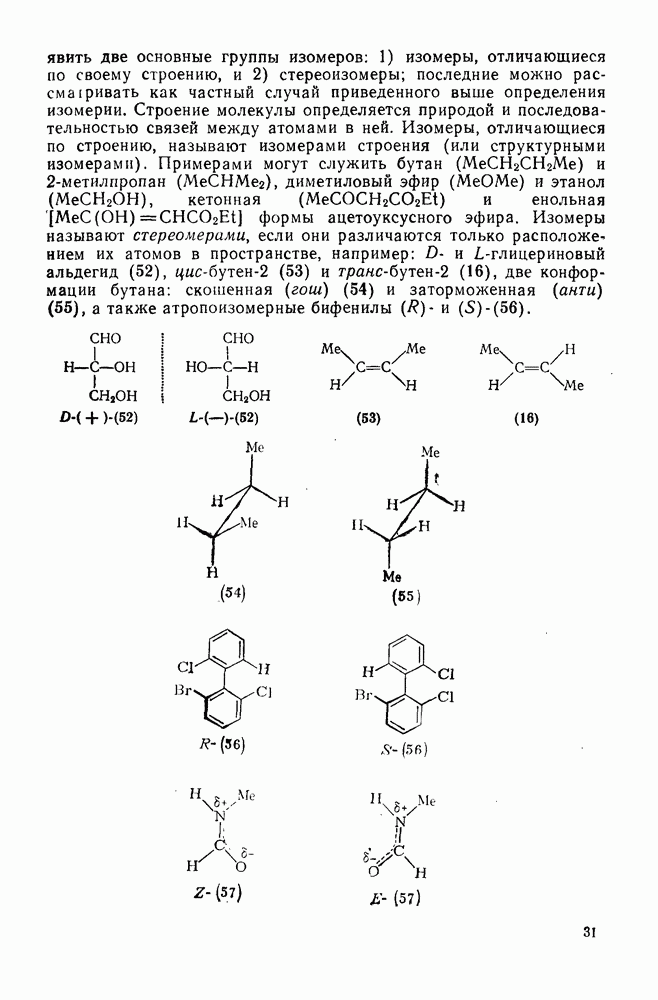
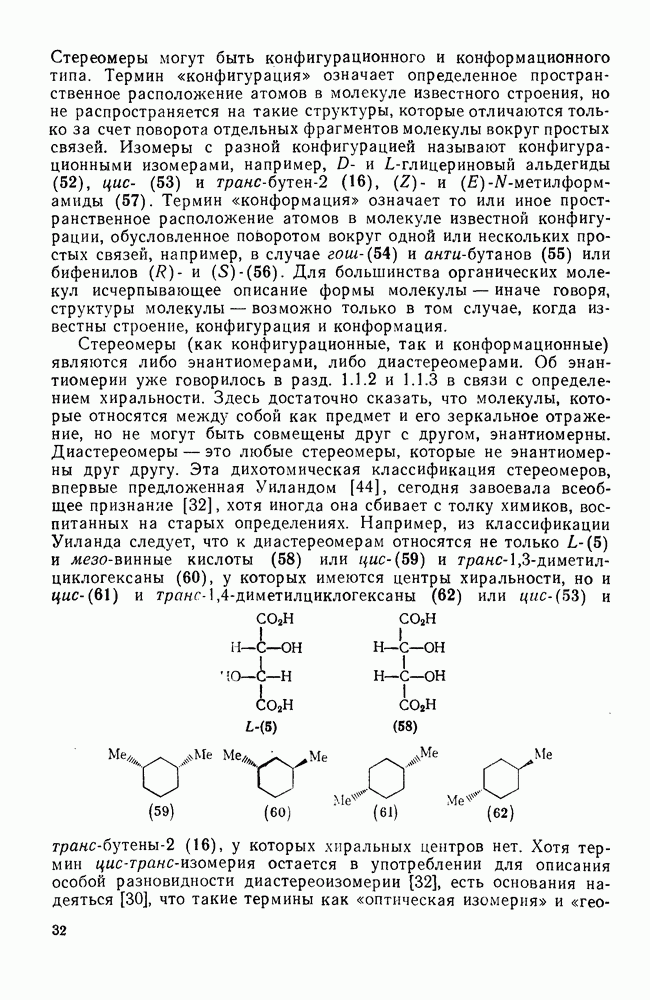
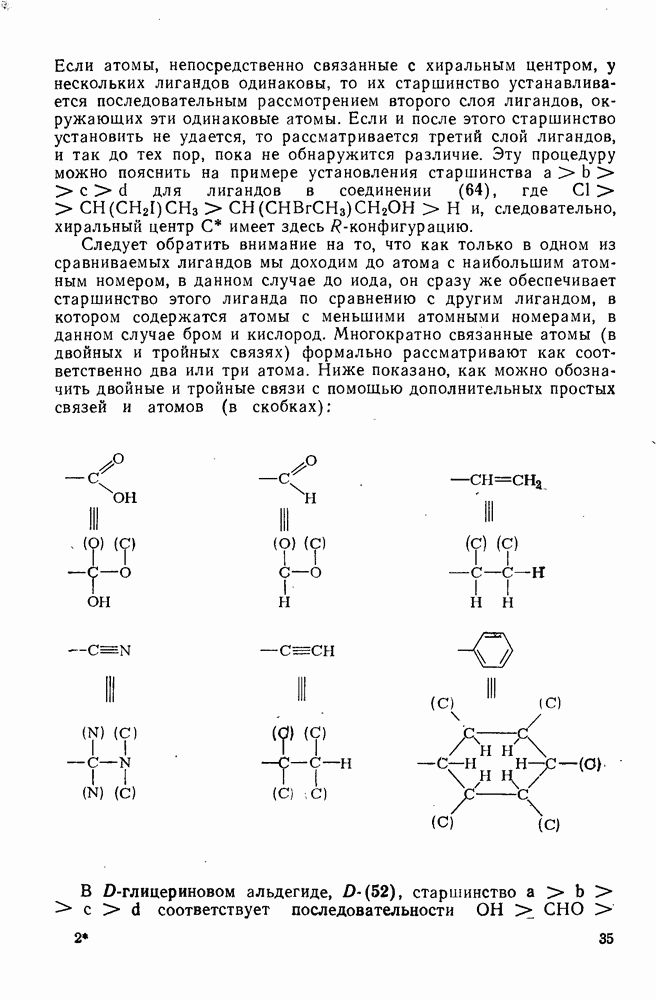
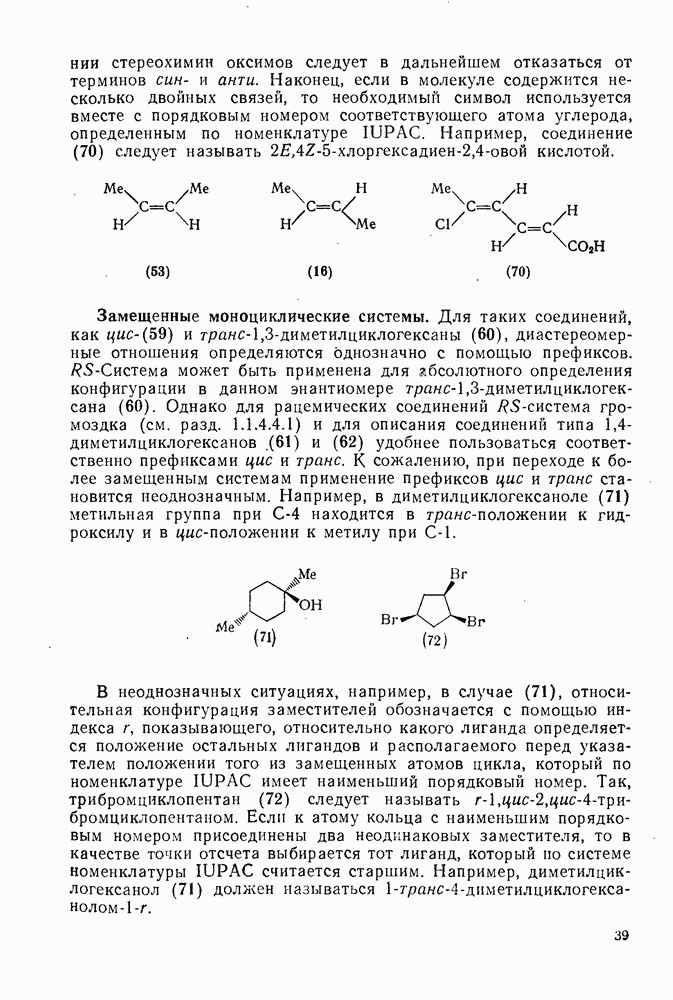
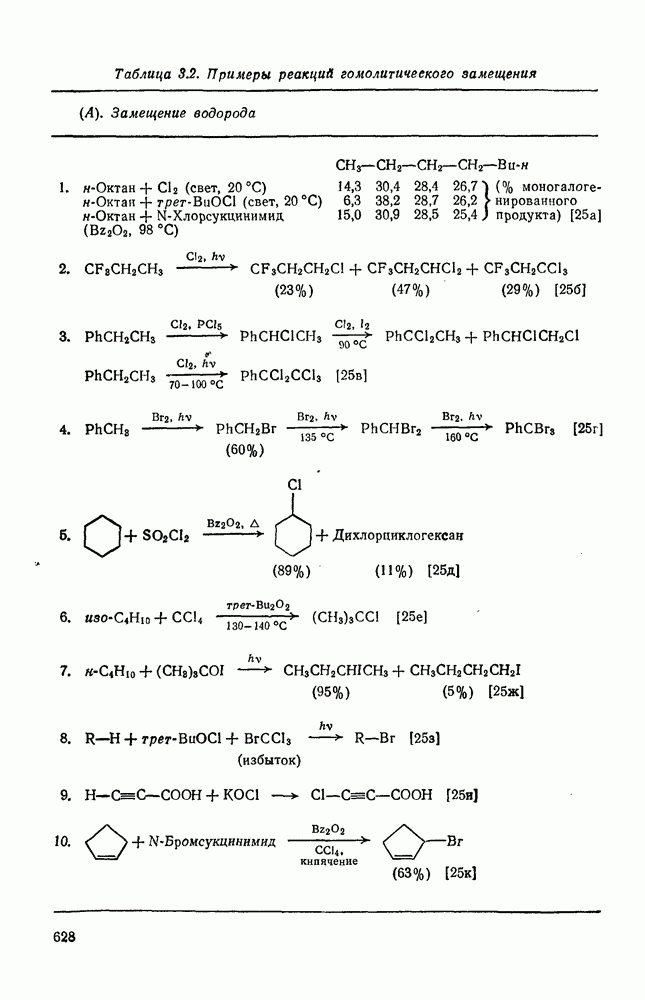
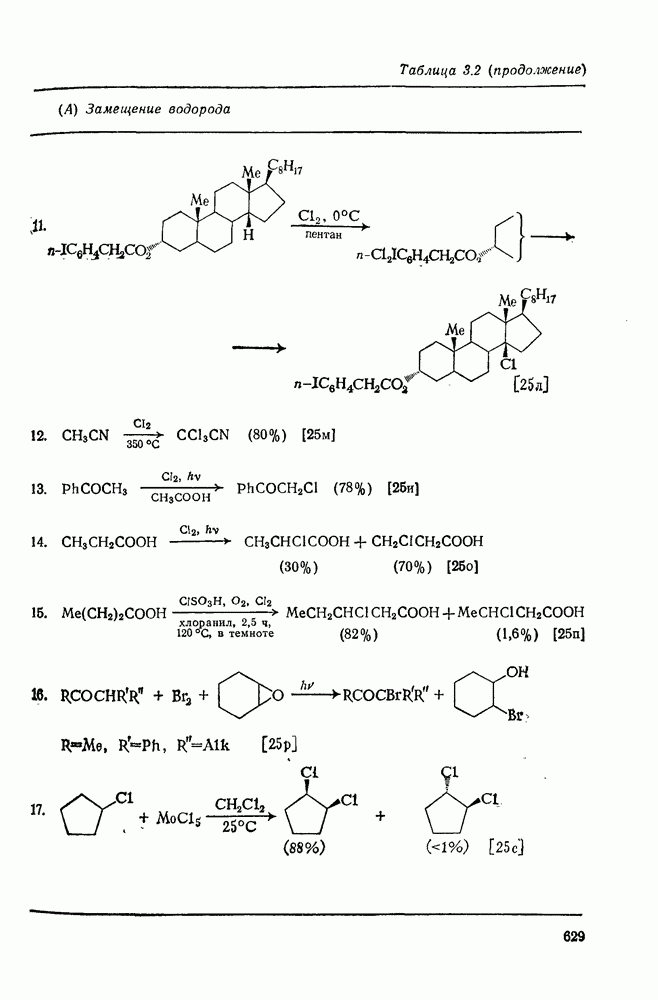
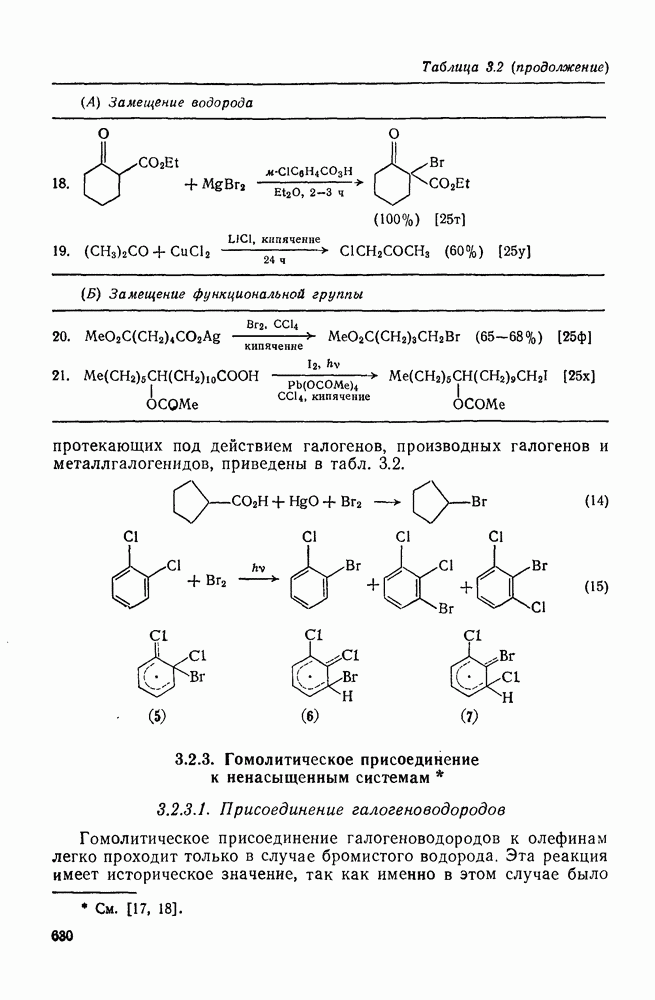
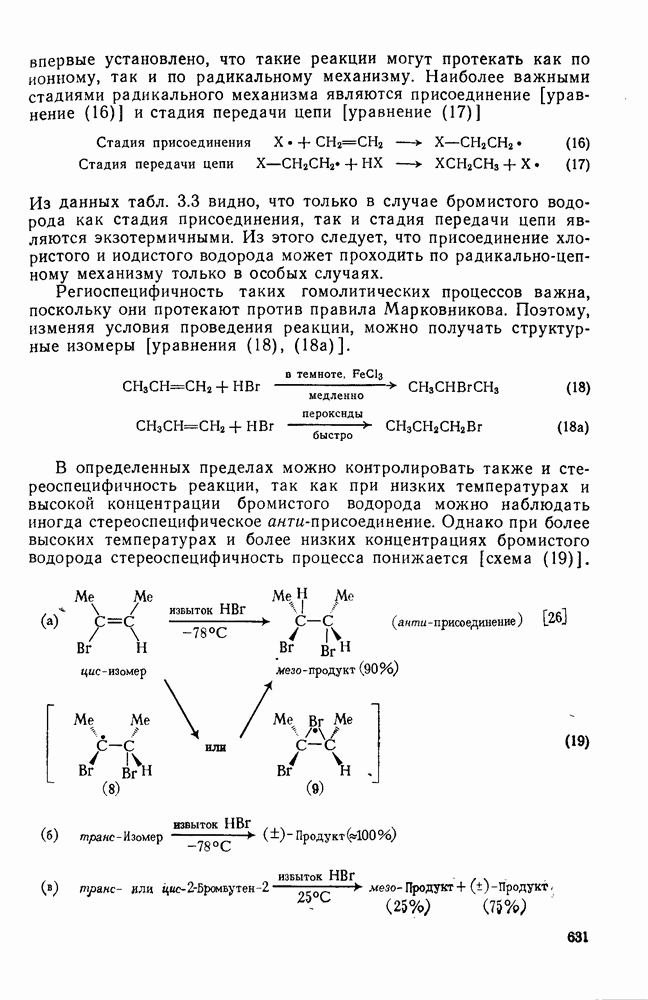
Пред.
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
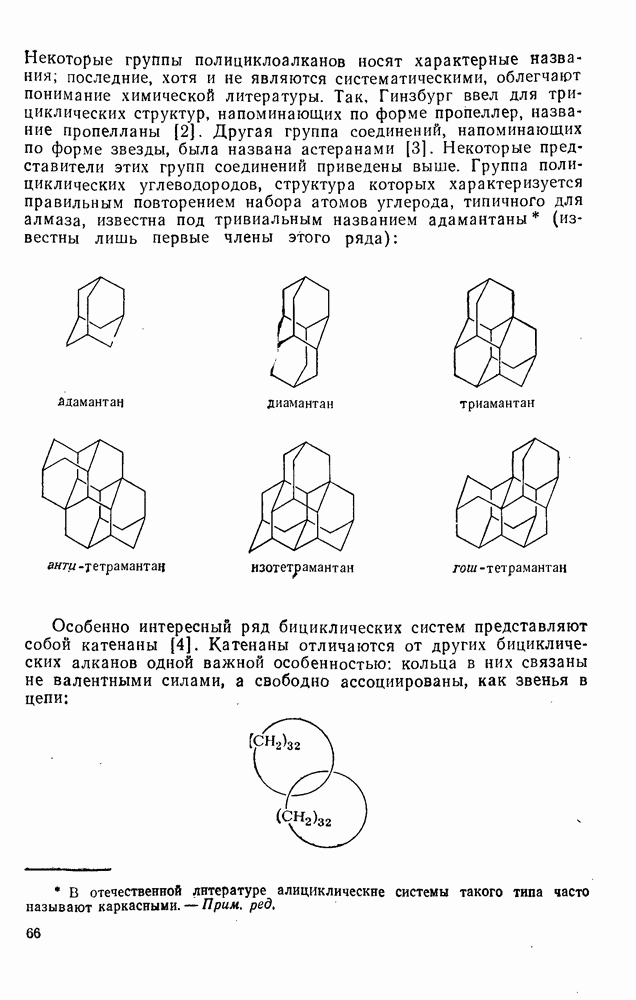
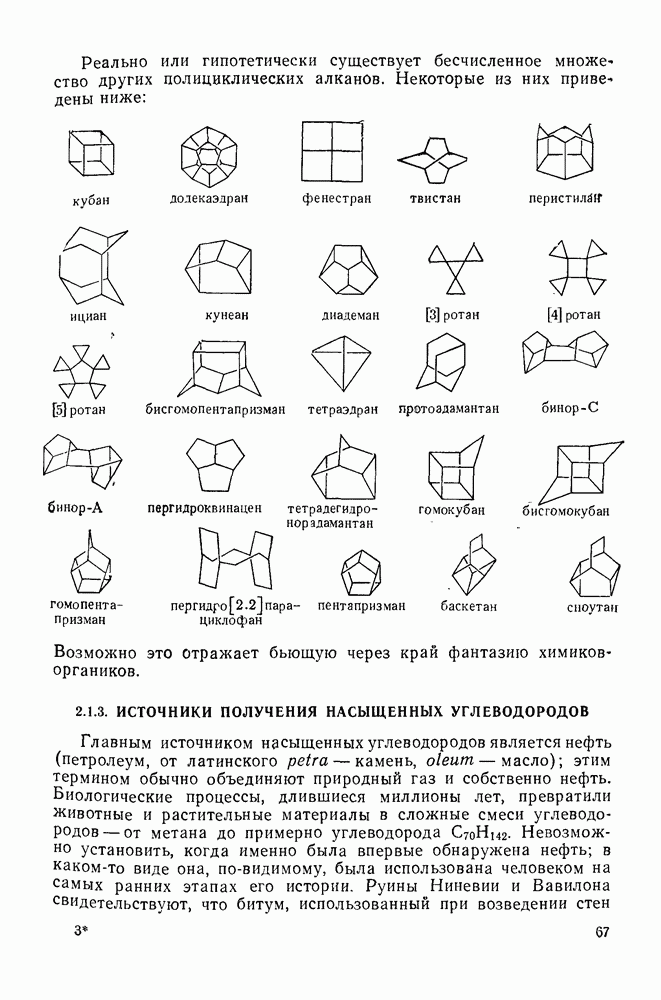
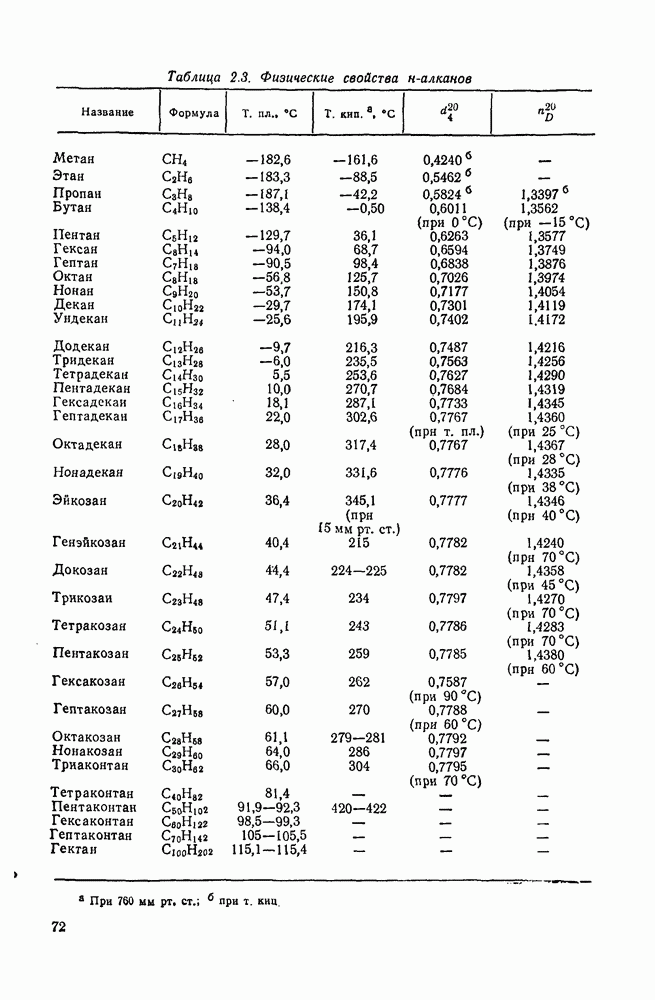
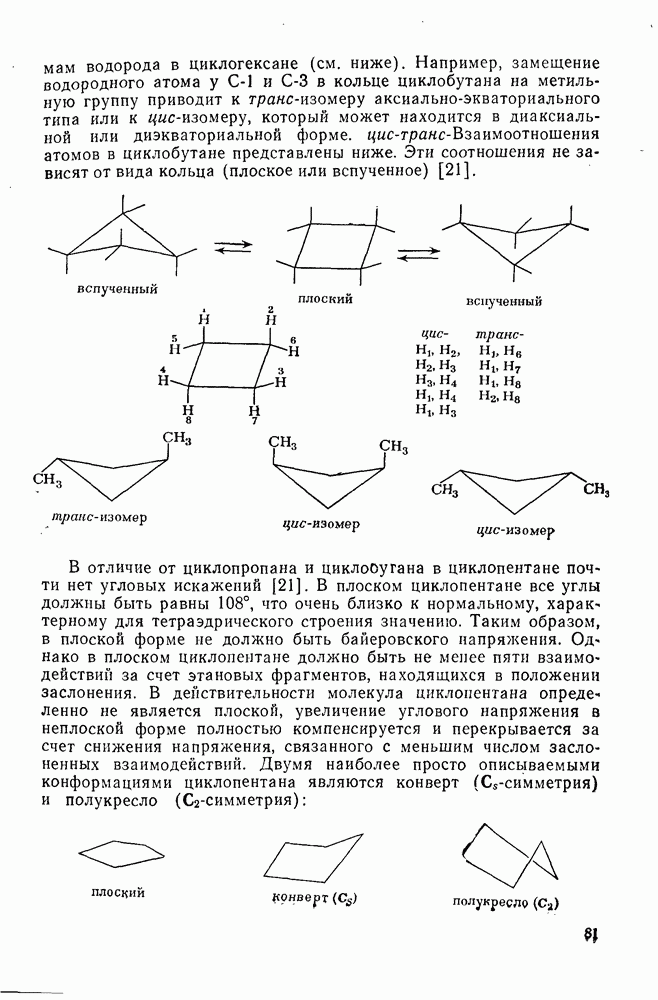
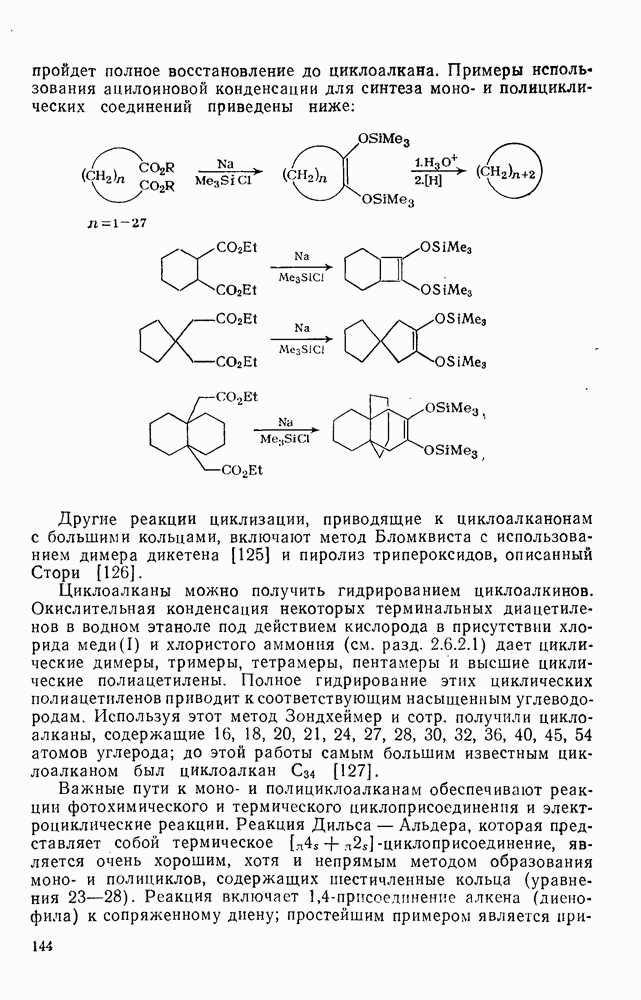
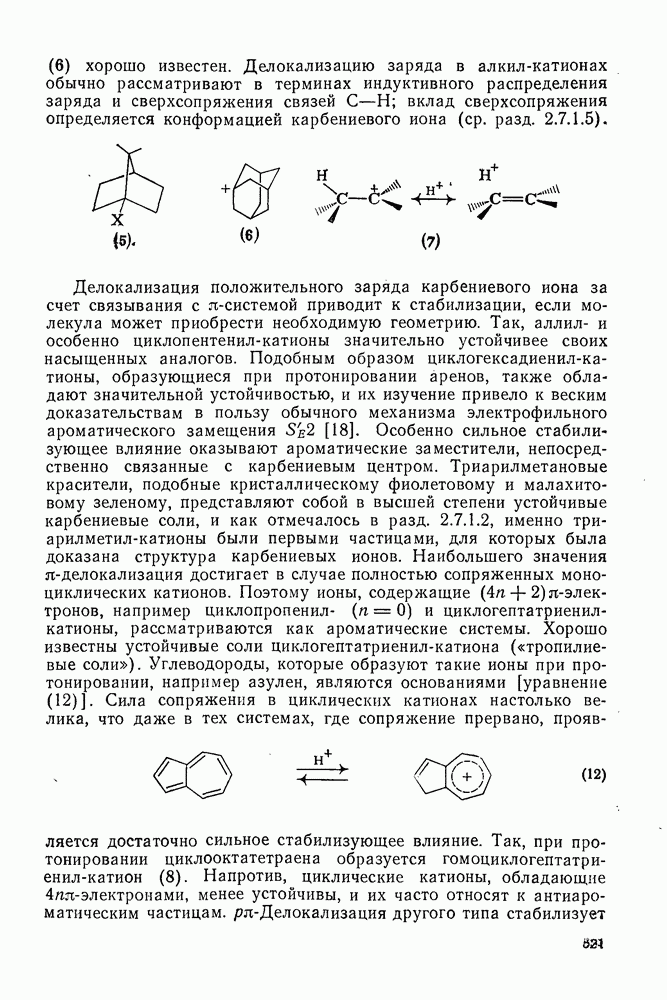
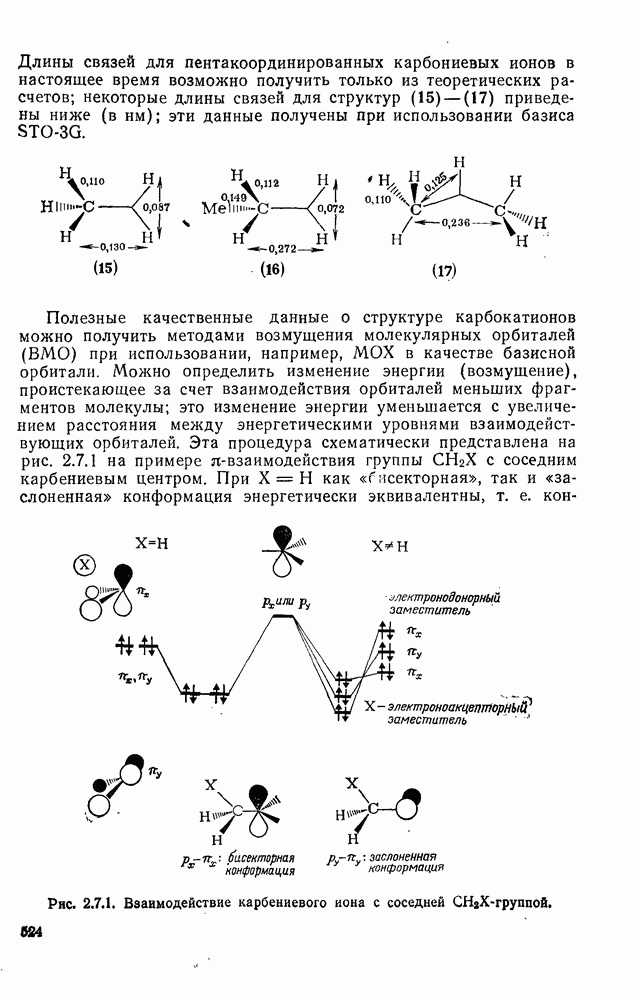
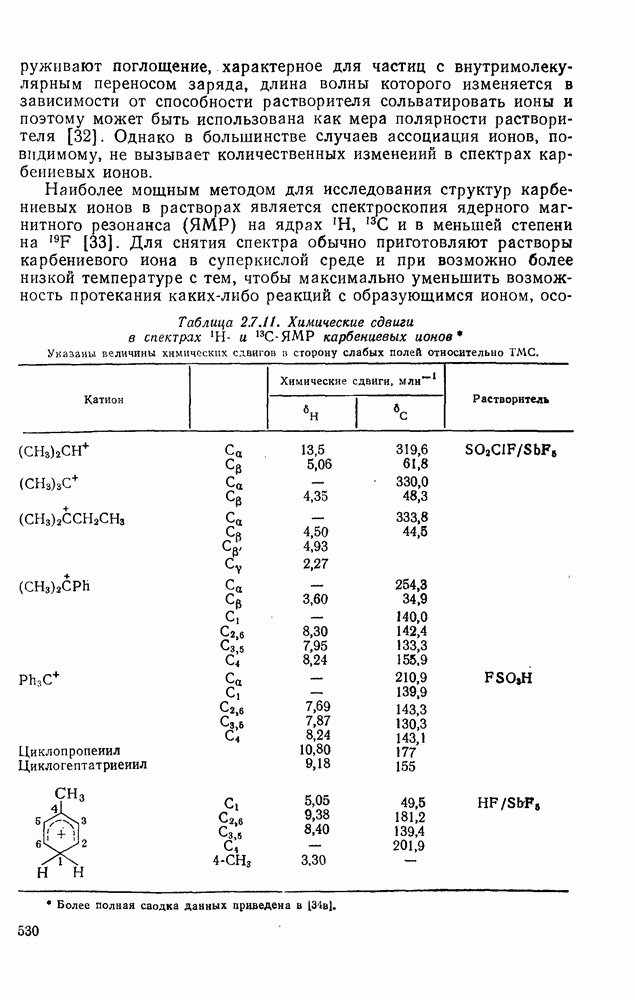
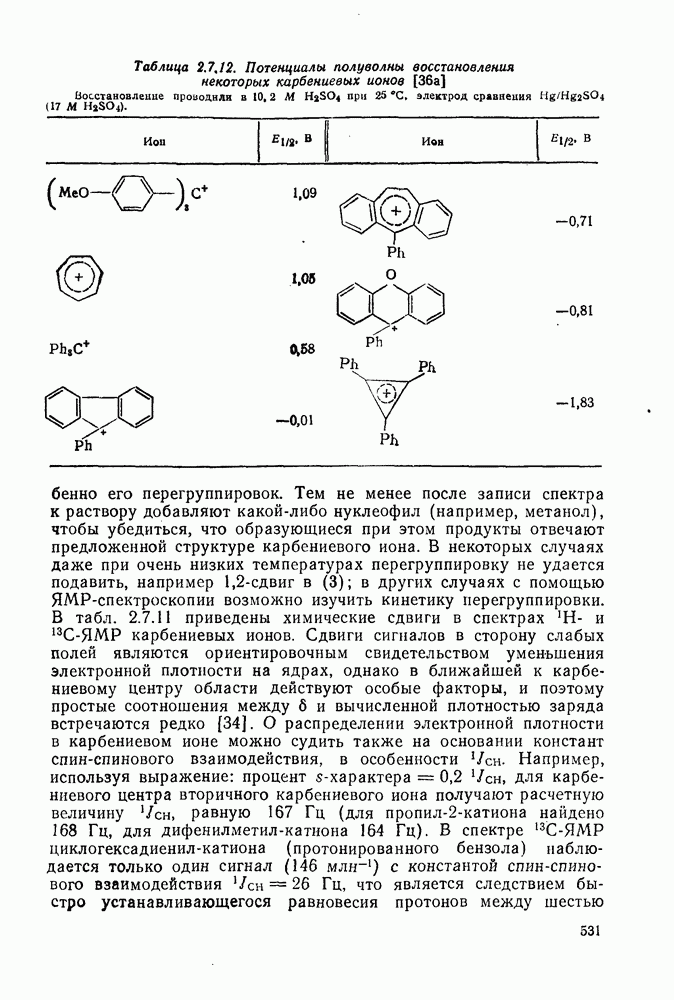
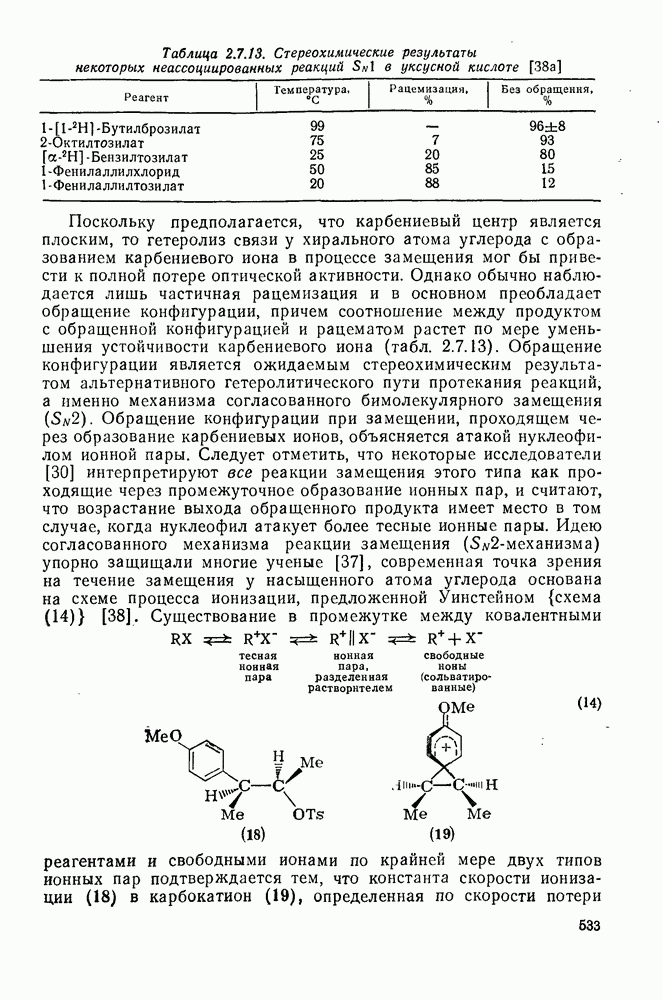
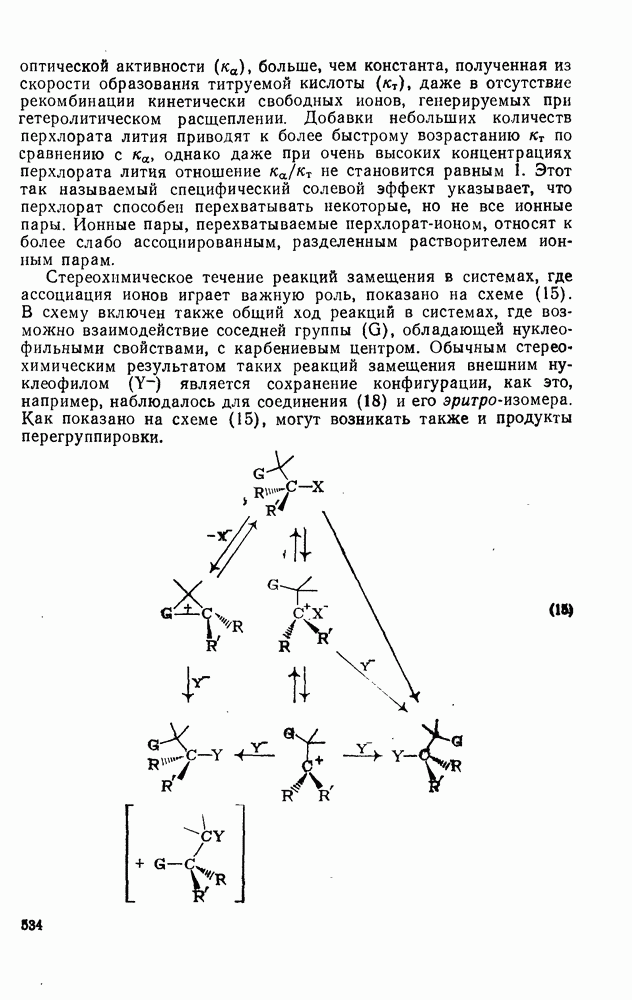
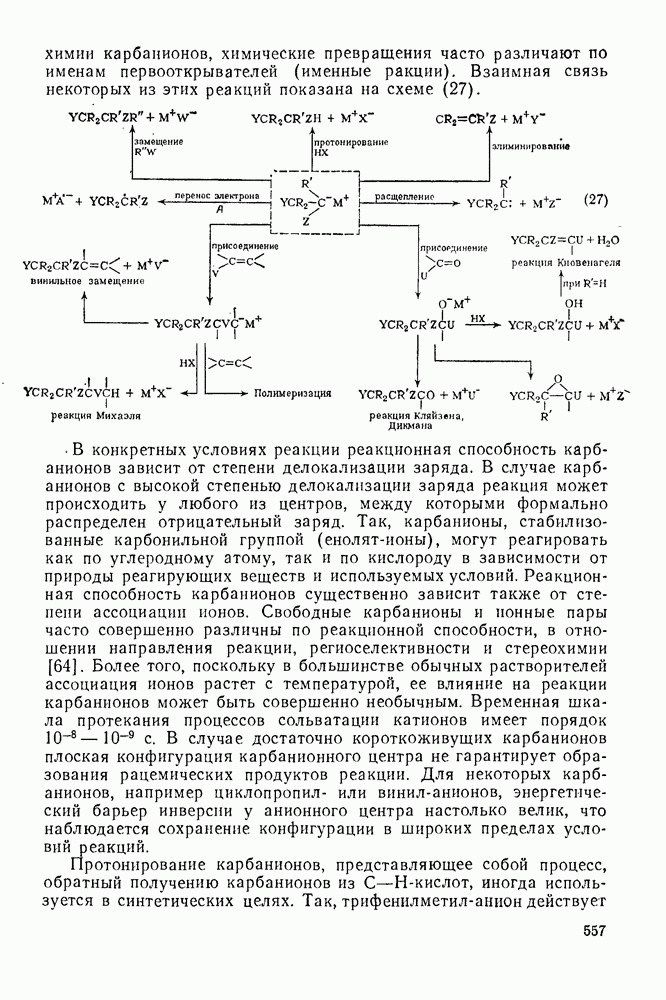
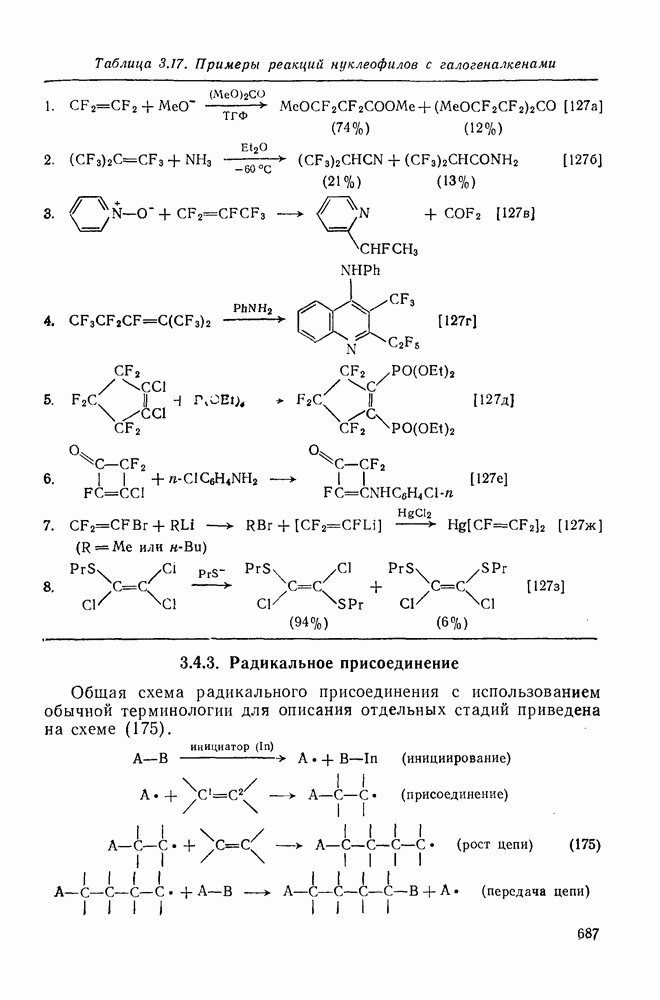
2.7.5.2. Реакции карбанионовКарбанионы проявляют основные и нуклеофильные свойства, а также могут выступать как одноэлектронные доноры. Главные реакции — протонирование, нуклеофильное замещение у насыщенного атома углерода, нуклеофильное присоединение к ненасыщенным функциям и перенос электрона — в обобщенном виде представлены на схеме (27). Отдельно рассмотрены перегруппировки карбанионов. Конечный результат реакций карбанионов зависит от структуры реагирующих веществ; многие важные в синтетическом отношении и тесно связанные между собой, с точки зрения химии карбанионов, химические превращения часто различают по именам первооткрывателей (именные ракции). Взаимная связь некоторых из этих реакций показана на схеме (27).
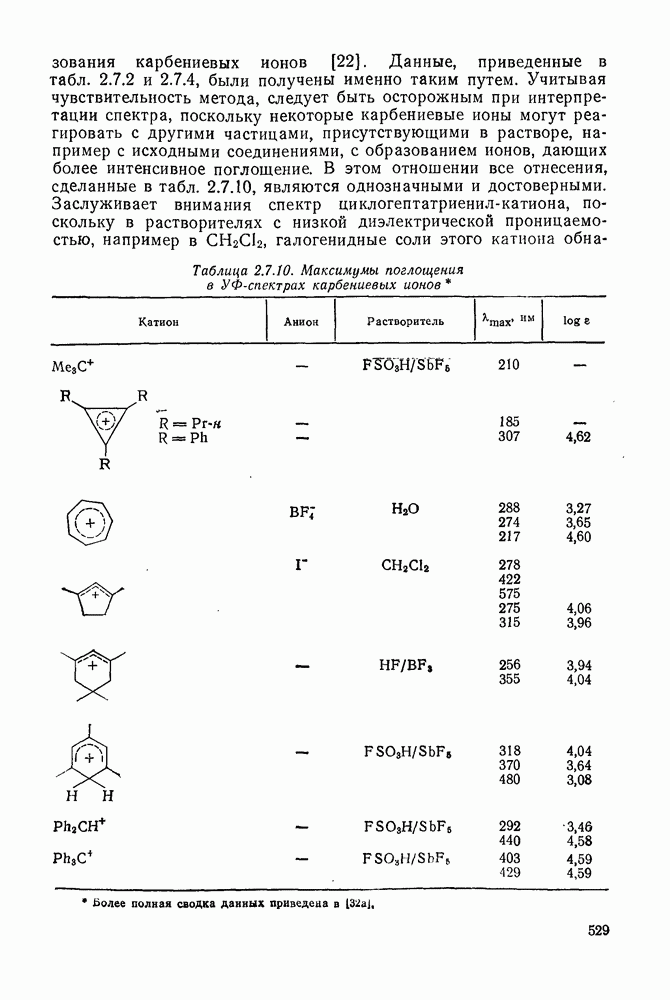
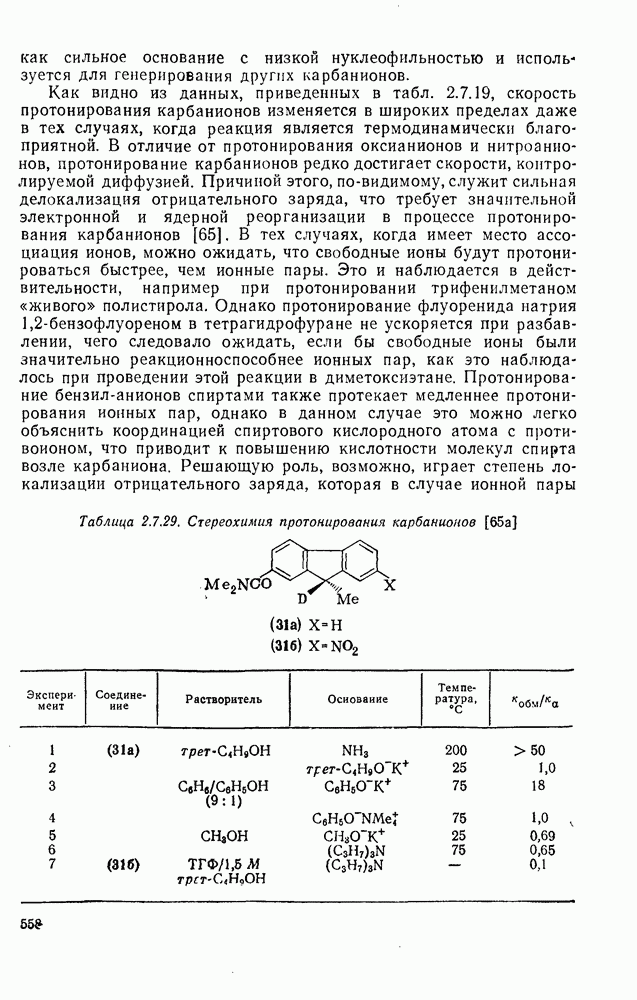
В конкретных условиях реакции реакционная способность карбанионов зависит от степени делокализации заряда. В случае карбанионов с высокой степенью делокализации заряда реакция может происходить у любого из центров, между которыми формально распределен отрицательный заряд. Так, карбанионы, стабилизованные карбонильной группой (енолят-ионы), могут реагировать как по углеродному атому, так и по кислороду в зависимости от природы реагирующих веществ и используемых условий. Реакционная способность карбанионов существенно зависит также от степени ассоциации ионов. Свободные карбанионы и ионные пары часто совершенно различны по реакционной способности, в отношении направления реакции, региоселективности и стереохимии [64]. Более того, поскольку в большинстве обычных растворителей ассоциация ионов растет с температурой, ее влияние на реакции карбанионов может быть совершенно необычным. Временная шкала протекания процессов сольватации катионов имеет порядок Протонирование карбанионов, представляющее собой процесс, обратный получению карбанионов из С—Н-кислот, иногда используется в синтетических целях. Так, трифенилметил-анион действует как сильное основание с низкой нуклеофильностью и используется для генерирования других карбанионов. Как видно из данных, приведенных в табл. 2.7.19, скорость протонирования карбанионов изменяется в широких пределах даже в тех случаях, когда реакция является термодинамически благоприятной. В отличие от протонирования оксианионов и нитроанионов, протонирование карбанионов редко достигает скорости, контролируемой диффузией. Причиной этого, по-видимому, служит сильная делокализация отрицательного заряда, что требует значительной электронной и ядерной реорганизации в процессе протонирования карбанионов [65]. В тех случаях, когда имеет место ассоциация ионов, можно ожидать, что свободные ионы будут протежироваться быстрее, чем ионные пары. Это и наблюдается в действительности, например при протонировании трифенилметаном «живого» полистирола. Однако протонирование флуоренида натрия Таблица 2.7.29. Стереохимия протонирования карбанионов [65а]
Стереохимия протонирования короткоживущих карбанионов широко изучена на примере изотопного водородного обмена в щелочных условиях у хиральных С—Н-кислот путем сравнения скорости обмена ( Некоторые иные аспекты влияния структуры карбанионов на реакционную способность были отмечены при обсуждении способов образования карбанионов. Однако общий конечный результат реакции обусловлен последующим участием образующегося аниона в тех или иных реакциях: протонирования, атаки других ненасыщенных молекул (как в случае анионной полимеризации), расщепления или замещения. Таблица 2.7.30. Константы скорости анионной гомополимеризации стирола на стадии роста при
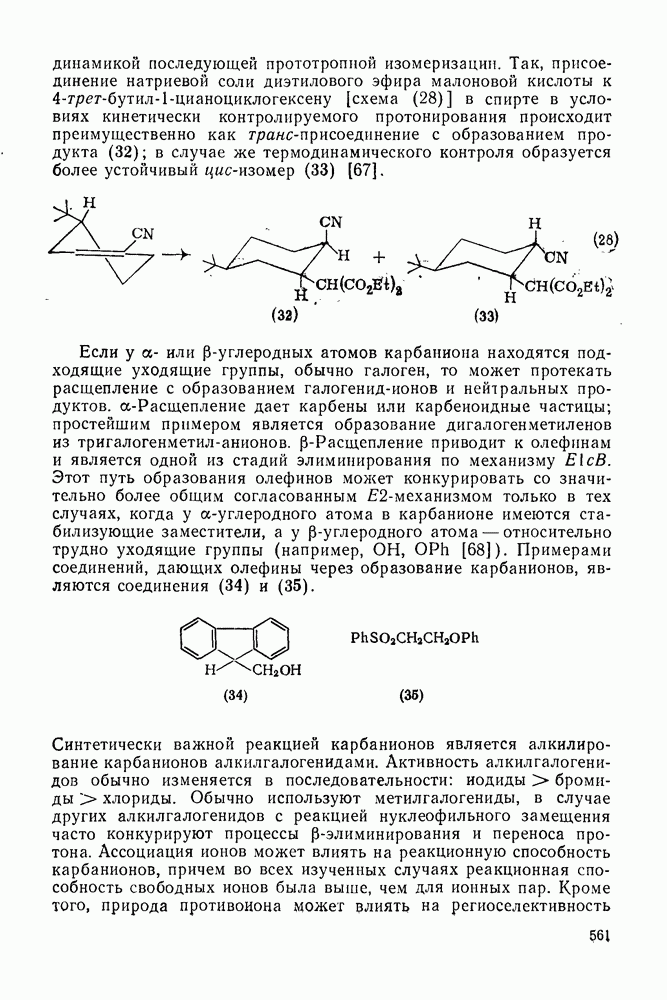
Взаимодействие карбанионов с диоксидом углерода непосредственно приводит к образованию карбокси-лат-иона, и эта реакция была использована для изучения распределения заряда в делокализованных ионах и стереохимии карбанионов. Важную роль в реакциях карбанионов играет ассоциация ионов, она была подробно изучена в связи с анионной полимеризацией [66]. В табл. 2.7.30 приведены данные о реакционной способности свободных карбанионов Карбанионы, образующиеся в реакциях присоединения по Михаэлю, отличаются от ионов реактанта, поэтому трактовка результатов здесь значительно сложнее, чем в случае полимеризации. Стереохимия реакции обычно определяется протонированием образующегося карбаниона и поэтому зависит от различных факторов, обсуждавшихся выше. Во многих случаях анион, образующийся при присоединениях по Михаэлю, стабилизуется заместителями, содержащими карбонильную группу. Тогда протонирование может быстрее всего протекать по атому кислорода и стереохимия нейтрального продукта реакции Михаэля будет обусловлена термодинамикой последующей прототропной изомеризации. Так, присоединение натриевой соли диэтилового эфира малоновой кислоты к
Если у
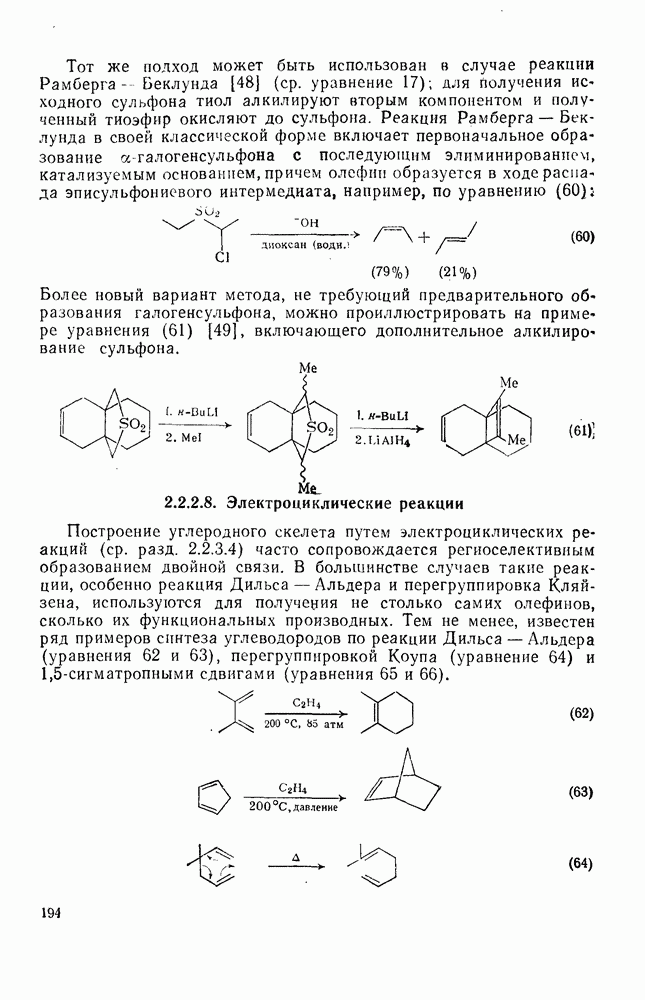
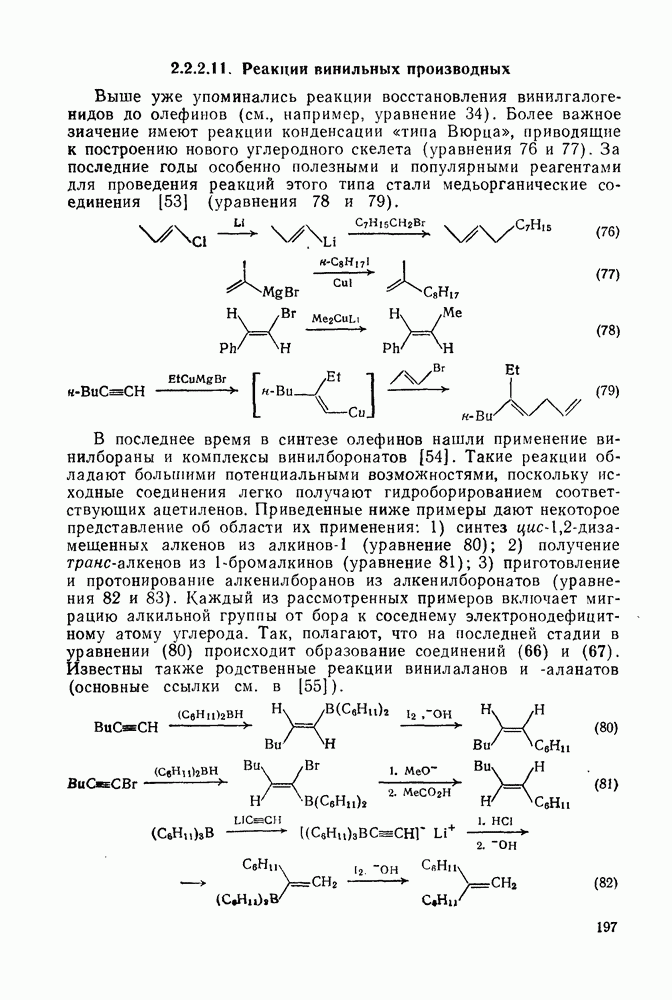
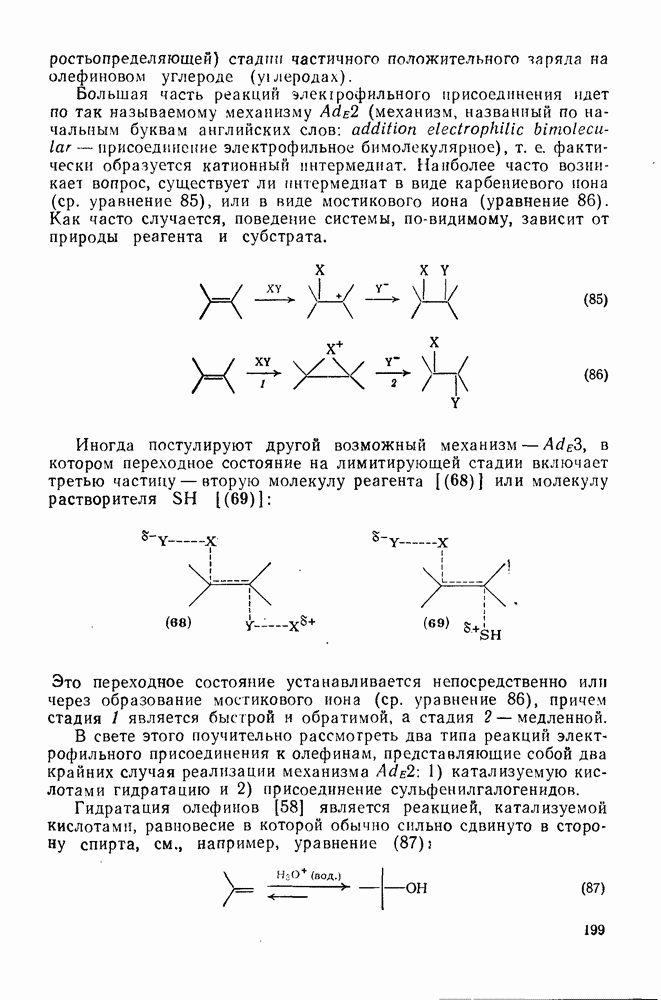
Синтетически важной реакцией карбанионов является алкилиро-вание карбанионов алкилгалогенидами. Активность алкилгалогенидов обычно изменяется в последовательности: йодиды > бромиды > хлориды. Обычно используют метилгалогениды, в случае других алкилгалогенидов с реакцией нуклеофильного замещения часто конкурируют процессы Иногда наблюдается внутримолекулярное алкилирование; например внутримолекулярное замещение галогена карб-анионом, стабилизованным карбонилом, которое протекает с промежуточным образованием циклопропаноиа, является ключевой стадией перегруппировки Фаворского. Аналогичное алкилирование карбанионом, стабилизованным сульфонильной группой, происходит при реакции Рамберга — Беклунда. Еще одним примером нуклеофильного замещения у насыщенного атома углерода является раскрытие кольца эпоксидов под действием карбанионов [уравнение (29)]. Эпоксиды хорошо координируются с катионами щелочных металлов и это облегчает раскрытие кольца. Именно по этой причине ионная пара карбанион — щелочной металл часто оказывается более реакционноспособной, чем свободный карбанион [64].
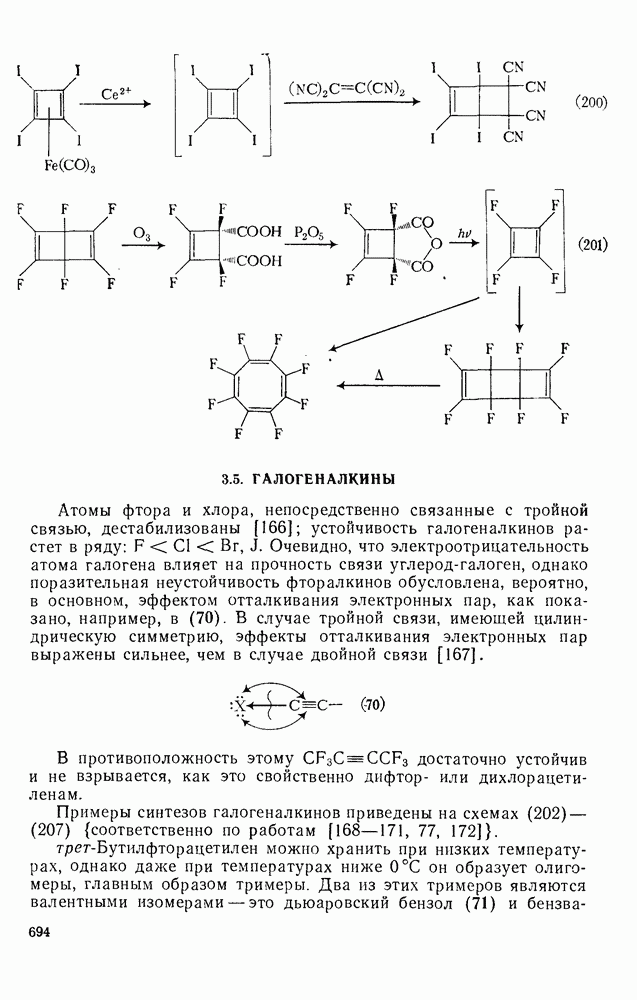
Сравнение сродства к электрону кислорода в его основном состоянии (0,43 эВ) с окислительно-восстановительными потенциалами карбанионов (см. табл. 2.7.28) показывает, что многие карбанионы должны легко окисляться молекулярным кислородом с образованием соответствующего радикала и пероксид-иона. Последующий тринлет-синглетный переход и распад приводят к гидропероксиду [схема (30)].
Первичные и вторичные алкилгидропероксиды в щелочных условиях реакции обычно отщепляют воду и превращаются в карбонильные соединения. В индуцируемом основанием аутоокислении флуорена перенос электрона от промежуточно образующегося флуоренил-аниона обычно является стадией, лимитирующей скорость всего процесса, однако в случае трифенилметана медленной стадией является образование карбаниона, а перенос электрона от менее устойчивого аниона происходит очень быстро [69]. Медленную стадию переноса электрона в случае флуоренил-анпонов можно устранить, используя синглетный
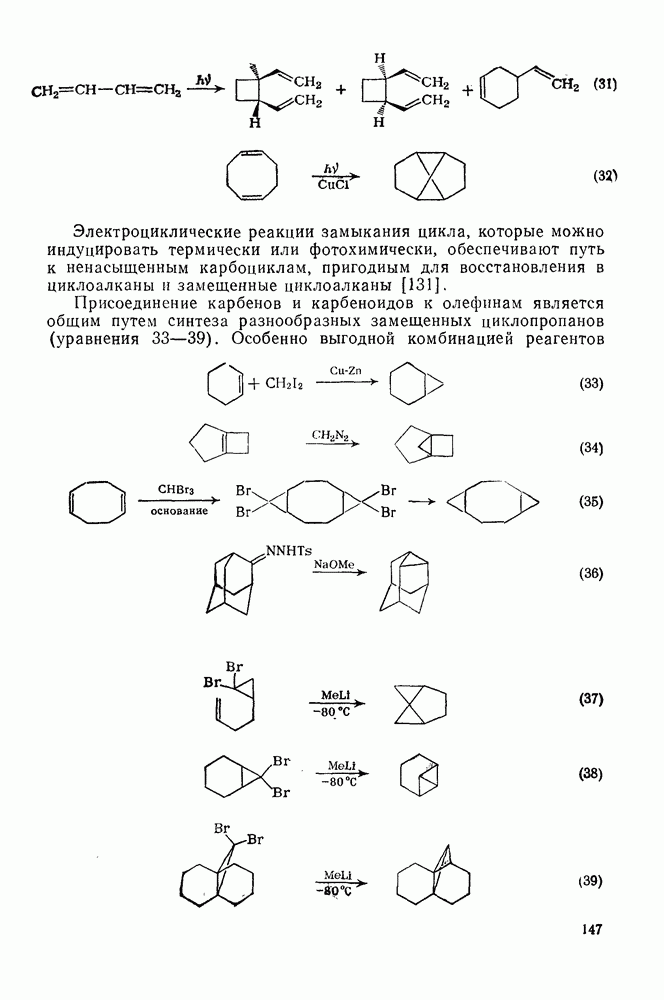
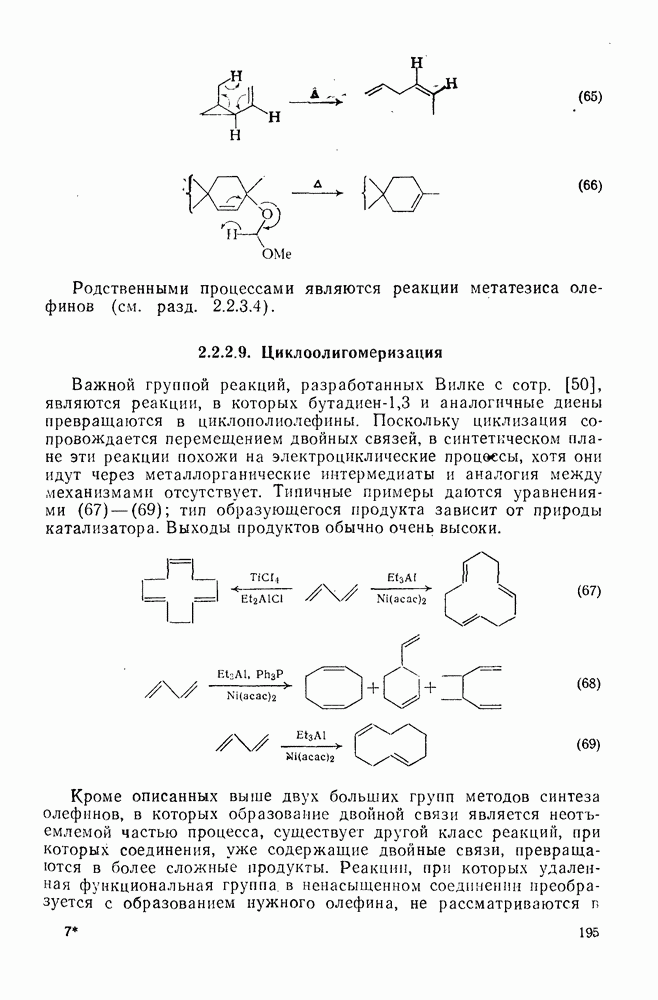
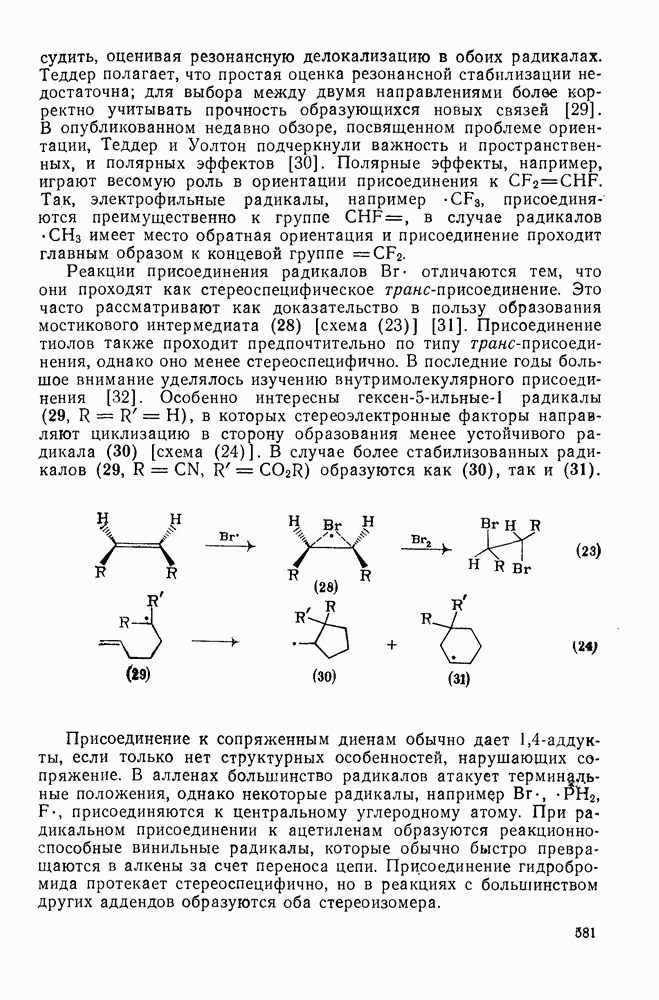
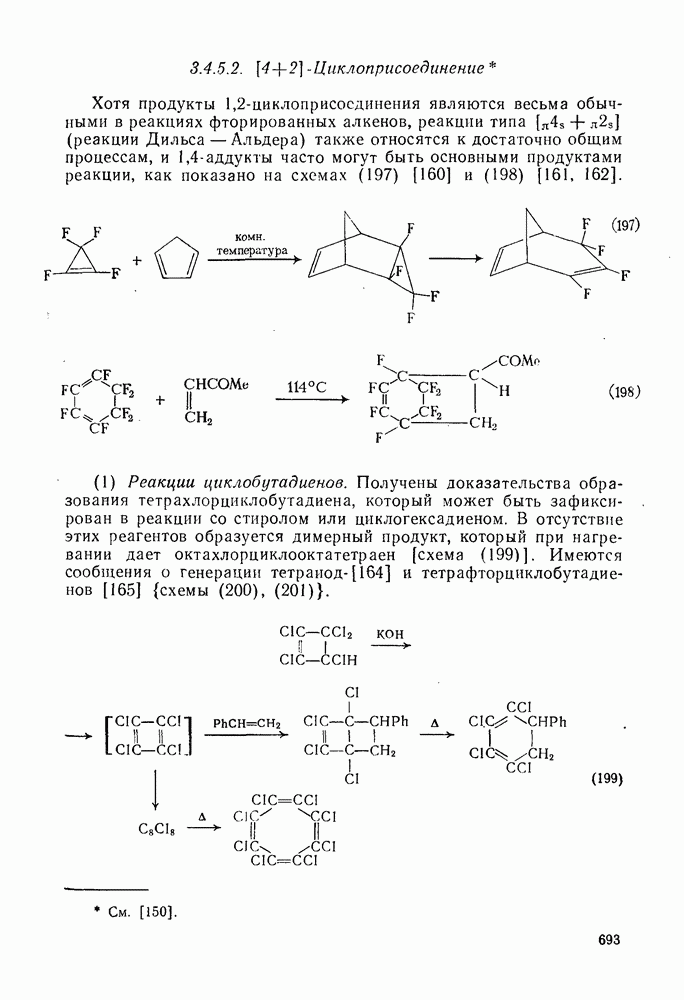
Схема перегруппировок карбанионов несколько отличается от таковой для карбениевых ионов. В случае карбанионов наблюдаются электроциклические реакции [например, схема (32], которые можно объяснить, используя правила сохранения орбитальной
симметрии [47], однако переходное состояние для супраповерхностного Для арильных групп возможен 1,2-сдвиг, как это показано для соединений типа (36) на схеме (33). Перегруппировка облегчается наличием в арильных группах электроноакцепторных заместителей;
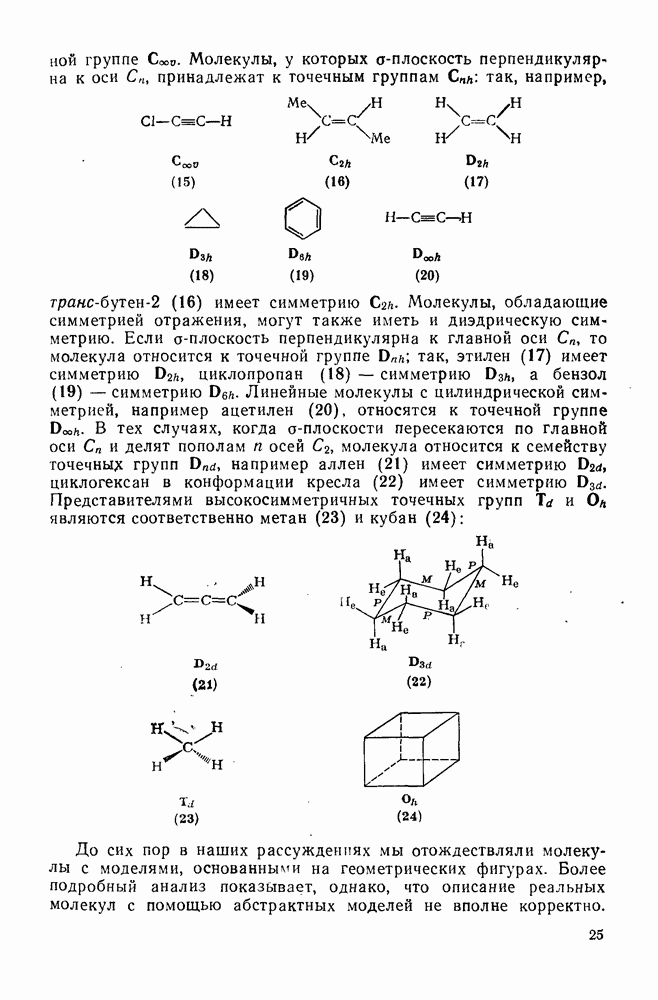
реакция, по-видимому, аналогична нуклеофильному ароматическому замещению [73]. Иногда перегруппировки могут проходить по механизму гетеролитического расщепления-рекомбинации [схема (34)].
ЛИТЕРАТУРА
Литература (продолжение) Литература (продолжение) Литература (продолжение) Литература (окончание)
|
 |
Оглавление
|






















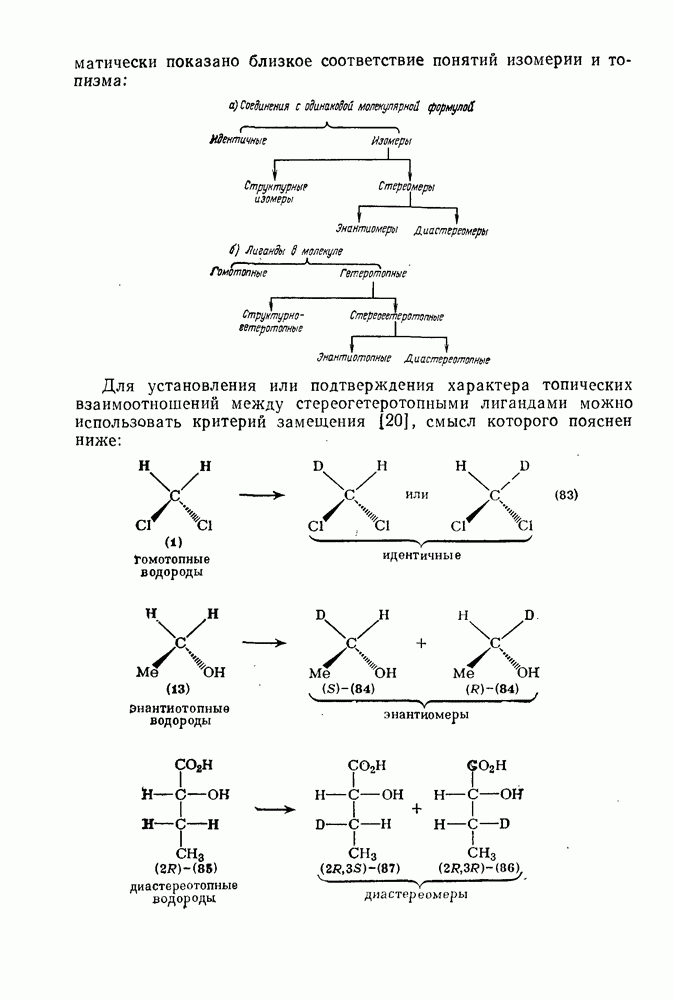




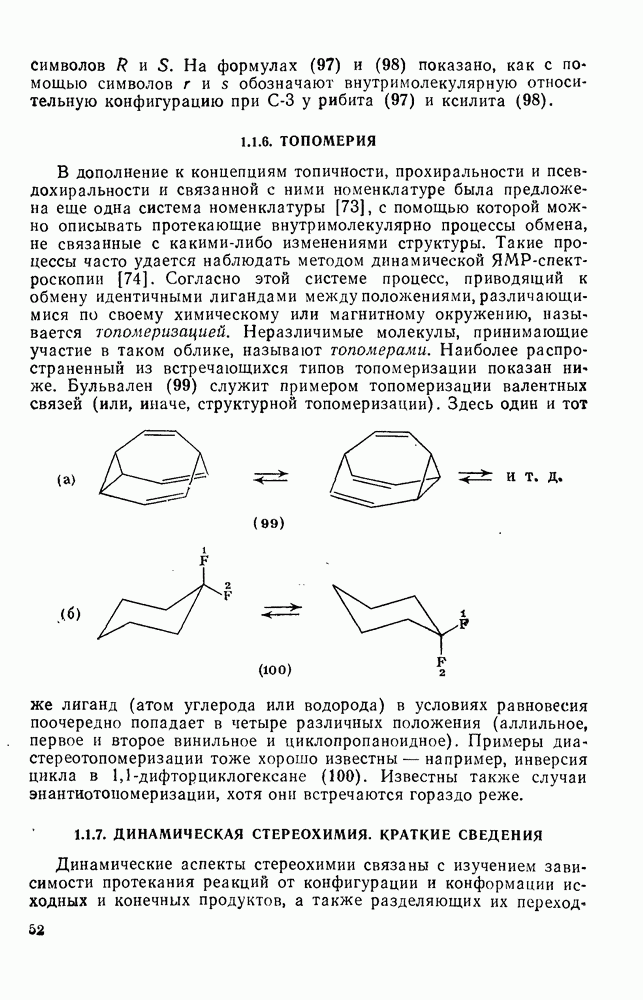

























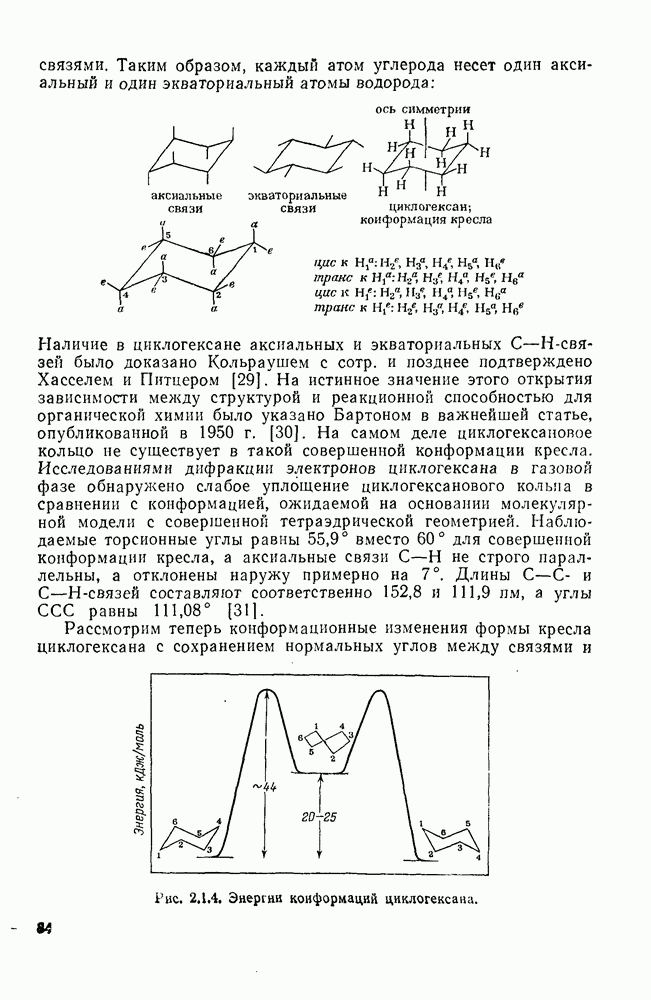


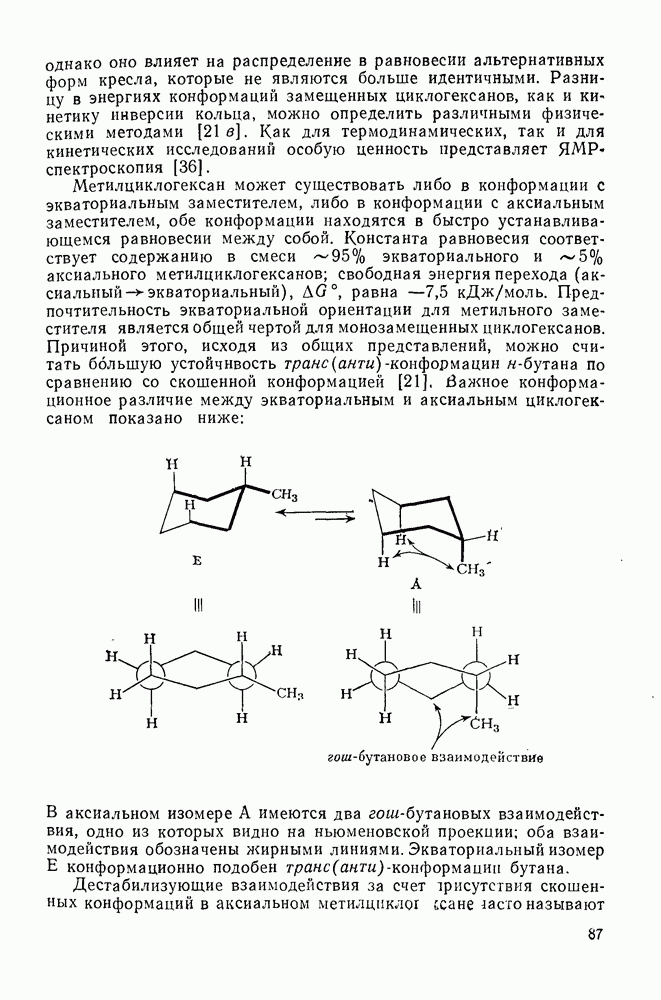


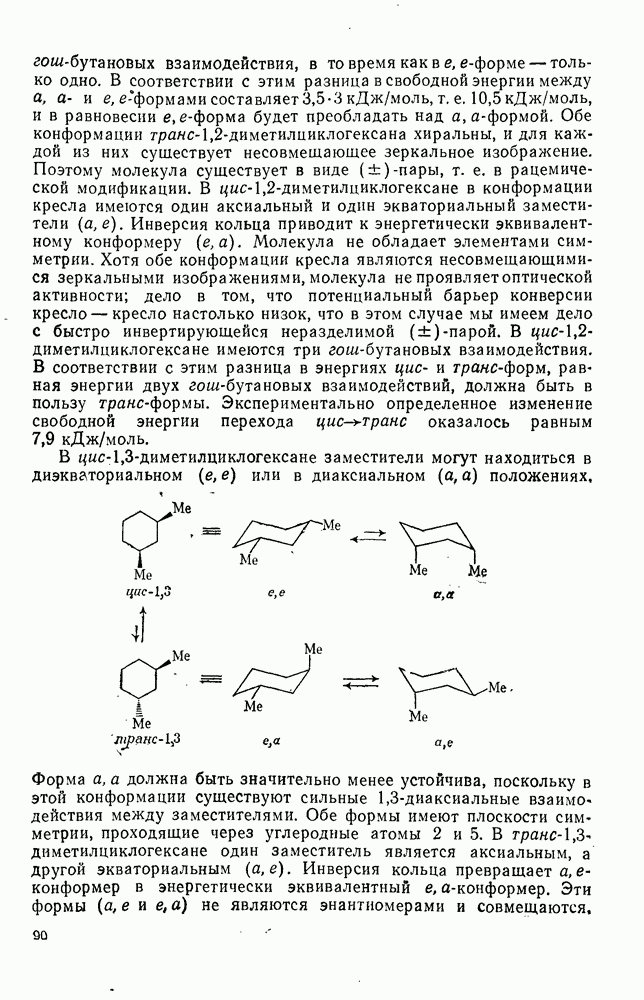
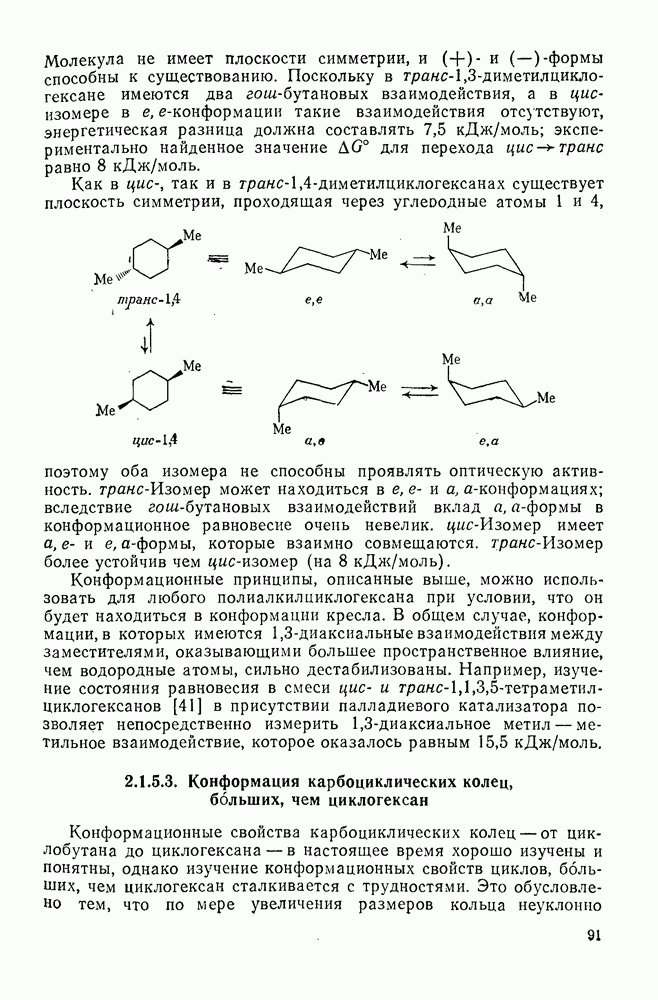

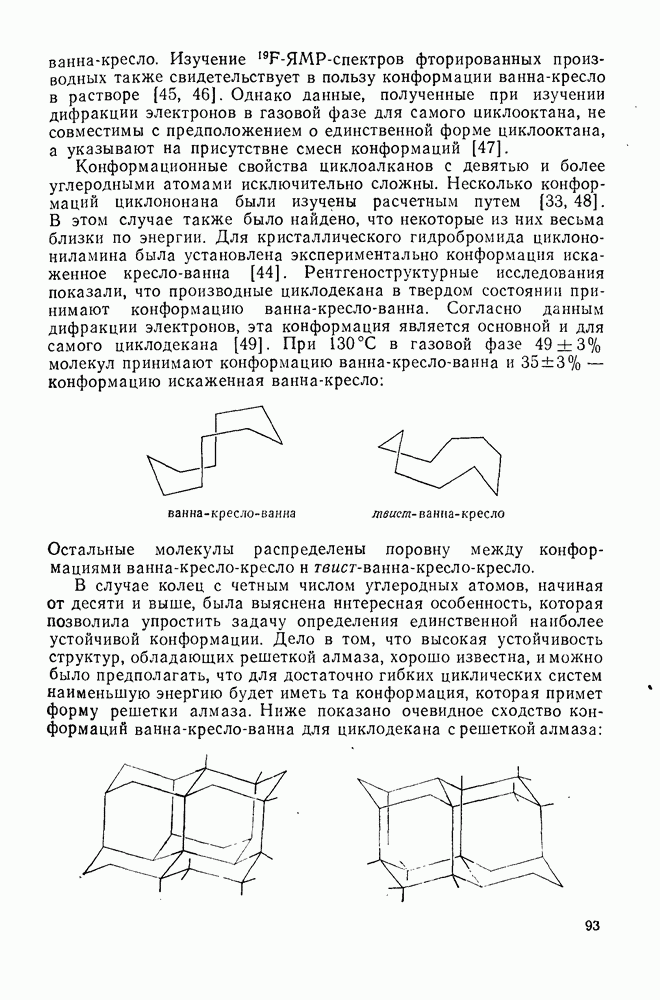






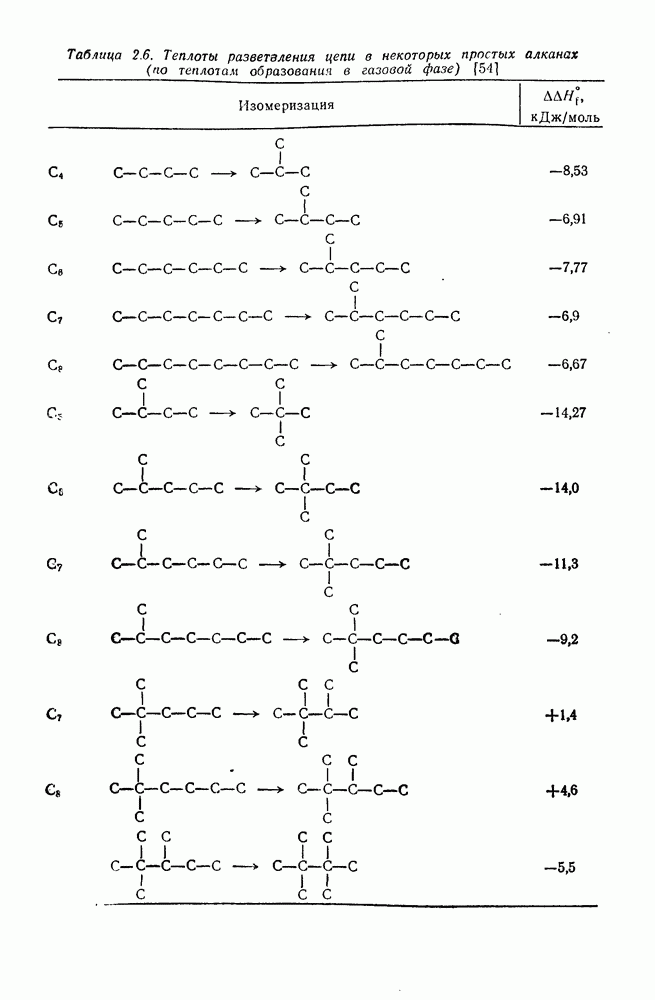







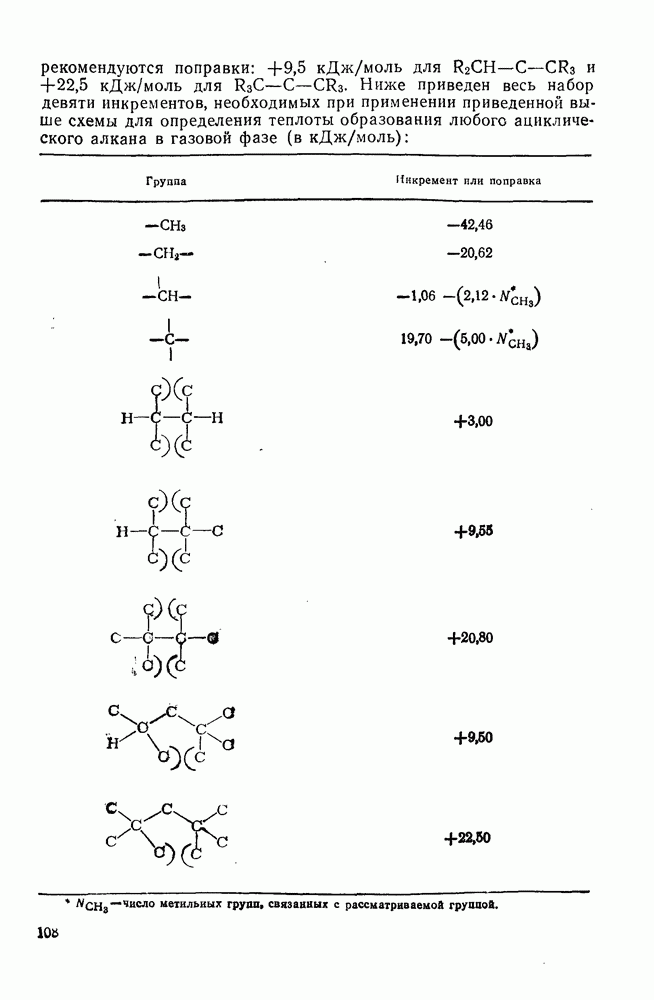

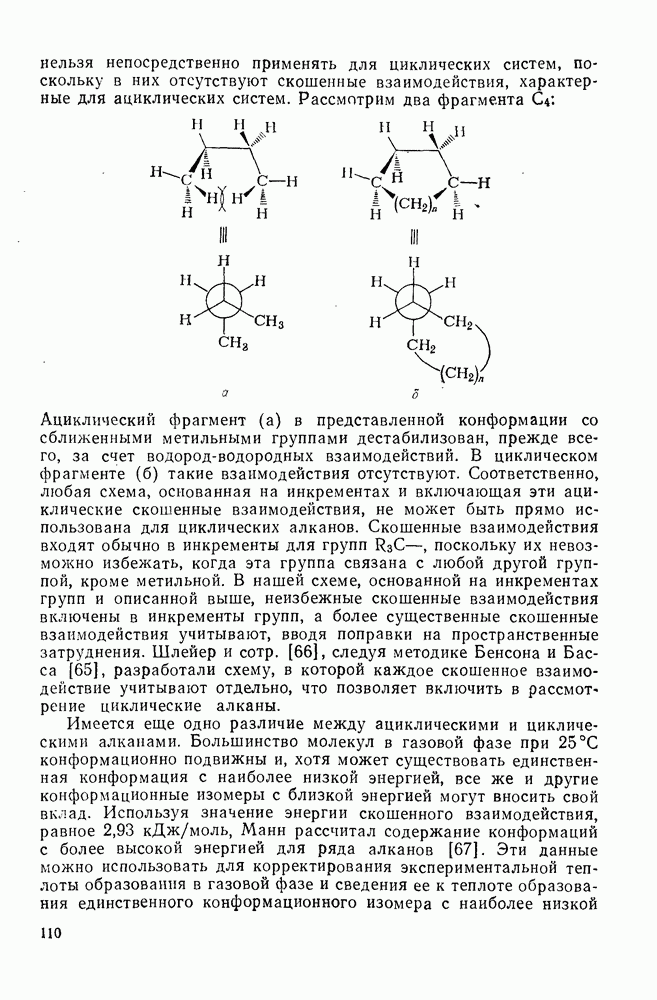









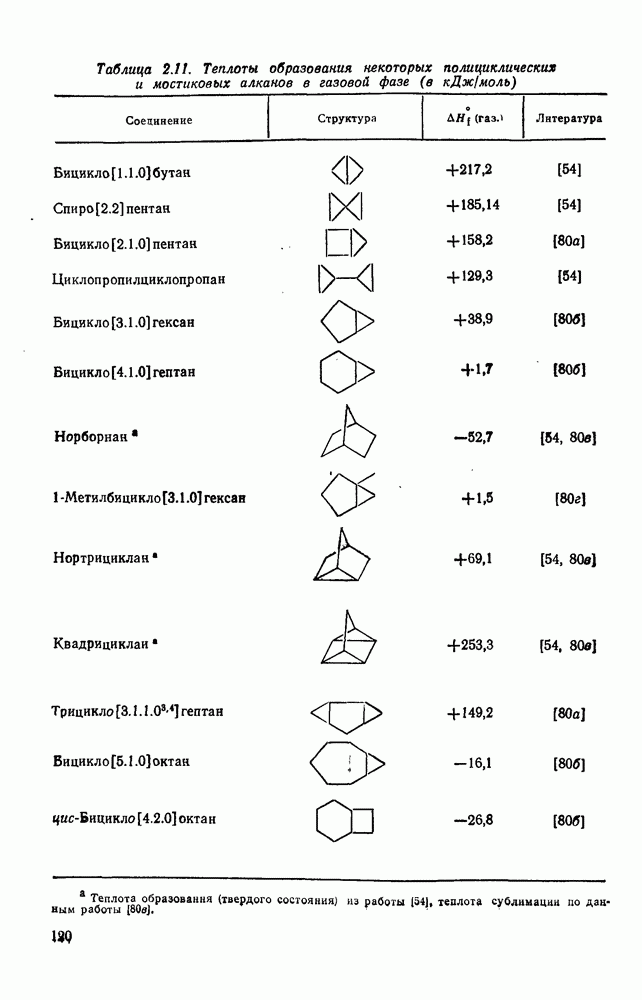

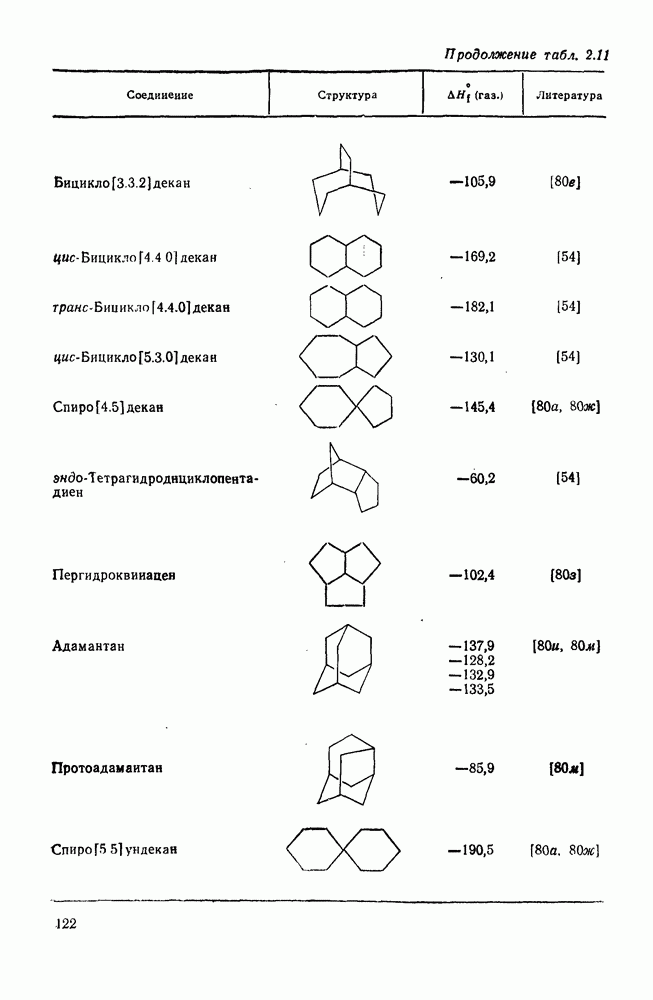
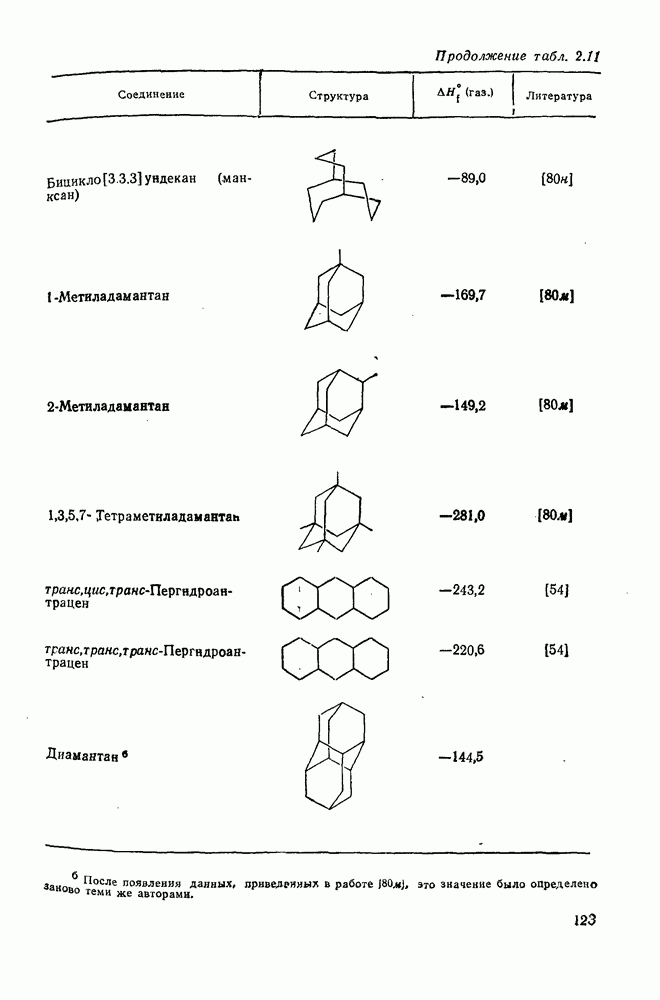
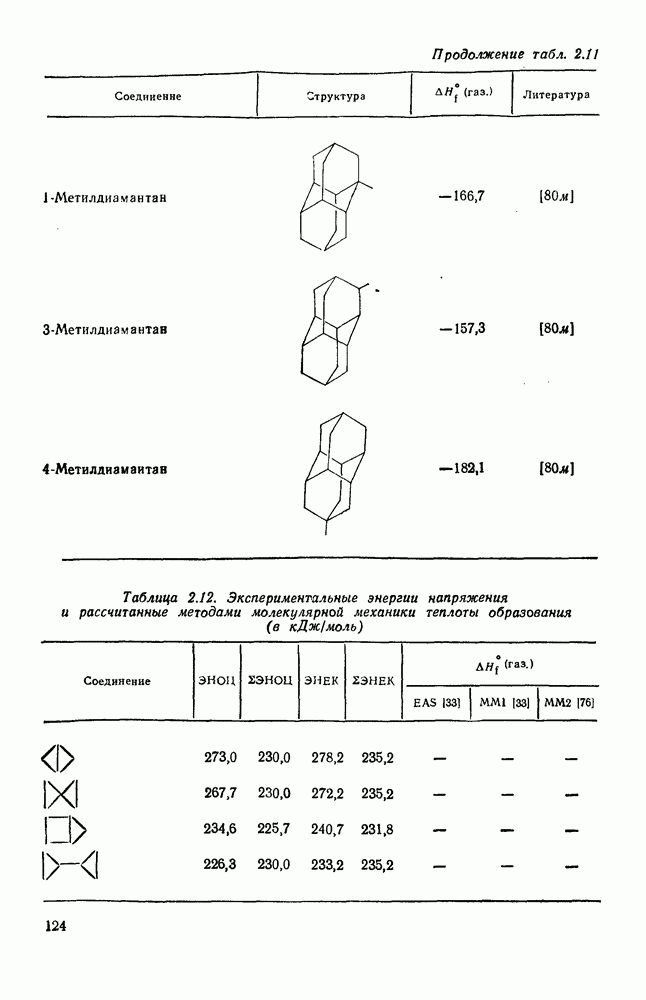



































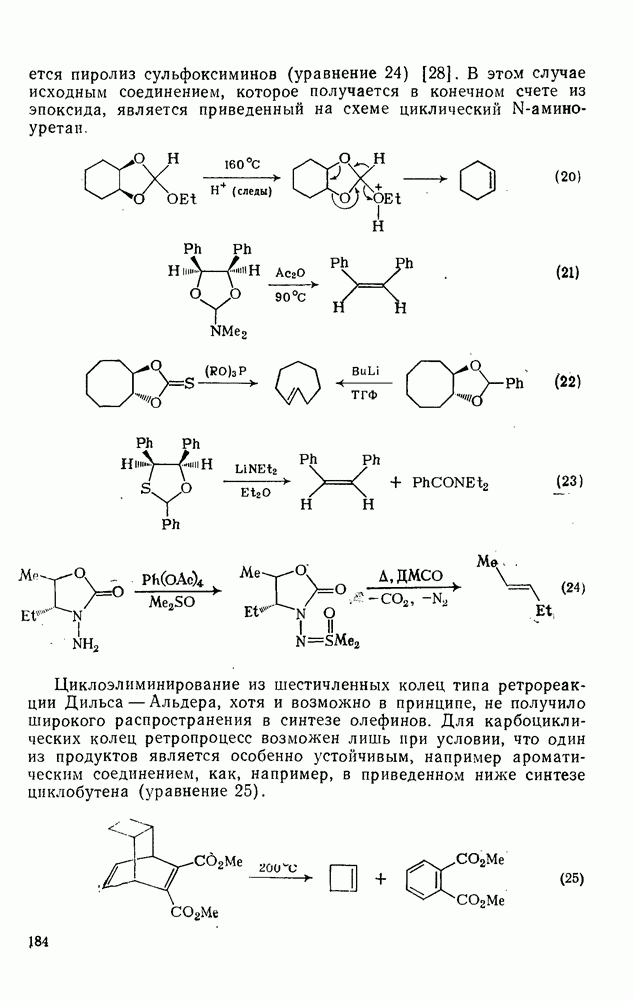























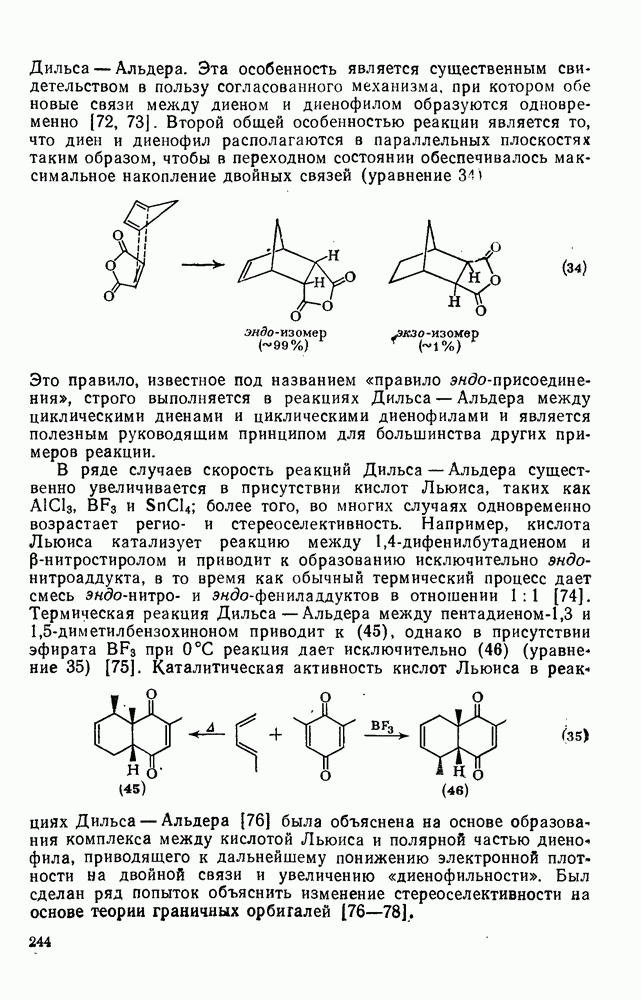
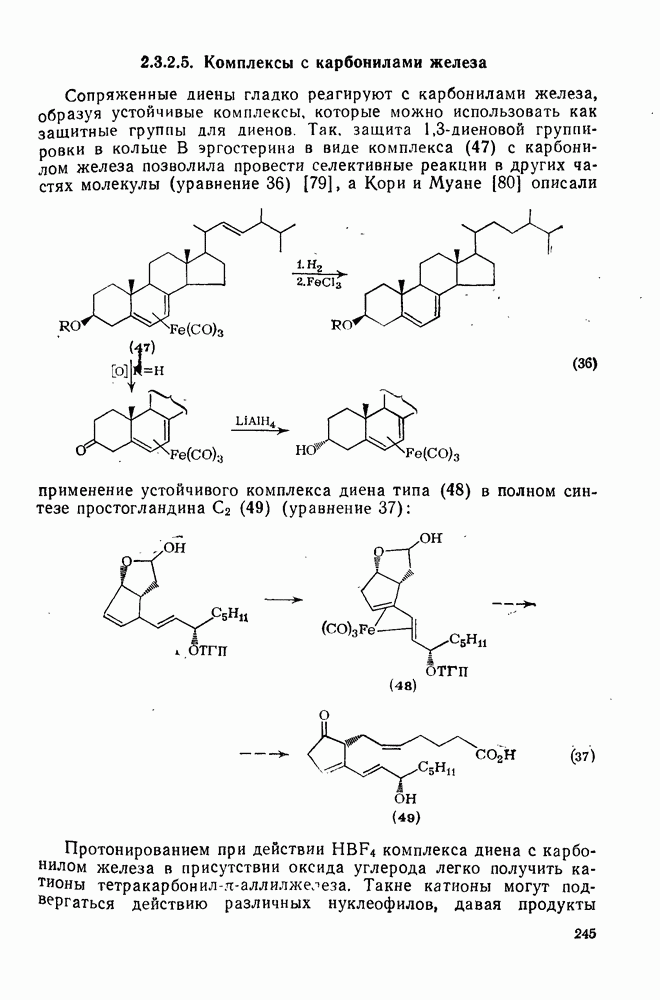
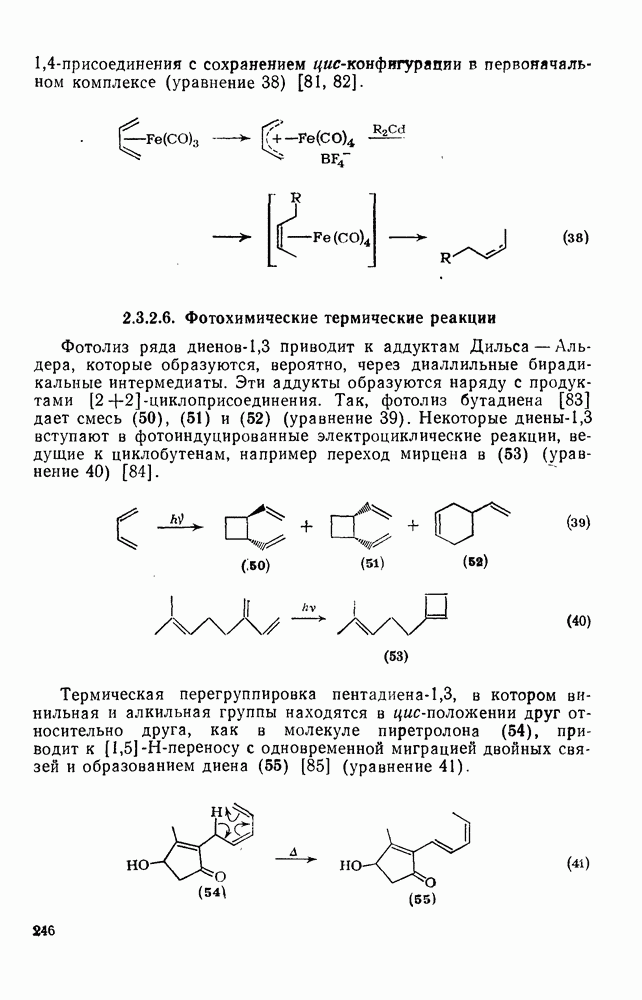
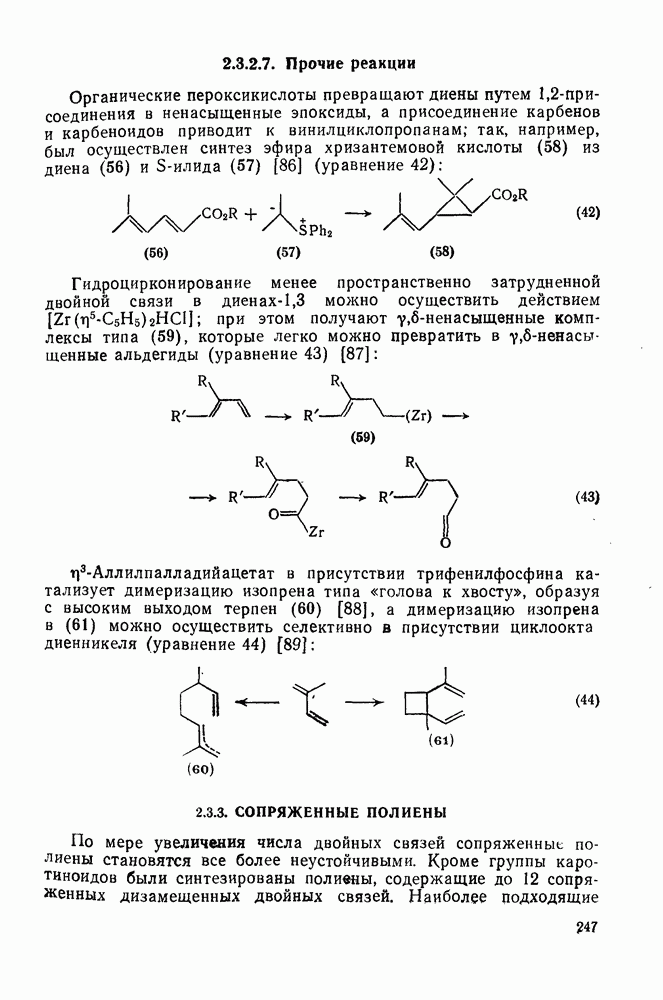

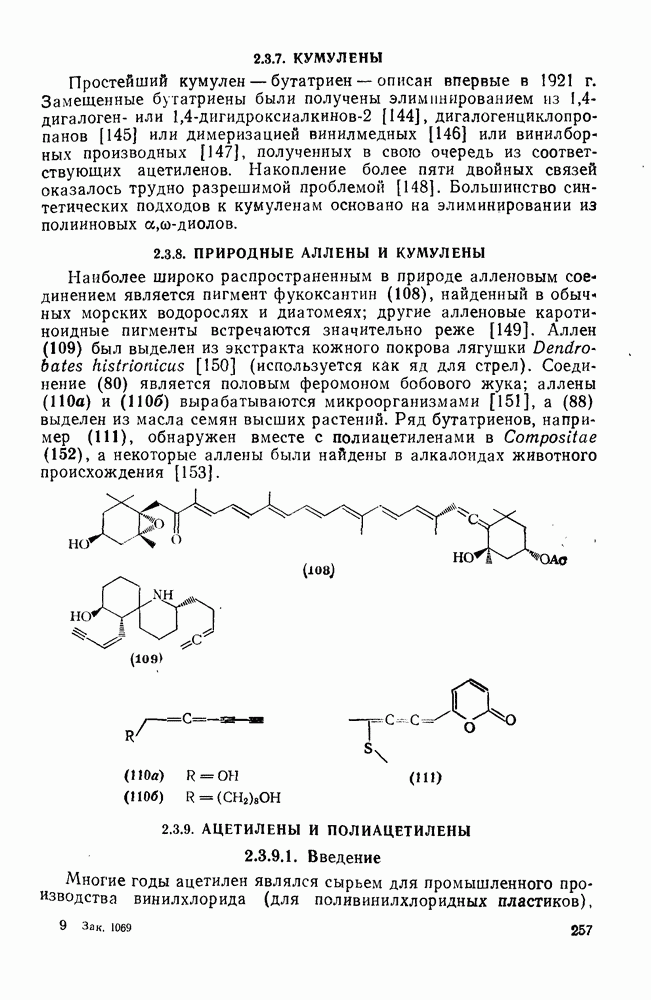

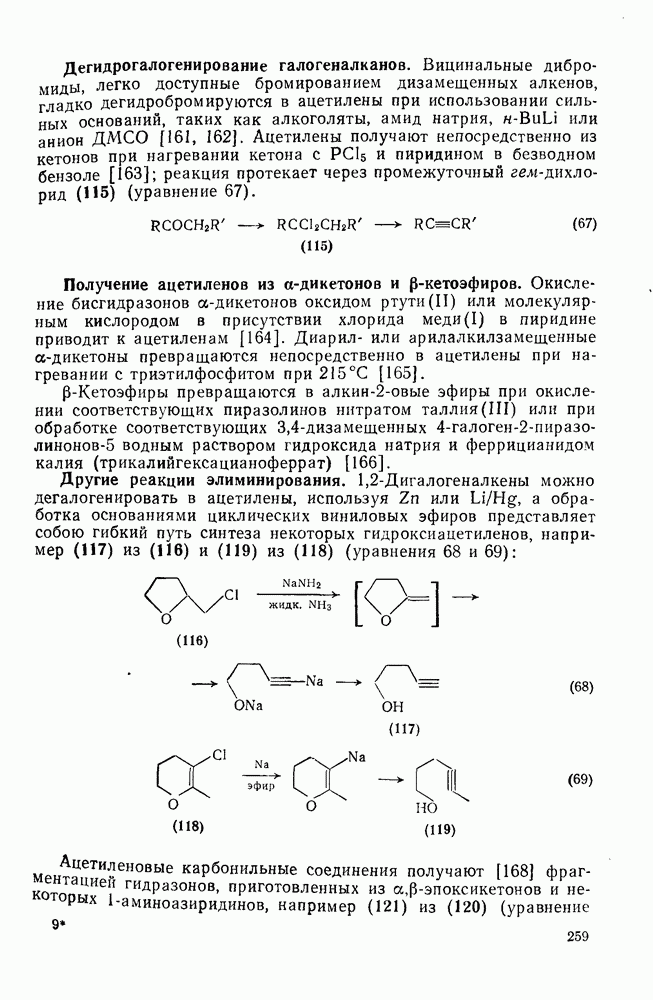
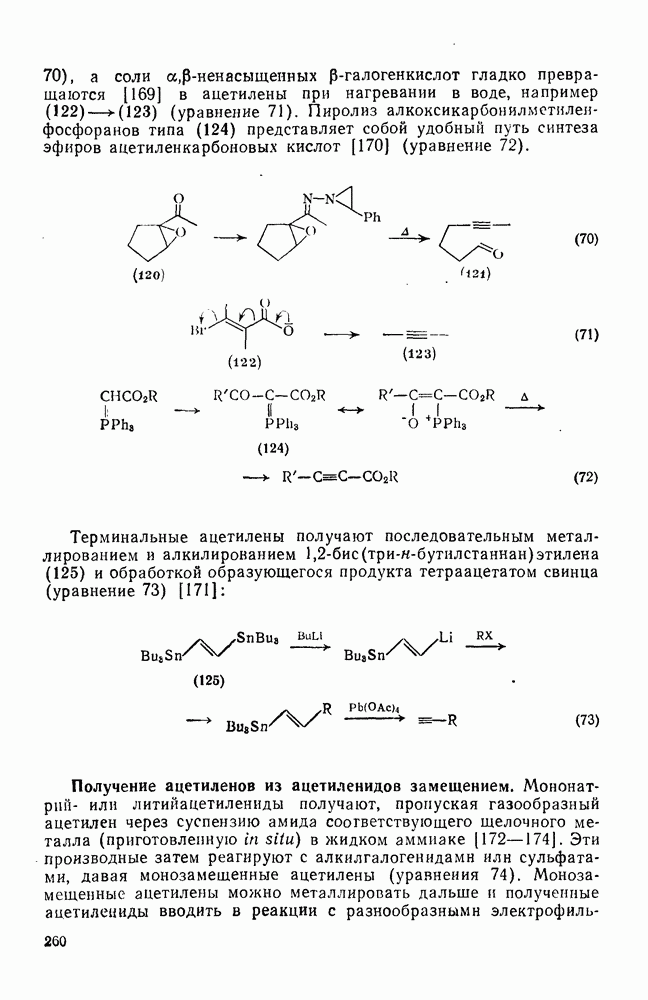
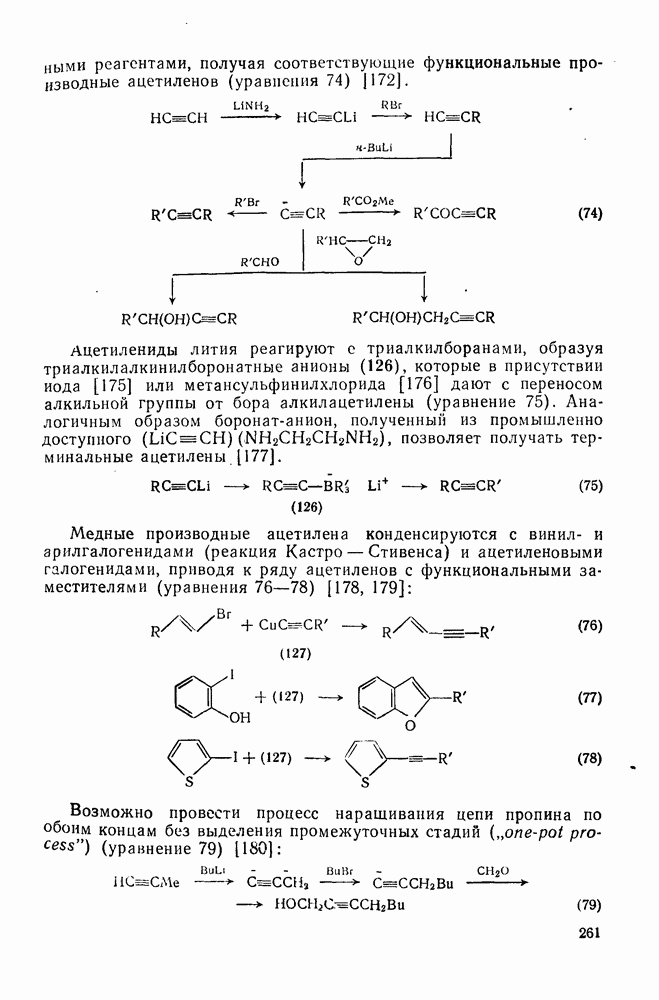































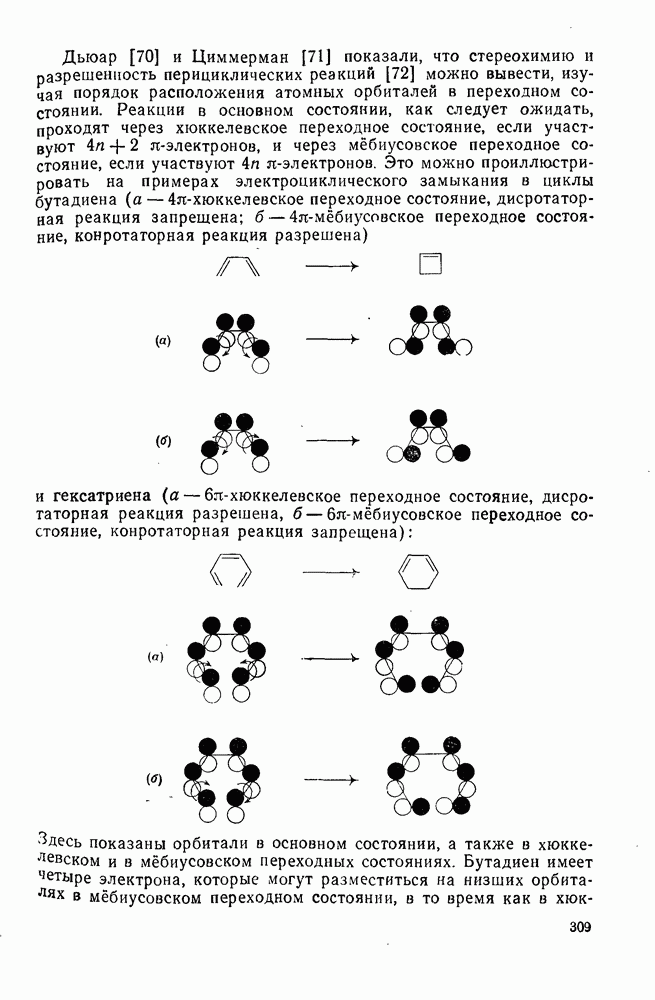

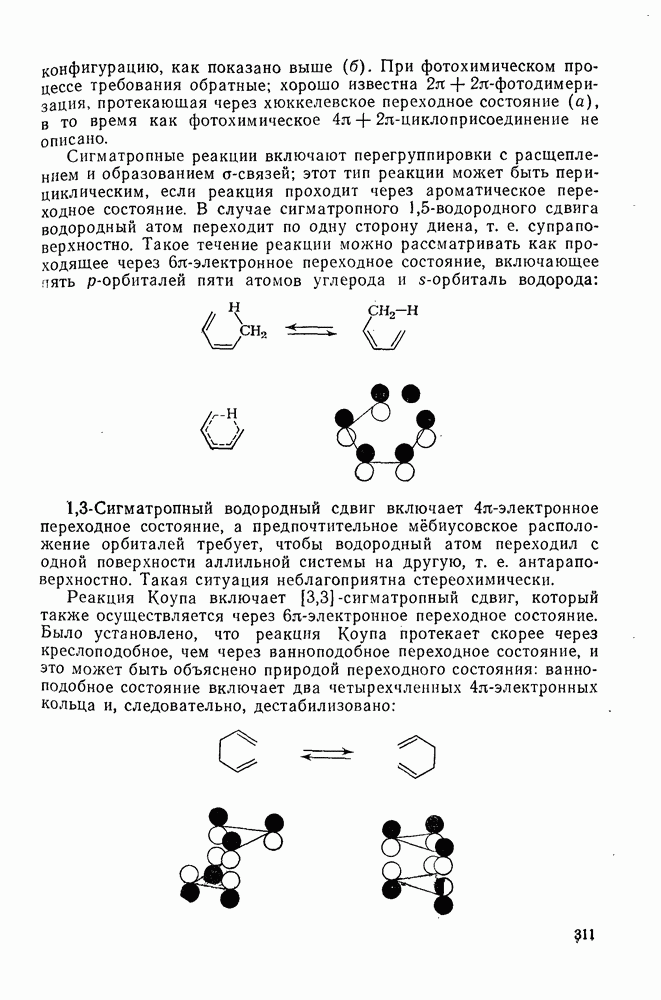



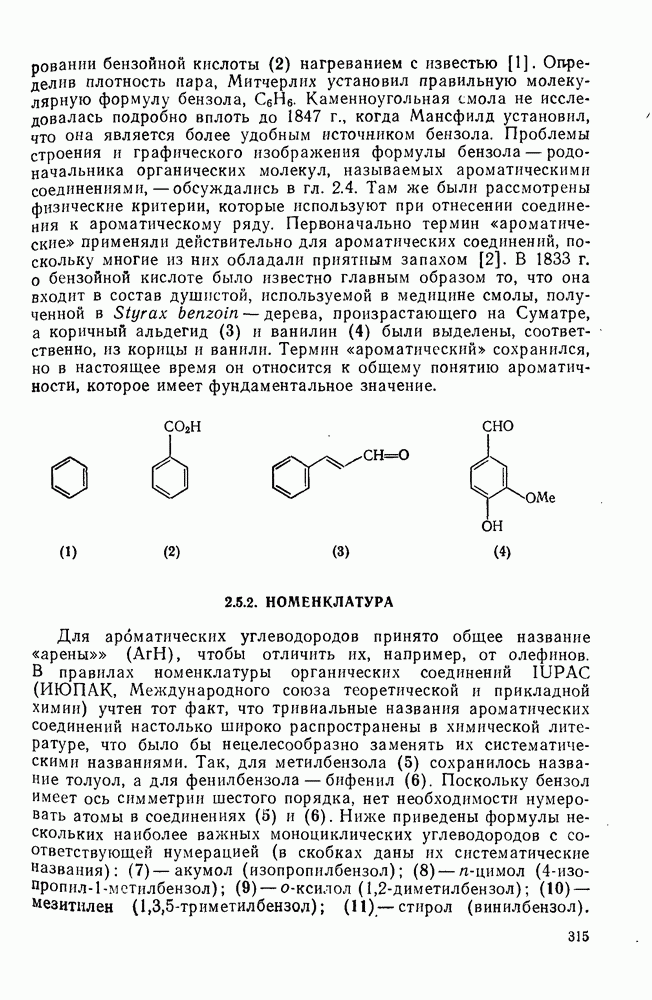
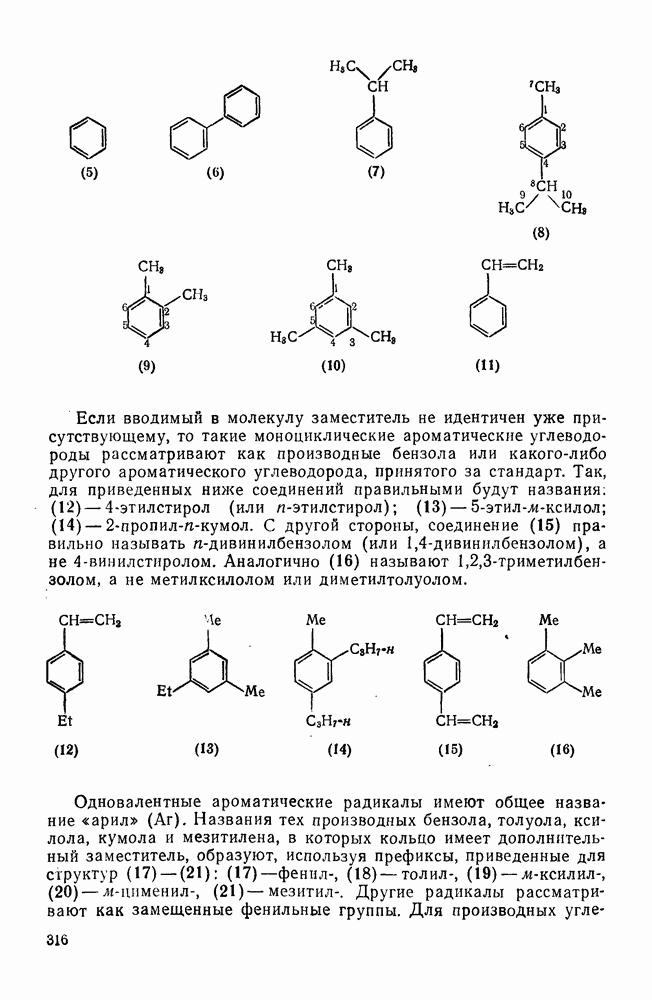

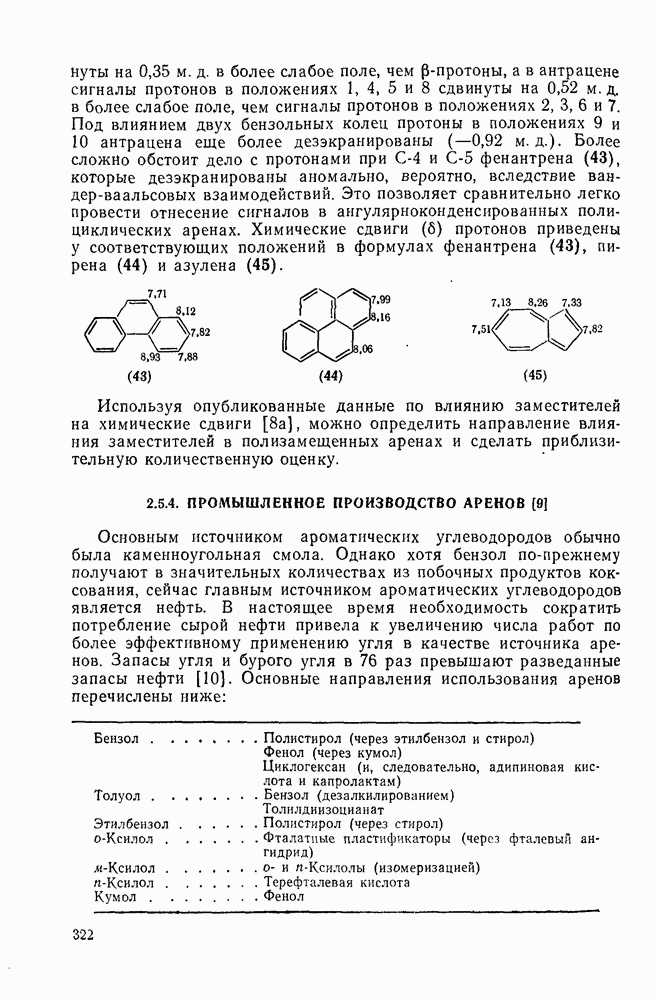



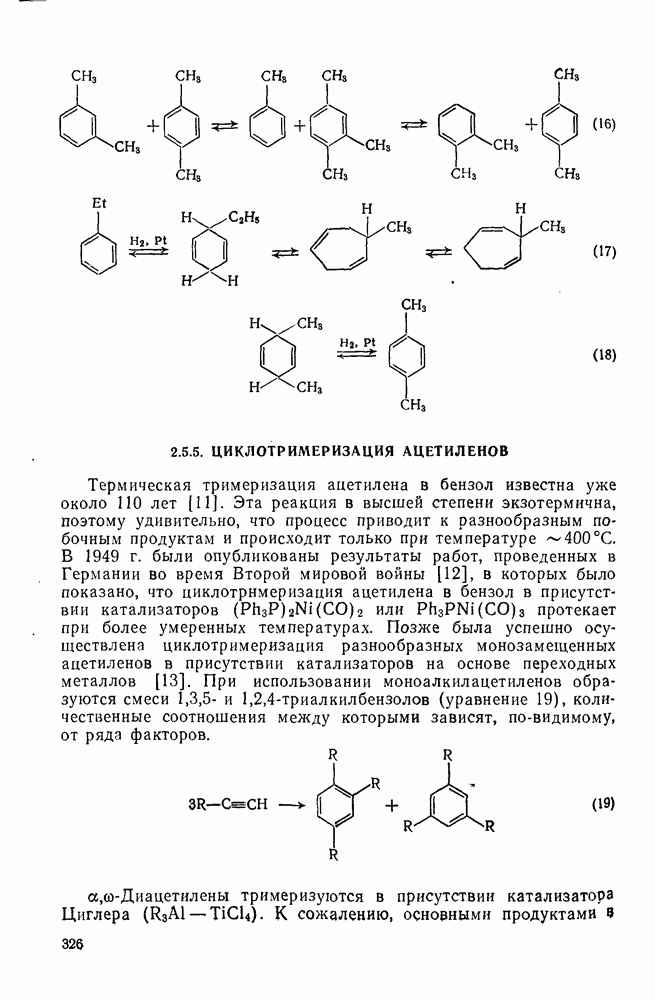
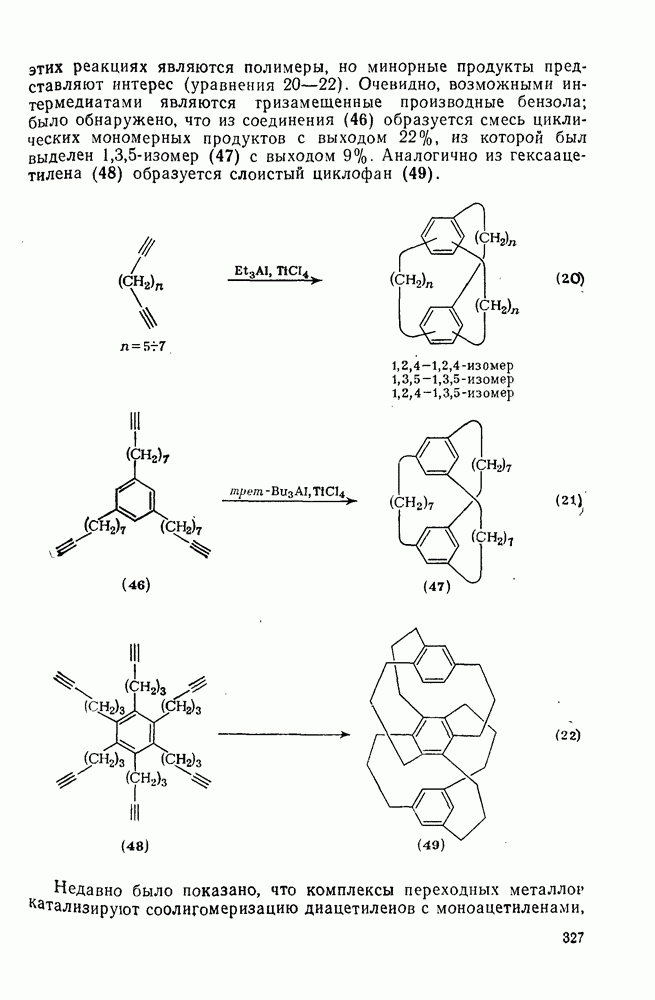
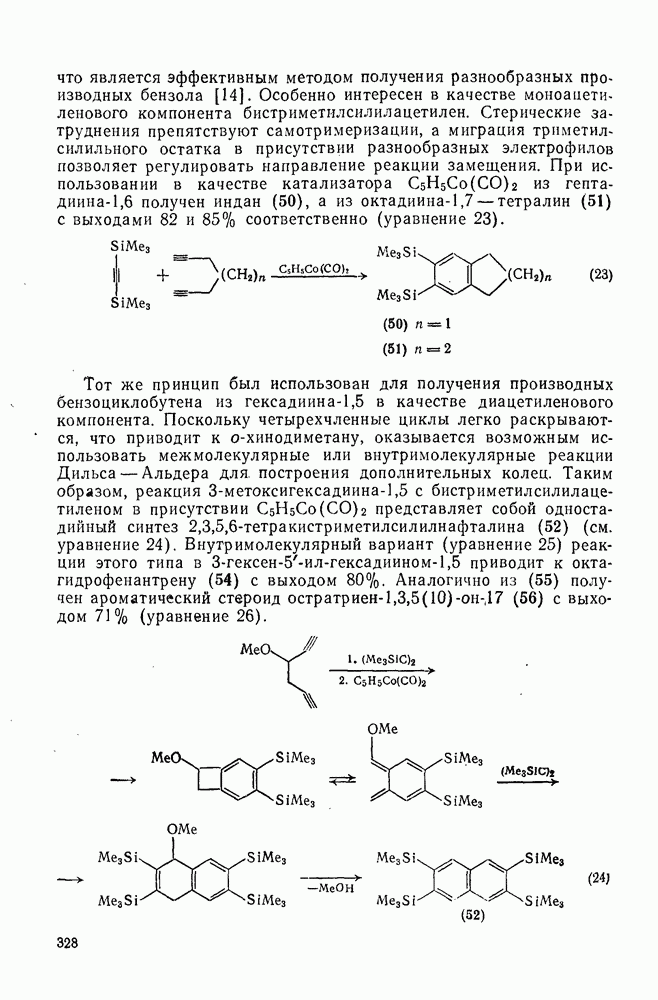







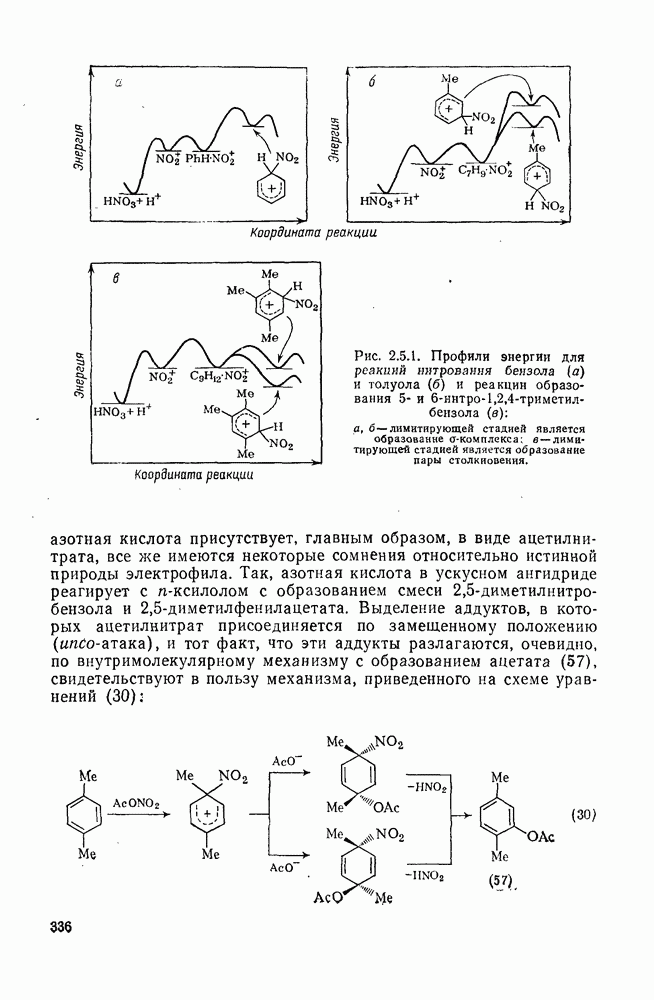
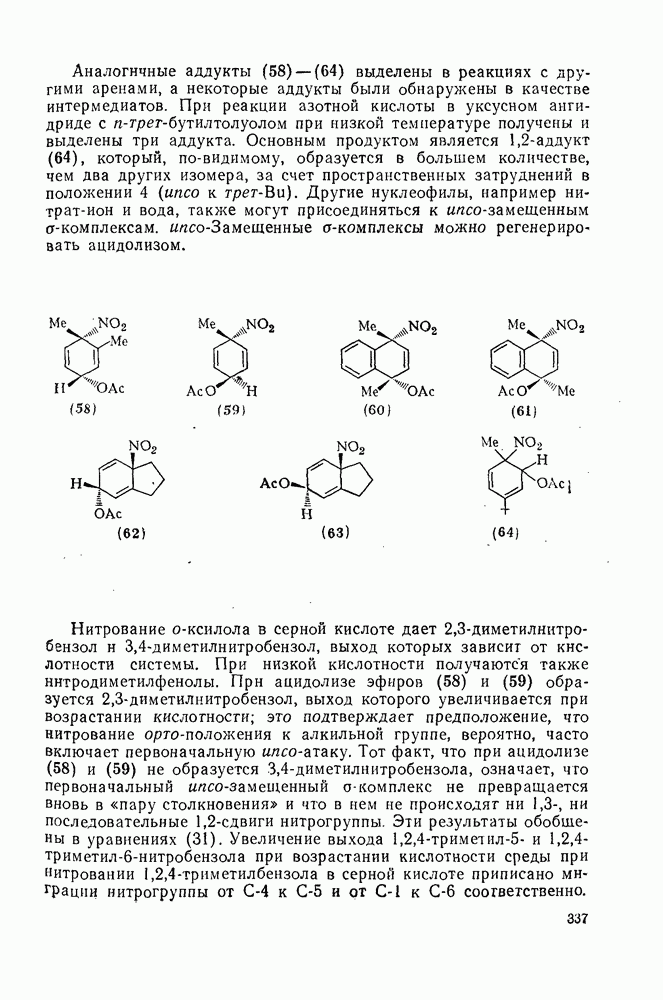
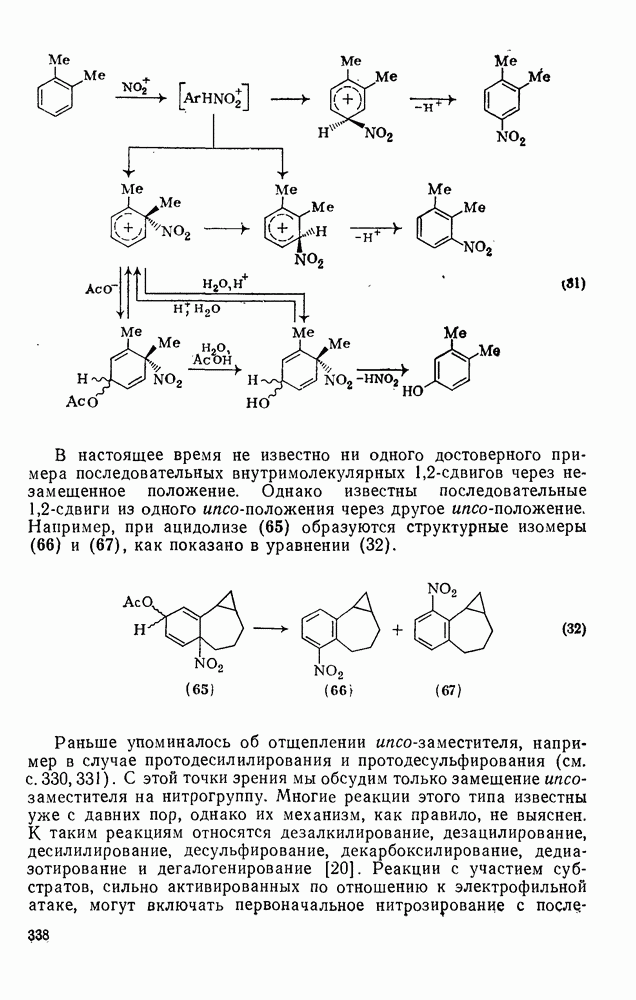
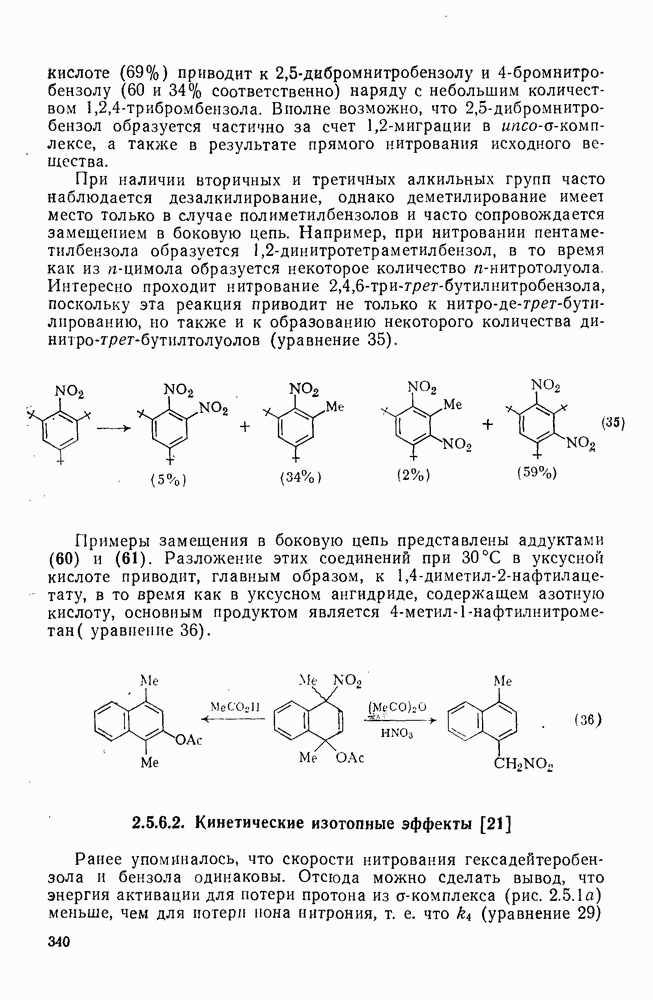

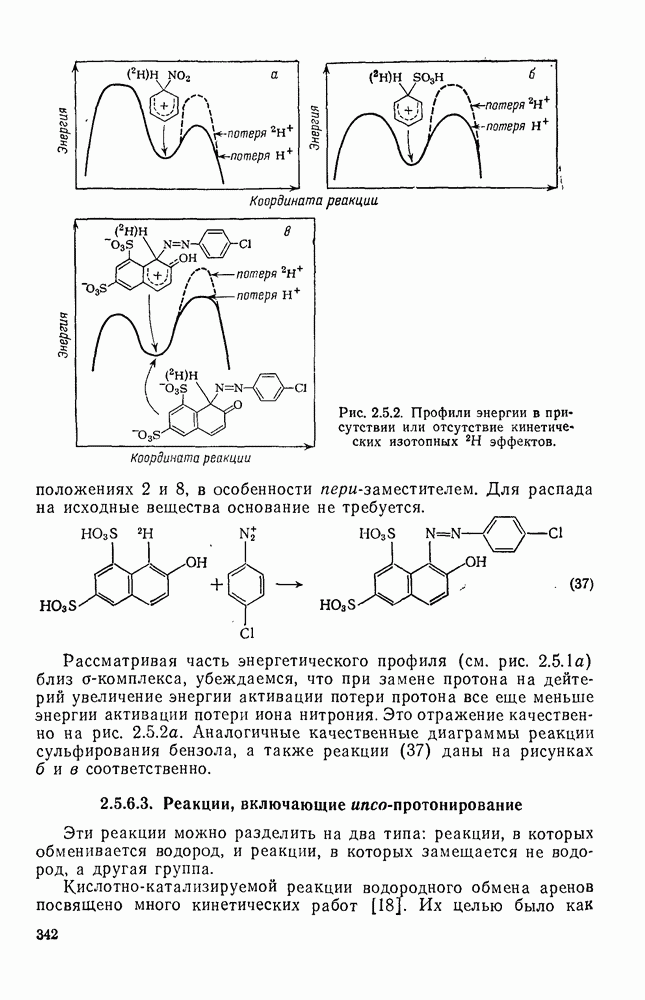



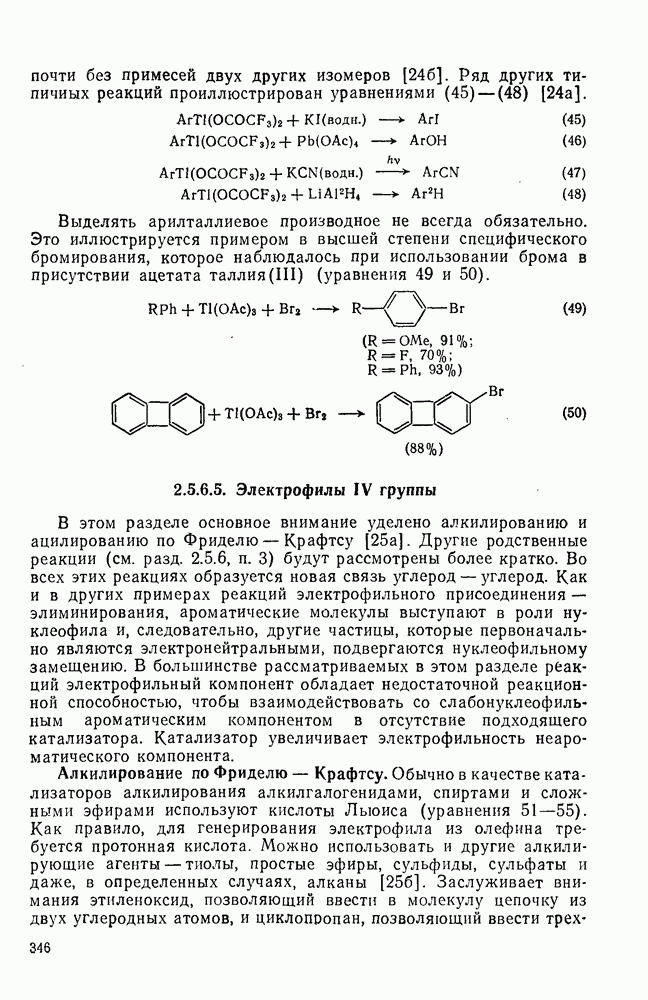



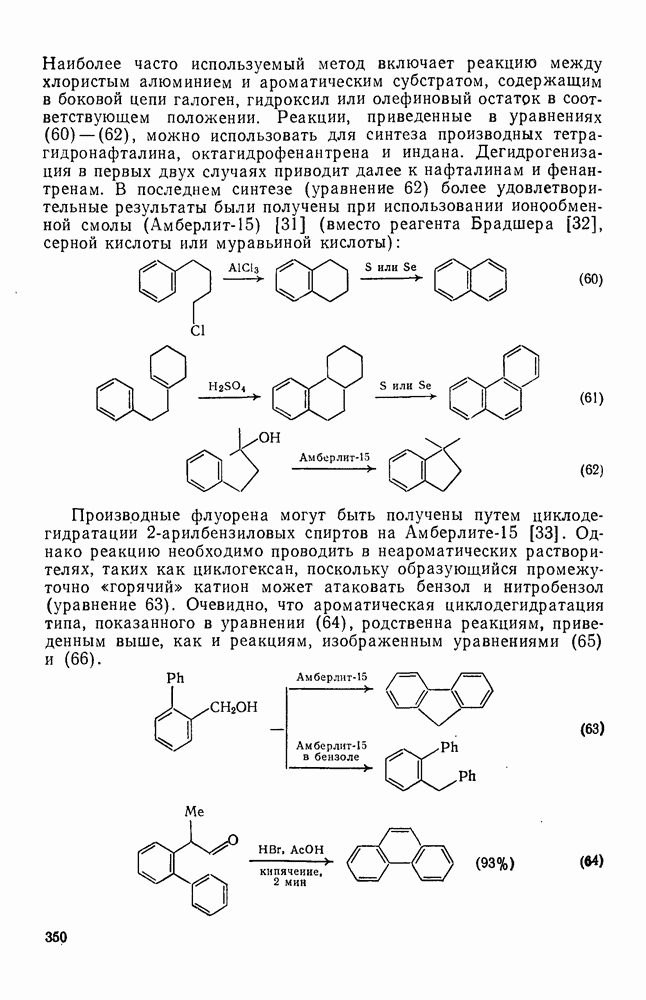









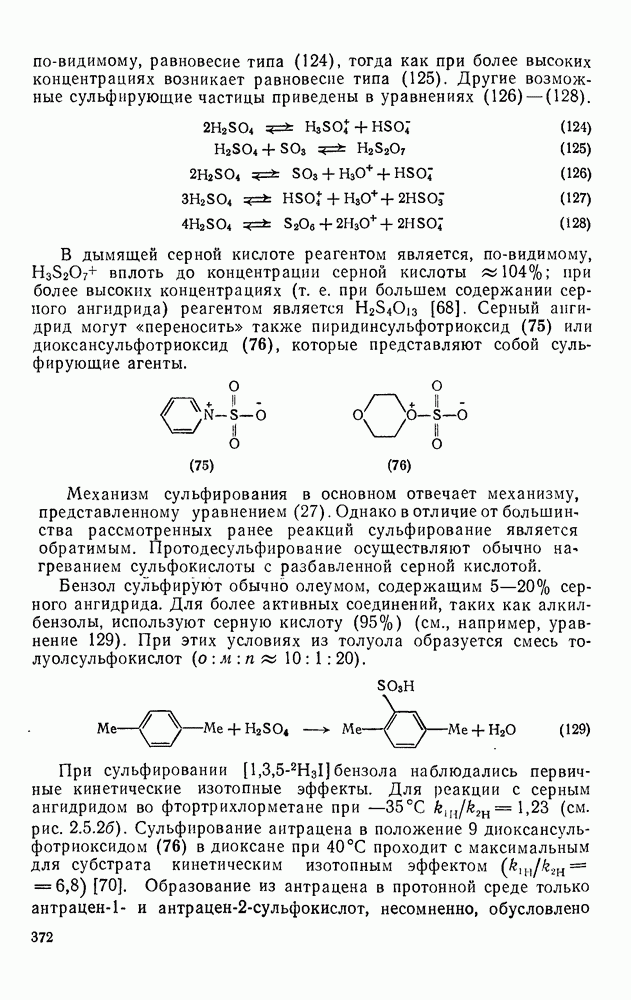
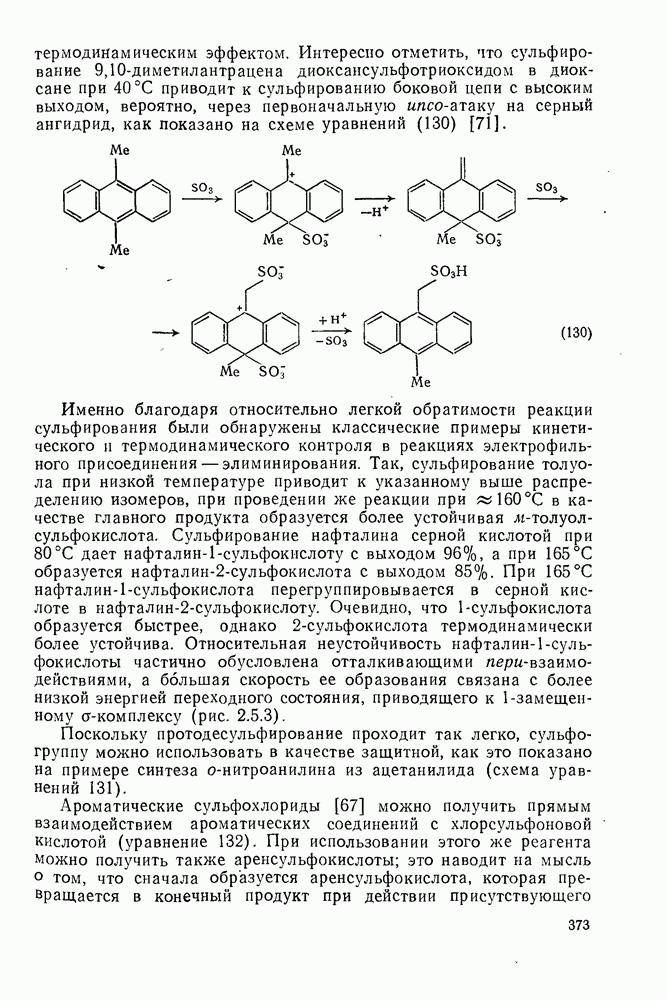
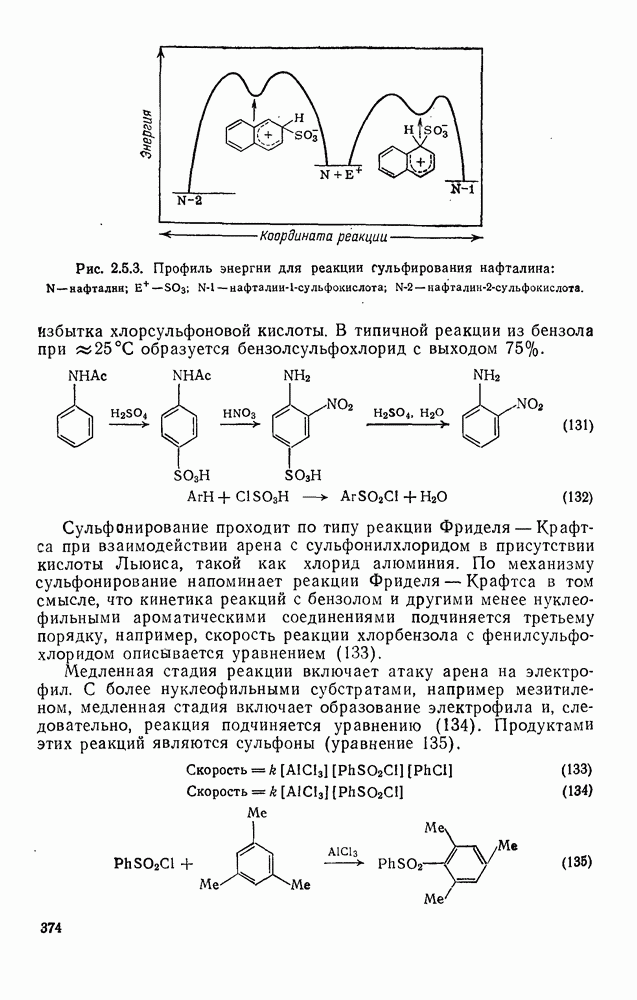

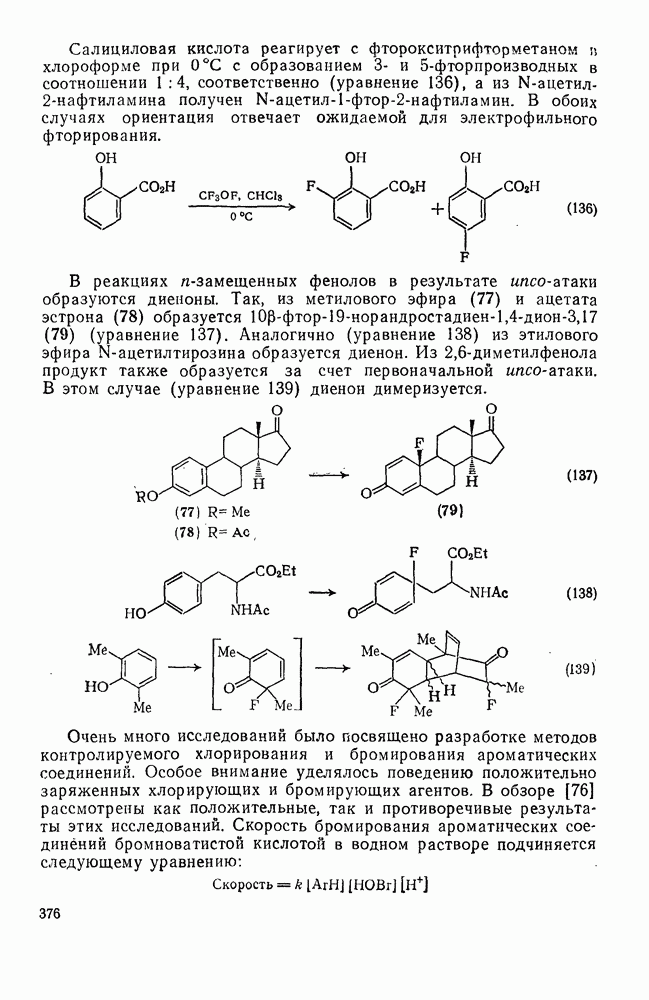












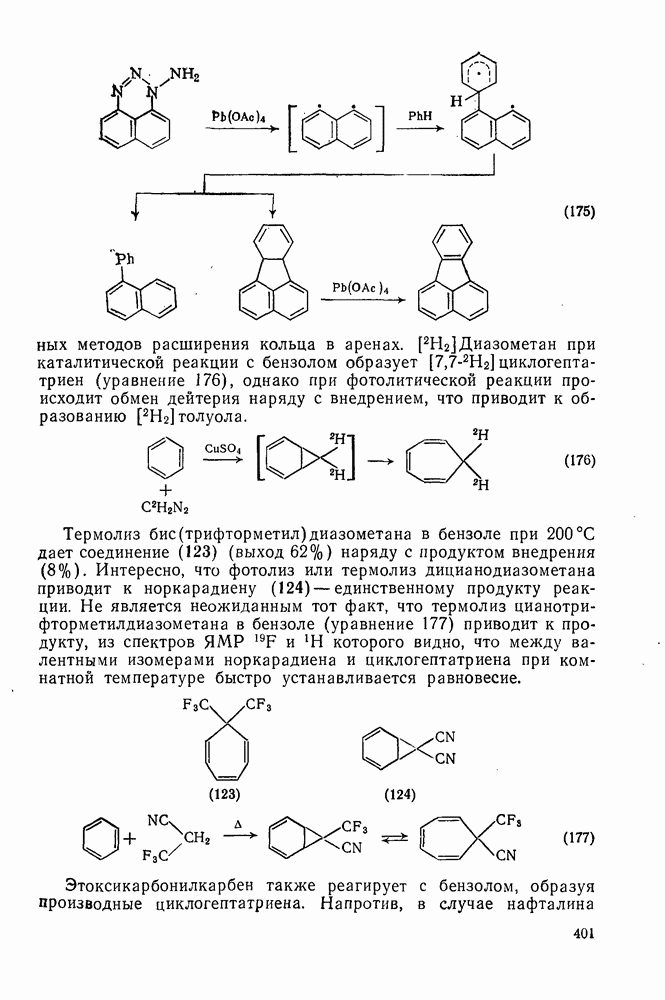
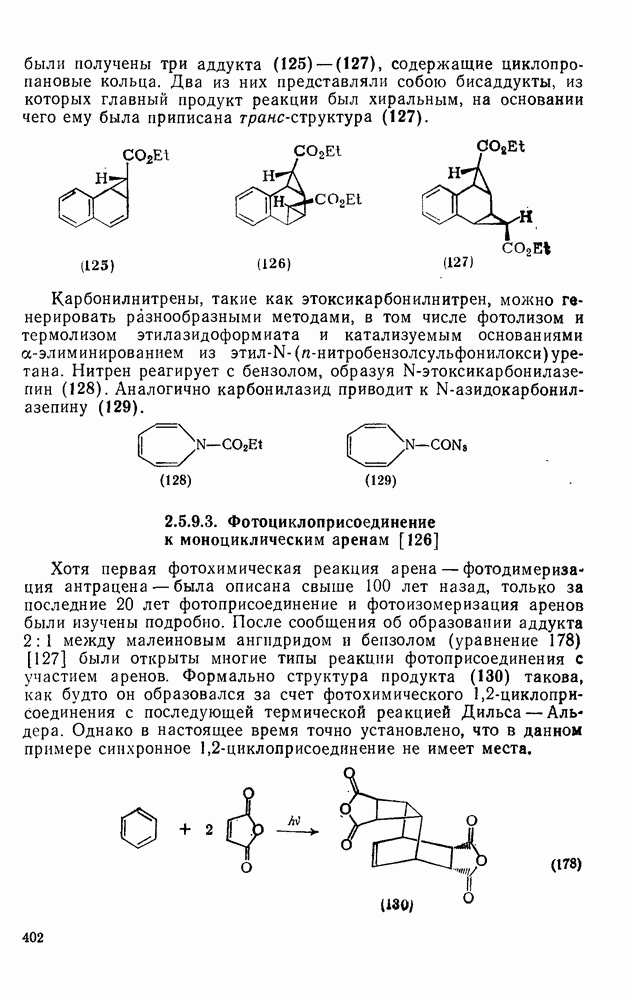
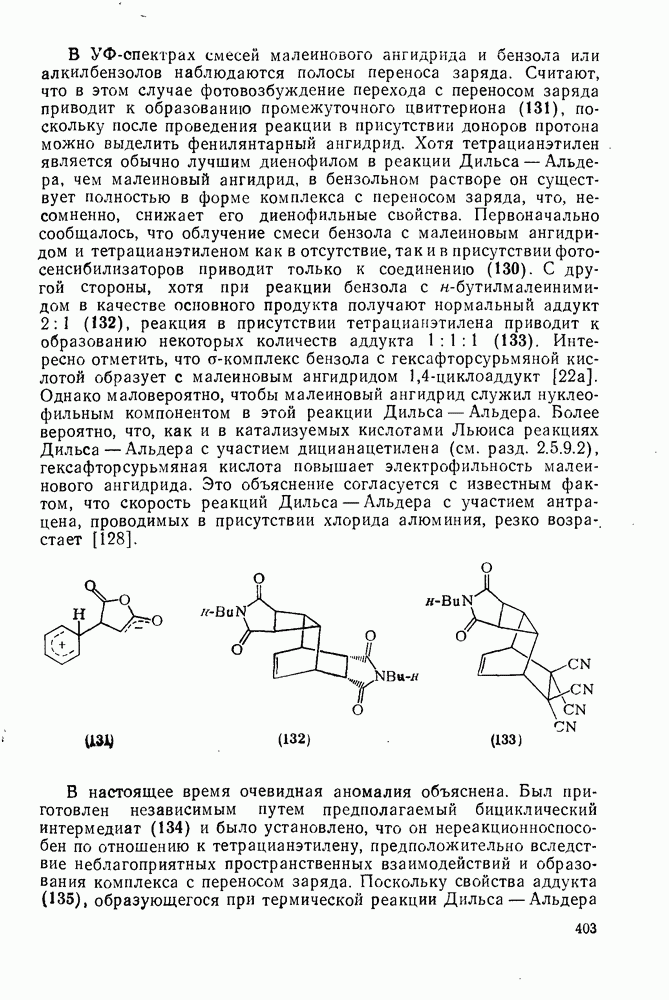
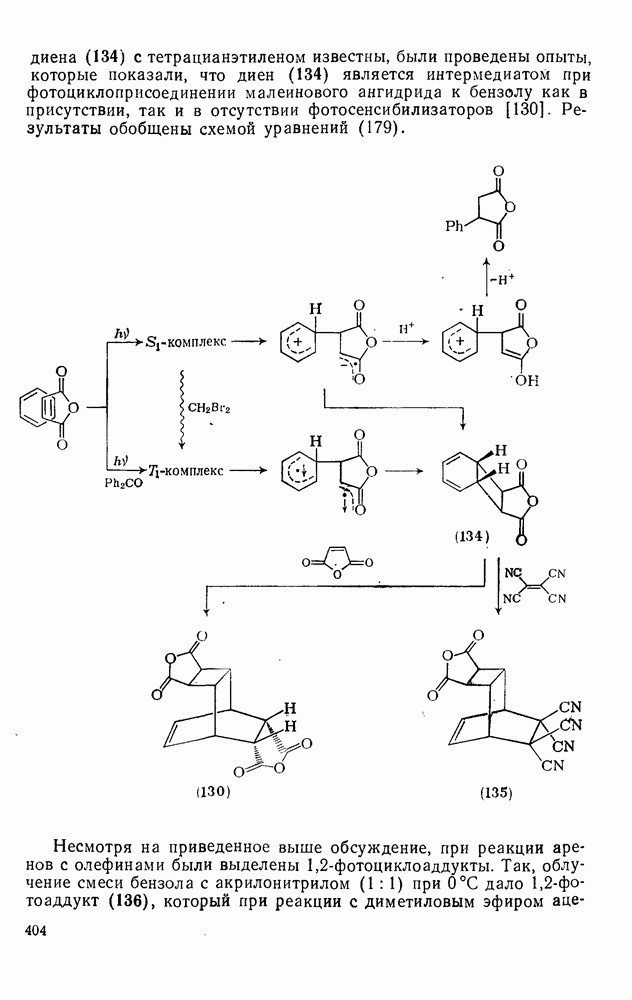

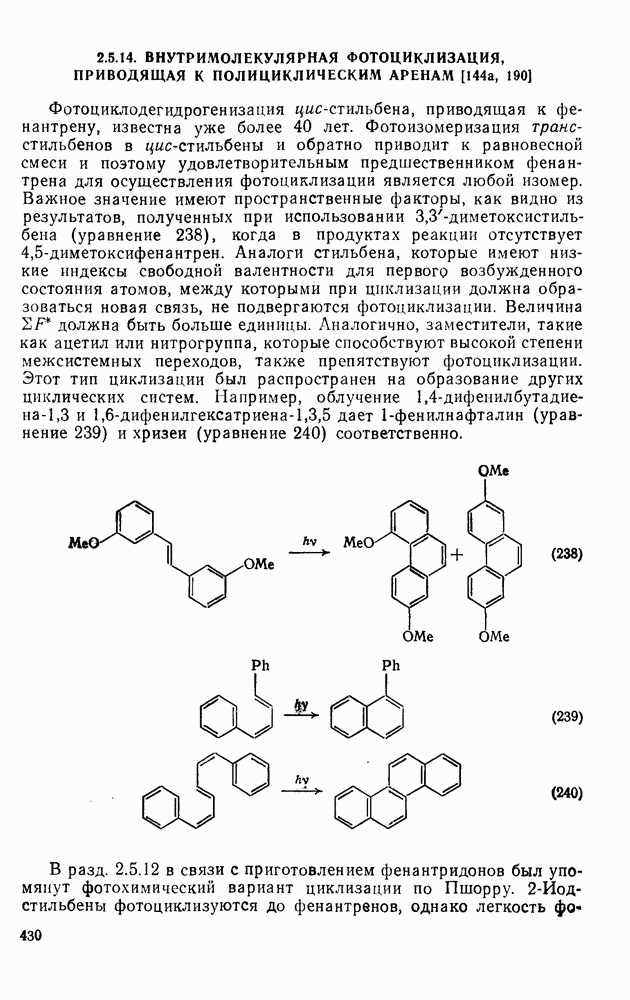
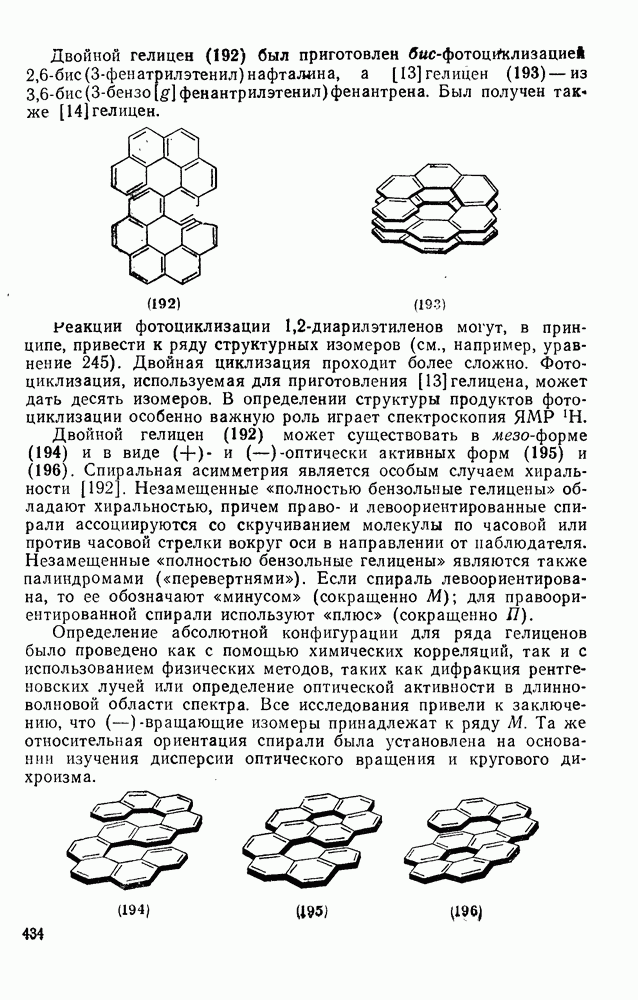
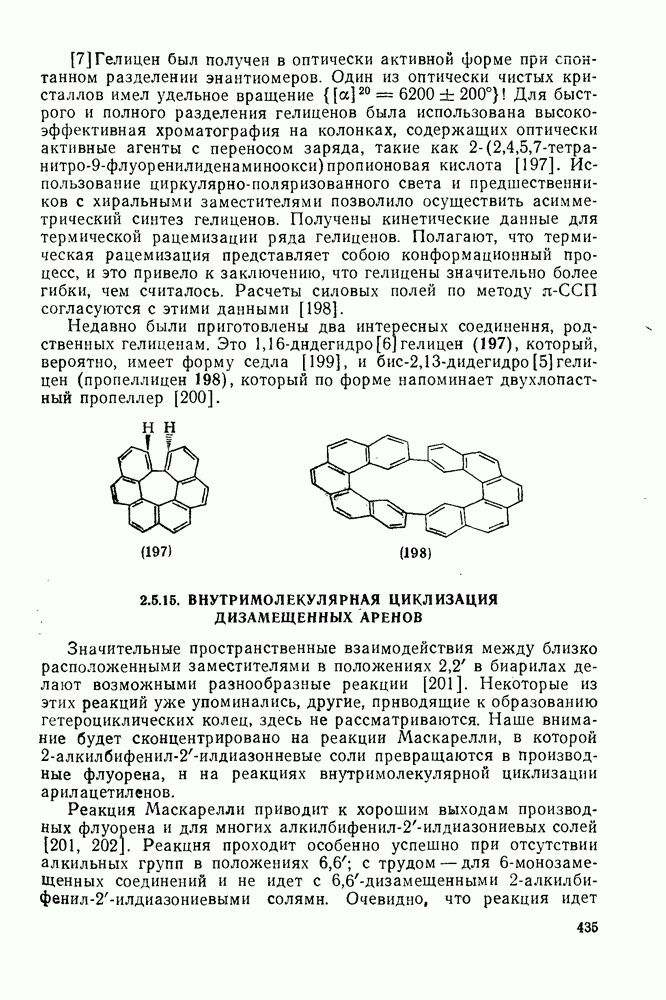
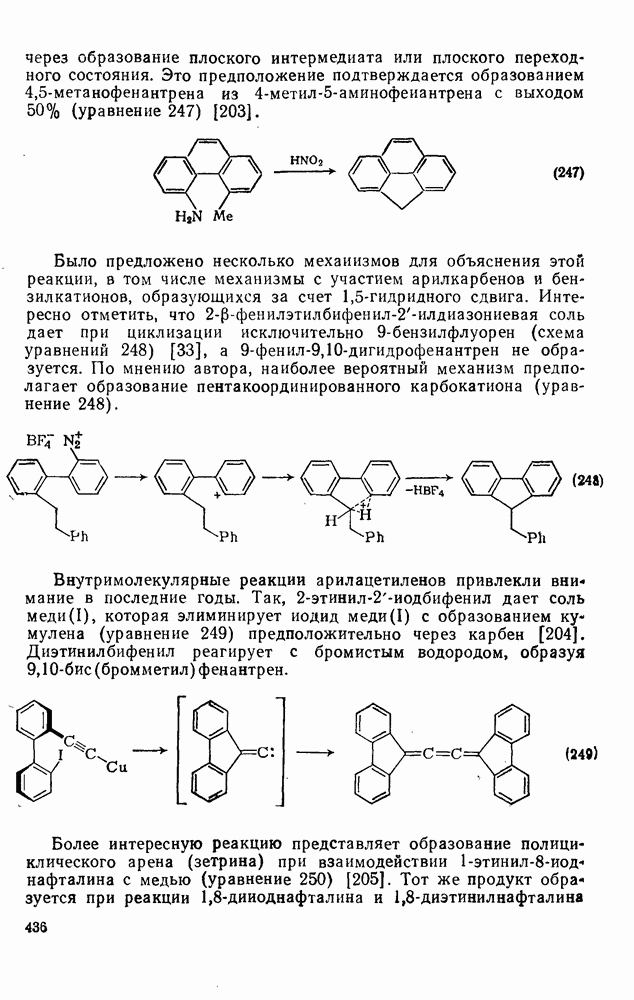
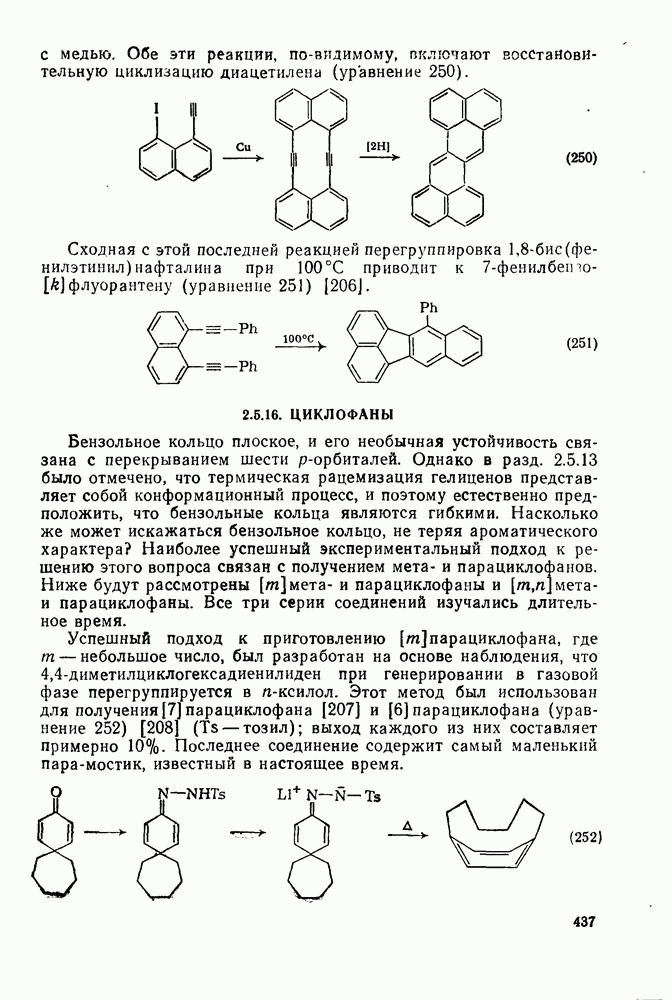

















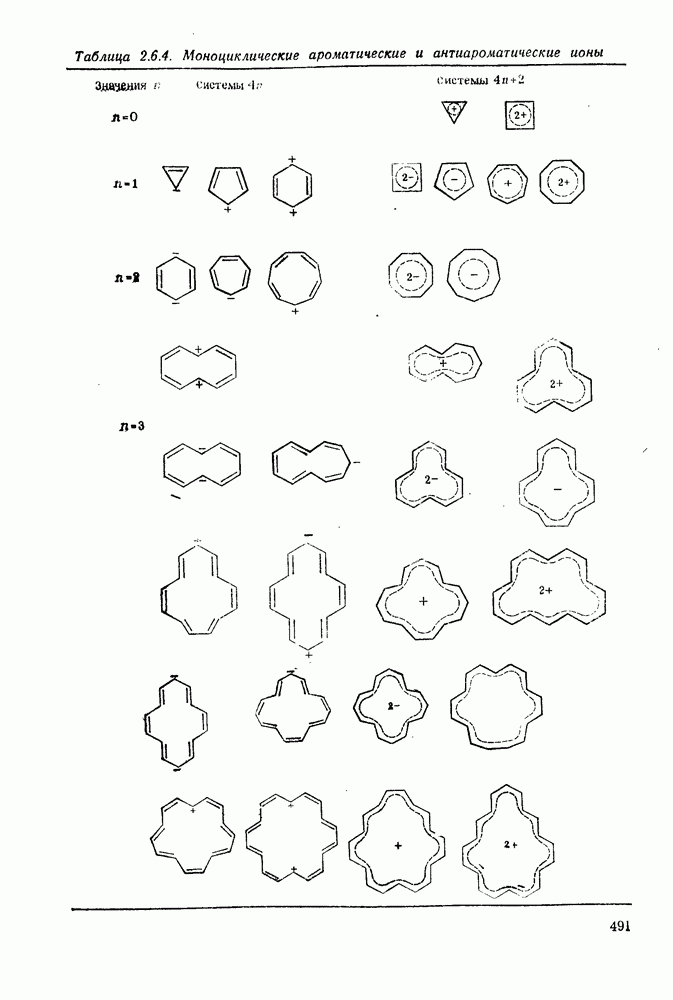
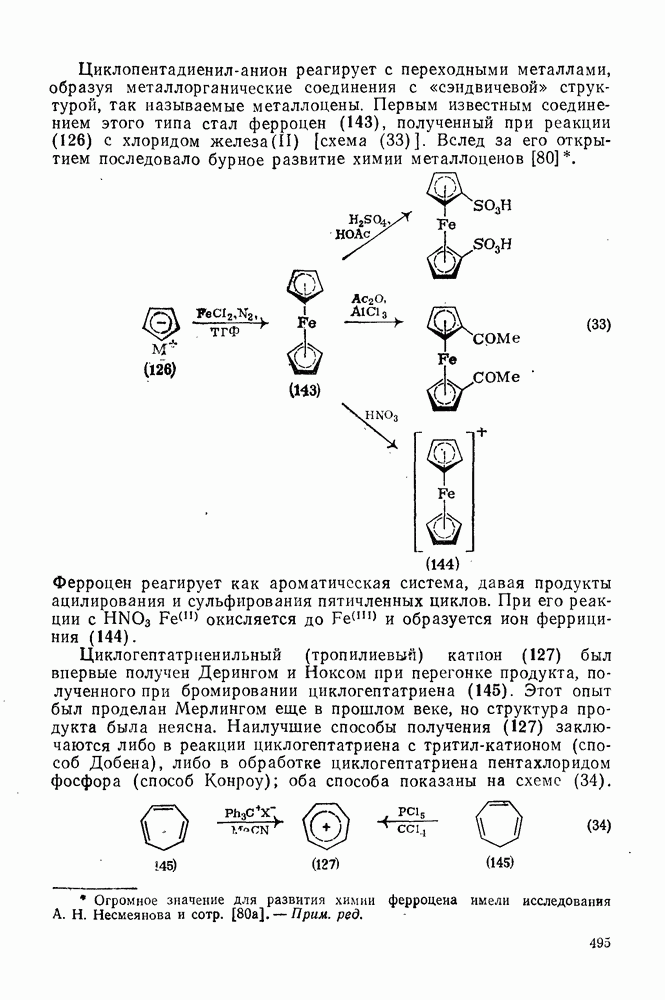
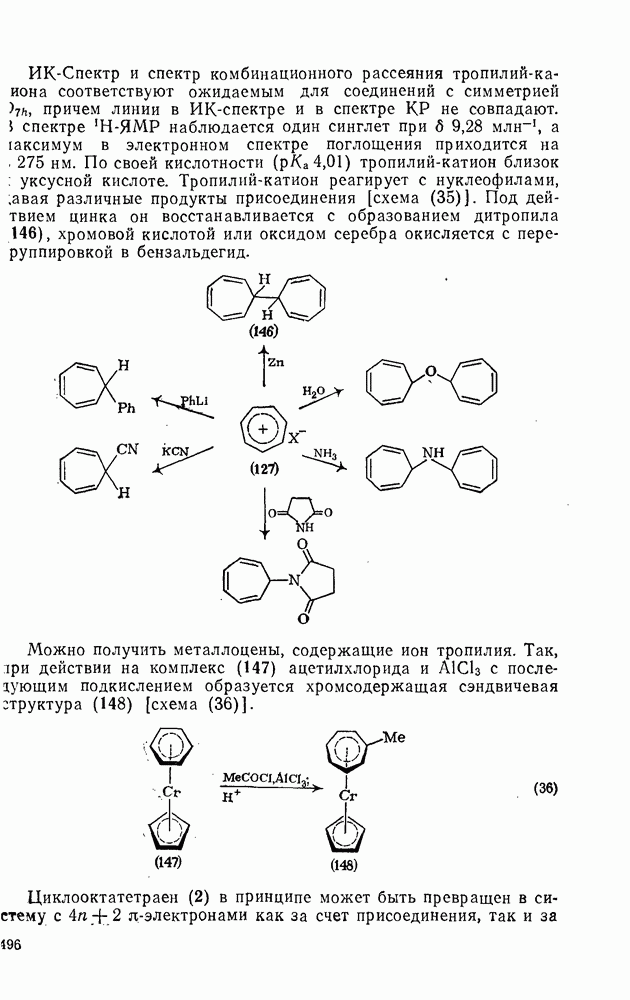
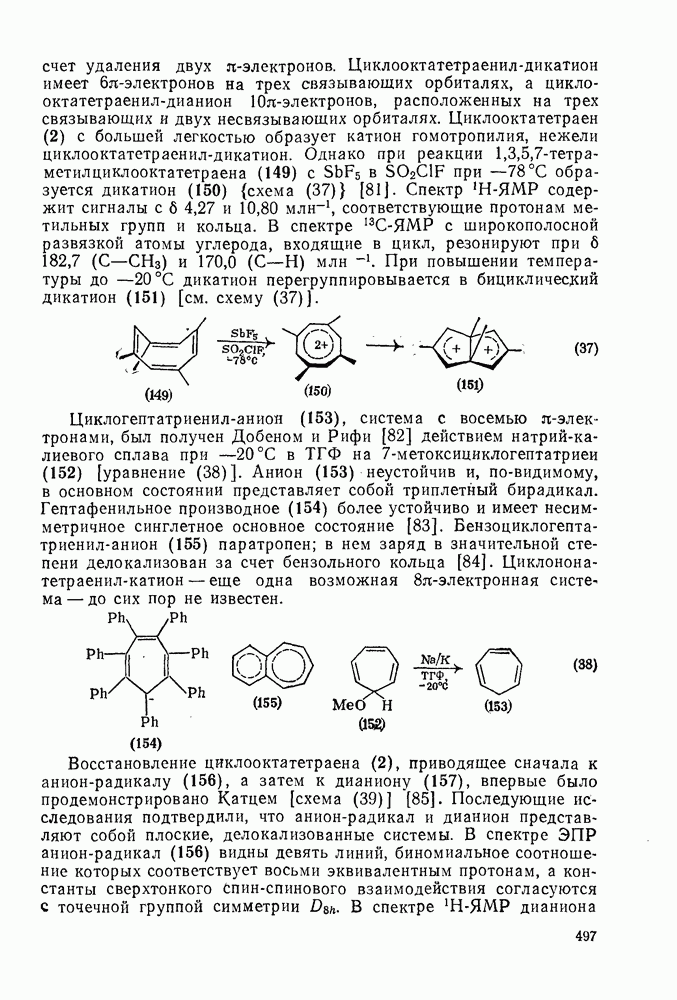
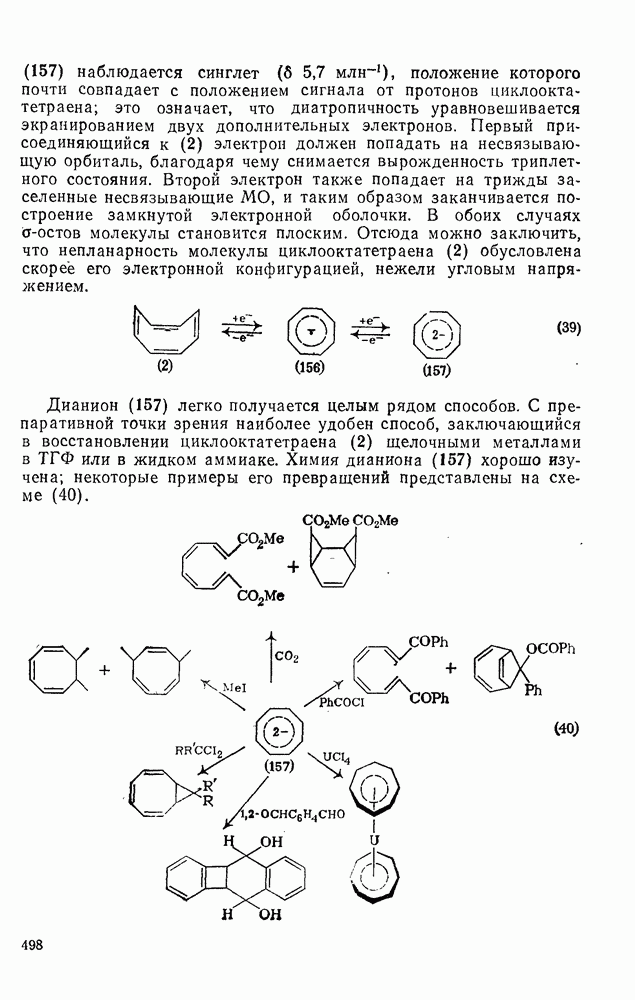

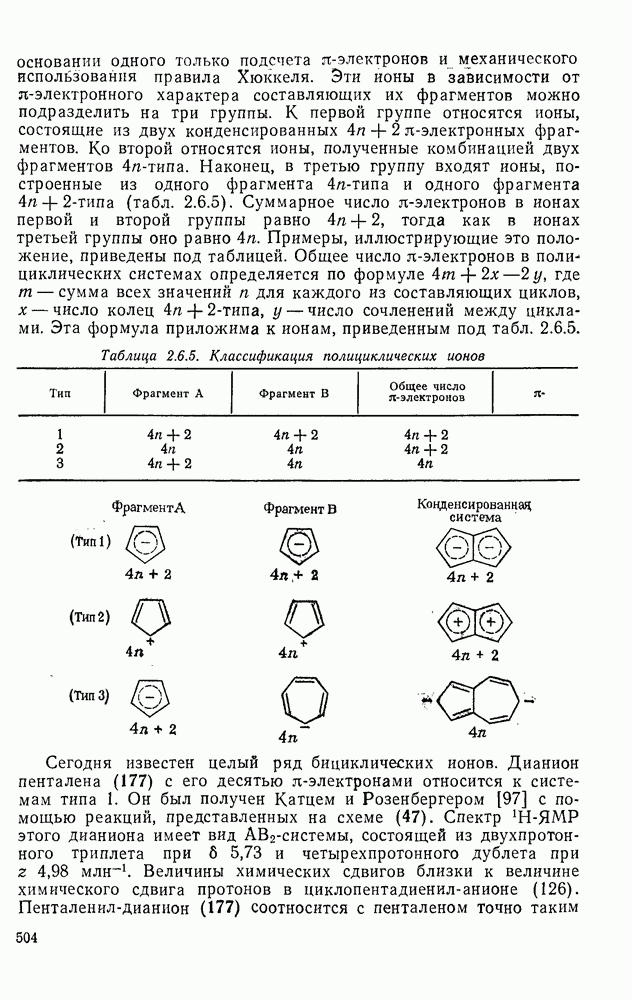
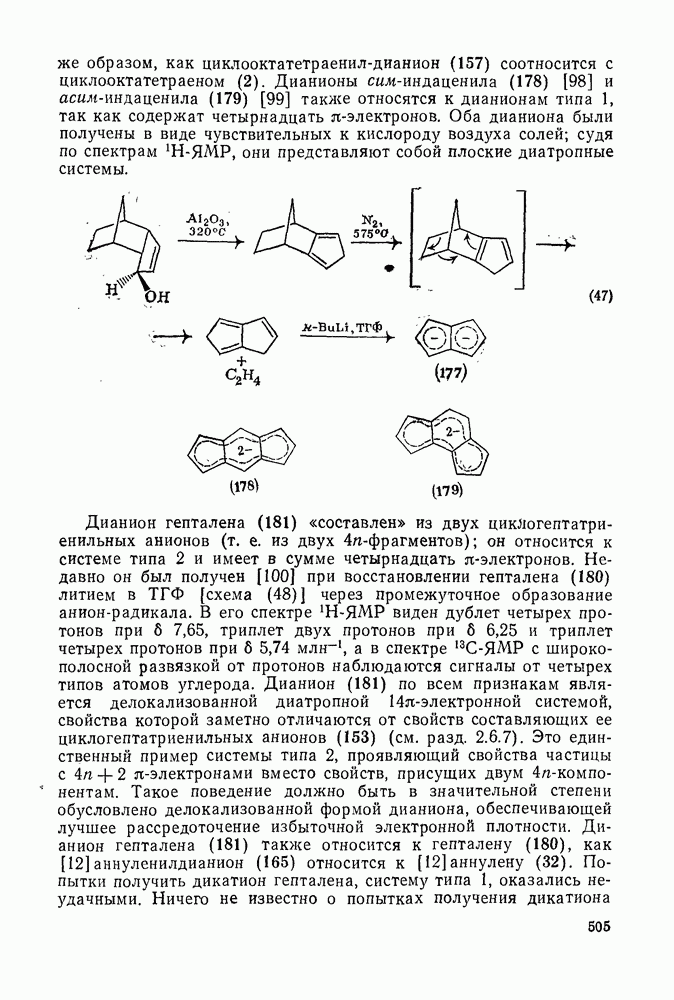











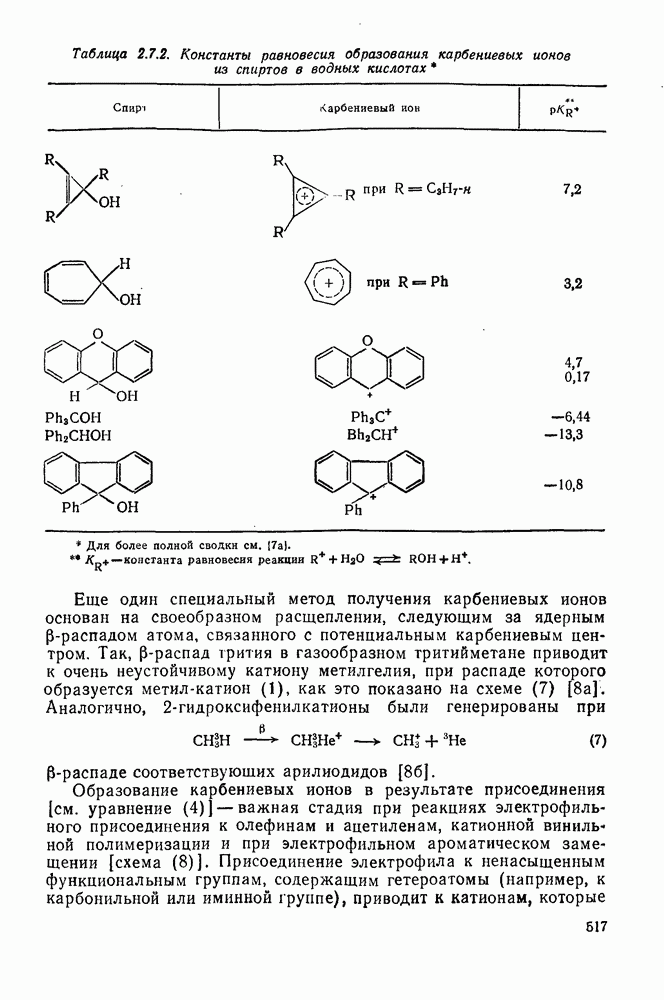

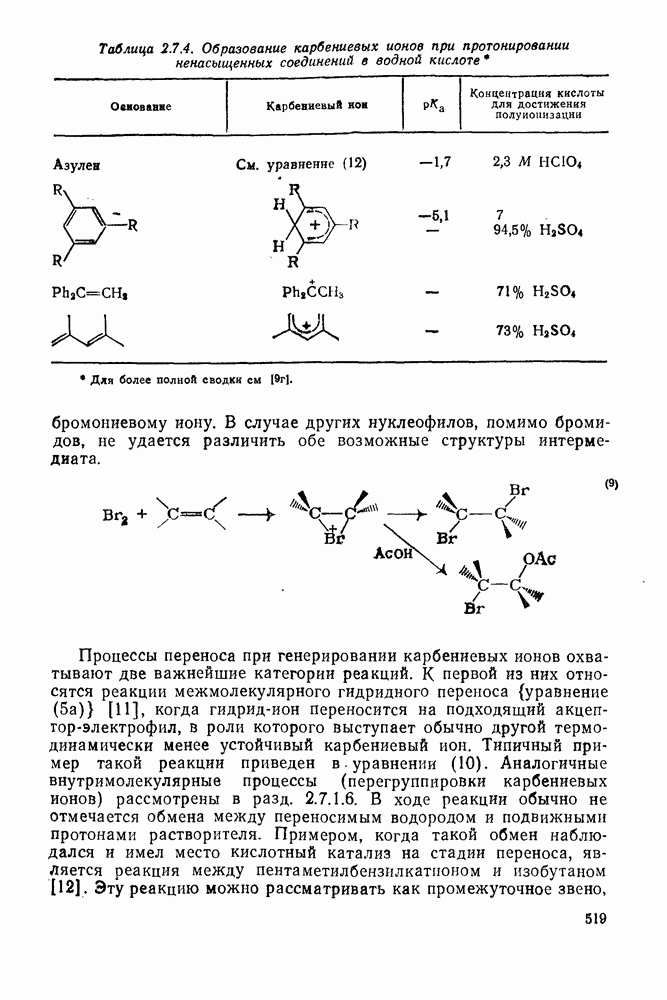





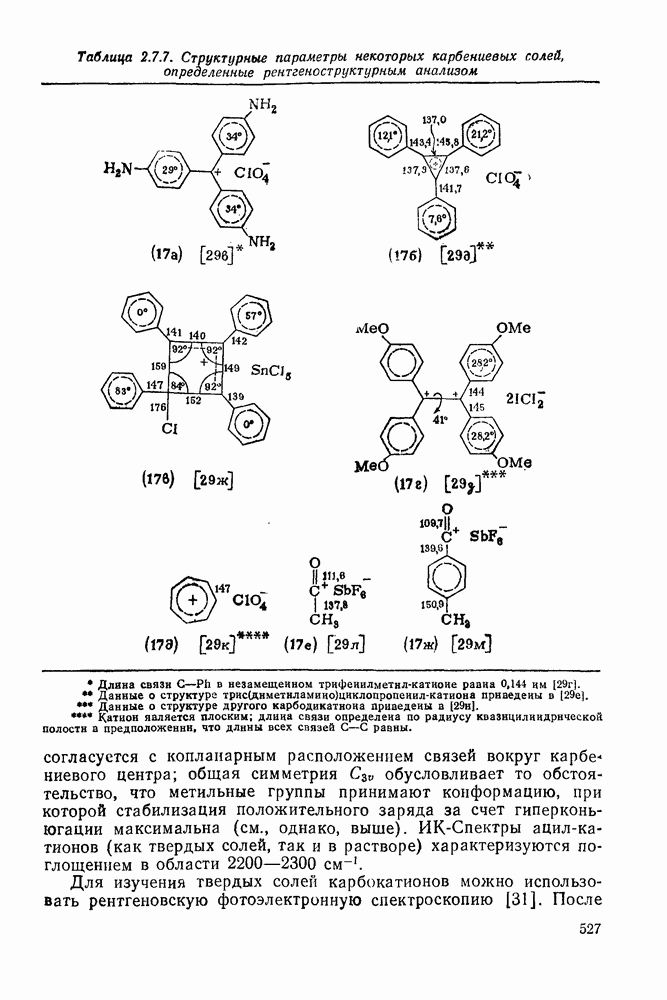















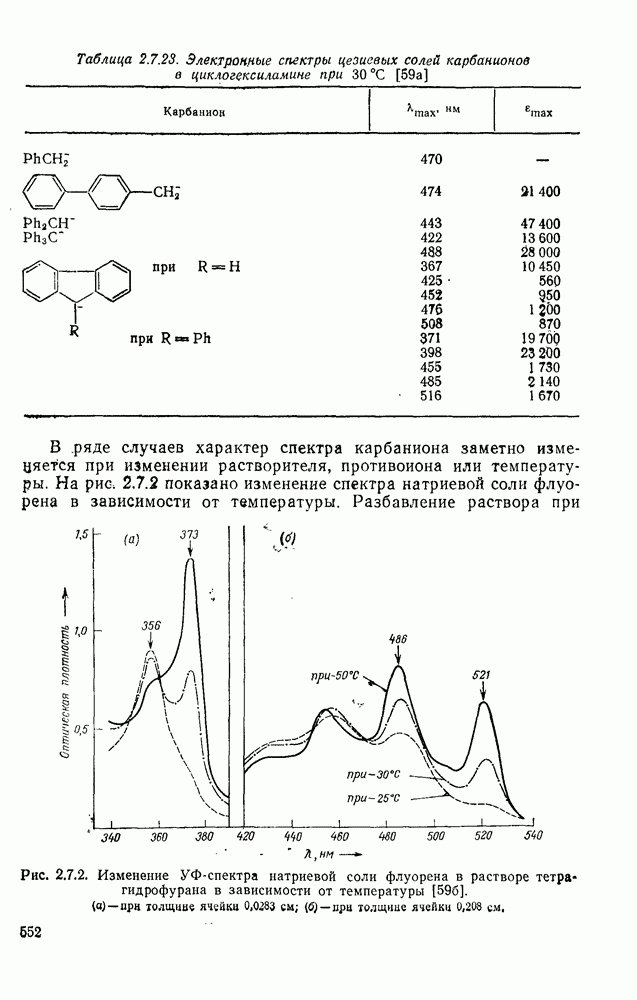

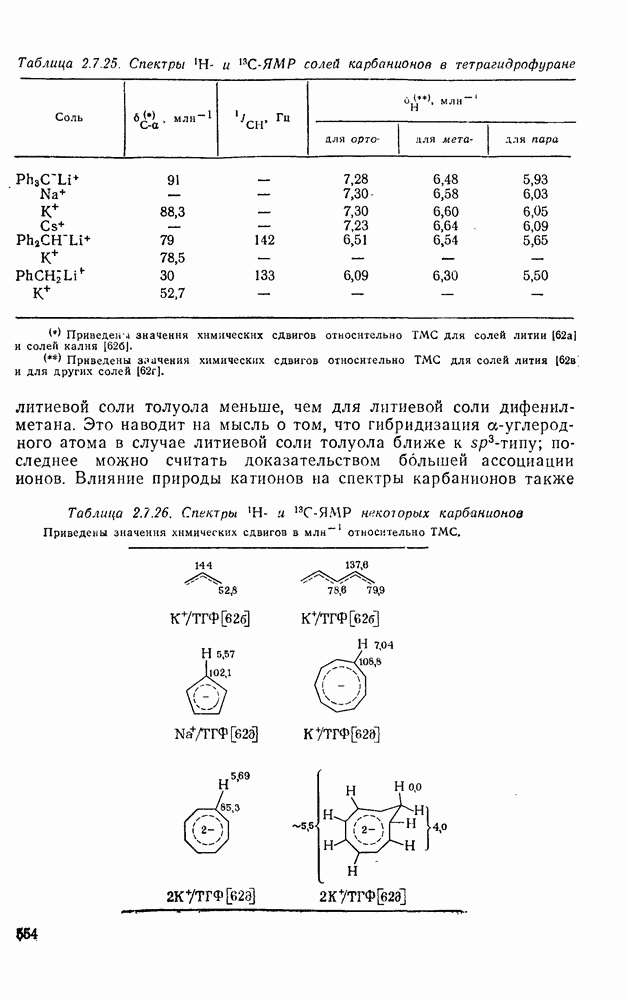

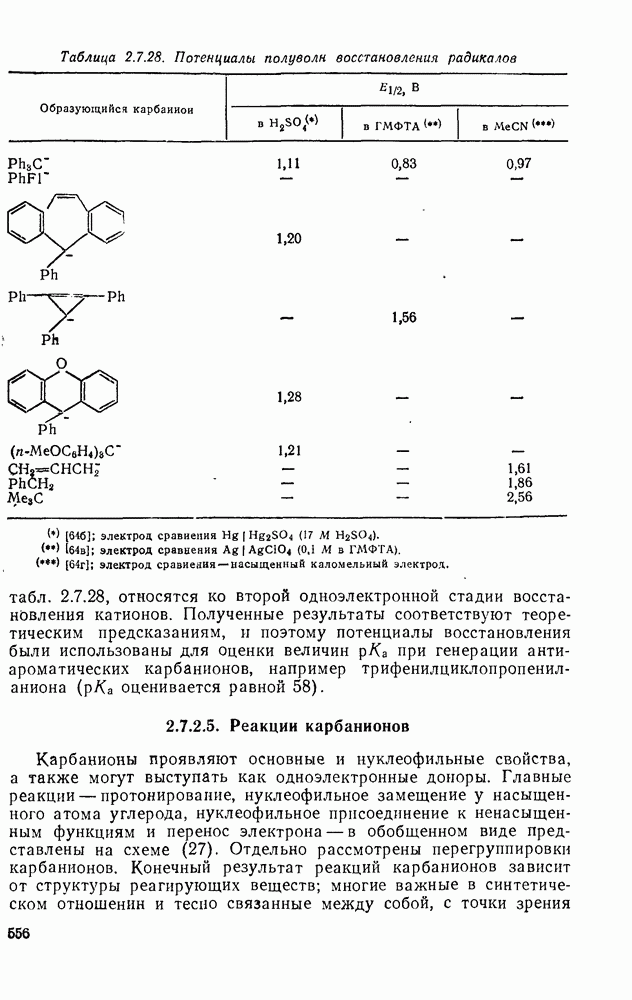













































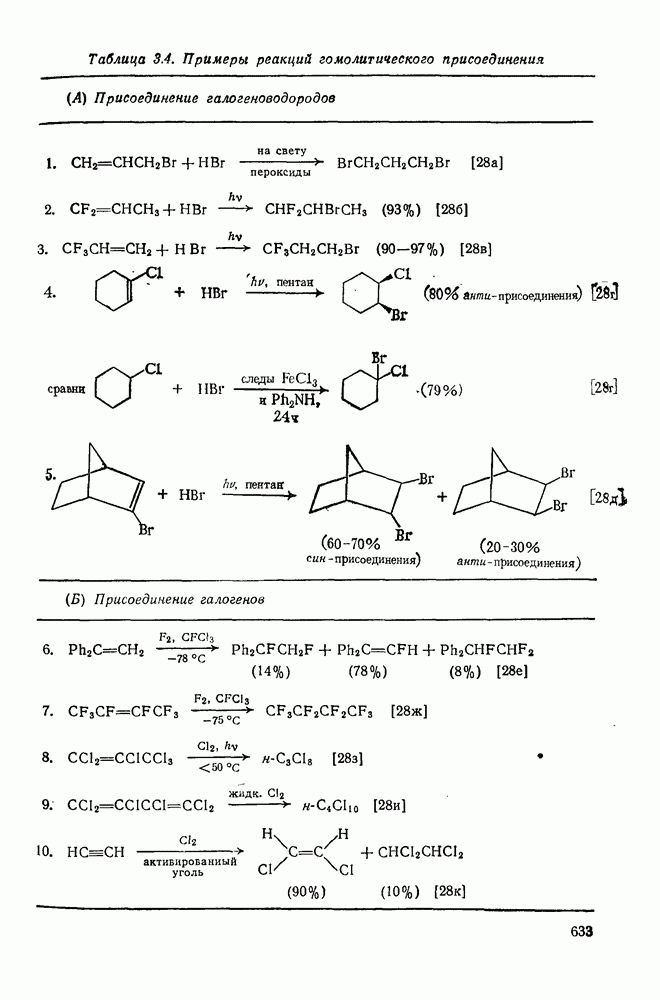
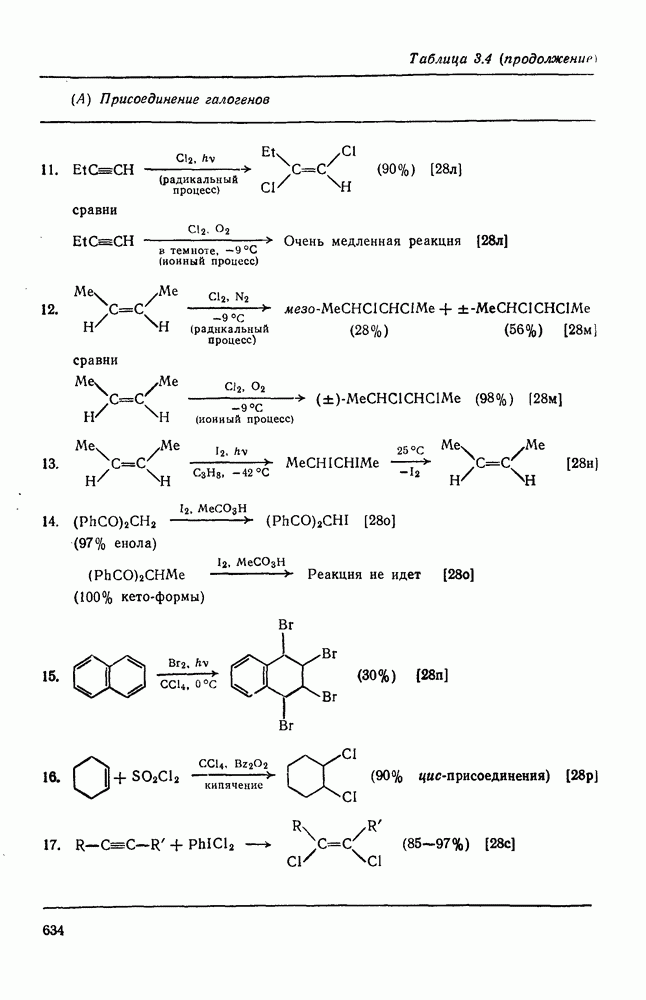
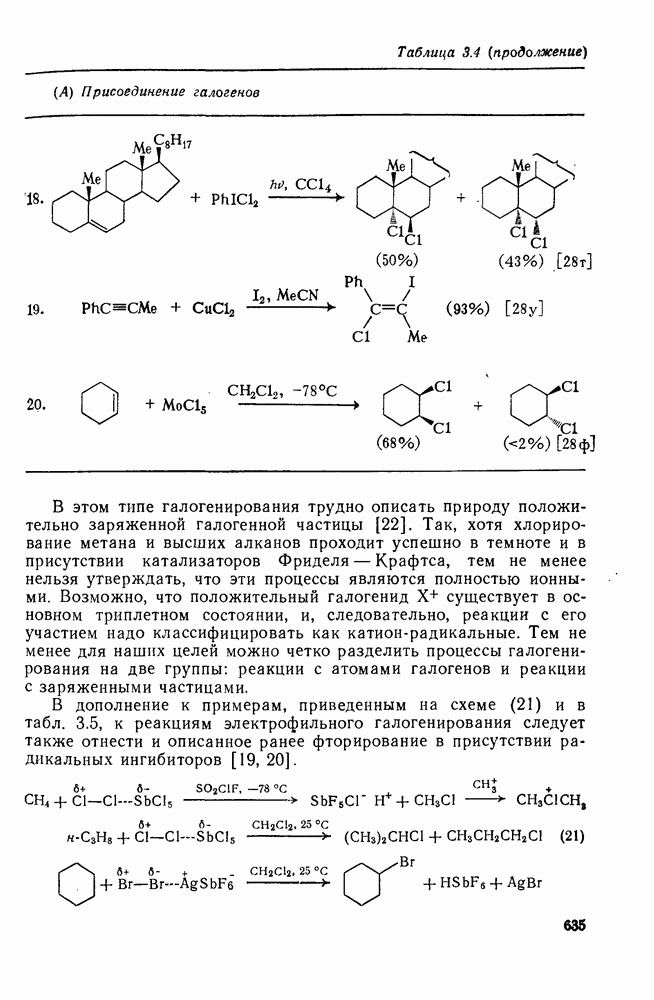
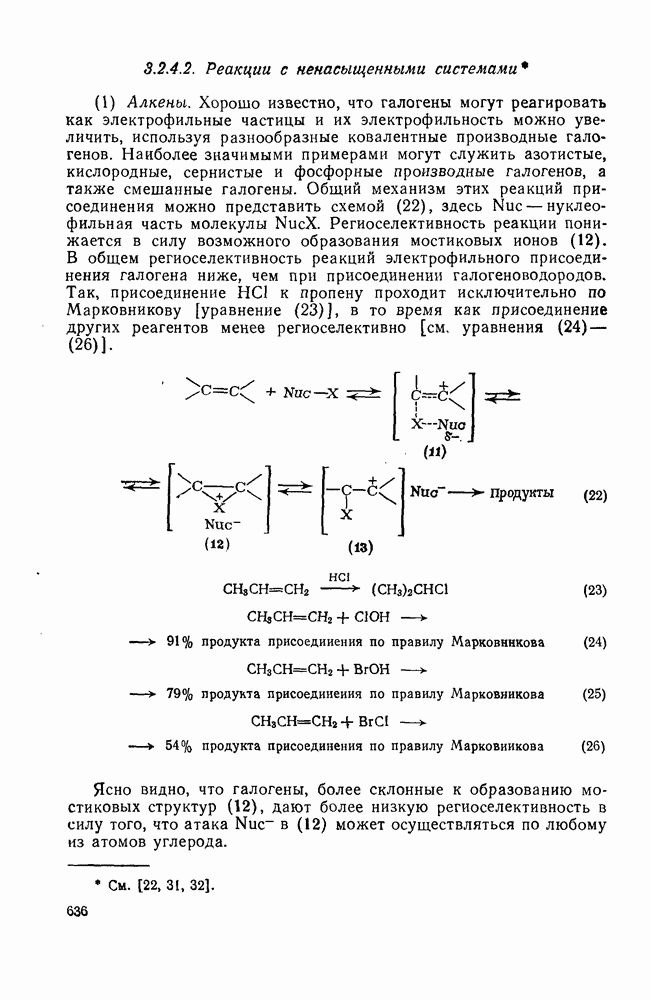
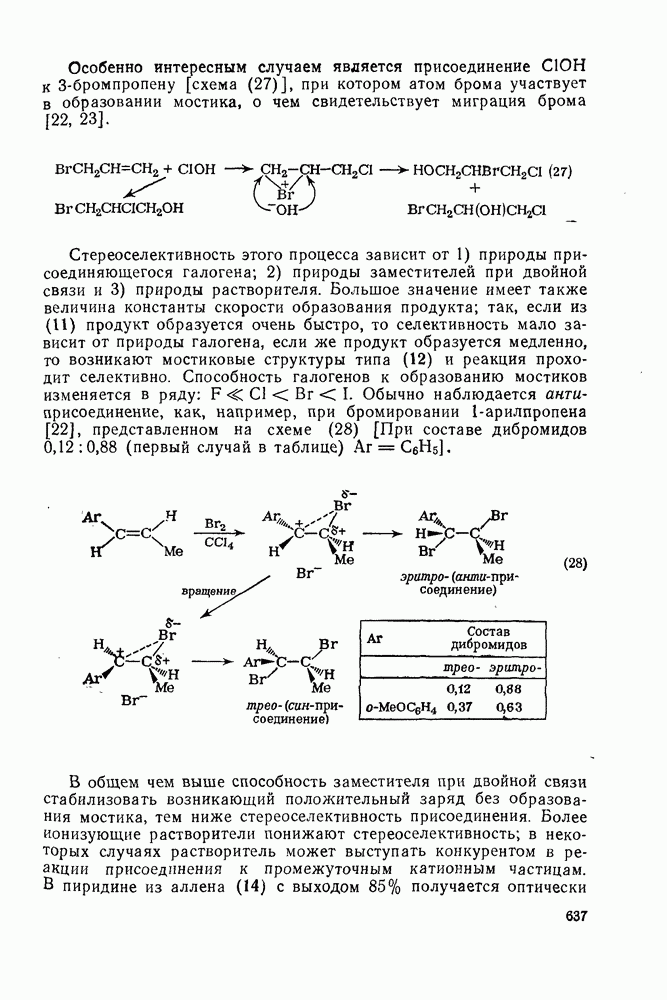
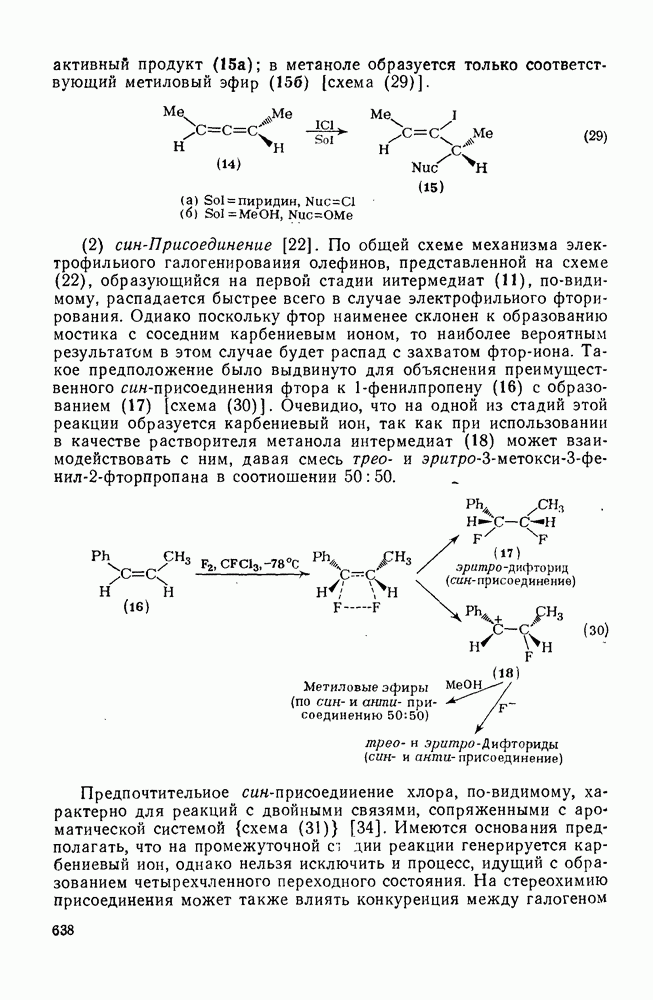
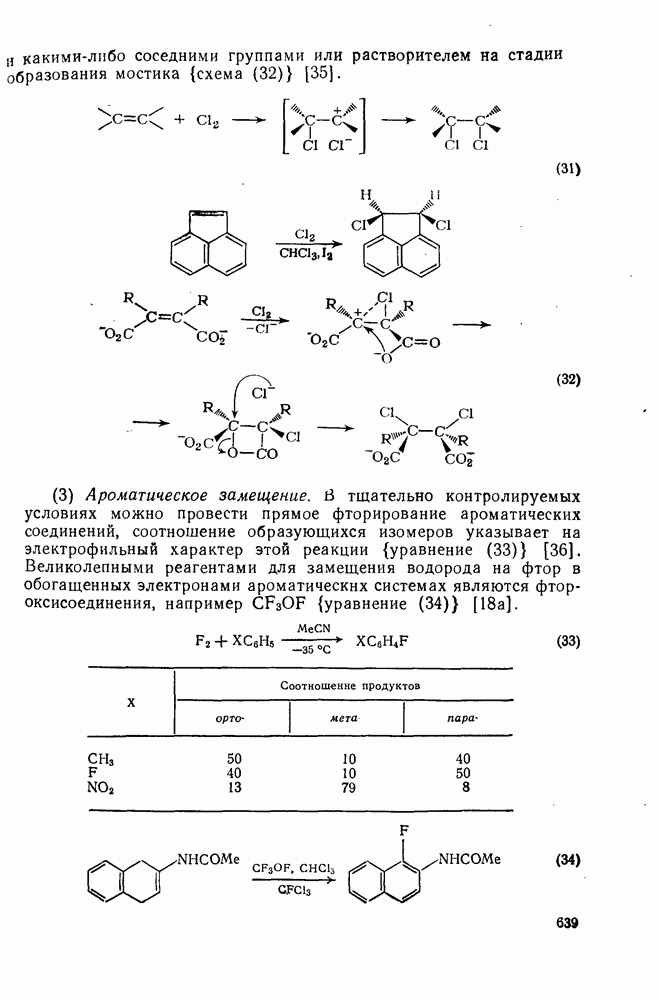

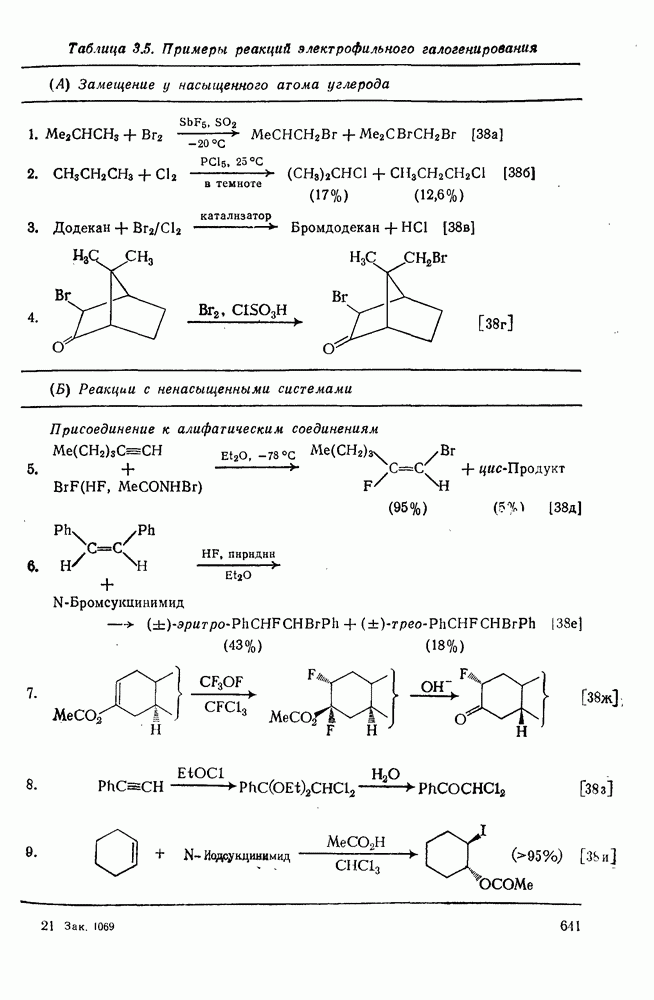
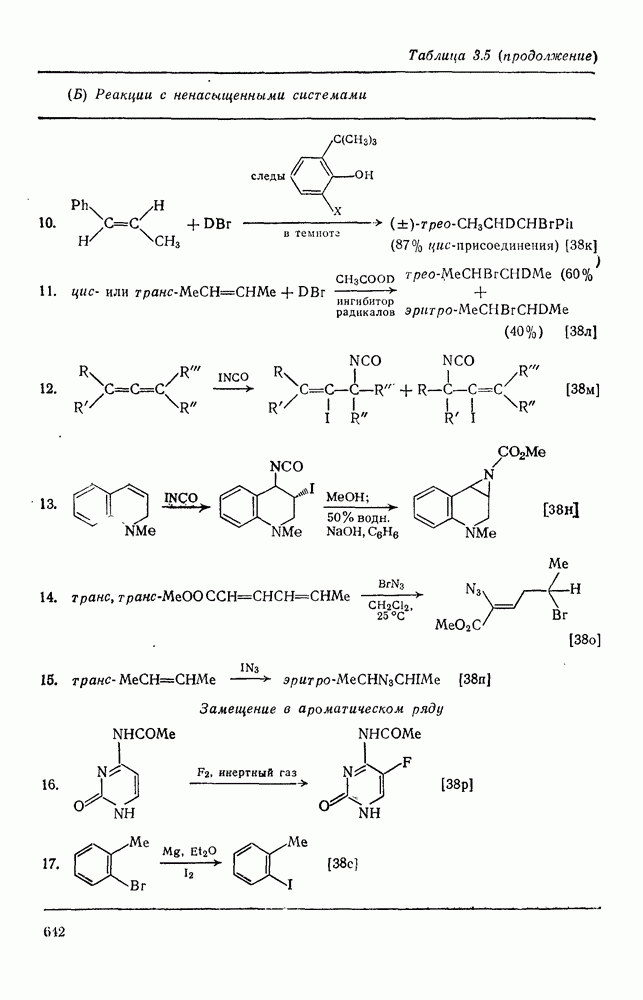
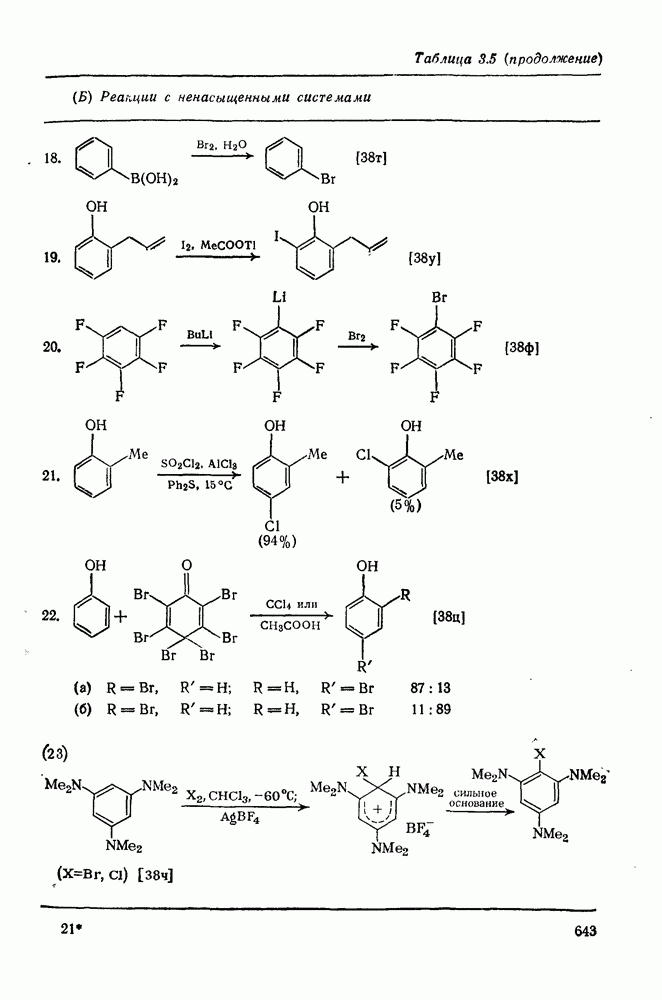
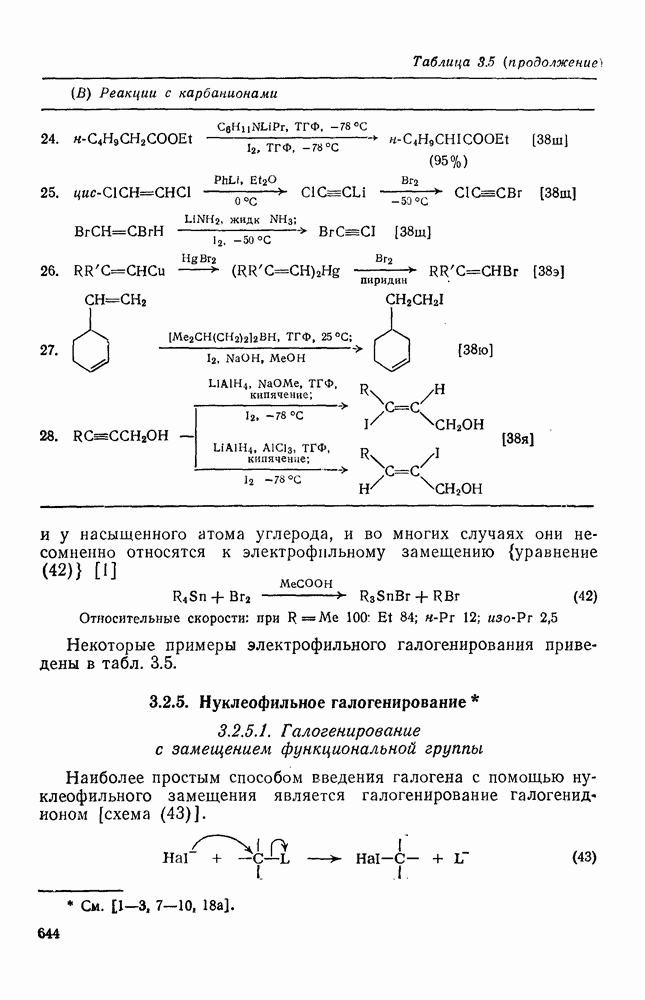
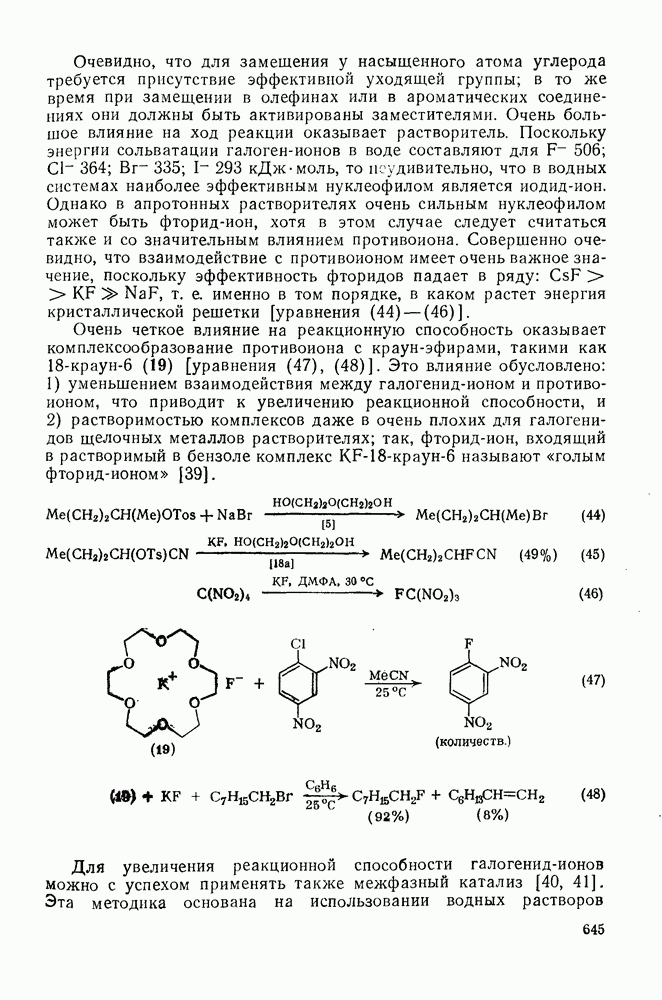
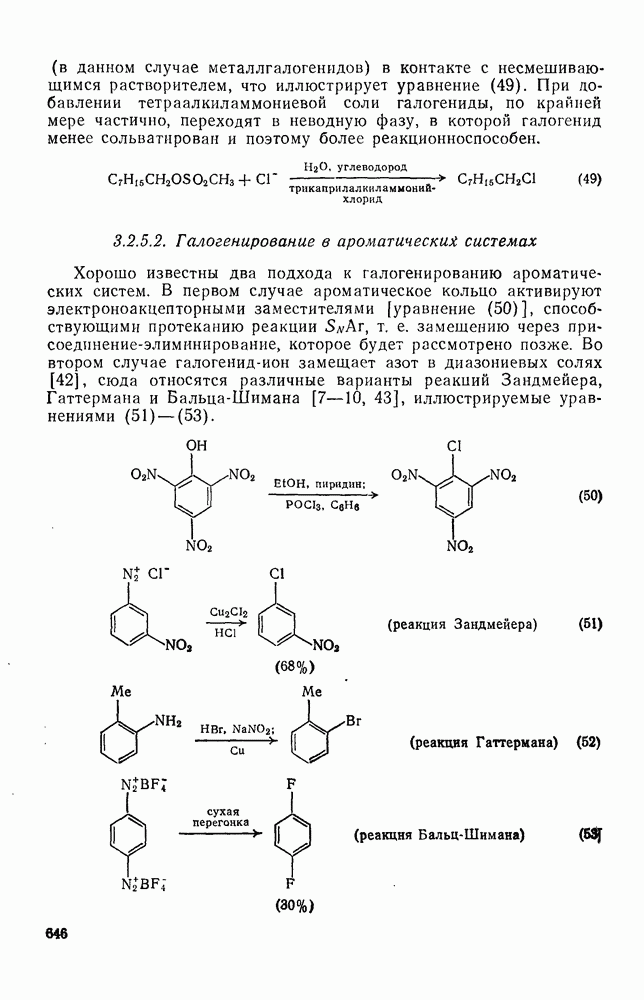
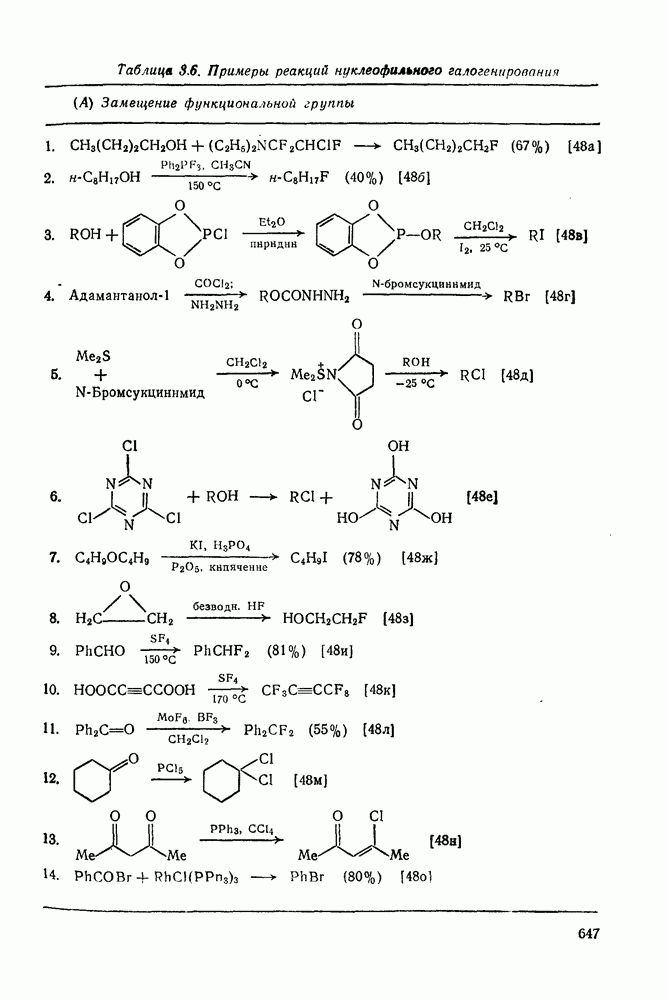
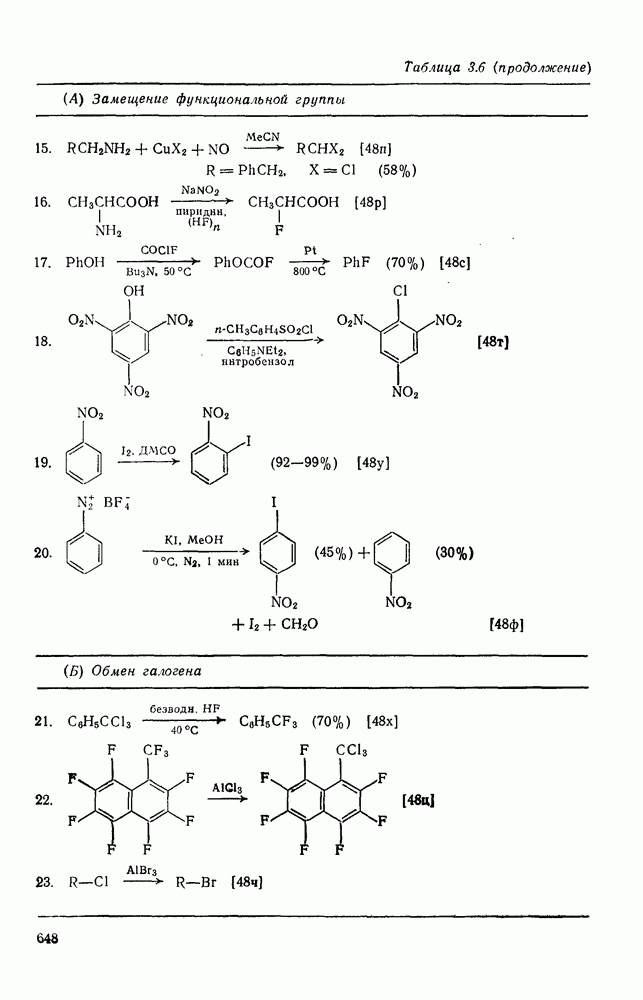
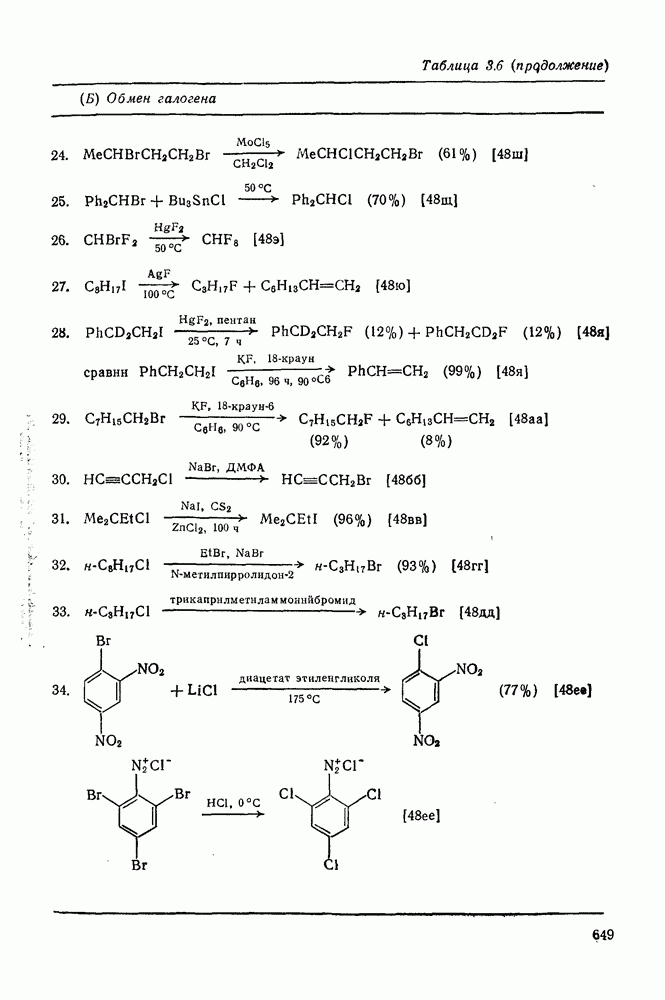
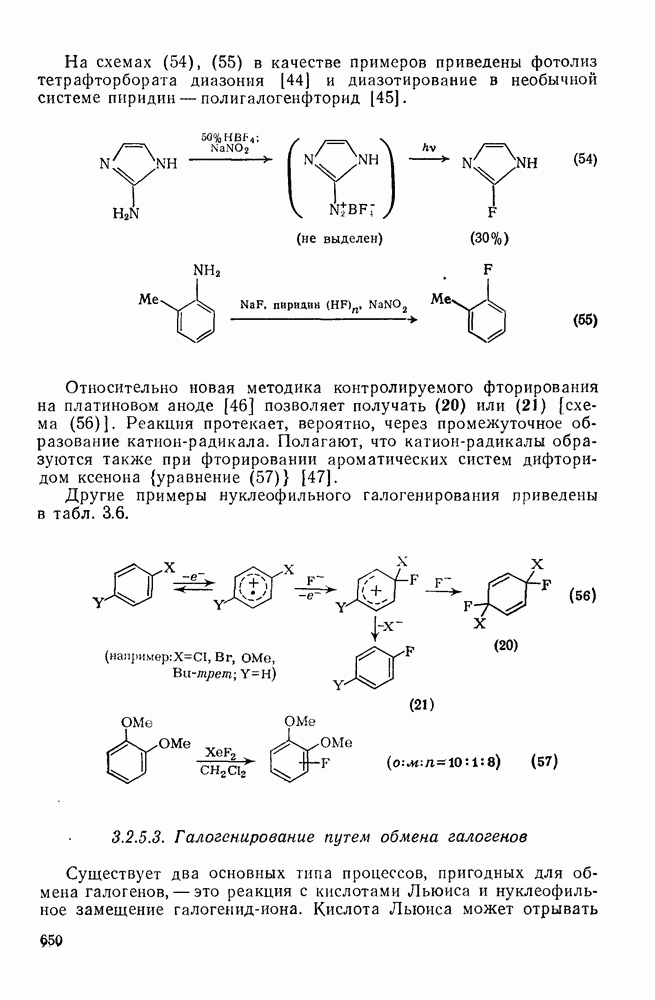

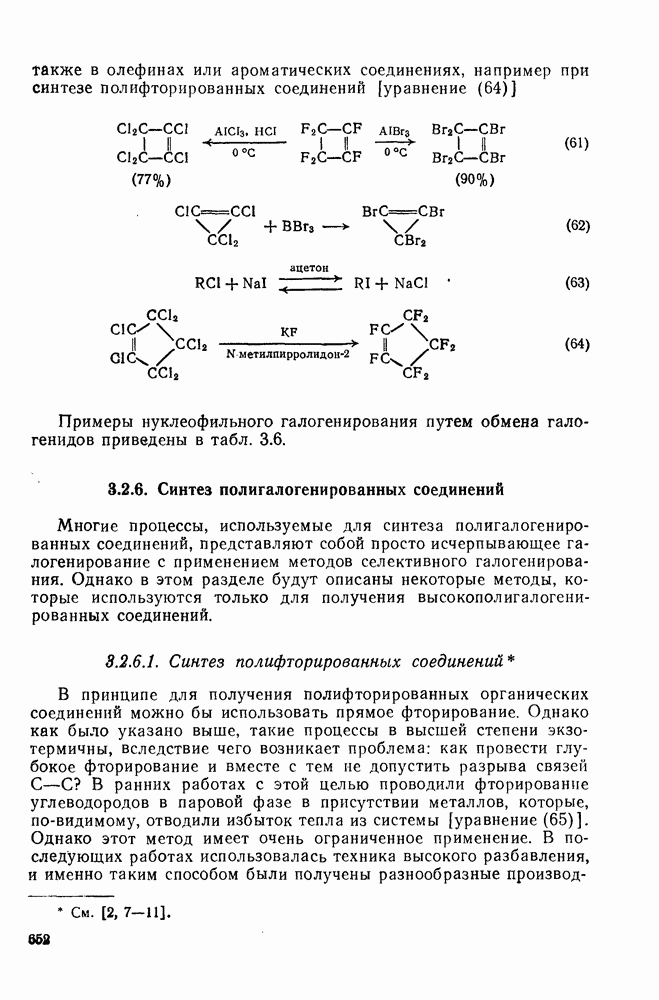
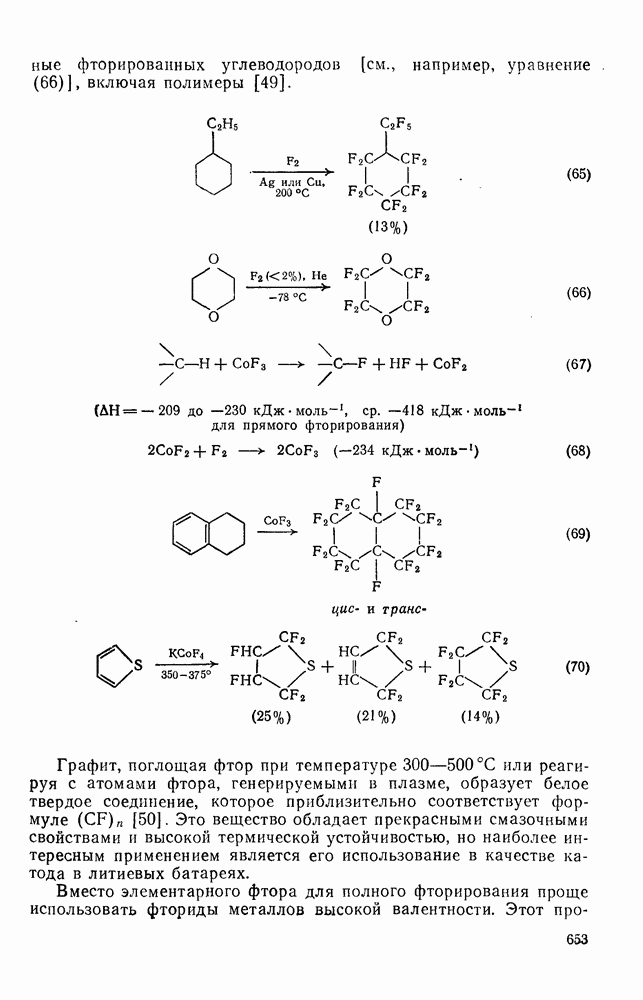
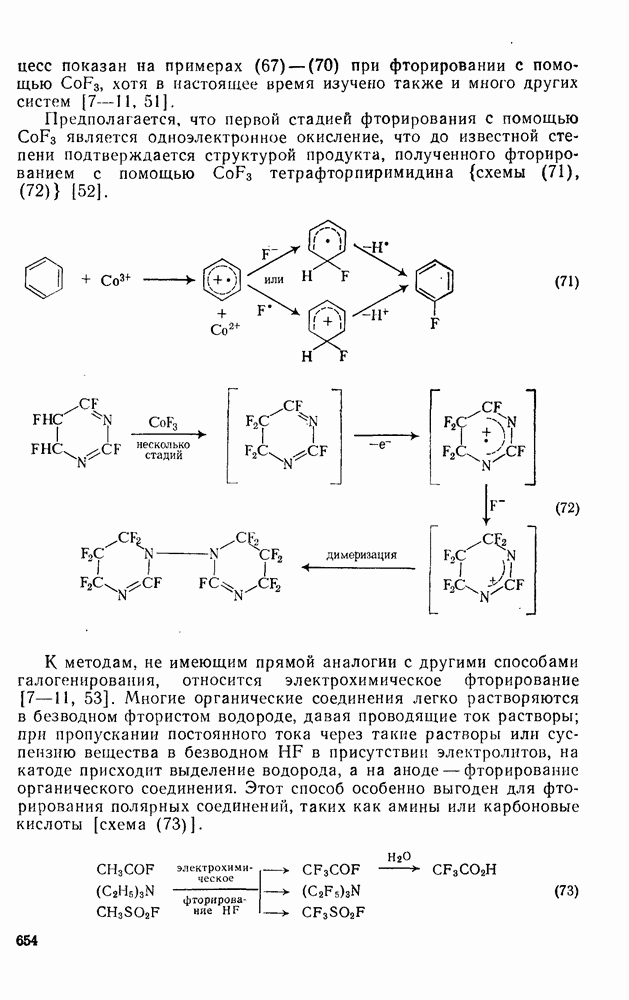









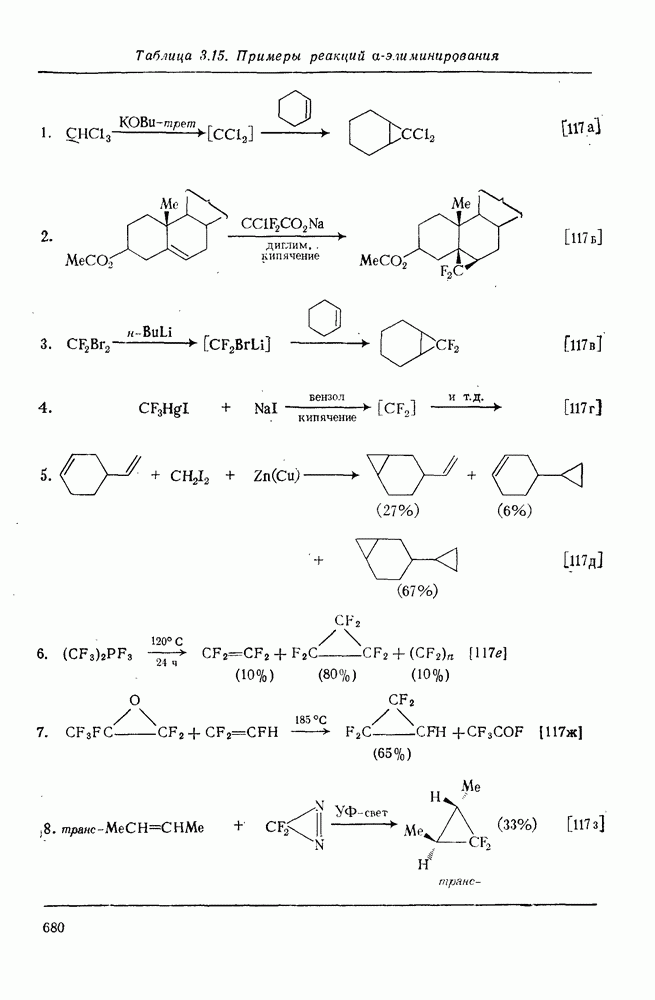



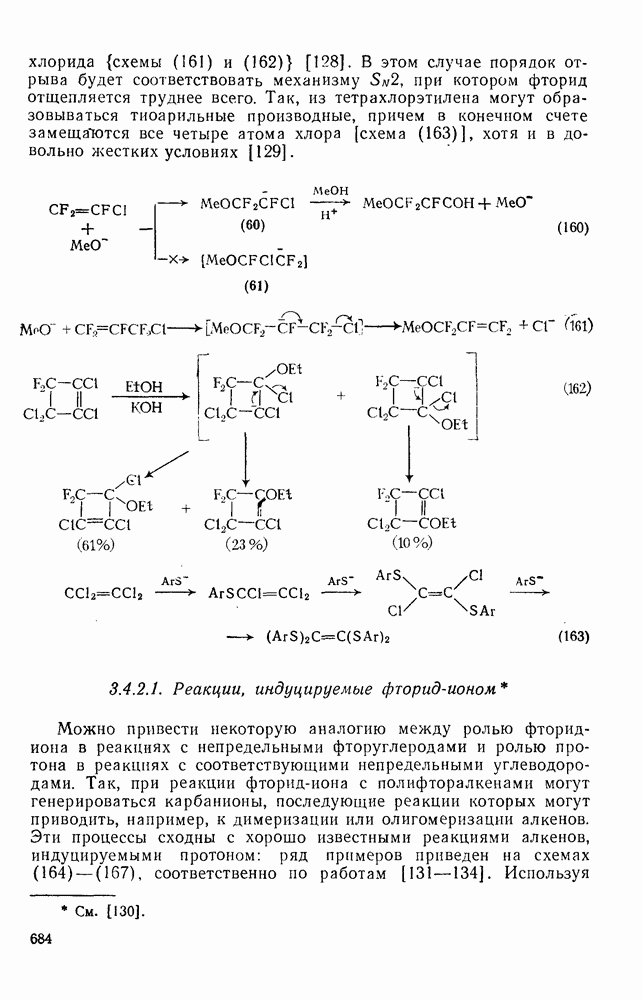
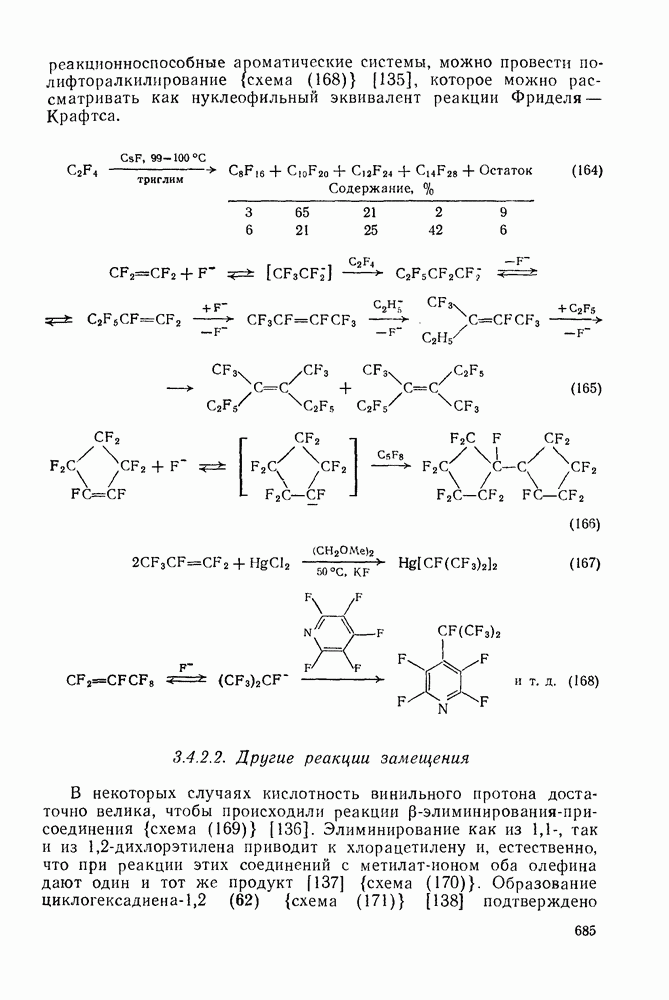
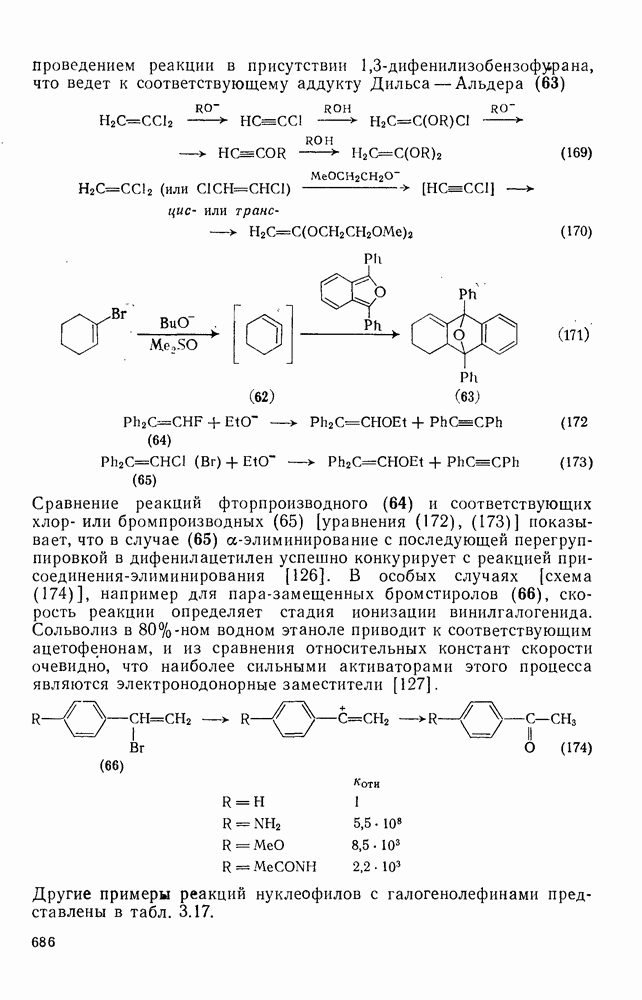






















 с. В случае достаточно короткоживущих карбанионов плоская конфигурация карбанионного центра не гарантирует образования рацемических продуктов реакции. Для некоторых карбанионов, например циклопропил- или винил-анионов, энергетический барьер инверсии у анионного центра настолько велик, что наблюдается сохранение конфигурации в широких пределах условий реакций.
с. В случае достаточно короткоживущих карбанионов плоская конфигурация карбанионного центра не гарантирует образования рацемических продуктов реакции. Для некоторых карбанионов, например циклопропил- или винил-анионов, энергетический барьер инверсии у анионного центра настолько велик, что наблюдается сохранение конфигурации в широких пределах условий реакций.
 -бензофлуореном в тетрагидрофуране не ускоряется при разбавлении, чего следовало ожидать, если бы свободные ионы были значительно реакционноспособнее ионных пар, как это наблюдалось при проведении этой реакции в диметоксиэтане. Протонирование бензил-анионов спиртами также протекает медленнее протонирования ионных пар, однако в данном случае это можно легко объяснить координацией спиртового кислородного атома с проти-воионом, что приводит к повышению кислотности молекул спирта возле карбаниона. Решающую роль, возможно, играет степень локализации отрицательного заряда, которая в случае ионной пары должна быть выше за счет поляризующего влияния ассоциированного катиона.
-бензофлуореном в тетрагидрофуране не ускоряется при разбавлении, чего следовало ожидать, если бы свободные ионы были значительно реакционноспособнее ионных пар, как это наблюдалось при проведении этой реакции в диметоксиэтане. Протонирование бензил-анионов спиртами также протекает медленнее протонирования ионных пар, однако в данном случае это можно легко объяснить координацией спиртового кислородного атома с проти-воионом, что приводит к повышению кислотности молекул спирта возле карбаниона. Решающую роль, возможно, играет степень локализации отрицательного заряда, которая в случае ионной пары должна быть выше за счет поляризующего влияния ассоциированного катиона.


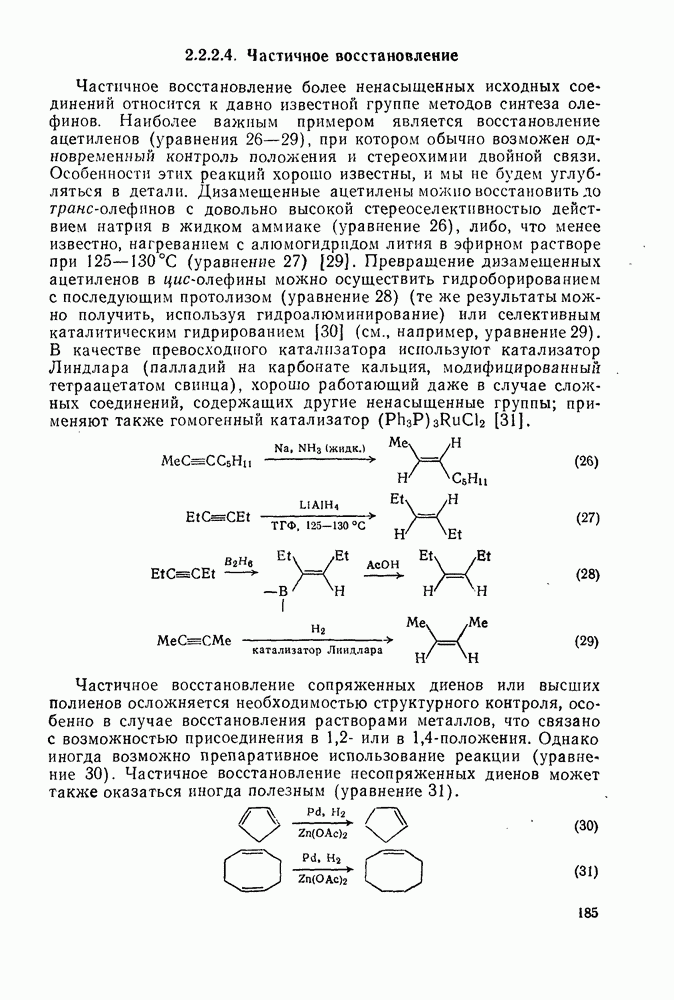
 ) со скоростью потери оптической активности
) со скоростью потери оптической активности  . В данном случае отношение
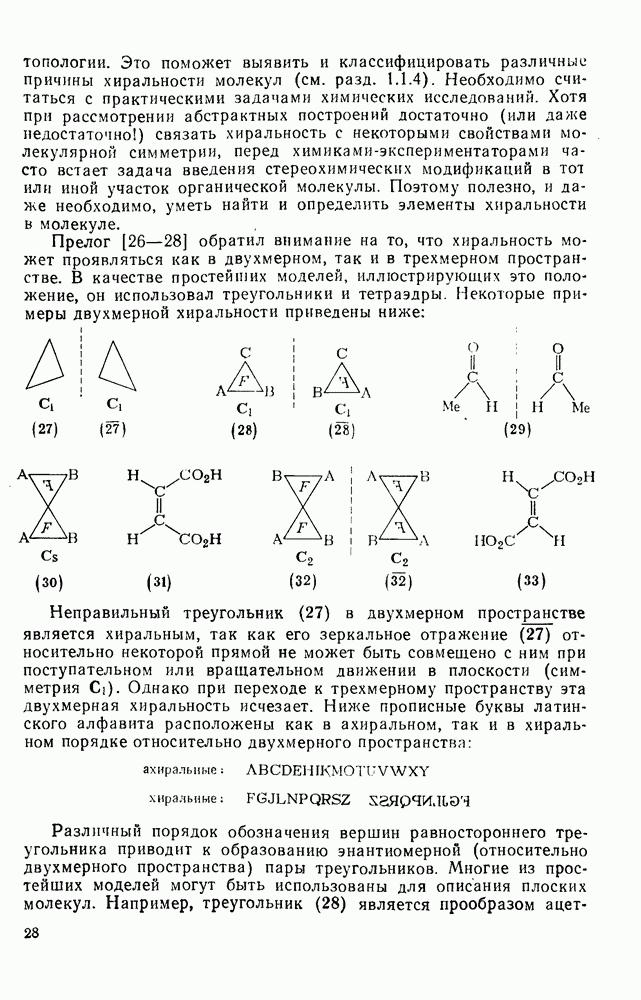
. В данном случае отношение  отражает стереохимию протонирования промежуточно образующихся карбанионов: большие величины
отражает стереохимию протонирования промежуточно образующихся карбанионов: большие величины  указывают на протонирование с сохранением конфигурации;
указывают на протонирование с сохранением конфигурации;  отвечает полной рацемизации;
отвечает полной рацемизации;  свидетельствует о полном обращении конфигурации;
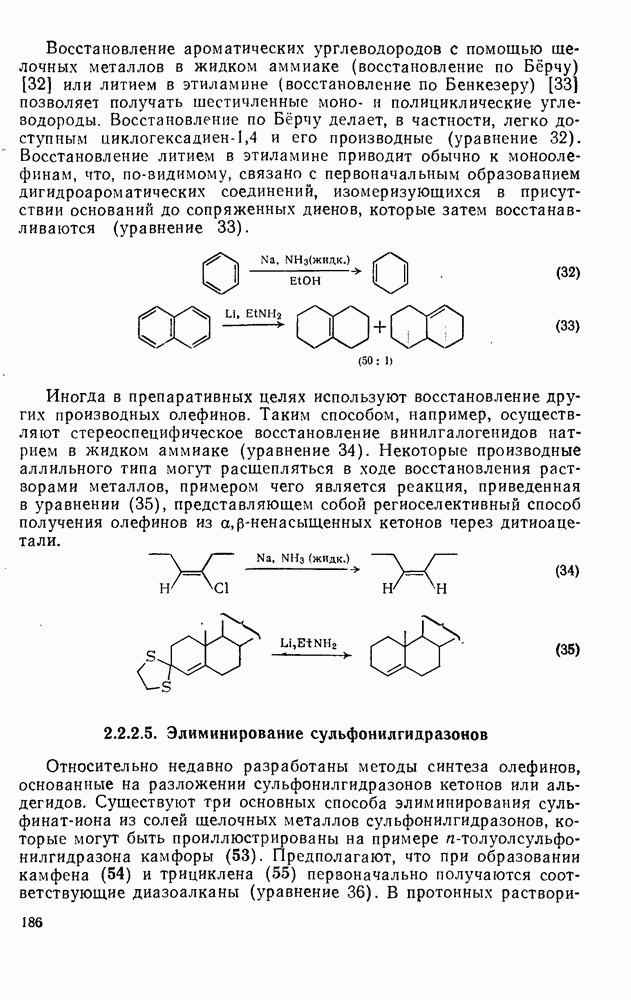
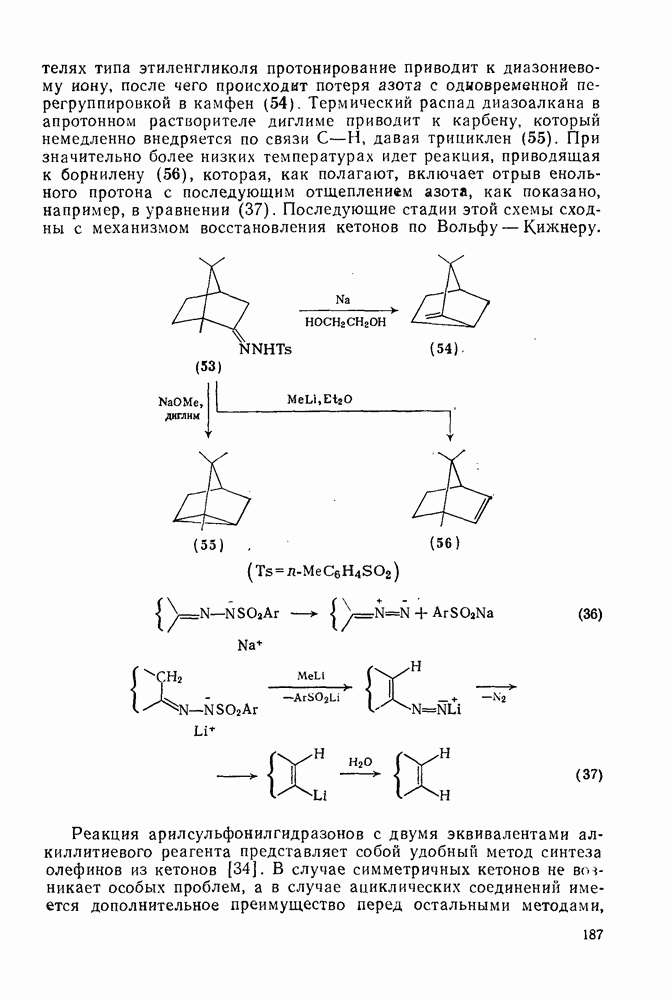
свидетельствует о полном обращении конфигурации;  характерно для процесса рацемизации без обмена (такой процесс называют изорацемизацией). В табл. 2.7.29 приведены некоторые данные для соединений (31). Сохранение конфигурации наблюдается, когда амин, использованный как основание, которое отщепляет дейтерий и дает катион, способен вновь протонировать карбанион еще до разделения ионной пары (эксп. 1) или когда протонирование осуществляется внутри ионной пары за счет молекул растворителя, координированных с противоионом (катионом) (ср. эксп. 3 и 4). Если протонирование по временной шкале сольватации протекает медленно, то промежуточно образующийся карбанион может оказаться в симметричном окружении, что приводит в результате к полной рацемизации (эксп. 2). Протонирование карбанионов в более полярной среде и с использованием более кислых спиртов, таких как метанол или гликоль, может протекать быстро и со стороны, противоположной той, с которой осуществляется атака основанием (эксп. 5 и 6). Изорацемизация (например, эксп. 7) может идти двумя различными путями. Первый путь включает вращение одного из компонентов ионной пары относительно другого с последующим репротонированием третичным аммониевым противоионом. Второй путь, так называемый механизм направленного переноса, может осуществляться в тех случаях, когда в молекуле карбаниона имеются другие основные центры, с которыми третичный аммониевый противоион может образовать водородную связь. В этом случае карбанионный центр может принять плоскую конфигурацию, не теряя связи с противоионом, который далее вновь протонирует анион. Как и следовало ожидать, добавление веществ, образующих комплексы с катионами, значительно ослабляет взаимодействие катиона с молекулами растворителя, что подавляет протонирование с сохранением конфигурации, и в таких случаях величина
характерно для процесса рацемизации без обмена (такой процесс называют изорацемизацией). В табл. 2.7.29 приведены некоторые данные для соединений (31). Сохранение конфигурации наблюдается, когда амин, использованный как основание, которое отщепляет дейтерий и дает катион, способен вновь протонировать карбанион еще до разделения ионной пары (эксп. 1) или когда протонирование осуществляется внутри ионной пары за счет молекул растворителя, координированных с противоионом (катионом) (ср. эксп. 3 и 4). Если протонирование по временной шкале сольватации протекает медленно, то промежуточно образующийся карбанион может оказаться в симметричном окружении, что приводит в результате к полной рацемизации (эксп. 2). Протонирование карбанионов в более полярной среде и с использованием более кислых спиртов, таких как метанол или гликоль, может протекать быстро и со стороны, противоположной той, с которой осуществляется атака основанием (эксп. 5 и 6). Изорацемизация (например, эксп. 7) может идти двумя различными путями. Первый путь включает вращение одного из компонентов ионной пары относительно другого с последующим репротонированием третичным аммониевым противоионом. Второй путь, так называемый механизм направленного переноса, может осуществляться в тех случаях, когда в молекуле карбаниона имеются другие основные центры, с которыми третичный аммониевый противоион может образовать водородную связь. В этом случае карбанионный центр может принять плоскую конфигурацию, не теряя связи с противоионом, который далее вновь протонирует анион. Как и следовало ожидать, добавление веществ, образующих комплексы с катионами, значительно ослабляет взаимодействие катиона с молекулами растворителя, что подавляет протонирование с сохранением конфигурации, и в таких случаях величина  обычно колеблется в пределах от 0,5 до 1.
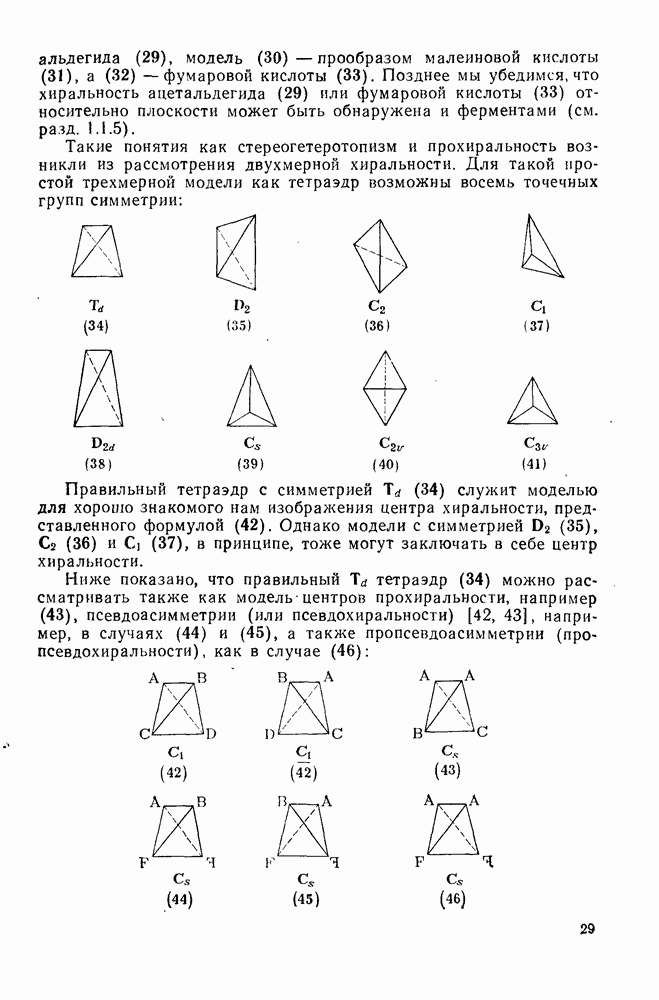
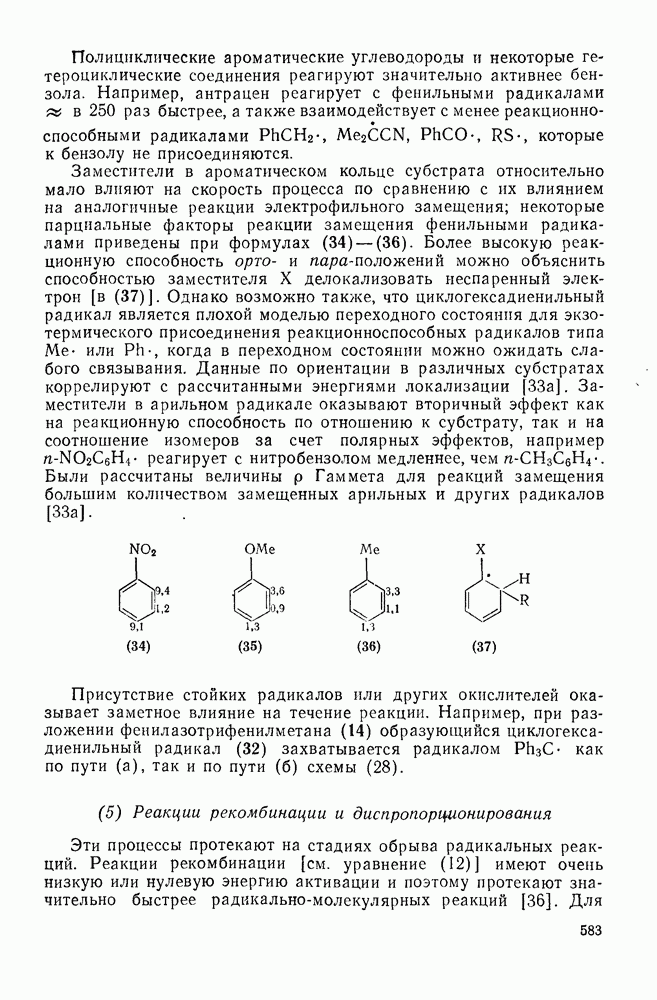
обычно колеблется в пределах от 0,5 до 1.
 [66а]
[66а]

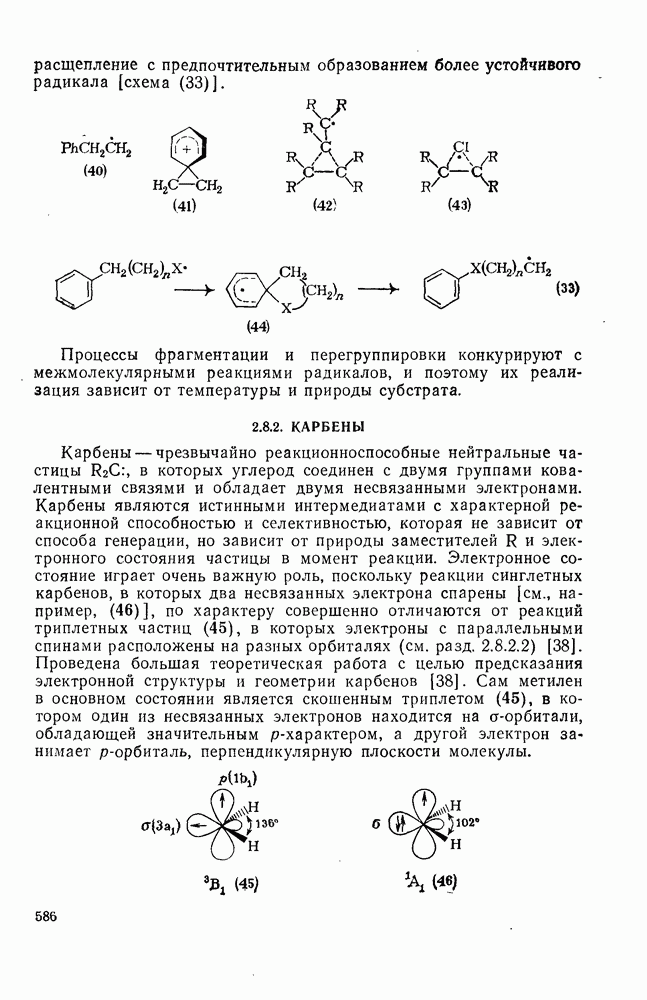
 и ионных пар
и ионных пар  «живых» анионов полистирола на стадии роста цепи при полимеризации стирола в различных растворителях. Разделенные ионные пары и свободные карбанионы реагируют приблизительно с одинаковыми скоростями, однако реакционная способность тесных ионных пар значительно ниже; одновременно наблюдается сложная зависимость от противо-иона и от растворителя. Стереохимия анионной полимеризации также изменчива. Так, метилметакрилат в присутствии литийорганиче-ских инициаторов в растворителях с низкой диэлектрической проницаемостью, например в толуоле, полимеризуется с образованием изотактического полимера. Однако добавление небольших количеств тетрагидрофурана или диметоксиэтана при низких температурах приводит к преимущественному образованию синдиотактического полимера.
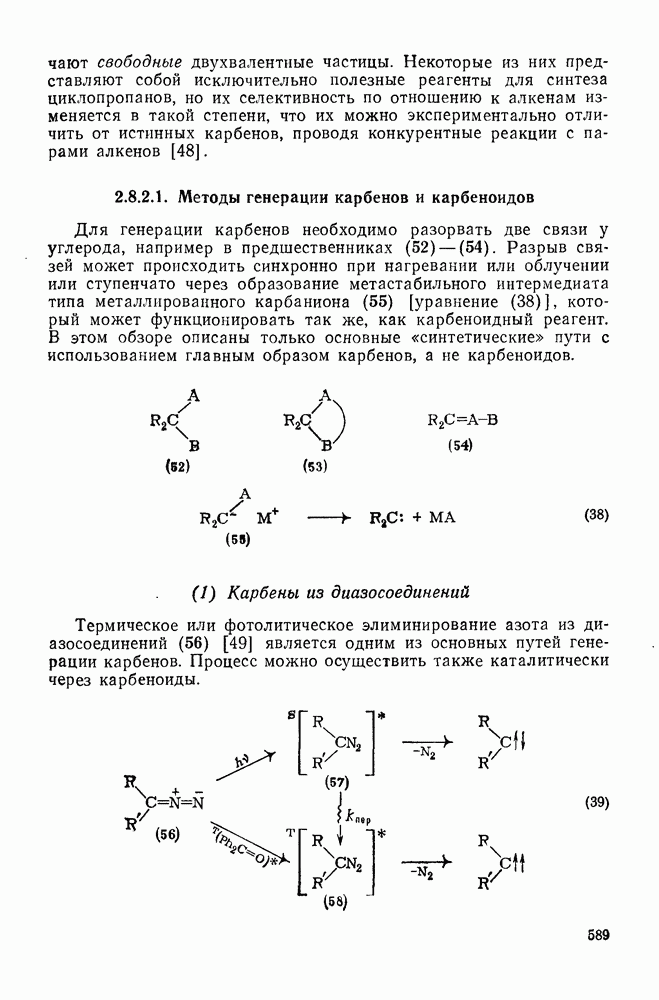
«живых» анионов полистирола на стадии роста цепи при полимеризации стирола в различных растворителях. Разделенные ионные пары и свободные карбанионы реагируют приблизительно с одинаковыми скоростями, однако реакционная способность тесных ионных пар значительно ниже; одновременно наблюдается сложная зависимость от противо-иона и от растворителя. Стереохимия анионной полимеризации также изменчива. Так, метилметакрилат в присутствии литийорганиче-ских инициаторов в растворителях с низкой диэлектрической проницаемостью, например в толуоле, полимеризуется с образованием изотактического полимера. Однако добавление небольших количеств тетрагидрофурана или диметоксиэтана при низких температурах приводит к преимущественному образованию синдиотактического полимера.
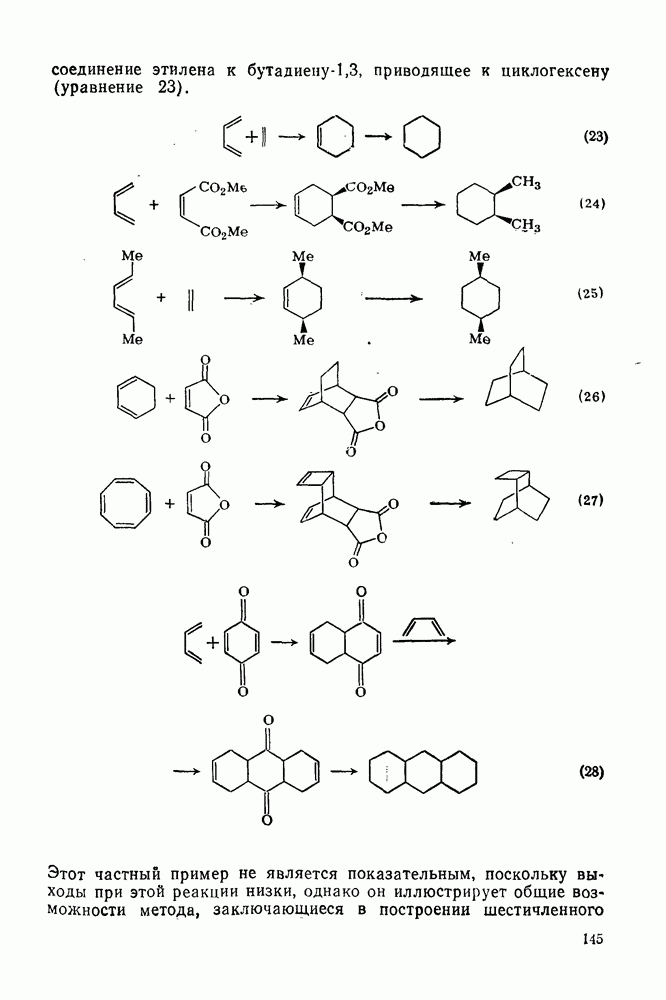
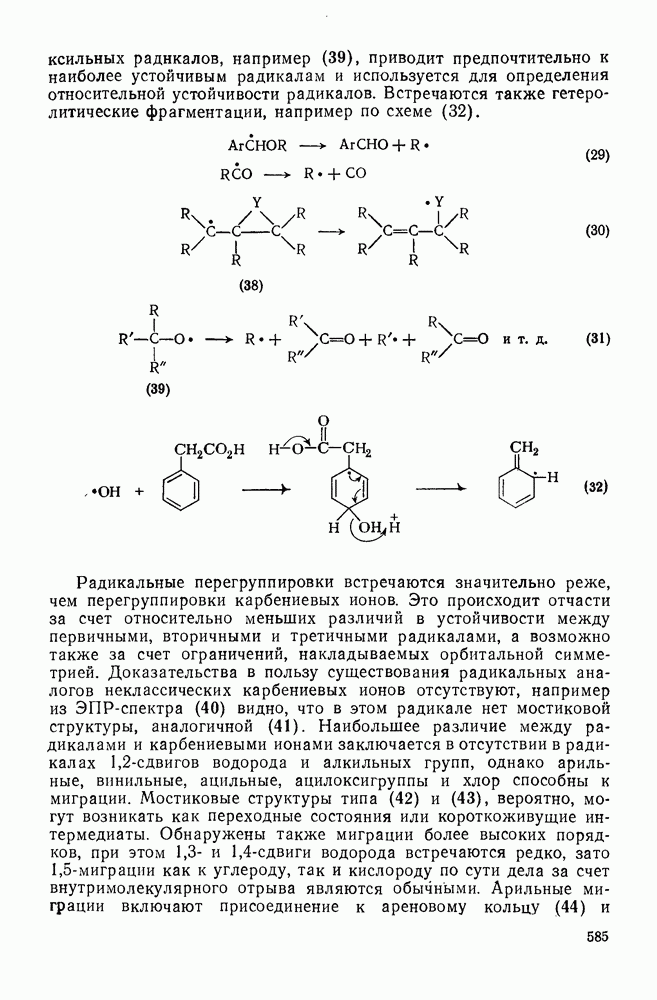
 [схема (28)] в спирте в условиях кинетически контролируемого протонирования происходит преимущественно как транс-присоединение с образованием продукта (32); в случае же термодинамического контроля образуется более устойчивый цис-изомер (33) [67].
[схема (28)] в спирте в условиях кинетически контролируемого протонирования происходит преимущественно как транс-присоединение с образованием продукта (32); в случае же термодинамического контроля образуется более устойчивый цис-изомер (33) [67].

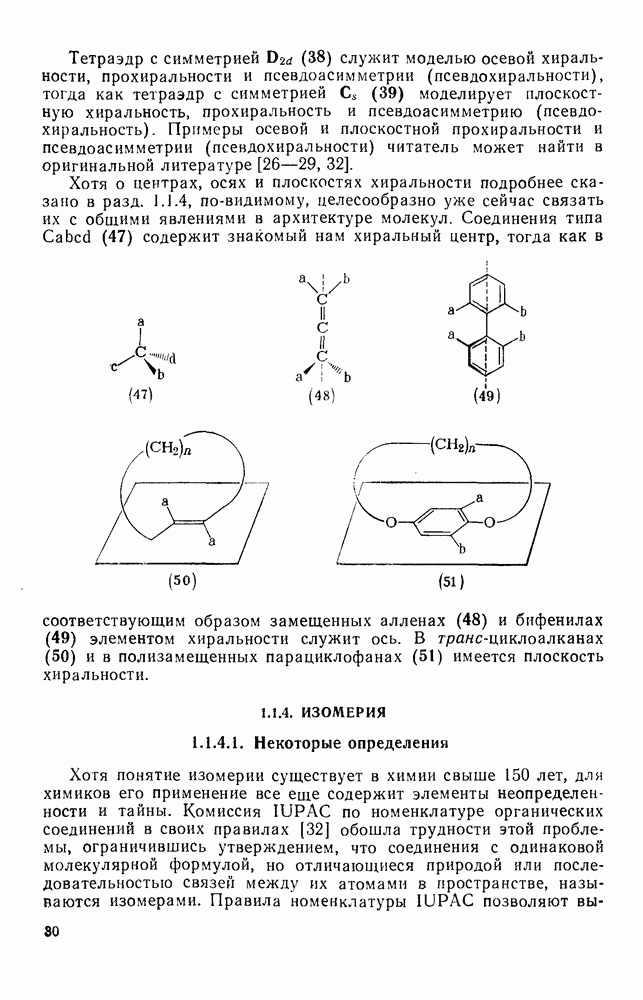
 или
или  -углеродных атомов карбаниона находятся подходящие уходящие группы, обычно галоген, то может протекать расщепление с образованием галогенид-ионов и нейтральных продуктов.
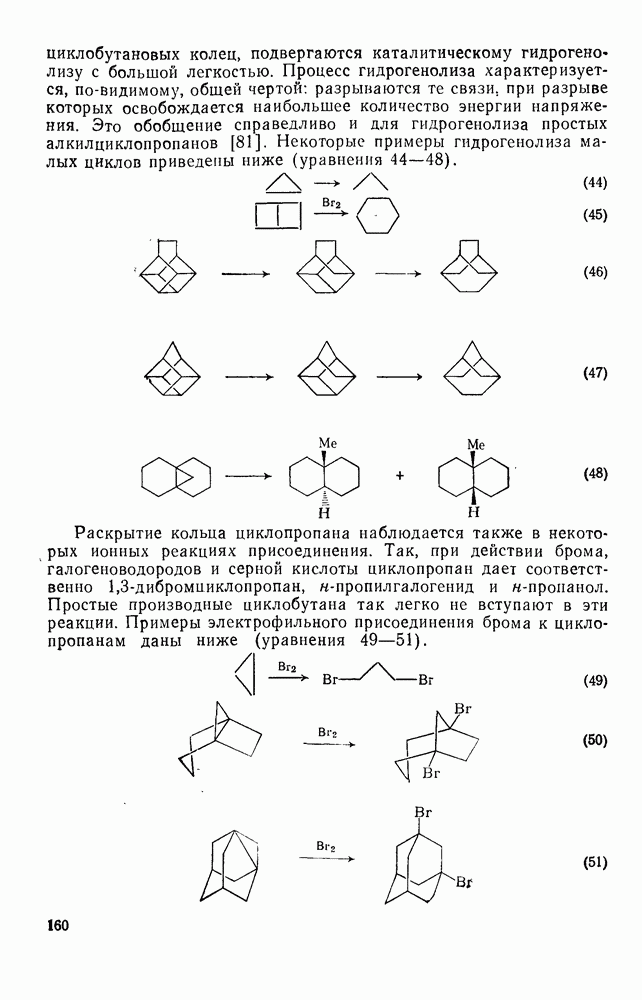
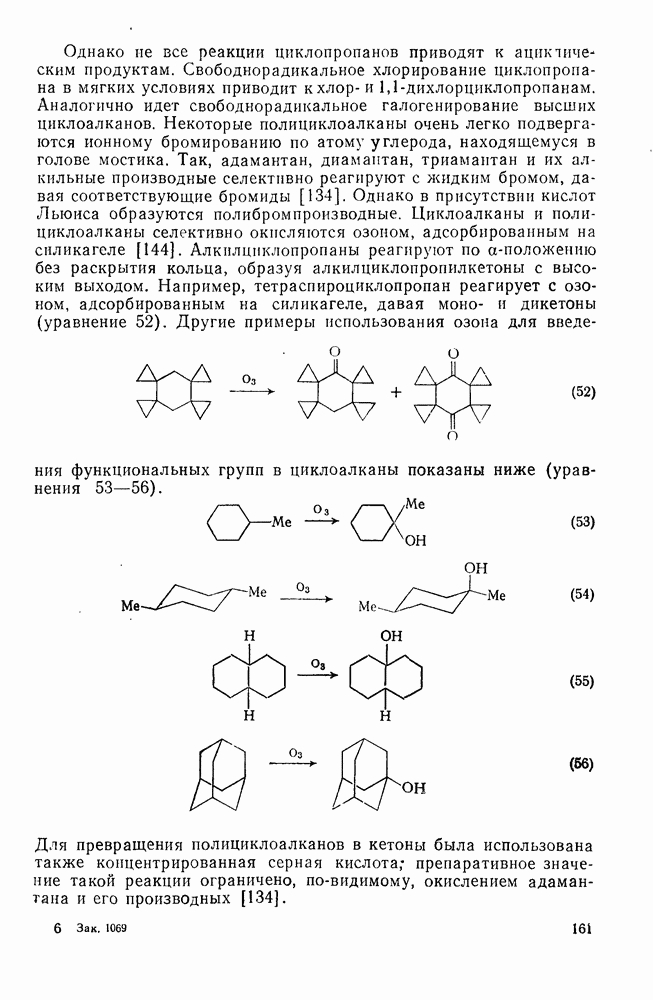
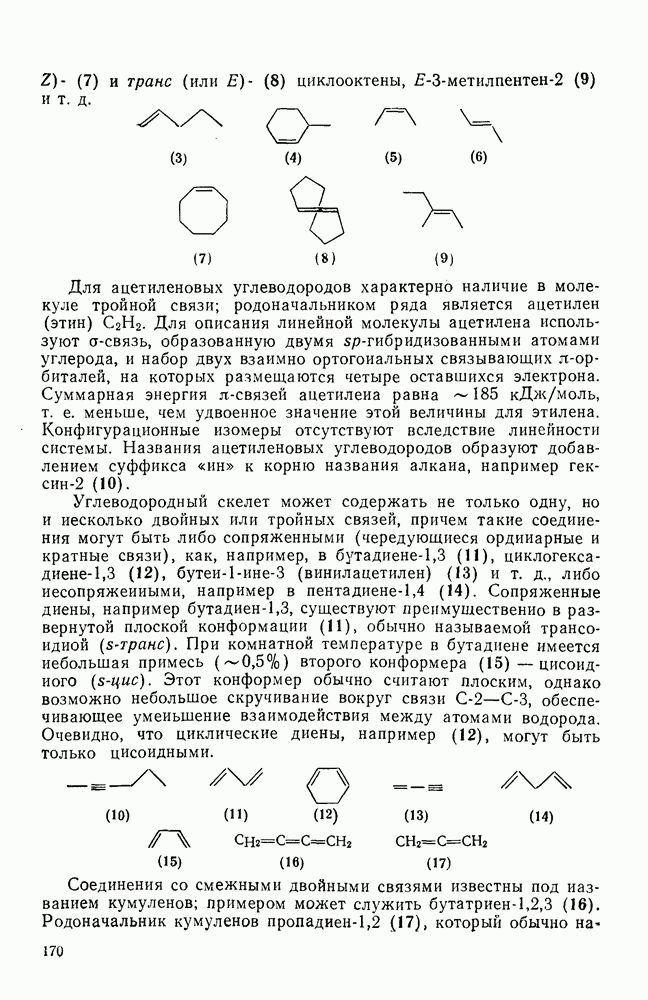
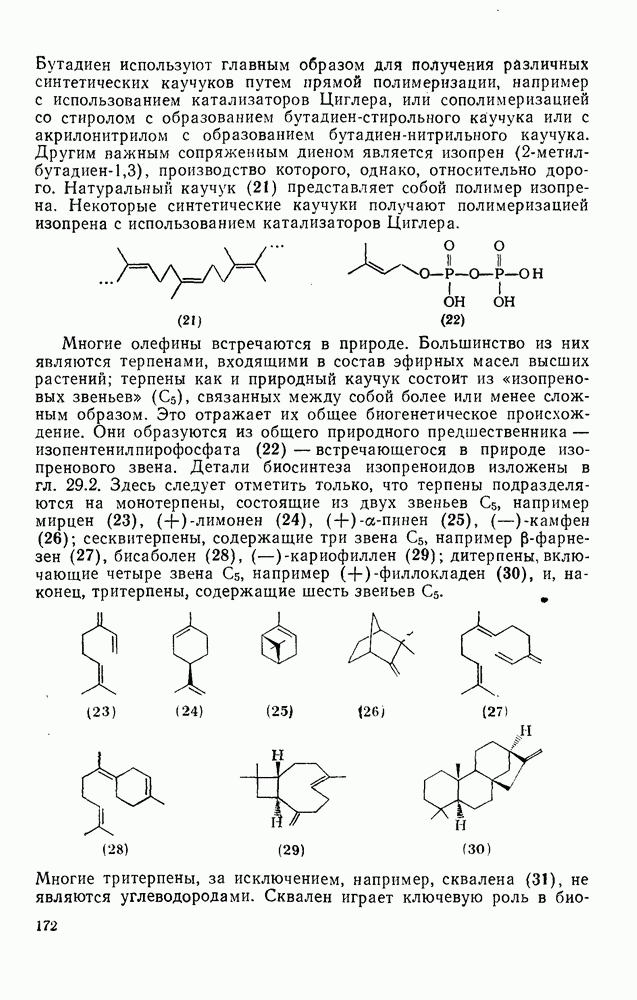
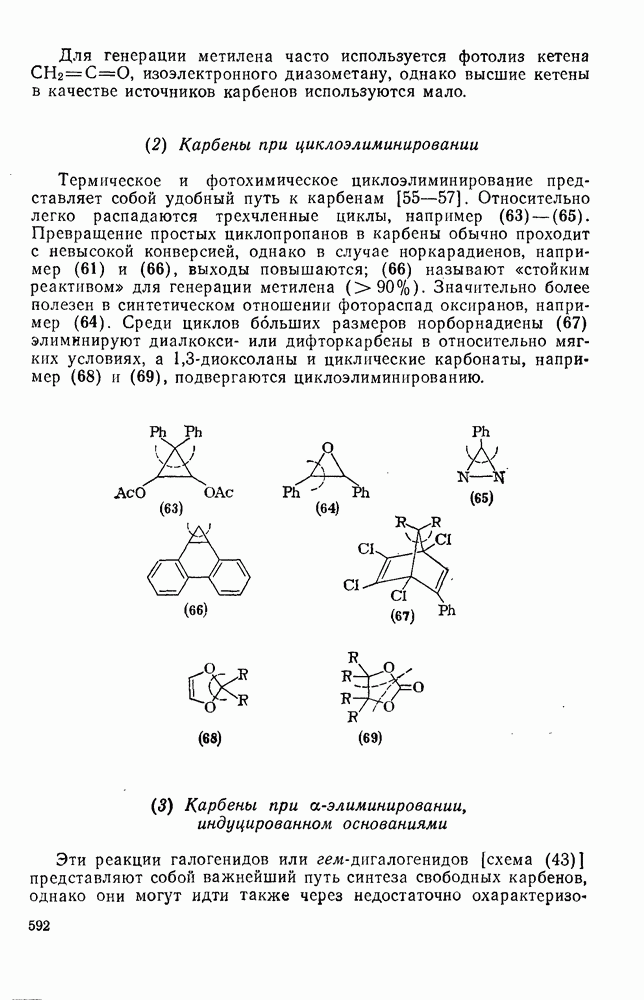
-углеродных атомов карбаниона находятся подходящие уходящие группы, обычно галоген, то может протекать расщепление с образованием галогенид-ионов и нейтральных продуктов.  -Расщепление дает карбены или карбеиоидные частицы; простейшим примером является образование дигалогенметиленов из тригалогенметил-анионов. р-Расщепление приводит к олефинам и является одной из стадий элиминирования по механизму
-Расщепление дает карбены или карбеиоидные частицы; простейшим примером является образование дигалогенметиленов из тригалогенметил-анионов. р-Расщепление приводит к олефинам и является одной из стадий элиминирования по механизму  . Этот путь образования олефинов может конкурировать со значительно более общим согласованным
. Этот путь образования олефинов может конкурировать со значительно более общим согласованным  -механизмом только в тех случаях, когда у
-механизмом только в тех случаях, когда у  -углеродного атома в карбанионе имеются стабилизующие заместители, а у
-углеродного атома в карбанионе имеются стабилизующие заместители, а у  -углеродного атома — относительно трудно уходящие группы (например, ОН,
-углеродного атома — относительно трудно уходящие группы (например, ОН,  [68]). Примерами соединений, дающих олефины через образование карбанионов, являются соединения (34) и (35).
[68]). Примерами соединений, дающих олефины через образование карбанионов, являются соединения (34) и (35).

 -элиминирования и переноса протона. Ассоциация ионов может влиять на реакционную способность карбанионов, причем во всех изученных случаях реакционная способность свободных ионов была выше, чем для ионных пар. Кроме того, природа противоиона может влиять на региоселективность алкилирования, в особенности заслуживает внимания влияние иона таллия.
-элиминирования и переноса протона. Ассоциация ионов может влиять на реакционную способность карбанионов, причем во всех изученных случаях реакционная способность свободных ионов была выше, чем для ионных пар. Кроме того, природа противоиона может влиять на региоселективность алкилирования, в особенности заслуживает внимания влияние иона таллия.


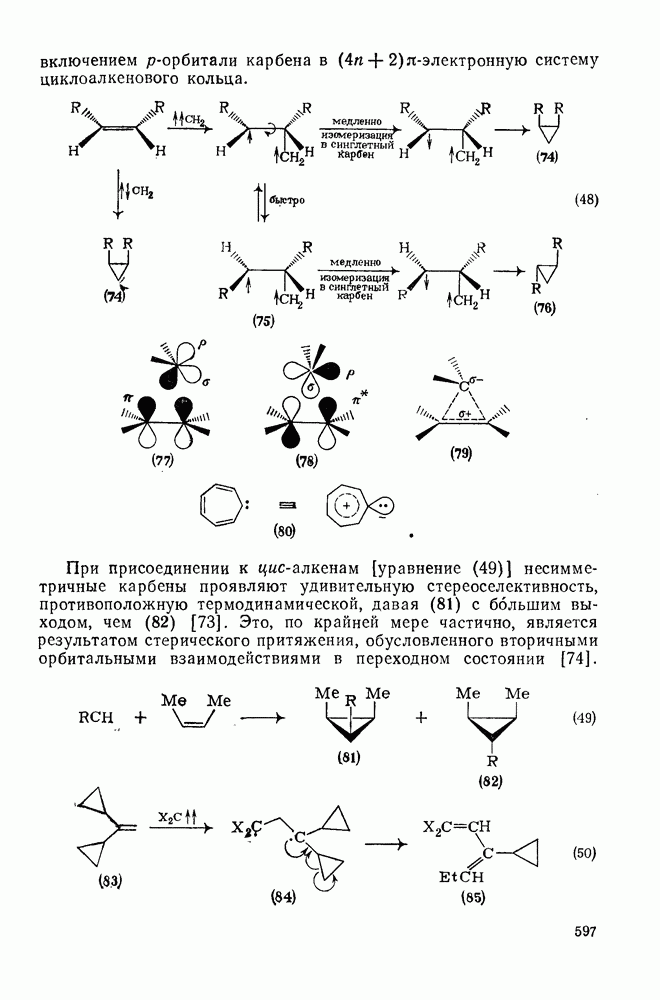
 молекулярный кислород, с которым они реагируют очень быстро [70]. Ароматические нитросоединения могут конкурировать с кислородом в качестве акцепторов электронов; образующиеся радикалы далее димеризуются [например, уравнение (31)] [71]. Димерные радикальные продукты образуются также при фотохимическом возбуждении карбанионов; так, циклопентадиенил-анион в трет-бутиловом спирте дает мезо- и
молекулярный кислород, с которым они реагируют очень быстро [70]. Ароматические нитросоединения могут конкурировать с кислородом в качестве акцепторов электронов; образующиеся радикалы далее димеризуются [например, уравнение (31)] [71]. Димерные радикальные продукты образуются также при фотохимическом возбуждении карбанионов; так, циклопентадиенил-анион в трет-бутиловом спирте дает мезо- и  [72].
[72].


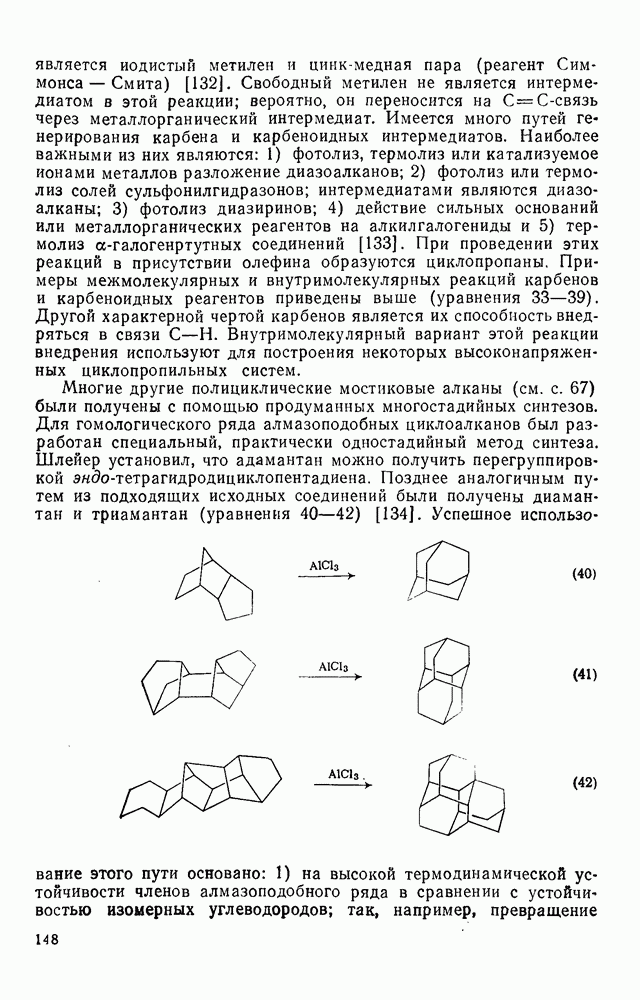
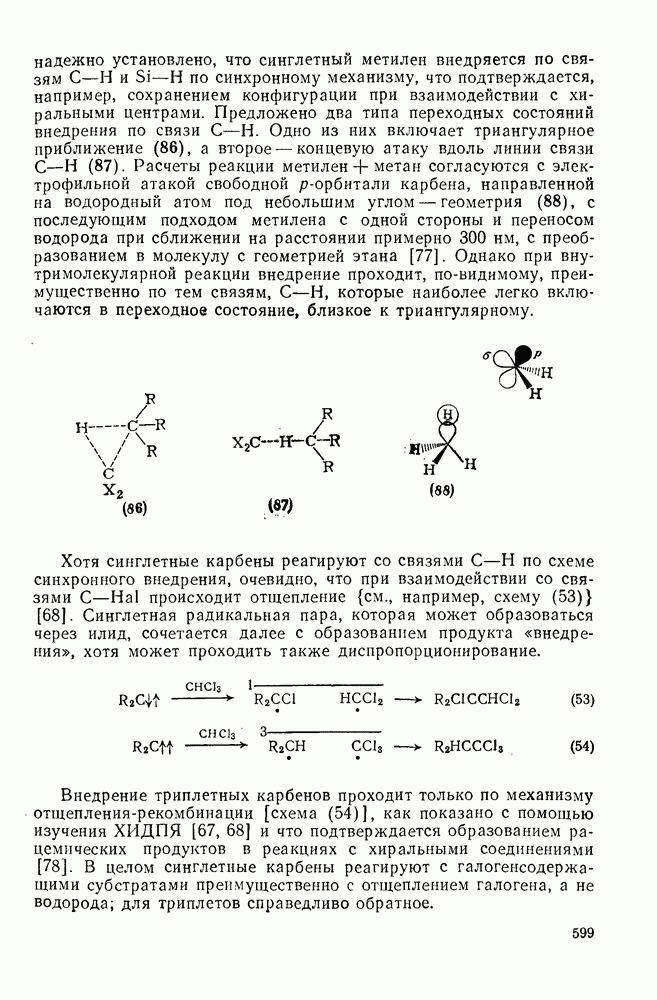
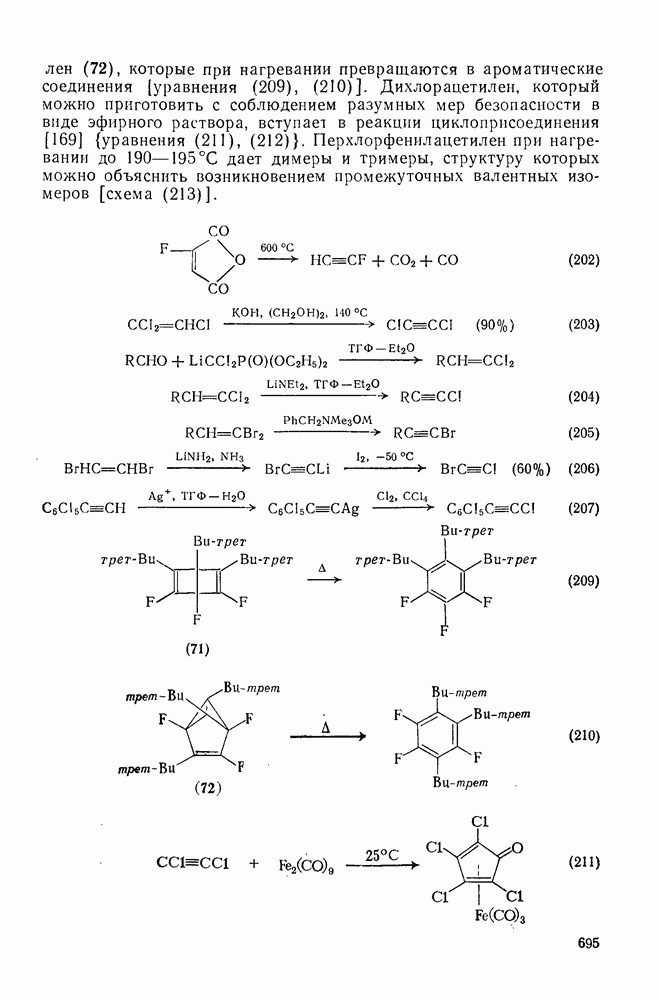
 -сдвига алкильной группы с сохранением конфигурации аналогично циклопропенил-аниону и представляет собой антиароматическую систему Хюккеля с
-сдвига алкильной группы с сохранением конфигурации аналогично циклопропенил-аниону и представляет собой антиароматическую систему Хюккеля с  -электронами, и следовательно, такая перегруппировка термически запрещена. Антраповерхностные
-электронами, и следовательно, такая перегруппировка термически запрещена. Антраповерхностные  -сдвиги с инверсией мигрирующей группы можно исключить по пространственным причинам. Для илидов обнаружены
-сдвиги с инверсией мигрирующей группы можно исключить по пространственным причинам. Для илидов обнаружены  -сдвиги алкильных групп с сохранением конфигурации, например в случае перегруппировки Стивенса. Однако образование в этой реакции димеров радикалов и обнаружение химически индуцируемой динамической поляризации ядер в перегруппированном продукте свидетельствует в пользу радикального механизма диссоциации-рекомбинации [58]. По аналогичному механизму протекает, вероятно, и перегруппировка Виттига
-сдвиги алкильных групп с сохранением конфигурации, например в случае перегруппировки Стивенса. Однако образование в этой реакции димеров радикалов и обнаружение химически индуцируемой динамической поляризации ядер в перегруппированном продукте свидетельствует в пользу радикального механизма диссоциации-рекомбинации [58]. По аналогичному механизму протекает, вероятно, и перегруппировка Виттига  -алкоксикарбанионов:
-алкоксикарбанионов:  . Миграция аллильных групп может осуществляться путем разрешенного согласованного [2,3] -сигматропного сдвига, включающего инверсию аллильной группы.
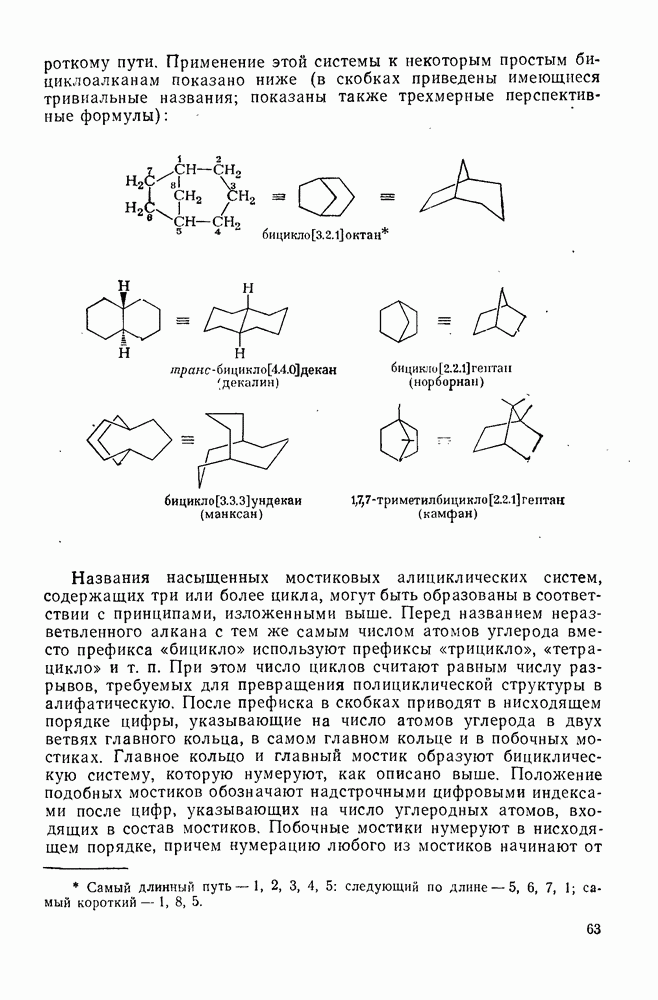
. Миграция аллильных групп может осуществляться путем разрешенного согласованного [2,3] -сигматропного сдвига, включающего инверсию аллильной группы.


