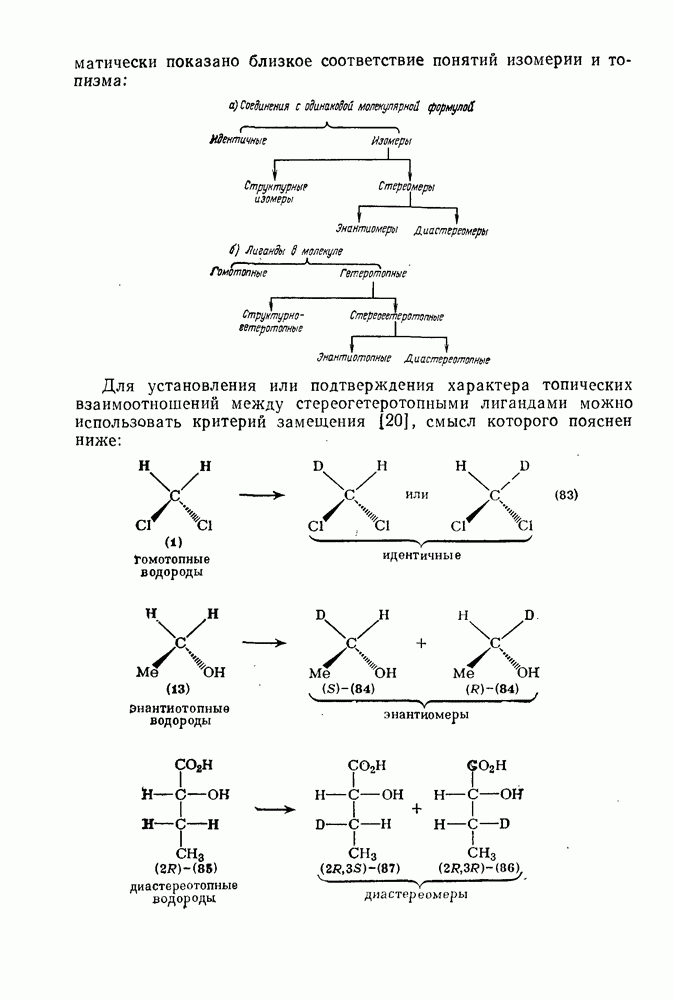
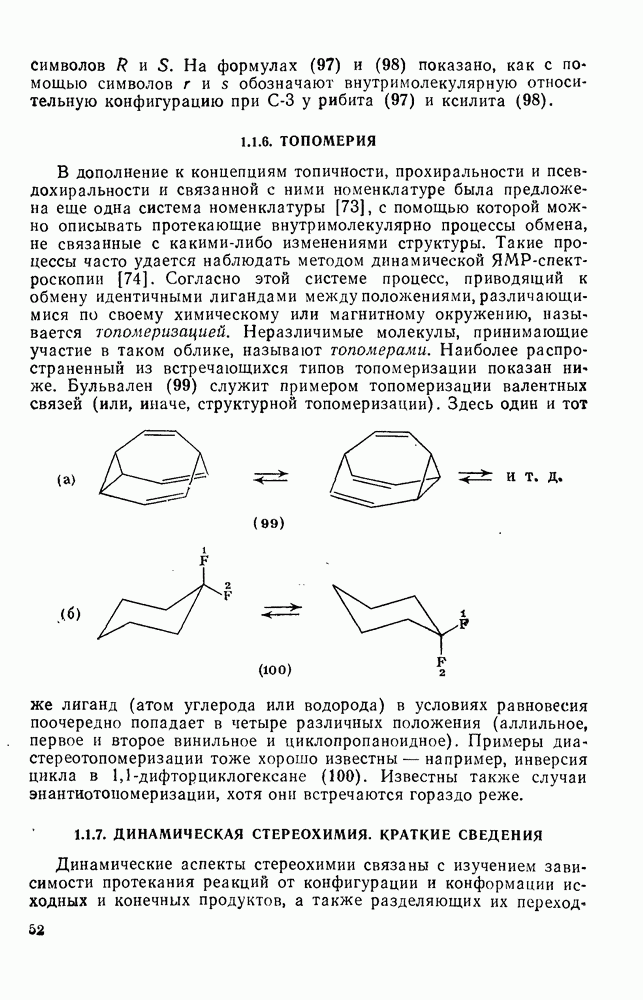
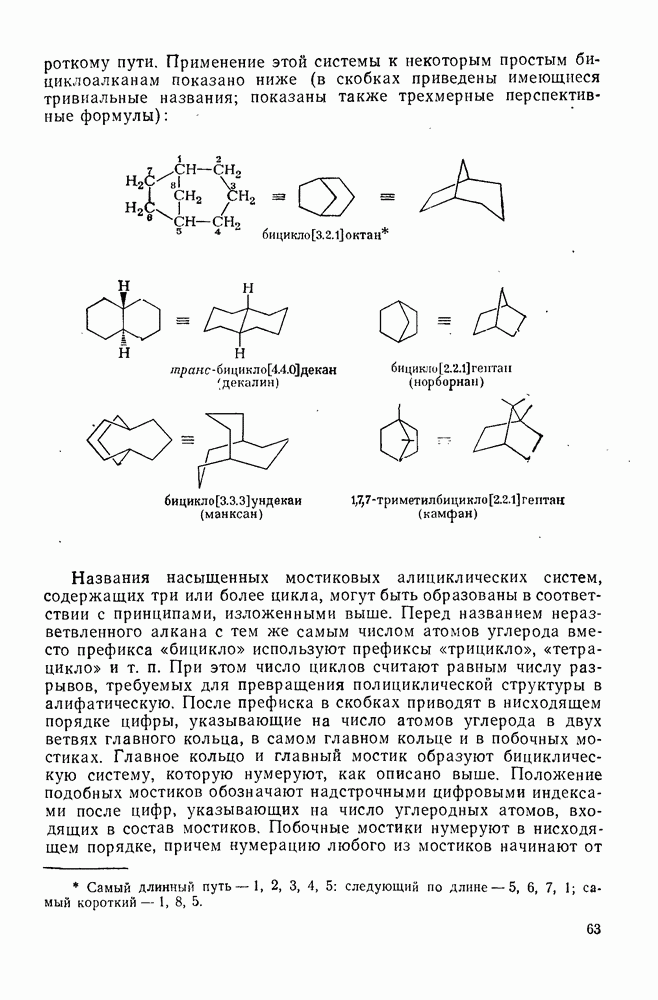
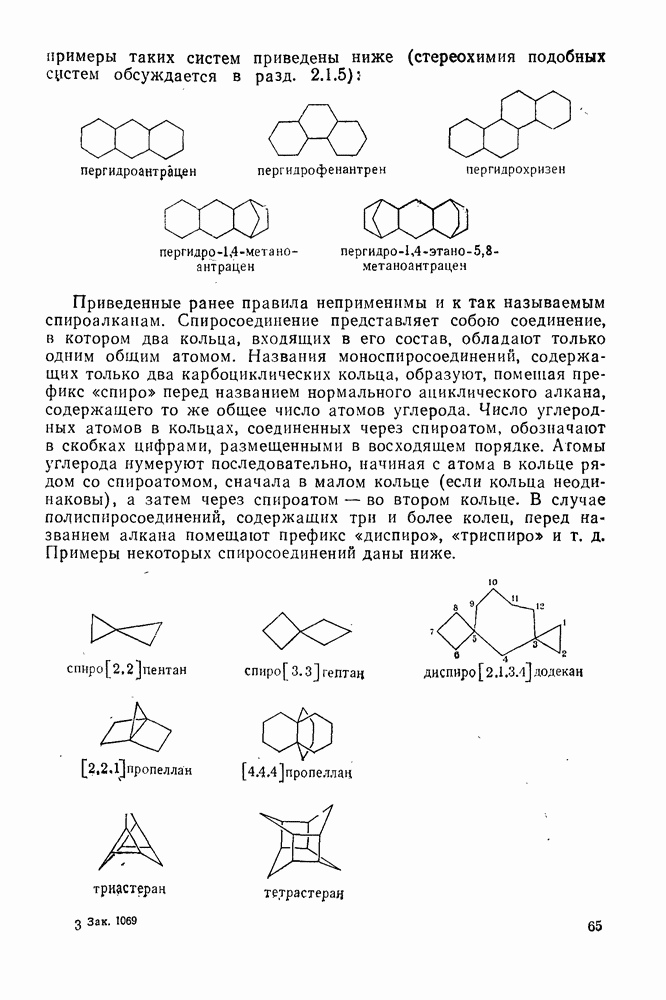
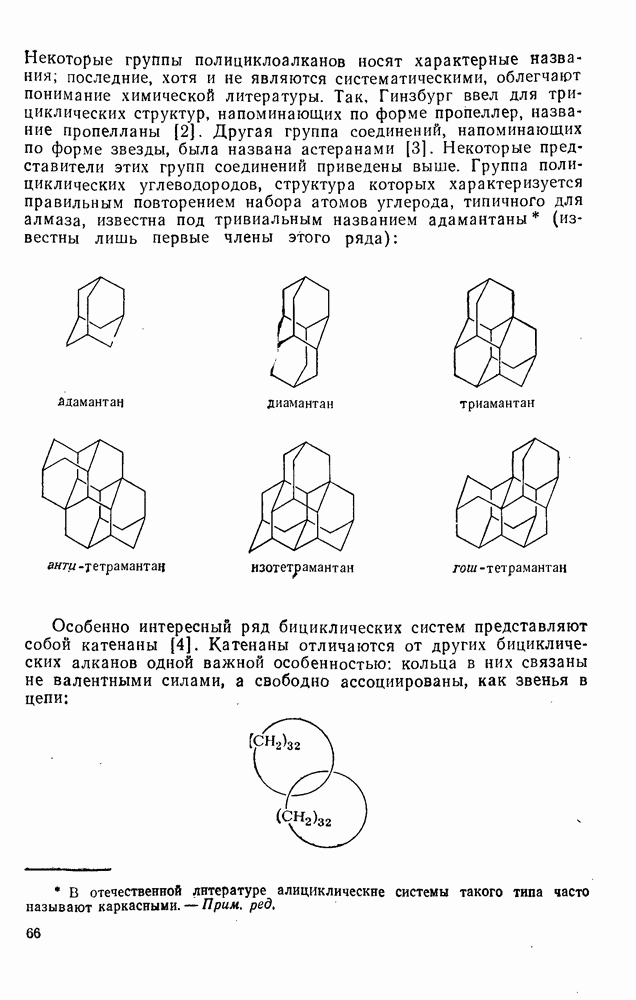
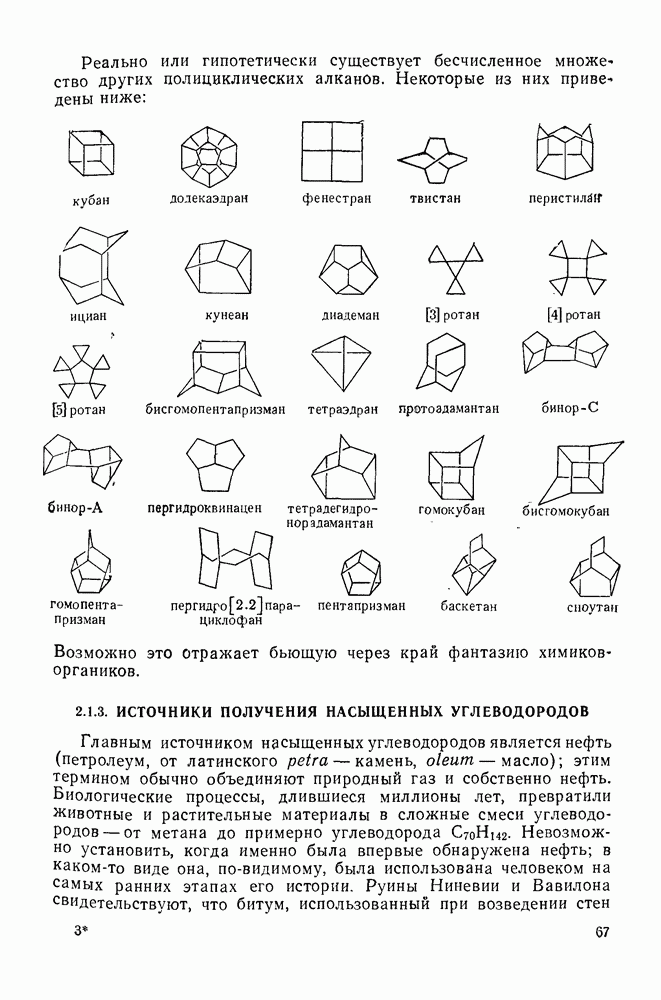
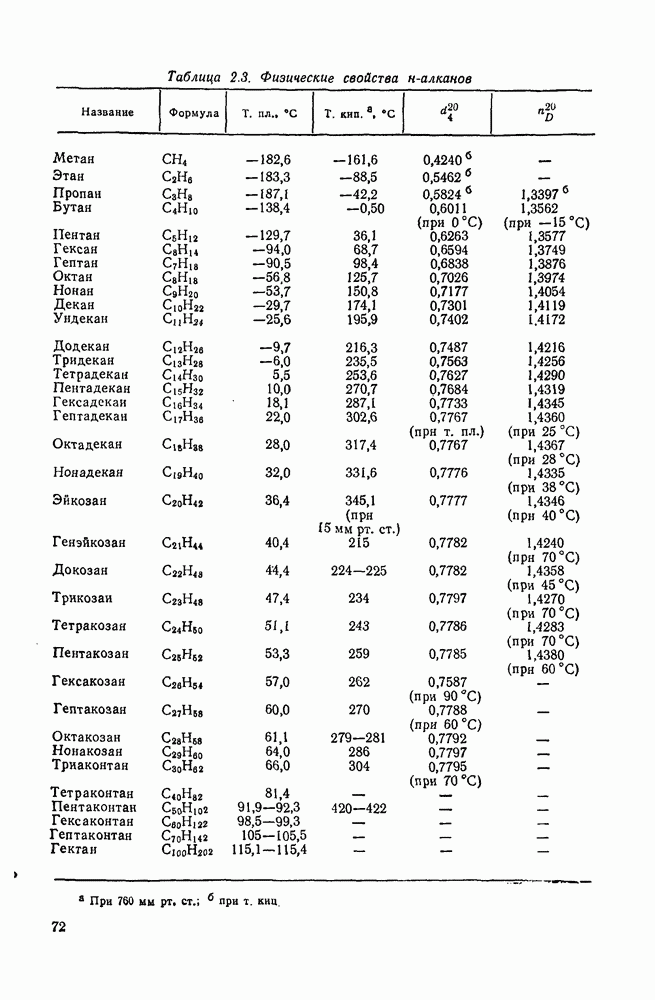
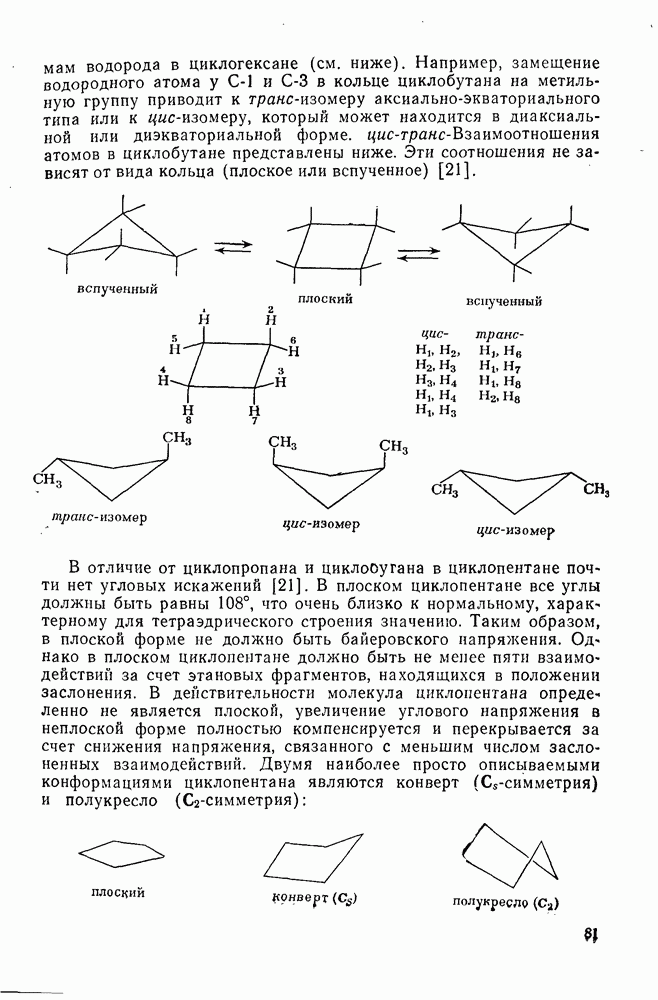
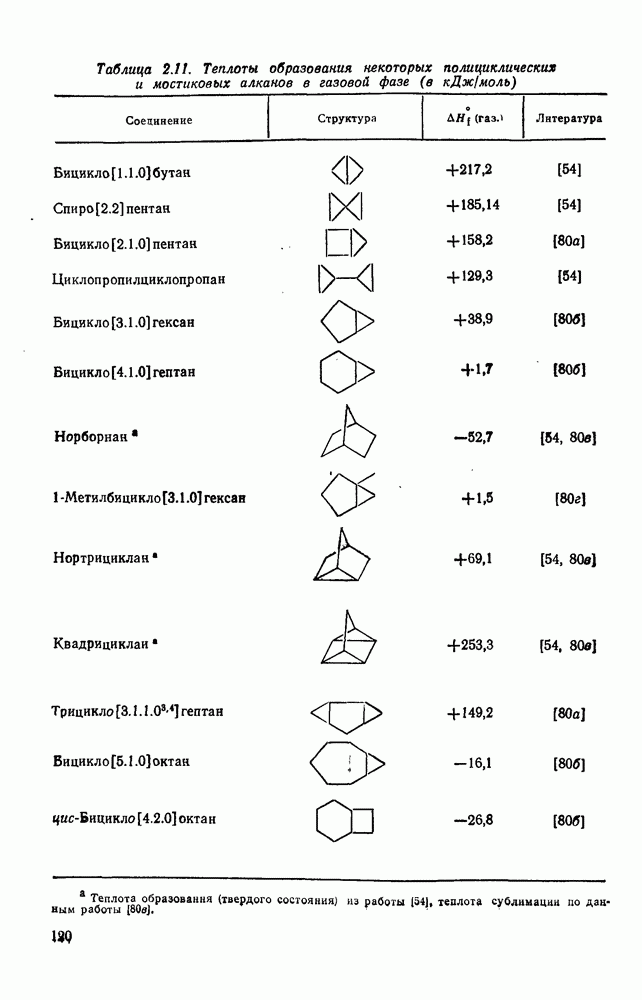
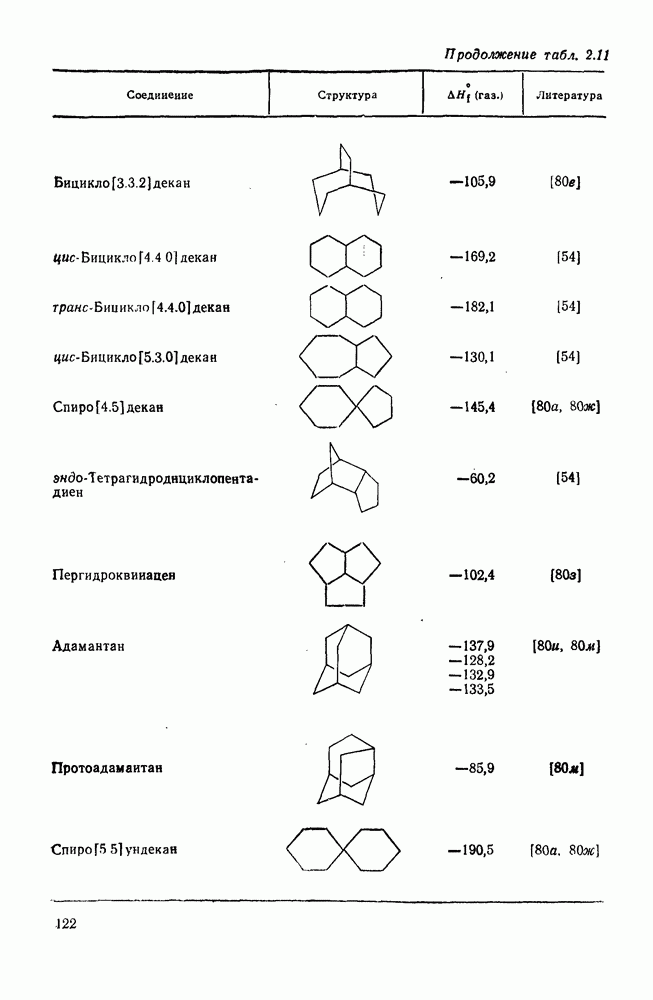
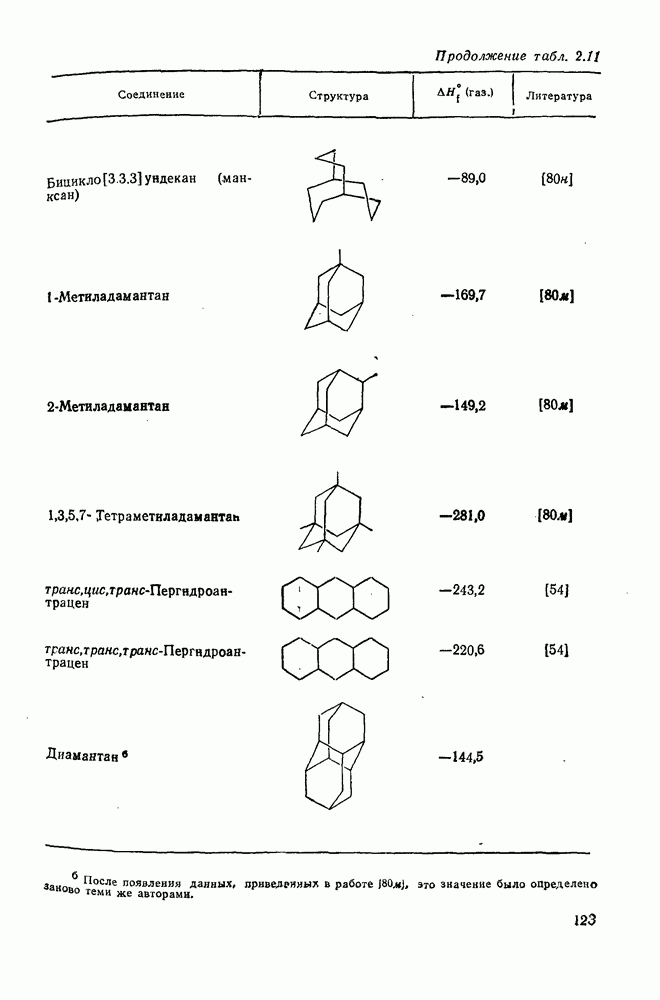
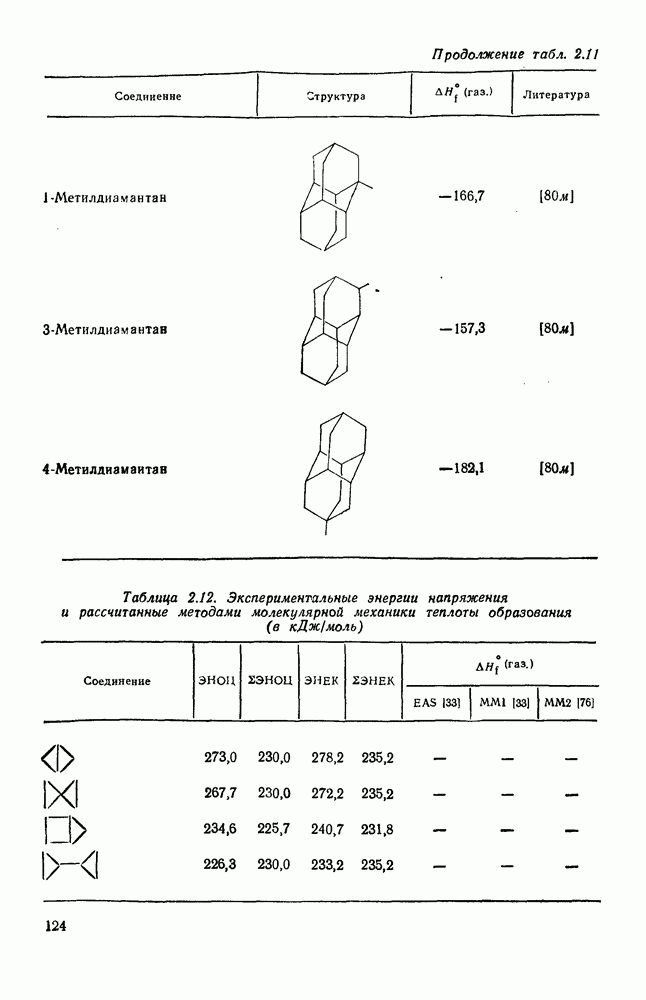
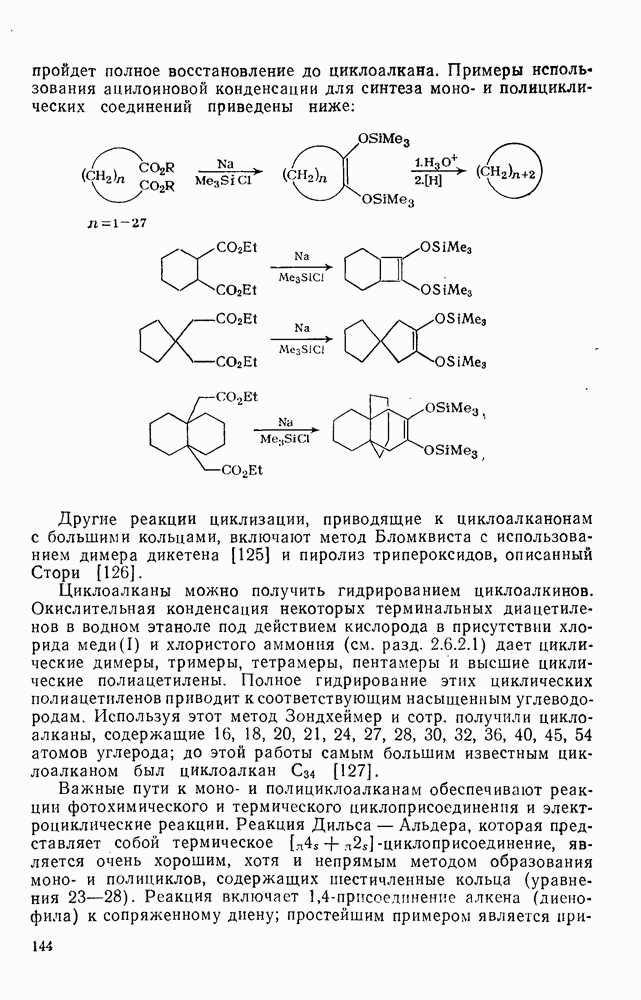
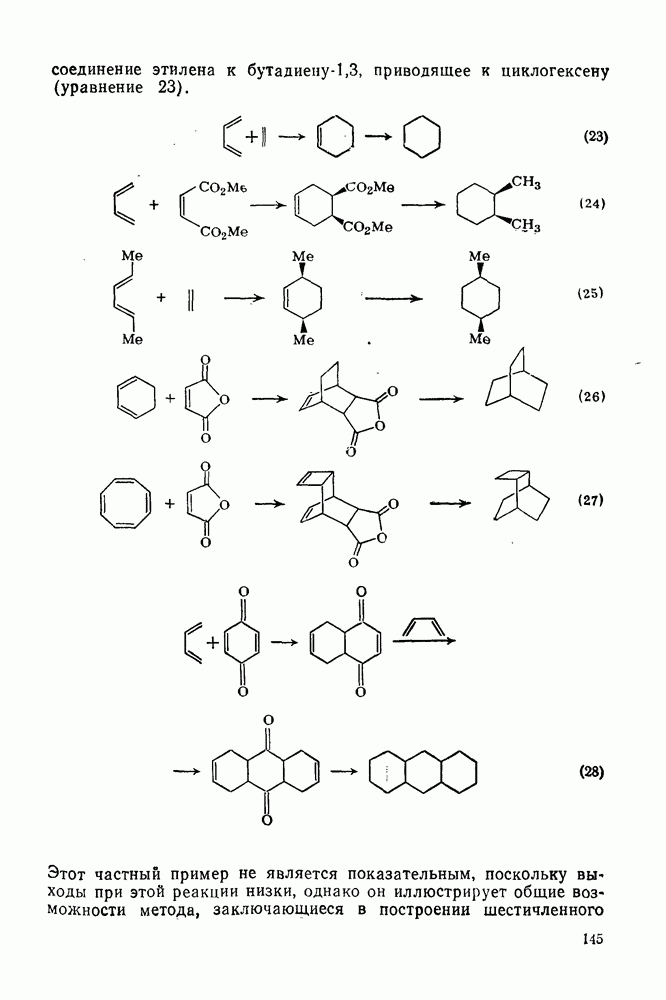
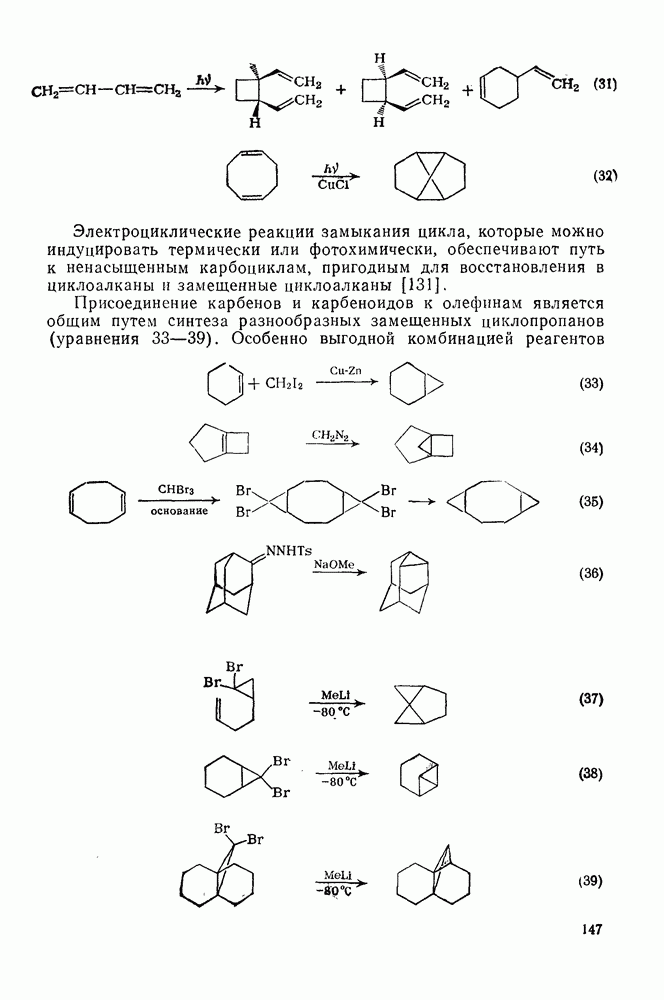
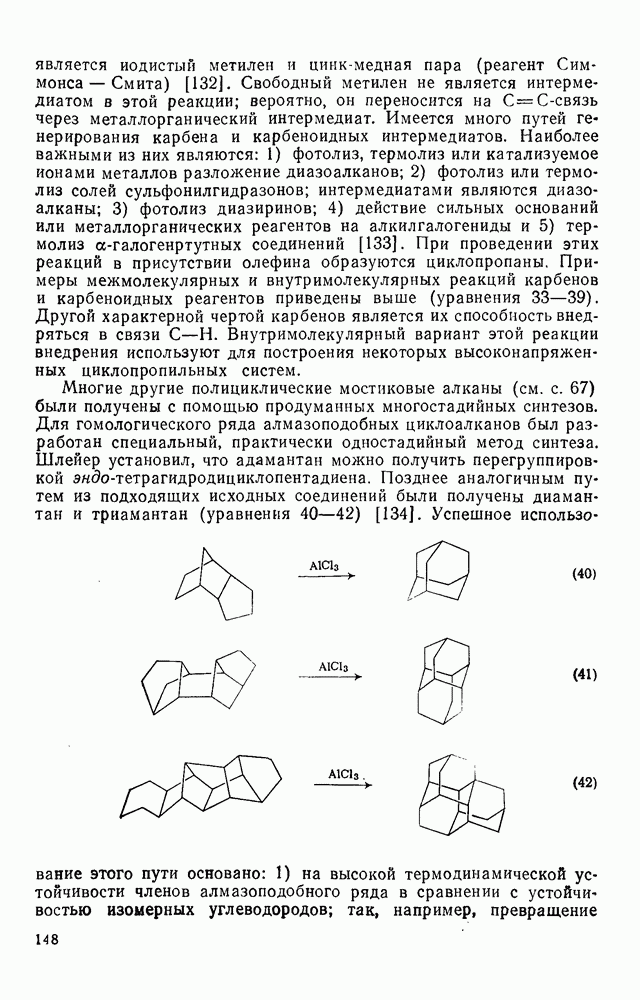
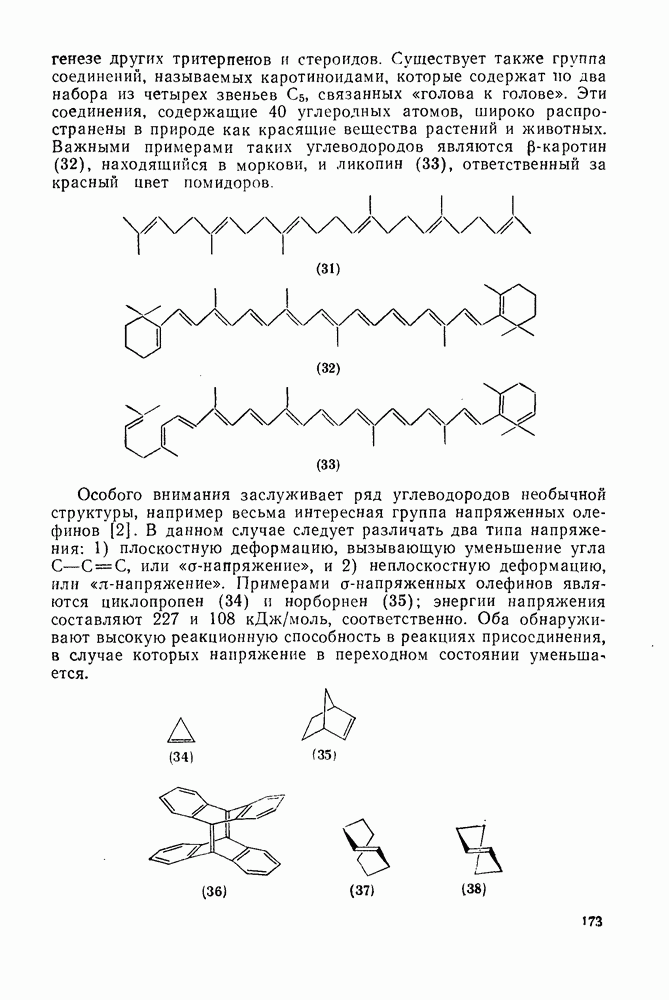
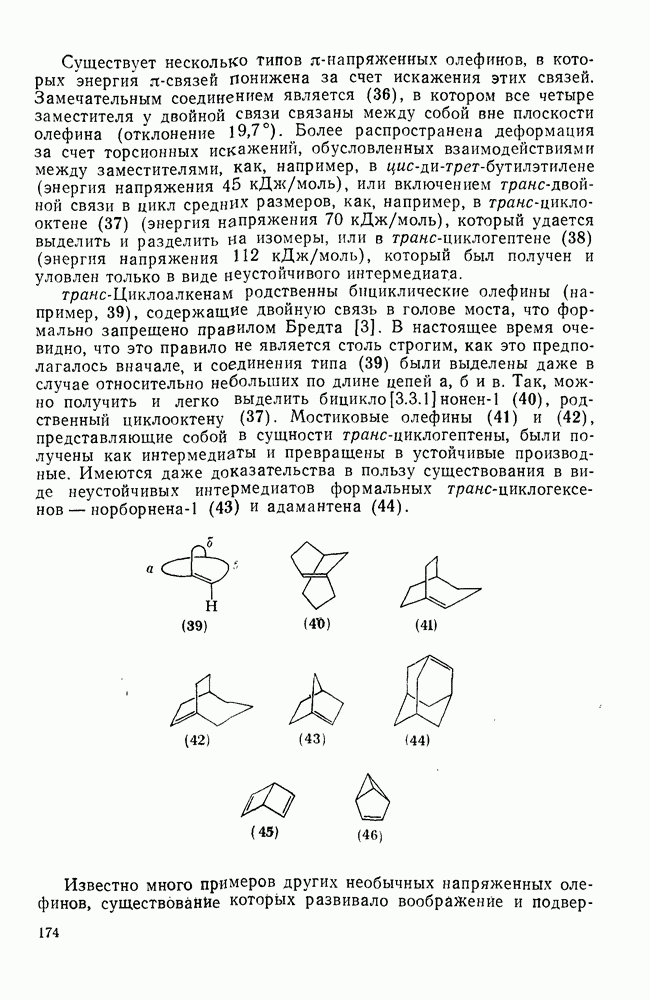
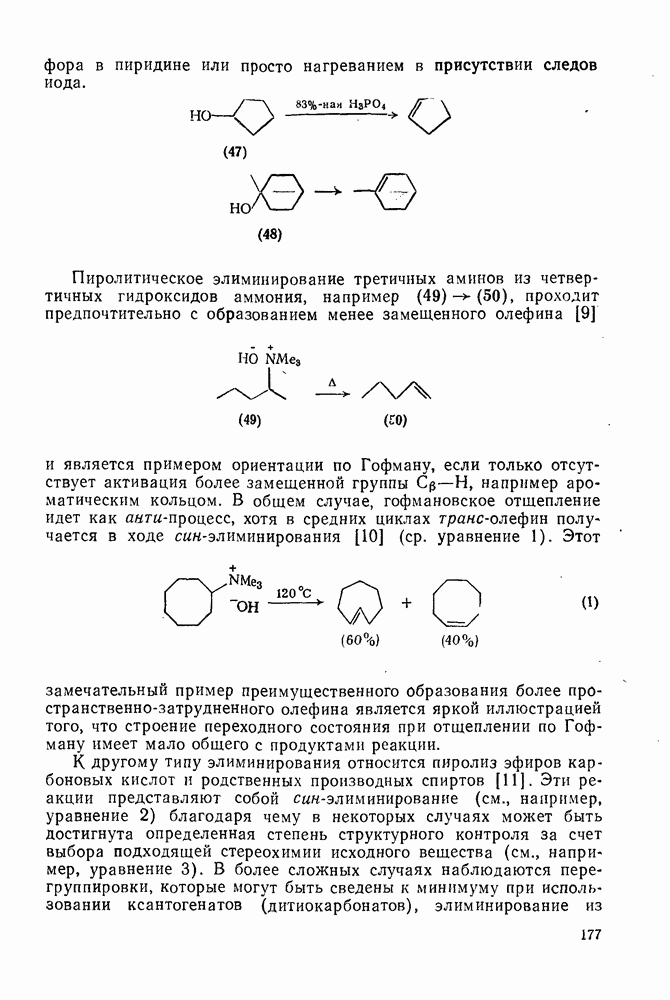
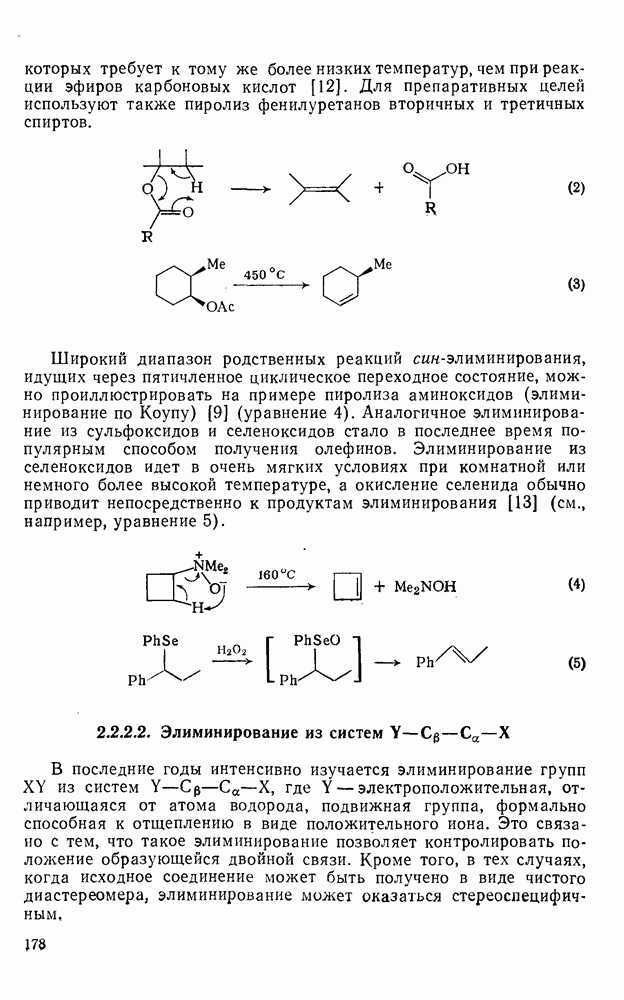
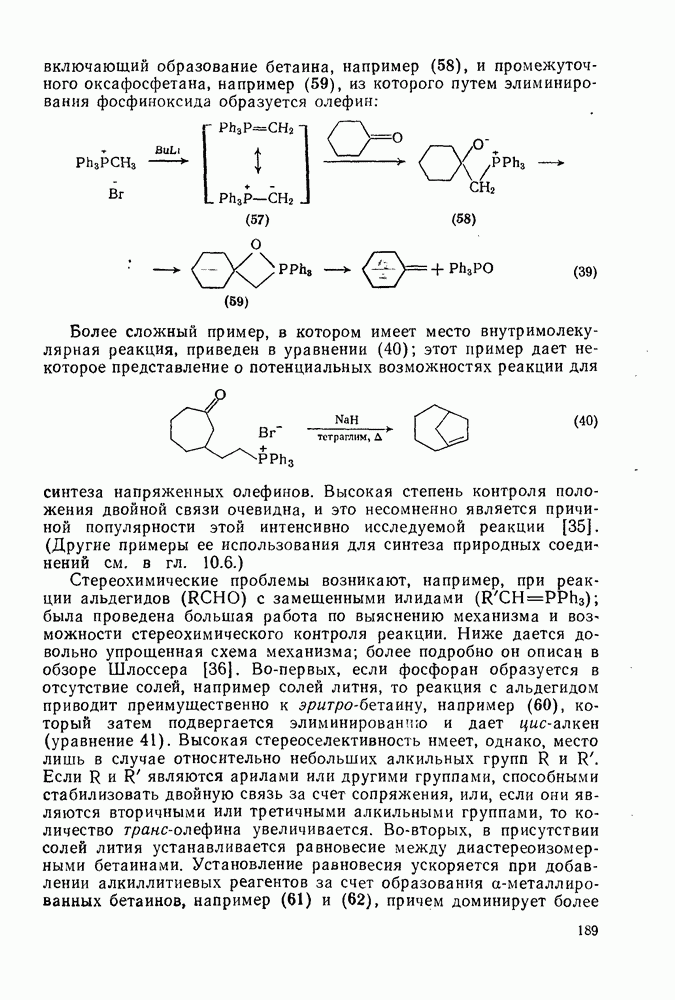
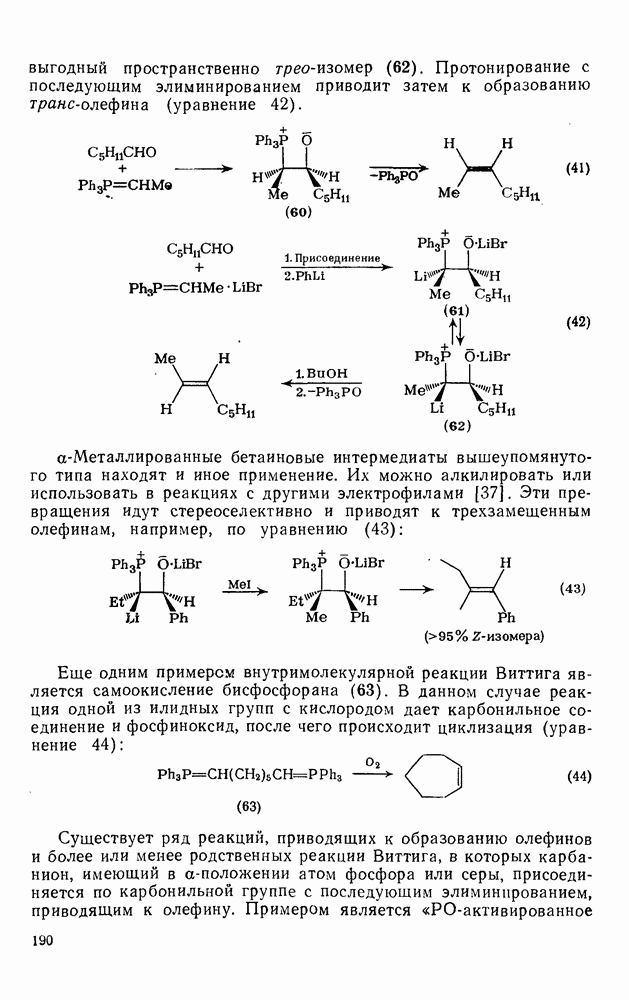
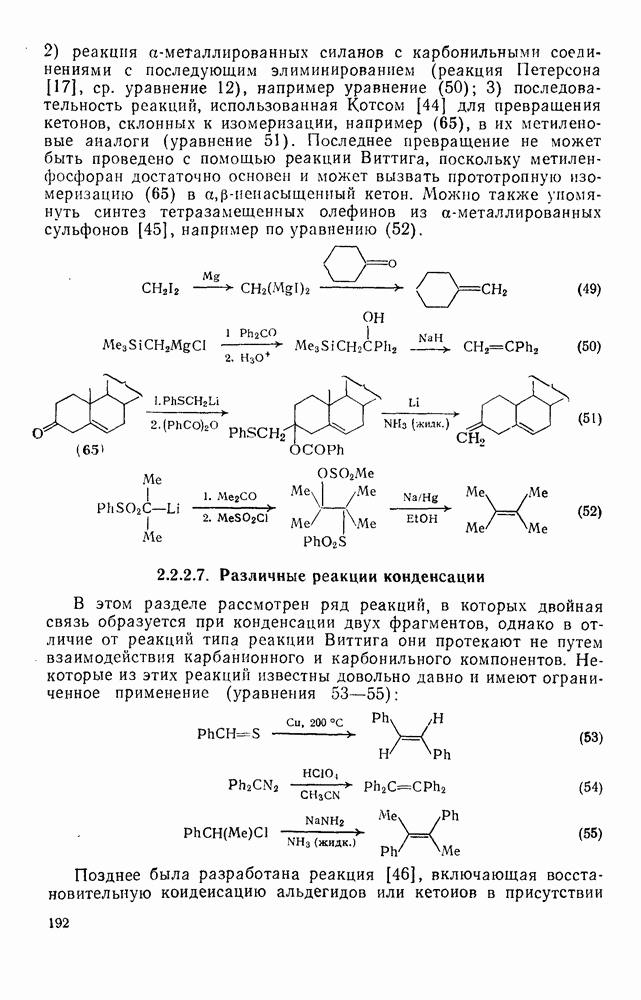
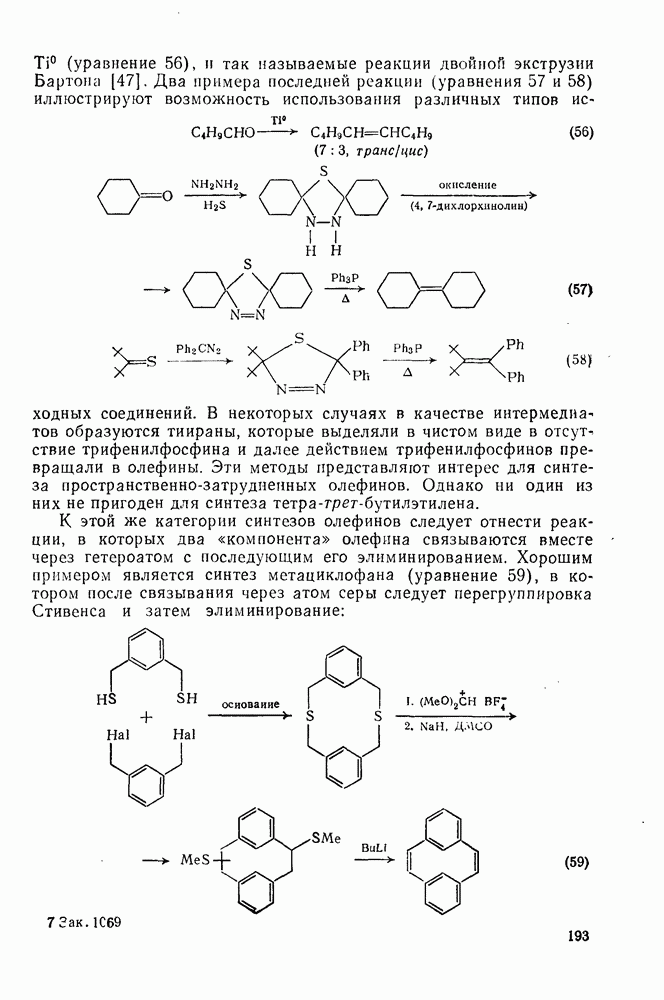
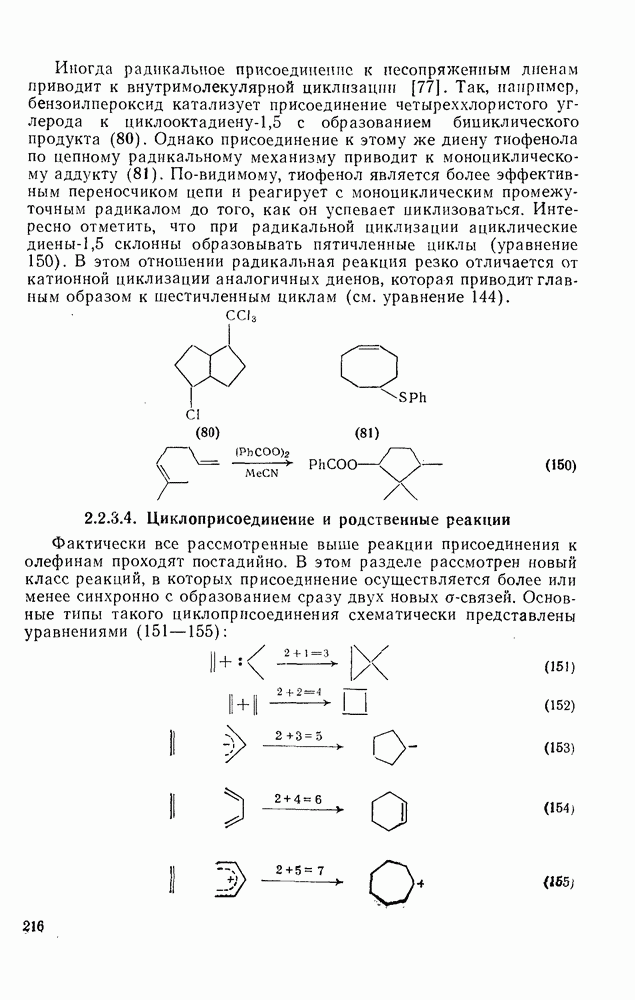
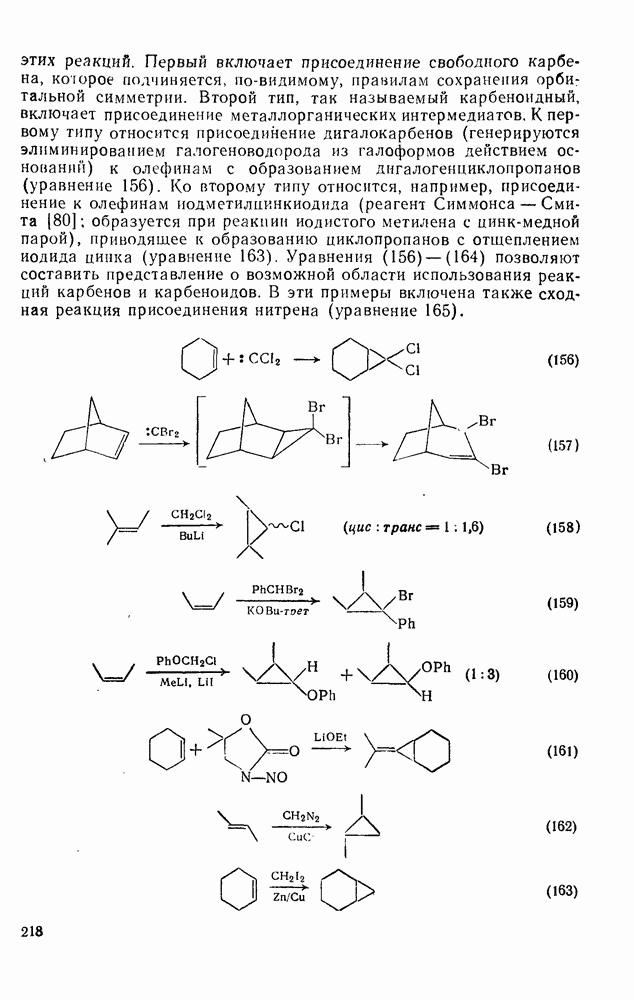
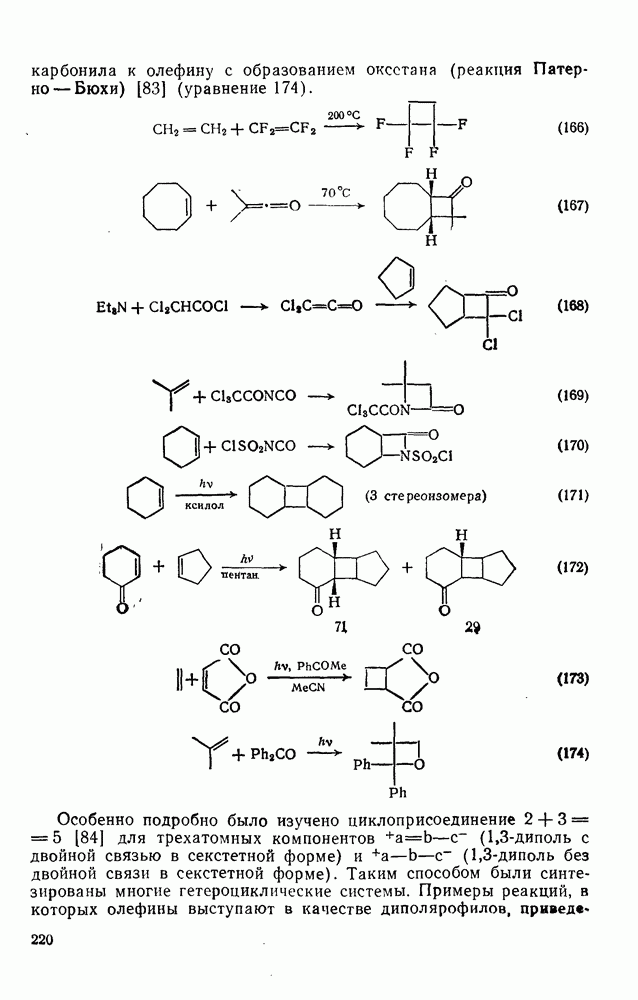
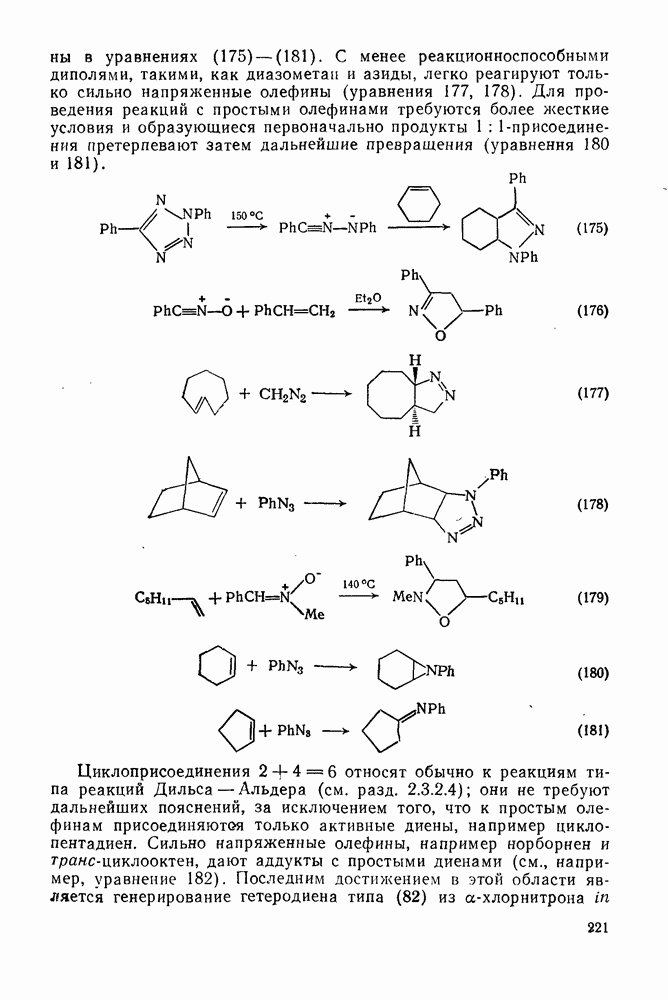
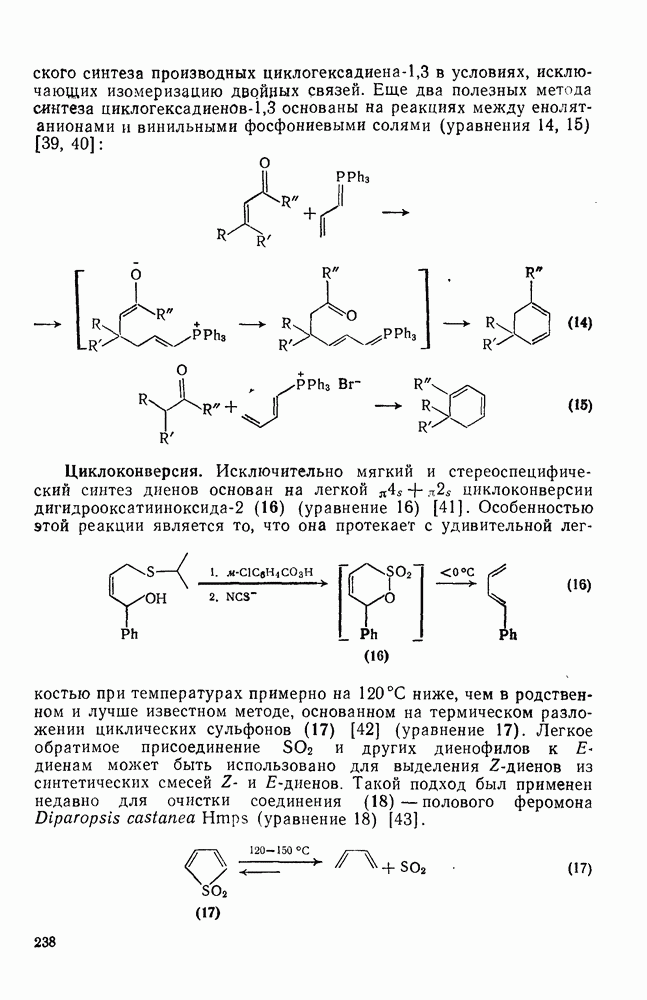
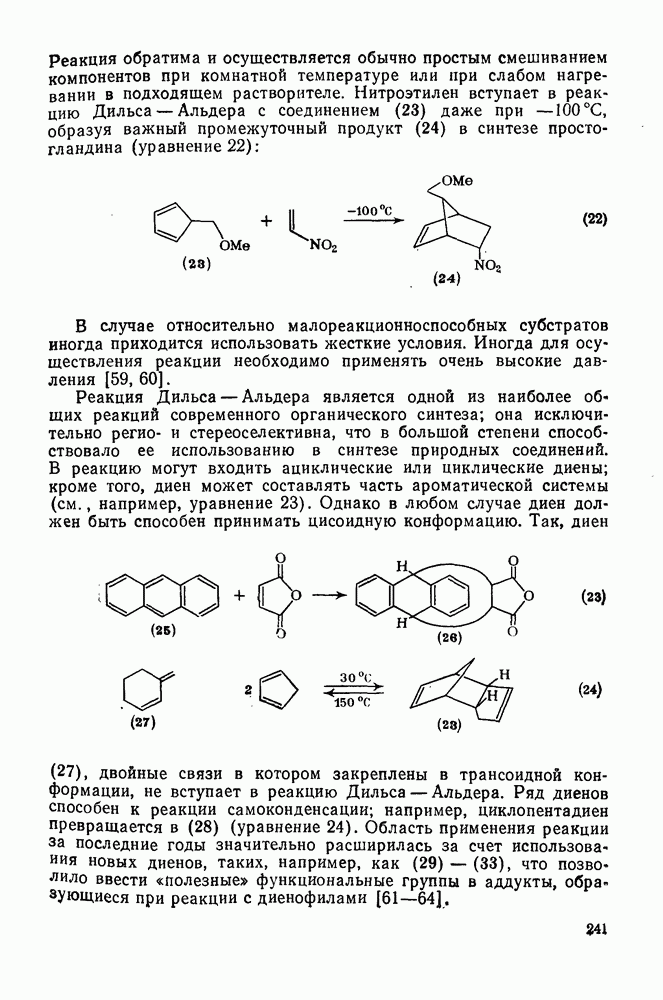
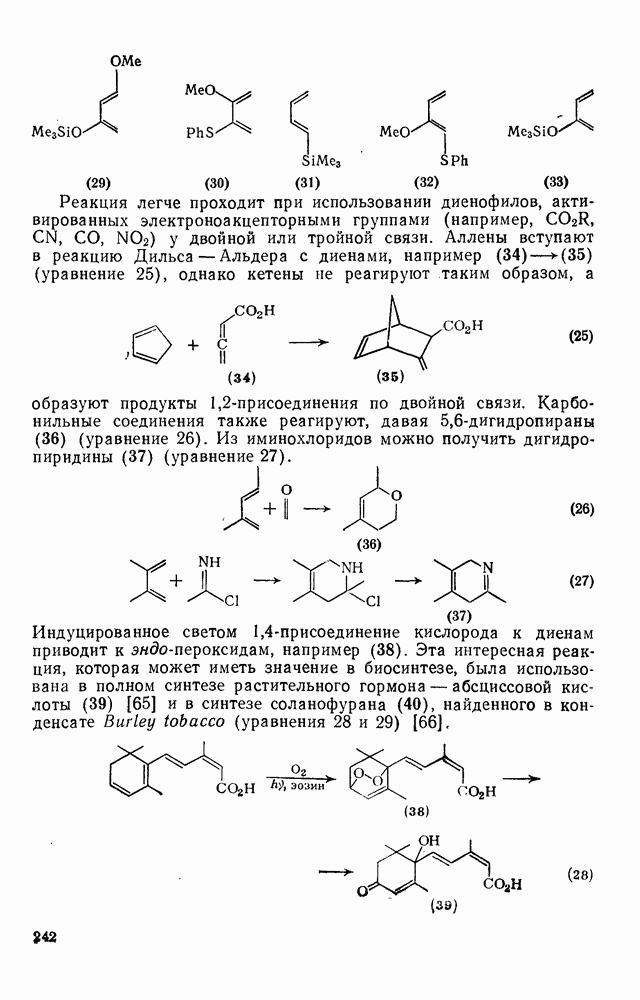
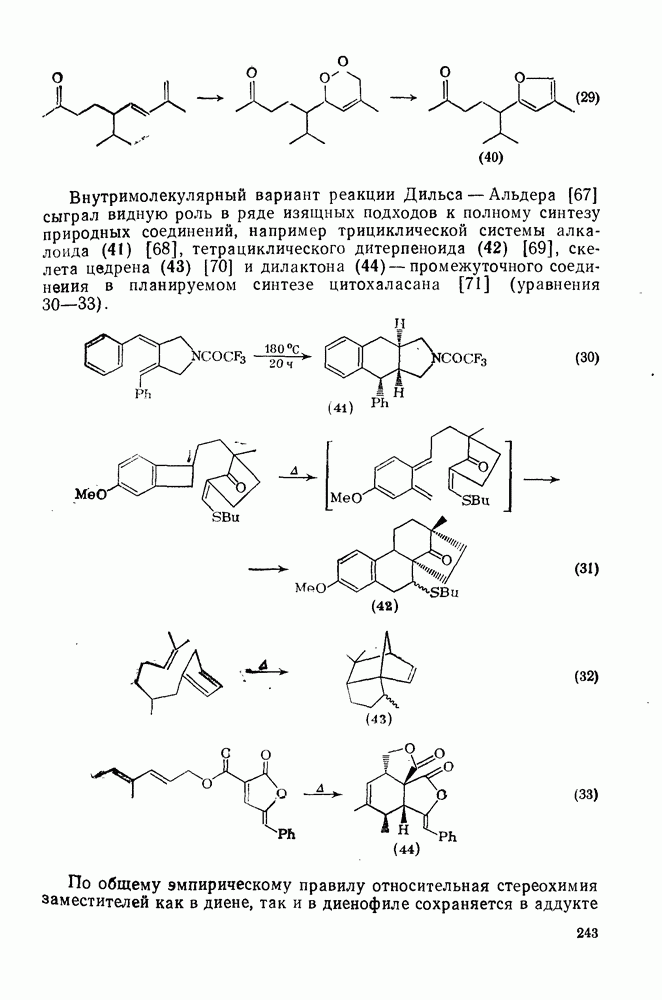
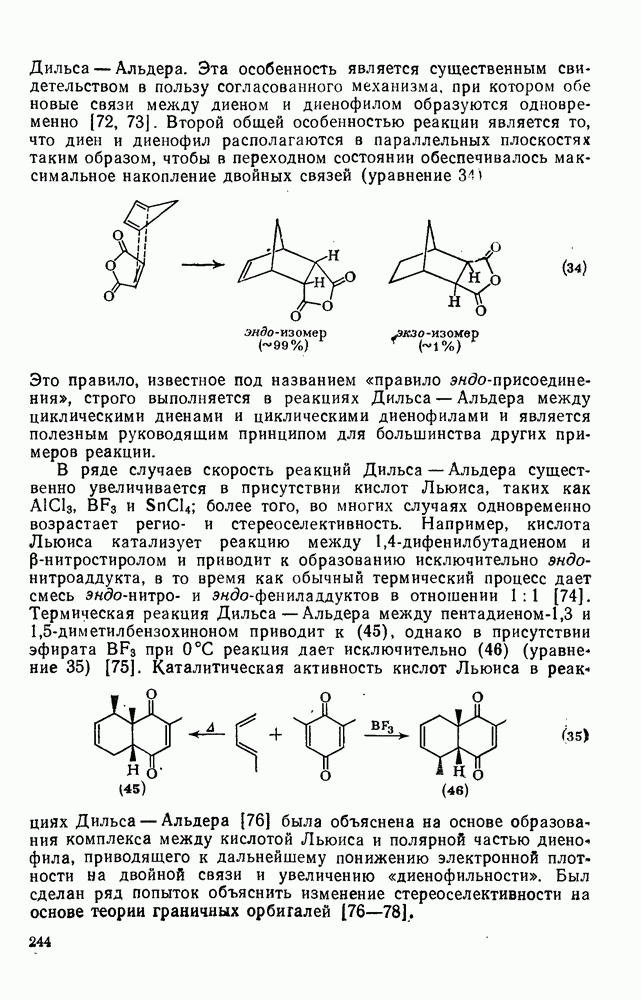
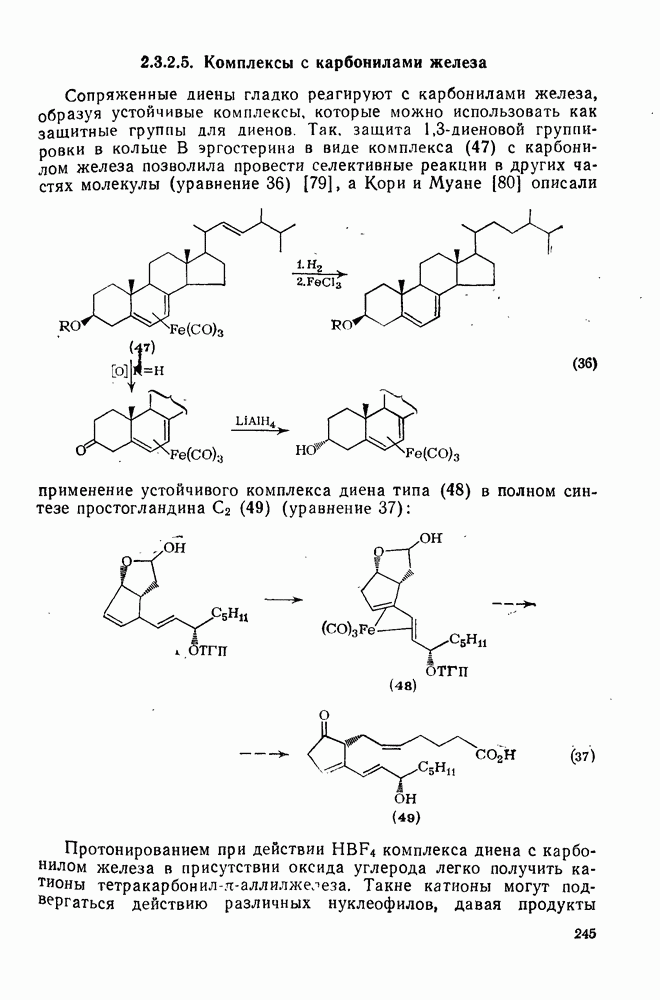
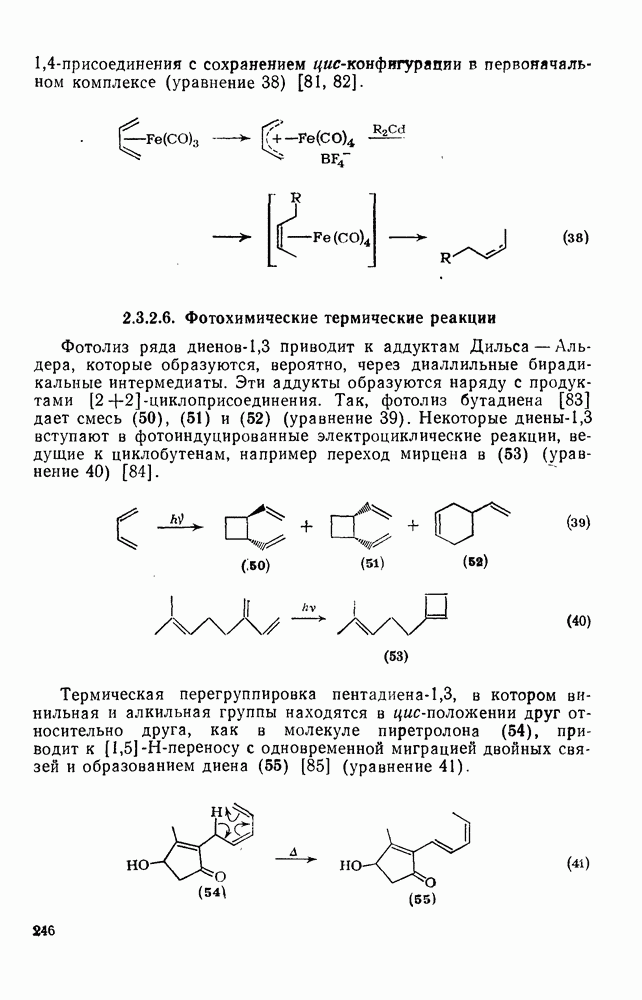
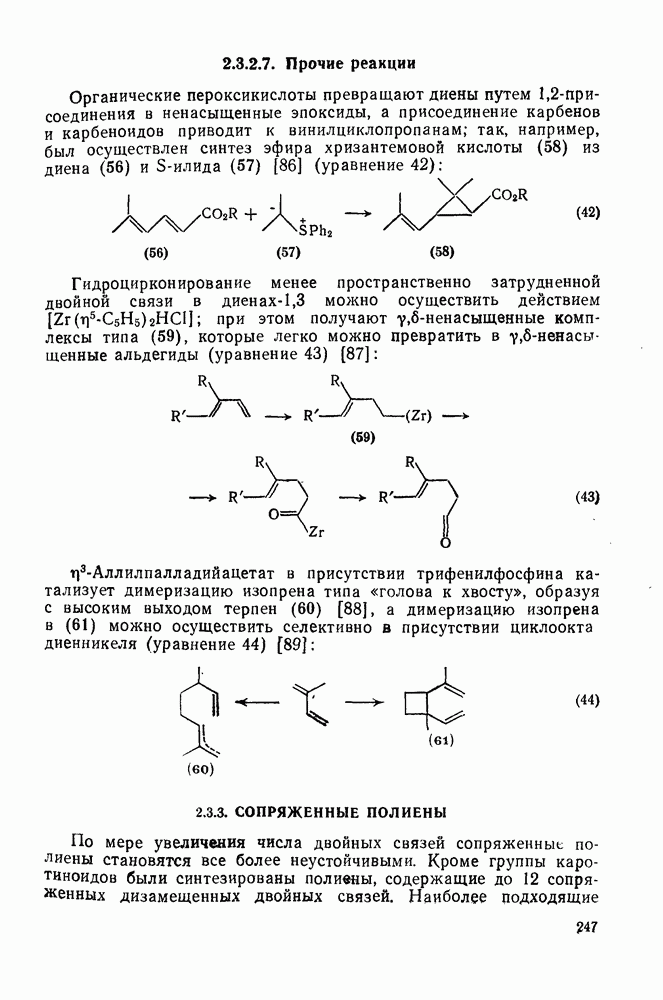
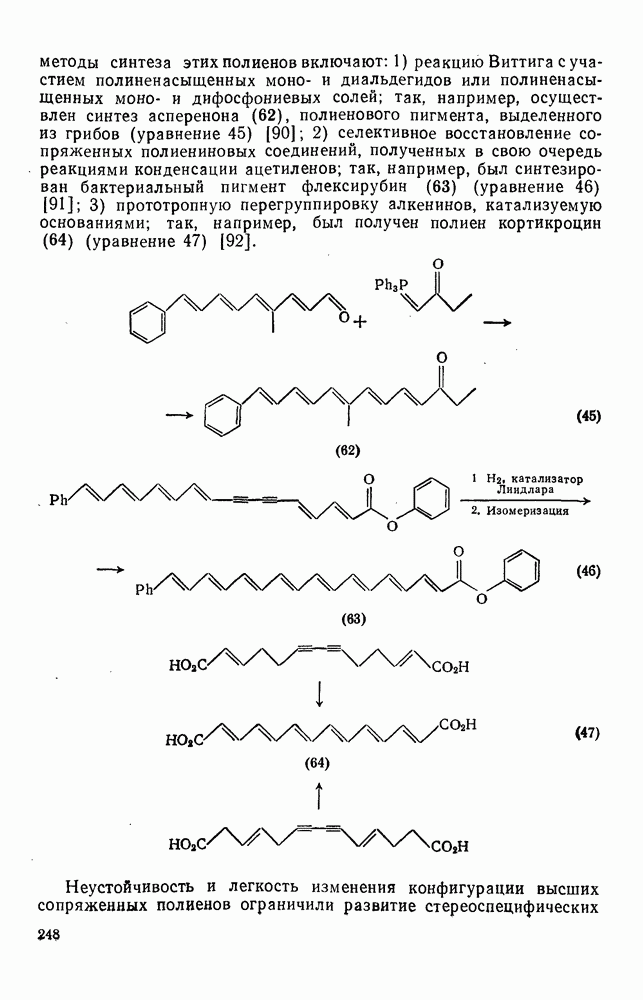
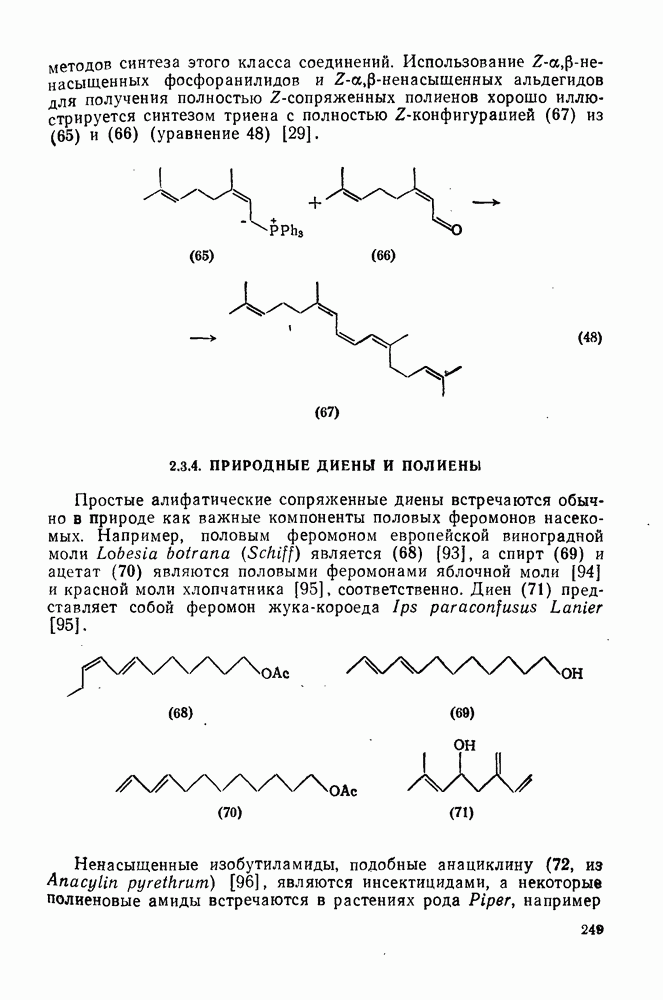
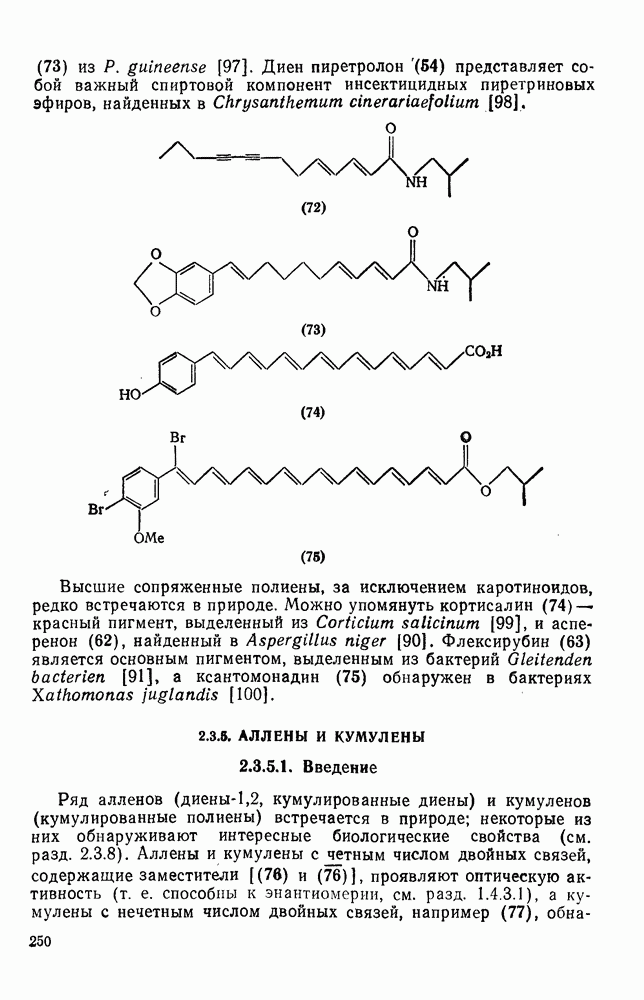
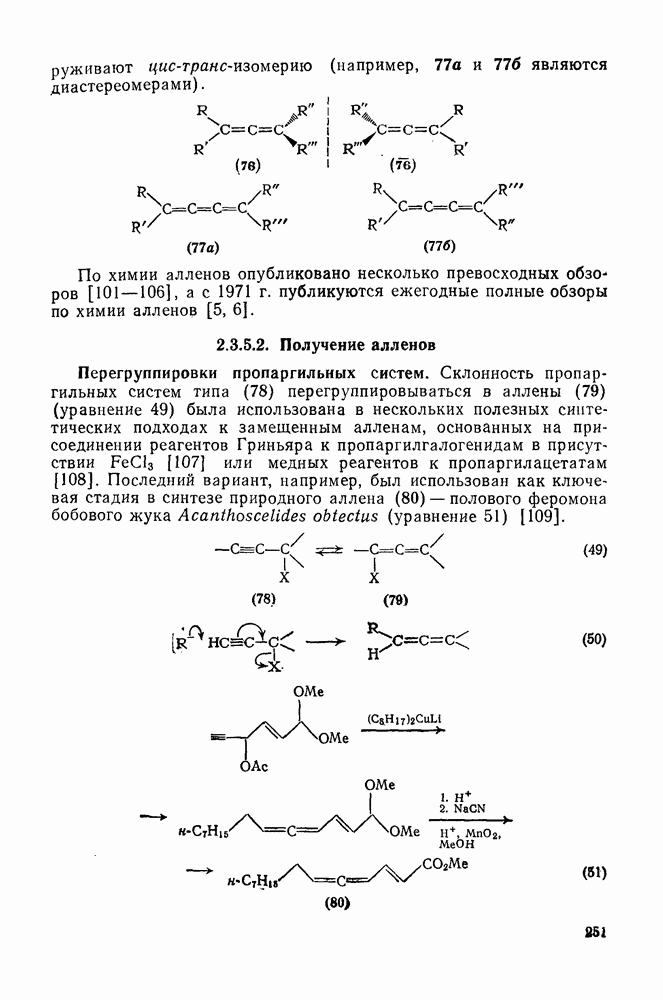
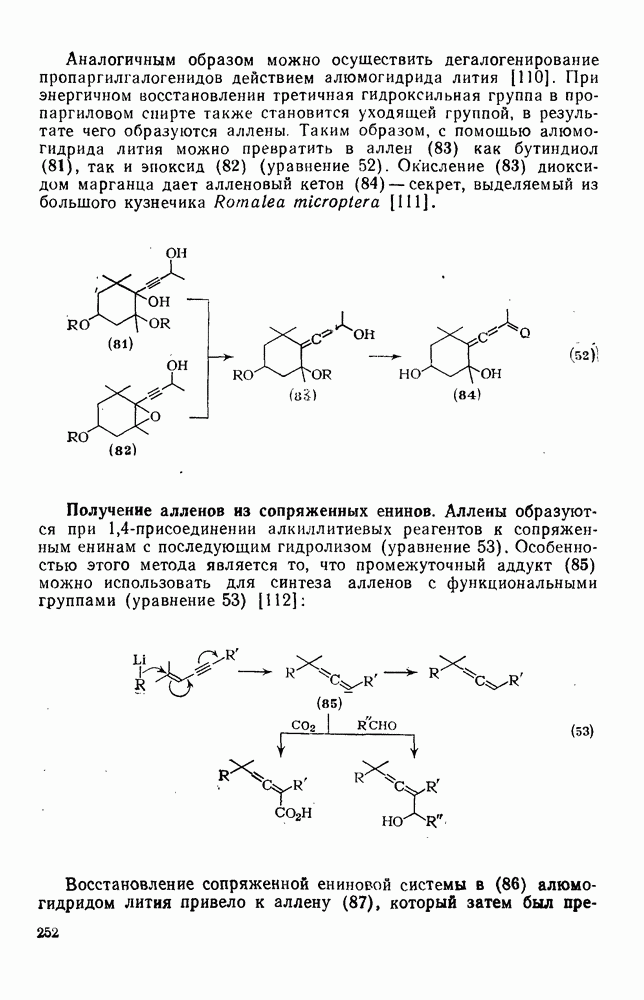
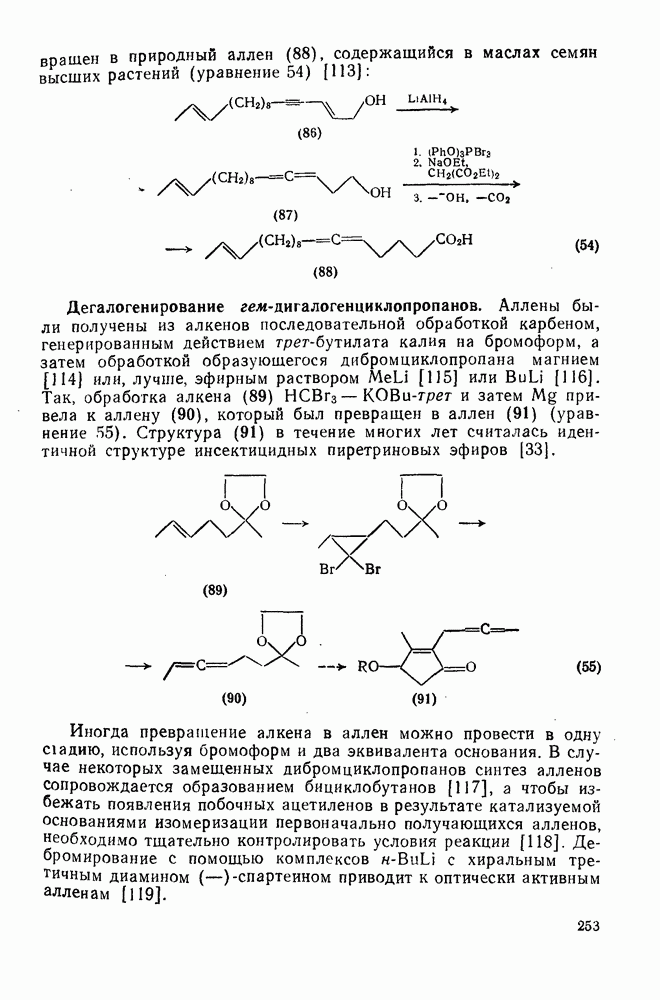
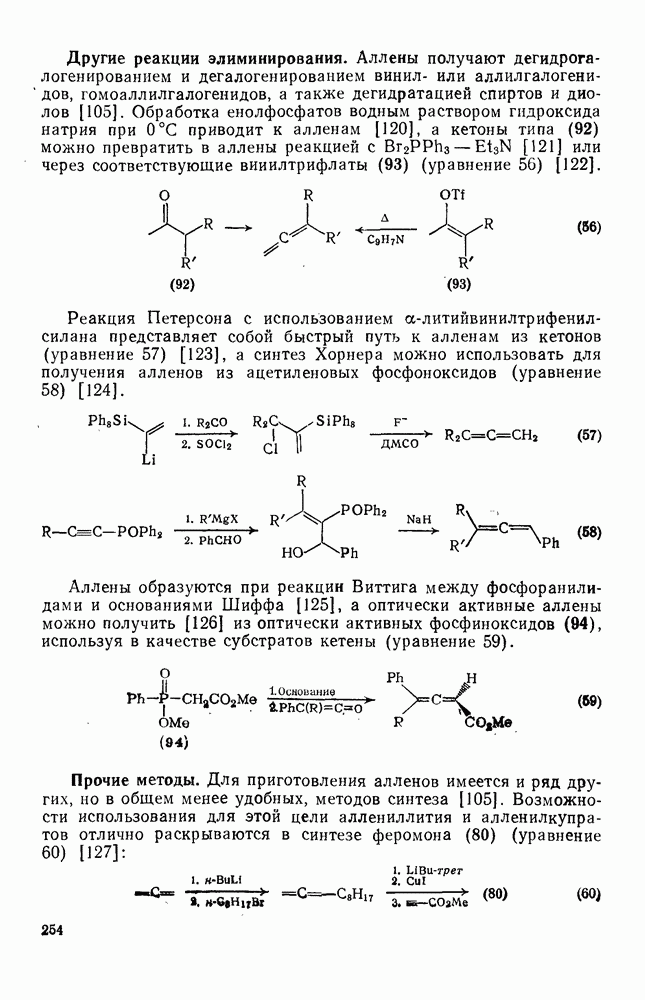
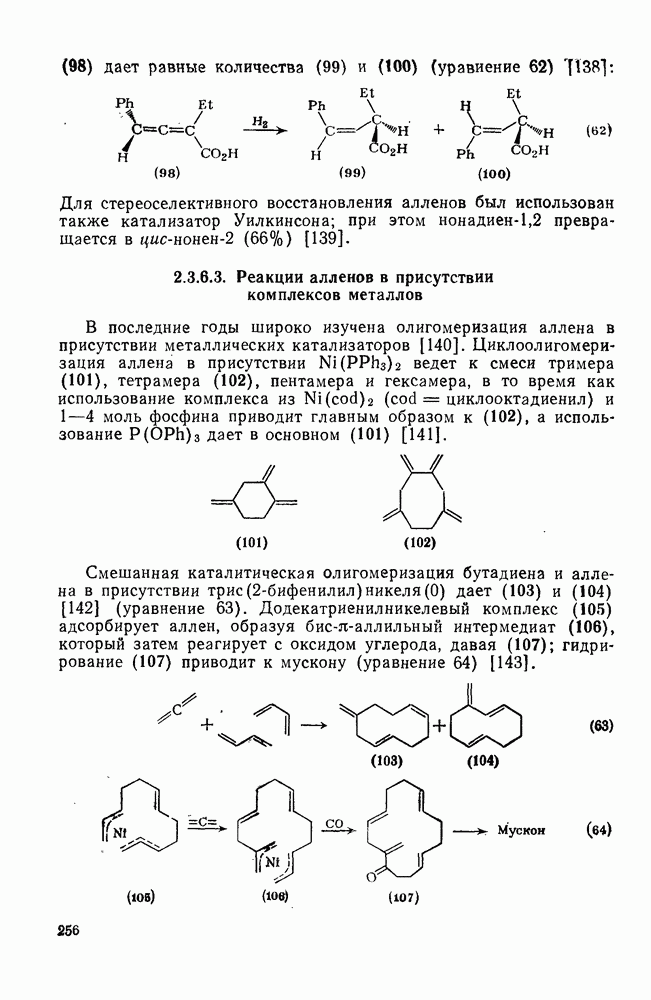
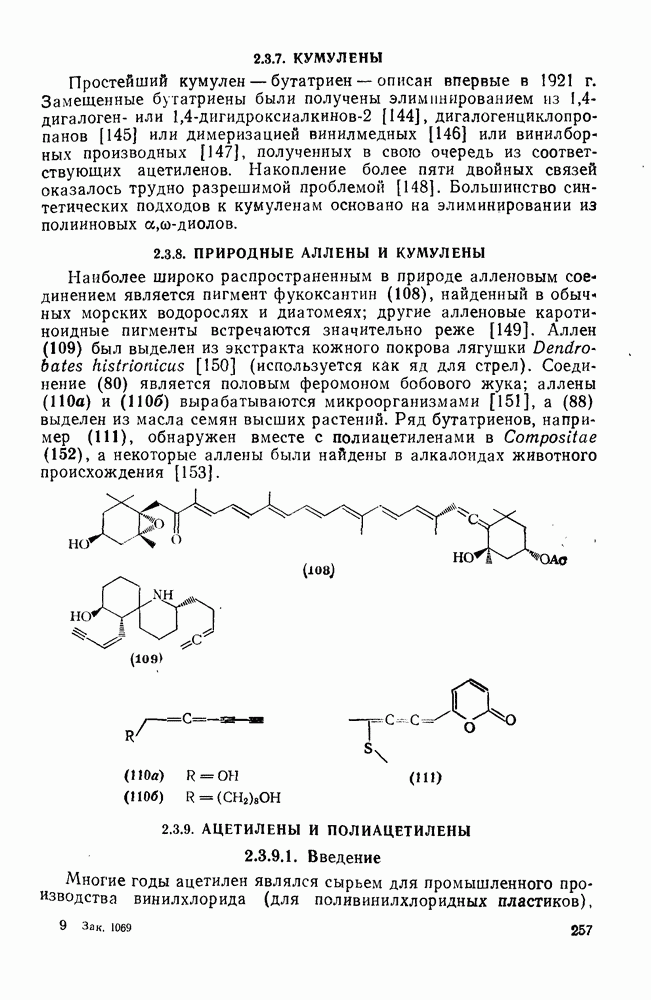
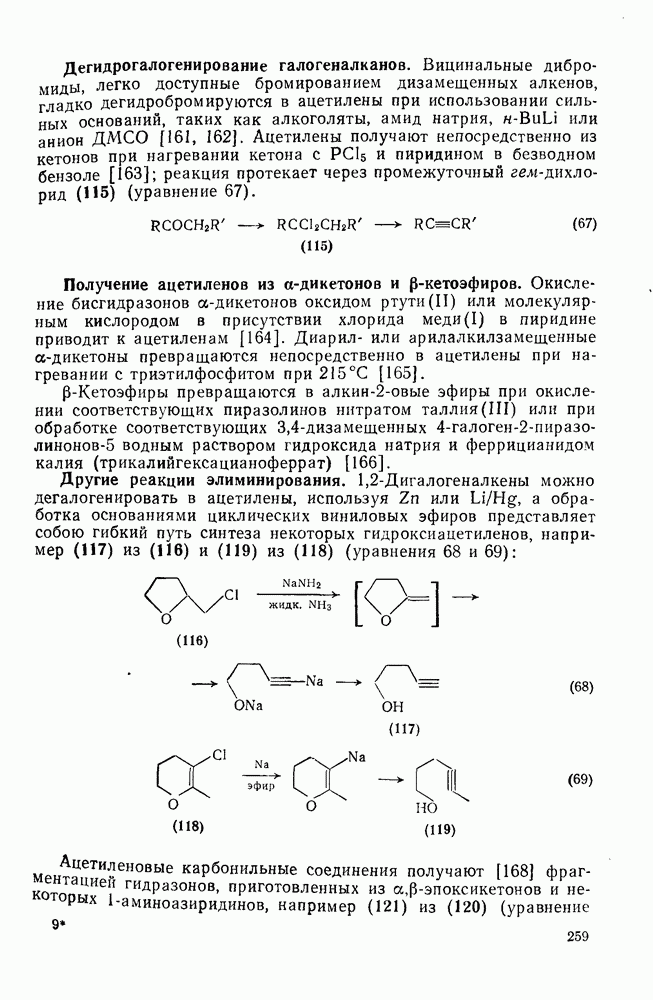
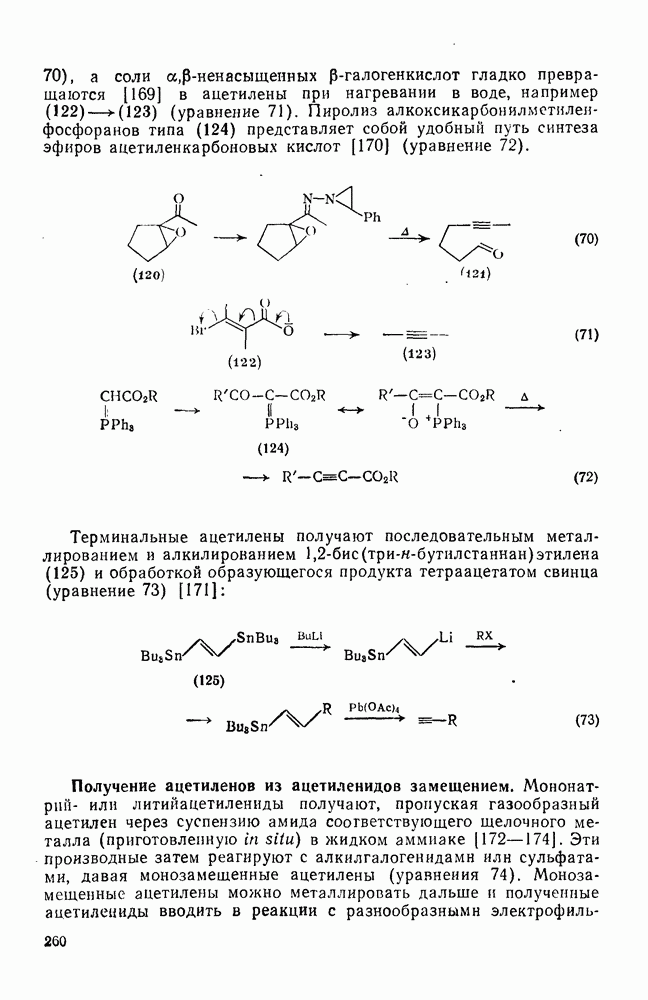
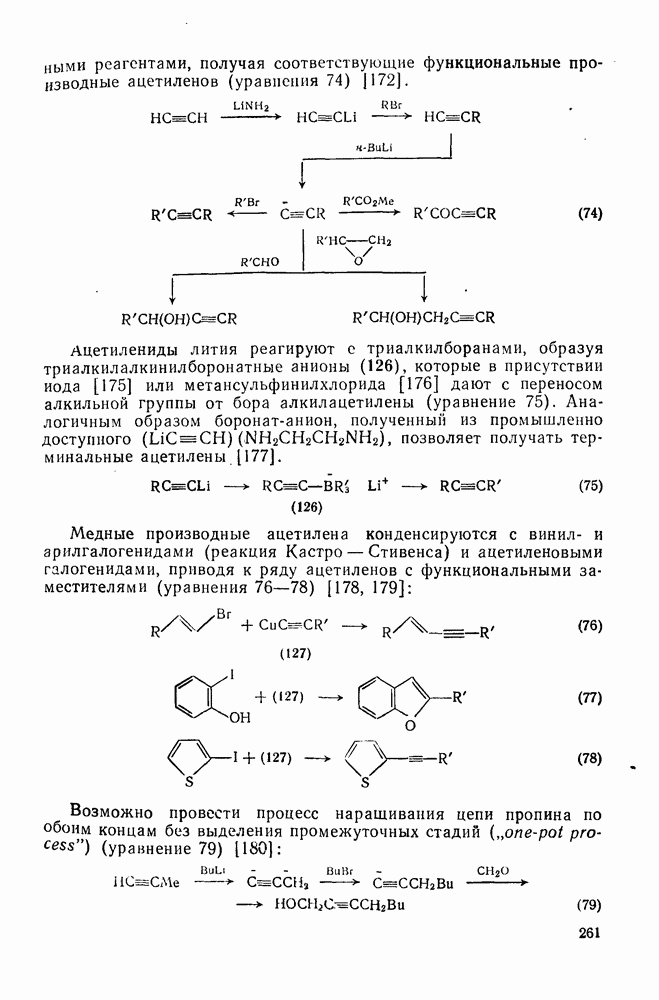
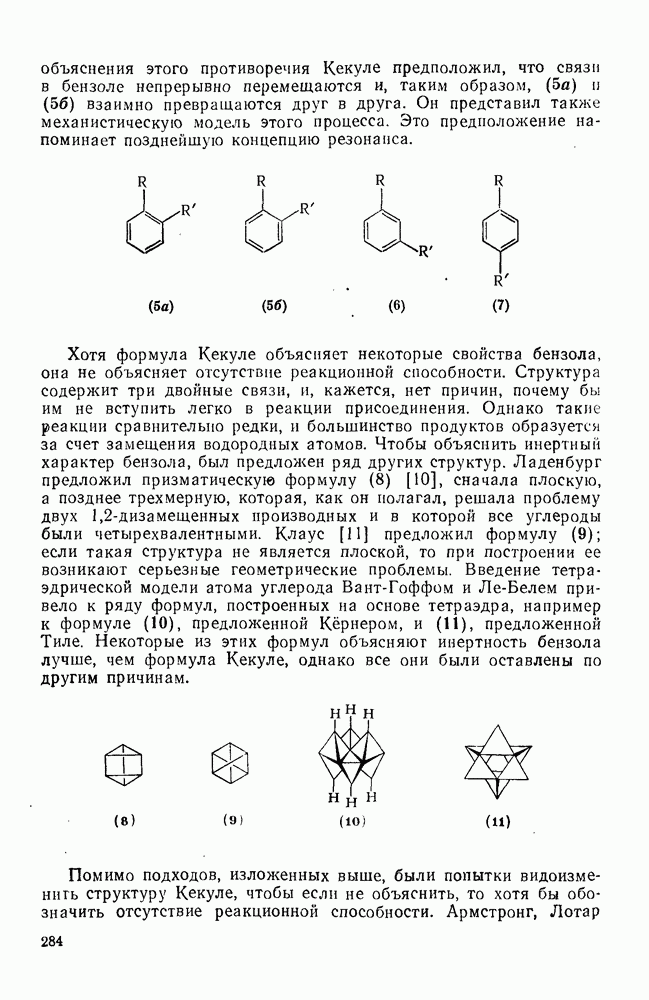
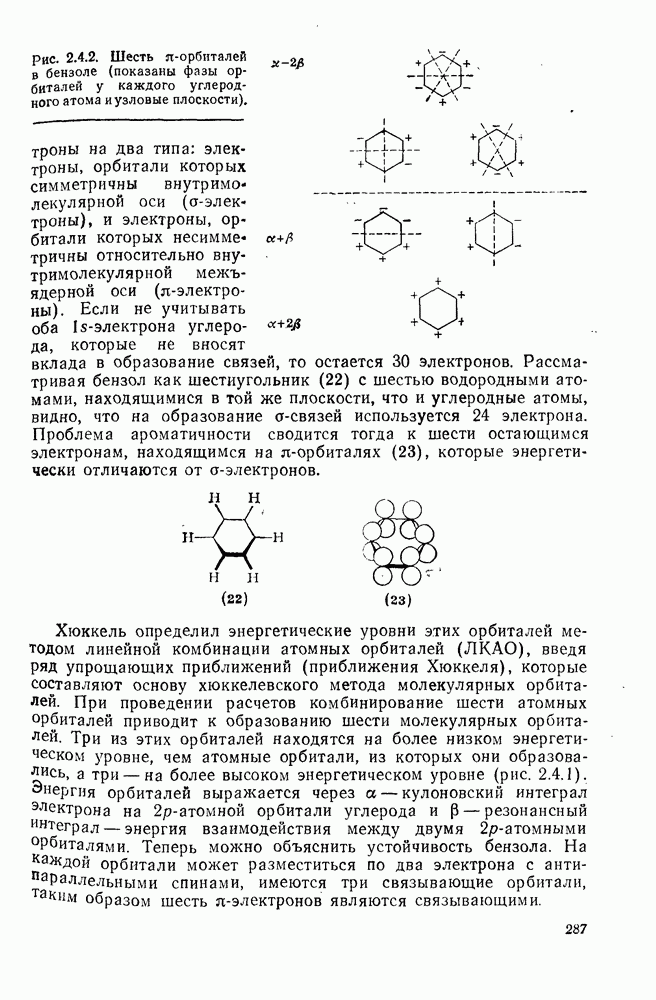
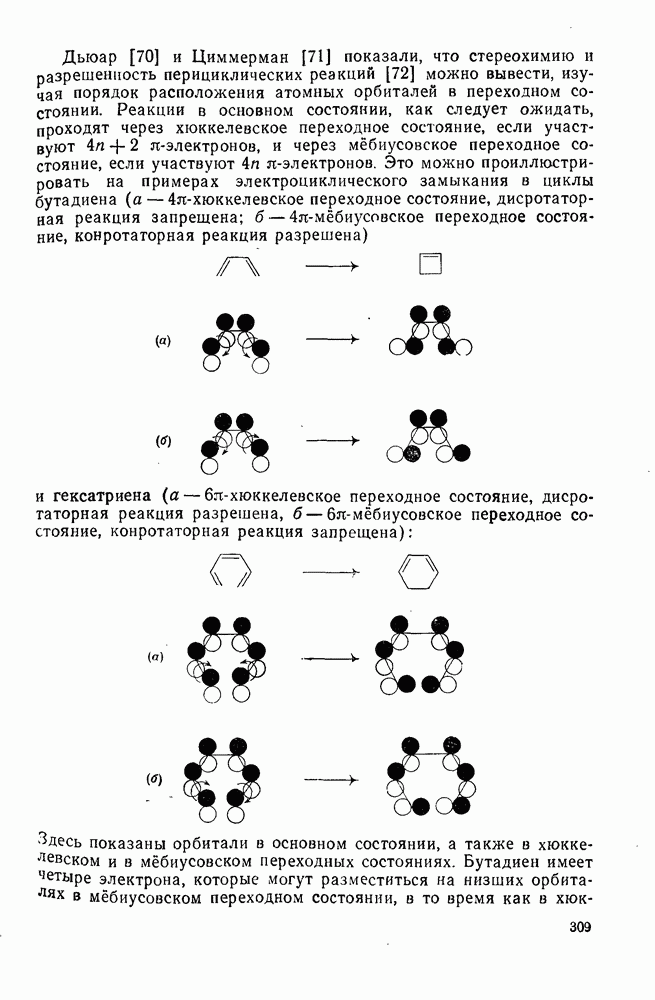
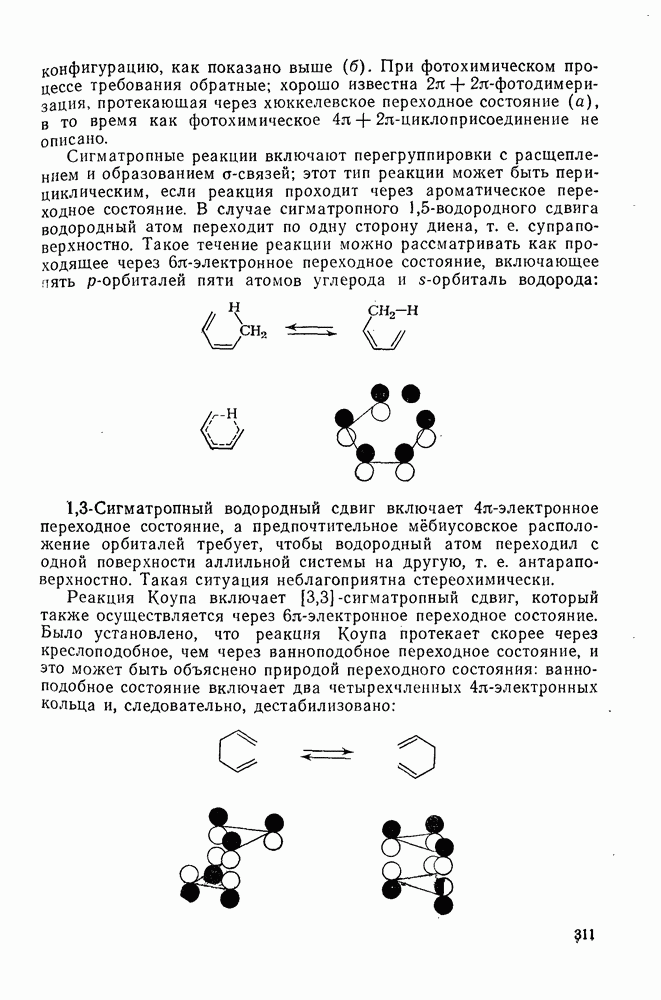
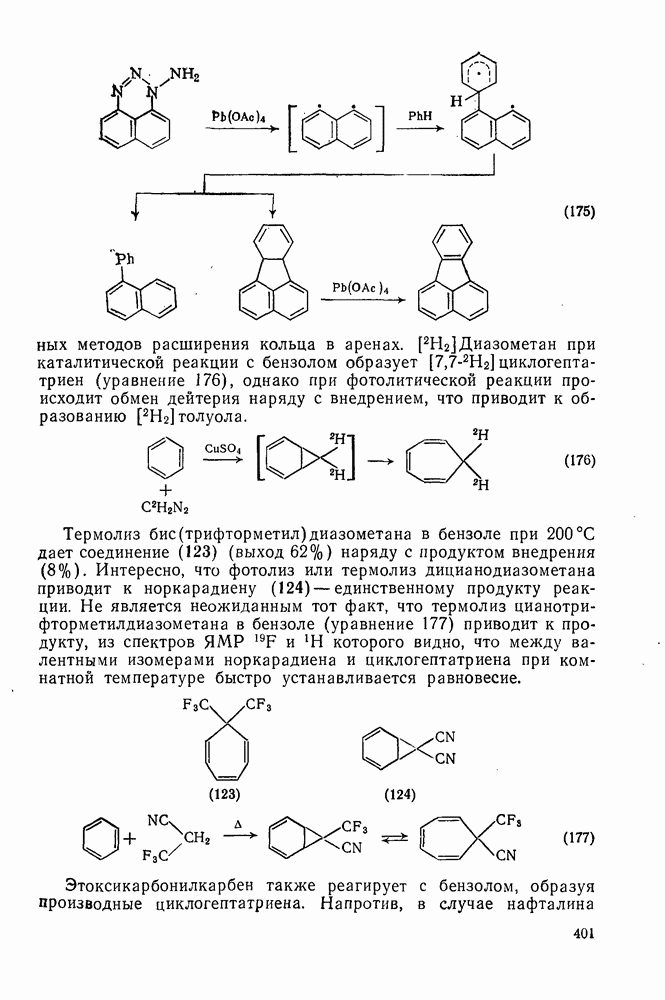
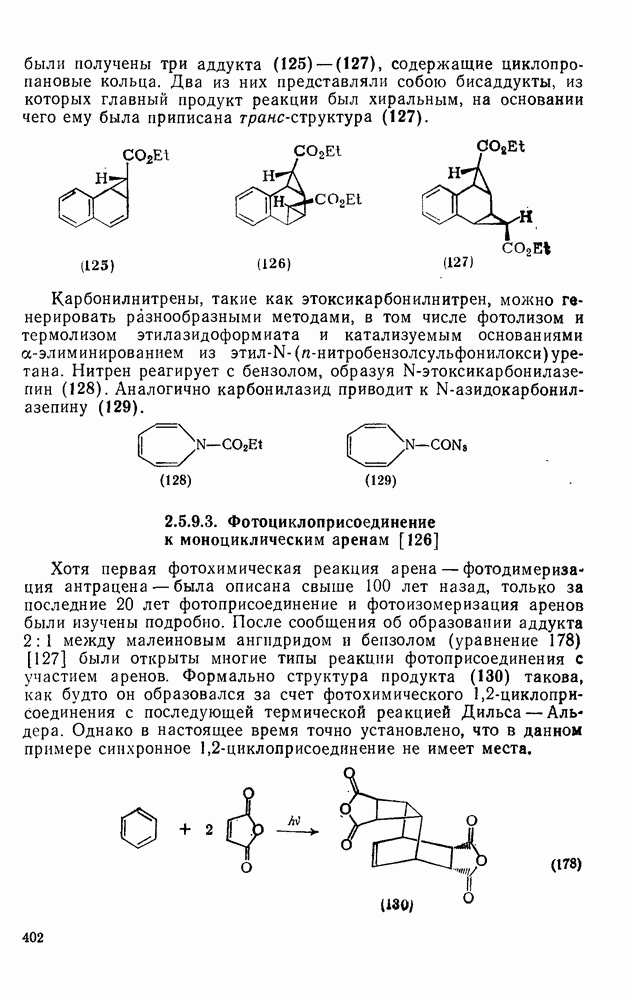
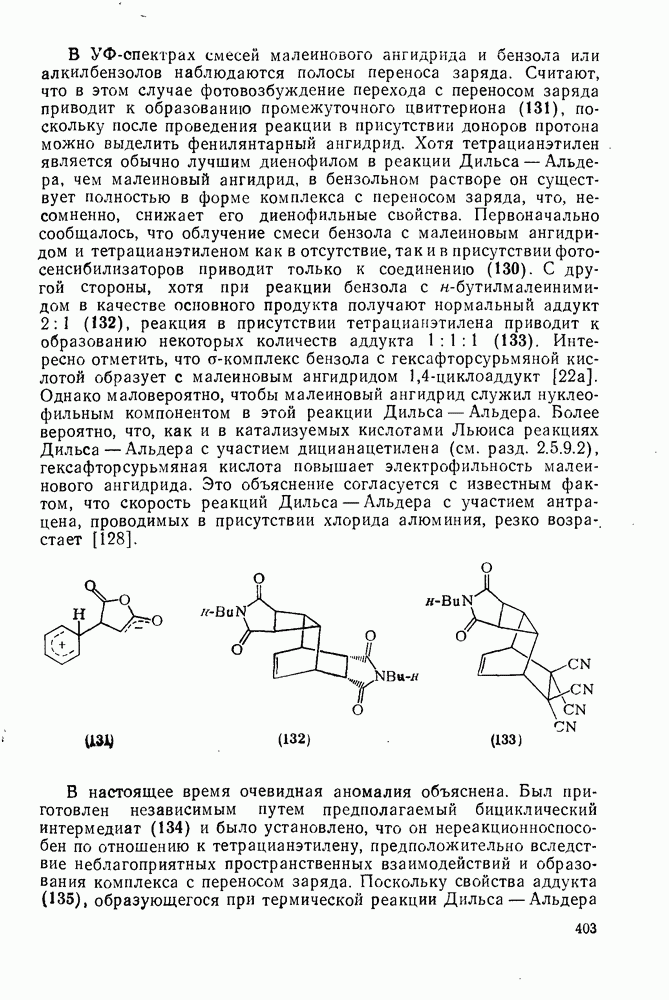
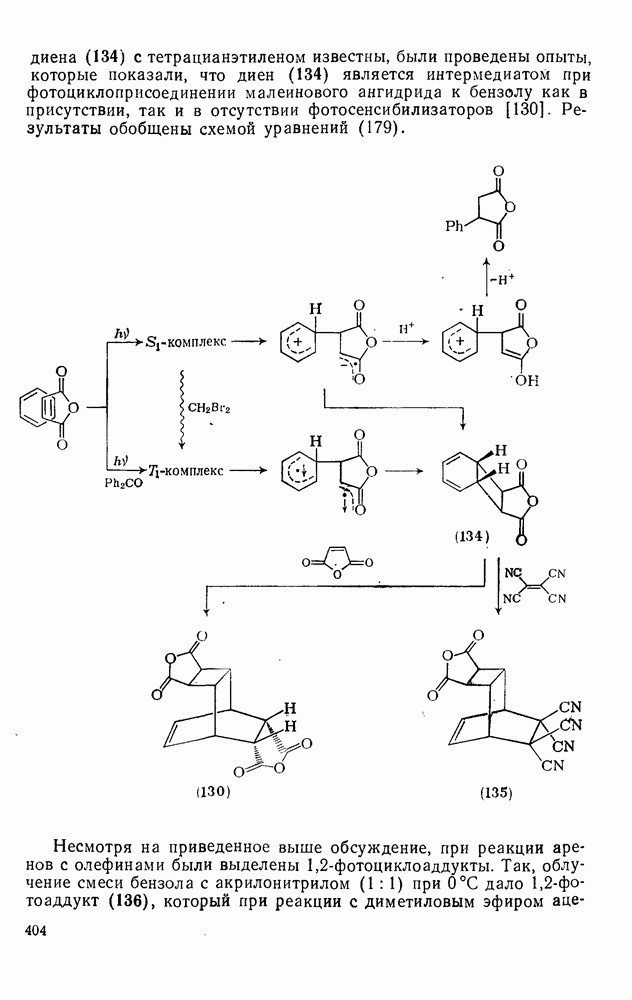
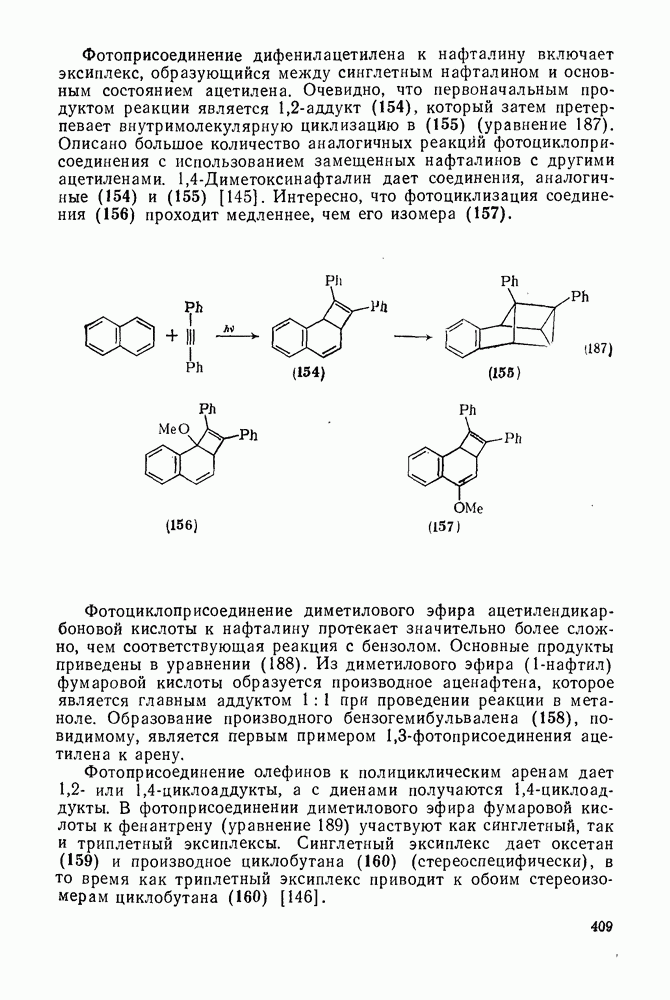
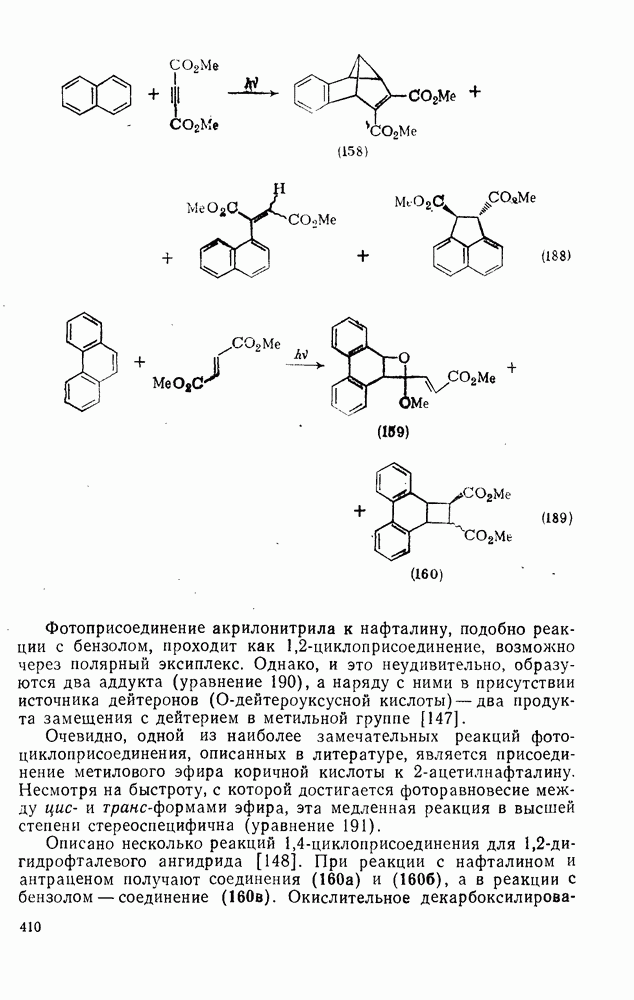
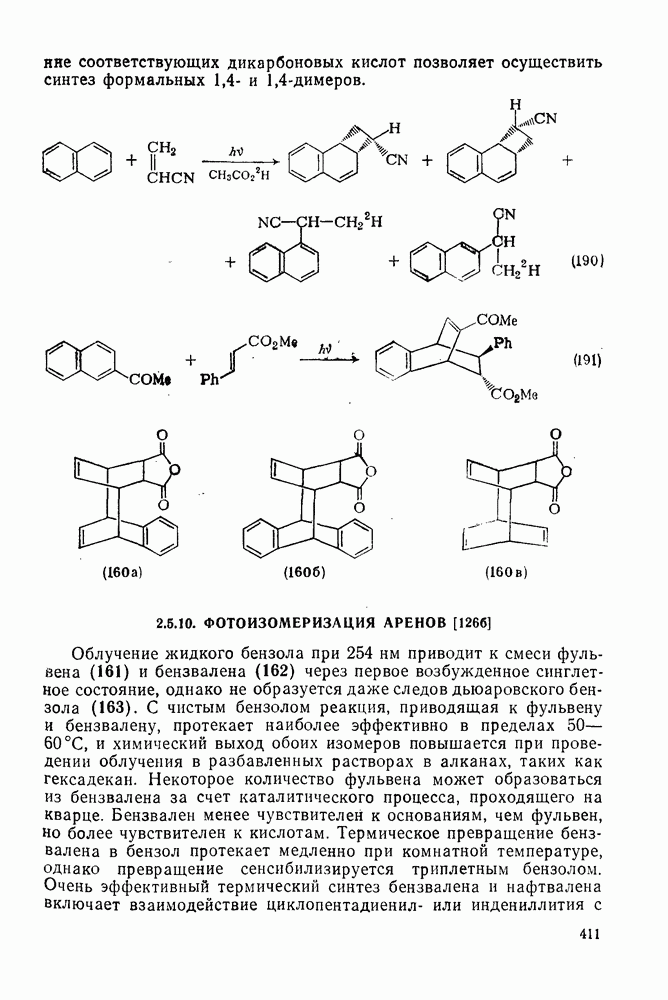
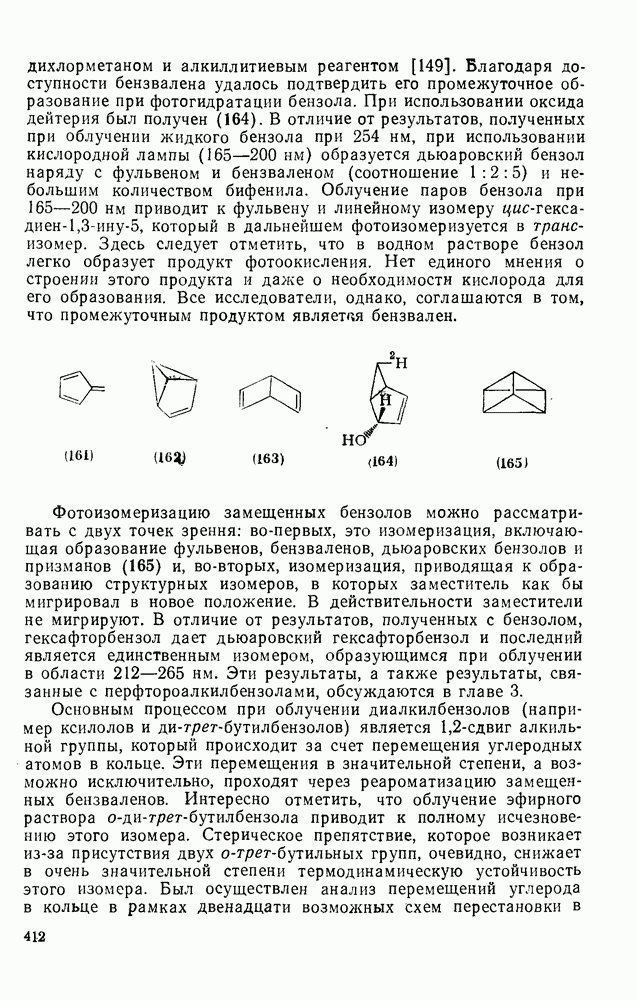
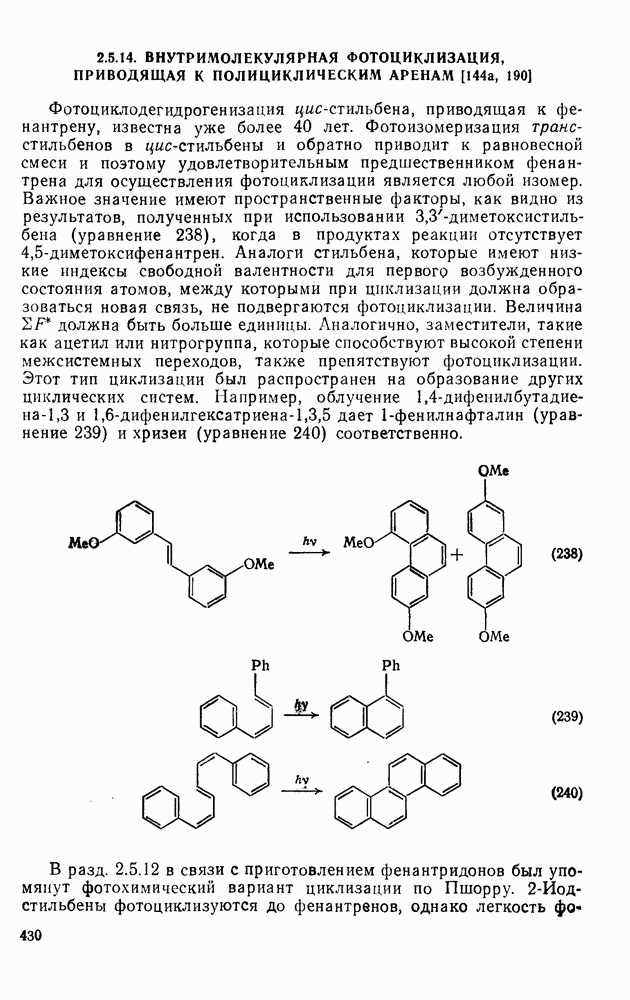
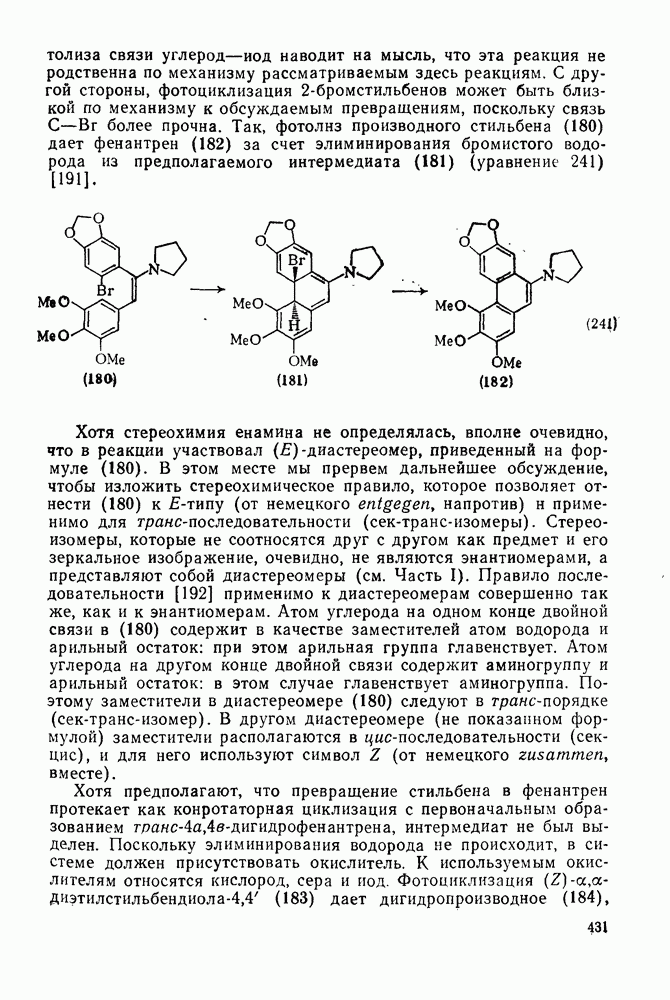
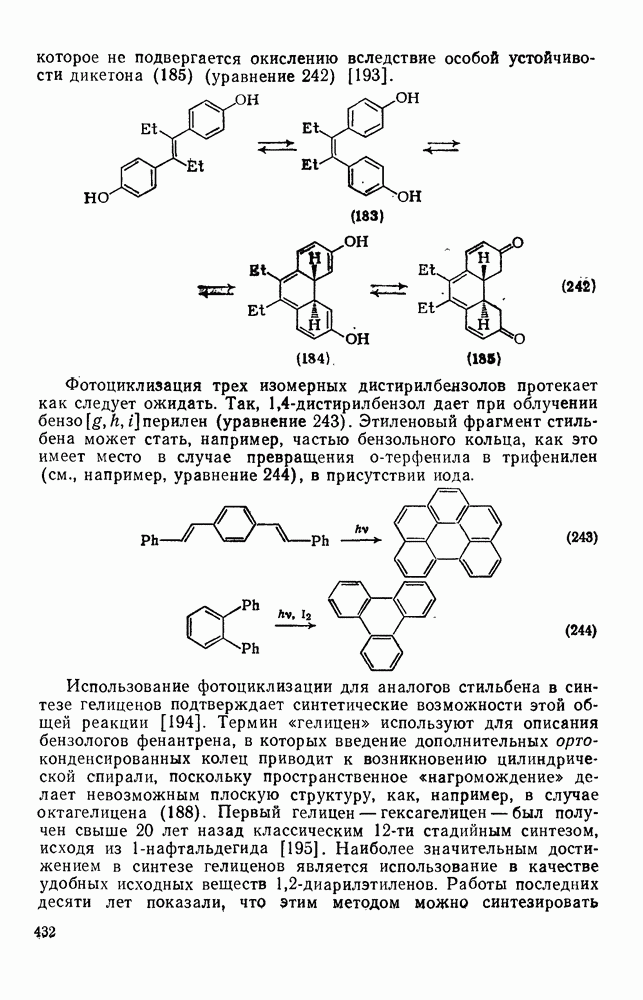
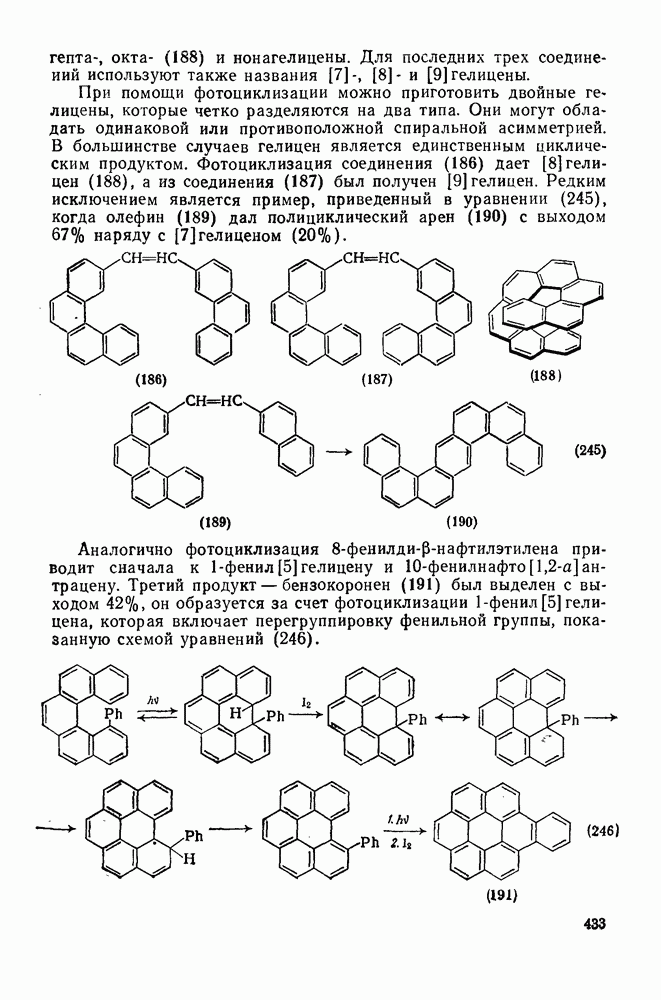
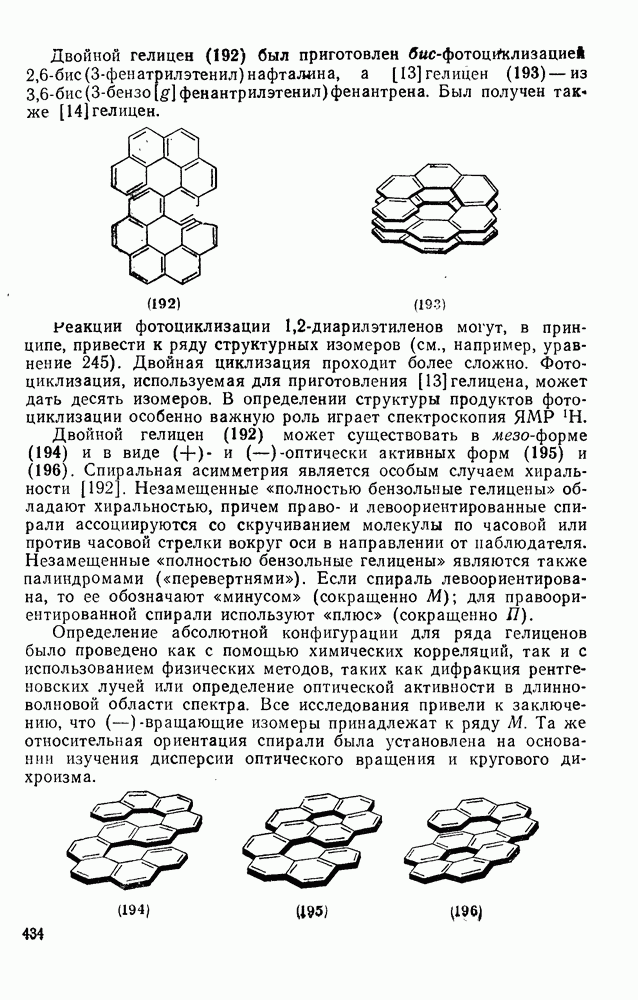
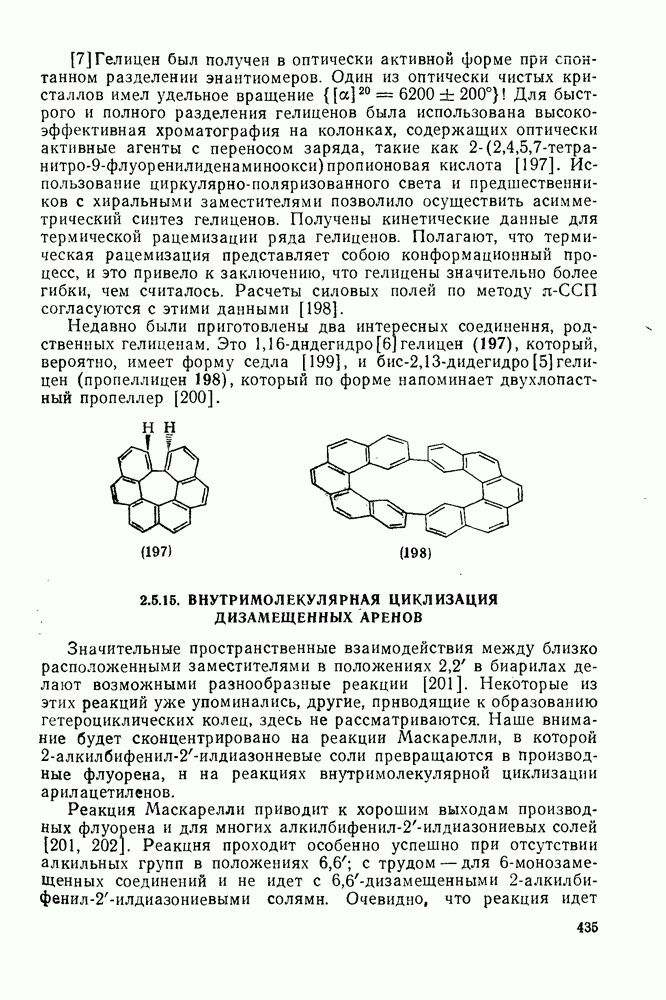
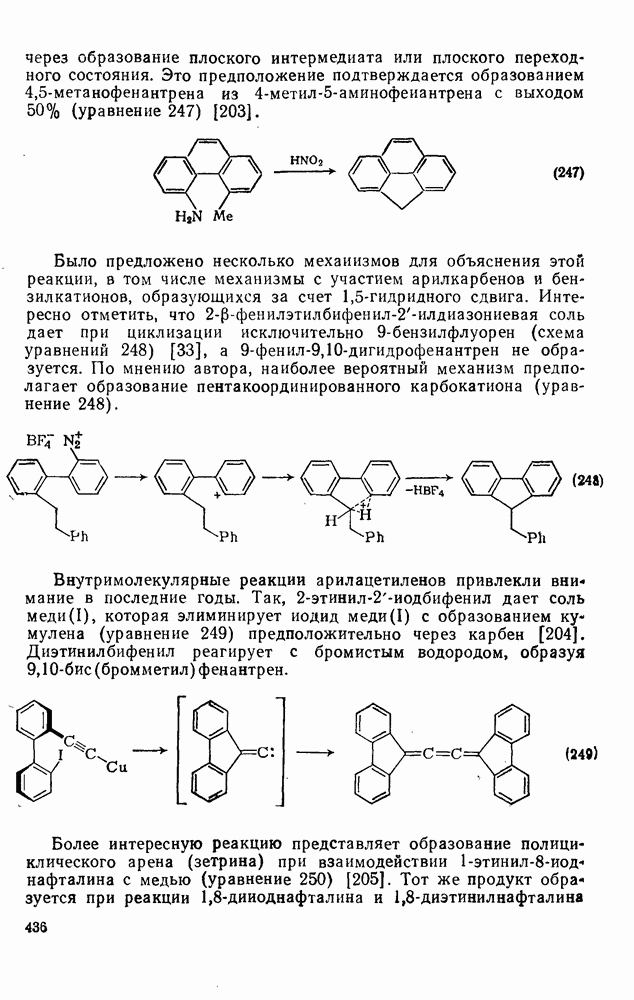
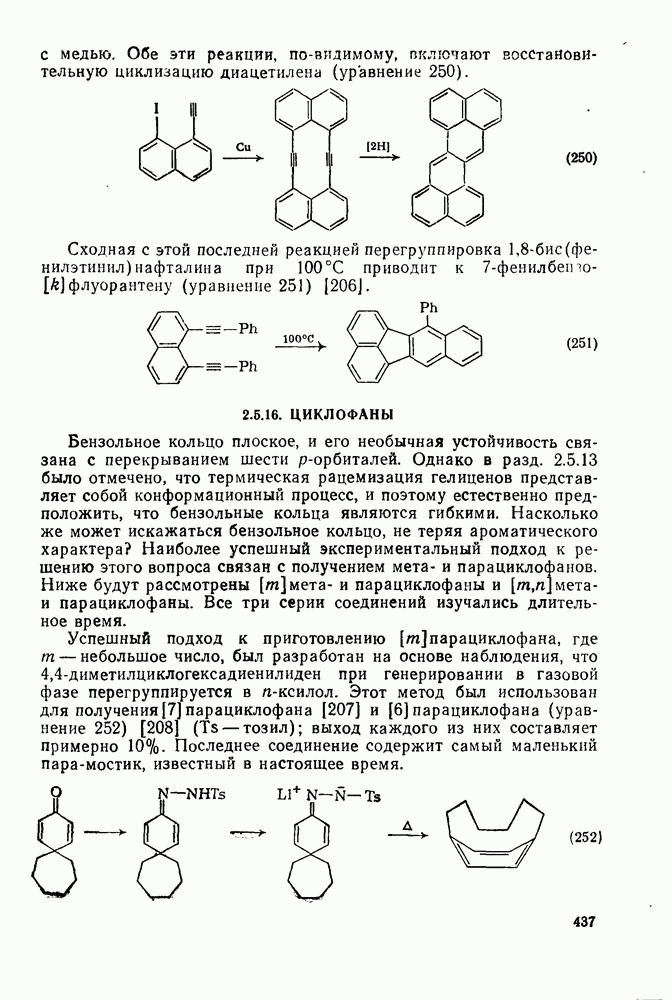
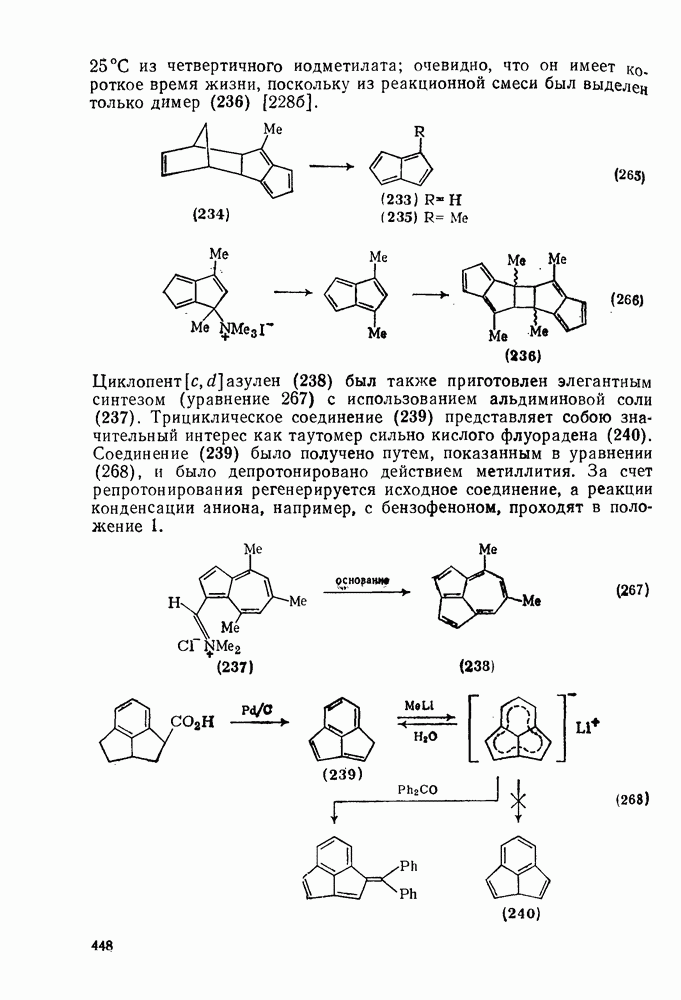
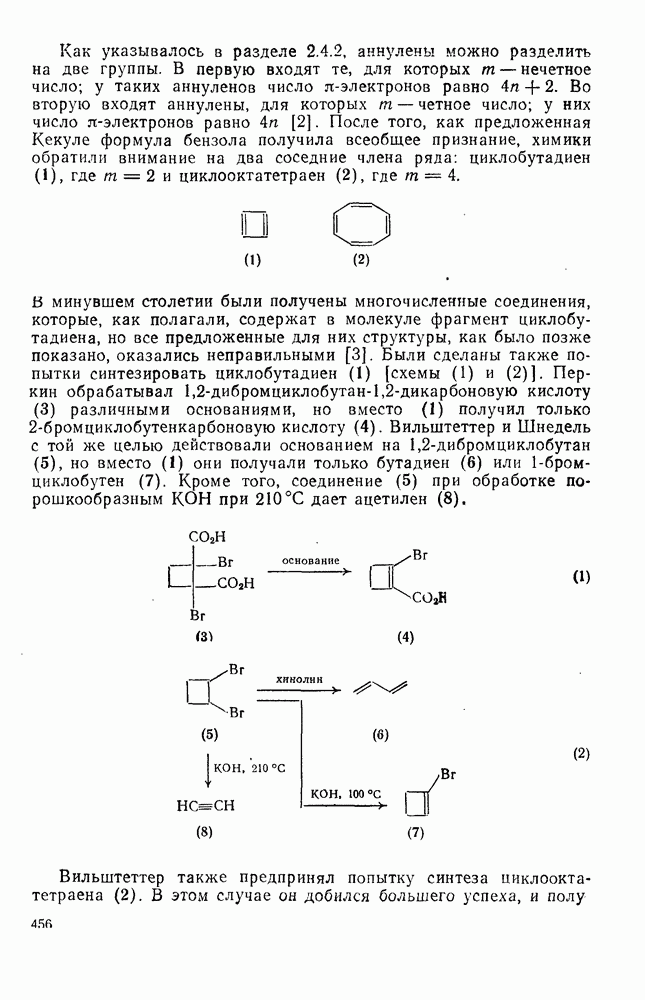
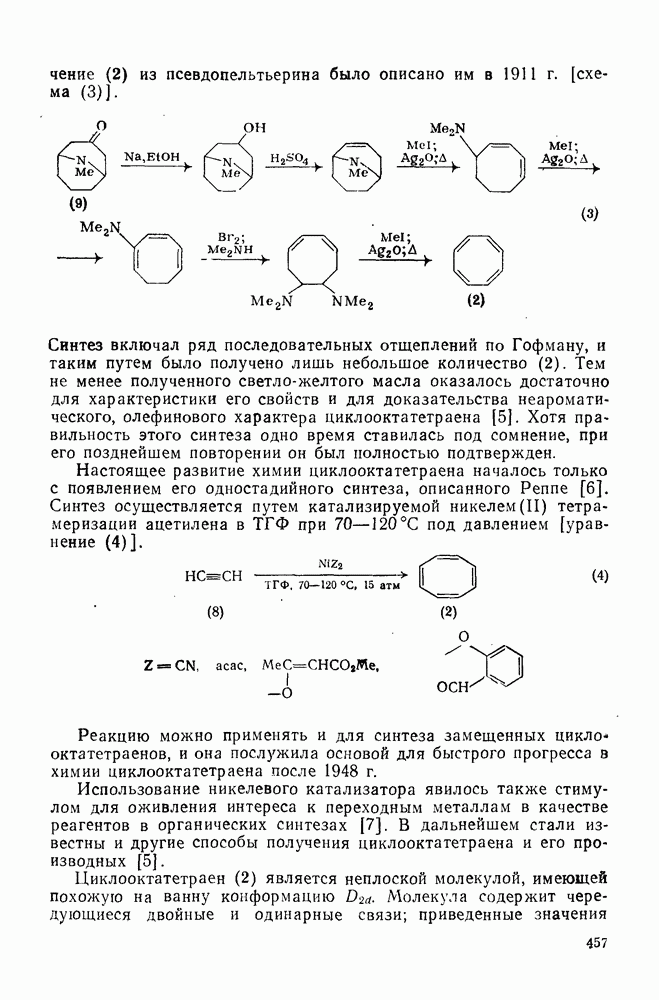
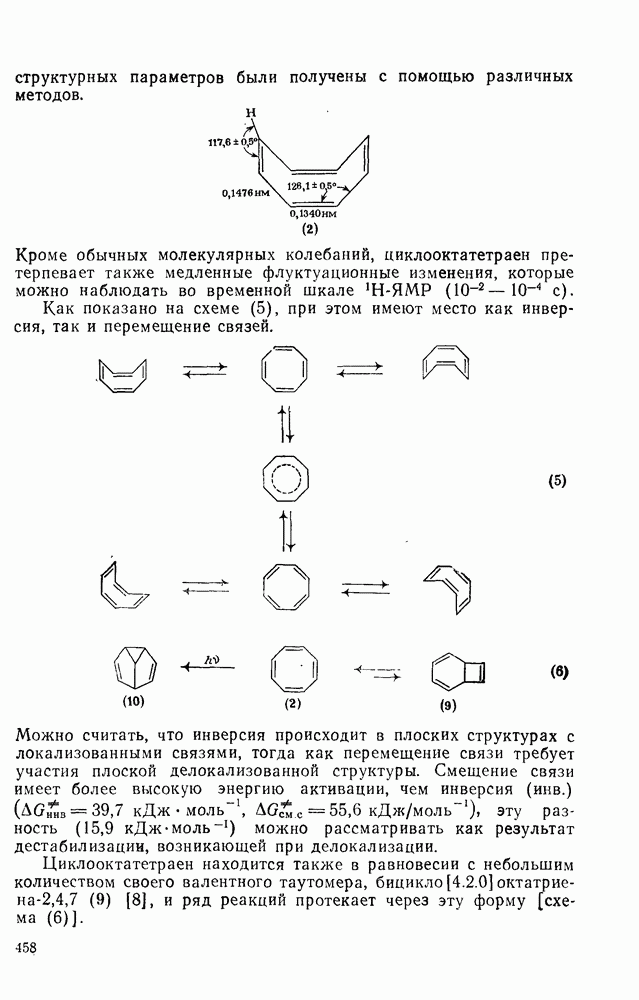
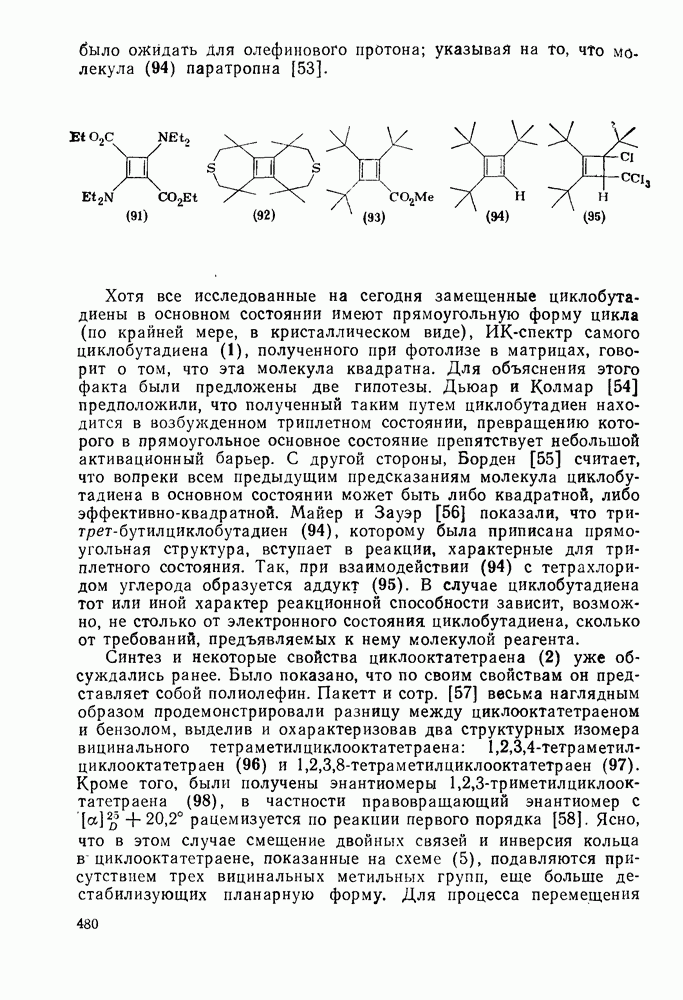
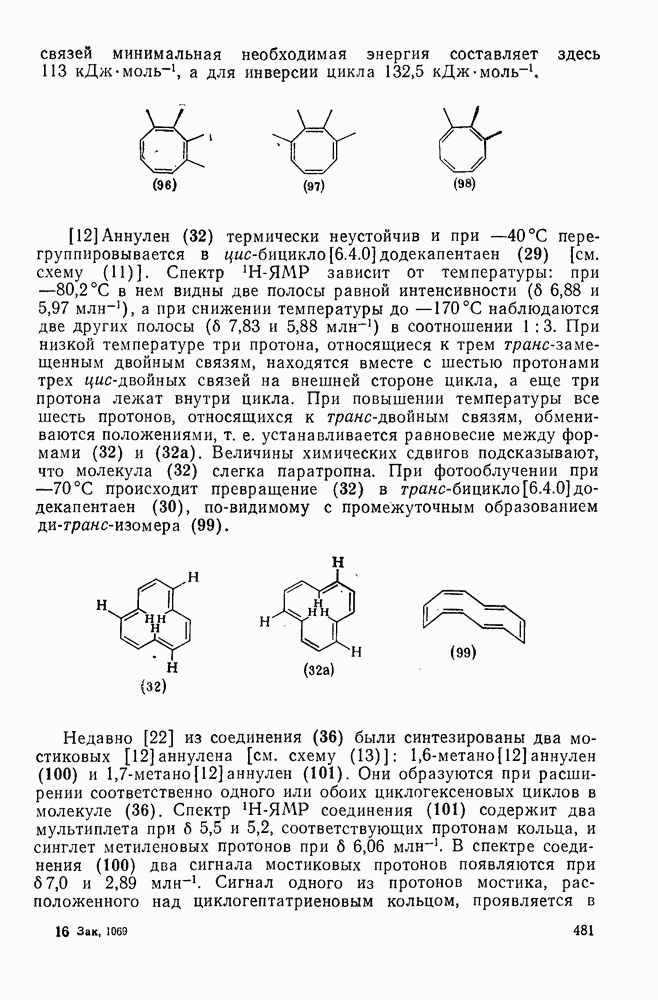
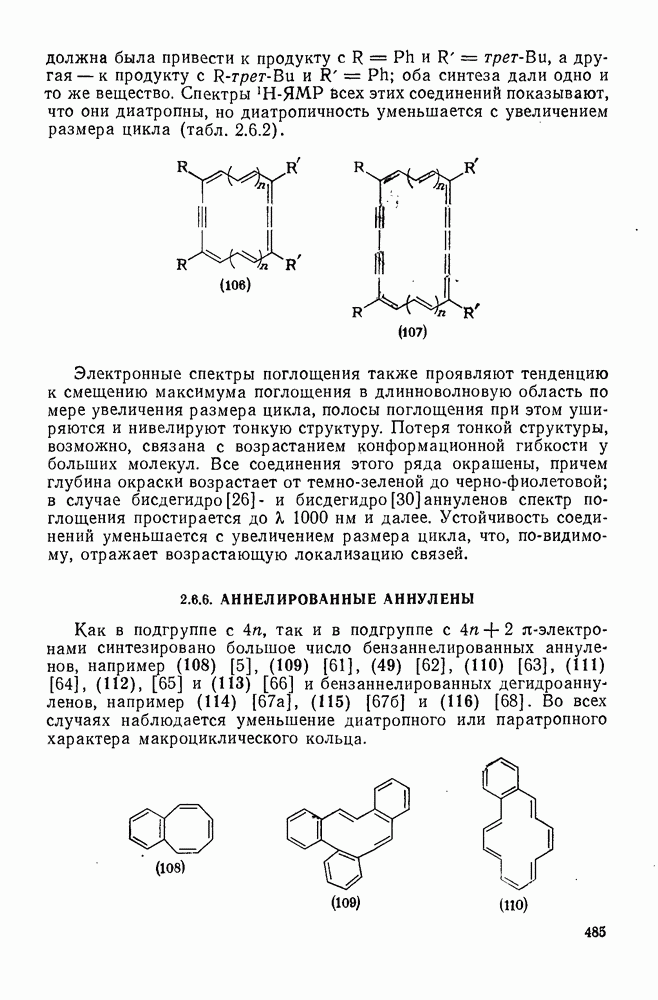
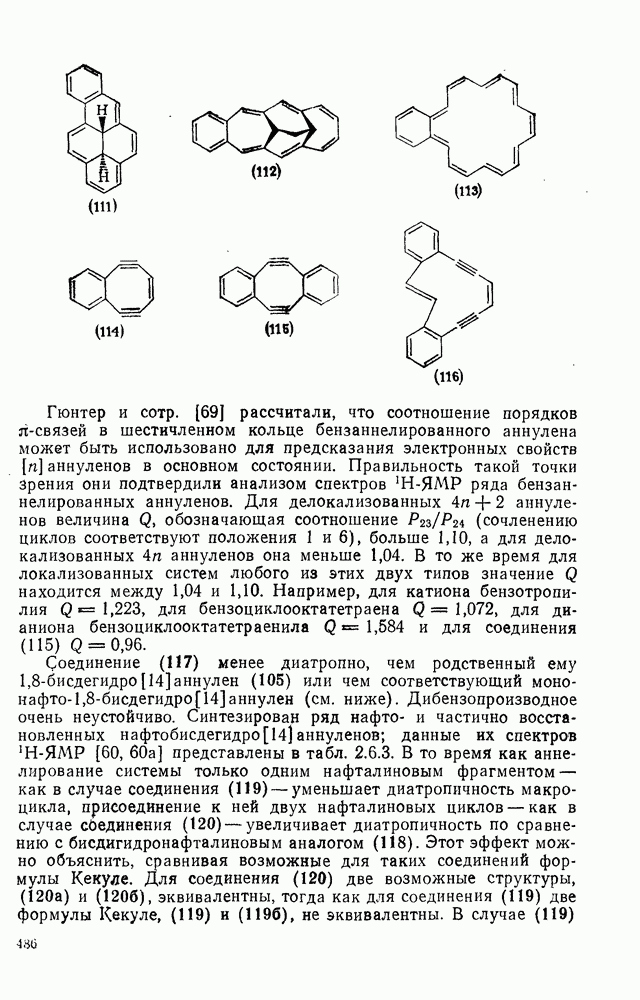
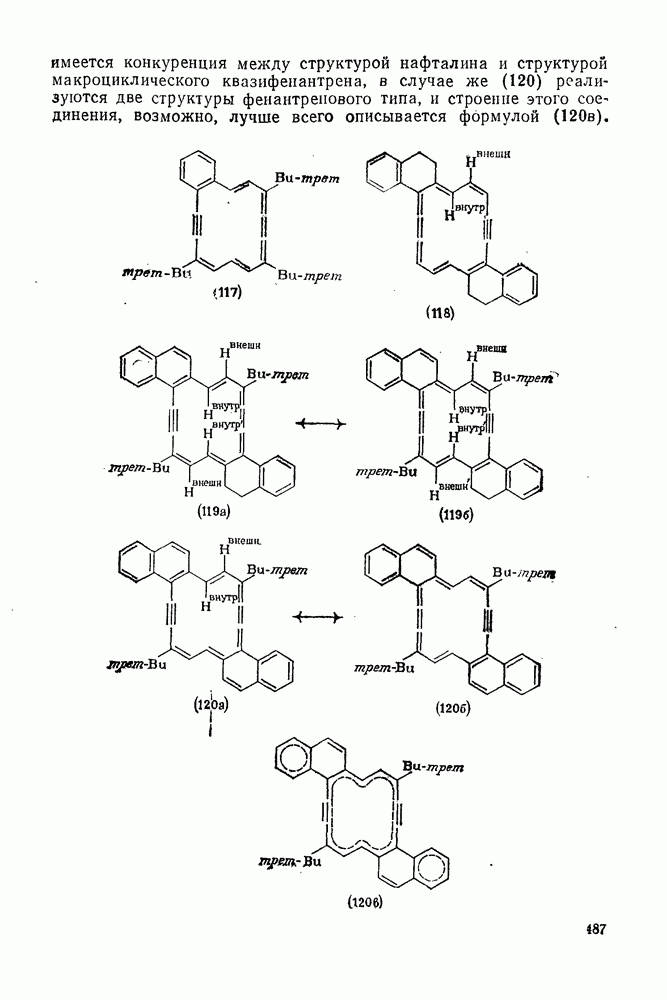
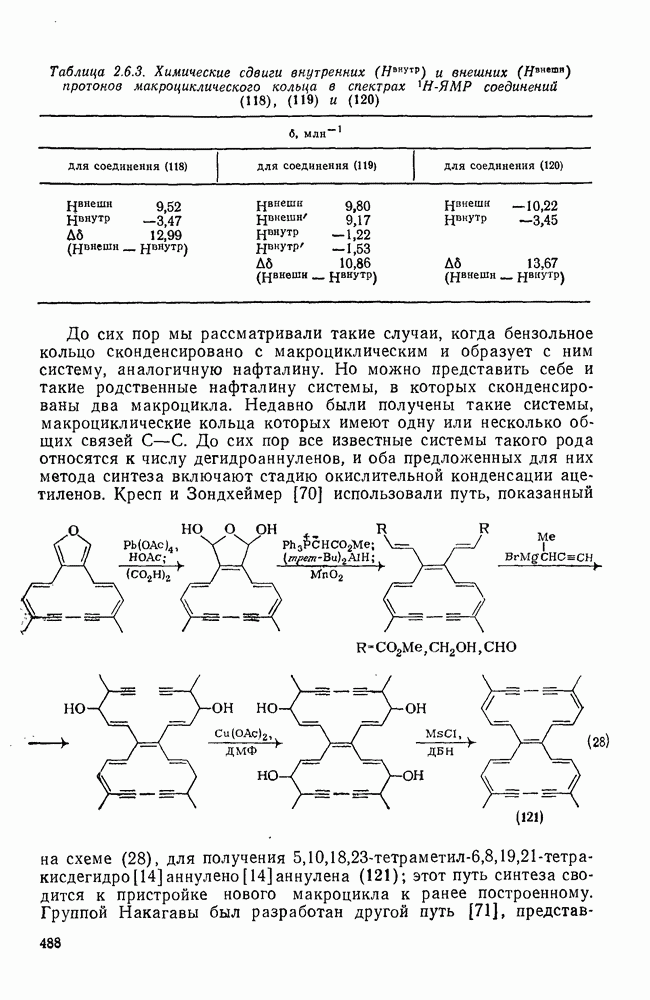
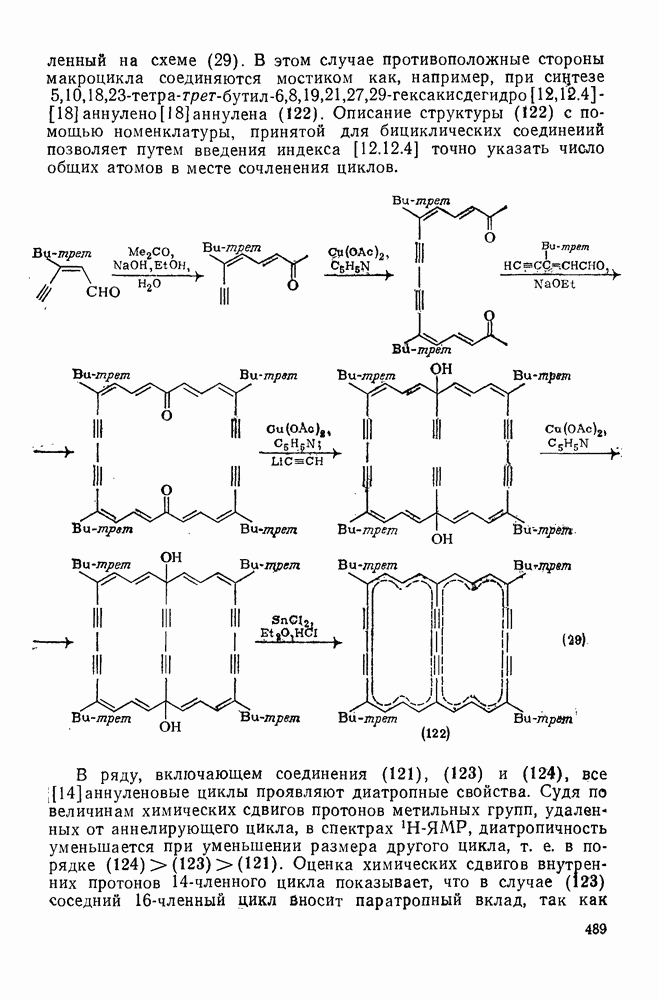
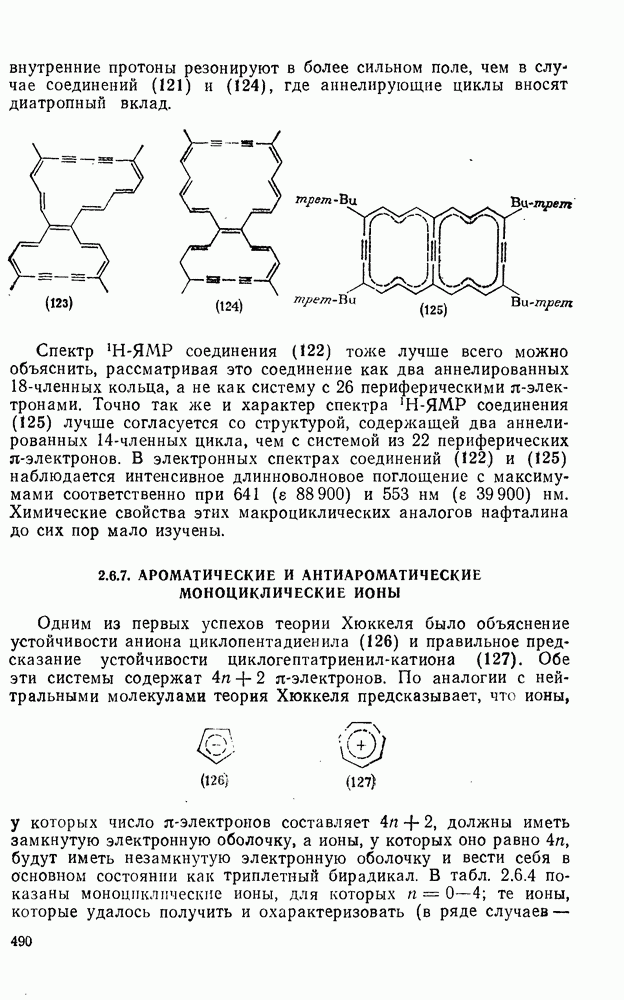
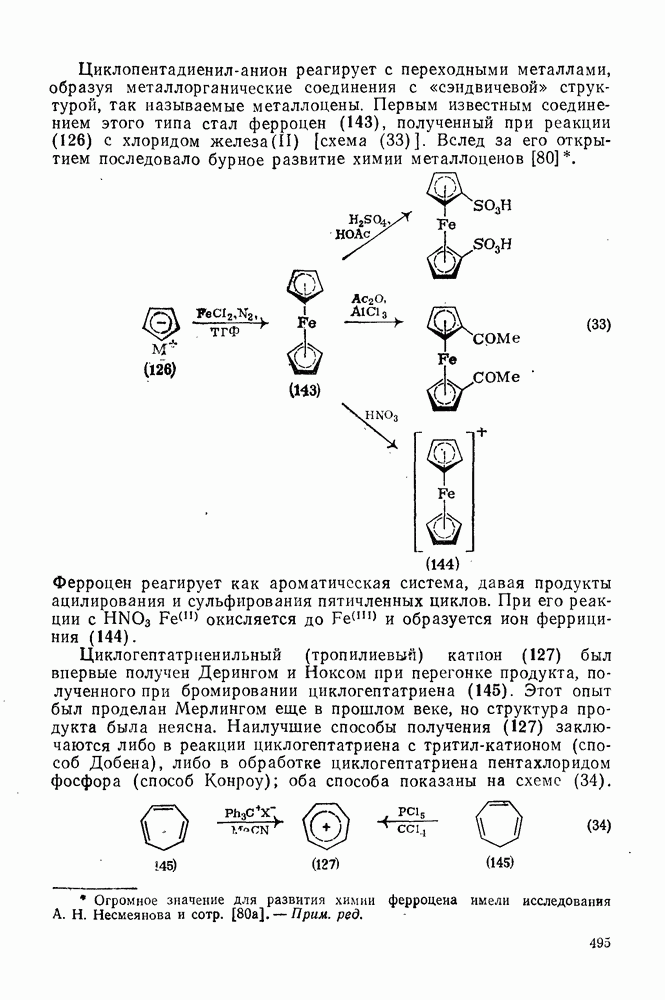
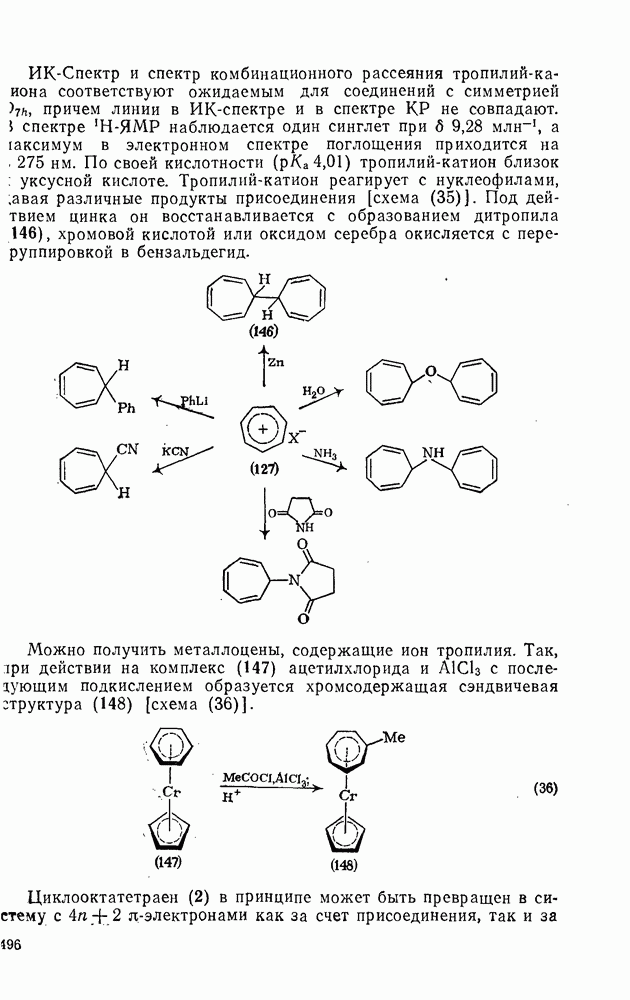
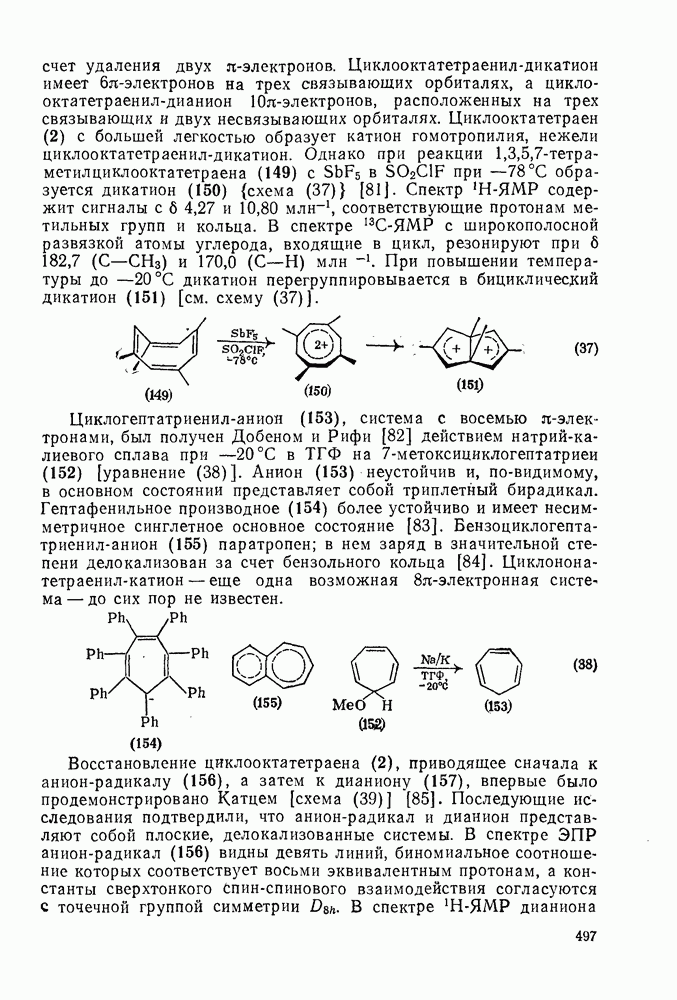
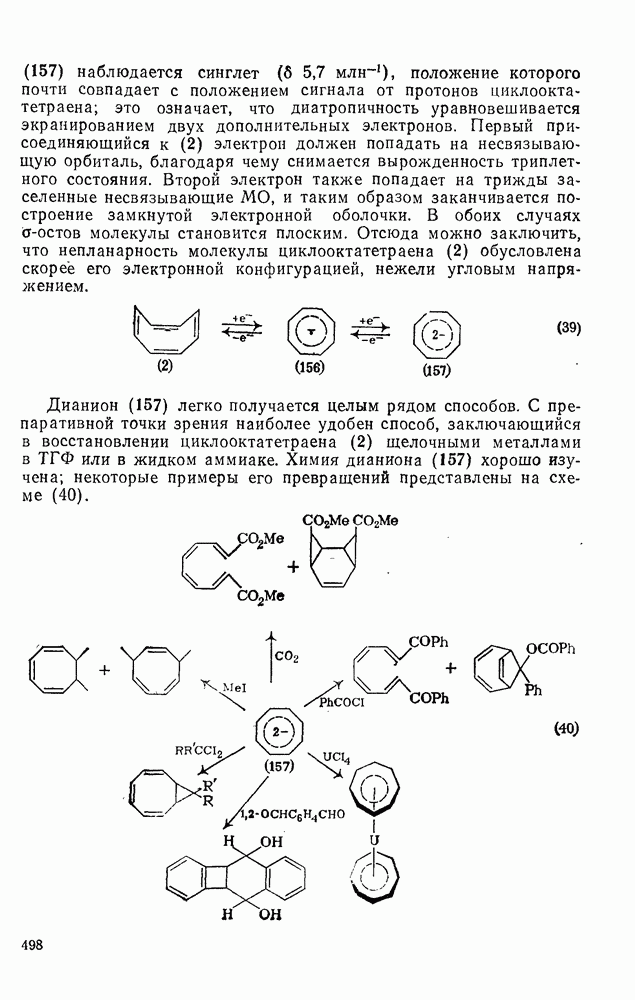
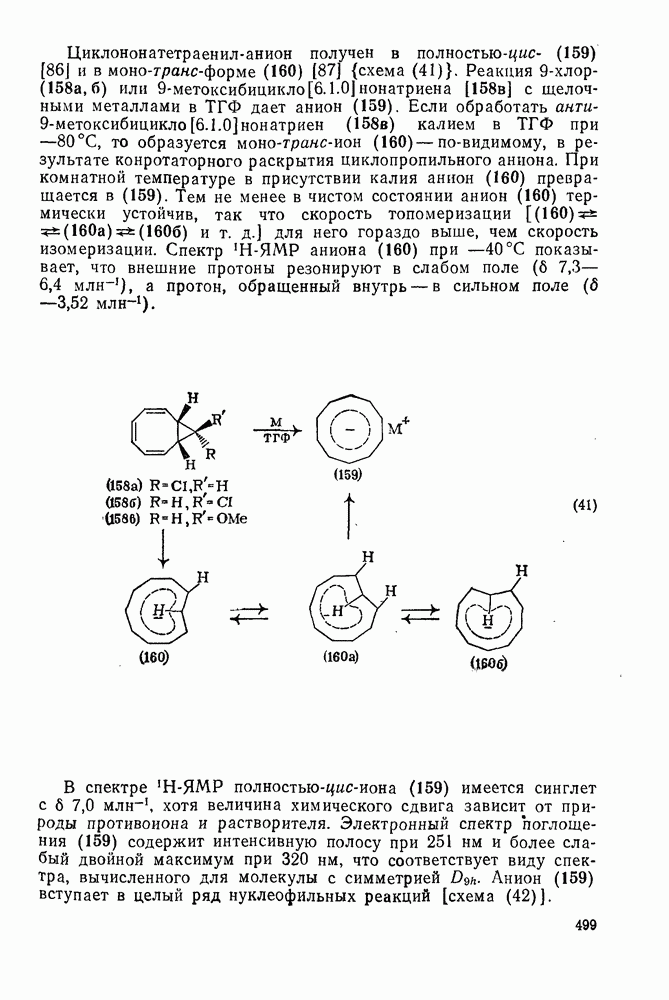
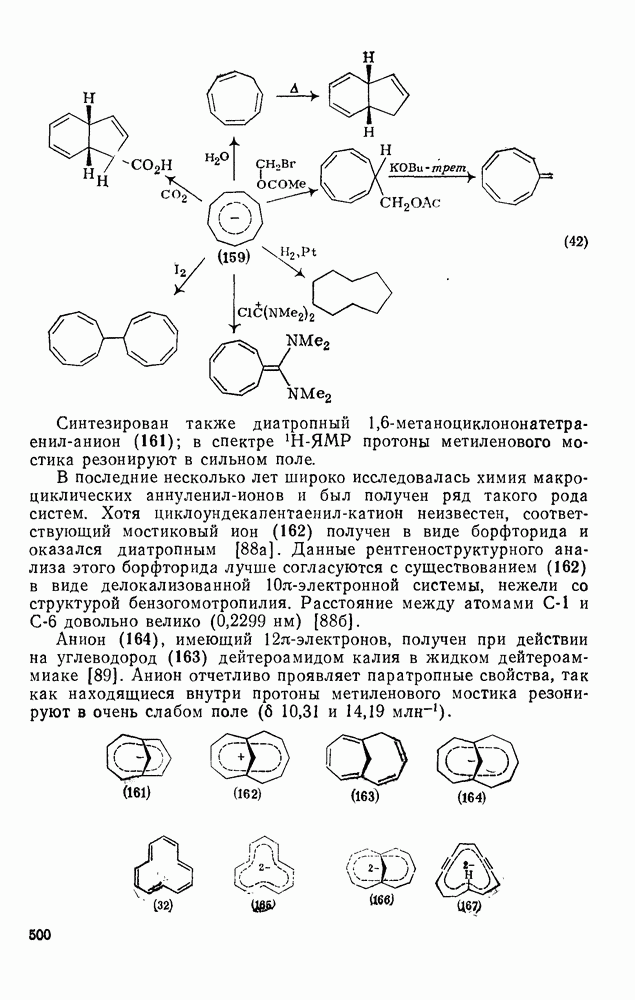
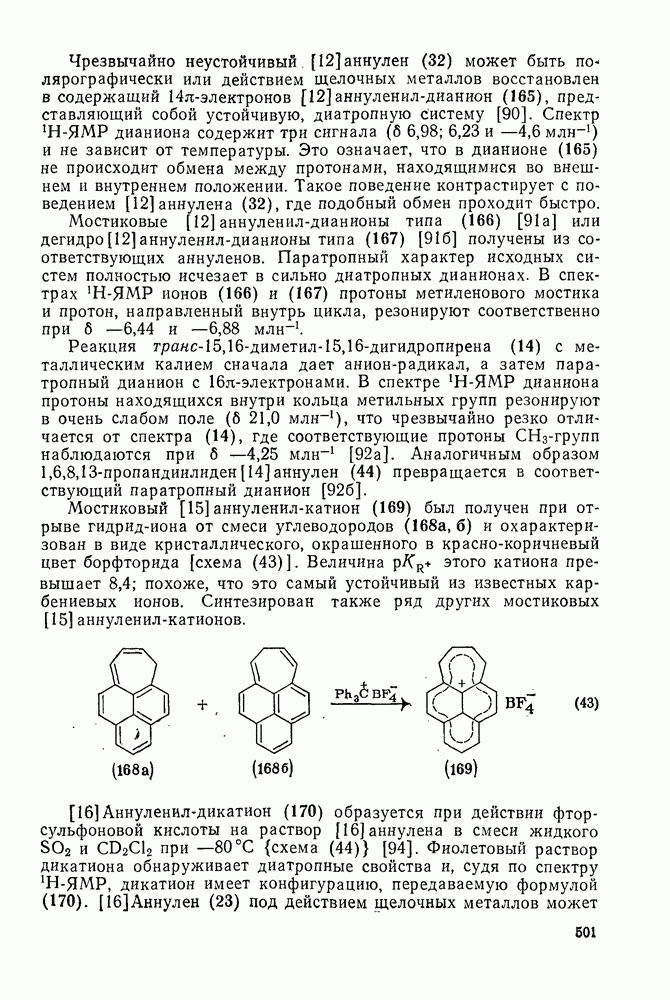
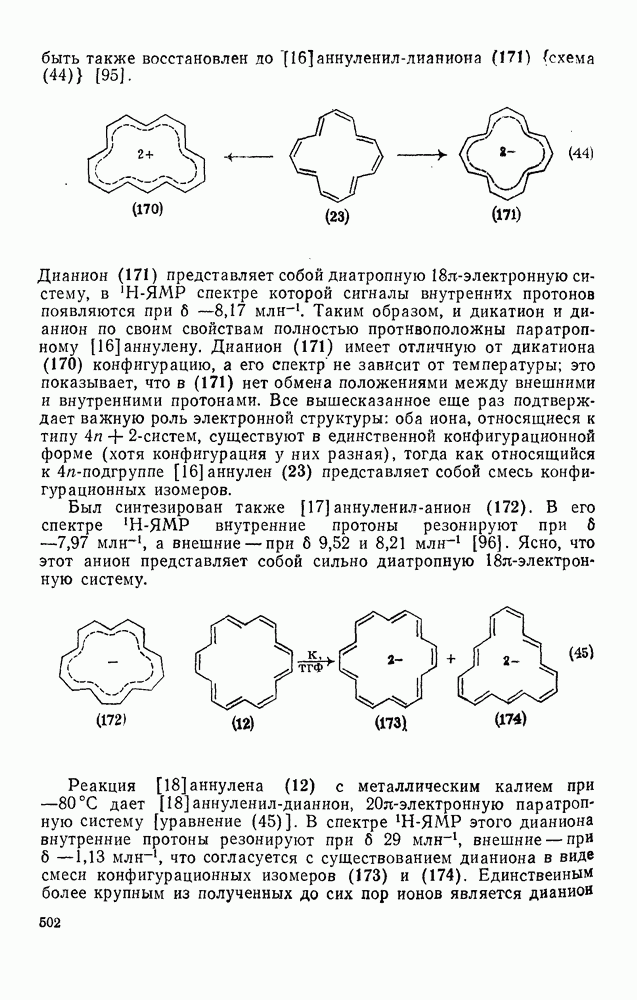
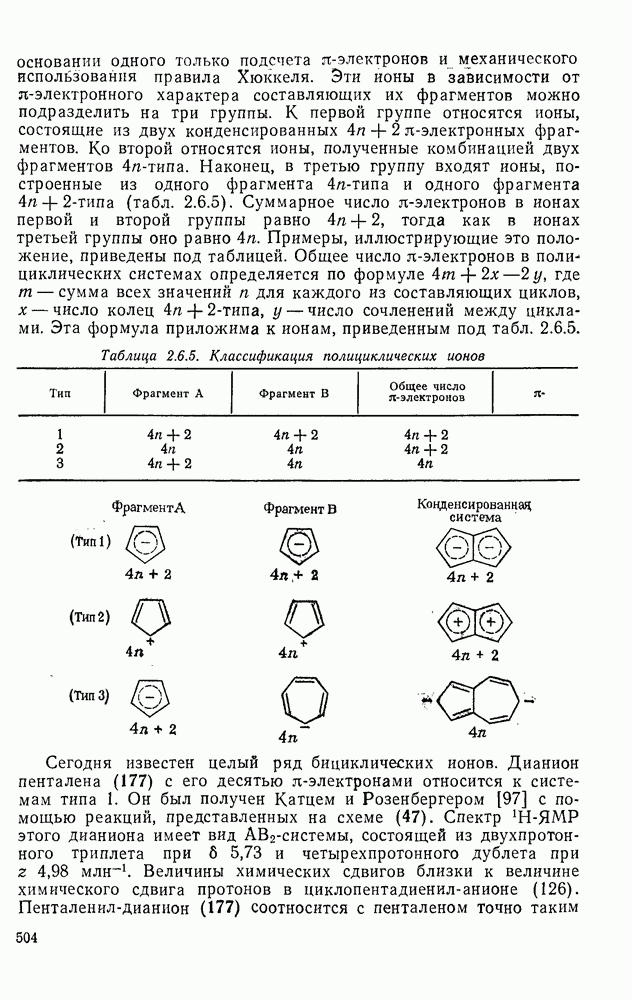
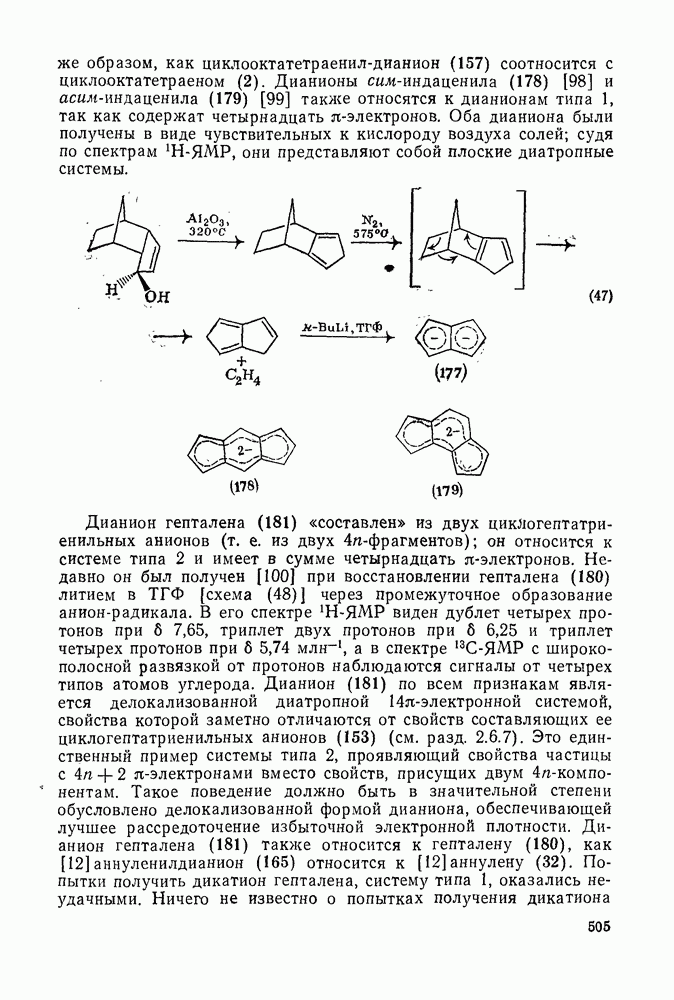
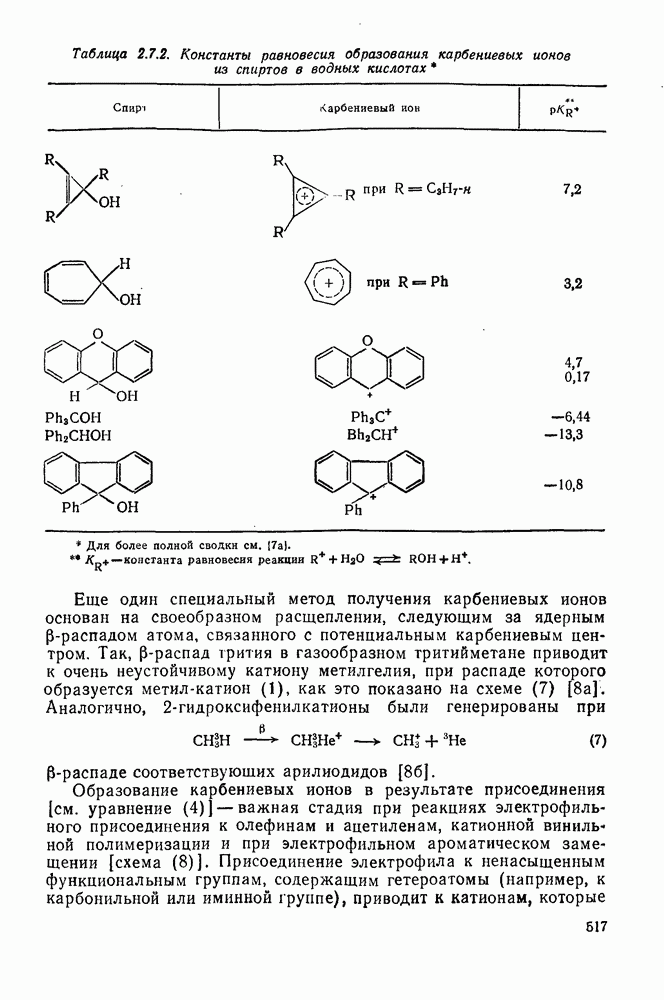
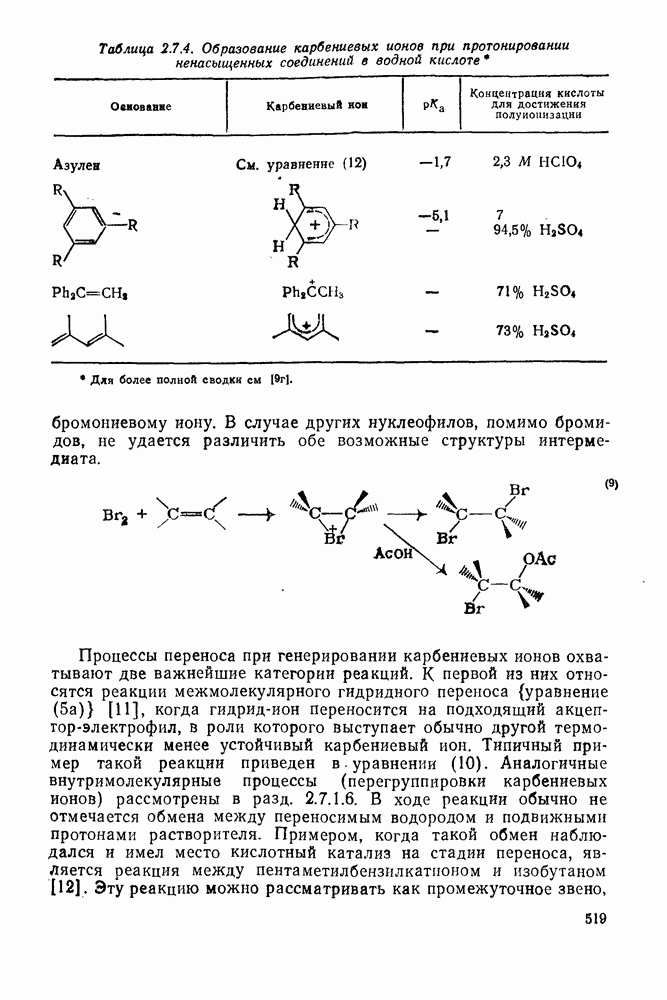
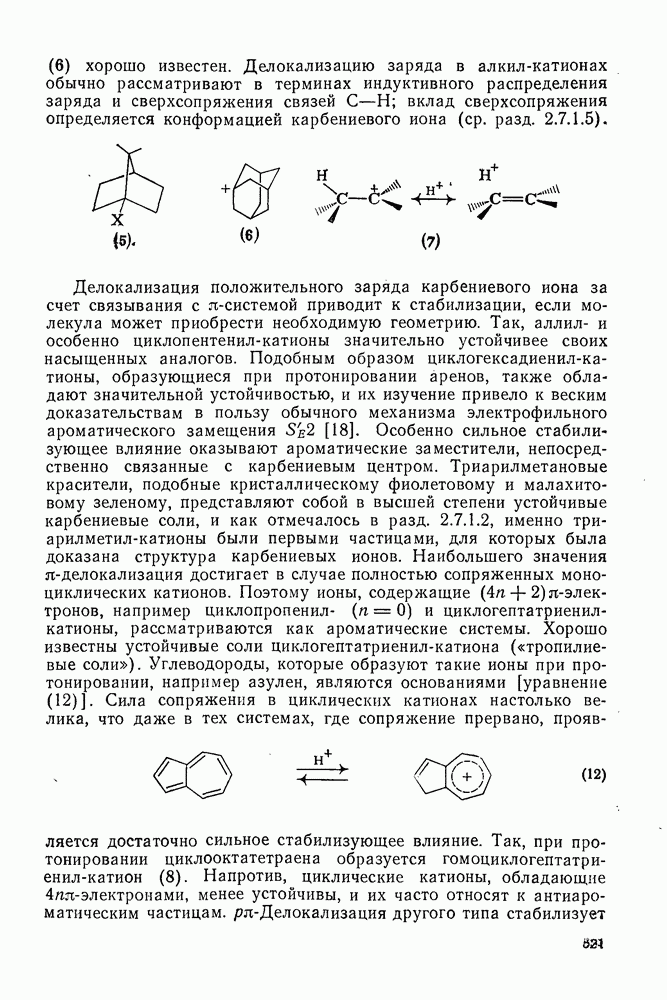
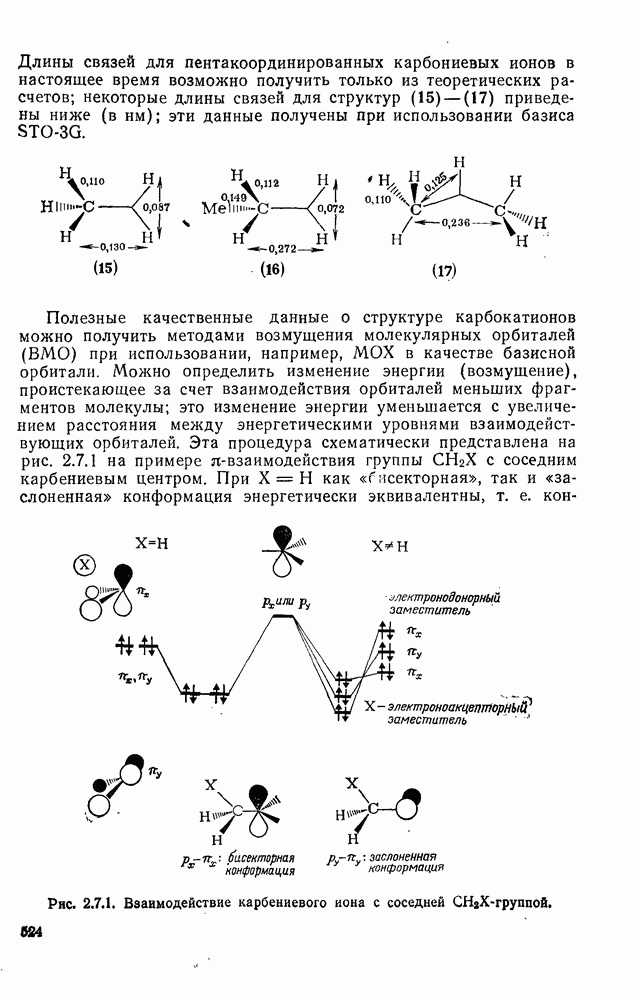
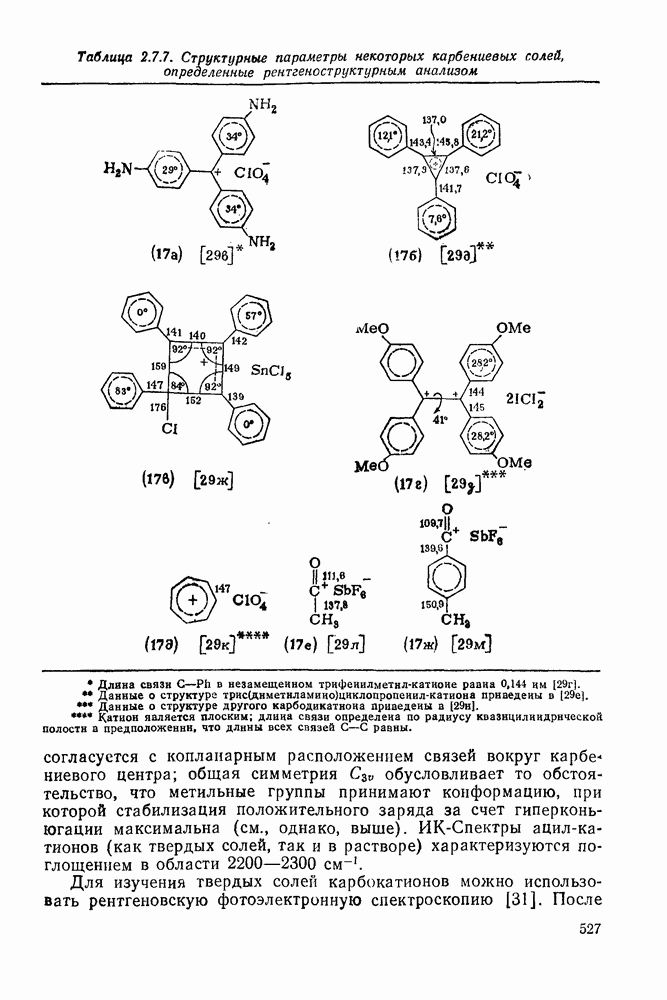
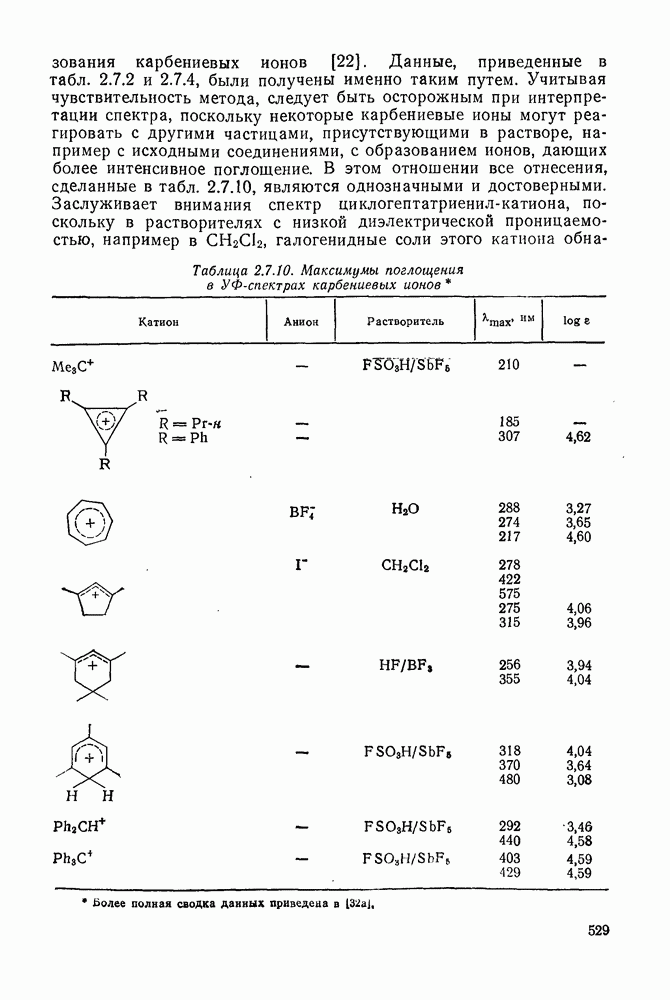
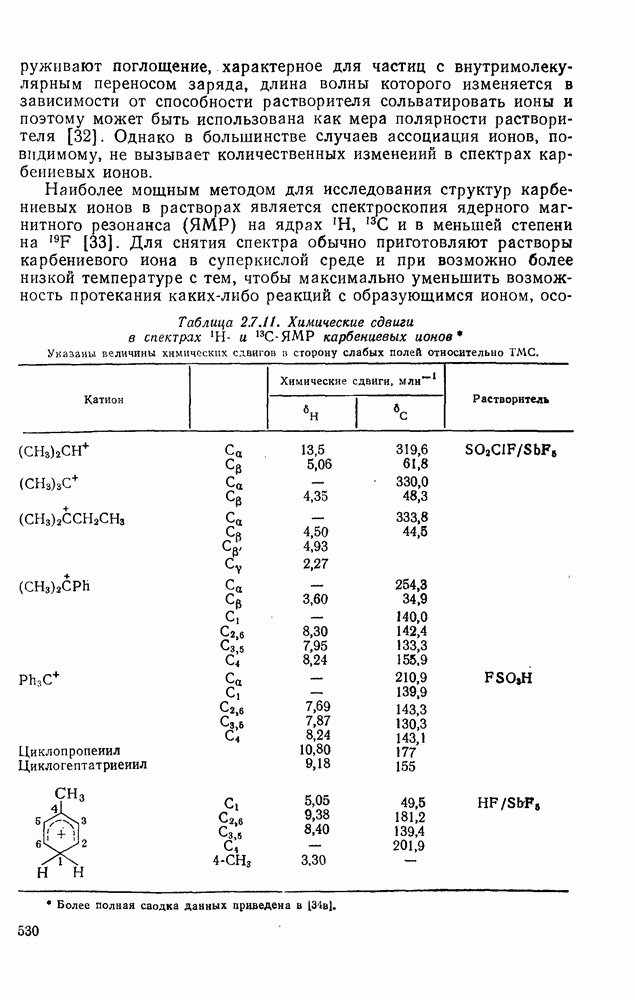
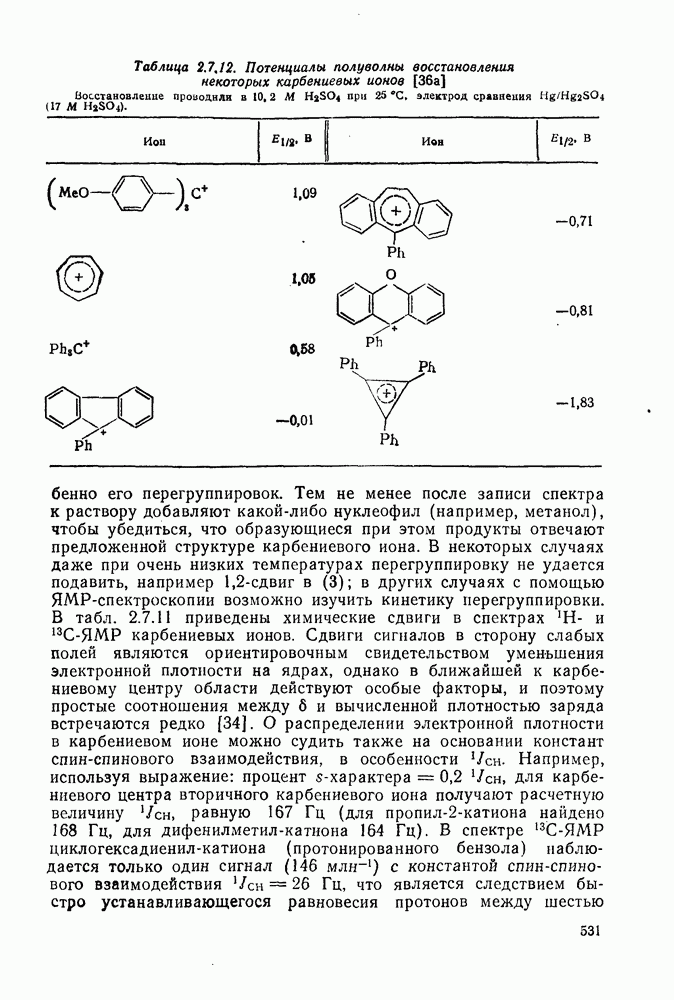
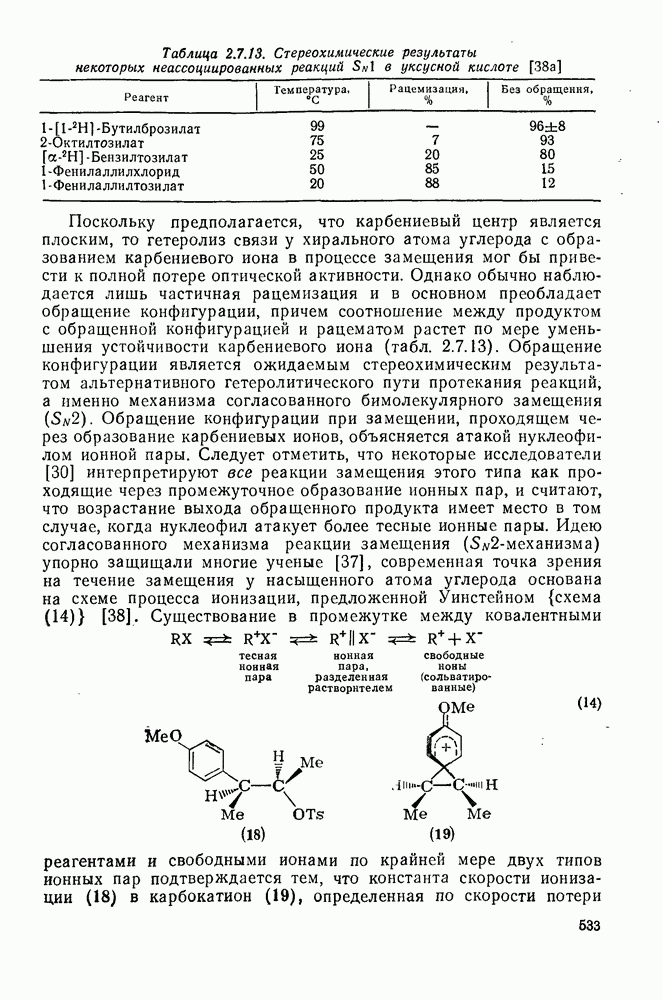
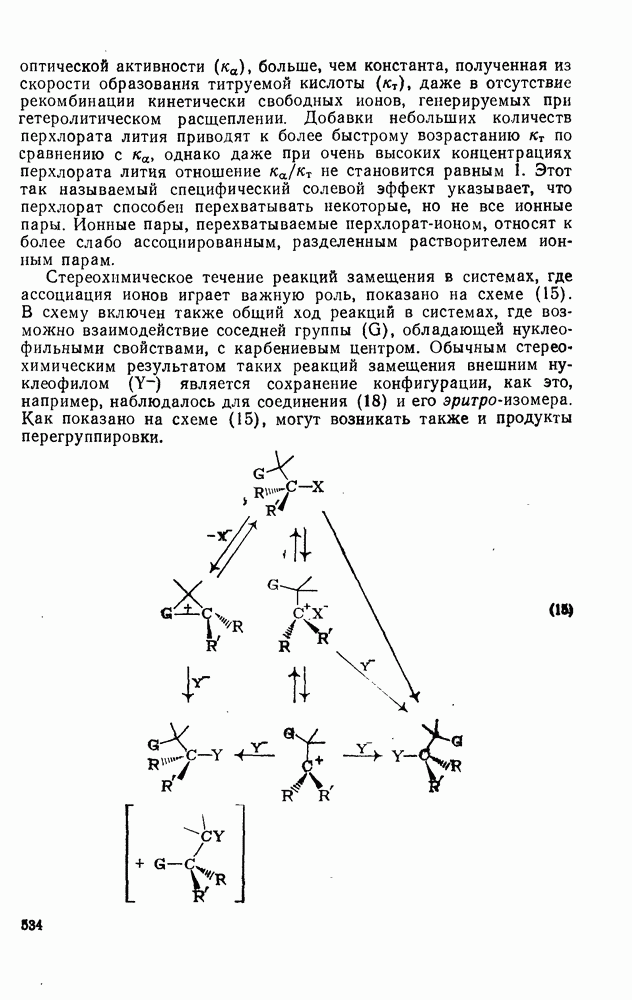
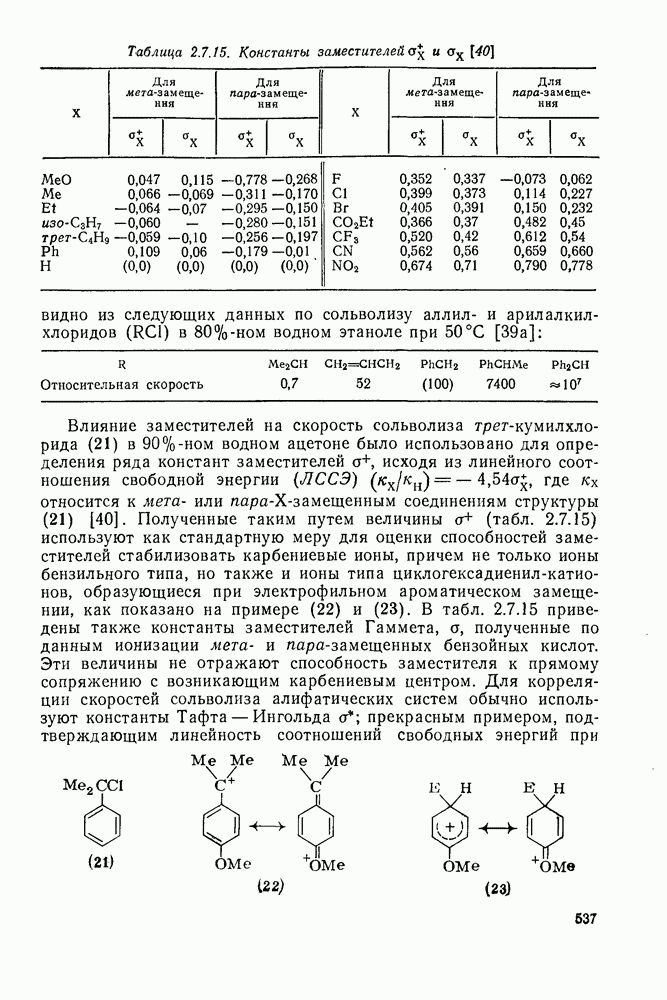
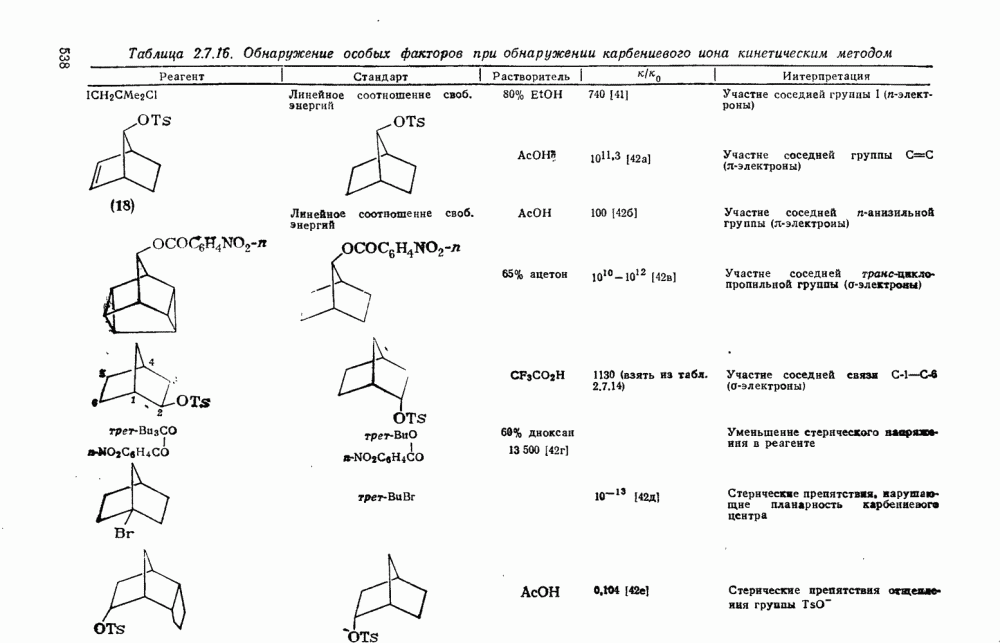
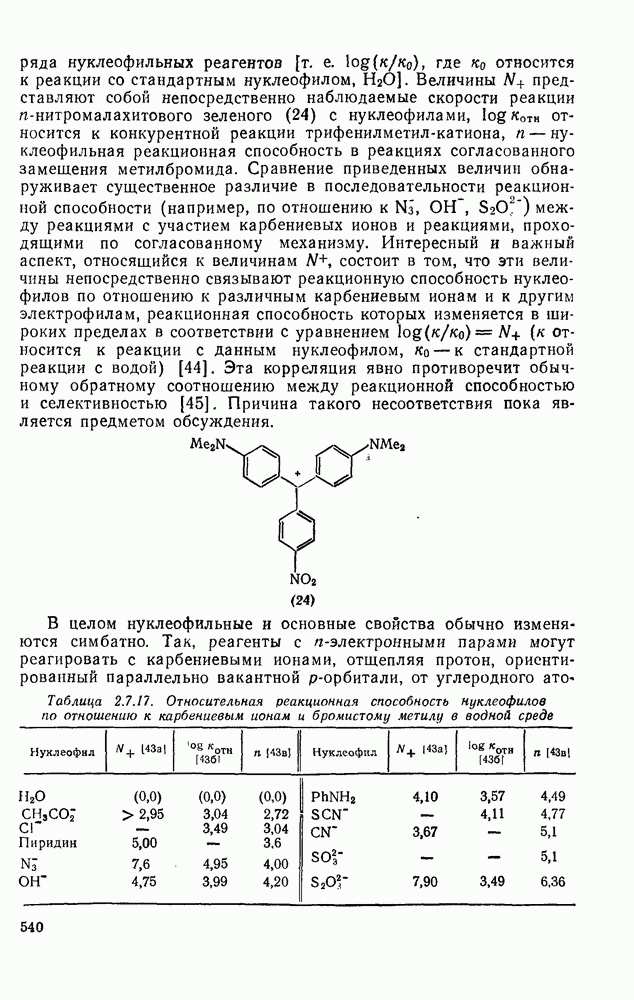
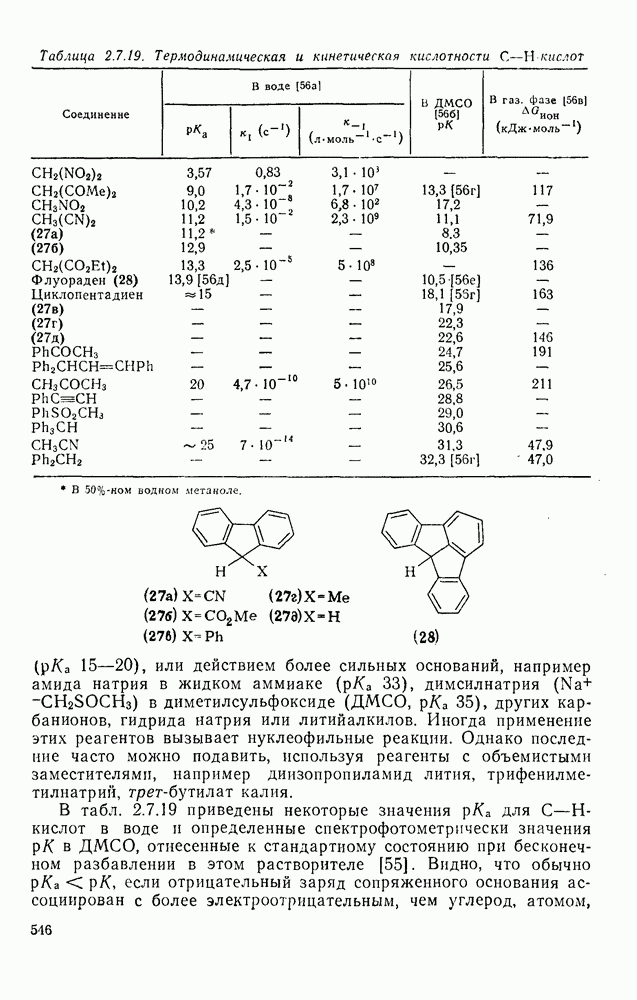
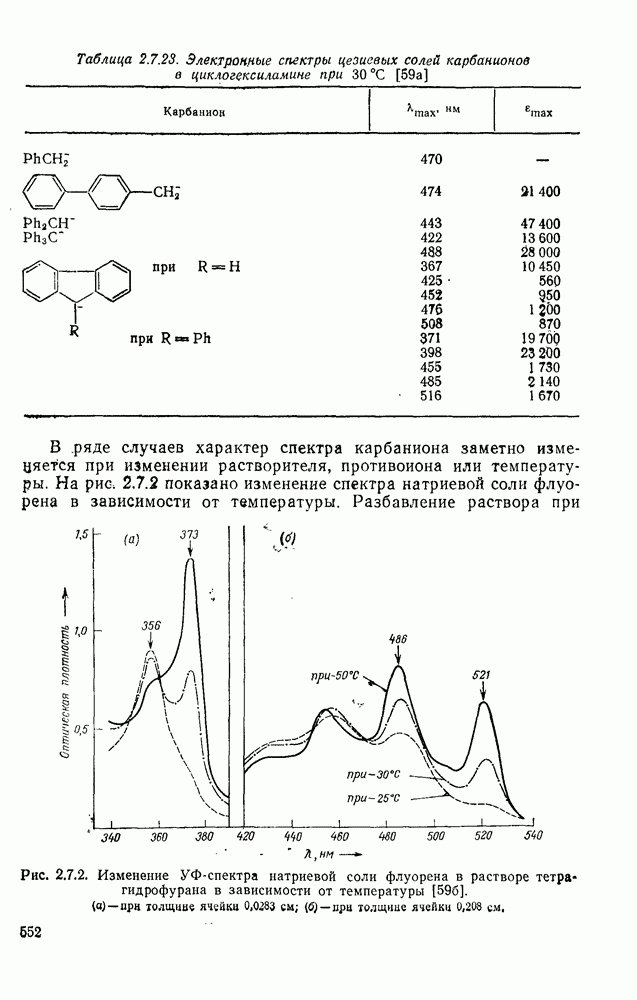
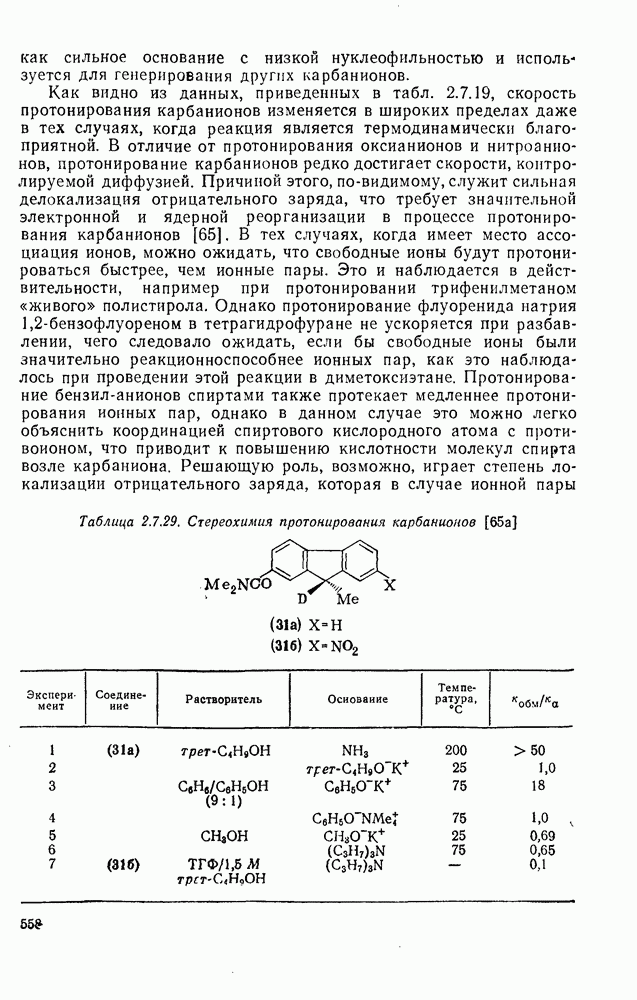
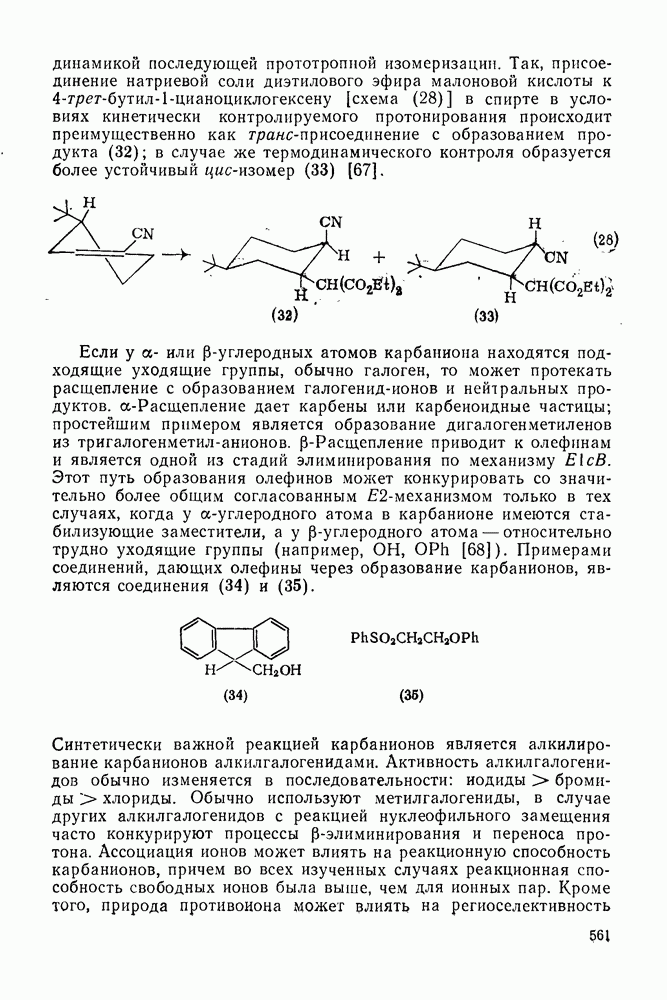
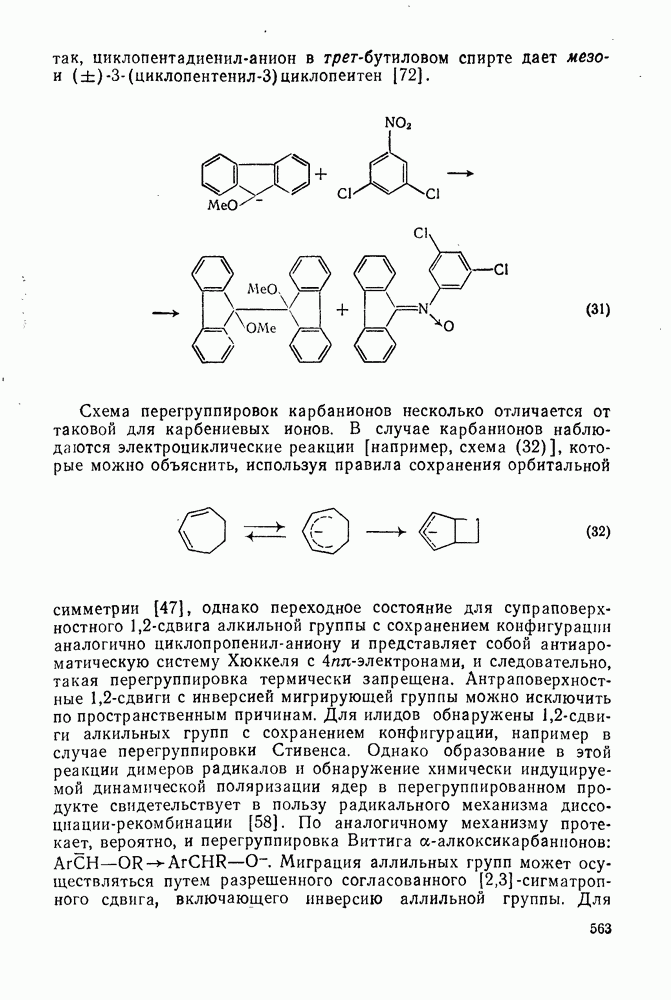
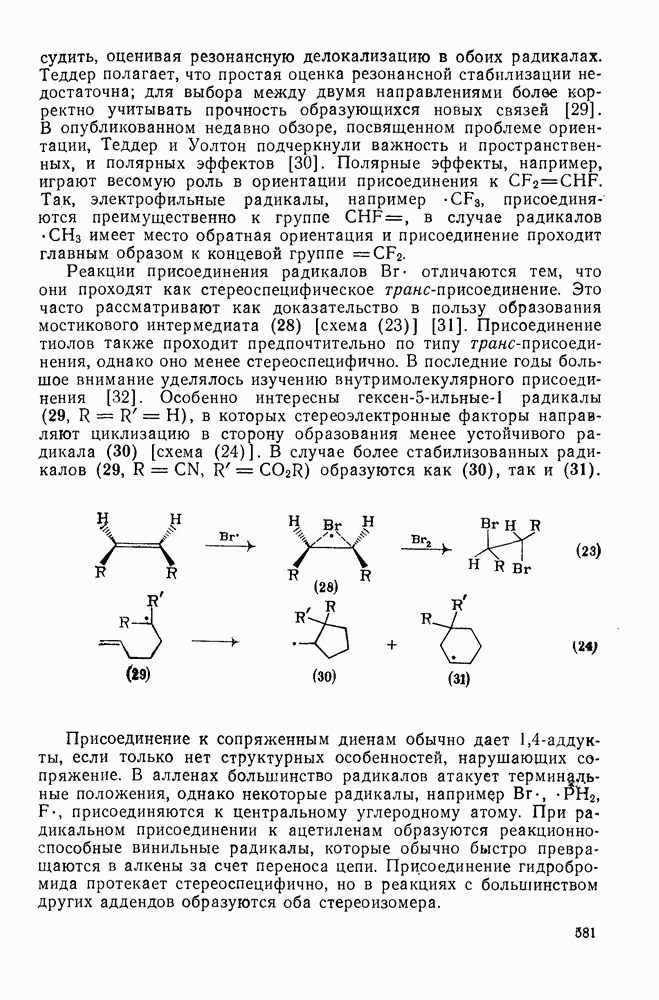
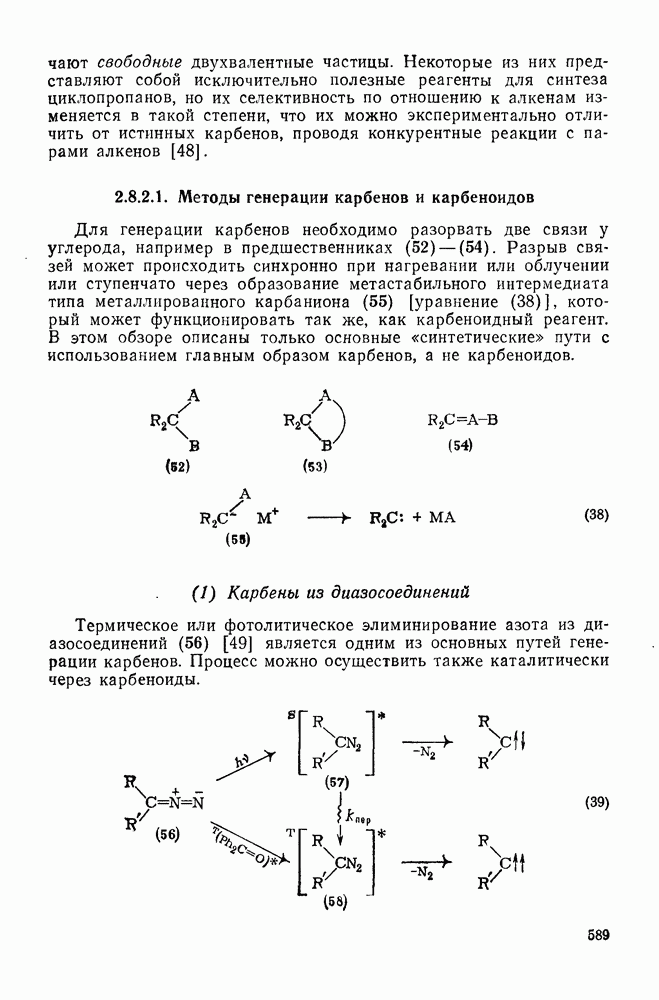
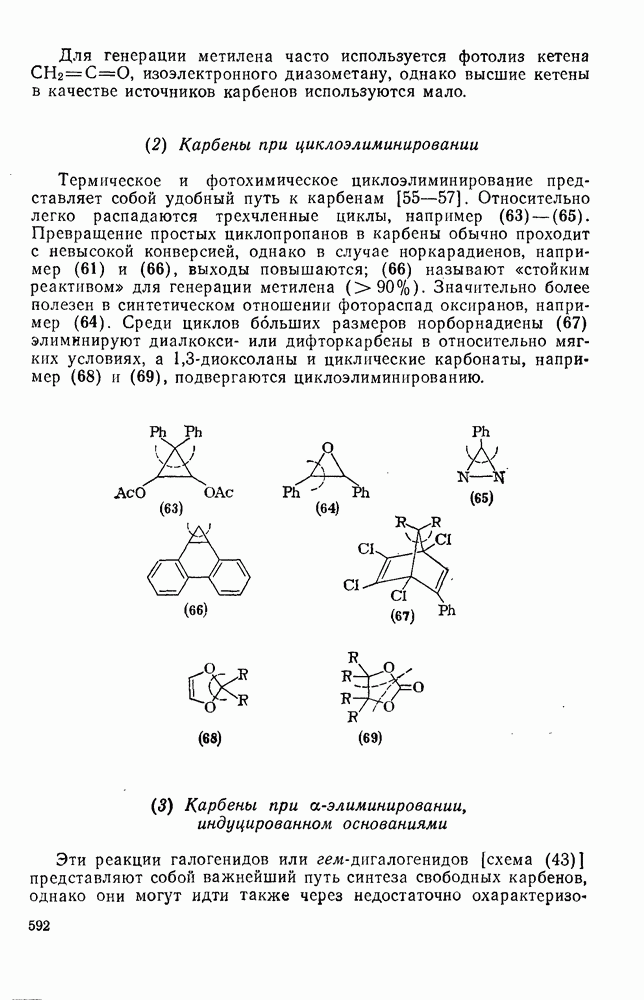
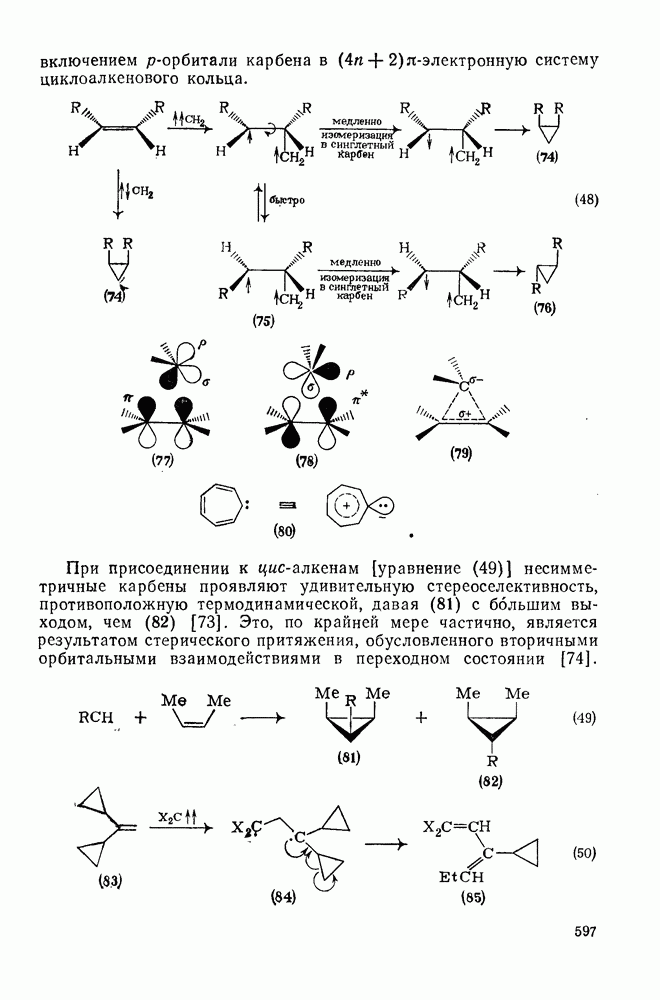
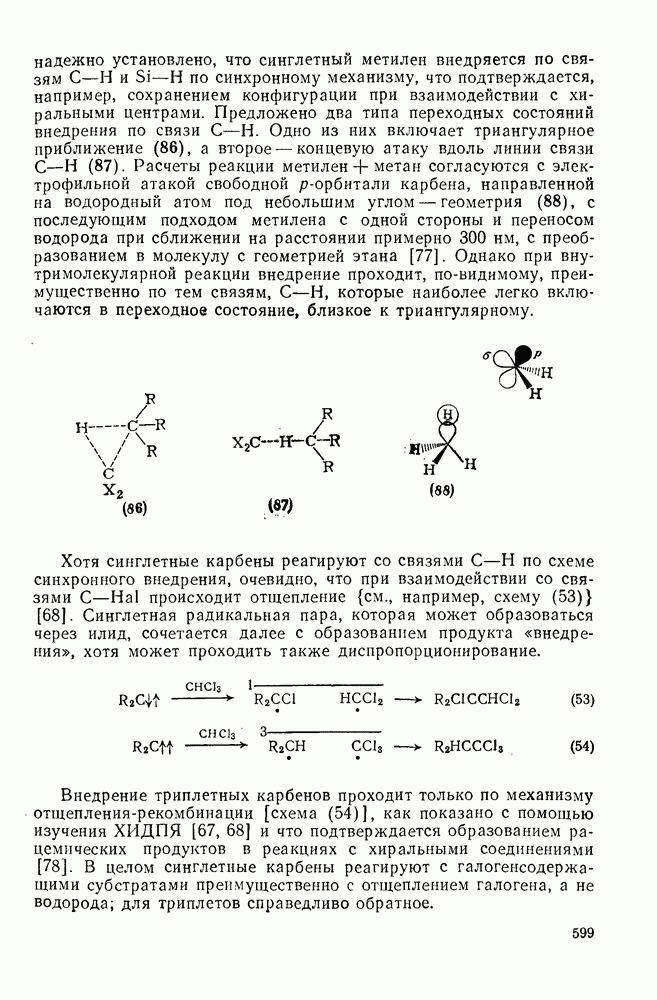
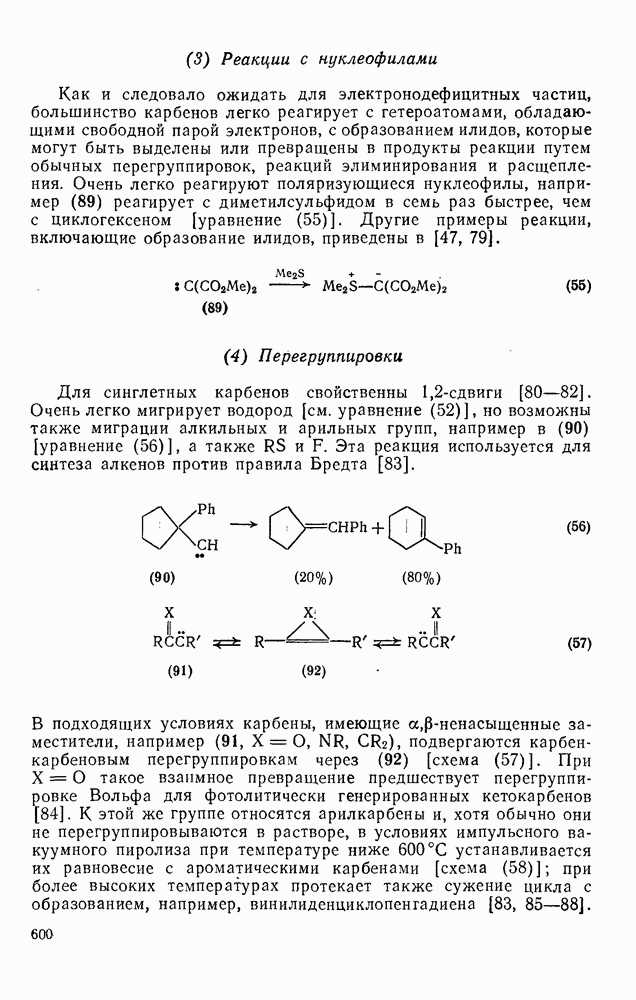
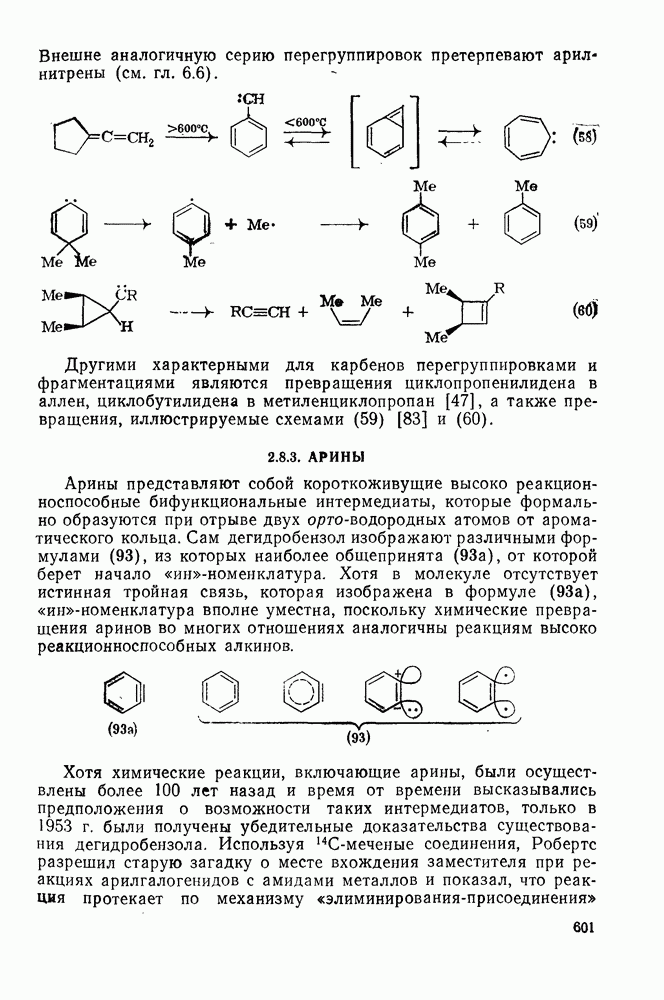
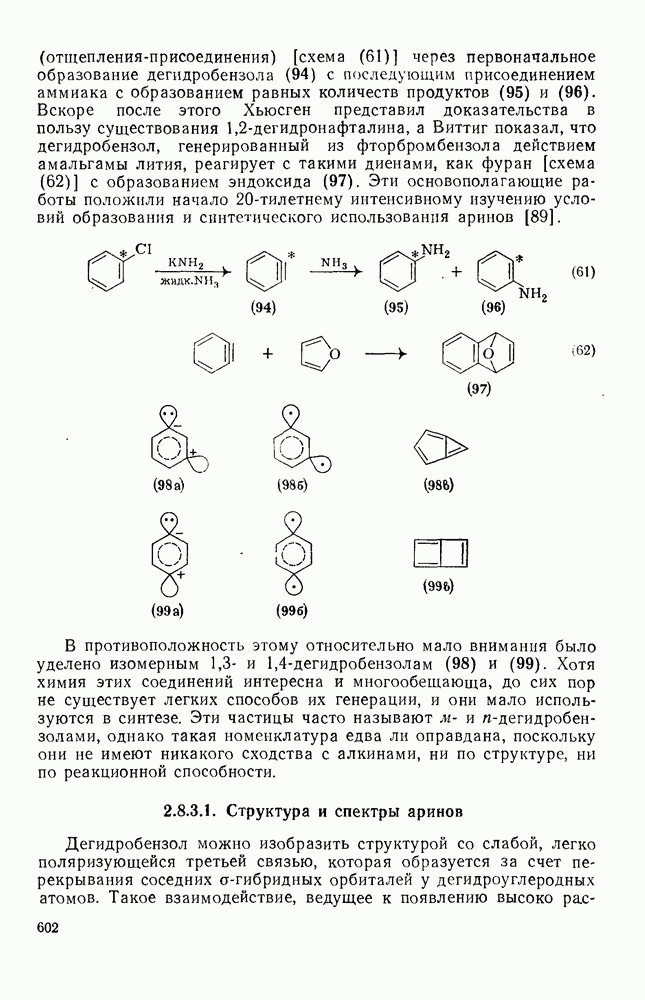
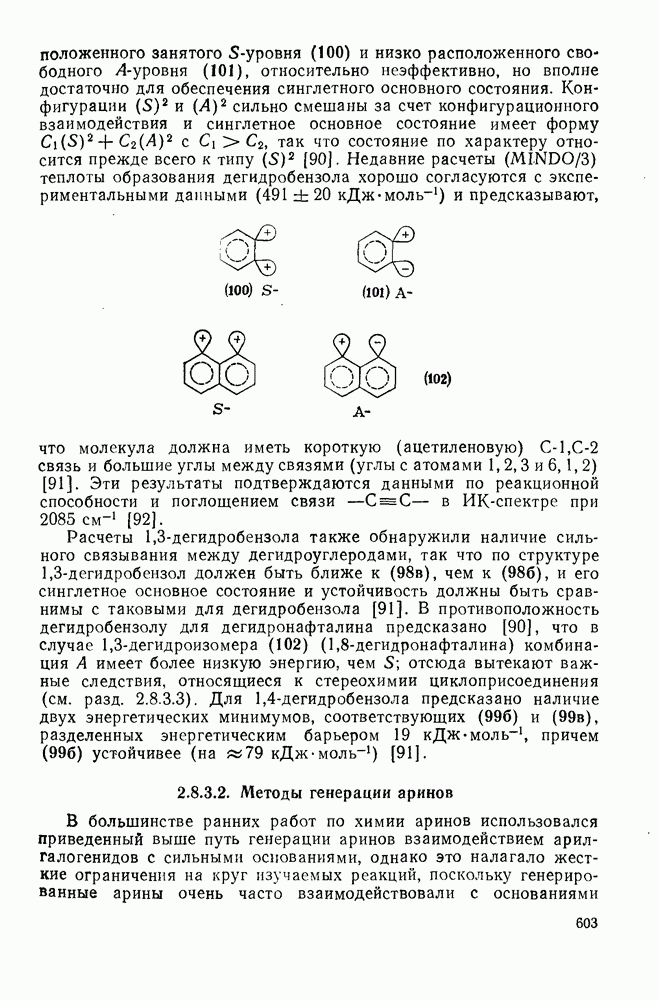
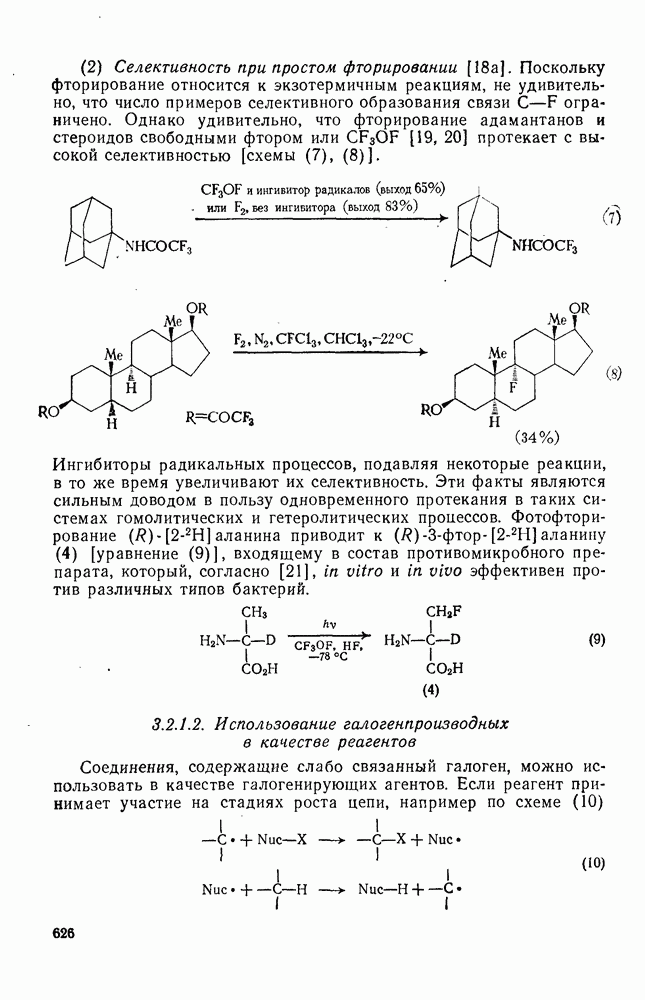
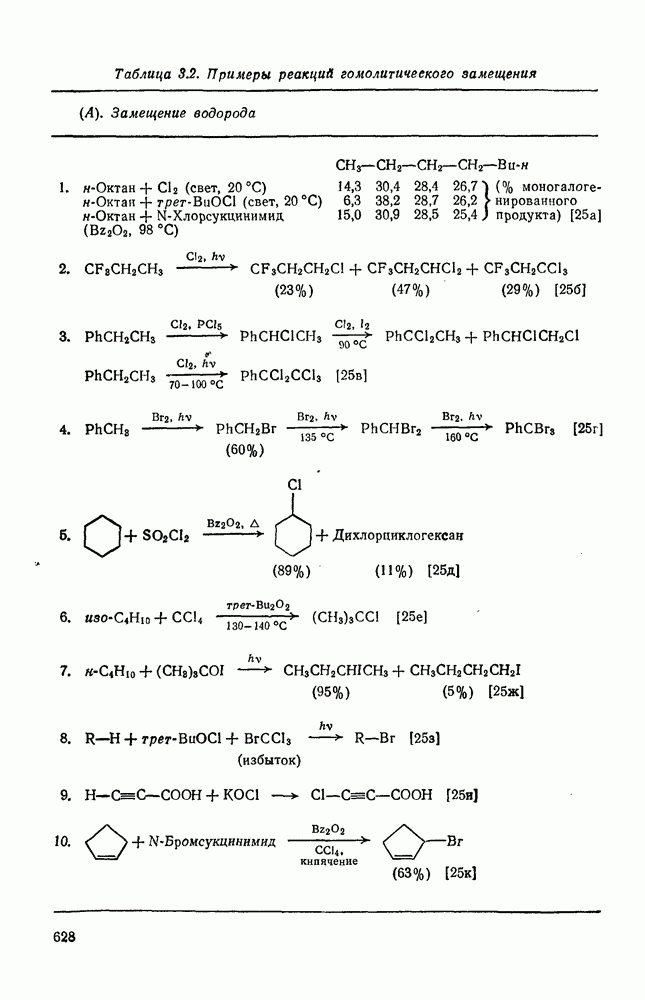
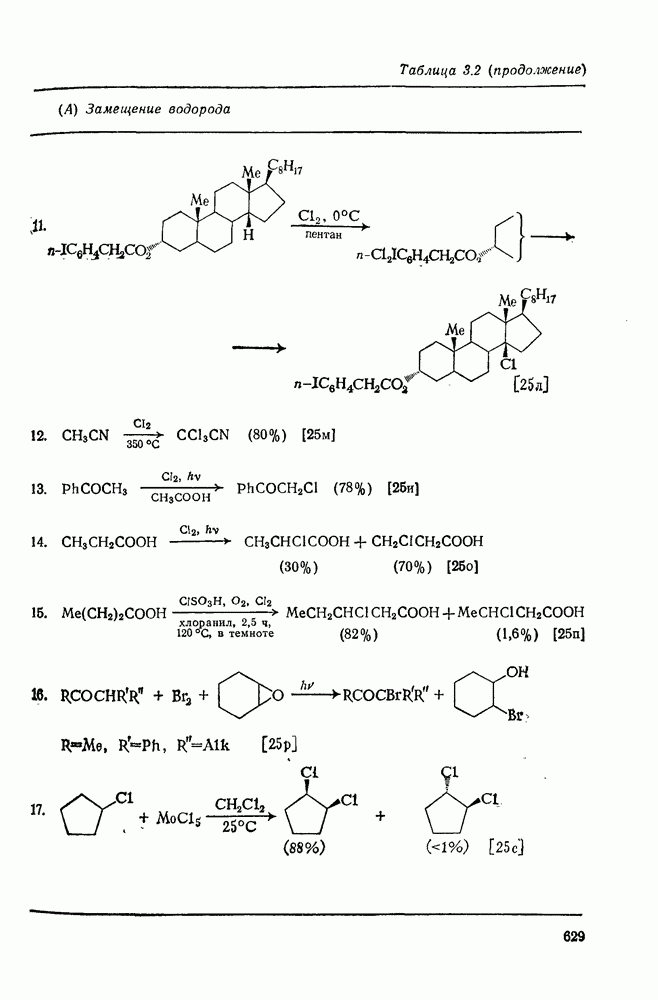
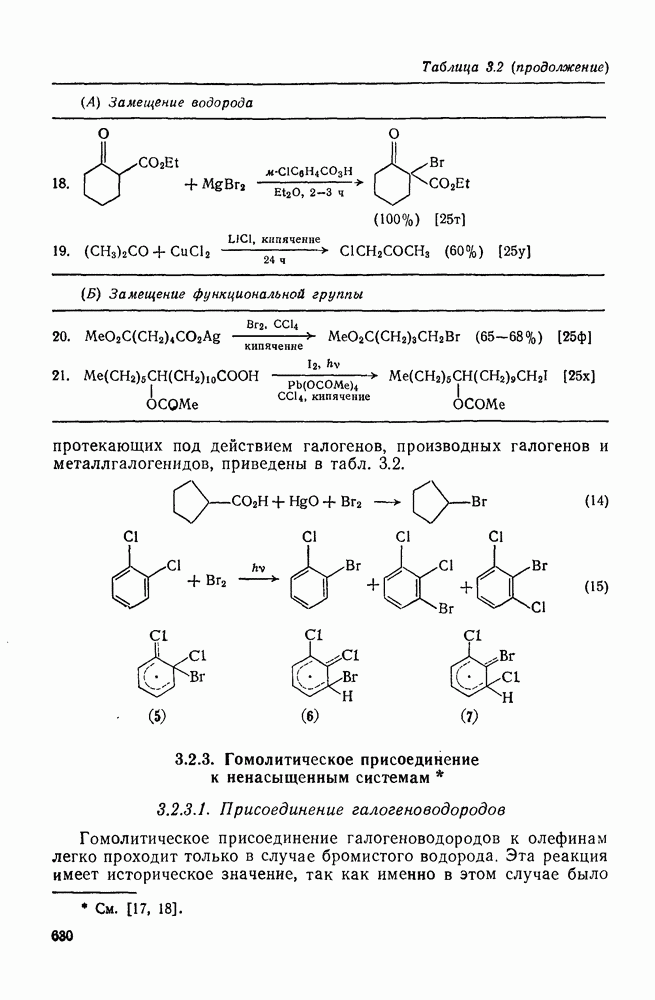
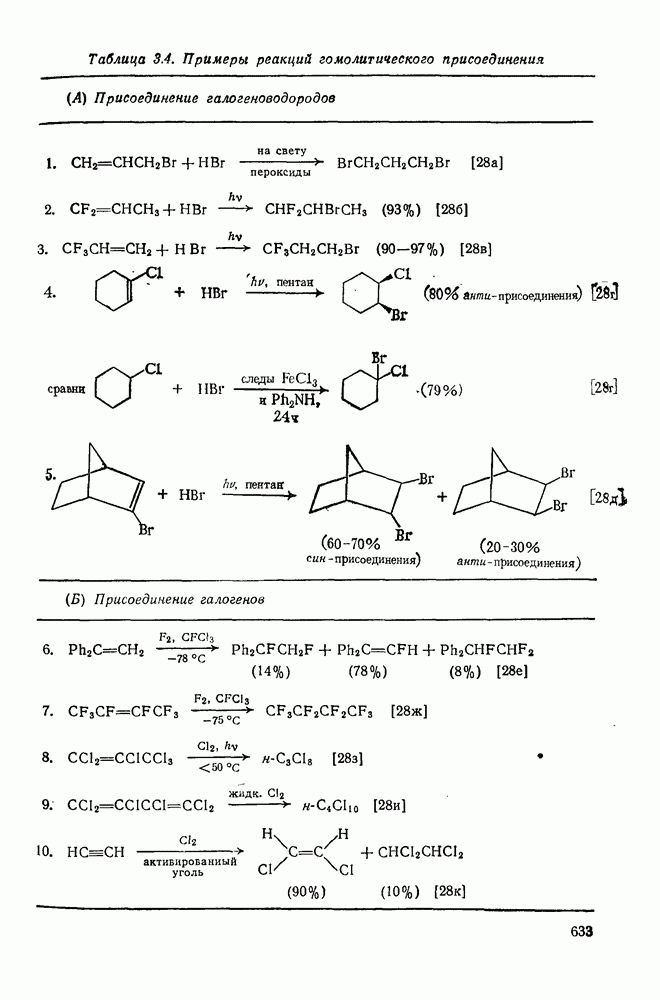
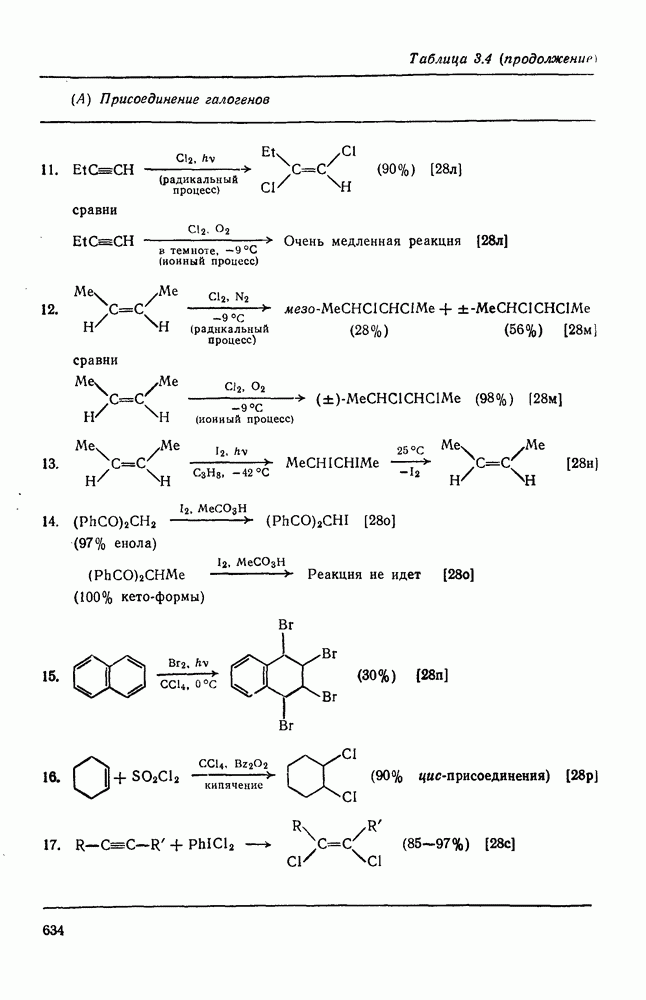
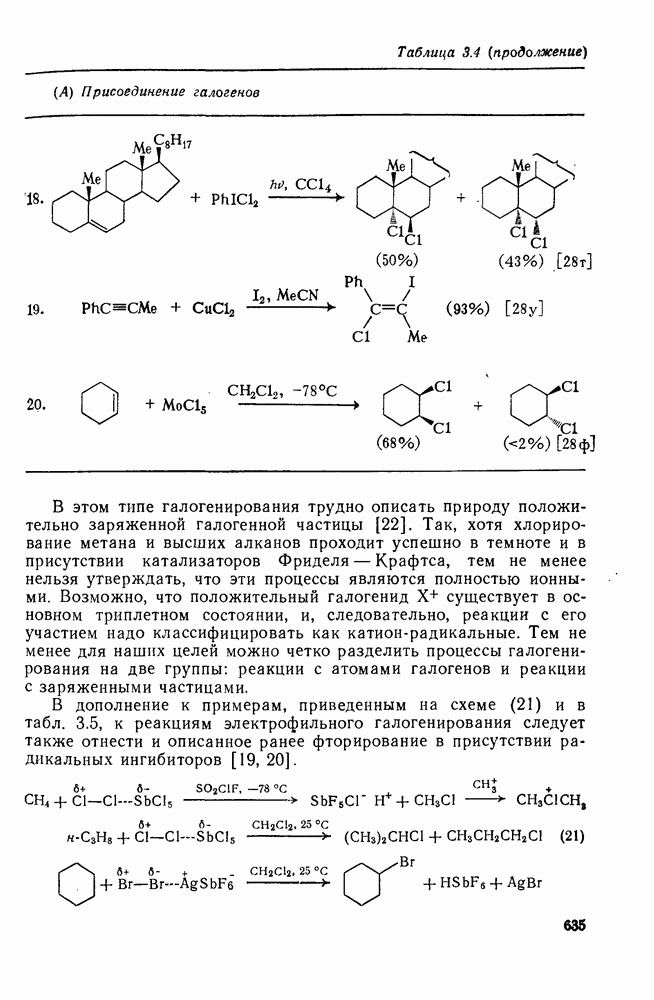
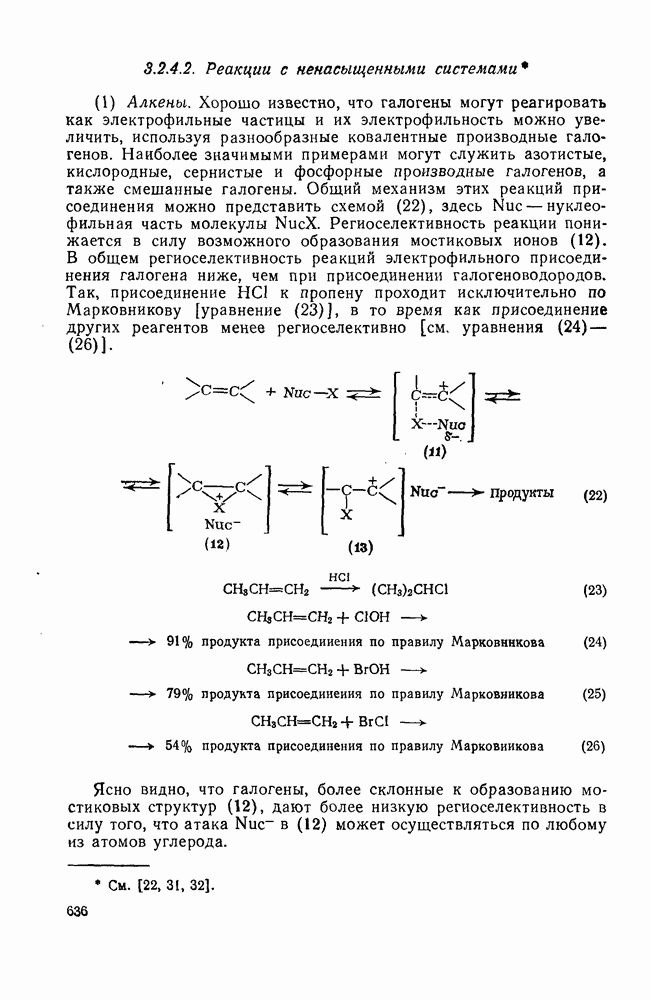
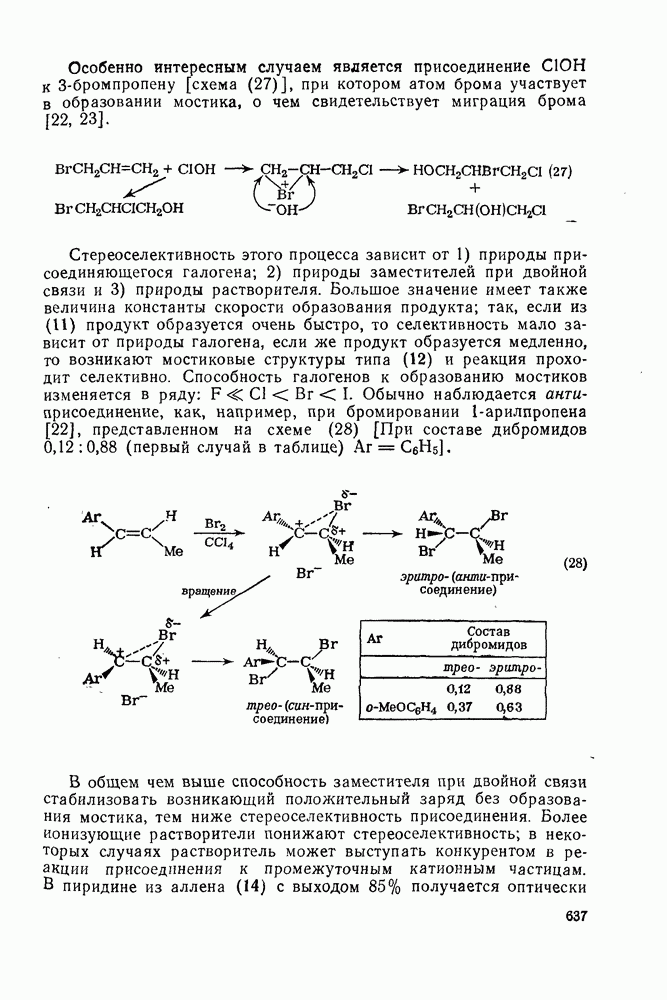
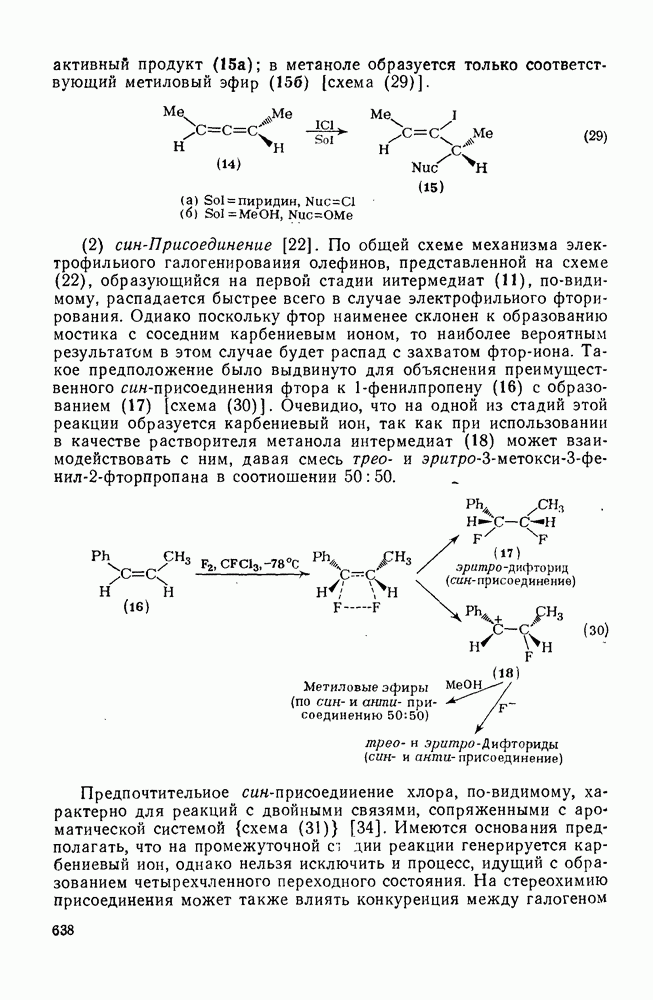
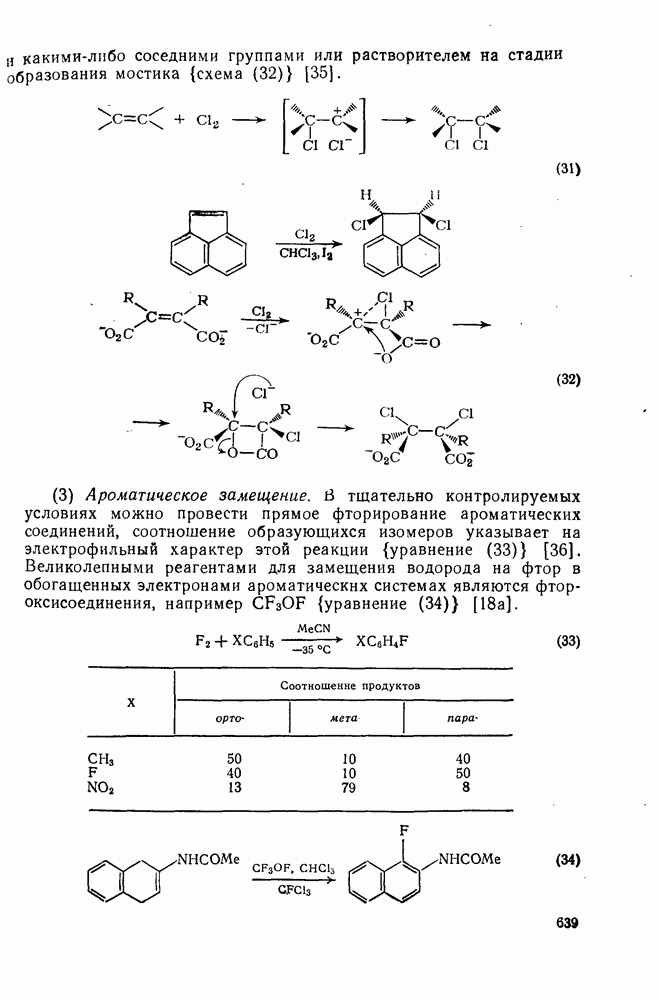
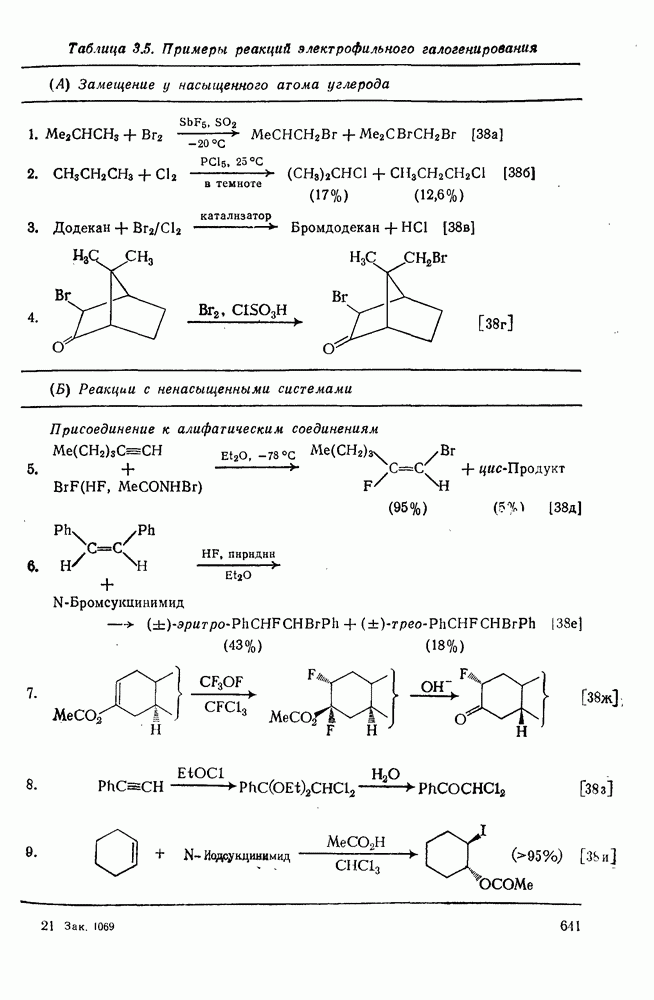
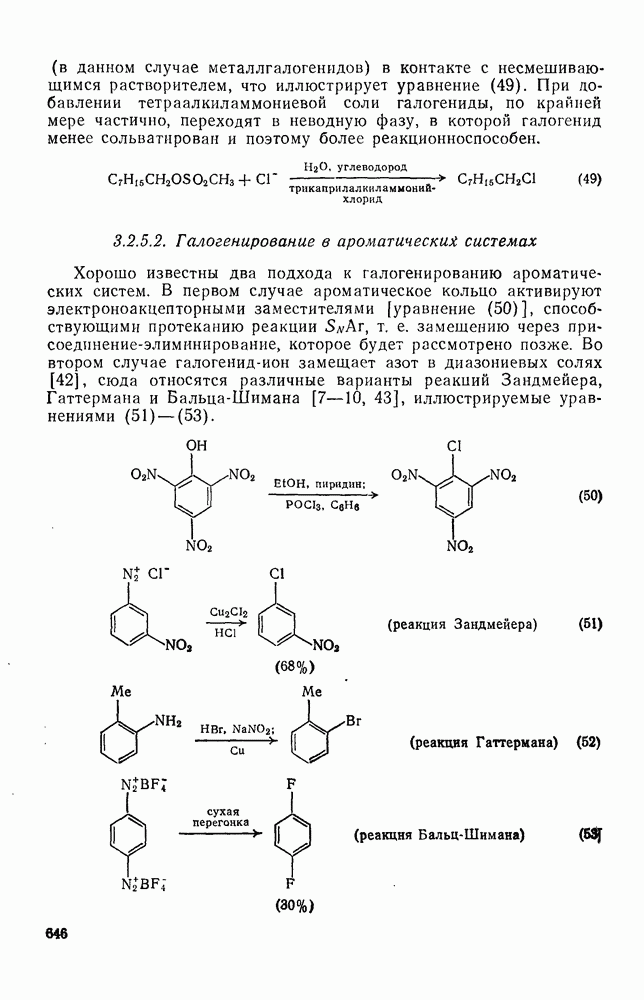
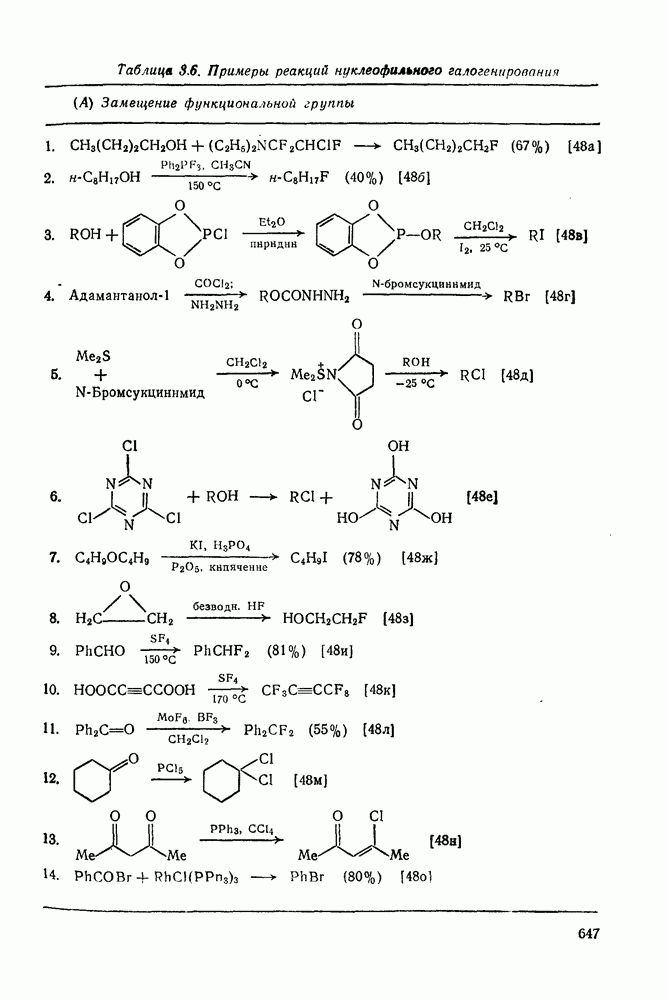
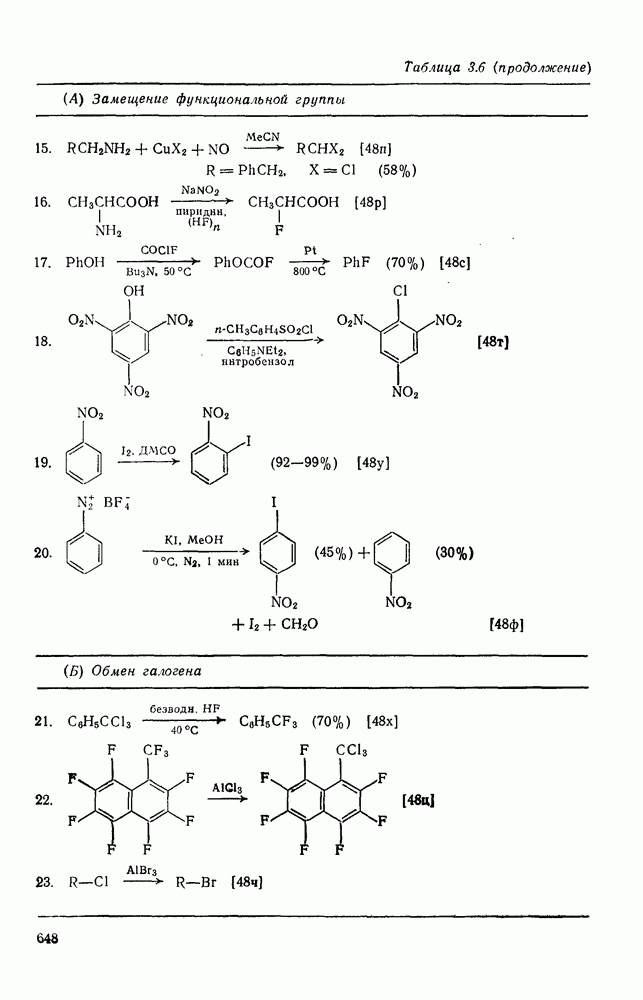
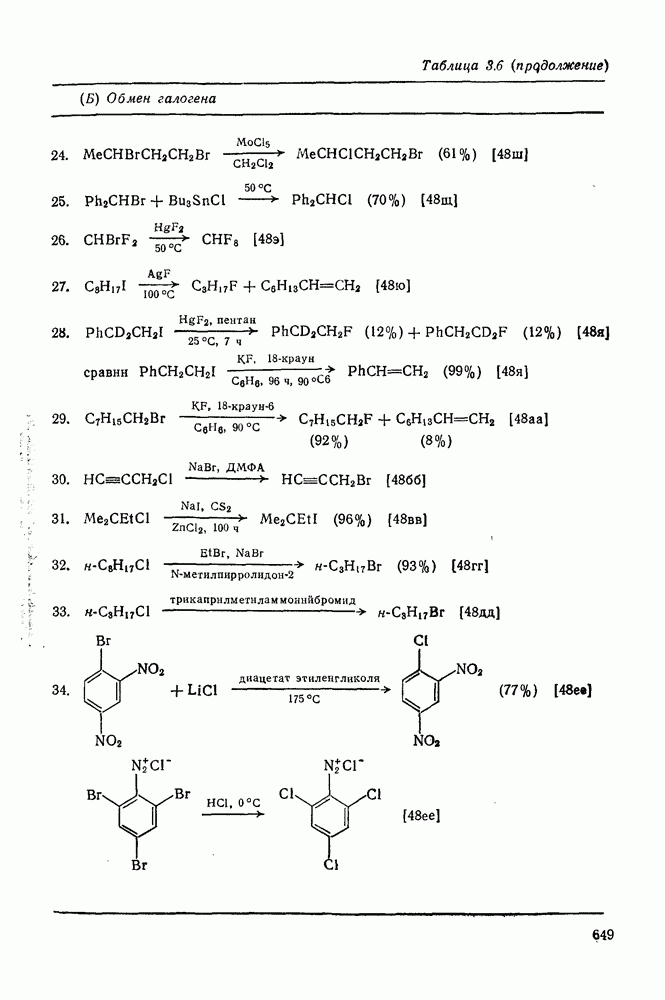
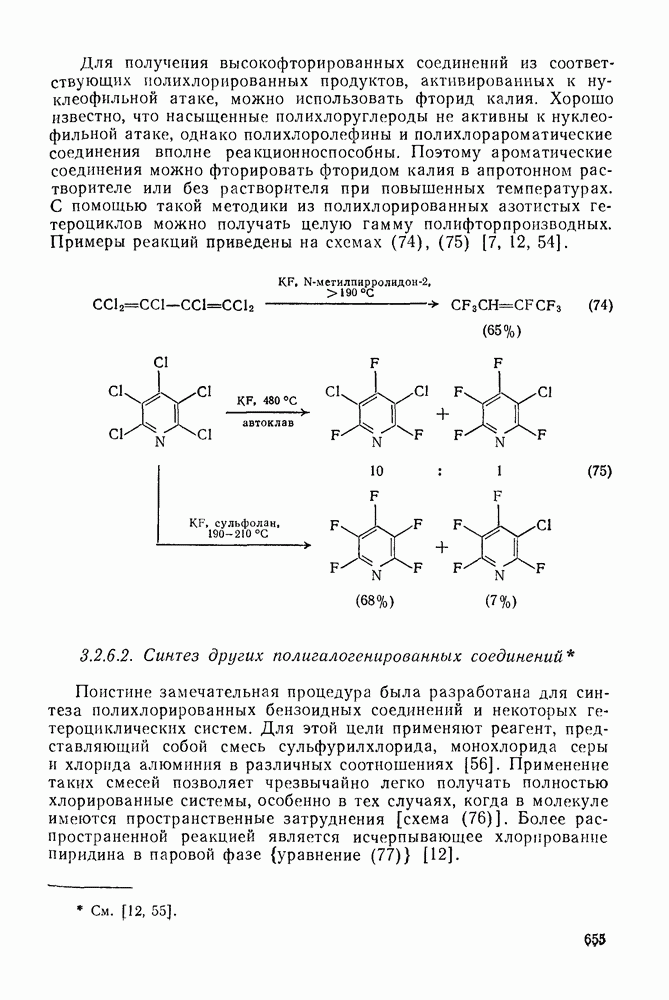
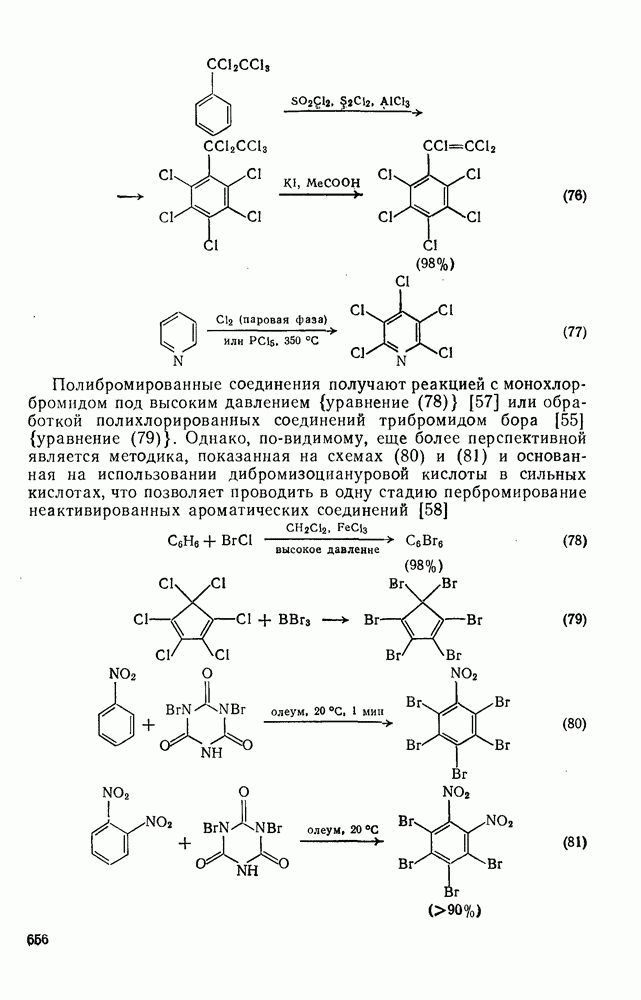
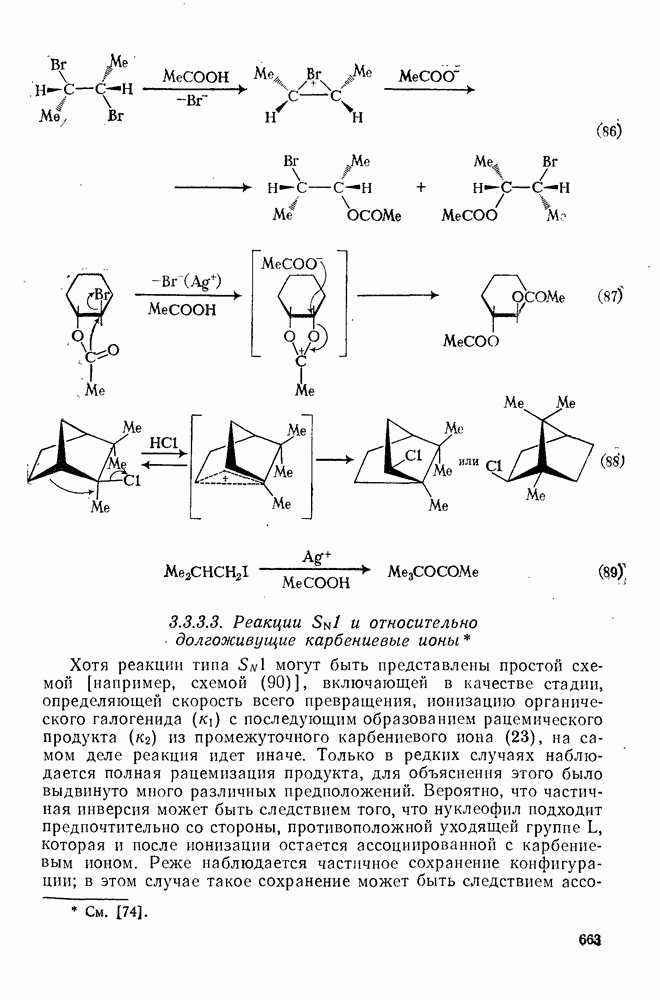
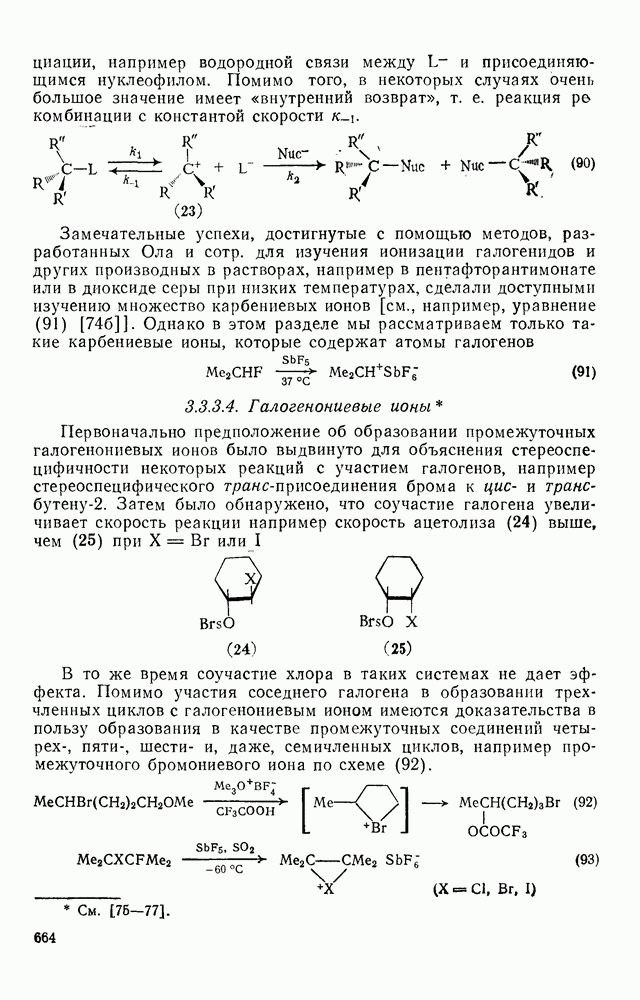
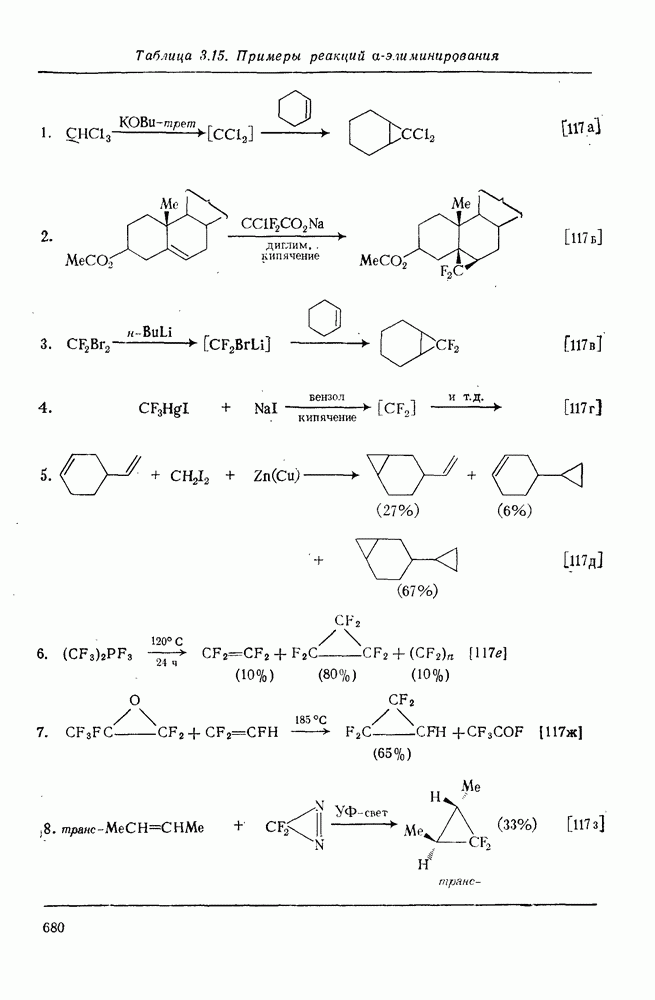
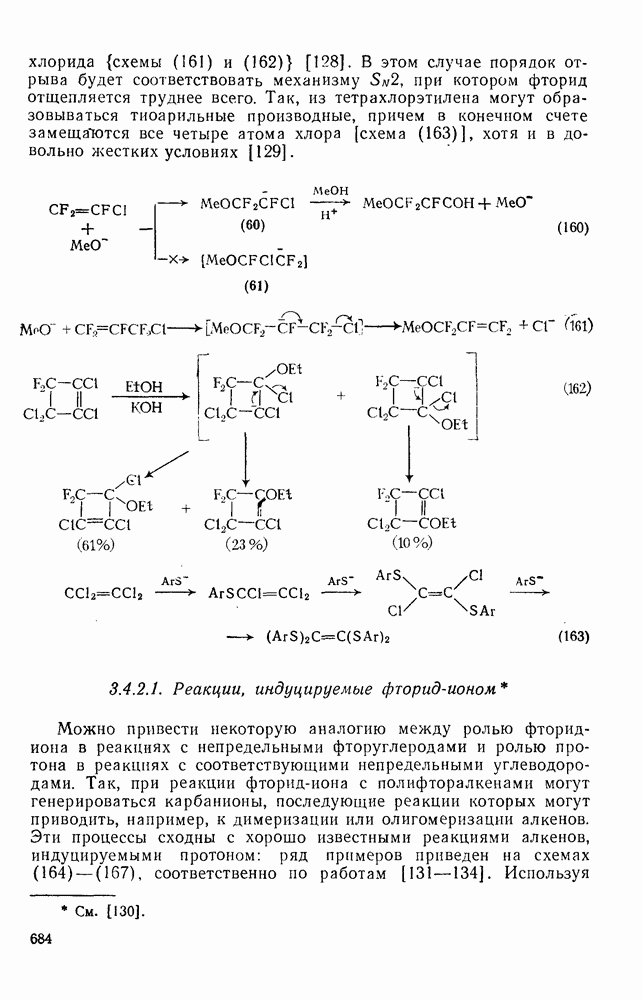
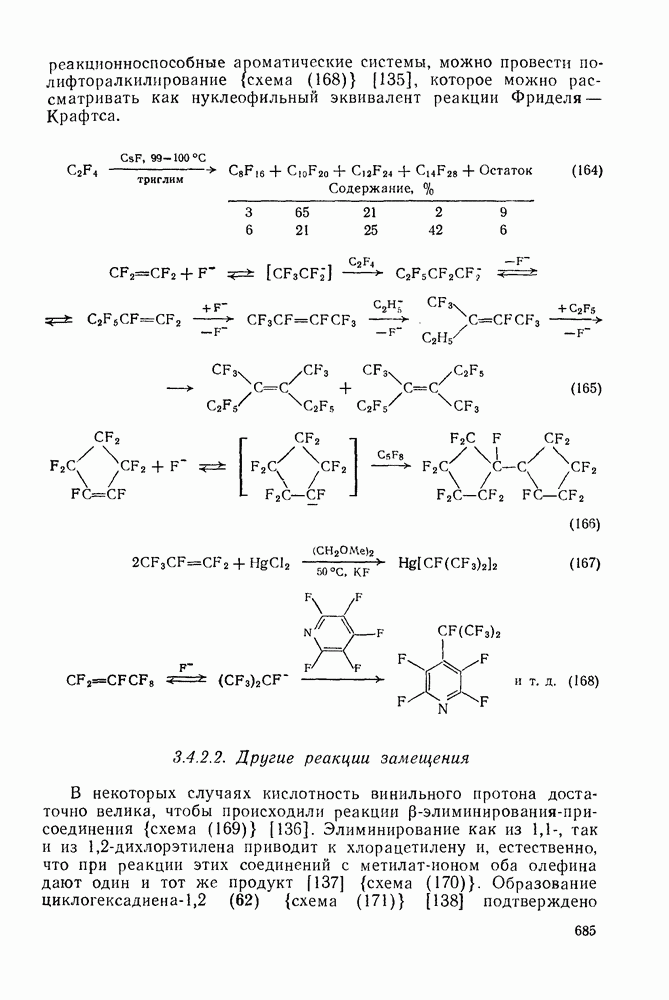
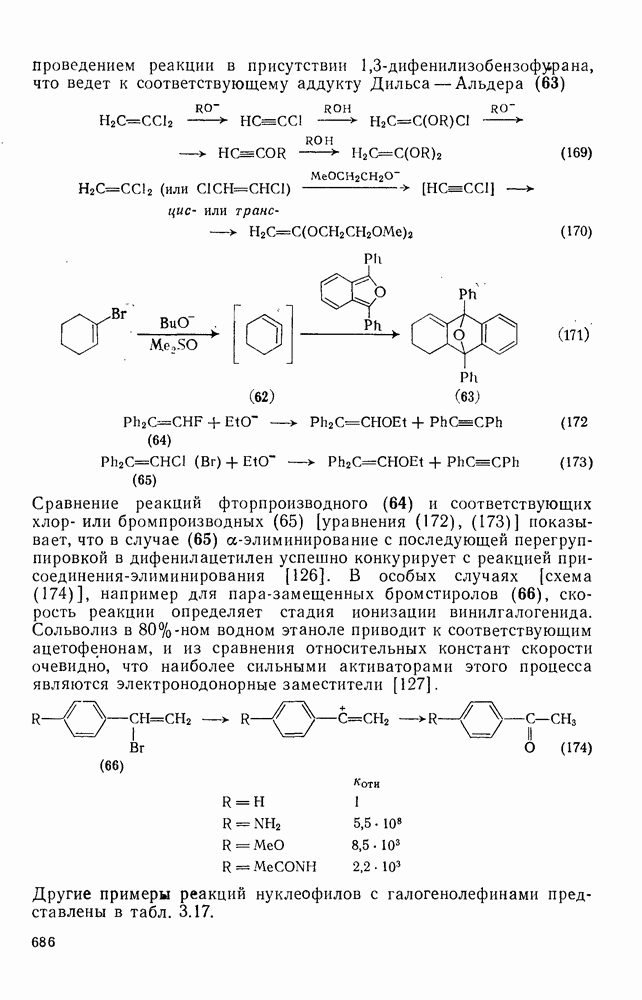
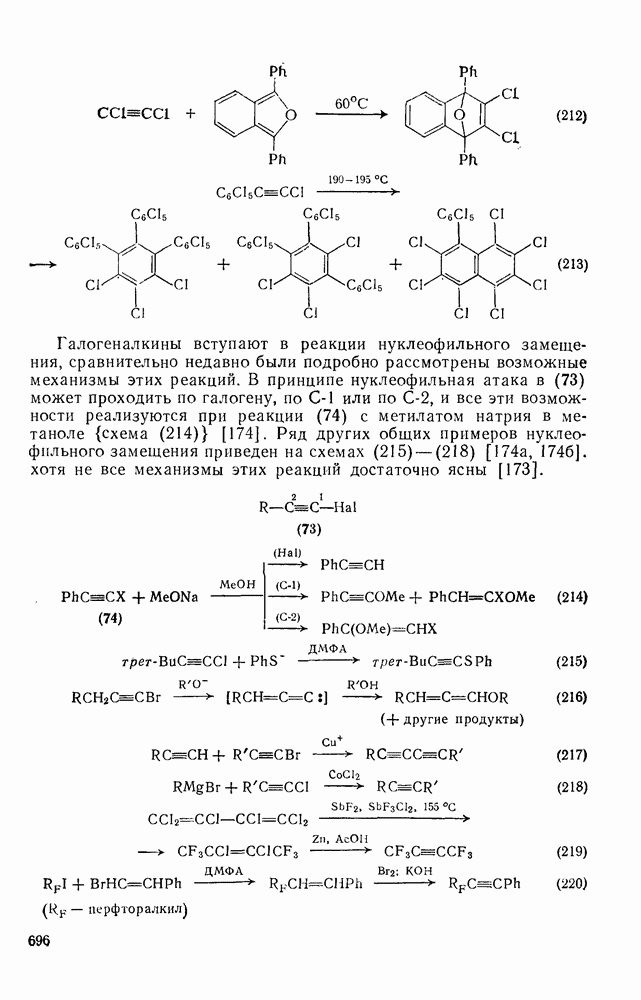
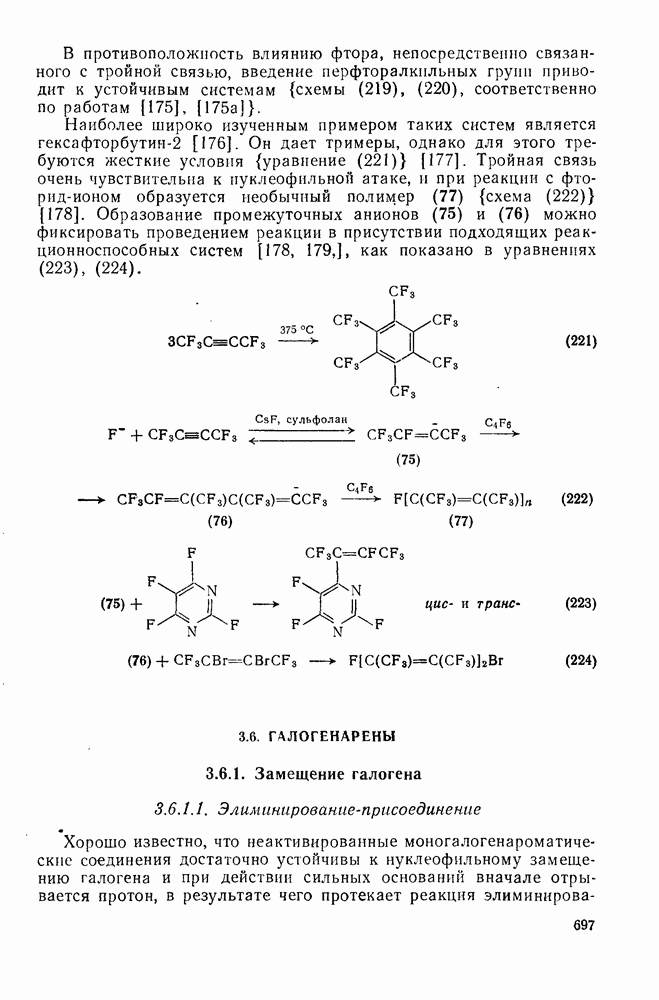
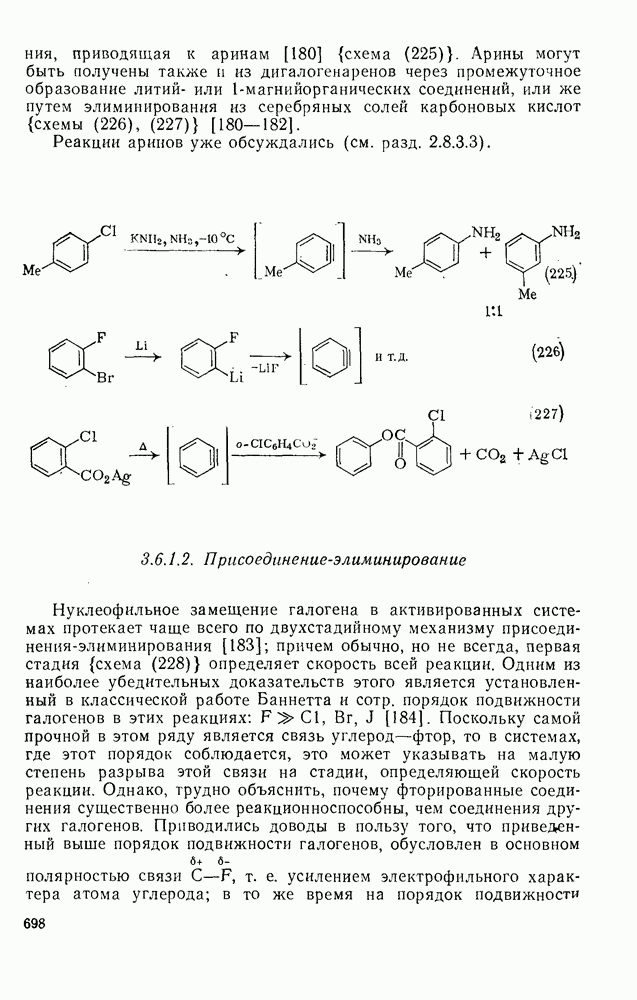
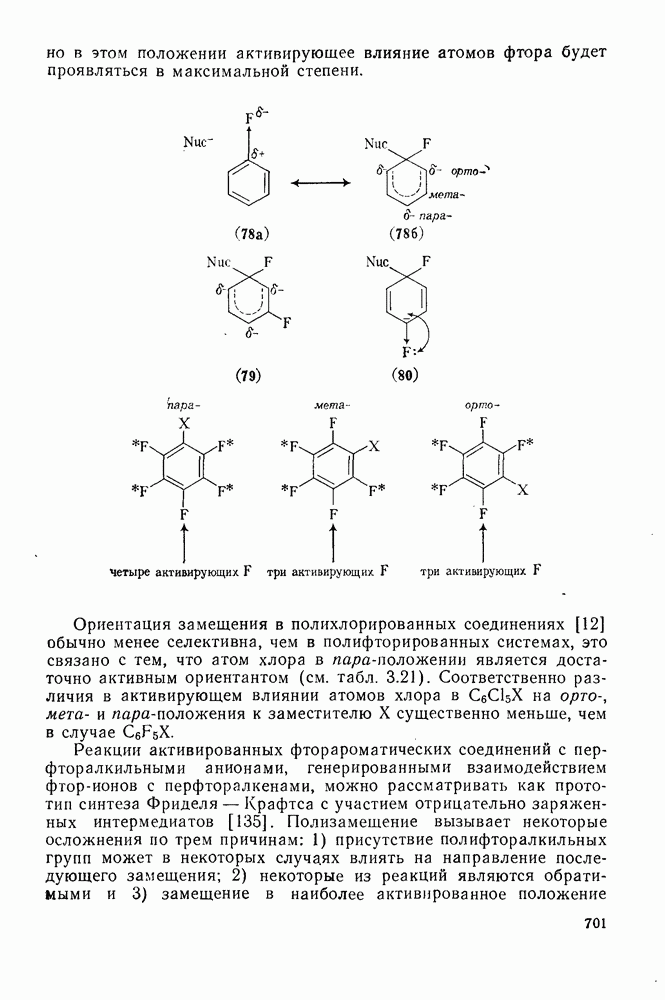
2.5.6.1. Механизм ароматического нитрования [15]
Существование  -комплексов, как устойчивых интермедиатов, Доказано прямыми опытами. Это не обязательно указывает на то, что переходное состояние реакции электрофильного присоединения — элиминирования сходно по структуре с
-комплексов, как устойчивых интермедиатов, Доказано прямыми опытами. Это не обязательно указывает на то, что переходное состояние реакции электрофильного присоединения — элиминирования сходно по структуре с  -комплексом. В самом деле, вопрос о роли комплексов в этих реакциях все еще остается предметом дискуссии [16]. В этом разделе основное внимание будет уделено механизму ароматического нитрования, который вызывает большой интерес [17]. Затем будут рассмотрены Другие из перечисленных выше реакций электрофильного присоединения — элиминирования.
-комплексом. В самом деле, вопрос о роли комплексов в этих реакциях все еще остается предметом дискуссии [16]. В этом разделе основное внимание будет уделено механизму ароматического нитрования, который вызывает большой интерес [17]. Затем будут рассмотрены Другие из перечисленных выше реакций электрофильного присоединения — элиминирования.
Результаты всех кинетических, спектроскопических и криоскопических работ, опубликованных до 1960 г., подтверждают точку зрения, что ион нитрония  является эффективным электрофилом в реакциях нитрования аренов. С помощью спектроскопических и криоскопических методов были обнаружены низкие концентрации иона нитрония в безводной азотной кислоте. Надежно установлено также, что в присутствии большого избытка серной кислоты азотная кислота полностью превращается в нитронийбисульфат. Однако нитрование ароматических соединений, сходных по реакционной способности с бензолом, проходит в присутствии таких количеств воды, что ионы нитрония обнаружить невозможно. Вместе с тем участие ионов нитрония в нитровании также строго показано путем сравнения скорости нитрования со скоростью обмена 180 между средой и азотной кислотой. Реакции нитрования некоторых реакционноспособных субстратов в присутствии воды имеют нулевой кинетический порядок. Это указывает на то, что нитрующий агент образуется из азотной кислоты в медленной (лимитирующей) стадии, предшествующей атаке электрофила ароматическим кольцом. В отсутствие ароматического субстрата скорость обмена 180 между водой и азотной кислотой имеет нулевой порядок, как и при нитровании. Эти результаты лучше всего объясняются следующей схемой (уравнение 28):
является эффективным электрофилом в реакциях нитрования аренов. С помощью спектроскопических и криоскопических методов были обнаружены низкие концентрации иона нитрония в безводной азотной кислоте. Надежно установлено также, что в присутствии большого избытка серной кислоты азотная кислота полностью превращается в нитронийбисульфат. Однако нитрование ароматических соединений, сходных по реакционной способности с бензолом, проходит в присутствии таких количеств воды, что ионы нитрония обнаружить невозможно. Вместе с тем участие ионов нитрония в нитровании также строго показано путем сравнения скорости нитрования со скоростью обмена 180 между средой и азотной кислотой. Реакции нитрования некоторых реакционноспособных субстратов в присутствии воды имеют нулевой кинетический порядок. Это указывает на то, что нитрующий агент образуется из азотной кислоты в медленной (лимитирующей) стадии, предшествующей атаке электрофила ароматическим кольцом. В отсутствие ароматического субстрата скорость обмена 180 между водой и азотной кислотой имеет нулевой порядок, как и при нитровании. Эти результаты лучше всего объясняются следующей схемой (уравнение 28):

Выделение нитрониевых солей, содержащих перхлорат-, тетра-фторборат- и гексафторфосфат-анионы, и тот факт, что эти соли являются эффективными нитрующими агентами, также свидетельствует в пользу ранее сделанных выводов.
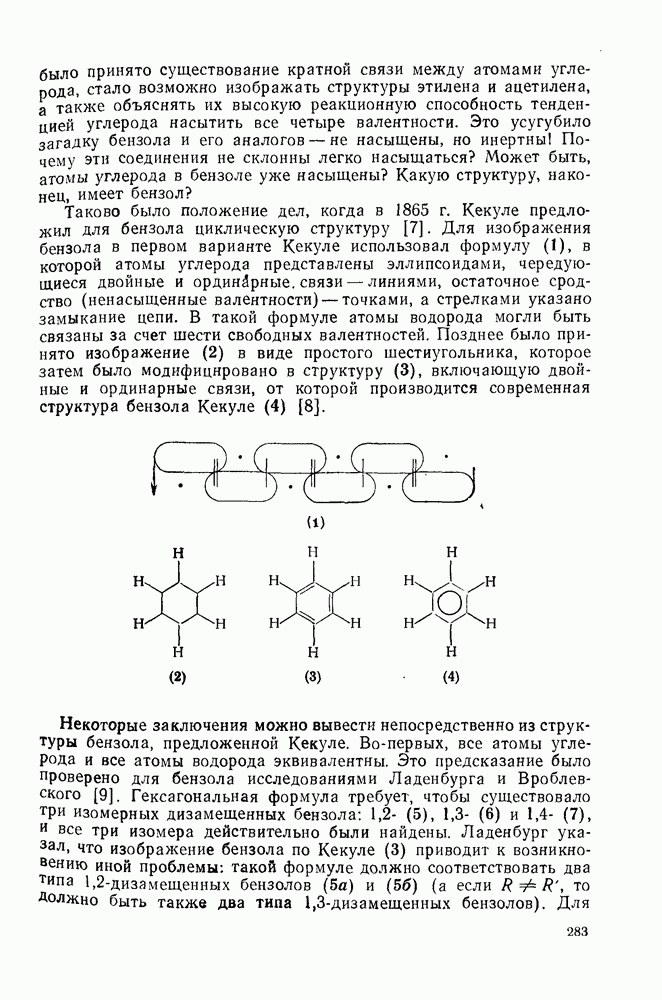
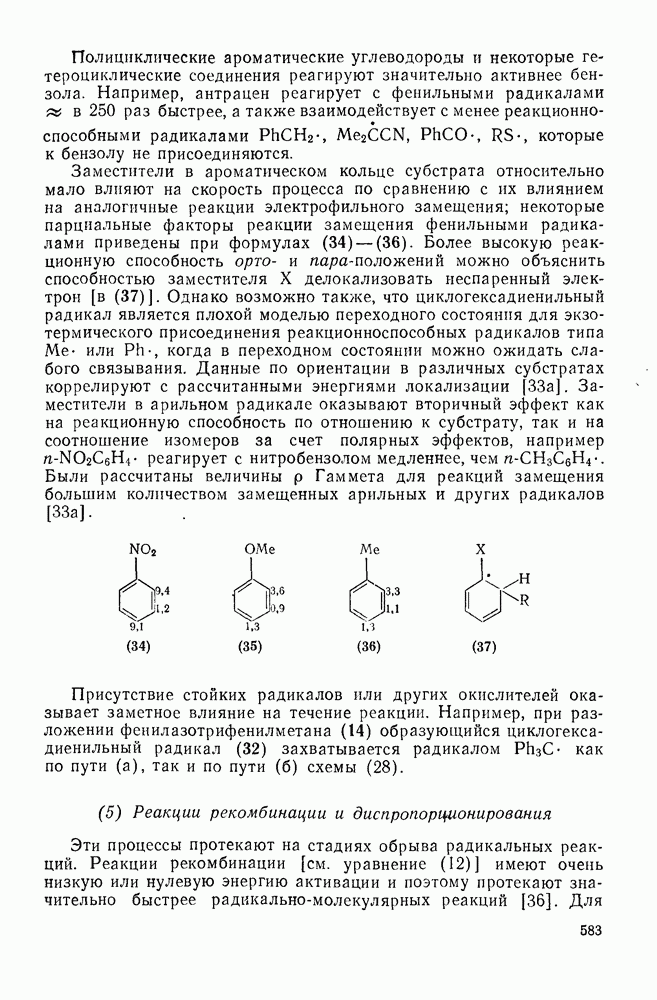
Нитрование было использовано для установления относительной реакционной способности большого числа ароматических соединений. Относительные скорости замещения в  и
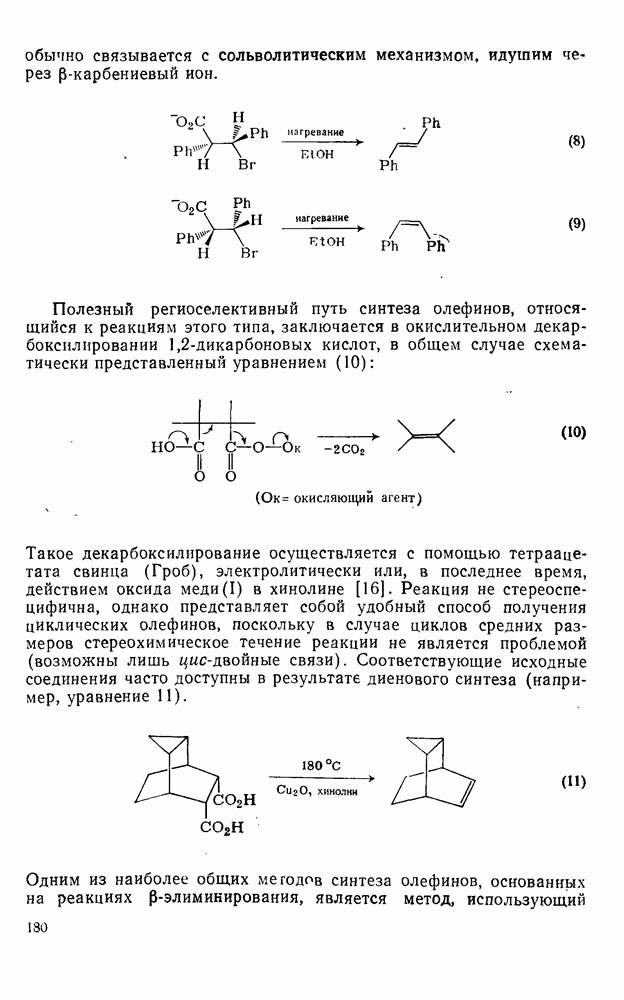
и и
и  -положения со статистическими поправками, так называемые парциальные факторы скорости, были рассчитаны для большого числа монозамещенных бензолов. Расчеты проводили, исходя из относительных скоростей
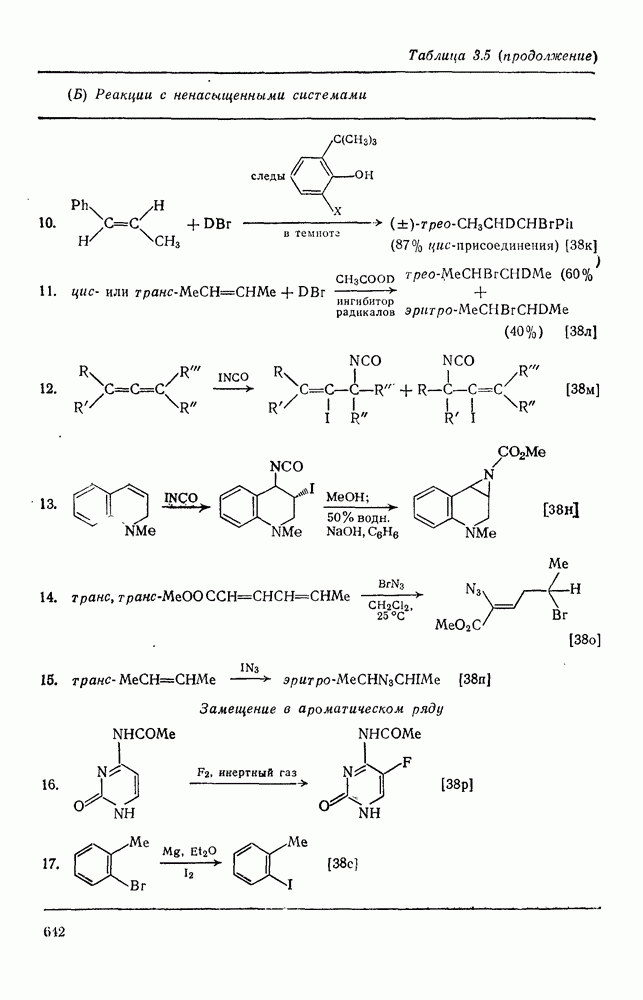
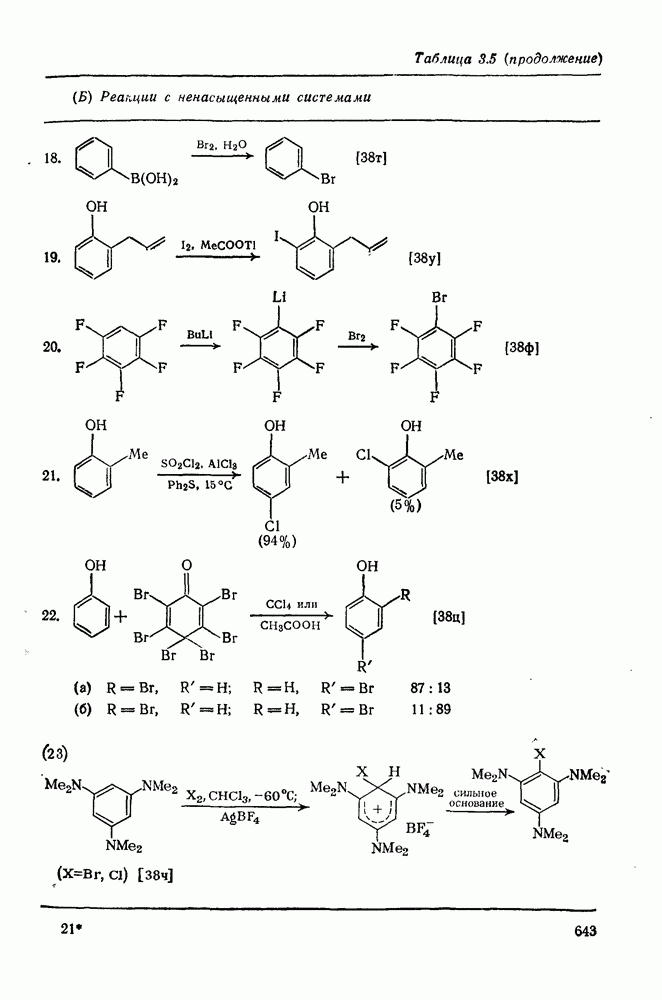
-положения со статистическими поправками, так называемые парциальные факторы скорости, были рассчитаны для большого числа монозамещенных бензолов. Расчеты проводили, исходя из относительных скоростей  по кинетическим данным или методом конкурентных реакций путем определения изомерного состава продукта реакции в тех же экспериментальных условиях. Очень близкие результаты были получены при нитровании эквимольных смесей бензола и толуола азотной кислотой в нитрометане, ацетонитриле, уксусном ангидриде или кислотных растворителях. Эти результаты также подтверждают универсальный характер механизма с участием нитроний-катиона. В табл. 2.5.2 приведены типичные примеры. Очевидно, что хотя распределение изомеров в реакциях с предварительно приготовленными нитрониевыми солями близко, к полученным ранее результатам, относительные скорости в реакциях с солями значительно ближе подходят друг к другу и, следовательно, парциальный фактор скорости, например для атаки метаположения, по-видимому, меньше, чем для отдельных положений бензола.
по кинетическим данным или методом конкурентных реакций путем определения изомерного состава продукта реакции в тех же экспериментальных условиях. Очень близкие результаты были получены при нитровании эквимольных смесей бензола и толуола азотной кислотой в нитрометане, ацетонитриле, уксусном ангидриде или кислотных растворителях. Эти результаты также подтверждают универсальный характер механизма с участием нитроний-катиона. В табл. 2.5.2 приведены типичные примеры. Очевидно, что хотя распределение изомеров в реакциях с предварительно приготовленными нитрониевыми солями близко, к полученным ранее результатам, относительные скорости в реакциях с солями значительно ближе подходят друг к другу и, следовательно, парциальный фактор скорости, например для атаки метаположения, по-видимому, меньше, чем для отдельных положений бензола.
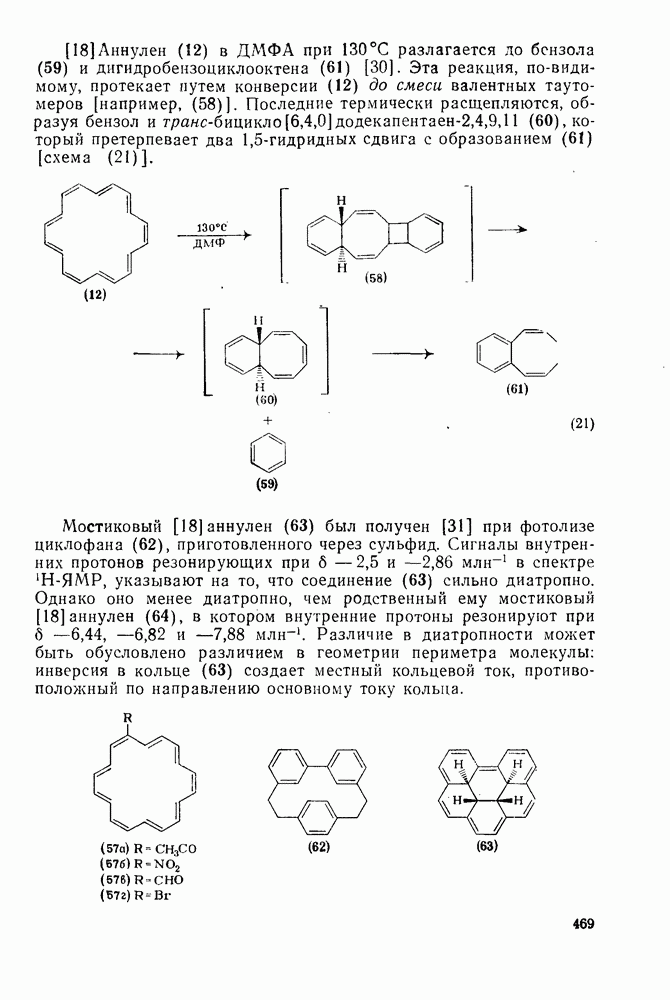
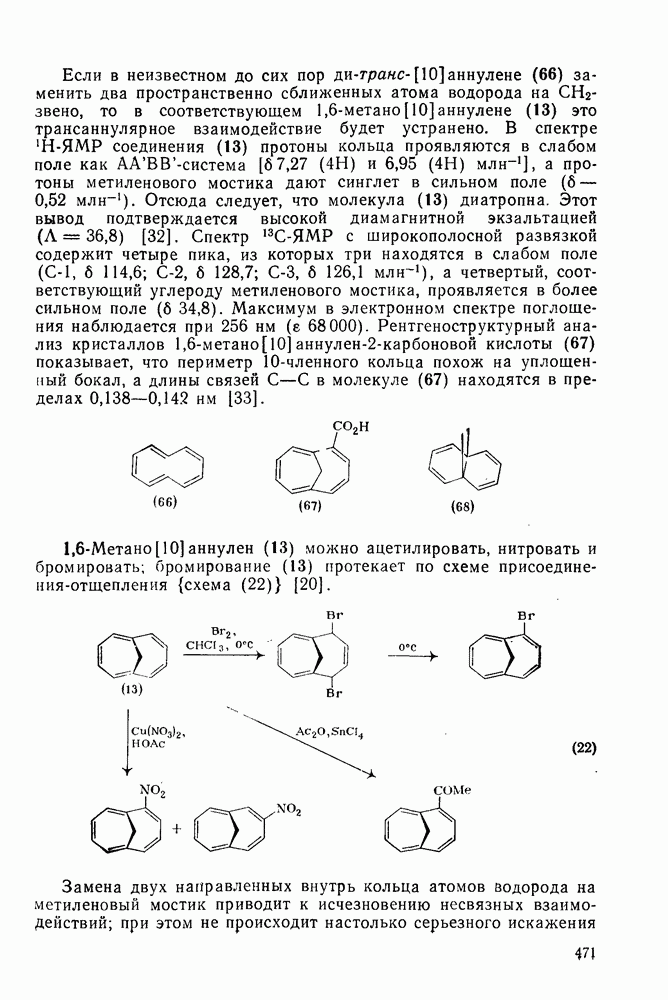
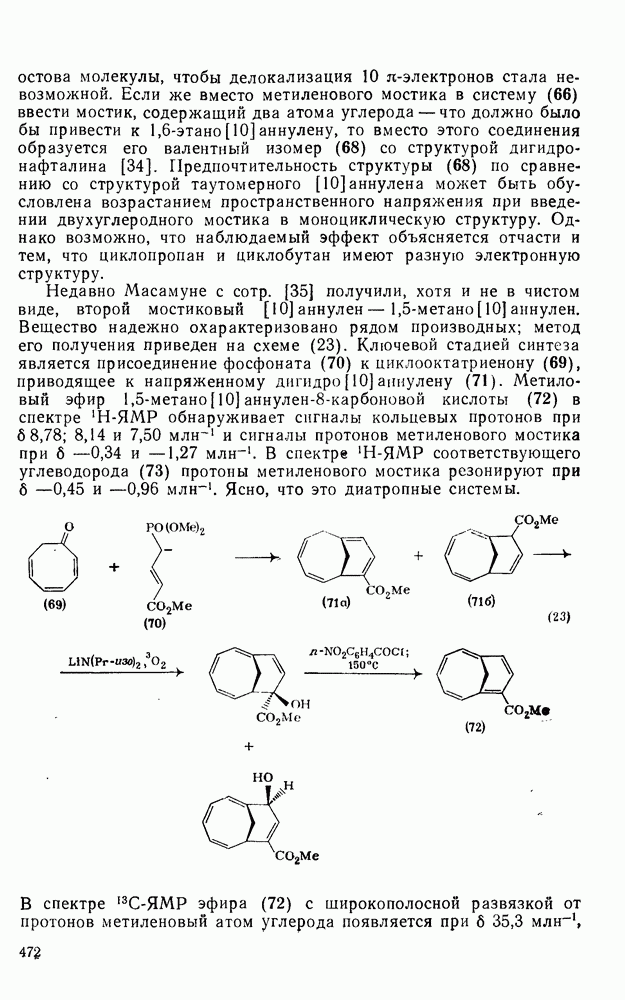
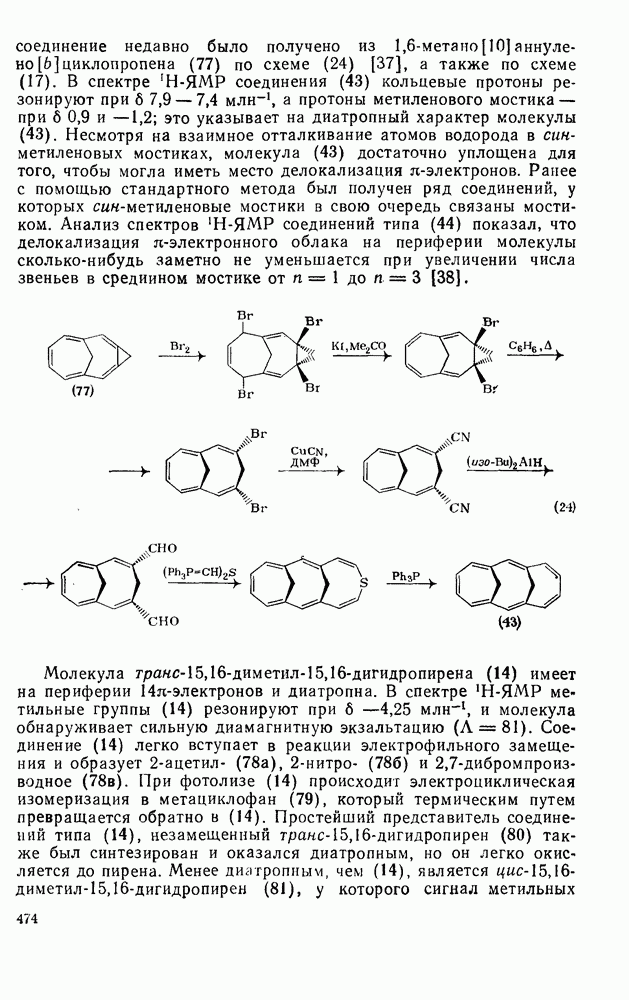
Таблица 2.5.2. Ориентация, относительная реакционная способность и парциальные факторы скорости при нитровании некоторых типичных молекул

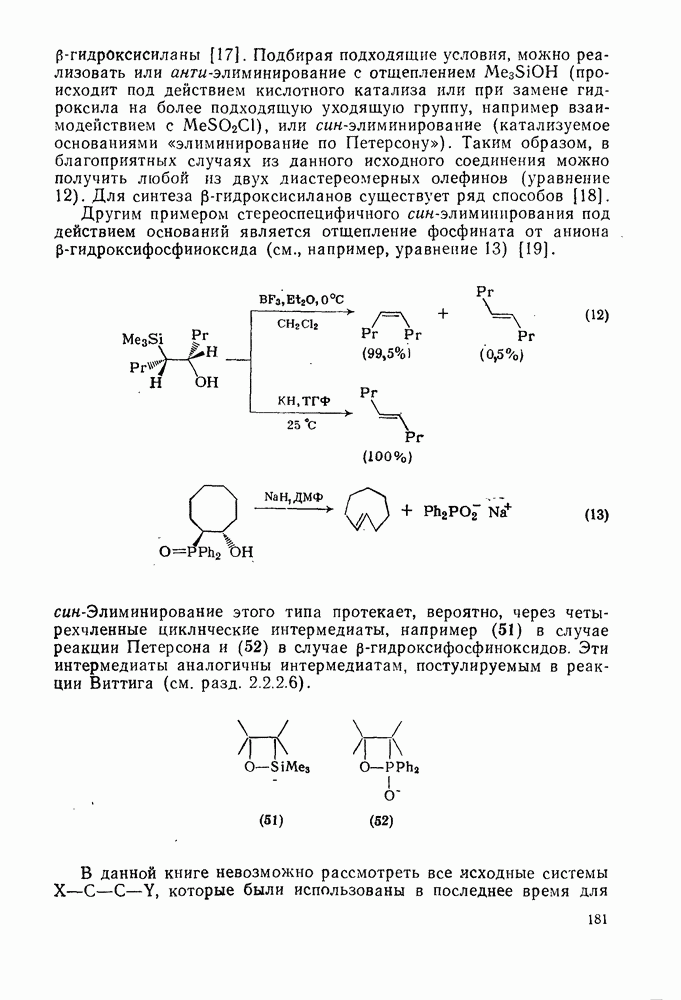
Ниже приведен метод расчета на примере нитрования толуола сравнительно с бензолом в уксусной кислоте при  . Напомним, что в бензоле имеется шесть эквивалентных положений, а в толуоле-одно пара-положение и по два орто- и мета-положений:
. Напомним, что в бензоле имеется шесть эквивалентных положений, а в толуоле-одно пара-положение и по два орто- и мета-положений:

Изучение нитрования смесей бензола с толуолом с использованием предварительно приготовленных нитрониевых солей (см. табл. 2.5.2) и аналогичные эксперименты с м-ксилолом и мезитиленом, относительная реакционная способность которых по сравнению с бензолом оказалась равной 1,7 и 2,7 соответственно, увеличили число неясных вопросов. Выше уже упоминалось о непонятных результатах, полученных при расчете парциального фактора скорости для мета-положения. Кроме того, отмечалось, что в том случае, когда реакция успевает пройти в основном до того, как реагенты образуют достаточно однородную смесь, результаты определения реакционной способности из опытов по конкурентному замещению могут быть ошибочными.
Очевидно, что эта аномальная реакционная способность не подтверждается при определении констант скоростей нитрования нитрониевыми солями с помощью кинетических методов. Тем не менее были сделаны попытки обеспечить достаточное смешивание реагентов проведением реакции при различных их концентрациях. Эти результаты привели к предположению, что скорость ряда реакций аренов с электрофилами, в том числе и скорость нитрования нитрониевыми солями, связана скорее с  -основностью, а не
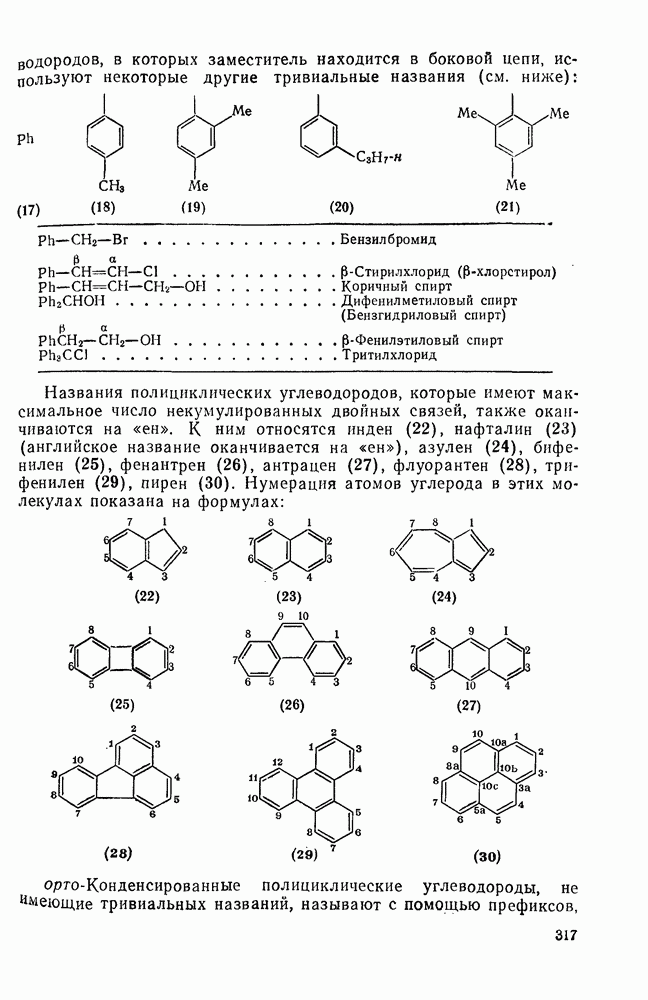
-основностью, а не  -основностью аренов. Иными словами, образование
-основностью аренов. Иными словами, образование  -комплекса является лимитирующей стадией. Однако место вхождения электрофила контролируется
-комплекса является лимитирующей стадией. Однако место вхождения электрофила контролируется  -основностью, как это показывают имеющиеся данные по распределению изомеров [16а]. Эта точка зрения на ароматическое нитрование была вновь подробно исследована [166]. Было отмечено, что корреляция между соотношением продуктов нитрования нитрониевыми солями и относительной устойчивостью протонированных метилбензолов, по-видимому, ничуть не хуже, чем корреляция с устойчивостью
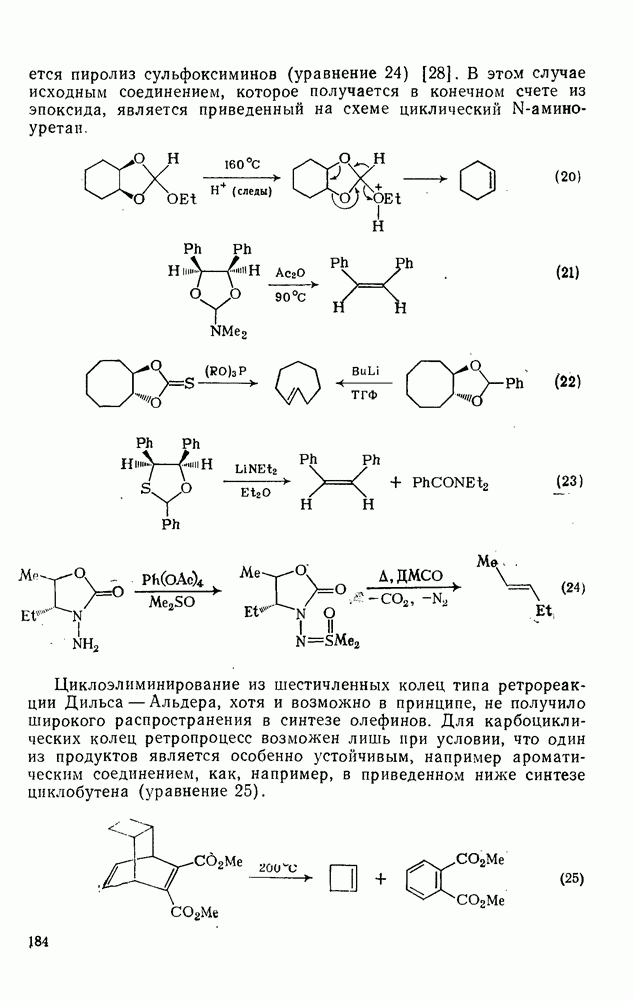
-основностью, как это показывают имеющиеся данные по распределению изомеров [16а]. Эта точка зрения на ароматическое нитрование была вновь подробно исследована [166]. Было отмечено, что корреляция между соотношением продуктов нитрования нитрониевыми солями и относительной устойчивостью протонированных метилбензолов, по-видимому, ничуть не хуже, чем корреляция с устойчивостью  -комплексов. Соотношение продуктов конкурентного нитрования полиметилбензолов смесью кислот в сульфолане также плохо коррелирует с устойчивостью
-комплексов. Соотношение продуктов конкурентного нитрования полиметилбензолов смесью кислот в сульфолане также плохо коррелирует с устойчивостью  -комплексов.
-комплексов.
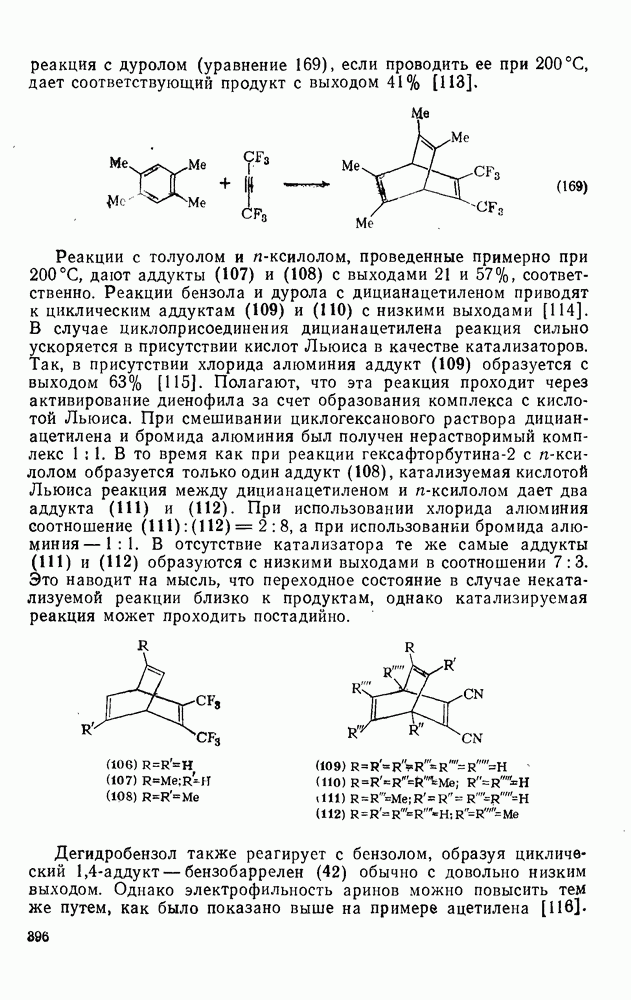
Вопрос, связанный со степенью прохождения реакции во время смешивания реагентов, изучен на примере нитрования бибензила, в котором каждое кольцо по реакционной способности аналогично толуолу, и, кроме того, передача влияния заместителя между кольцами минимальна. При использовании эквимольных концентраций бибензила и нитрониевой соли и полном смешении реагентов до начала реакции должны образоваться мононитробибензил (50%) и динитробибензил  . С другой стороны, в случае неполного смешения реагентов до реакции должно увеличиться количество непрореагировавшего субстрата и динитробибеизила и уменьшиться количество мононитробибензила. Однако такое представление является слишком упрощенным, что со всей очевидностью следует из результатов нитрования бибензила азотной кислотой в уксусном ангидриде. Полнота смешивания не имеет значения для этой системы, но количество динитробибеизила составляет только 55% от ожидаемого. При нитровании бибензила в сульфолане нитронийтетрафторборатом с использованием различных концентраций и условий смешивания количество дизамещенного продукта всегда было значительно выше, чем это получается по приведенному расчету. Таким образом, простой метод конкурентных реакций не пригоден для определения относительной реакционной способности ароматических соединений при нитровании нитрониевыми солями. Очевидно, основное влияние оказывает скорость смешивания.
. С другой стороны, в случае неполного смешения реагентов до реакции должно увеличиться количество непрореагировавшего субстрата и динитробибеизила и уменьшиться количество мононитробибензила. Однако такое представление является слишком упрощенным, что со всей очевидностью следует из результатов нитрования бибензила азотной кислотой в уксусном ангидриде. Полнота смешивания не имеет значения для этой системы, но количество динитробибеизила составляет только 55% от ожидаемого. При нитровании бибензила в сульфолане нитронийтетрафторборатом с использованием различных концентраций и условий смешивания количество дизамещенного продукта всегда было значительно выше, чем это получается по приведенному расчету. Таким образом, простой метод конкурентных реакций не пригоден для определения относительной реакционной способности ароматических соединений при нитровании нитрониевыми солями. Очевидно, основное влияние оказывает скорость смешивания.
Интересные результаты были получены также при изучении кинетики [18] нитрования аренов, реакционная способность которых выше, чем у бензола и толуола.
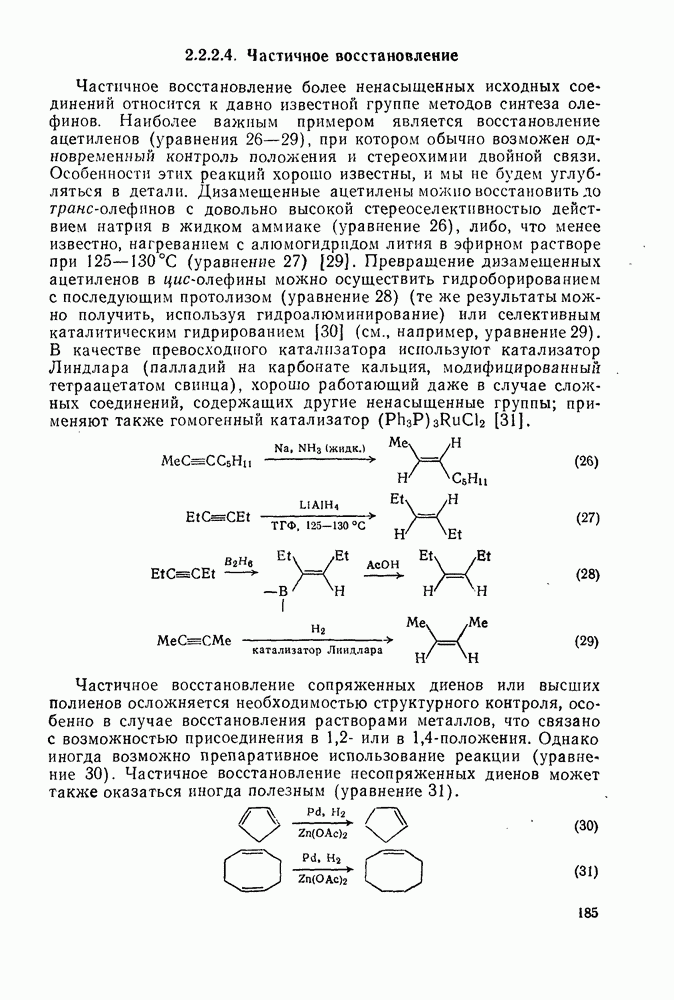
Можно было бы ожидать, что реакционная способность  -ксилола,
-ксилола,  -ксилола и мезитилена значительно больше, чем у бензола (факторы около
-ксилола и мезитилена значительно больше, чем у бензола (факторы около  и
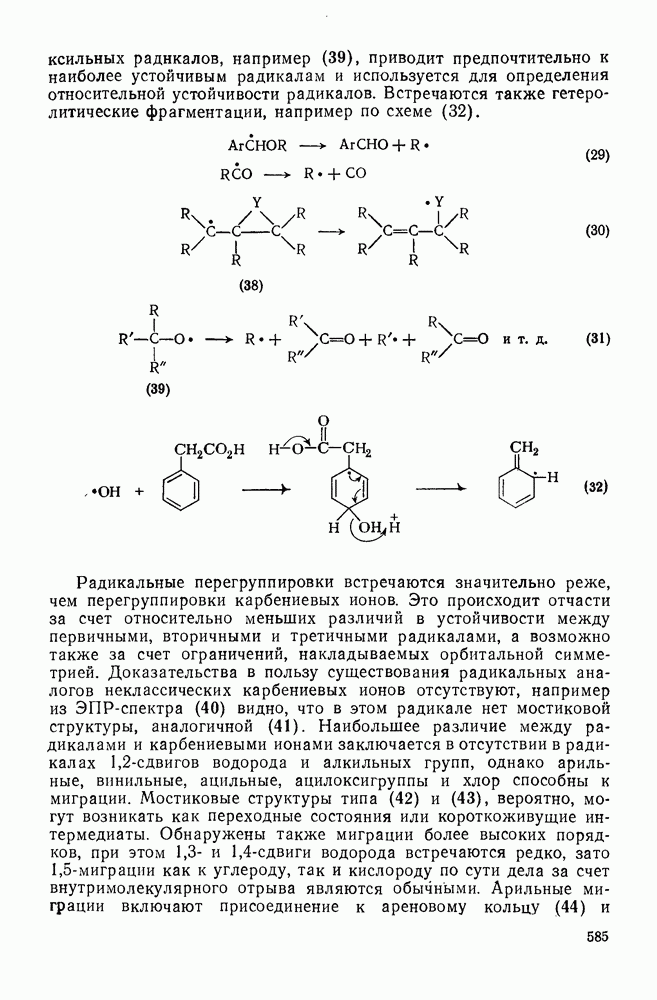
и  ), однако при рассмотрении результатов нитрования в водной серной кислоте (68,3%) оказывается, что этот фактор ограничен значением 40. Предполагают, что лимитирующей стадией в этих реакциях является образование пары при столкновении между ионом нитрония и ароматическим субстратом. В чем различие между этим предположением и предположением о лимитирующем скорость образовании
), однако при рассмотрении результатов нитрования в водной серной кислоте (68,3%) оказывается, что этот фактор ограничен значением 40. Предполагают, что лимитирующей стадией в этих реакциях является образование пары при столкновении между ионом нитрония и ароматическим субстратом. В чем различие между этим предположением и предположением о лимитирующем скорость образовании  -комплексе? Ранее было отмечено, что соотношение продуктов плохо коррелирует с устойчивостью
-комплексе? Ранее было отмечено, что соотношение продуктов плохо коррелирует с устойчивостью  -комплексов. Эти факты можно объяснить, не прибегая к притягивающим взаимодействиям в пределах «пары столкновения». Энергетический барьер диссоциации «пары столкновения» на компоненты может превышать
-комплексов. Эти факты можно объяснить, не прибегая к притягивающим взаимодействиям в пределах «пары столкновения». Энергетический барьер диссоциации «пары столкновения» на компоненты может превышать  в кислой среде, этой энергии вполне достаточно для селективного образования
в кислой среде, этой энергии вполне достаточно для селективного образования  -комплекса. Поскольку кинетические данные согласуются с диффузионной теорией соударений, то следует предпочесть термин "пара столкновения". Обсуждение всех приведенных выше данных приводит к заключению, что схему уравнений (28) следует видоизменить так, как это показано в уравнении (29), где
-комплекса. Поскольку кинетические данные согласуются с диффузионной теорией соударений, то следует предпочесть термин "пара столкновения". Обсуждение всех приведенных выше данных приводит к заключению, что схему уравнений (28) следует видоизменить так, как это показано в уравнении (29), где  определяет скорость только в исключительных обстоятельствах. Различные константы скорости
определяет скорость только в исключительных обстоятельствах. Различные константы скорости  атаки в орто-, мета- или пара-положения указывают, что распределение изомеров не зависит от лимитирующей стадии, поскольку в «пару столкновения» входит вся молекула арена, а не отдельные ее атомы.
атаки в орто-, мета- или пара-положения указывают, что распределение изомеров не зависит от лимитирующей стадии, поскольку в «пару столкновения» входит вся молекула арена, а не отдельные ее атомы.

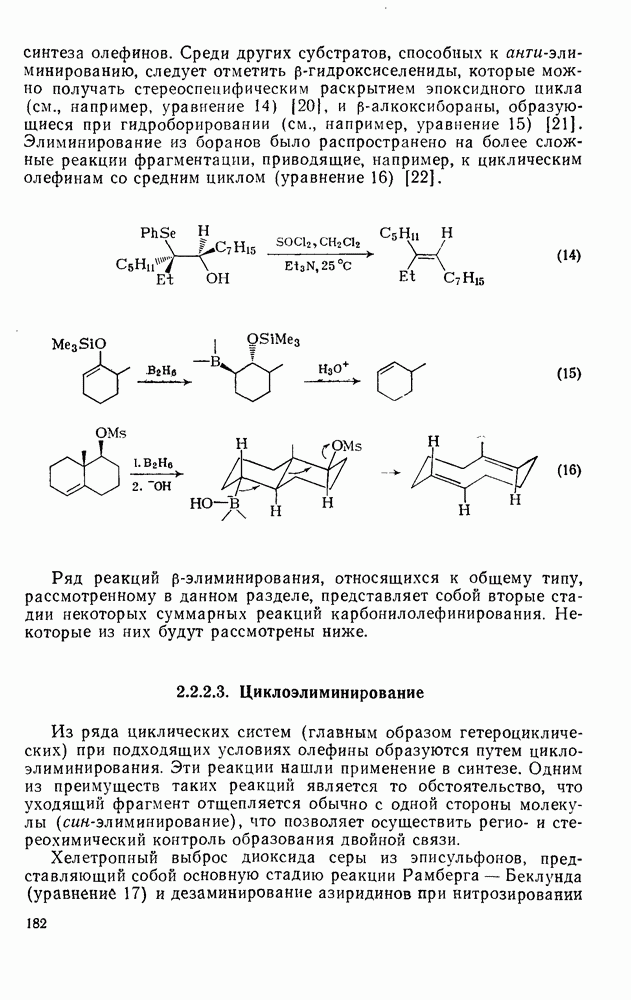
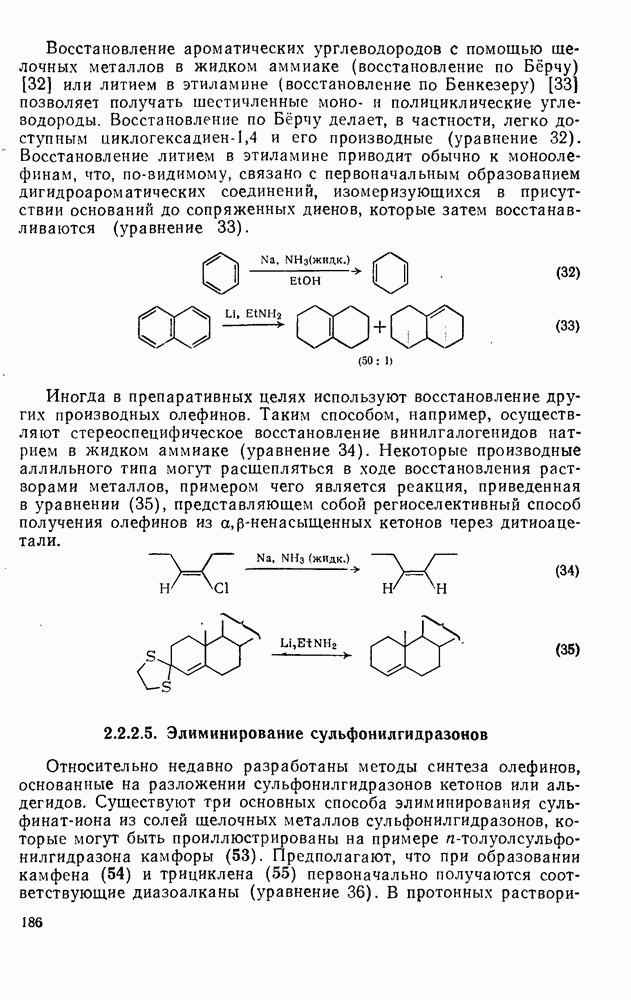
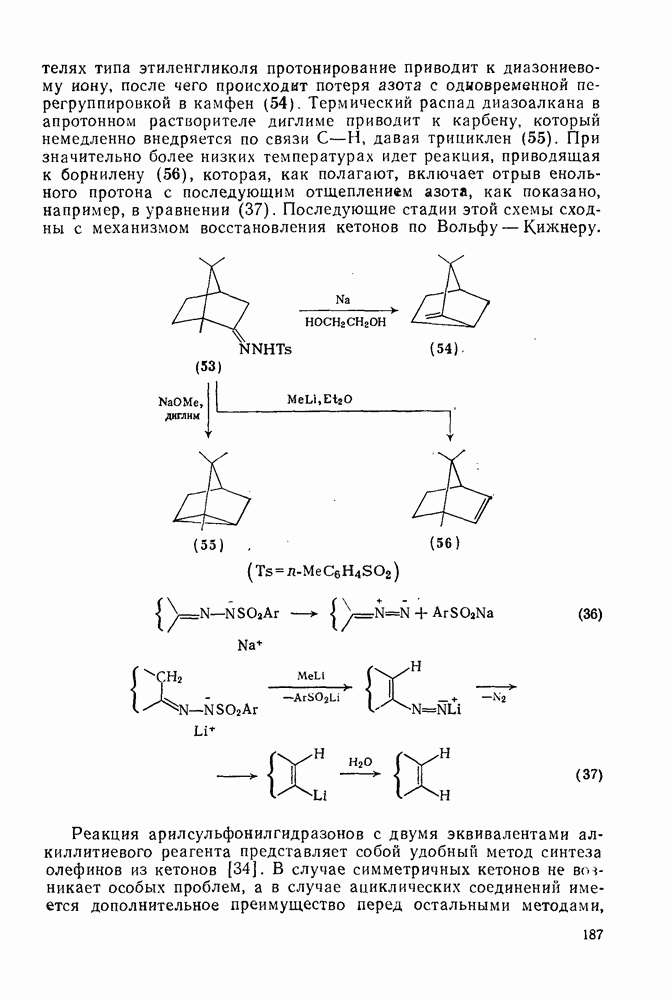
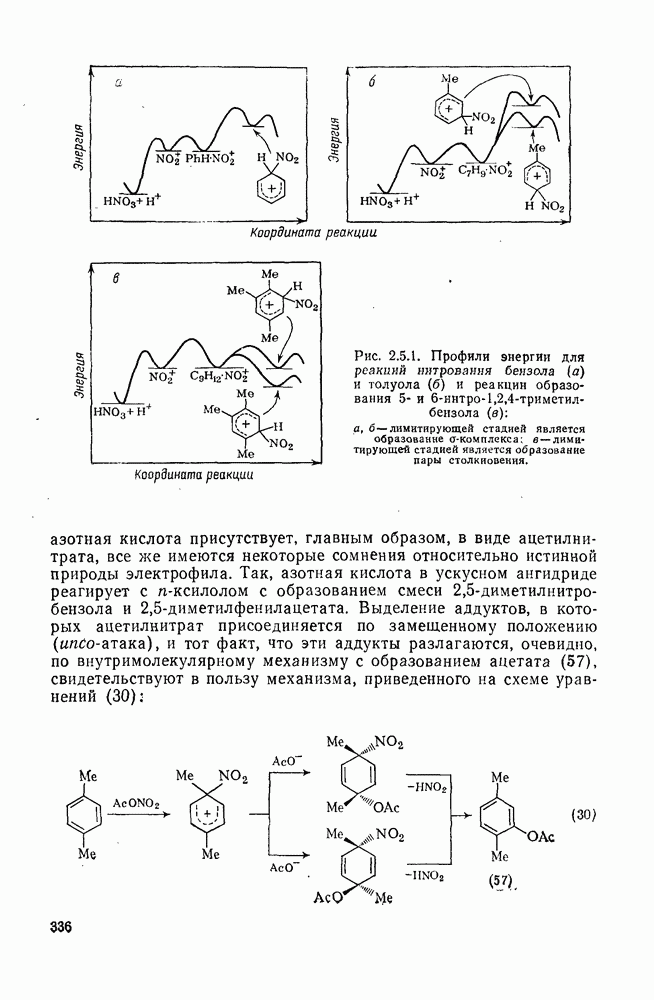
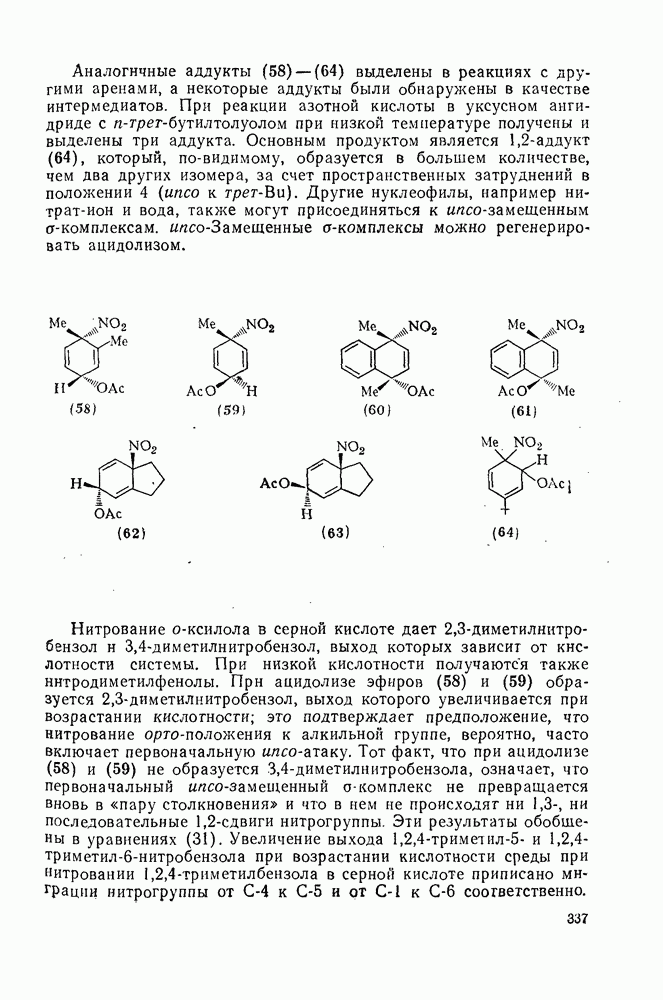
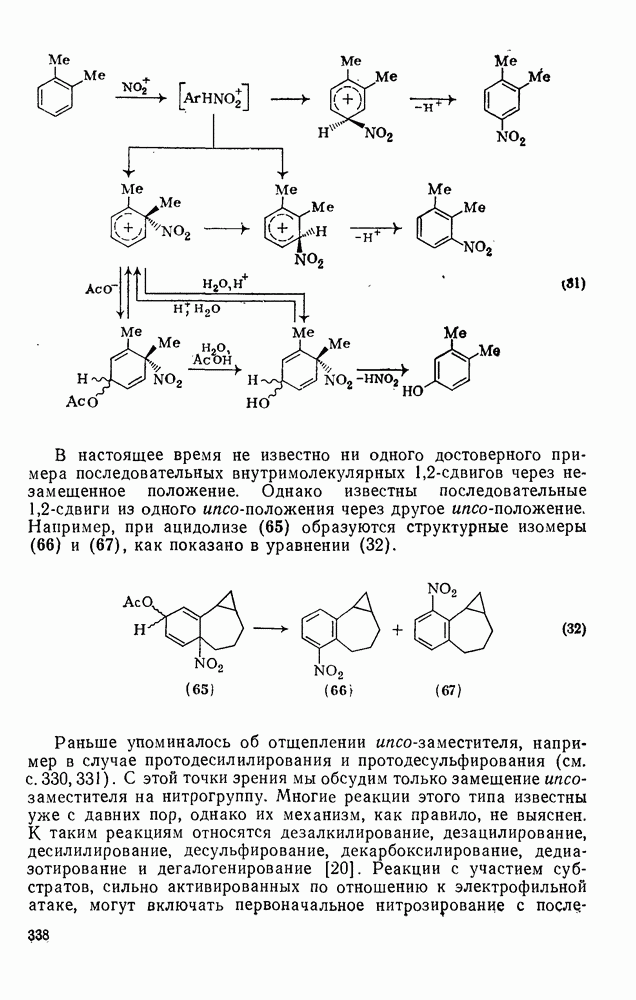
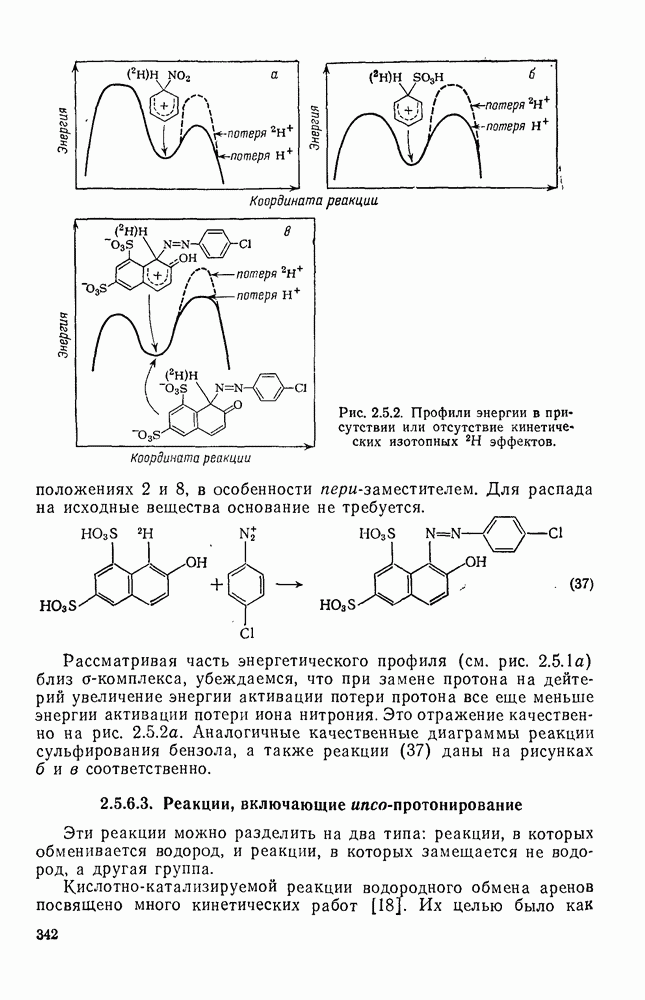
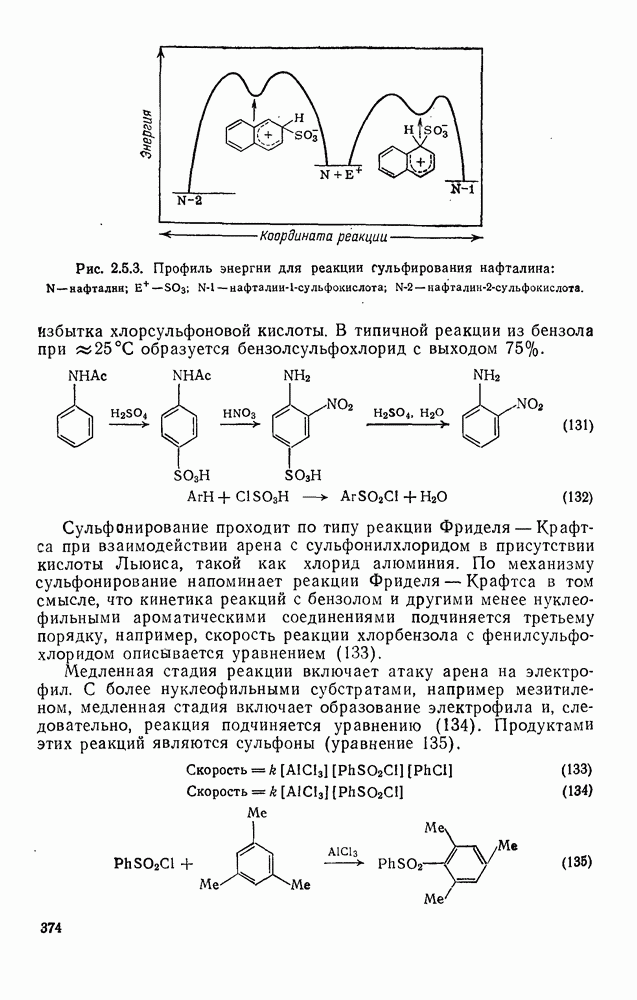
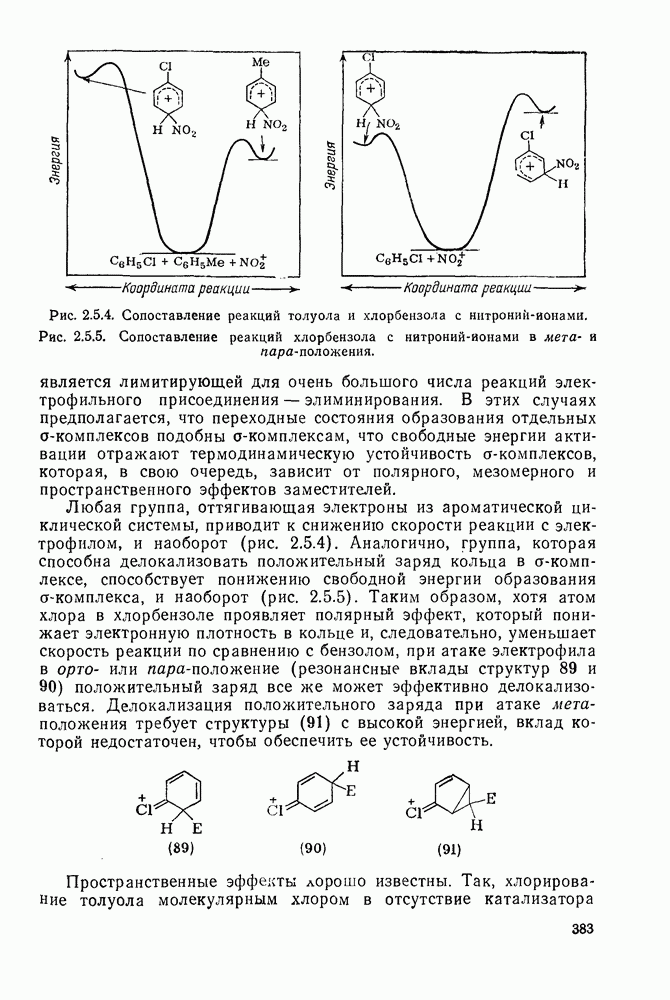
Обсуждение нитрования можно удобно обобщить в виде энергетических профилей, как это сделано на рис. 2.5.1. Экспериментальные результаты показывают, что в случае нитрования бензола и толуола скорость определяющей стадией является образование  -комплекса (рис. а и б соответственно), а образование «пары столкновения» определяет скорость нитрования псевдокумола (
-комплекса (рис. а и б соответственно), а образование «пары столкновения» определяет скорость нитрования псевдокумола ( -триметилбензола) (рис. в).
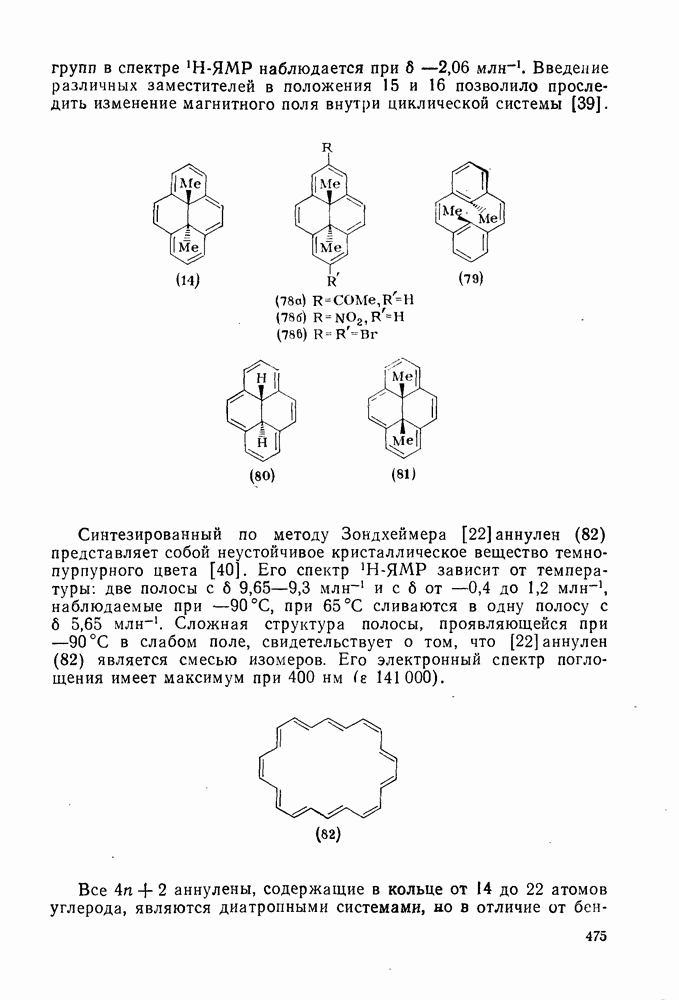
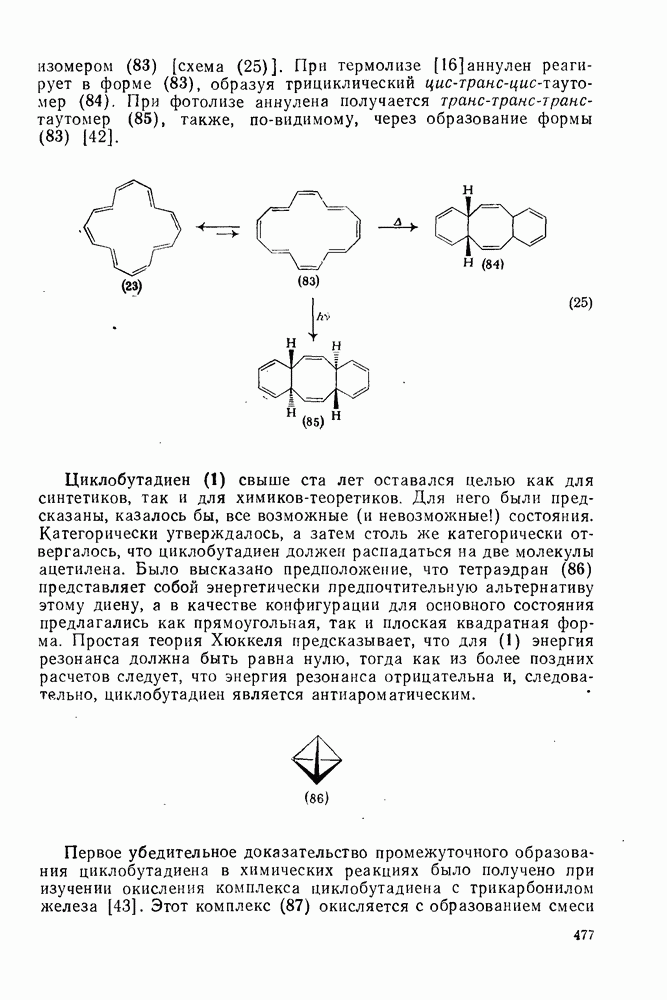
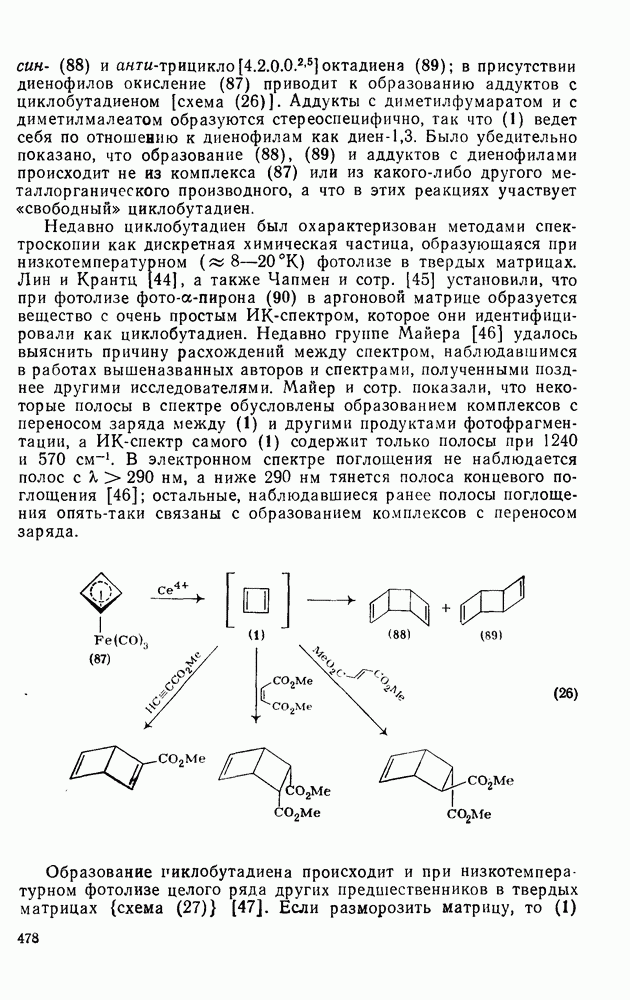
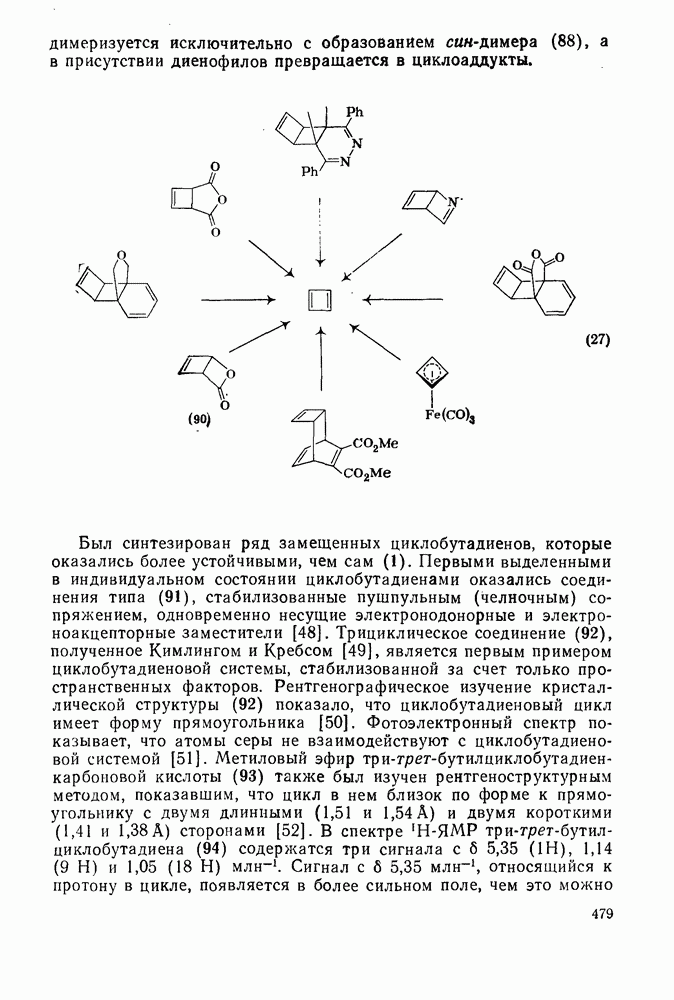
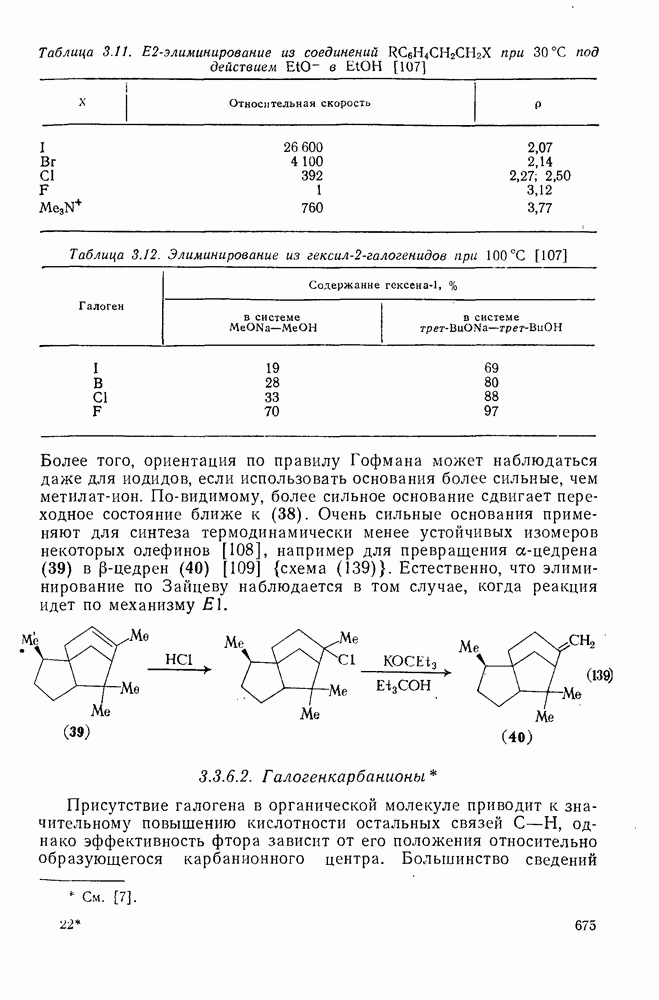
-триметилбензола) (рис. в).
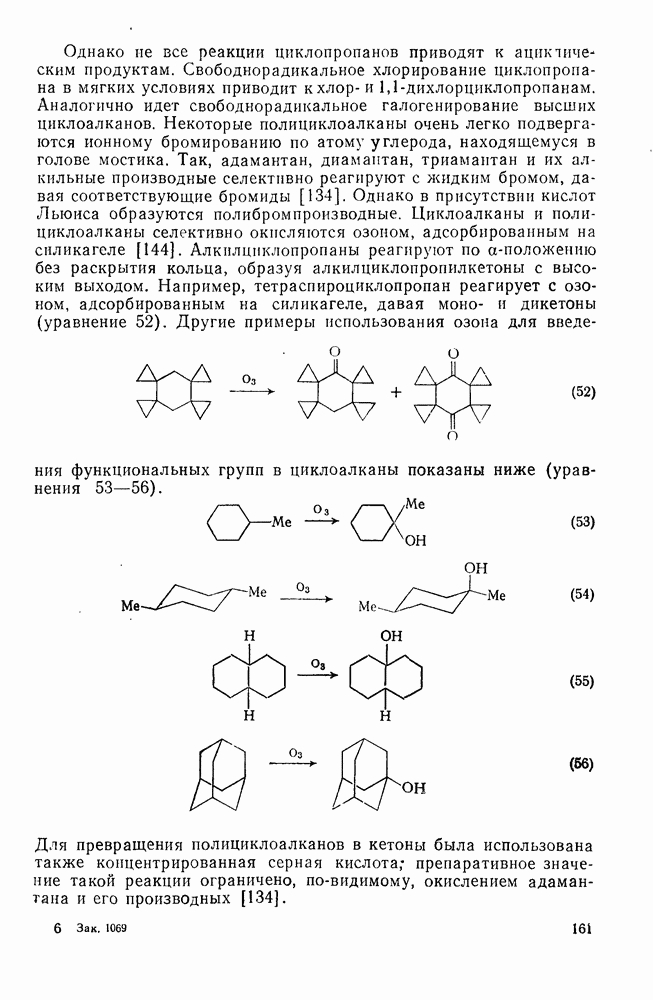
Распределение изомеров, которое было обнаружено при нитровании толуола (см. табл. 2.5.2.), в основном не зависит от условий реакции, исключая обычное влияние температуры. Однако необычные результаты, полученные при изучении некоторых полиметилбензолов, осложняют понимание реакции нитрования [19]. Так, нитрование азотной кислотой в уксусном ангидриде приводит к продуктам ацетоксилирования. Это смешивает все карты, заставляя предположить, что проходит электрофильное ацетоксилирование. В настоящее время известно, что это не имеет места.


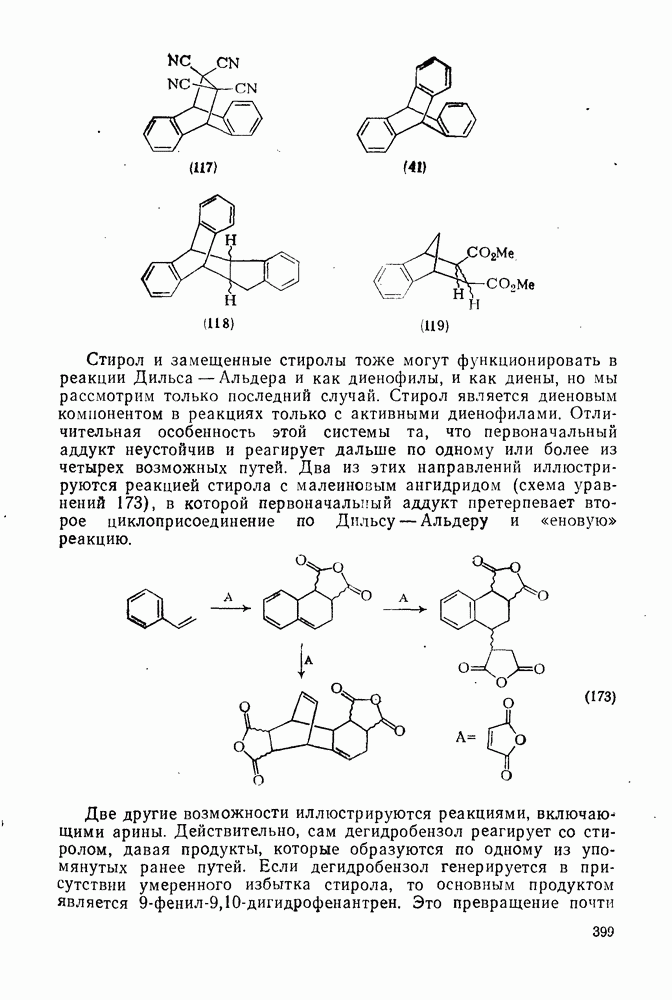
Рис. 2.5.1. Профили энергии для реакций нитрования бензола (а) и толуола (б) и реакции образования 5- и  : а, б—лимитирующей стадией является образование
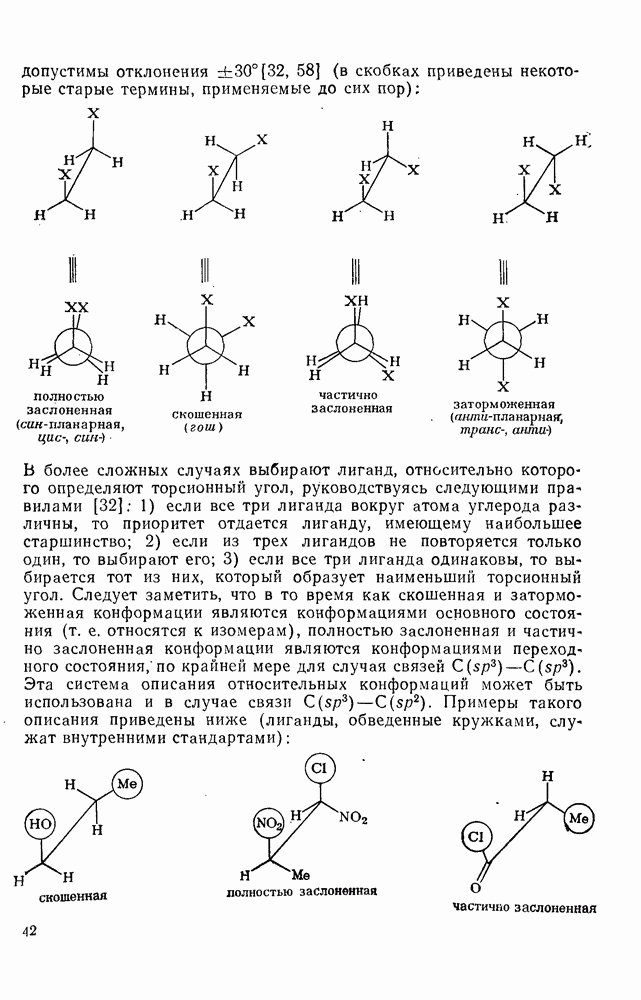
: а, б—лимитирующей стадией является образование  -комплекса; в — лимитирующей стадией является образование пары столкновения.
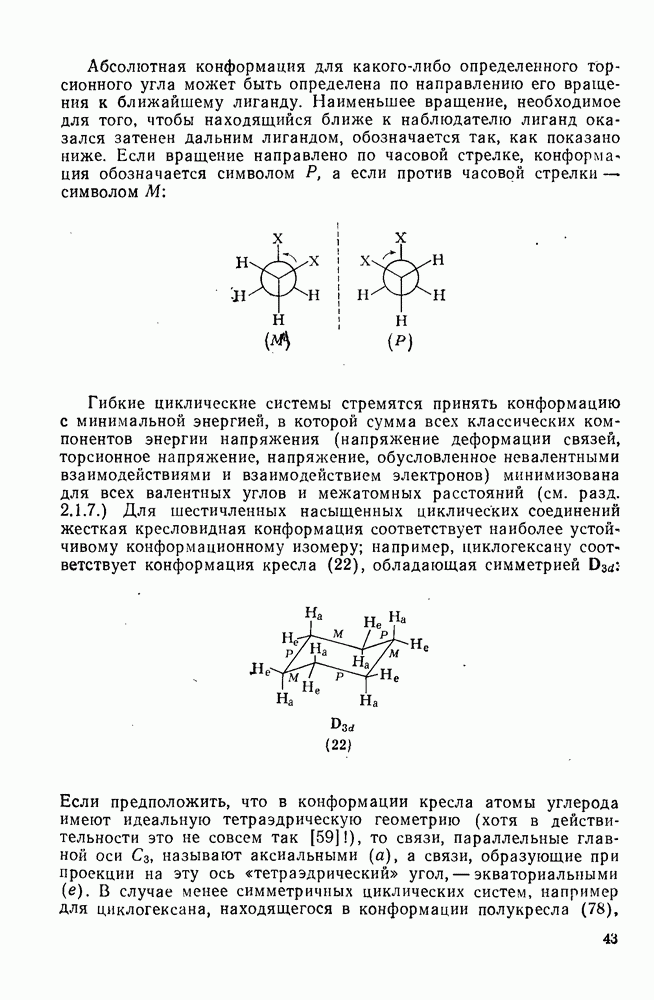
-комплекса; в — лимитирующей стадией является образование пары столкновения.
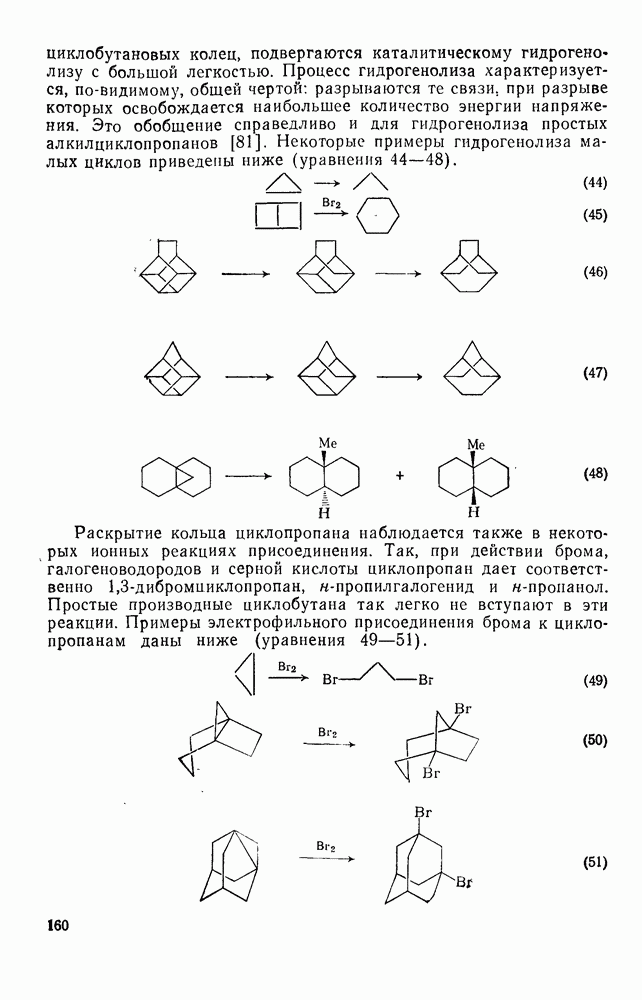
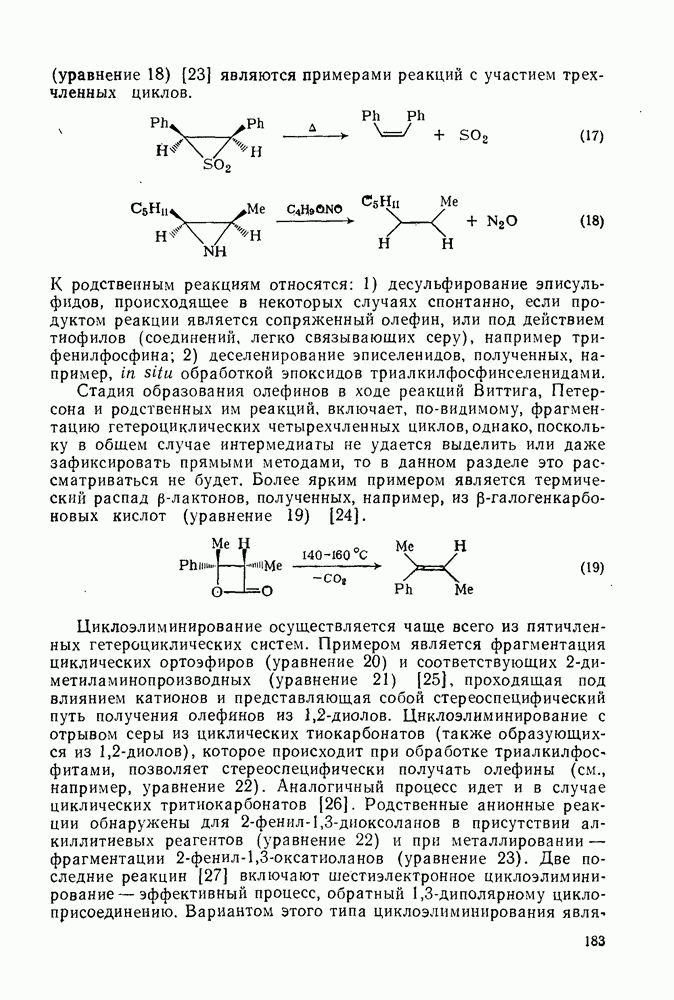
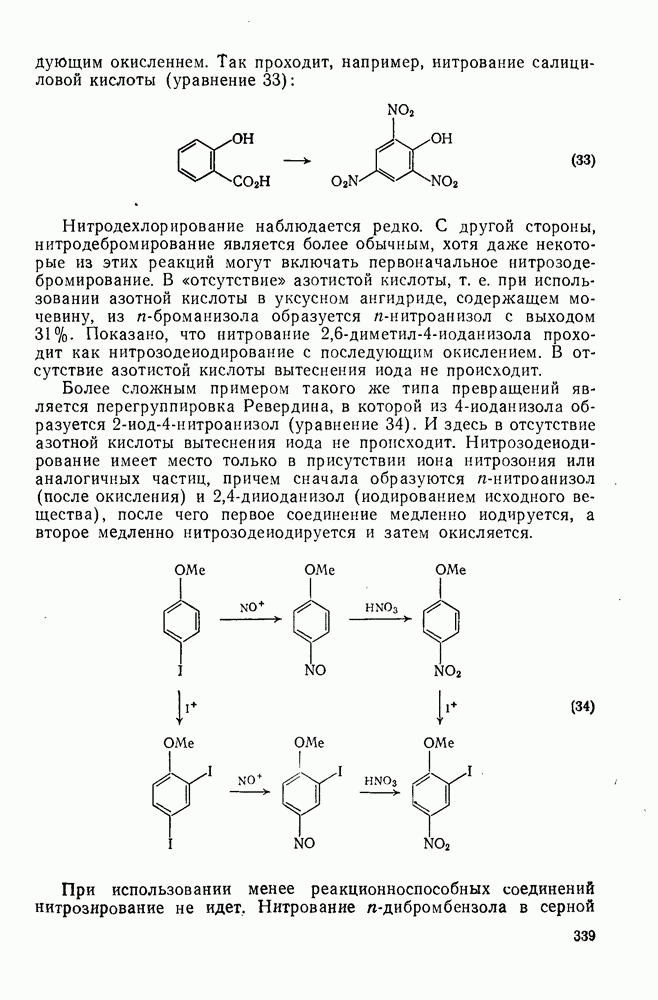
Хотя азотная кислота присутствует, главным образом, в виде ацетилнитрата, все же имеются некоторые сомнения относительно истинной природы электрофила. Так, азотная кислота в ускусном ангидриде реагирует с  -ксилолом с образованием смеси
-ксилолом с образованием смеси  -диметилнитробензола и
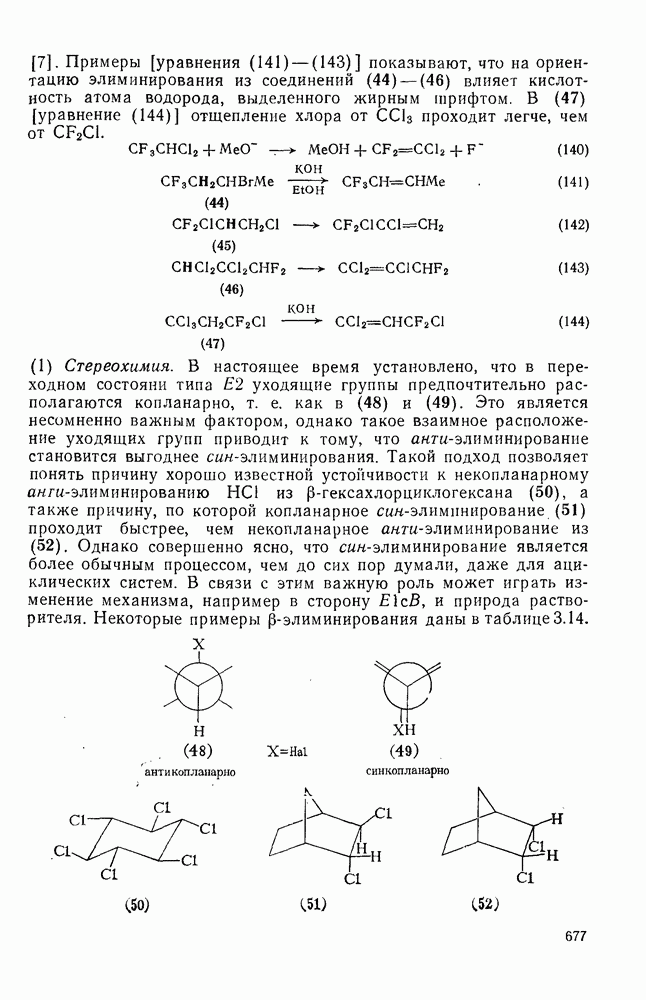
-диметилнитробензола и  -диметилфенилацетата. Выделение аддуктов, в которых ацетилнитрат присоединяется по замещенному положению (ипсо-атака), и тот факт, что эти аддукты разлагаются, очевидно, по внутримолекулярному механизму с образованием ацетата (57), свидетельствуют в пользу механизма, приведенного на схеме уравнений (30):
-диметилфенилацетата. Выделение аддуктов, в которых ацетилнитрат присоединяется по замещенному положению (ипсо-атака), и тот факт, что эти аддукты разлагаются, очевидно, по внутримолекулярному механизму с образованием ацетата (57), свидетельствуют в пользу механизма, приведенного на схеме уравнений (30):

Аналогичные аддукты (58)-(64) выделены в реакциях с другими аренами, а некоторые аддукты были обнаружены в качестве интермедиатов. При реакции азотной кислоты в уксусном ангидриде с n-трет-бутилтолуолом при низкой температуре получены и выделены три аддукта. Основным продуктом является  -аддукт (64), который, по-видимому, образуется в большем количестве, чем два других изомера, за счет пространственных затруднений в положении 4 (ипсо к трет-Ви). Другие нуклеофилы, например нитрат-ион и вода, также могут присоединяться к ипсо-замещенным
-аддукт (64), который, по-видимому, образуется в большем количестве, чем два других изомера, за счет пространственных затруднений в положении 4 (ипсо к трет-Ви). Другие нуклеофилы, например нитрат-ион и вода, также могут присоединяться к ипсо-замещенным  -комплекса
-комплекса  . Ипсо-Замещенные
. Ипсо-Замещенные  -комплексы можно регенерировать ацидолизом.
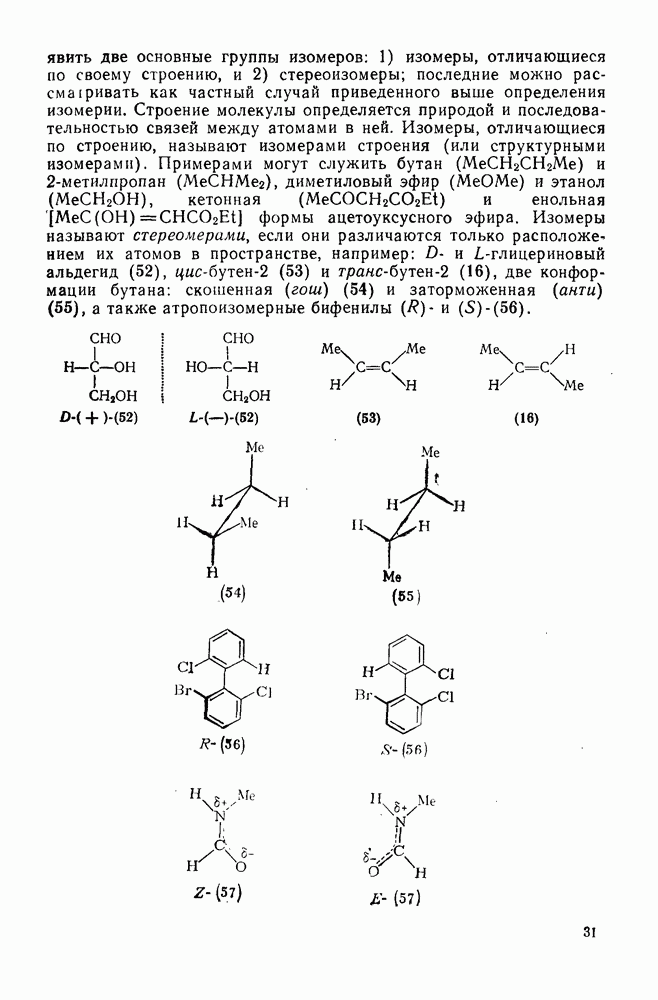
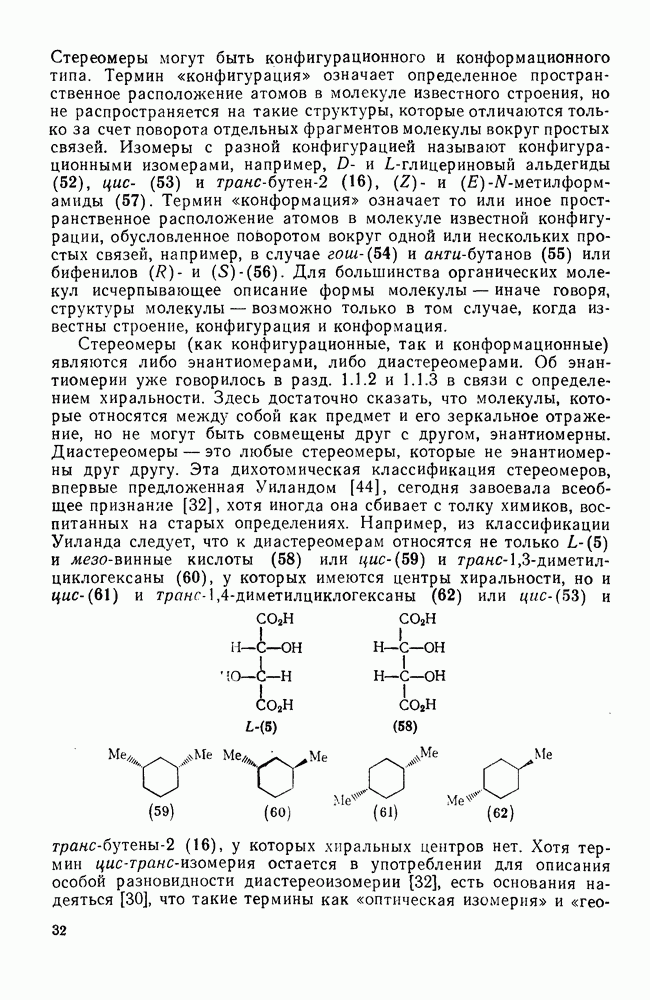
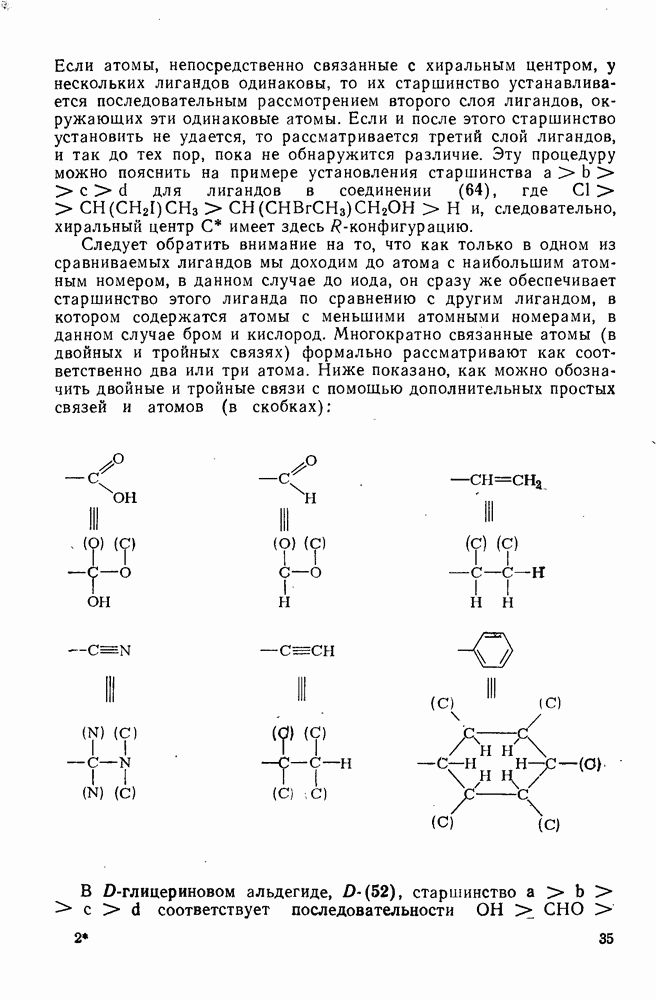
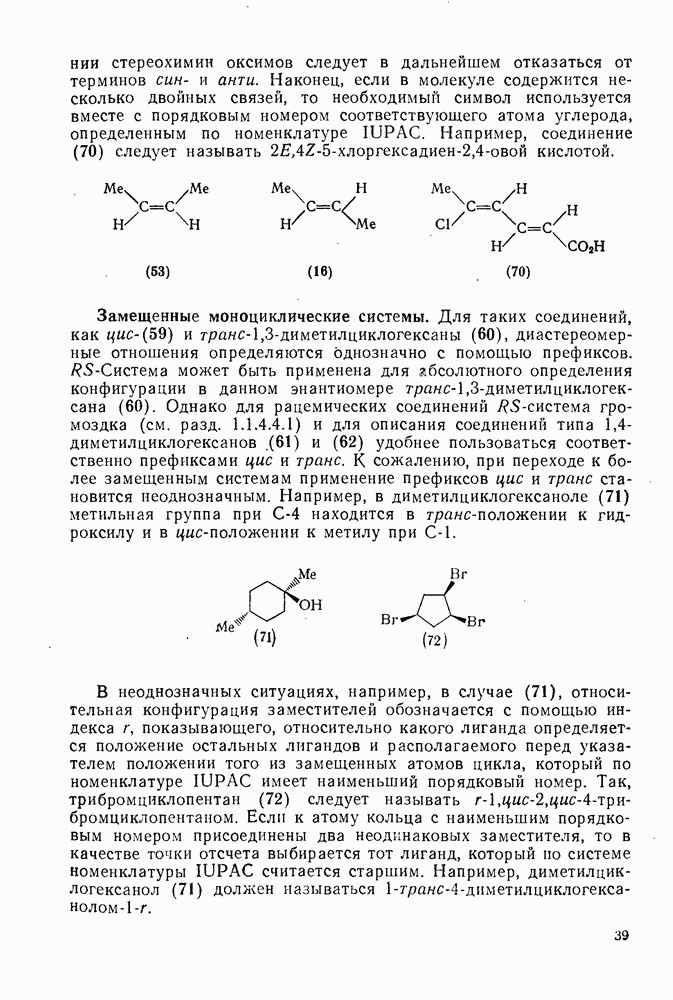
-комплексы можно регенерировать ацидолизом.

Нитрование о-ксилола в серной кислоте дает  -диметилнитробензол и
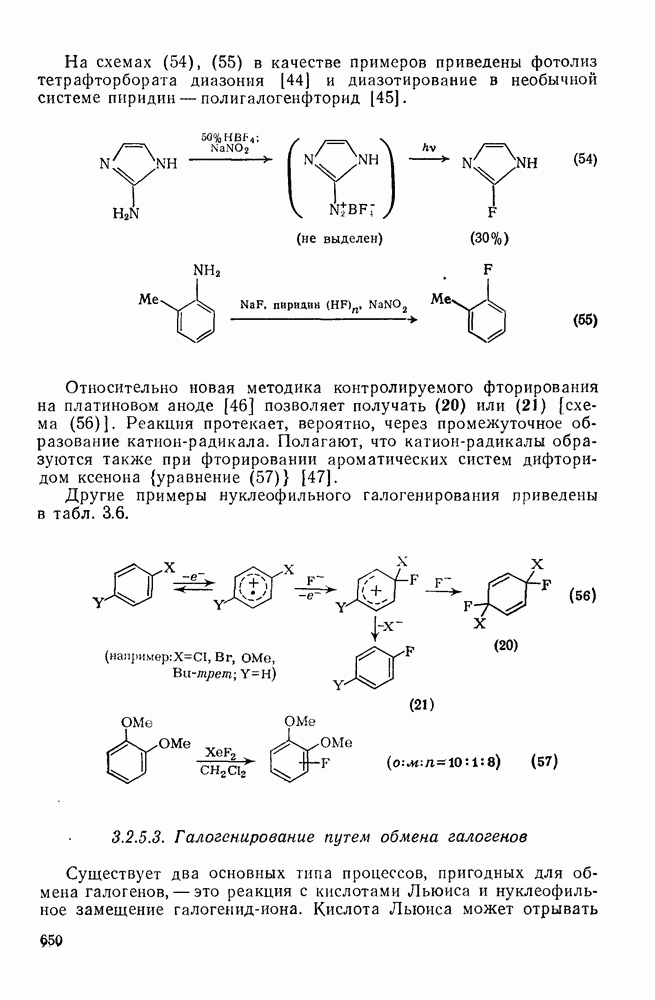
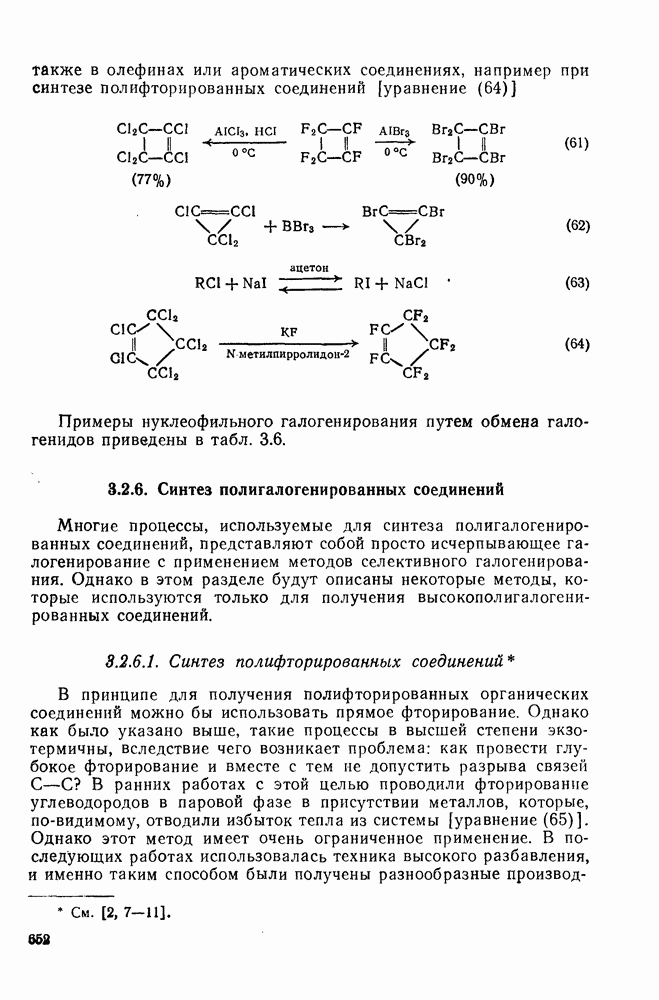
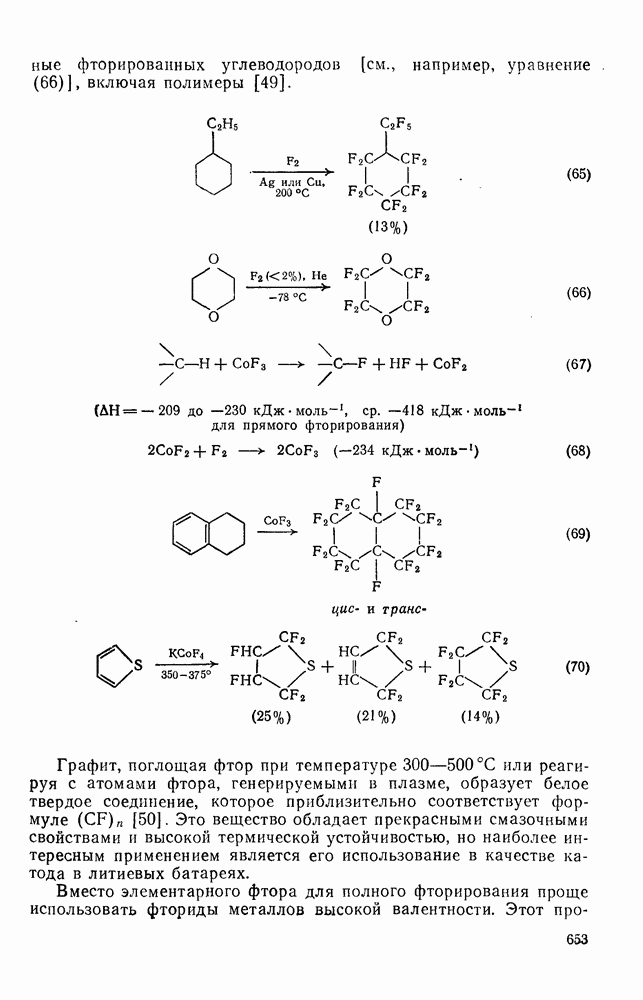
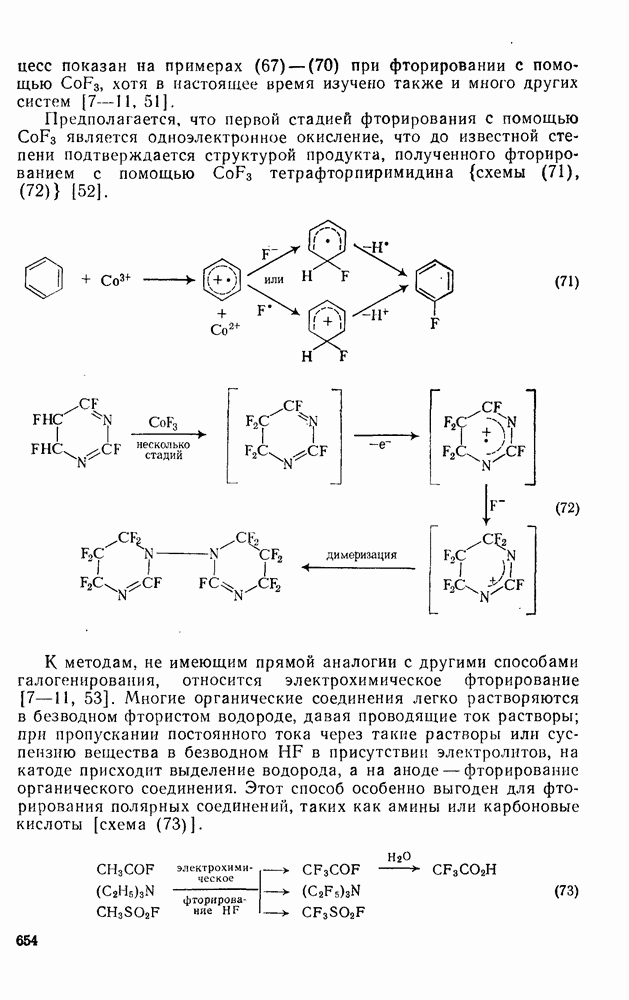
-диметилнитробензол и  -диметилнитробензол, выход которых зависит от кислотности системы. При низкой кислотности получаются также нитродиметилфенолы. При ацидолизе эфиров (58) и (59) образуется
-диметилнитробензол, выход которых зависит от кислотности системы. При низкой кислотности получаются также нитродиметилфенолы. При ацидолизе эфиров (58) и (59) образуется  -диметилнитробензол, выход которого увеличивается при возрастании кислотности; это подтверждает предположение, что нитрование ортоположения к алкильной группе, вероятно, часто включает первоначальную ипсо-атаку. Тот факт, что при ацидолизе (58) и (59) не образуется
-диметилнитробензол, выход которого увеличивается при возрастании кислотности; это подтверждает предположение, что нитрование ортоположения к алкильной группе, вероятно, часто включает первоначальную ипсо-атаку. Тот факт, что при ацидолизе (58) и (59) не образуется  -диметилнитробензола, означает, что первоначальный ипсо-замещенный о-комплекс не превращается вновь в «пару столкновения» и что в нем не происходят ни
-диметилнитробензола, означает, что первоначальный ипсо-замещенный о-комплекс не превращается вновь в «пару столкновения» и что в нем не происходят ни  , ни последовательные
, ни последовательные  -сдвиги нитрогруппы. Эти результаты обобщены в уравнениях (31). Увеличение выхода
-сдвиги нитрогруппы. Эти результаты обобщены в уравнениях (31). Увеличение выхода  и
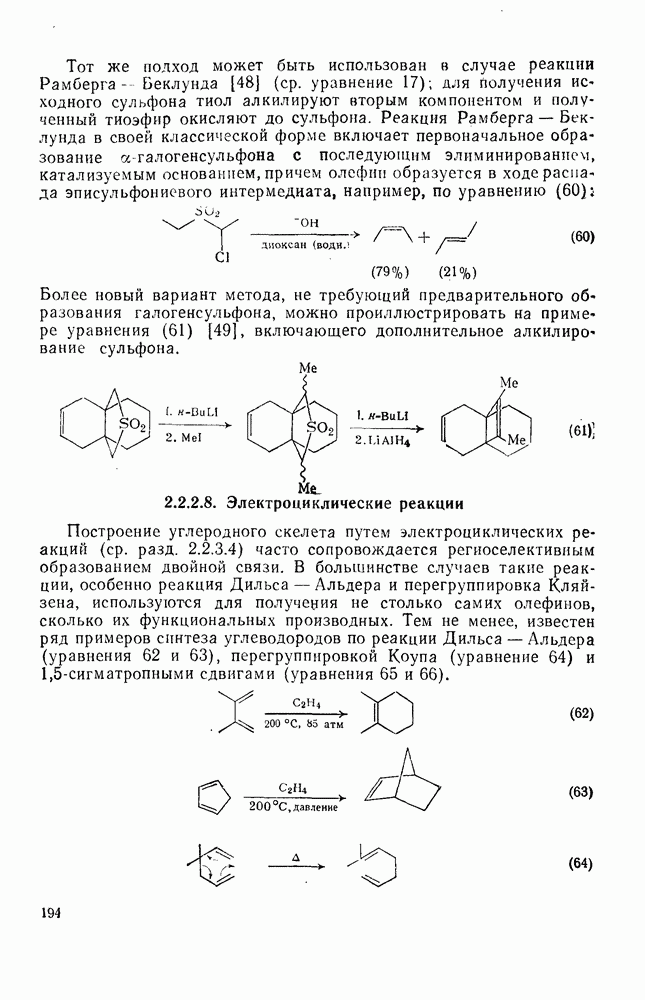
и  при возрастании кислотности среды при нитровании
при возрастании кислотности среды при нитровании  -триметилбензола в серной кислоте приписано миграции нитрогруппы от
-триметилбензола в серной кислоте приписано миграции нитрогруппы от  к
к  и от
и от  к
к  соответственно.
соответственно.

В настоящее время не известно ни одного достоверного примера последовательных внутримолекулярных  -сдвигов через незамещенное положение. Однако известны последовательные
-сдвигов через незамещенное положение. Однако известны последовательные  -сдвиги из одного ипсо-положения через другое ипсо-положение. Например, при ацидолизе (65) образуются структурные изомеры (66) и (67), как показано в уравнении (32).
-сдвиги из одного ипсо-положения через другое ипсо-положение. Например, при ацидолизе (65) образуются структурные изомеры (66) и (67), как показано в уравнении (32).

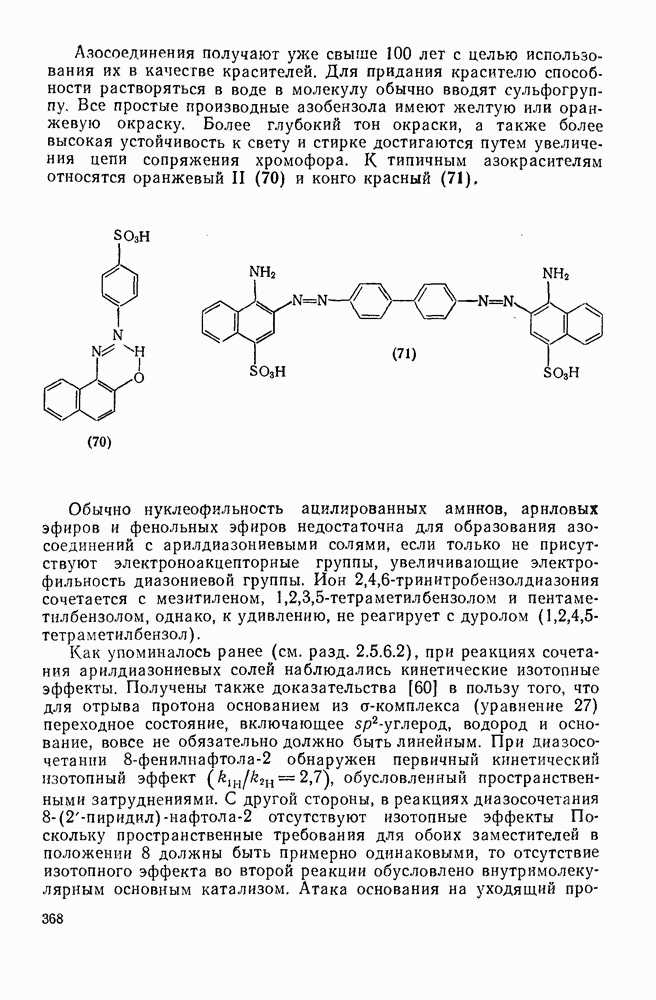
Раньше упоминалось об отщеплении ипсо-заместителя, например в случае протодесилилирования и протодесульфирования (см. с. 330,331). С этой точки зрения мы обсудим только замещение ипсо-заместителя на нитрогруппу. Многие реакции этого типа известны уже с давних пор, однако их механизм, как правило, не выяснен. К таким реакциям относятся дезалкилирование, дезацилирование, десилилирование, десульфирование, декарбоксилирование, дедиазотирование и дегалогенирование [20]. Реакции с участием субстратов, сильно активированных по отношению к электрофильной атаке, могут включать первоначальное нитрозированце с последующим окислением.
Так проходит, например, нитрование салициловой кислоты (уравнение 33):

Нитродехлорирование наблюдается редко. С другой стороны, нитродебромирование является более обычным, хотя даже некоторые из этих реакций могут включать первоначальное нитрозодебромирование. В «отсутствие» азотистой кислоты, т. е. при использовании азотной кислоты в уксусном ангидриде, содержащем мочевину, из  -броманизола образуется
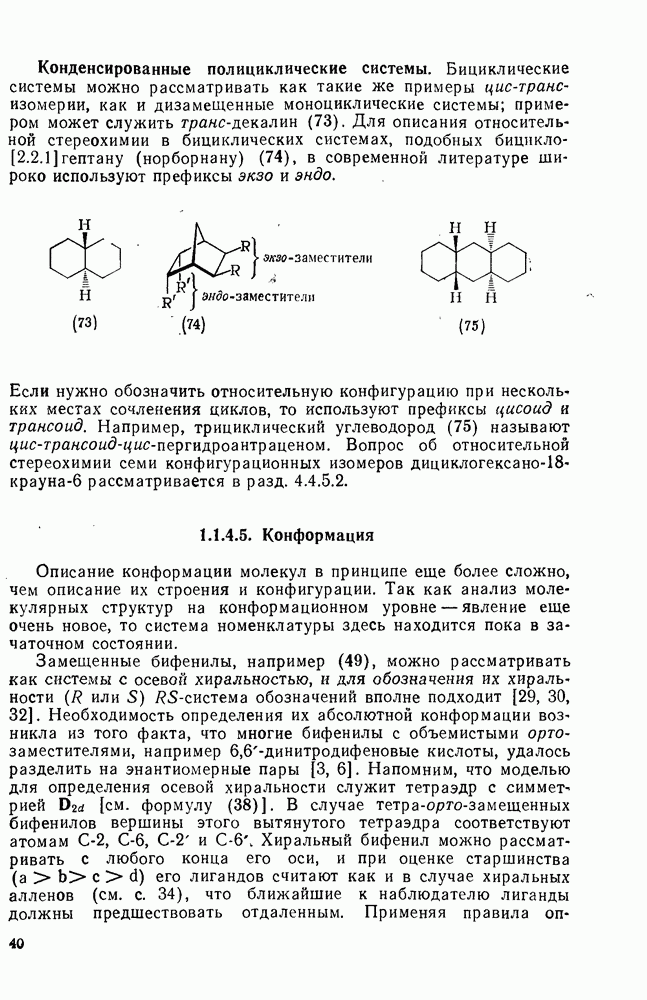
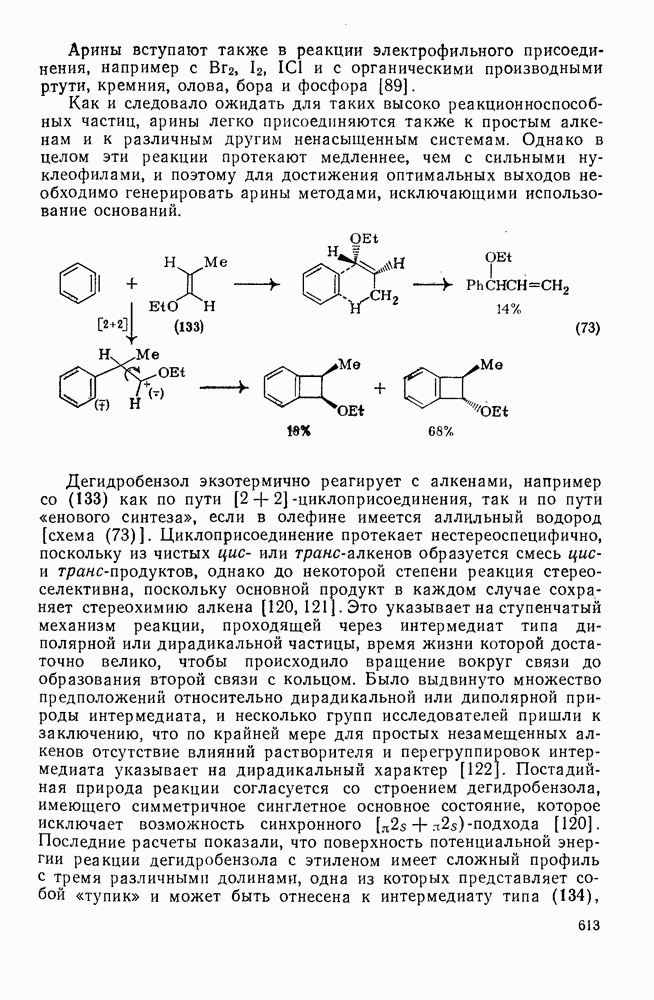
-броманизола образуется  -нитроанизол с выходом
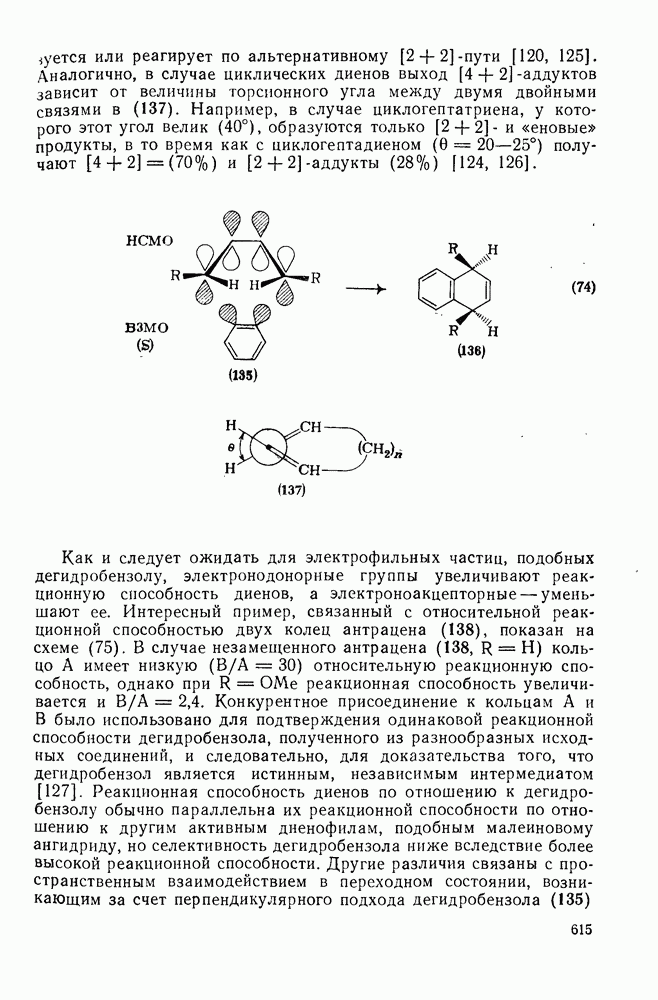
-нитроанизол с выходом  . Показано, что нитрование
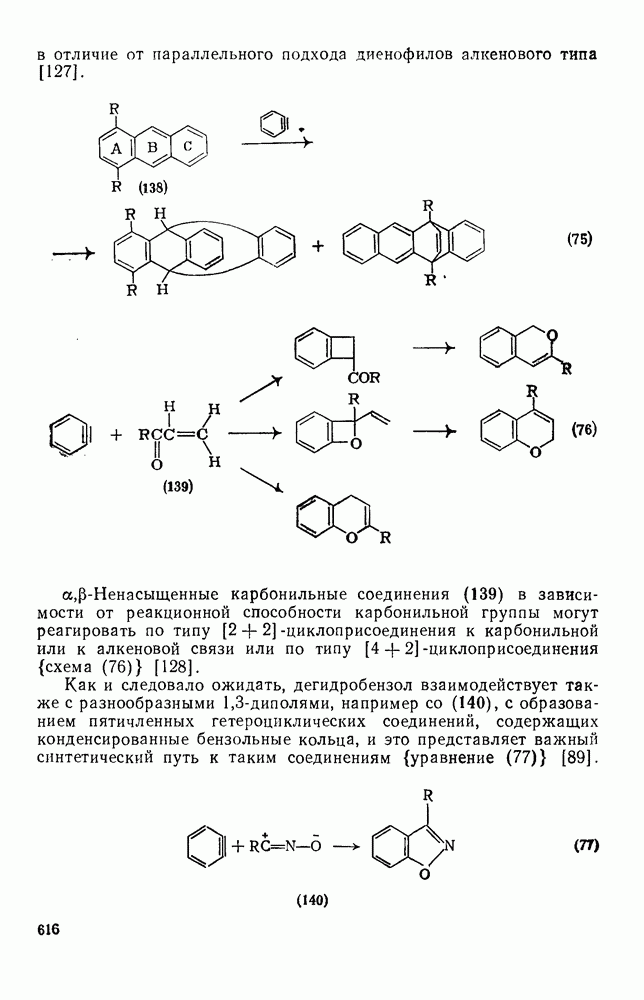
. Показано, что нитрование  проходит как нитрозодеиодирование с последующим окислением. В отсутствие азотистой кислоты вытеснения иода не происходит.
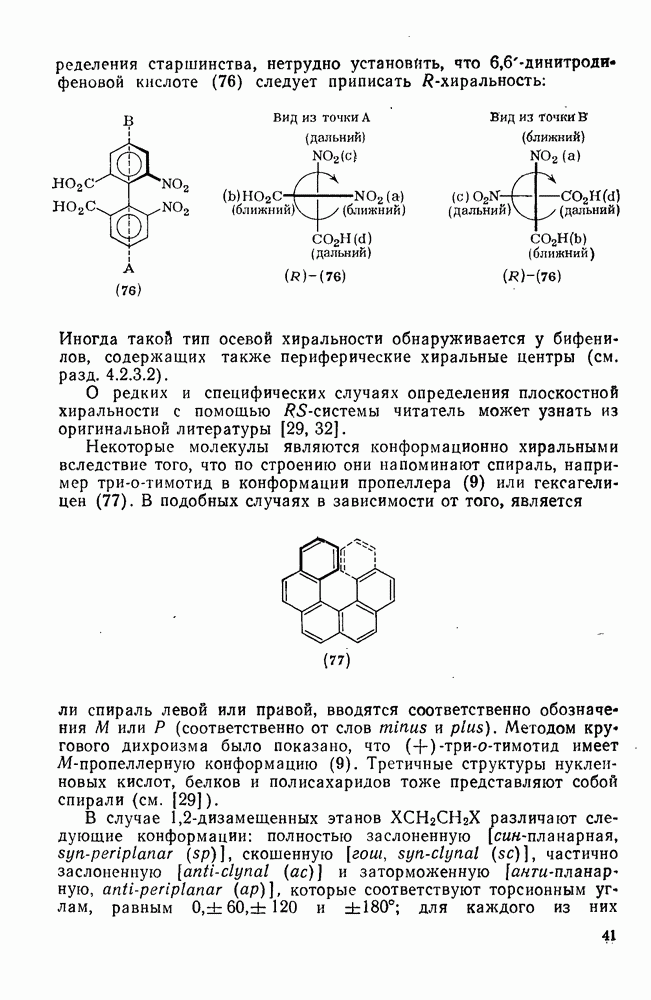
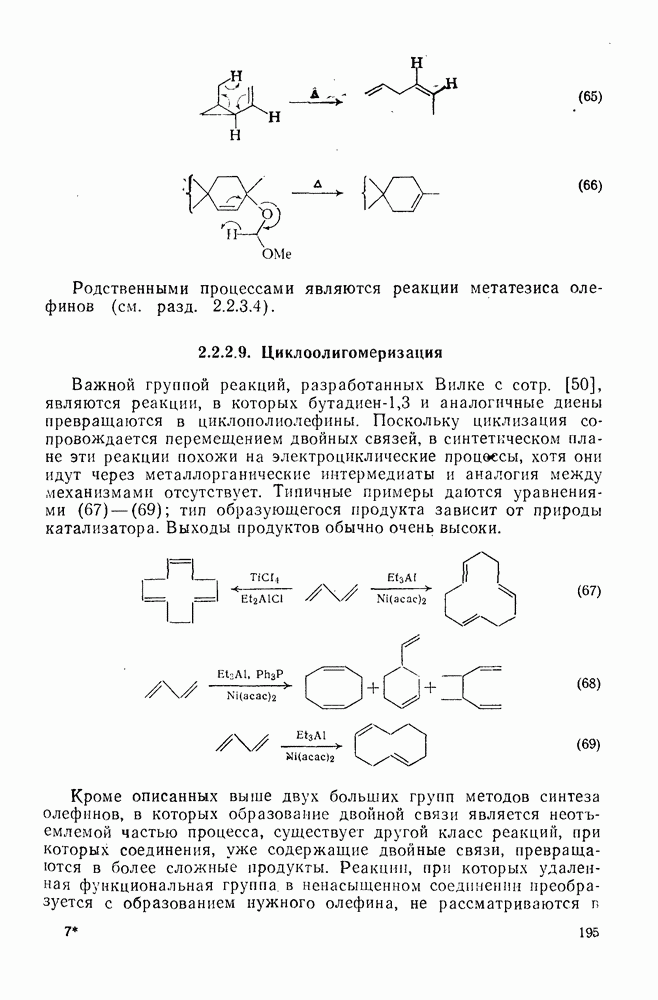
проходит как нитрозодеиодирование с последующим окислением. В отсутствие азотистой кислоты вытеснения иода не происходит.
Более сложным примером такого же типа превращений является перегруппировка Ревердина, в которой из  -иоданизола образуется
-иоданизола образуется  (уравнение 34). И здесь в отсутствие азотной кислоты вытеснения иода не происходит. Нитрозодеиодирование имеет место только в присутствии иона нитрозония или аналогичных частиц, причем сначала образуются
(уравнение 34). И здесь в отсутствие азотной кислоты вытеснения иода не происходит. Нитрозодеиодирование имеет место только в присутствии иона нитрозония или аналогичных частиц, причем сначала образуются  -нитроанизол (после окисления) и
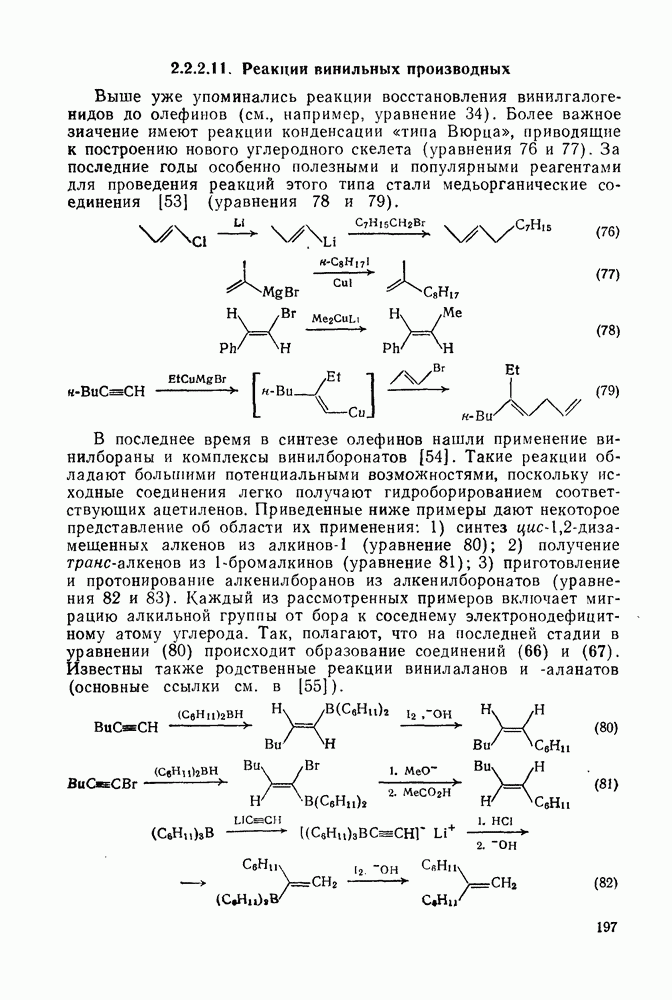
-нитроанизол (после окисления) и  -дииоданизол (иодированием исходного вещества), после чего первое соединение медленно иодируется, а второе медленно нитрозодеиодируется и затем окисляется.
-дииоданизол (иодированием исходного вещества), после чего первое соединение медленно иодируется, а второе медленно нитрозодеиодируется и затем окисляется.

При использовании менее реакционноспособных соединений нитрозирование не идет. Нитрование  -дибромбензола в серной кислоте (69%) приводит к
-дибромбензола в серной кислоте (69%) приводит к  -дибромнитробензолу и
-дибромнитробензолу и  -бромнитробензолу (60 и 34% соответственно) наряду с небольшим количеством
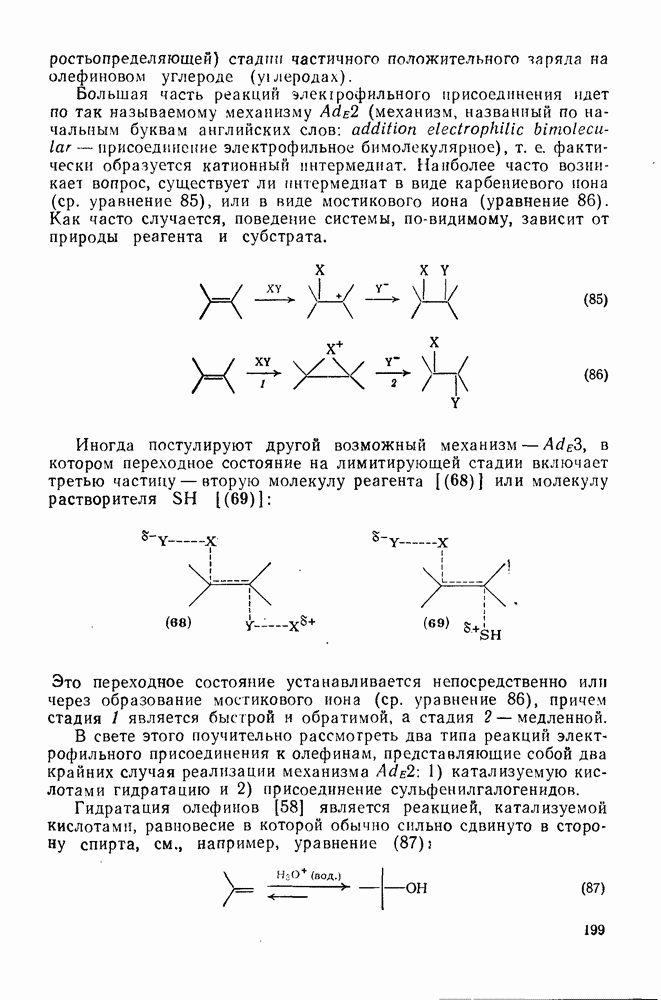
-бромнитробензолу (60 и 34% соответственно) наряду с небольшим количеством  -трибромбензола.
-трибромбензола.
Вполне возможно, что  -дибромнитробензол образуется частично за счет
-дибромнитробензол образуется частично за счет  -миграции в ипсо-
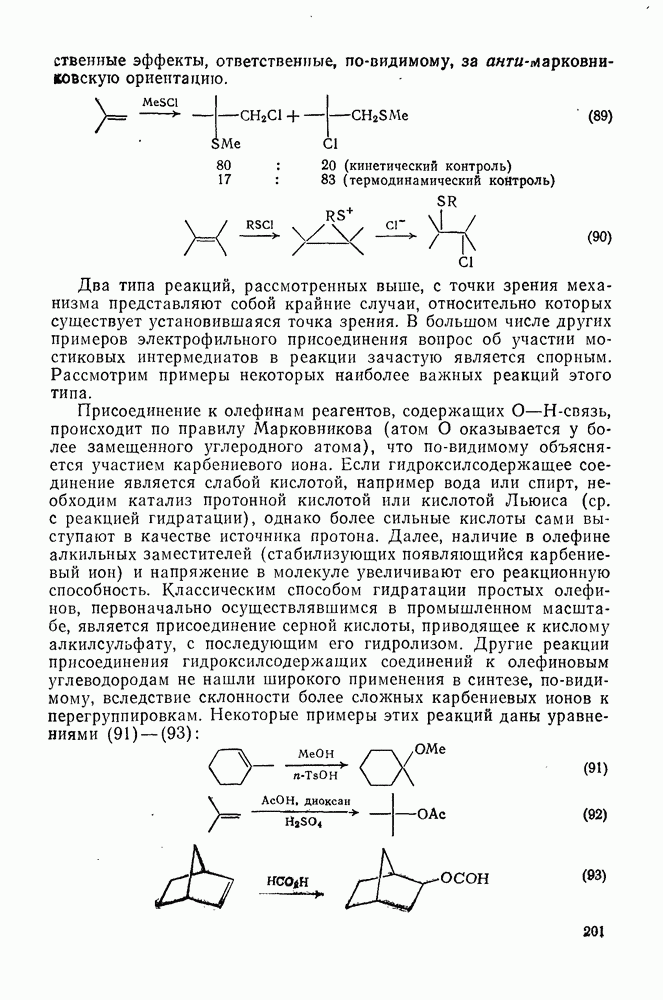
-миграции в ипсо- -комплексе, а также в результате прямого нитрования исходного вещества.
-комплексе, а также в результате прямого нитрования исходного вещества.
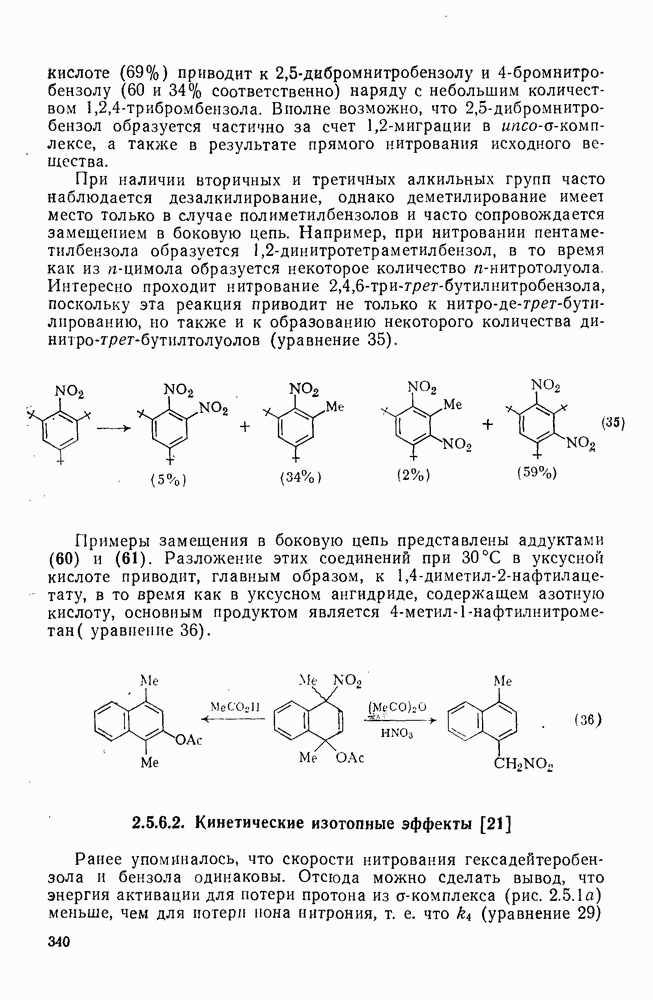
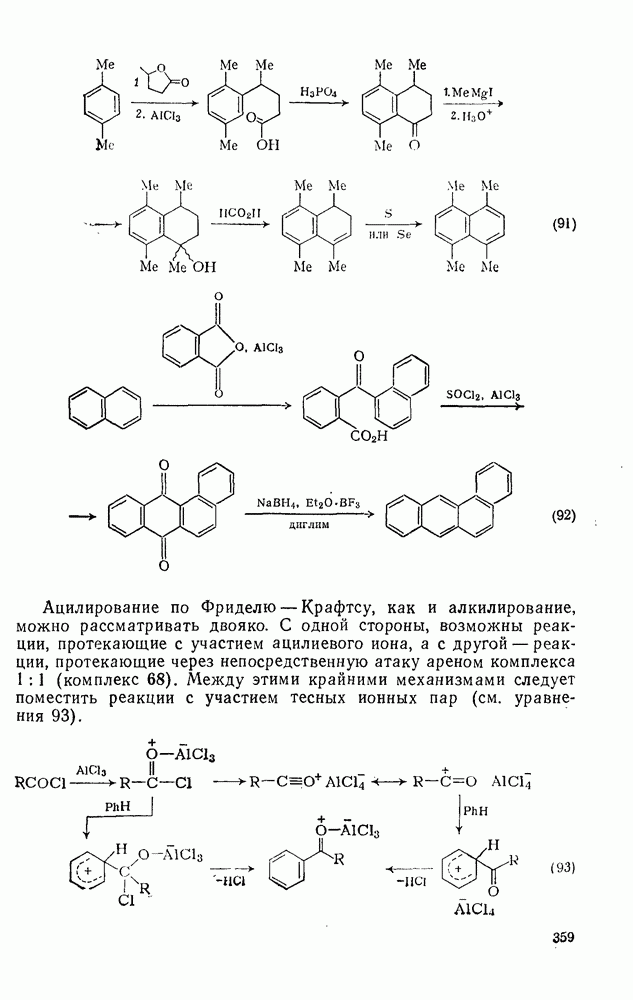
При наличии вторичных и третичных алкильных групп часто наблюдается дезалкилирование, однако деметилирование имеет место только в случае полиметилбензолов и часто сопровождается замещением в боковую цепь. Например, при нитровании пентаметилбензола образуется  -динитротетраметилбензол, в то время как из
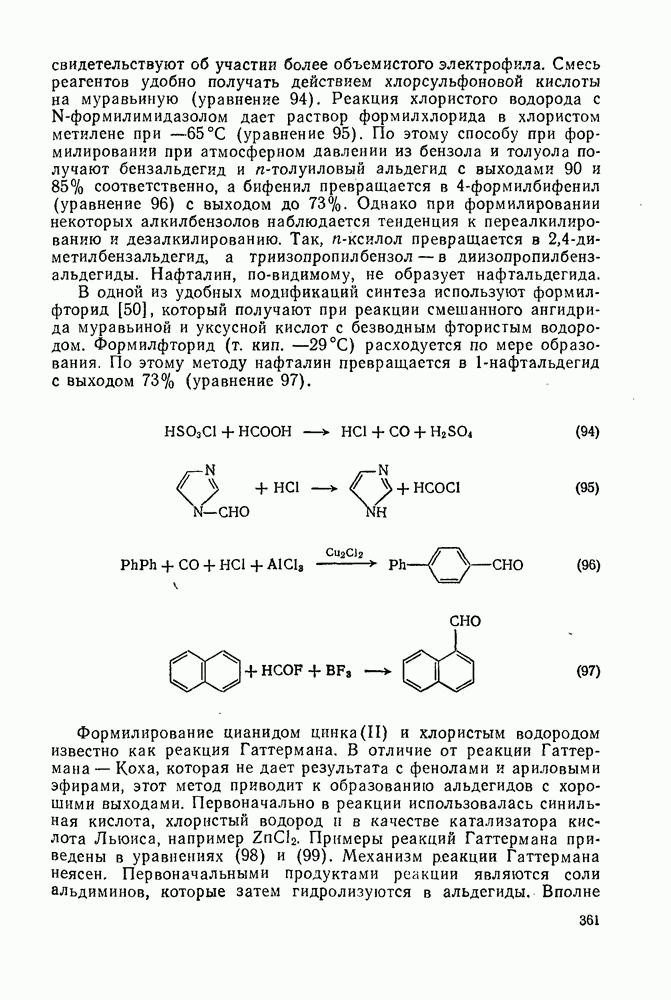
-динитротетраметилбензол, в то время как из  -цимола образуется некоторое количество n-нитротолуола. Интересно проходит нитрование
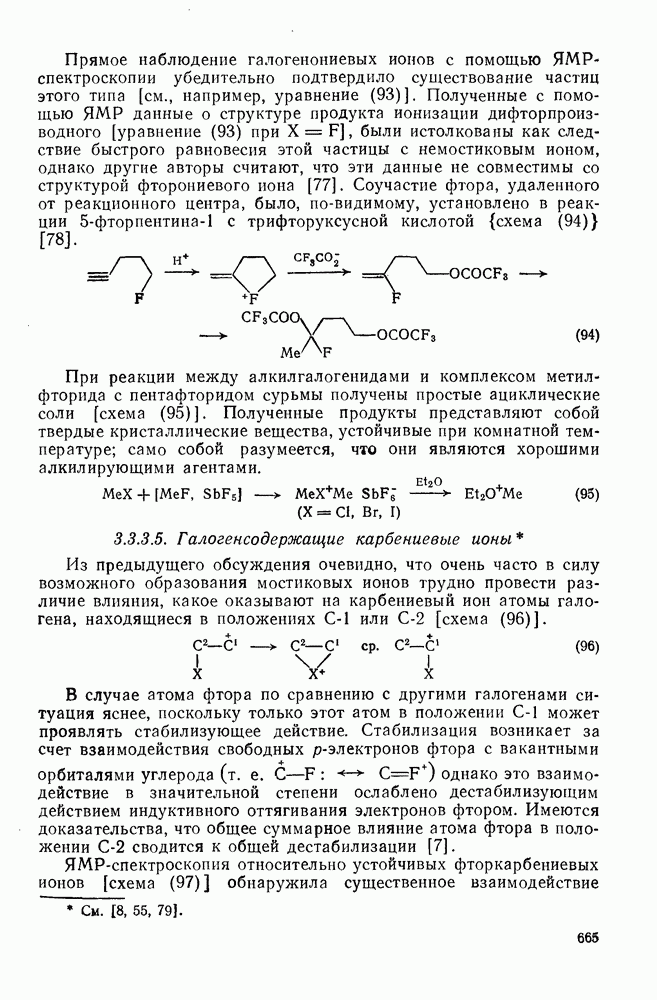
-цимола образуется некоторое количество n-нитротолуола. Интересно проходит нитрование  -три-трет-бутилнитробензола, поскольку эта реакция приводит не только к нитро-де-трет-бутилированию, но также и к образованию некоторого количества ди-нитро-трет-бутилтолуолов (уравнение 35).
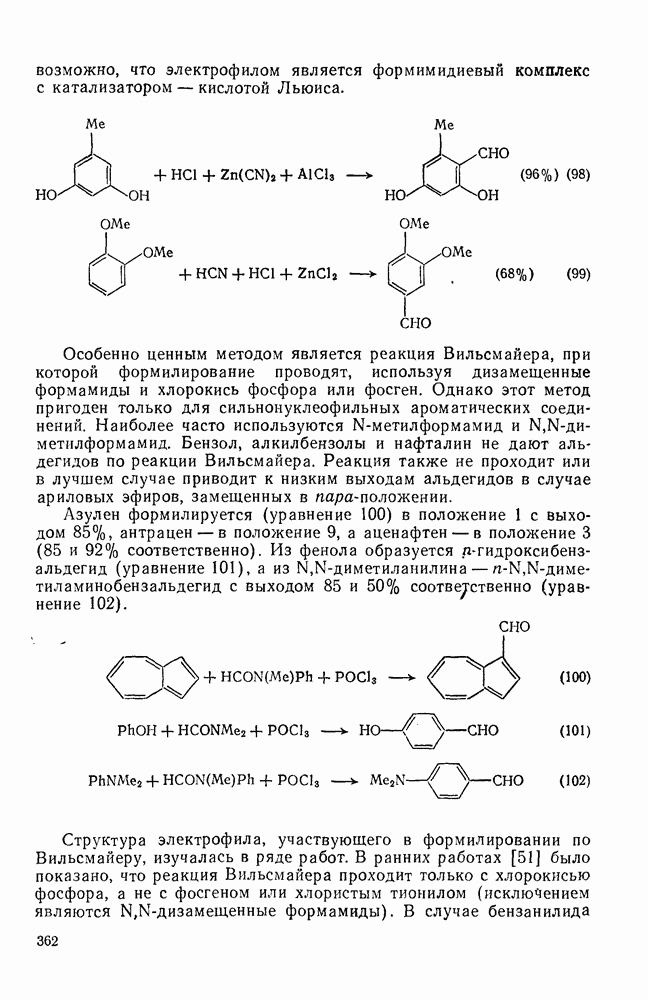
-три-трет-бутилнитробензола, поскольку эта реакция приводит не только к нитро-де-трет-бутилированию, но также и к образованию некоторого количества ди-нитро-трет-бутилтолуолов (уравнение 35).

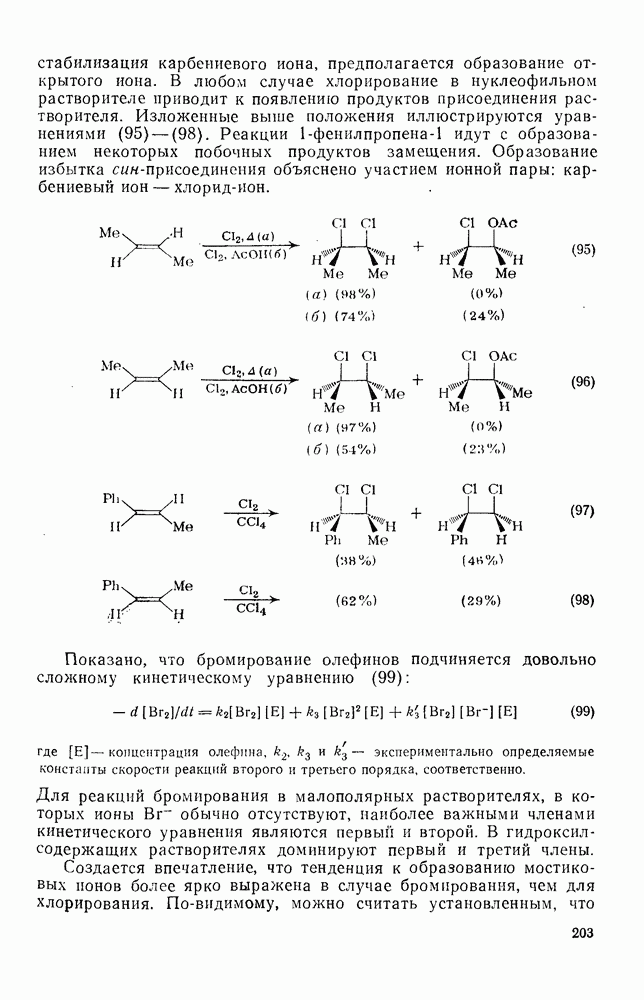
Примеры замещения в боковую цепь представлены аддуктами (60) и (61). Разложение этих соединений при  в уксусной кислоте приводит, главным образом, к
в уксусной кислоте приводит, главным образом, к  , в то время как в уксусном ангидриде, содержащем азотную кислоту, основным продуктом является
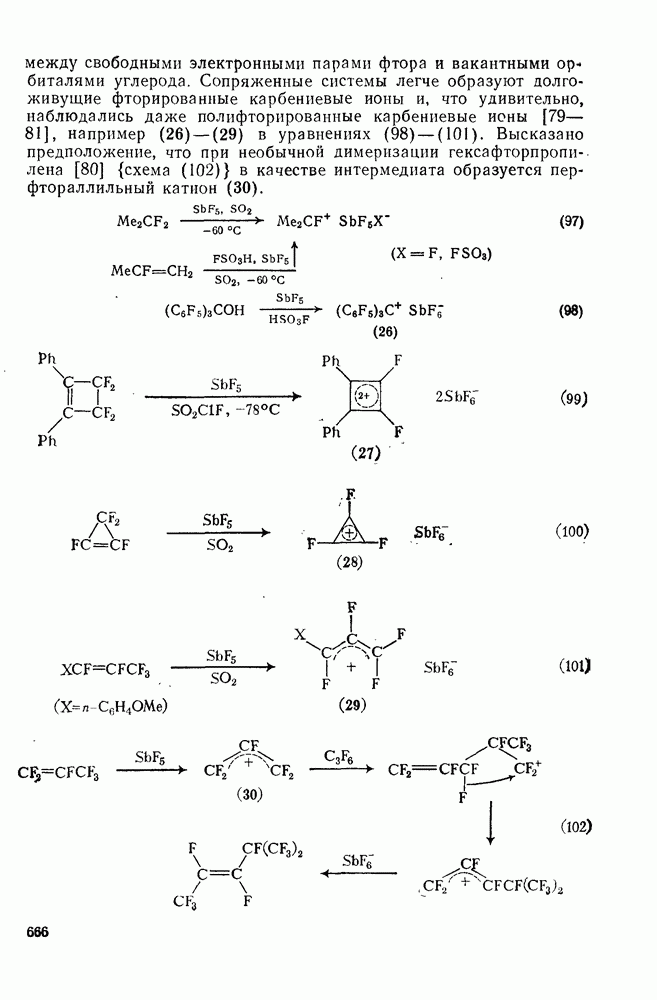
, в то время как в уксусном ангидриде, содержащем азотную кислоту, основным продуктом является  -метил-
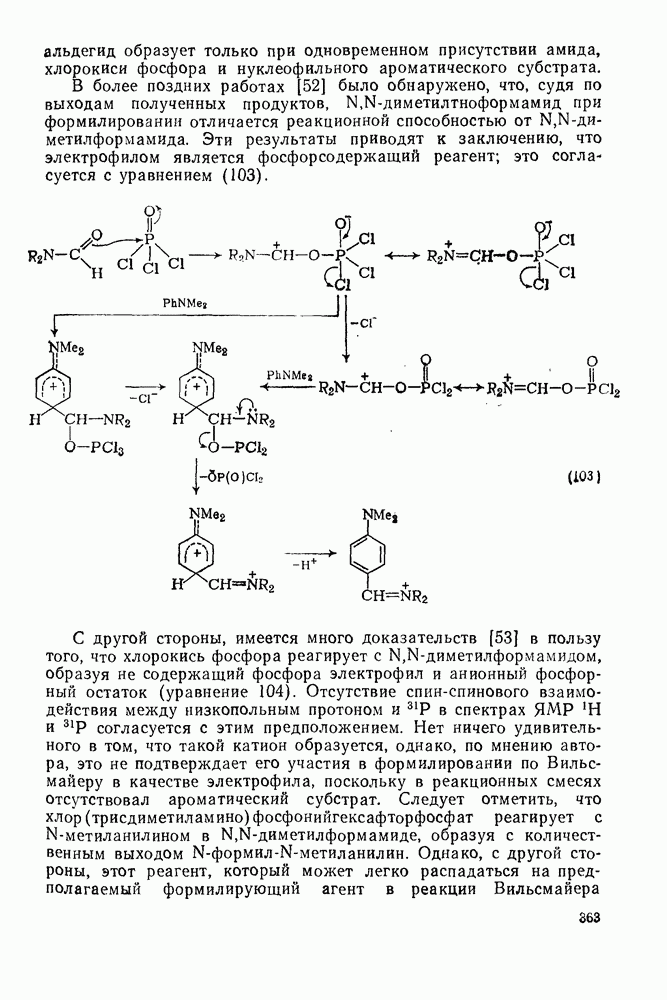
-метил-  уравнение 36).
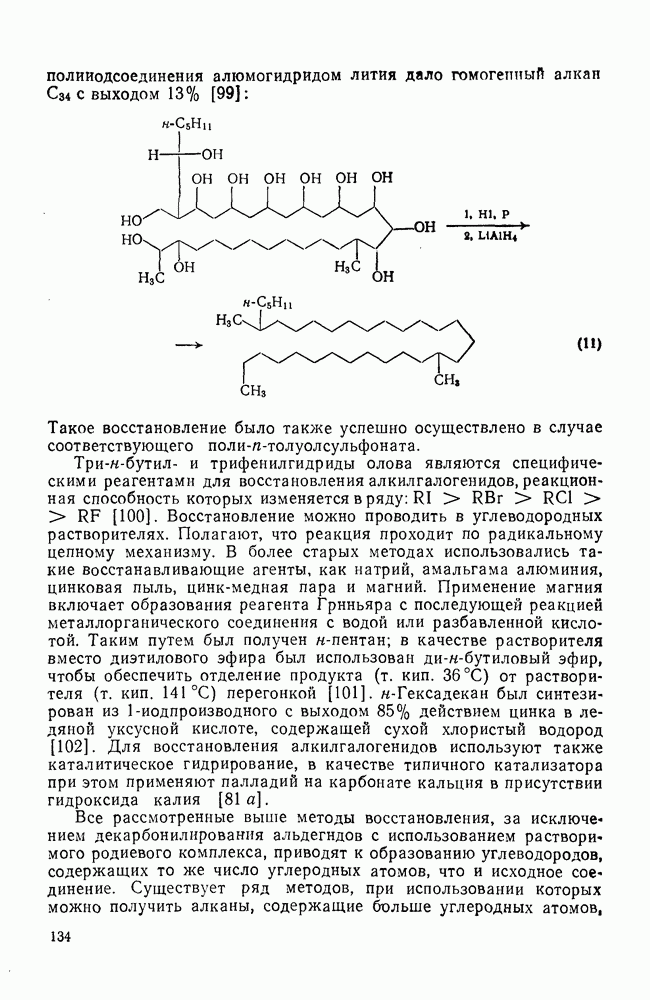
уравнение 36).