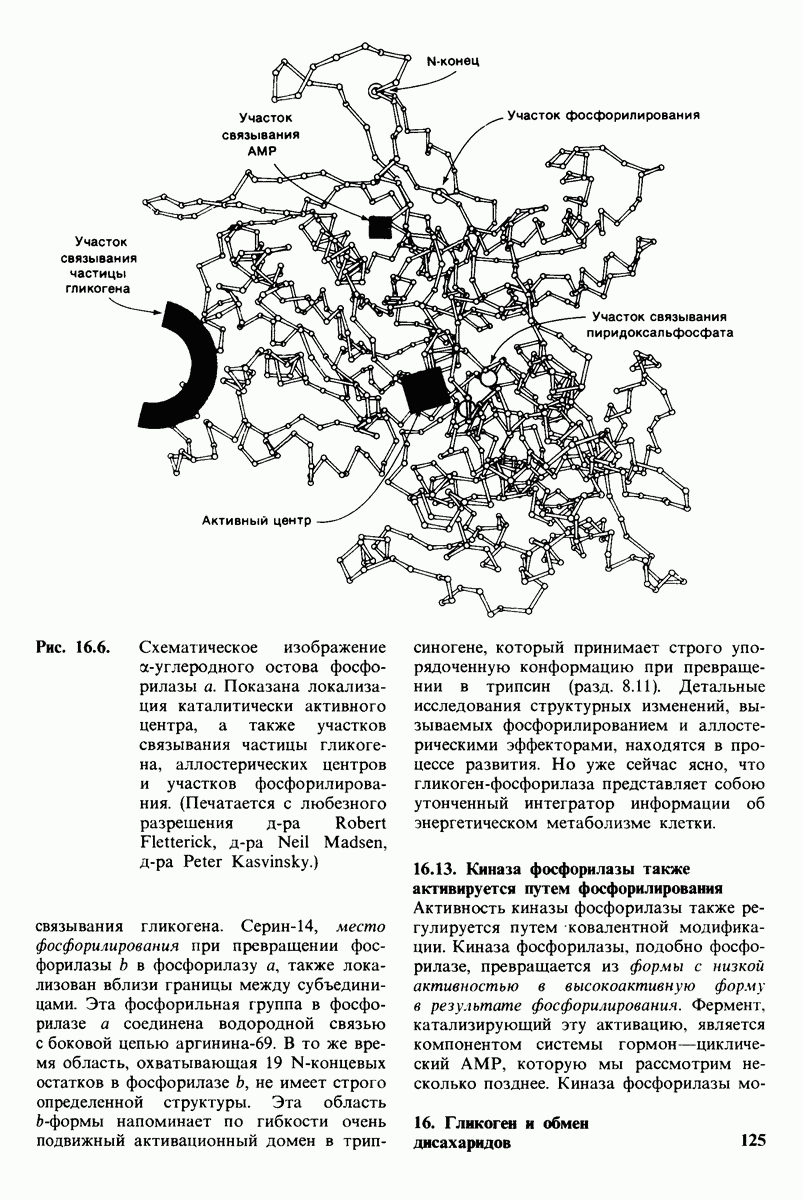
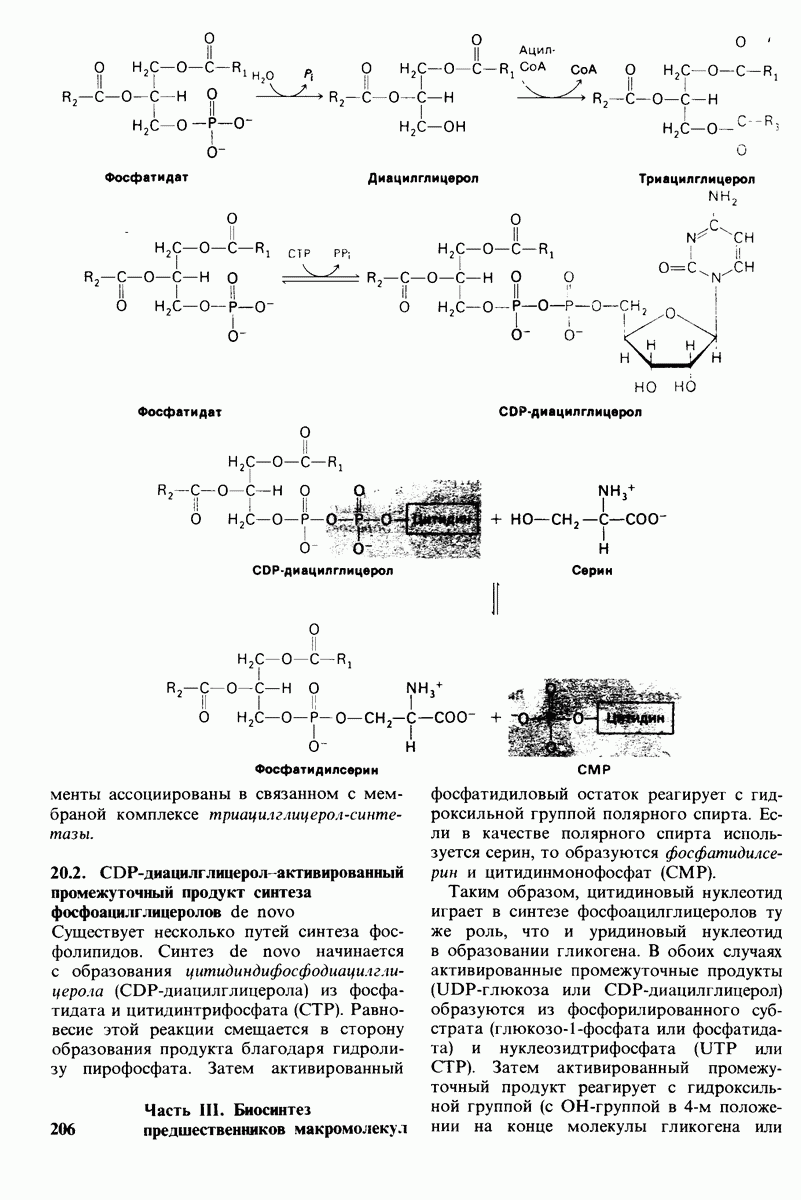
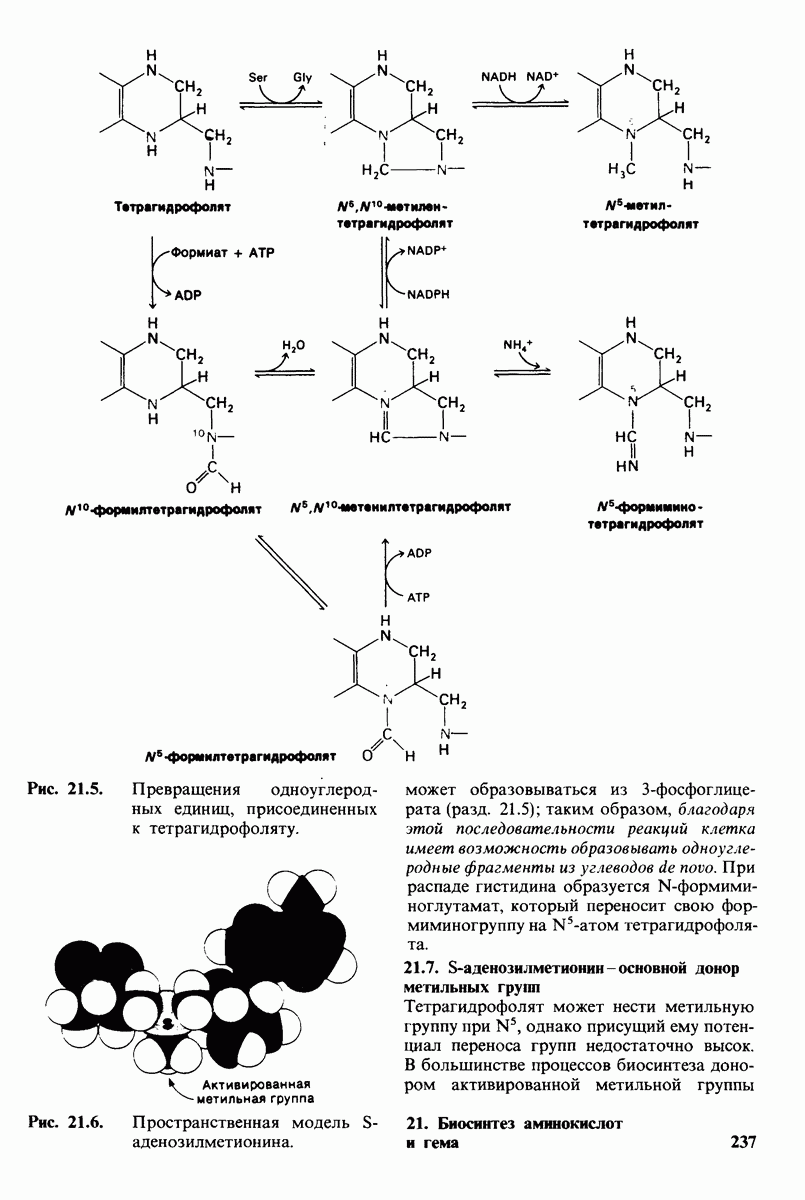
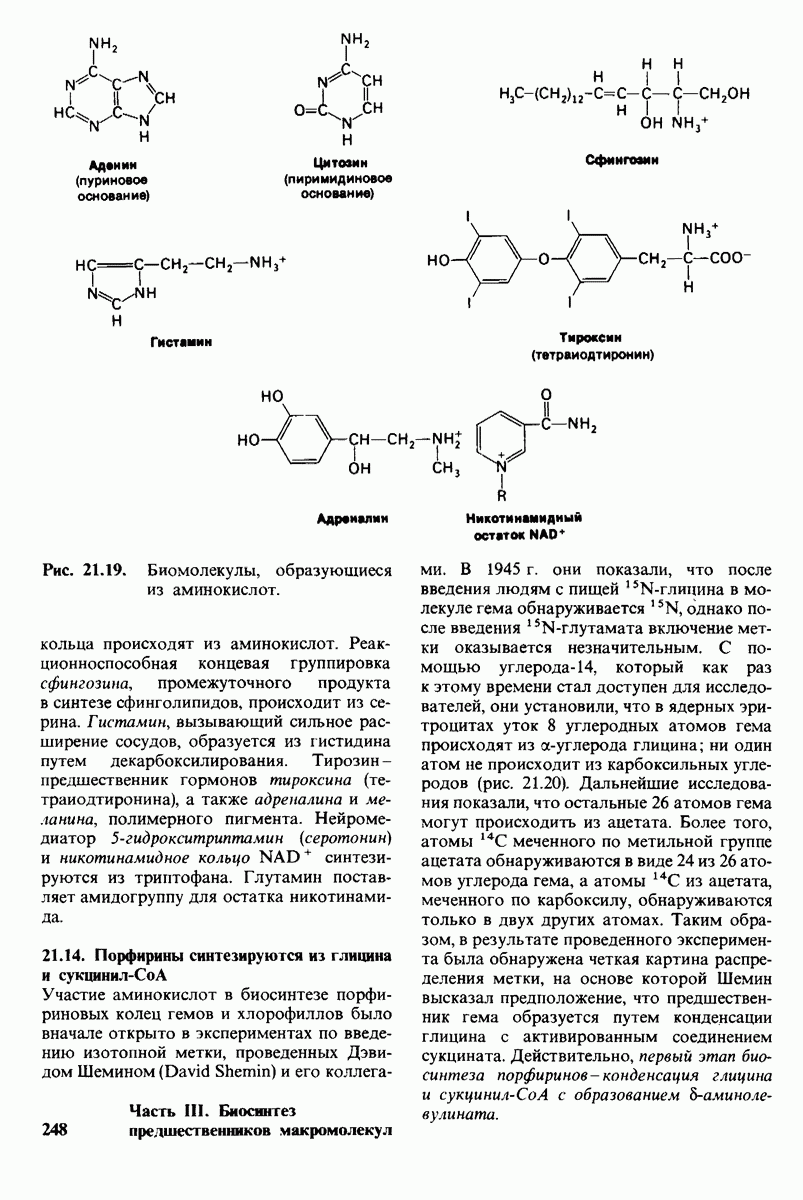
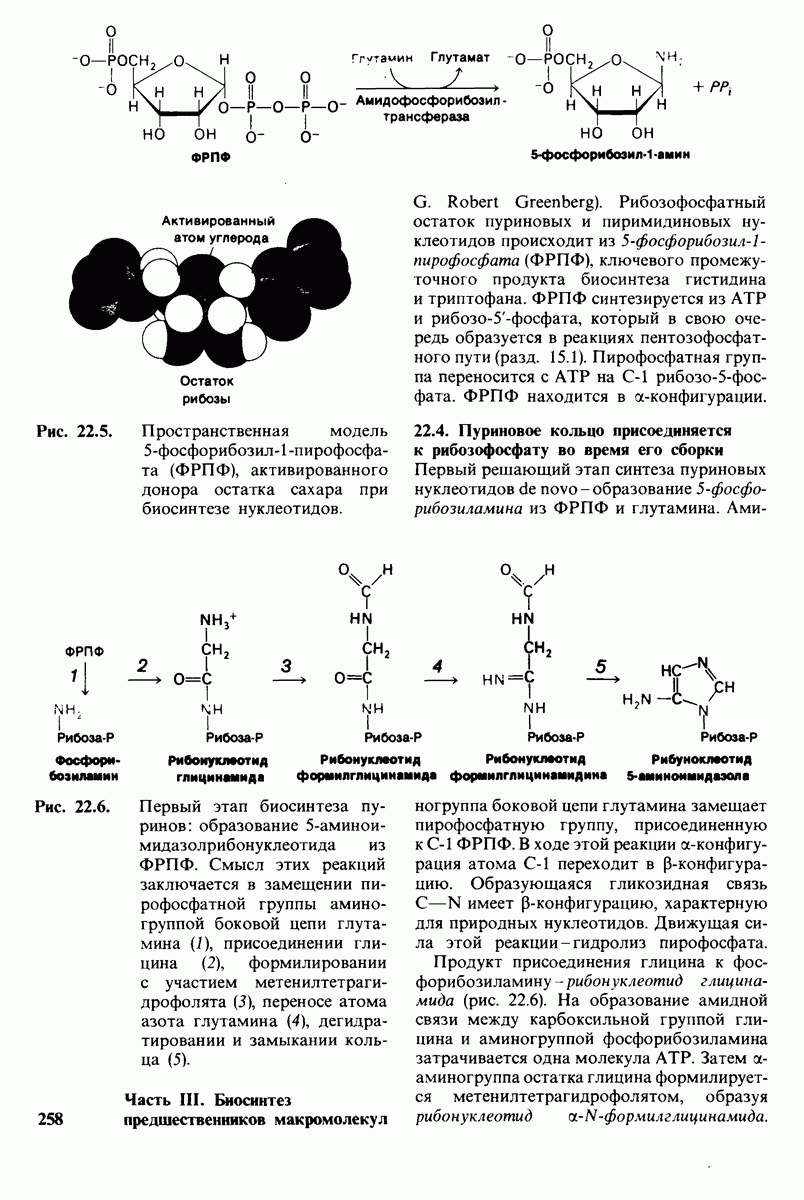
18.18. Блокирование гидроксилировання фенилаланина может привести к резко выраженной умственной отсталости
Фенилкетонурия, врожденное нарушение обмена фенилаланина, в отличие от алкаптонурии может оказывать сильное разрушительное действие на организм. Нелеченные больные с фенилкетонурией почти всегда отличаются резко выраженной умственной отсталостью. Действительно, примерно у 1% пациентов психиатрических клиник обнаруживается фенилкетонурия. Вес мозга

таких больных ниже нормы, у них нарушена миелинизация нервов, отмечается гиперактивность рефлексов. Продолжительность жизни нелеченных больных резко сокращена. Половина из них умирает в возрасте 20 лет, три четверти - в возрасте 30 лет.
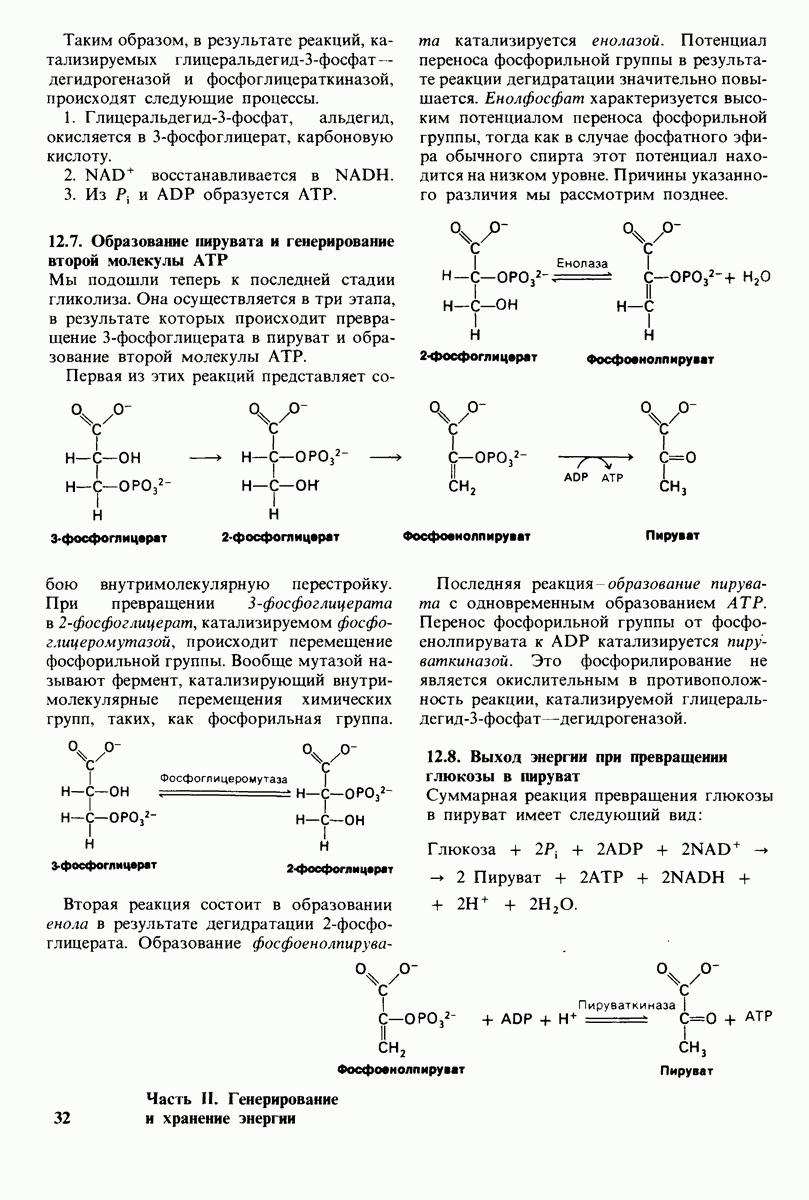
Фенилкетонурия вызывается отсутствием или недостатком фенилаланин-гидроксилазы или, реже, - ее тетрагидробиоптеринового кофактора. Фенилаланин не может превратиться в тирозин и поэтому накапливается во всех жидкостях тела. Некоторые превращения фенилаланина, количественно несущественные в норме, становятся заметными при фенилкетонурии. Наиболее значительным из них является трансаминирование фенилаланина с образованием фенилпирувата. В основе самого названия болезни лежит высокое содержание этого фенилкетона в моче. Из фенилпирувата образуются фениллактат, фенилацетат и  -гидроксифенилацетат. а-Аминогруппа глутамина образует амидную связь с карбоксильной группой фенилацетата, давая фенилацетилглутамин.
-гидроксифенилацетат. а-Аминогруппа глутамина образует амидную связь с карбоксильной группой фенилацетата, давая фенилацетилглутамин.
Существует множество других видов нарушения аминокислотного обмена, связанных с фенилкетонурией, в особенности это касается обмена ароматических соединений. Кожа и волосы у больных фенилкетонурией светлее  чем у их сибсов. Гидроксилирование тирозина - это первый этап в образовании пигмента меланина. При фенилкетонурии эта реакция конкурентно ингибируется высоким содержанием фенилаланина, так что образование меланина уменьшается. Биохимическая основа задержки умственного развития при не леченной фенилкетонурии представляет собой загадку.
чем у их сибсов. Гидроксилирование тирозина - это первый этап в образовании пигмента меланина. При фенилкетонурии эта реакция конкурентно ингибируется высоким содержанием фенилаланина, так что образование меланина уменьшается. Биохимическая основа задержки умственного развития при не леченной фенилкетонурии представляет собой загадку.
При рождении дети с фенилкетонурией производят впечатление здоровых, однако при отсутствии лечения уже в возрасте одного года появляются тяжелые нарушения. Лечение фенилкетонурии сводится к приему пищи с низким содержанием фенилаланина. Цель заключается в том, чтобы поступление фенилаланина в организм не превышало потребности в нем для роста и замещения. Белки, исходно содержащие малые количества фенилаланина, такие, как казеин молока, гидролизуют и удаляют фенилаланин путем адсорбции. Бедную фенилаланином пищу надо начинать давать сразу после рождения, чтобы предотвратить развитие необратимых повреждений мозга. В одном из исследований в группе больных фенилкетонурией, лечение которых было начато в течение нескольких недель после рождения, среднее значение  (коэффициента интеллектуальности) составляло 93, а в контрольной группе сибсов, которых начали лечить в возрасте одного года,
(коэффициента интеллектуальности) составляло 93, а в контрольной группе сибсов, которых начали лечить в возрасте одного года,  равнялся в среднем 53.
равнялся в среднем 53.
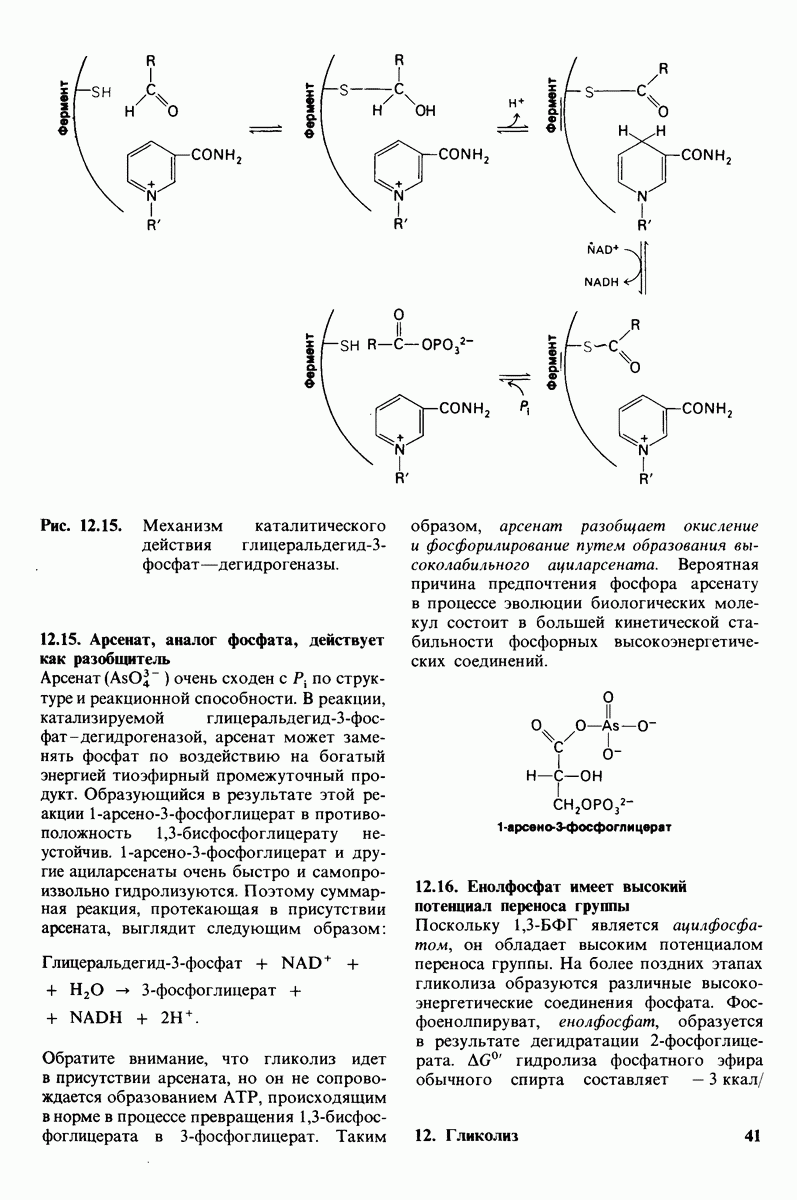
Ранняя диагностика фенилкетонурии имеет большое значение и осуществляется по программам массового скрининга. В последние годы мочу новорожденных исследуют, добавляя в нее  который в присутствии фенилпирувата дает оливково-зеленое окрашивание. Предпочтительным диагностическим критерием из-за его большей надежности служит в настоящее время содержание фенилаланина в крови. Частота возникновения фенилкетонурии составляет около 1 случая на 20000 новорожденных.
который в присутствии фенилпирувата дает оливково-зеленое окрашивание. Предпочтительным диагностическим критерием из-за его большей надежности служит в настоящее время содержание фенилаланина в крови. Частота возникновения фенилкетонурии составляет около 1 случая на 20000 новорожденных.
«Природа никогда не раскрывает в такой степени свои секреты, как в тех случаях, когда она оставляет следы своей работы в стороне от проторенного пути. Нет лучшего способа для достижения успеха в практической медицине, чем обратить наш взгляд на раскрытие обычных законов природы путем внимательного исследования редких форм заболеваний».
Уильям Гарвей (1657)
Болезнь наследуется как аутосомный рецессивный признак. Гетерозиготы, составляющие примерно 1,5% типичной популяции, не обнаруживают видимых отклонений от нормы. У носителей гена фенилкетонурии снижено содержание фенилаланин-гидроксилазы и соответственно повышено содержание фенилаланина в крови. Эти критерии, однако, не являются абсолютными, потому что уровень содержания в крови фенилаланина у носителей дефектного гена и у нормальных лиц в некоторой степени перекрывают друг друга. Более точным тестом на носительство данного гена является измерение кинетики исчезновения внутривенно введенного фенилаланина. Следует отметить, что высокое содержание фенилаланина в крови беременной женщины может привести к нарушениям в развитии плода. Это - яркий пример взаимоотношений материи и плода на молекулярном уровне.
Заключение
Избыточные аминокислоты используются как метаболическое топливо. Деградация большинства избыточных аминокислот начинается с удаления  -аминогруппы путем трансаминирования с
-аминогруппы путем трансаминирования с  -оксокислотой. Ко-ферментом всех трансаминаз служит пиридоксальфосфат. Аминогруппы переносятся на
-оксокислотой. Ко-ферментом всех трансаминаз служит пиридоксальфосфат. Аминогруппы переносятся на  -оксоглутарат с образованием глута-мата, который затем подвергается окислительному дезаминированию под действием глутамат-дегидрогеназы, давая
-оксоглутарат с образованием глута-мата, который затем подвергается окислительному дезаминированию под действием глутамат-дегидрогеназы, давая  и
и  -оксоглутарат. Акцепторами электронов в этой реакции служат NAD+ или NADP+. У наземных позвоночных
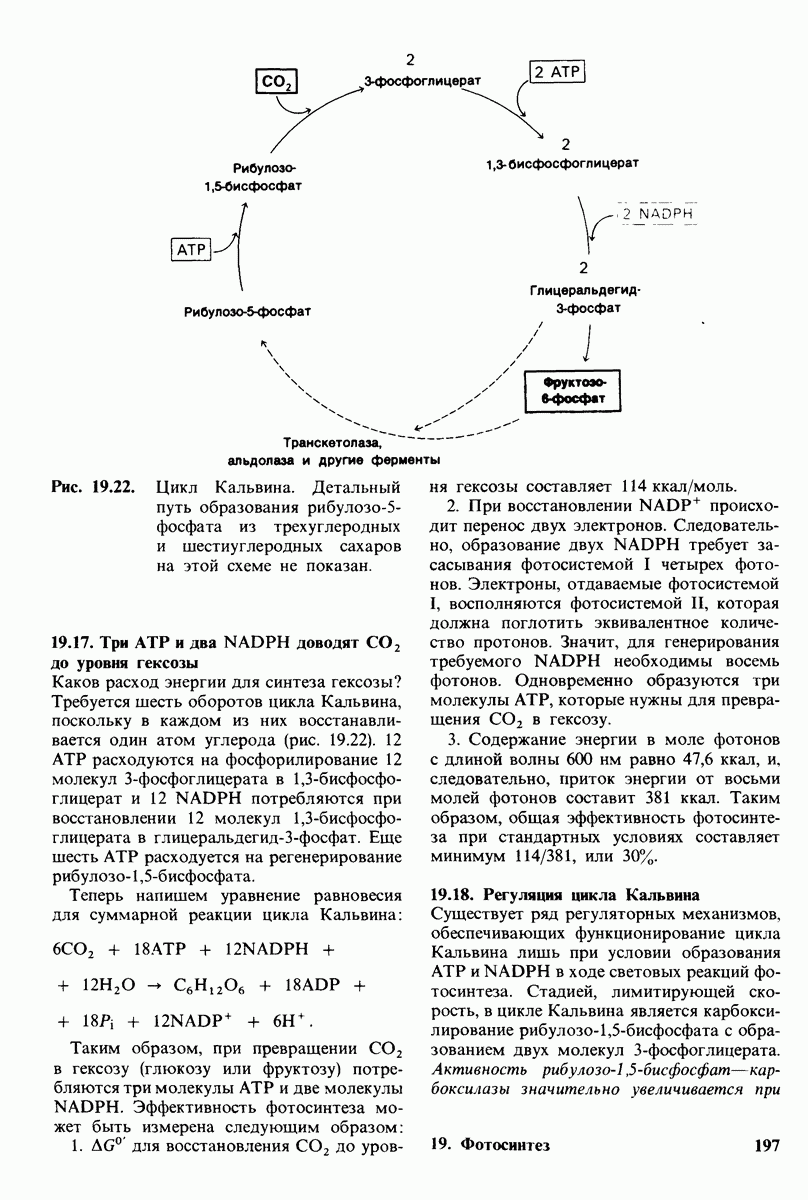
-оксоглутарат. Акцепторами электронов в этой реакции служат NAD+ или NADP+. У наземных позвоночных  превращается в мочевину в цикле мочевины. Мочевина образуется в результате гидролиза аргинина. В ходе последующих реакций цикла мочевины происходит синтез аргинина из орнитина, другого продукта реакции гидролиза. Сначала орнитин подвергается карбамоилированию в цитруллин при участии карбамоилфосфата. Цитруллин затем конденсируется с аспартатом, образуя аргининосукцинат, который расщепляется на аргинин и фумарат. Атом углерода и один атом азота мочевины происходят из карбамоилфосфата, синтезирующегося из
превращается в мочевину в цикле мочевины. Мочевина образуется в результате гидролиза аргинина. В ходе последующих реакций цикла мочевины происходит синтез аргинина из орнитина, другого продукта реакции гидролиза. Сначала орнитин подвергается карбамоилированию в цитруллин при участии карбамоилфосфата. Цитруллин затем конденсируется с аспартатом, образуя аргининосукцинат, который расщепляется на аргинин и фумарат. Атом углерода и один атом азота мочевины происходят из карбамоилфосфата, синтезирующегося из  и АТР. Другой атом азота мочевины происходит из аспартата. При синтезе одной молекулы мочевины используются четыре высокоэнергетические фосфатные связи.
и АТР. Другой атом азота мочевины происходит из аспартата. При синтезе одной молекулы мочевины используются четыре высокоэнергетические фосфатные связи.
Углеродные атомы распавшихся аминокислот превращаются в пируват, ацетил-СоА, ацетоацетат или промежуточный продукт цикла трикарбоновых кислот. Большинство аминокислот являются только глюкогенными, одна - исключительно кетогенной и несколько могут быть и кето- и глюкогенными. При расщеплении аланина, серина, цистеина, глицина и треонина образуется пируват. Аспарагин и аспартат превращаются в оксалоацетат.  представляет собою «пункт входа» (в цикл трикарбоновых кислот) для глутамата и четырех аминокислот (глутамина, гистидина, пролина и аргинина), которые могут быть превращены в глутамат.
представляет собою «пункт входа» (в цикл трикарбоновых кислот) для глутамата и четырех аминокислот (глутамина, гистидина, пролина и аргинина), которые могут быть превращены в глутамат.  входа» для некоторых углеродных атомов еще четырех аминокислот (метионина, изолейцина, треонина и валина), распад которых идет через метилмалонил-СоА. Для изомеризации
входа» для некоторых углеродных атомов еще четырех аминокислот (метионина, изолейцина, треонина и валина), распад которых идет через метилмалонил-СоА. Для изомеризации  требуется дезоксиаденозилкобаламин, производное витамина
требуется дезоксиаденозилкобаламин, производное витамина  Лейцин распадается до ацетоацетил-СоА и ацетил-СоА. Ароматические кольца тирозина и фенилаланина распадаются под действием оксигеназ. Фенилаланин-гидроксилаза, монооксигеназа, использует в качестве восстановителя тетрагидробиоптерин. Некоторые углеродные атомы фенилаланина и тирозина превращаются в фумарат, тогда как другие появляются в ацетоацетате.
Лейцин распадается до ацетоацетил-СоА и ацетил-СоА. Ароматические кольца тирозина и фенилаланина распадаются под действием оксигеназ. Фенилаланин-гидроксилаза, монооксигеназа, использует в качестве восстановителя тетрагидробиоптерин. Некоторые углеродные атомы фенилаланина и тирозина превращаются в фумарат, тогда как другие появляются в ацетоацетате.