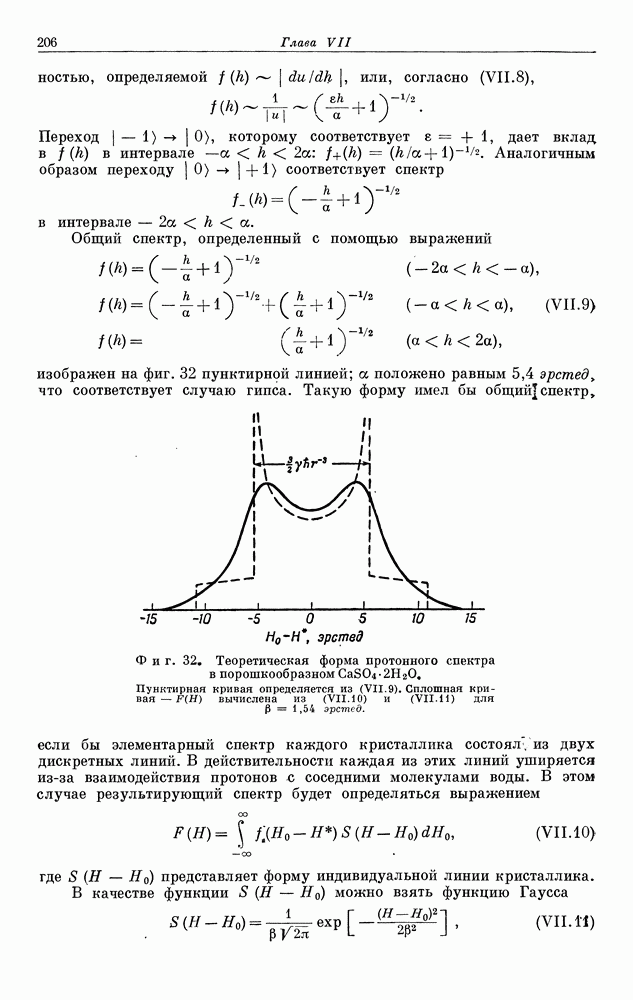
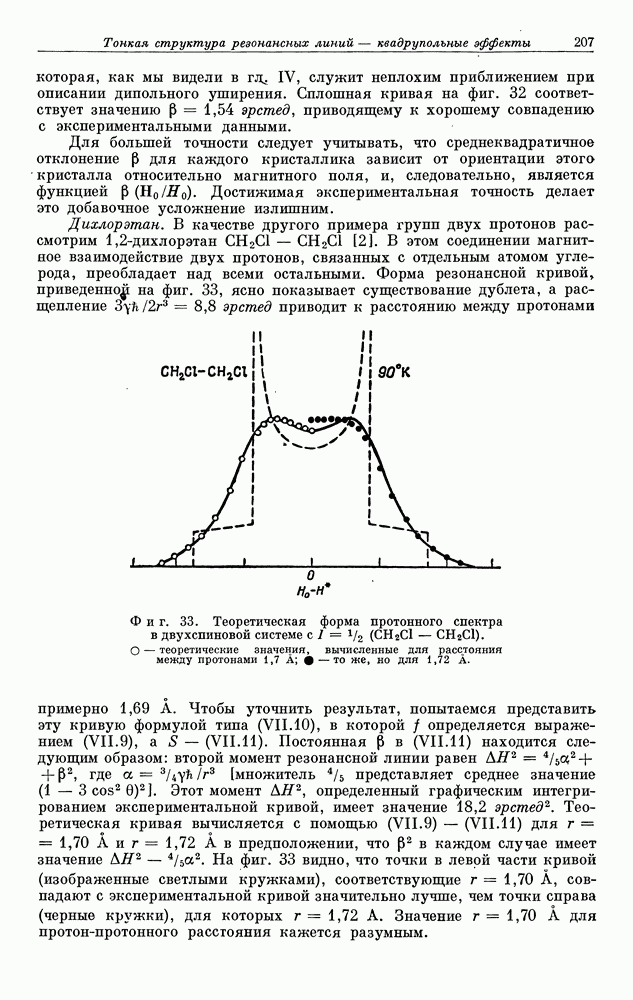
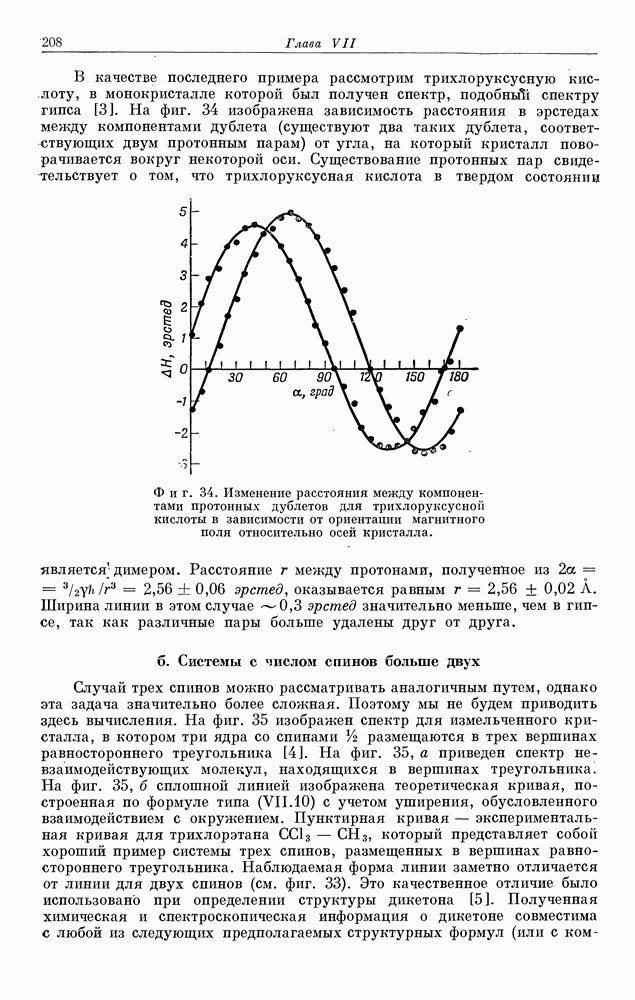
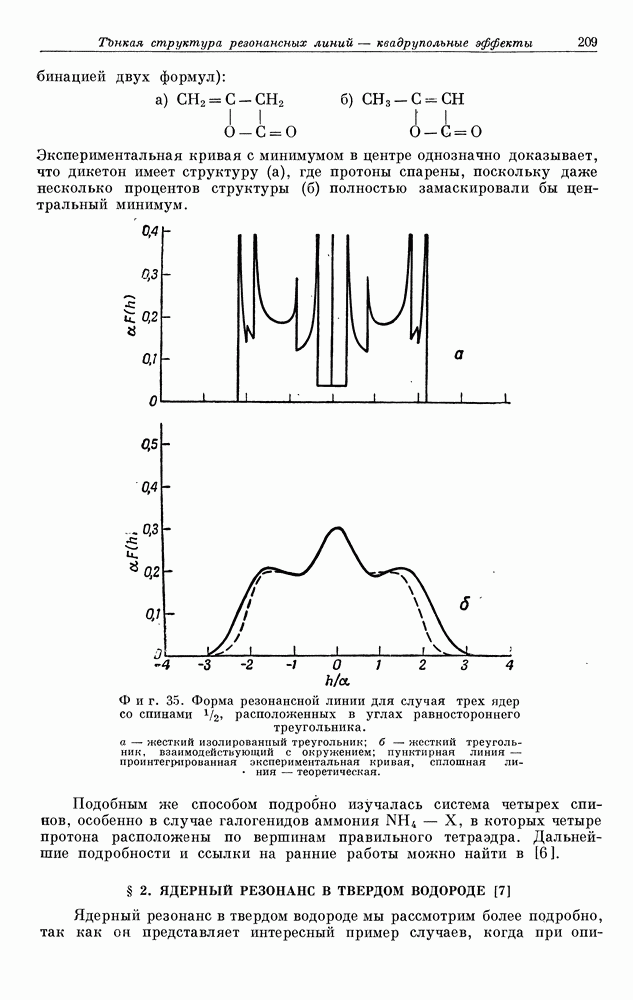
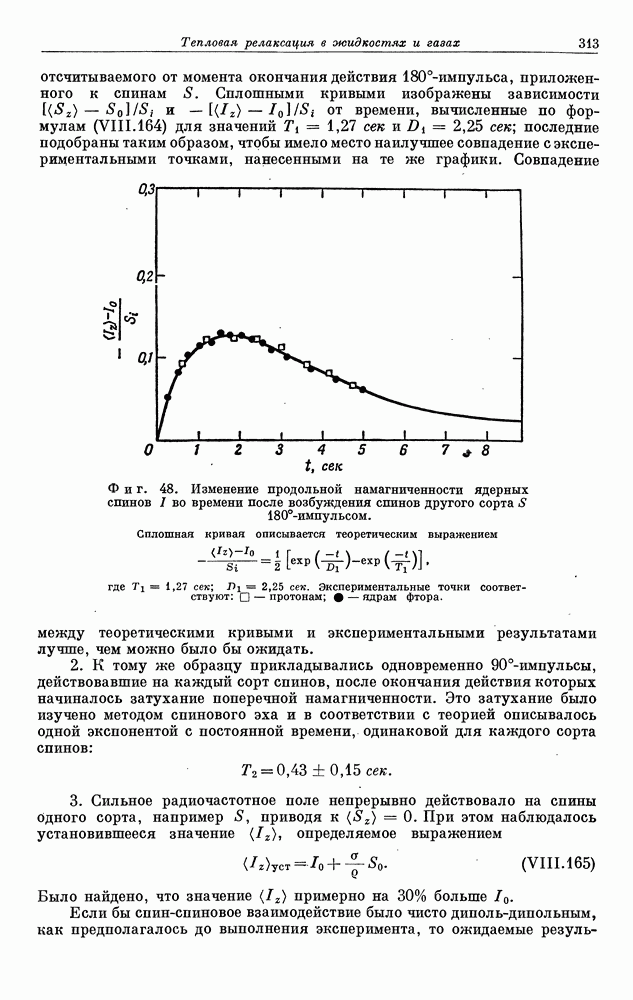
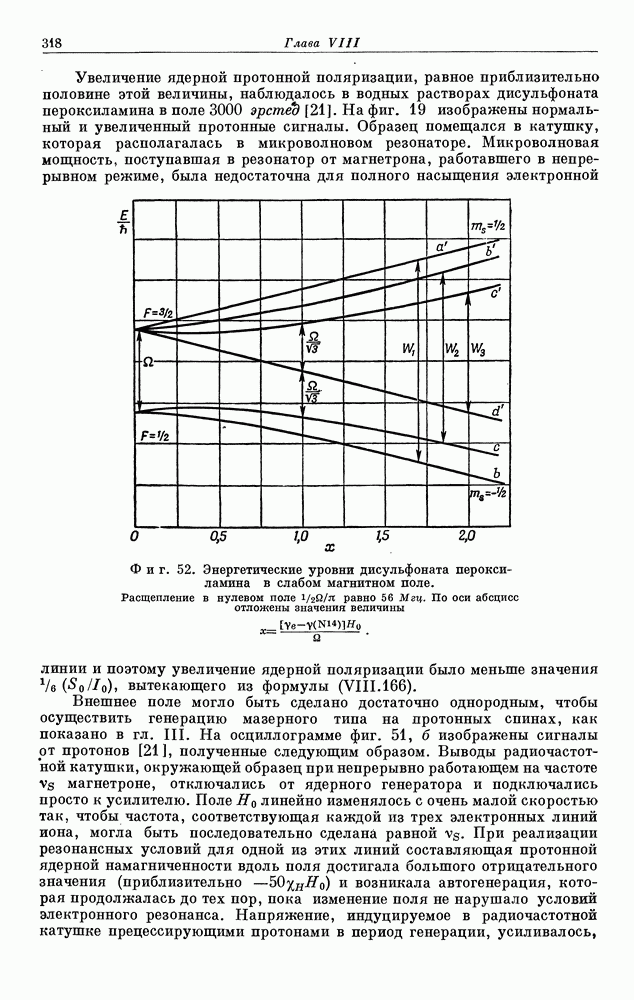
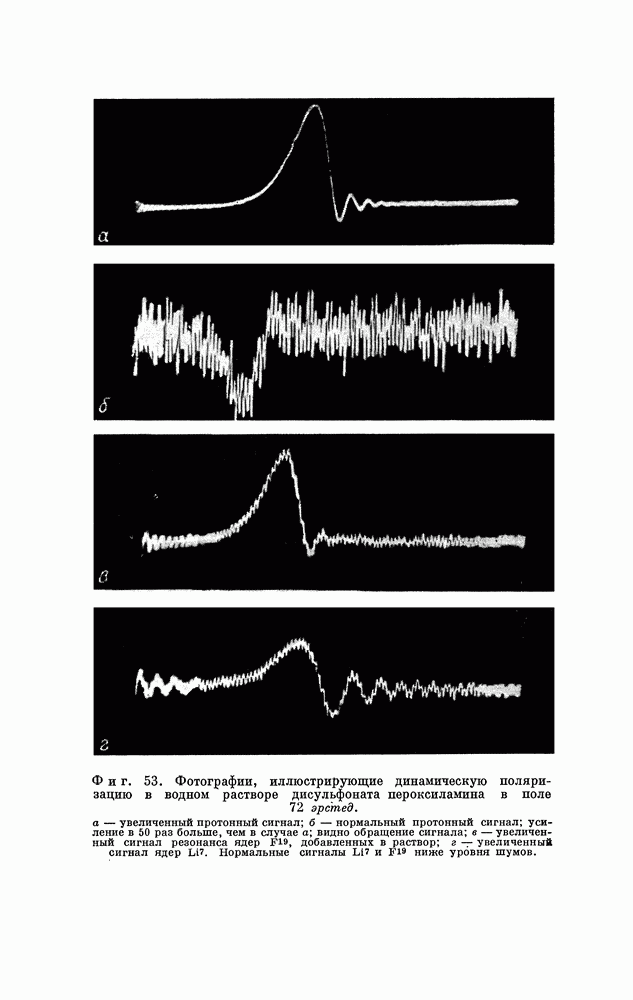
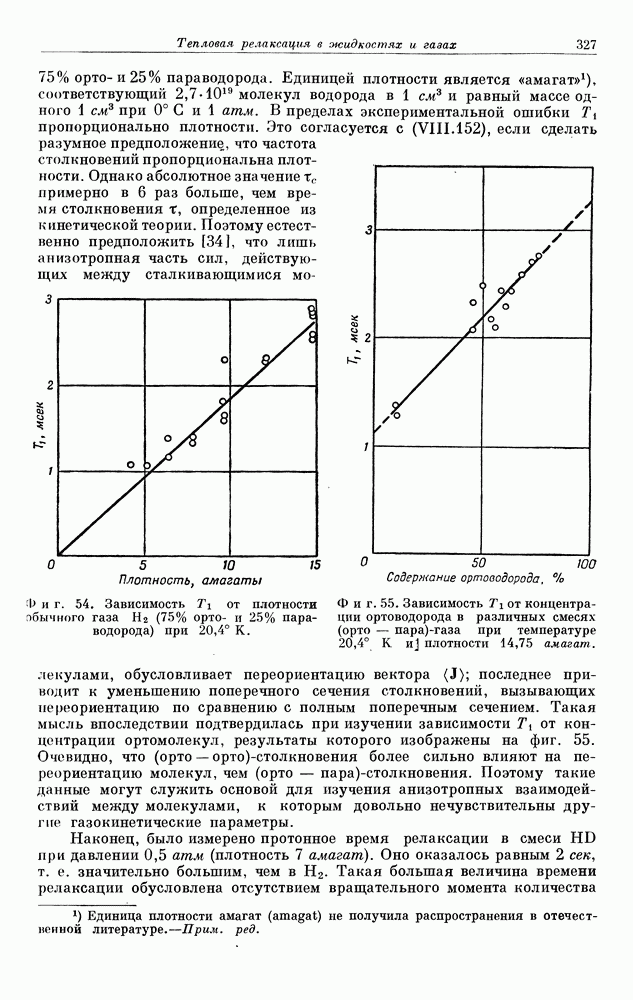
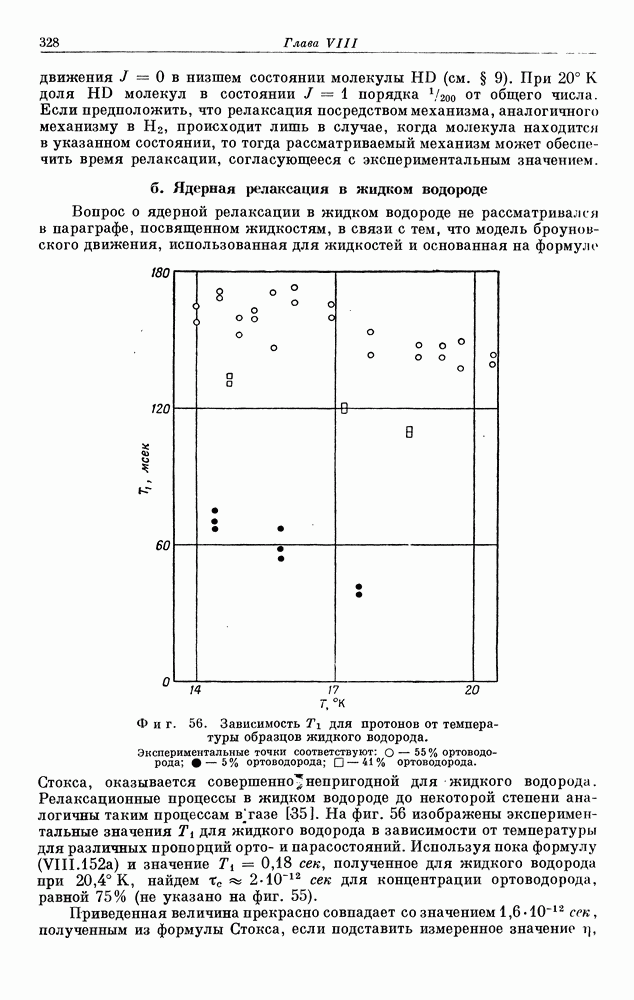
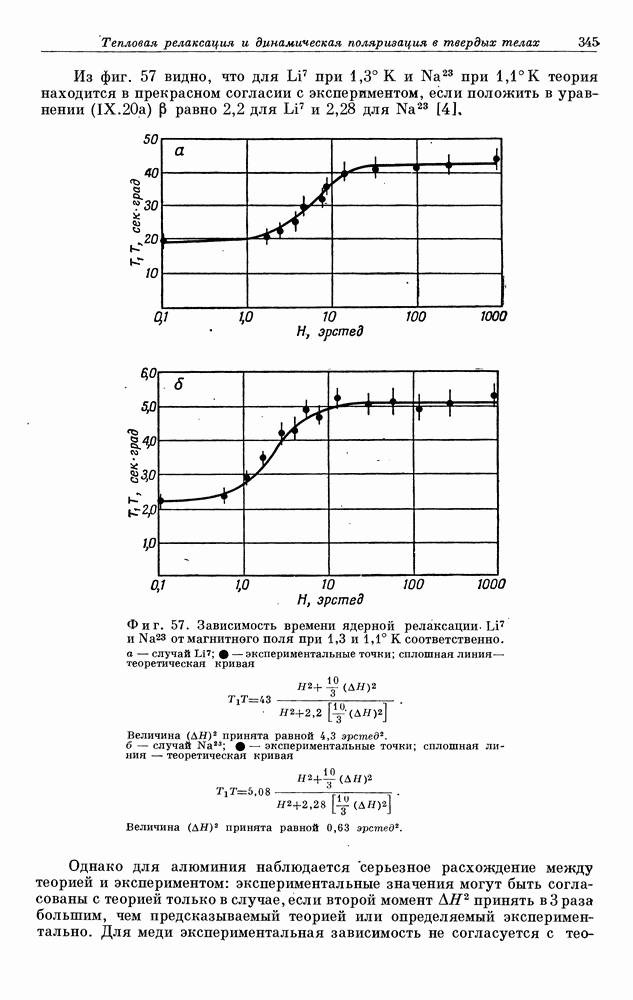
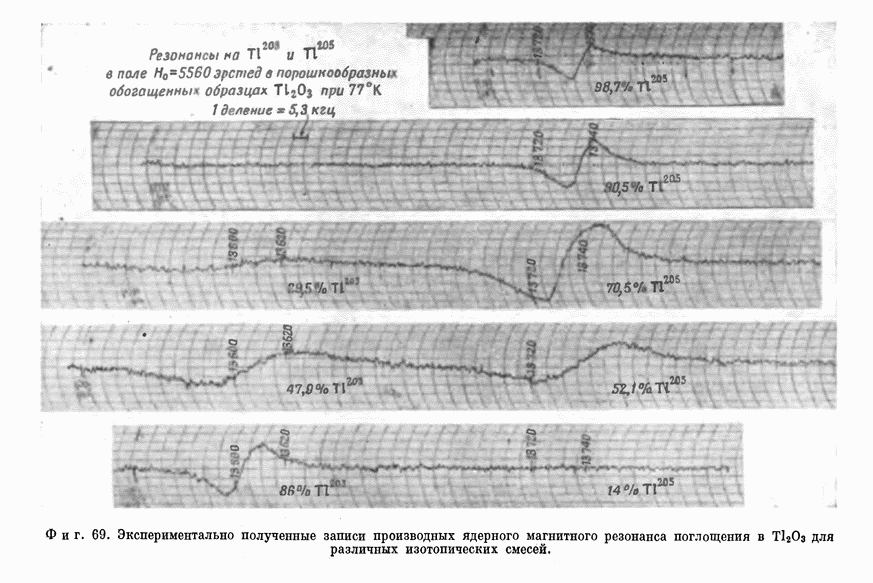
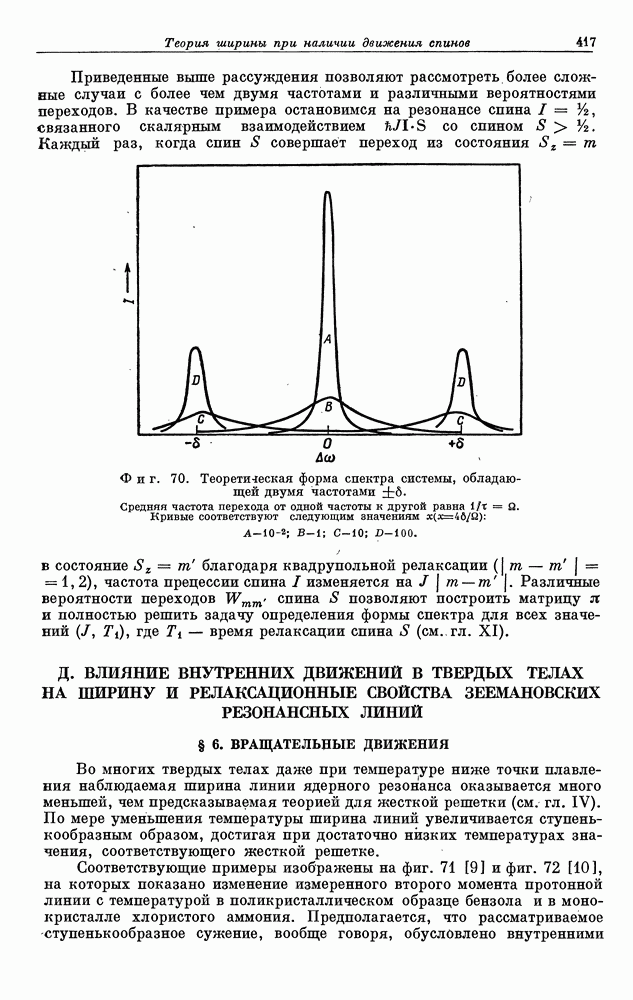
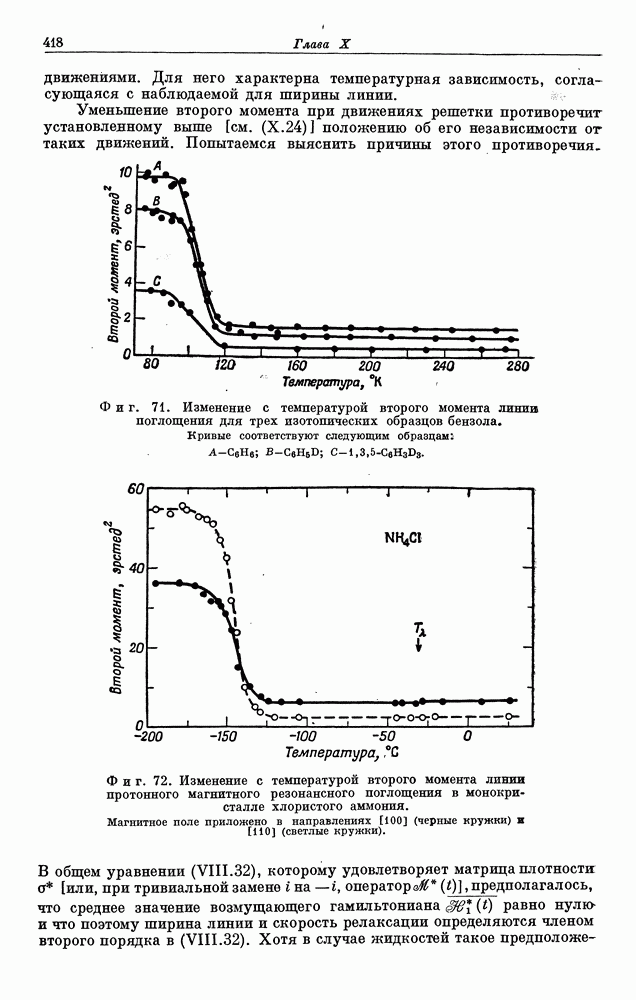
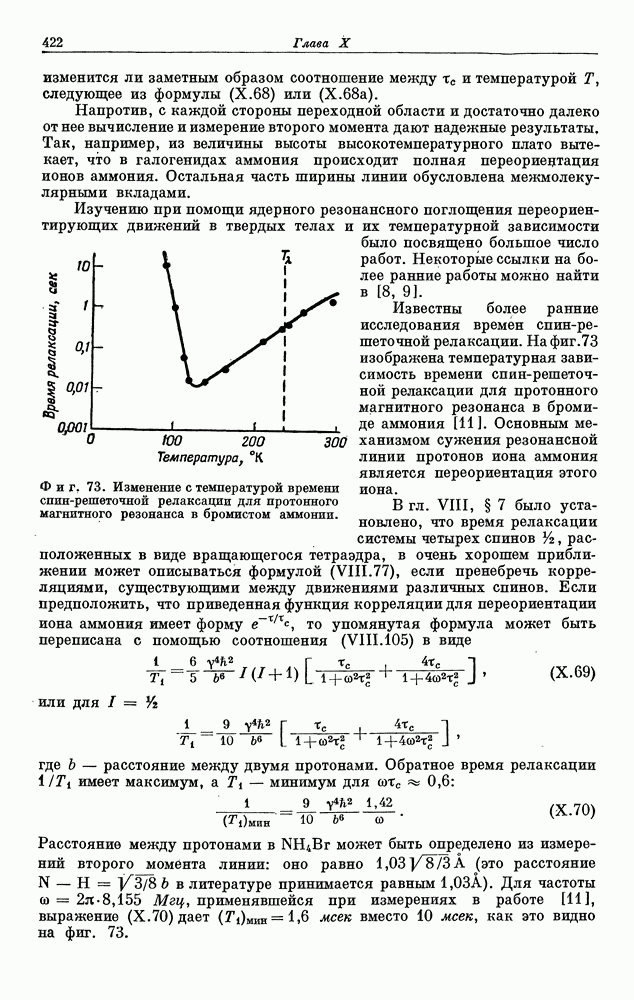
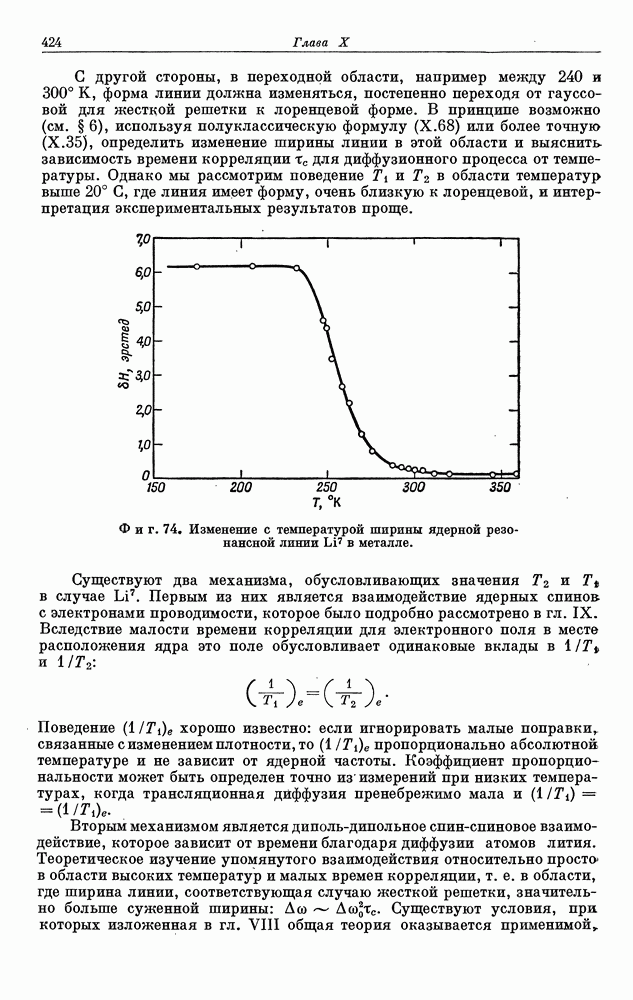
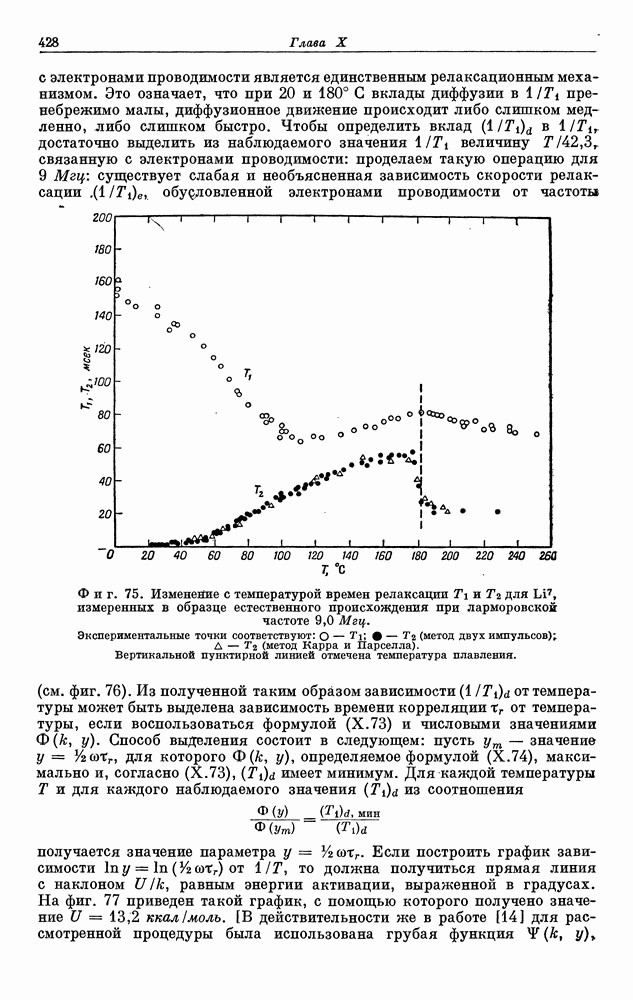
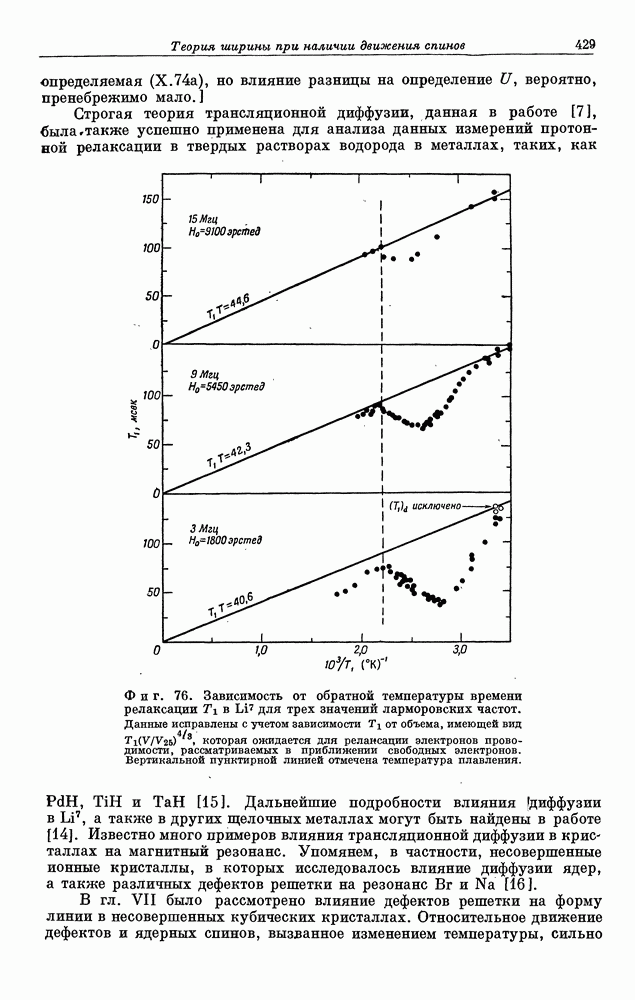
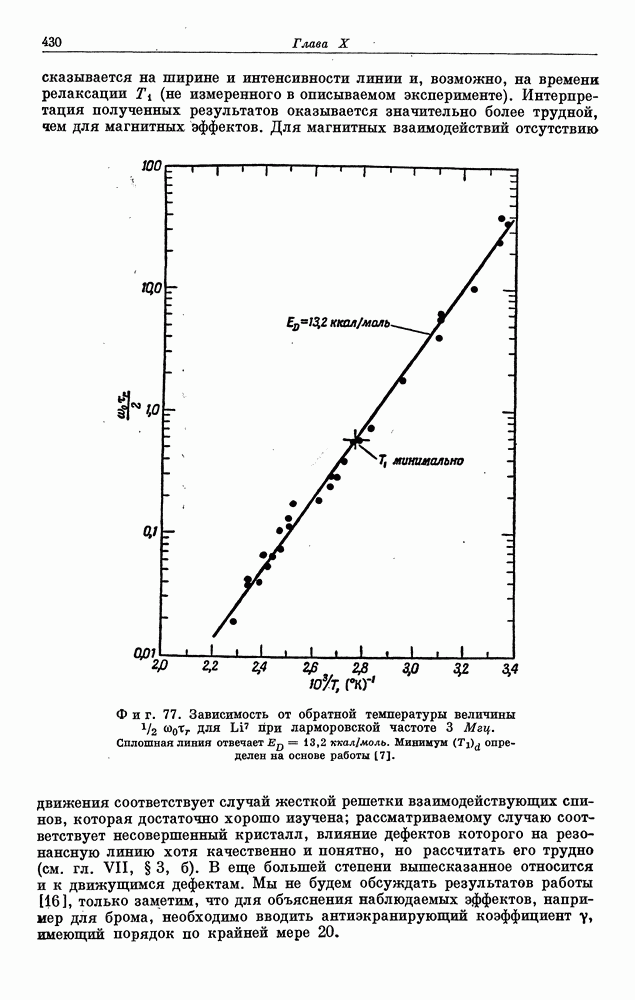
§ 5. ЭЛЕКТРОННО-ЯДЕРНОЕ ВЗАИМОДЕЙСТВИЕ В ДИАМАГНИТНЫХ ВЕЩЕСТВАХ
а. Общие положения
Подавляющее большинство опытов по ядерному резонансу проводится на диамагнитных образцах, т. е. на веществах, не обладающих спиновым или орбитальным электронным парамагнетизмом. Отсутствие электронного парамагнетизма, например, у молекулы, в молекулярном кристалле, или у иона, находящегося в кристалле или растворе, обусловлено равенством нулю ожидаемых значений  для всех составляющих полного орбитального или спинового момента молекулы или иона, где
для всех составляющих полного орбитального или спинового момента молекулы или иона, где  — функция как орбитальных, так и спиновых координат, описывающая основное состояние.
— функция как орбитальных, так и спиновых координат, описывающая основное состояние.
Несмотря на очевидную аналогию свойств орбитальных и спиновых моментов в диамагнитных веществах между их поведением и, следовательно, между их влиянием на характер резонанса ядерных спинов, имеется существенное различие. Силы, действующие между электронами, атомами и молекулами, по существу являются электростатическими силами, а магнитные силы, зависящие от спинов, пренебрежимо малы по сравнению с ними. Отсюда следует, что общий спин электронной системы  можно считать хорошим квантовым числом и что
можно считать хорошим квантовым числом и что  подсостояний состояния с данным
подсостояний состояния с данным  имеют приблизительно одинаковую энергию. Следовательно, если ожидаемые значения трех компонент вектора
имеют приблизительно одинаковую энергию. Следовательно, если ожидаемые значения трех компонент вектора  в невырожденном основном состоянии равны нулю, то для этого состояния обязательно
в невырожденном основном состоянии равны нулю, то для этого состояния обязательно 
В отношении орбитального момента, который для молекулы или иона в плотном веществе не является, вообще говоря, хорошим квантовым числом, подобное заключение сделать нельзя. Равенство нулю  в невырожденном электронном основном состоянии необязательно означает, что общий орбитальный момент
в невырожденном электронном основном состоянии необязательно означает, что общий орбитальный момент  равен нулю, или какому-либо определенному значению в этом состоянии. Напротив, легко показать, что отсутствие орбитального вырождения представляет собой достаточное условие равенства нулю значений
равен нулю, или какому-либо определенному значению в этом состоянии. Напротив, легко показать, что отсутствие орбитального вырождения представляет собой достаточное условие равенства нулю значений  или, как говорят, «замораживания» орбитального момента [9]. Пусть
или, как говорят, «замораживания» орбитального момента [9]. Пусть  волновая функция невырожденного состояния электронной системы. Гамильтониан, являющийся суммой кинетической и электростатической энергии электронов, будет вещественным, и
волновая функция невырожденного состояния электронной системы. Гамильтониан, являющийся суммой кинетической и электростатической энергии электронов, будет вещественным, и  также можно считать вещественной функцией. В противном случае ее действительная и мнимая части были бы по отдельности собственными функциями гамильтониана для одного и того же значения энергии, а это было бы несовместимо с предположением об отсутствии вырождения. Поскольку функция
также можно считать вещественной функцией. В противном случае ее действительная и мнимая части были бы по отдельности собственными функциями гамильтониана для одного и того же значения энергии, а это было бы несовместимо с предположением об отсутствии вырождения. Поскольку функция  вещественная, то величина
вещественная, то величина

обязательно мнимая. С другой стороны, она одновременно должна быть вещественной, ибо  — эрмитов оператор. Таким образом, величина
— эрмитов оператор. Таким образом, величина  должна быть равна нулю.
должна быть равна нулю.
Прежде чем обсуждать различные проявления электронно-ядерного взаимодействия, вернемся к вопросу, кратко обсуждавшемуся в гл. 1, а именно, к вопросу о том, что ядерное орбитальное движение не вызывает появления ядерного парамагнетизма (в отличие от положения, существующего в теории электронного парамагнетизма, для которого орбитальное движение электрона существенно).
Прежде всего следует отметить, что орбитальный ядерный парамагнетизм не всегда мал. Когда ядерное орбитальное движение не замораживается (как, например, в случае молекулы водорода в пучке, когда столкновений почти нет), ядерный орбитальный и спиновый парамагнетизм сравнимы по величине.
В плотном веществе орбитальный ядерный момент замораживается так же, как и электронный орбитальный момент. Основное различие между ядрами и электронами с этой точки зрения состоит в том, что для части электронов орбитальные моменты могут оказаться незамороженными, восстанавливаясь с помощью спин-орбитального взаимодействия. Относительная малость спин-орбитального взаимодействия при ядерном орбитальном движении, вызванная большими ядерными массами, делает такое неполное замораживание в ядерном магнетизме пренебрежимо малым.
В диамагнитном веществе в первом приближении электронно-ядер-ное взаимодействие, представленное гамильтонианом (VI.32) (или суммой таких гамильтонианов для различных электронов, окружающих ядро), исчезает вследствие замораживания орбитального момента и вследствие равенства нулю полного спина  Однако электронно-ядерное взаимодействие проявляется в виде следующих эффектов.
Однако электронно-ядерное взаимодействие проявляется в виде следующих эффектов.
1) Резонансная частота различна для ядра, находящегося в плотном веществе и для «голого» ядра. Природа такого сдвига частоты двоякая: во-первых в присутствии магнитного поля  ларморовская прецессия электронных зарядов вокруг него эквивалентна электрическим токам
ларморовская прецессия электронных зарядов вокруг него эквивалентна электрическим токам
создающим в месте расположения ядра магнитное поле  которое добавляется к внешнему полю и пропорционально ему; во-вторых, внешнее поле
которое добавляется к внешнему полю и пропорционально ему; во-вторых, внешнее поле  поляризует электронные оболочки. Оболочки искажаются, и тем самым создают в месе расположения ядра магнитное поле
поляризует электронные оболочки. Оболочки искажаются, и тем самым создают в месе расположения ядра магнитное поле  также пропорциональное
также пропорциональное 
Общее поле, которое действует на ядро, равно  , где а — относительный сдвиг резонансной частоты, не зависящий от величины
, где а — относительный сдвиг резонансной частоты, не зависящий от величины  Этот сдвиг зависит от распределения электронов вокруг ядра и, конечно, имеет разные значения в различных химических соединениях, поэтому его и называют химическим сдвигом.
Этот сдвиг зависит от распределения электронов вокруг ядра и, конечно, имеет разные значения в различных химических соединениях, поэтому его и называют химическим сдвигом.
2) Косвенные спин-спиновые взаимодействия, отличные от обычных дипольных взаимодействий, могут возникнуть за счет электронов следующим образом. Ядерный момент  создает поле, которое искажает электронные оболочки. Искаженные таким путём оболочки создают поле Н, пропорциональное величине
создает поле, которое искажает электронные оболочки. Искаженные таким путём оболочки создают поле Н, пропорциональное величине  в месте расположения другого ядерного момента
в месте расположения другого ядерного момента  Взаимодействие этого поля с
Взаимодействие этого поля с  приводит в результате к взаимодействию, билинейному относительно
приводит в результате к взаимодействию, билинейному относительно 
Ниже будут приведены основы расчета химического сдвига и косвенных спин-спиновых взаимодействий.
б. Вычисление химического сдвига
Метод вычисления основывается главным образом на работе [10], где впервые была дана полная теория химического сдвига. Для данной молекулы, содержащей  ядерных моментов и
ядерных моментов и  электронов, гамильтониан в присутствии внешнего магнитного поля
электронов, гамильтониан в присутствии внешнего магнитного поля  имеет вид
имеет вид

В этой формуле  — значение векторного потенциала внешнего поля
— значение векторного потенциала внешнего поля  в месте
в месте  электрона, а
электрона, а

является значением векторного потенциала в той же самой точке, созданного ядерным моментом  расположенным в точке с
расположенным в точке с  Начало векторов
Начало векторов  остается неопределенным. Ясно, что все физические результаты не должны зависеть от выбора начала координат; это следует из общего принципа калибровочной инвариантности в электромагнетизме. Для изолированного атома естественно выбрать в качестве начала координат ядро этого атома. В молекуле удобно выбрать в качестве начала координат отдельное ядро
остается неопределенным. Ясно, что все физические результаты не должны зависеть от выбора начала координат; это следует из общего принципа калибровочной инвариантности в электромагнетизме. Для изолированного атома естественно выбрать в качестве начала координат ядро этого атома. В молекуле удобно выбрать в качестве начала координат отдельное ядро  для которого будет рассчитываться частотный сдвиг. Пусть V — электростатическая энергия системы. Для простоты опустим магнитные взаимодействия между электронами, так как они не имеют отношения к рассматриваемой задаче; опустим также зеемановские взаимодействия ядер с внешним полем
для которого будет рассчитываться частотный сдвиг. Пусть V — электростатическая энергия системы. Для простоты опустим магнитные взаимодействия между электронами, так как они не имеют отношения к рассматриваемой задаче; опустим также зеемановские взаимодействия ядер с внешним полем  а также их дипольные взаимодействия, поскольку они хорошо известны из предыдущих глав.
а также их дипольные взаимодействия, поскольку они хорошо известны из предыдущих глав.
Расписывая (VI.36), получаем следующие члены:

где

— кинетическая энергия электронов, 

— их диамагнитная энергия. Эти члены не играют роли в последующем рассуждении. Член  представляет собой зеемановскую энергию электронов во внешнем поле, а
представляет собой зеемановскую энергию электронов во внешнем поле, а

— орбитальную зеемановскую энергию электронов. Сумма  описывает магнитные взаимодействия (VI.32) между
описывает магнитные взаимодействия (VI.32) между  электронами и
электронами и  ядрами
ядрами

где

Не встречавшиеся раньше члены  билинейны, первый по отношению к
билинейны, первый по отношению к  а второй — к
а второй — к 

Член  представляет взаимодействие (упомянутое выше) между ядерными моментами и магнитным полем токов, наведенных ларморовской прецессией электронов во внешнем поле
представляет взаимодействие (упомянутое выше) между ядерными моментами и магнитным полем токов, наведенных ларморовской прецессией электронов во внешнем поле 

Химический сдвиг (а также косвенные взаимодействия, которые будут рассмотрены ниже) соответствует малым изменениям энергии системы и вычисляется с помощью теории возмущений. Более того, химический сдвиг настолько мал, что он обычно не наблюдаем, за исключением жидких (или газообразных) образцов, в которых имеет место быстрое вращение
молекул. Не уточняя способ описания этого вращения (квантовомеханический в газах или классический в большинстве жидкостей), будем обозначать основное состояние молекулы символом  где X относится к ориентации молекулы, и
где X относится к ориентации молекулы, и  к ее другим степеням свободы (электронному и, возможно, колебательному состояниям). Для диамагнитного вещества единственными членами в правой части уравнения (VI.37), для которых ожидаемое значение
к ее другим степеням свободы (электронному и, возможно, колебательному состояниям). Для диамагнитного вещества единственными членами в правой части уравнения (VI.37), для которых ожидаемое значение  не равно нулю, являются
не равно нулю, являются  Первый член, билинейный по и
Первый член, билинейный по и  дает вклад в химический сдвиг. Если выбрать в качестве начала координат отдельный ядерный момент ядра
дает вклад в химический сдвиг. Если выбрать в качестве начала координат отдельный ядерный момент ядра  для которого вычисляется сдвиг, так что
для которого вычисляется сдвиг, так что  становится равным просто
становится равным просто  то в первом приближении изменение будет равно
то в первом приближении изменение будет равно

Это выражение можно переписать в виде  (значок
(значок  обозначает «диамагнитный»). В (VI.38) тензор
обозначает «диамагнитный»). В (VI.38) тензор  может быть разложен на часть с нулевым шпуром
может быть разложен на часть с нулевым шпуром

и скалярную часть  определяемую выражением
определяемую выражением  где
где

не зависит от вращательного состояния молекулы, которое нет необходимости конкретизировать и может быть записано в виде

Ясно, что среднее значение части с нулевым шпуром (VI.38а), взятое по различным вращательным состояниям  ), равно нулю. В жидкости молекула очень быстро переходит из одного состояния
), равно нулю. В жидкости молекула очень быстро переходит из одного состояния  в другое так что наблюдается только среднее по состояниям, поэтому 2 не дает вклада в химический сдвиг, и изменение зеемановской энергии ядра будет равно
в другое так что наблюдается только среднее по состояниям, поэтому 2 не дает вклада в химический сдвиг, и изменение зеемановской энергии ядра будет равно  . В противоположность этому, в опытах с молекулярными пучками, где число столкновений незначительно, и молекулы находятся во вполне определенных вращательных состояниях
. В противоположность этому, в опытах с молекулярными пучками, где число столкновений незначительно, и молекулы находятся во вполне определенных вращательных состояниях  ), наблюдается влияние анизотропной части 2. Постоянная а а всегда положительна и уменьшает внешнее поле
), наблюдается влияние анизотропной части 2. Постоянная а а всегда положительна и уменьшает внешнее поле  на величину
на величину  поэтому ее называют постоянной экранирования. Грубую оценку порядка величины
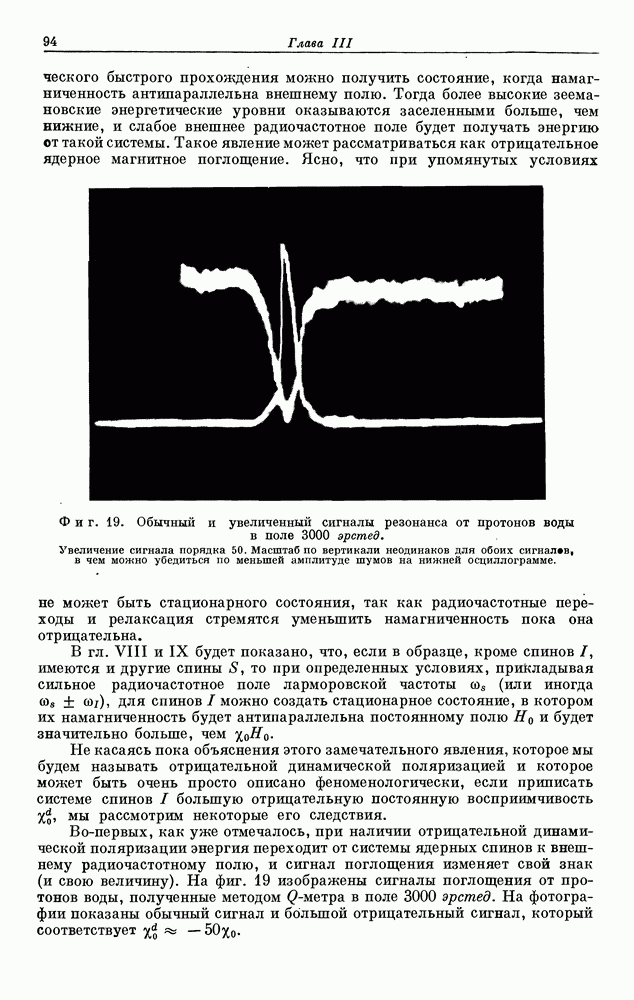
поэтому ее называют постоянной экранирования. Грубую оценку порядка величины  можно сделать, если учесть, что
можно сделать, если учесть, что  — классический радиус электрона, где
— классический радиус электрона, где  — радиус первой боровской орбиты водородного атома. Учитывая, что
— радиус первой боровской орбиты водородного атома. Учитывая, что  имеет порядок
имеет порядок  найдем для
найдем для  значения, лежащие между
значения, лежащие между 
Можно получить другой энергетический член  билинейный по
билинейный по  , следовательно, дающий вклад в химический сдвиг, комбинируя с помощью теории возмущения второго порядка член А из (VI.37), пропорциональный
, следовательно, дающий вклад в химический сдвиг, комбинируя с помощью теории возмущения второго порядка член А из (VI.37), пропорциональный  с членом В, пропорциональным
с членом В, пропорциональным 
Согласно формуле

как А, так и В, должны иметь не равные нулю матричные элементы для перехода между основным состоянием  и возбужденным электронным состоянием
и возбужденным электронным состоянием  (для краткости индекс ориентации К опущен). Поскольку существует три члена, пропорциональных
(для краткости индекс ориентации К опущен). Поскольку существует три члена, пропорциональных  а именно
а именно  и два члена, пропорциональных
и два члена, пропорциональных  а именно
а именно  и
и  то имеется a priori шесть возможных комбинаций, которые могут использоваться в (VI.40). Однако легко показать, что отличной от нуля будет только комбинация
то имеется a priori шесть возможных комбинаций, которые могут использоваться в (VI.40). Однако легко показать, что отличной от нуля будет только комбинация 
Отсутствие вклада от  обусловлено тем, что основное состояние представляет собой собственное состояние
обусловлено тем, что основное состояние представляет собой собственное состояние  Таким образом, оператор В в (VI.40) может быть только, оператором
Таким образом, оператор В в (VI.40) может быть только, оператором  Определим оператор
Определим оператор

Если взаимодействия, а следовательно, и энергии не зависят от спинов, то С — чисто орбитальный оператор. Более того, если считать, что энергетические уровни возбужденных состояний не зависят от ориентации молекулы, то ясно, что оператор С инвариантен относительно вращения. Тогда вклад в (VI.40) за счет  можно записать в виде
можно записать в виде

Поскольку  — собственное состояние
— собственное состояние  — линейные функции составляющих электронных спинов, то (VI.42) становится равным нулю. Если снова ввести индекс ориентации X для основного состояния и выбрать в качестве начала координат рассматриваемый ядерный момент
— линейные функции составляющих электронных спинов, то (VI.42) становится равным нулю. Если снова ввести индекс ориентации X для основного состояния и выбрать в качестве начала координат рассматриваемый ядерный момент  то
то  становится просто равным
становится просто равным  и
и

Выражение (VI.43) можно также переписать в виде тензорного взаимодействия  (индекс
(индекс  обозначает парамагнитный), причем часть
обозначает парамагнитный), причем часть  с нулевым шпуром равна
с нулевым шпуром равна

а скалярная часть имеет вид  где
где

Индекс 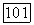 в выражении (VI.436) опущен, поскольку скаляр
в выражении (VI.436) опущен, поскольку скаляр

от него не зависит.
В жидкости вклад анизотропной части в частотный сдвив равен нулю по той же причине, что и для диамагнитной поправки.
Вычисление постоянной парамагнитного экранирования 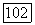 значительно сложнее, чем постоянной так как благодаря оператору
значительно сложнее, чем постоянной так как благодаря оператору 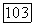 определяемому (VI.41), требуется знание возбужденных состояний молекулы. Величину
определяемому (VI.41), требуется знание возбужденных состояний молекулы. Величину 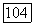 можно грубо оценить, используя так называемое близкое приближение, при котором оператор
можно грубо оценить, используя так называемое близкое приближение, при котором оператор 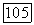 заменяется одной постоянной
заменяется одной постоянной 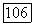 где
где 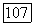 имеет значение средней энергии возбуждения молекулы. Близкое приближение становится точным, если вклад отдельного состояния в сумму (VI.40) значительно превосходит все остальные вклады, например, в случае, когда это состояние очень близко к основному состоянию. В последнем случае такое приближение имеет более глубокий смысл, чем простое обозначение. Оценку порядка величины
имеет значение средней энергии возбуждения молекулы. Близкое приближение становится точным, если вклад отдельного состояния в сумму (VI.40) значительно превосходит все остальные вклады, например, в случае, когда это состояние очень близко к основному состоянию. В последнем случае такое приближение имеет более глубокий смысл, чем простое обозначение. Оценку порядка величины

можно сделать, учитывая, что 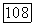 имеет порядок
имеет порядок 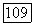 где
где 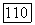 — радиус первой боровской орбиты, и что энергия возбуждения порядка энергии ионизации
— радиус первой боровской орбиты, и что энергия возбуждения порядка энергии ионизации 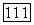 водородного атома. Оказывается, что
водородного атома. Оказывается, что 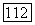
Такую оценку можно считать реальной, потому что хотя 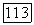 и является поправкой второго порядка, нет причины ожидать, что
и является поправкой второго порядка, нет причины ожидать, что 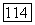 по порядку величины должна быть меньше, чем поправка первого порядка.
по порядку величины должна быть меньше, чем поправка первого порядка.
Если вернуться к выражению (VI.39) для 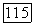 то может показаться странным, что вклад в него от разных электронов спадает довольно медленно как
то может показаться странным, что вклад в него от разных электронов спадает довольно медленно как 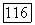 так что даже электроны, локализованные в атомах молекулы, значительно удаленных от рассматриваемого ядра, заметно влияют на химический сдвиг. Это объясняется тем, что парамагнитный член
так что даже электроны, локализованные в атомах молекулы, значительно удаленных от рассматриваемого ядра, заметно влияют на химический сдвиг. Это объясняется тем, что парамагнитный член 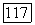 также содержит вклады от удаленных атомов, которые частично погашают вклады, содержащиеся в
также содержит вклады от удаленных атомов, которые частично погашают вклады, содержащиеся в 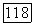 Как
Как 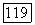 так и
так и 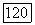 зависят от выбора начала системы координат, в которой заданы векторы
зависят от выбора начала системы координат, в которой заданы векторы 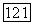 в выражении
в выражении 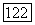 для векторного потенциала
для векторного потенциала 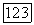 в то время как их сумма
в то время как их сумма 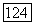 являющаяся измеряемой величиной, естественно не зависит от выбора координат. Если вместо ядра
являющаяся измеряемой величиной, естественно не зависит от выбора координат. Если вместо ядра 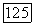 выбрать другое начало координат (например, центр масс молекулы), то выражения для
выбрать другое начало координат (например, центр масс молекулы), то выражения для 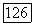 будут иметь вид
будут иметь вид

Было бы желательно так видоизменить выражения для 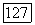 в (VI.39) и (VI.436) или
в (VI.39) и (VI.436) или 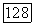 чтобы их сумма не являлась малой разностью двух больших чисел. Ключ к таким вычислениям
чтобы их сумма не являлась малой разностью двух больших чисел. Ключ к таким вычислениям 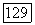 лежит в достижении удовлетворительного описания электронных состояний молекул. Однако эта задача относится к области теоретической химии и выходит за рамки настоящей книги. Для детального ознакомления с методами вычисления а на основе теории молекулярных орбит и сравнением с экспериментом мы отсылаем читателя к оригинальной литературе, незначительная часть которой представлена в ссылках [11—14]. Вклад
лежит в достижении удовлетворительного описания электронных состояний молекул. Однако эта задача относится к области теоретической химии и выходит за рамки настоящей книги. Для детального ознакомления с методами вычисления а на основе теории молекулярных орбит и сравнением с экспериментом мы отсылаем читателя к оригинальной литературе, незначительная часть которой представлена в ссылках [11—14]. Вклад 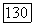 в сдвиг частоты особенно велик, когда молекула имеет возбужденные
в сдвиг частоты особенно велик, когда молекула имеет возбужденные
электронные состояния, близкие к основному состоянию. Если их расстояние от основного состояния сравнимо с 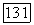 и если тепловые переходы между этими состояниями происходят сравнительно быстро, то будет наблюдаться зависящий от температуры средний химический сдвиг. Критерий необходимой для этого скорости переходов будет дан в гл. X. Описанные эффекты наблюдались в диамагнитных соединениях кобальта [15, 16].
и если тепловые переходы между этими состояниями происходят сравнительно быстро, то будет наблюдаться зависящий от температуры средний химический сдвиг. Критерий необходимой для этого скорости переходов будет дан в гл. X. Описанные эффекты наблюдались в диамагнитных соединениях кобальта [15, 16].
Для линейных молекул в состояниях с моментом 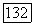 постоянную парамагнитного экранирования
постоянную парамагнитного экранирования 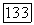 можно связать с магнитным полем
можно связать с магнитным полем 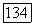 создаваемым в месте расположения ядерного момента
создаваемым в месте расположения ядерного момента 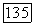 молекулы при ее вращении и приводящем к взаимодействию —
молекулы при ее вращении и приводящем к взаимодействию —  Поле
Поле 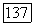 имеет составляющую
имеет составляющую 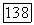 обусловленную вращением ядерных зарядов и составляющую
обусловленную вращением ядерных зарядов и составляющую 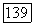 обусловленную электронами.
обусловленную электронами.
Вычисление поля 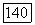 не вызывает затруднений, если исходить из выражения
не вызывает затруднений, если исходить из выражения 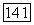 , где
, где 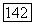 — скорости всех других ядер
— скорости всех других ядер

молекулы относительно рассматриваемого ядра 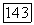 — электростатические поля, создаваемые их зарядами
— электростатические поля, создаваемые их зарядами 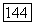 в месте расположения ядра
в месте расположения ядра 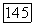 Представим векторы
Представим векторы 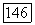 в виде паи где
в виде паи где 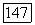 — единичный вектор оси линейной молекулы. Тогда
— единичный вектор оси линейной молекулы. Тогда

где 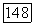 — вектор угловой скорости вращения молекулы,
— вектор угловой скорости вращения молекулы,  ее момент инерции. Таким образом,
ее момент инерции. Таким образом,

Вычисление электронной части 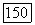 оказывается более сложным. Пусть
оказывается более сложным. Пусть 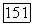 — оператор электронного момента количества движения молекулы (центр масс выбирается в качестве начала координат) и
— оператор электронного момента количества движения молекулы (центр масс выбирается в качестве начала координат) и 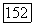 момент, возникающий при вращении молекулы как твердого тела. Энергия этого вращения соответствует гамильтониану
момент, возникающий при вращении молекулы как твердого тела. Энергия этого вращения соответствует гамильтониану
Ожидаемое значение перекрестного члена 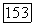 в основном состоянии равно нулю, в то время как матричные элементы для перехода между этим состоянием и возбужденными электронными состояниями молекулы (точно так же, как и зеемановского гамильтониана
в основном состоянии равно нулю, в то время как матричные элементы для перехода между этим состоянием и возбужденными электронными состояниями молекулы (точно так же, как и зеемановского гамильтониана 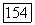 отличны от нуля. Поэтому во втором приближении теории, возмущений можно получить вклады в энергию молекулы, билинейные по
отличны от нуля. Поэтому во втором приближении теории, возмущений можно получить вклады в энергию молекулы, билинейные по 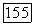 . Совершенно аналогично формуле (VI.43) такой вклад можно записать в виде
. Совершенно аналогично формуле (VI.43) такой вклад можно записать в виде

Второму равенству (VI.46) можно придать вид

где V — векторный оператор. Для основного состояния вектор V можна заменить вектором — 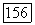 пропорциональным
пропорциональным 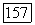 . С помощью метода у
. С помощью метода у
неоднократно использованного ранее, можно получить для Н в выражение

Поскольку величина Не не зависит от молекулярной ориентации Я, можно взять среднее по всем ориентациям молекулы

Внутри символа 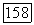 в выражении (VI.48) пояэляются члены вида
в выражении (VI.48) пояэляются члены вида 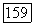 Если фиксировать оси
Если фиксировать оси 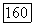 по отношению к остову молекулы
по отношению к остову молекулы 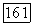 выбрать вдоль оси линейной молекулы), то хорошо известно [17], что
выбрать вдоль оси линейной молекулы), то хорошо известно [17], что 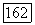 коммутируют с составляющими всех электронных операторов, например, таких, как
коммутируют с составляющими всех электронных операторов, например, таких, как 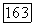 вдоль тех же осей. Среднее в (VI.48) по составляющим
вдоль тех же осей. Среднее в (VI.48) по составляющим 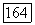 можно вычислять независимо, поэтому перекрестные произведения
можно вычислять независимо, поэтому перекрестные произведения 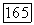 равны нулю. Более того, поскольку
равны нулю. Более того, поскольку 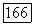 перпендикулярно оси молекулы, то
перпендикулярно оси молекулы, то 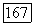 Теперь (VI.48а) можно переписать в виде
Теперь (VI.48а) можно переписать в виде

Поскольку 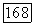 коммутирует с электронным гамильтонианом в линейной молекуле, то
коммутирует с электронным гамильтонианом в линейной молекуле, то 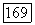 равно нулю. Поэтому, не изменяя результата, в (VI.49) можно ввести добавочный член, пропорциональный
равно нулю. Поэтому, не изменяя результата, в (VI.49) можно ввести добавочный член, пропорциональный

В результате получим выражение

которое при сравнении с (VI.44) дает 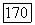 . Учитывая вклад
. Учитывая вклад 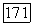 определяемый (VI.45), найдем
определяемый (VI.45), найдем

Следует подчеркнуть, что для справедливости выражения (VI.51) величины 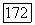 и соответственно а, определенные (VI.44), должны вычисляться по отношению к центру масс молекулы, а не по отношению к ядру
и соответственно а, определенные (VI.44), должны вычисляться по отношению к центру масс молекулы, а не по отношению к ядру 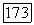
В случае молекулы водорода для вычисления а а по (VI.44) использовались волновая функция, найденная Нордсиком, и значение 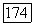 определенное из величины
определенное из величины 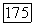 эрстед, найденной методами молекулярных пучков; в результате было получено [10]:
эрстед, найденной методами молекулярных пучков; в результате было получено [10]:

Наконец, отметим, что для вычисления 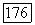 применялись вариационные методы вполне аналогично тому, как это делалось для вычисления наведенных квадрупольных градиентов. Используется возмущенная функция
применялись вариационные методы вполне аналогично тому, как это делалось для вычисления наведенных квадрупольных градиентов. Используется возмущенная функция 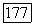 где малая пробная возмущающая функция, содержащая параметры, определяемые из условия минимума энергии молекулы при наличии внешнего поля Н. Затем подсчитывается ожидаемое значение энергии взаимодействия электронов, описываемых с помощью этой возмущенной волновой функции, с ядерным спином. При специальном выборе пробной функции [18] для молекулы водорода было найдено значение
где малая пробная возмущающая функция, содержащая параметры, определяемые из условия минимума энергии молекулы при наличии внешнего поля Н. Затем подсчитывается ожидаемое значение энергии взаимодействия электронов, описываемых с помощью этой возмущенной волновой функции, с ядерным спином. При специальном выборе пробной функции [18] для молекулы водорода было найдено значение 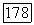 порядка
порядка 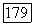 хорошо согласующееся со значением, вытекающим из (VI.51).
хорошо согласующееся со значением, вытекающим из (VI.51).
в. Косвенное взаимодействие между ядерными спинами в диамагнитных веществах
Существование билинейных взаимодействий между ядерными спинами, отличающихся от их дипольных взаимодействий, впервые было предположено после открытия, сделанного несколькими группами исследователей в 1950 г. Было установлено, что в жидких образцах некоторые линии ядерного резонанса имеют мультиплетную структуру, причем расстояния между различными компонентами не зависят от поля. Независимость расщепления от поля показывает, что различные компоненты мультиплета не могут быть приписаны ядрам с различными химическими окружениями, а следовательно, с различными химическими сдвигами, ибо в этом случае интервалы между линиями были бы прямо пропорциональны полю. Поскольку такие структуры наблюдались в жидкостях, в которых существует очень быстрое хаотическое вращение молекулы, то в противоположность случаю твердых образцов дипольное спин-спи-новое взаимодействие не может быть ответственным за наблюдавшуюся структуру. Действительно, любое билинейное взаимодействие между двумя ядерными спинами 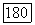 можно записать в виде
можно записать в виде 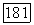 , где
, где 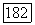 — некоторый
— некоторый 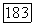 тензор (удобно ввести постоянную
тензор (удобно ввести постоянную 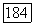 , чтобы компоненты тензора
, чтобы компоненты тензора 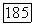 имели размерности частоты). Тензор
имели размерности частоты). Тензор 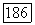 может быть разложен на две части: часть со шпуром, равным нулю, где
может быть разложен на две части: часть со шпуром, равным нулю, где 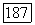 и диагональную часть
и диагональную часть  где
где 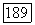 Выше уже отмечалось, что при условиях быстрого вращения молекул, которое преобладает в жидкости, часть тензора с равным нулю шпуром отсутствует. Так как дипольное взаимодействие точно описывается тензором с равным нулю шпуром, то
Выше уже отмечалось, что при условиях быстрого вращения молекул, которое преобладает в жидкости, часть тензора с равным нулю шпуром отсутствует. Так как дипольное взаимодействие точно описывается тензором с равным нулю шпуром, то 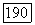 не влияет на энергию уровней ядерных спинов и не может быть причиной наблюдавшейся структуры.
не влияет на энергию уровней ядерных спинов и не может быть причиной наблюдавшейся структуры.
Так же как и в случае квадрупольных взаимодействий, рассмотренных в разделе А, анализ косвенных спин-спиновых взаимодействий можно разделить на две части. К первой части отнесем вычисление тензоров 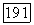 на основании общих принципов, исходя из электронной структуры молекул; ко второй — вычисление с помощью этих тензоров (или, по крайней мере для жидкостей, их диагональных частей
на основании общих принципов, исходя из электронной структуры молекул; ко второй — вычисление с помощью этих тензоров (или, по крайней мере для жидкостей, их диагональных частей 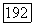 относительных положений интенсивностей, а иногда, возможно, и ширин линий компонент мультиплетной структуры, вызванной этими взаимодействиями (вообще говоря, в присутствии химических сдвигов). В настоящей главе будет рассмотрена только первая часть этого анализа; мультиплетныеструктуры будут рассмотрены в гл. XI.
относительных положений интенсивностей, а иногда, возможно, и ширин линий компонент мультиплетной структуры, вызванной этими взаимодействиями (вообще говоря, в присутствии химических сдвигов). В настоящей главе будет рассмотрена только первая часть этого анализа; мультиплетныеструктуры будут рассмотрены в гл. XI.
С тех пор как было выяснено, что косвенные взаимодействия объясняются с помощью искаженной электронной волновой функции [19]
(отсюда и название косвенных взаимодействий), было сделано много попыток связать величину этих взаимодействий с электронной структурой молекул. Хотя эти расчеты в принципе просты, в них мы встречаемся практически с теми же трудностями, что и при вычислении химических сдвигов, а именно с отсутствием удовлетворительных методов описания возбужденных состояний молекул (а также иногда и их основного состояния). Как и выше, рассмотрим основы теории, опуская подробности, которые относятся к области теоретической химии и которые можно найти в далеко не исчерпывающем списке работ [20—23].
1. Орбитальная связь. Гамильтониан (VI.37) содержит член 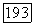 билинейный по отношению к ядерным моментам молекул, а ожидаемое значение энергии соответствующего ядерного взаимодействия равно
билинейный по отношению к ядерным моментам молекул, а ожидаемое значение энергии соответствующего ядерного взаимодействия равно 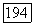 где
где 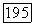 — волновад функция основного состояния молекулы.
— волновад функция основного состояния молекулы.
Хотя 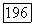 — член первого порядка, он очень мал. Последнее по существу обусловлено тем, что
— член первого порядка, он очень мал. Последнее по существу обусловлено тем, что 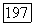 представляет собой сумму одноэлектронных операторов вида
представляет собой сумму одноэлектронных операторов вида

Когда первый множитель 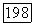 относительно велик
относительно велик  электрон находится в непосредственной близости от ядерного момента
электрон находится в непосредственной близости от ядерного момента  второй множитель обязательно мал. Более того, вклад в
второй множитель обязательно мал. Более того, вклад в  от внутренних атомных оболочек, для которых соответствующий множитель очень велик, существенно уменьшается благодаря его угловой зависимости. Пусть
от внутренних атомных оболочек, для которых соответствующий множитель очень велик, существенно уменьшается благодаря его угловой зависимости. Пусть  соответствует внутренней орбите атома А с линейными размерами, значительно меньшими, чем междуядерное расстояние
соответствует внутренней орбите атома А с линейными размерами, значительно меньшими, чем междуядерное расстояние 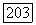 Расстояние от электрона на этой орбите до ядра В практически постоянно и равно
Расстояние от электрона на этой орбите до ядра В практически постоянно и равно 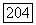 поэтому
поэтому

ибо орбита 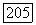 имеет вполне определенную четнрсть по отношению к ядру А. Во втором приближении теории возмущений, кроме члена первого порядка
имеет вполне определенную четнрсть по отношению к ядру А. Во втором приближении теории возмущений, кроме члена первого порядка 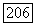 можно получить другие вклады в спин-спиновое взаимодействие, комбинируя члены
можно получить другие вклады в спин-спиновое взаимодействие, комбинируя члены 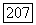 по два. Полученные таким способом члены имеют вид
по два. Полученные таким способом члены имеют вид 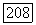 , где
, где 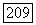 — один из трех операторов
— один из трех операторов  оператор, уже определенный выше,
оператор, уже определенный выше,

Из шести комбинаций, которые можно образовать таким способом, комбинации, смешивающие орбитальные и спиновые члены, а именно 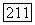 не дают вклада. Доказательство этого утверждения аналогично уже приведенному выше для случая химического сдвига: если зависящие от спинов силы (например, спин-орбитальные) малы, то С будет чисто орбитальным оператором, и так как основное состояние
не дают вклада. Доказательство этого утверждения аналогично уже приведенному выше для случая химического сдвига: если зависящие от спинов силы (например, спин-орбитальные) малы, то С будет чисто орбитальным оператором, и так как основное состояние 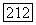 — собственное состояние полного спина с равным нулю собственным значением, член вида
— собственное состояние полного спина с равным нулю собственным значением, член вида 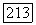 равен нулю. Кроме того, если опыт проводится в жидкости и наблюдается только скалярная часть взаимодействия, то смешанные члены
равен нулю. Кроме того, если опыт проводится в жидкости и наблюдается только скалярная часть взаимодействия, то смешанные члены 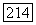 также равны нулю благодаря различию трансформационных свойств
также равны нулю благодаря различию трансформационных свойств 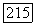
Грубую оценку порядка величины полного вклада орбитальных членов 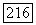 можно сделать следующим образом [23]. Будем считать, что размеры двух атомов А и В, содержащих ядерные спины
можно сделать следующим образом [23]. Будем считать, что размеры двух атомов А и В, содержащих ядерные спины 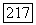 значительно меньше, чем расстояние
значительно меньше, чем расстояние 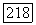 тогда поле, созданное спином
тогда поле, созданное спином 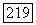 приблизительно однородно в области атома В и имеет составляющие
приблизительно однородно в области атома В и имеет составляющие

где

Вводя тензор экранирования Для атома В, получаем для энергии взаимодействия ядерного спина с этим полем выражение

Отсюда для косвенного взаимодействия найдем

К этому выражению должен быть прибавлен аналогичный член, содержащий тензор экранирования  Для атома А. Таким образом, отношение энергии орбитального косвенного и прямого взаимодействий
Для атома А. Таким образом, отношение энергии орбитального косвенного и прямого взаимодействий 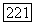 имеет порядок постоянной экранирования а, которая всегда оказывается очень малым числом. Поскольку в определенных случаях косвенное взаимодействие может даже превосходить прямое, то ясно, что орбитальное взаимодействие не будет его преобладающей частью. Ниже рассмотрены некоторые приближенные способы оценки косвенных взаимодействий.
имеет порядок постоянной экранирования а, которая всегда оказывается очень малым числом. Поскольку в определенных случаях косвенное взаимодействие может даже превосходить прямое, то ясно, что орбитальное взаимодействие не будет его преобладающей частью. Ниже рассмотрены некоторые приближенные способы оценки косвенных взаимодействий.
2. Приближение Гайтлера — Лондона. В приближении Гайтлера — Лондона химическая связь между двумя атомами А и В описывается двухэлектронной волновой функцией

Здесь 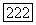 — нормированные атомные орбиты, относящиеся соответственно к атомам А и В, или линейные комбинации таких орбит, а
— нормированные атомные орбиты, относящиеся соответственно к атомам А и В, или линейные комбинации таких орбит, а 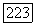 — функция двух электронных спинов и
— функция двух электронных спинов и 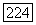 описывающая синглетное спиновое состояние [заметим, что (VI.55) не слетеровский детерминант, а сумма двух таких детерминантов]. Постоянная
описывающая синглетное спиновое состояние [заметим, что (VI.55) не слетеровский детерминант, а сумма двух таких детерминантов]. Постоянная

определяет степень перекрытия двух орбит. В близком приближении, когда оператор С заменяется постоянной, косвенное взаимодействие между двумя ядерными спинами 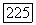 определяется выражением
определяется выражением

(индекс X для ориентации молекулы опущен), где 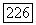 содержат соответственно
содержат соответственно 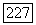 в сумме
в сумме 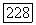 Естественно, что из (VI.56) должны быть выбраны только члены, билинейные по
Естественно, что из (VI.56) должны быть выбраны только члены, билинейные по 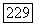
Раскрывая (VI.56) и учитывая (VI.55), получаем шесть типов членов

где все матричные элементы являются одноэлектронными интегралами. Гамильтониан 36 к велик, если электрон находится вблизи ядра атома А, и мал, если электрон находится где-нибудь в другом месте. Поэтому разумно ожидать, что первый член будет значительно больше остальных, и в первом приближении можно сохранить только этот член. Если орбиты 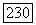 представляются вещественными волновыми функциями, так что орбитальный момент замораживается (как это бывает обычно в случае диамагнитных веществ), то
представляются вещественными волновыми функциями, так что орбитальный момент замораживается (как это бывает обычно в случае диамагнитных веществ), то 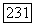 , и остаются только спиновые члены. Тогда ожидаемая величина
, и остаются только спиновые члены. Тогда ожидаемая величина

обязательно имеет вид 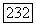 где
где 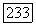 - некоторый тензор, который описывал бы сверхтонкую структуру атома А, если бы можно было пренебречь искажением орбиты
- некоторый тензор, который описывал бы сверхтонкую структуру атома А, если бы можно было пренебречь искажением орбиты 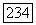 присутствием атома В. В этом случае взаимодействие между двумя спинами
присутствием атома В. В этом случае взаимодействие между двумя спинами 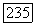 можно записать в виде
можно записать в виде 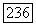 где симметричный тензор
где симметричный тензор 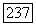 определяется выражением
определяется выражением

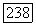 — симметризованное произведение, равное
— симметризованное произведение, равное 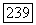 Поскольку
Поскольку 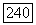 — синглетная спиновая функция, справедливы следующие соотношения:
— синглетная спиновая функция, справедливы следующие соотношения:

откуда

где 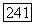 — симметризованное тензорное произведение тензоров
— симметризованное тензорное произведение тензоров 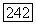
Из (VI.58) видно, что 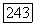 — величина порядка
— величина порядка  где
где 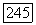 и
и 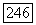 -постоянные сверхтонкого расщепления для каждого атома в единицах частоты,
-постоянные сверхтонкого расщепления для каждого атома в единицах частоты, 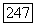 — среднее расстояние (в тех же единицах) между основным и возбужденным состояниями молекулы. Поэтому следует ожидать значительных
— среднее расстояние (в тех же единицах) между основным и возбужденным состояниями молекулы. Поэтому следует ожидать значительных 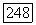 -взаимодействий, когда атомы А и В являются тяжелыми атомами с большими постоянными сверхтонкого расщепления или когда молекула имеет весьма низко расположенные возбужденные состояния.
-взаимодействий, когда атомы А и В являются тяжелыми атомами с большими постоянными сверхтонкого расщепления или когда молекула имеет весьма низко расположенные возбужденные состояния.
Изложенную выше теорию можно применить к молекуле 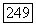 в этом случае изотропная часть
в этом случае изотропная часть 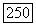 представляет собой единственную часть
представляет собой единственную часть
взаимодействия, которая измерялась в опыте, выполненном с газообразным образцом под давлением [24]. Эта величина оказалась равной 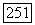 гц (причина, по которой не мог быть поставлен опыт с
гц (причина, по которой не мог быть поставлен опыт с 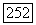 будет рассмотрена в гл. XI). Лучше всего полученный результат сравнивать с теорией, если считать
будет рассмотрена в гл. XI). Лучше всего полученный результат сравнивать с теорией, если считать 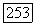 неизвестным и с помощью (VI.58) проверять, получится ли для него разумное значение. Если для
неизвестным и с помощью (VI.58) проверять, получится ли для него разумное значение. Если для 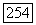 взять
взять  -орбиты Н и
-орбиты Н и 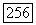 то
то 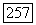 становятся скалярами, и (VI.58) принимает вид
становятся скалярами, и (VI.58) принимает вид

где 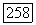 — величина сверхтонкого расщепления для водородного атома. Принимая
— величина сверхтонкого расщепления для водородного атома. Принимая

найдем, что

Для описания основного состояния молекулы 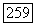 и для вычисления
и для вычисления 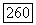 по выражению (VI.56) значительно лучше использовать вместо приближения Гайтлера — Лондона волновую функцию Джеймса и Кулиджа. Значение
по выражению (VI.56) значительно лучше использовать вместо приближения Гайтлера — Лондона волновую функцию Джеймса и Кулиджа. Значение 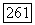 полученное таким способом, равно
полученное таким способом, равно 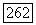 Для расстояния между возбужденным триплетным и основным состояниями Джеймс и Кулидж нашли
Для расстояния между возбужденным триплетным и основным состояниями Джеймс и Кулидж нашли 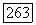 Хотя для описания основного состояния волновая функция Джеймса и Кулиджа, несомненно, лучше, чем приближение Гайтлера — Лондона, неопределенности, обусловленные использованием близкого приближения, таковы, что любое дальнейшее обсуждение этого расхождения, по-видимому, бесполезно.
Хотя для описания основного состояния волновая функция Джеймса и Кулиджа, несомненно, лучше, чем приближение Гайтлера — Лондона, неопределенности, обусловленные использованием близкого приближения, таковы, что любое дальнейшее обсуждение этого расхождения, по-видимому, бесполезно.
Интересно выяснить природу косвенного спин-спинового взаимодействия в другом веществе, а именно в твердом ацетате меди, однако отметим, что благодаря экспериментальным трудностям косвенное спин-спиновое взаимодействие в нем до сих пор не наблюдалось.
Опыты по электронному резонансу [25] показали, что в кристалле ацетата меди ионы меди 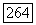 с электронными спинами
с электронными спинами 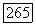 объединяются в пары и что между этими спинами существует электронное обменное взаимодействие
объединяются в пары и что между этими спинами существует электронное обменное взаимодействие 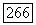 Знак А положителен, а его величина
Знак А положителен, а его величина 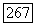 Таким образом, каждая пара ионов имеет диамагнитное основное состояние
Таким образом, каждая пара ионов имеет диамагнитное основное состояние 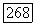 и парамагнитное возбужденное состояние
и парамагнитное возбужденное состояние 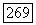 в котором можно наблюдать парамагнитный резонанс. Очевидная аналогия с молекулой
в котором можно наблюдать парамагнитный резонанс. Очевидная аналогия с молекулой 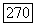 позволяет фазу вычислить косвенное взаимодействие с помощью формулы (VI.58), принимая
позволяет фазу вычислить косвенное взаимодействие с помощью формулы (VI.58), принимая 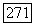 Сравнивая со случаем
Сравнивая со случаем 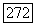 можно сделать следующие выводы.
можно сделать следующие выводы.
1) При 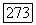 вместо
вместо 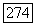 (при равенстве остальных величин) косвенное взаимодействие должно быть в 200 раз больше. На основании известных значений сверхтонких взаимодействий для изолированных ионов
(при равенстве остальных величин) косвенное взаимодействие должно быть в 200 раз больше. На основании известных значений сверхтонких взаимодействий для изолированных ионов 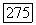 измеренных в различных солях меди, должны ожидаться значения
измеренных в различных солях меди, должны ожидаться значения 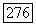 порядка
порядка 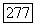 гц, т. е. почти в 20 раз больше, чем величина обычного дипольного взаимодействия между ядерными спинами пары.
гц, т. е. почти в 20 раз больше, чем величина обычного дипольного взаимодействия между ядерными спинами пары.
2) Поскольку основной вклад во взаимодействие 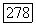 вносится отдельным возбужденным состоянием, то близкое приближение хорошо подтверждается.
вносится отдельным возбужденным состоянием, то близкое приближение хорошо подтверждается.
3) Тензоры 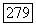 описывающие сверхтонкую структуру индивидуальных ионов меди, обладают большой анизотропией. Отношение между наибольшим и наименьшим главными значениями тензора порядка
описывающие сверхтонкую структуру индивидуальных ионов меди, обладают большой анизотропией. Отношение между наибольшим и наименьшим главными значениями тензора порядка 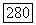 . Оси двух тензоров
. Оси двух тензоров 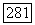 в Для двух ионов пары параллельны, и соответствующая величина (анизотропия) для тензора
в Для двух ионов пары параллельны, и соответствующая величина (анизотропия) для тензора 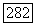 определенного формулой (VI.58), может быть порядка 50.
определенного формулой (VI.58), может быть порядка 50.
В качестве третьего примера, в котором может быть использовано приближение Гайтлера — Лондона, проведем анализ [26] тонкой структуры спектра квадрупольного резонанса в кристаллическом йоде [27].
Орбиты 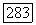 предполагаются линейными комбинациями
предполагаются линейными комбинациями  -атомных орбит атомов двухатомной молекулы йода
-атомных орбит атомов двухатомной молекулы йода

где постоянная 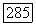 — мера
— мера 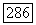 -характера волновой функции. Тензор
-характера волновой функции. Тензор 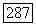 описывающий косвенное взаимодействие, можно вычислить по (VI.58). Тензоры сверхтонкого взаимодействия
описывающий косвенное взаимодействие, можно вычислить по (VI.58). Тензоры сверхтонкого взаимодействия  а и
а и 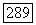 в легко найти, вычисляя ожидаемые значения гамильтониана (VI.32) с помощью волновых функций (VI.60). Если ось
в легко найти, вычисляя ожидаемые значения гамильтониана (VI.32) с помощью волновых функций (VI.60). Если ось 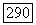 направить вдоль оси молекулы йода, то
направить вдоль оси молекулы йода, то

кроме того, та (VI.58) имеем

где пренебрегается перекрытием А двух орбит  Несколько неопределенные экспериментальные данные приблизительно соответствуют
Несколько неопределенные экспериментальные данные приблизительно соответствуют

Значение 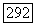 точно известно из наблюдения спектра атома йода,
точно известно из наблюдения спектра атома йода, 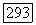 можно подсчитать с помощью волновых функций экранированных атомов, а
можно подсчитать с помощью волновых функций экранированных атомов, а 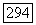 -характер связи определяется либо по анизотропии, либо по абсолютной величине главных значений тензора
-характер связи определяется либо по анизотропии, либо по абсолютной величине главных значений тензора 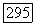 Изотропия
Изотропия 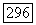 потребовала бы
потребовала бы 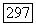 что привело бы, если принять
что привело бы, если принять 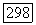 к неприемлемому значению
к неприемлемому значению 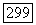
Так как величина главных значений тензора 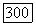 более чувствительна к значению чем его анизотропия, то авторы [26] выбрали значение которое при сделанных ими предположениях должно давать
более чувствительна к значению чем его анизотропия, то авторы [26] выбрали значение которое при сделанных ими предположениях должно давать

Это значение представляет собой компромисс между значениями, полученными из наблюдавшейся анизотропии и абсолютной величины взаимодействия. Совпадение с другими химическими данными вполне приемлемо.
3. Метод молекулярных орбит. Метод молекулярных орбит, позволяющий подходить к проблеме химической связи другим, отличным от приближения Гайтлера — Лондона путем, также дает
возможность рассмотреть проблему косвенных спин-спиновых взаимодействий [22].
В принципе метод молекулярных орбит состоит в распространении метода самосогласованного поля для атомов на молекулы. Вместо центрального самосогласованного поля, как в случае атомов, рассматривается самосогласованное поле, обладающее симметрией молекулы. Осуществить практически такое обобщение слишком трудно, и часто, пренебрегая самосогласованностью, довольствуются представлением каждой молекулярной орбиты в виде линейной комбинации орбит, относящихся к различным атомам молекулы. В случае диамагнитной молекулы в основном состоянии можно принять, что имеются ортогональных орбит населенных электронами. Учитывая спин, можно построить слетеровский детерминант одноэлектронных состояний два на орбиту). Затем можно подсчитать косвенное взаимодействие второго порядка с помощью соотношения

где — гамильтониан (VI.32) электрона относительно ядра Если используется близкое приближение, то (VI.62) принимает вид

Выражение (VI.63) можно записать в явной форме, пользуясь обычными правилами раскрытия слетеровских детерминантов. При этом получаются члены следующих типов: одночастичные матричные элементы

прямые двухчастичные матричные элементы

обменные двухчастичные матричные элементы

Орбиты не обязательно локализованы вблизи некоторого отдельного атома, поэтому не существует простого критерия оценки относительной величины этих членов, как в приближении Гайтлера — Лондона. Поскольку каждая молекулярная орбита является линейной комбинацией

где — атомные орбиты атома то (VI.63) может быть вычислено, вели известны Дальнейшее развитие этого метода можно найти в [22]. В разделе о металлах (см. гл. VI, § 6, б), где орбиты электронов проводимости простираются на весь образец, будет рассмотрено важное лриложение описанного метода.