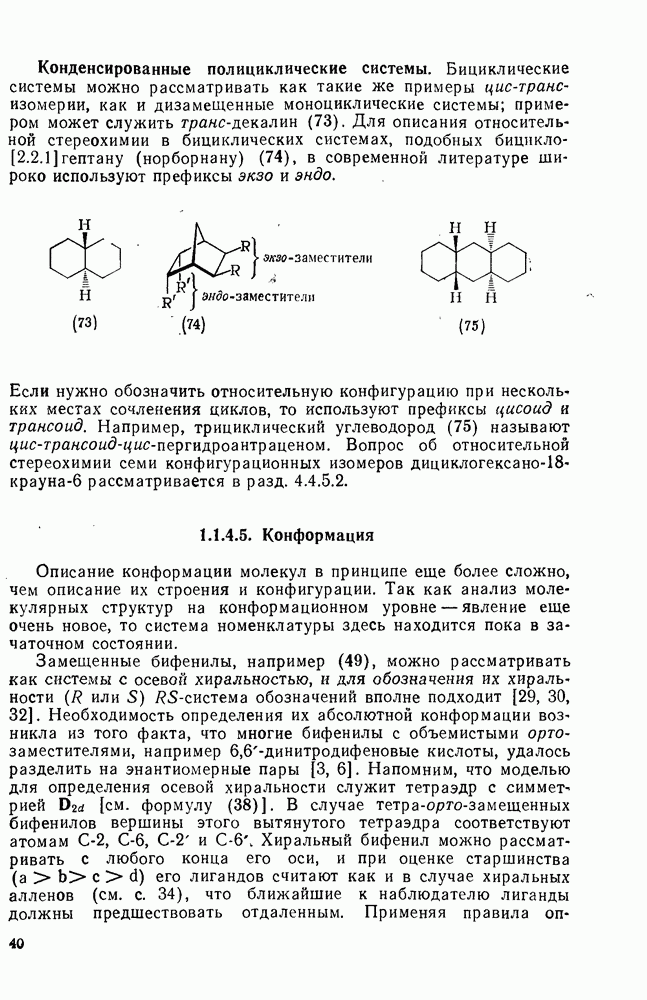
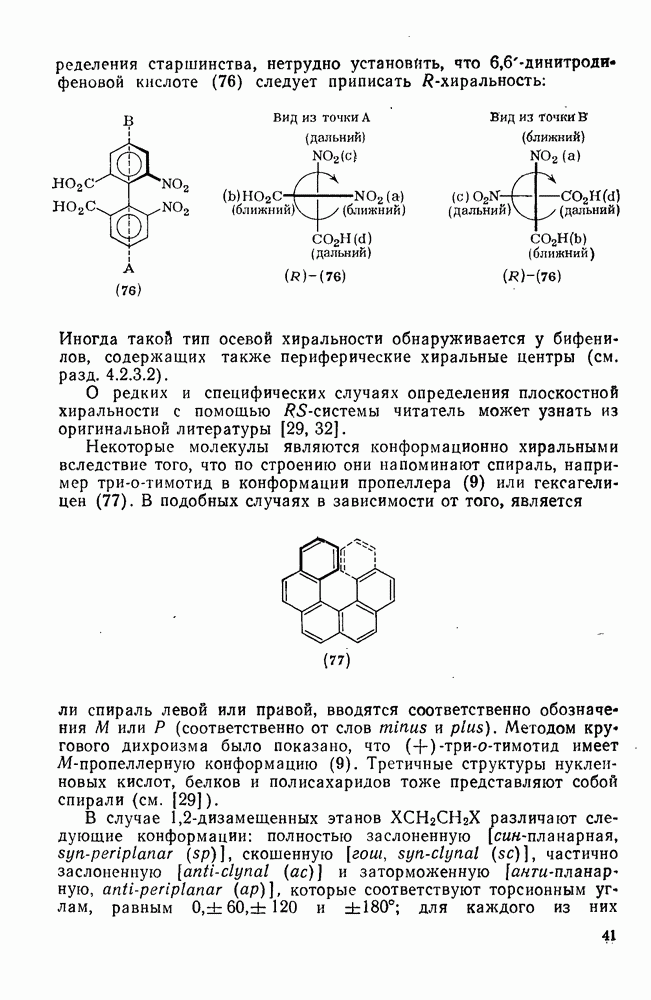
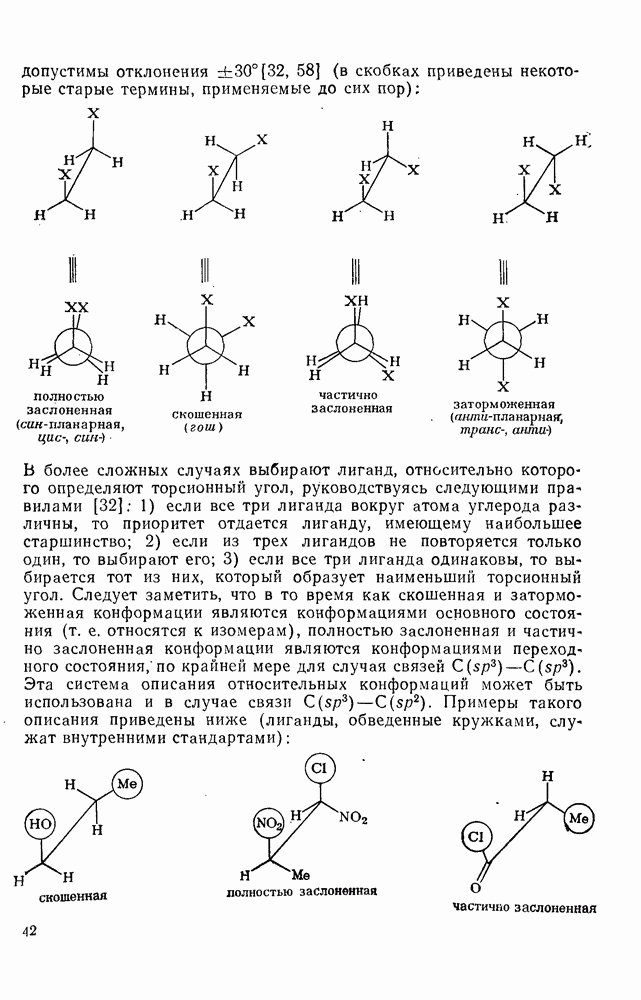
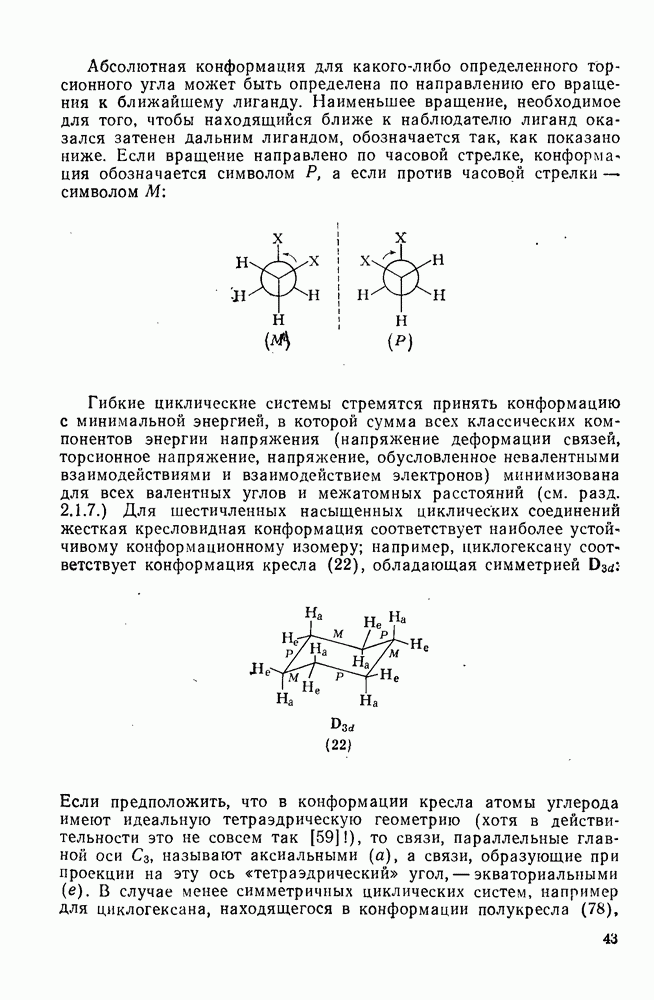
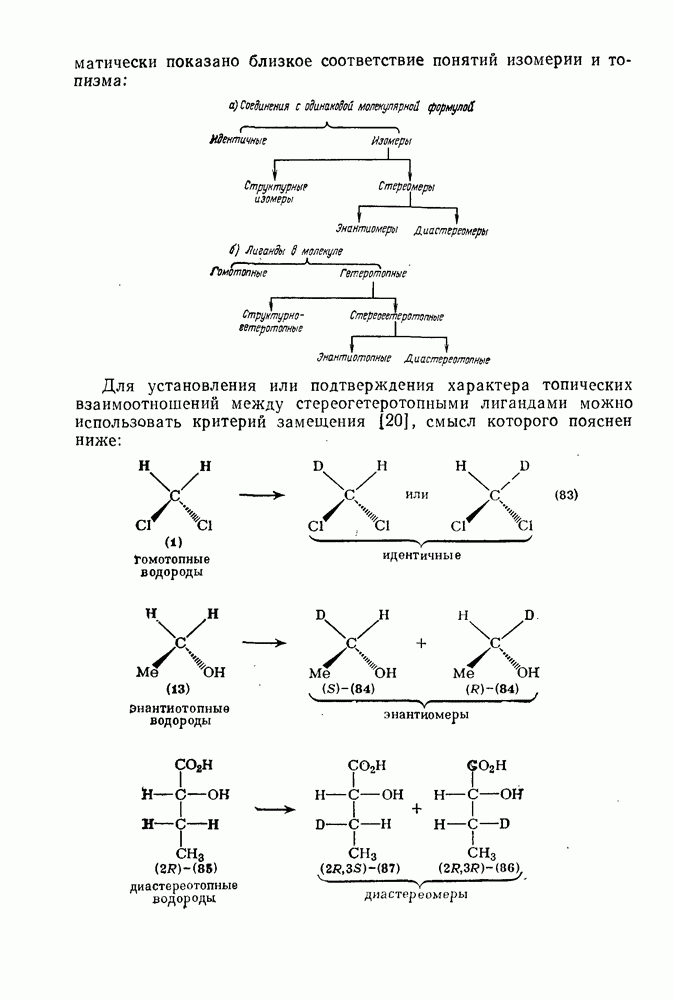
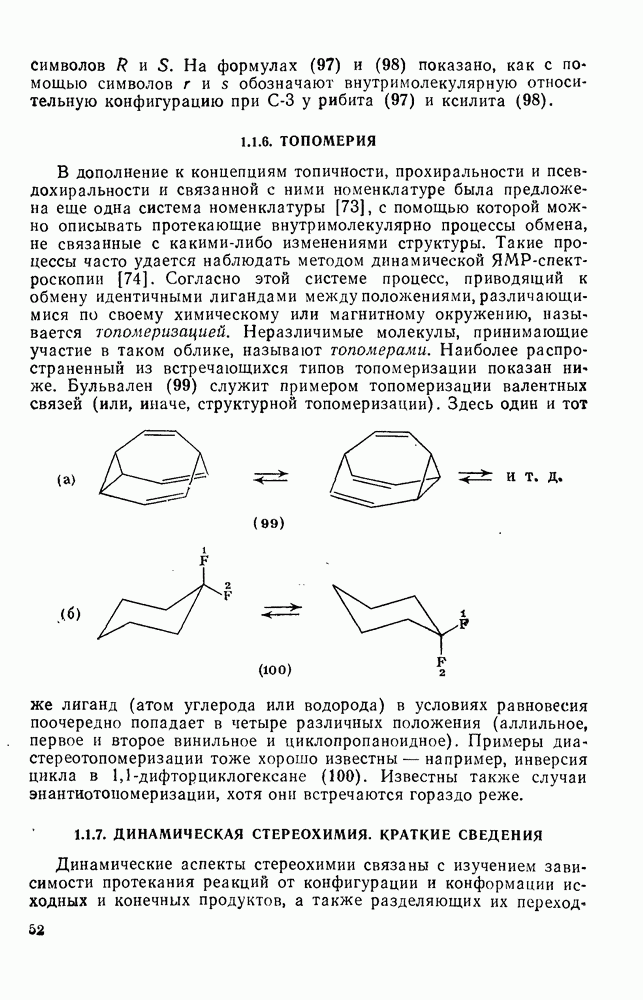
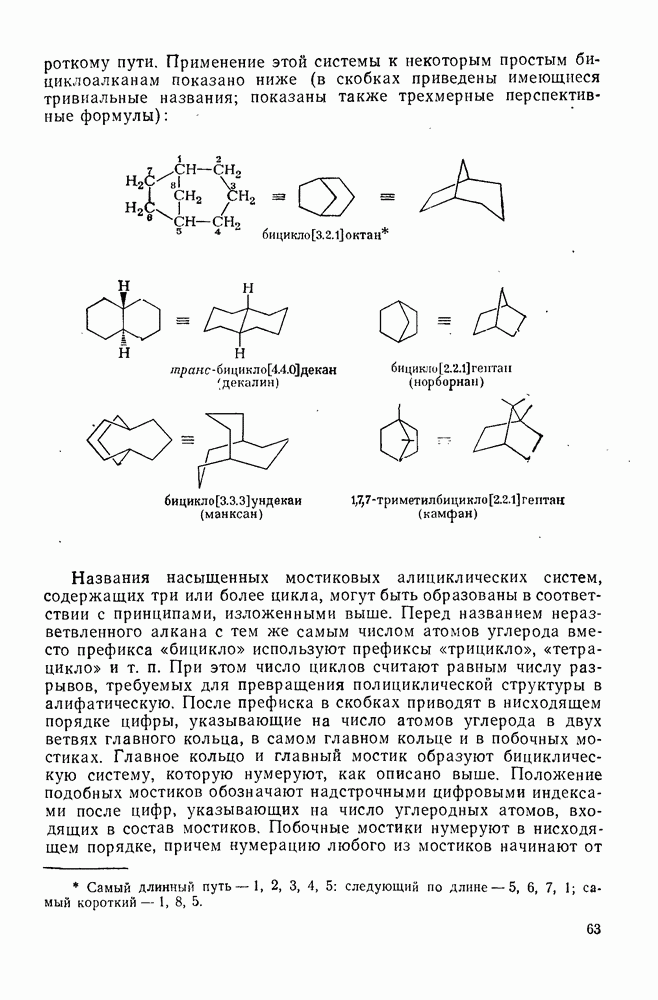
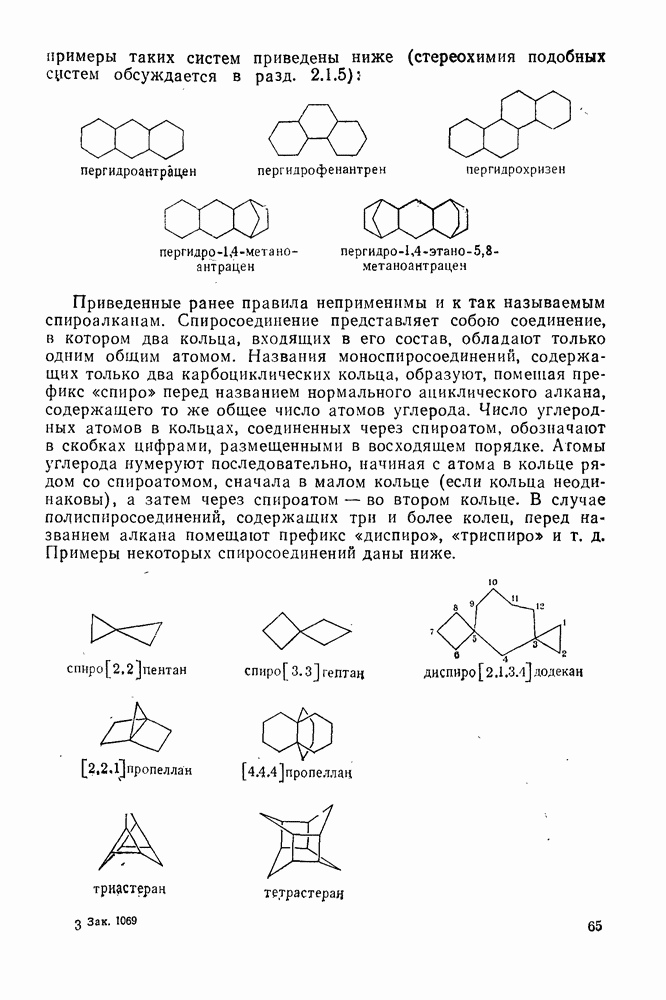
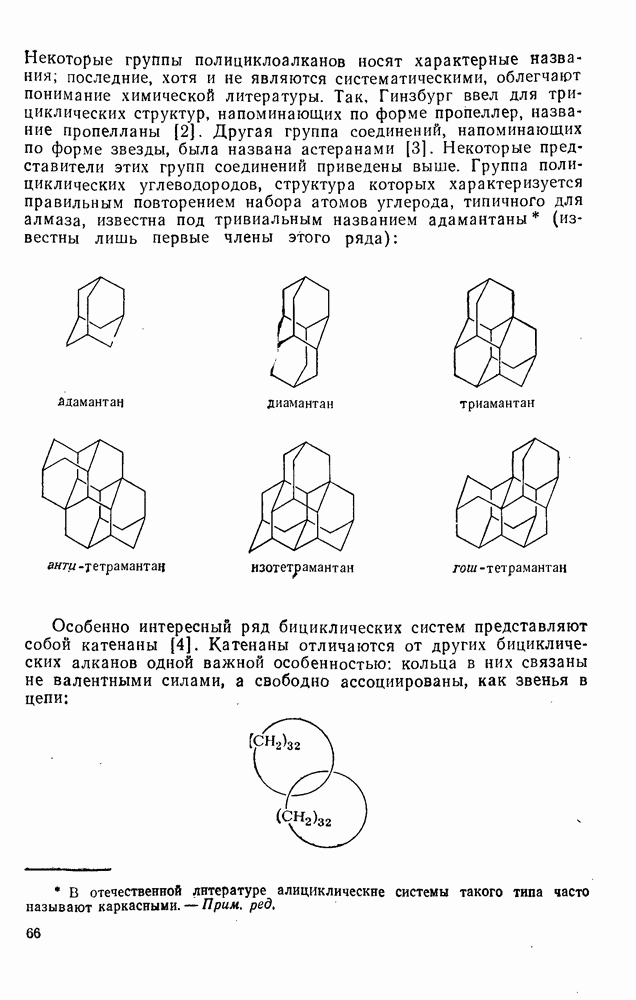
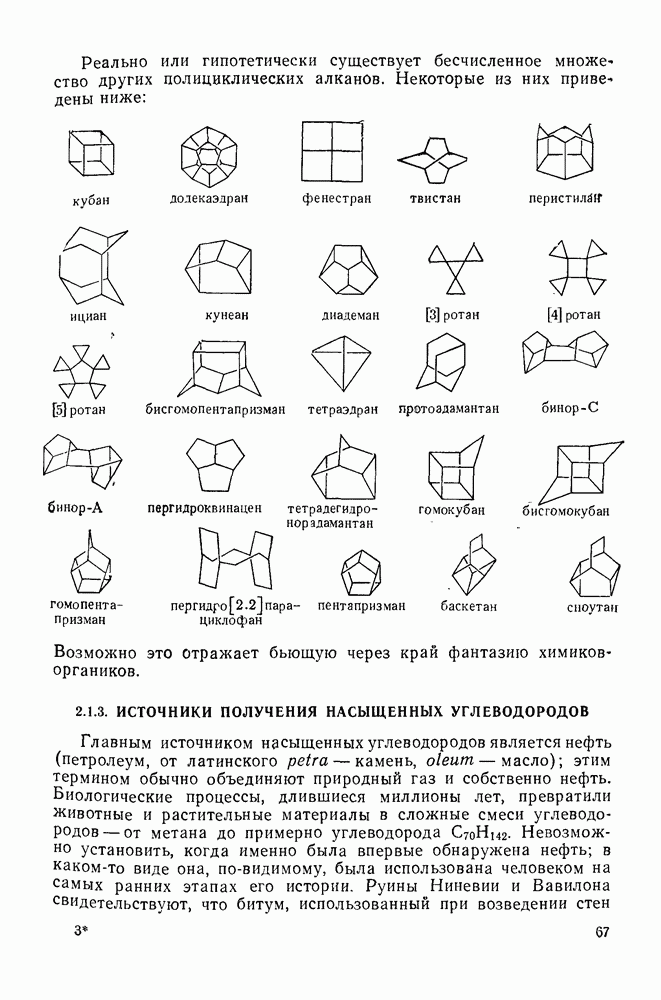
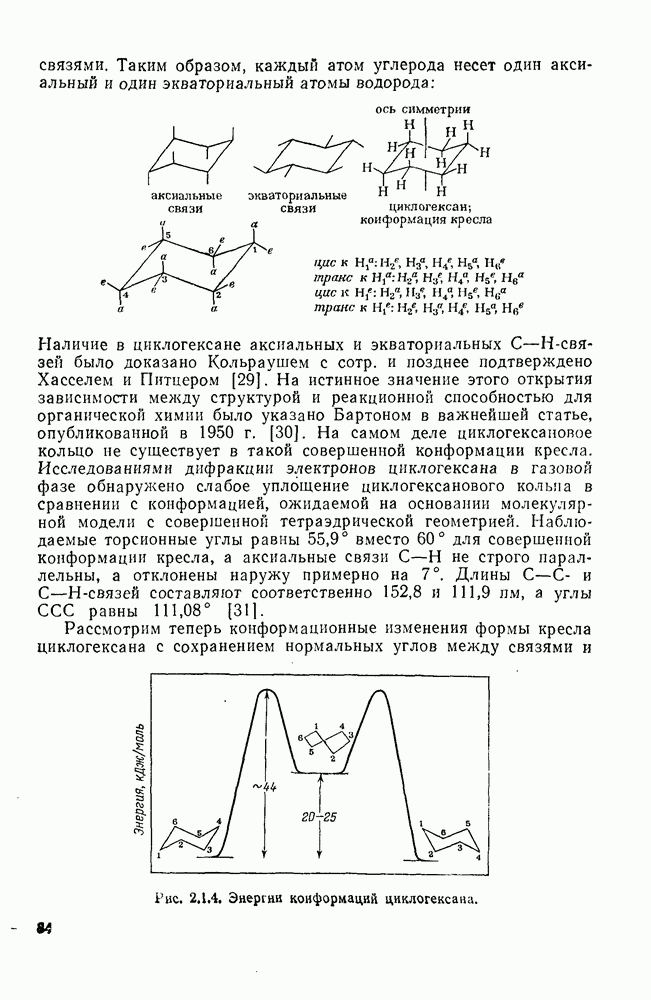
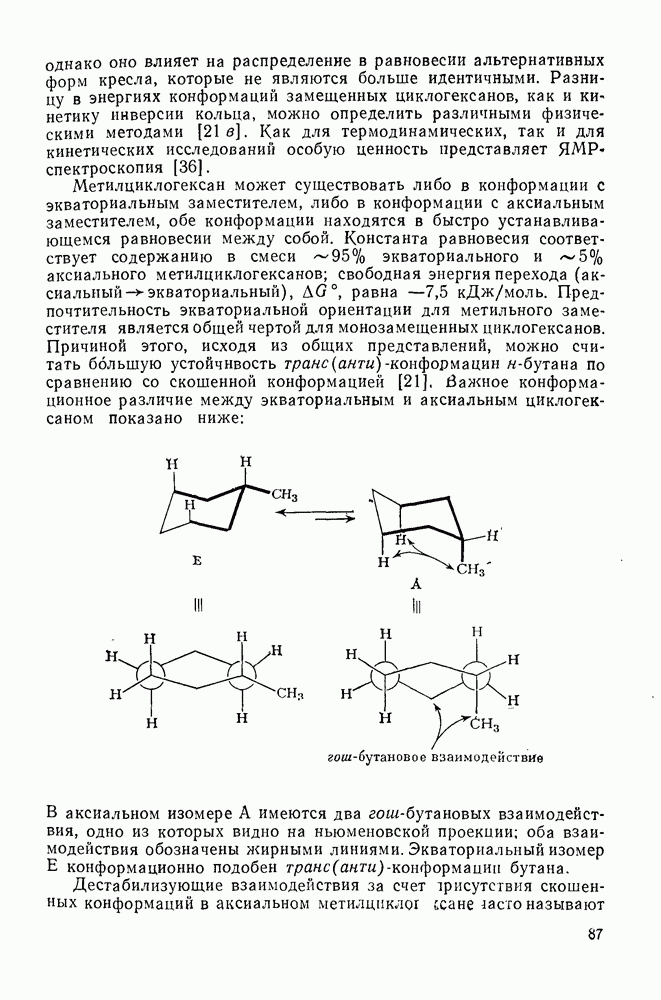
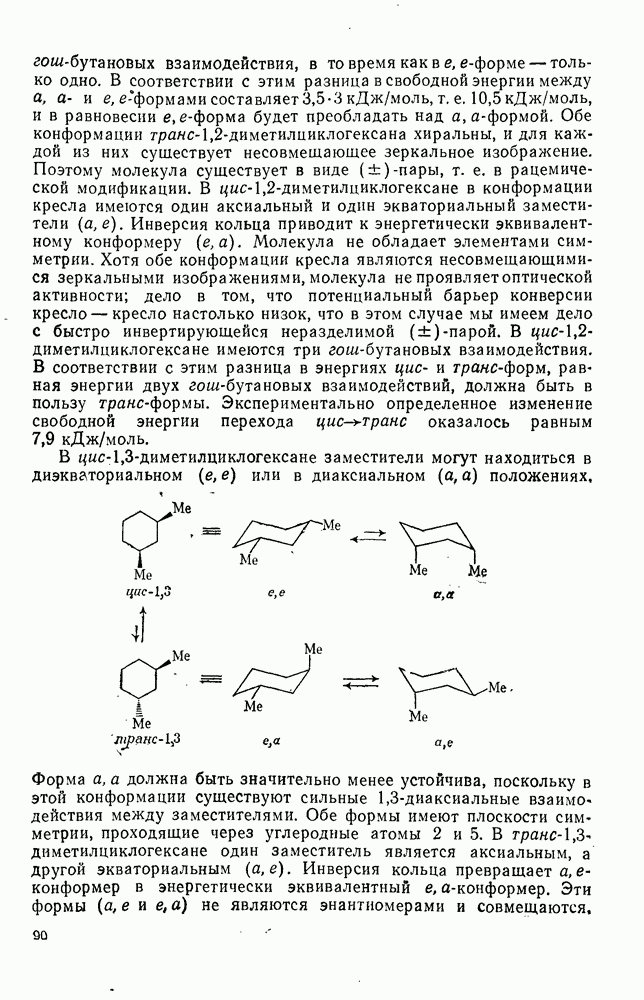
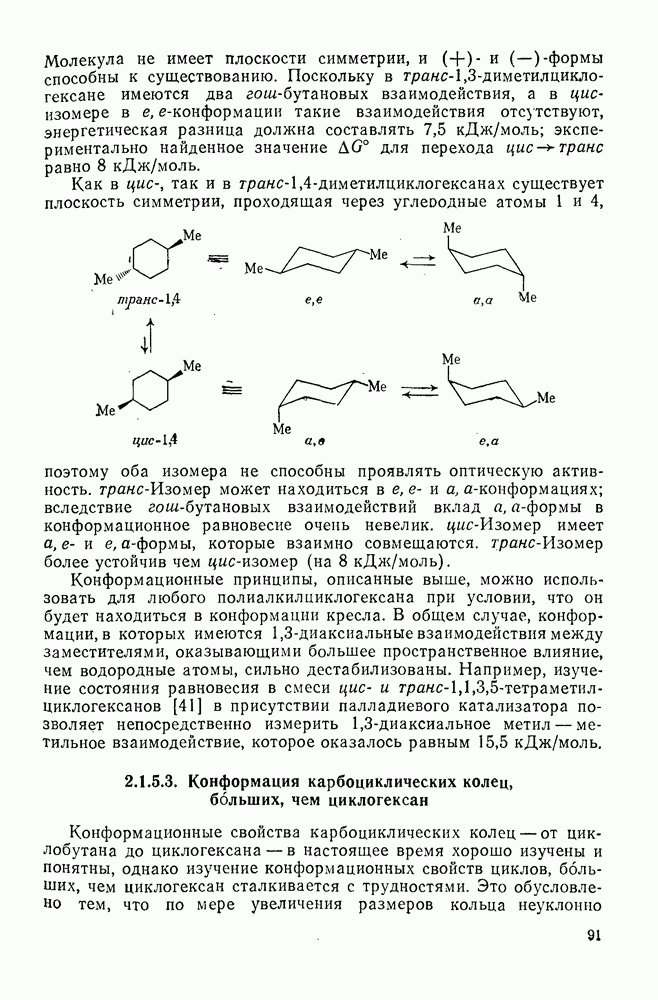
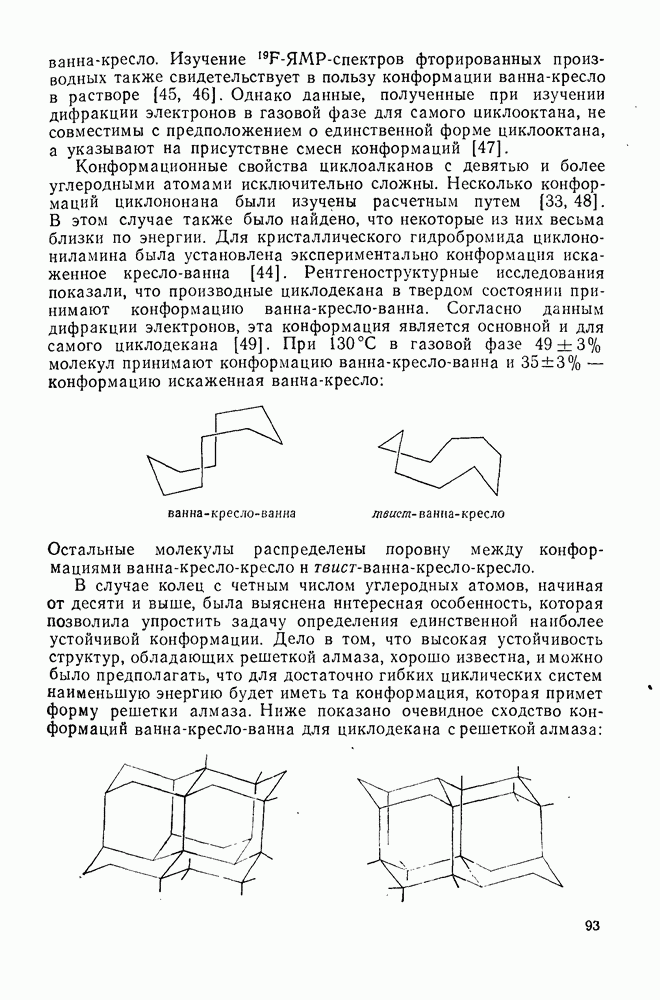
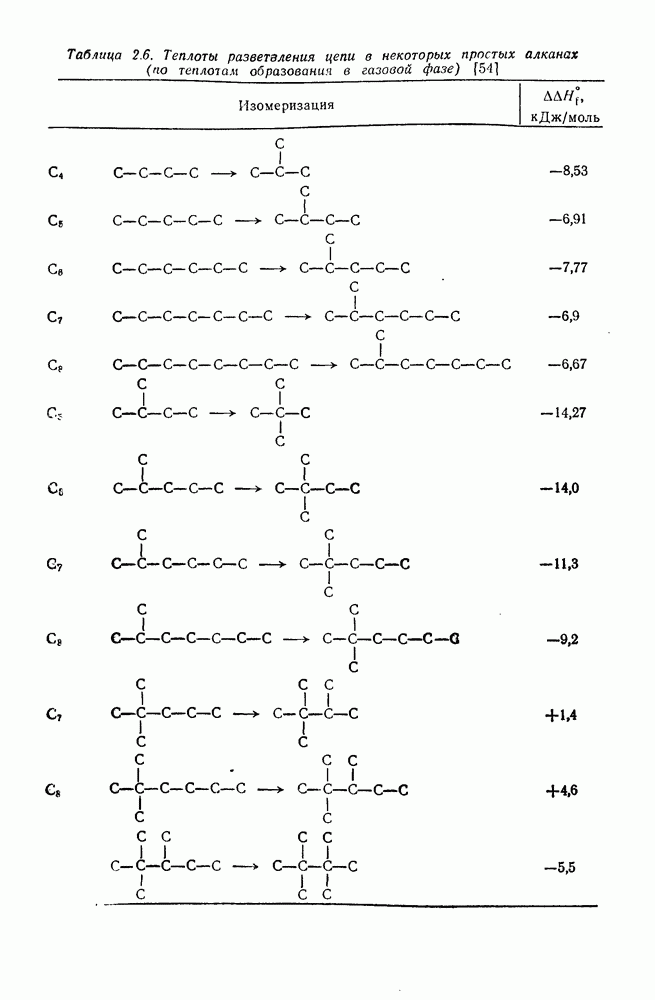
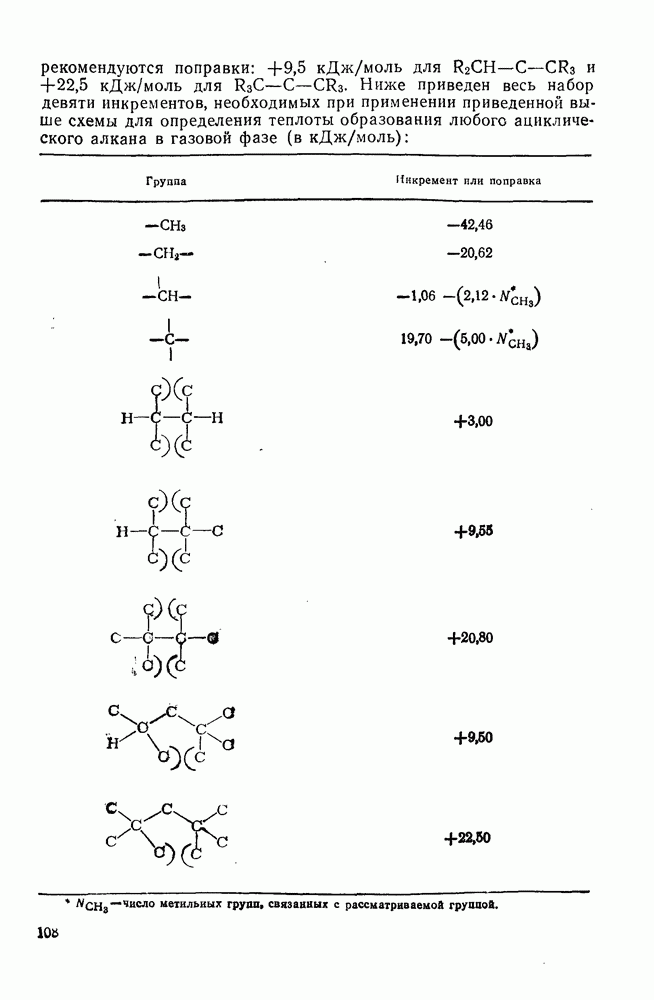
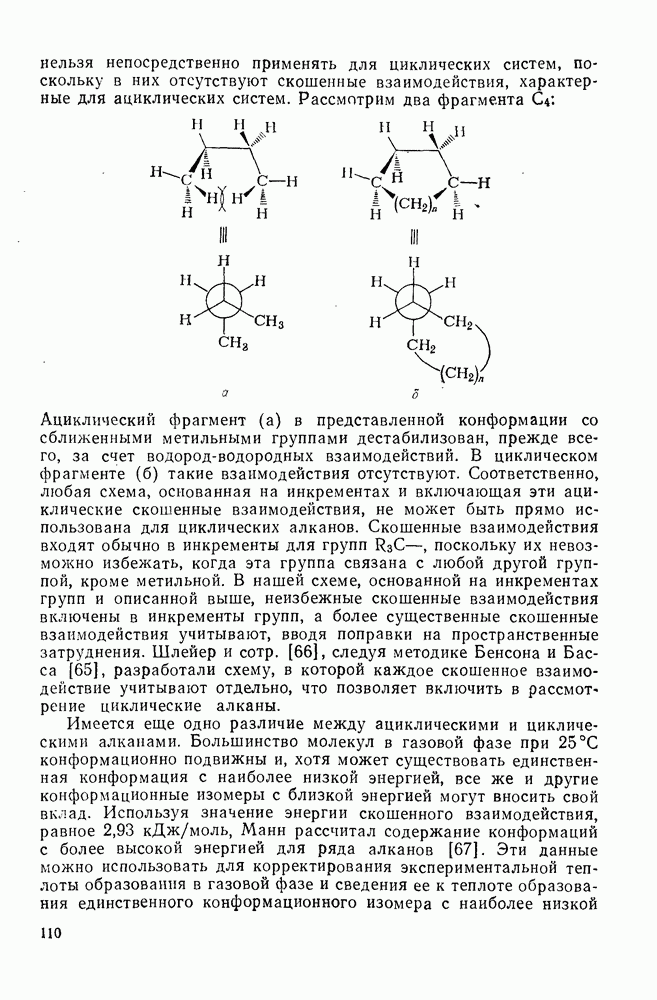
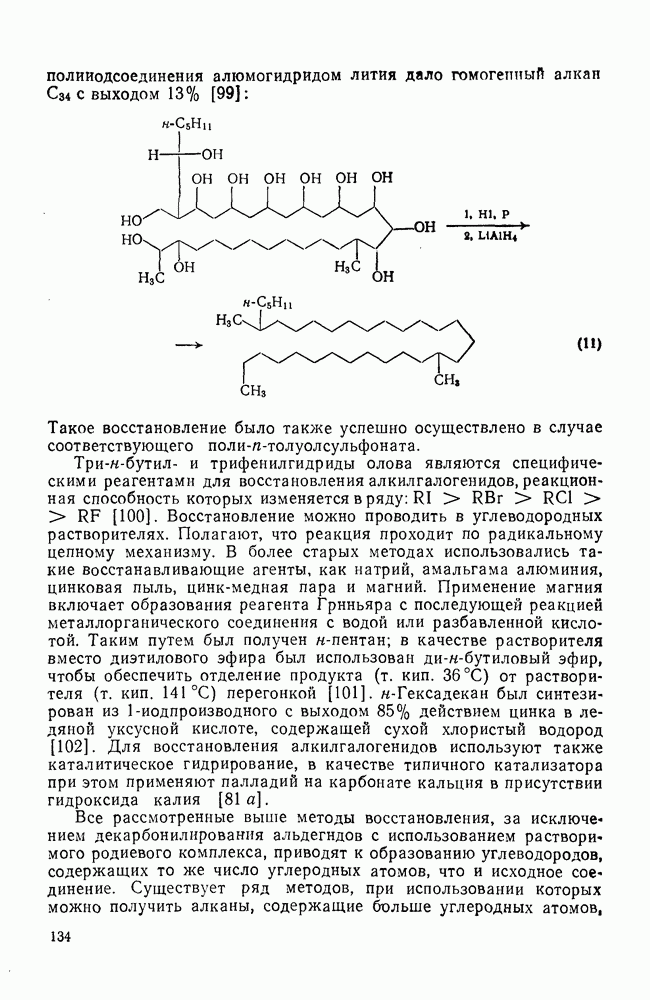
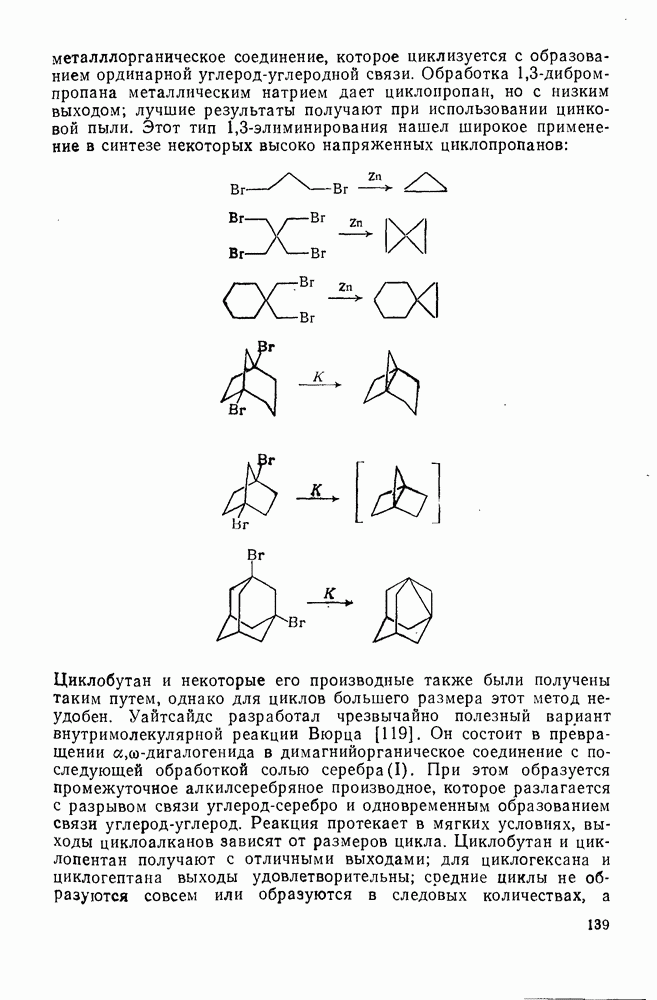
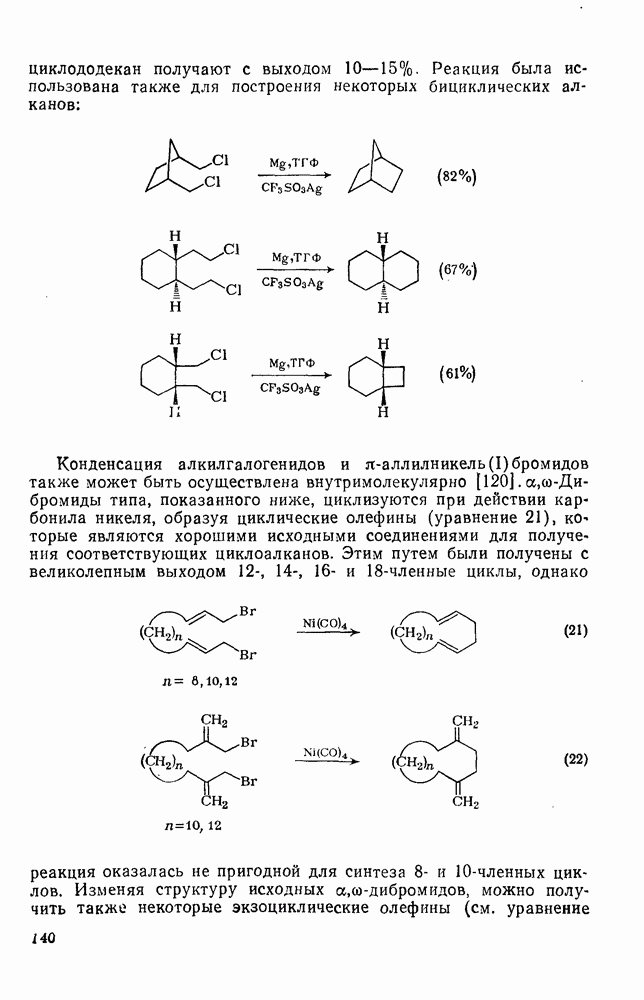
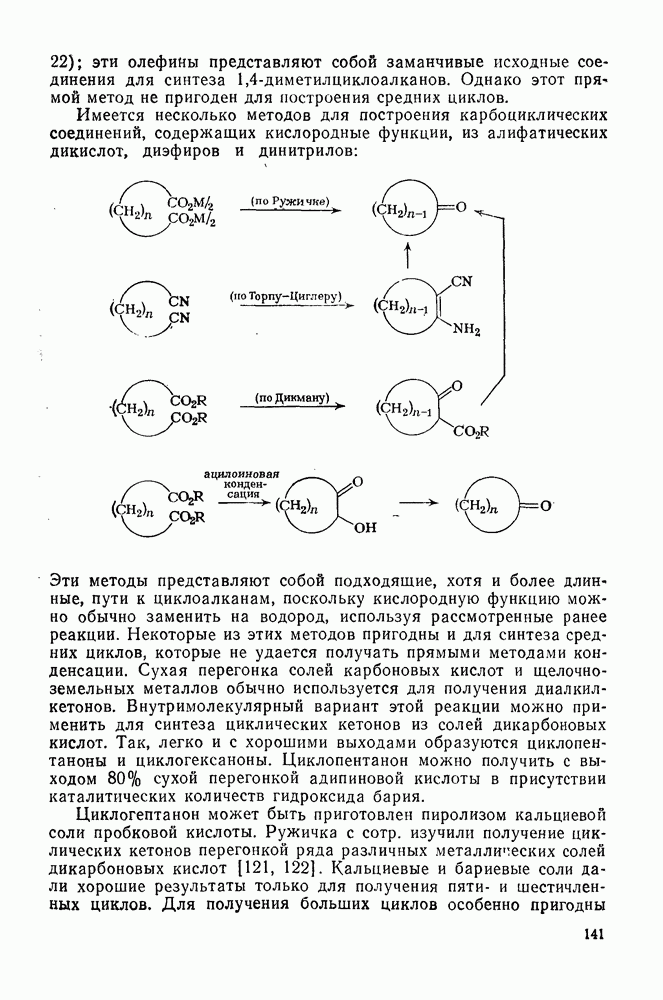
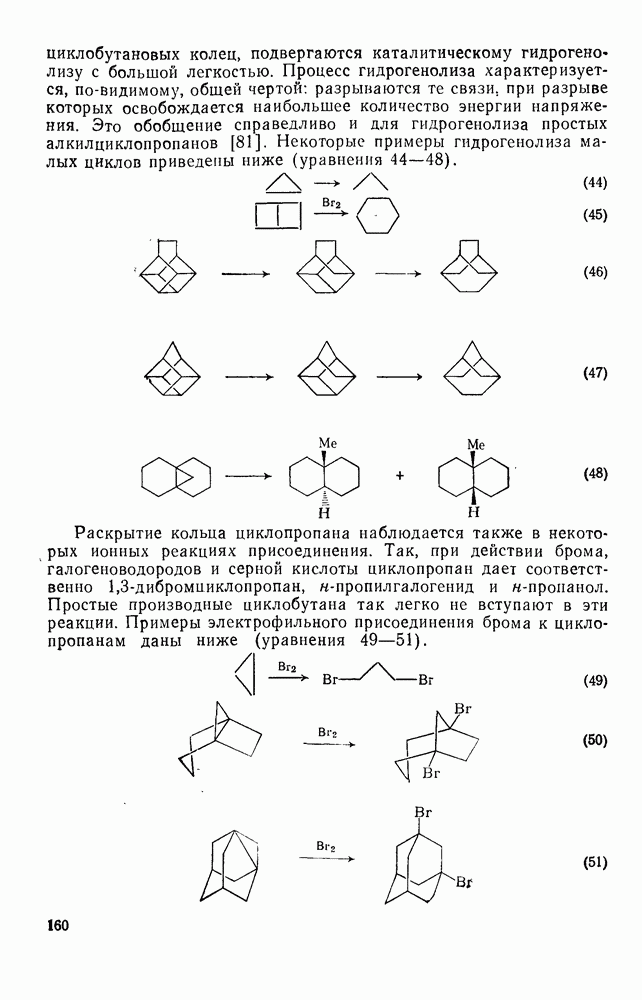
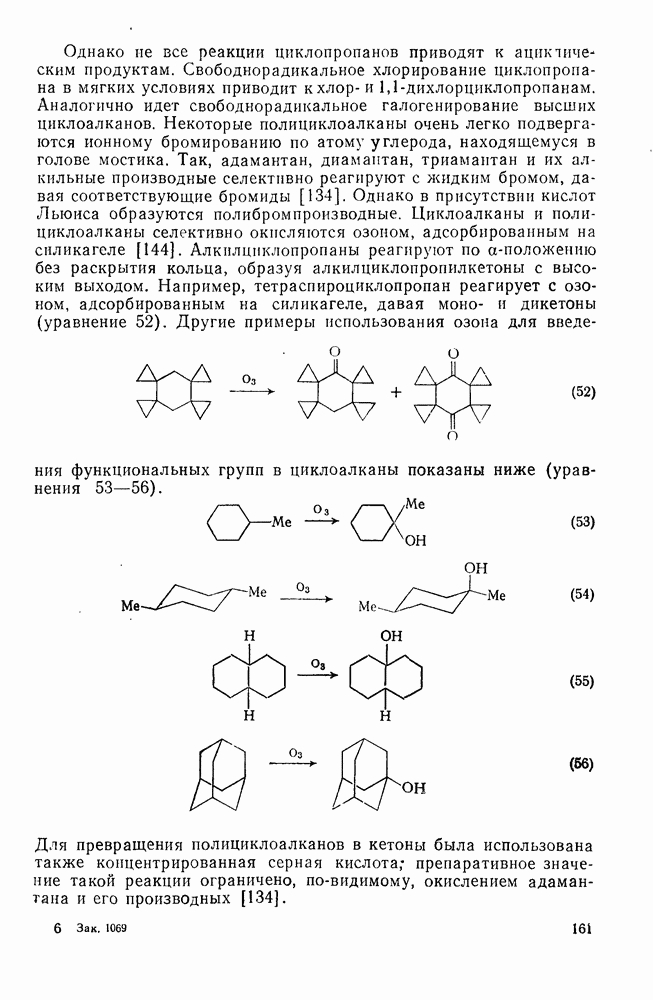
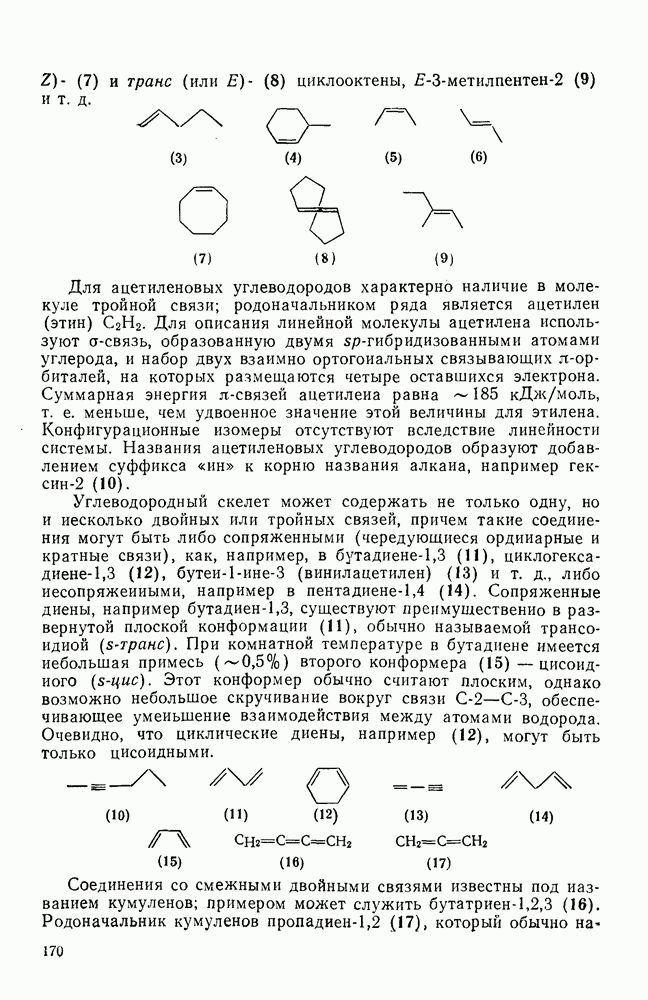
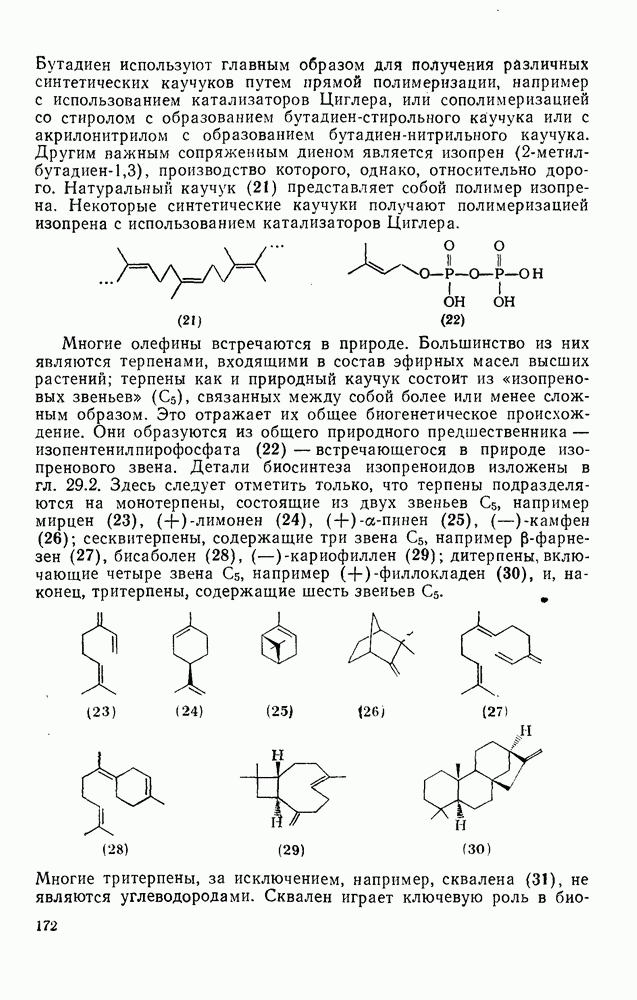
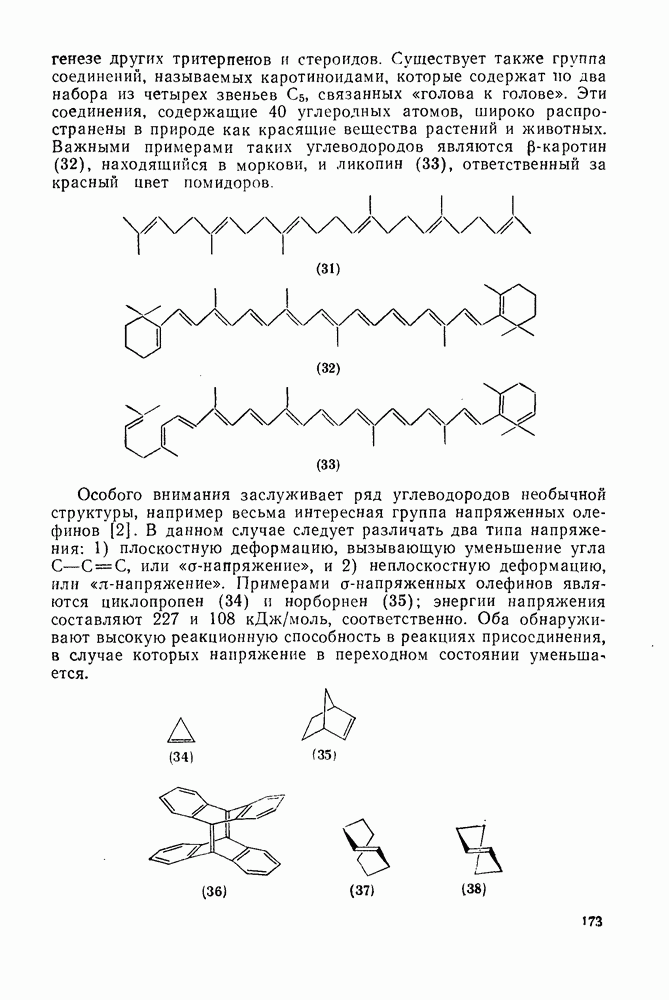
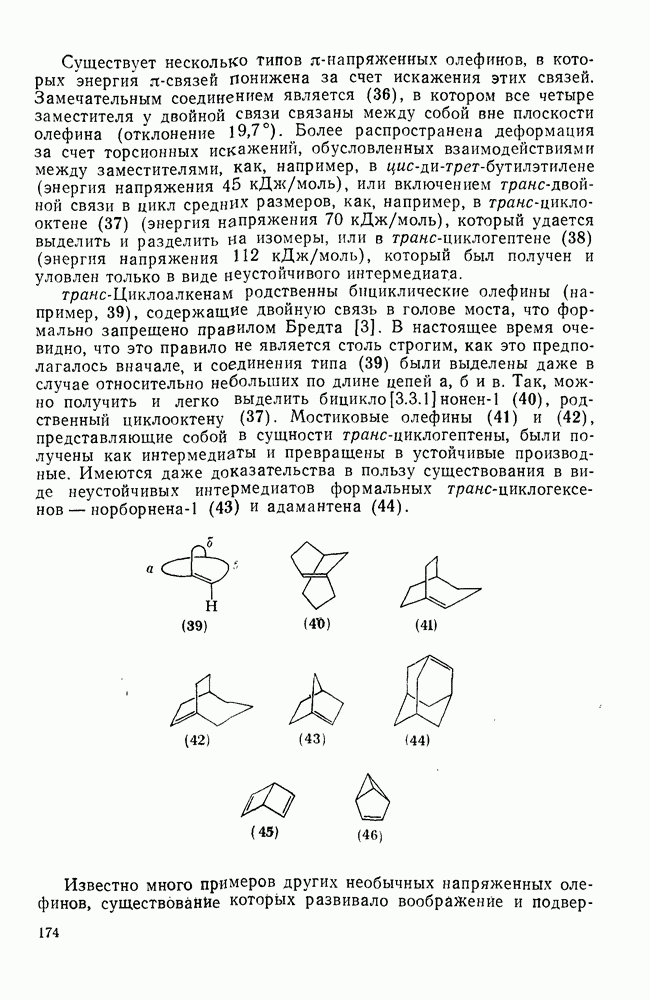
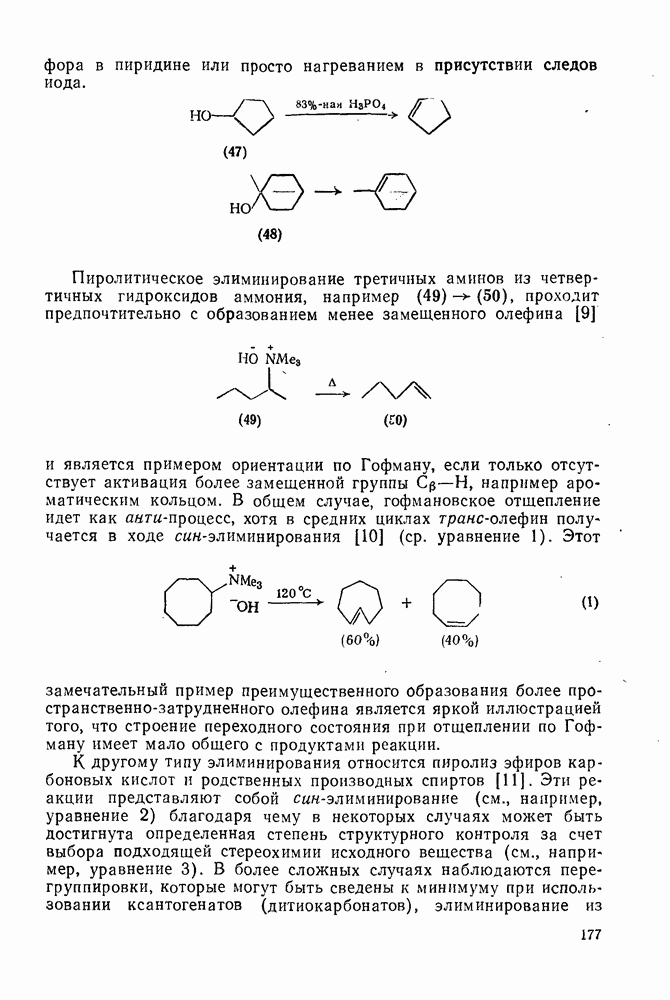
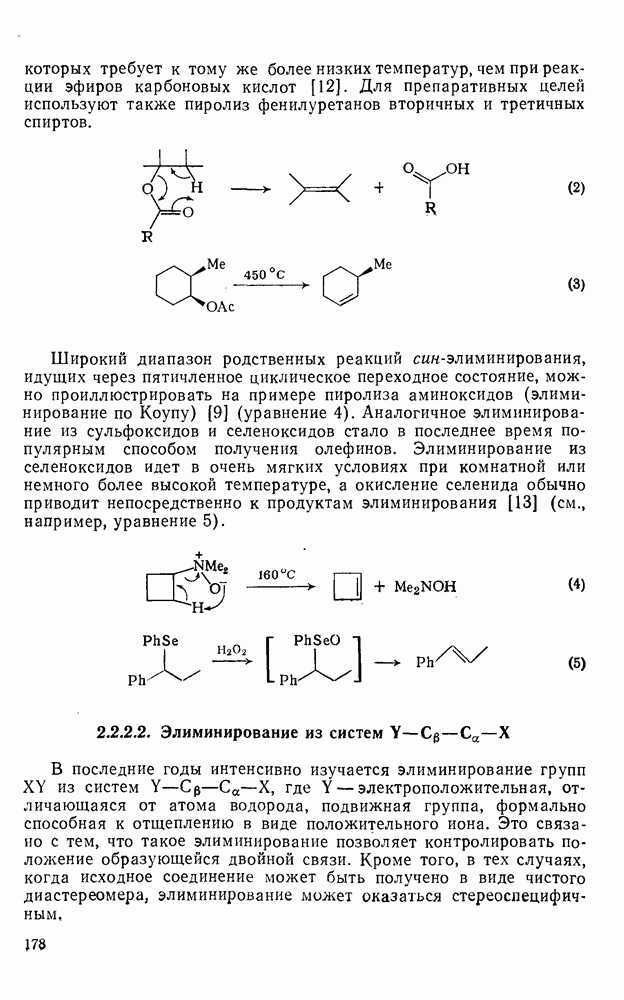
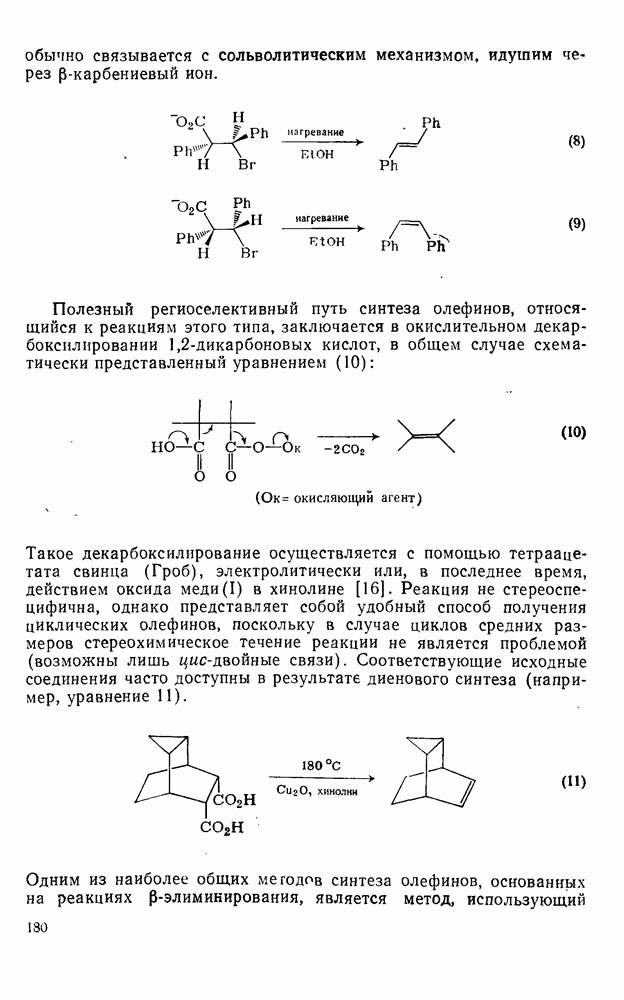
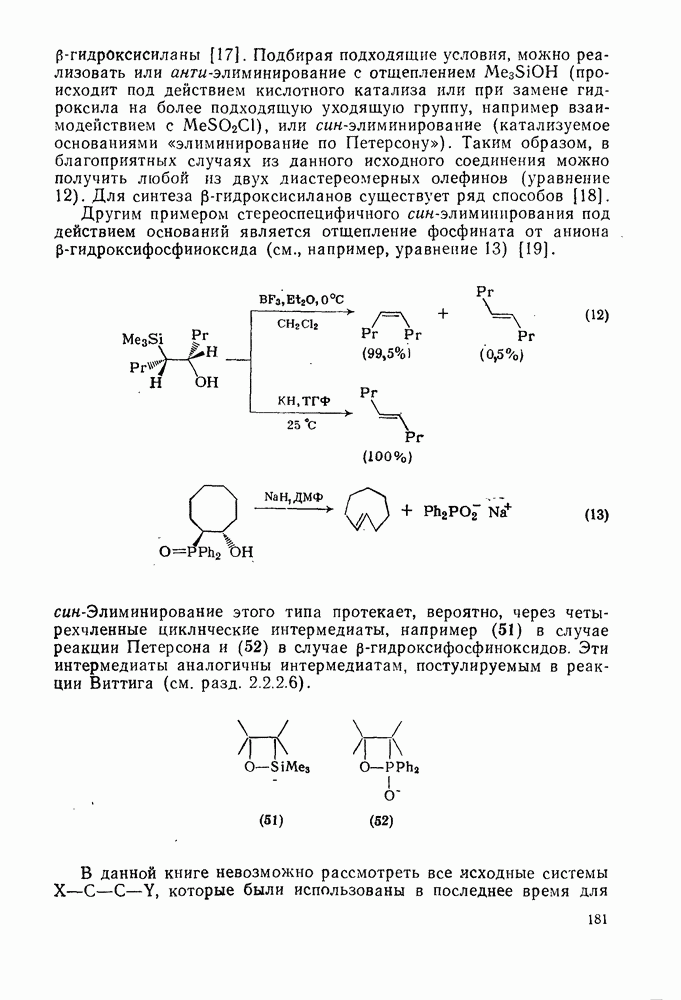
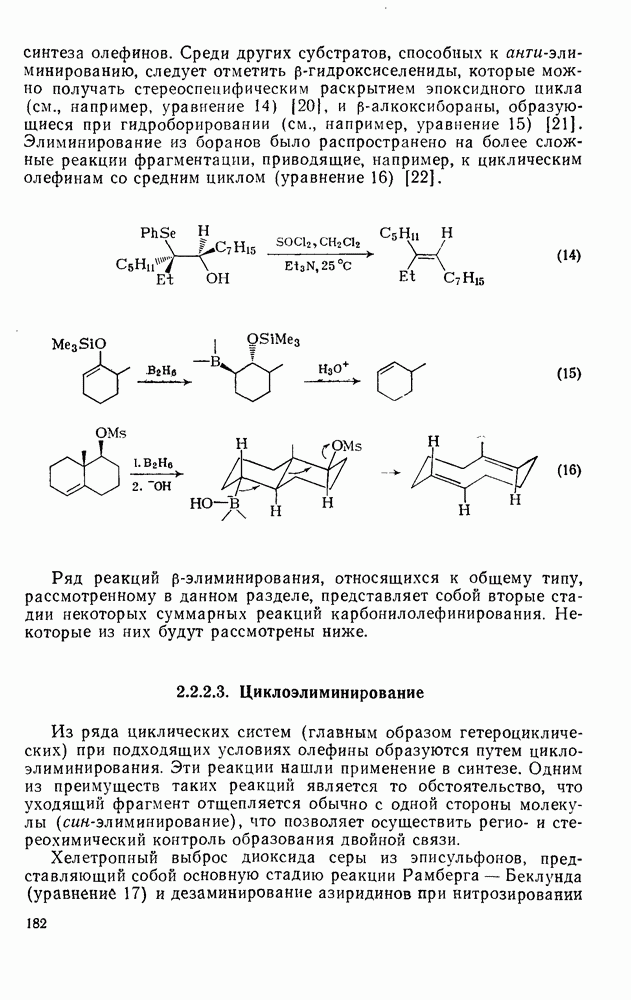
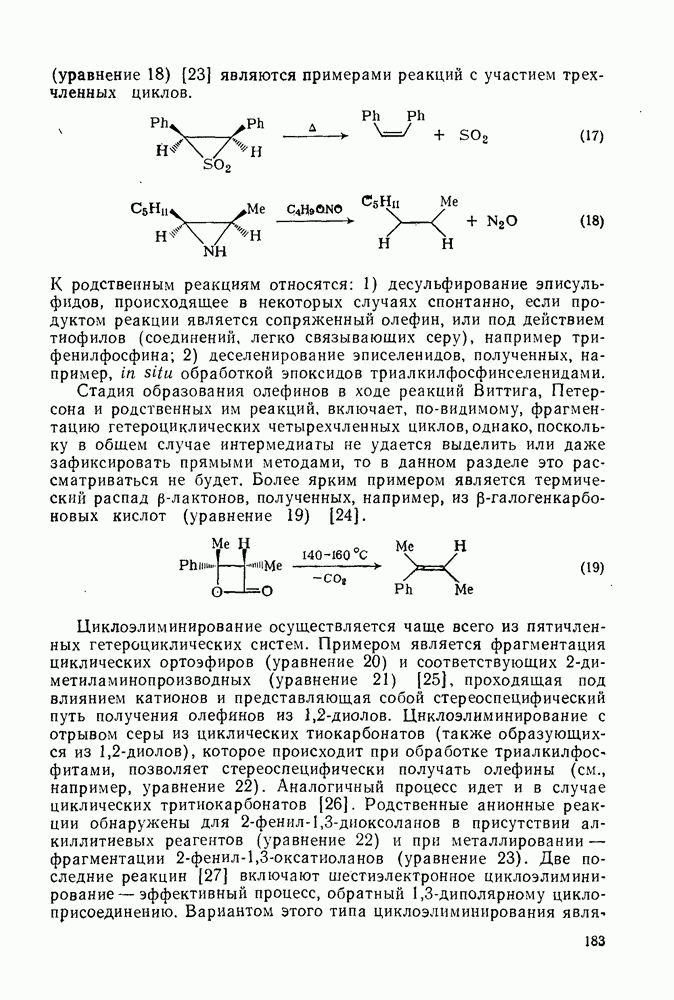
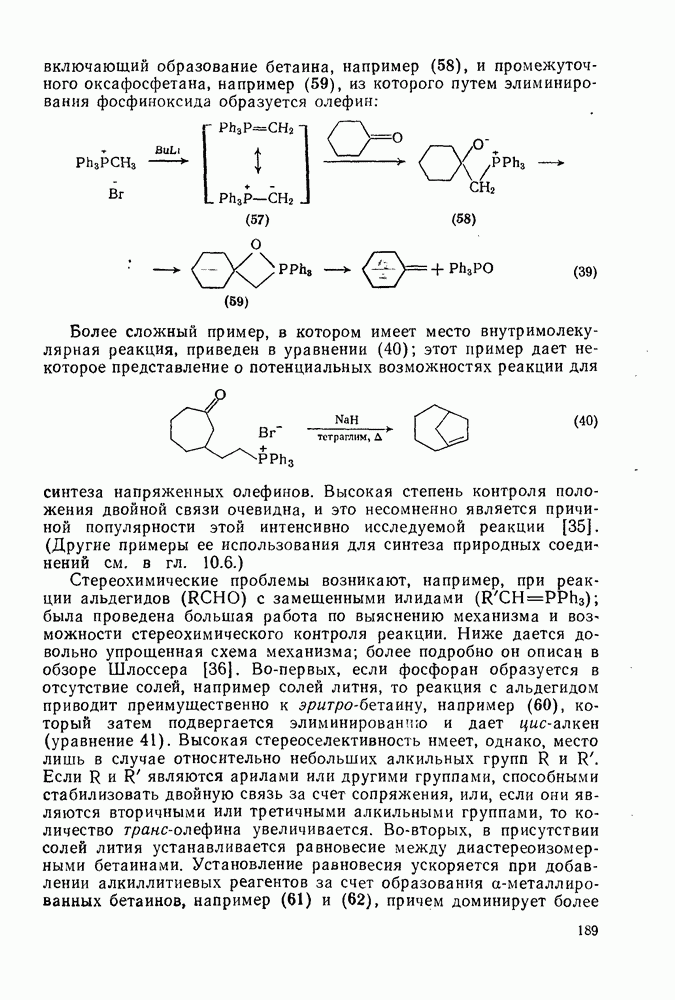
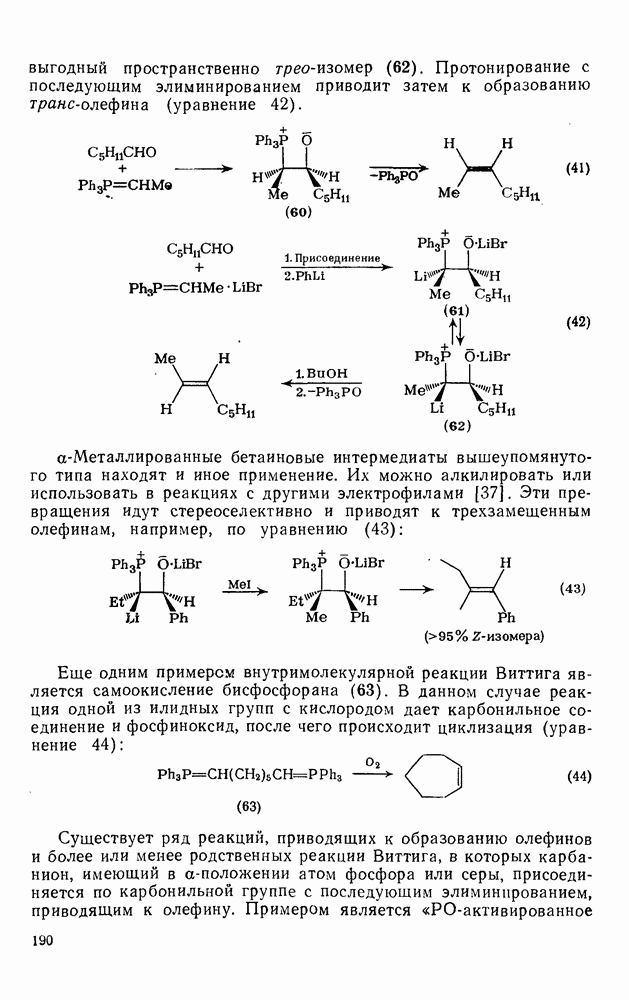
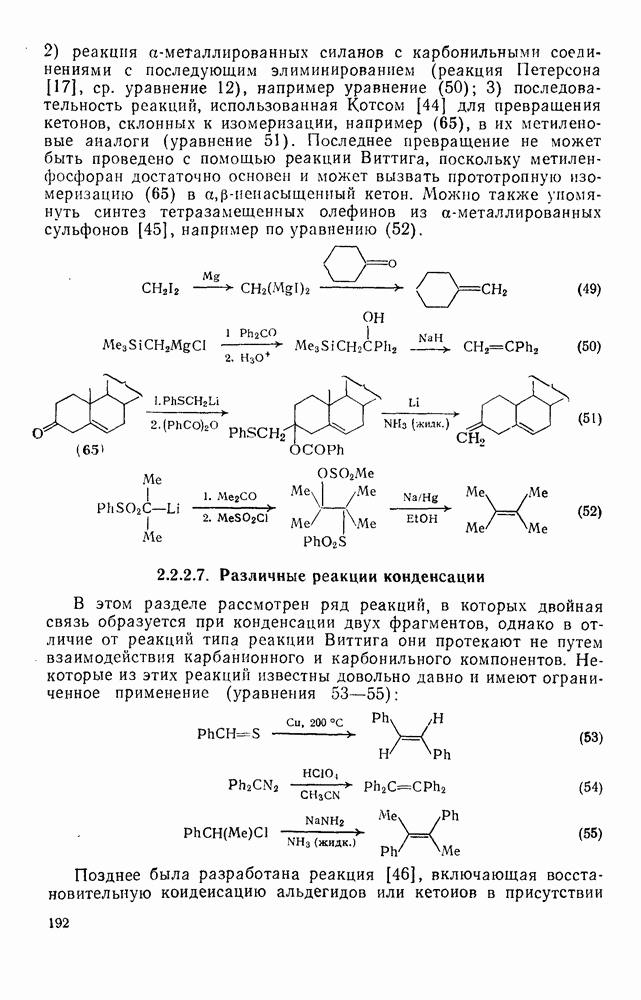
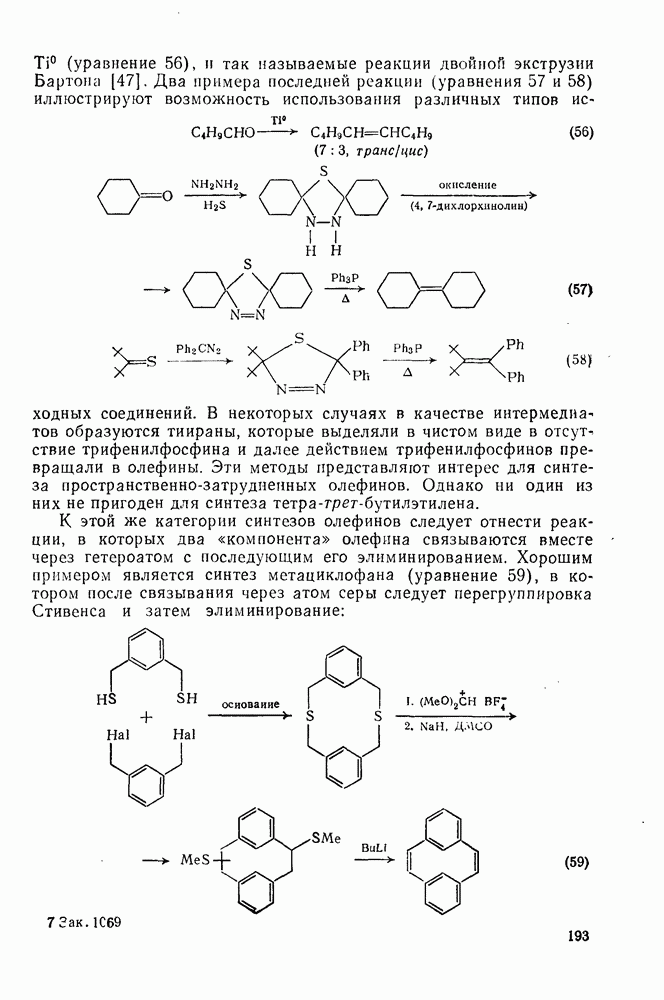
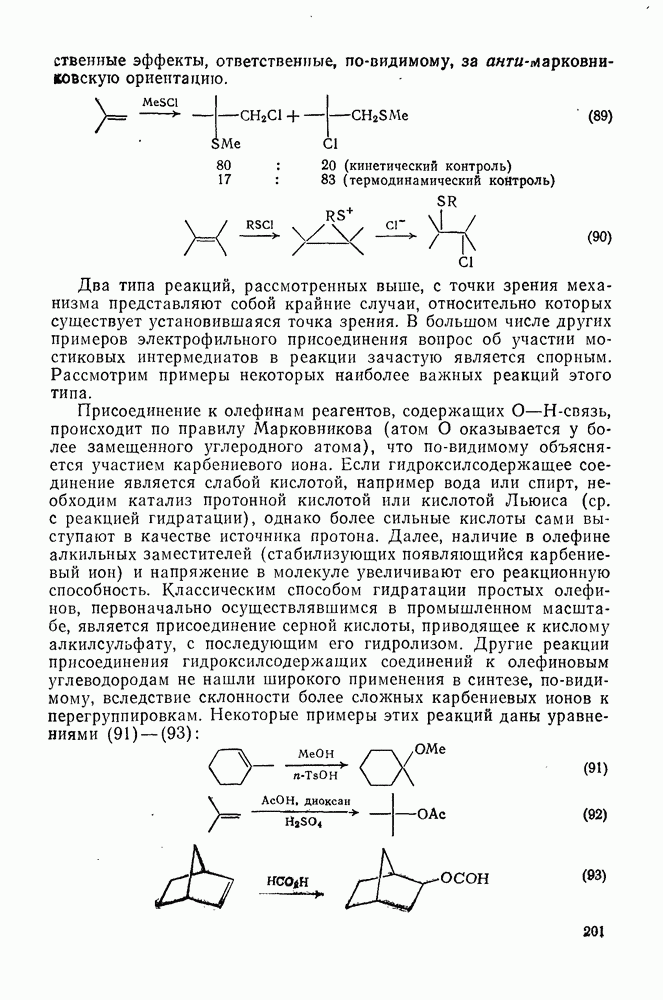
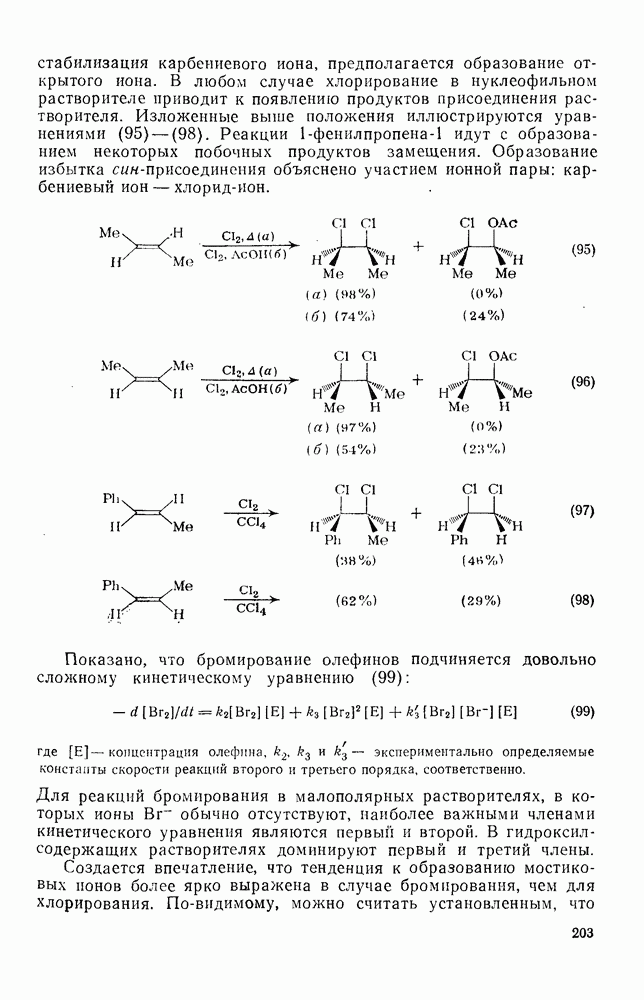
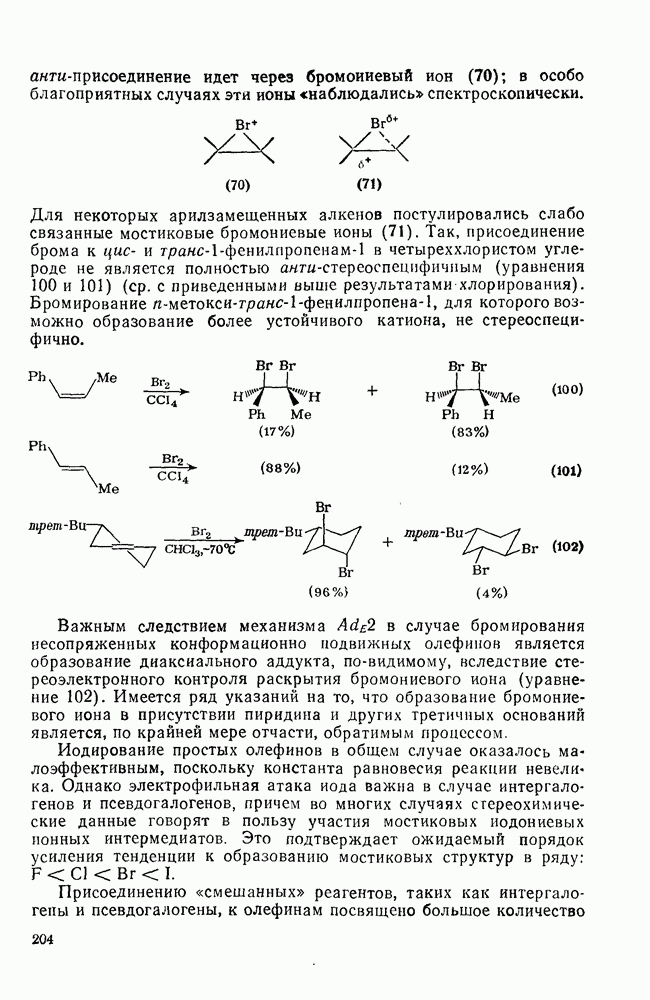
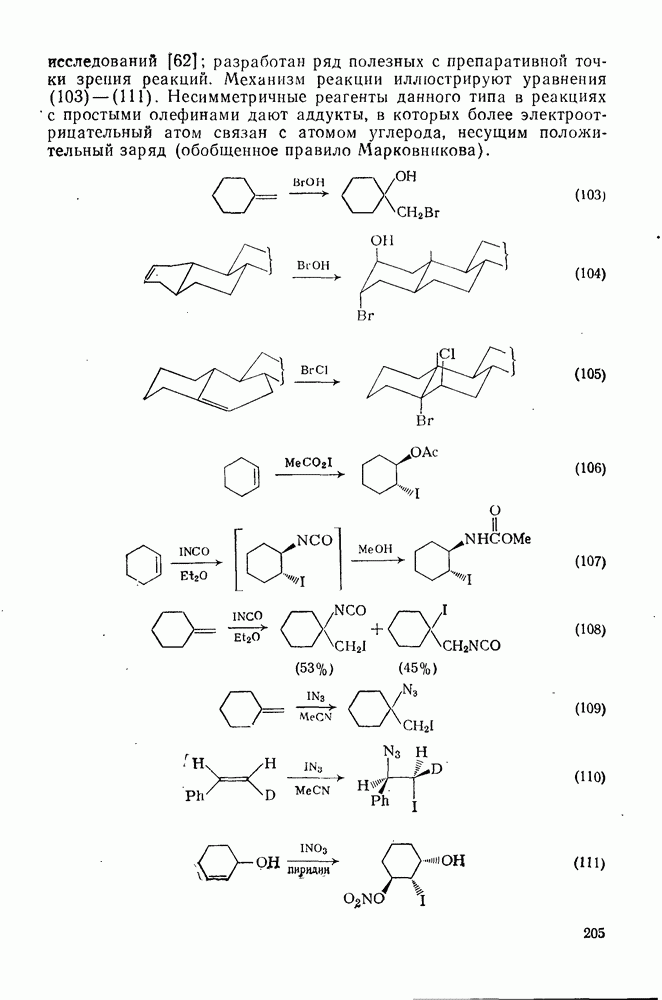
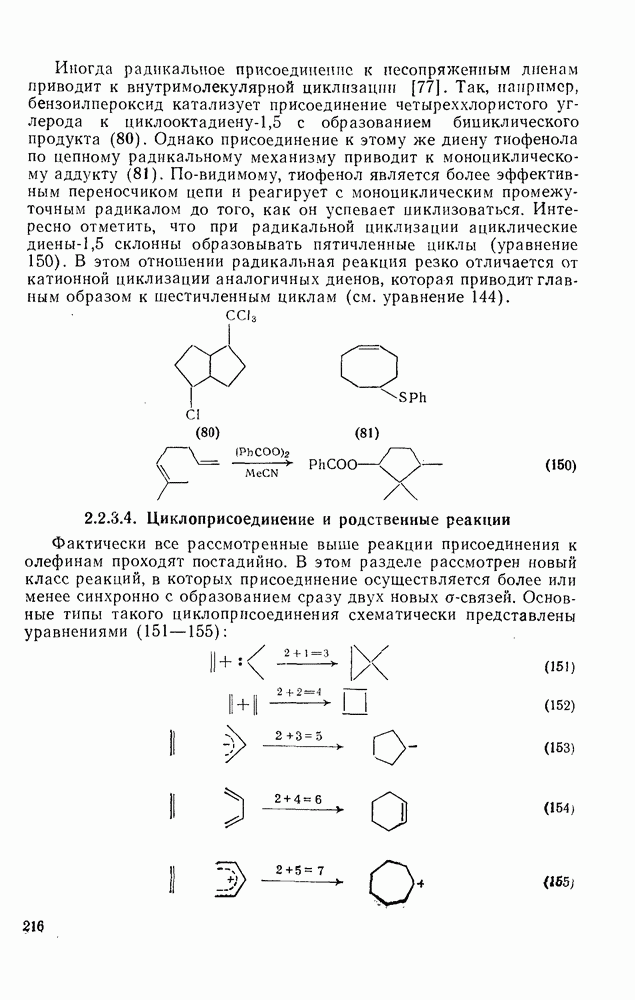
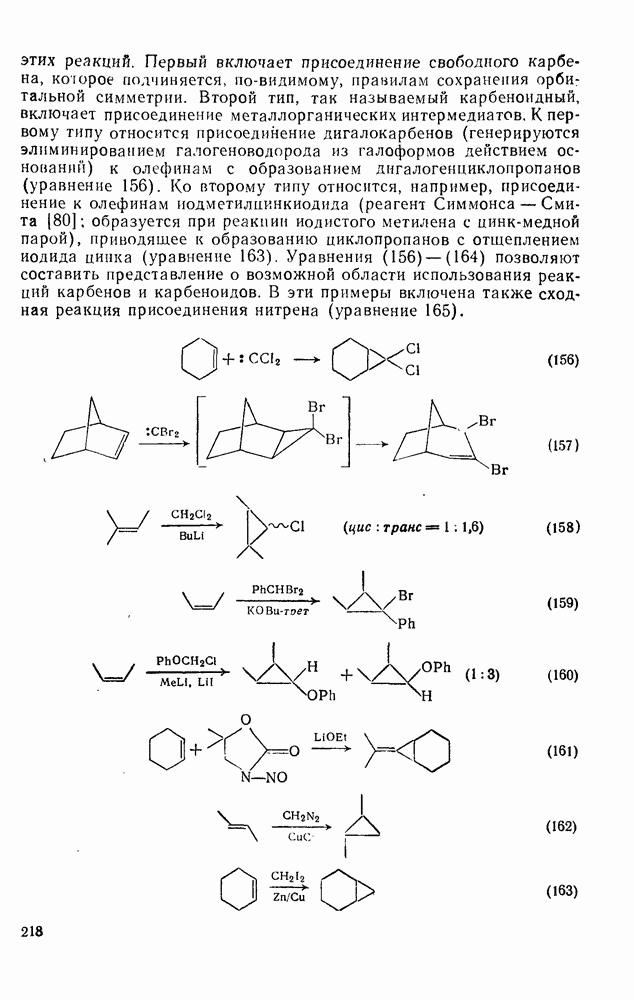
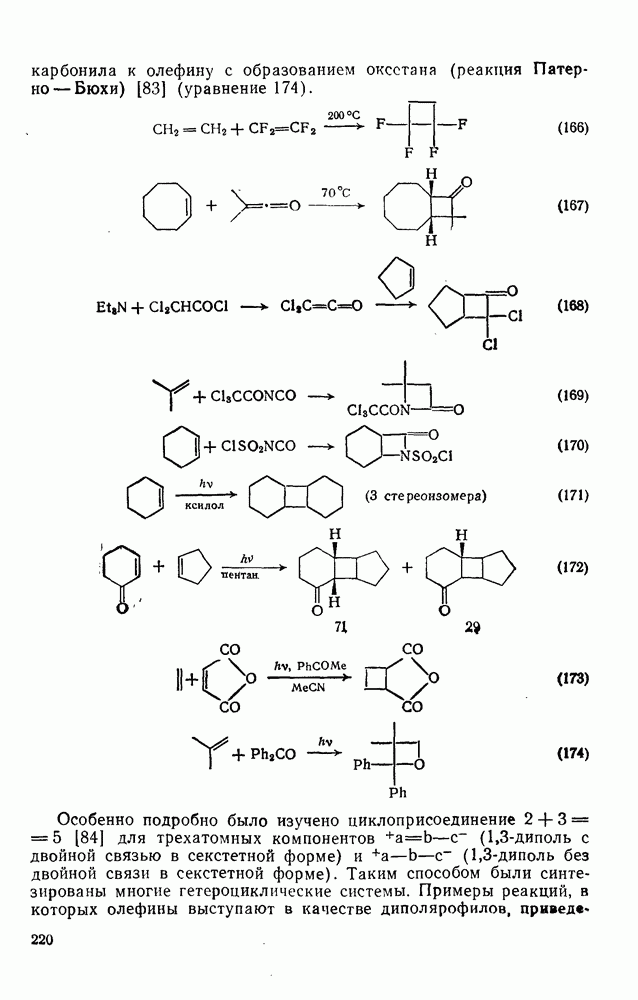
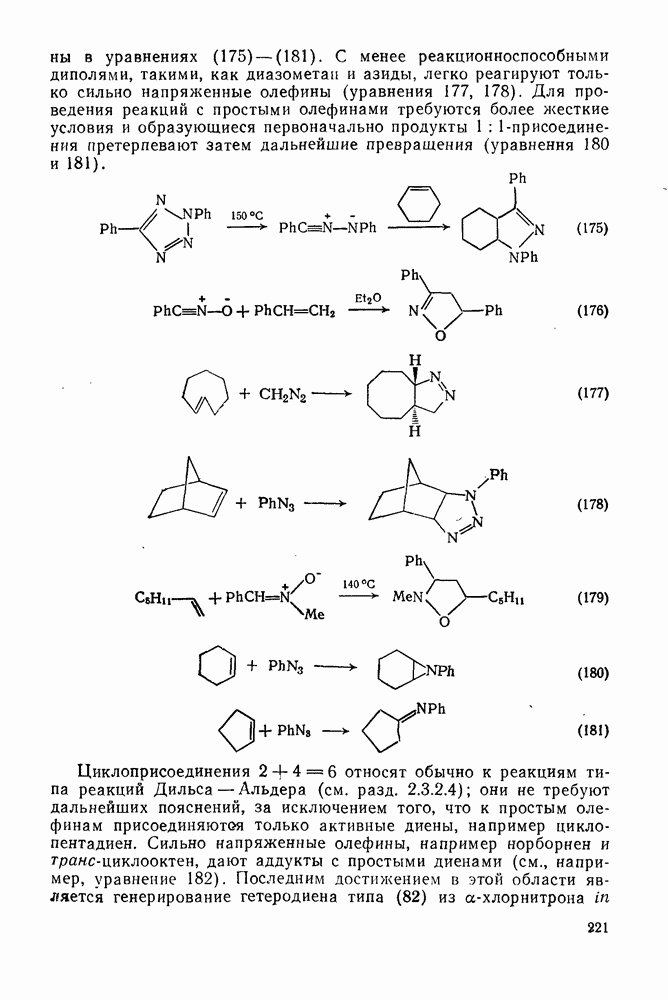
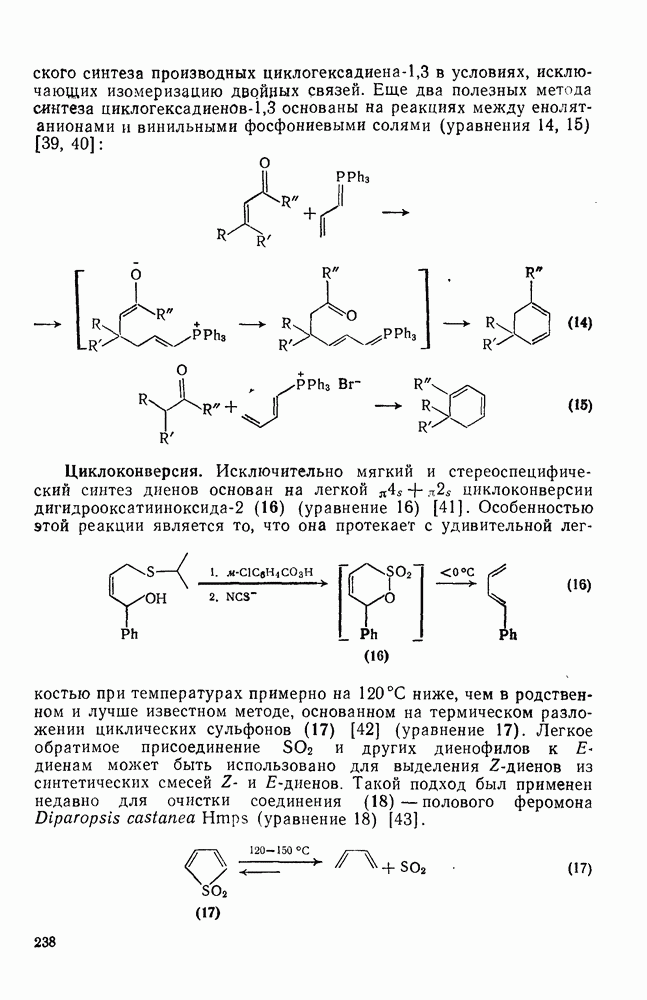
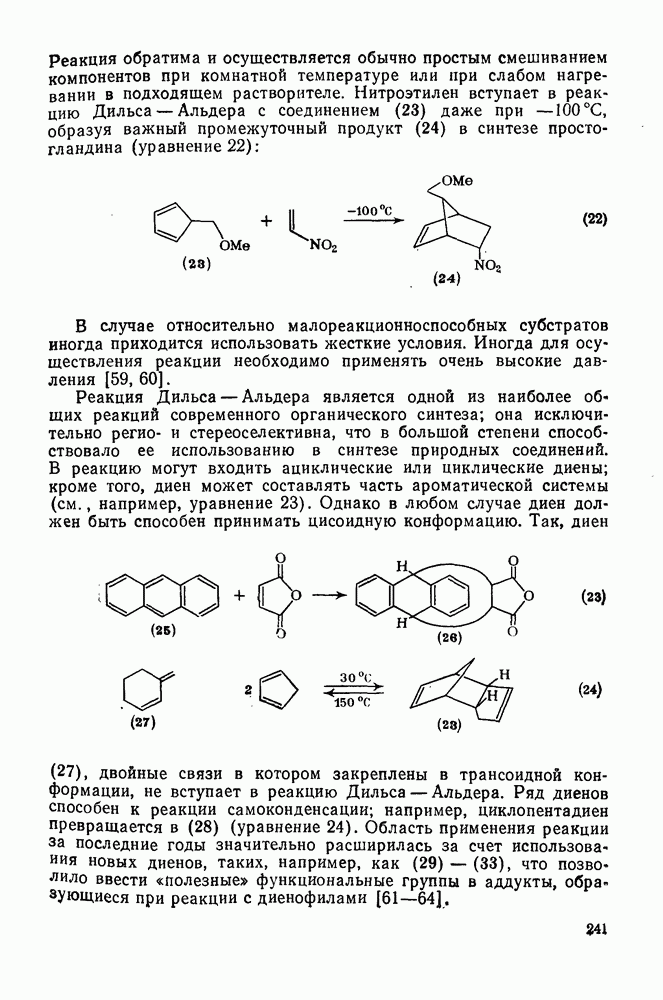
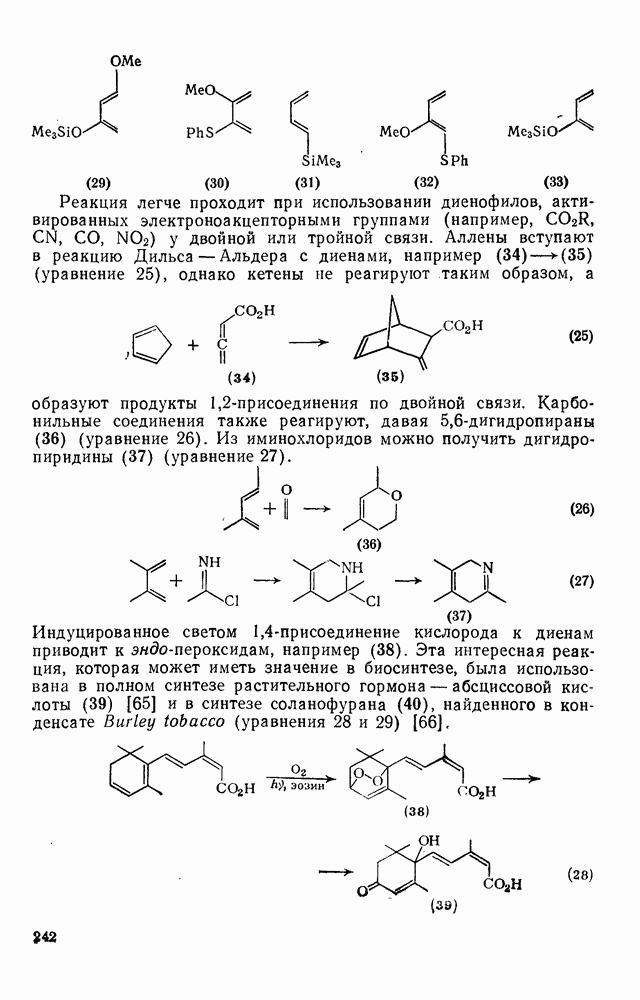
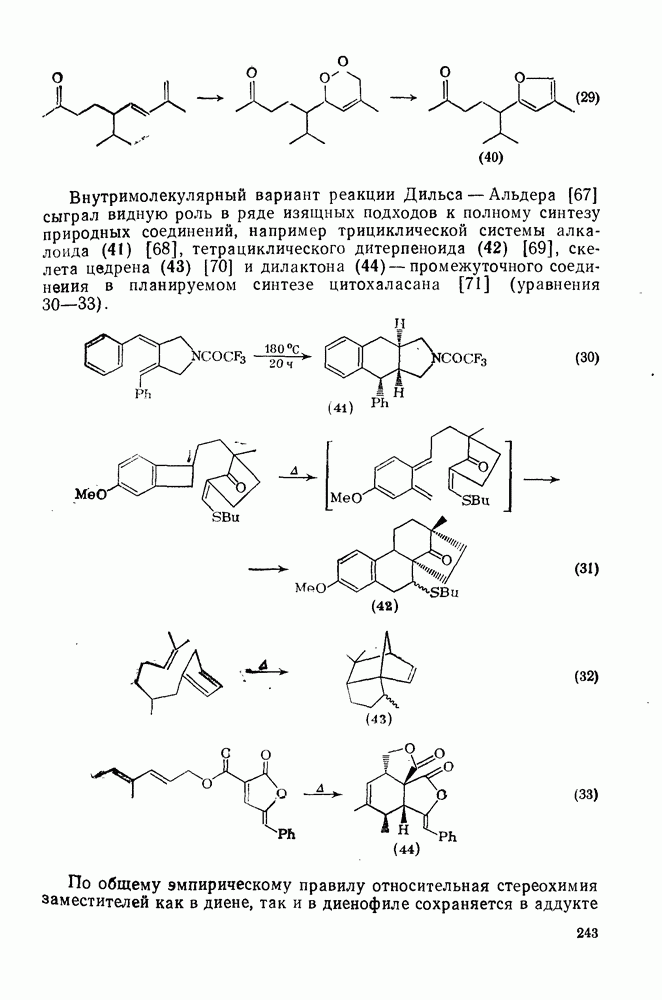
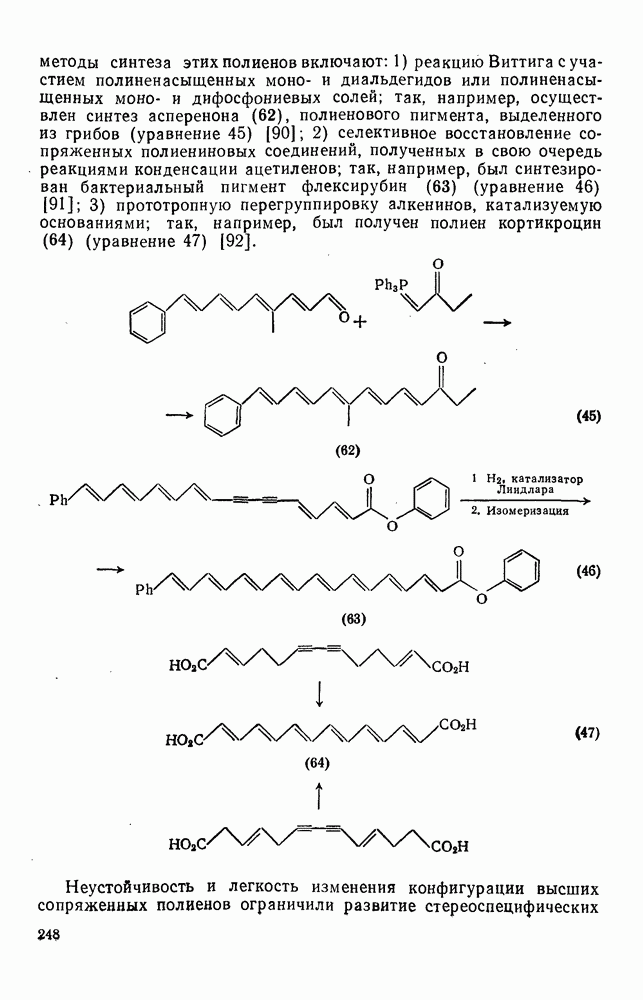
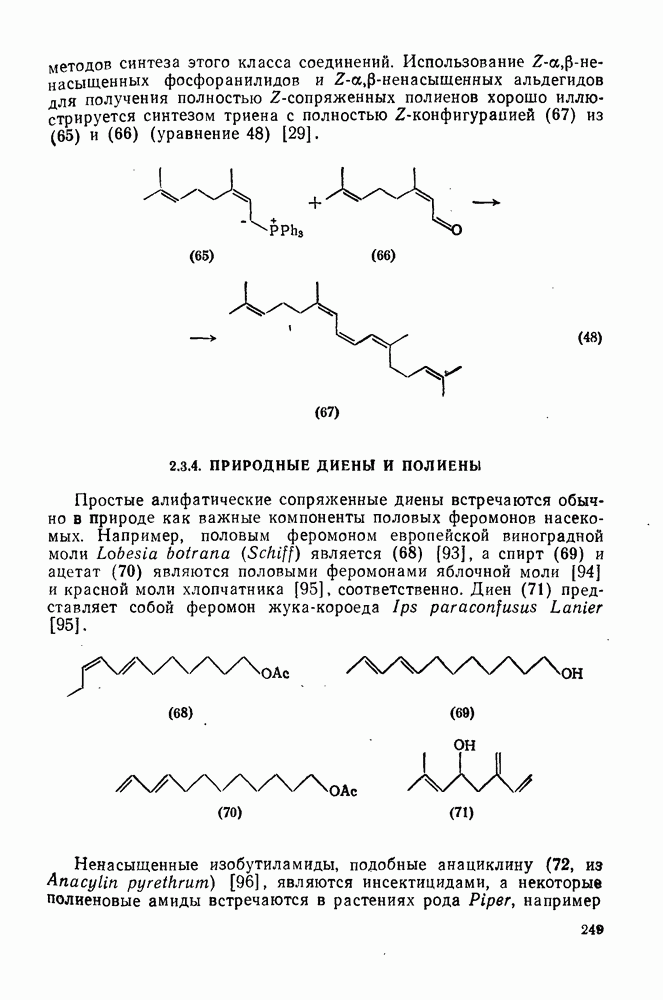
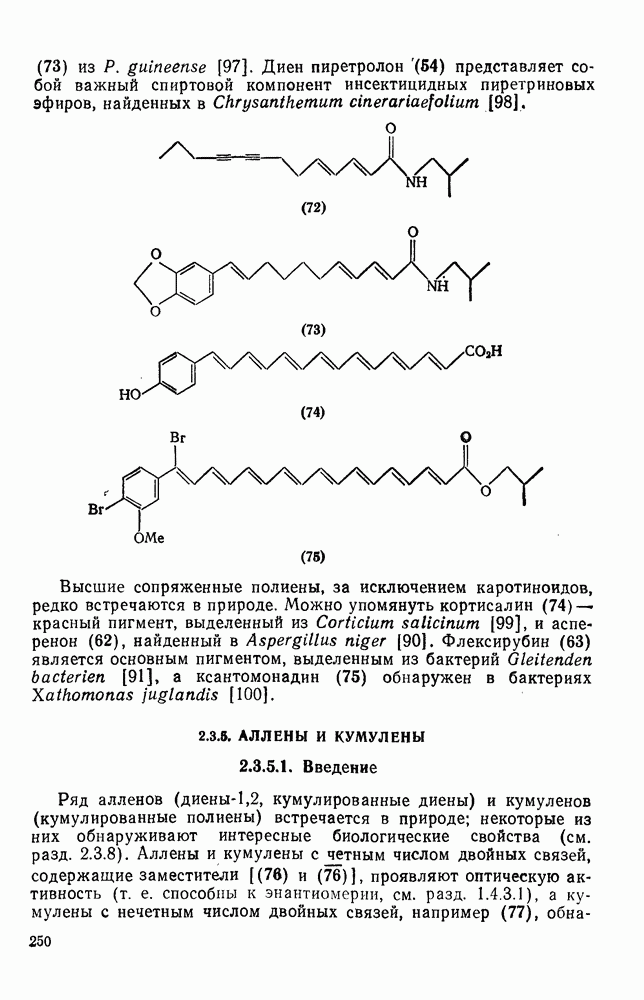
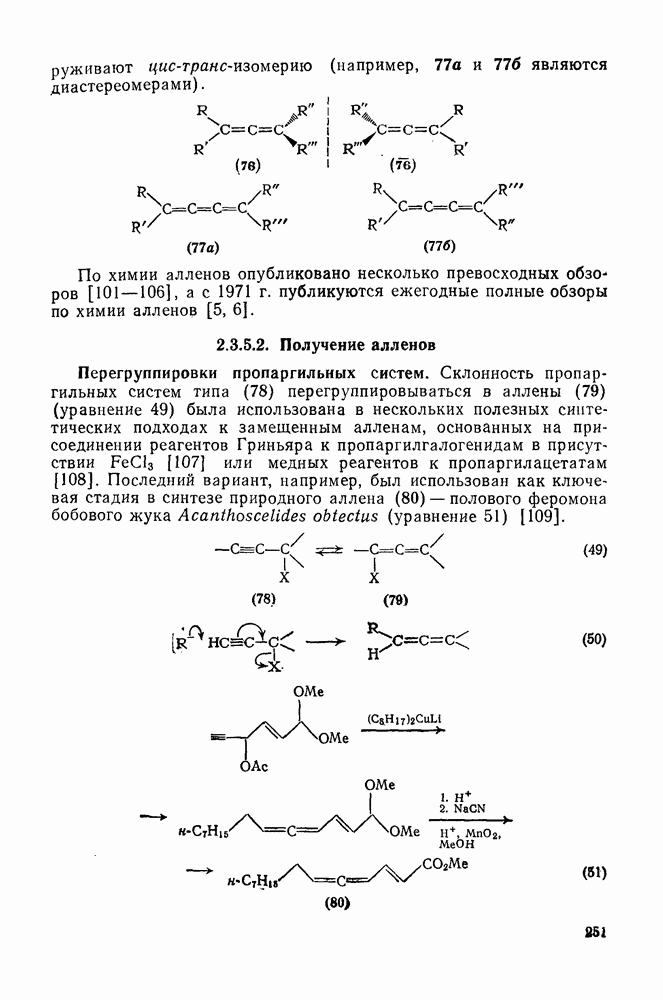
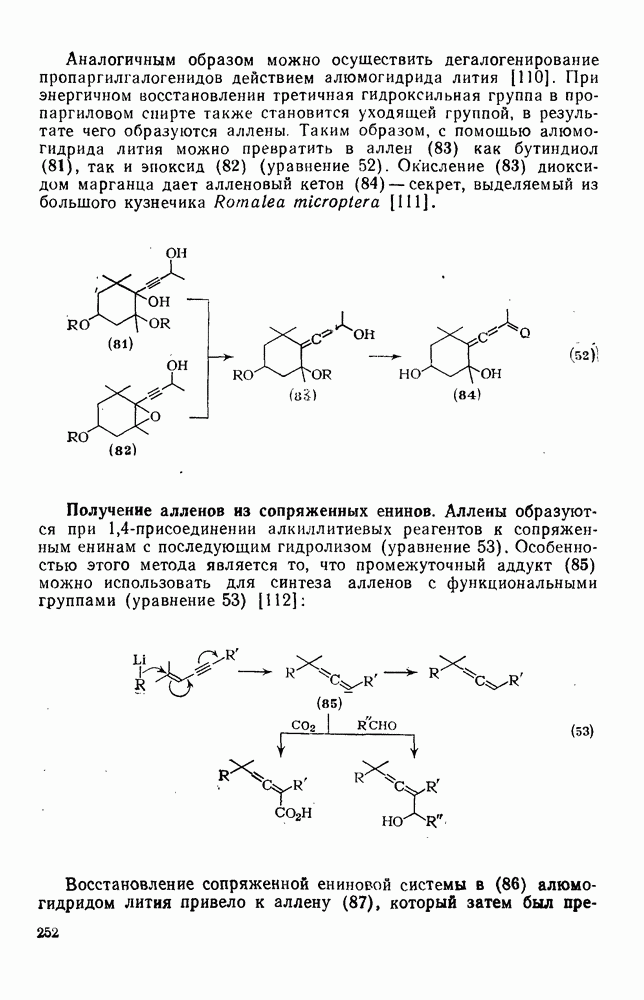
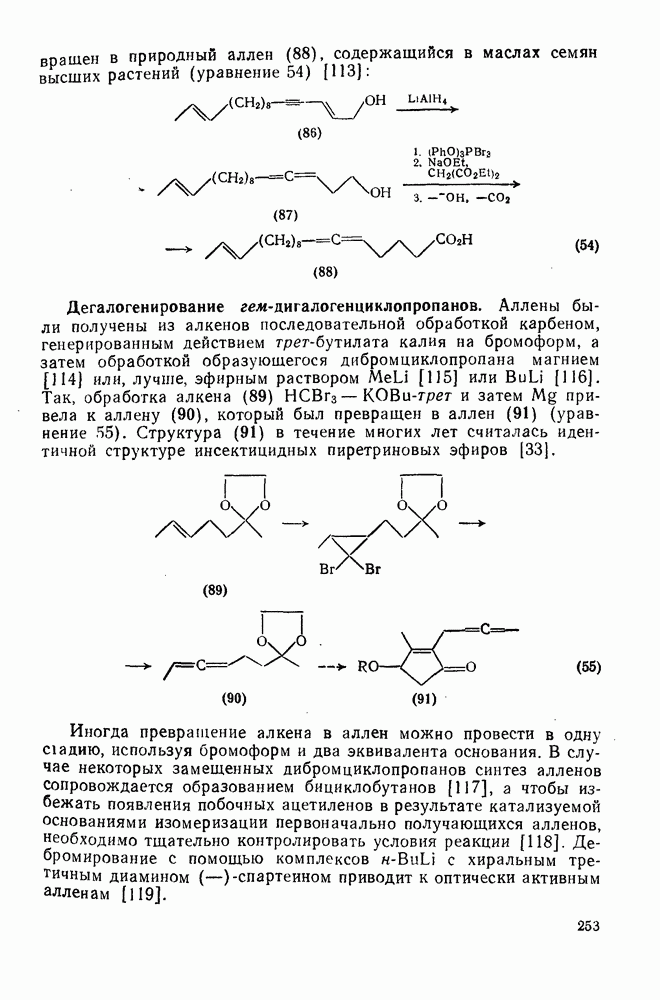
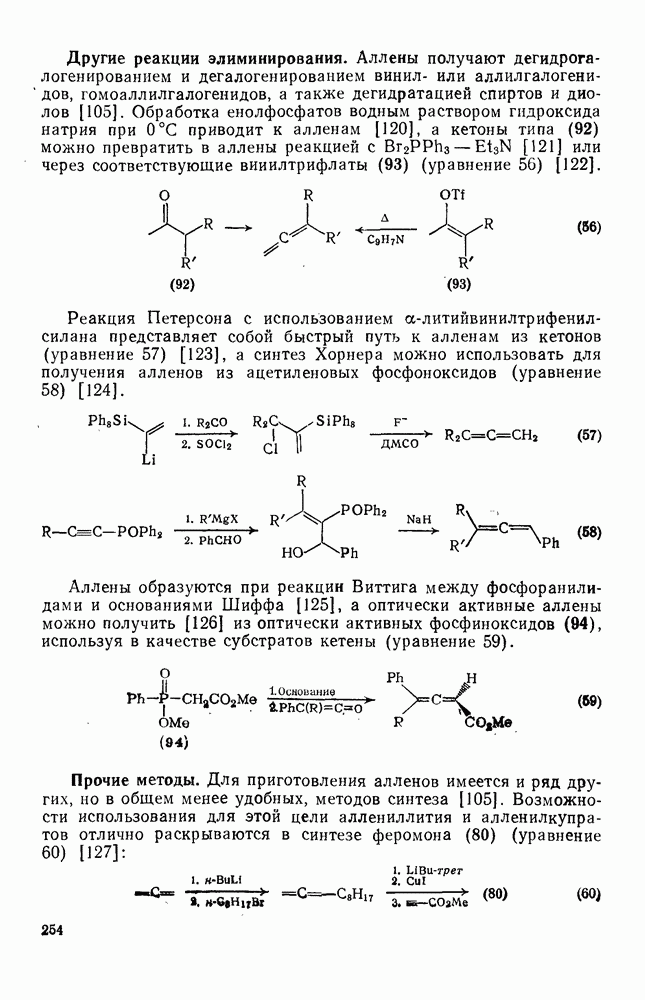
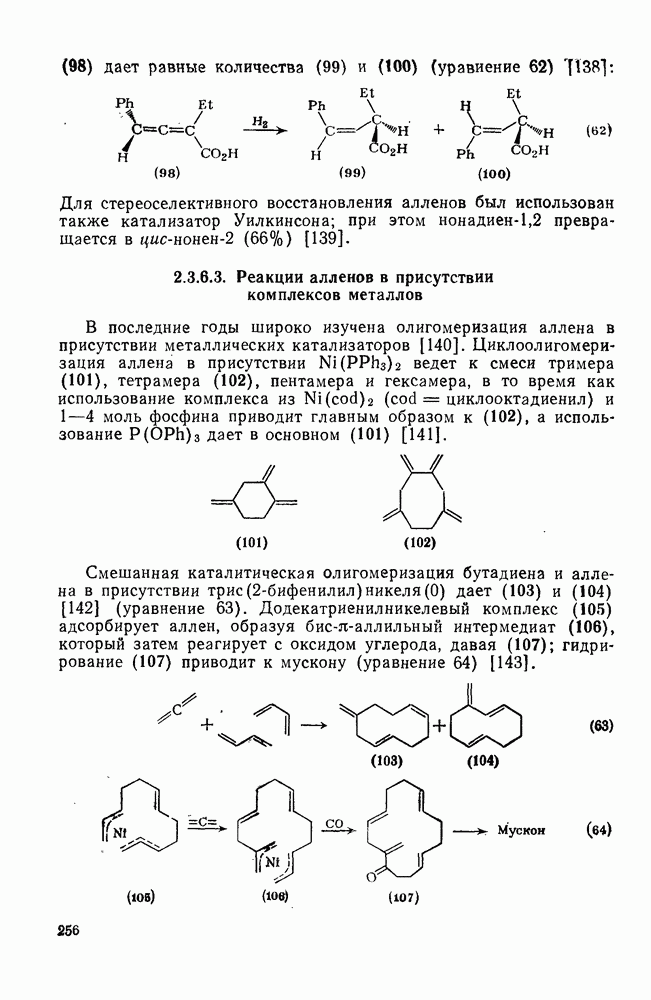
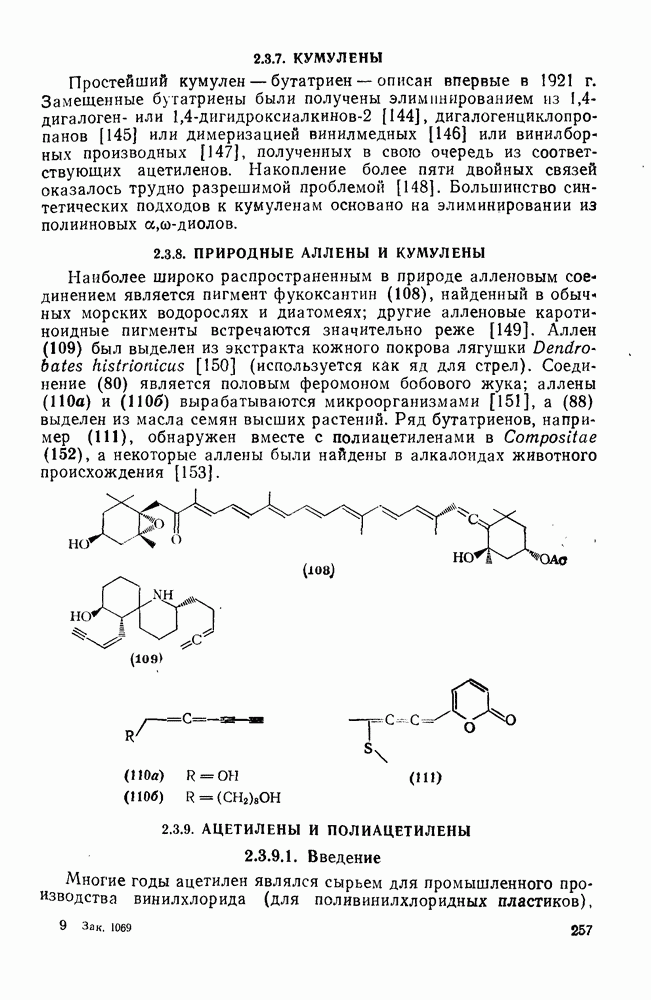
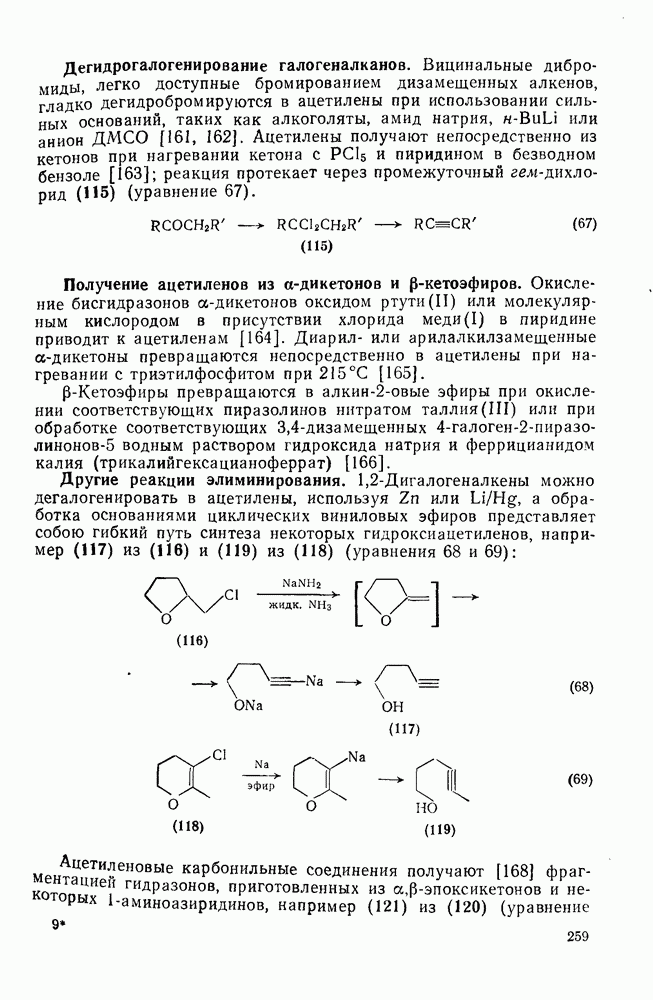
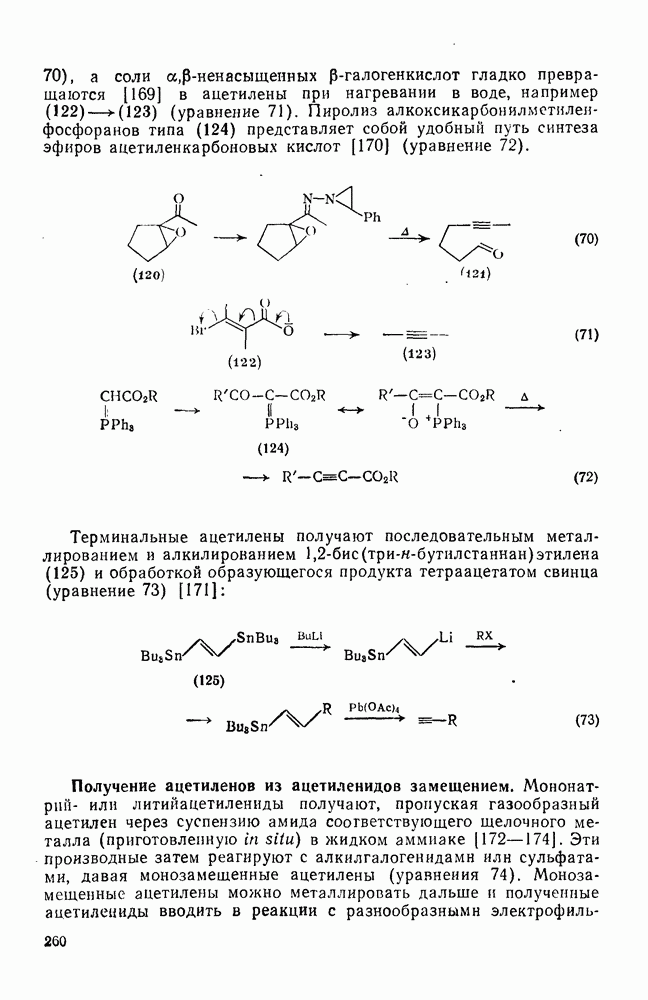
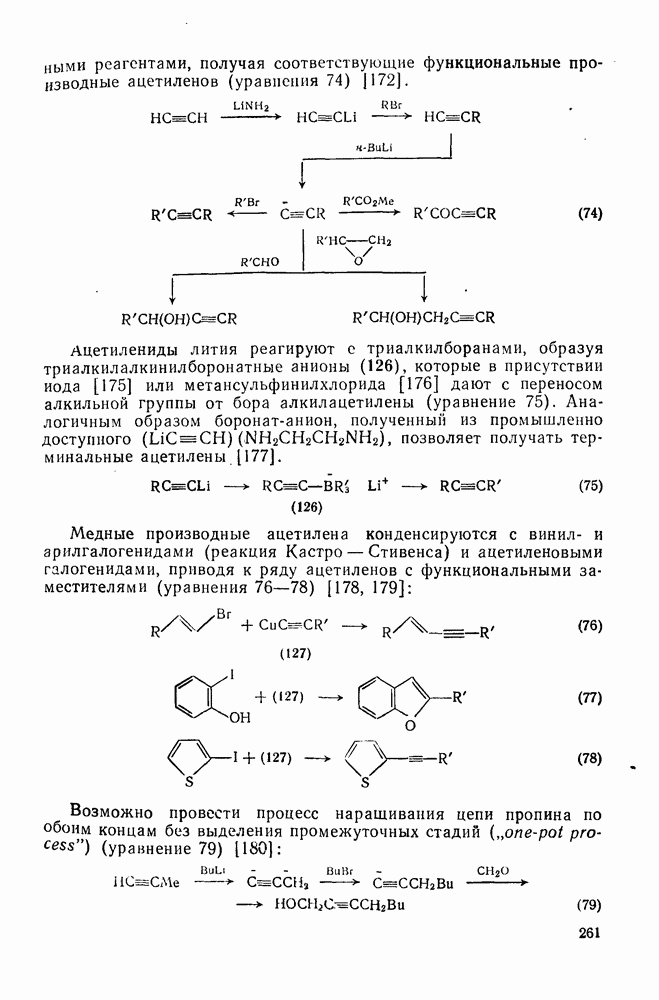
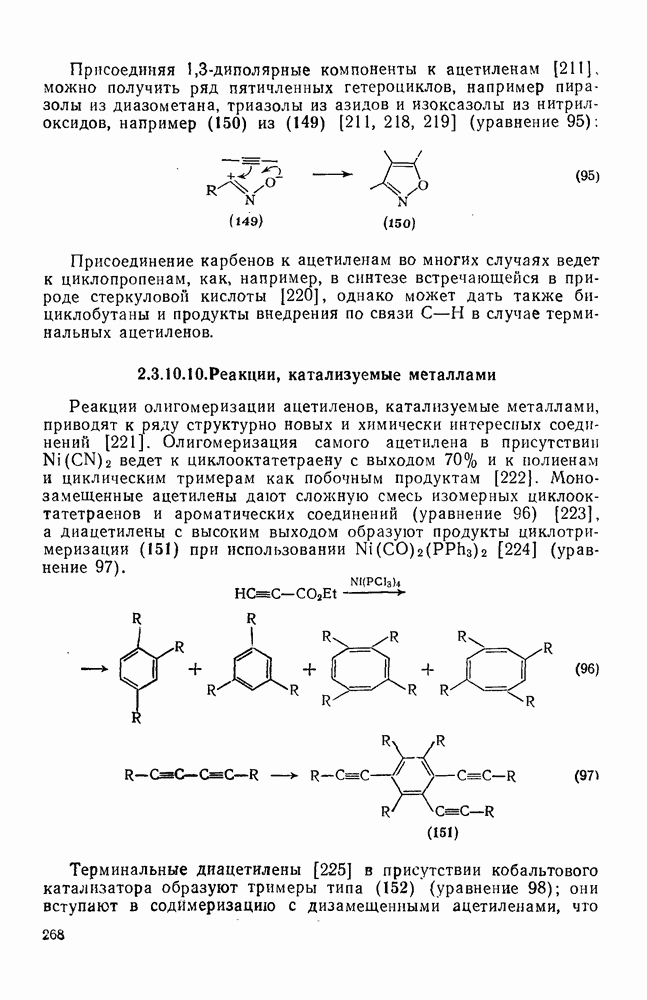
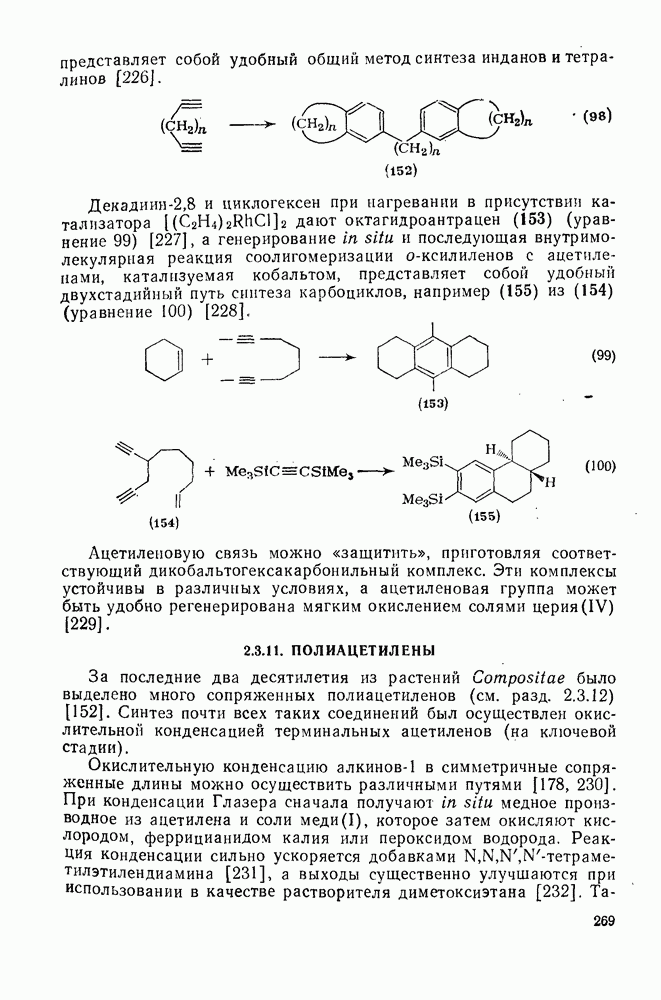
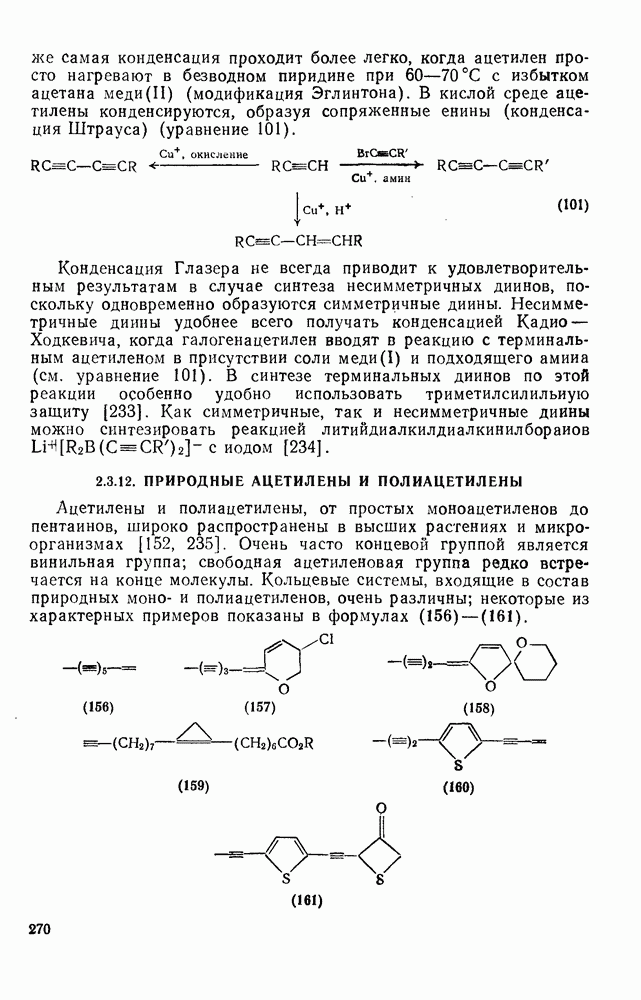
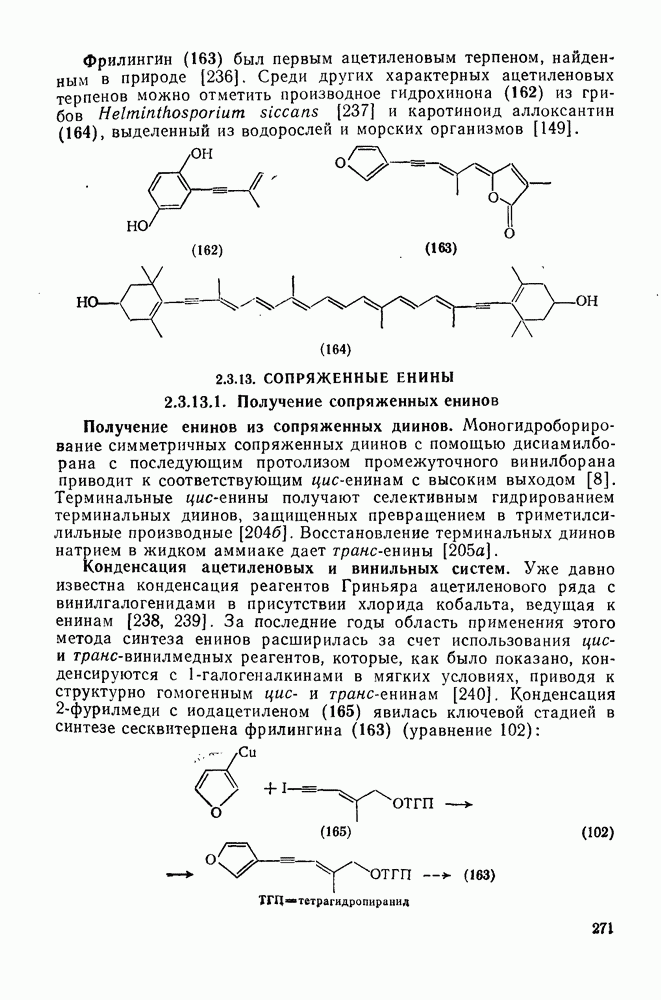
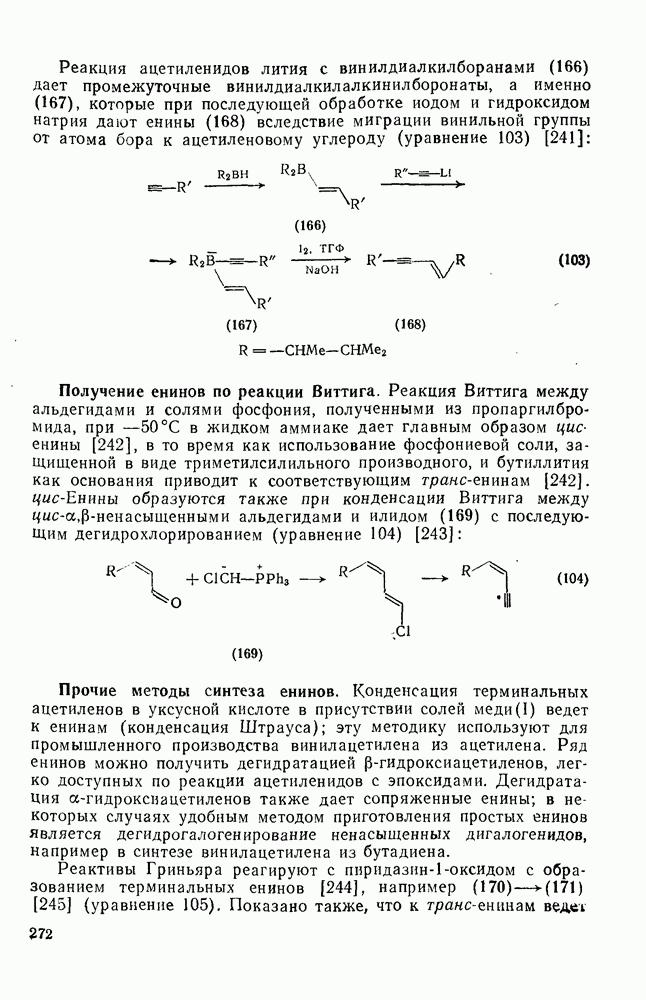
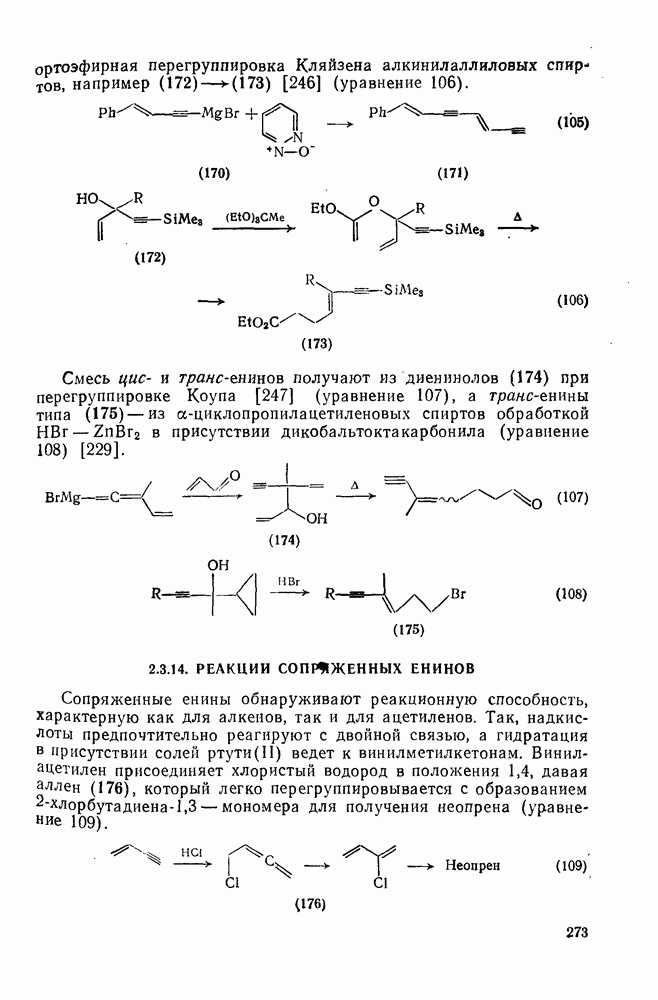
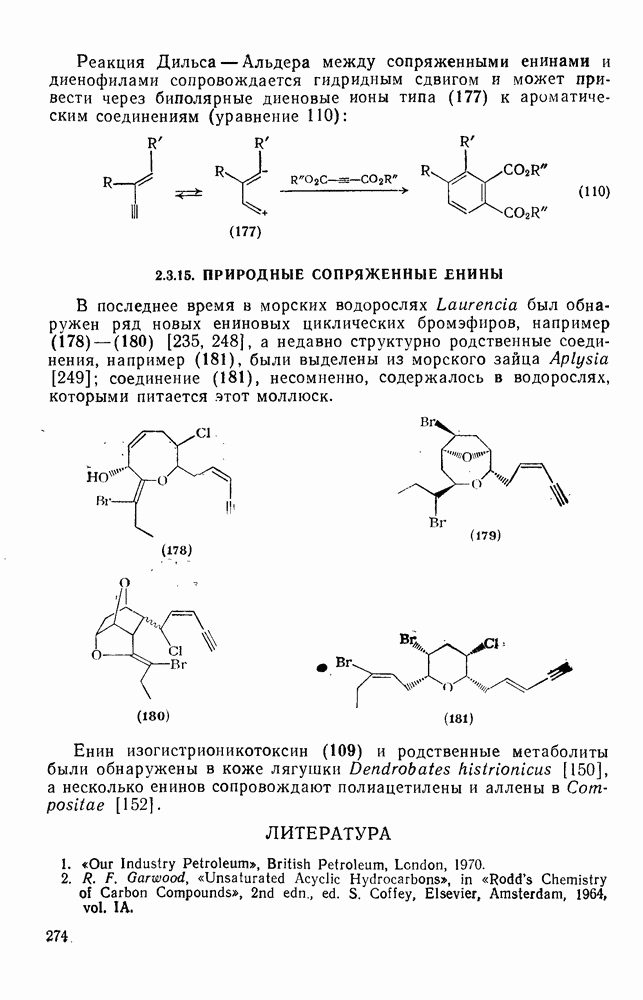
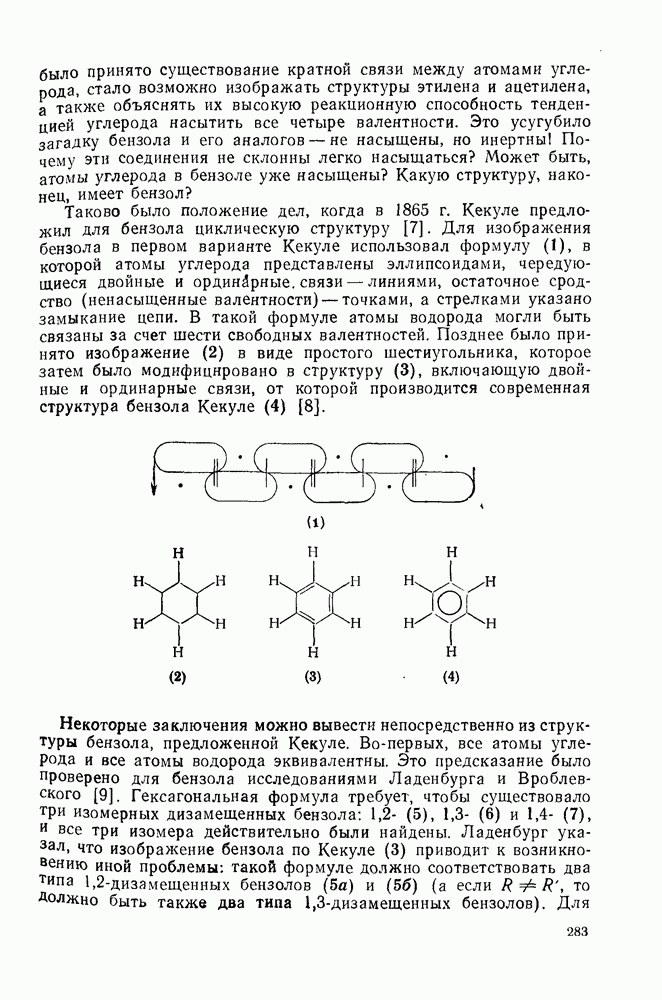
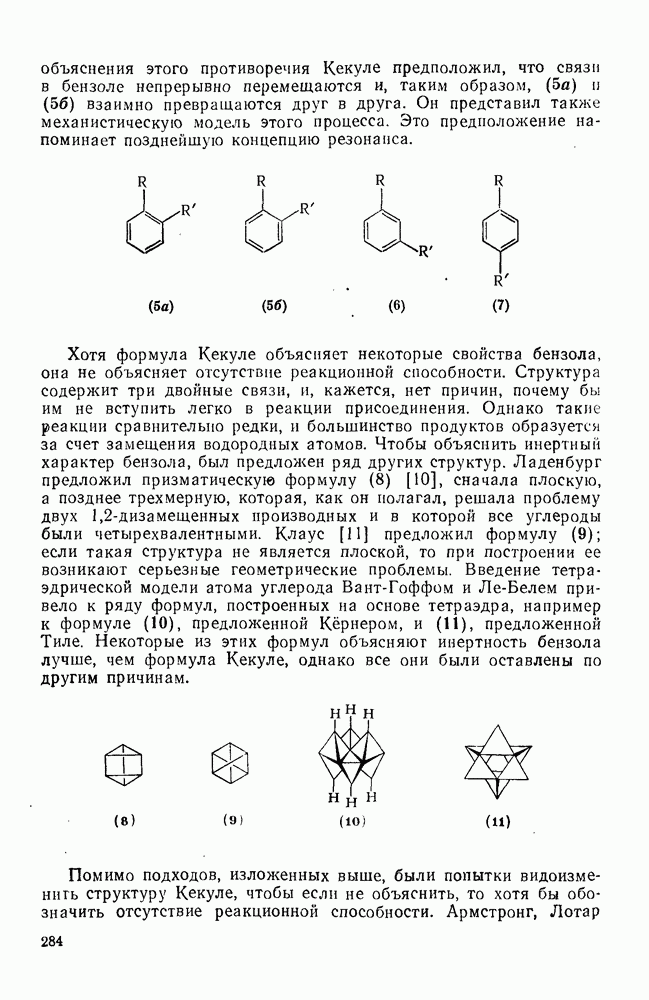
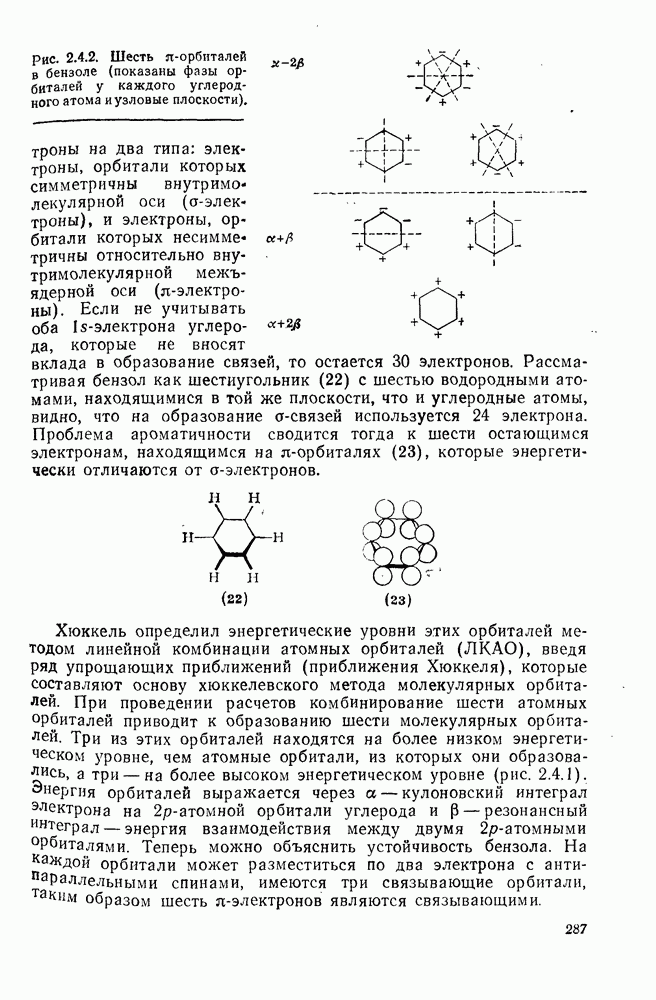
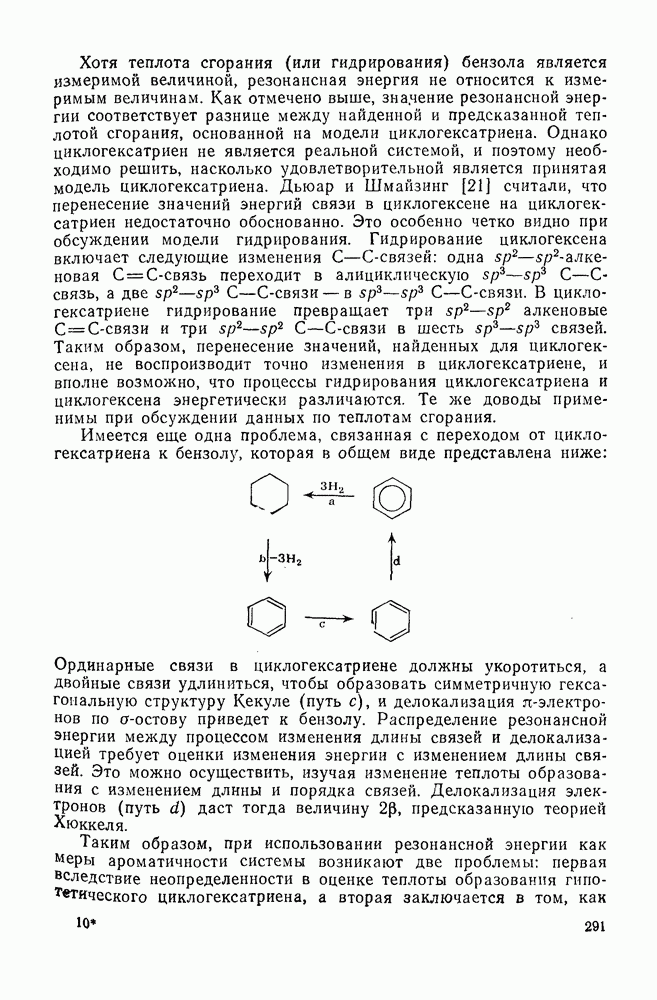
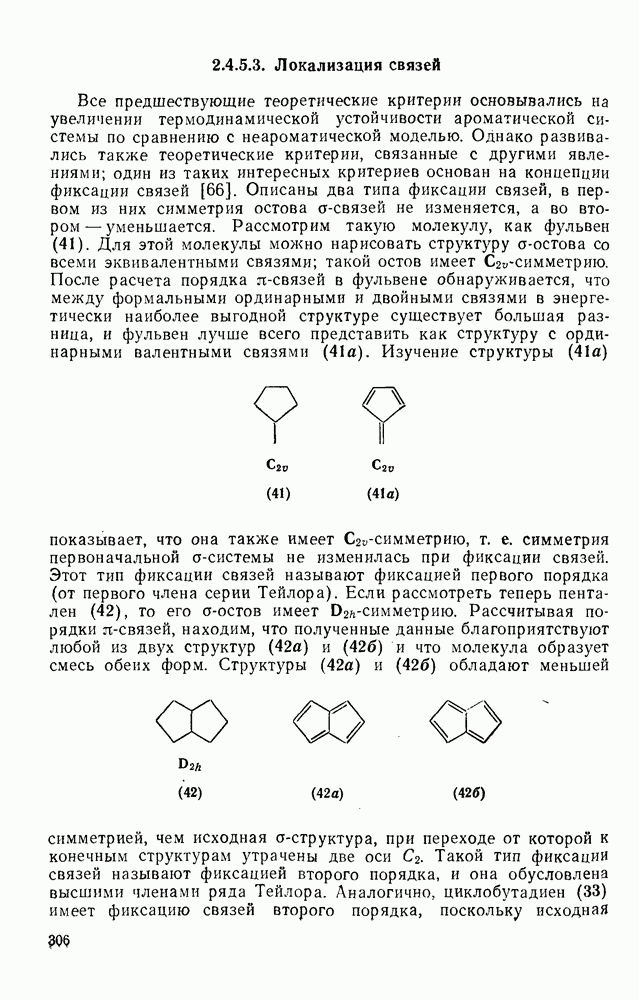
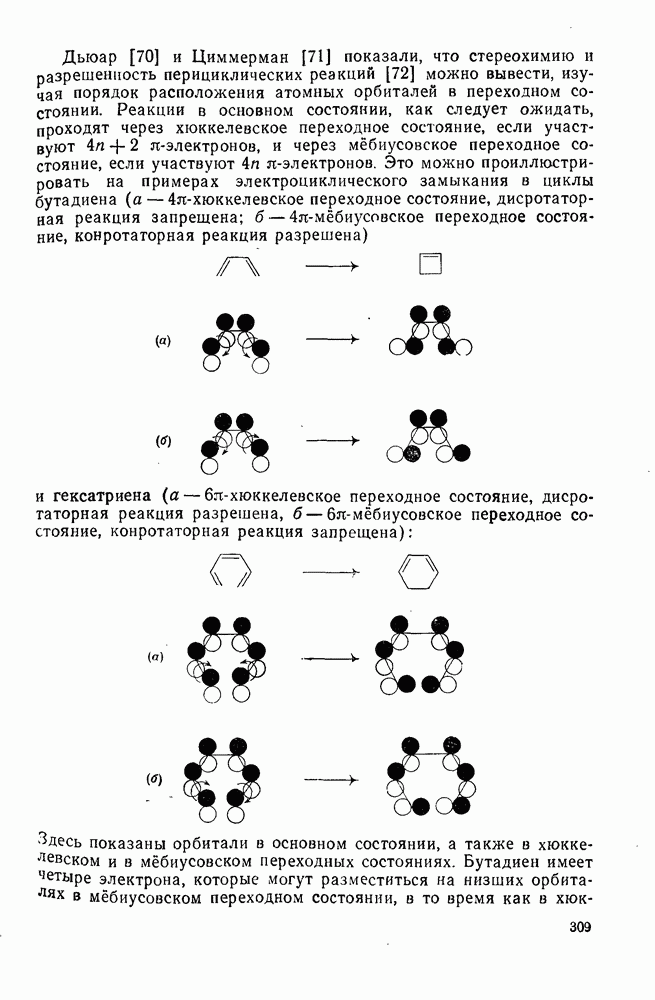
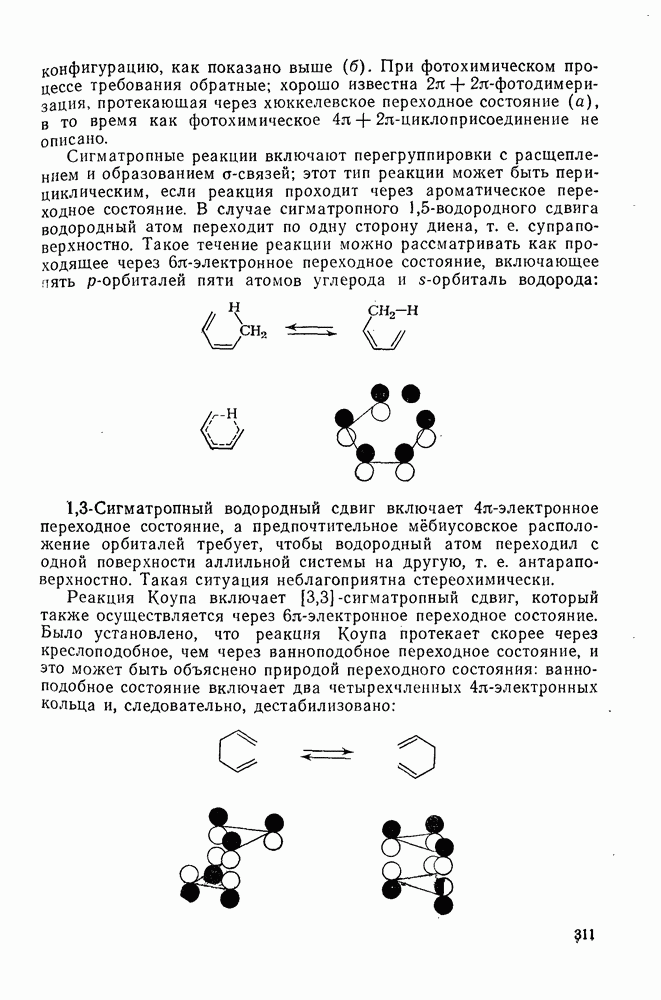
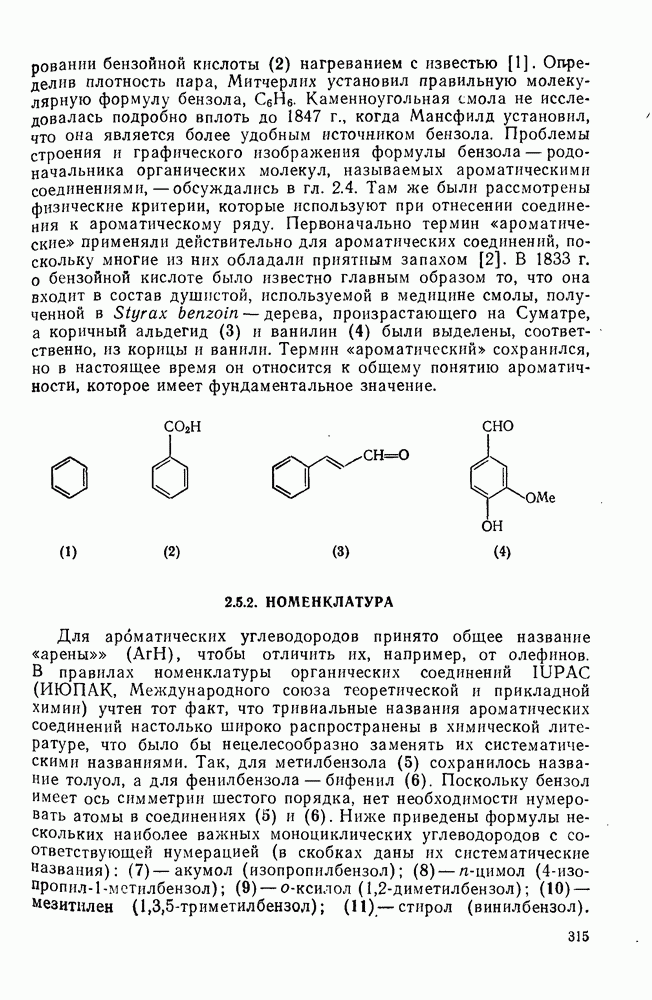
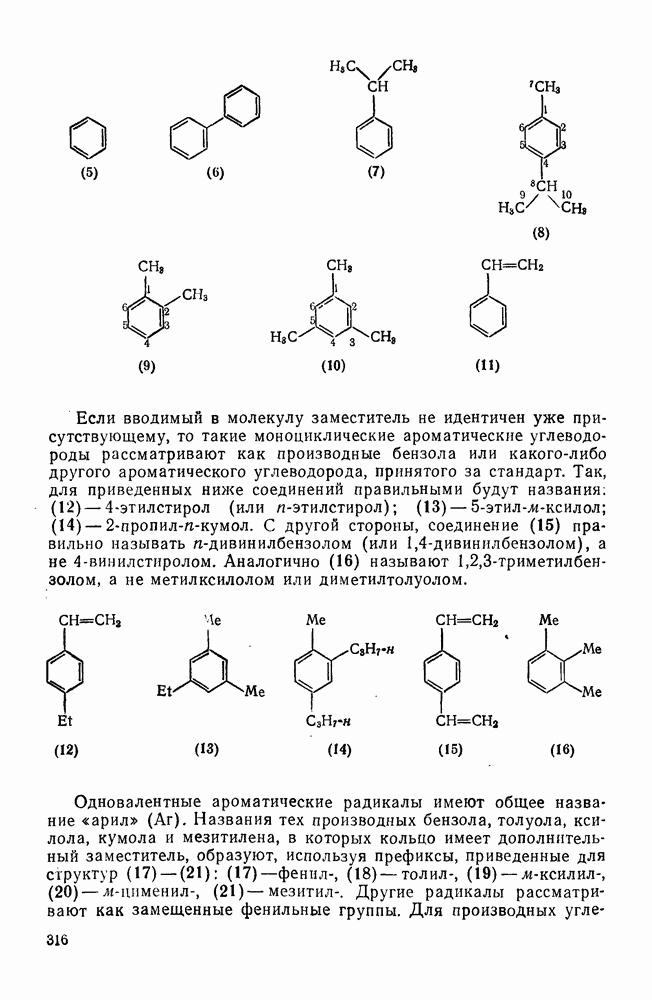
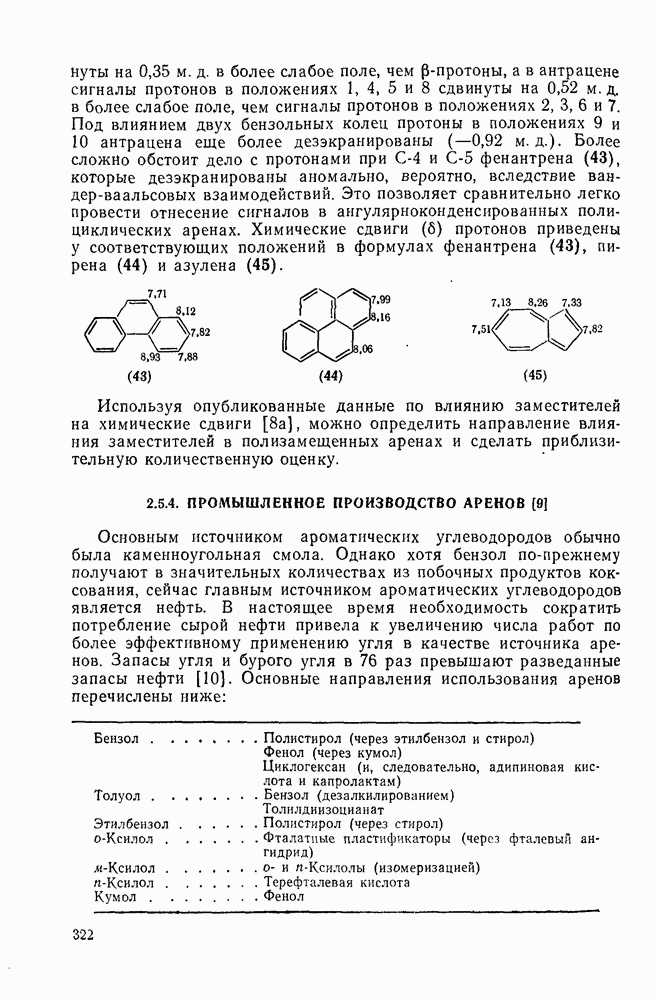
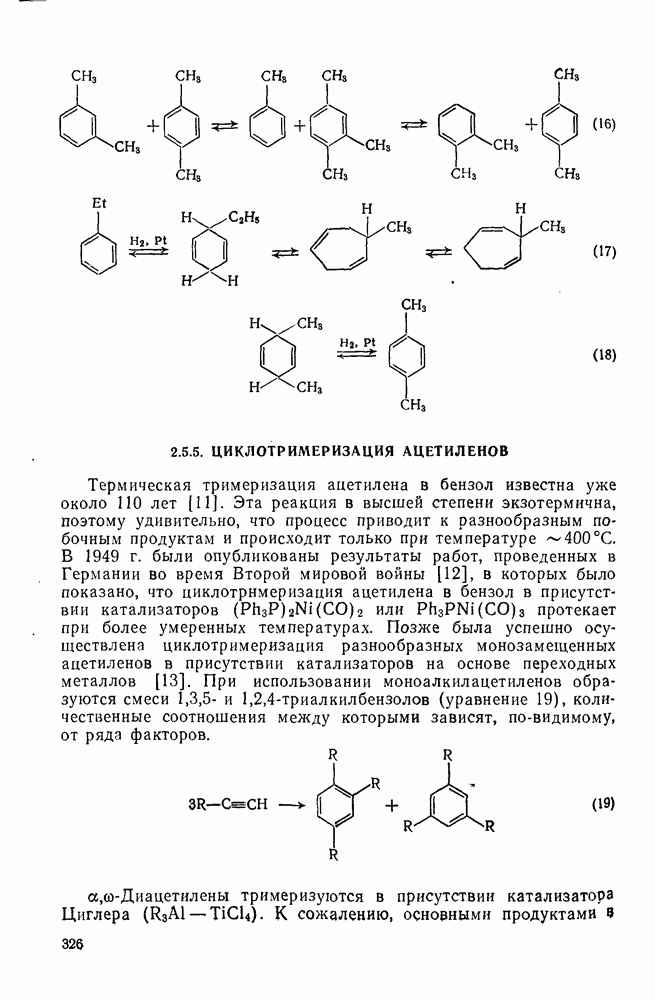
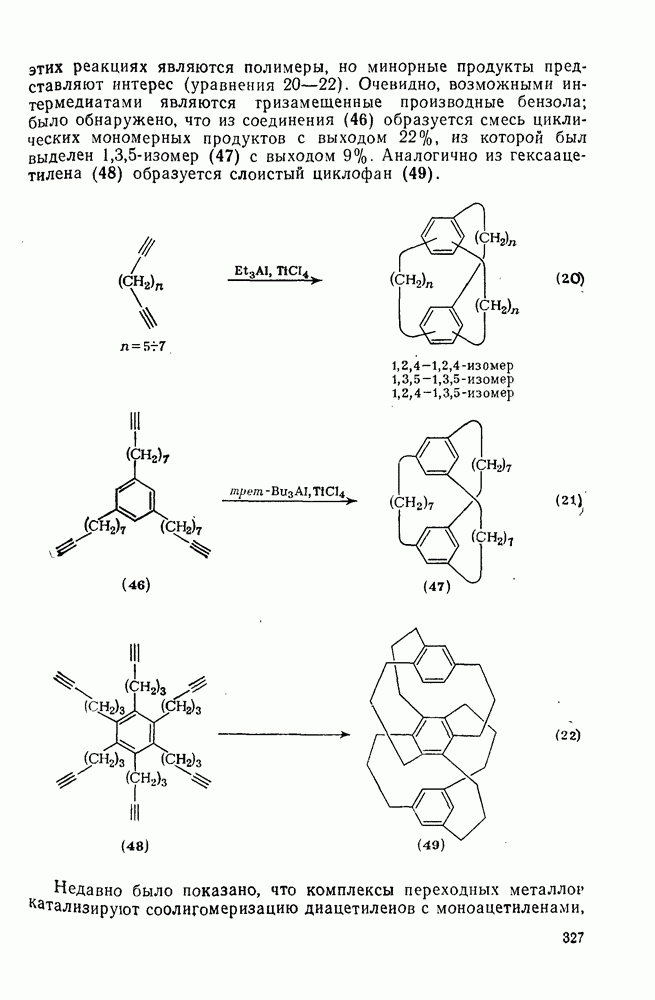
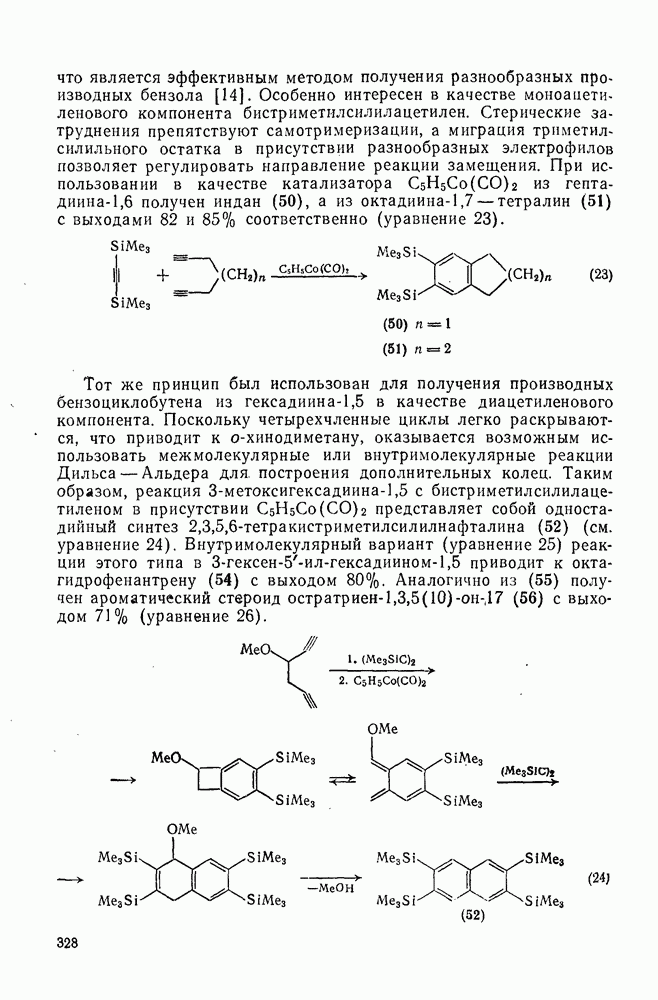
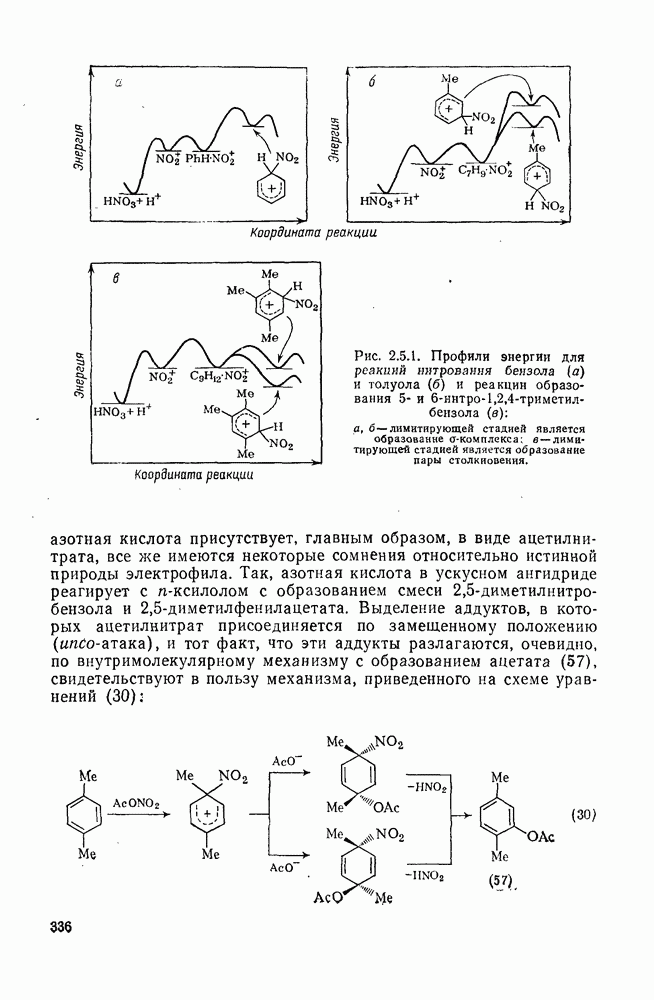
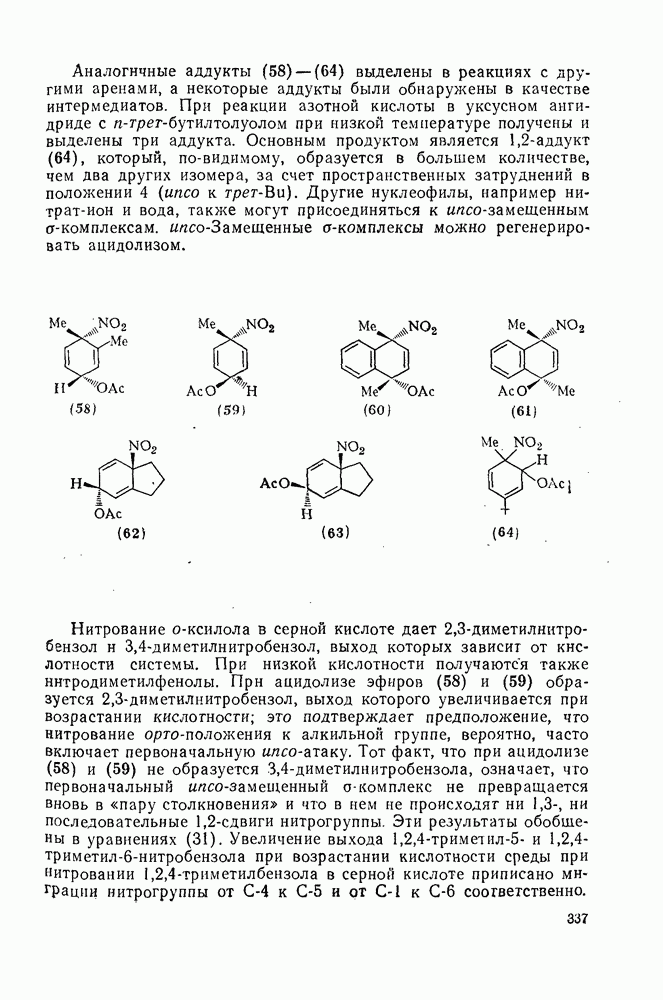
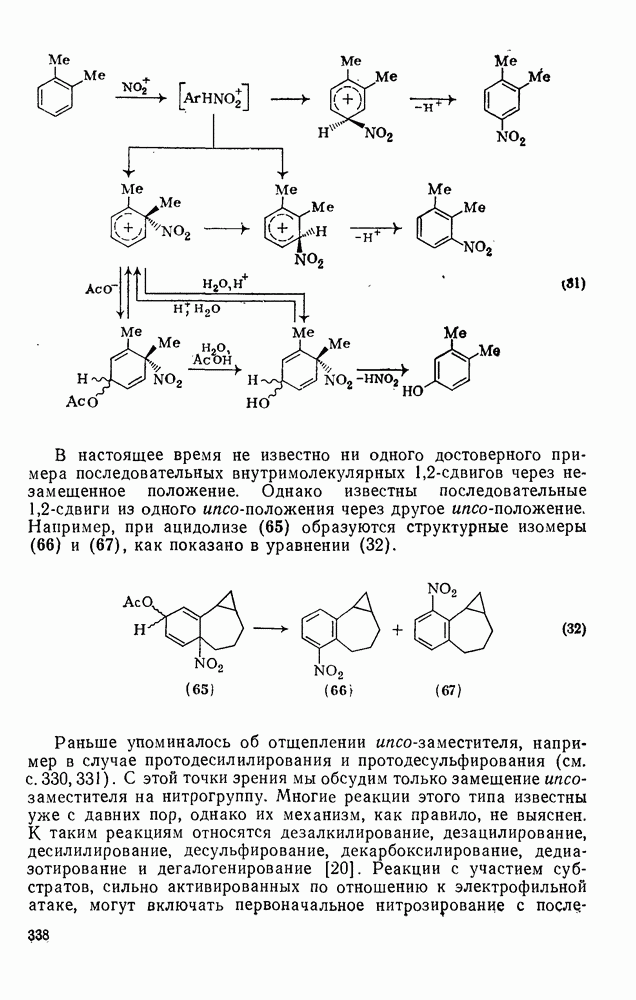
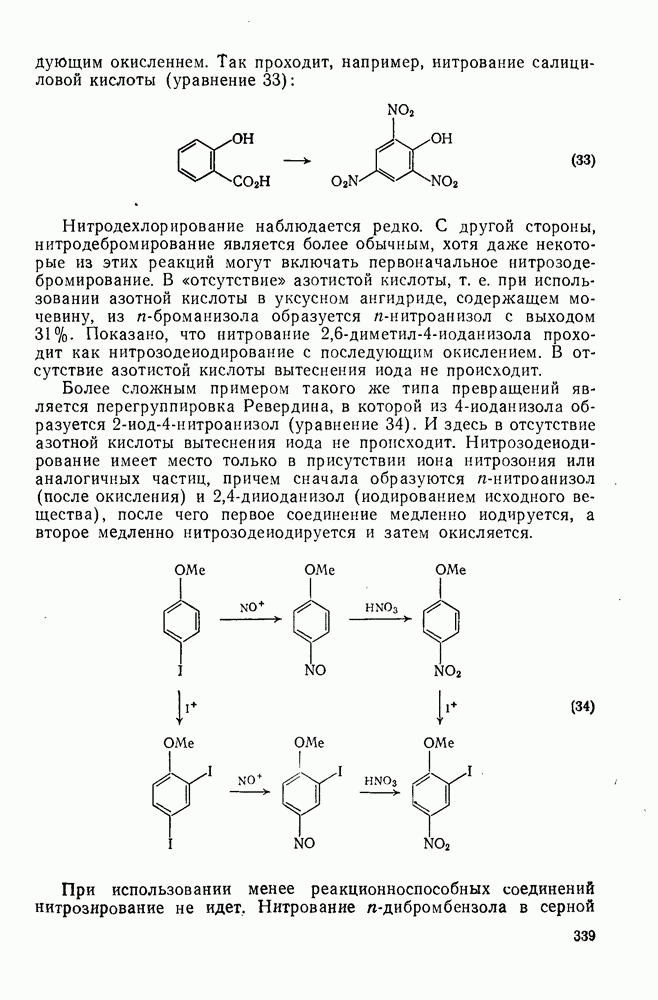
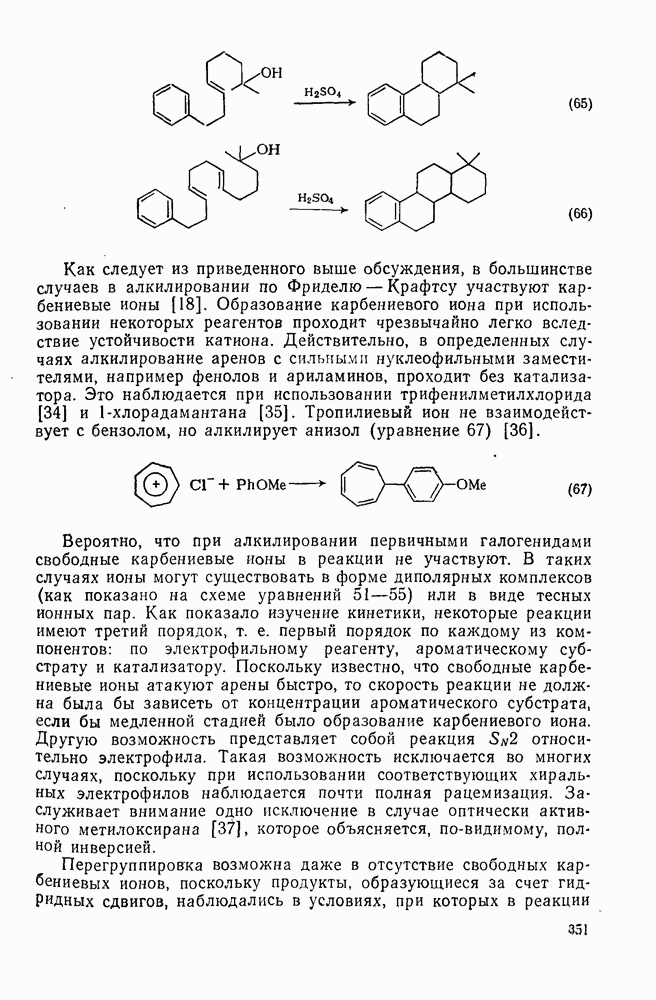
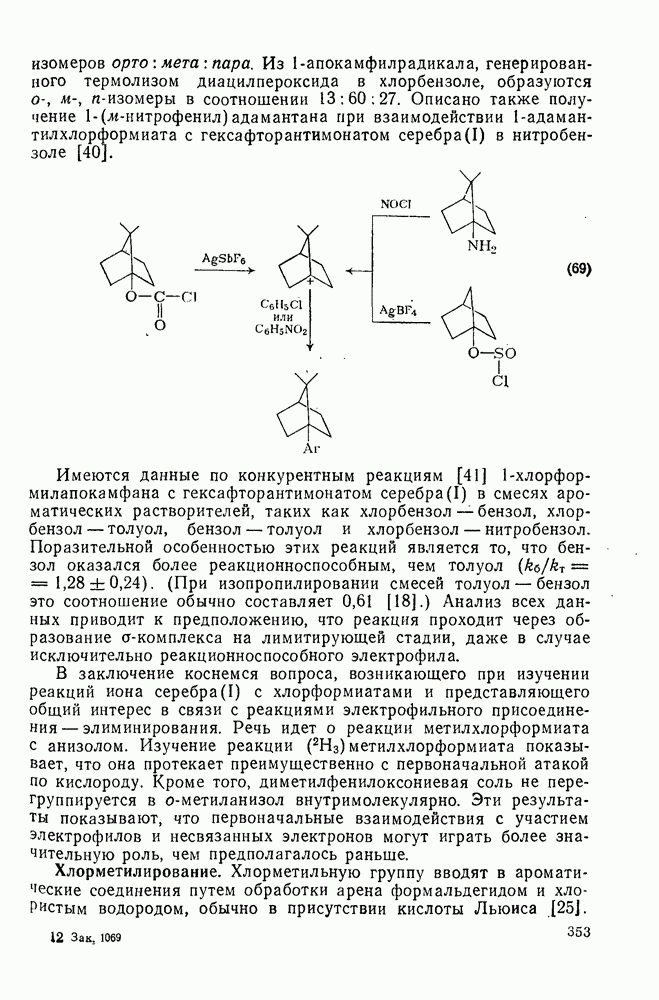
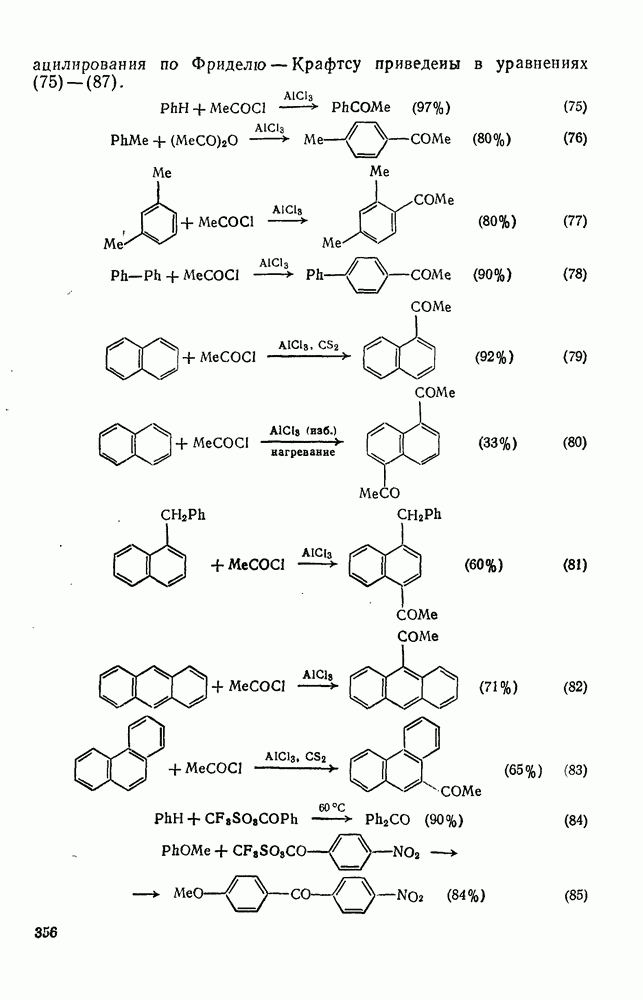
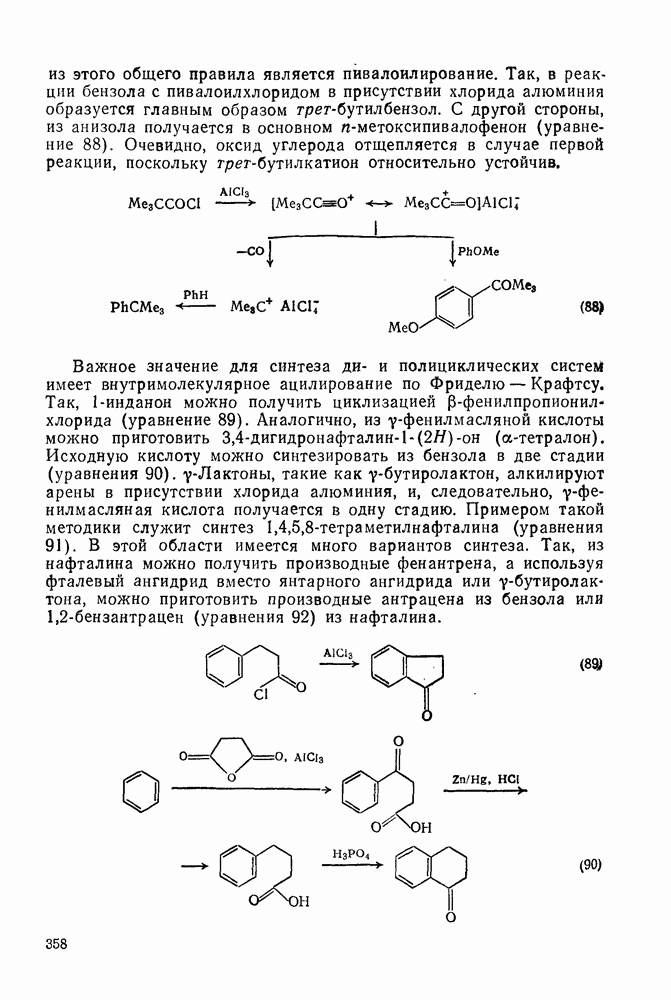
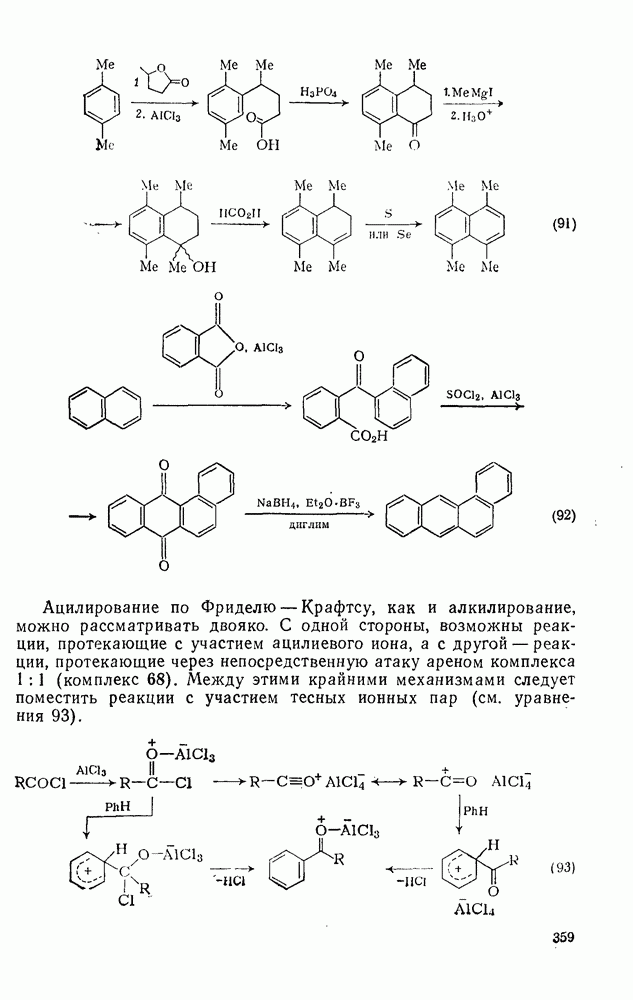
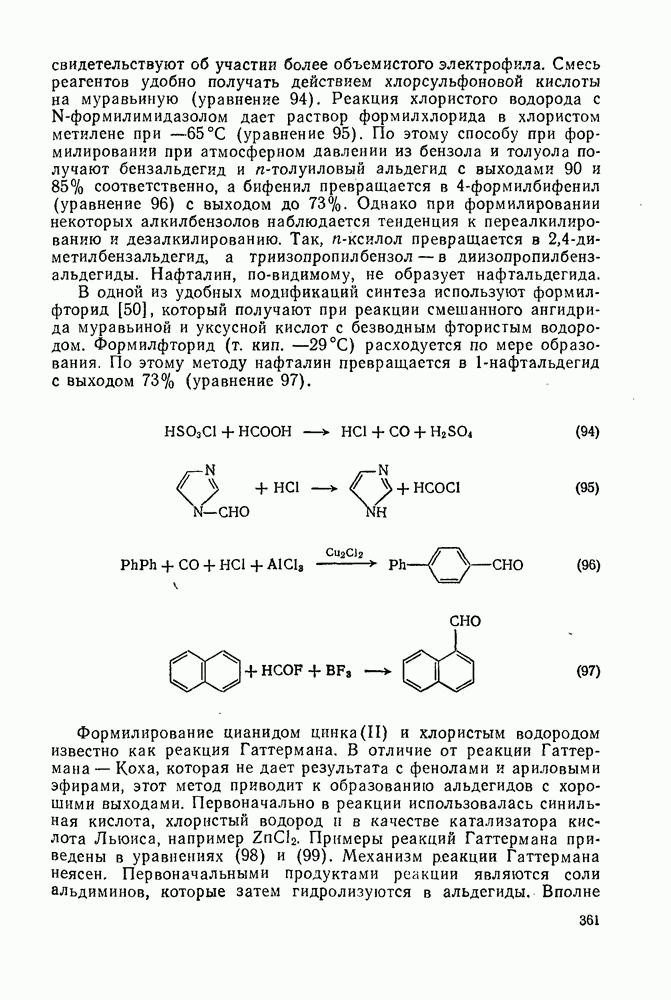
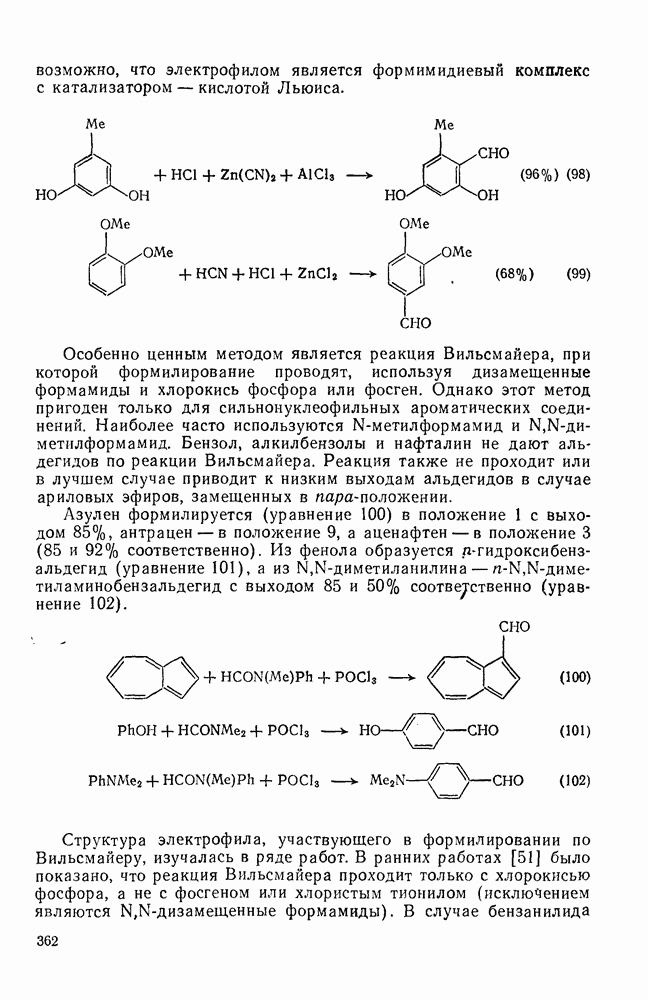
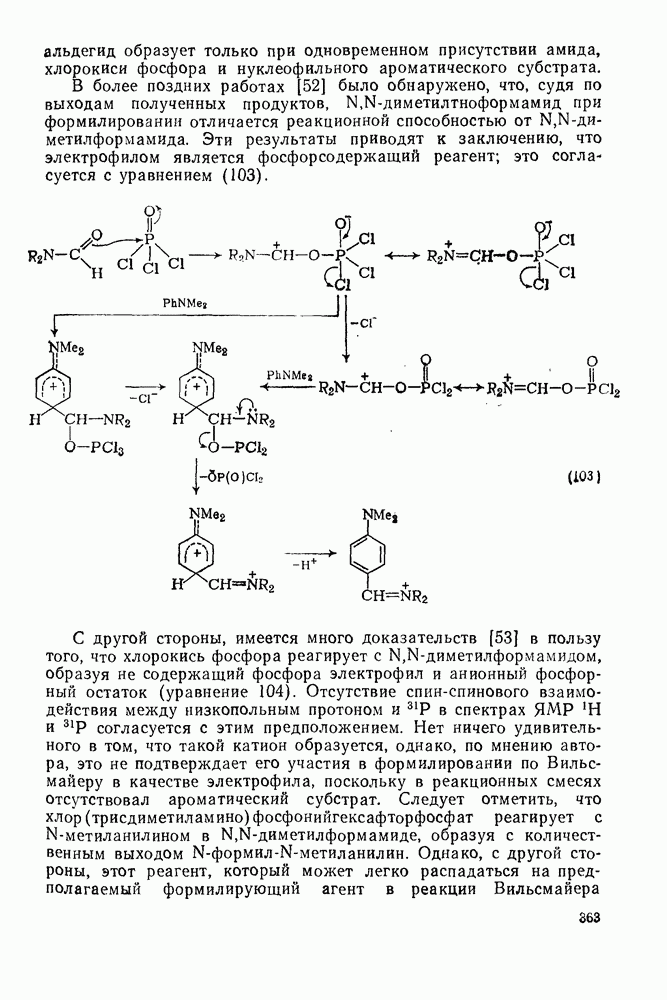
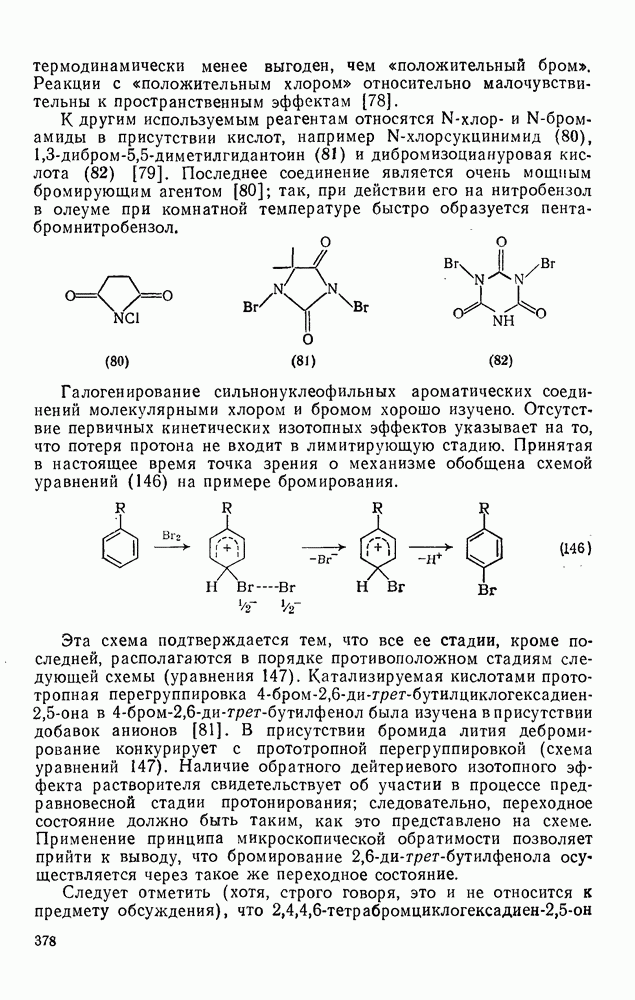
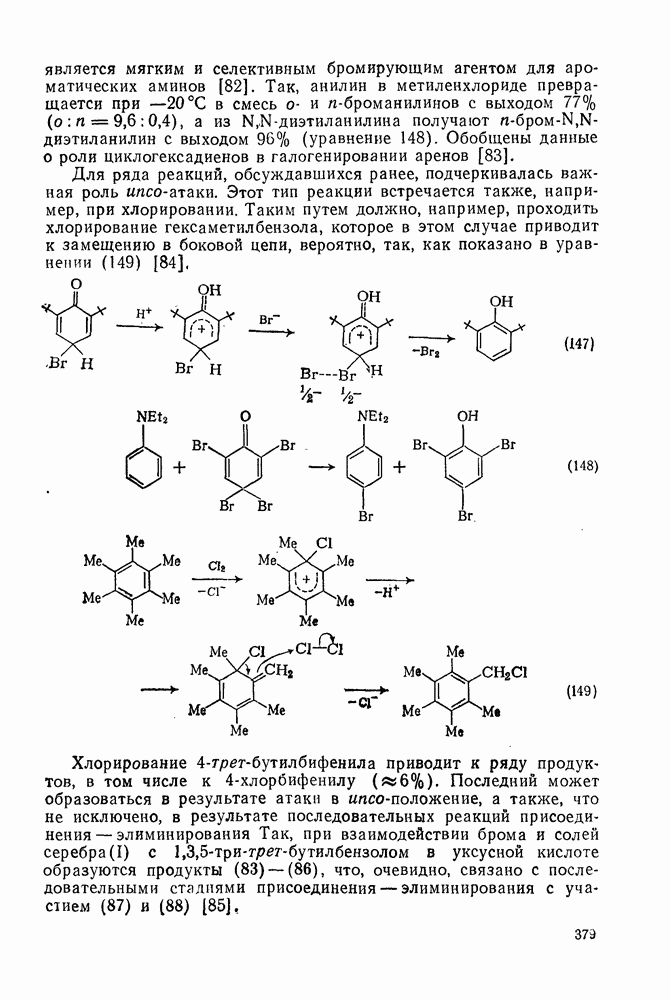
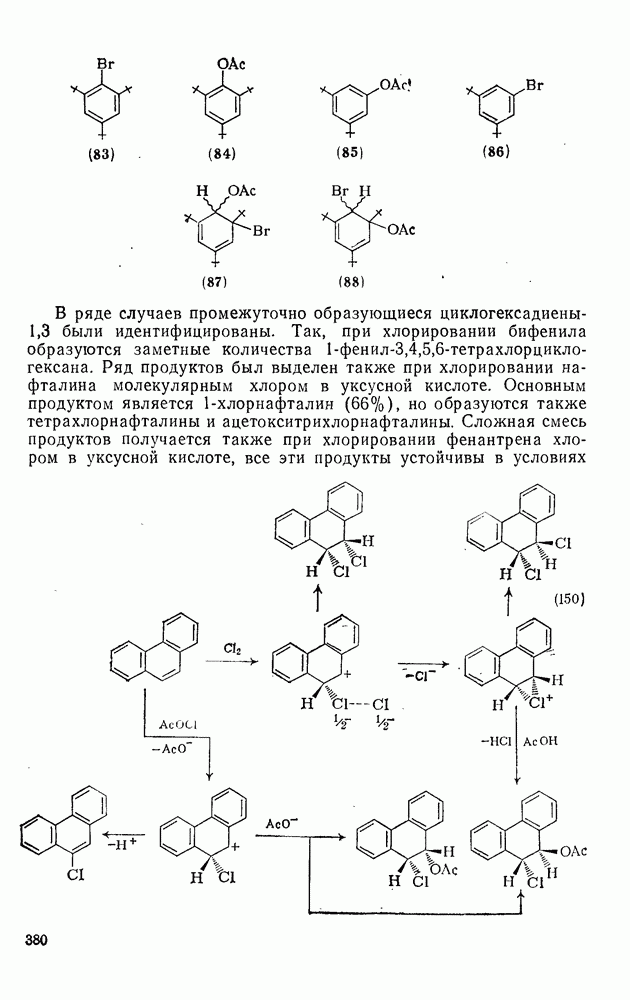
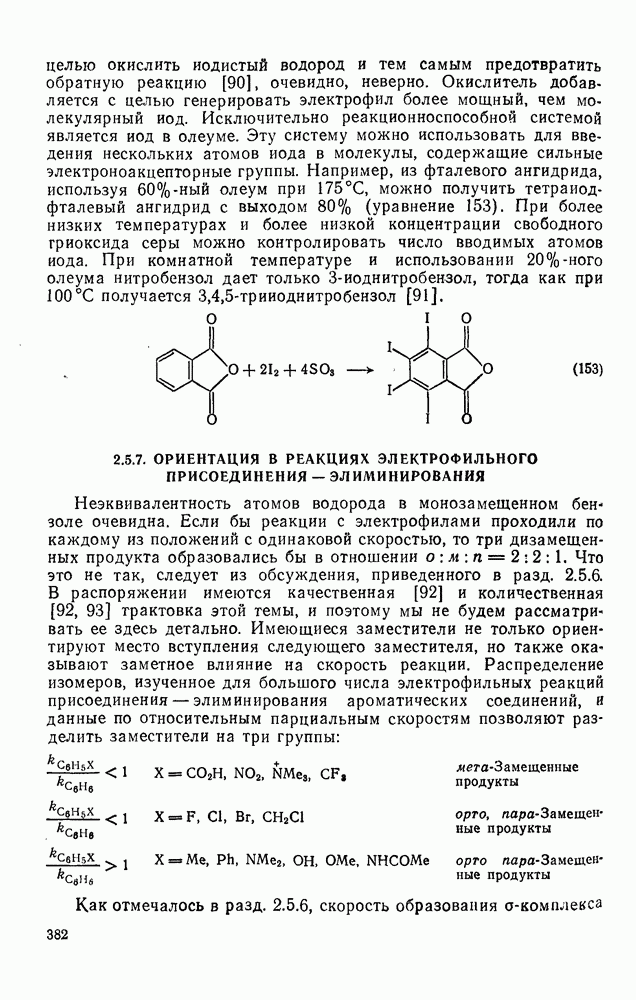
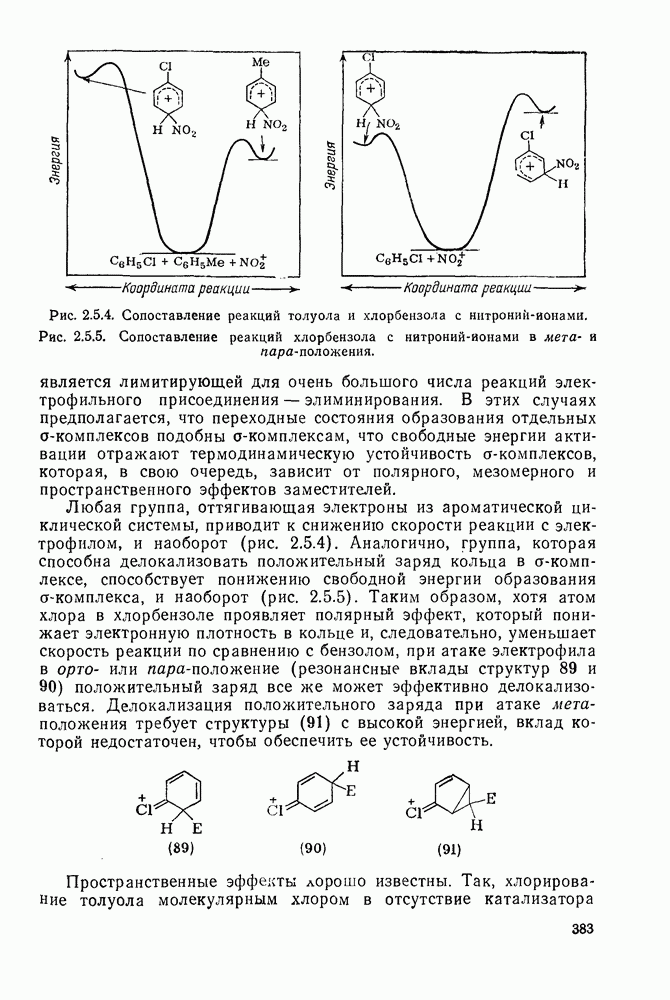
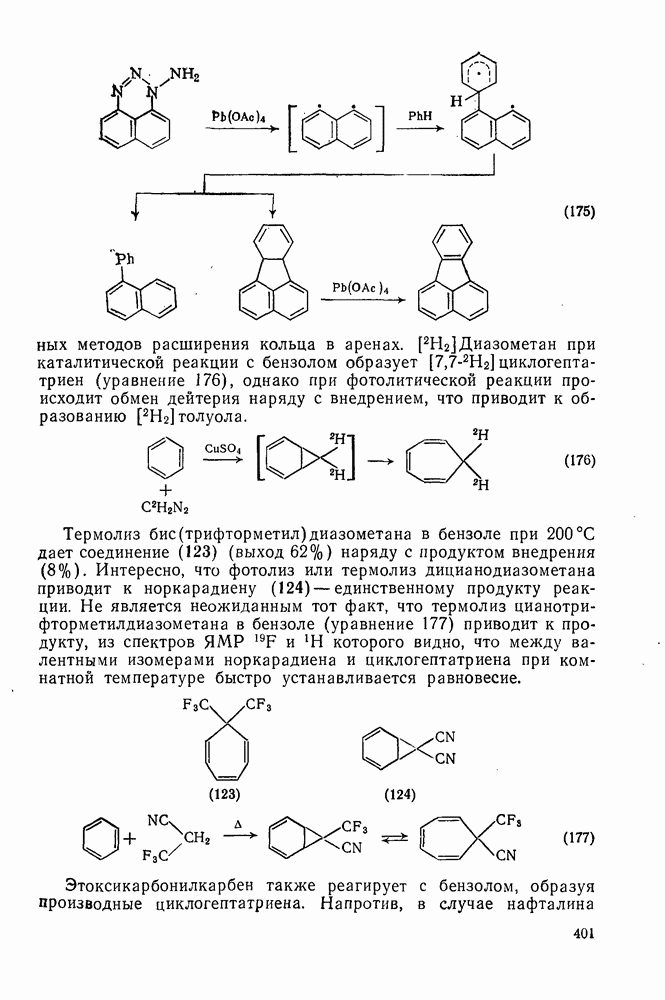
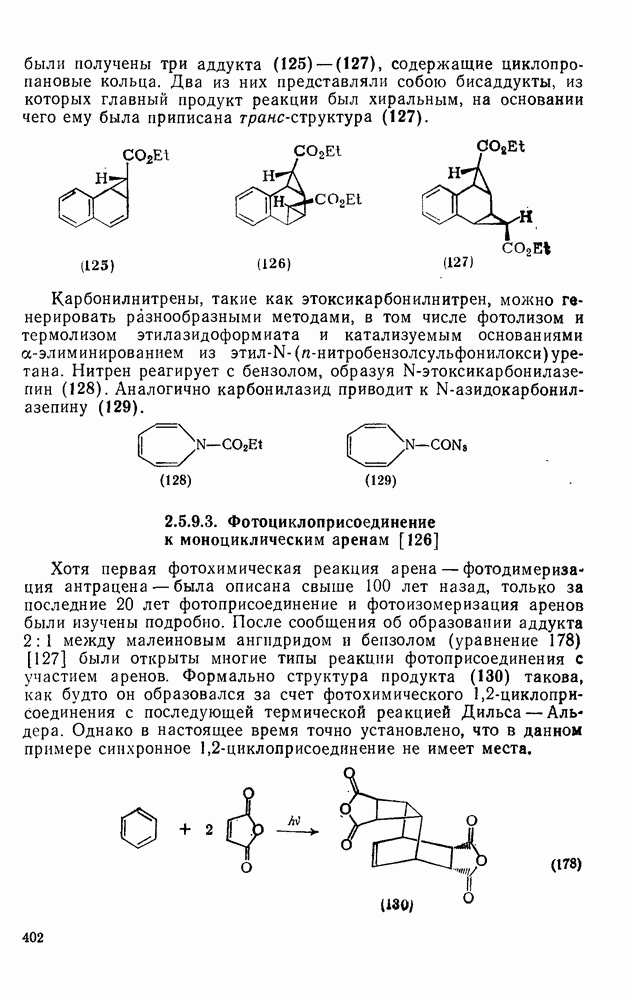
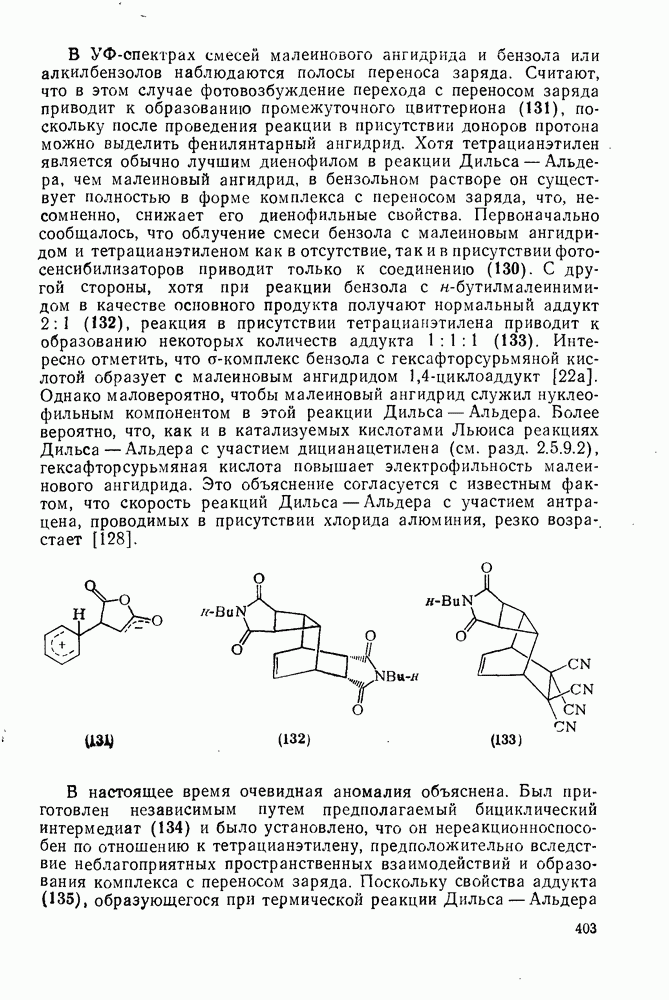
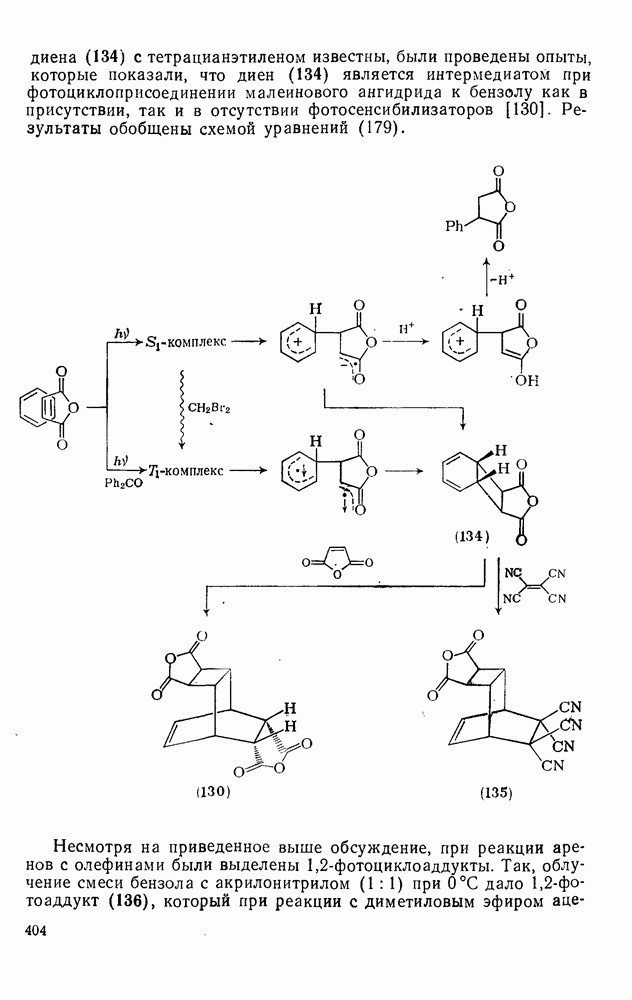
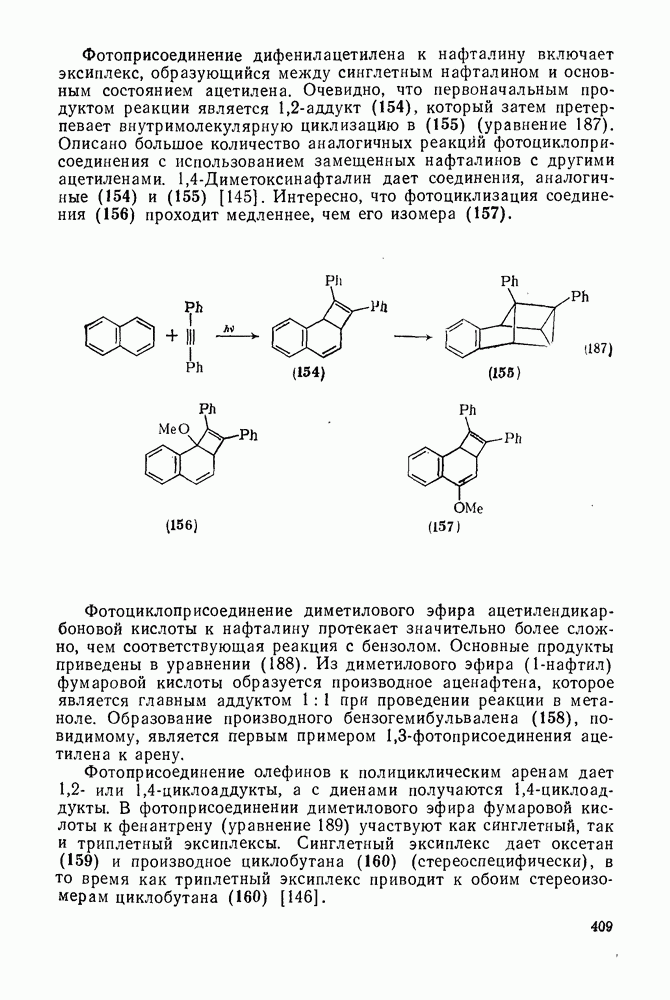
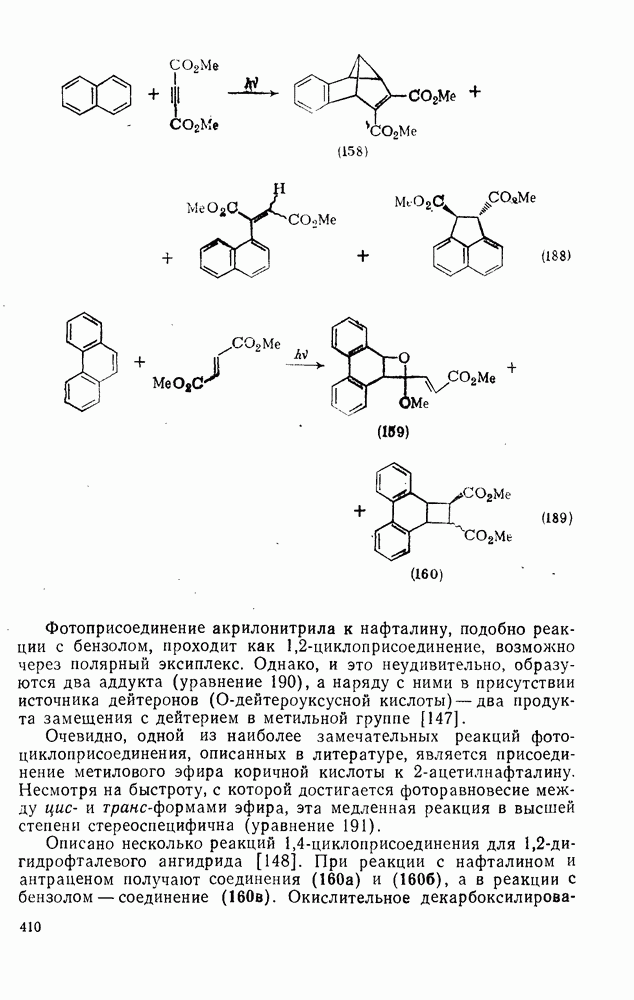
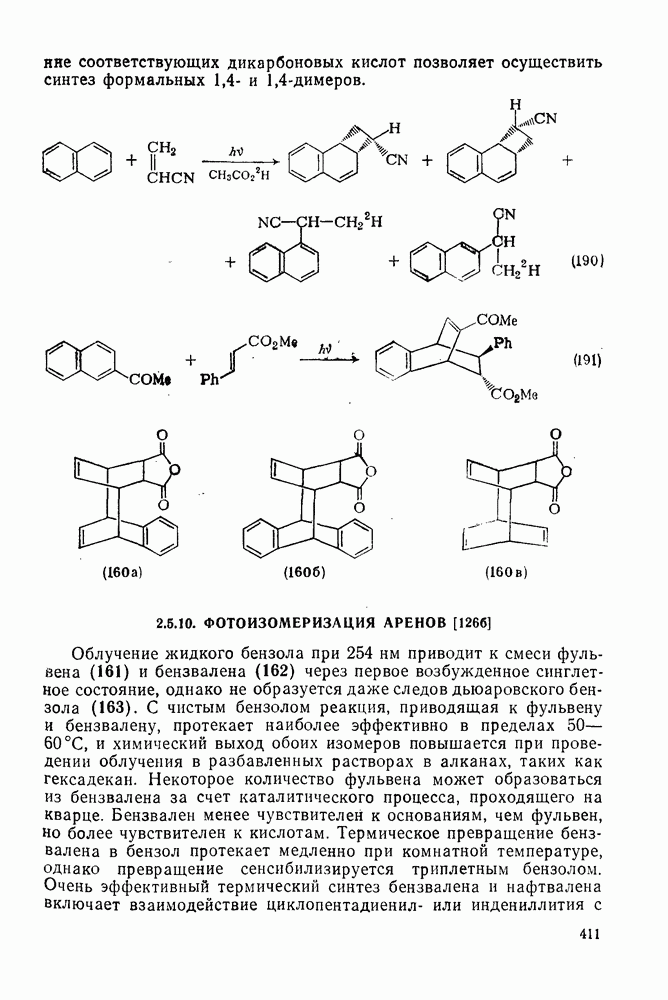
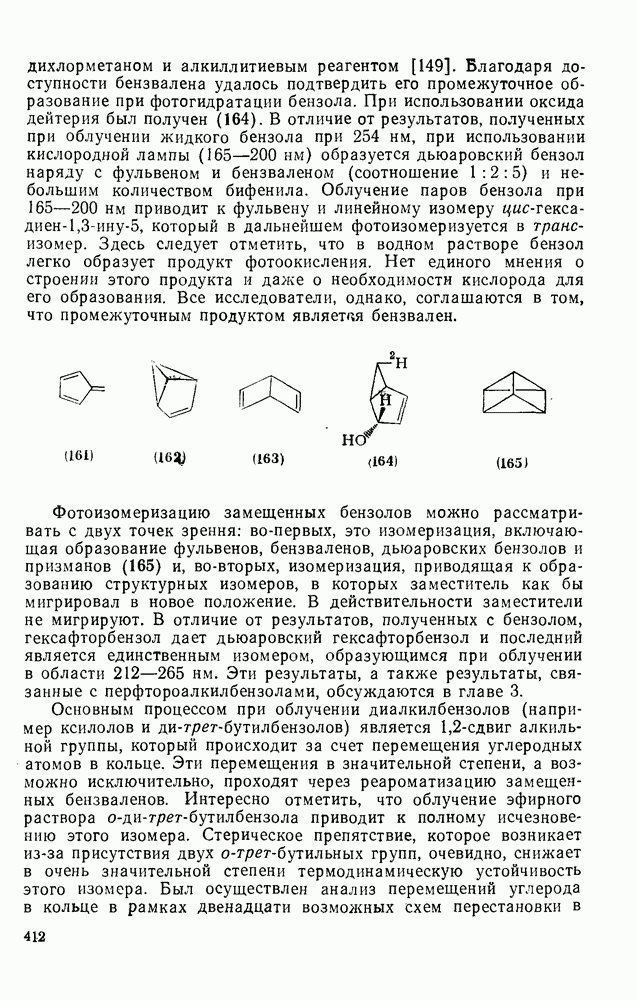
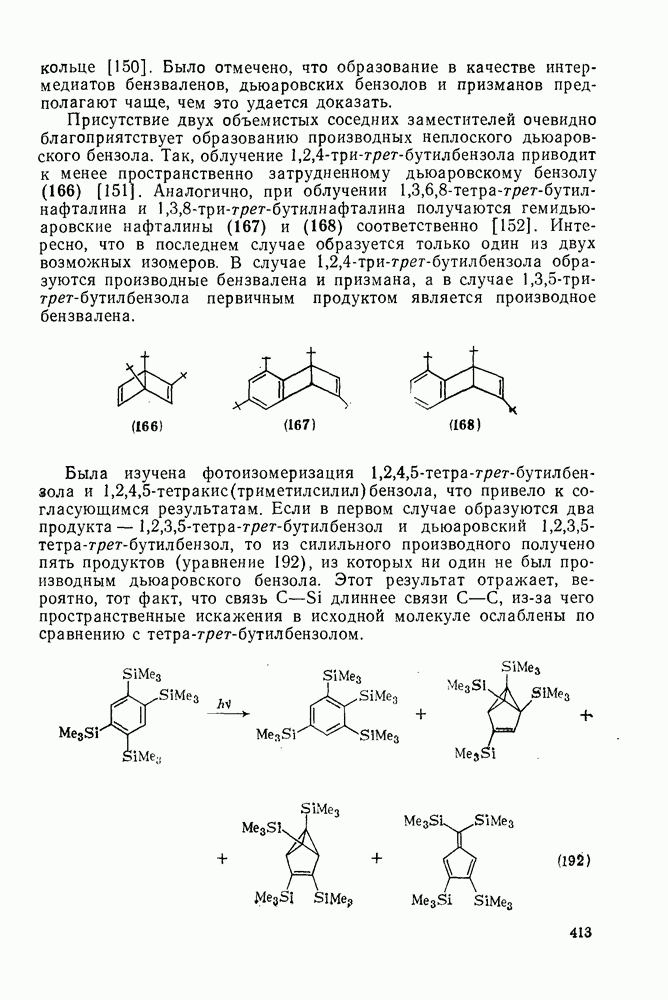
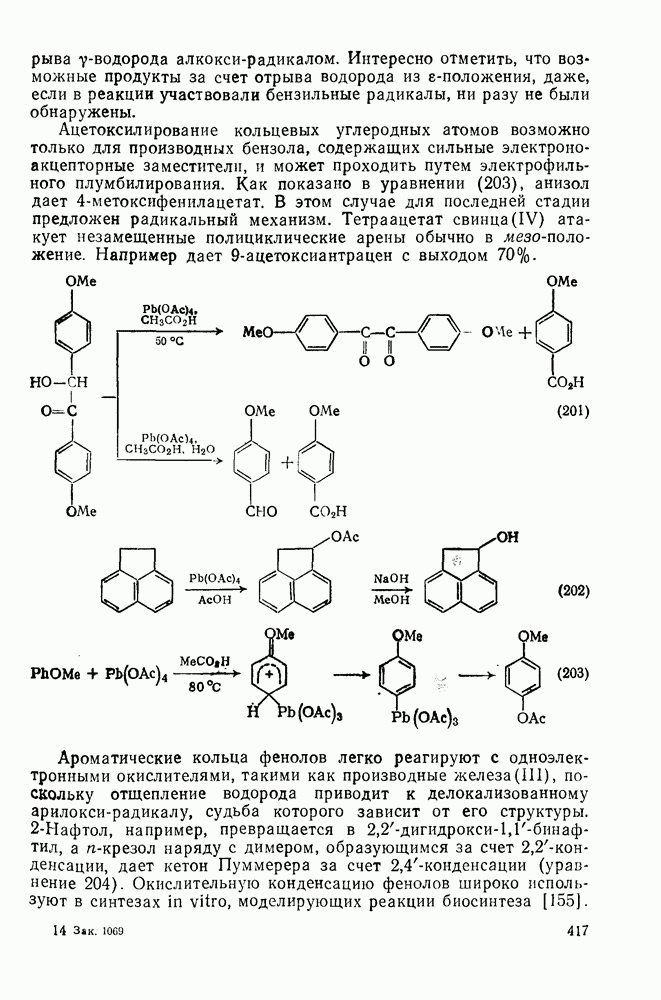
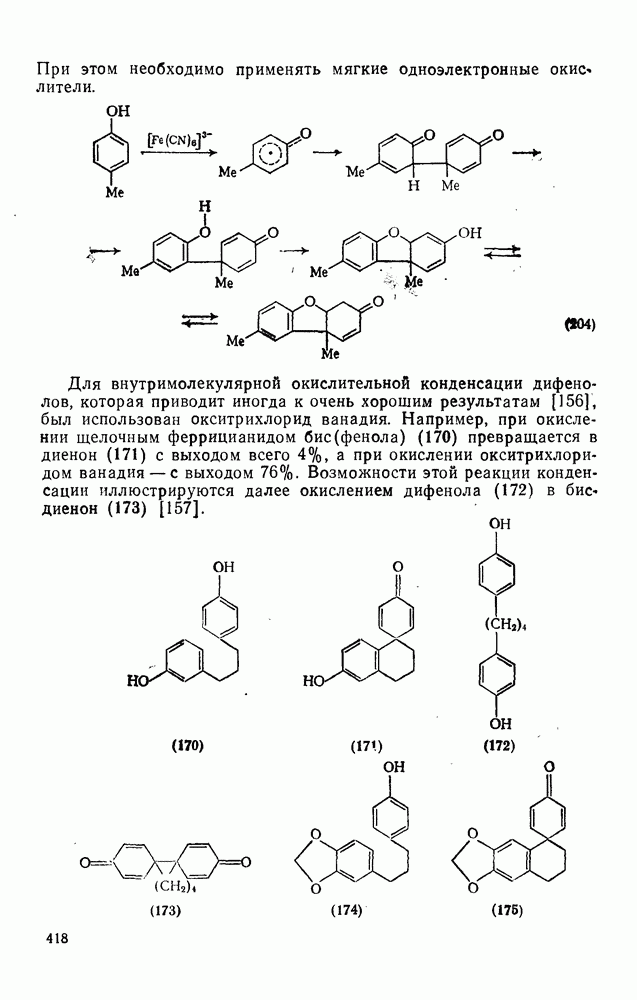
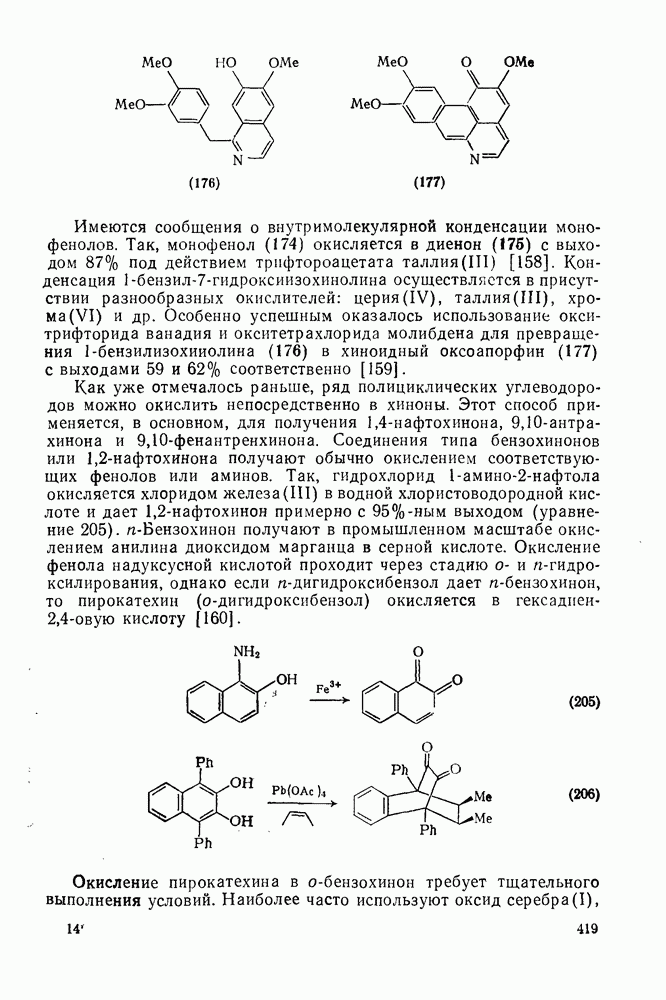
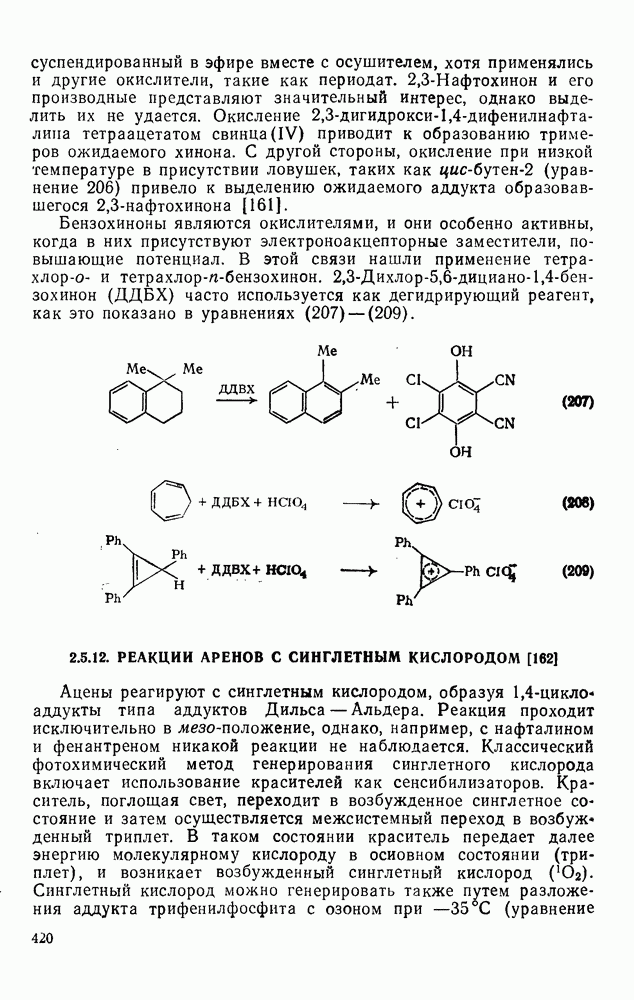
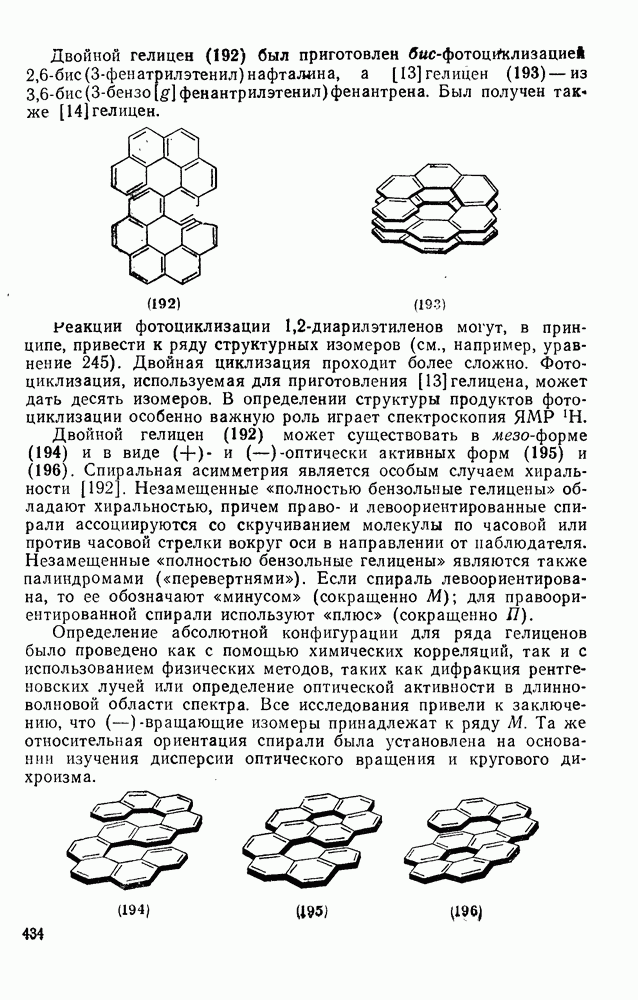
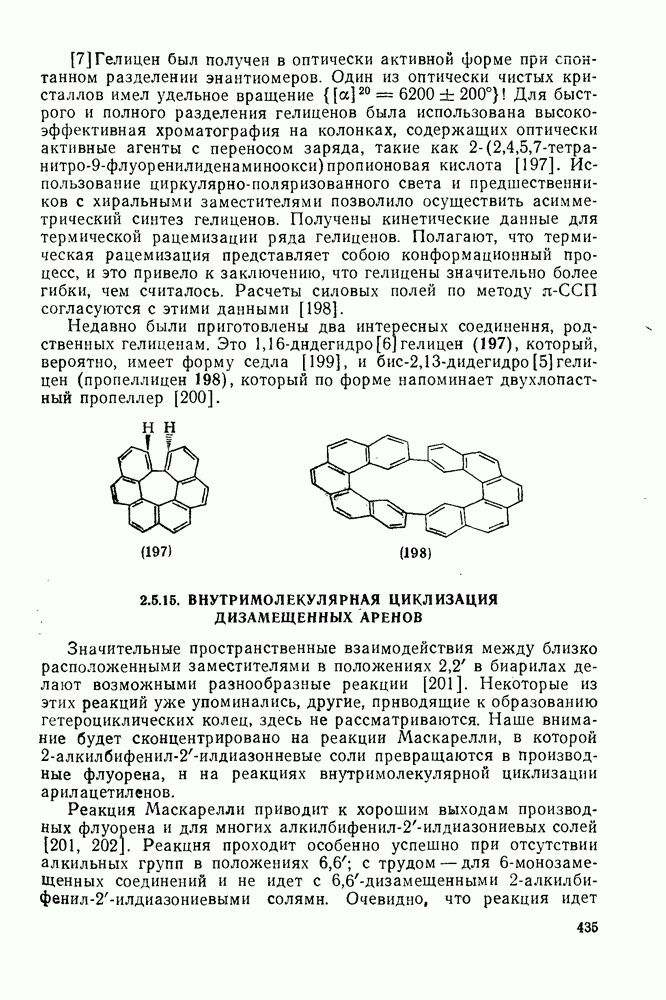
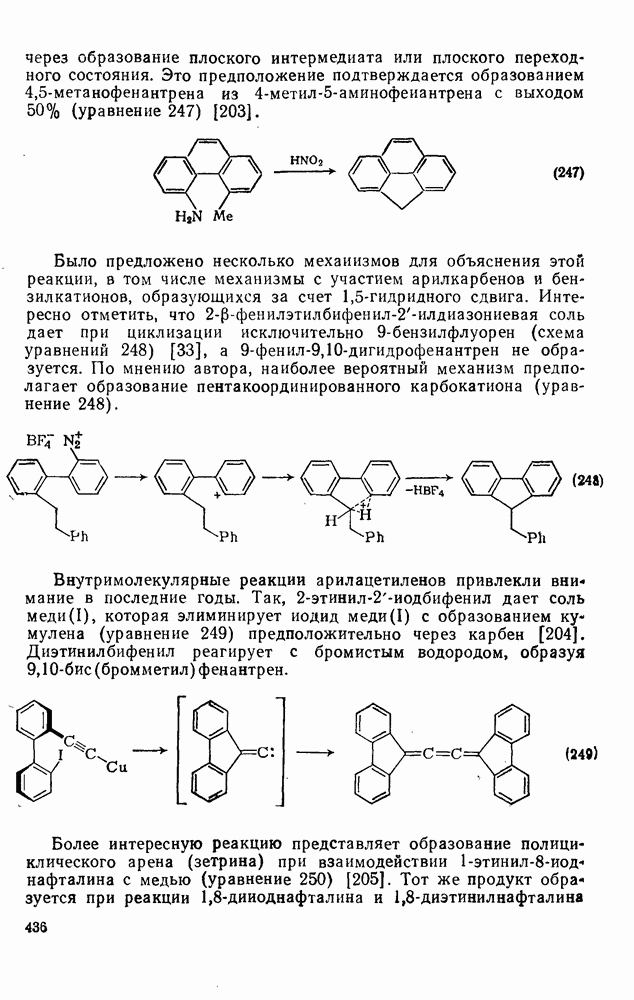
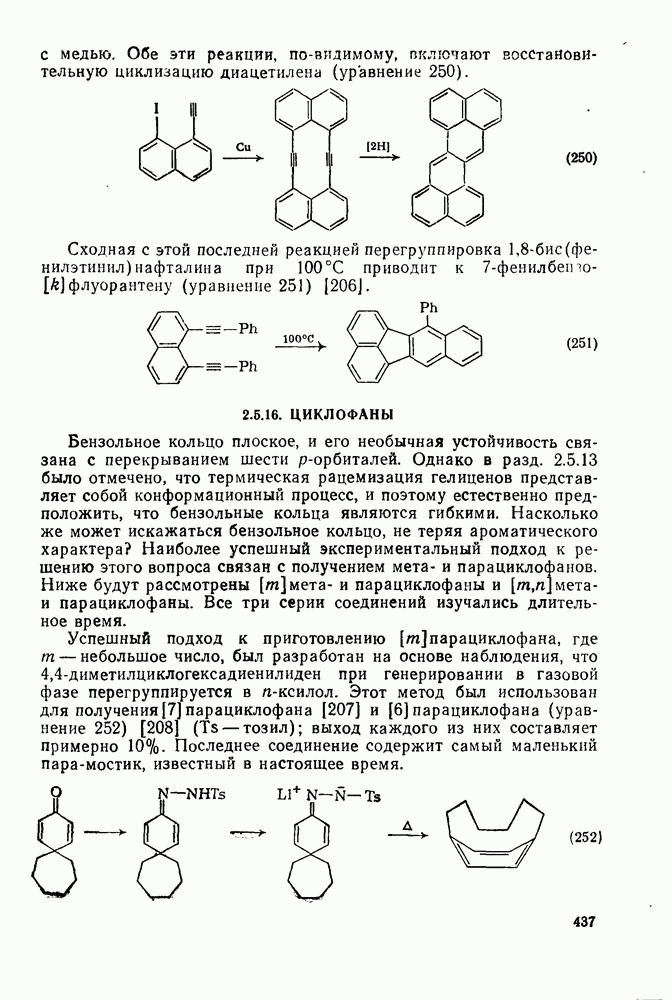
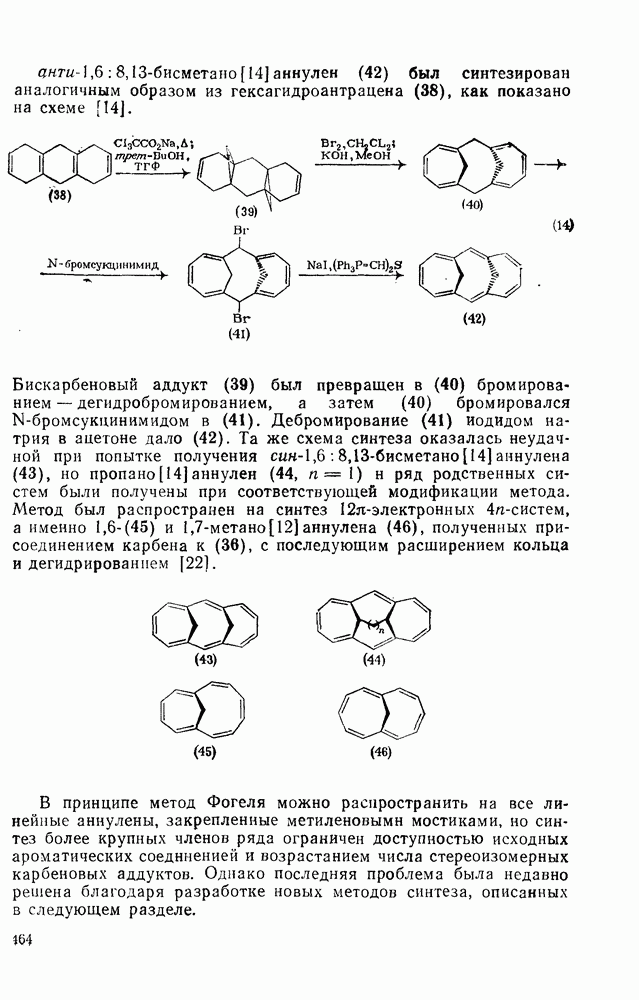
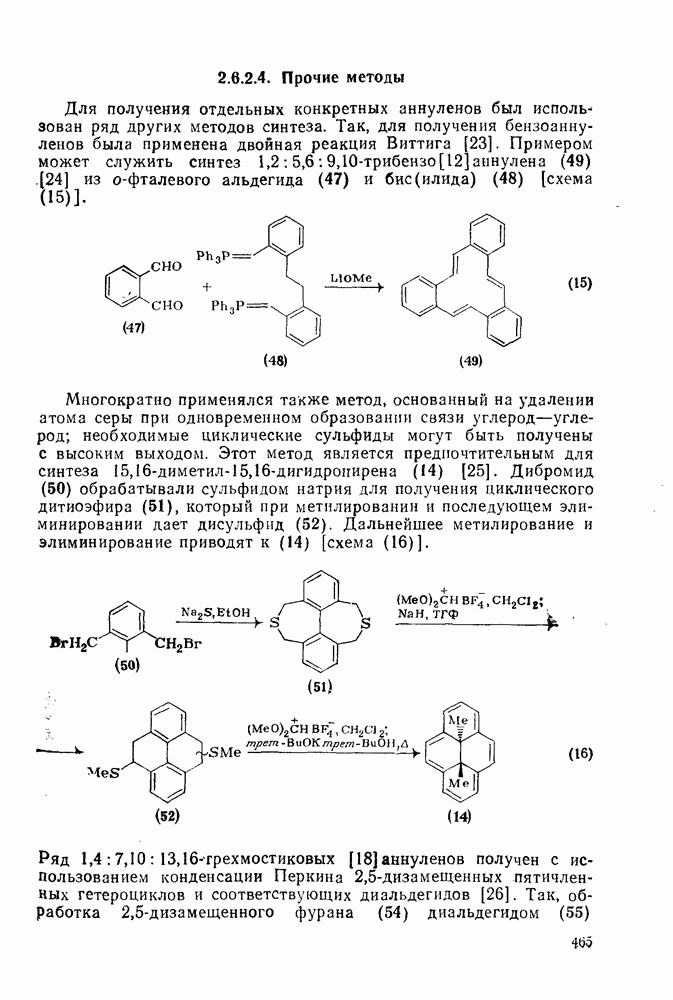
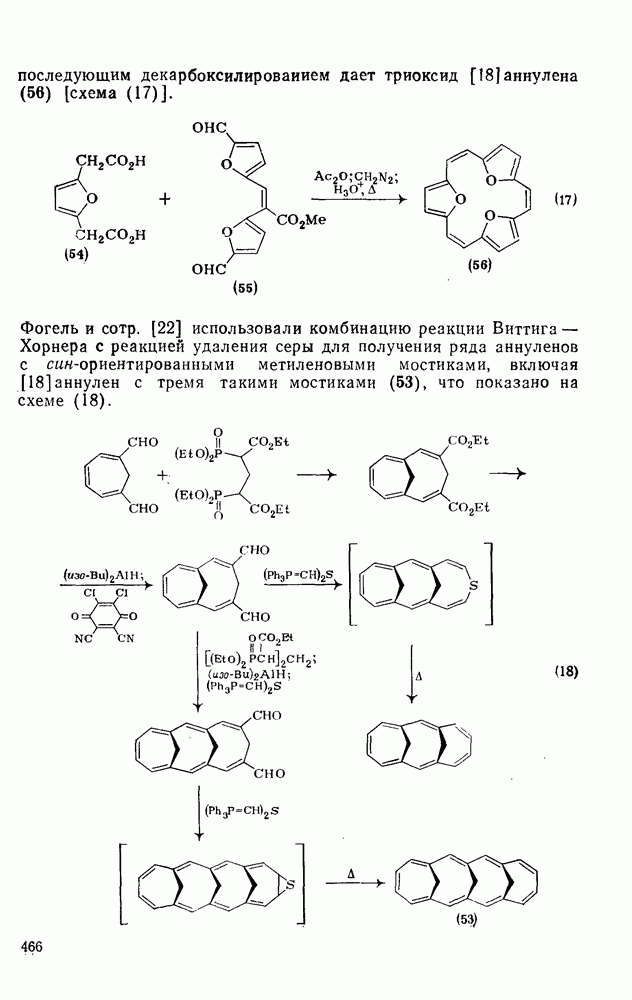
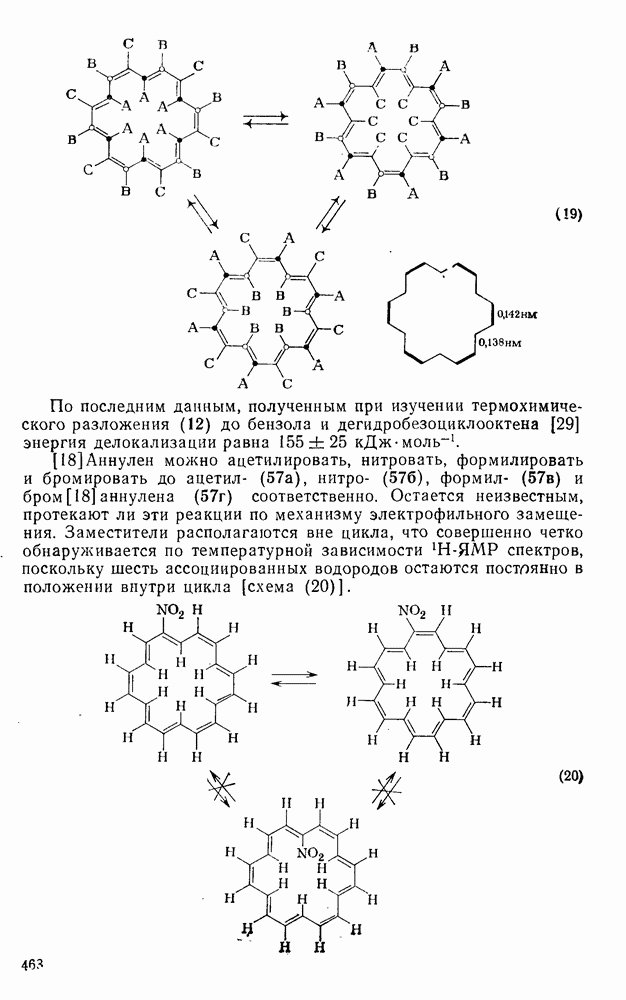
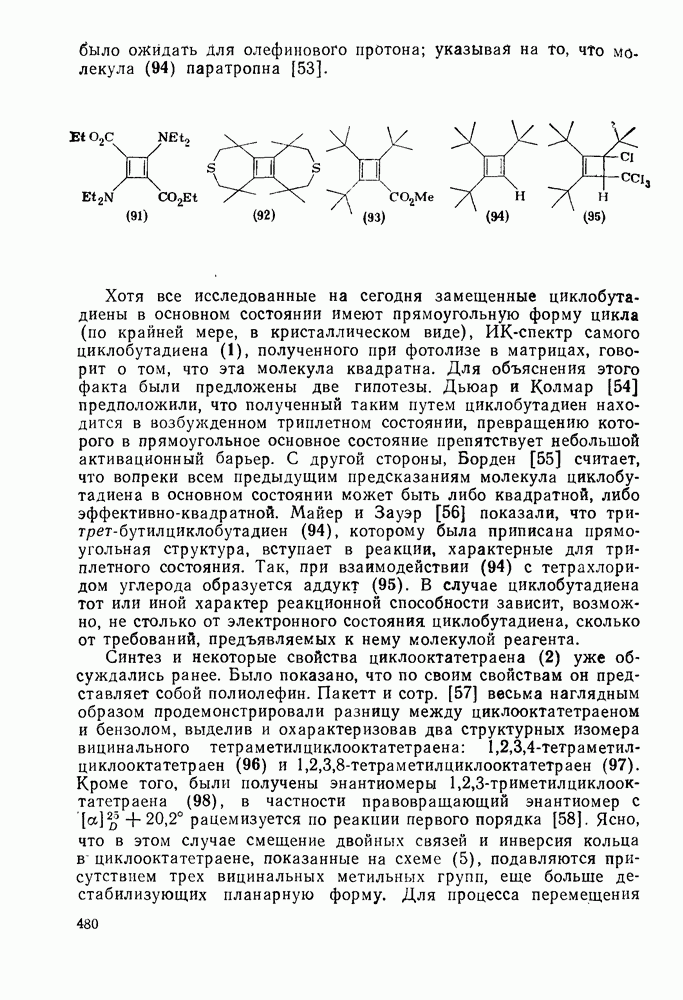
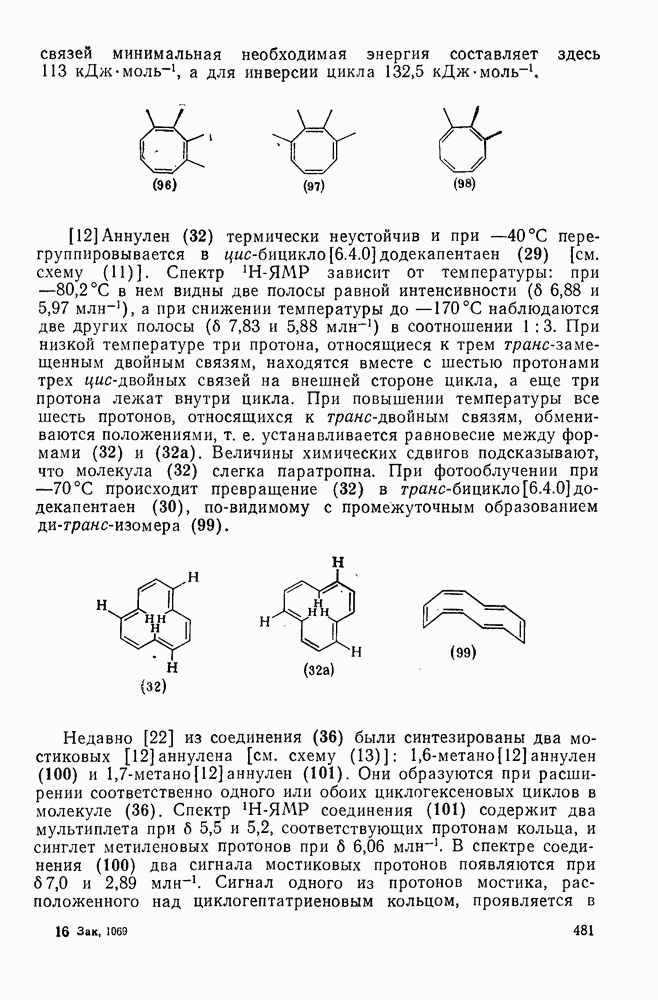
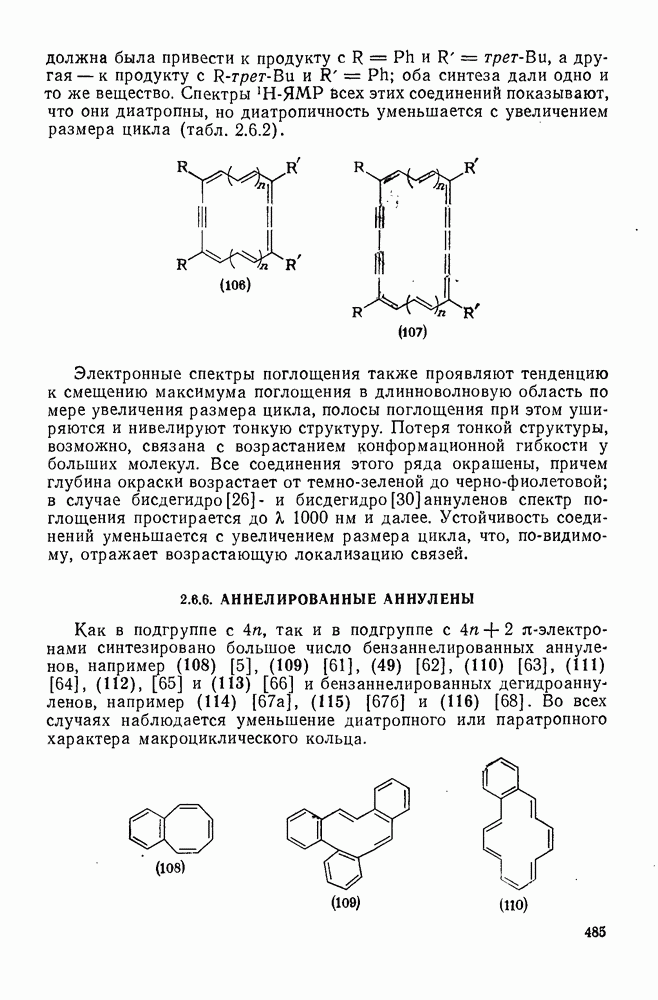
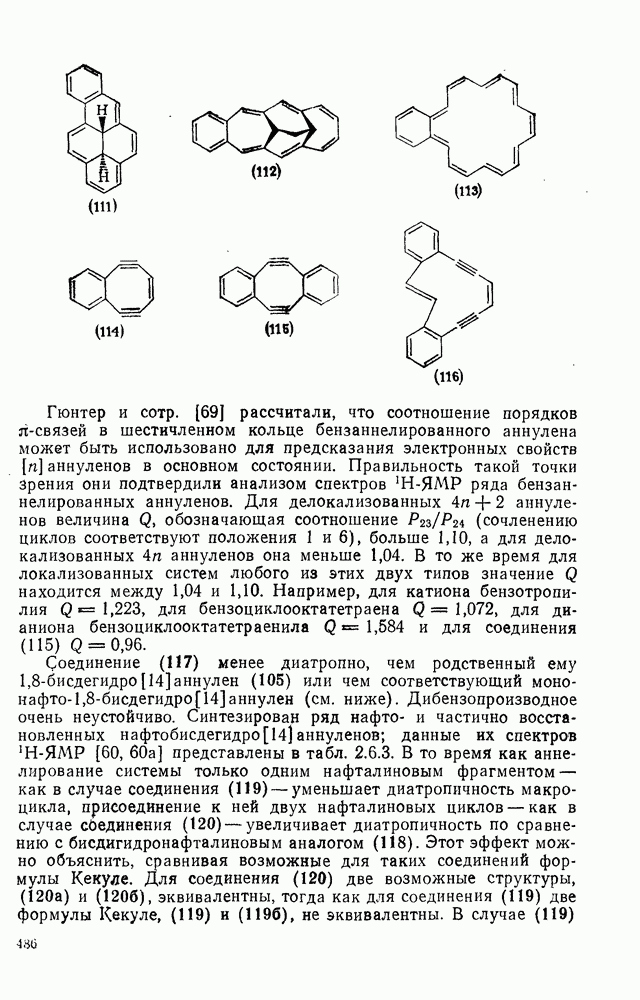
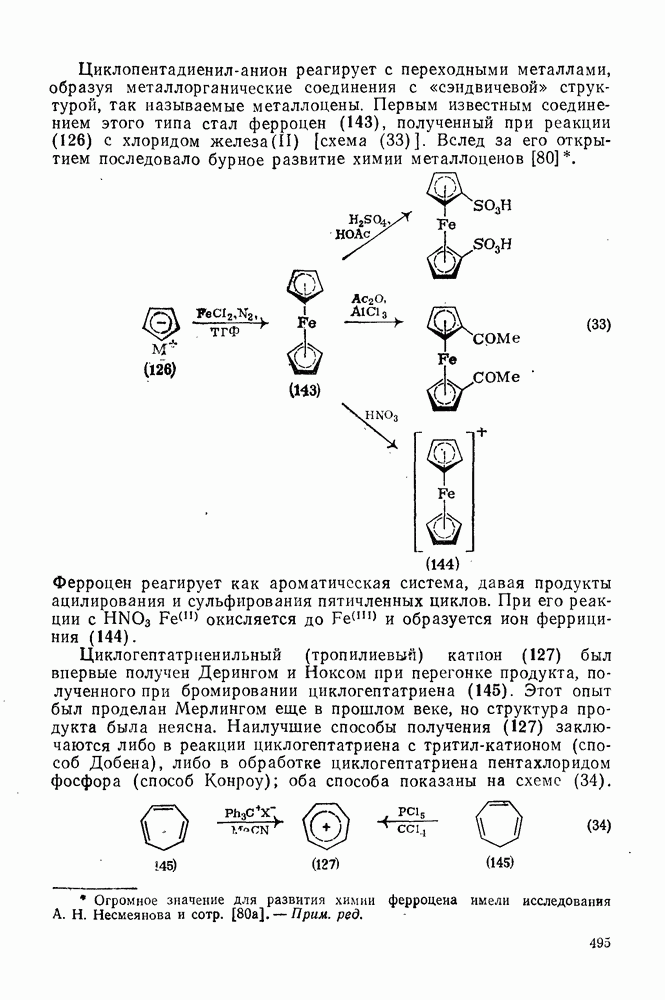
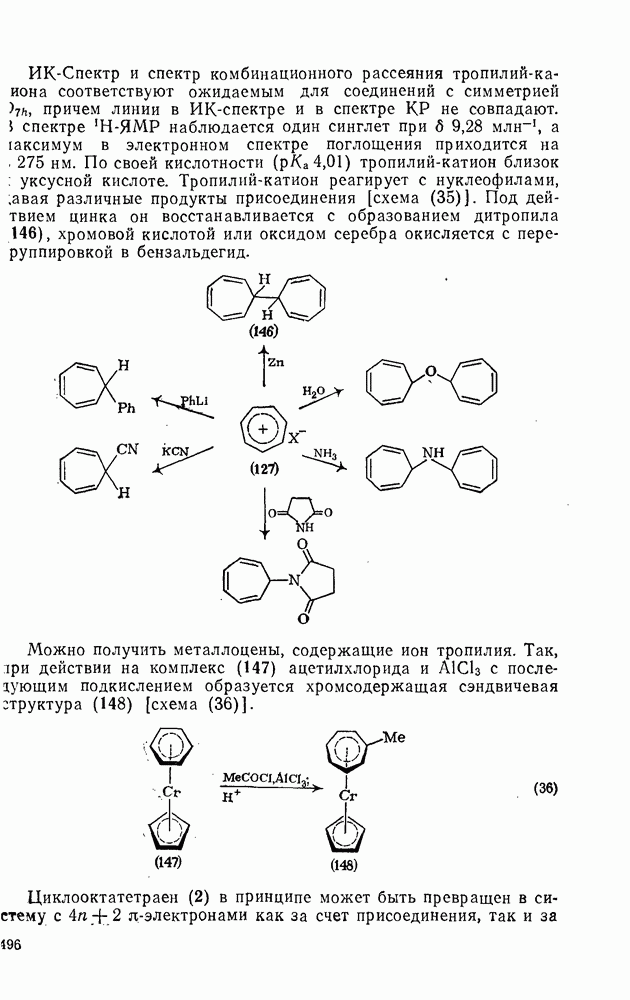
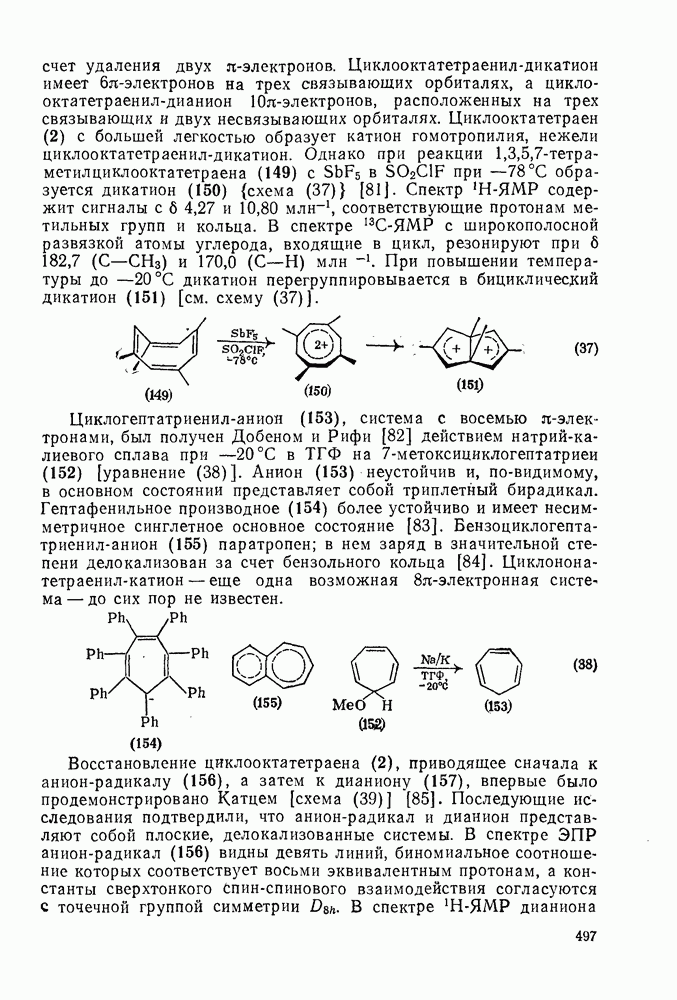
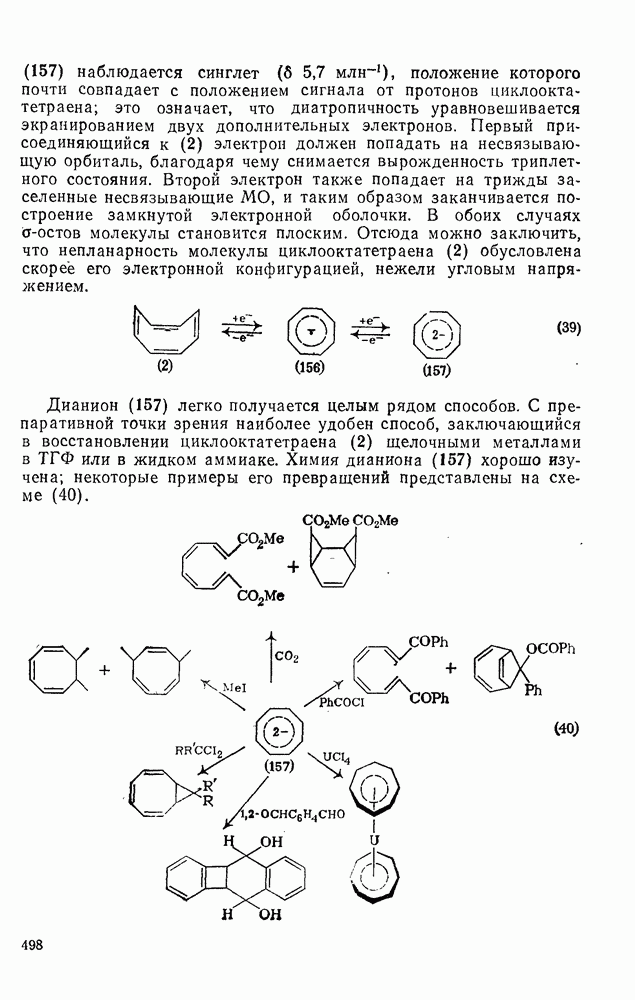
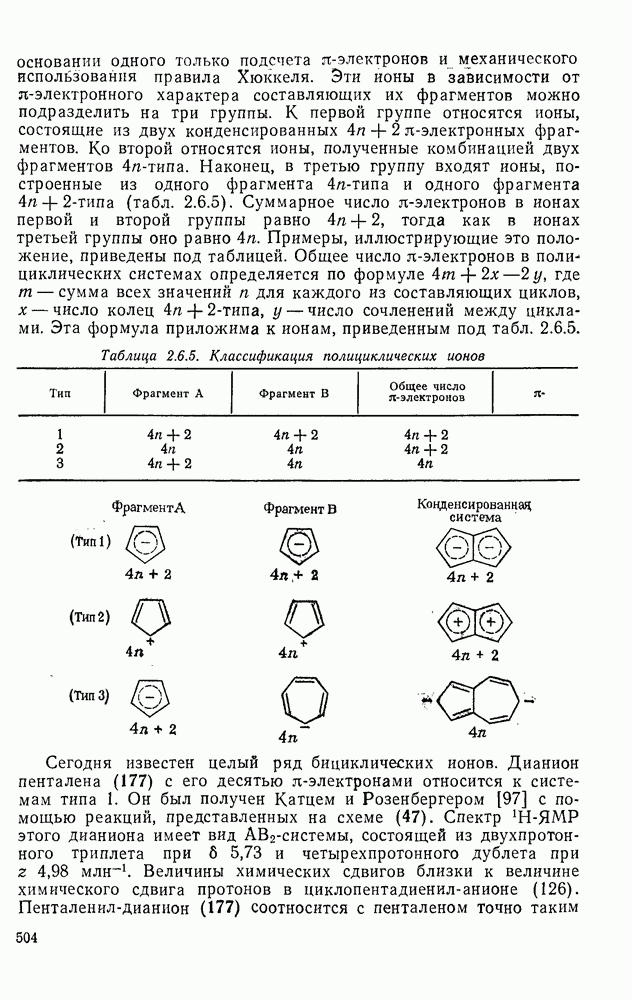
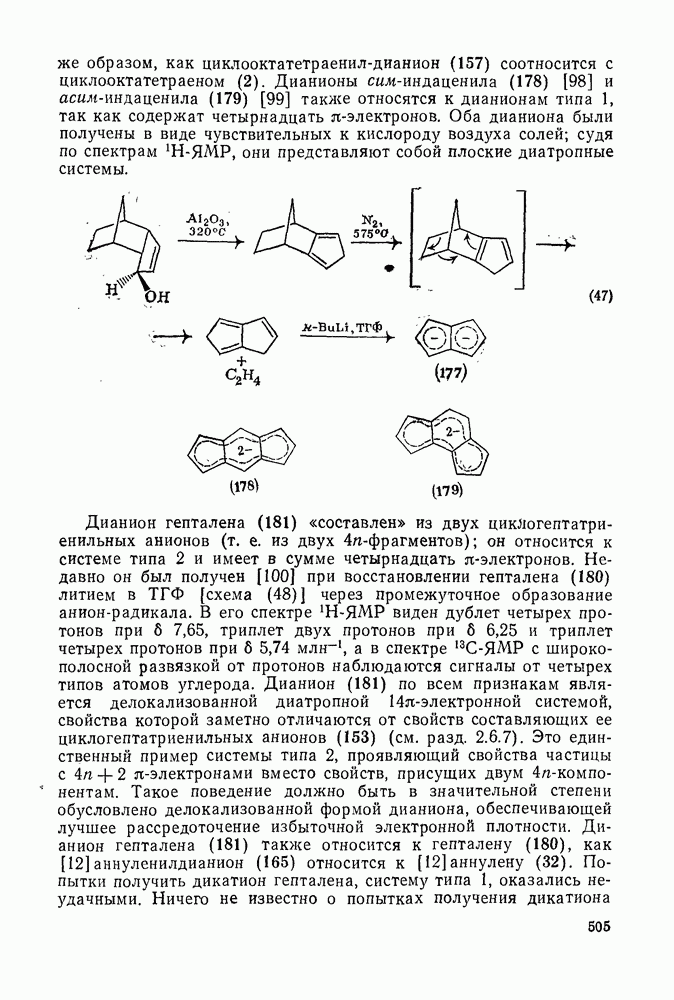
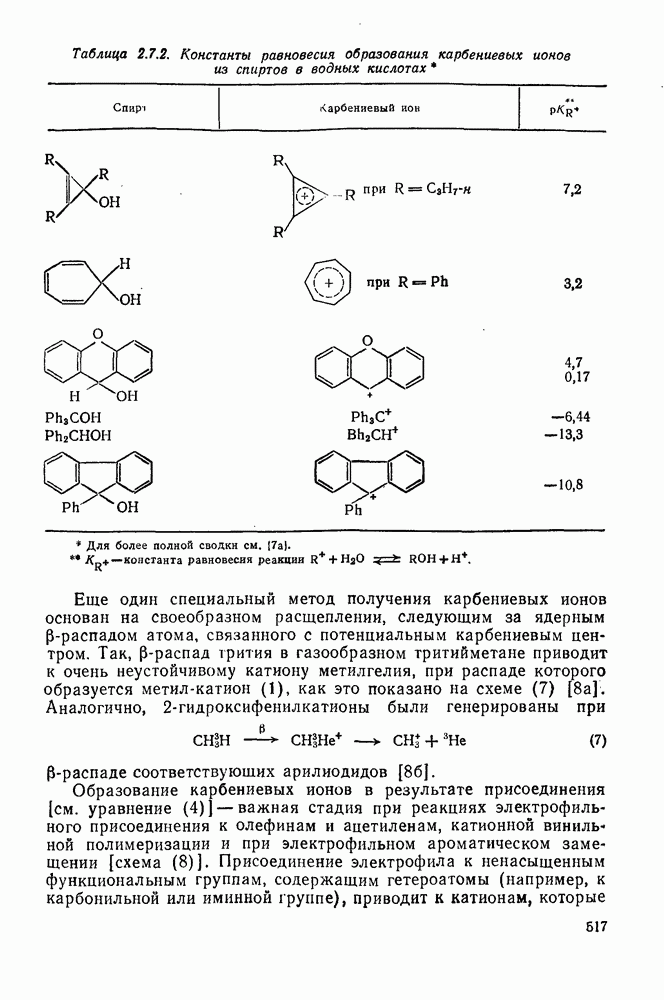
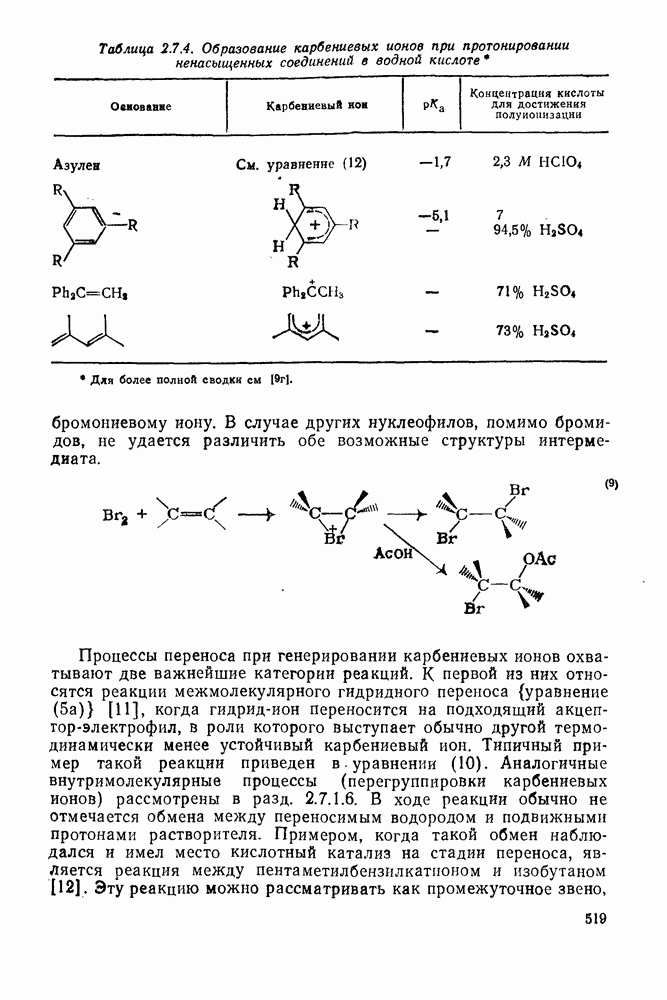
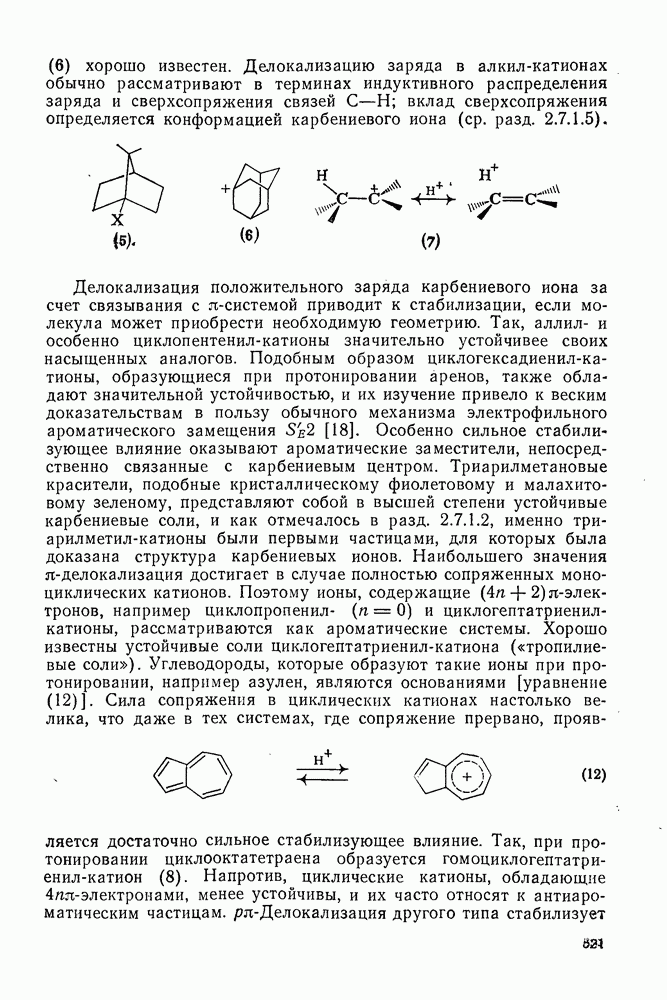
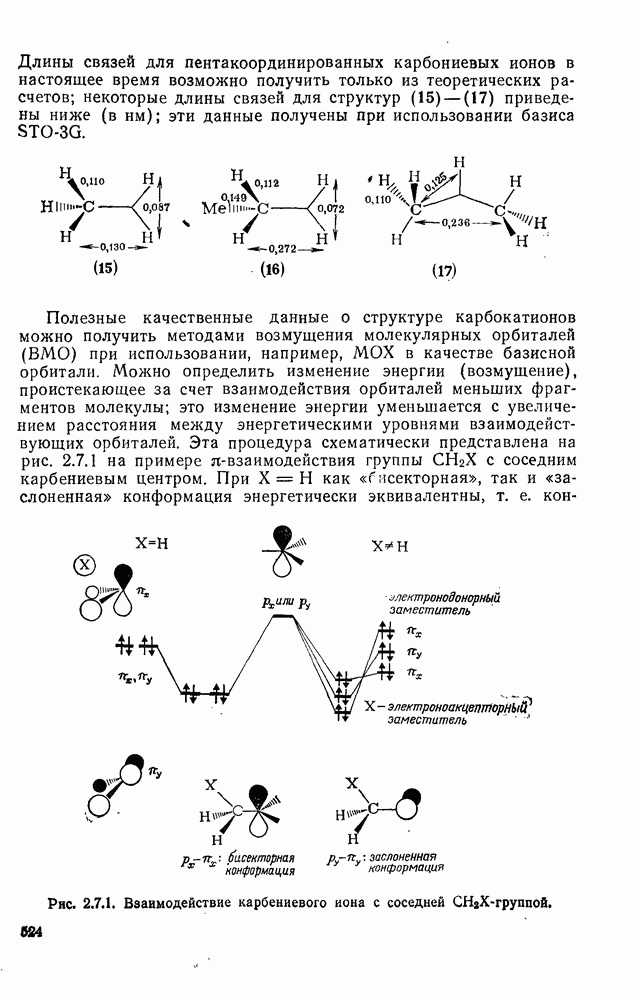
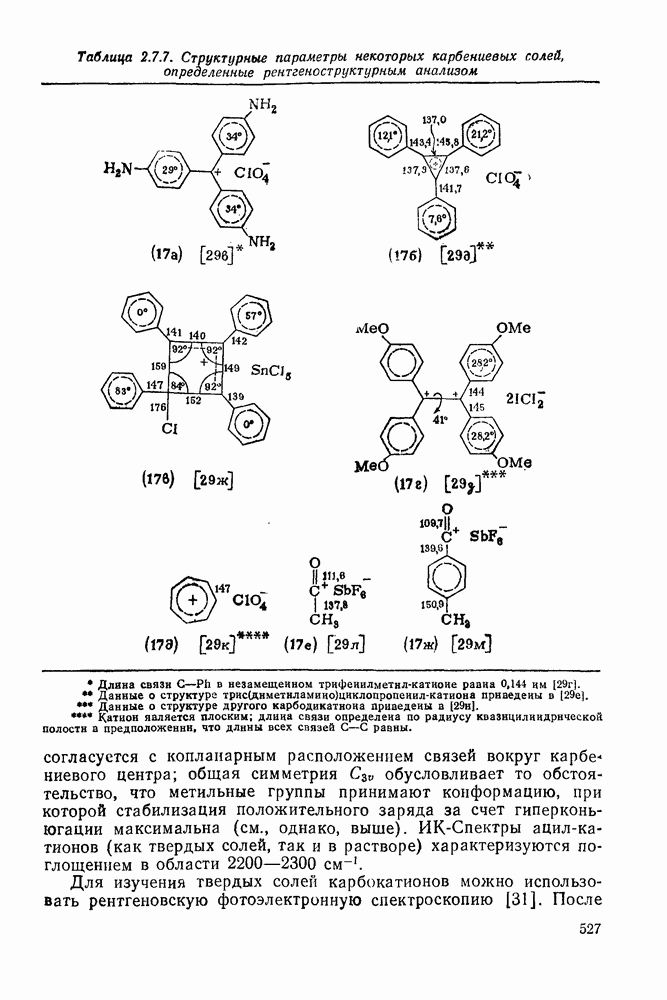
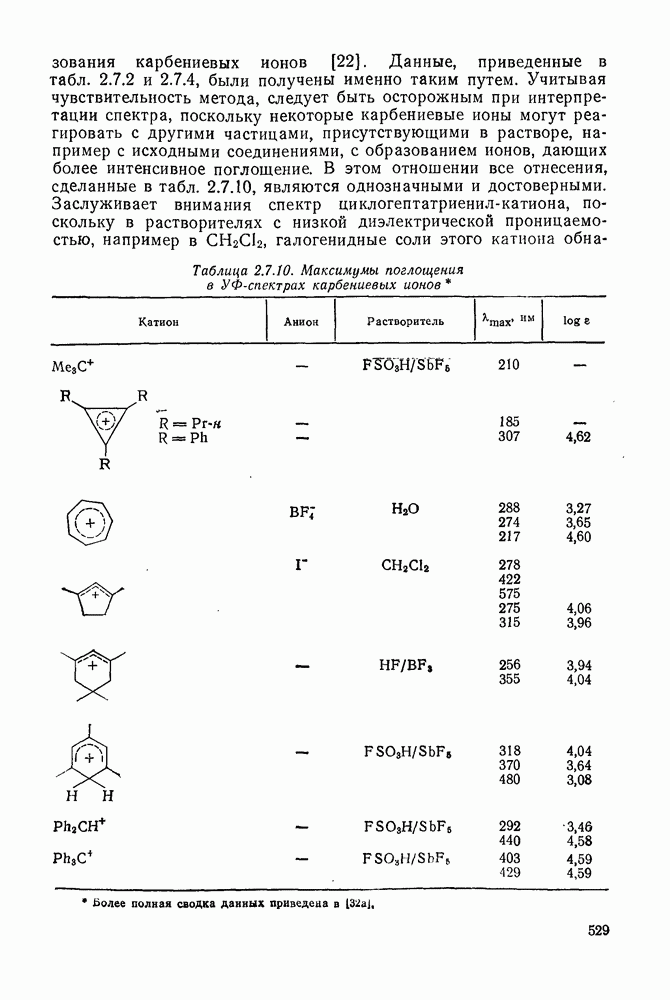
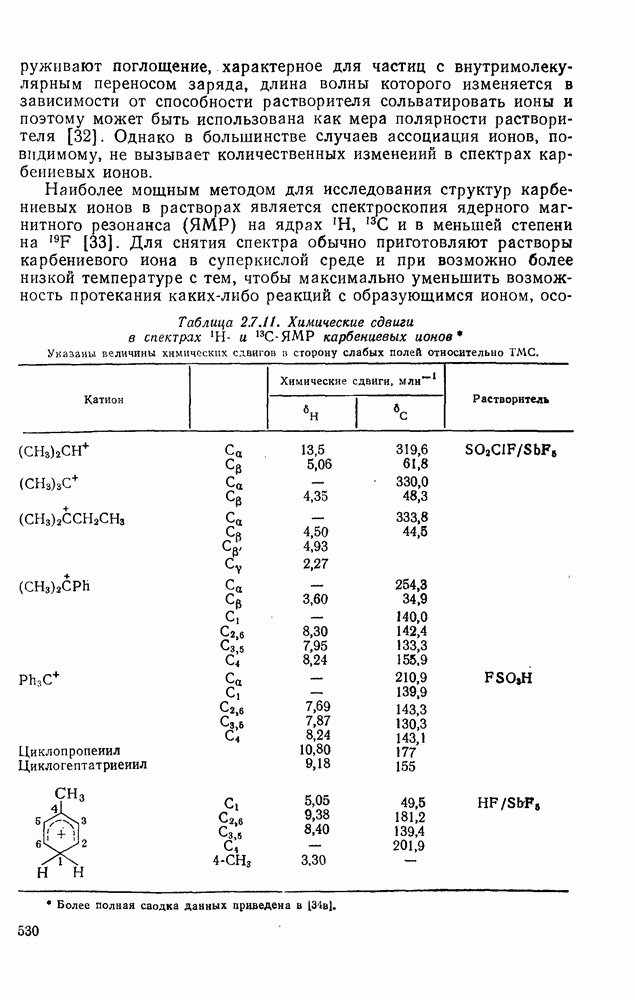
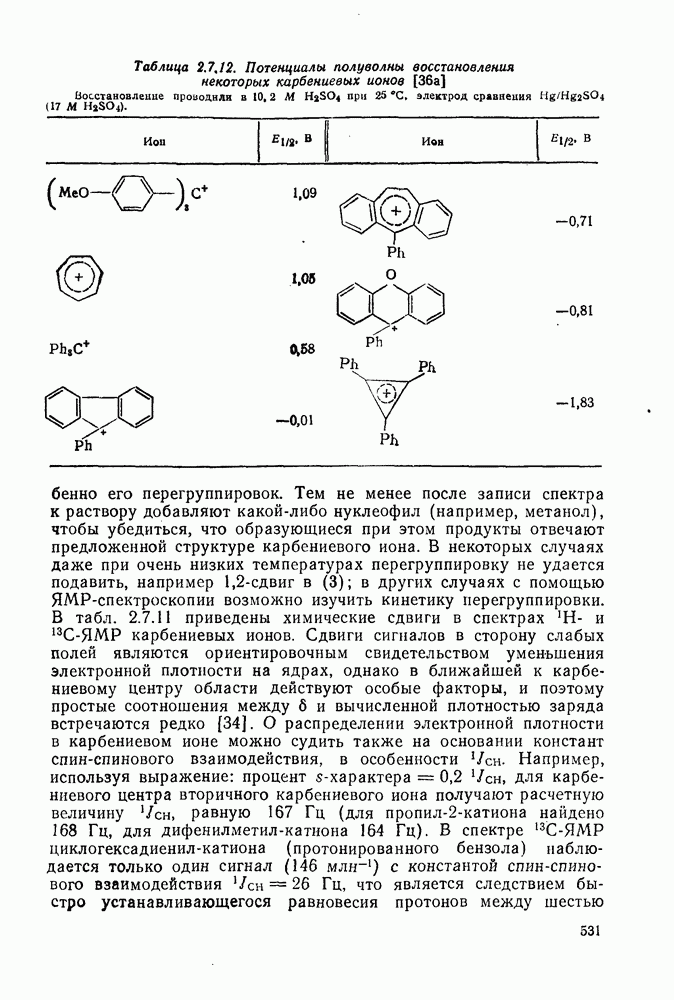
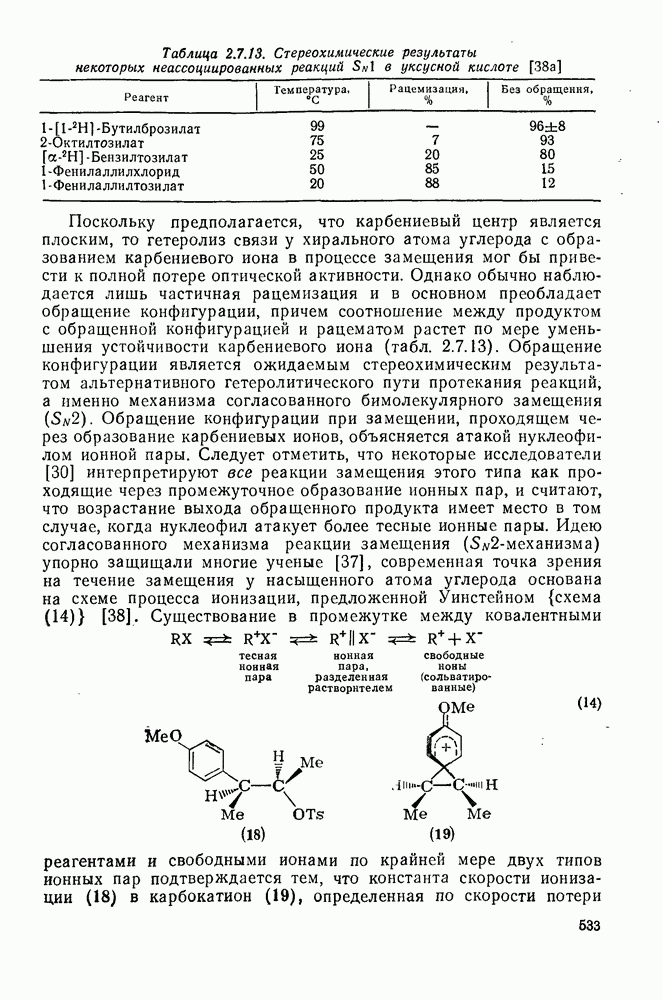
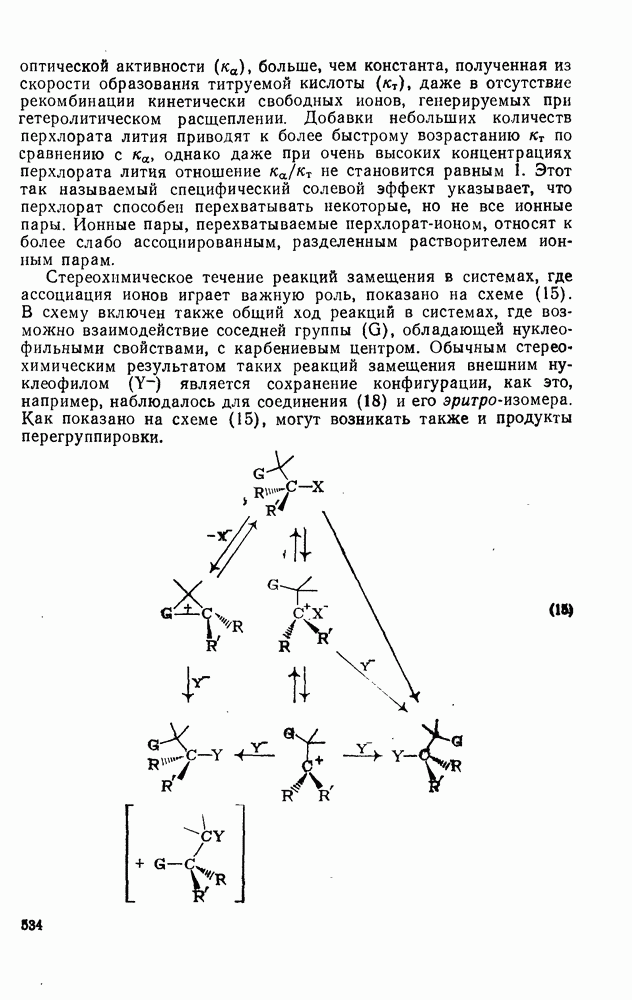
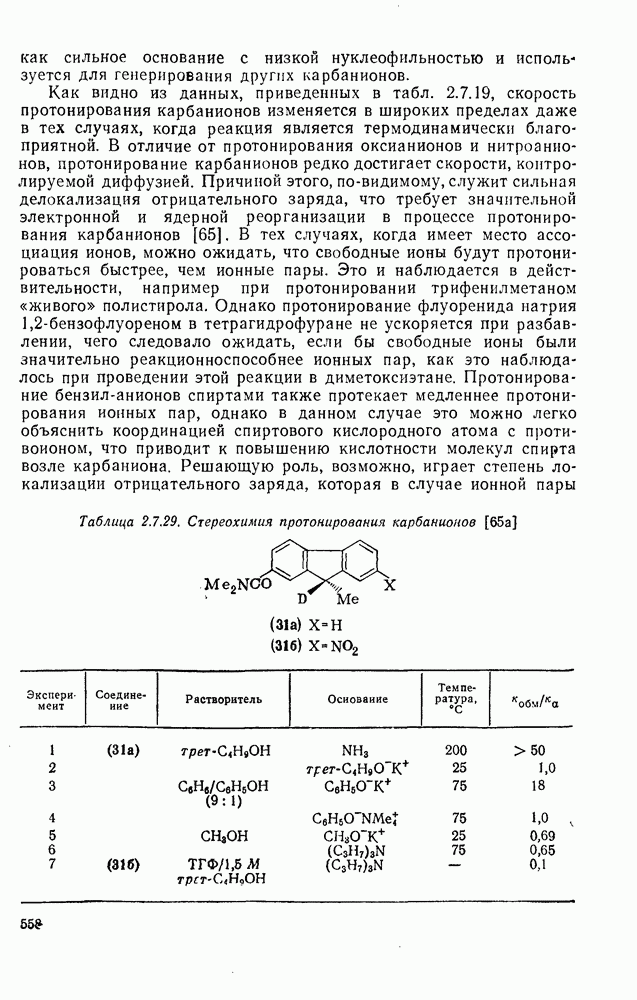
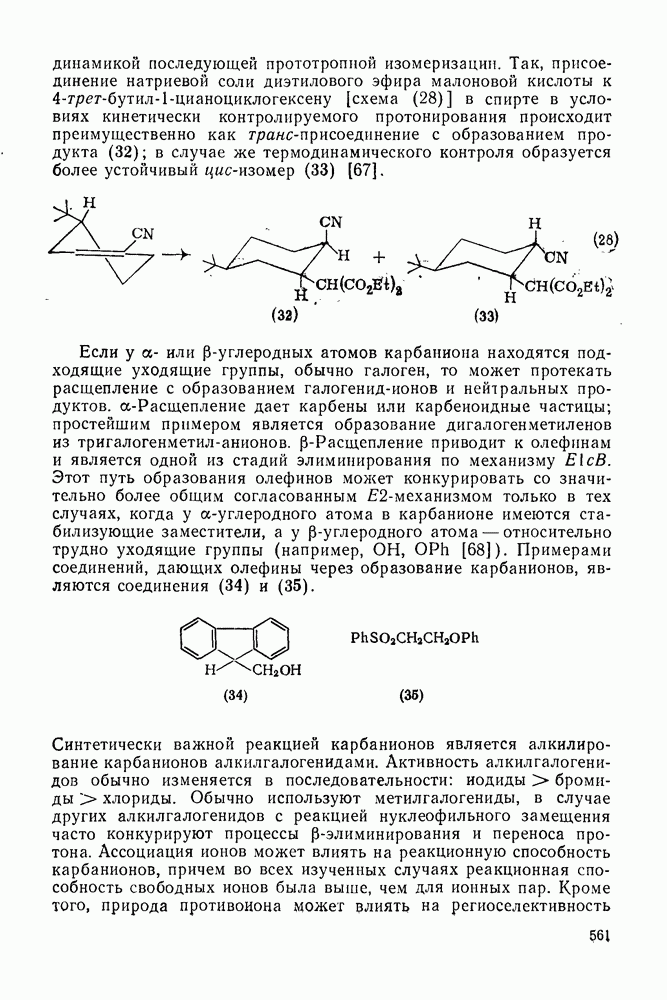
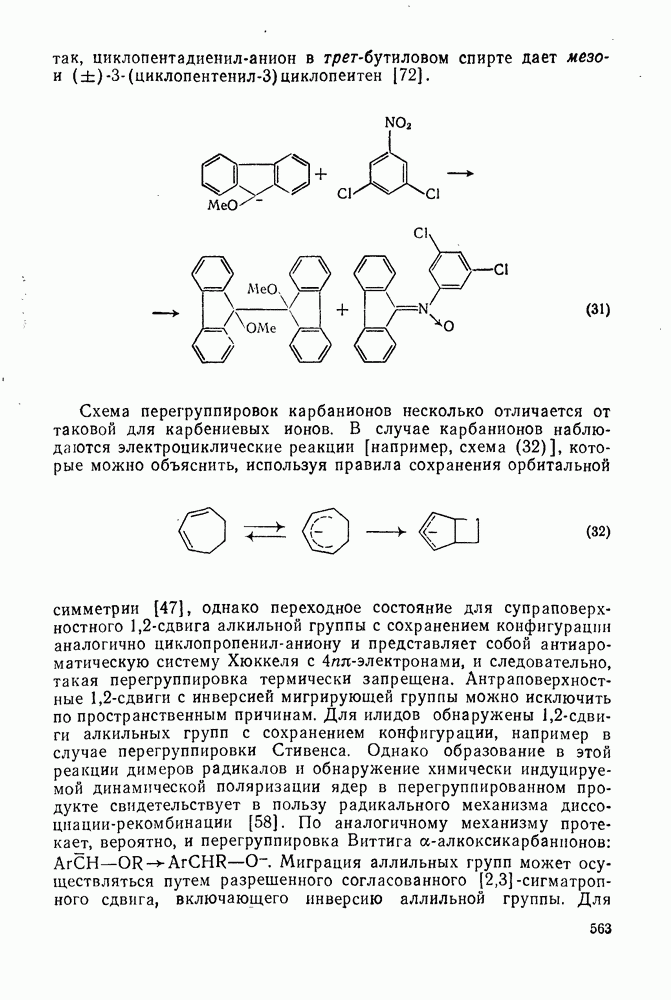
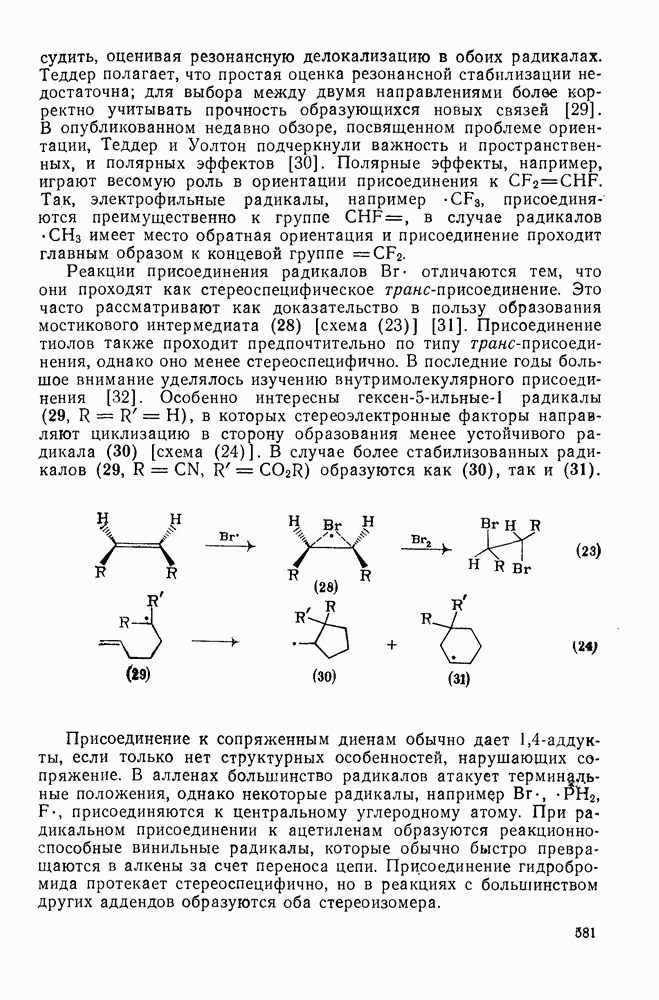
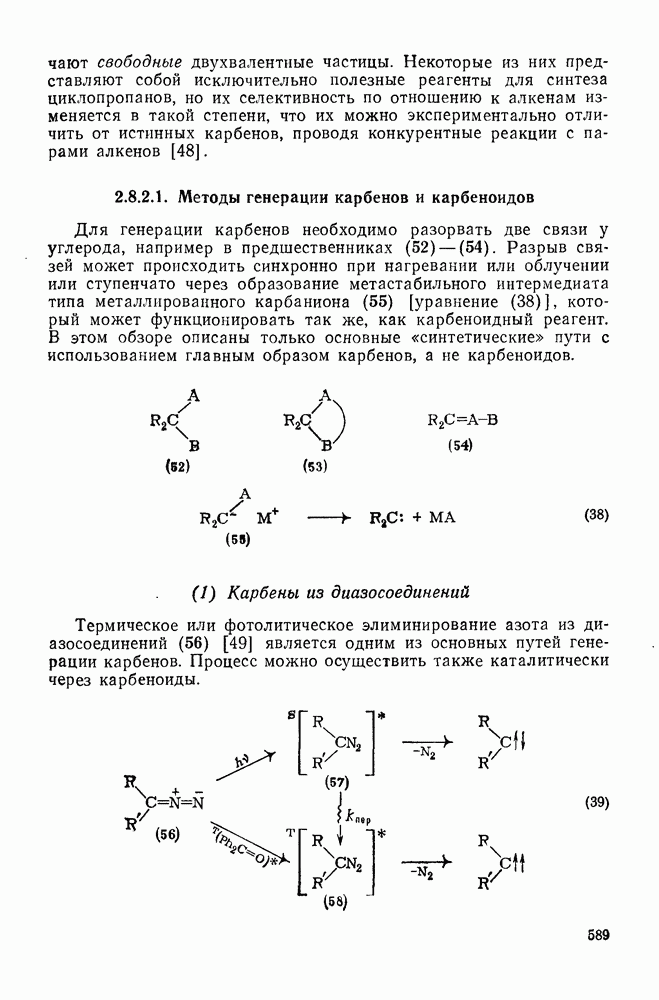
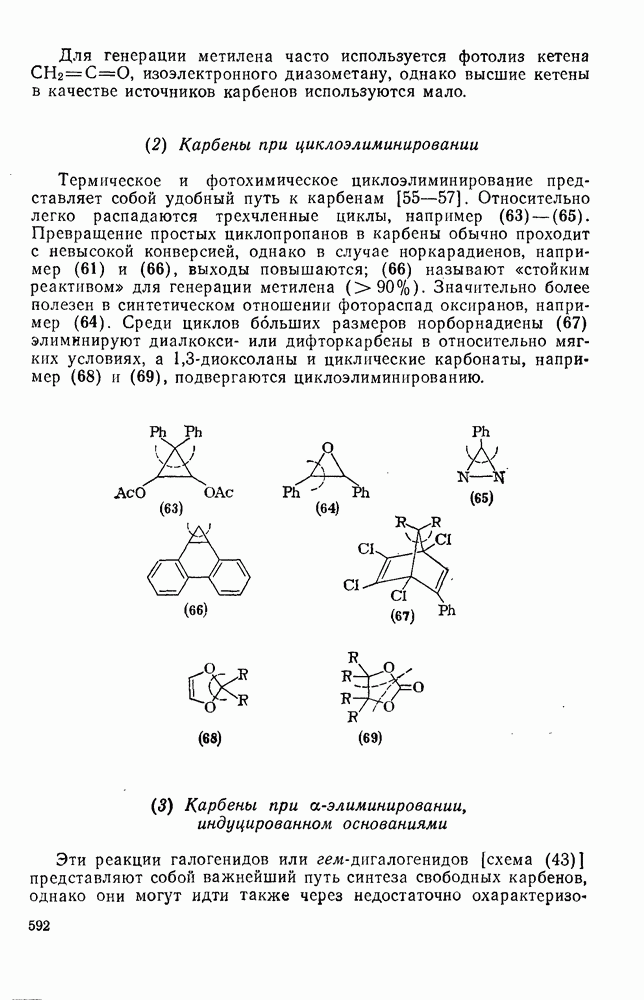
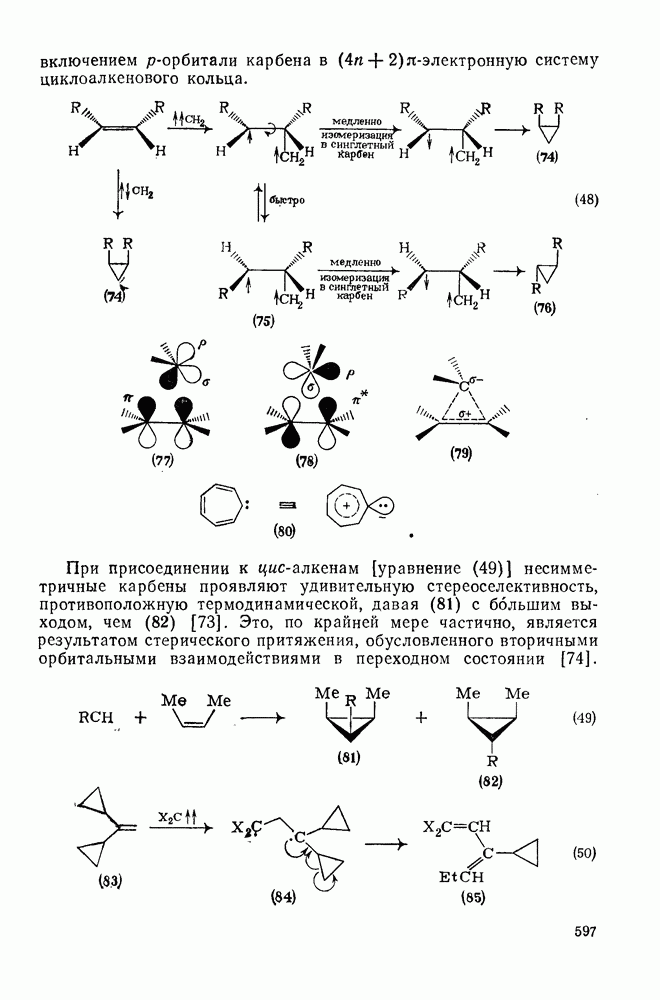
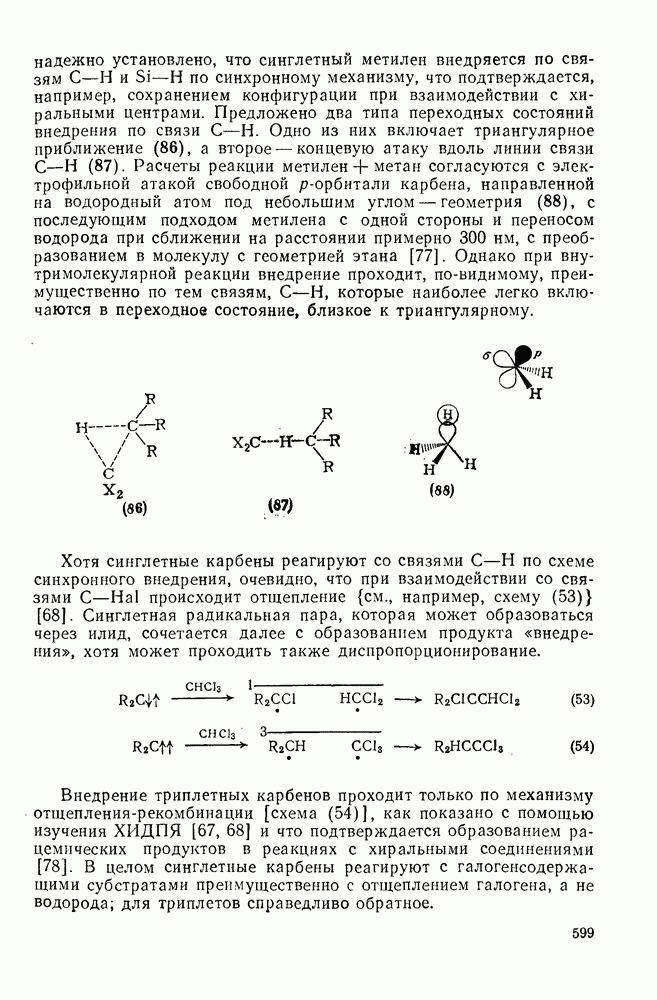
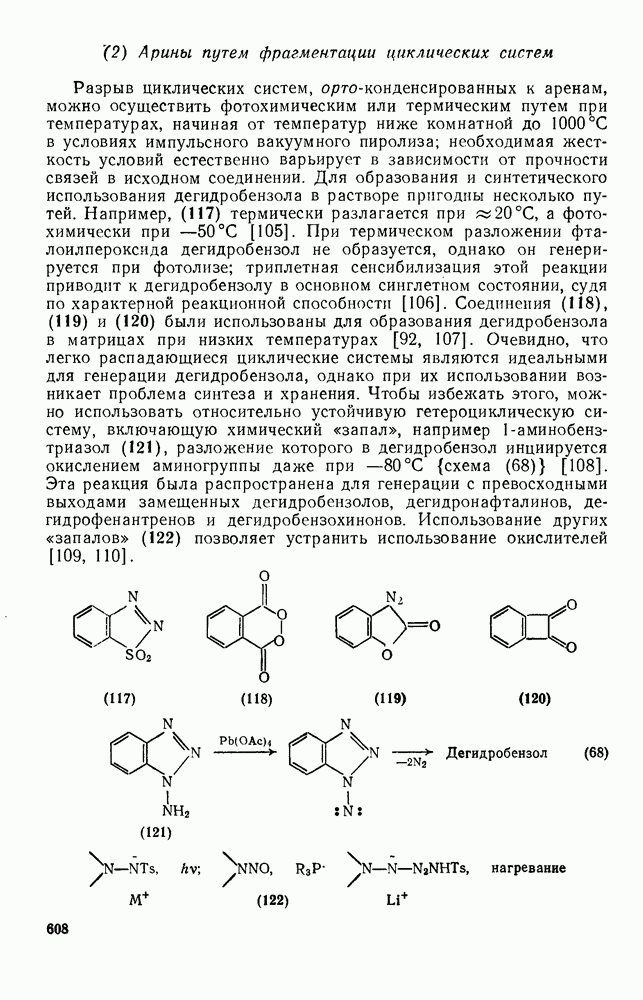
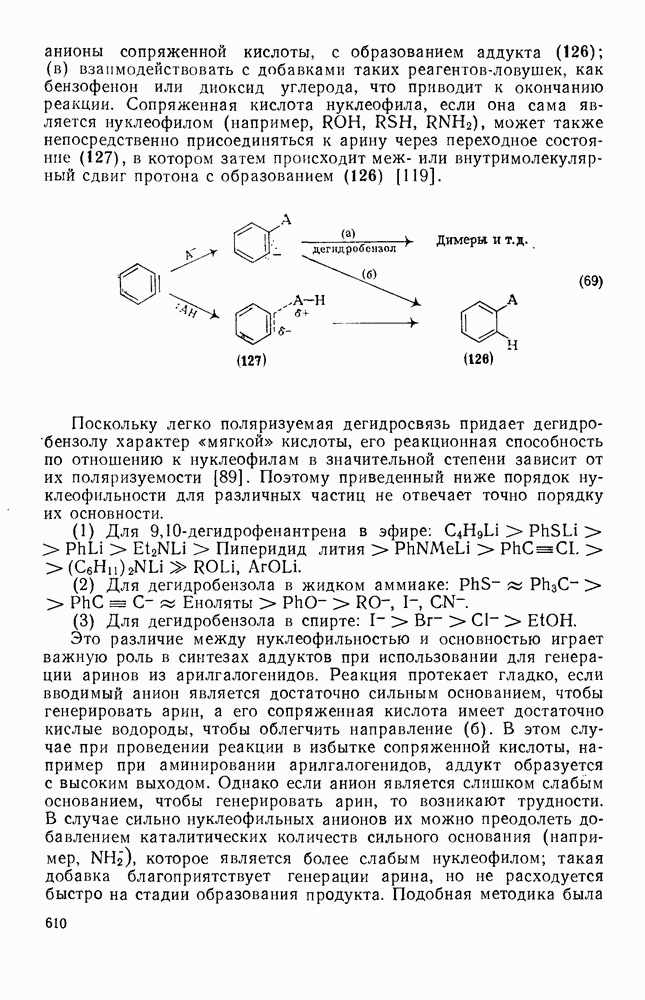
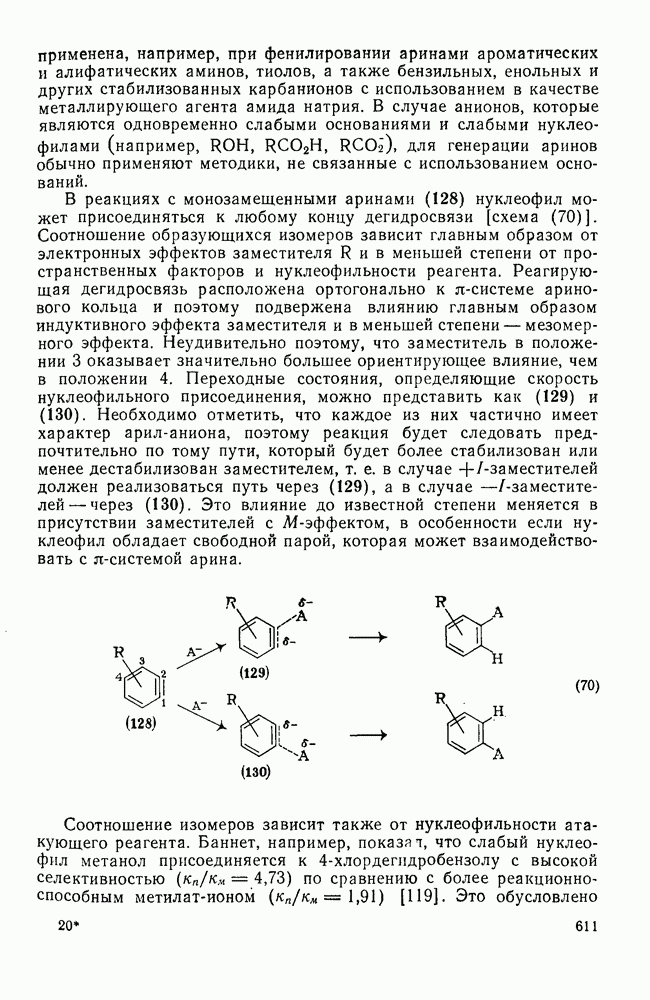
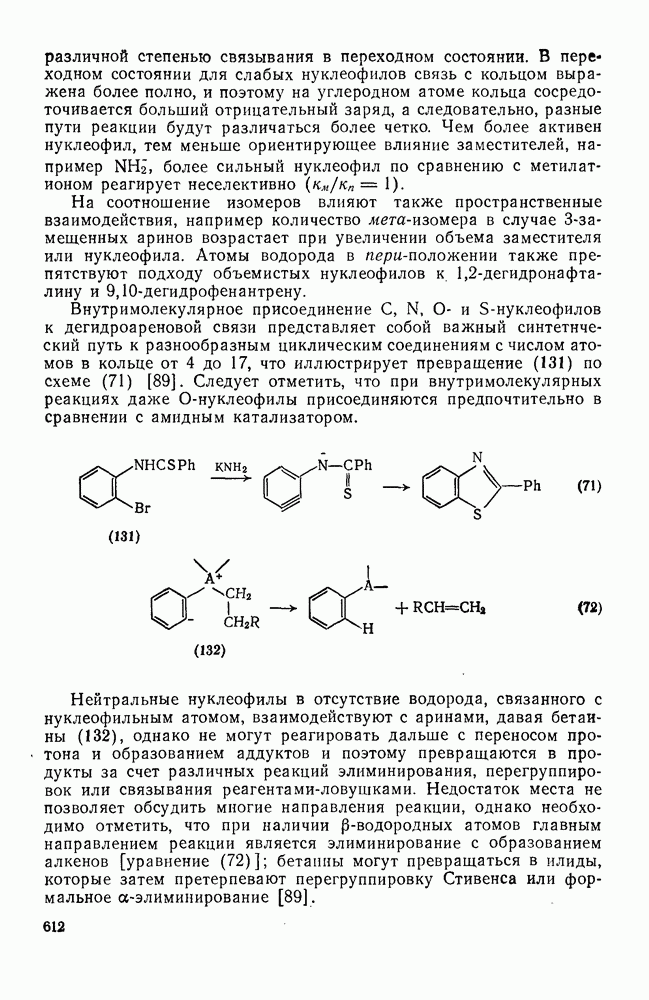
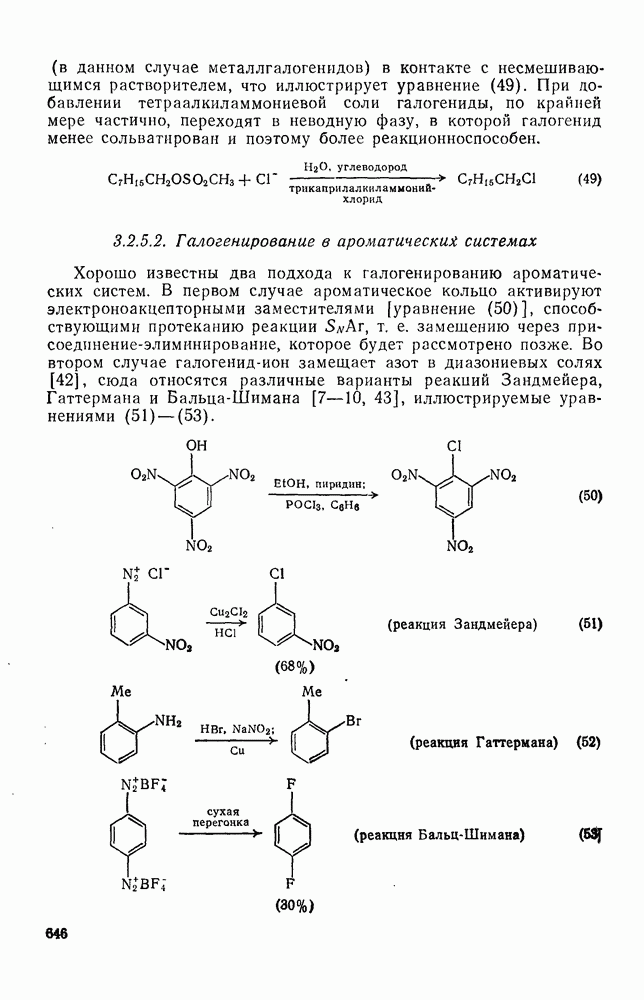
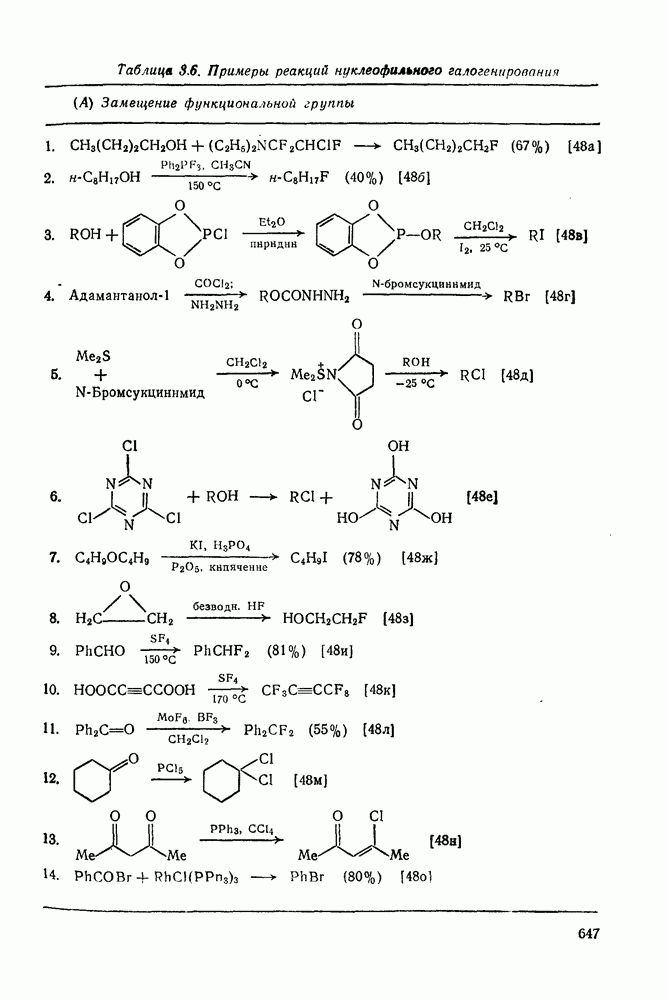
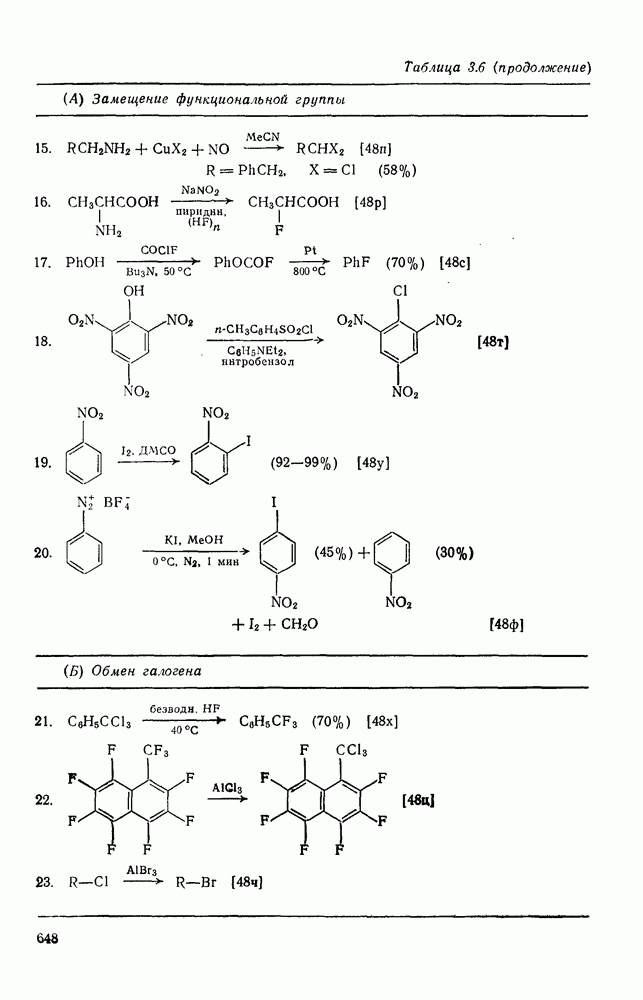
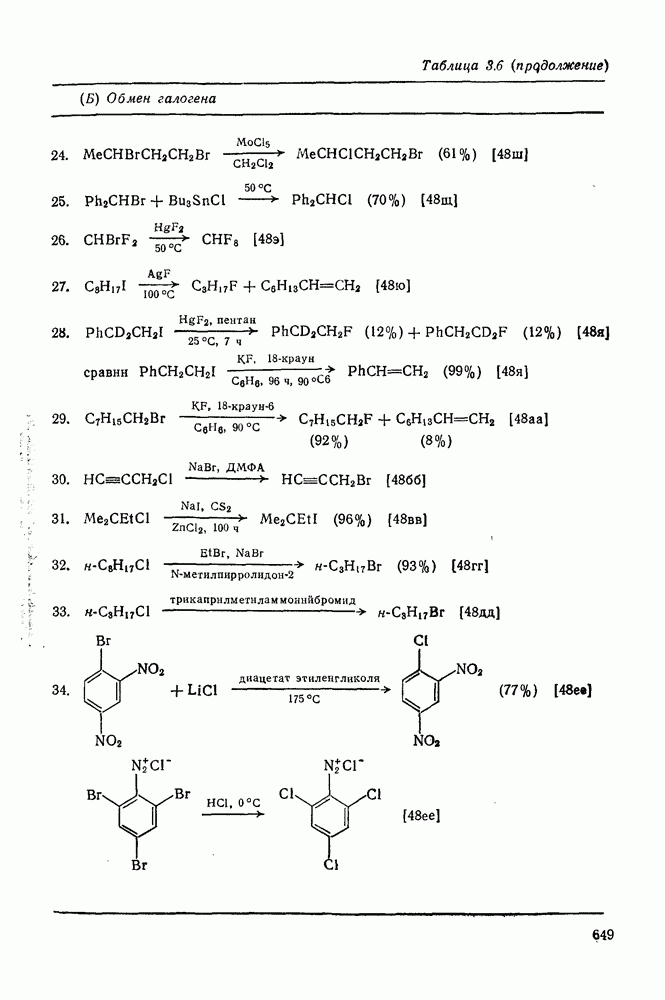
2.7.1.6. Реакции карбокатионов
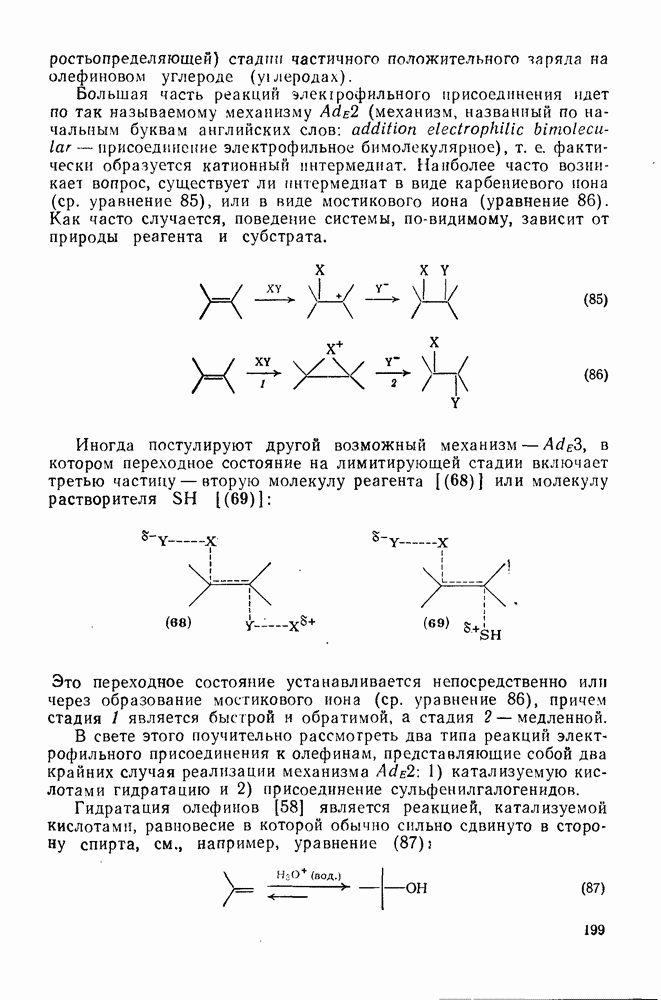
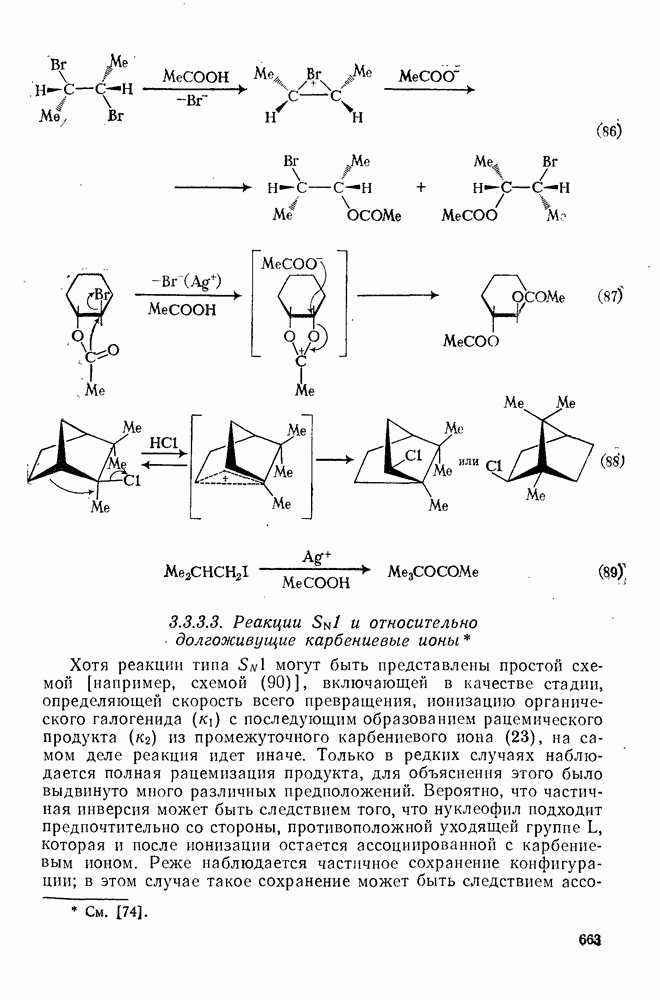
Вообще говоря, реакции карбокатионов являются обратными реакциями их образования. Пентакоординированные карбониевые ионы обычно отщепляют протон, давая нейтральный углеводород или в случае карбоциклических систем превращаются в карбениевые ионы за счет расщепления связи  . Почти все реакции карбениевых ионов можно классифицировать по типу электронной пары (
. Почти все реакции карбениевых ионов можно классифицировать по типу электронной пары ( или
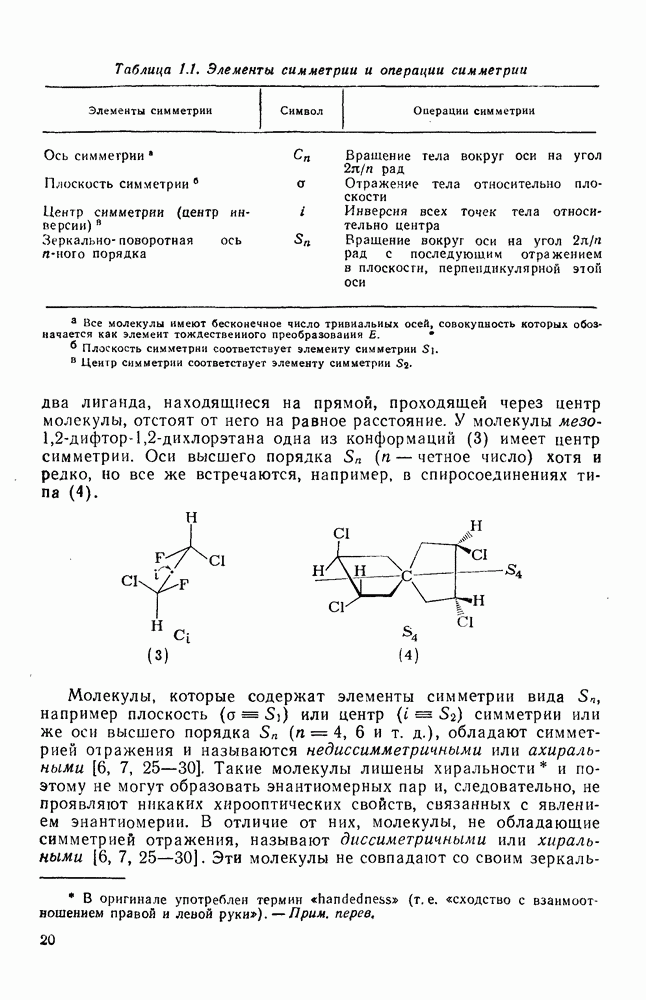
или  ), с которой взаимодействует катионный центр, причем имеются примеры как внутримолекулярных, так и межмолекулярных реакций. В случае внутримолекулярных превращений в переходном состоянии могут возникать взаимодействия, ведущие к образованию промежуточного карбениевого иона, что ведет к возрастанию скорости реакции (участие соседней группы) и характерной стереохимии.
), с которой взаимодействует катионный центр, причем имеются примеры как внутримолекулярных, так и межмолекулярных реакций. В случае внутримолекулярных превращений в переходном состоянии могут возникать взаимодействия, ведущие к образованию промежуточного карбениевого иона, что ведет к возрастанию скорости реакции (участие соседней группы) и характерной стереохимии.
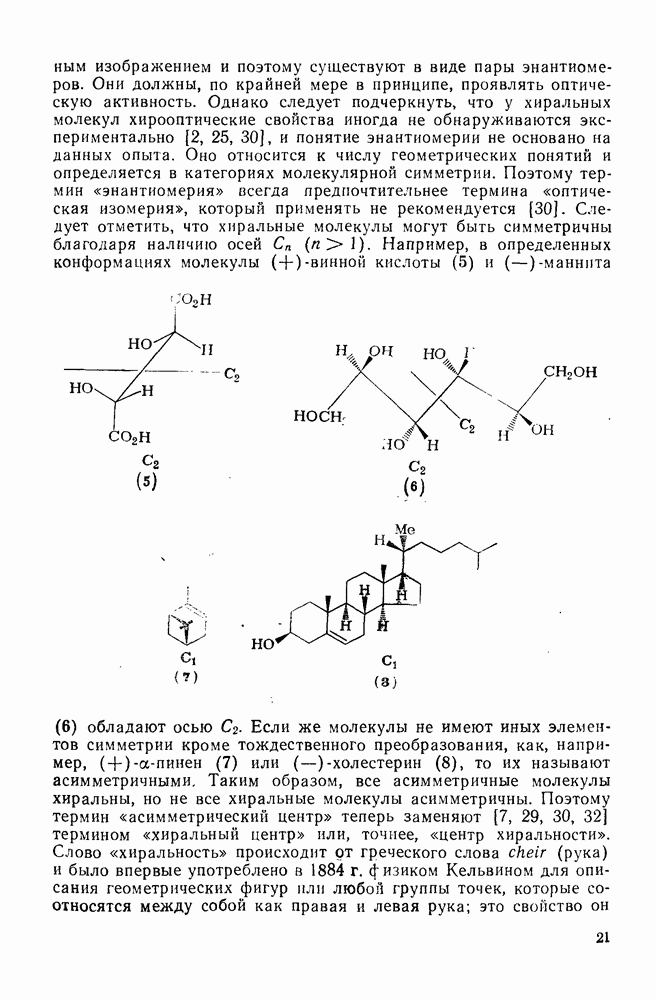
Одной из наиболее обычных реакций карбениевых ионов, широко представленной в процессах  , является взаимодействие с неподеленной
, является взаимодействие с неподеленной  парой электронов нуклеофила. В качестве нуклеофилов могут выступать анионы, например
парой электронов нуклеофила. В качестве нуклеофилов могут выступать анионы, например  ,
,  ,
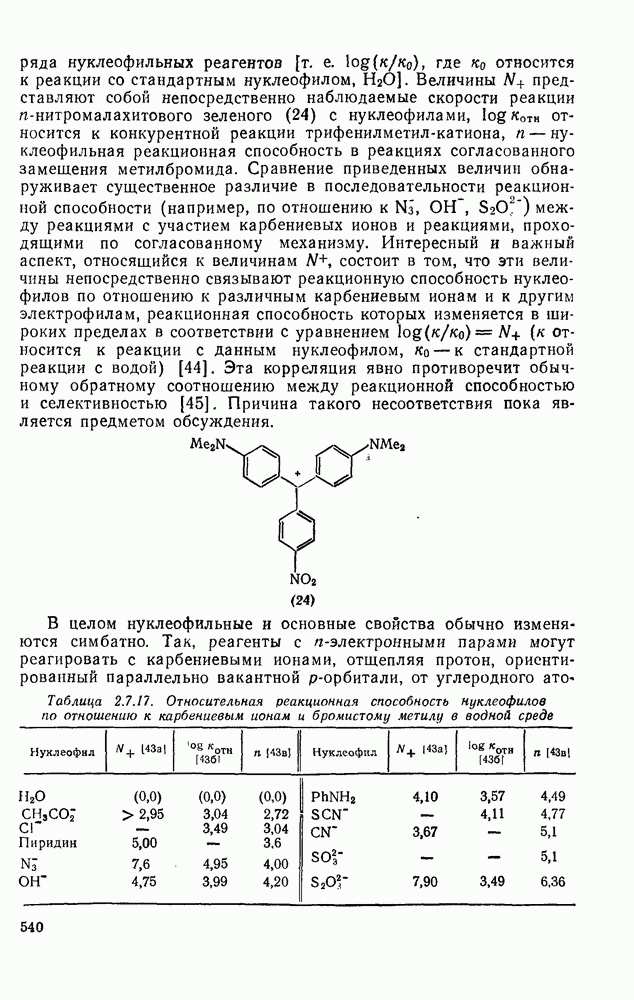
,  , или нейтральные молекулы (например, ROH, СО); в последнем случае устойчивые, электрически нейтральные молекулы могут образоваться только за счет последующей реакции, такой как перенос протона. Эти реакции проходят обычно очень быстро, и их скорости могут быть надежно измерены только в случае очень устойчивых карбениевых ионов типа триарилметила. Однако бензил-, ди-фенилметил- и трифенилметил-катионы, генерированные импульсным радиолизным методом в дихлорэтане [43], реагируют с галогенид-ионами с диффузно-контролируемыми скоростями. Чаще всего для оценки реакционной способности нуклеофилов используют метод конкурентных реакций. В табл. 2.7.17 приведены логарифмические величины относительной реакционной способности ряда нуклеофильных реагентов (т. е.
, или нейтральные молекулы (например, ROH, СО); в последнем случае устойчивые, электрически нейтральные молекулы могут образоваться только за счет последующей реакции, такой как перенос протона. Эти реакции проходят обычно очень быстро, и их скорости могут быть надежно измерены только в случае очень устойчивых карбениевых ионов типа триарилметила. Однако бензил-, ди-фенилметил- и трифенилметил-катионы, генерированные импульсным радиолизным методом в дихлорэтане [43], реагируют с галогенид-ионами с диффузно-контролируемыми скоростями. Чаще всего для оценки реакционной способности нуклеофилов используют метод конкурентных реакций. В табл. 2.7.17 приведены логарифмические величины относительной реакционной способности ряда нуклеофильных реагентов (т. е.  , где
, где  относится к реакции со стандартным нуклеофилом,
относится к реакции со стандартным нуклеофилом,  ).
).
Величины  представляют собой непосредственно наблюдаемые скорости реакции
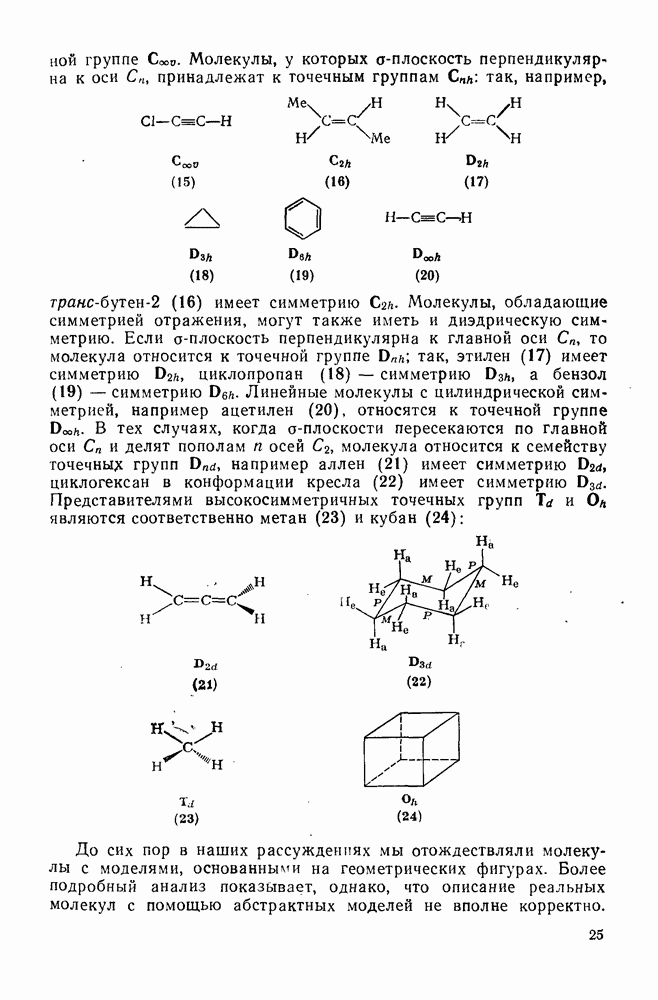
представляют собой непосредственно наблюдаемые скорости реакции  -нитромалахитового зеленого (24) с нуклеофилами,
-нитромалахитового зеленого (24) с нуклеофилами,  носится к конкурентной реакции трифенилметил-катиона, n — нуклеофильная реакционная способность в реакциях согласованного замещения метилбромида. Сравнение приведенных величин обнаруживает существенное различие в последовательности реакционной способности (например, по отношению к
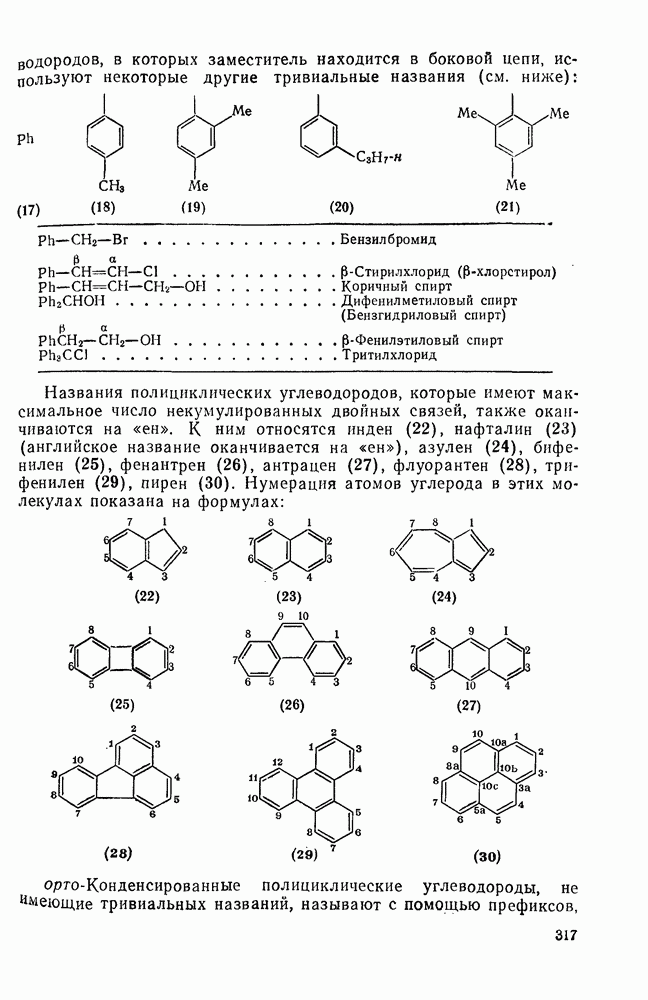
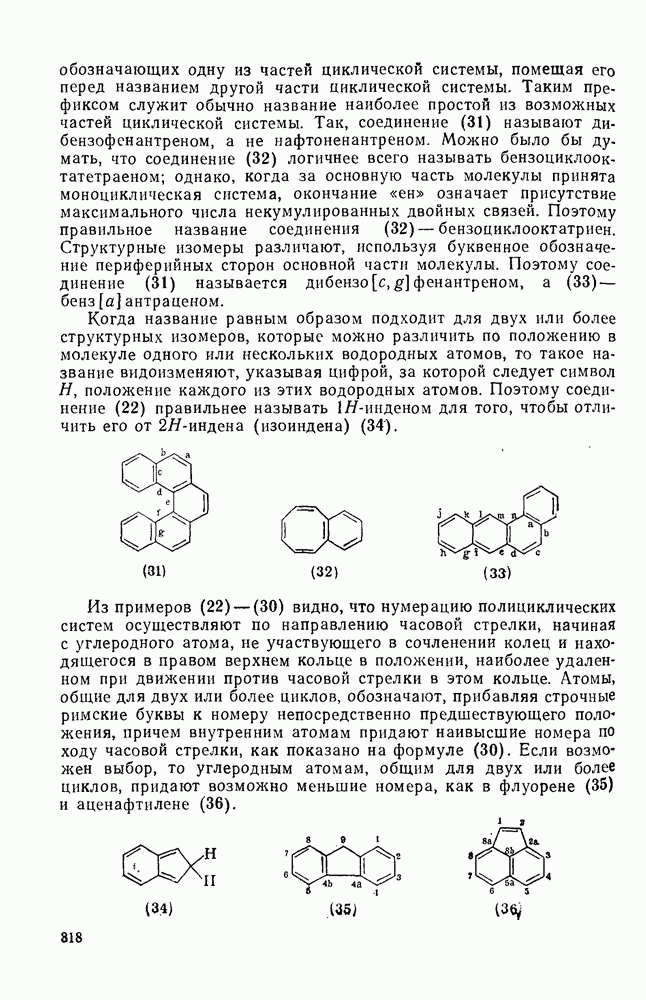
носится к конкурентной реакции трифенилметил-катиона, n — нуклеофильная реакционная способность в реакциях согласованного замещения метилбромида. Сравнение приведенных величин обнаруживает существенное различие в последовательности реакционной способности (например, по отношению к  ,
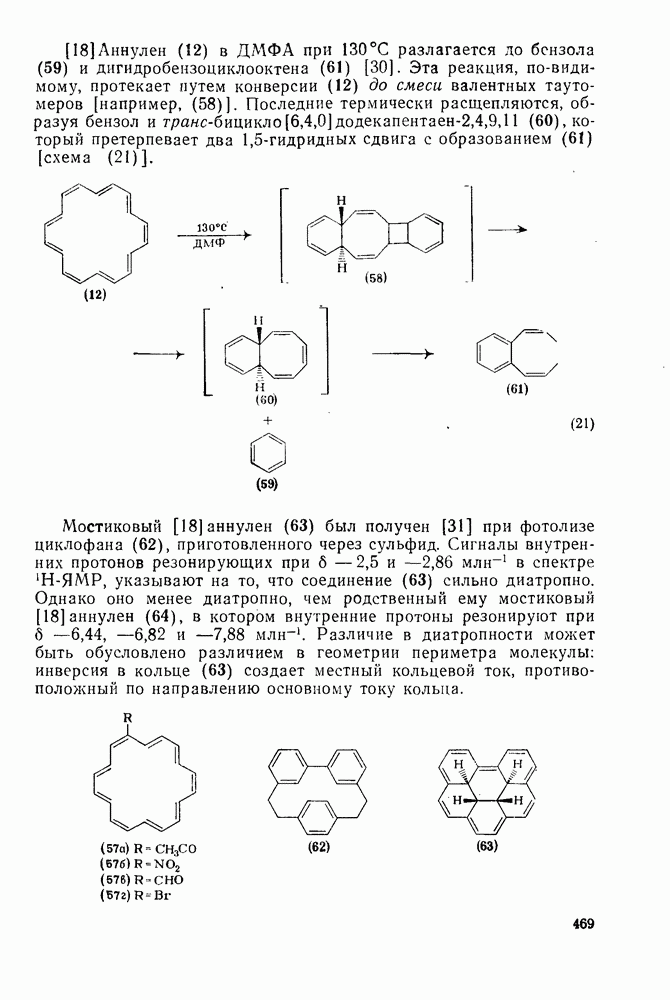
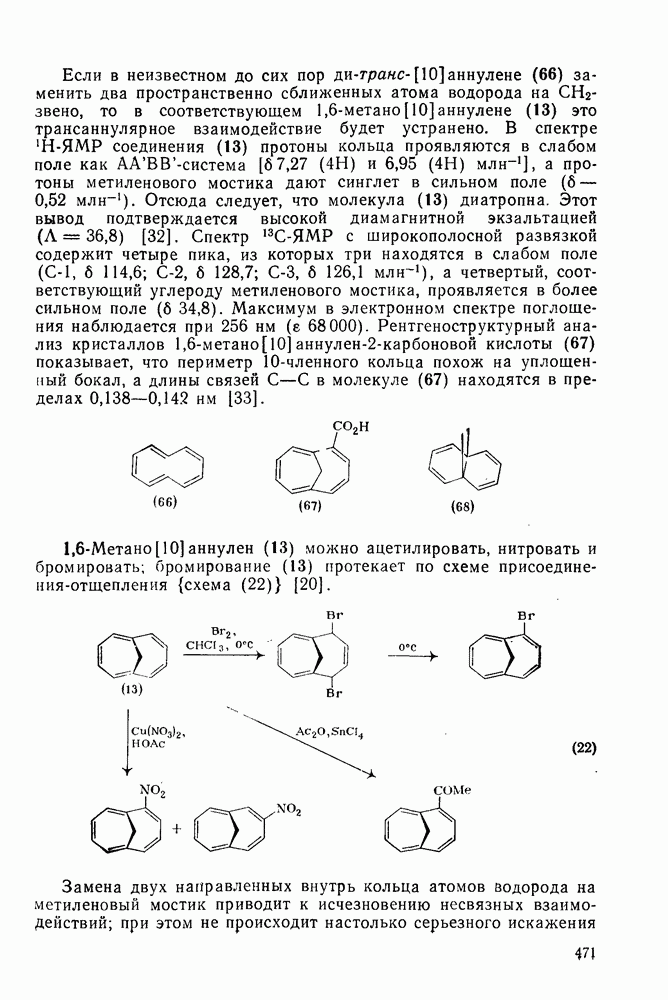
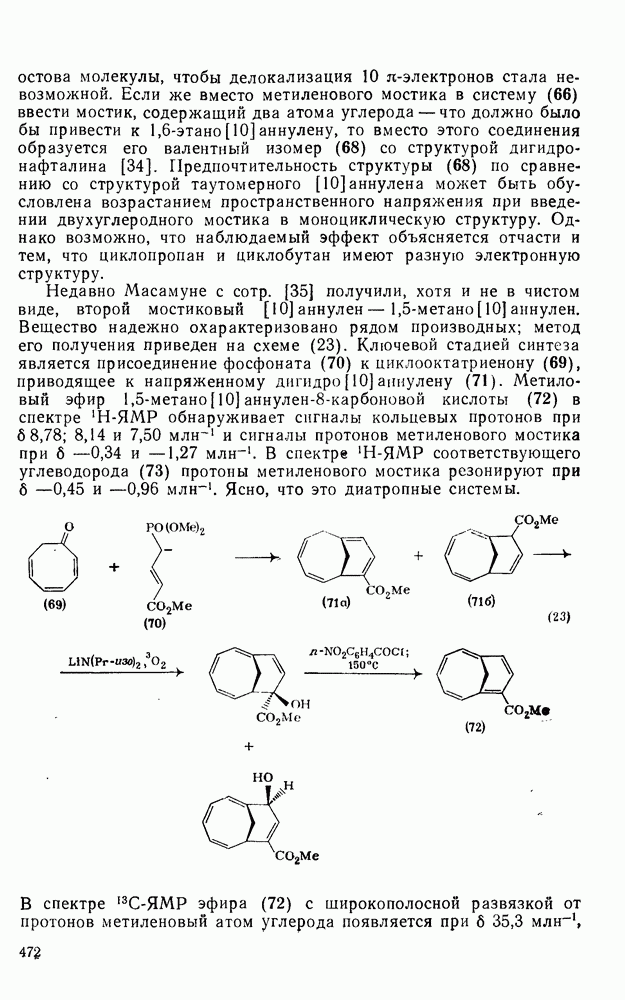
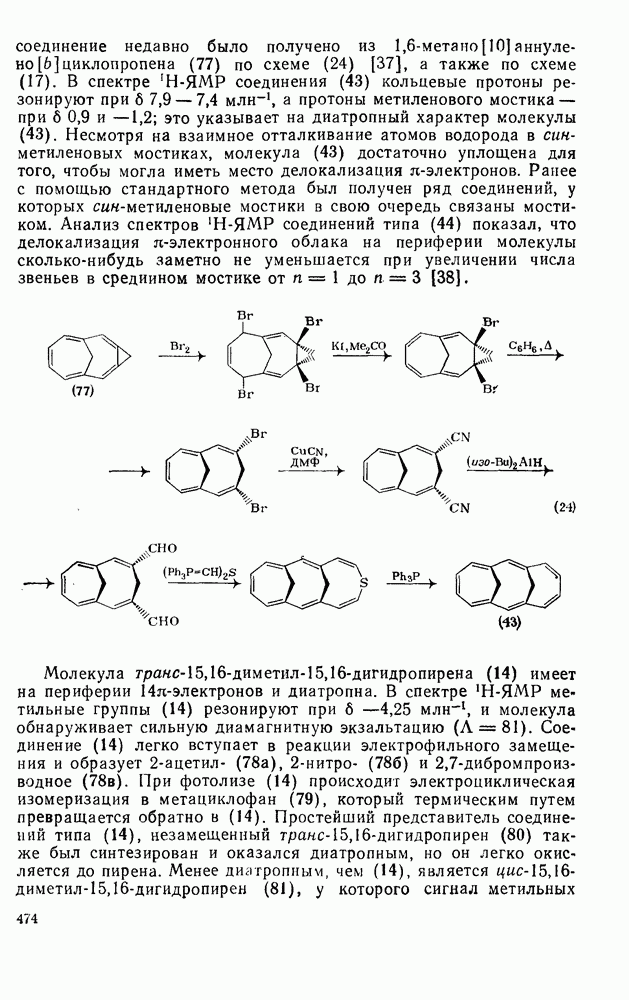
,  ) между реакциями с участием карбениевых ионов и реакциями, проходящими по согласованному механизму. Интересный и важный аспект, относящийся к величинам
) между реакциями с участием карбениевых ионов и реакциями, проходящими по согласованному механизму. Интересный и важный аспект, относящийся к величинам  , состоит в том, что эти величины непосредственно связывают реакционную способность нуклеофилов по отношению к различным карбениевым ионам и к другим электрофилам, реакционная способность которых изменяется в широких пределах в соответствии с уравнением
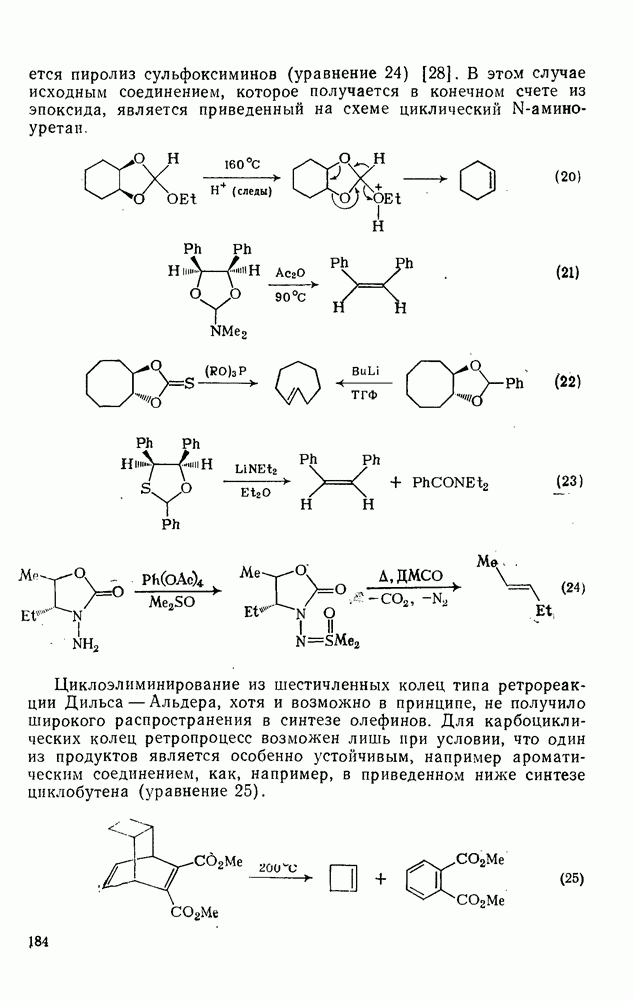
, состоит в том, что эти величины непосредственно связывают реакционную способность нуклеофилов по отношению к различным карбениевым ионам и к другим электрофилам, реакционная способность которых изменяется в широких пределах в соответствии с уравнением  (
( относится к реакции с данным нуклеофилом,
относится к реакции с данным нуклеофилом,  — к стандартной реакции с водой) [44]. Эта корреляция явно противоречит обычному обратному соотношению между реакционной способностью и селективностью [45]. Причина такого несоответствия пока является предметом обсуждения.
— к стандартной реакции с водой) [44]. Эта корреляция явно противоречит обычному обратному соотношению между реакционной способностью и селективностью [45]. Причина такого несоответствия пока является предметом обсуждения.

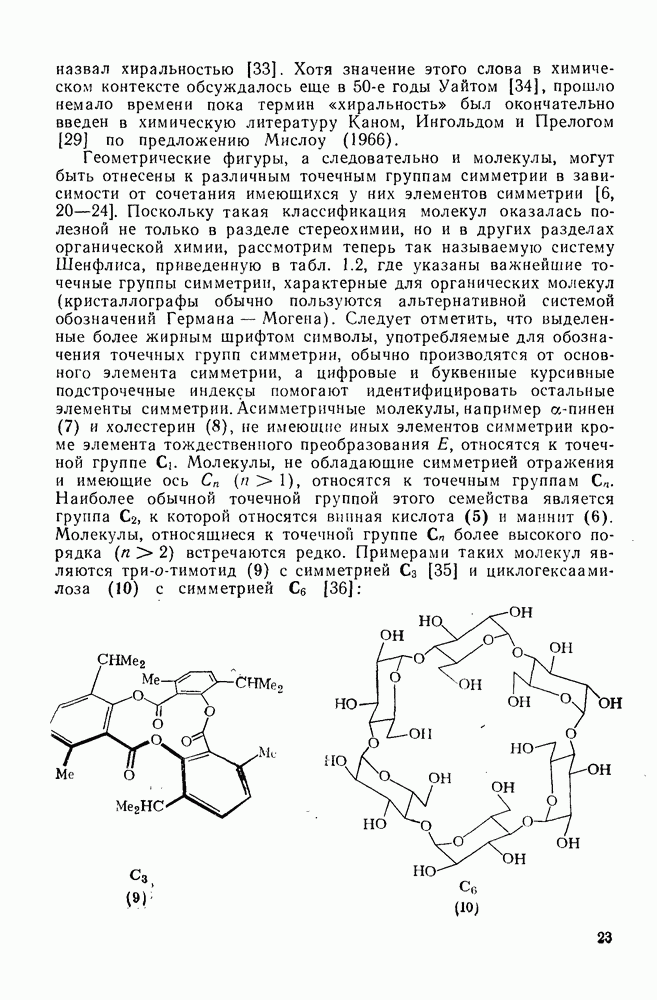
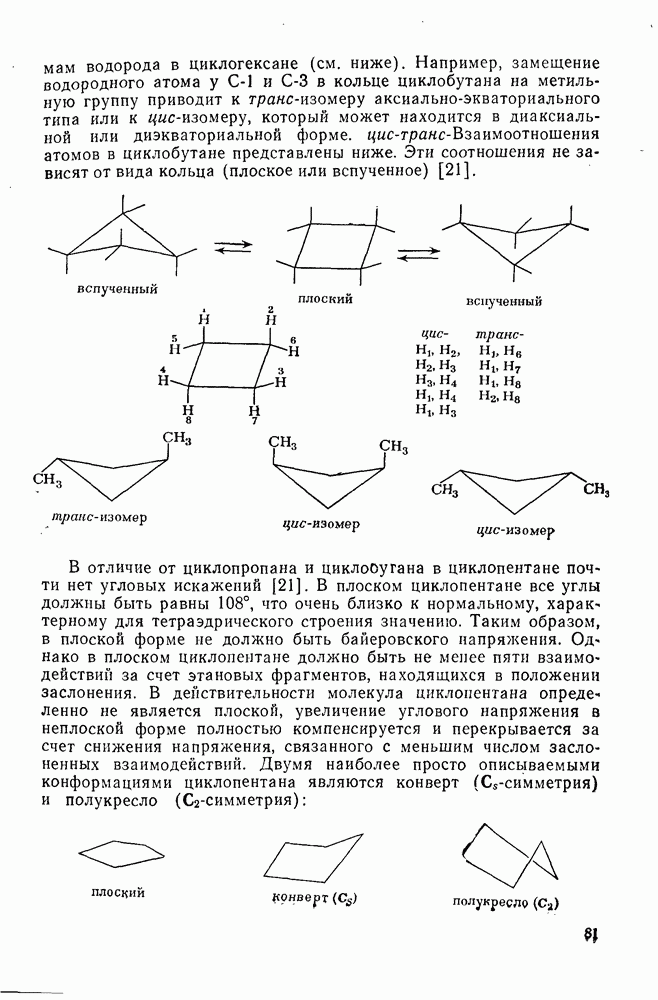
В целом нуклеофильные и основные свойства обычно изменяются симбатно. Так, реагенты с  -электронными парами могут реагировать с карбениевыми ионами, отщепляя протон, ориентированный параллельно вакантной р-орбитали, от углеродного атома в
-электронными парами могут реагировать с карбениевыми ионами, отщепляя протон, ориентированный параллельно вакантной р-орбитали, от углеродного атома в  -положении к катионному центру с образованием олефина.
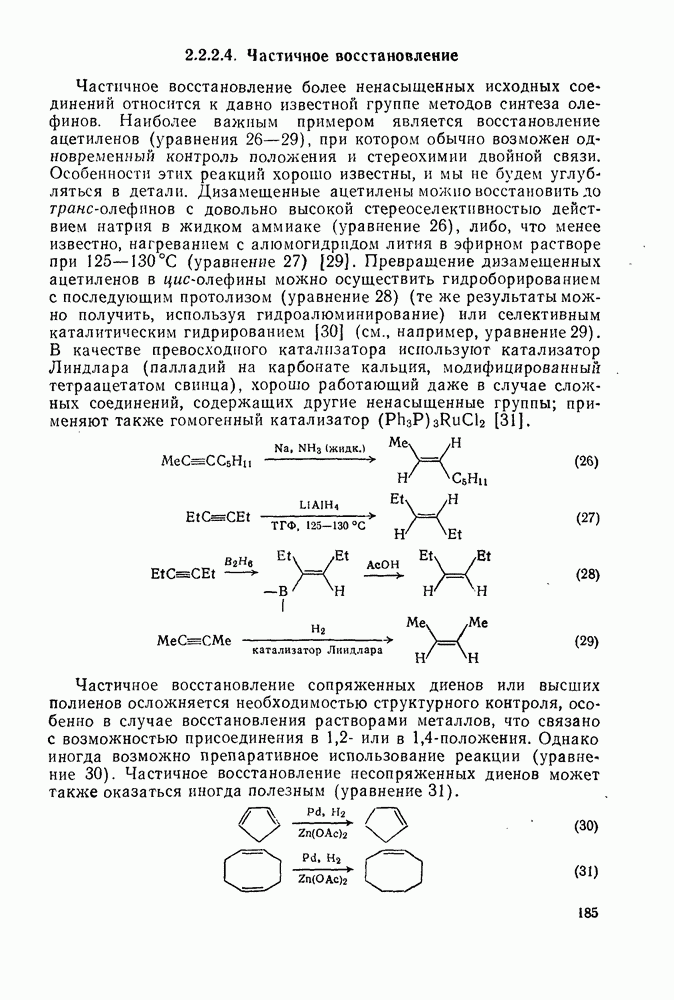
-положении к катионному центру с образованием олефина.
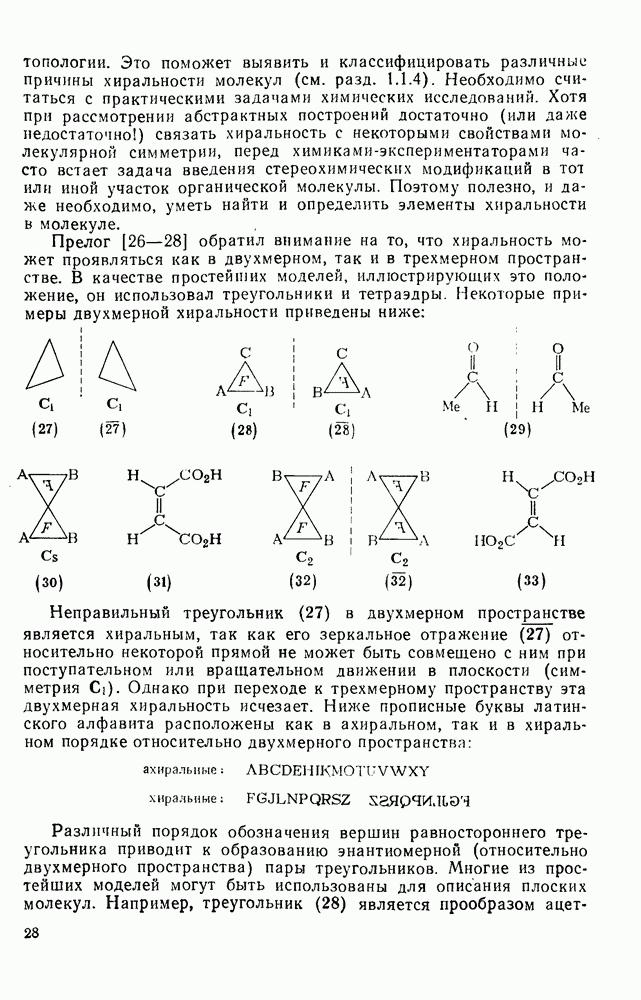
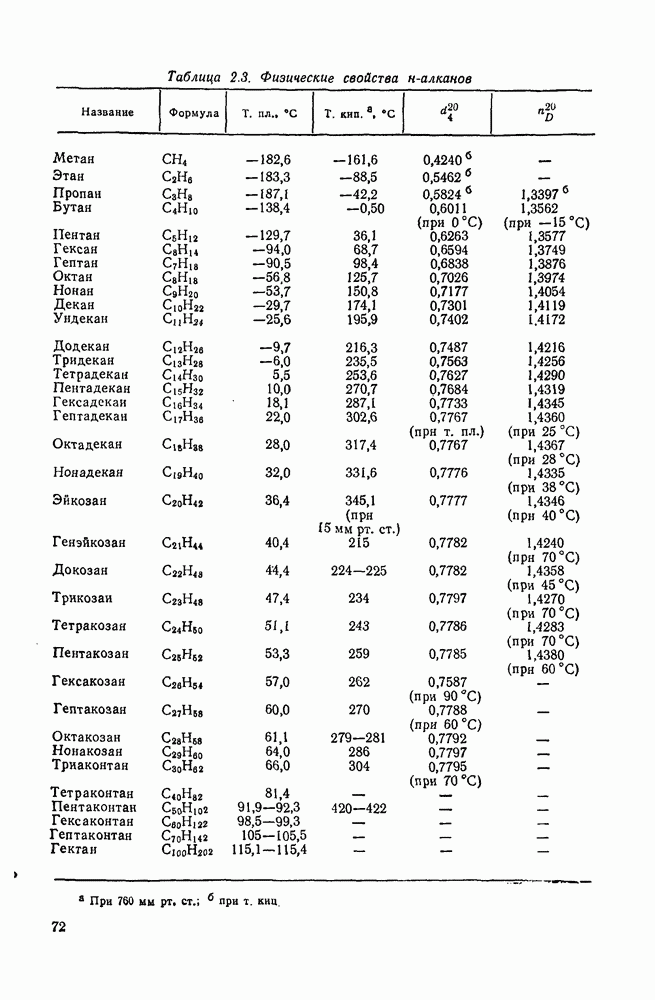
Таблица 2.7.17. Относительная реакционная способность нуклеофилов по отношению к карбениевым ионам и бромистому метилу в водной среде

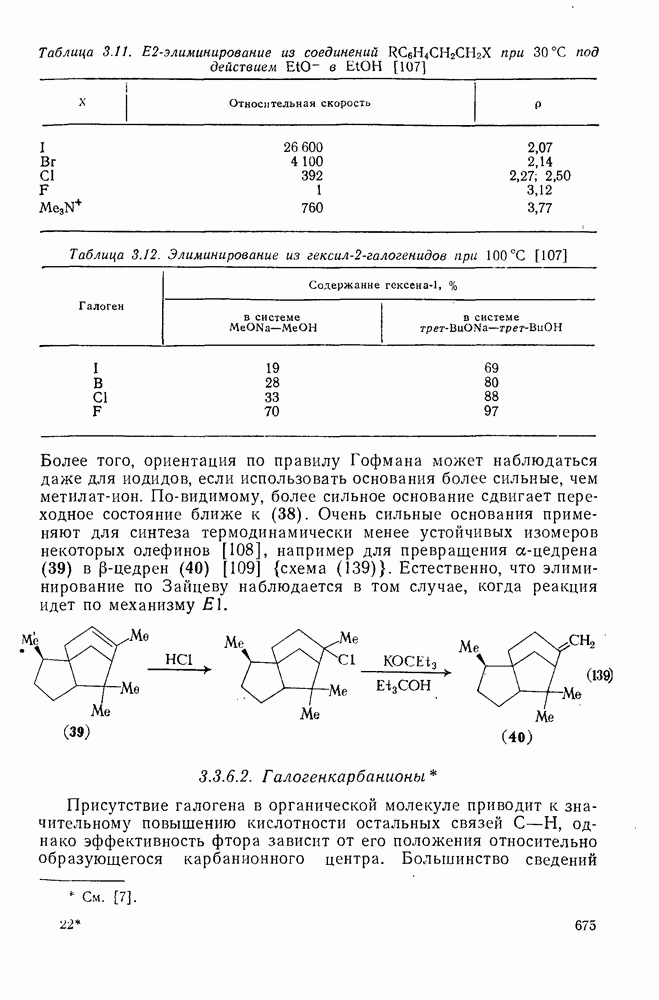
Однако в случае простых алкил-катионов преобладает координация нуклеофила с карбениевым центром. В тех случаях, когда реакция может привести к нескольким олефинам, в отсутствие пространственных затруднений преимущественно образуется олефин с наиболее замещенной двойной связью. Соотношение образующихся диастереоизомеров контролируется обычно конформационными особенностями карбениевого иона и возможностью возникновения заслоненной конформации в переходном состоянии на стадии удаления протона. Однако карбениевые ионы, генерируемые из ионов диазония, с потерей азота ведут себя иначе. Они образуются при экзотермическом процессе, имеют короткое время жизни, и направление их дальнейших реакций в значительной степени определяется конформацией аминов, из которых они были генерированы. Образование олефина может также проходить путем  -элиминирования не только протона, но и других катионов. Типичными подобными катионами являются
-элиминирования не только протона, но и других катионов. Типичными подобными катионами являются  -стабилизованные карбениевые ионы [схема (16)].
-стабилизованные карбениевые ионы [схема (16)].

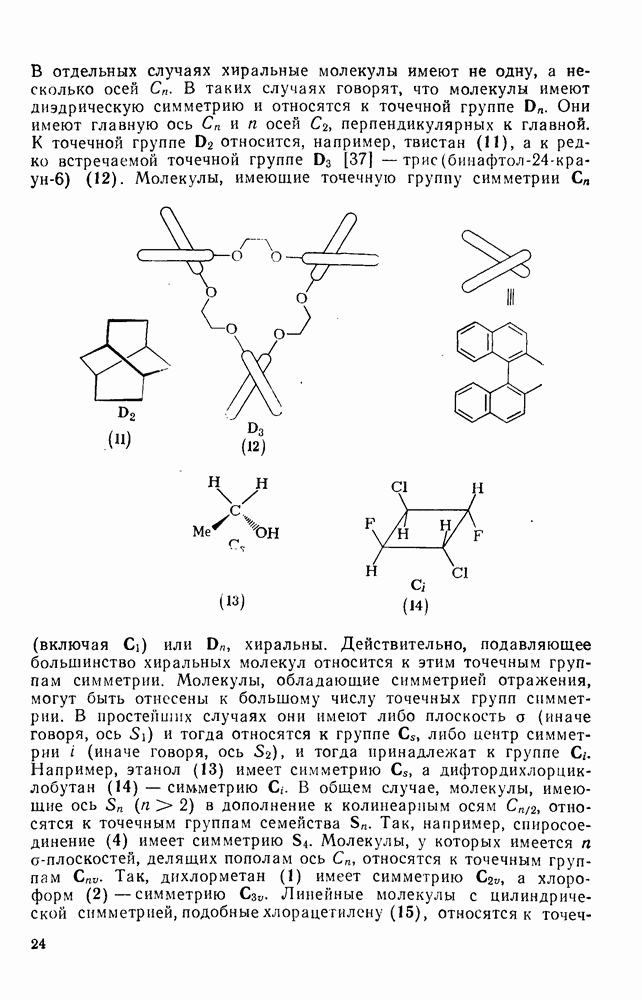
Обсуждение реакций карбениевых ионов с  -электронными парами будет ограничено здесь рассмотрением реакций с олефинами и бензоидными ароматическими соединениями. В обоих случаях первоначальным продуктом является другой карбениевый ион, который далее реагирует с образованием устойчивых продуктов. Среди реакций циклогексадиенил-катионов, генерируемых электрофильной атакой на бензоидиые соединения, преобладает реакция, ведущая к восстановлению ароматического секстета обычно за счет потери протона. Карбениевые ионы, образующиеся при взаимодействии карбениевых ионов с олефинами, могут претерпевать дальнейшие превращения по нескольким конкурирующим направлениям, одним из которых является атака на другую молекулу олефина, что приводит к образованию полимерных продуктов. Из простых
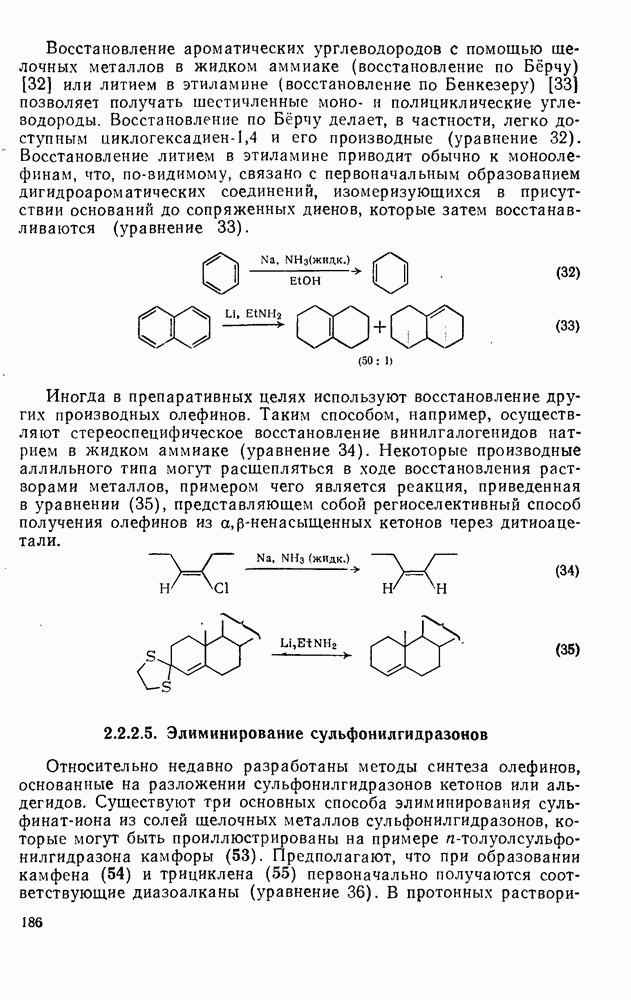
-электронными парами будет ограничено здесь рассмотрением реакций с олефинами и бензоидными ароматическими соединениями. В обоих случаях первоначальным продуктом является другой карбениевый ион, который далее реагирует с образованием устойчивых продуктов. Среди реакций циклогексадиенил-катионов, генерируемых электрофильной атакой на бензоидиые соединения, преобладает реакция, ведущая к восстановлению ароматического секстета обычно за счет потери протона. Карбениевые ионы, образующиеся при взаимодействии карбениевых ионов с олефинами, могут претерпевать дальнейшие превращения по нескольким конкурирующим направлениям, одним из которых является атака на другую молекулу олефина, что приводит к образованию полимерных продуктов. Из простых  -олефинов при катионной полимеризации образуются продукты с низкой молекулярной массой, поскольку в таких системах процессы переноса преобладают над процессами роста цепи. Полимеры с высокой молекулярной массой образуются обычно из таких олефинов как виниловые эфиры и стиролы. Типичные величины относительной реакционной способности виниловых мономеров, определенные при изучении сополимеризации в нитробензоле, следующие [46]: бутадиен 0,02, изопрен 0,12, винилацетат 0,4, стирол (1,0), изобутен 4; виниловые эфиры реагируют очень быстро. Иногда катионная полимеризация протекает стереорегулярно.
-олефинов при катионной полимеризации образуются продукты с низкой молекулярной массой, поскольку в таких системах процессы переноса преобладают над процессами роста цепи. Полимеры с высокой молекулярной массой образуются обычно из таких олефинов как виниловые эфиры и стиролы. Типичные величины относительной реакционной способности виниловых мономеров, определенные при изучении сополимеризации в нитробензоле, следующие [46]: бутадиен 0,02, изопрен 0,12, винилацетат 0,4, стирол (1,0), изобутен 4; виниловые эфиры реагируют очень быстро. Иногда катионная полимеризация протекает стереорегулярно.
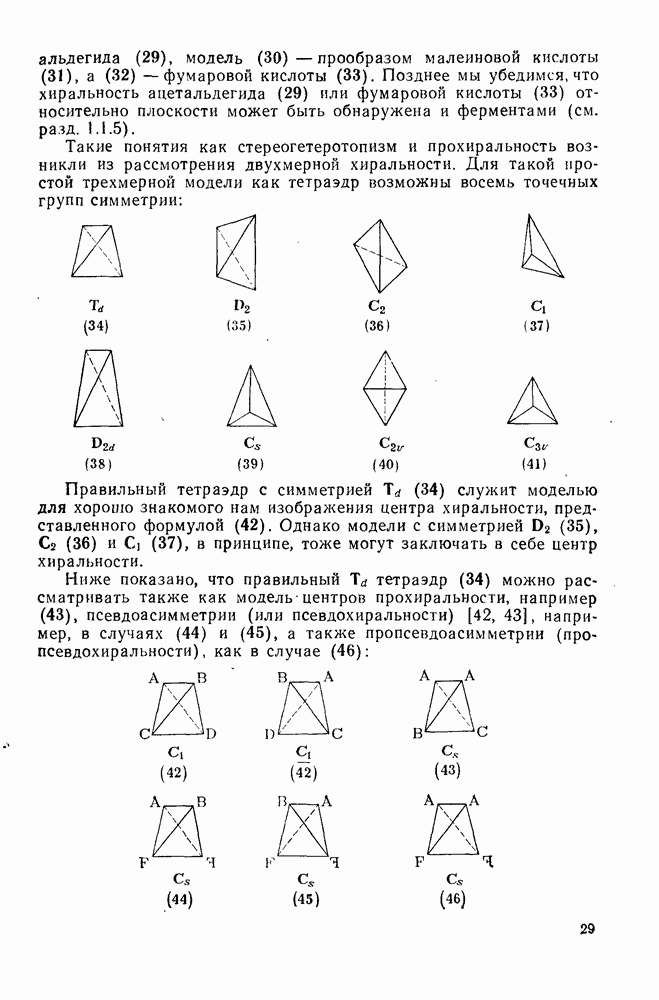
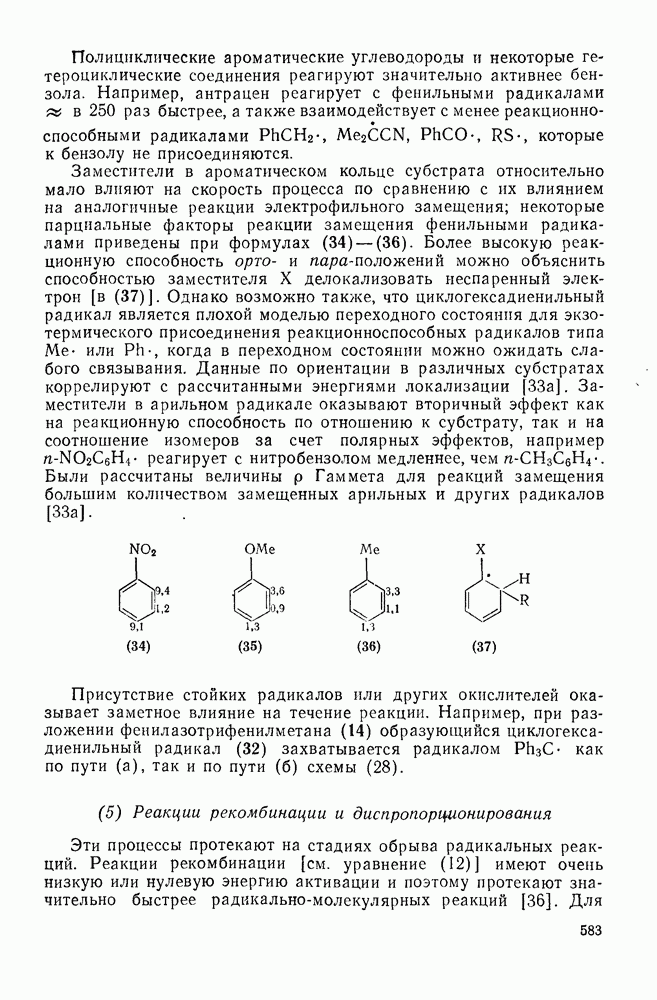
Таблица 2.7.18. Селективность в реакциях электрофильного ароматического замещения (40)

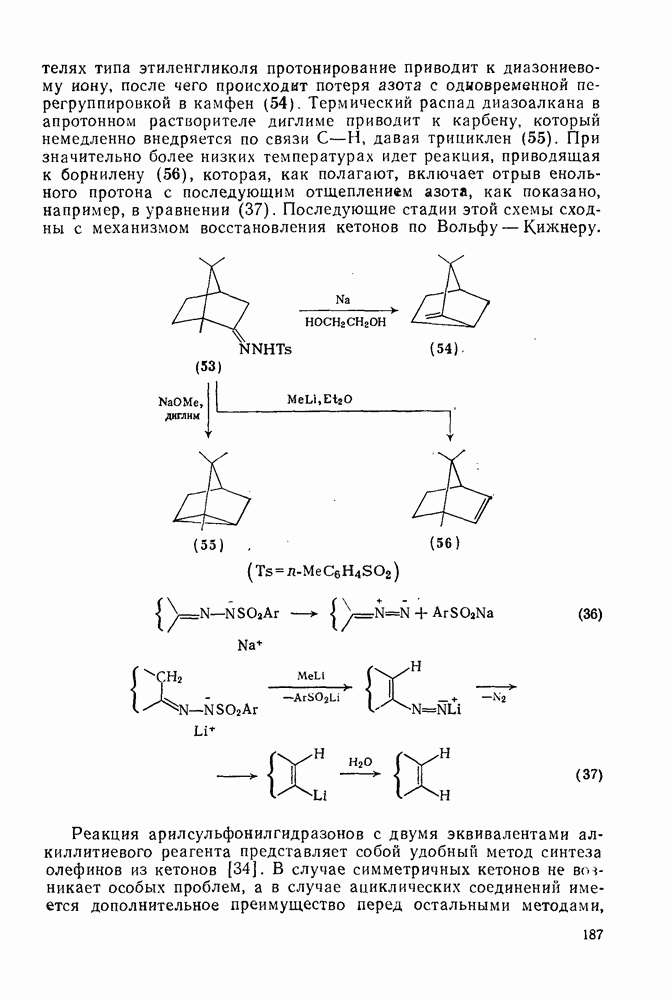
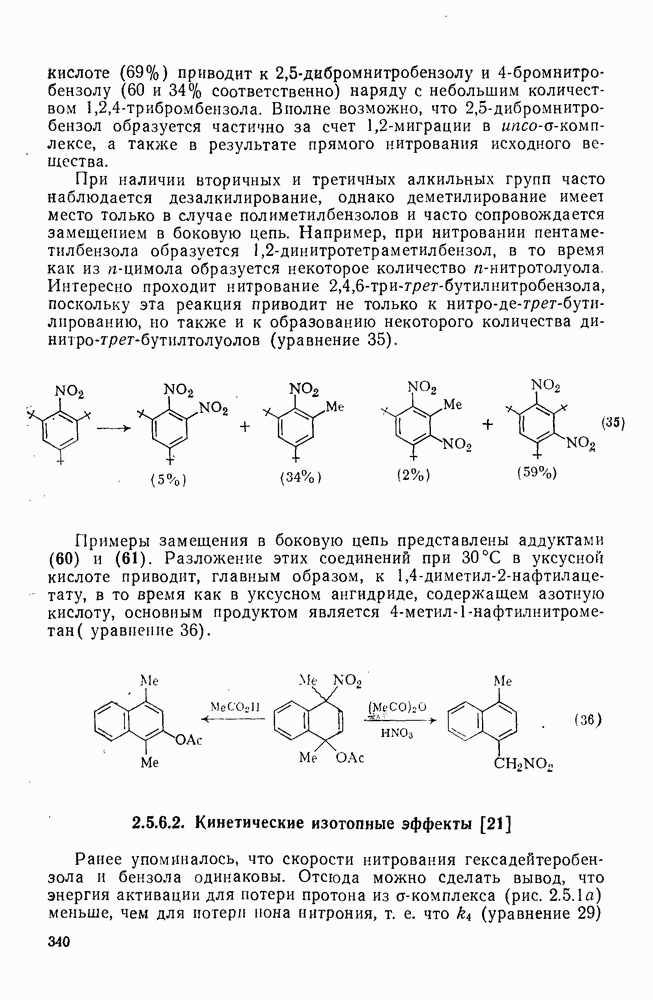
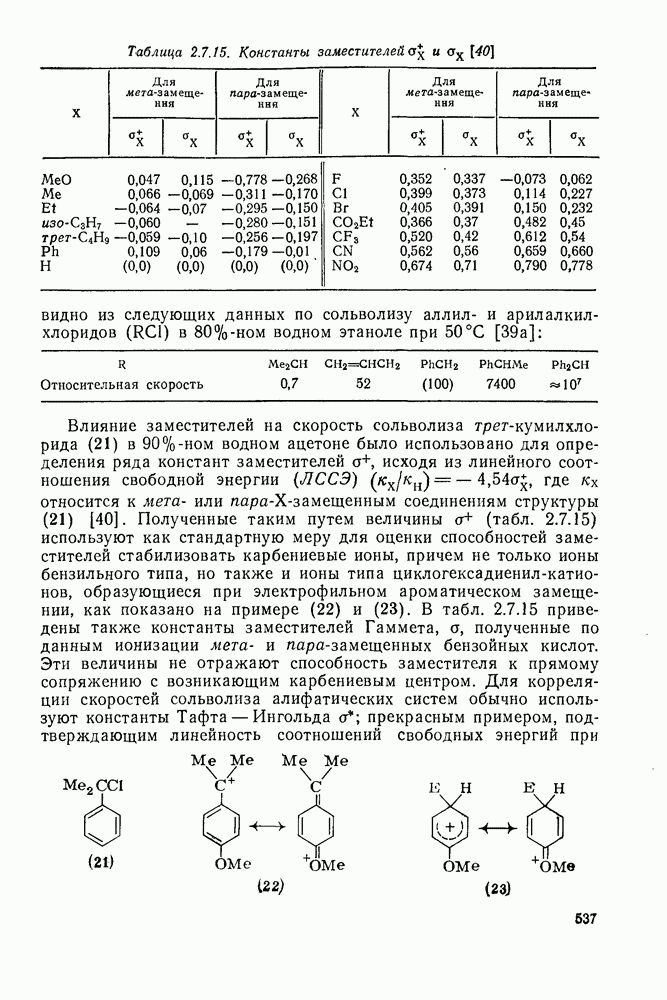
Атака ароматического ядра любым электрофилом приводит к возникновению карбениевого иона. Таким образом, константы скоростей этой атаки представляют собой константы скоростей образования делокализованных карбениевых ионов. Последующий перенос протона является лимитирующей стадией только в некоторых специальных случаях, например если этот перенос очень сильно пространственно затруднен. Реакционную способность различных положений в монозамещенных бензолах обычно рассматривают относительно замещения одного из шести равноценных положений бензола (парциальные факторы скорости,  ,
,  ). Для данного реагента
). Для данного реагента  и
и  пропорциональны величине
пропорциональны величине  для мета- и пара-заместителя; при этом коистаита пропорциональности р, которая имеет отрицательные значения для электрофильного замещения, является мерой селективности электрофила [40]. Некоторые значения констант
для мета- и пара-заместителя; при этом коистаита пропорциональности р, которая имеет отрицательные значения для электрофильного замещения, является мерой селективности электрофила [40]. Некоторые значения констант  приведены в табл. 2.7.18. Из приведенных данных заслуживает внимания очень низкая селективность этилирования по Фриделю—Крафтсу, включая использование очень мощного реагента, по-видимому, этил-катиона, и значительно более высокая селективность при электрофильном ацилировании по Фриделю — Крафтсу.
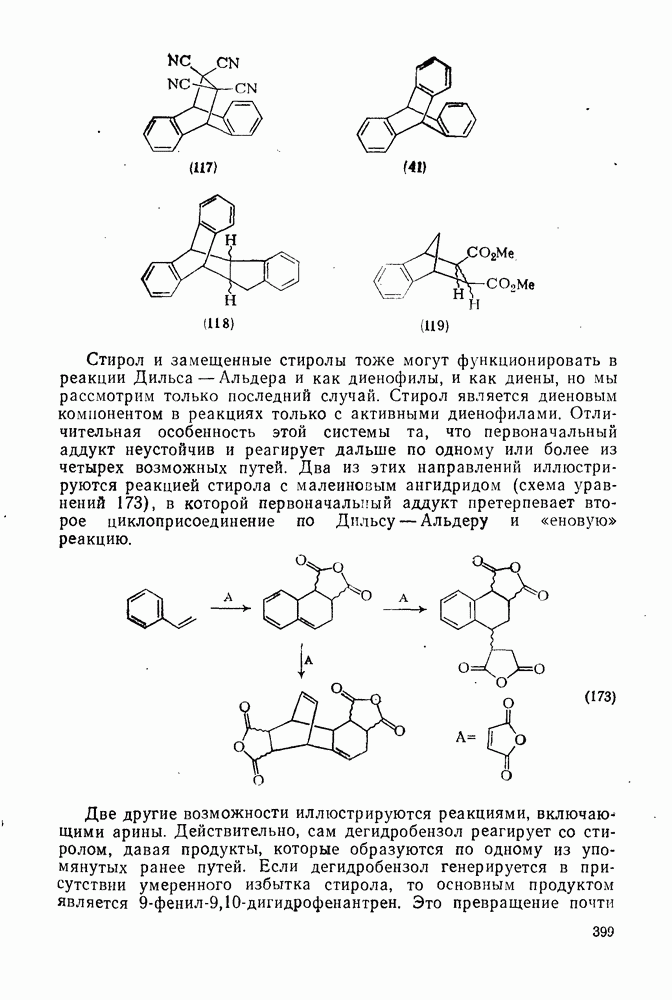
приведены в табл. 2.7.18. Из приведенных данных заслуживает внимания очень низкая селективность этилирования по Фриделю—Крафтсу, включая использование очень мощного реагента, по-видимому, этил-катиона, и значительно более высокая селективность при электрофильном ацилировании по Фриделю — Крафтсу.
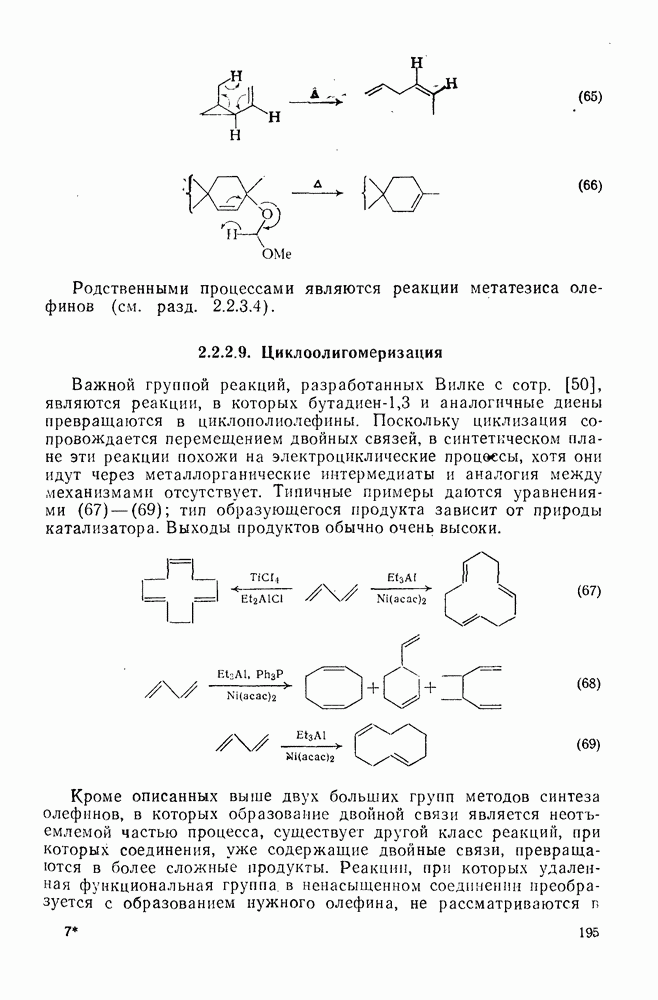
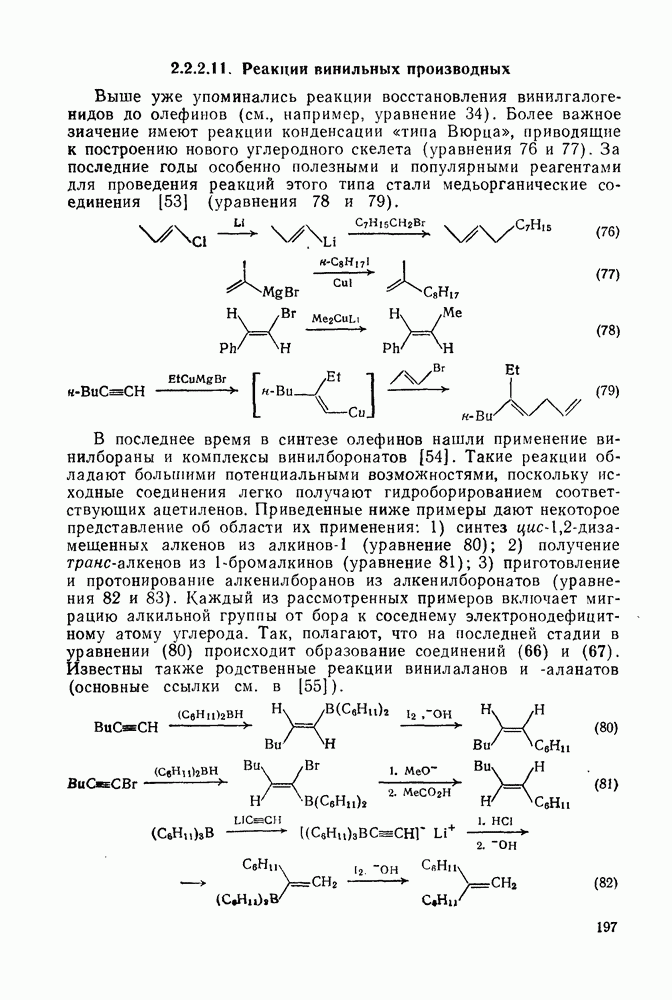
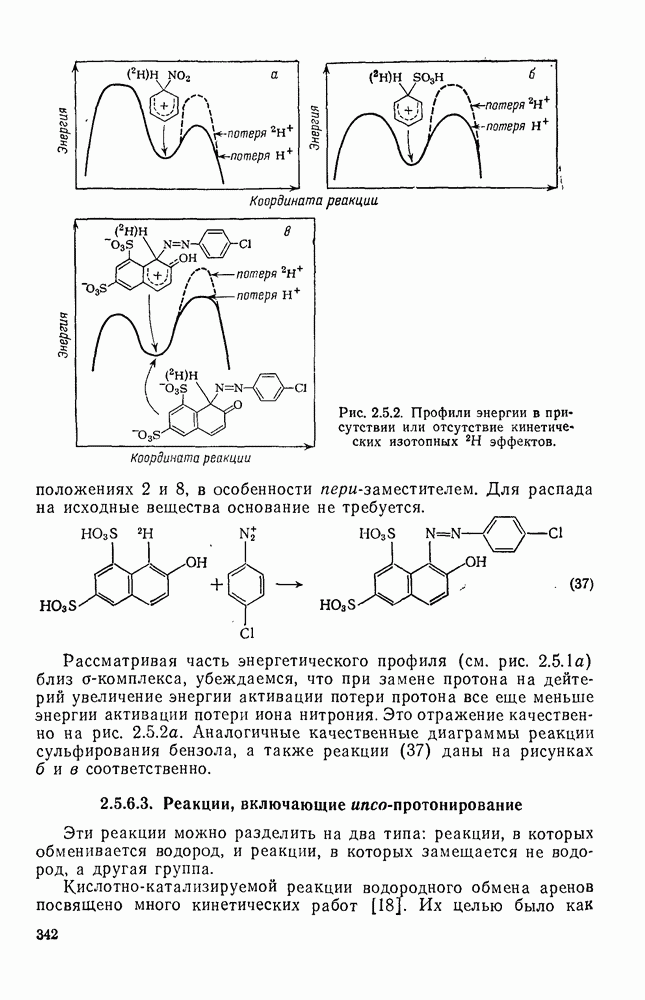
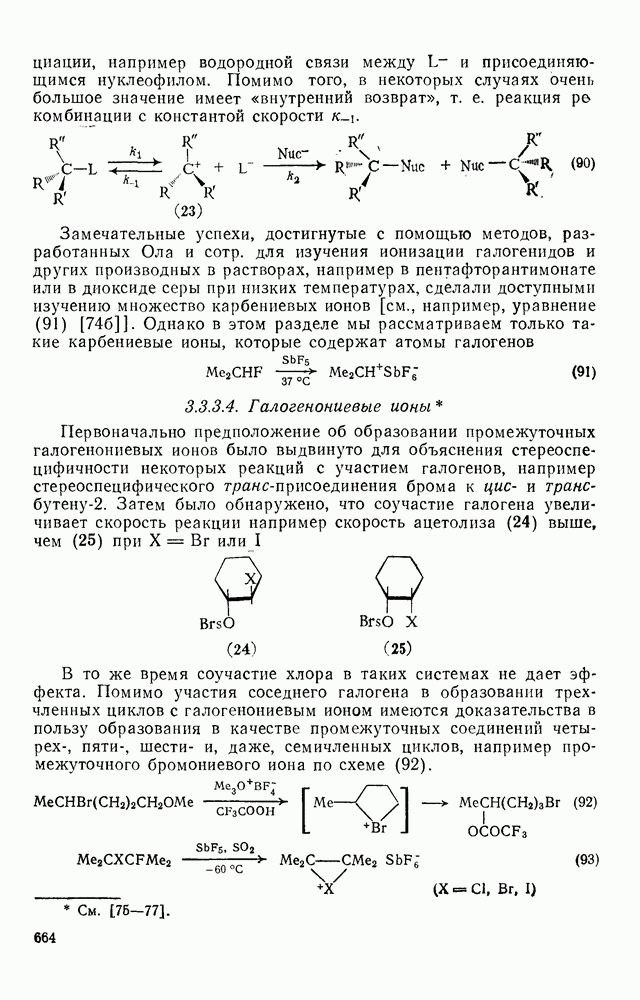
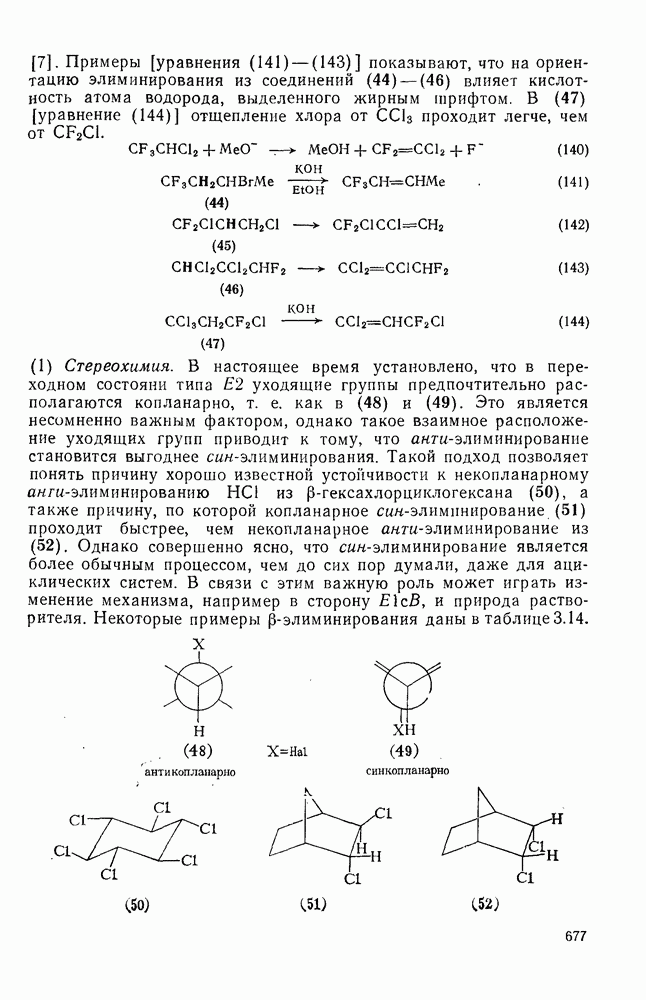
Хорошо известны межмолекулярные реакции карбениевых ионов с электронами  -связей, когда электроны осуществляют связь водорода с потенциально более стабильным карбениевым центром; в целом такие реакции представляют собой гидридный перенос [см., например, уравнение (10)]. Аналогичное прямое межмолеку-лярное перемещение алкильного остатка с его электронной парой к карбениевому центру однозначно не доказано [16], однако образование пентакоординированных карбоииевых ионов координацией карбениевых ионов со связями
-связей, когда электроны осуществляют связь водорода с потенциально более стабильным карбениевым центром; в целом такие реакции представляют собой гидридный перенос [см., например, уравнение (10)]. Аналогичное прямое межмолеку-лярное перемещение алкильного остатка с его электронной парой к карбениевому центру однозначно не доказано [16], однако образование пентакоординированных карбоииевых ионов координацией карбениевых ионов со связями  и
и  при газофазных реакциях хорошо известно [15].
при газофазных реакциях хорошо известно [15].
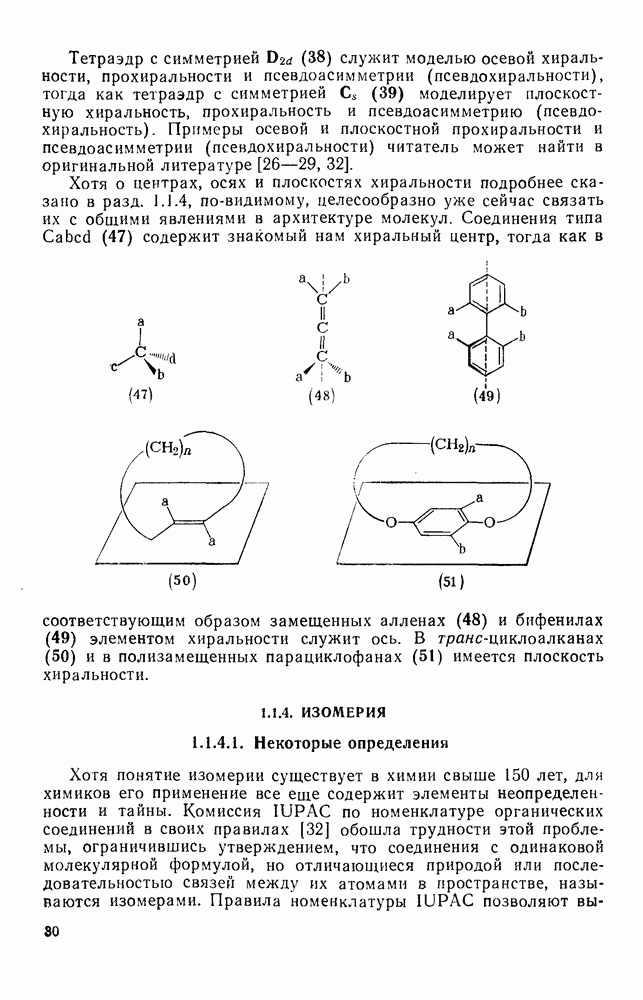
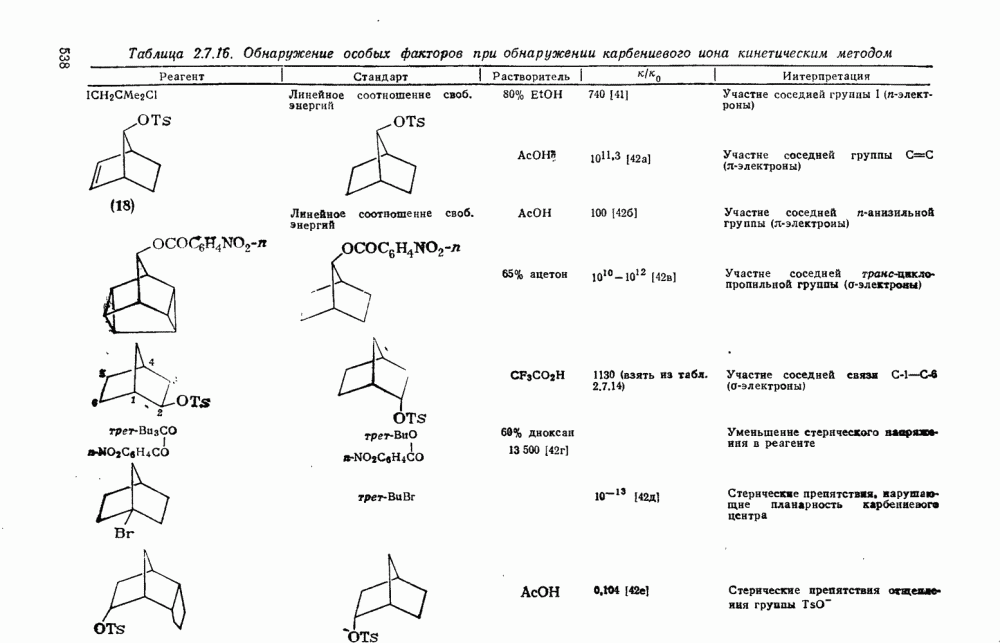
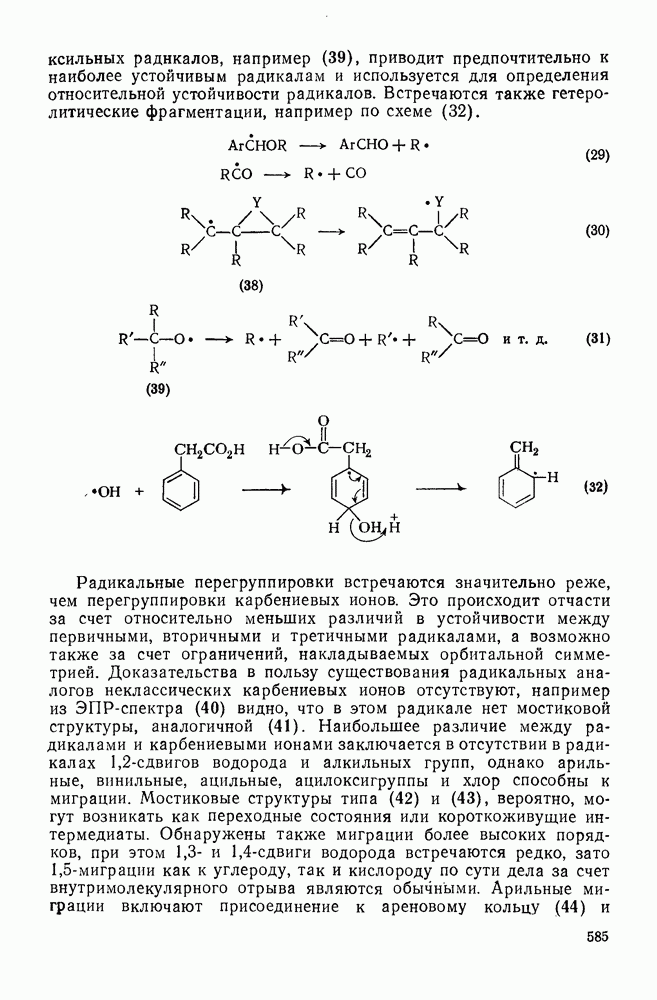
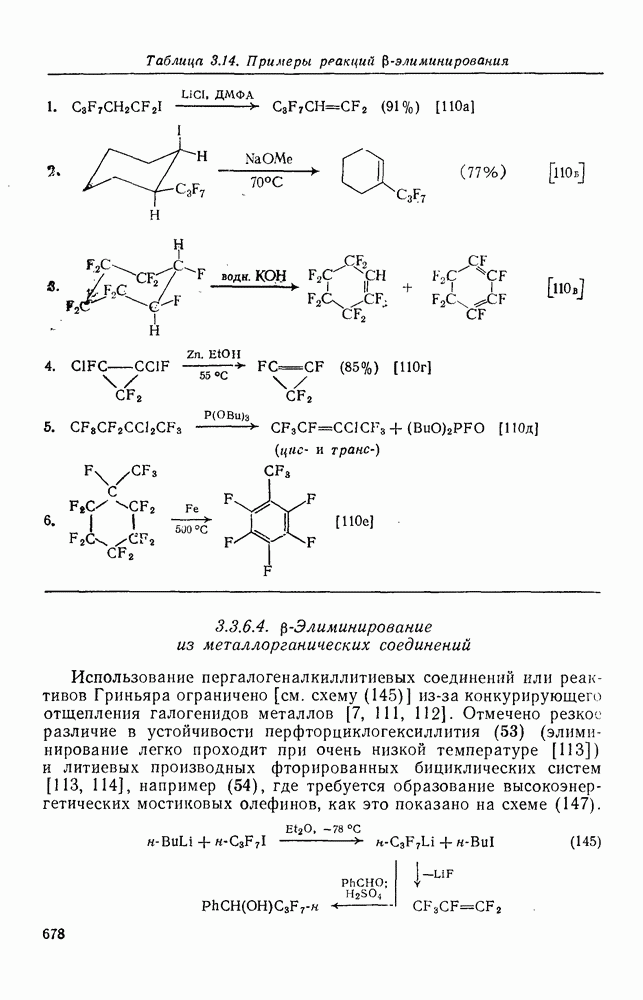
Более важными и характерными для карбениевых ионов являются их внутримолекулярные реакции с электронами  -связей. Обычным результатом является
-связей. Обычным результатом является  -гидридный или
-гидридный или  -алкильный сдвиг, приводящий к образованию перегруппированного, термодинамически более устойчивого карбениевого иона [см. схемы (17) и (18)]. Значительно реже встречаются перегруппировки с
-алкильный сдвиг, приводящий к образованию перегруппированного, термодинамически более устойчивого карбениевого иона [см. схемы (17) и (18)]. Значительно реже встречаются перегруппировки с  -сдвигом или со сдвигами более высоких порядков. Следует обратить внимание на то, что переходное состояние для
-сдвигом или со сдвигами более высоких порядков. Следует обратить внимание на то, что переходное состояние для  -алкильного сдвига, представленное формулой (25), эквивалентно пентакоординированному карбониевому иону.
-алкильного сдвига, представленное формулой (25), эквивалентно пентакоординированному карбониевому иону.
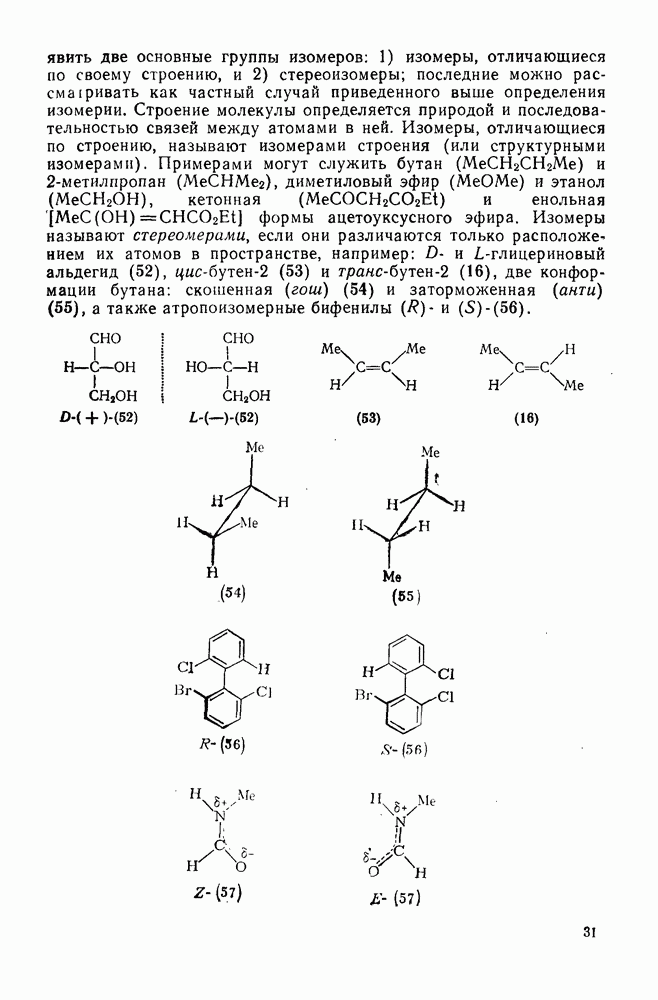
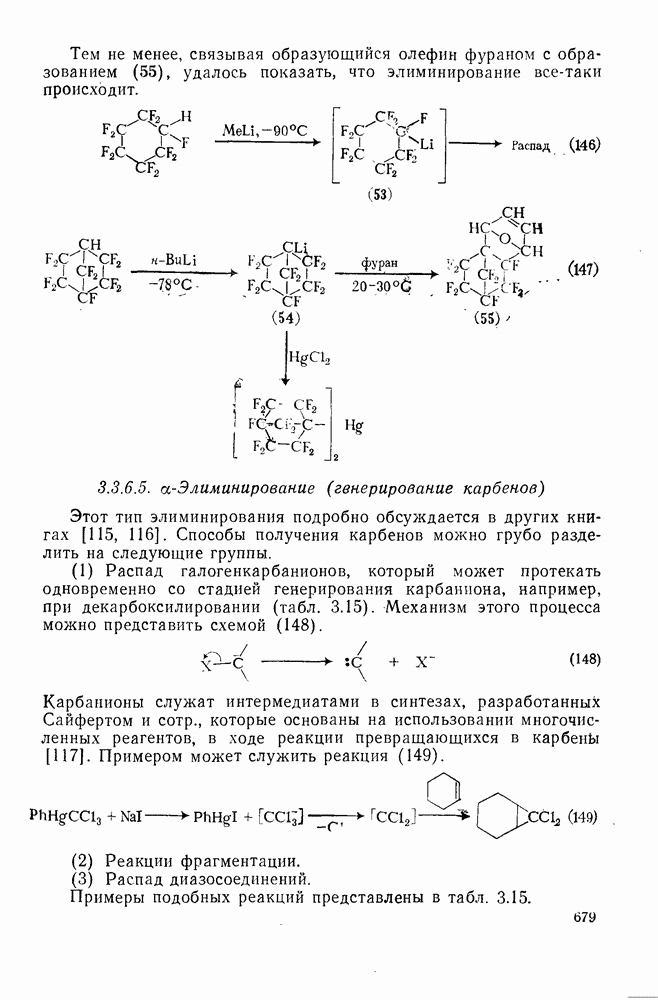
Особенно широко обсуждался вопрос, следует ли рассматривать пентакоординированный карбонатной в реакциях, идущих со скелетной перегруппировкой при наличии кинетических или стереохимических доказательств участия соседней группы за счет  -связей, в качестве единственного (амбидеитного) интермедиата, из которого далее образуются как перегруппированный, так и неперегруппированный продукты реакции. В любом случае структура (25) является устойчивой при небольших смещениях вдоль всех молекулярных координат, за исключением координаты, определяющей перегруппировку родственных карбениевых ионов; две точки зрения различаются только тем, следует ли структуру (25) относить к максимуму или к минимуму вдоль этой координаты перегруппировки.
-связей, в качестве единственного (амбидеитного) интермедиата, из которого далее образуются как перегруппированный, так и неперегруппированный продукты реакции. В любом случае структура (25) является устойчивой при небольших смещениях вдоль всех молекулярных координат, за исключением координаты, определяющей перегруппировку родственных карбениевых ионов; две точки зрения различаются только тем, следует ли структуру (25) относить к максимуму или к минимуму вдоль этой координаты перегруппировки.

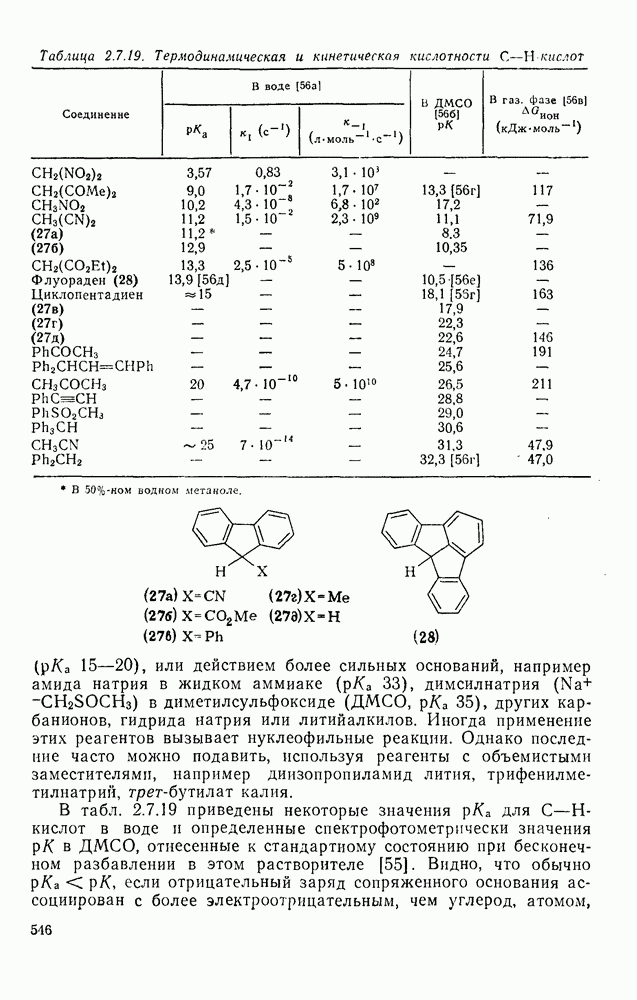
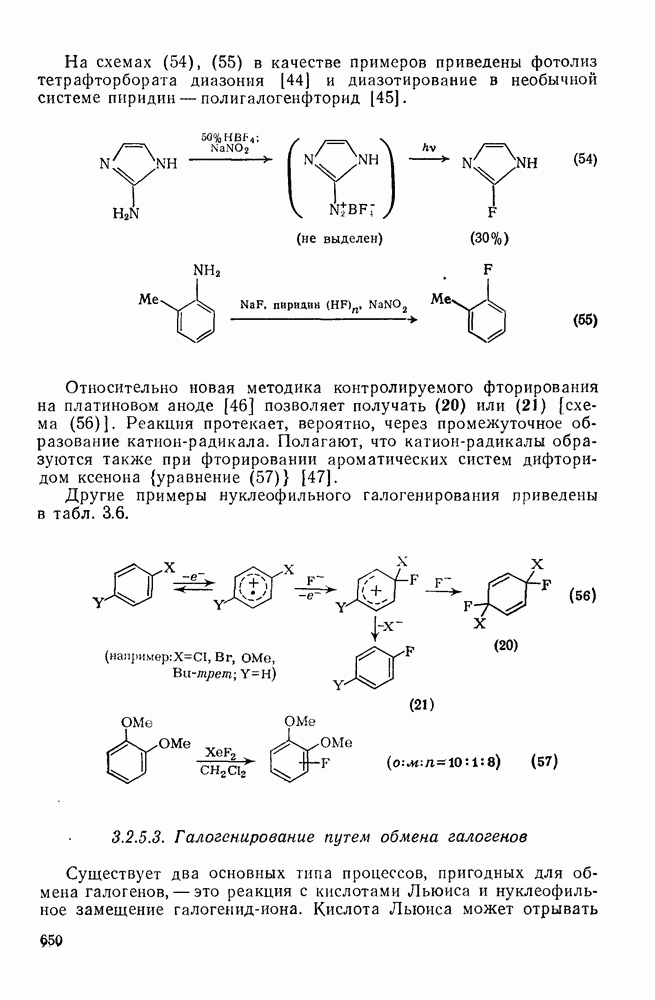
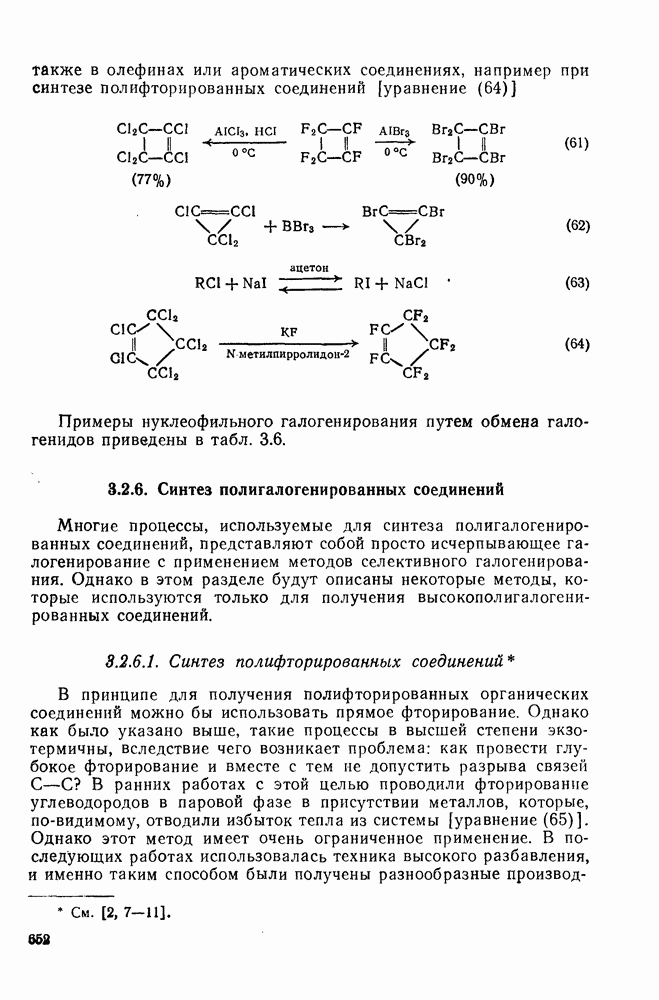
1,2-Сдвиги в карбениевых ионах могут быть отнесены к перициклическим (конкретно, сигматропным) реакциям [47]. Орбитали, вовлеченные в супраповерхностные сдвиги, показанные на схемах (17) и (18), топологически аналогичны  -орбиталям цикло-пропенил-катиона — карбениевого иона, который в соответствии с обычными критериями Хюккеля является ароматическим. По этой причине 1,2-гидридные сдвиги представляют собою термически разрешенные согласованные процессы, так же как и алкидные сдвиги с сохранением конфигурации в мигрирующей группе; этот стереохимический результат установлен экспериментально [48].
-орбиталям цикло-пропенил-катиона — карбениевого иона, который в соответствии с обычными критериями Хюккеля является ароматическим. По этой причине 1,2-гидридные сдвиги представляют собою термически разрешенные согласованные процессы, так же как и алкидные сдвиги с сохранением конфигурации в мигрирующей группе; этот стереохимический результат установлен экспериментально [48].
Внутримолекулярные 1,2-сдвиги при комнатной температуре обычно протекают быстро, однако во многих случаях их можно проследить кинетически (обычно с помощью ЯМР-спектроскопии) при низких температурах.
Однако существует ряд примеров, когда перегруппировки карбокатионов протекают слишком быстро (относительно временной шкалы ЯМР-спектрометра) даже при температуре  , что отвечает энергии активации перегруппировки менее
, что отвечает энергии активации перегруппировки менее  в этом случае трудно отличить пару быстро взаимопревращающихся карбениевых ионов от единственного карбокатиона промежуточной структуры.
в этом случае трудно отличить пару быстро взаимопревращающихся карбениевых ионов от единственного карбокатиона промежуточной структуры.
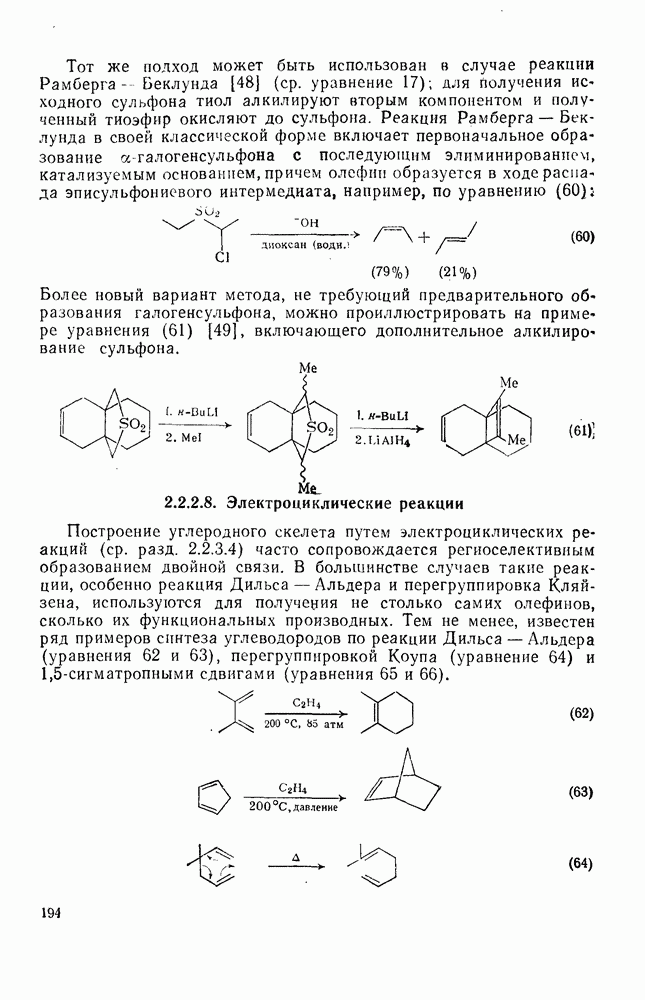
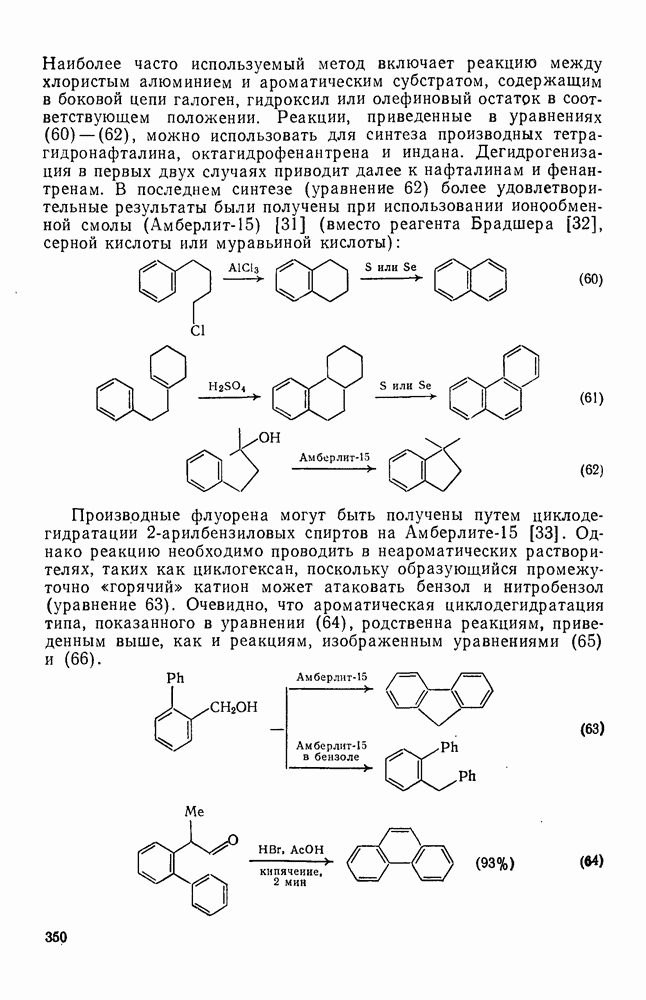
Легкость миграции различных групп при перегруппировках карбениевых ионов зависит от ряда факторов. Одним из основных факторов является необходимая конформация исходного карбокатиона, при которой связь, присоединяющая мигрирующую группу к исходному пункту миграции, должна лежать в той же плоскости, что и вакантная р-орбиталь карбениевого центра. Достижение такой конформации зависит от природы других групп, находящихся у исходного и конечного пункта миграции, которые будут определять также термодинамику перегруппировки и ее скорость за счет напряжения заслонения в переходном состоянии, например типа структуры (25). Кроме того, на перегруппировку влияет также мигрирующая способность, присущая данной группе; обычная последовательность легкости миграции:  .
.
Сложные перегруппировки, особенно в полициклических карбениевых ионах, можно представить в виде последовательно проходящих  -сдвигов. Важным примером является превращение эпоксида сквалена в ланостерин. В ряде случаев выбор между альтернативными последовательностями
-сдвигов. Важным примером является превращение эпоксида сквалена в ланостерин. В ряде случаев выбор между альтернативными последовательностями  -сдвигов можно сделать на основании теории графов в сочетании с теоретическими квантовыми или молекулярно-механическими расчетами энергий возможных промежуточных катионов [49].
-сдвигов можно сделать на основании теории графов в сочетании с теоретическими квантовыми или молекулярно-механическими расчетами энергий возможных промежуточных катионов [49].
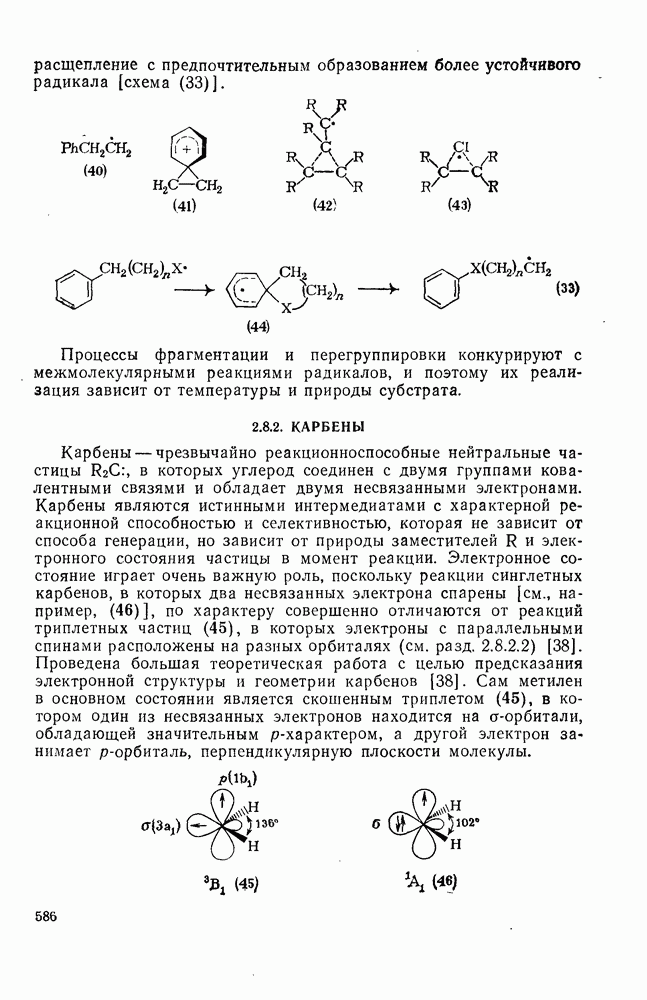
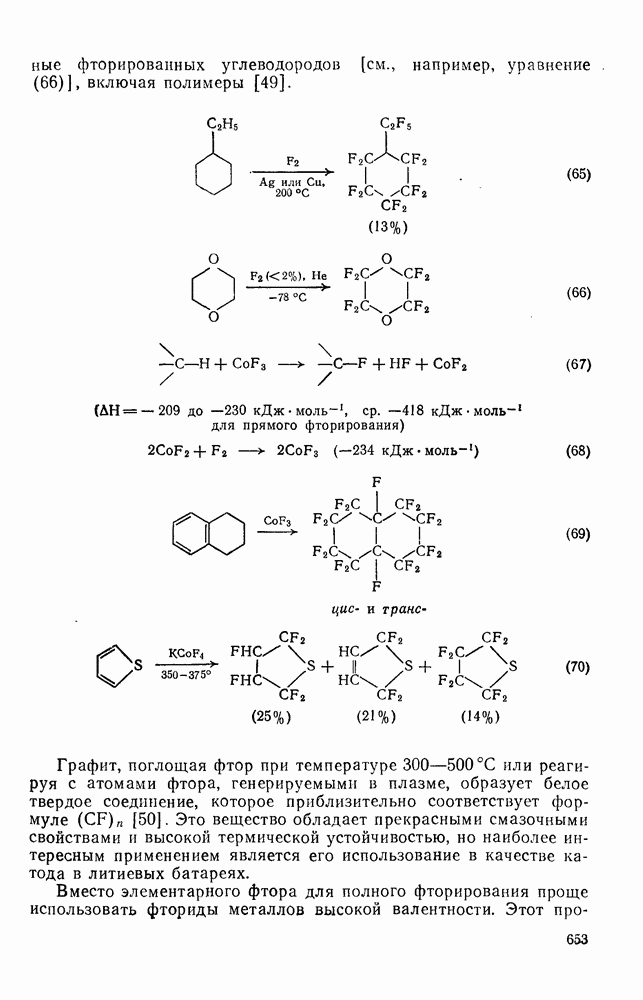
Фотохимические реакции карбениевых ионов [50] разделяются на две категории: 1) валентные изомеризации и 2) процессы переноса электрона. Примером валентной изомеризации является превращение пентаметилциклогексадиенил-катиона в соответствующий бицикло [3.1.0] гексенил-катион [схема (19)]. Такое превращение представляет собой фотохимически разрешенную согласованную дисротаторную электроциклическую циклизацию пентадиенильной части исходного карбокатиона. Обратная реакция протекает, однако, в удивительно мягких условиях для такого термически запрещенного дисротаторного раскрытия; для объяснения этого факта был предложен постадийный механизм.
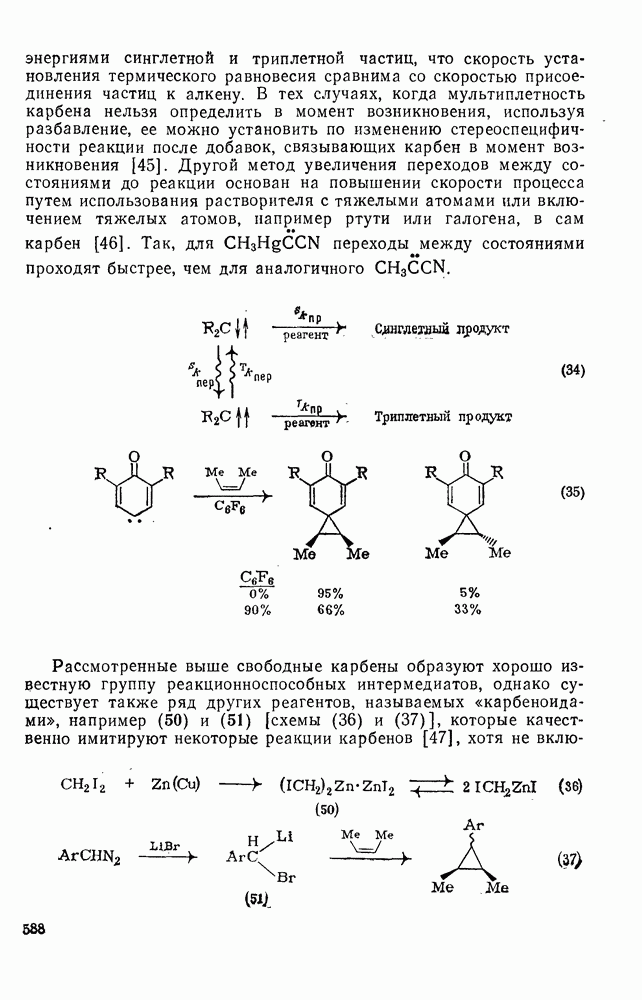
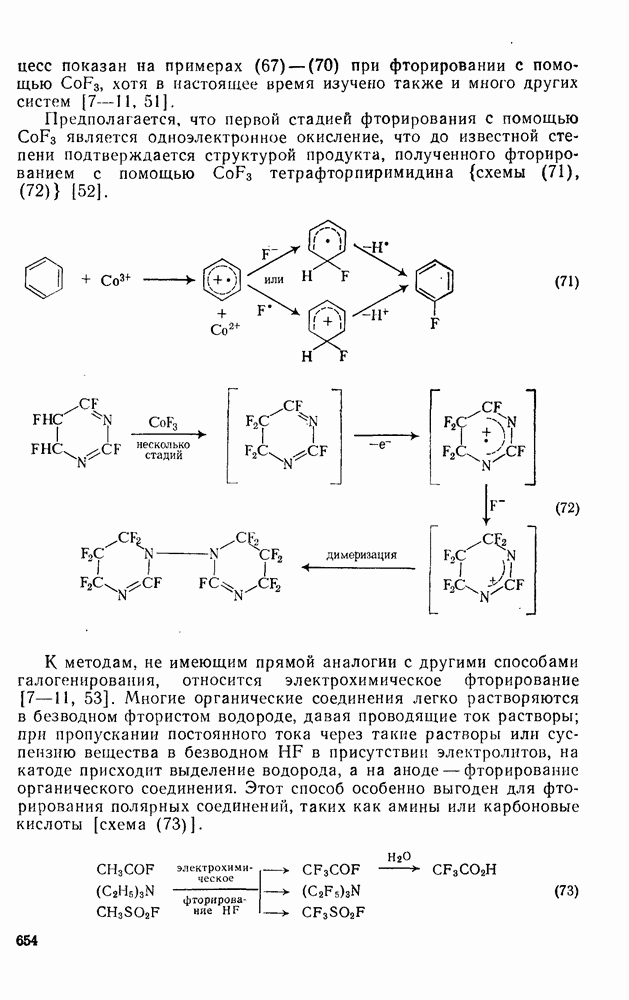
При облучении трифенилциклопропенил-катиона в  -ной
-ной  образуется гексафенилбензол. Предполагают, что при поглощении света происходит перенос электрона с образованием циклопропенил-радикала, который, как известно, димеризуется. Образовавшийся димер далее фотохимически перегруппировывается в гексафенилбензол. Считают, что ключевой стадией сенсибилизированного фотоокисления триарилметанов [51] является отщепление электрона фотогенерированным триплетным трифенил-метил-катионом.
образуется гексафенилбензол. Предполагают, что при поглощении света происходит перенос электрона с образованием циклопропенил-радикала, который, как известно, димеризуется. Образовавшийся димер далее фотохимически перегруппировывается в гексафенилбензол. Считают, что ключевой стадией сенсибилизированного фотоокисления триарилметанов [51] является отщепление электрона фотогенерированным триплетным трифенил-метил-катионом.