаях раствор нагревают до кипения, кипятят 5— 10 мин до появления осадка, разбавляют хлористоводородной кислотой ( до объема
до объема  и оставляют на ночь. Далее, как описано при определении селена в индии, но при анализе антимонида индия вместо
и оставляют на ночь. Далее, как описано при определении селена в индии, но при анализе антимонида индия вместо
 вводят
вводят  раствора
раствора  -диамииобензидина.
-диамииобензидина.
Определение теллура в висмуте
Хлоридный комплекс теллура (IV) с диантипирилпропилметаном количественно экстрагируется дихлорэтаном из сильносолянокислых растворов. Таким путем удается легко отделять любые количества теллура от селена, меди, свинца и висмута (от последних двух в присутствии комплексона III).
Для определения теллура хлоридный комплекс переводят в бромидный комплекс с диантипирилпропилметаном или диантипирилметаном и измеряют оптическую плотность экстракта. Молярный коэффициент поглощения  при 450 нм равен 4200.
при 450 нм равен 4200.
Метод очень прост в выполнении, дает хорошо воспроизводимые результаты, отнимает немного времени и может быть использован для определения Те в товарном селене, висмуте, меди и свинце.
Калибровочный график. В серию делительных воронок емкостью  вводят от 10 до
вводят от 10 до  Те в виде раствора теллуристой кислоты с содержанием
Те в виде раствора теллуристой кислоты с содержанием  . Прибавляют воду до объема
. Прибавляют воду до объема  , затем
, затем  бромистоводородной кислоты и оставляют на 1—2 мин. Вводят
бромистоводородной кислоты и оставляют на 1—2 мин. Вводят  -ного раствора диантипирилпропилметана и
-ного раствора диантипирилпропилметана и  дихлорэтана, встряхивают 1 мин. Отделяют экстракты, фильтруют их через сухой фильтр в кювету с толщиной слоя 1 см, измеряют оптическую плотность на фотоэлектроколориметре (или спектрофотометре) относительно дихлорэтана и строят калибровочную кривую.
дихлорэтана, встряхивают 1 мин. Отделяют экстракты, фильтруют их через сухой фильтр в кювету с толщиной слоя 1 см, измеряют оптическую плотность на фотоэлектроколориметре (или спектрофотометре) относительно дихлорэтана и строят калибровочную кривую.
Окраска экстрактов устойчива длительное время.
Методика определения. Навеску  г свинца или висмута растворяют в
г свинца или висмута растворяют в  азотной кислоты
азотной кислоты  . Раствор выпаривают почти досуха, обмывают стенки колбы водой и вновь выпаривают досуха. Сухой остаток растворяют в
. Раствор выпаривают почти досуха, обмывают стенки колбы водой и вновь выпаривают досуха. Сухой остаток растворяют в  раствора комплексона III при нагревании. Добавляют концентрированную хлористоводородную кислоту в объеме, равном объему введенного раствора комплексона III. Раствор переносят в делительную воронку емкостью 100 или
раствора комплексона III при нагревании. Добавляют концентрированную хлористоводородную кислоту в объеме, равном объему введенного раствора комплексона III. Раствор переносят в делительную воронку емкостью 100 или  , добавляют
, добавляют  -ного раствора диантипирилпропилметана в уксусной кислоте
-ного раствора диантипирилпропилметана в уксусной кислоте  , перемешивают и добавляют
, перемешивают и добавляют  дихлорэтана. Встряхивают 1 мин.
дихлорэтана. Встряхивают 1 мин.
После разделения фаз экстракт сливают в чистую делительную воронку емкостью  . К оставшейся водной фазе прибавляют еще
. К оставшейся водной фазе прибавляют еще  дихлорэтана и снова встряхивают 1 мин. Дихлорэтанные экстракты объединяют. Водные растворы отбрасывают. Экстракты промывают двумя порциями по
дихлорэтана и снова встряхивают 1 мин. Дихлорэтанные экстракты объединяют. Водные растворы отбрасывают. Экстракты промывают двумя порциями по  хлористоводородной кислоты
хлористоводородной кислоты  .
.
Теллур извлекают, встряхивая экстракт с  раствора комплексона III. Эту операцию проводят 2 раза. После разделения фаз сливают дихлорэтан. Водные растворы объединяют, переводят в мерную колбу емкостью 50 или
раствора комплексона III. Эту операцию проводят 2 раза. После разделения фаз сливают дихлорэтан. Водные растворы объединяют, переводят в мерную колбу емкостью 50 или  и доводят до метки 0,02 М раствором комплексона III.
и доводят до метки 0,02 М раствором комплексона III.
Отбирают аликвотную часть раствора  , добавляют
, добавляют  раствора комплексона III,
раствора комплексона III,  бромистоводородной кислоты,
бромистоводородной кислоты,  -ного раствора диантипирилпропилметана,
-ного раствора диантипирилпропилметана,  дихлорэтана и встряхивают 1 мин.
дихлорэтана и встряхивают 1 мин.
После разделения органическую фазу фильтруют через сухой фильтр в кювету с толщиной слоя 1 см. Измеряют оптическую плотность на фотоэлектроколориметре ФЭК-Н-57 или на спектрофотометре относительно дихлорэтана.
Содержание теллура находят по калибровочному графику.
Определение теллура в селене
Хлоридный комплекс теллура (IV) с диантипирилпропилметаном количественно экстрагируется дихлорэтаном из солянокислых растворов, а тот же комплекс селена (IV) при этом полностью остается вводной фазе (вне зависимости от концентрации кислоты). Таким путем удается легко отделять любые количества теллура от селена.
Для определения теллура хлоридный комплекс переводят в бромидный комплекс с диантипирилпропилметаном и измеряют оптическую плотность полученного раствора.
Калибровочный график. В серию делительных воронок емкостью  вводят от 10 до
вводят от 10 до  Те в виде раствора теллуристой кислоты, добавляют воду до объема
Те в виде раствора теллуристой кислоты, добавляют воду до объема  , по
, по  перегнанной бромистоводородной кислоты и оставляют на 1—2 мин, затем добавляют по
перегнанной бромистоводородной кислоты и оставляют на 1—2 мин, затем добавляют по  раствора аскорбиновой кислоты,
раствора аскорбиновой кислоты,  -ного раствора диантипирилпропилметана,
-ного раствора диантипирилпропилметана,  дихлорэтана и встряхивают 1 мин.
дихлорэтана и встряхивают 1 мин.
После разделения фаз экстракты отделяют, фильтруют через сухие фильтры в кювету с толщиной слоя 1 см и измеряют оптическую плотность на фотоэлектроколориметре или спектрофотометре относительно дихлорэтана.
Окраска экстрактов устойчива длительное время.
Методика определения. Навеску 0,5 г металлического селена растворяют в  концентрированной азотной кислоты, добавляют
концентрированной азотной кислоты, добавляют  серной кислоты
серной кислоты  и выпаривают до появления паров
и выпаривают до появления паров  . После охлаждения осторожно прибавляют
. После охлаждения осторожно прибавляют  воды и снова выпаривают до появления паров
воды и снова выпаривают до появления паров  , охлаждают и остаток переводят в мерную колбу емкостью
, охлаждают и остаток переводят в мерную колбу емкостью  хлористоводородной кислотой
хлористоводородной кислотой  .
.














































































































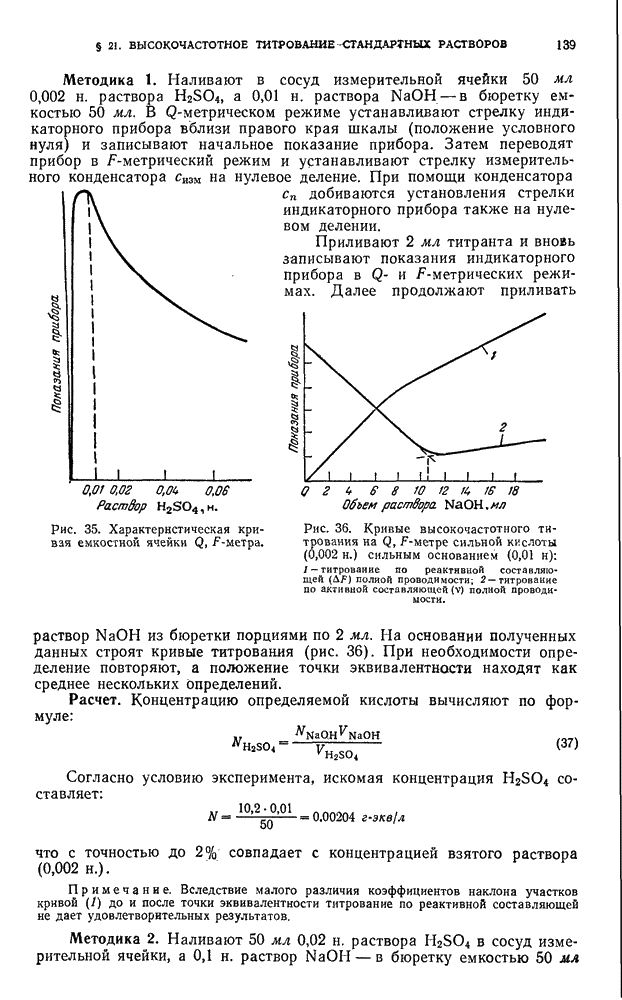
















































































































































































































































































 (в зависимости от содержания теллура) полученного раствора в делительную воронку емкостью
(в зависимости от содержания теллура) полученного раствора в делительную воронку емкостью  , вводят
, вводят  -ного раствора диантипирилпропилметана в уксусной кислоте
-ного раствора диантипирилпропилметана в уксусной кислоте  прибавляют хлористоводородную кислоту
прибавляют хлористоводородную кислоту  до объема
до объема  дихлорэтана и встряхивают 1 мин.
дихлорэтана и встряхивают 1 мин.
 . К оставшейся водной фазе добавляют еще
. К оставшейся водной фазе добавляют еще  дихлорэтана и снова встряхивают 1 мин, экстракт отделяют. Дихлорэтанные экстракты объединяют (водный раствор отбрасывают) и промывают 2 раза хлористоводородной кислотой
дихлорэтана и снова встряхивают 1 мин, экстракт отделяют. Дихлорэтанные экстракты объединяют (водный раствор отбрасывают) и промывают 2 раза хлористоводородной кислотой  порциями по
порциями по  .
.
 раствора комплексона III. После разделения фаз дихлорэтан сливают. К оставшемуся водному раствору добавляют
раствора комплексона III. После разделения фаз дихлорэтан сливают. К оставшемуся водному раствору добавляют  бромистоводородной кислоты и дают постоять 1 мин, затем прибавляют 0,
бромистоводородной кислоты и дают постоять 1 мин, затем прибавляют 0, раствора аскорбиновой кислоты,
раствора аскорбиновой кислоты,  -ного раствора диантипирилпропилметана и
-ного раствора диантипирилпропилметана и  дихлорэтана, встряхивают 1 мин. Объединенные экстракты фильтруют через сухой фильтр в кювету с толщиной слоя 1 см. Оптическую плотность измеряют относительно дихлорэтана на фотоэлектроколориметре ФЭК-Н-57 или спектрофотометре.
дихлорэтана, встряхивают 1 мин. Объединенные экстракты фильтруют через сухой фильтр в кювету с толщиной слоя 1 см. Оптическую плотность измеряют относительно дихлорэтана на фотоэлектроколориметре ФЭК-Н-57 или спектрофотометре.