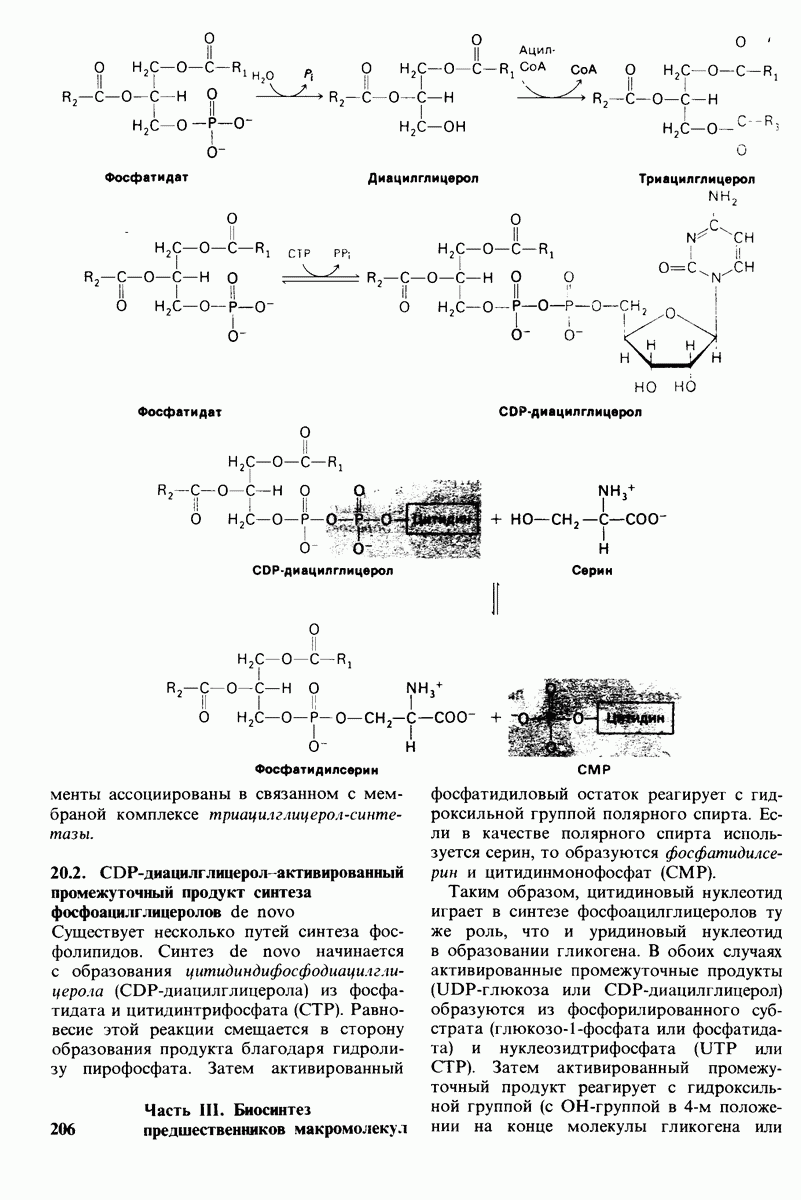
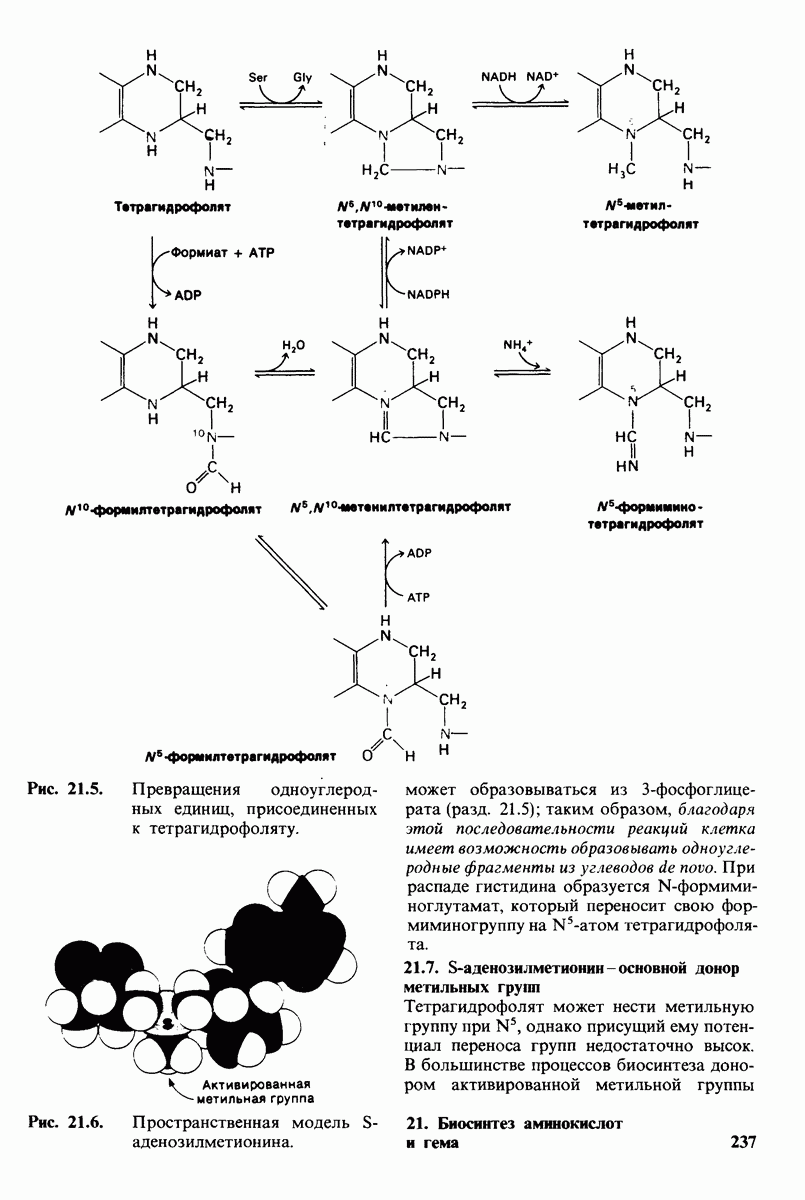
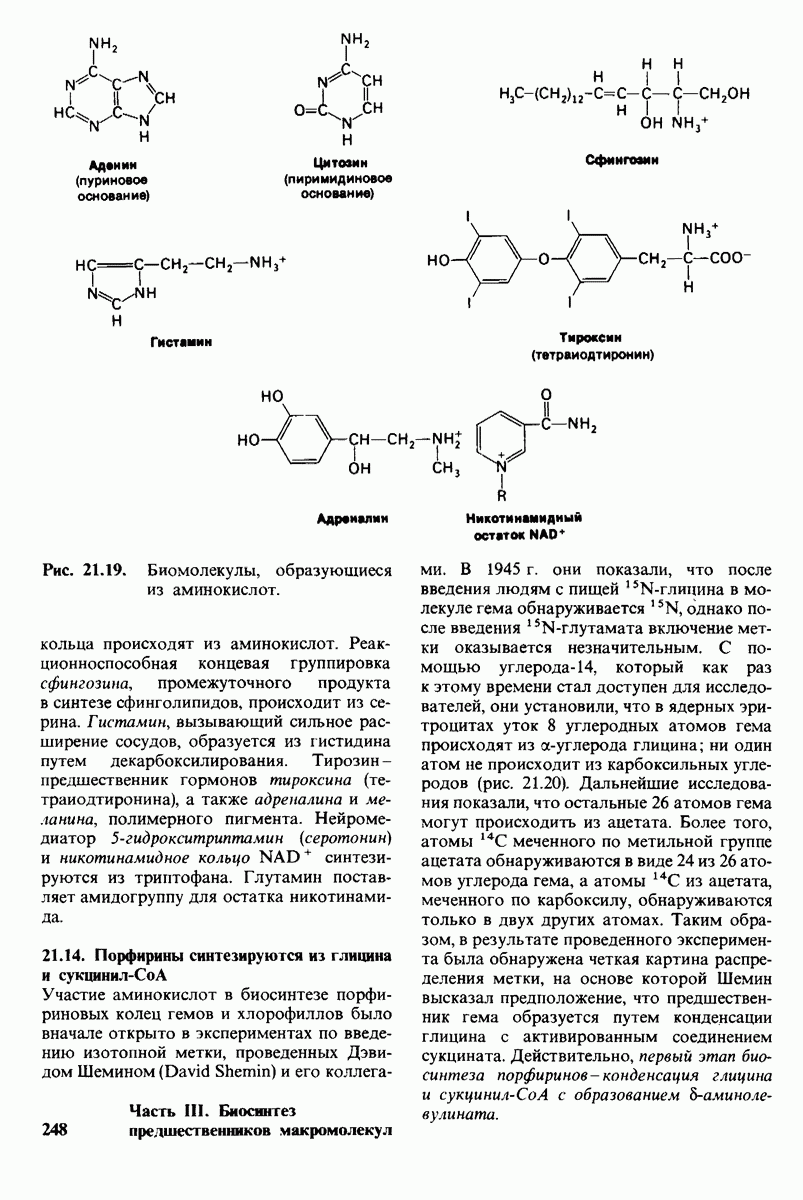
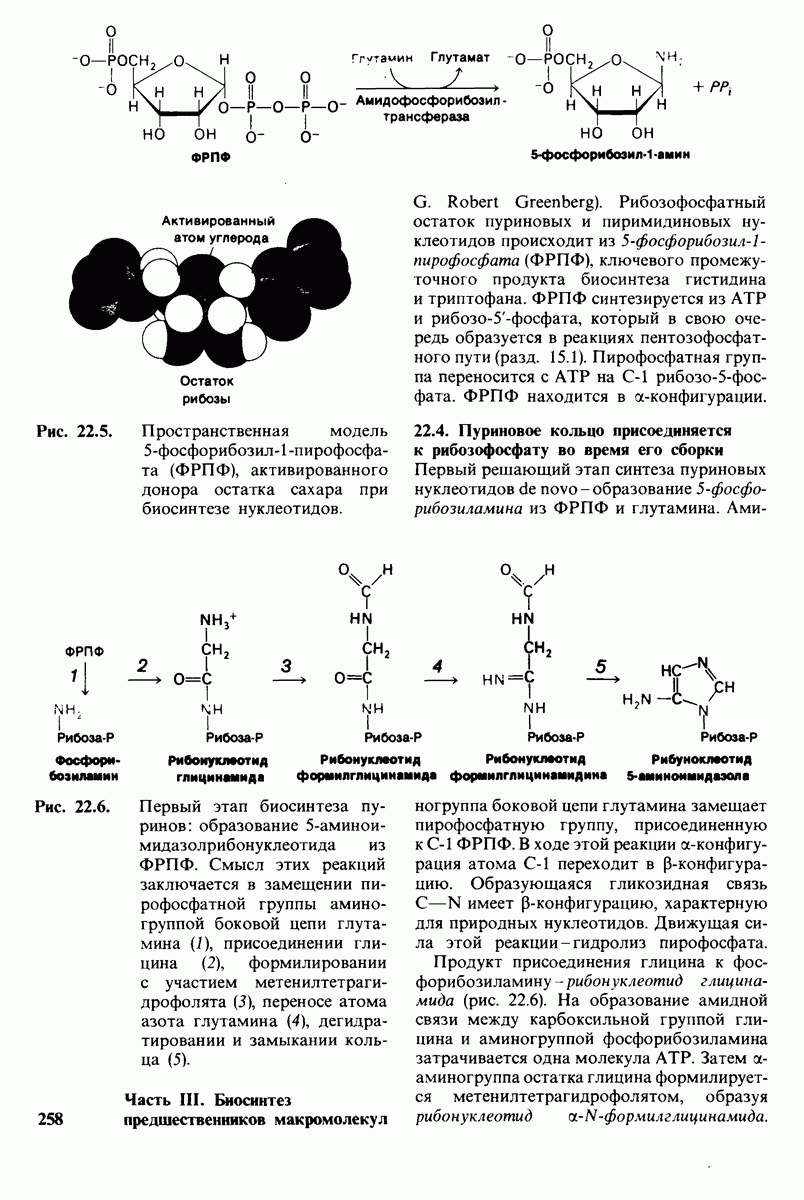
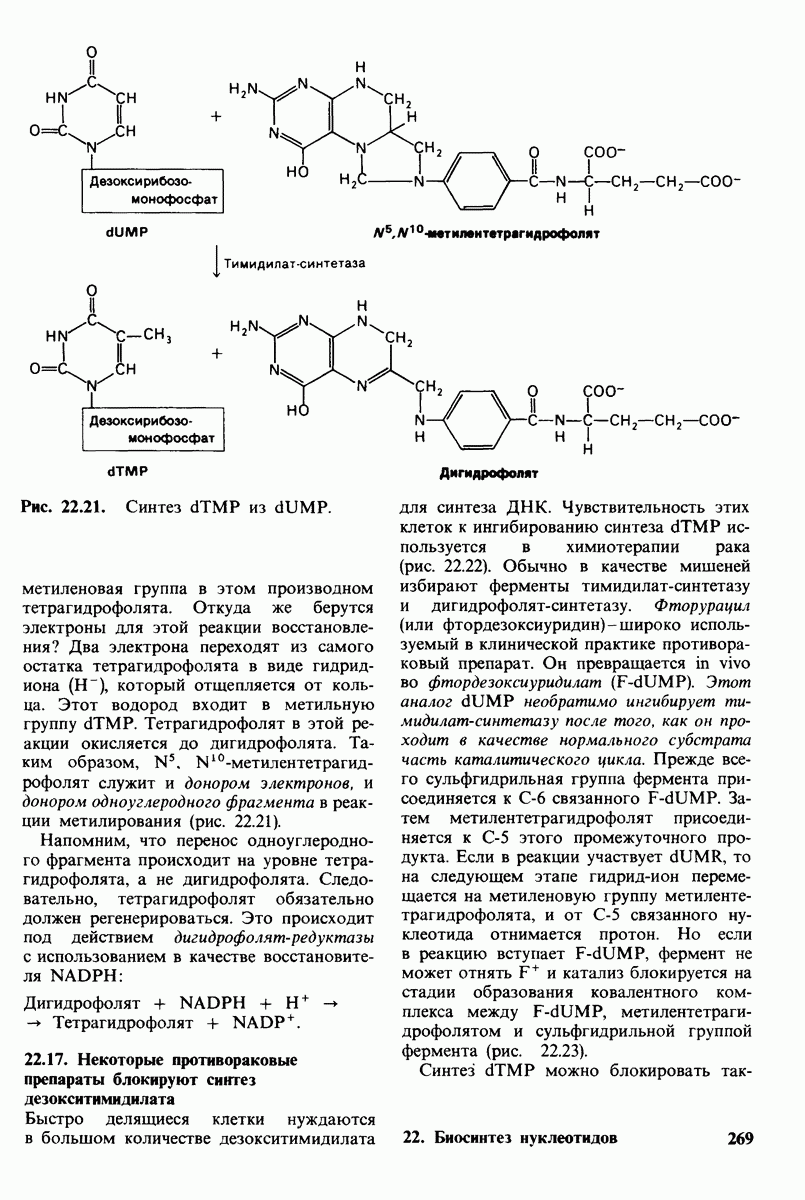
20.8. Болезнь Тея-Сакса: наследуемое нарушение расщепления ганглиозидов
Ганглиозиды содержатся в наибольшей концентрации в нервной системе, особенно в сером веществе, где они составляют 6% всех липидов. Ганглиозиды постоянно синтезируются и расщепляются путем последовательного удаления концевых остатков сахара. Катализирующие эти реакции гликозид-гидролазы высокоспецифичны. Расщепление ганглиозидов происходит в лизосомах. Эти органеллы содержат самые разнообразные ферменты расщепления и предназначены для упорядоченного разрушения компонентов клетки.
Нарушения в способности клетки к расщеплению ганглиозидов могут привести к серьезным клиническим последствиям.

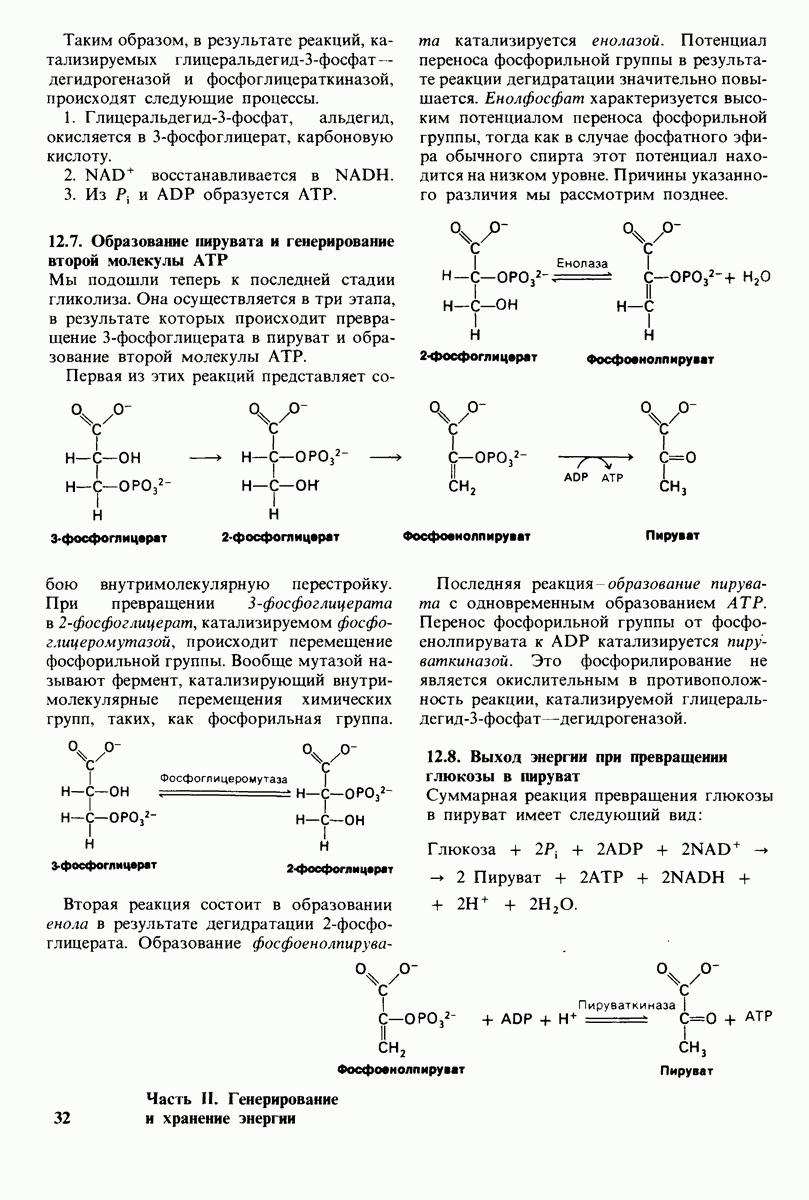
Рис. 20.5. Строение ганглиозида 
Использованные сокращения:  галактоза;
галактоза;  ацетилгалактозамин;
ацетилгалактозамин;  глюкоза
глюкоза  ацетилнейраминовая кислота. Над линиями, обозначающими связи между сахарами, указан тип этих связей, например (31,4.
ацетилнейраминовая кислота. Над линиями, обозначающими связи между сахарами, указан тип этих связей, например (31,4.
Симптомы болезни  Сакса обычно проявляются у ребенка в возрасте до одного года. К характерным ранним симптомам относятся слабость, отставание в развитии, затруднения при кормлении. Через несколько месяцев обычно наступает слепота. Летальный исход при болезни
Сакса обычно проявляются у ребенка в возрасте до одного года. К характерным ранним симптомам относятся слабость, отставание в развитии, затруднения при кормлении. Через несколько месяцев обычно наступает слепота. Летальный исход при болезни  Сакса обычно наступает до 3 лет. Эта болезнь сопровождается разительными патологическими изменениями в нервной системе: ганглиозные клетки коры головного мозга и некоторых других участков мозга чрезмерно разбухают. Кроме того, на сетчатке видны отчетливые вишнево-красные пятна.
Сакса обычно наступает до 3 лет. Эта болезнь сопровождается разительными патологическими изменениями в нервной системе: ганглиозные клетки коры головного мозга и некоторых других участков мозга чрезмерно разбухают. Кроме того, на сетчатке видны отчетливые вишнево-красные пятна.
Содержание ганглиозидов в мозгу ребенка, страдающего болезнью  сильно повышено. Особенно сильно, во много раз по сравнению с нормой, повышается концентрация ганглиозида
сильно повышено. Особенно сильно, во много раз по сравнению с нормой, повышается концентрация ганглиозида  Аномально высокое содержание этого ганглиозида обусловлено недостаточностью фермента, отщепляющего концевой остаток
Аномально высокое содержание этого ганглиозида обусловлено недостаточностью фермента, отщепляющего концевой остаток  -ацет ил галактоз амина.
-ацет ил галактоз амина.

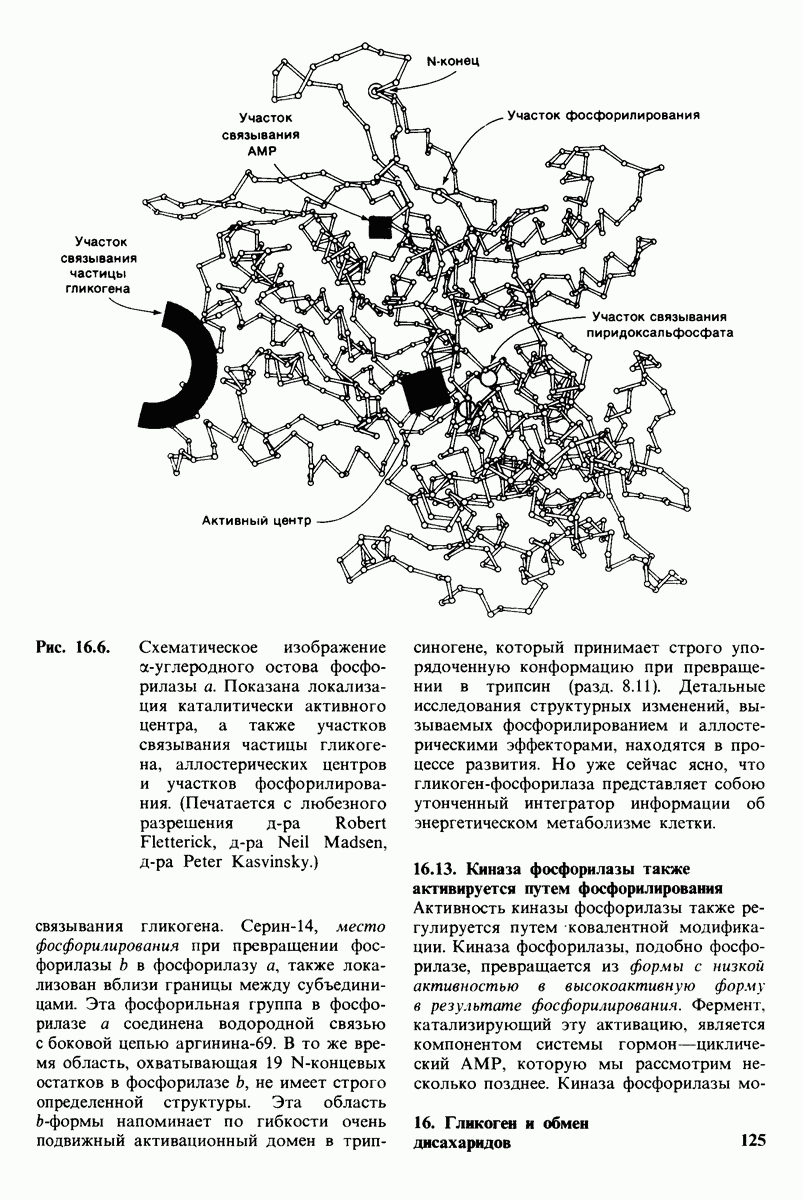
Рис. 20.6. Электронная микрофотография лизосомы. (Печатается с любезного разрешения 
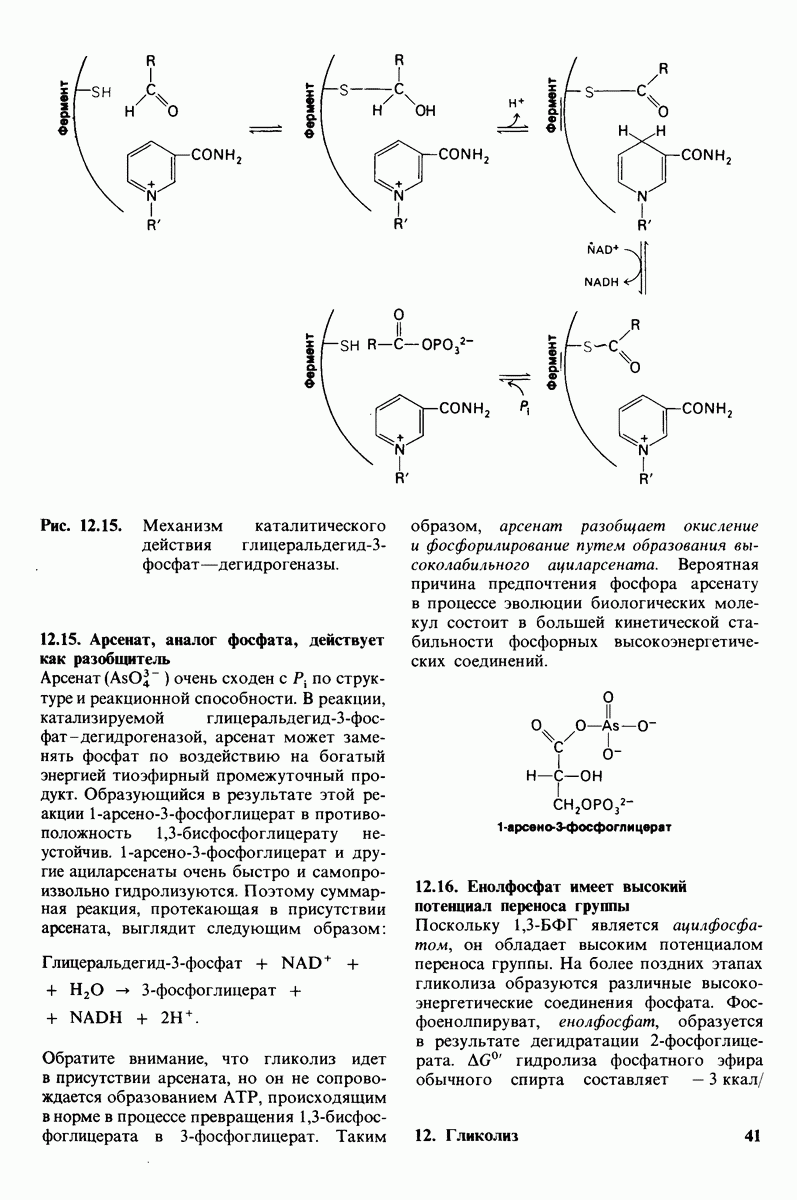
Фермент, отсутствие (или недостаточная активность) которого обусловливают эту болезнь, - специфическая - ацетилгексозаминидаза.
Болезнь  наследуется как аутосомный рецессивный признак. Частота носителей составляет 1/30 среди американцев еврейского происхождения и 1/300 среди прочих американцев. Следовательно, частота болезни среди американцев еврейского происхождения выше примерно в 100 раз. Болезнь
наследуется как аутосомный рецессивный признак. Частота носителей составляет 1/30 среди американцев еврейского происхождения и 1/300 среди прочих американцев. Следовательно, частота болезни среди американцев еврейского происхождения выше примерно в 100 раз. Болезнь  можно диагностировать еще во время эмбрионального развития. Для этого берут пробу
можно диагностировать еще во время эмбрионального развития. Для этого берут пробу


Рис. 20.7. Пространственная модель холестерола.
амниотической жидкости с помощью амниоцентеза и определяют в ней активность  -ацетилгексозаминидазы.
-ацетилгексозаминидазы.