Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
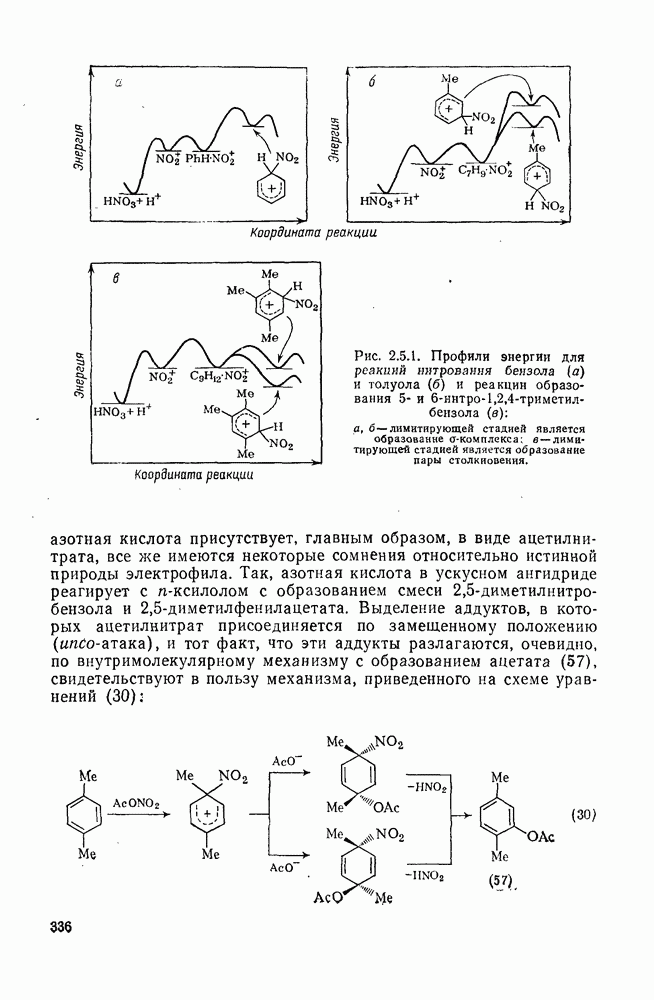
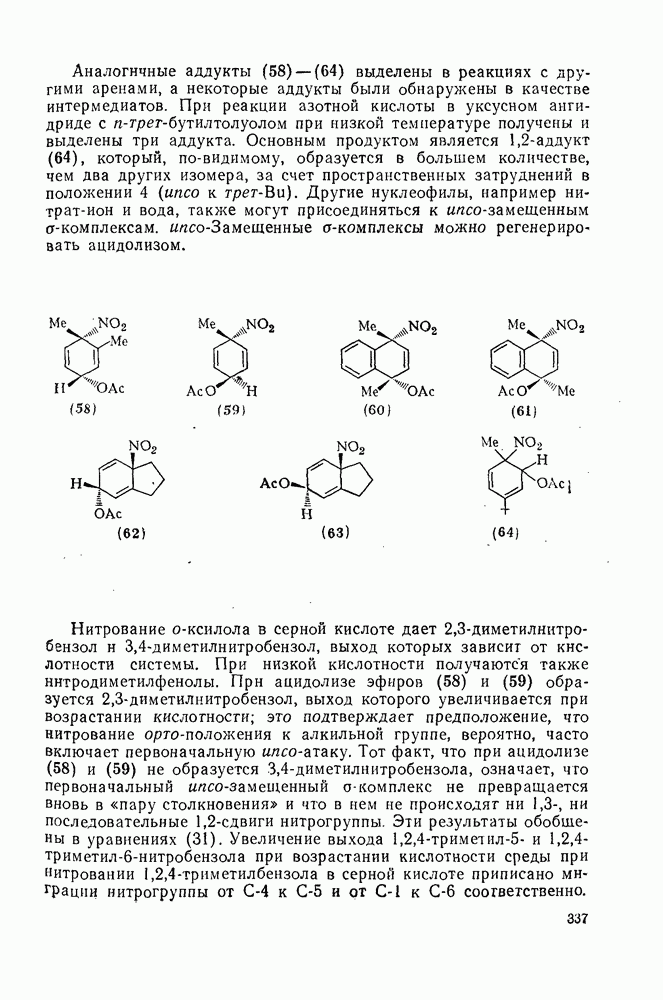
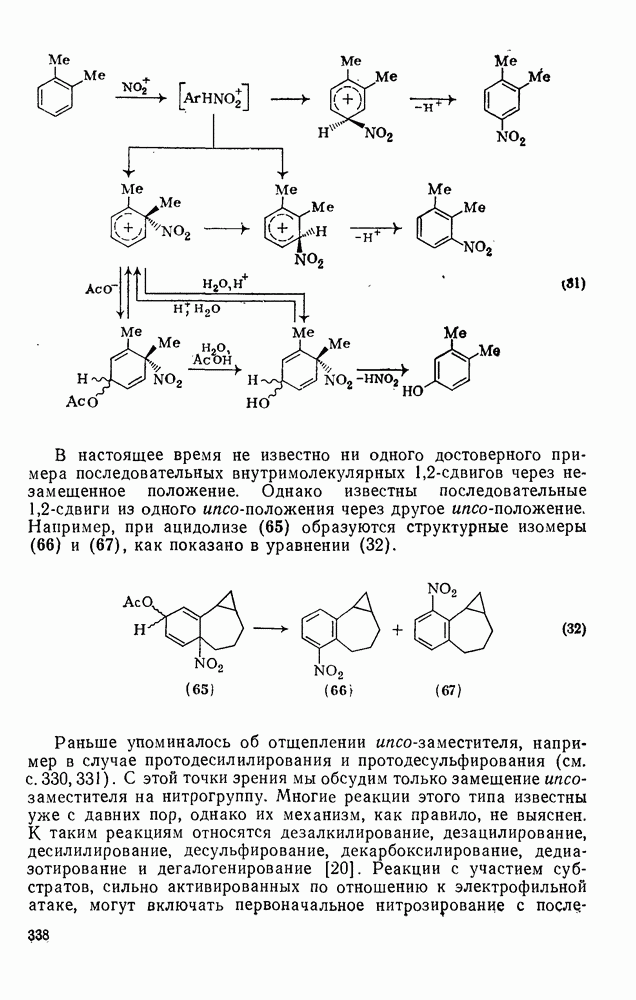
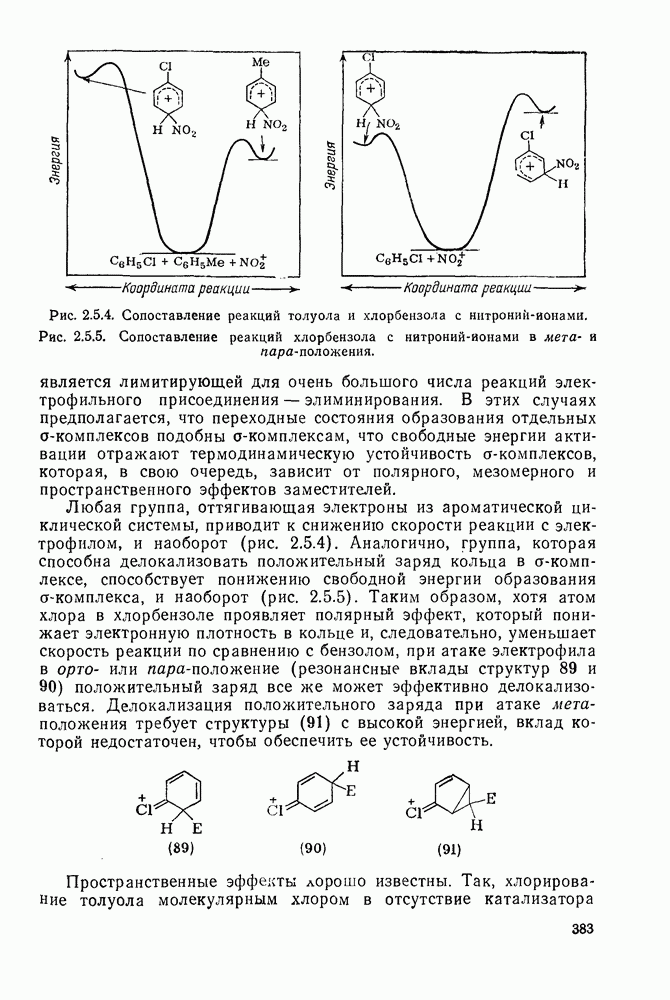
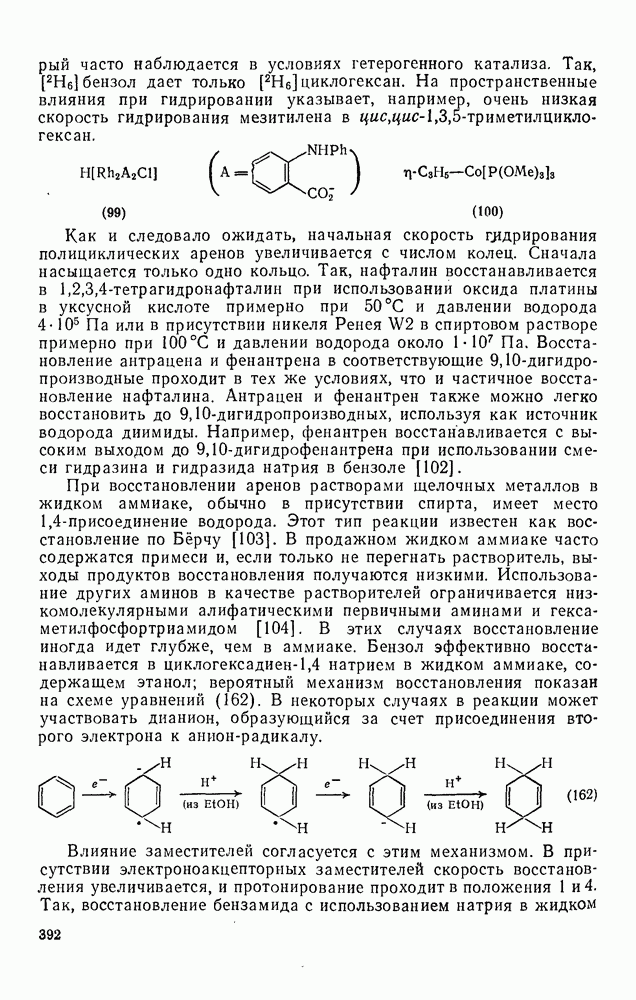
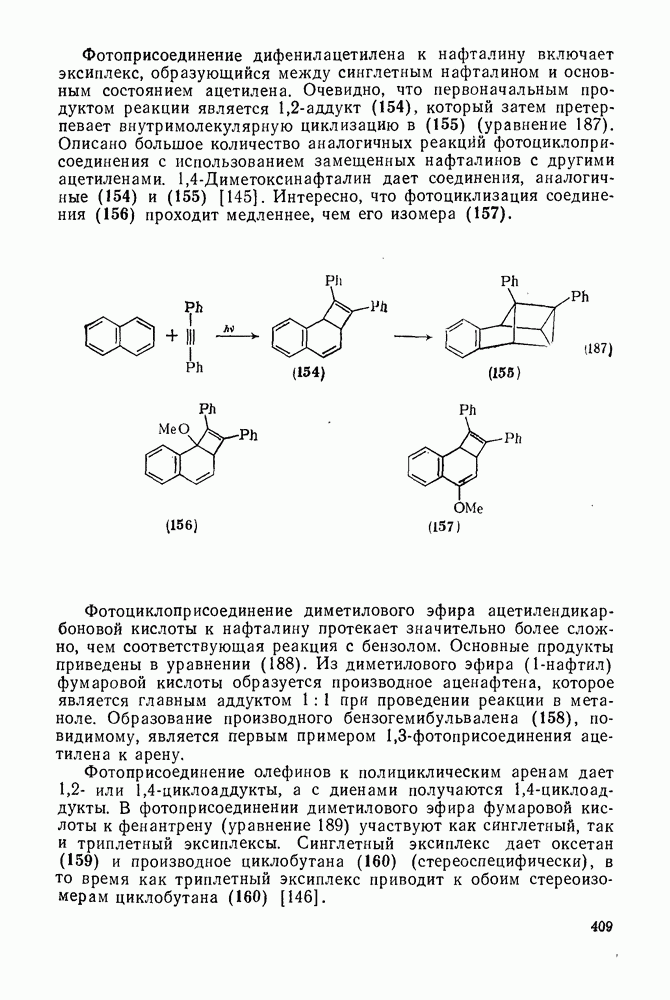
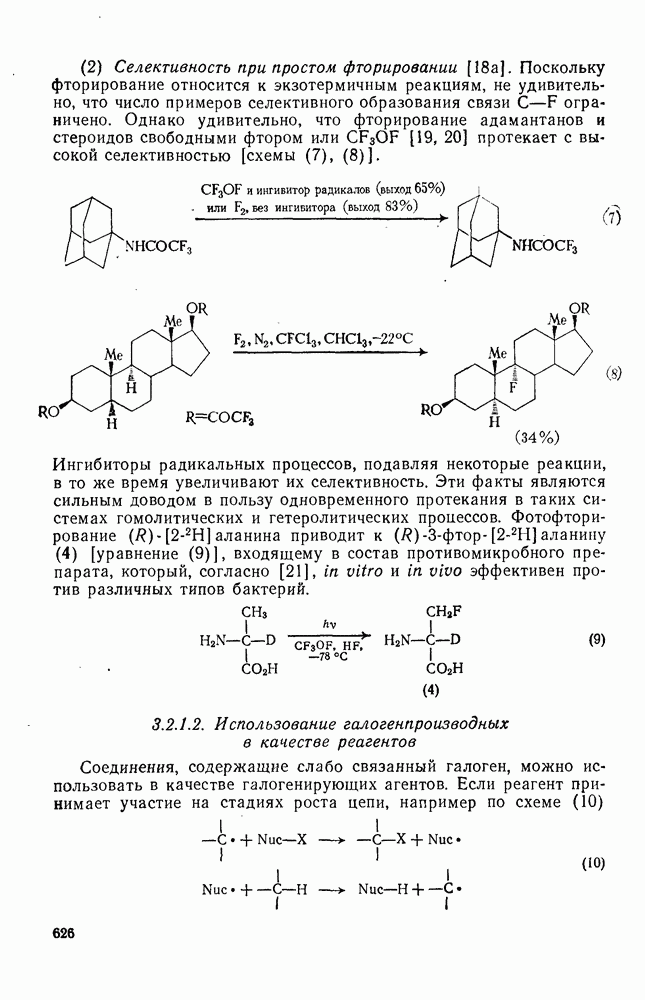
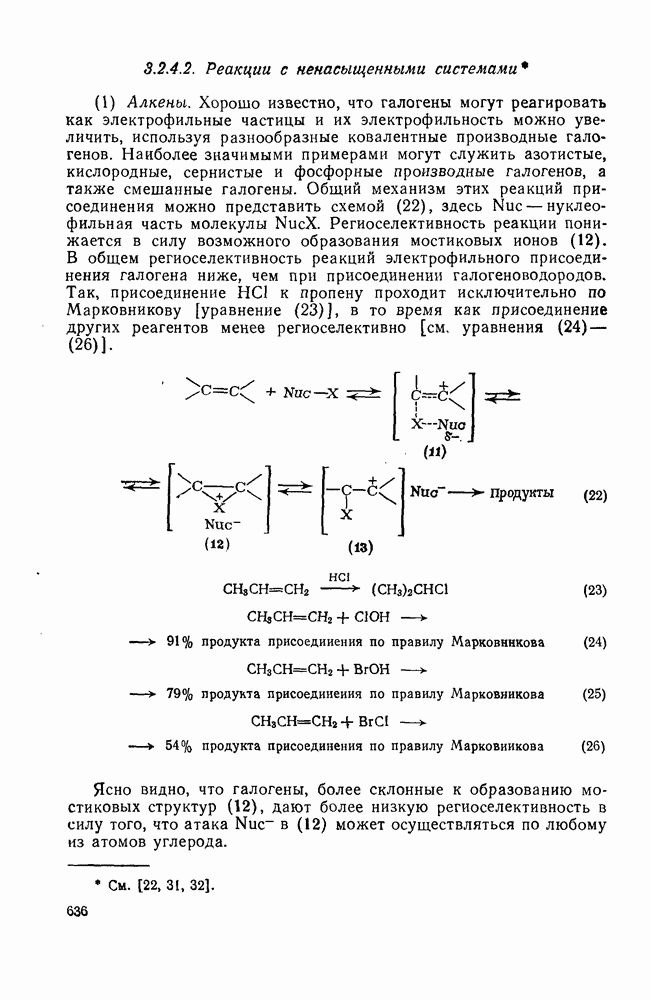
2.5.6.5. Электрофилы IV группыВ этом разделе основное внимание уделено алкилированию и ацилированию по Фриделю — Крафтсу [25а]. Другие родственные реакции (см. разд. 2.5.6, п. 3) будут рассмотрены более кратко. Во всех этих реакциях образуется новая связь углерод — углерод. Как и в других примерах реакций электрофильного присоединения — элиминирования, ароматические молекулы выступают в роли нуклеофила и, следовательно, другие частицы, которые первоначально являются электронейтральными, подвергаются нуклеофильному замещению. В большинстве рассматриваемых в этом разделе реакций электрофильный компонент обладает недостаточной реакционной способностью, чтобы взаимодействовать со слабонуклеофильным ароматическим компонентом в отсутствие подходящего катализатора. Катализатор увеличивает электрофильность неароматического компонента. Алкилирование по Фриделю — Крафтсу.Обычно в качестве катализаторов алкилирования алкилгалогенидами, спиртами и сложными эфирами используют кислоты Льюиса (уравнения 51—55). Как правило, для генерирования электрофила из олефина требуется протонная кислота. Можно использовать и другие алкилирующие агенты — тиолы, простые эфиры, сульфиды, сульфаты и даже, в определенных случаях, алканы [256]. Заслуживает внимания этиленоксид, позволяющий ввести в молекулу цепочку из двух углеродных атомов, и циклопропан, позволяющий ввести трех-углеродную цепочку.Как и в случае реакции обычного нуклеофильного замещения (см. гл. 3), следует рассмотреть два крайних механизма.
В реакциях алкилирования по Фриделю — Крафтсу в качестве катализатора чаще всего используют хлорид алюминия. Вероятно, что единого порядка активности катализаторов, который можно было бы установить опытным путем, не существует, поскольку вполне возможно, что активность катализатора будет зависеть от природы арена, электрофила и условий реакции. Предложен [26] следующий ряд активностей катализаторов:
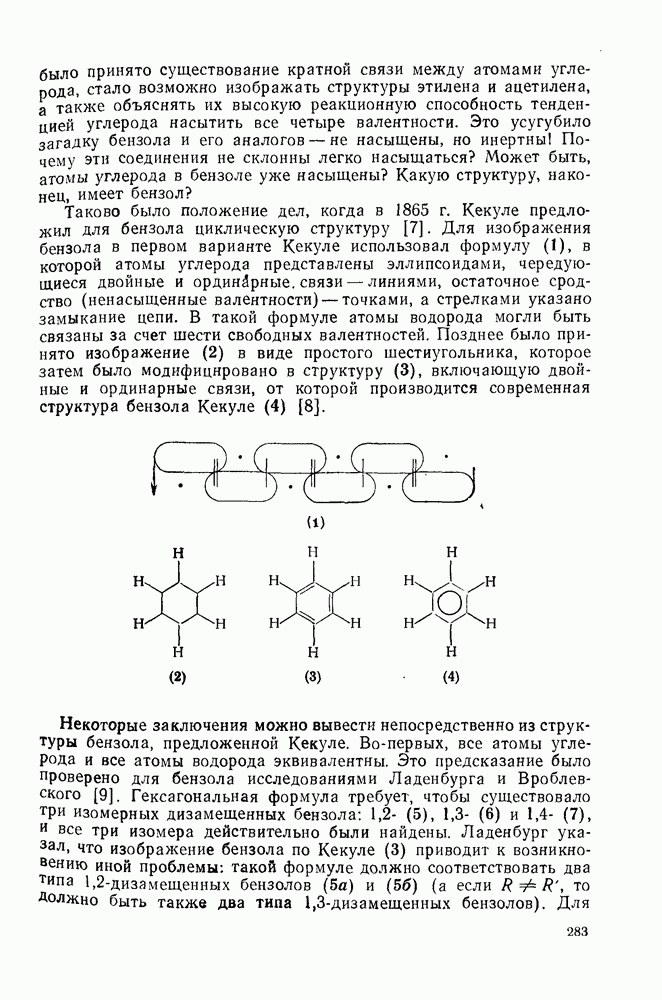
Для алкилирования в качестве электрофилов часто выбирают алкилгалогениды, и в случае достаточно нуклеофильных ароматических субстратов и реакционноспособных алкилгалогенидов с успехом можно использовать мягкий катализатор — хлористый Алкилирование по Фриделю — Крафтсу относится к группе наиболее сложных реакций электрофильного присоединения — элиминирования. Не только не существует единого механизма (см. уравнения 51—55), подходящего для всех случаев реакции, но возникают также и многие другие осложнения. Вступающая в ядро алкильная группа является естественно, электронодонорной, и поэтому продукт реакции более склонен к реакции с электрофилом, чем исходный субстрат. В связи с этим часто наблюдается ди- и полиалкилирование. Однако если использовать в качестве растворителя сам арен и проводить реакцию при энергичном перемешивании, часто удается получить продукты моноалкилирования с хорошим выходом. Эти два фактора имеют особенно большое значение. Относительные скорости алкилирования по Фриделю — Крафтсу для простых алкилбензолов в Присутствие электроноакцепторных групп в ароматическом кольце в большинстве случаев снижает нуклеофильность арена до такой степени, что алкилирование по Фриделю — Крафтсу в обычных условиях становится невозможным. Поэтому вместо сероуглерода в качестве растворителя часто используют нитробензол. Проведение реакций с субстратами, содержащими Двойственность механизма алкилирования по Фриделю — Крафтсу сильно ограничивает синтетическое использование этой реакции. Так, часто наблюдаются перегруппировки алкильного остатка [27]. Если алкилирование проходит по механизму, который можно формально отнести к типу
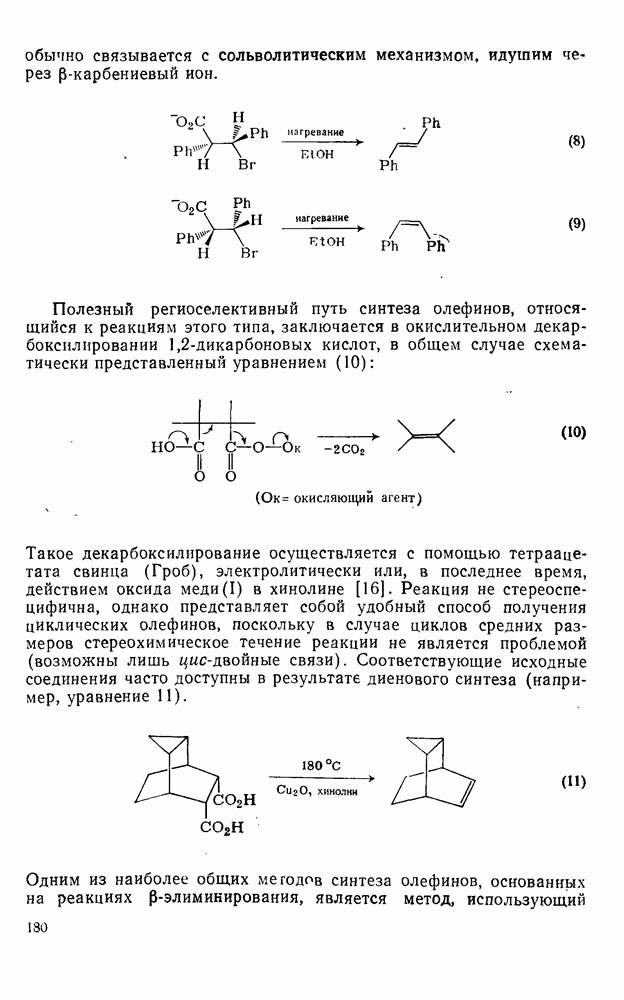
Перегруппировка алкила происходит обычно в порядке от первичного ко вторичному и далее третичному и осуществляется, как правило, путем миграции гидрид-иона. Возможна также миграция анионного фрагмента. Примеры этих типов перегруппировок приведены в уравнениях (57) и (58).
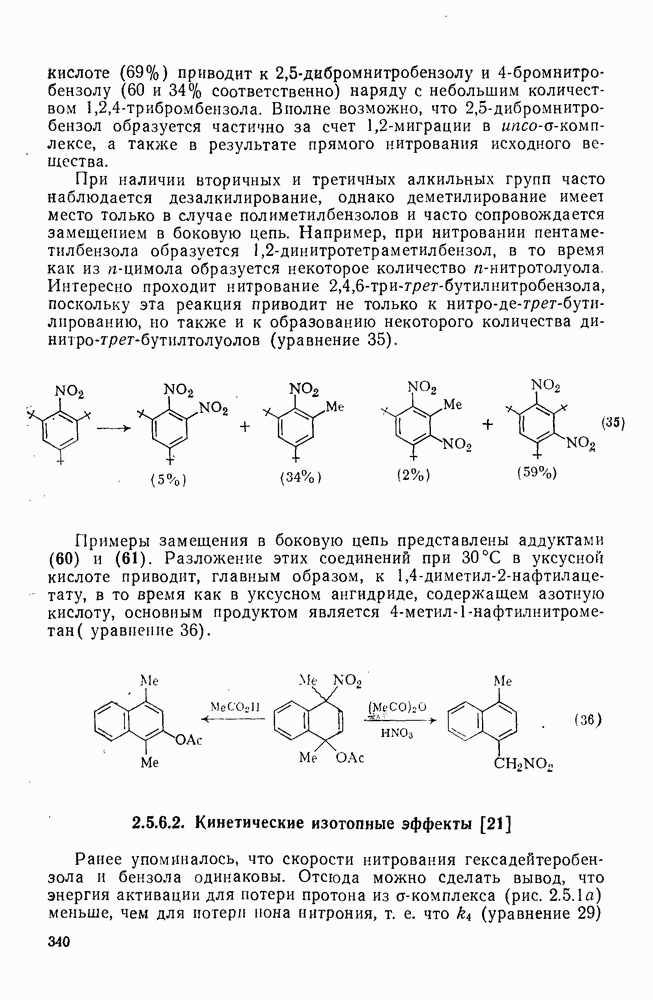
Если катализируемую трифторидом бора реакцию бензола с Однако при проведении реакции в нитрометане этого не наблюдается. Эти примеры приведены с целью подтвердить промежуточное образование карбениевых ионов в первом случае [28]. В отличие от этого при реакции Проблемы, связанные с перегруппировками алкильного остатка, не единственная причина использования этого альтернативного пути. Переалкилирование продукта, образующегося при кинетическом контроле, может приводить в конечном счете к термодинамически наиболее устойчивому продукту. Наиболее устойчивыми термодинамически являются мета-алкильные производные. Например, было найдено, что при реакции толуола с этилбромидом в присутствии бромида
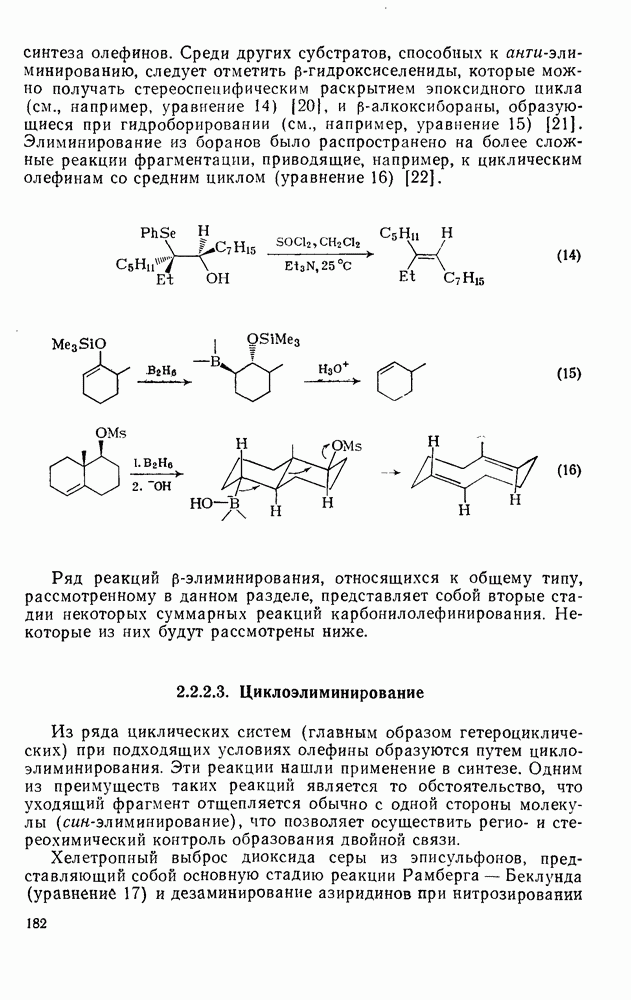
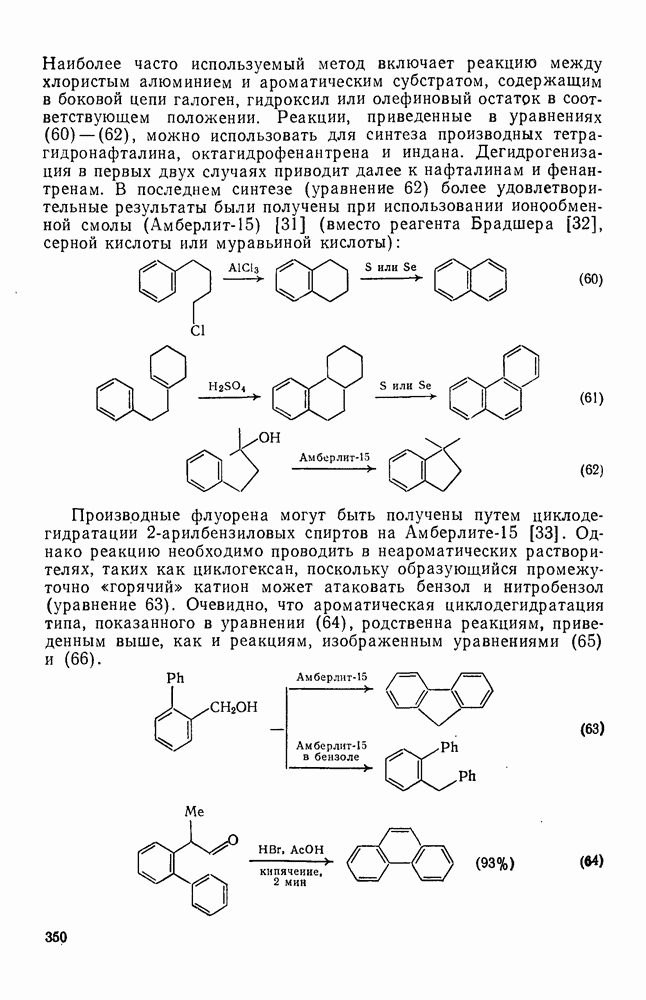
Одним из наиболее важных синтетических приложений алкилирования по Фриделю — Крафтсу являются реакции циклизации. Наиболее часто используемый метод включает реакцию между хлористым алюминием и ароматическим субстратом, содержащим в боковой цепи галоген, гидроксил или олефиновый остаток в соответствующем положении. Реакции, приведенные в уравнениях
Производные флуорена могут быть получены путем циклодегидратации
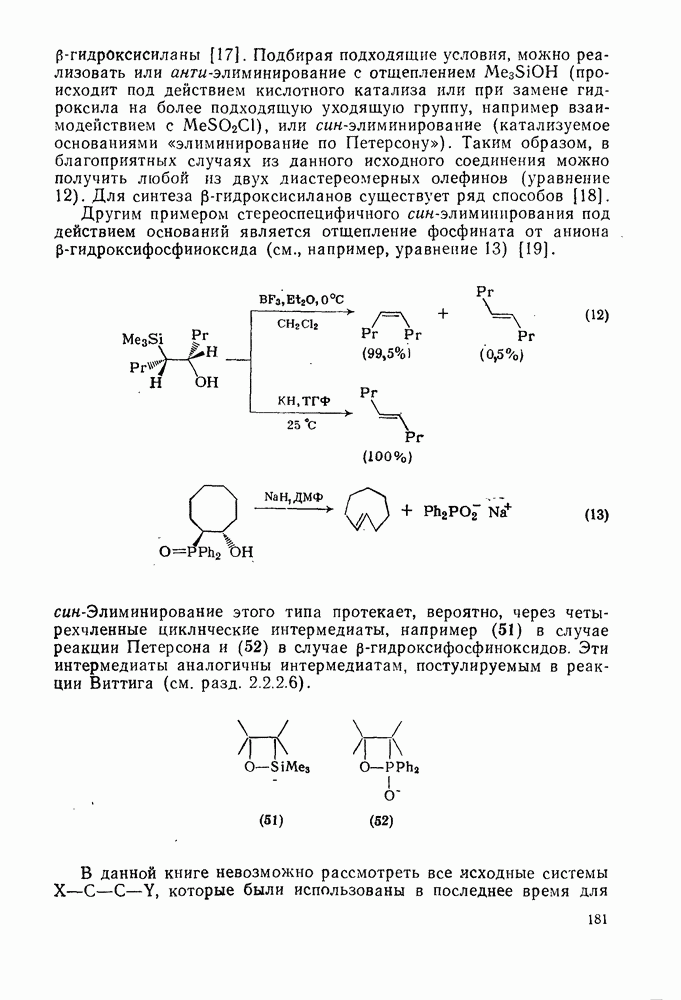
Как следует из приведенного выше обсуждения, в большинстве случаев в алкилировании по Фриделю — Крафтсу участвуют кар-бениевые ионы [18]. Образование карбениевого иона при использовании некоторых реагентов проходит чрезвычайно легко вследствие устойчивости катиона. Действительно, в определенных случаях алкилирование аренов с сильными нуклеофильными заместителями, например фенолов и ариламинов, проходит без катализатора. Это наблюдается при использовании трифенилметилхлорида [34] и 1 -хлорадамантана [35]. Тропилиевый ион не взаимодействует с бензолом, но алкилирует анизол (уравнение 67) [36].
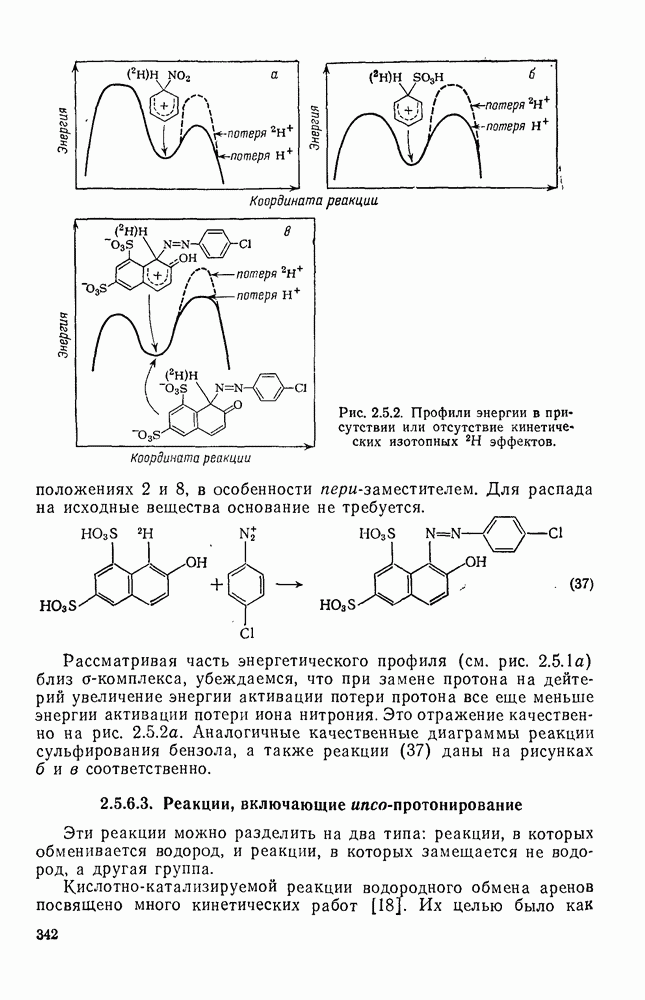
Вероятно, что при алкилировании первичными галогенидами свободные карбениевые ионы в реакции не участвуют. В таких случаях ионы могут существовать в форме диполярных комплексов (как показано на схеме уравнений 51—55) или в виде тесных ионных пар. Как показало изучение кинетики, некоторые реакции имеют третий порядок, т. е. первый порядок по каждому из компонентов: по электрофильному реагенту, ароматическому субстрату и катализатору. Поскольку известно, что свободные карбениевые ионы атакуют арены быстро, то скорость реакции не должна была бы зависеть от концентрации ароматического субстрата, если бы медленной стадией было образование карбениевого иона. Другую возможность представляет собой реакция Перегруппировка возможна даже в отсутствие свободных кар-бениевых ионов, поскольку продукты, образующиеся за счет гидридных сдвигов, наблюдались в условиях, при которых в реакции участвуют тесные ионные пары. Помимо возможности перегруппировки иона до атаки ареном, в силу обратимости алкилирования по Фриделю — Крафтсу не исключена перегруппировка и после образования первоначального алкиларена. Здесь следует отметить, что изотопное перераспределение при взаимодействии
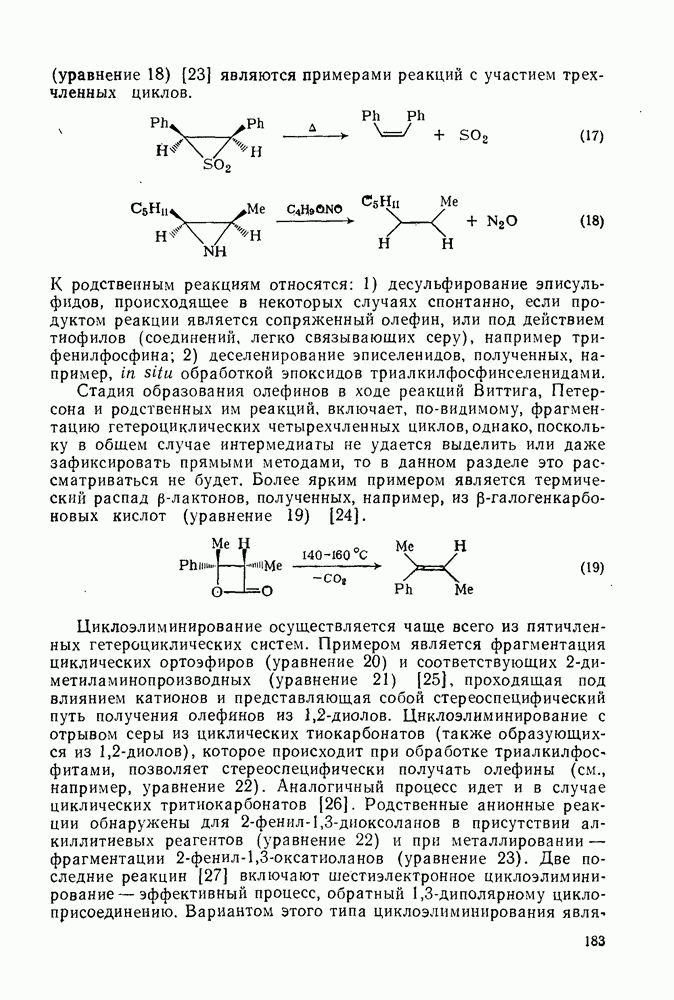
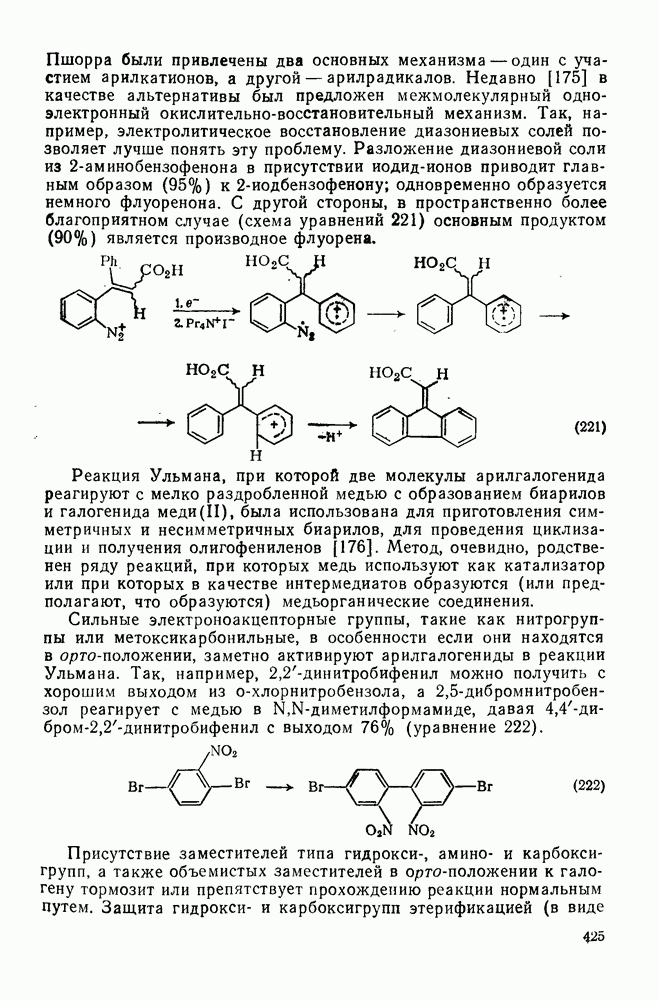
До сих пор мы совсем не упоминали реакций карбениевых ионов, образующихся при диазотировании первичных аминов. Как известно, этот метод приводит к образованию «горячих», или «несольватированных», карбениевых ионов. Были получены сходные результаты Хорошо известна инертность положения в вершине мостиковой группы бицикло Из
Имеются данные по конкурентным реакциям [41] В заключение коснемся вопроса, возникающего при изучении реакций иона серебра(I) с хлорформиатами и представляющего общий интерес в связи с реакциями электрофильного присоединения— элиминирования. Речь идет о реакции метилхлорформиата с анизолом. Изучение реакции Хлорметилирование. Хлорметильную группу вводят в ароматические соединения путем обработки арена формальдегидом и хлористым водородом, обычно в присутствии кислоты Льюиса [25]. Например, бензилхлорид можно получить с хорошим выходом, пропуская сухой хлористый водород в суспензию хлористого цинка (II) и параформа в бензоле (уравнение 70):
Фторметилирование, бромметилирование и иодметилирование осуществляют при использовании соответствующих галогеноводородных кислот. Для галогеналкилирования ароматических субстратов можно использовать также другие альдегиды. Реакция проходит успешно в присутствии некоторых электронодонорных заместителей (например, алкил- и алкоксигрупп) и электроноакцепторных групп (например, нитрогруппы). В отличие от нитробензола м-динитробензол не вступает в реакцию хлорметилирования. Амины и фенолы имеют слишком высокую реакционную способность и, если не содержат других электроноакцепторных групп, то образуют полимерные продукты. Так же легко хлорметилируются полициклические арены. Так, нафталин хлорметилируется преимущественно в а-положение. Хлорметилирование антрацена трудно задержать на стадии монохлорметильного производного, и в результате образуется
Изучение механизма хлорметилирования показало, что первоначально образуется бензиловый спирт, который затем превращается в конечный продукт. Атакующим электрофилом является гидроксиметилкатион (схема уравнений 72).
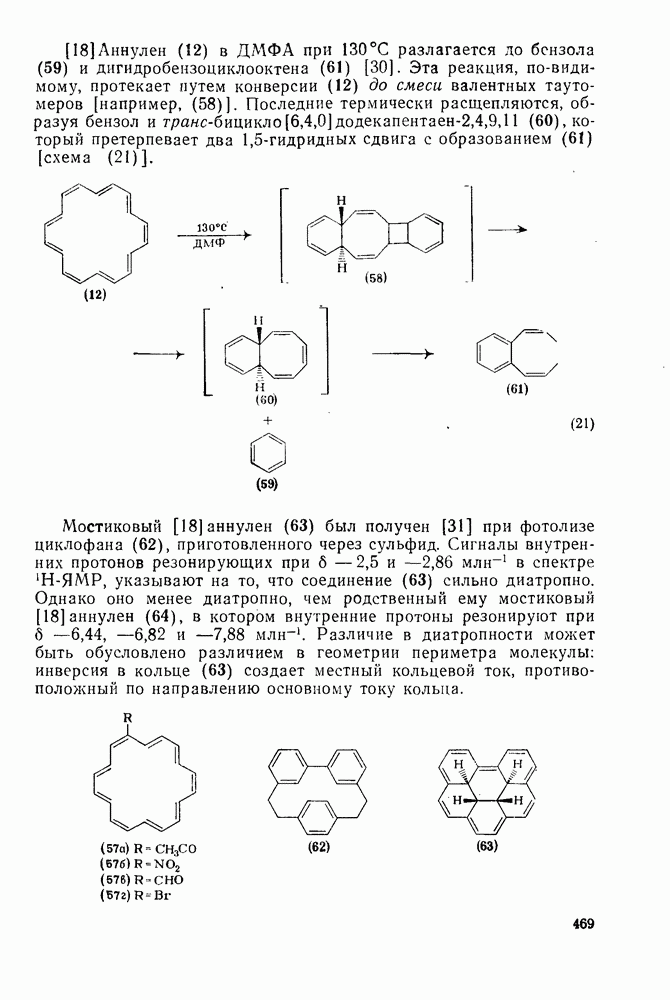
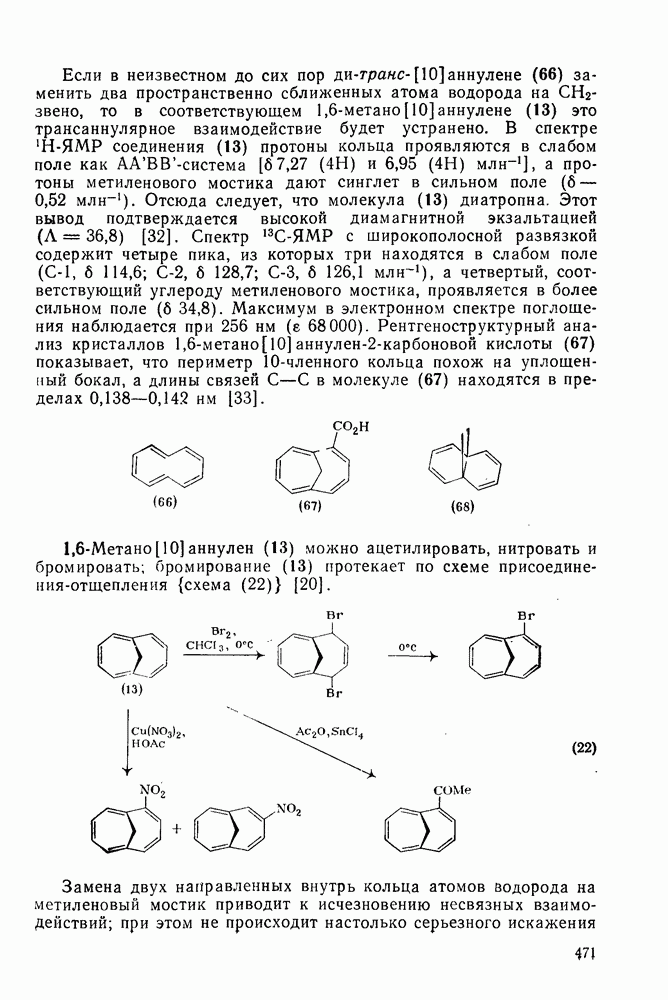
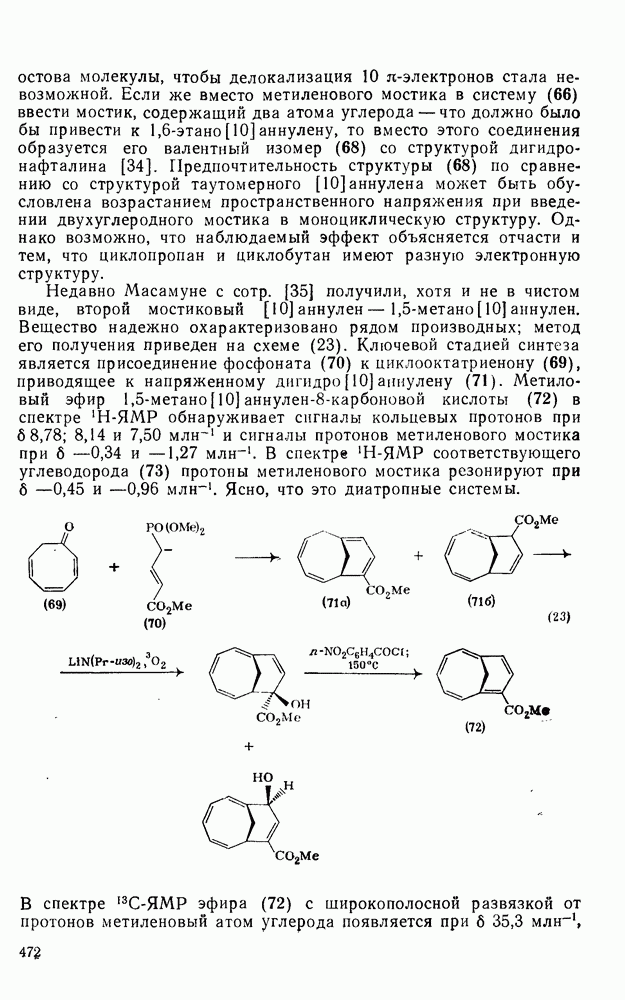
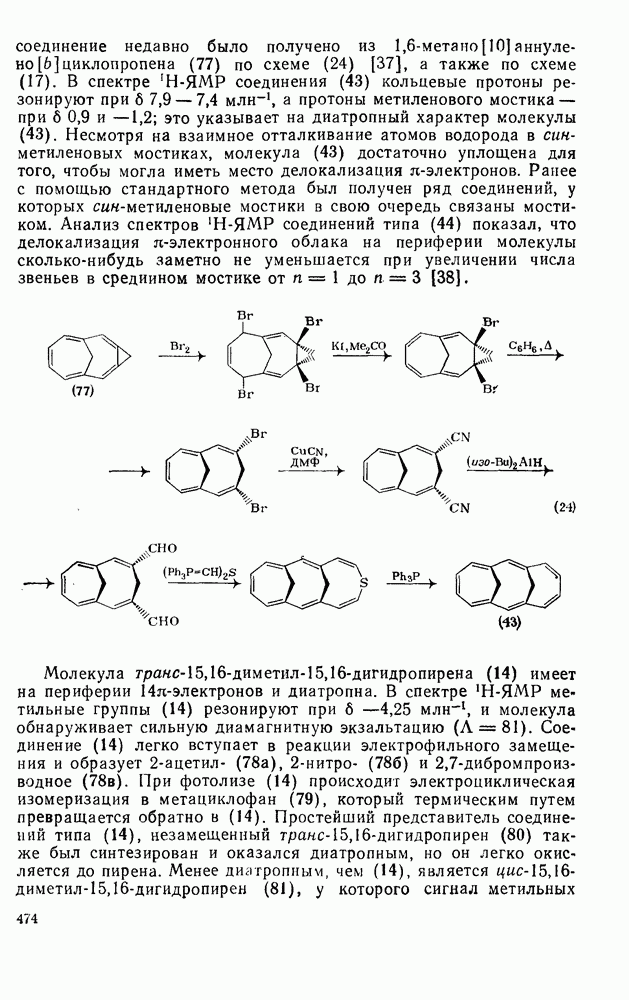
При хлорметилировании могут возникнуть два осложнения, которых уже упоминалось выше. Во-первых, хлорметильная группа является, хотя и в меньшей степени, чем метильная, электронодонорным заместителем, поэтому обычно трудно избежать дальнейшего хлорметилирования. Эта проблема, впрочем, не является столь же серьезной, как в случае алкилирования по Фриделю — Крафтсу. Во-вторых, продукт хлоралкилирования представляет собой бензилгалогенид, который в присутствии кислоты Льюиса может сам алкилировать по реакции Фриделя — Крафтса другую молекулу начального субстрата (уравнение 73). Эта реакция играет заметную роль, если в молекуле субстрата имеются электронодонорные заместители; так, в случае реакции с фенолами и ариламинами образуются полимерные продукты.
Аминометилирование.Введение аминометильной группы в ароматическое кольцо, очевидно, сходно с хлорметилированием и представляет собой особый случай реакции Манниха. Поскольку аминоалкильная группа — недостаточно реакционноспособный электрофил, реакция проходит только с сильнонуклеофильиыми аренами. Реакция довольно широко используется для таких соединений, как пиррол и индол, однако аминоалкилирование карбоциклических систем в настоящее время ограничено фенолами (уравнения 74), а также вторичными и третичными ариламинами [44]. Возможно, что использование предварительно приготовленных электрофилов, таких как диметил (метилен) аммонийтрифторацетат, приведет к расширению сферы применения реакции аминометилирования.
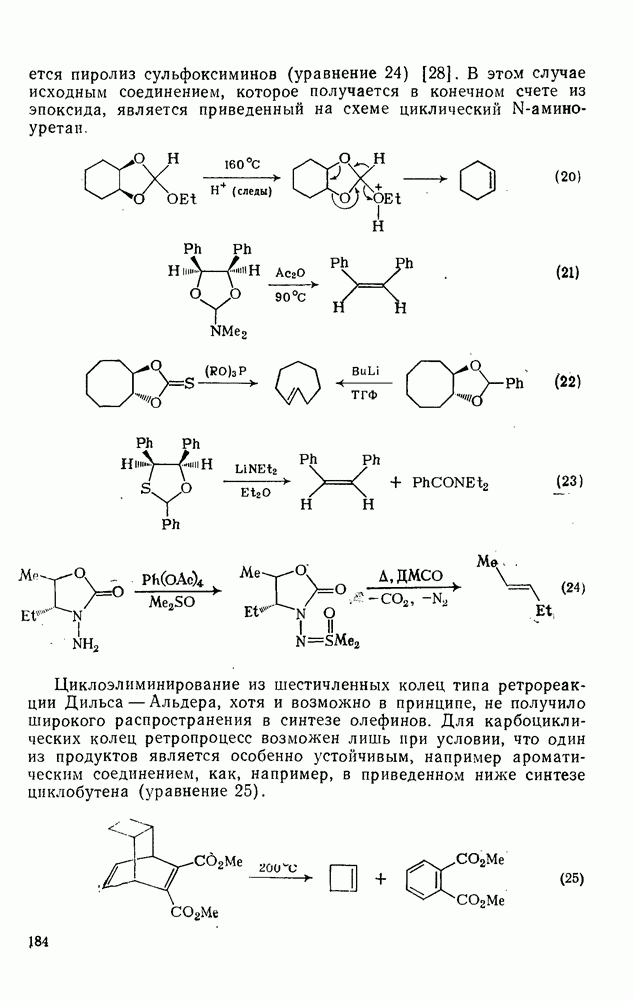
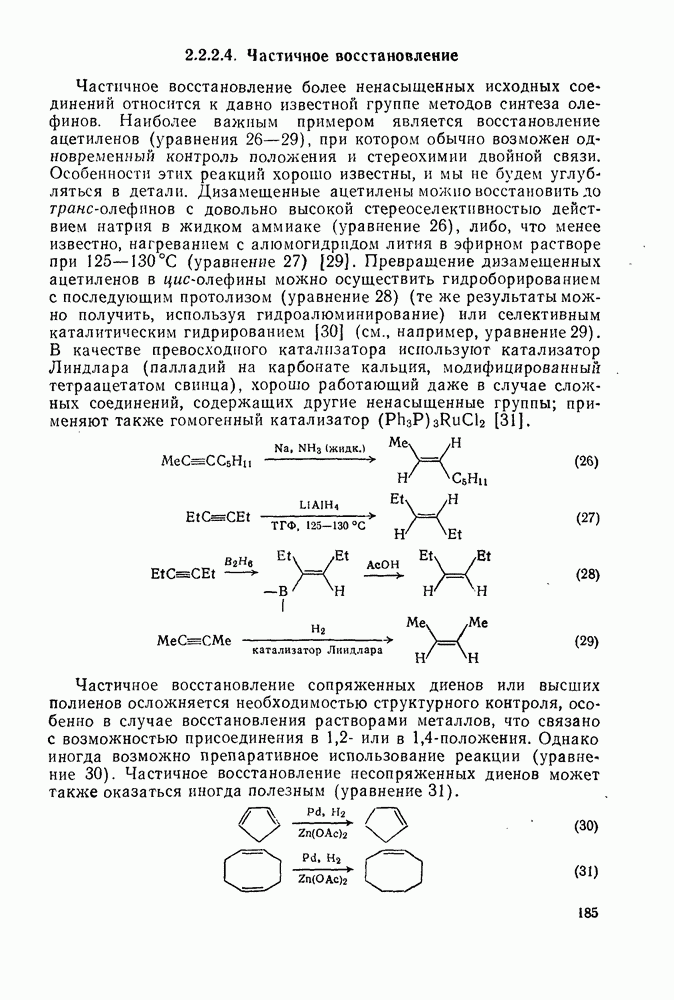
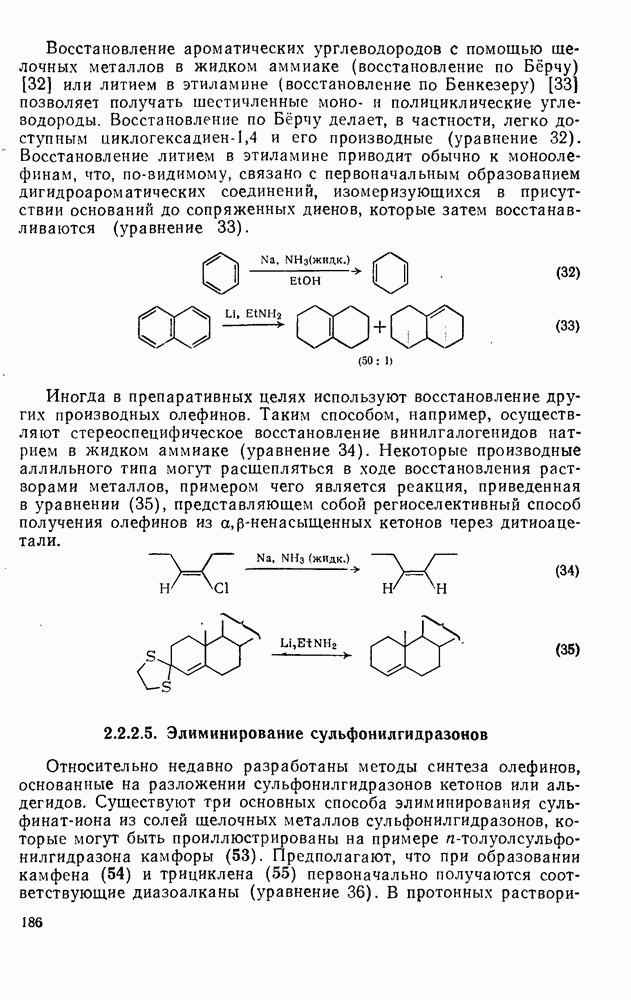
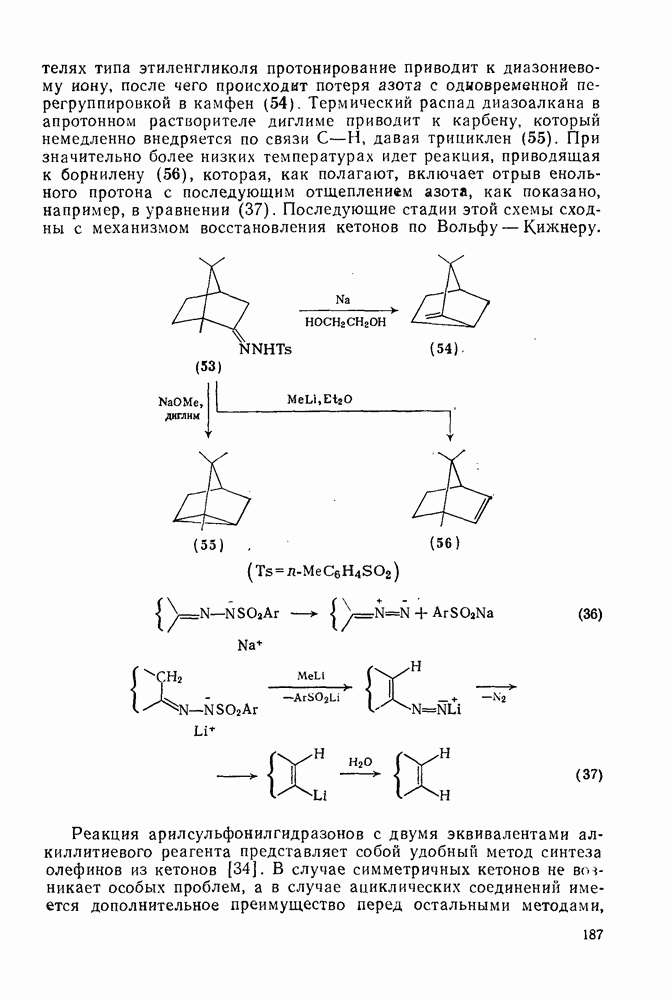
Ацилирование по Фриделю — Крафтсу.Наиболее важный общий метод получения арилкетонов включает реакции галогенангидридов или ангидридов карбоновых кислот с аренами в присутствии кислот Льюиса, а также реакции карбоновых кислот с аренами в присутствии протонных кислот [25]. Смешанные ангидриды карбоновой кислоты и сульфокислоты [46] и в особенности производные трифторметансульфокислоты являются наиболее активными ацилирующими агентами и гладко ацилируют бензол в отсутствие катализатора. Смешанные ангидриды можно получать также situ реакцией галогенангидрида с трифгормегансульфокислотой; последнюю после проведения реакции можно регенерировать в виде бариевой соли почти количественно. situ реакцией галогенангидрида с трифгормегансульфокислотой; последнюю после проведения реакции можно регенерировать в виде бариевой соли почти количественно.
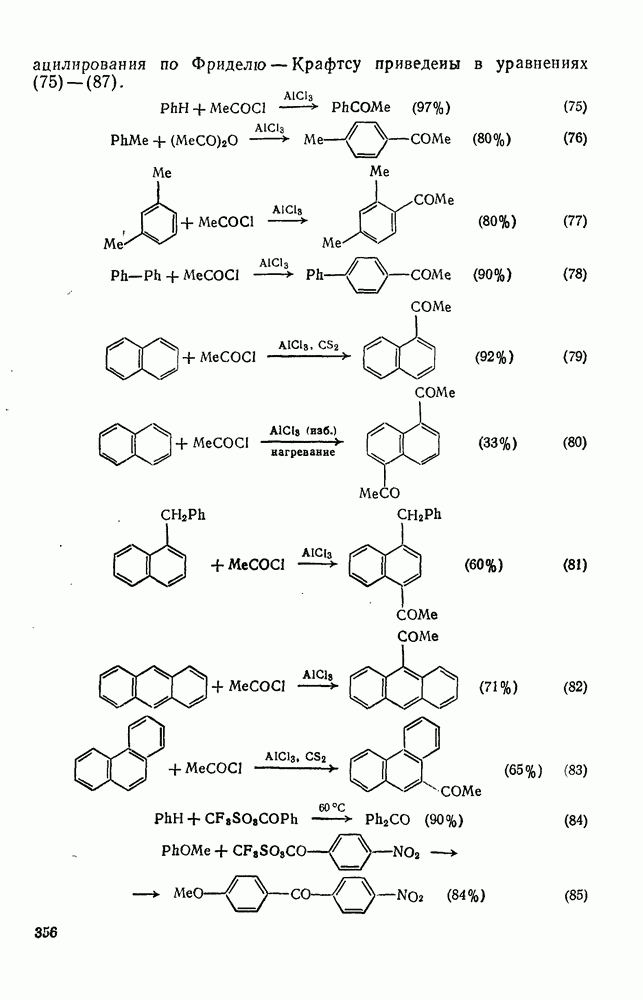
Типичные примеры реакций ацилирования по Фриделю — Крафтсу приведены в уравнениях (75)-(87).
В качестве ацилирующих агентов обычно применяют хлорангид-риды. Можно использовать и другие галогенангидриды, которые располагаются по своей реакционной способности в следующий ряд:
Ацилирование ди- и полиалкилбензолов иногда протекает сложно. Так, хотя при ацетилировании Еще одним преимуществом ацилирования по сравнению с алкилированием является отсутствие перегруппировки алкильного остатка в ацилирующем реагенте. Кроме того, при ацилировании Редко имеют место реакции диспропорционирования. Исключением из этого общего правила является пивалоилирование. Так, в реакции бензола с пивалоилхлоридом в присутствии хлорида алюминия образуется главным образом трет-бутилбензол. С другой стороны, из анизола получается в основном n-метоксипивалофенон (уравнение 88). Очевидно, оксид углерода отщепляется в случае первой реакции, поскольку трет-бутилкатион относительно устойчив.
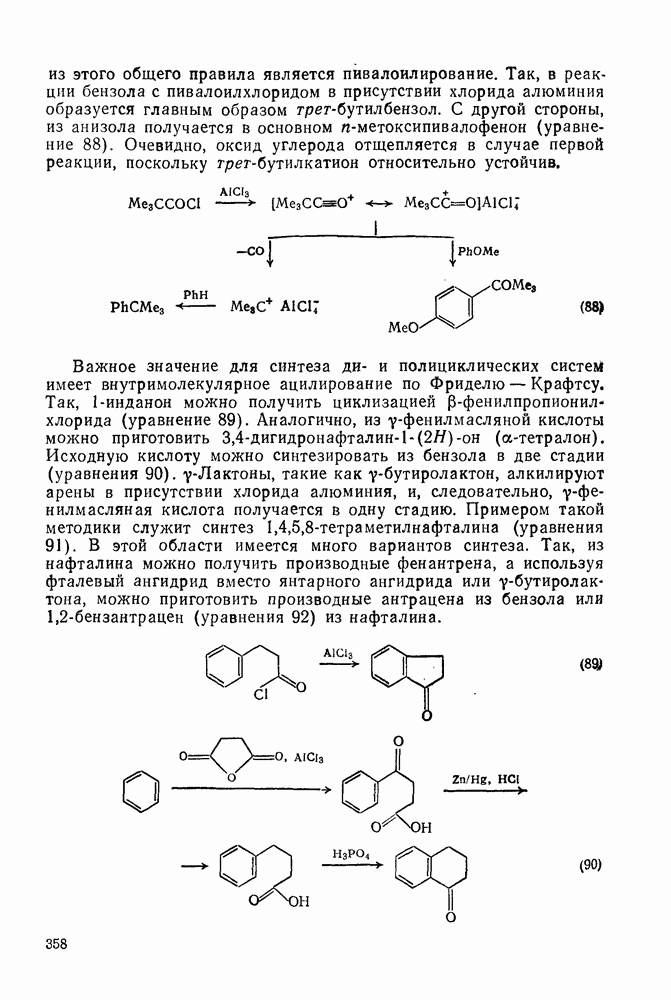
Важное значение для синтеза ди- и полициклических систем имеет внутримолекулярное ацилирование по Фриделю — Крафтсу. Так,
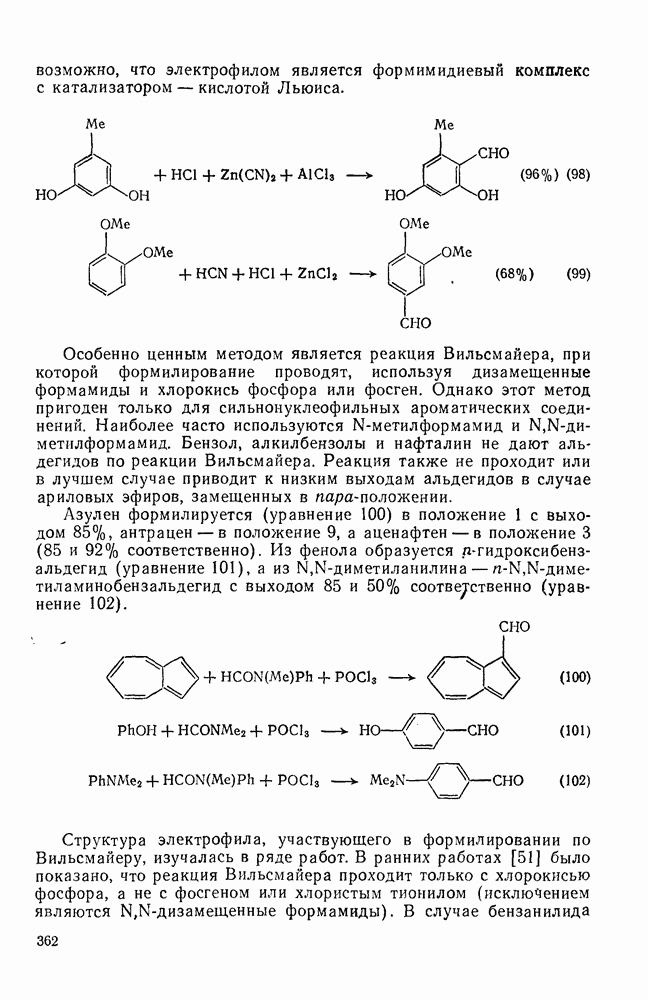
Ацилирование по Фриделю — Крафтсу, как и алкилирование, можно рассматривать двояко. С одной стороны, возможны реакции, протекающие с участием ацилиевого иона, а с другой — реакции, протекающие через непосредственную атаку ареном комплекса 1: 1 (комплекс 68). Между этими крайними механизмами следует поместить реакции с участием тесных ионных пар (см. уравнения 93).
По какому именно механизму будет проходить данная реакция, зависит от ряда факторов, таких как нуклеофильность арена, структура ацилирующего агента, характер катализатора и природа растворителя. Так, ацетилий-ион обнаружен с помощью ИК-спектроскопии в жидком комплексе хлористого ацетила с хлоридом алюминия в полярных растворителях, таких как нитробензол. С другой стороны, в хлороформе обнаружен только комплекс (68), а не ацетилий-ион. В последнее время было выполнено много работ [18], позволяющих в известной степени объяснить ситуацию. Прежде всего следует отметить пространственные затруднения при ацилировании толуола по сравнению, например, с его нитрованием (см. табл. 2.5.2). Так, используя ацетилхлорид и хлорид алюминия, получают Формилирование [25].В этом разделе будут рассмотрены реакции из перечня, приведенного в разд. 2.5.6, а также формилирование с участием формилфторида, дихлорметилового эфира и реакция Раймера — Тимана. из перечня, приведенного в разд. 2.5.6, а также формилирование с участием формилфторида, дихлорметилового эфира и реакция Раймера — Тимана.
Формилирование бензола и простых алкилбензолов можно осуществить с хорошим выходом, используя оксид углерода, хлористый водород и хлористый алюминий под высоким давлением Смесь реагентов удобно получать действием хлорсульфоновой кислоты на муравьиную (уравнение 94). Реакция хлористого водорода с В одной из удобных модификаций синтеза используют формил-фторид [50], который получают при реакции смешанного ангидрида муравьиной и уксусной кислот с безводным фтористым водородом. Формилфторид (т. кип.
Формилирование цианидом цинка(II) и хлористым водородом известно как реакция Гаттермана. В отличие от реакции Гаттермана — Коха, которая не дает результата с фенолами и ариловыми эфирами, этот метод приводит к образованию альдегидов с хорошими выходами. Первоначально в реакции использовалась синильная кислота, хлористый водород и в качестве катализатора кислота Льюиса, например Вполне возможно, что электрофилом является формимидиевый комплекс с катализатором — кислотой Льюиса.
Особенно ценным методом является реакция Вильсмайера, при которой формилирование проводят, используя дизамещенные формамиды и хлорокись фосфора или фосген. Однако этот метод пригоден только для сильнонуклеофильных ароматических соединений. Наиболее часто используются Азулен формилируется (уравнение 100) в положение 1 с выходом 85%, антрацен — в положение 9, а аценафтен — в положение 3 (85 и 92% соответственно). Из фенола образуется n-гидроксибензальдегид (уравнение 101), а из
Структура электрофила, участвующего в формилировании по Вильсмайеру, изучалась в ряде работ. В ранних работах [51] было показано, что реакция Вильсмайера проходит только с хлорокисью фосфора, а не с фосгеном или хлористым тионилом (исключением являются В случае бензанилида альдегид образует только при одновременном присутствии амида, хлорокиси фосфора и нуклеофильного ароматического субстрата. В более поздних работах [52] было обнаружено, что, судя по выходам полученных продуктов,
С другой стороны, имеется много доказательств [53] в пользу того, что хлорокись фосфора реагирует с
Арены, такие как бензол, бифенил, нафталин, формилируются дихлорметилалкиловыми эфирами в присутствии кислоты Льюиса в качестве катализатора. Эти эфиры канцерогенны, и поэтому использовать эту реакцию обычно избегают. Однако дихлорметилалкиловые эфиры легко получаются реакцией пентахлорида фосфора с алкилформиатами. В качестве катализатора наиболее часто используют хлориды титана (IV) и олова (IV). Вероятно, реакция проходит так, как показано на схеме (106):
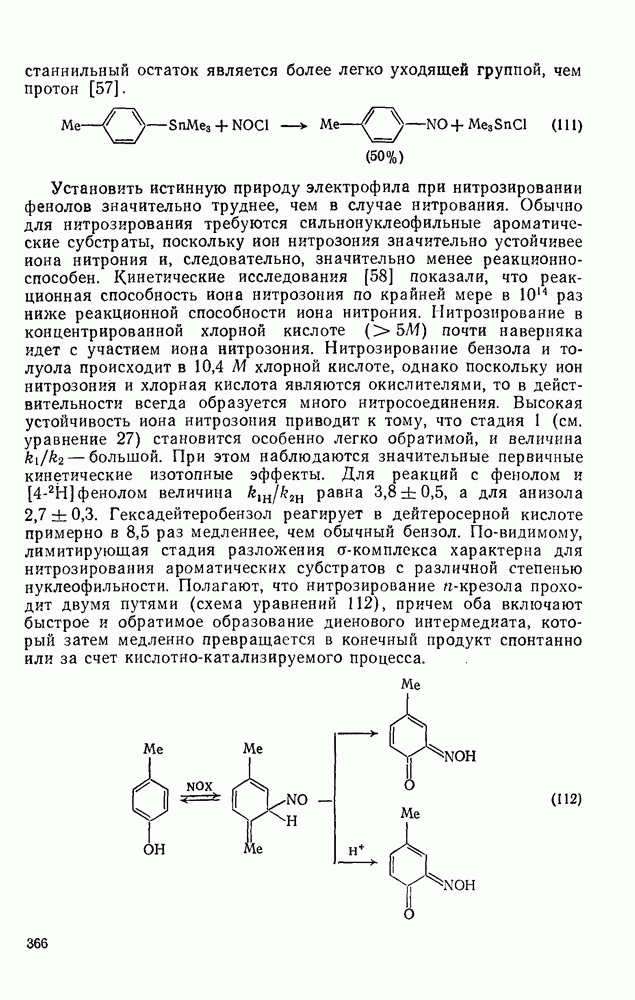
В реакции Раймера — Тимана, которая пригодна только для формилирования фенолов, а также некоторых нуклеофильных гетероциклических систем, таких как пиррол и индол, выходы редко превышают 50% [55]. Реакция приводится в щелочном растворе, а электрофил представляет собой нейтральный дихлоркарбен (уравнения 107 и 108).
Фенолы с заместителем в пара-положении, кроме производных салицилового альдегида, образуют Два атома хлора можно удалить гидрогенолизом (уравнение 53).
Арены можно ацилировать, используя модифицированную реакцию Гаттермана, в которой синильная кислота заменена на алкил- или арилцианид [25]. Эта реакция известна как реакция Губена — Гёша. Для ее проведения в большинстве случаев необходим катализатор - кислота Льюиса; наиболее часто используют хлорид цинка(II). Реакция пригодна для аренов только при использовании активированных нитрилов, например, таких как дихлорацето- нитрил, который реагирует с бензолом в присутствии
В этой реакции электрофил может быть протонированным нитрилом.
|
 |
Оглавление
|





































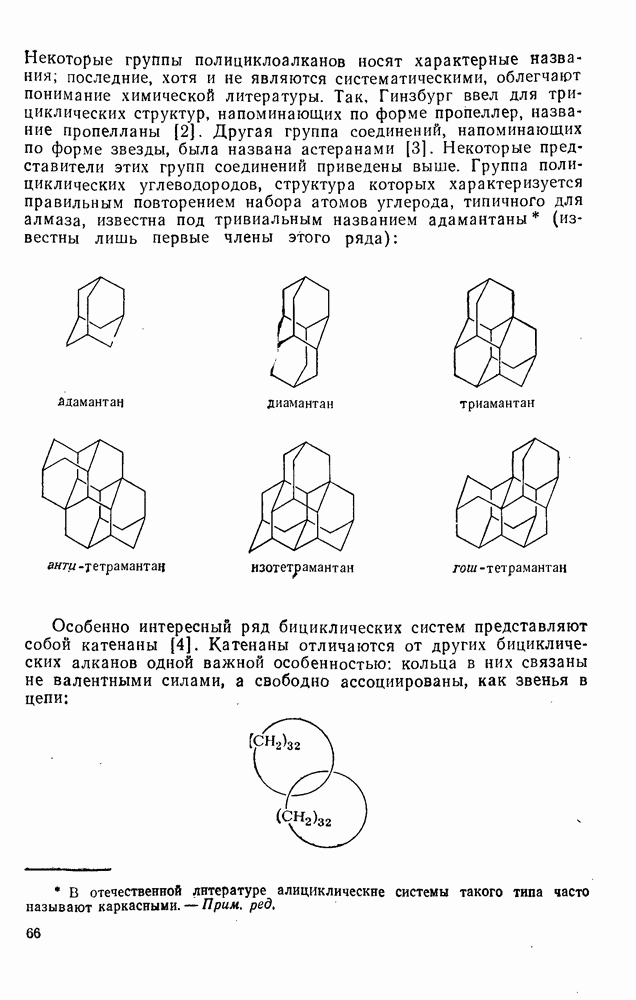


































































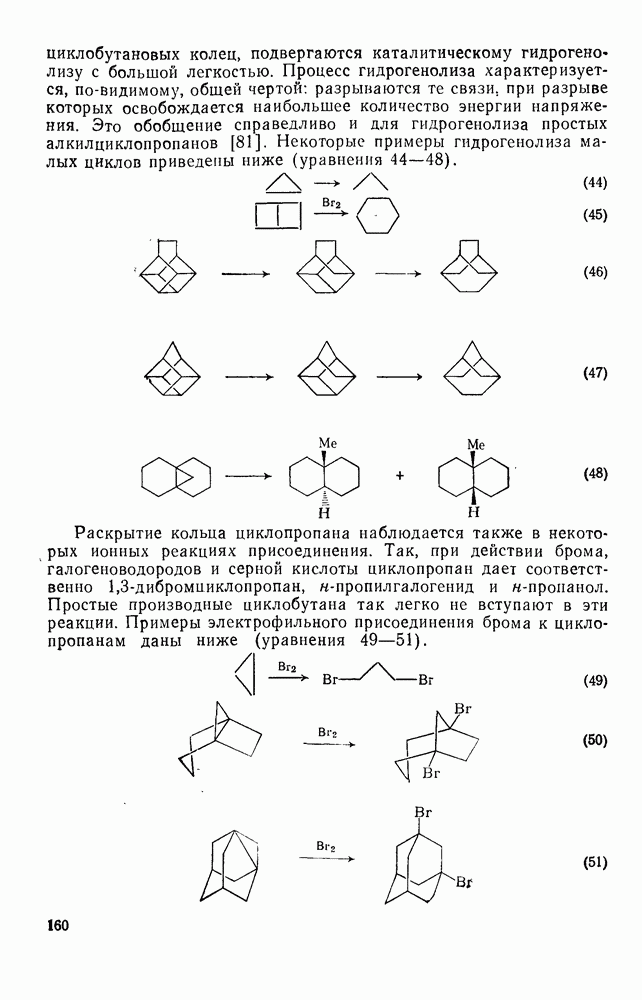



















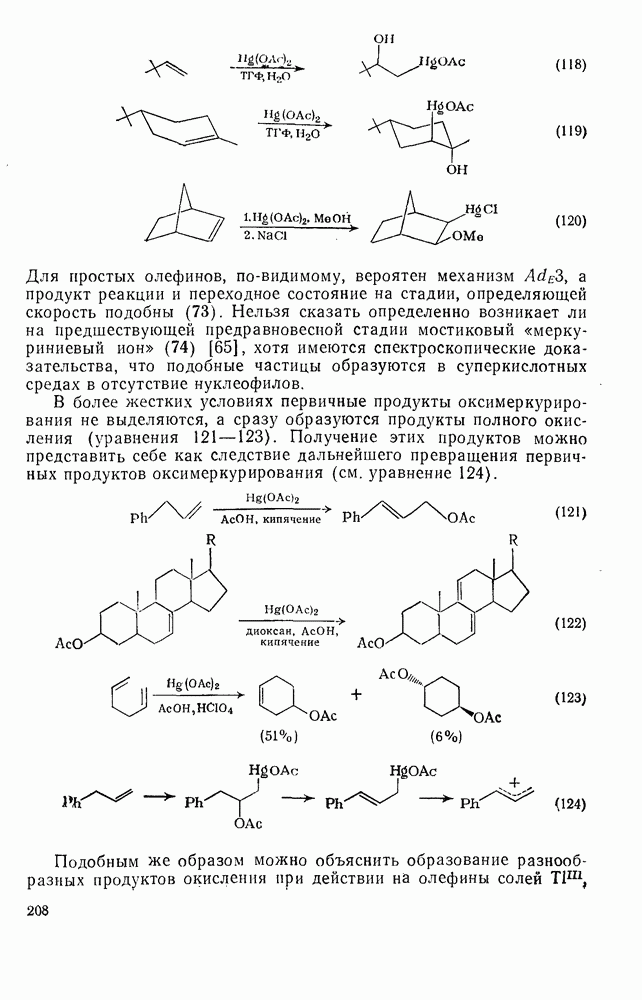
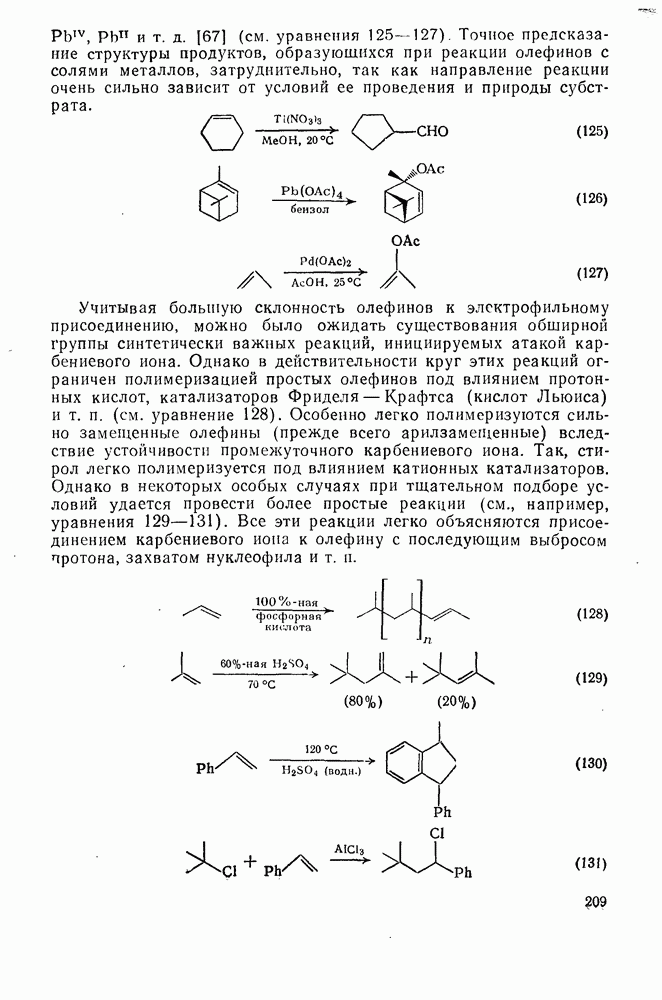
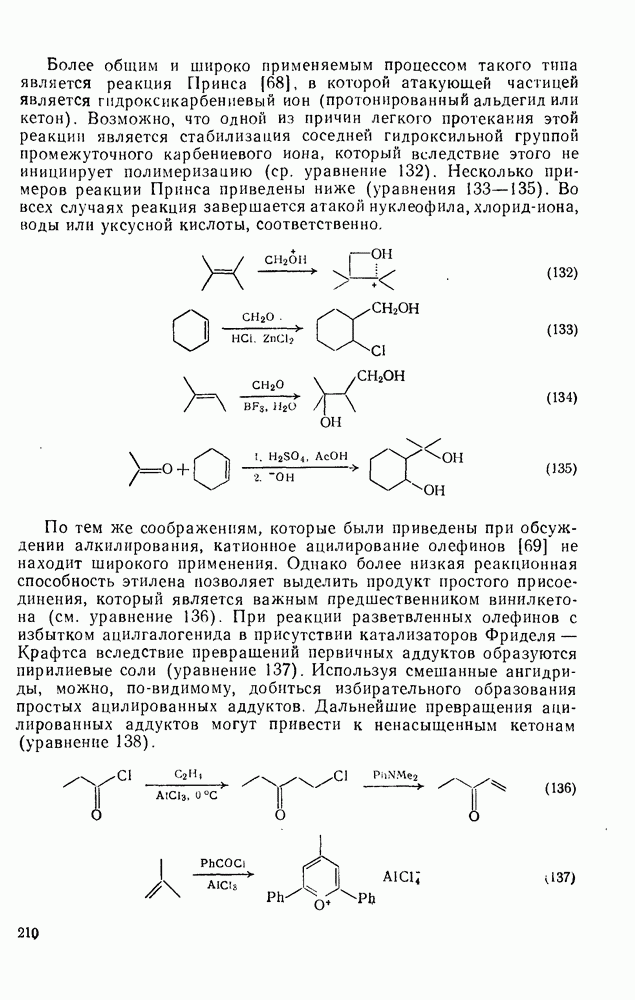




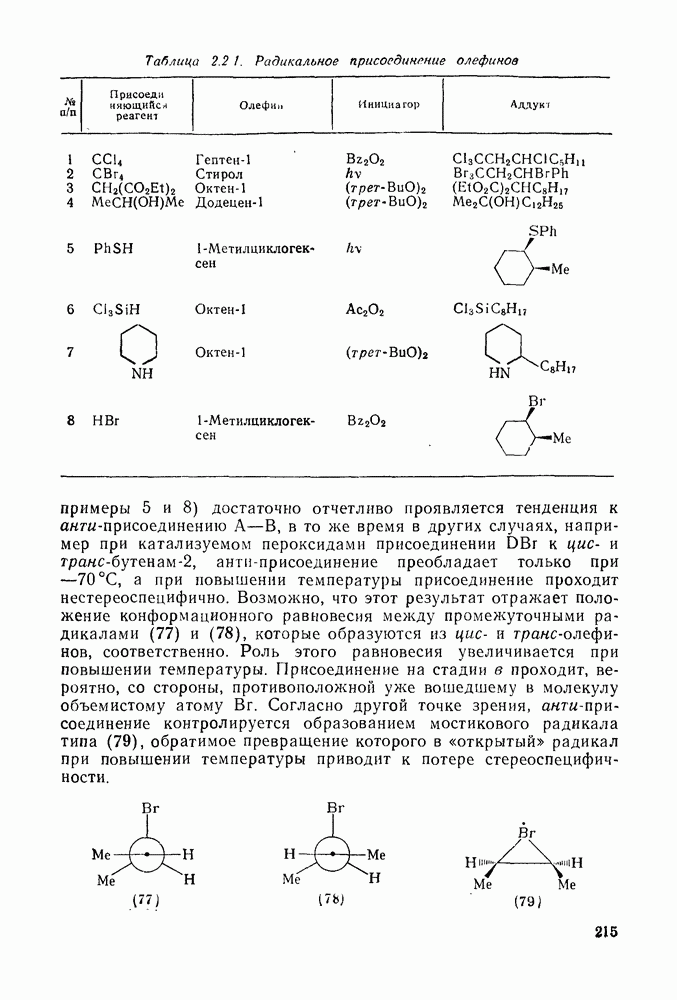











































































































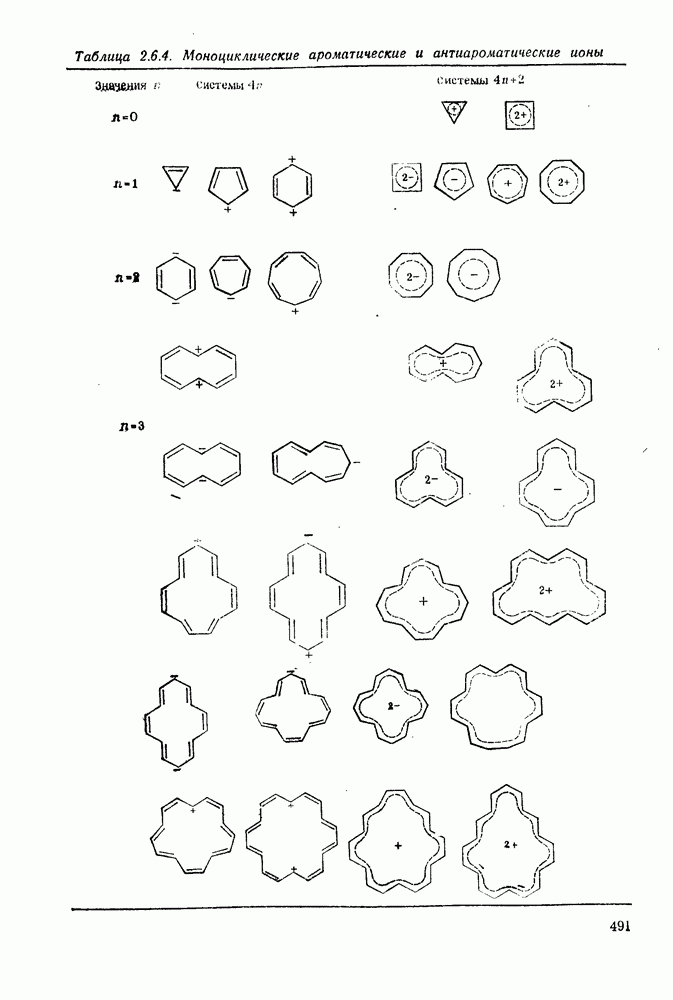
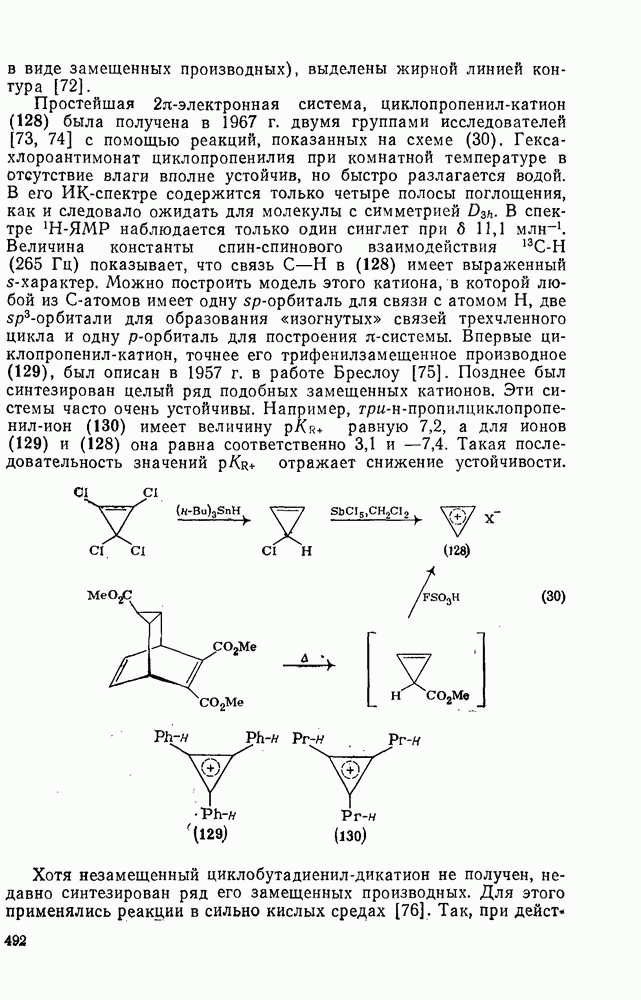
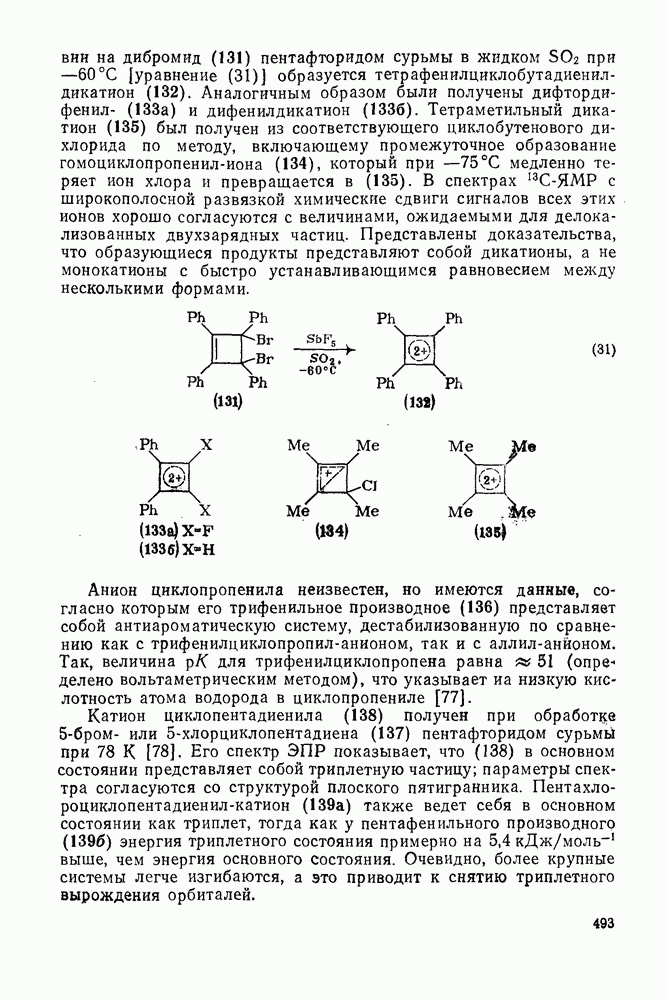
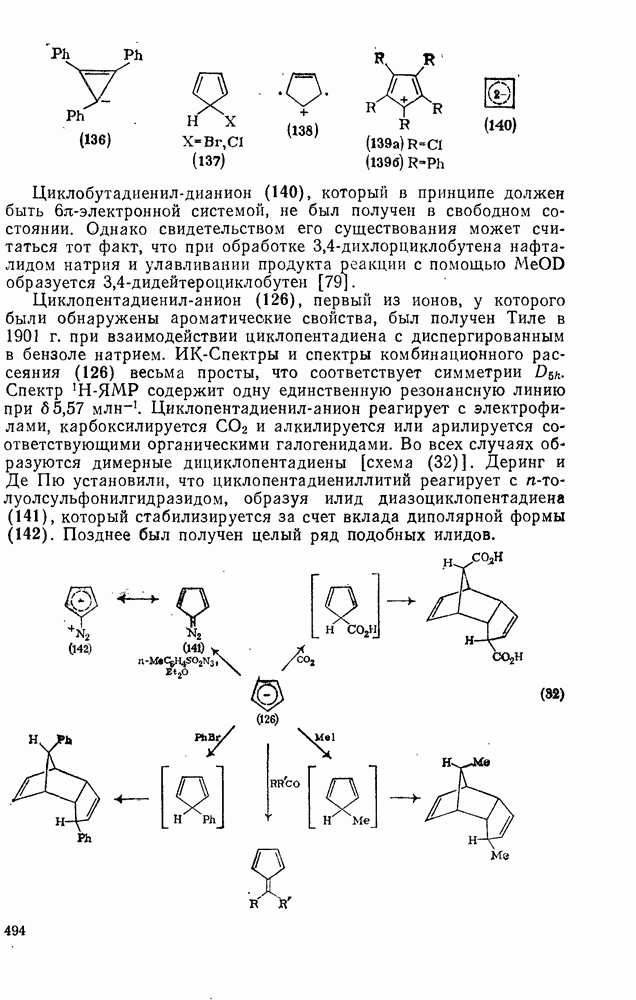



































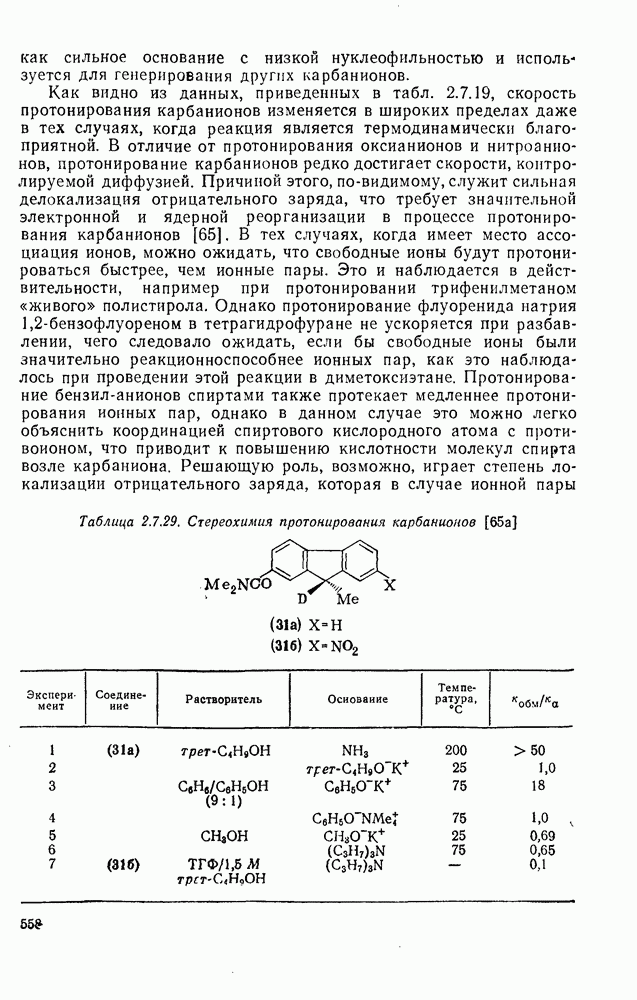


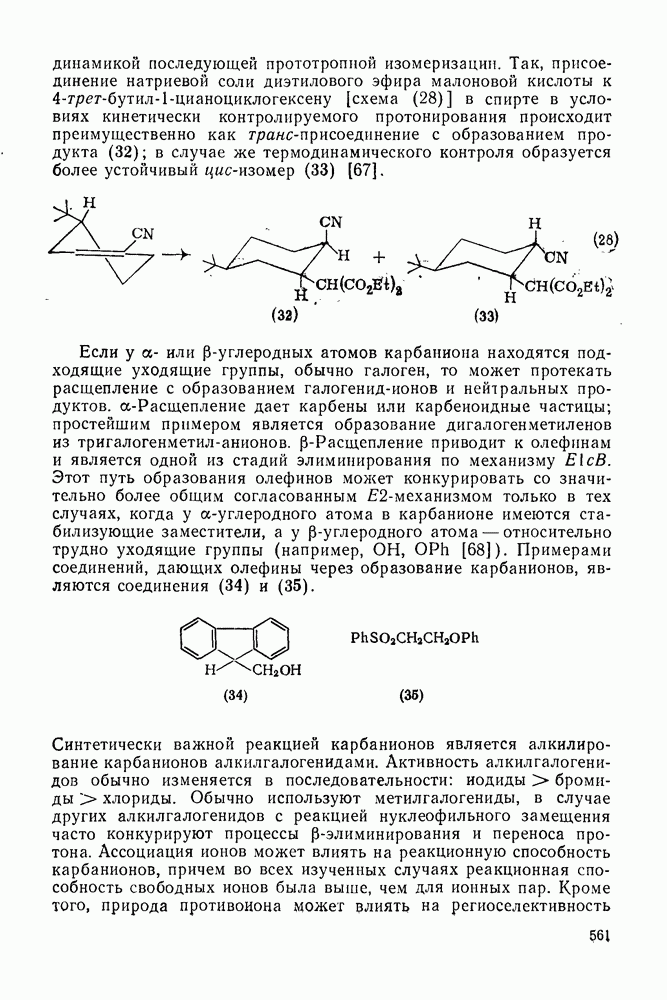

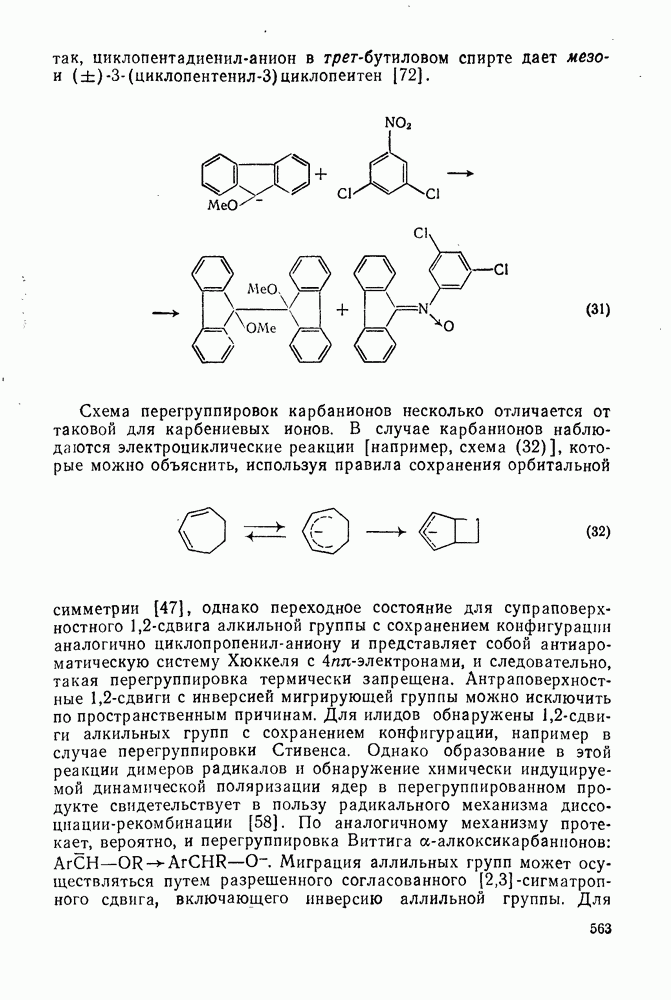















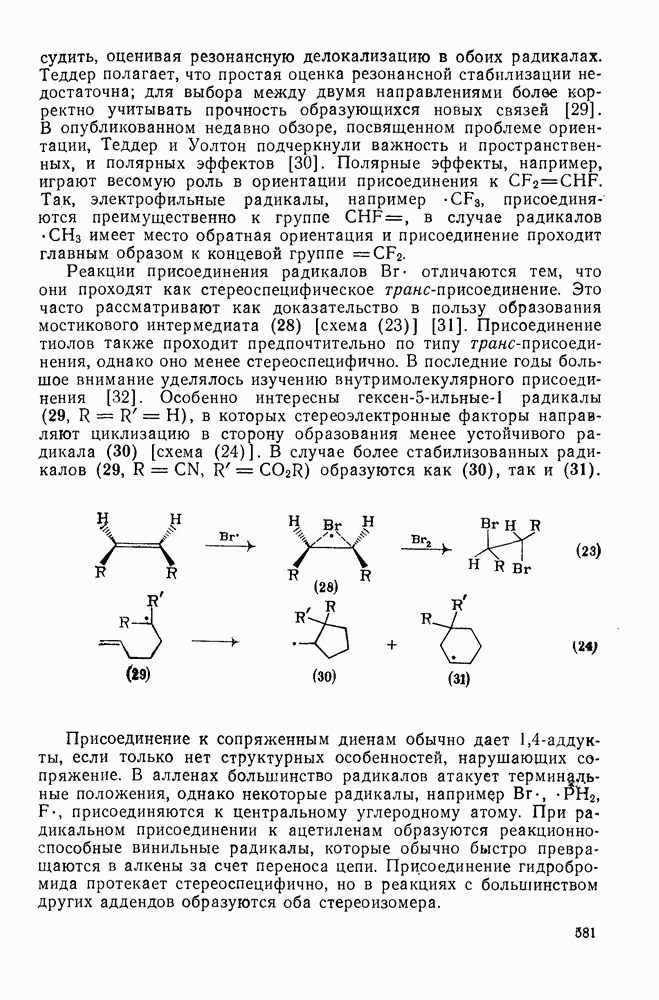


























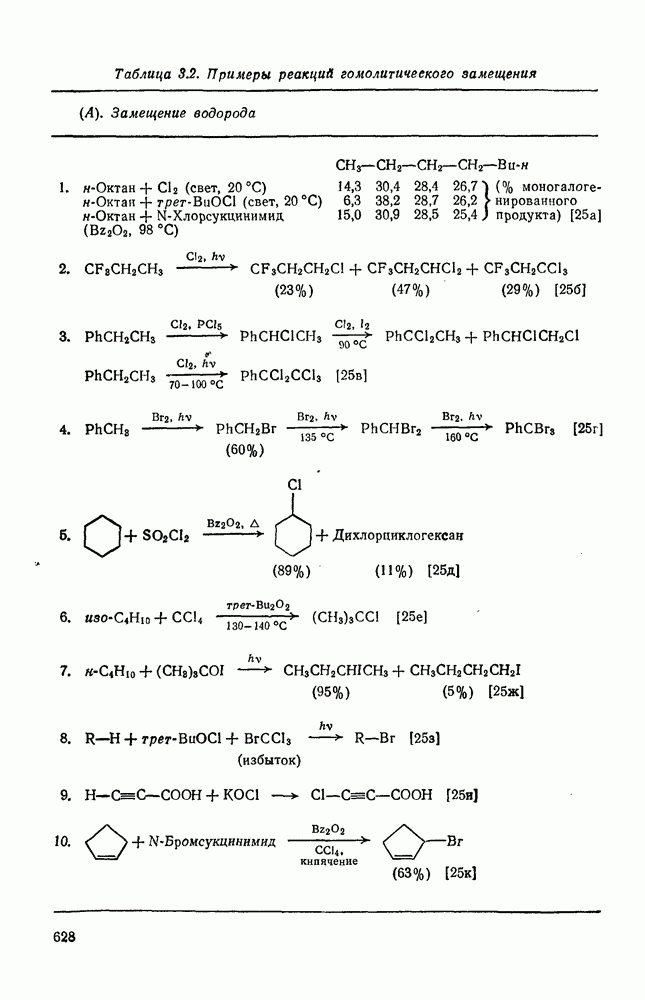
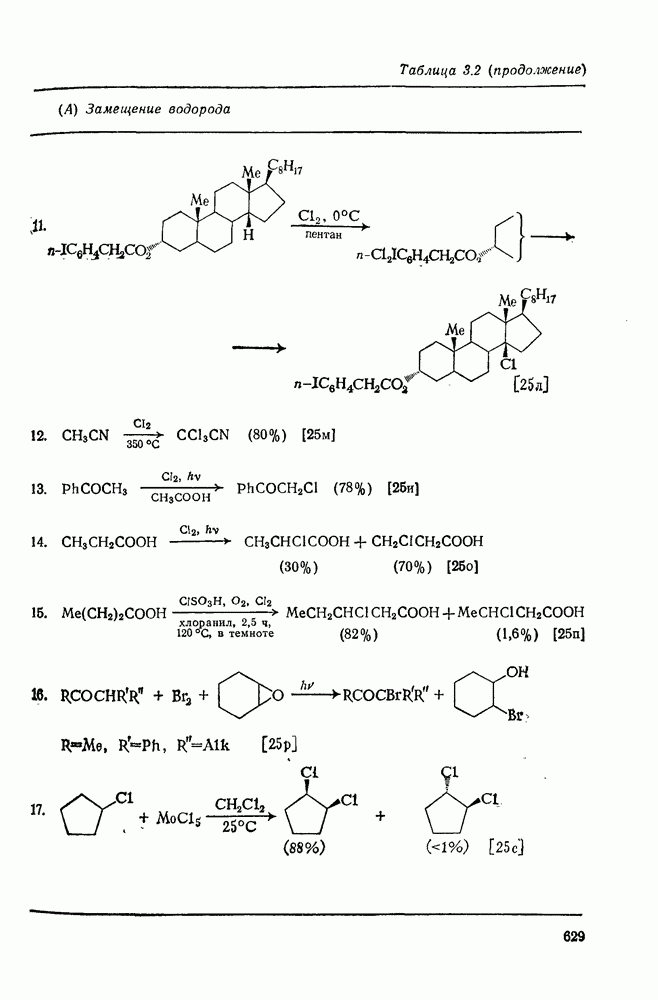
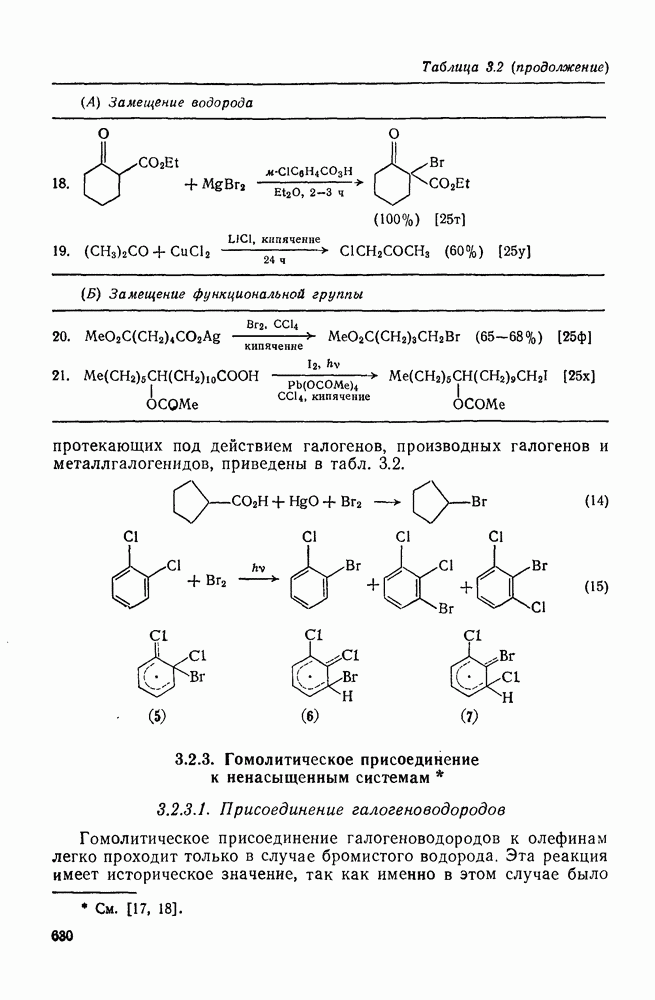
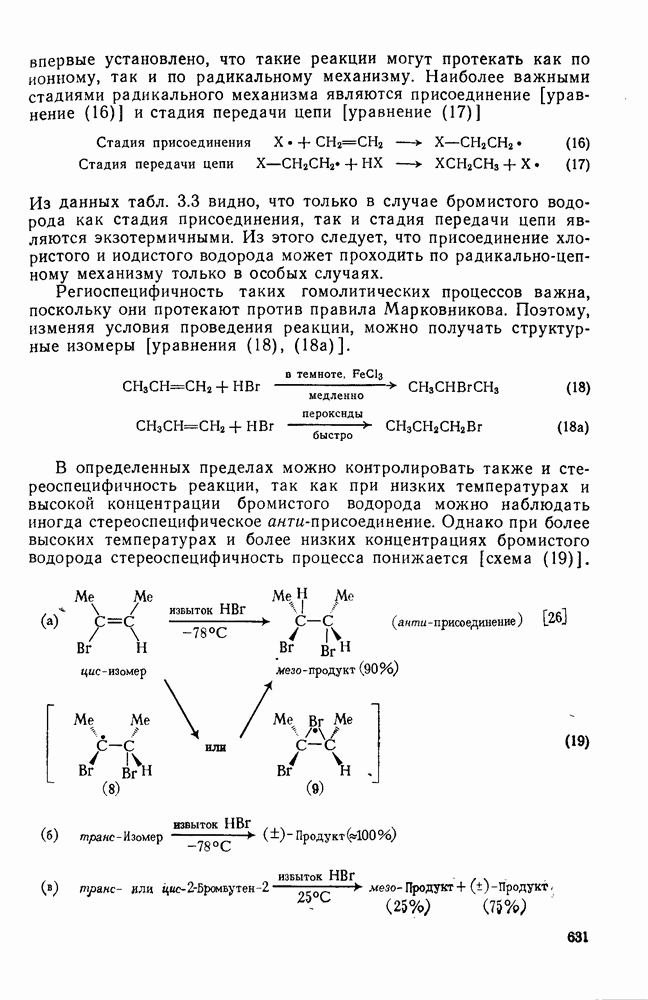


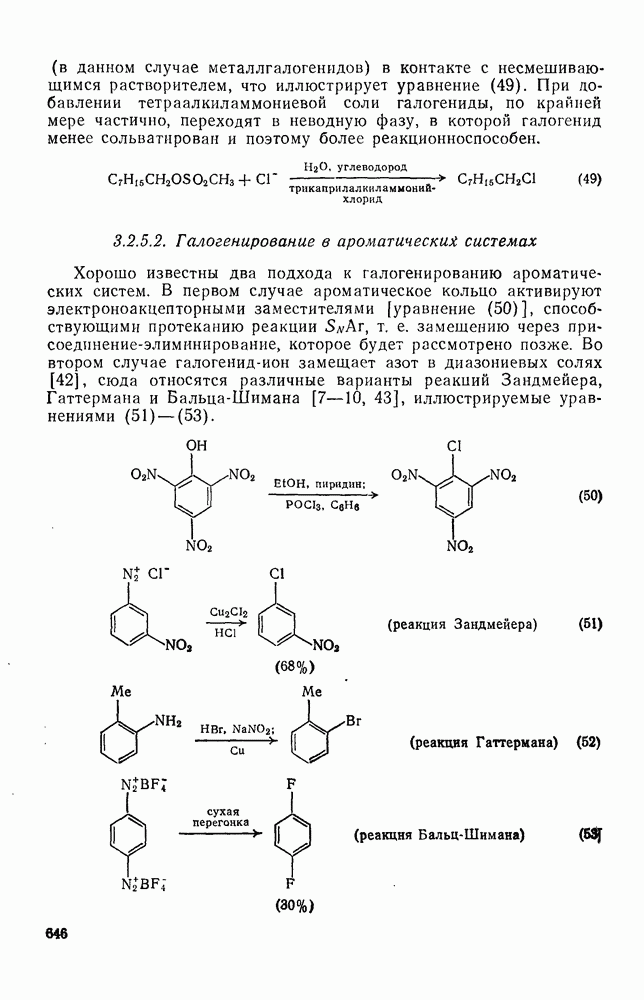
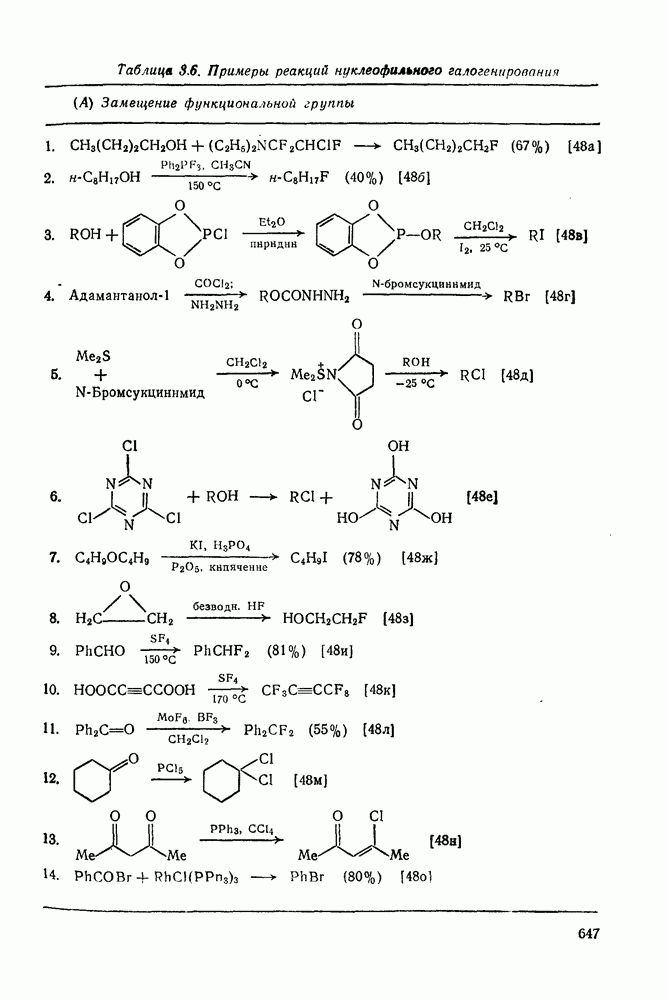
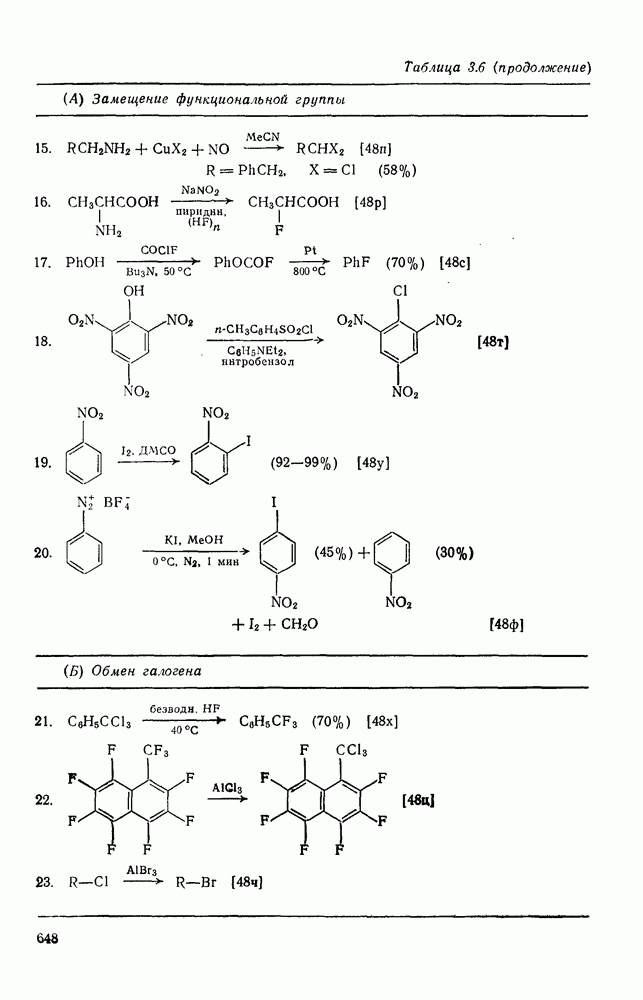
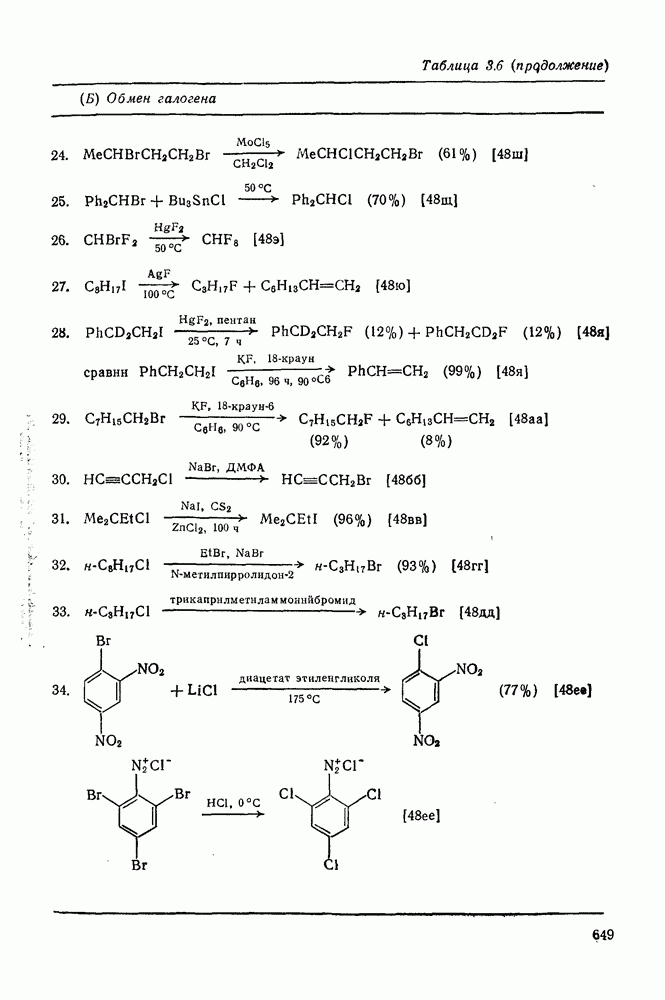








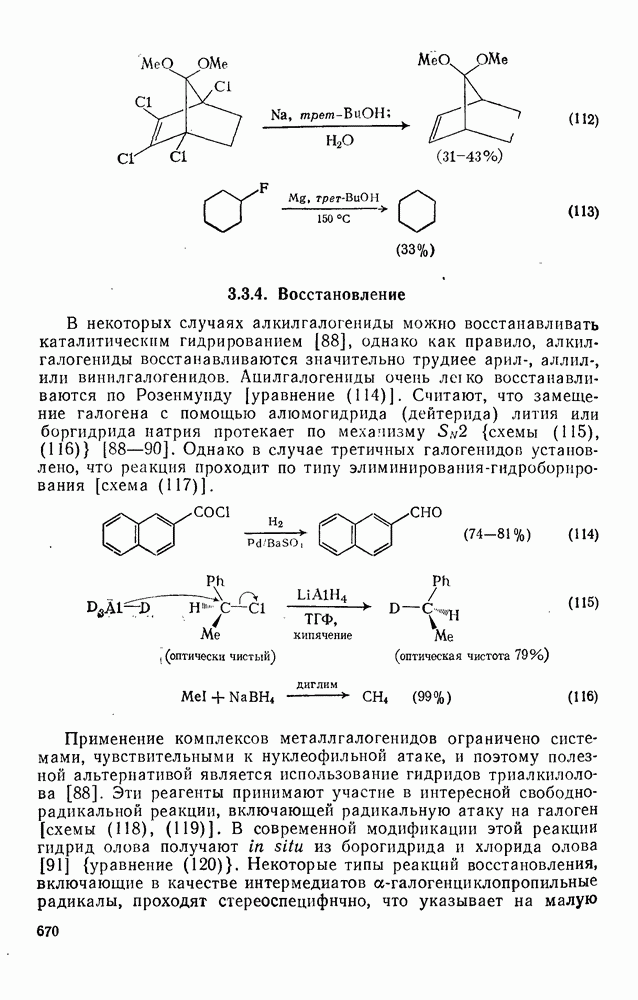
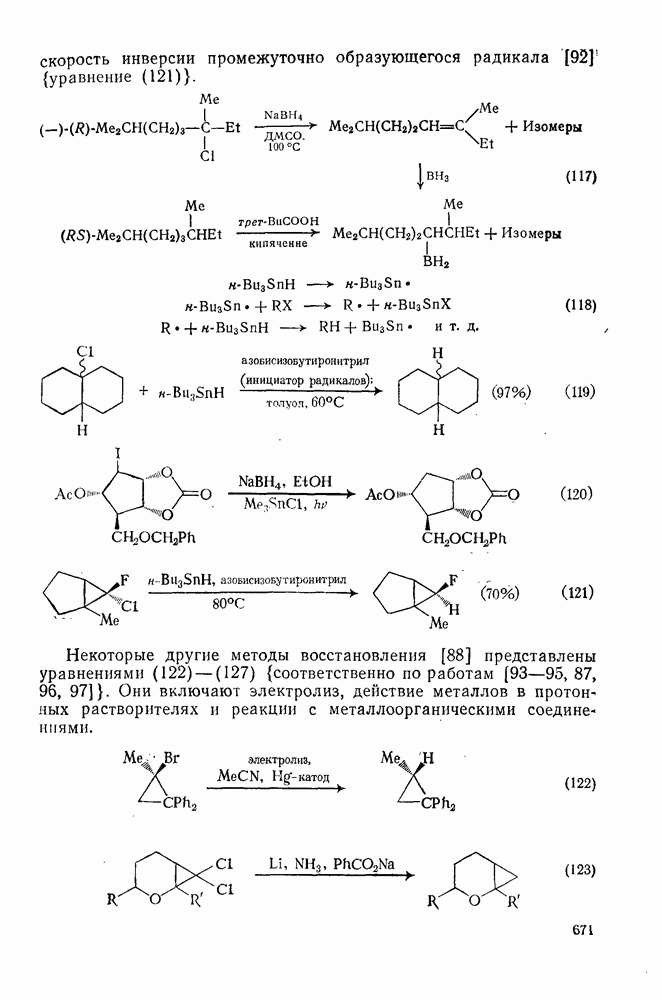

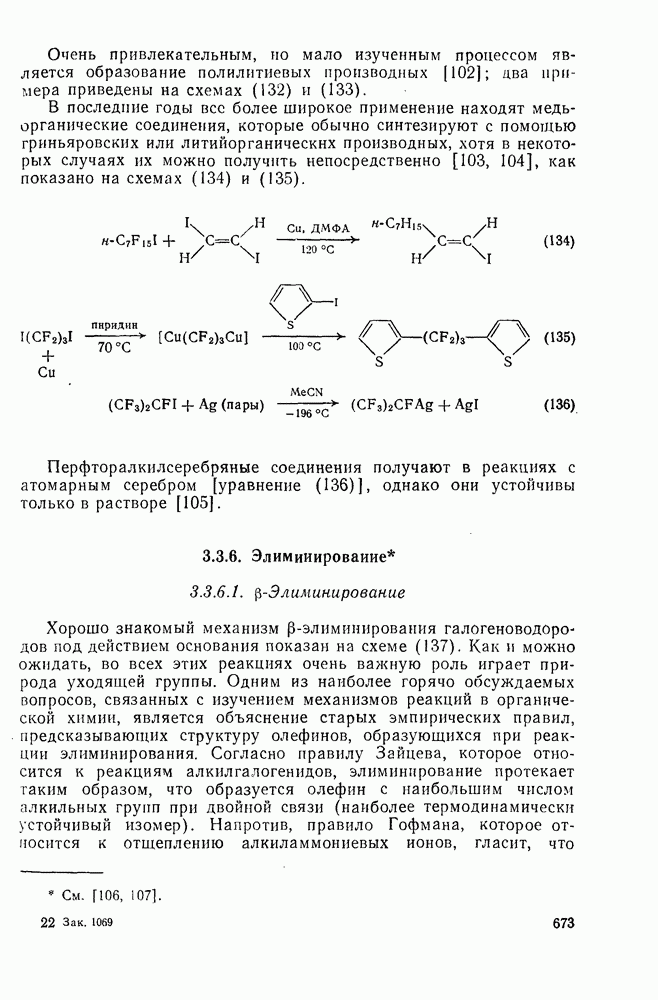
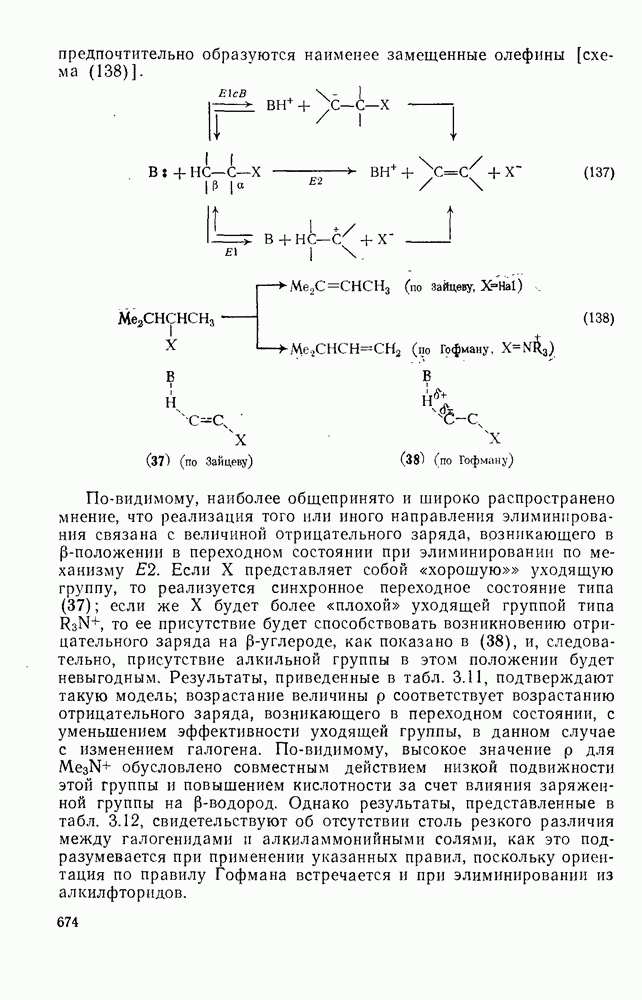



























 . По реакционной способности галогениды располагаются в следующий ряд:
. По реакционной способности галогениды располагаются в следующий ряд:  . Так,
. Так,  -хлор-3-фторпропан реагирует с бензолом в присутствии трихлорида бора с образованием
-хлор-3-фторпропан реагирует с бензолом в присутствии трихлорида бора с образованием  -хлор-3-фенилпропана. По реакционной способности все алкили-рующие агенты размещаются в следующей последовательности: аллил
-хлор-3-фенилпропана. По реакционной способности все алкили-рующие агенты размещаются в следующей последовательности: аллил  бензил > третичный алкил > вторичный алкил > первичных алкил.
бензил > третичный алкил > вторичный алкил > первичных алкил.
 раза больше, чем для бензола. При эффективном перемешивании снижается до минимума влияние преимущественного растворения алкилбензолов в слое катализатора, где происходит реакция.
раза больше, чем для бензола. При эффективном перемешивании снижается до минимума влияние преимущественного растворения алкилбензолов в слое катализатора, где происходит реакция.
 ,
,  и алкоксигруппы, вызывает затруднения. Дело в том, что эти группы обладают относительно высокой основностью и координируются с катализатором — кислотой Льюиса, эффективно выводя катализатор из системы. Так, ариламины легко алкилируются олефинами только при использовании в качестве катализаторов анилидов алюминия.
и алкоксигруппы, вызывает затруднения. Дело в том, что эти группы обладают относительно высокой основностью и координируются с катализатором — кислотой Льюиса, эффективно выводя катализатор из системы. Так, ариламины легко алкилируются олефинами только при использовании в качестве катализаторов анилидов алюминия.
 , такая перегруппировка не создает трудностей. Неразветвленные спирты обычно не претерпевают перегруппировки при использовании в качестве катализатора хлорида алюмнния, но перегруппировываются в присутствии трифторида бора или серной кислоты. На результат часто влияют условия проведения реакции. Так, при комнатной температуре бензол реагирует с н-пропилхлоридом в присутствии хлорида алюминия с образованием главным образом н-пропилбензола, тогда как при более высоких температурах образуются значительные количества кумола (уравнение 56).
, такая перегруппировка не создает трудностей. Неразветвленные спирты обычно не претерпевают перегруппировки при использовании в качестве катализатора хлорида алюмнния, но перегруппировываются в присутствии трифторида бора или серной кислоты. На результат часто влияют условия проведения реакции. Так, при комнатной температуре бензол реагирует с н-пропилхлоридом в присутствии хлорида алюминия с образованием главным образом н-пропилбензола, тогда как при более высоких температурах образуются значительные количества кумола (уравнение 56).


 проводить в неполярных растворителях, то происходит перераспределение метки в образующемся этилбензоле.
проводить в неполярных растворителях, то происходит перераспределение метки в образующемся этилбензоле.
 с бензолом наблюдалось лишь незначительное перераспределение метки [29]. Чтобы произошло перераспределение метки в последней реакции при изомеризации н-пропилкарбениевого иона в другой н-пропил-карбениевый ион, должны были бы осуществиться два гидридных сдвига или один метильный сдвиг. Порядок термодинамической устойчивости алкилбензолов изменяется в ряду: третичный < вторичный < первичный, и поэтому реакции, которые на первый взгляд проходят без перегруппировки, в действительности протекают в условиях термодинамического контроля, причем первоначально образующийся втор-алкилбензол (кинетический контроль) затем подвергается перегруппировке с образованием н-алкилбензола. Такое течение реакции возможно только потому, что алкилирование по Фриделю — Крафтсу обратимо. Поэтому, чтобы избежать осложнений, первичные алкиларены часто получают ацилированием по Фриделю — Крафтсу с последующим восстановлением карбонильной группы по Клемменсену или Вольфу — Кижнеру.
с бензолом наблюдалось лишь незначительное перераспределение метки [29]. Чтобы произошло перераспределение метки в последней реакции при изомеризации н-пропилкарбениевого иона в другой н-пропил-карбениевый ион, должны были бы осуществиться два гидридных сдвига или один метильный сдвиг. Порядок термодинамической устойчивости алкилбензолов изменяется в ряду: третичный < вторичный < первичный, и поэтому реакции, которые на первый взгляд проходят без перегруппировки, в действительности протекают в условиях термодинамического контроля, причем первоначально образующийся втор-алкилбензол (кинетический контроль) затем подвергается перегруппировке с образованием н-алкилбензола. Такое течение реакции возможно только потому, что алкилирование по Фриделю — Крафтсу обратимо. Поэтому, чтобы избежать осложнений, первичные алкиларены часто получают ацилированием по Фриделю — Крафтсу с последующим восстановлением карбонильной группы по Клемменсену или Вольфу — Кижнеру.
 образуются о-этилтолуол
образуются о-этилтолуол  , м-этилтолуол (21%) и n-этилтолуол
, м-этилтолуол (21%) и n-этилтолуол  . По сравнению с соотношением изомеров, получаемым при нитровании толуола, в данном случае выход м-изомера высок. Этот факт может быть успешно использован в синтетических целях. Так, при реакции трет-бутилбензола с трет-бутилхлоридом в присутствии хлористого алюминия (в начальной стадии при
. По сравнению с соотношением изомеров, получаемым при нитровании толуола, в данном случае выход м-изомера высок. Этот факт может быть успешно использован в синтетических целях. Так, при реакции трет-бутилбензола с трет-бутилхлоридом в присутствии хлористого алюминия (в начальной стадии при  ) образуется
) образуется  -три-трет-бутилбензол с выходом
-три-трет-бутилбензол с выходом  [30]. Тот же продукт получается при трет-бутилировании
[30]. Тот же продукт получается при трет-бутилировании  -ди-трет-бутилбензола. Опыты с меченными тритием соединениями обнаружили, что преобладающим, если не совершенно полным, является межмолекулярный перенос трет-бутильных групп. Результаты согласуются с механизмом, приведенным ниже (уравнение (59):
-ди-трет-бутилбензола. Опыты с меченными тритием соединениями обнаружили, что преобладающим, если не совершенно полным, является межмолекулярный перенос трет-бутильных групп. Результаты согласуются с механизмом, приведенным ниже (уравнение (59):

 , можно использовать для синтеза производных тетрагидронафталина, октагидрофенантрена и индана. Дегидрогенизация в первых двух случаях приводит далее к нафталинам и фенантренам. В последнем синтезе (уравнение 62) более удовлетворительные результаты были получены при использовании ионообменной смолы
, можно использовать для синтеза производных тетрагидронафталина, октагидрофенантрена и индана. Дегидрогенизация в первых двух случаях приводит далее к нафталинам и фенантренам. В последнем синтезе (уравнение 62) более удовлетворительные результаты были получены при использовании ионообменной смолы  (вместо реагента Брадшера [32], серной кислоты или муравьиной кислоты):
(вместо реагента Брадшера [32], серной кислоты или муравьиной кислоты):

 -арилбензиловых спиртов на
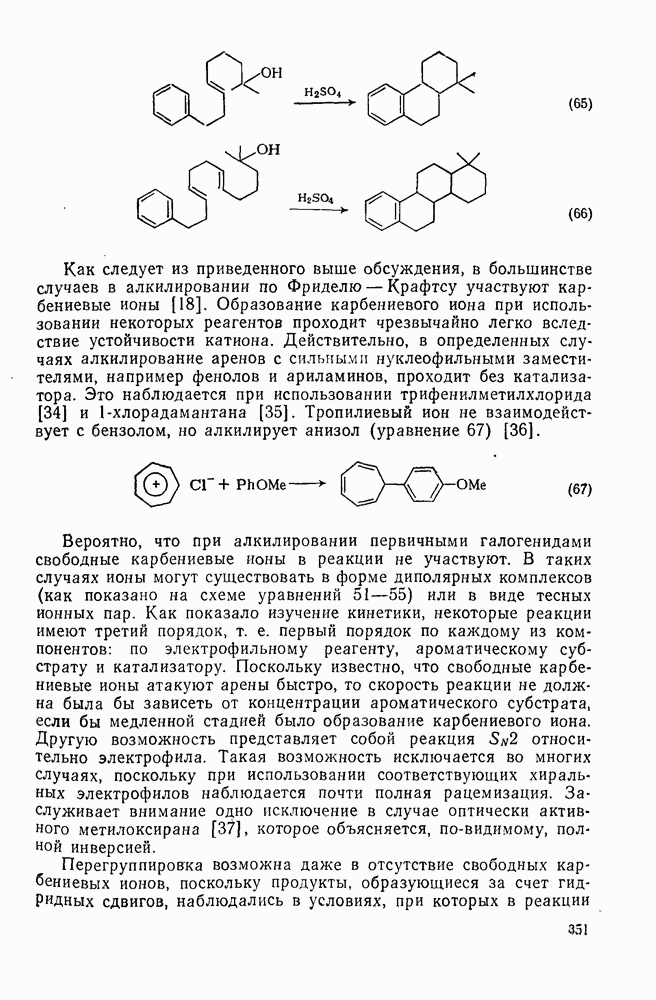
-арилбензиловых спиртов на  [33]. Однако реакцию необходимо проводить в неароматических растворителях, таких как циклогексан, поскольку образующийся промежуточно «горячий» катион может атаковать бензол и нитробензол (уравнение 63). Очевидно, что ароматическая циклодегидратация типа, показанного в уравнении (64), родственна реакциям, приведенным выше, как и реакциям, изображенным уравнениями (65) и (66).
[33]. Однако реакцию необходимо проводить в неароматических растворителях, таких как циклогексан, поскольку образующийся промежуточно «горячий» катион может атаковать бензол и нитробензол (уравнение 63). Очевидно, что ароматическая циклодегидратация типа, показанного в уравнении (64), родственна реакциям, приведенным выше, как и реакциям, изображенным уравнениями (65) и (66).



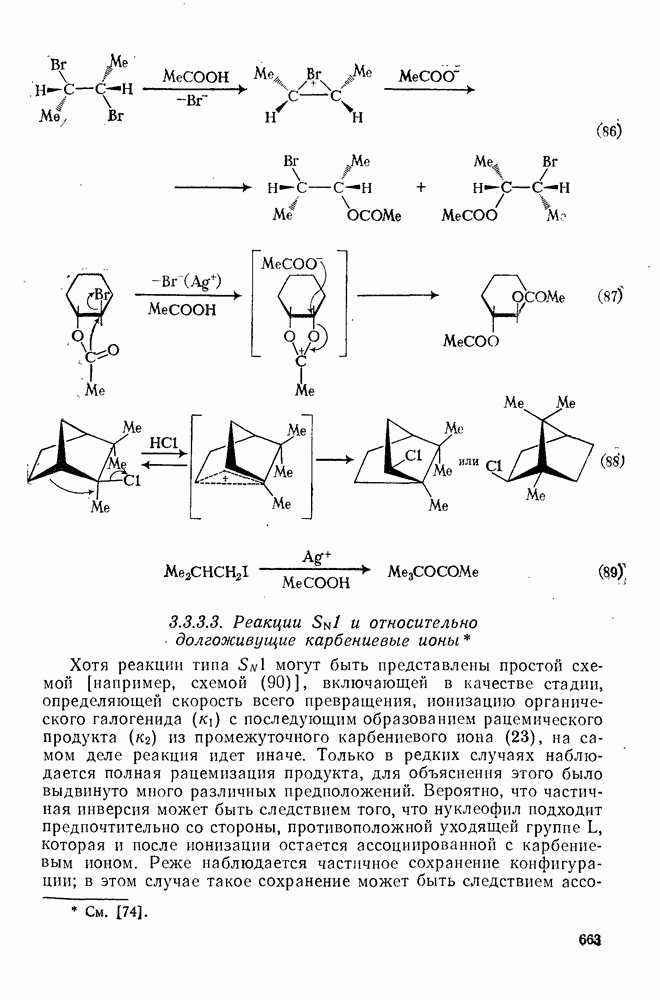
 относительно электрофила. Такая возможность исключается во многих случаях, поскольку при использовании соответствующих хиральных электрофилов наблюдается почти полная рацемизация. Заслуживает внимание одно исключение в случае оптически активного метилоксирана [37], которое объясняется, по-видимому, полной инверсией.
относительно электрофила. Такая возможность исключается во многих случаях, поскольку при использовании соответствующих хиральных электрофилов наблюдается почти полная рацемизация. Заслуживает внимание одно исключение в случае оптически активного метилоксирана [37], которое объясняется, по-видимому, полной инверсией.
 с бромидом алюминия проходит в отсутствие арена, а перераспределение метки в
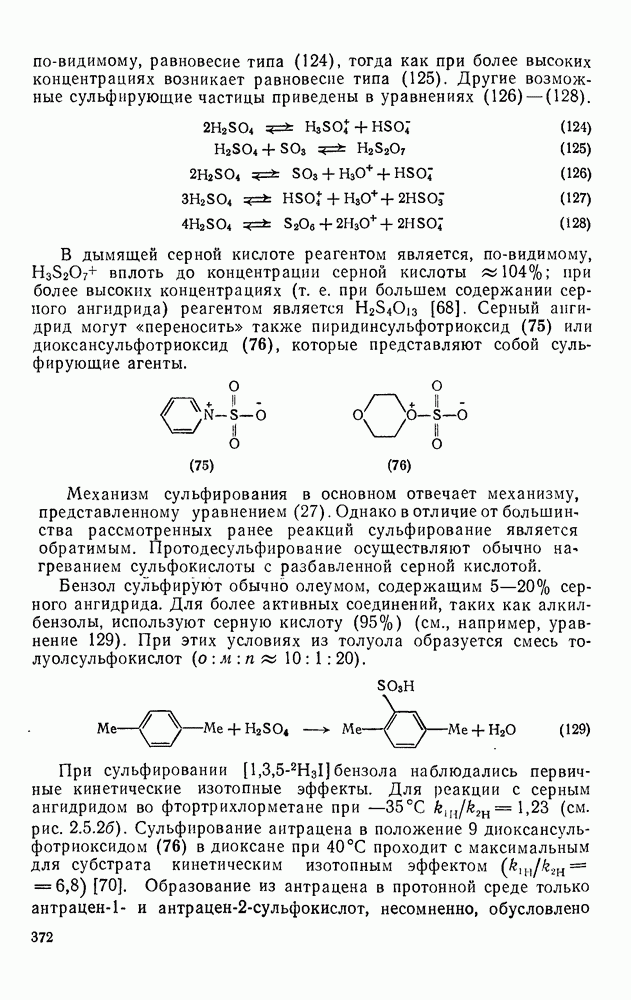
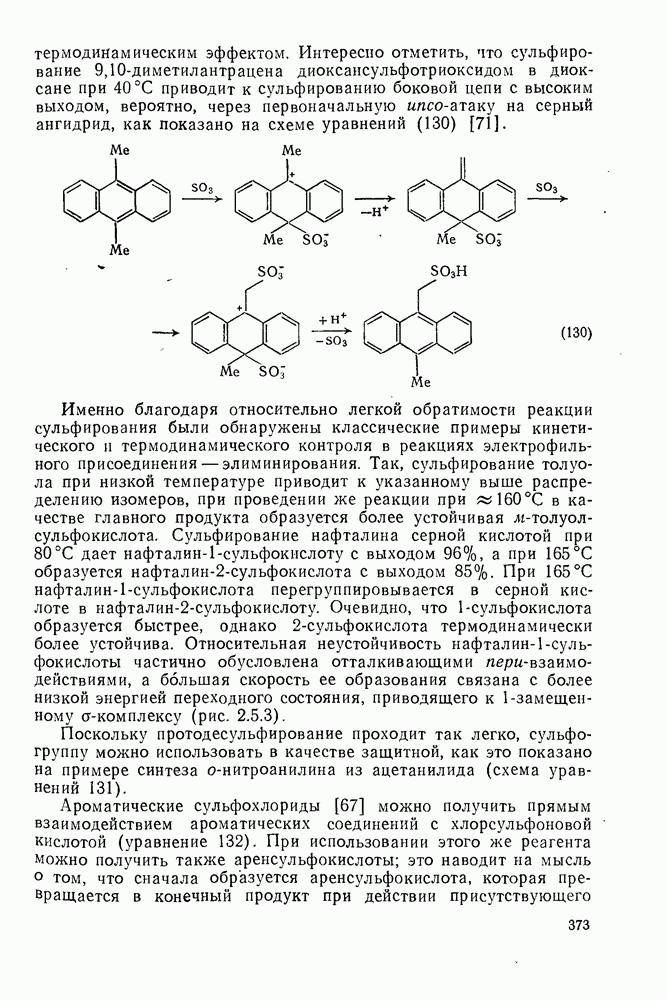
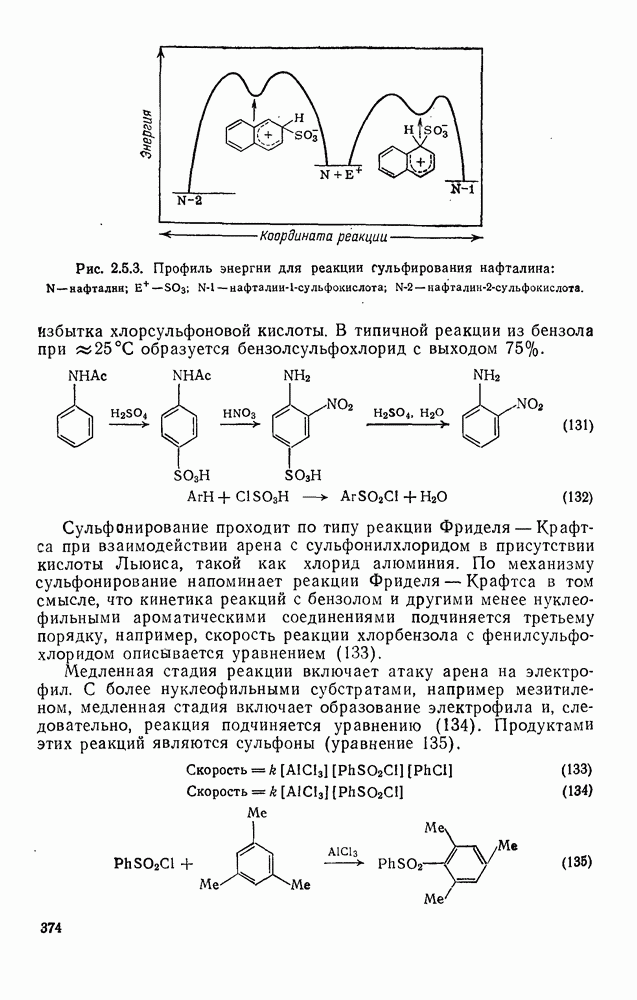
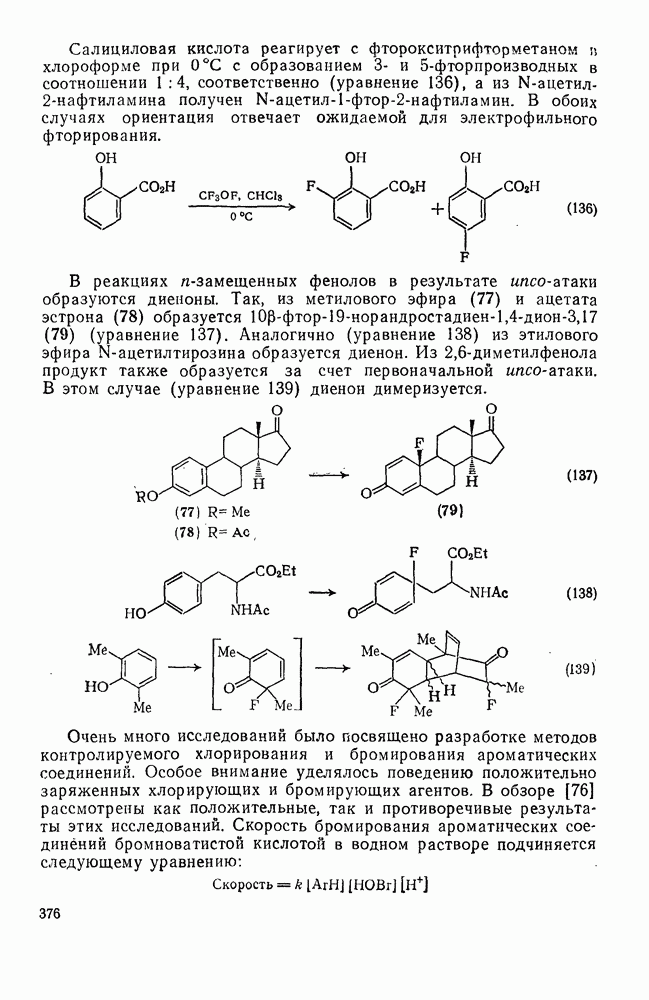
с бромидом алюминия проходит в отсутствие арена, а перераспределение метки в  -фенил
-фенил  этилбромиде осуществляется так быстро, что кинетику его можно проследить только при температурах ниже
этилбромиде осуществляется так быстро, что кинетику его можно проследить только при температурах ниже  . Это, конечно, особый случай, и, по-видимому, здесь осуществляется последовательность стадий, приведенная в уравнении (68).
. Это, конечно, особый случай, и, по-видимому, здесь осуществляется последовательность стадий, приведенная в уравнении (68).

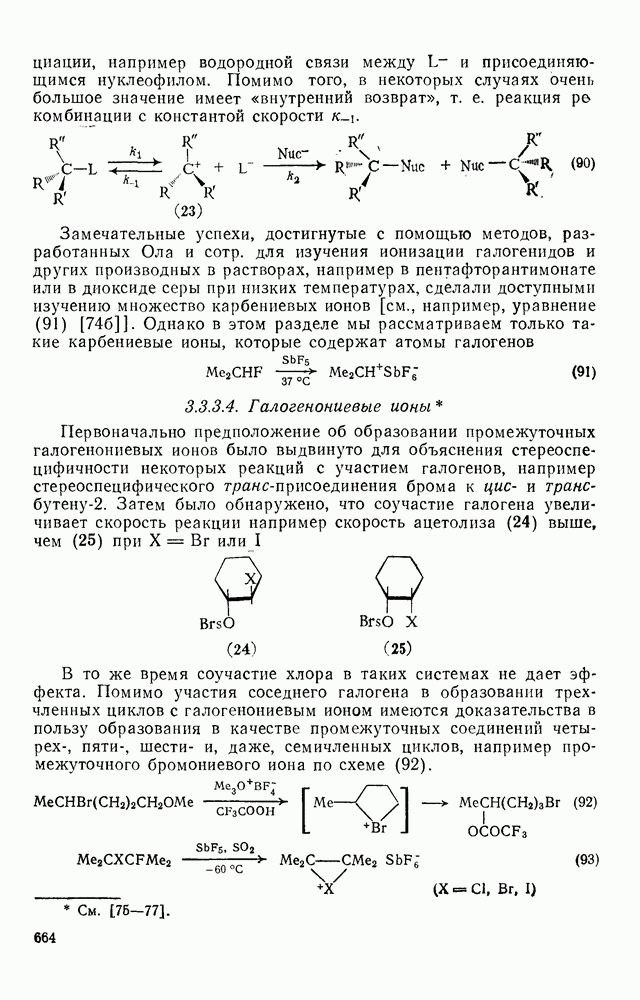
 для дезаминирования аминов и дехлордекарбоксили-рования хлорформиатов. Это обусловлено, несомненно, присутствием легкоудаляемых групп в соответствующих интермедиатах [ионов диазония
для дезаминирования аминов и дехлордекарбоксили-рования хлорформиатов. Это обусловлено, несомненно, присутствием легкоудаляемых групп в соответствующих интермедиатах [ионов диазония  и карбалкоксильных ионов
и карбалкоксильных ионов  ]. Мы рассмотрим реакции карбениевых ионов, генерированных из хлорформиатов под действием ионов серебра(I) [38], поскольку последние результаты в этой области представляют широкий интерес в связи с различными аспектами реакций электрофильного присоединения — элиминирования.
]. Мы рассмотрим реакции карбениевых ионов, генерированных из хлорформиатов под действием ионов серебра(I) [38], поскольку последние результаты в этой области представляют широкий интерес в связи с различными аспектами реакций электрофильного присоединения — элиминирования.
 при различных попытках генерировать катион [39]. Так,
при различных попытках генерировать катион [39]. Так,  -аминоапокамфан реагирует с нитрозилхлоридом в хлорбензоле с образованием смеси
-аминоапокамфан реагирует с нитрозилхлоридом в хлорбензоле с образованием смеси  -хлорфенилапокамфанов (выход
-хлорфенилапокамфанов (выход  ). Реакция
). Реакция  -хлорформилапокамфана с гекса-фторантимонатом серебра проходит более гладко с образованием
-хлорформилапокамфана с гекса-фторантимонатом серебра проходит более гладко с образованием  -хлорапокамфана (17%) и смеси
-хлорапокамфана (17%) и смеси  -хлорфенилапокамфанов
-хлорфенилапокамфанов  . Было найдено, что соотношение изомеров орто
. Было найдено, что соотношение изомеров орто  : мета
: мета  : пара идентично соотношению изомеров, образующихся в реакции дезаминирования (т. е.
: пара идентично соотношению изомеров, образующихся в реакции дезаминирования (т. е.  ). Следует отметить также, что
). Следует отметить также, что  -хлорапокамфан не реагирует с ионами
-хлорапокамфан не реагирует с ионами  . Эти реакции проходят через мостиковый катион, о чем свидетельствует образование единственного продукта
. Эти реакции проходят через мостиковый катион, о чем свидетельствует образование единственного продукта  апокамфана с выходом 24% при проведении аналогичной реакции хлорформиата в нитробензоле. Возможность осуществления этой реакции (уравнения 69) через радикальный центр в вершине мостиковой группы исключается в силу наблюдаемого соотношения изомеров орто : мета : пара.
апокамфана с выходом 24% при проведении аналогичной реакции хлорформиата в нитробензоле. Возможность осуществления этой реакции (уравнения 69) через радикальный центр в вершине мостиковой группы исключается в силу наблюдаемого соотношения изомеров орто : мета : пара.
 -апокамфилрадикала, генерированного термолизом диацилпероксида в хлорбензоле, образуются
-апокамфилрадикала, генерированного термолизом диацилпероксида в хлорбензоле, образуются  , n-изомеры в соотношении
, n-изомеры в соотношении  . Описано также получение
. Описано также получение  адамантана при взаимодействии
адамантана при взаимодействии  -адамантилхлорформиата с гексафторантимонатом серебра(I) в нитробензоле [40].
-адамантилхлорформиата с гексафторантимонатом серебра(I) в нитробензоле [40].

 -хлорфор-милапокамфана с гексафторантимонатом серебра(I) в смесях ароматических растворителей, таких как хлорбензол — бензол, хлорбензол— толуол, бензол — толуол и хлорбензол — нитробензол. Поразительной особенностью этих реакций является то, что бензол оказался более реакционноспособным, чем толуол
-хлорфор-милапокамфана с гексафторантимонатом серебра(I) в смесях ароматических растворителей, таких как хлорбензол — бензол, хлорбензол— толуол, бензол — толуол и хлорбензол — нитробензол. Поразительной особенностью этих реакций является то, что бензол оказался более реакционноспособным, чем толуол  . (При изопропилировании смесей толуол — бензол это соотношение обычно составляет
. (При изопропилировании смесей толуол — бензол это соотношение обычно составляет  ) Анализ всех данных приводит к предположению, что реакция проходит через образование
) Анализ всех данных приводит к предположению, что реакция проходит через образование  -комплекса на лимитирующей стадии, даже в случае исключительно реакционноспособного электрофила.
-комплекса на лимитирующей стадии, даже в случае исключительно реакционноспособного электрофила.
 метилхлорформиата показывает, что она протекает преимущественно с первоначальной атакой по кислороду. Кроме того, диметилфенилоксониевая соль не перегруппируется в
метилхлорформиата показывает, что она протекает преимущественно с первоначальной атакой по кислороду. Кроме того, диметилфенилоксониевая соль не перегруппируется в  -метиланизол внутримолекулярно. Эти результаты показывают, что первоначальные взаимодействия с участием электрофилов и несвязанных электронов могут играть более значительную роль, чем предполагалось раньше.
-метиланизол внутримолекулярно. Эти результаты показывают, что первоначальные взаимодействия с участием электрофилов и несвязанных электронов могут играть более значительную роль, чем предполагалось раньше.

 антрацен с хорошим выходом [42]. Восстановительное дехлорирование последнего до
антрацен с хорошим выходом [42]. Восстановительное дехлорирование последнего до  -диметилантрацена — диена, интересно для реакции Дильса — Альдера, который трудно получить иным путем, — успешно проходит при действии лигийалюми-нийгидрида в ТГФ (уравнение 71).
-диметилантрацена — диена, интересно для реакции Дильса — Альдера, который трудно получить иным путем, — успешно проходит при действии лигийалюми-нийгидрида в ТГФ (уравнение 71).






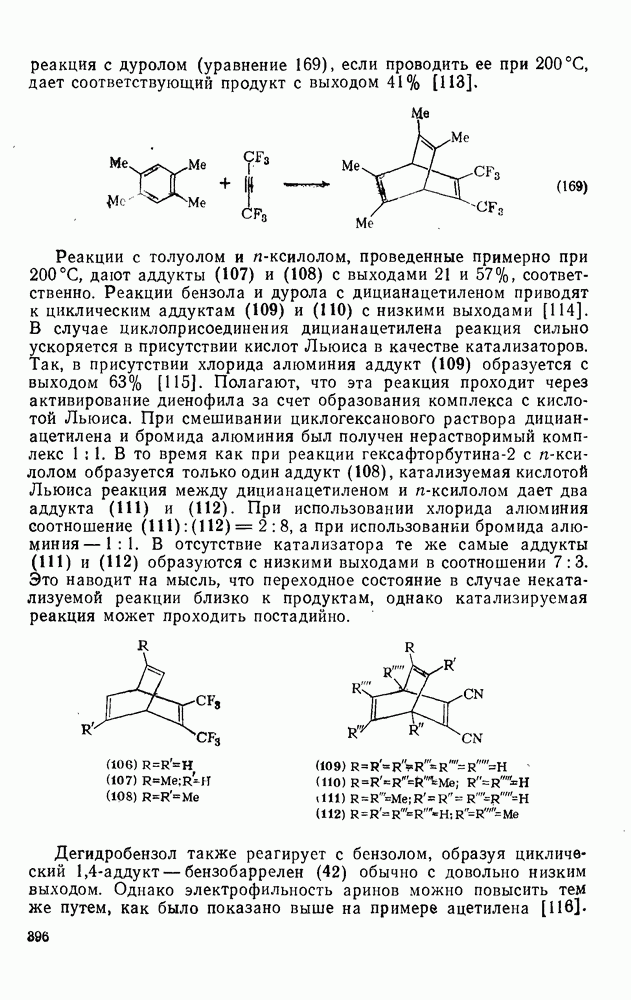
 . Катализаторами могут служить обычные катализаторы алкилирования по Фриделю — Крафтсу, однако чаще всего применяют хлорид алюминия. Основное отличие ацилирования от алкилирования заключается в том, что в случае галогенангидридов необходимо использовать немногим больше одного моля катализатора, а в случае ангидридов карбоновых кислот — немногим больше двух молей катализатора. Один моль катализатора «исключается» из системы за счет координации по карбонильным группам присутствующего карбонильного соединения. Иногда комплексы между хлорангидридом и хлоридом алюминия состава 1: 1 (68) получают до введения ароматического субстрата. Образующийся продукт реакции обычно также координируется с хлоридом алюминия (69).
. Катализаторами могут служить обычные катализаторы алкилирования по Фриделю — Крафтсу, однако чаще всего применяют хлорид алюминия. Основное отличие ацилирования от алкилирования заключается в том, что в случае галогенангидридов необходимо использовать немногим больше одного моля катализатора, а в случае ангидридов карбоновых кислот — немногим больше двух молей катализатора. Один моль катализатора «исключается» из системы за счет координации по карбонильным группам присутствующего карбонильного соединения. Иногда комплексы между хлорангидридом и хлоридом алюминия состава 1: 1 (68) получают до введения ароматического субстрата. Образующийся продукт реакции обычно также координируется с хлоридом алюминия (69).

 -ксилола в сероуглероде обычно образуется
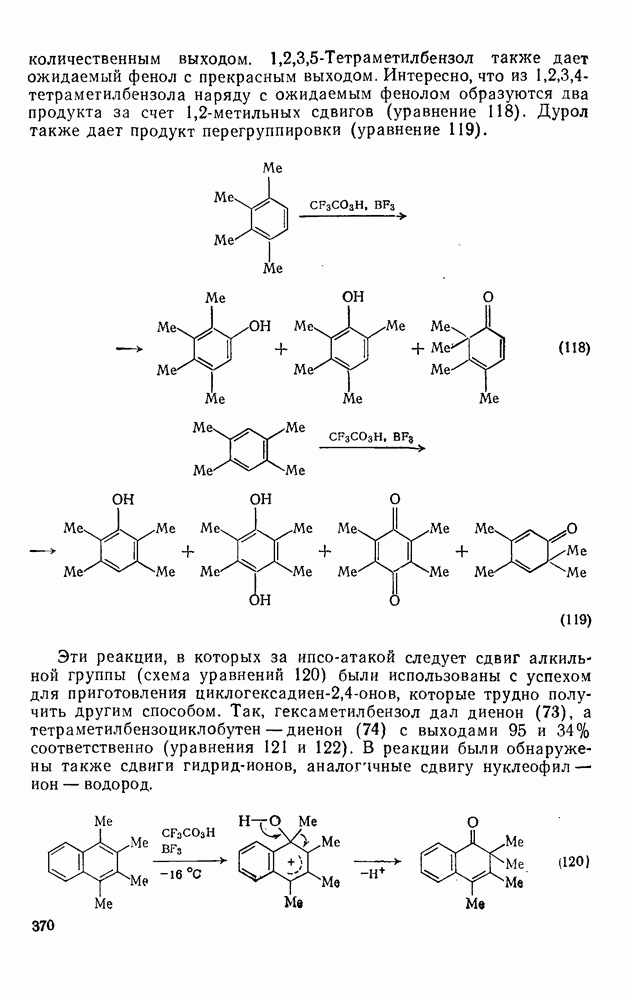
-ксилола в сероуглероде обычно образуется  , при проведений той же реакции в избытке арена как растворителя получается
, при проведений той же реакции в избытке арена как растворителя получается  -диметилацетофенон. Очевидно, при ацилировании происходит переалкилирование и в реакциях Фриделя — Крафтса возможна ипсо-атака. Однака в общем ацилировании по Фриделю — Крафтсу проходит более просто, чем алкилирование. Нуклеофильность продукта ацилирования ниже, чем исходного вещества, за счет электроноакцепторного эффекта карбонильной группы, и, следовательно, продукт обладает меньшей реакционной способностью.
-диметилацетофенон. Очевидно, при ацилировании происходит переалкилирование и в реакциях Фриделя — Крафтса возможна ипсо-атака. Однака в общем ацилировании по Фриделю — Крафтсу проходит более просто, чем алкилирование. Нуклеофильность продукта ацилирования ниже, чем исходного вещества, за счет электроноакцепторного эффекта карбонильной группы, и, следовательно, продукт обладает меньшей реакционной способностью.

 -инданон можно получить циклизацией
-инданон можно получить циклизацией  -фенилпропионилхлорида (уравнение 89). Аналогично, из
-фенилпропионилхлорида (уравнение 89). Аналогично, из  -фенилмасляной кислоты можно приготовить
-фенилмасляной кислоты можно приготовить  (
( -тетралон). Исходную кислоту можно синтезировать из бензола в две стадии (уравнения 90).
-тетралон). Исходную кислоту можно синтезировать из бензола в две стадии (уравнения 90).  -Лактоны, такие как
-Лактоны, такие как  -бутиролактон, алкилируют арены в присутствии хлорида алюминия, и, следовательно,
-бутиролактон, алкилируют арены в присутствии хлорида алюминия, и, следовательно,  -фенилмасляная кислота получается в одну стадию. Примером такой методики служит синтез
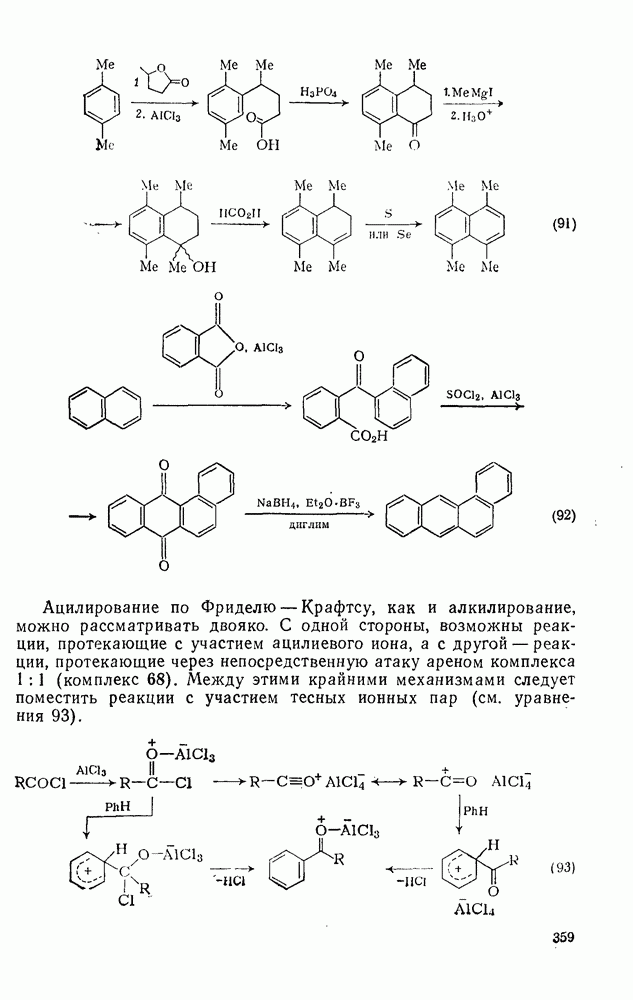
-фенилмасляная кислота получается в одну стадию. Примером такой методики служит синтез  -тетраметилнафталина (уравнения 91). В этой области имеется много вариантов синтеза. Так, из нафталина можно получить производные фенантрена, а используя фталевый ангидрид вместо янтарного ангидрида или у-бутиролактона, можно приготовить производные антрацена из бензола или
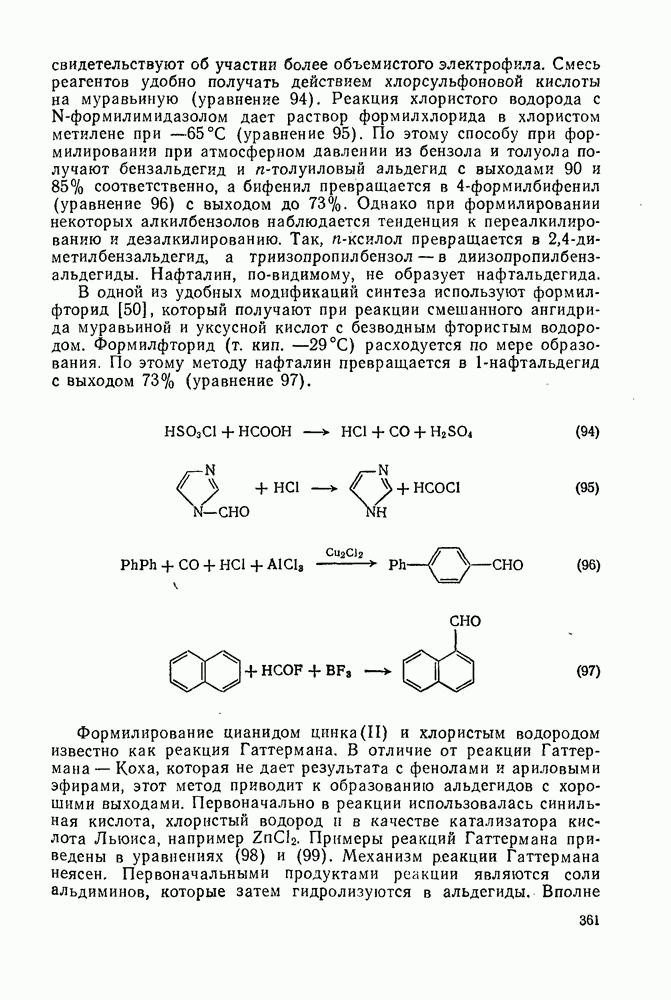
-тетраметилнафталина (уравнения 91). В этой области имеется много вариантов синтеза. Так, из нафталина можно получить производные фенантрена, а используя фталевый ангидрид вместо янтарного ангидрида или у-бутиролактона, можно приготовить производные антрацена из бензола или  -бензантрацен (уравнения 92) из нафталина.
-бензантрацен (уравнения 92) из нафталина.



 -метилацетофенон с очень высоким выходом (до
-метилацетофенон с очень высоким выходом (до  ), в то время как при нитровании основным изомером является
), в то время как при нитровании основным изомером является  -нитротолуол (см., однако, обсуждение ипсо-атаки). При ацетилировании в сероуглероде наблюдаются меньшие пространственные затруднения по сравнению с реакциями в нитробензоле. Нафталин образует в сероуглероде
-нитротолуол (см., однако, обсуждение ипсо-атаки). При ацетилировании в сероуглероде наблюдаются меньшие пространственные затруднения по сравнению с реакциями в нитробензоле. Нафталин образует в сероуглероде  -ацетилнафталин, однако при использовании в качестве растворителя нитробензола образуется в основном
-ацетилнафталин, однако при использовании в качестве растворителя нитробензола образуется в основном  -ацетилнафталин. Интересно, что бензоилирование менее чувствительно к пространственным затруднениям, чем ацетилирование. Аномальное изменение реакционной способности в реакциях ацилирования при переходе от бензола к мезитилену обусловлено, вероятно, изменением механизма реакции [47]. Аналогично, более низкая реакционная способность хлорацетилхлорида по сравнению с ацетил- и бензоилхлоридом в реакции с сильнонуклеофильными аренами [48] может быть отнесена за счет изменений отдельных деталей механизма. Для реакций с участием свободного ацилиевого иона
-ацетилнафталин. Интересно, что бензоилирование менее чувствительно к пространственным затруднениям, чем ацетилирование. Аномальное изменение реакционной способности в реакциях ацилирования при переходе от бензола к мезитилену обусловлено, вероятно, изменением механизма реакции [47]. Аналогично, более низкая реакционная способность хлорацетилхлорида по сравнению с ацетил- и бензоилхлоридом в реакции с сильнонуклеофильными аренами [48] может быть отнесена за счет изменений отдельных деталей механизма. Для реакций с участием свободного ацилиевого иона  скорость уменьшается в присутствии электронодонорного заместителя в группе R. Обратная картина имеет место для реакций с участием комплекса хлорангидрида с катализатором (комплекс 68). В связи с этим следует отметить, что удалось получить ацилийгексафторантимонаты и с успехом использовать их в ацилировании по Фриделю — Крафтсу [49].
скорость уменьшается в присутствии электронодонорного заместителя в группе R. Обратная картина имеет место для реакций с участием комплекса хлорангидрида с катализатором (комплекс 68). В связи с этим следует отметить, что удалось получить ацилийгексафторантимонаты и с успехом использовать их в ацилировании по Фриделю — Крафтсу [49].
 Па. При атмосферном давлении реакцию проводят в присутствии хлорида
Па. При атмосферном давлении реакцию проводят в присутствии хлорида  , который, по-видимому, обеспечивает высокую местную концентрацию СО за счет комплексообразования. Эта реакция известна как реакция Гаттермана — Коха. Реакция происходит таким образом, как если бы формилхлорид был ионизован до формилиона, который и является гипотетическим электрофилом. Однако высокие выходы
, который, по-видимому, обеспечивает высокую местную концентрацию СО за счет комплексообразования. Эта реакция известна как реакция Гаттермана — Коха. Реакция происходит таким образом, как если бы формилхлорид был ионизован до формилиона, который и является гипотетическим электрофилом. Однако высокие выходы  -формилалкилбензолов свидетельствуют об участии более объемистого электрофила.
-формилалкилбензолов свидетельствуют об участии более объемистого электрофила.
 -формилимидазолом дает раствор формилхлорида в хлористом метилене при
-формилимидазолом дает раствор формилхлорида в хлористом метилене при  (уравнение 95). По этому способу при формилировании при атмосферном давлении из бензола и толуола получают бензальдегид и л-толуиловый альдегид с выходами 90 и 85% соответственно, а бифенил превращается в
(уравнение 95). По этому способу при формилировании при атмосферном давлении из бензола и толуола получают бензальдегид и л-толуиловый альдегид с выходами 90 и 85% соответственно, а бифенил превращается в  -формилбифенил (уравнение 96) с выходом до 73%. Однако при формилировании некоторых алкилбензолов наблюдается тенденция к переалкилиро-ванию и дезалкилированию. Так, м-ксилол превращается в
-формилбифенил (уравнение 96) с выходом до 73%. Однако при формилировании некоторых алкилбензолов наблюдается тенденция к переалкилиро-ванию и дезалкилированию. Так, м-ксилол превращается в  -ди-метилбензальдегид, а триизопропилбензол — в диизопропилбензальдегиды. Нафталин, по-видимому, не образует нафтальдегида.
-ди-метилбензальдегид, а триизопропилбензол — в диизопропилбензальдегиды. Нафталин, по-видимому, не образует нафтальдегида.
 ) расходуется по мере образования. По этому методу нафталин превращается в
) расходуется по мере образования. По этому методу нафталин превращается в  -нафтальдегид с выходом 73% (уравнение 97).
-нафтальдегид с выходом 73% (уравнение 97).

 . Примеры реакций Гаттермана приведены в уравнениях (98) и (99). Механизм реакции Гаттермана неясен. Первоначальными продуктами реакции являются соли альдиминов, которые затем гидролизуются в альдегиды.
. Примеры реакций Гаттермана приведены в уравнениях (98) и (99). Механизм реакции Гаттермана неясен. Первоначальными продуктами реакции являются соли альдиминов, которые затем гидролизуются в альдегиды.

 -метилформамид и
-метилформамид и  -диметилформамид. Бензол, алкилбензолы и нафталин не дают альдегидов по реакции Вильсмайера. Реакция также не проходит или в лучшем случае приводит к низким выходам альдегидов в случае ариловых эфиров, замещенных в пара-положении.
-диметилформамид. Бензол, алкилбензолы и нафталин не дают альдегидов по реакции Вильсмайера. Реакция также не проходит или в лучшем случае приводит к низким выходам альдегидов в случае ариловых эфиров, замещенных в пара-положении.
 -диметиланилина —
-диметиланилина —  с выходом 85 и 50% соответственно (уравнение 102).
с выходом 85 и 50% соответственно (уравнение 102).

 -дизамещенные формамиды).
-дизамещенные формамиды).
 -диметилтноформамид при формилировании отличается реакционной способностью от
-диметилтноформамид при формилировании отличается реакционной способностью от  -диметилформамида. Эти результаты приводят к заключению, что электрофилом является фосфорсодержащий реагент; это согласуется с уравнением (103).
-диметилформамида. Эти результаты приводят к заключению, что электрофилом является фосфорсодержащий реагент; это согласуется с уравнением (103).

 -диметилформамидом, образуя не содержащий фосфора электрофил и анионный фосфорный остаток (уравнение 104). Отсутствие спин-спинового взаимодействия между низкопольным протоном и
-диметилформамидом, образуя не содержащий фосфора электрофил и анионный фосфорный остаток (уравнение 104). Отсутствие спин-спинового взаимодействия между низкопольным протоном и  в спектрах ЯМР
в спектрах ЯМР  и
и  согласуется с этим предположением. Нет ничего удивительного в том, что такой катион образуется, однако, по мнению автора, это не подтверждает его участия в формилировании по Вильсмайеру в качестве электрофила, поскольку в реакционных смесях отсутствовал ароматический субстрат. Следует отметить, что хлор (трисдиметиламино) фосфонийгексафторфосфат реагирует с
согласуется с этим предположением. Нет ничего удивительного в том, что такой катион образуется, однако, по мнению автора, это не подтверждает его участия в формилировании по Вильсмайеру в качестве электрофила, поскольку в реакционных смесях отсутствовал ароматический субстрат. Следует отметить, что хлор (трисдиметиламино) фосфонийгексафторфосфат реагирует с  -метиланилином в
-метиланилином в  -диметилформамиде, образуя с количественным выходом
-диметилформамиде, образуя с количественным выходом  -формил-
-формил- -метиланилин. Однако, с другой стороны, этот реагент, который может легко распадаться на предполагаемый формилирующий агент в реакции Вильсмайера (уравнение 105), не дает продукта С-формилирования в реакции с
-метиланилин. Однако, с другой стороны, этот реагент, который может легко распадаться на предполагаемый формилирующий агент в реакции Вильсмайера (уравнение 105), не дает продукта С-формилирования в реакции с  -диметиланилином [54].
-диметиланилином [54].



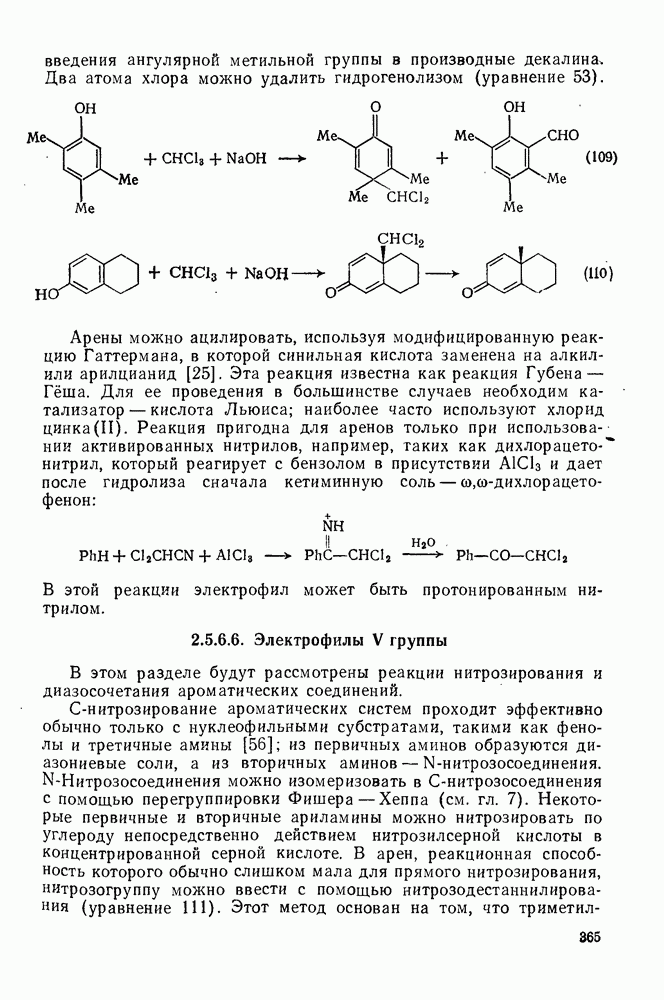
 -дизамещенные
-дизамещенные  . Два атома хлора неопентильного типа сохраняются вследствие пространственных затруднений для гидролиза по механизму
. Два атома хлора неопентильного типа сохраняются вследствие пространственных затруднений для гидролиза по механизму  (уравнение 109). Этот тип реакции используют для введения ангулярной метильной группы в производные декалина.
(уравнение 109). Этот тип реакции используют для введения ангулярной метильной группы в производные декалина.

 и дает после гидролиза сначала кетиминную соль —
и дает после гидролиза сначала кетиминную соль —  -дихлор ацетофенон:
-дихлор ацетофенон:
