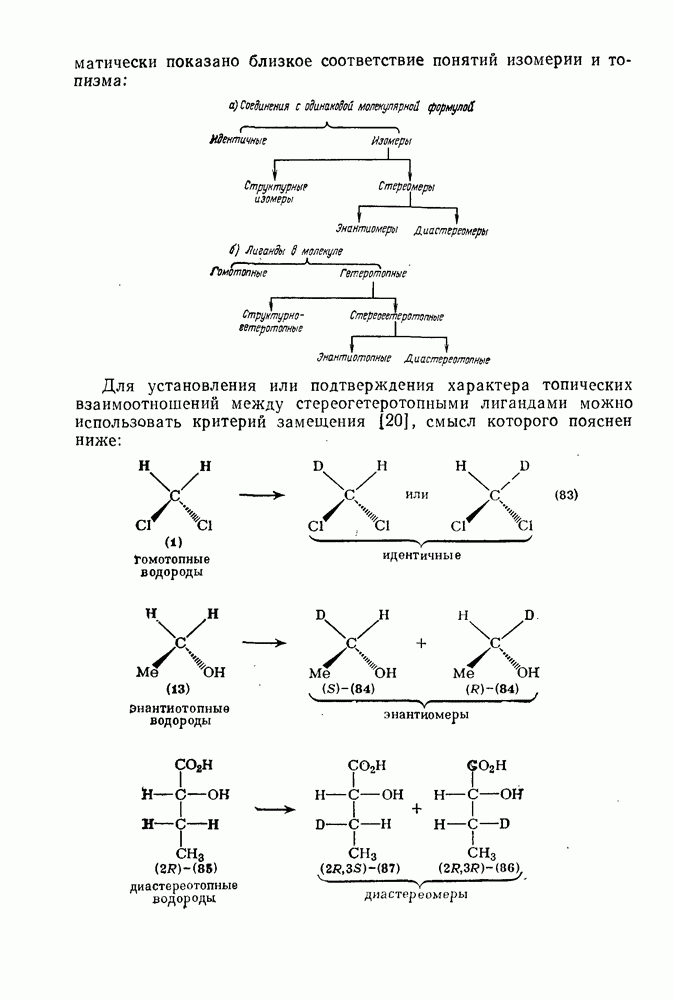
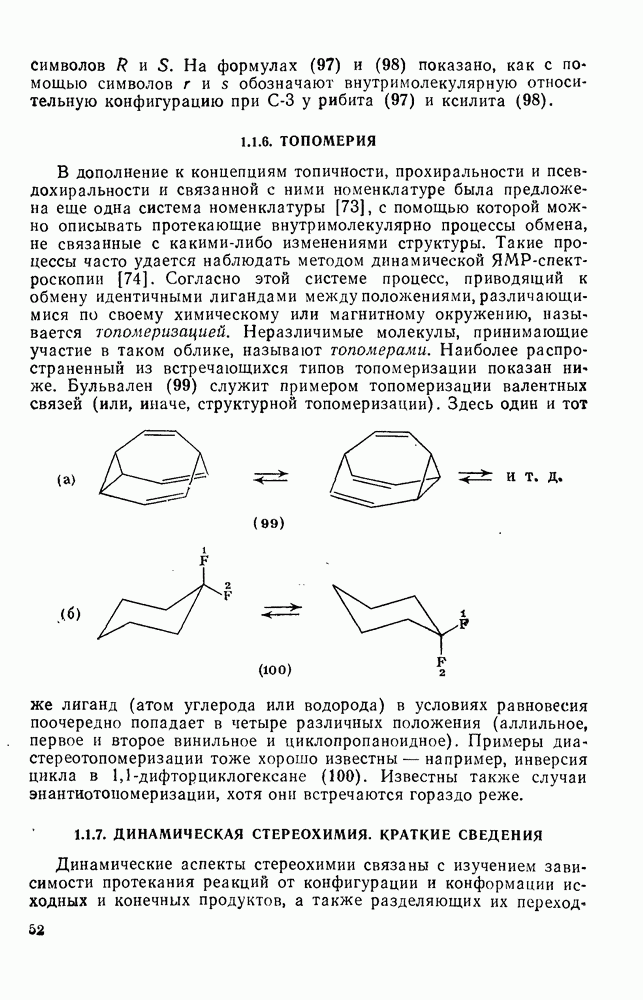
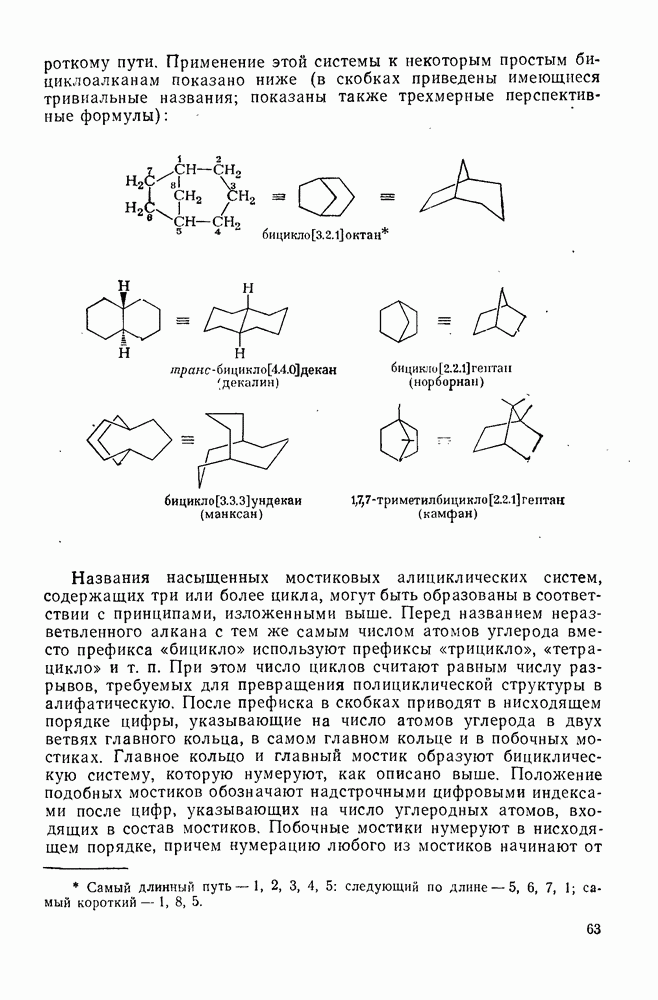
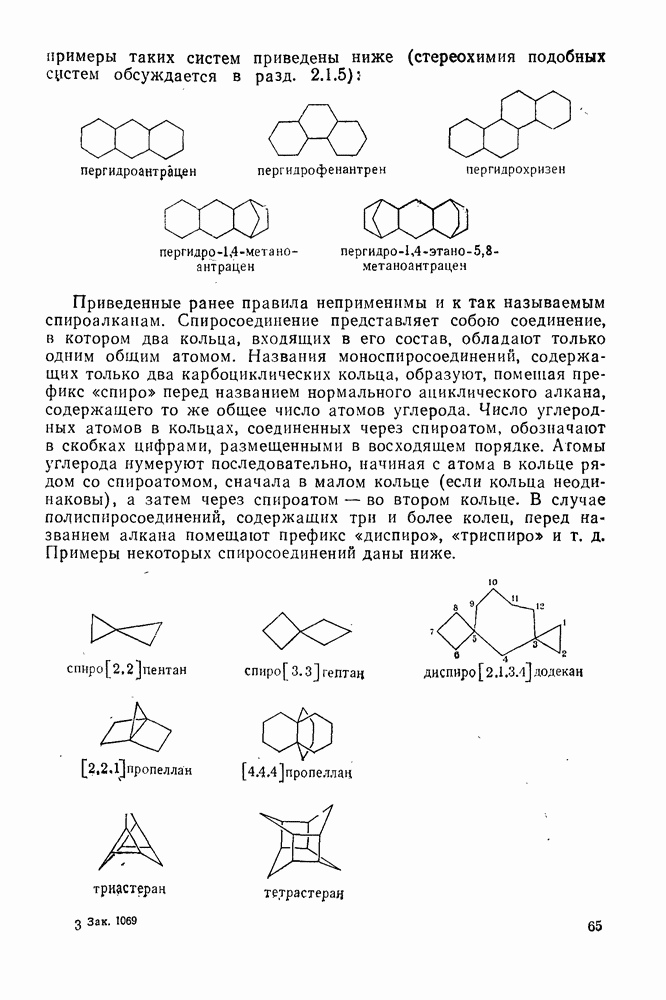
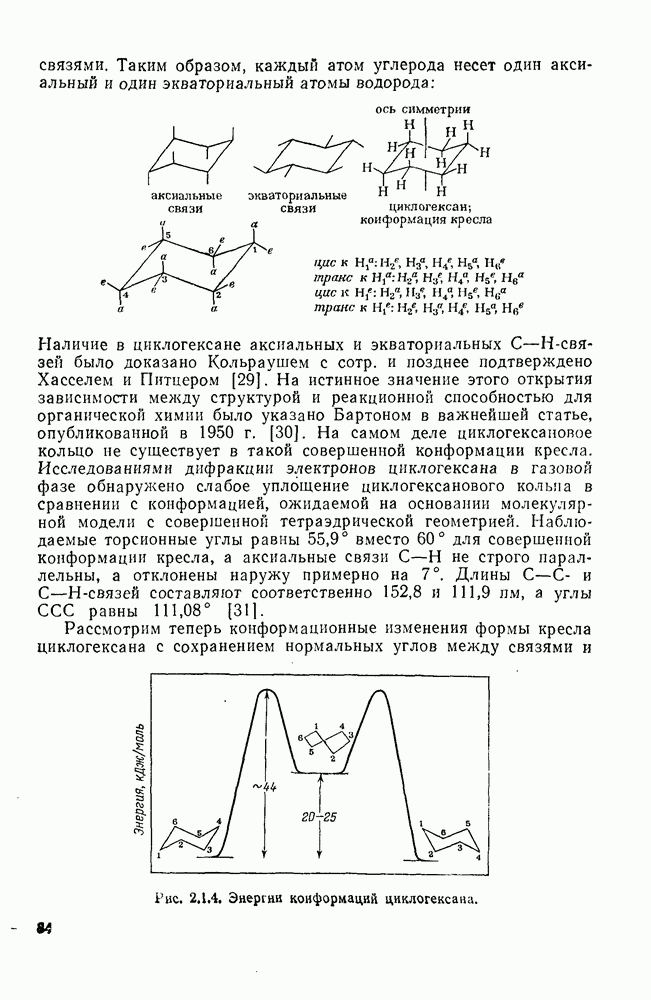
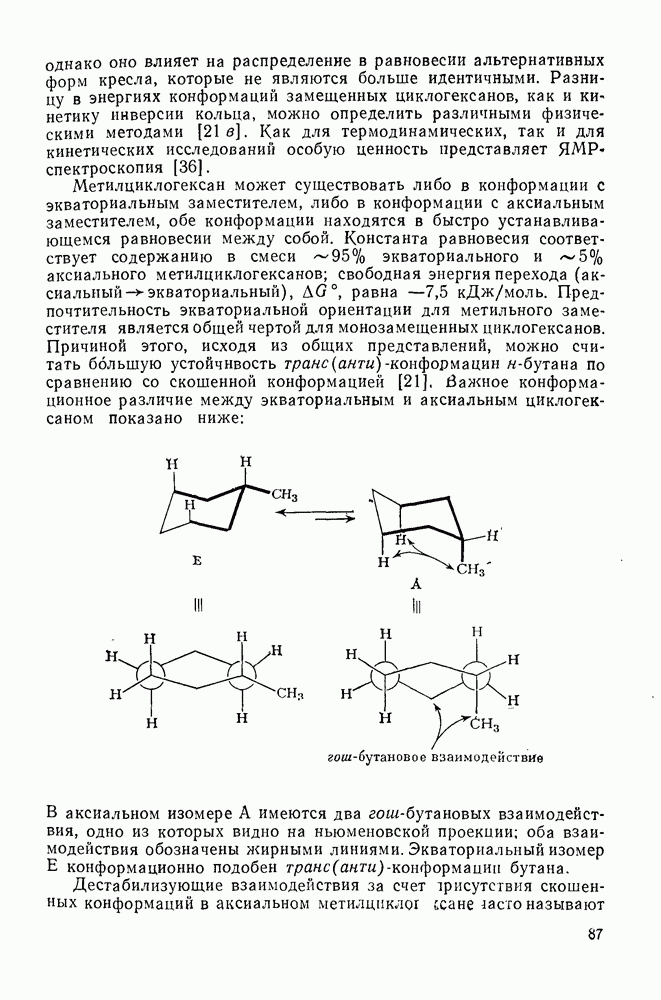
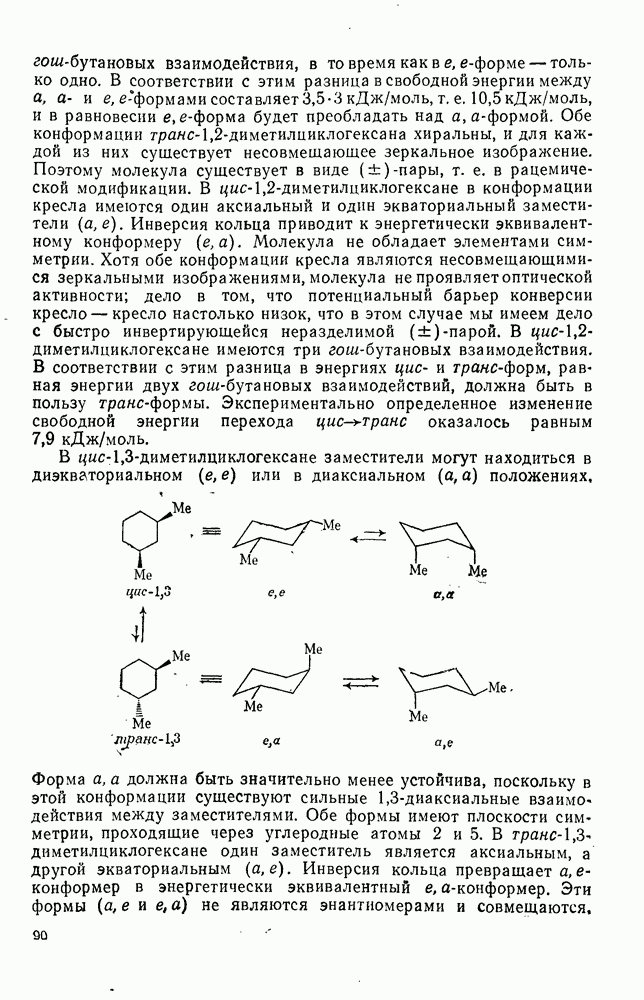
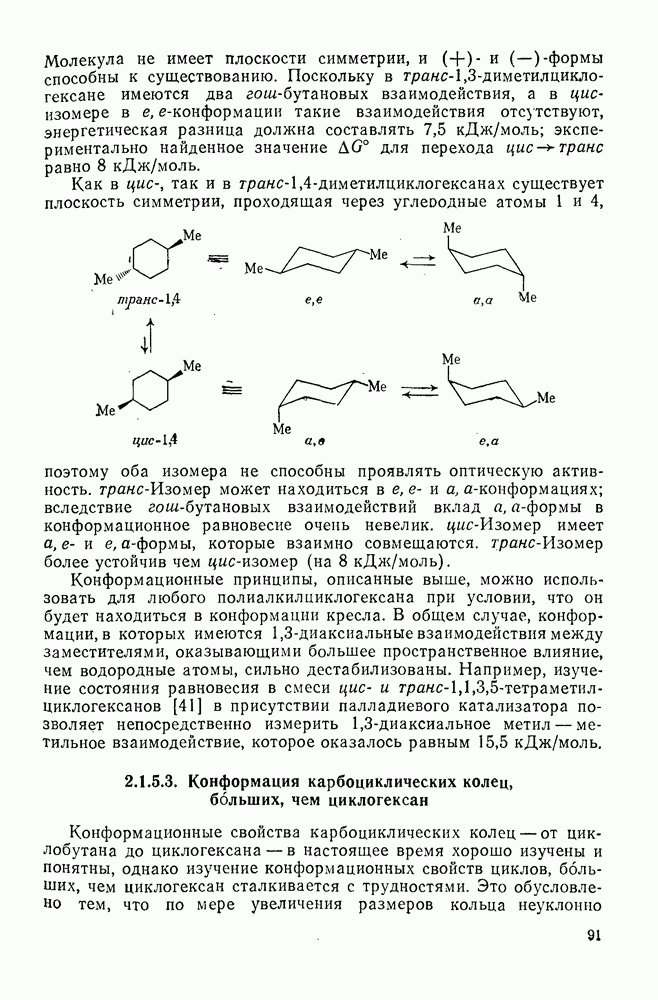
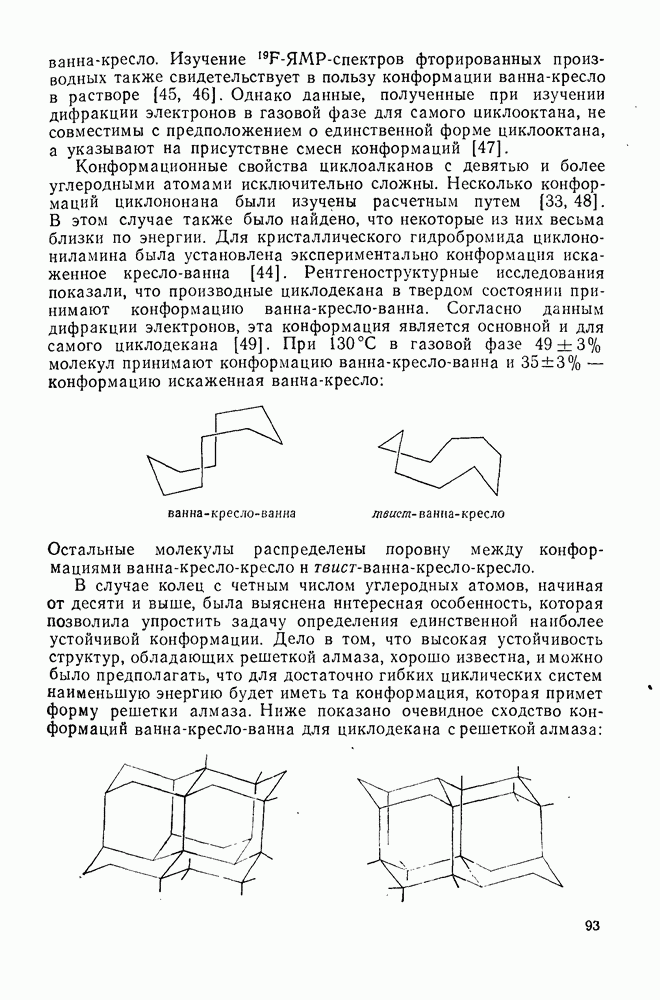
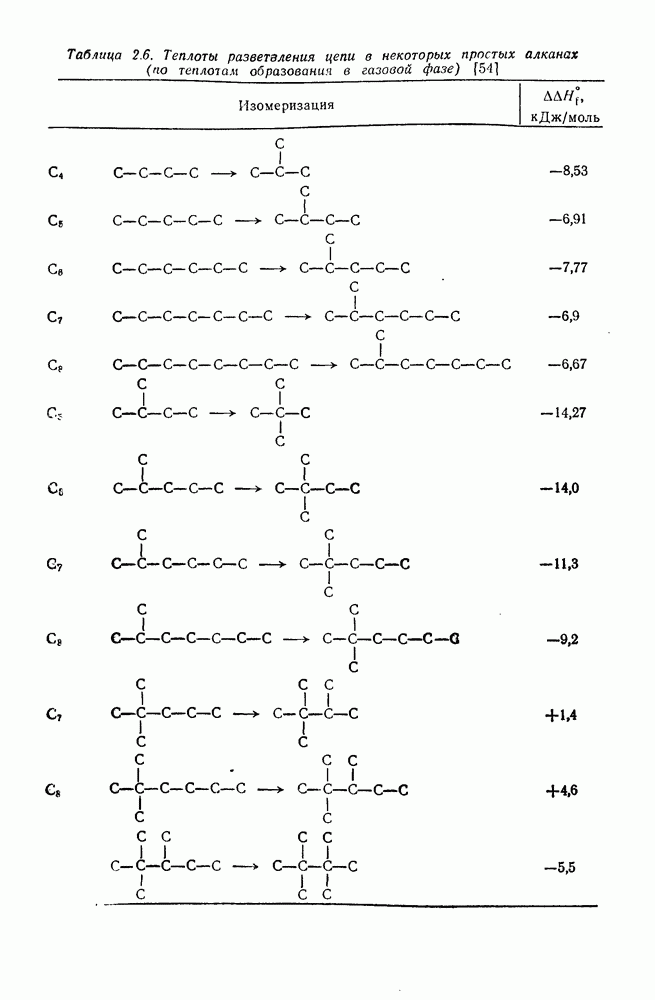
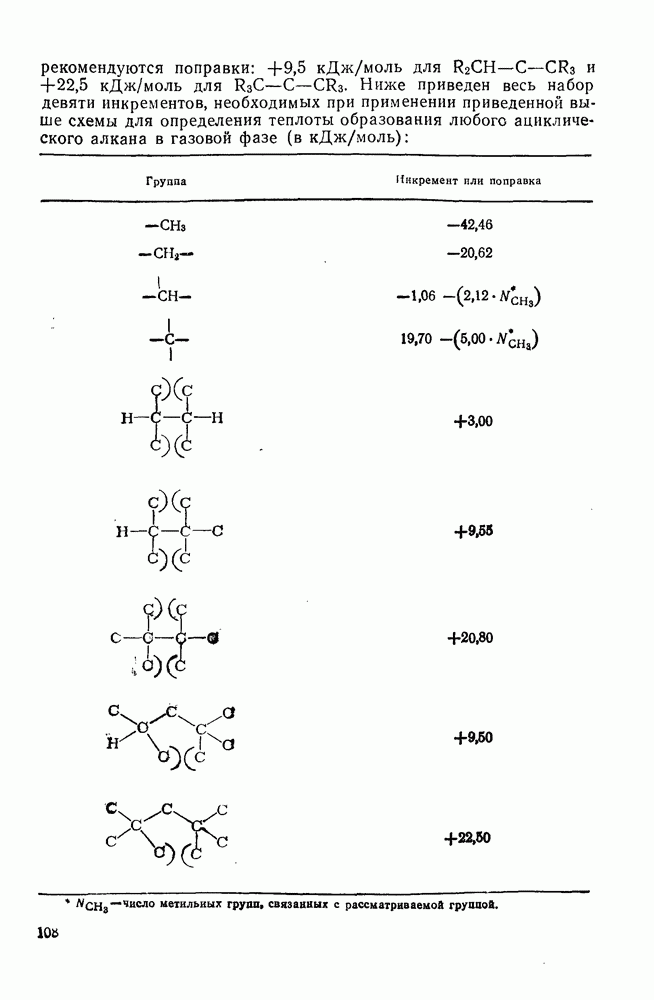
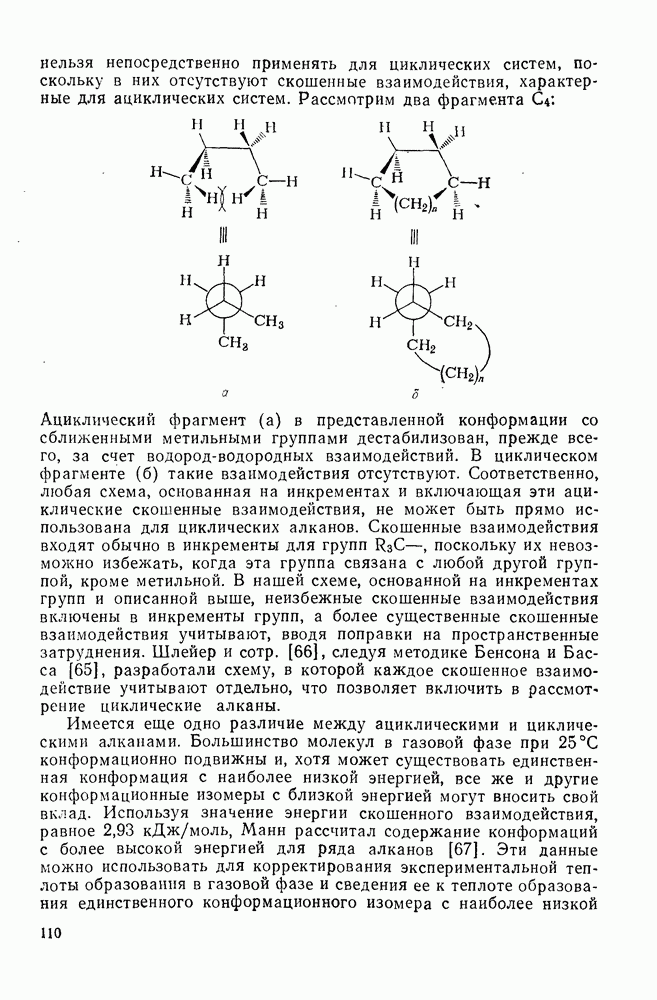
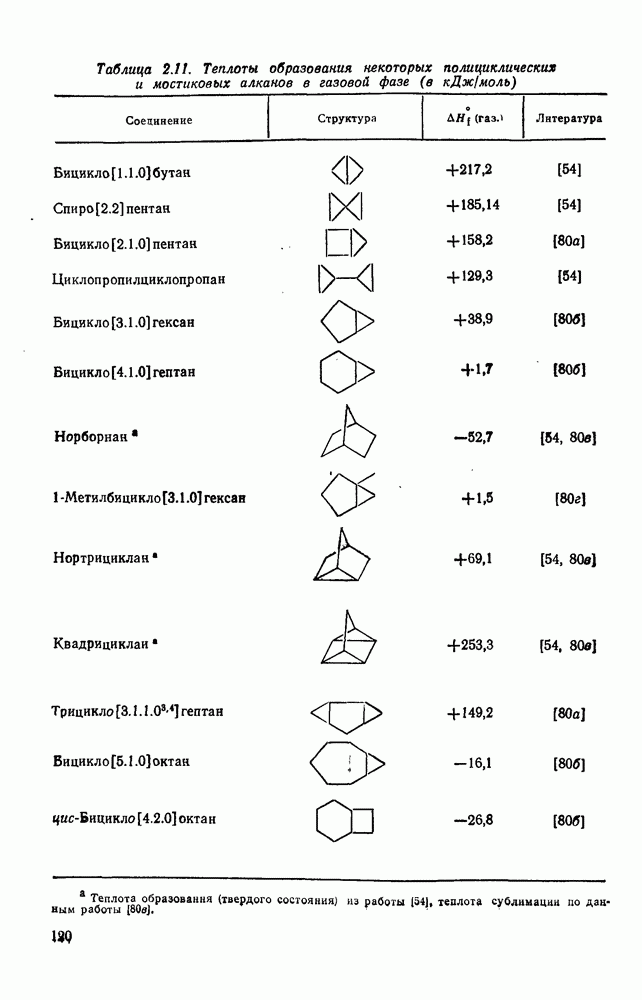
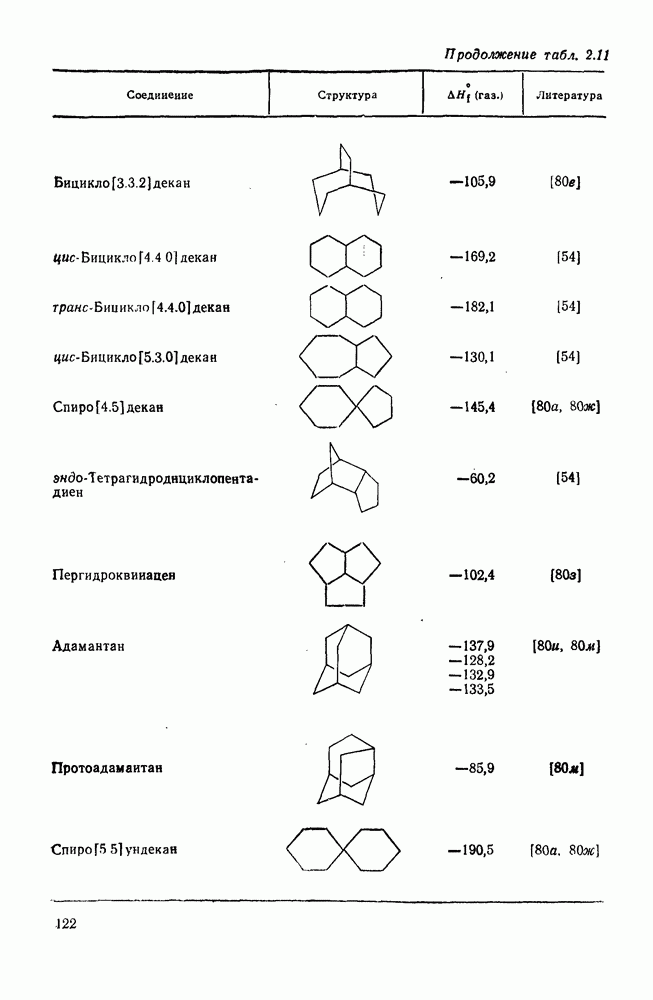
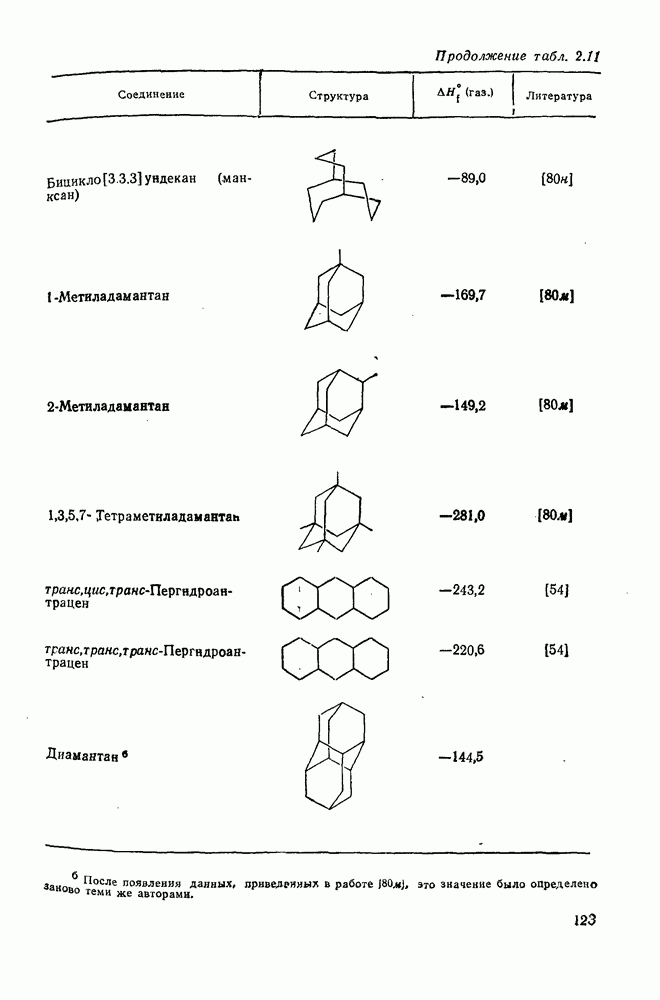
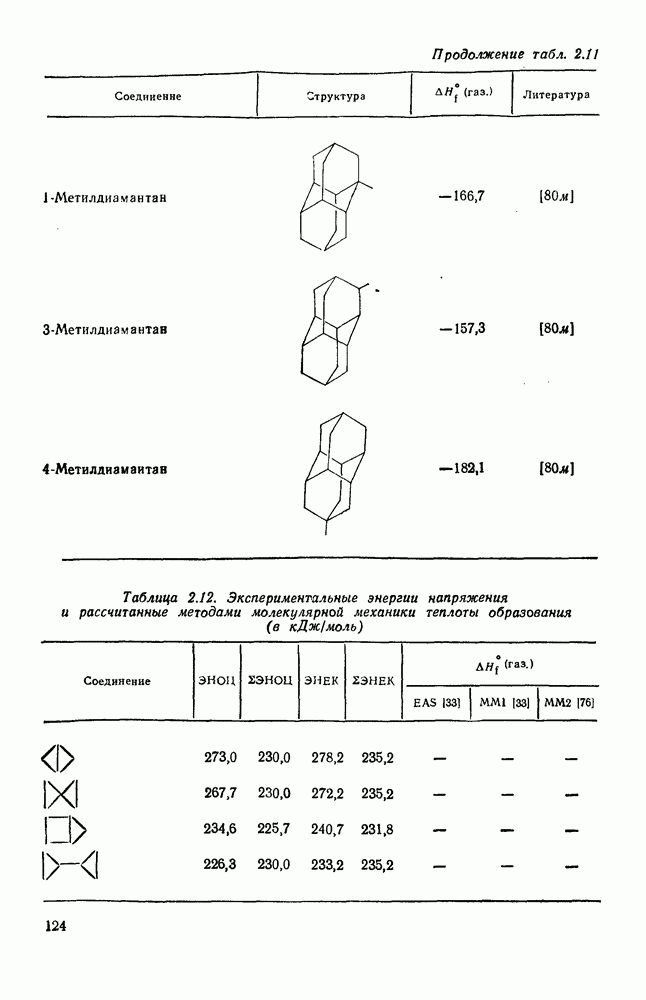
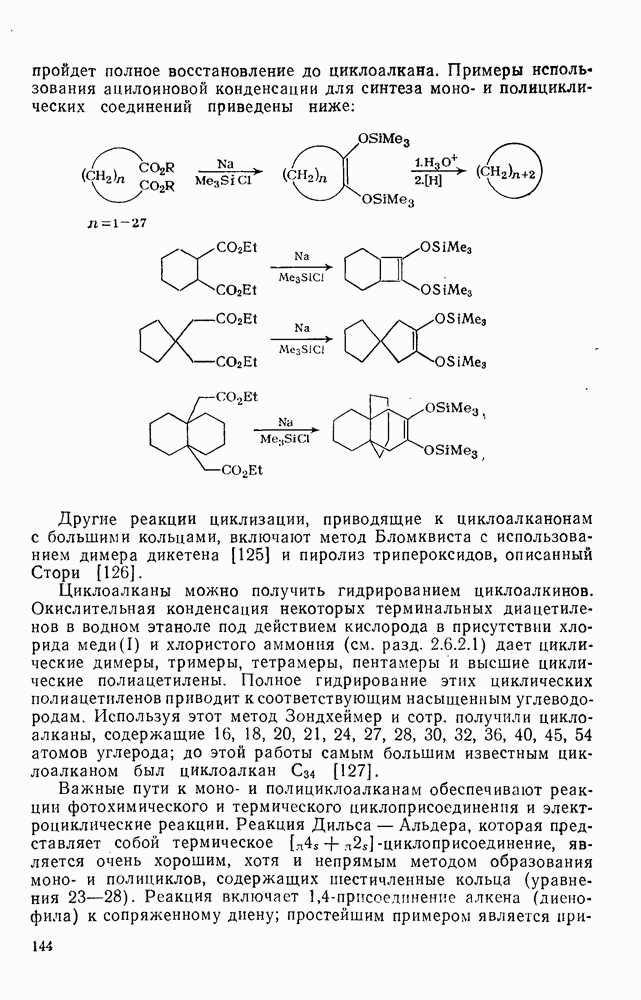
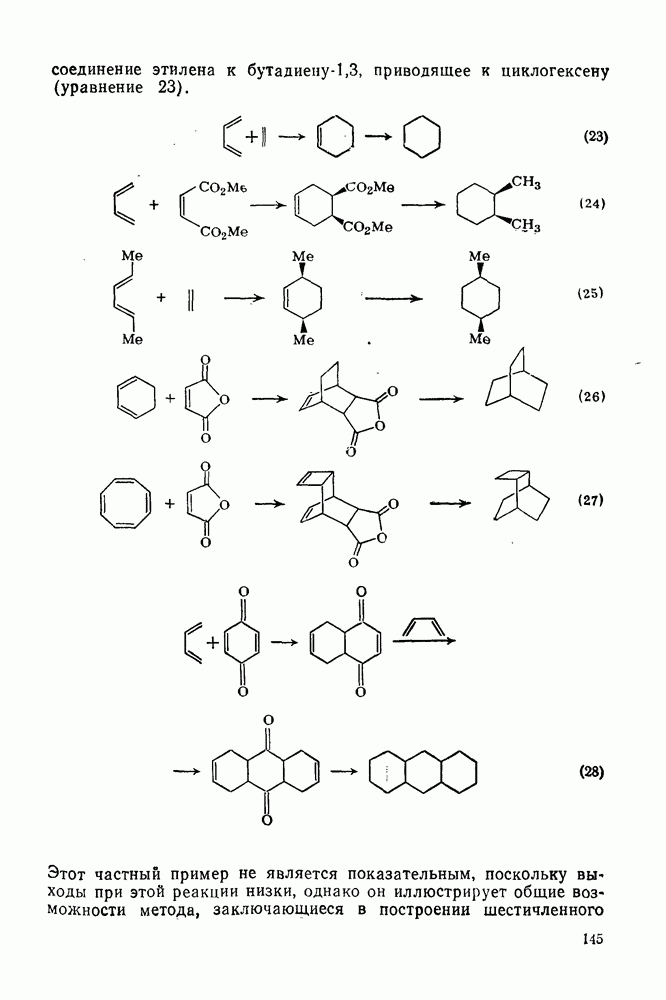
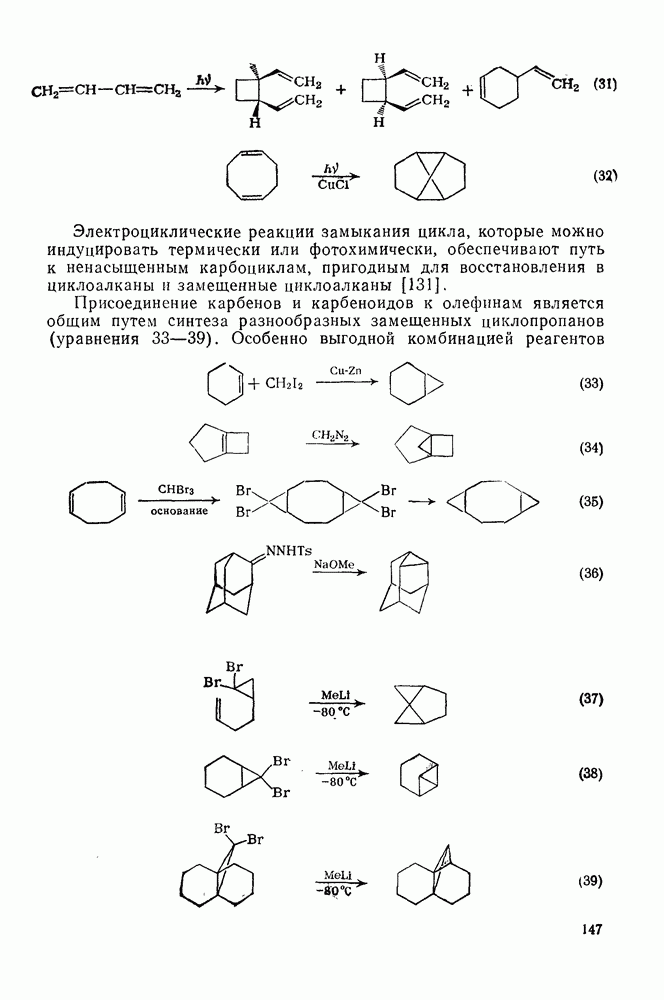
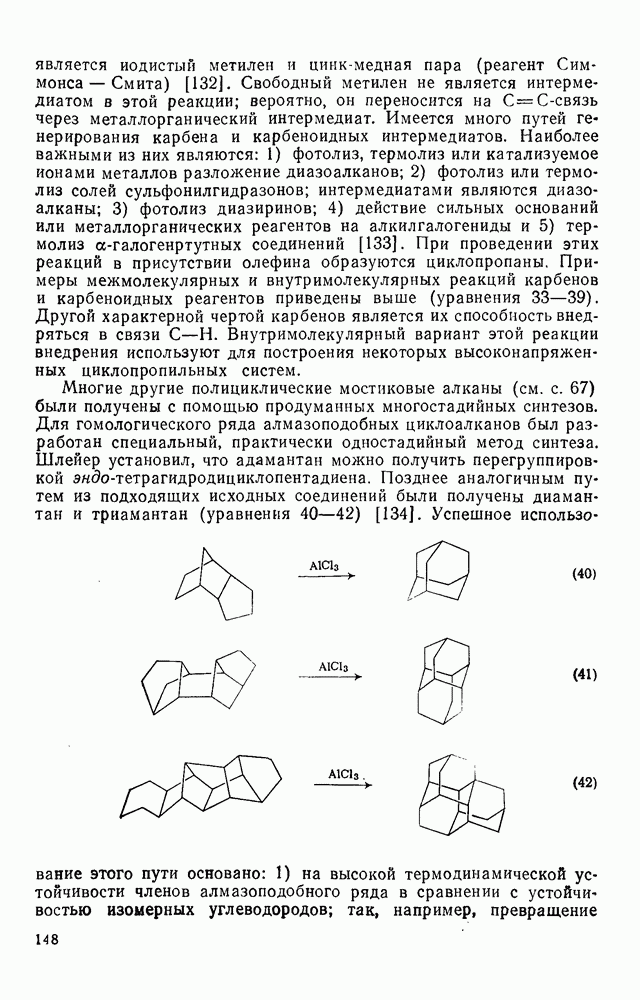
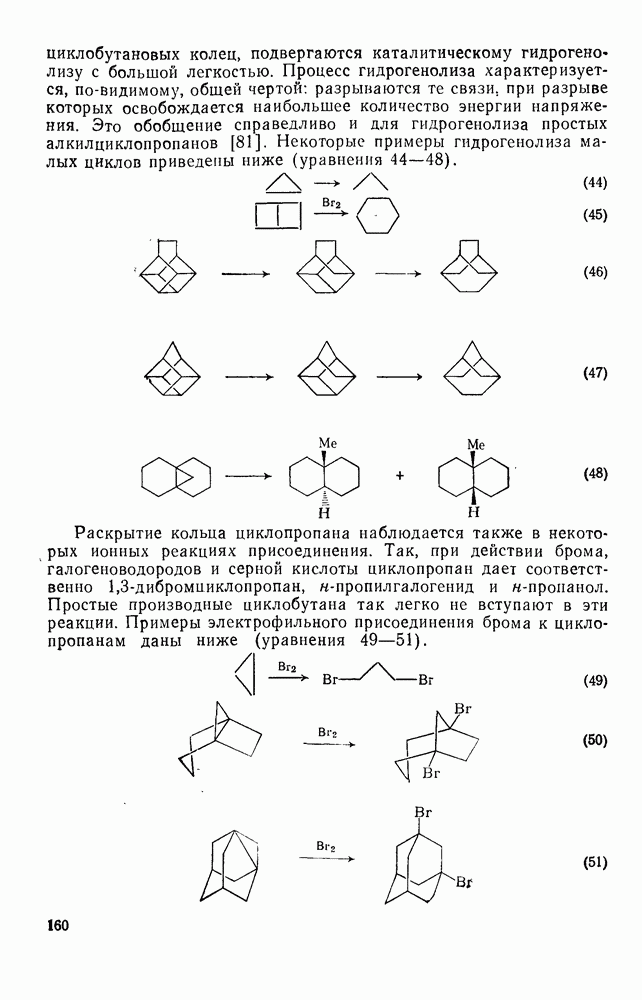
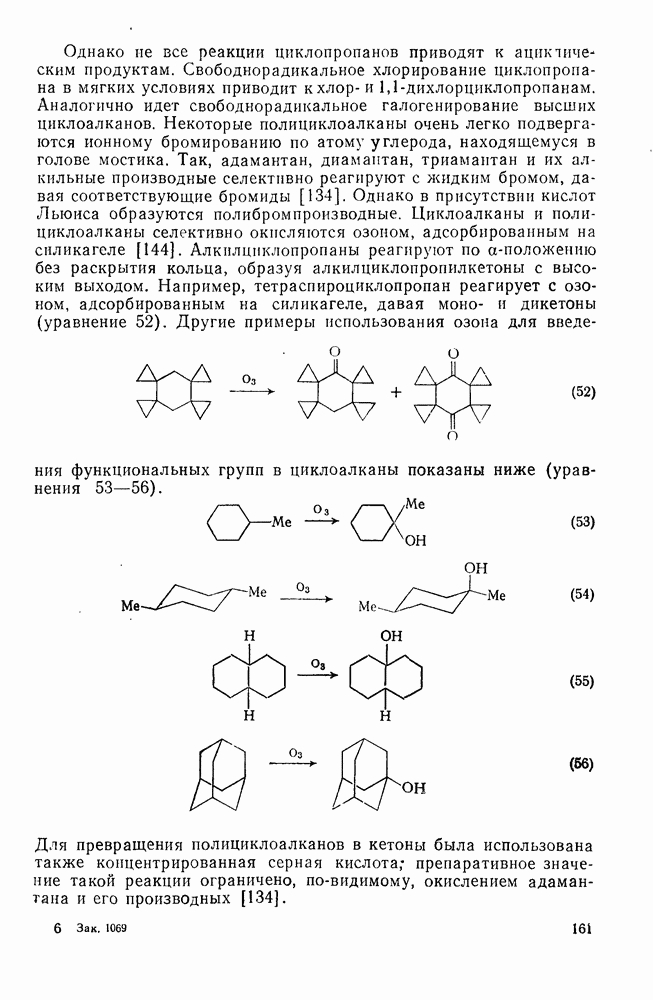
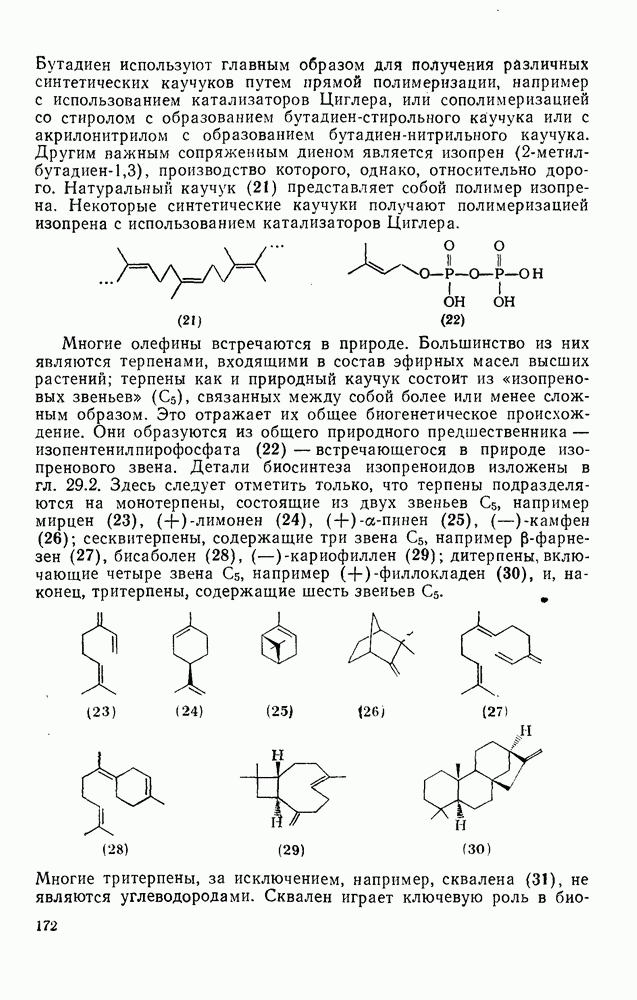
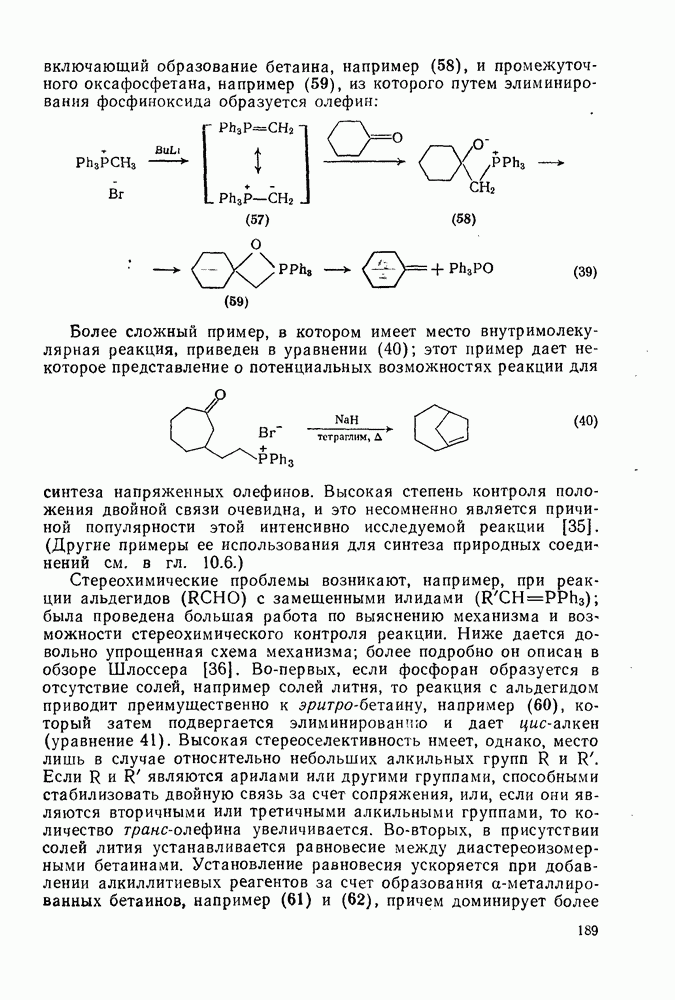
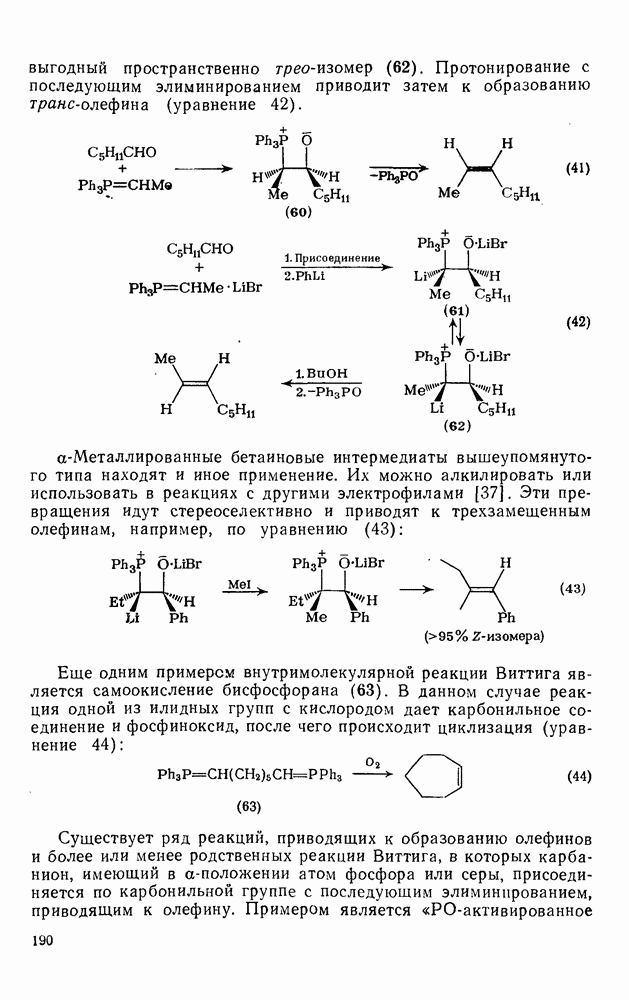
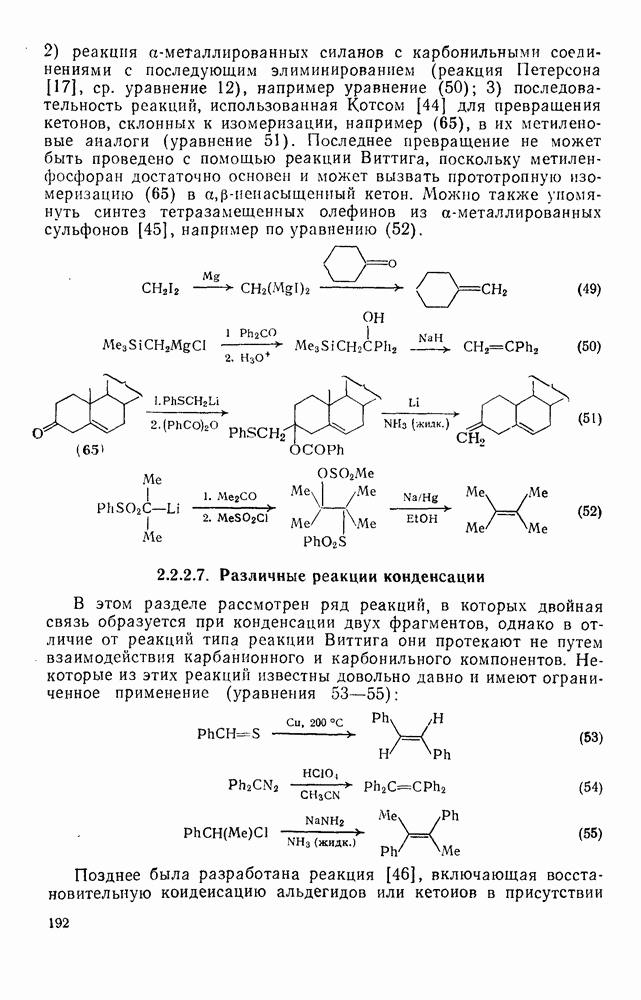
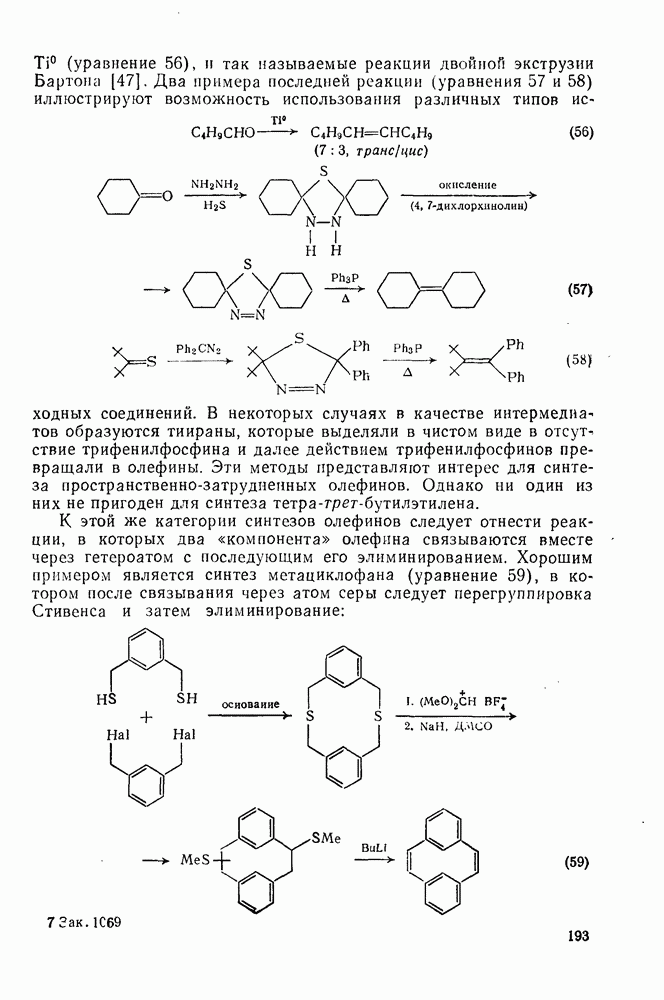
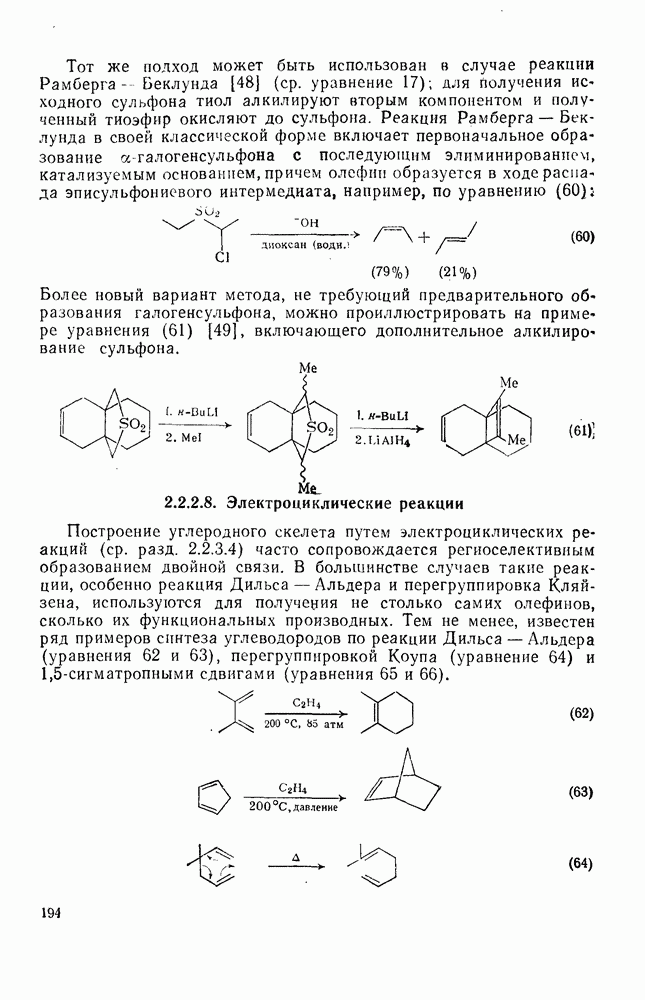
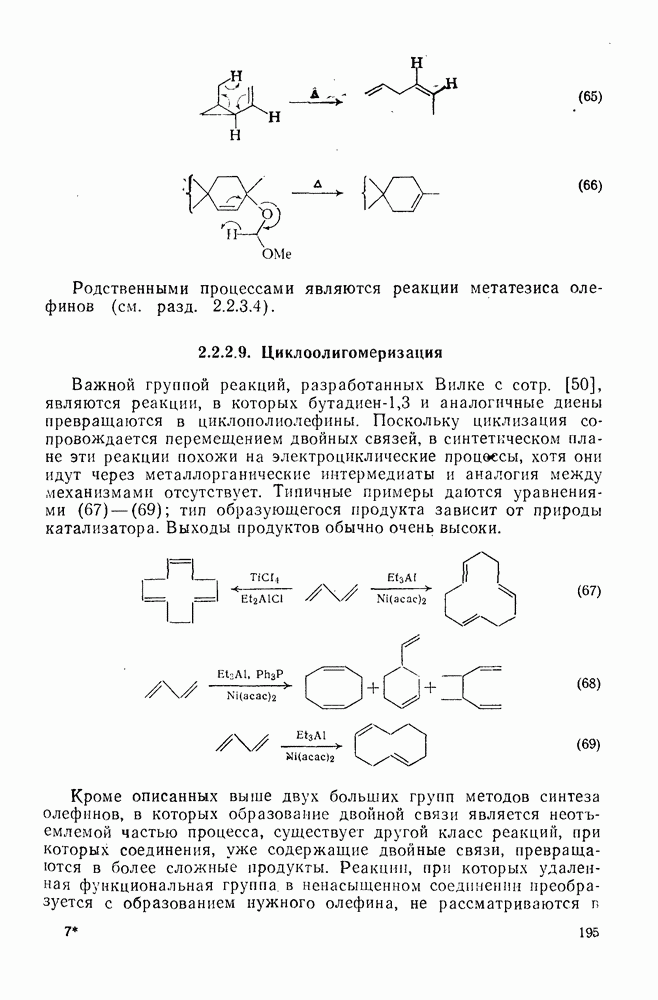
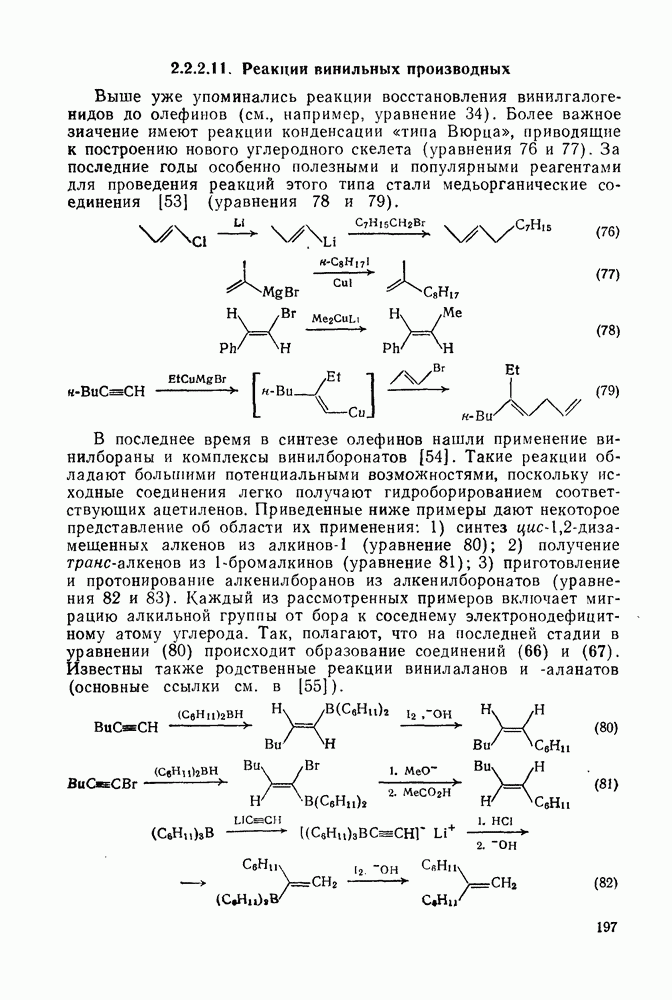
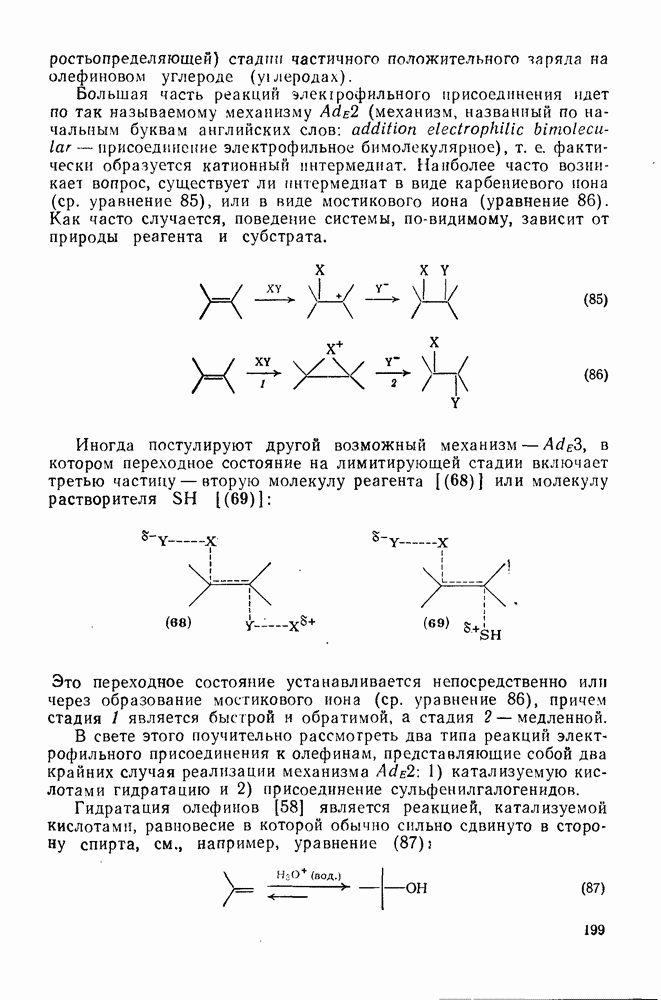
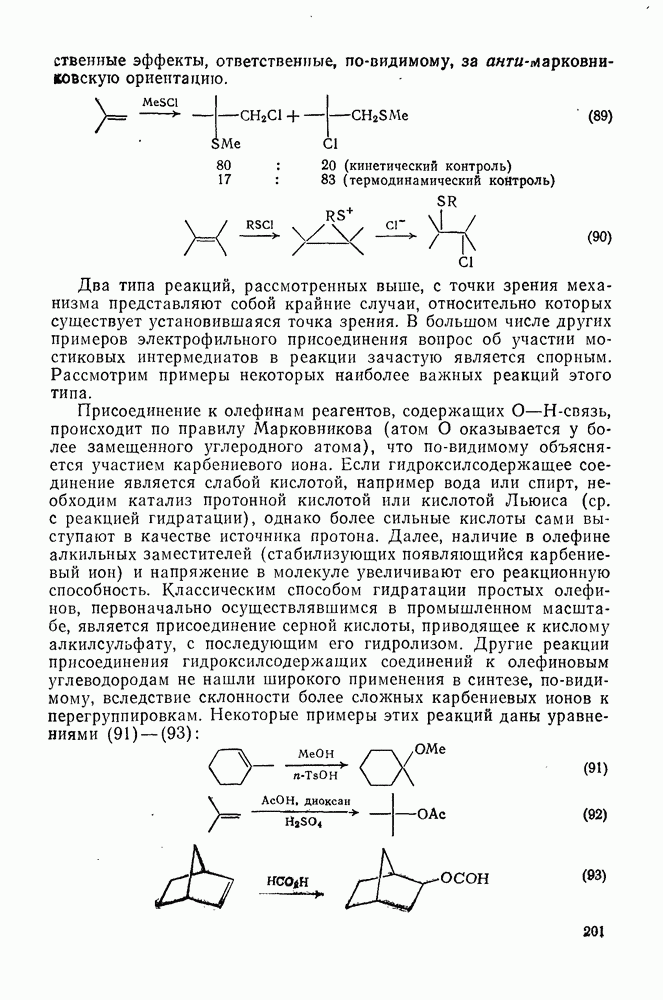
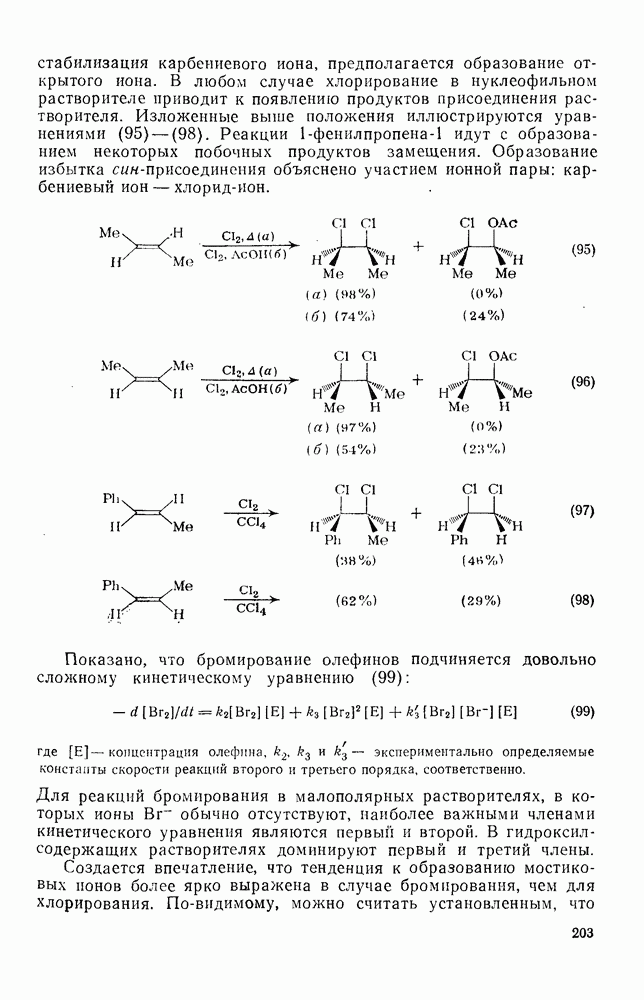
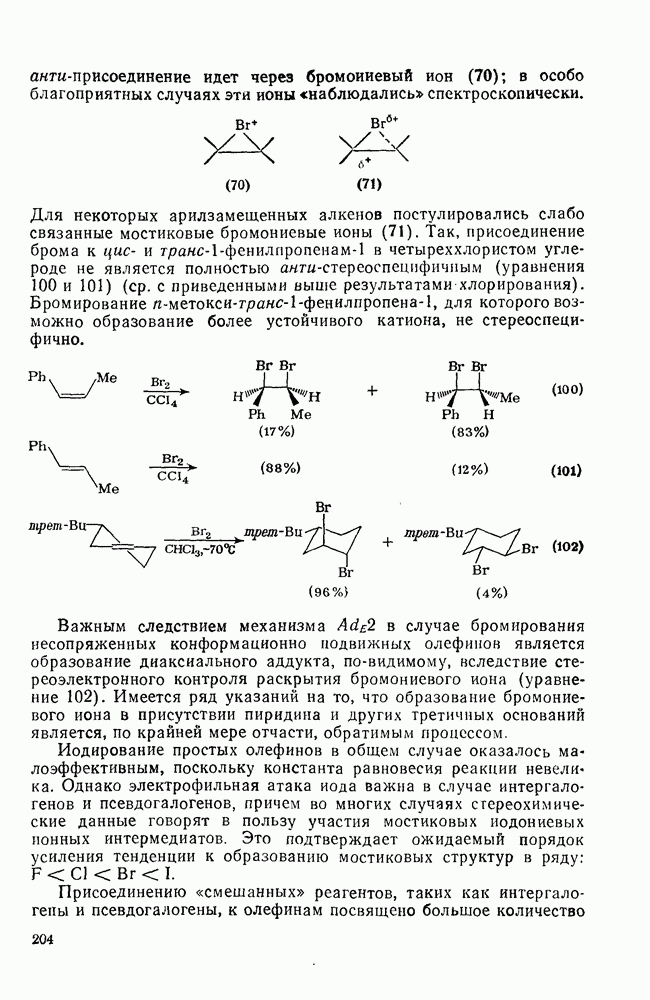
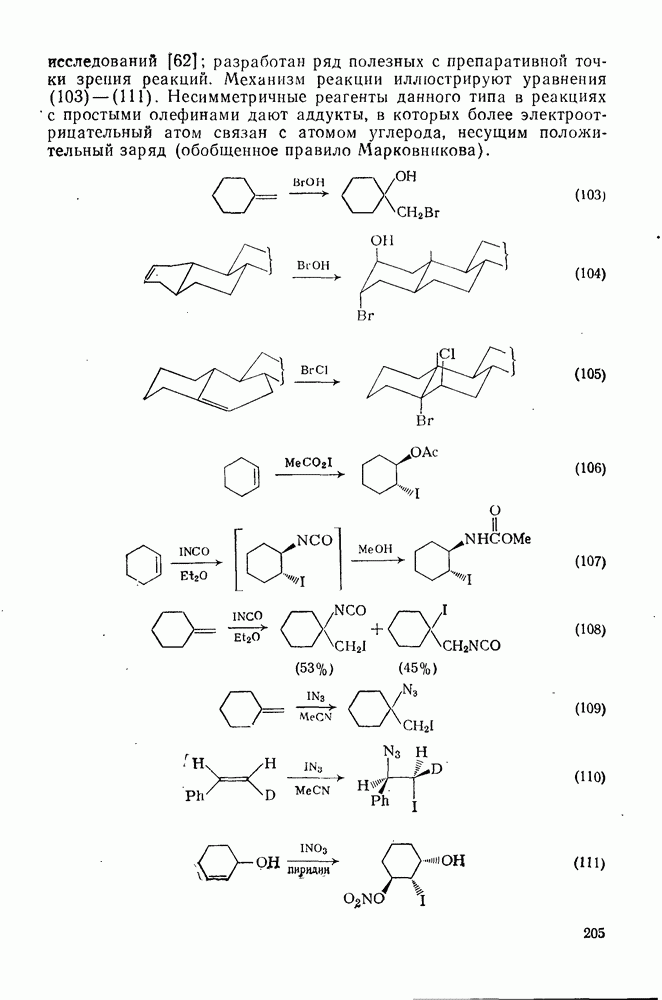
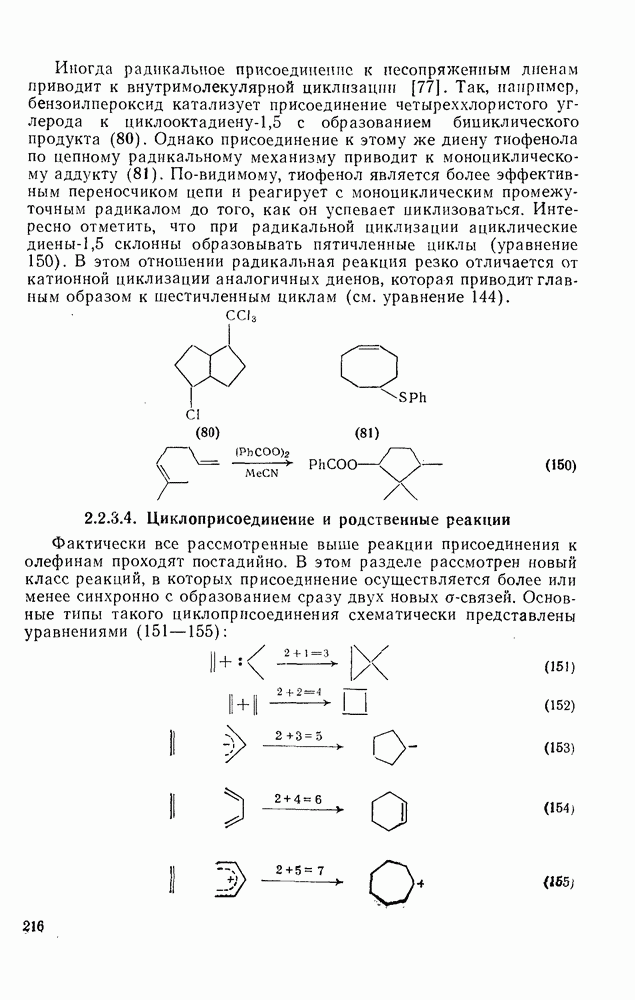
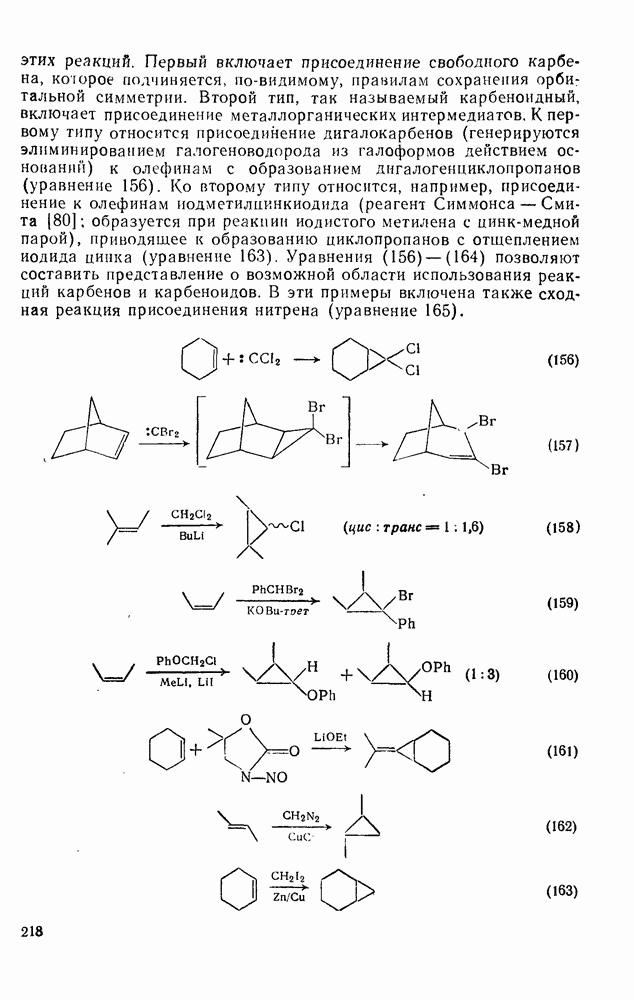
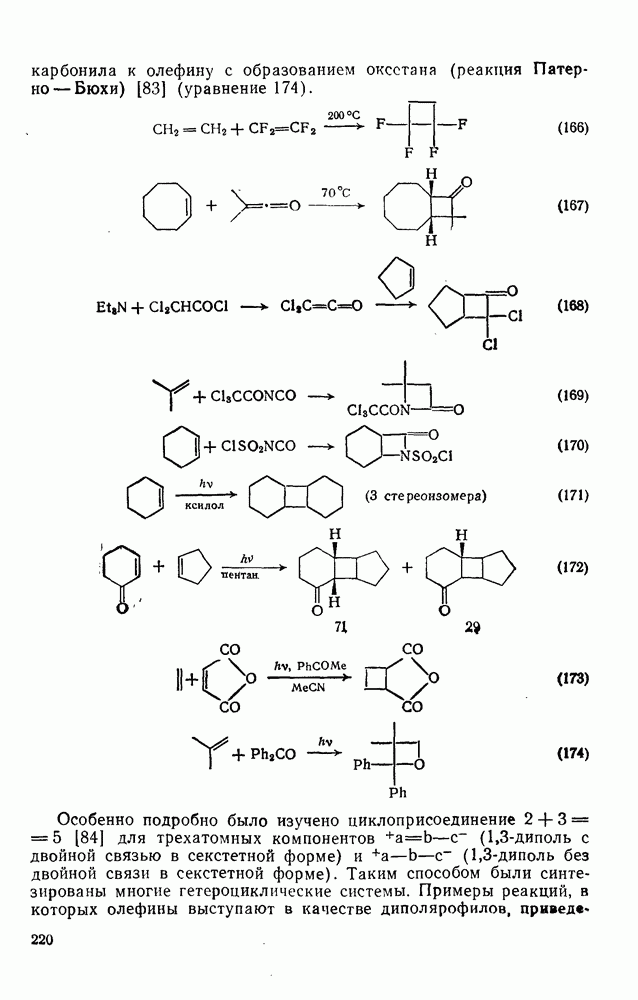
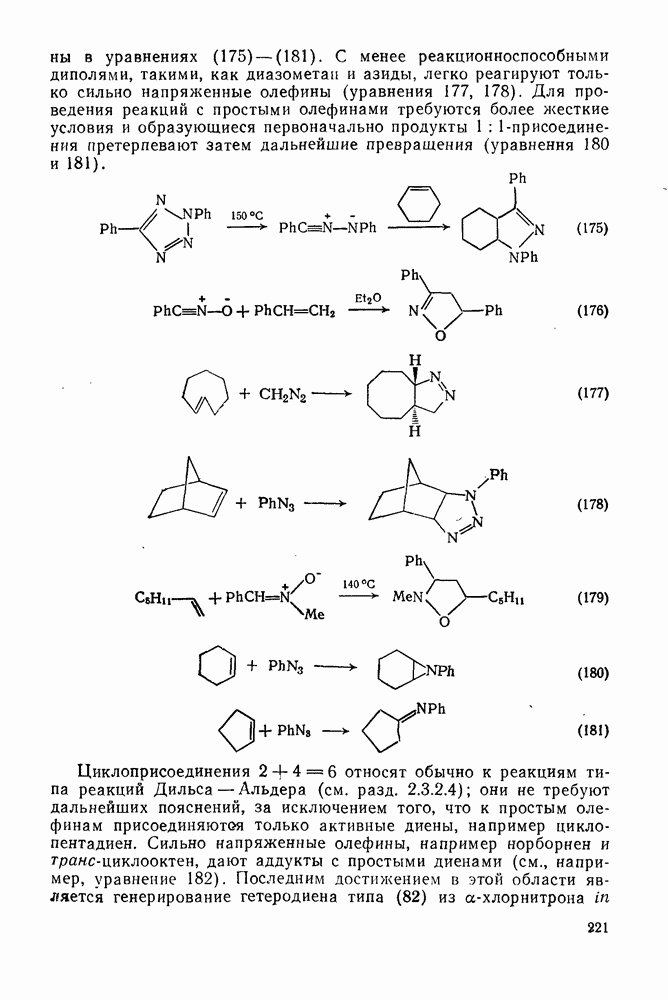
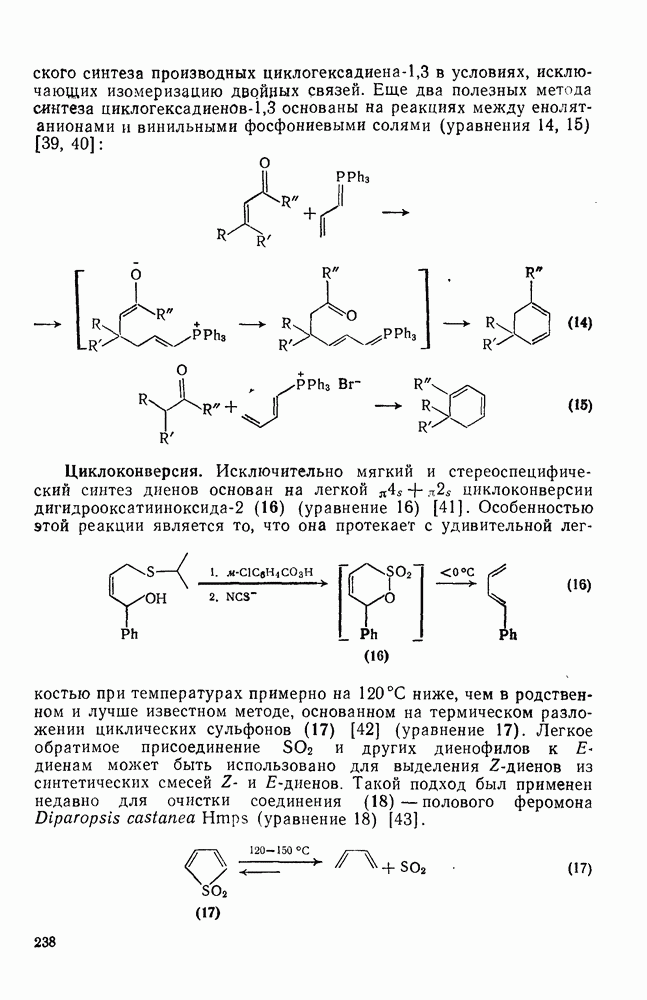
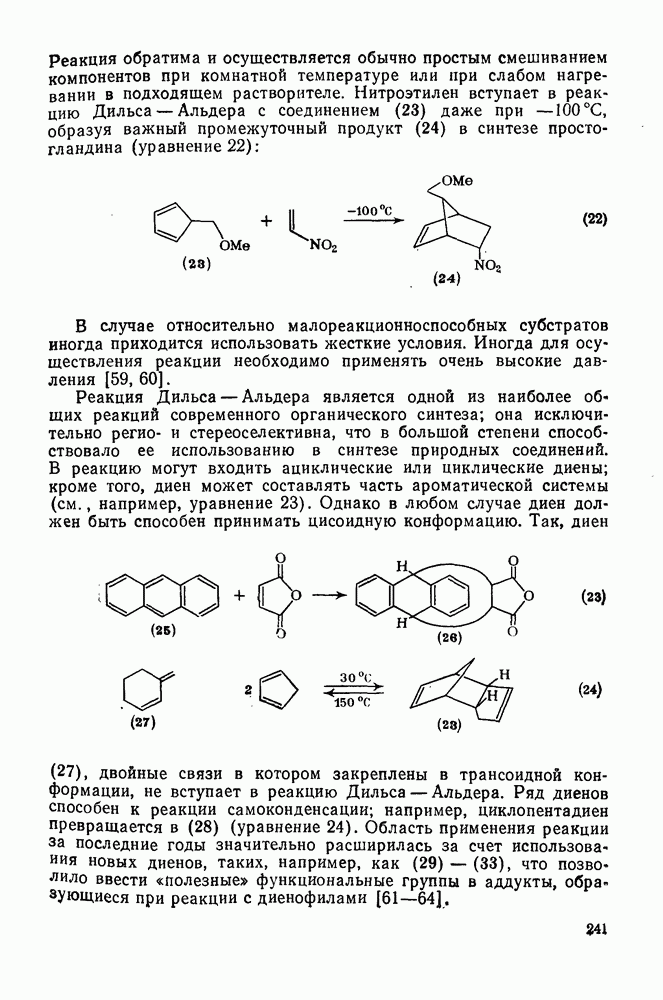
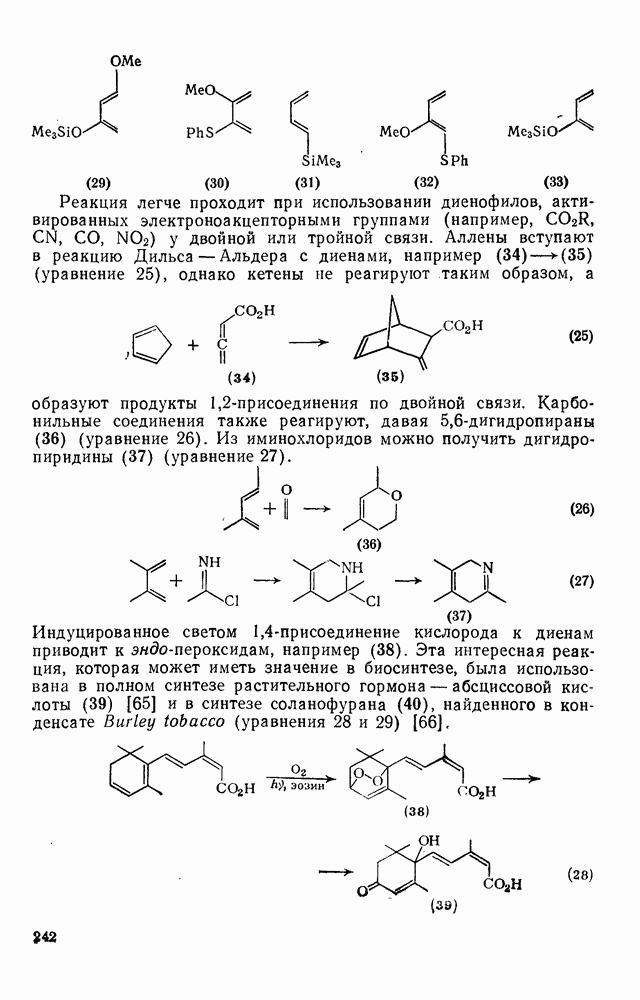
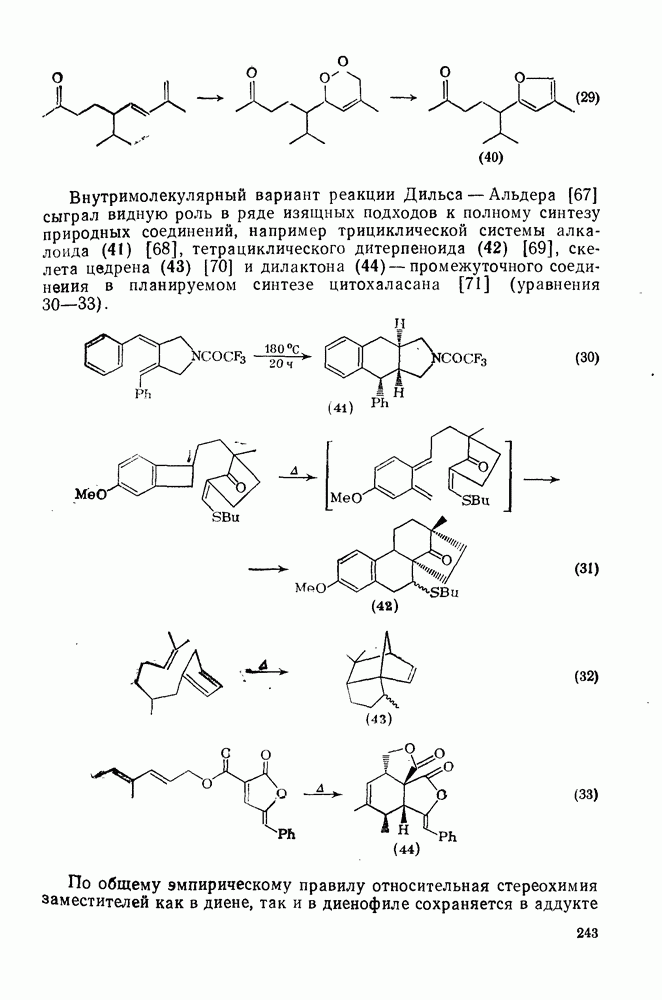
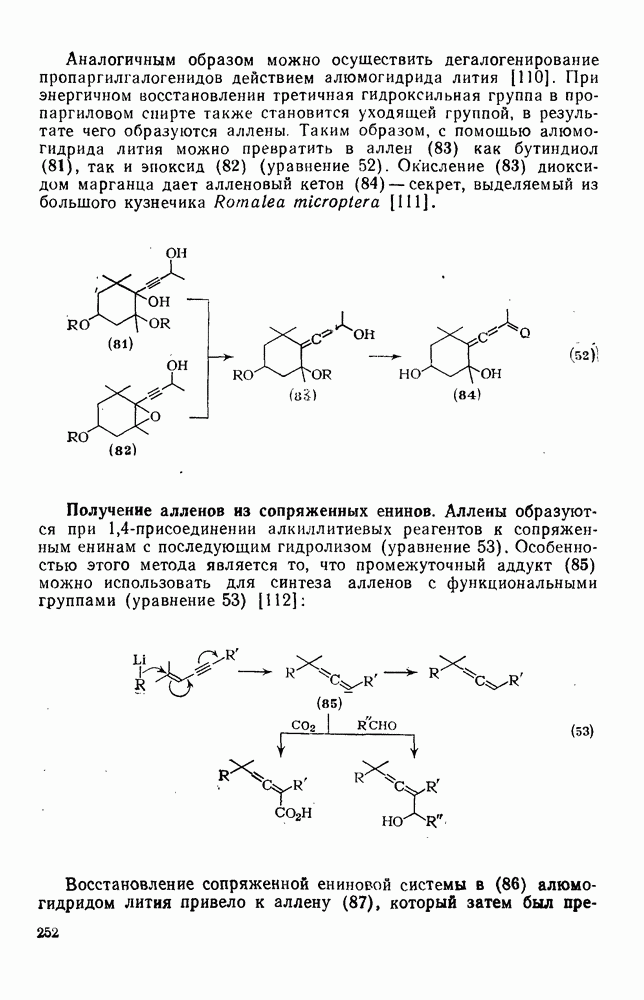
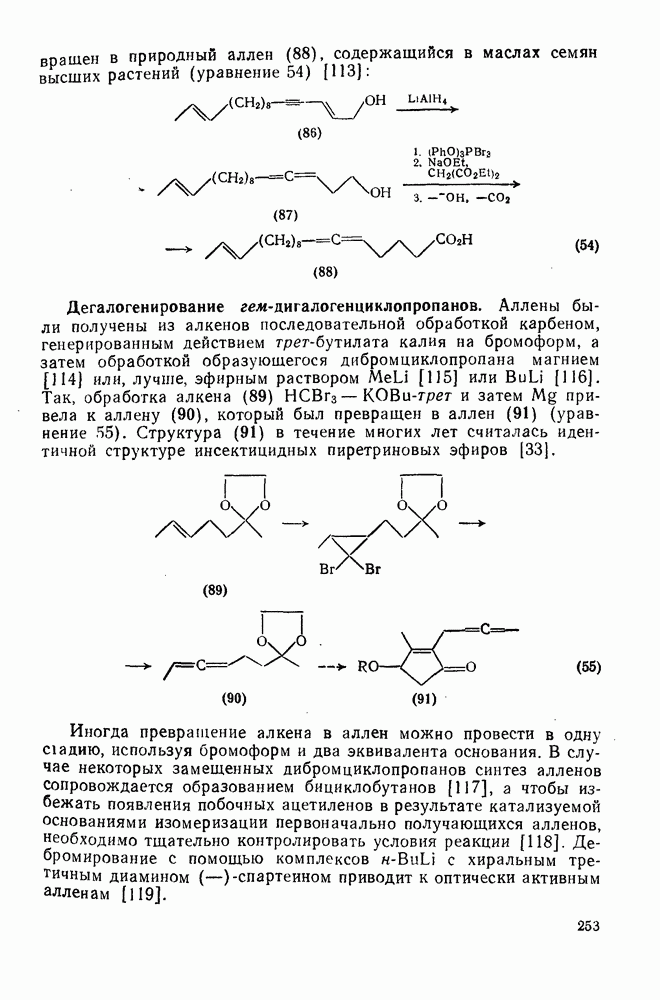
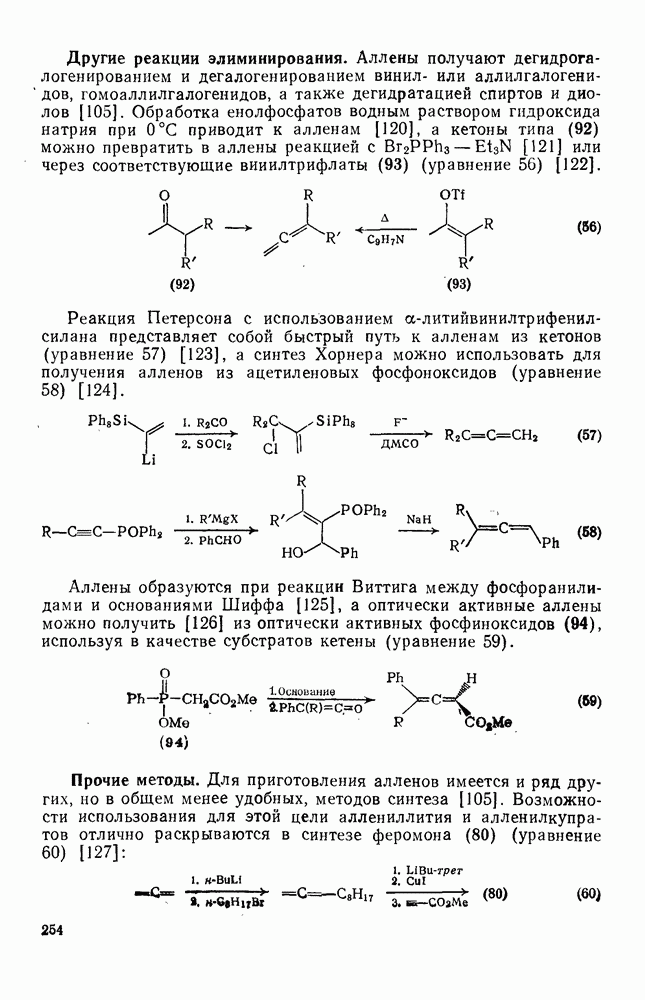
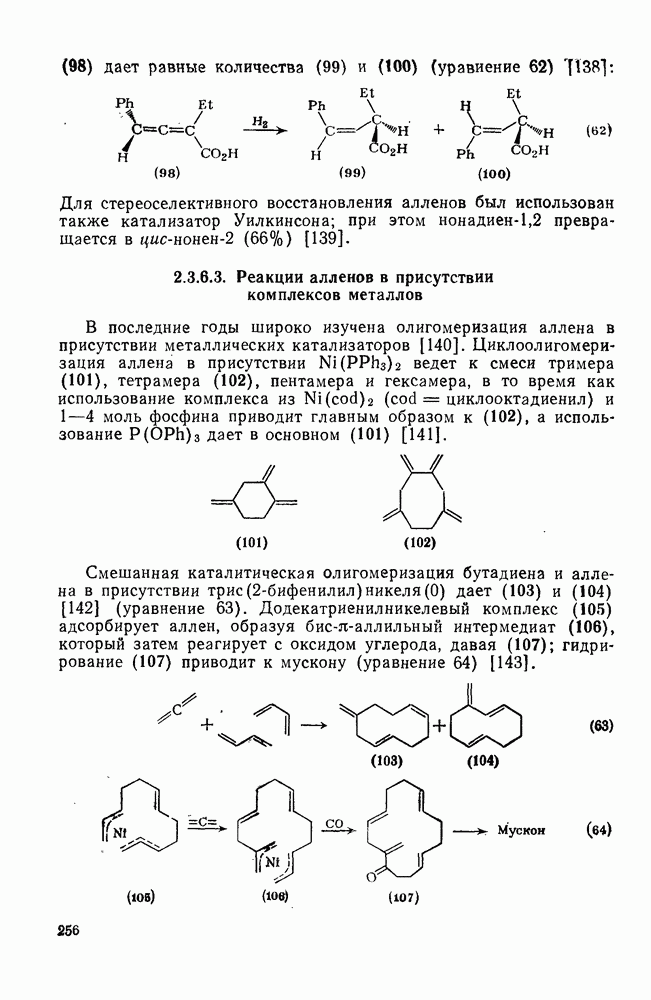
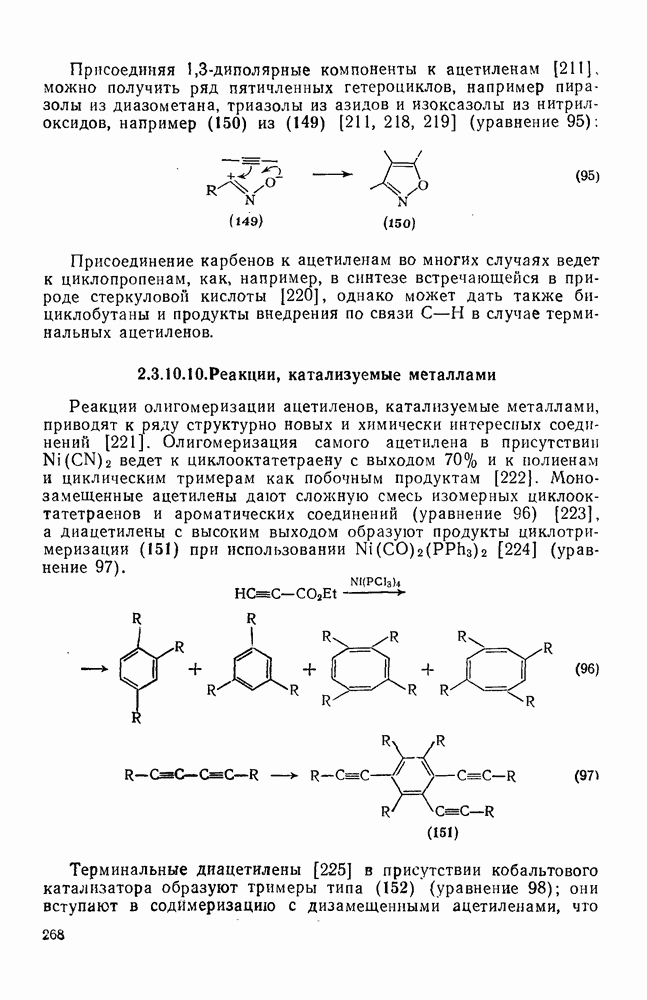
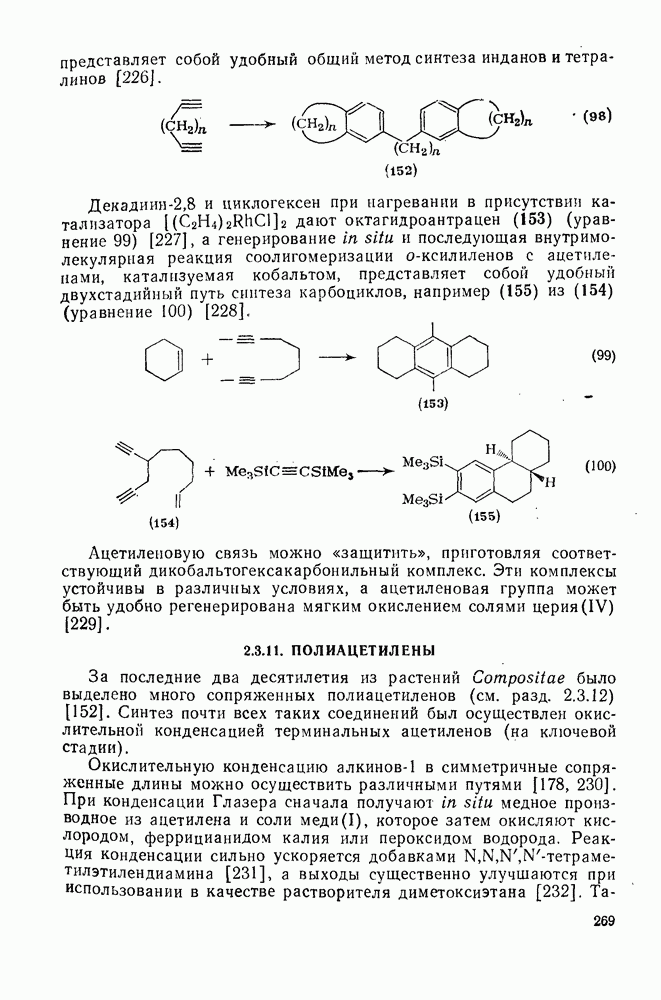
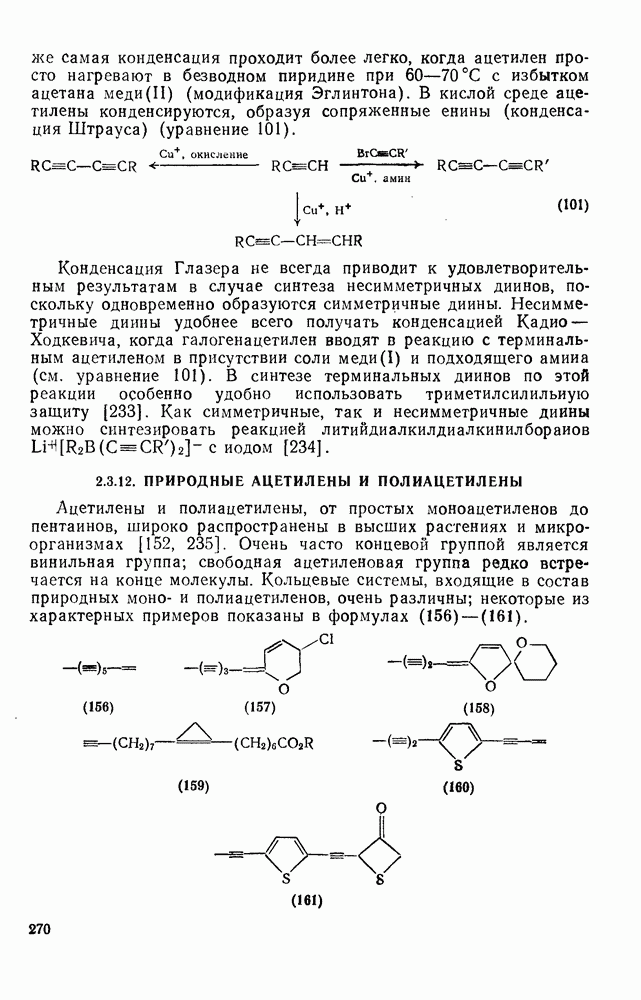
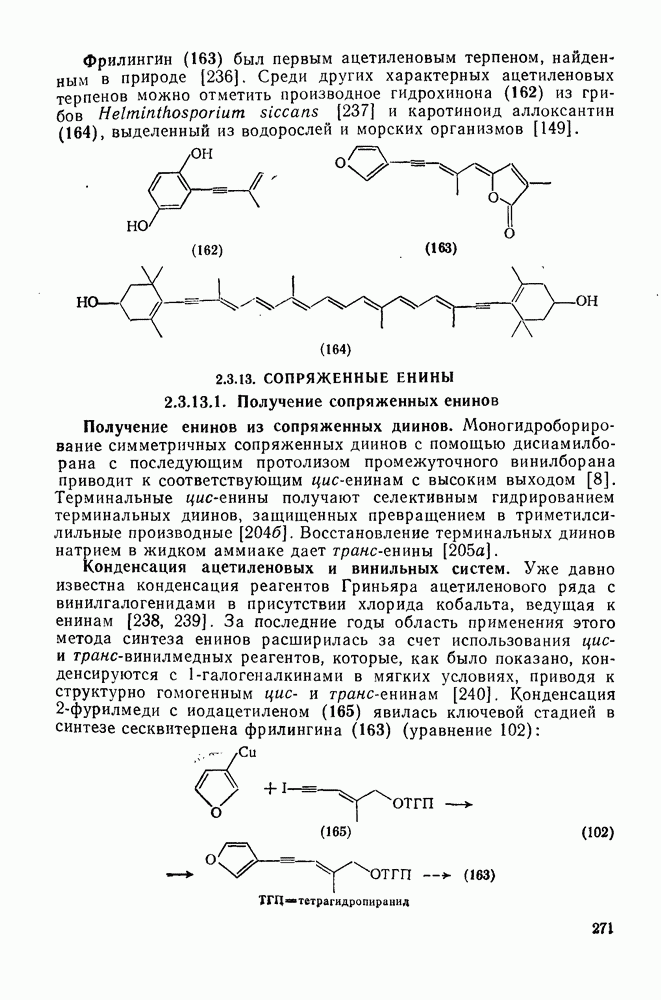
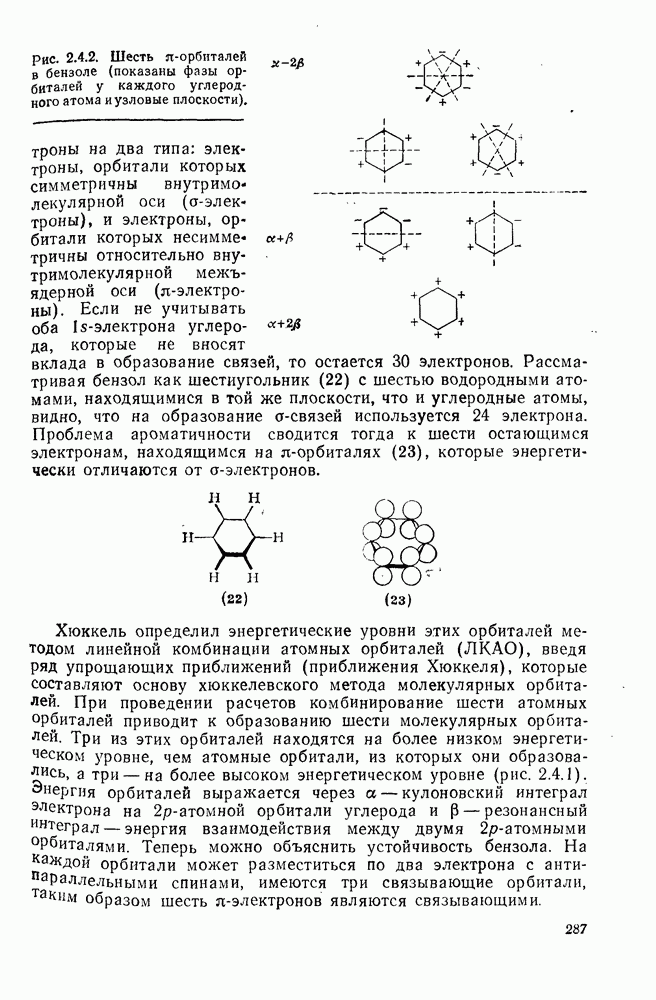
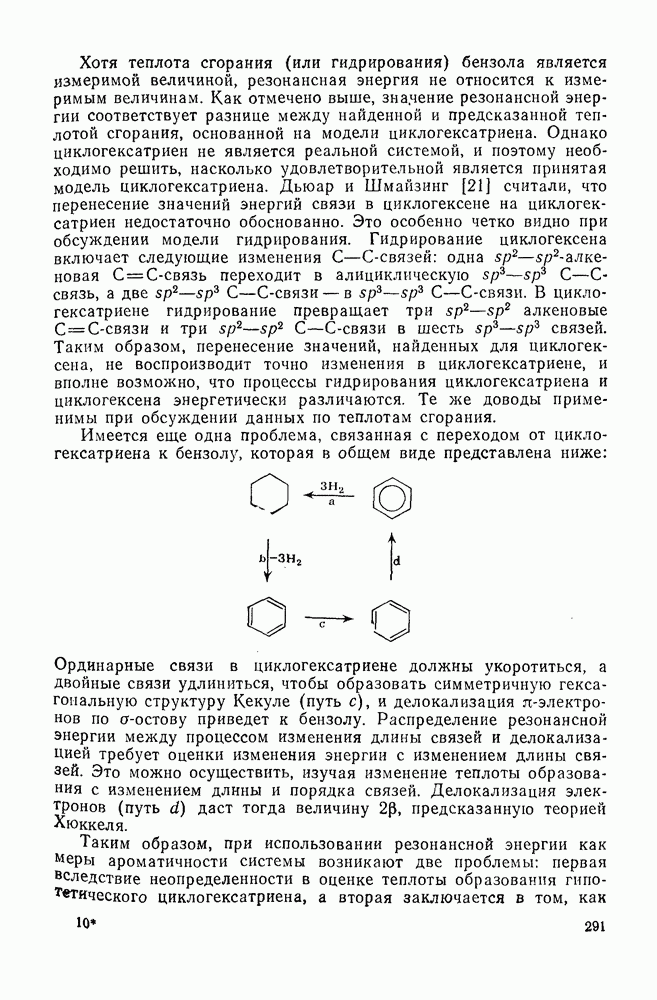
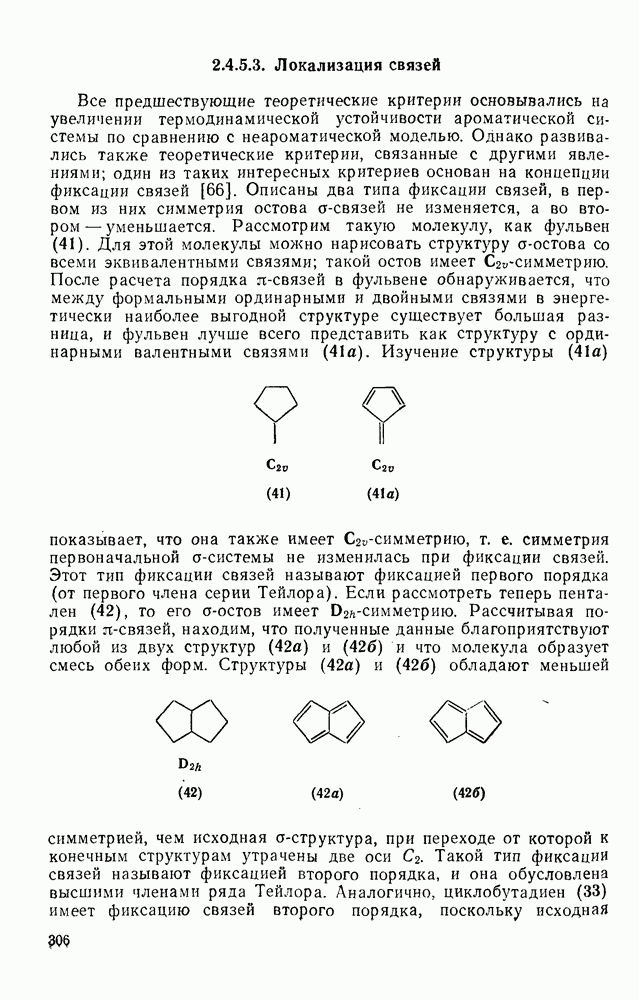
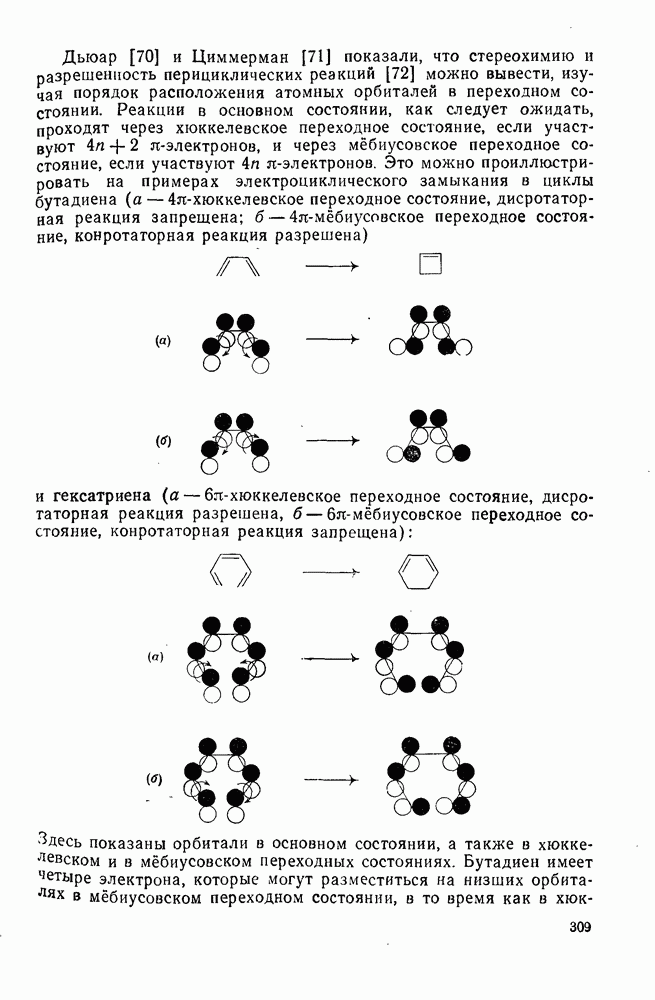
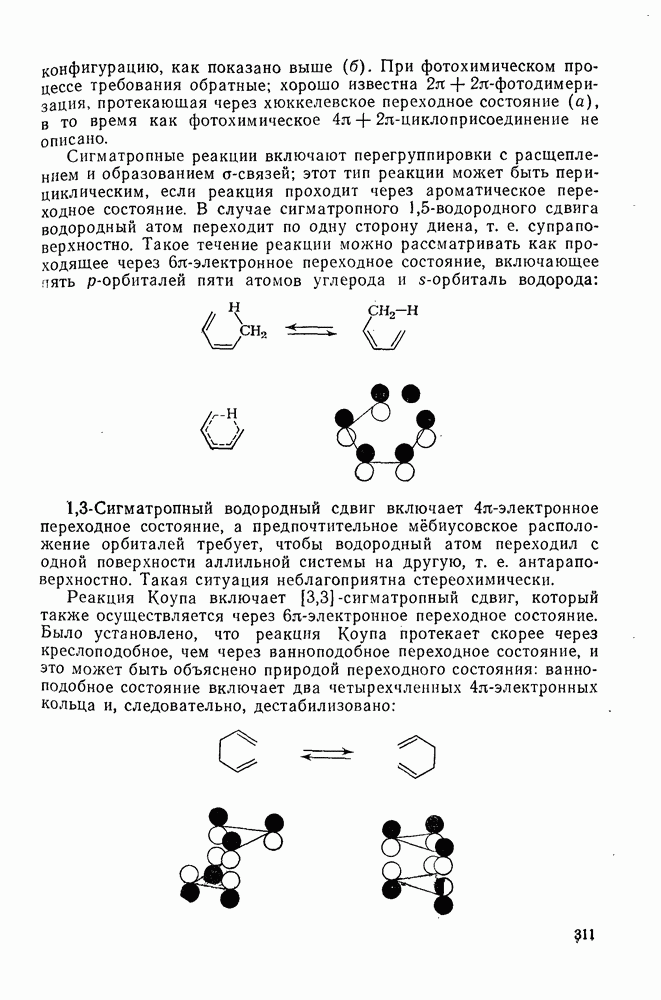
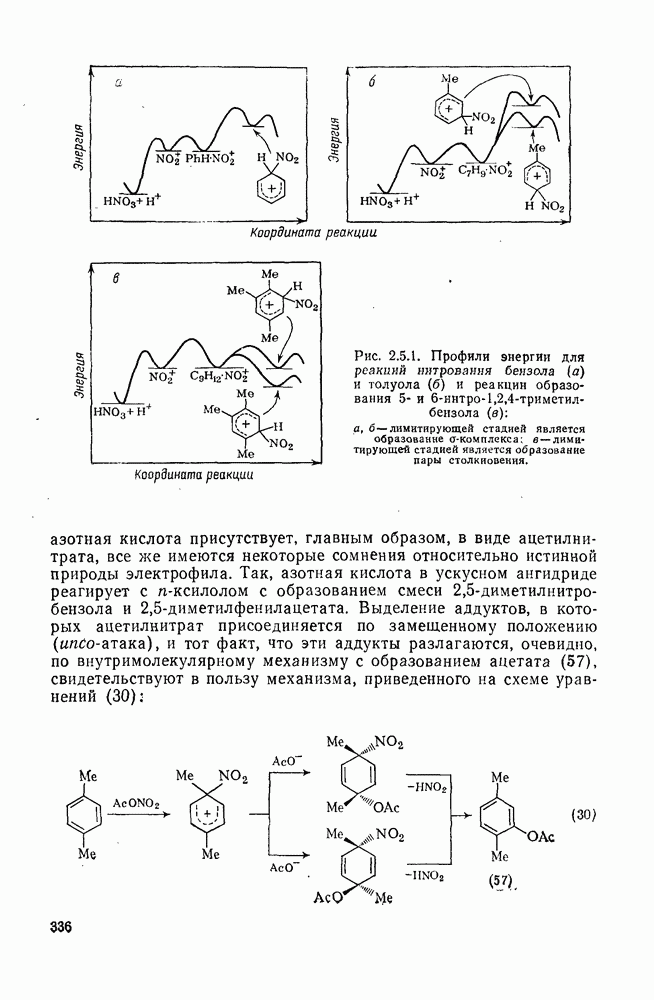
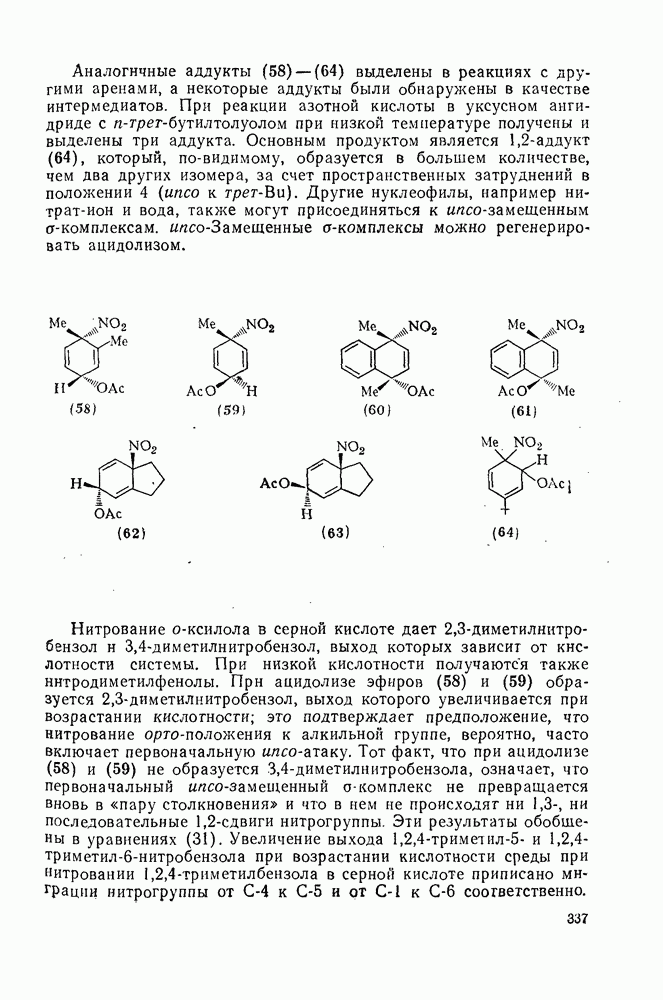
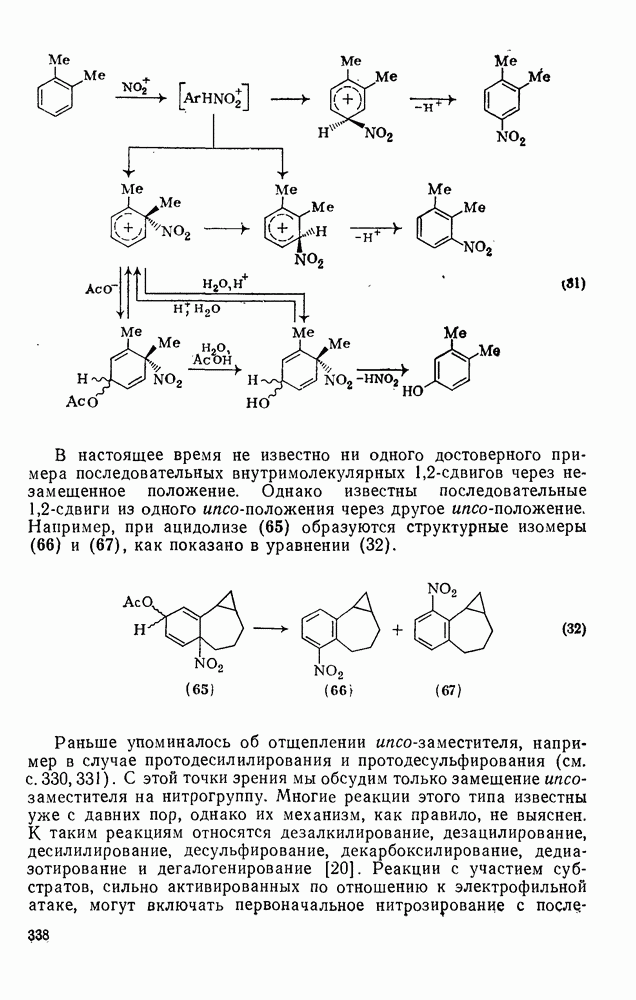
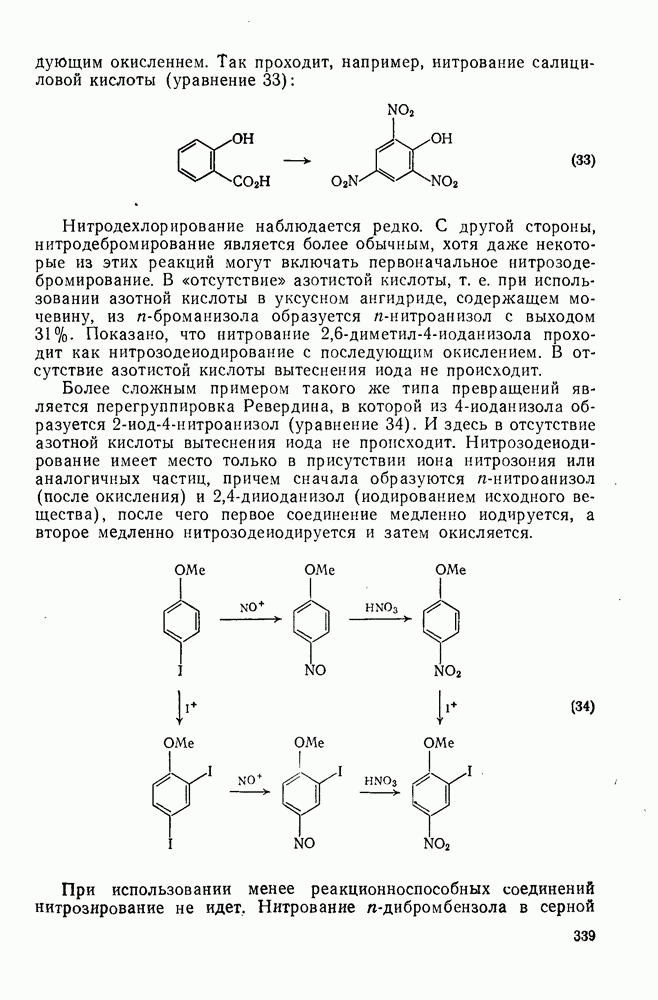
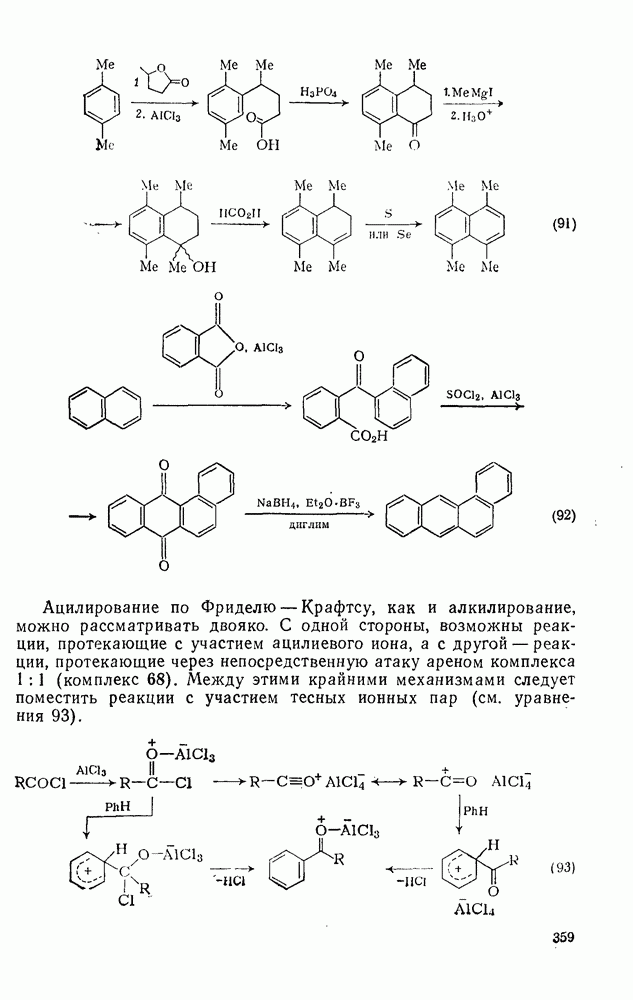
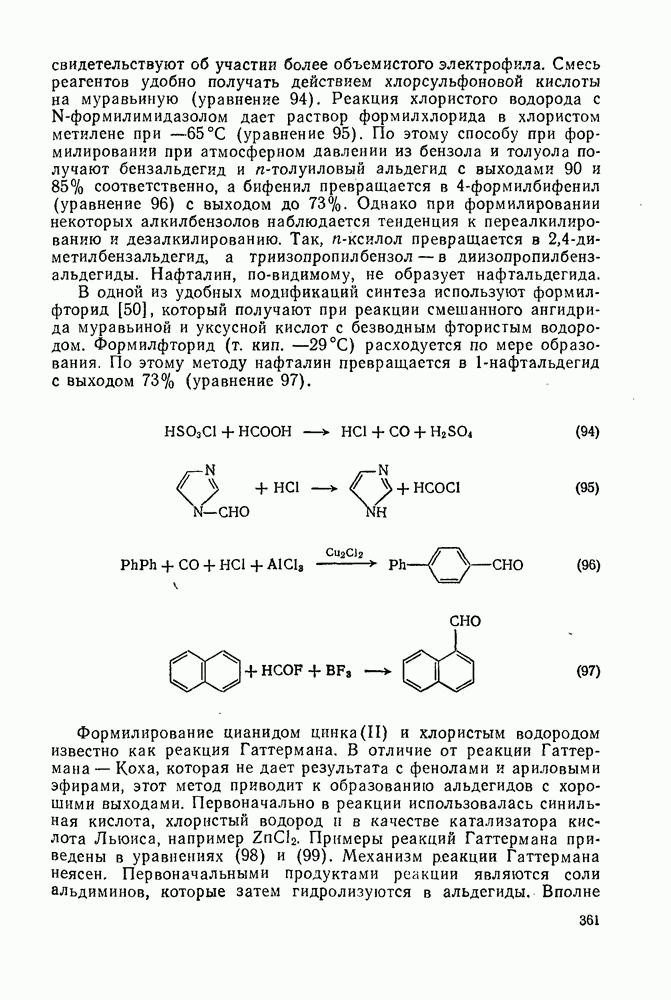
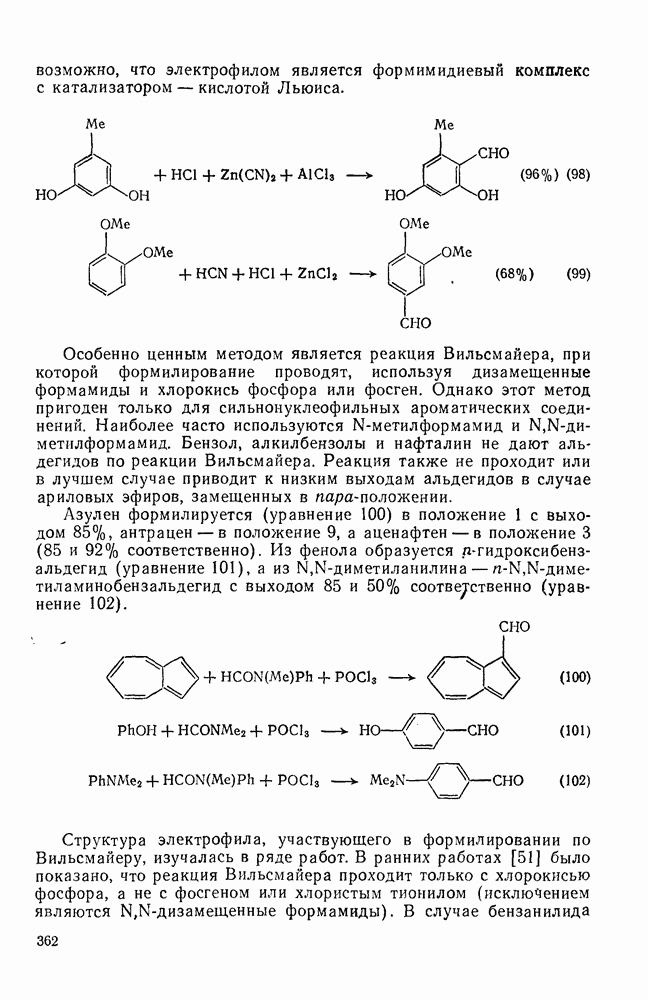
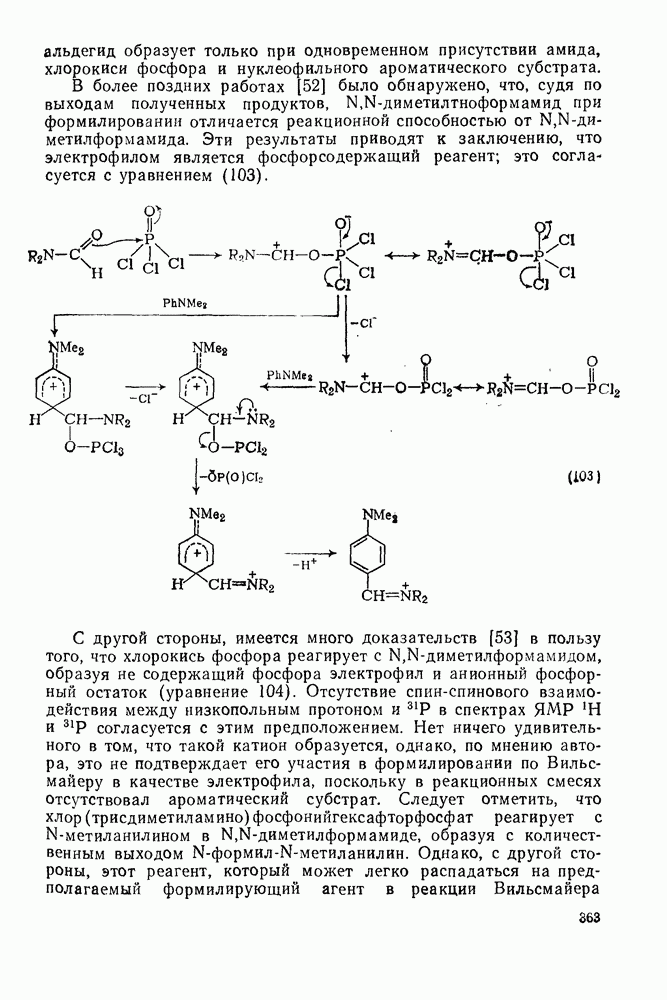
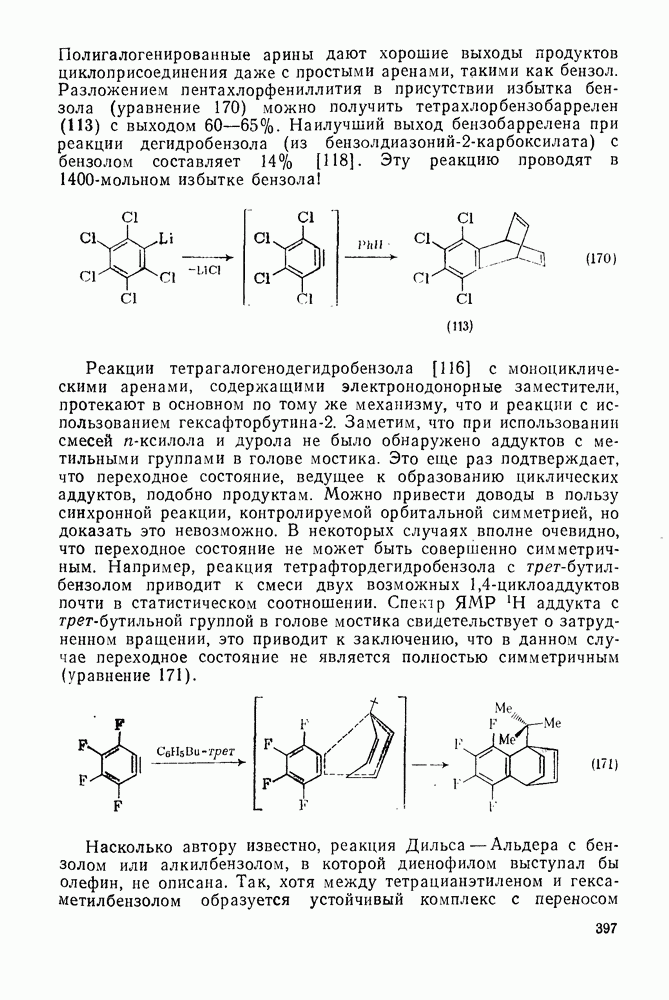
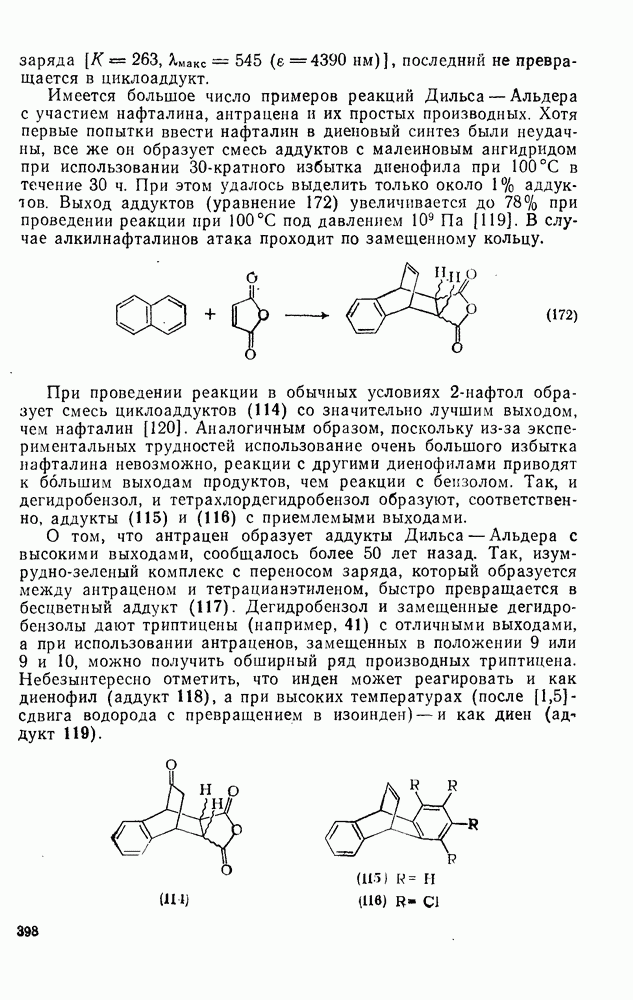
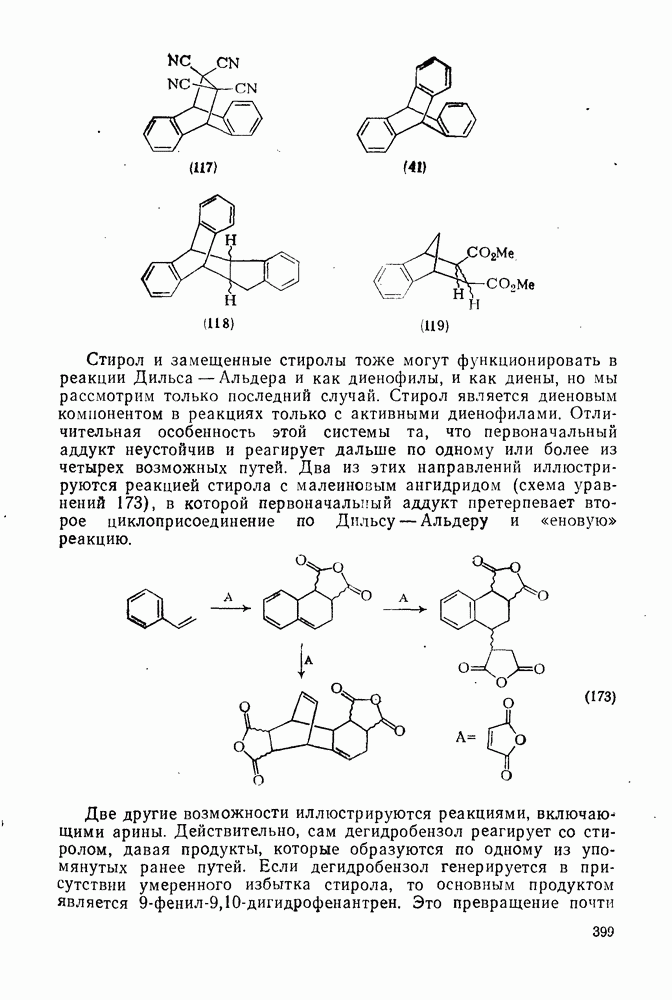
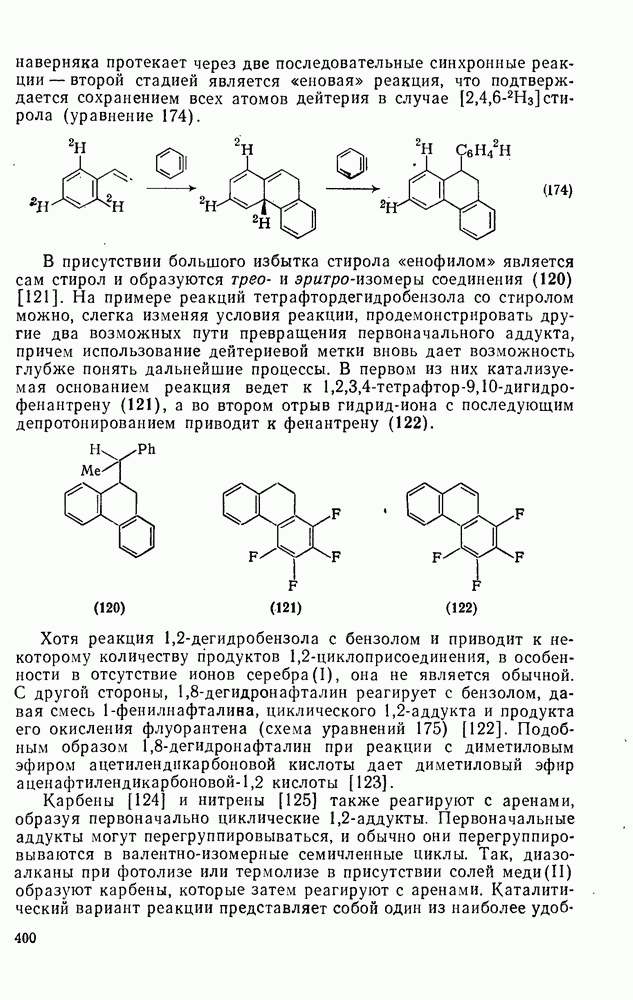
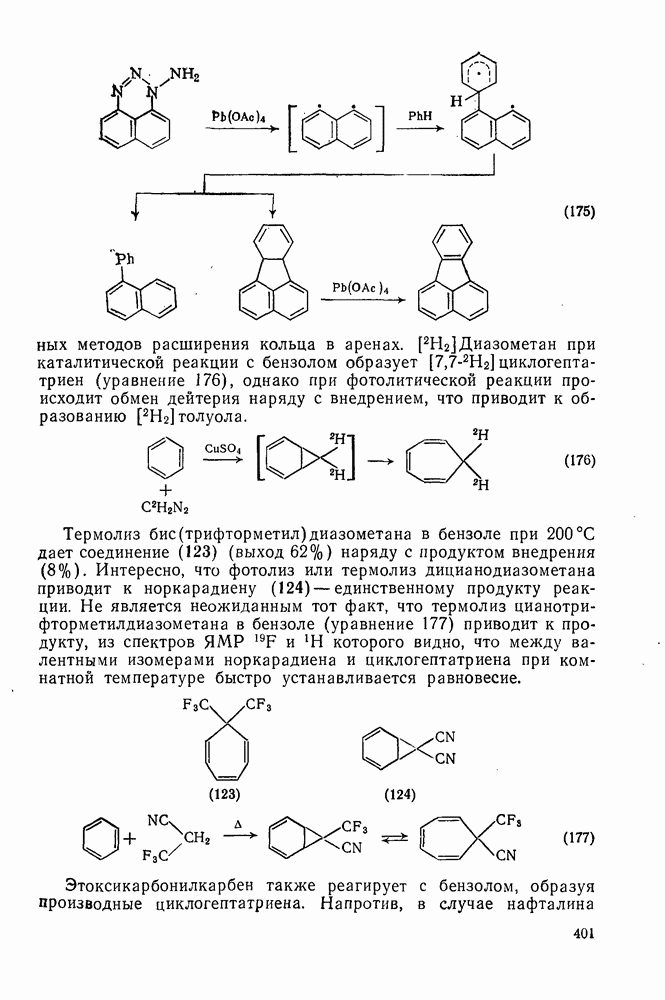
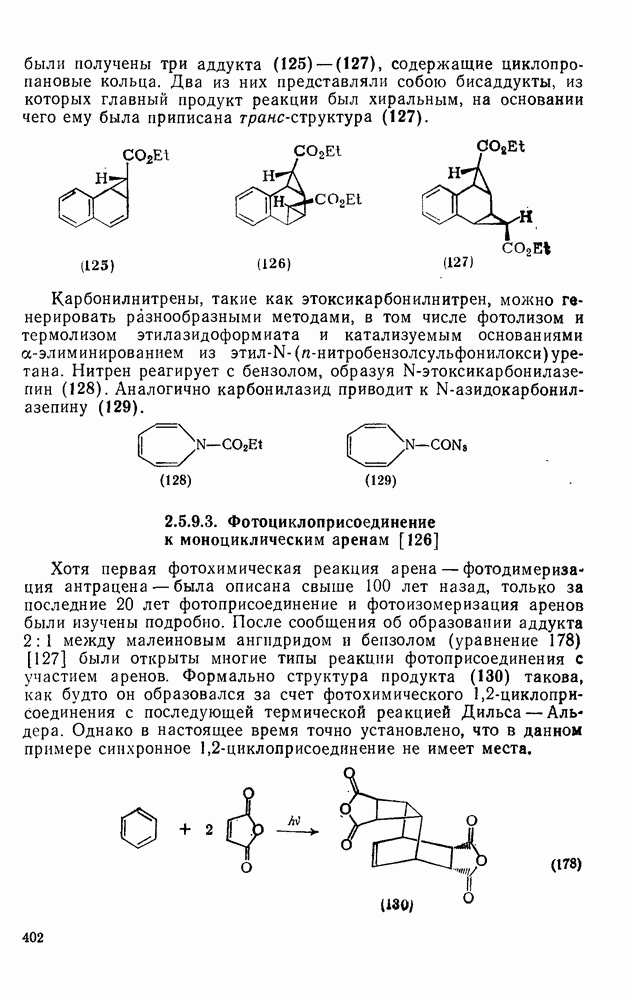
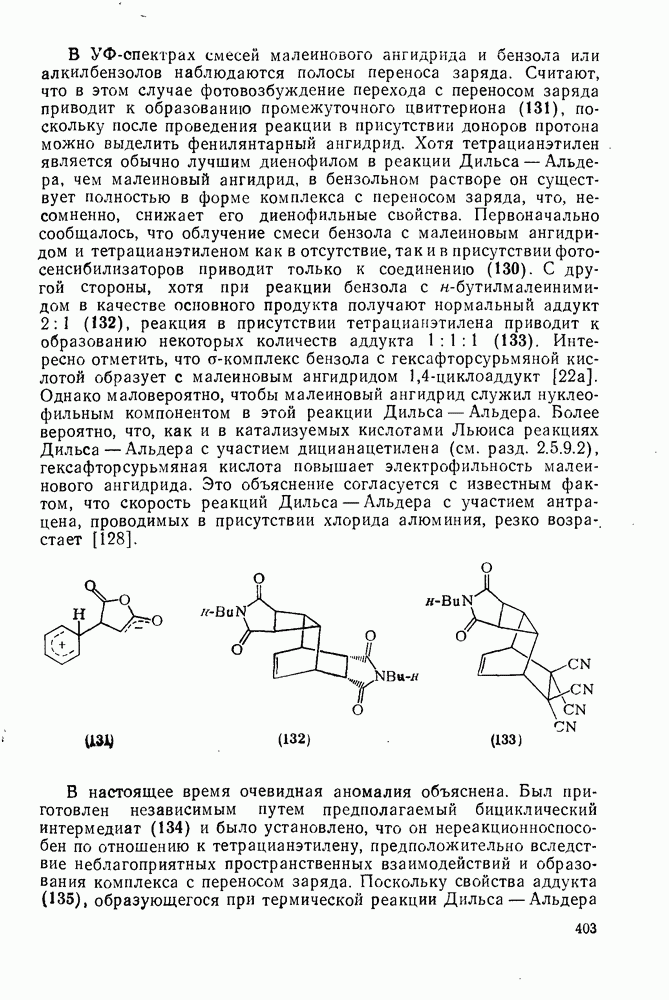
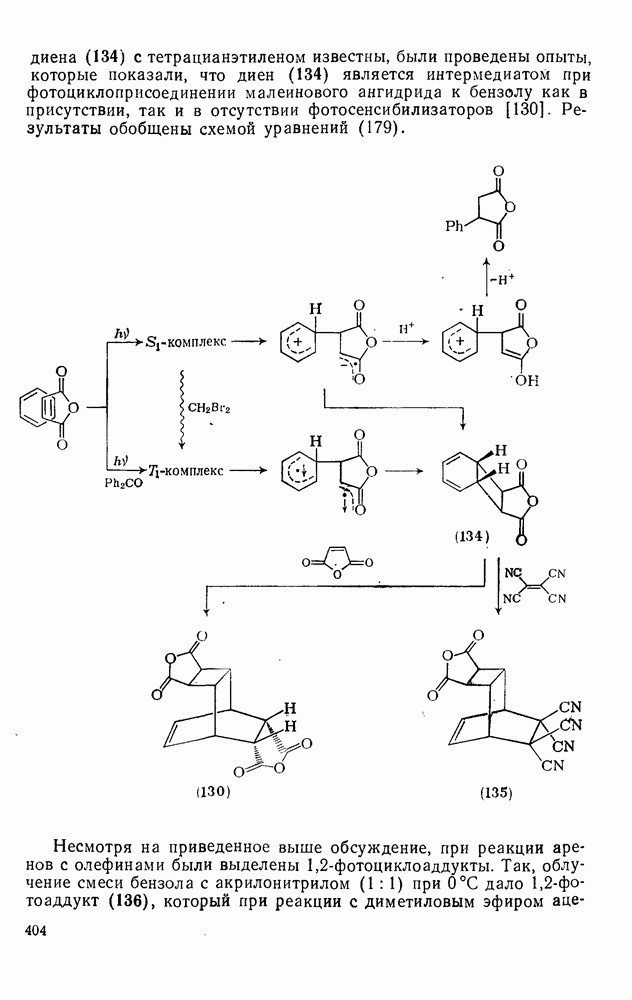
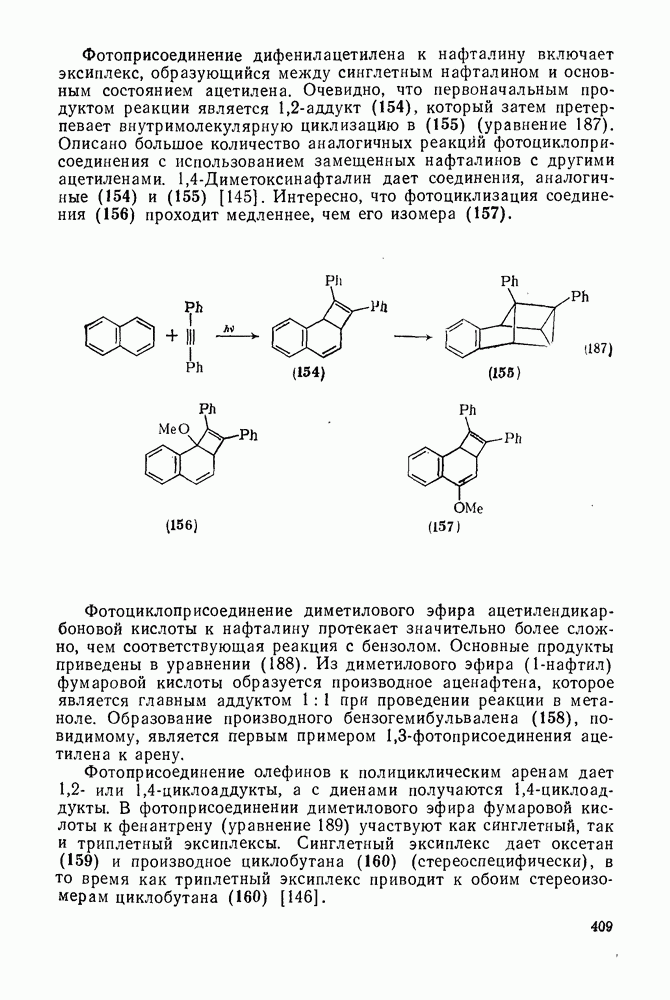
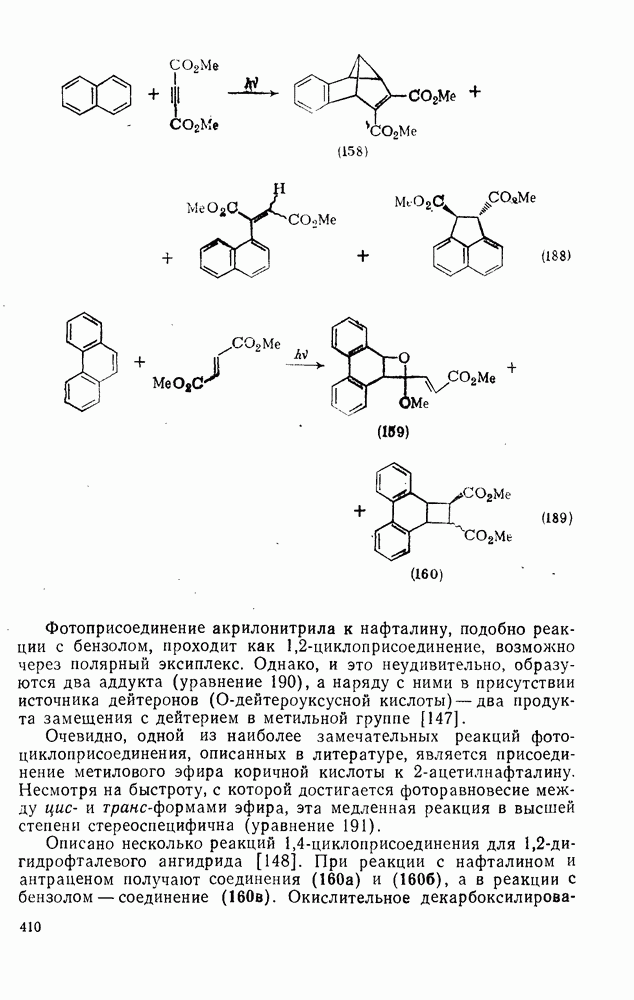
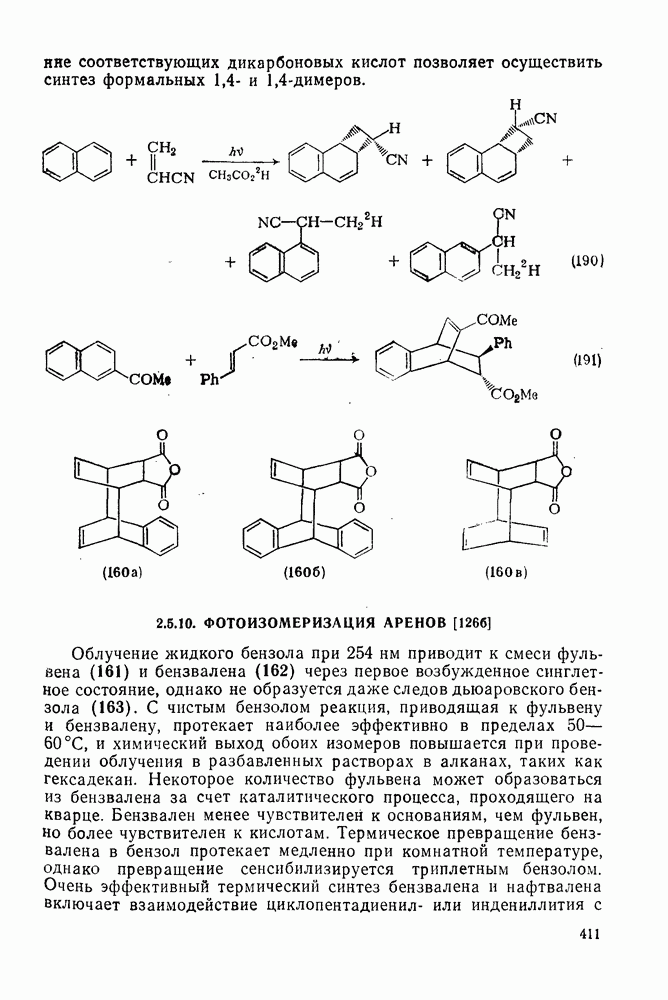
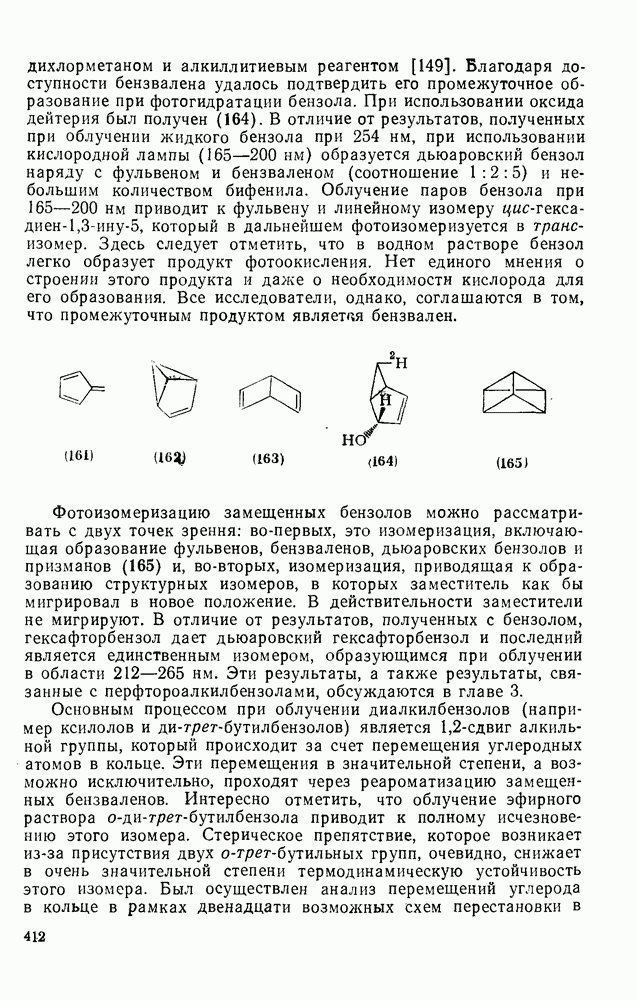
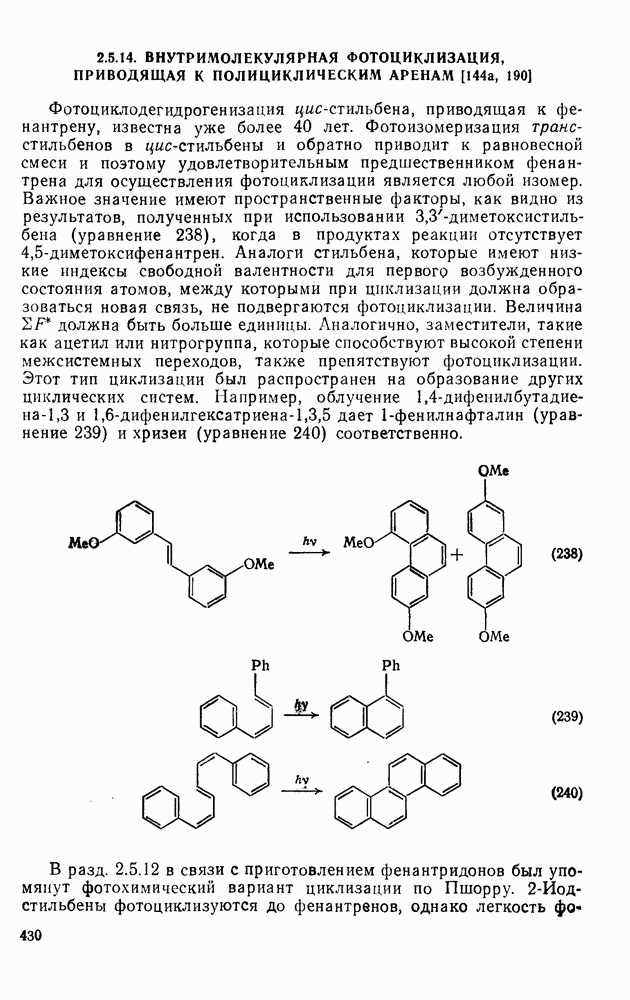
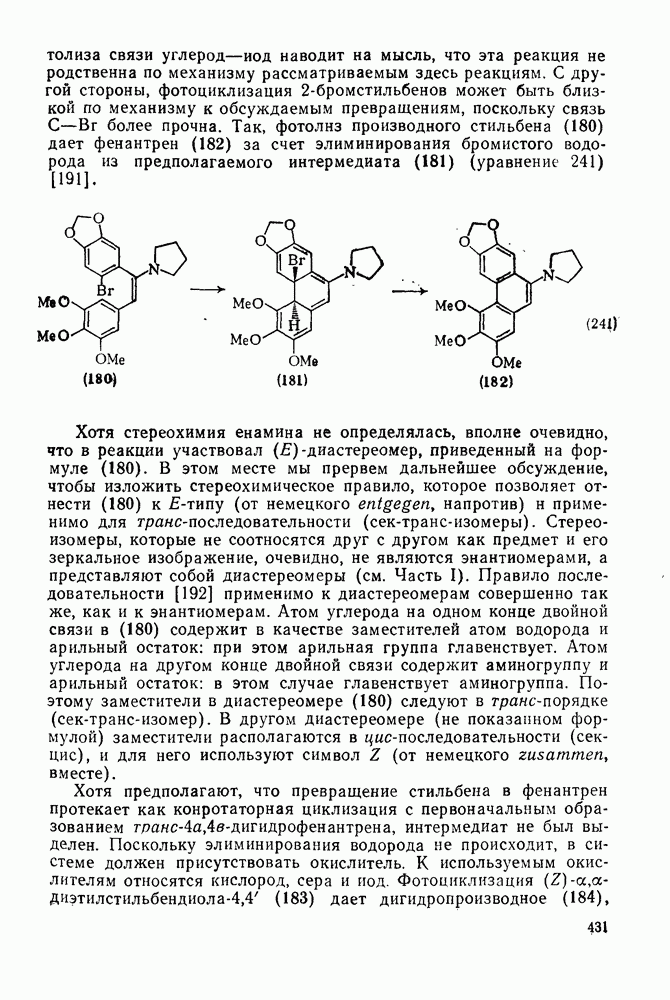
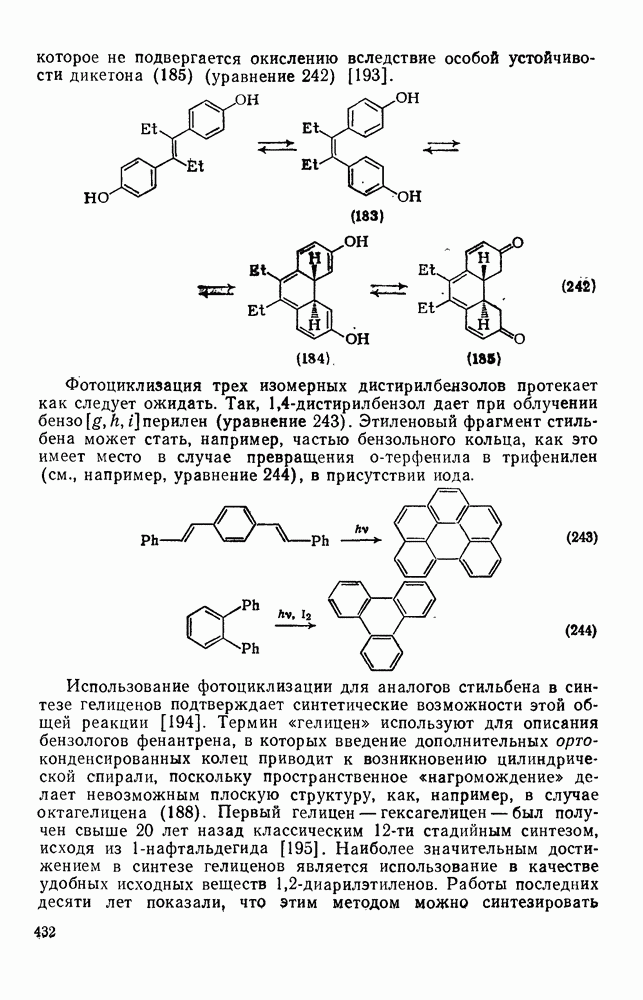
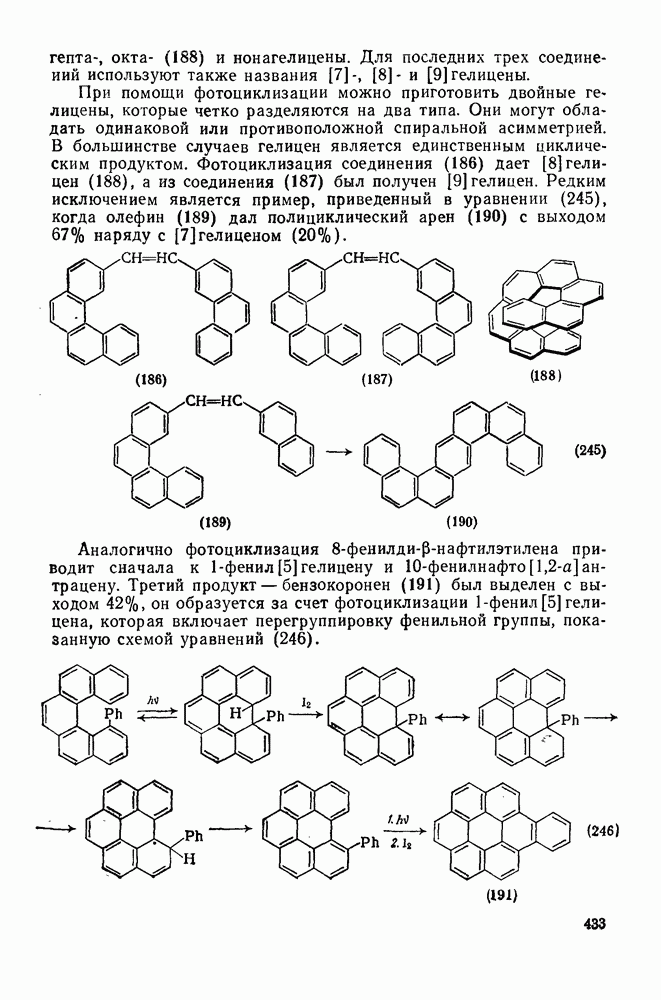
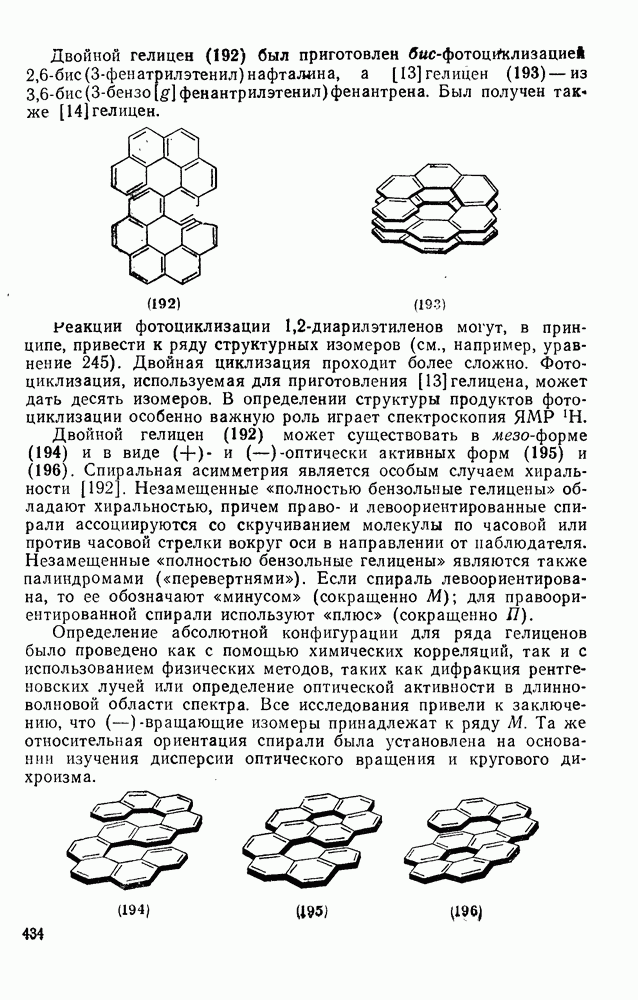
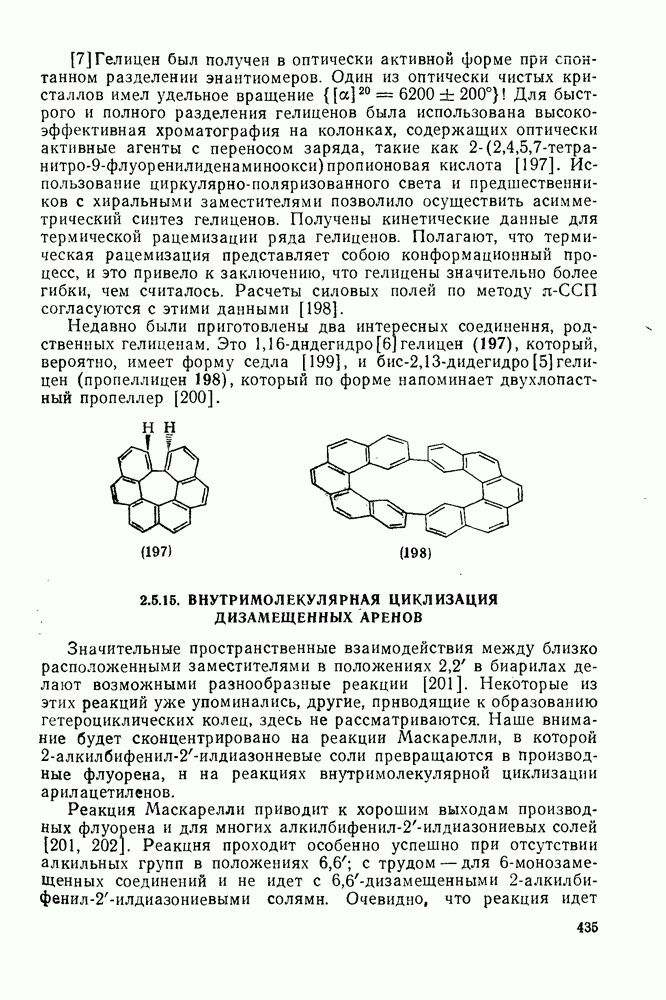
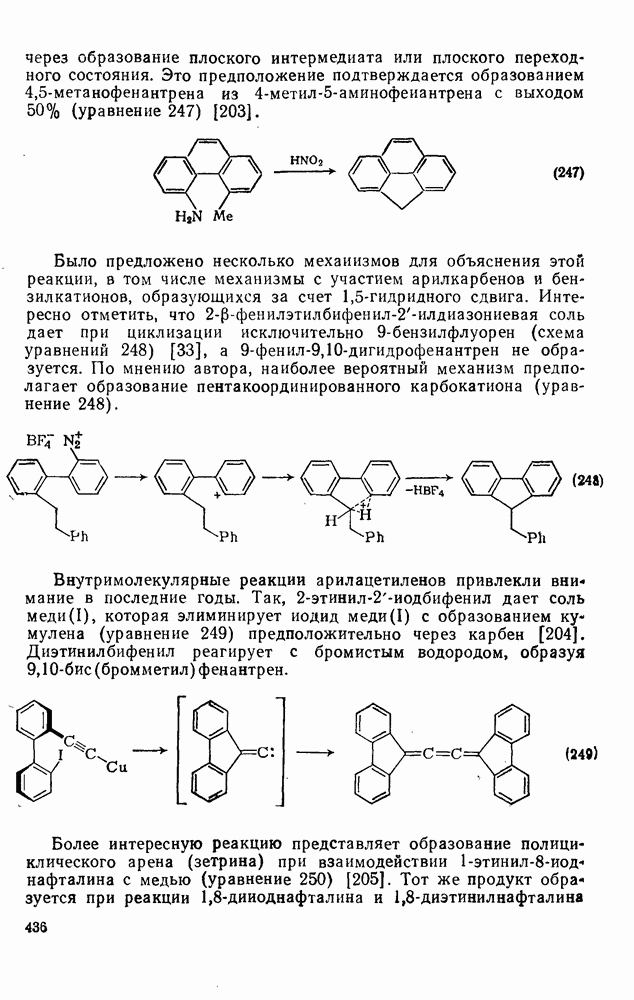
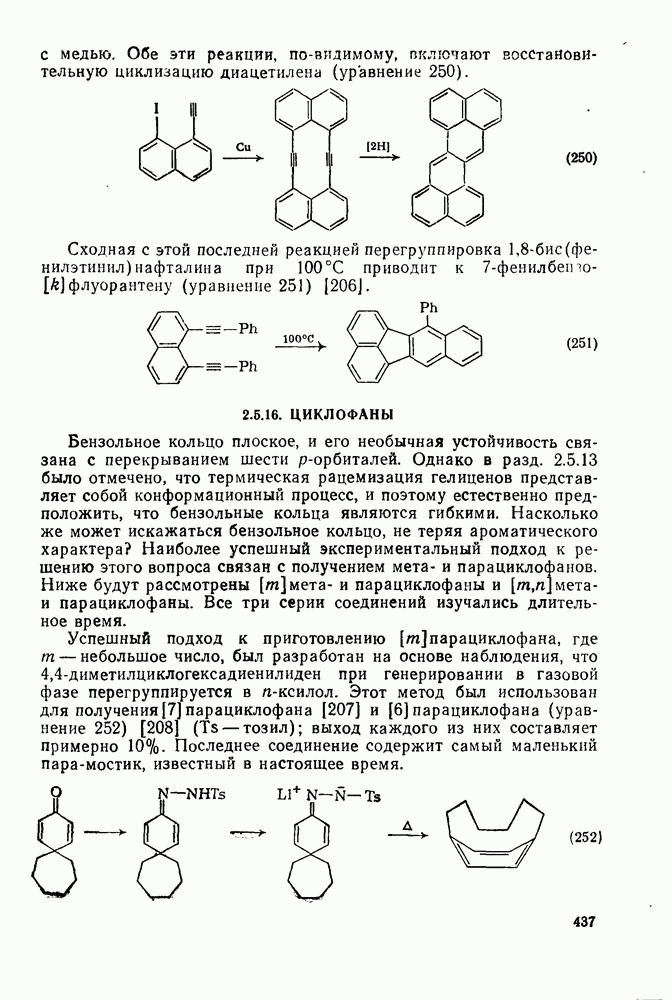
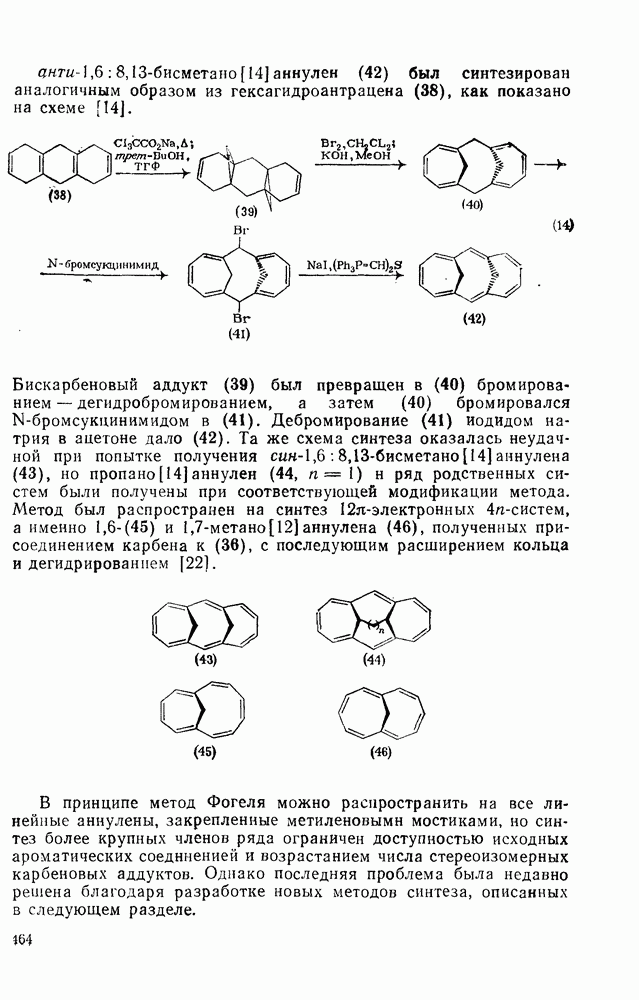
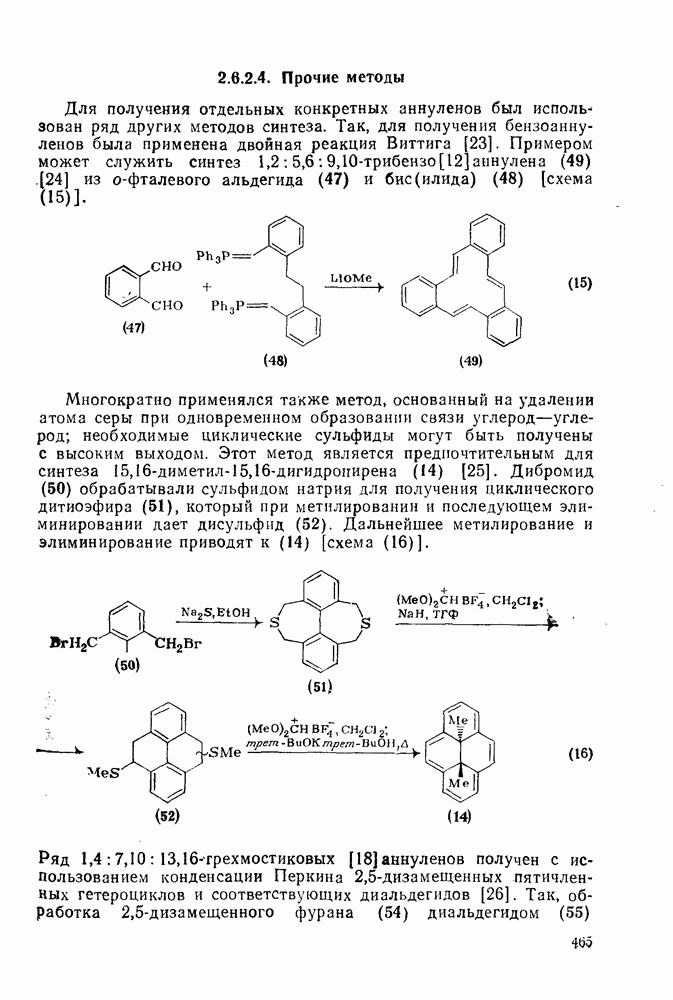
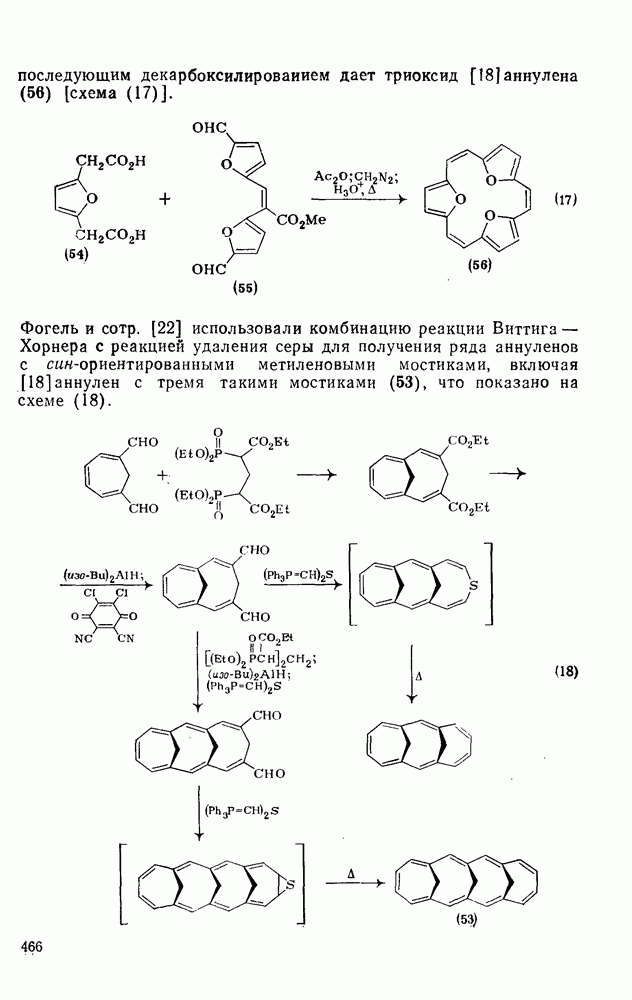
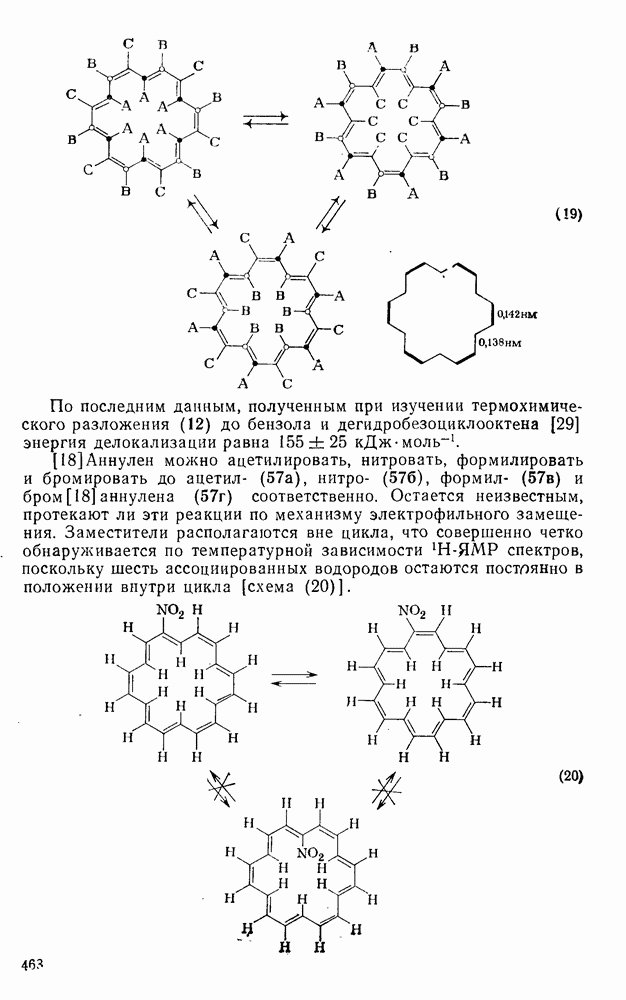
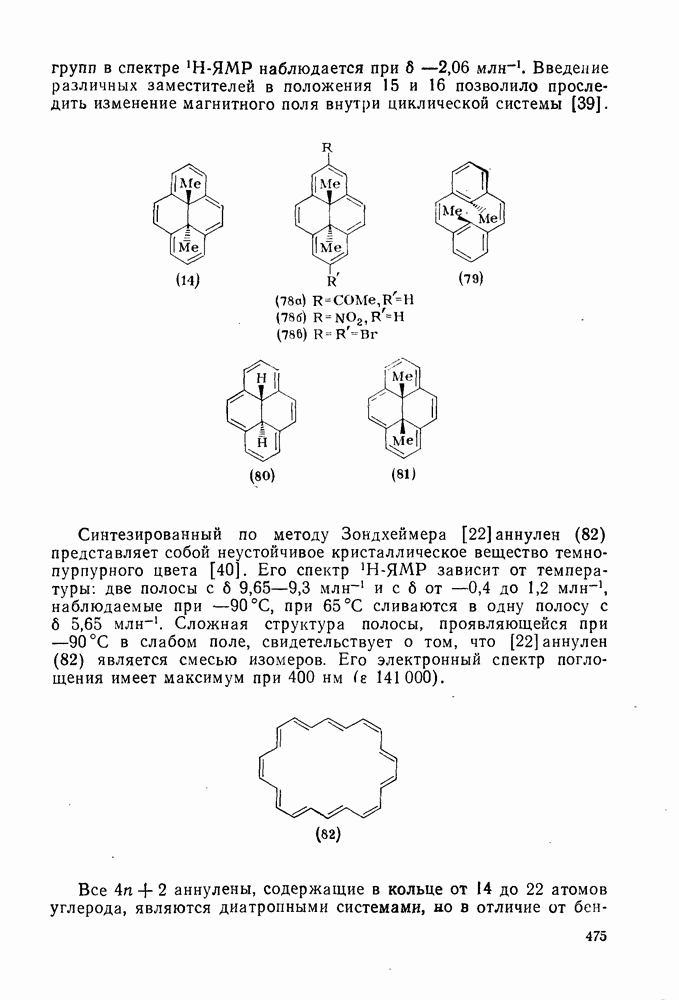
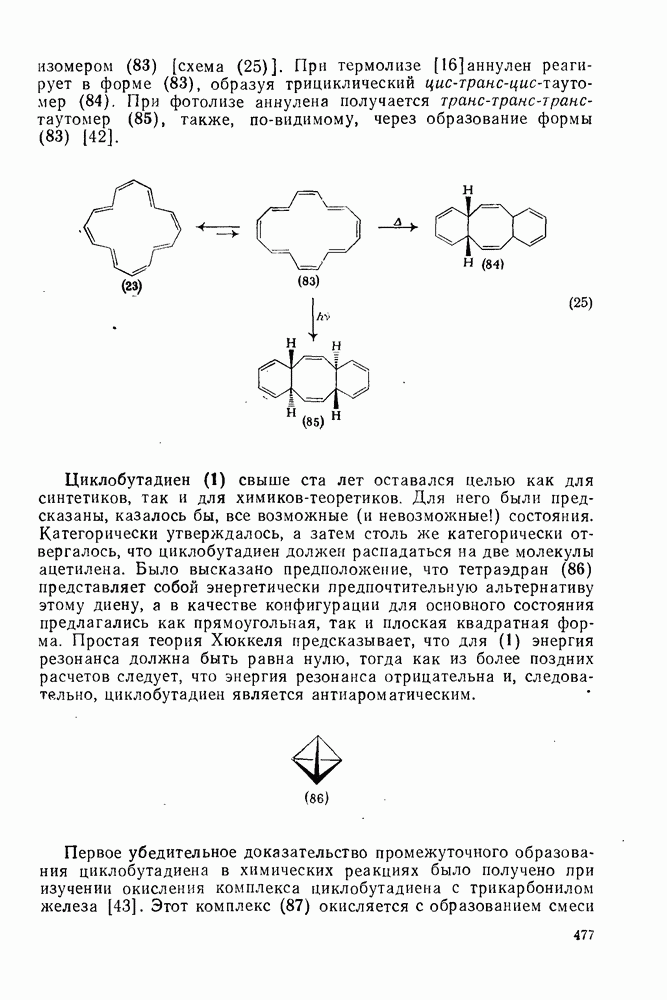
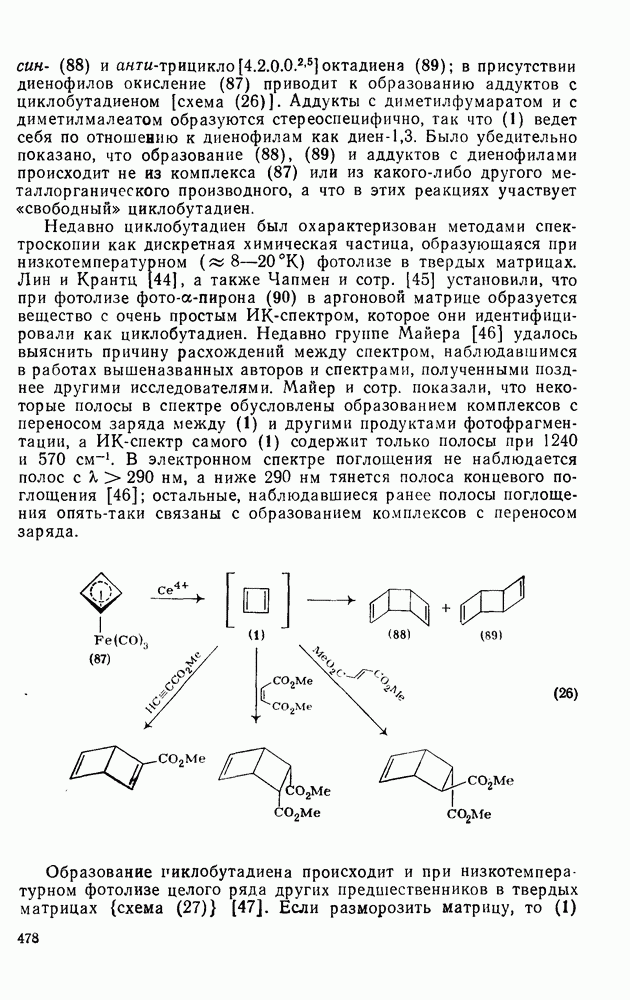
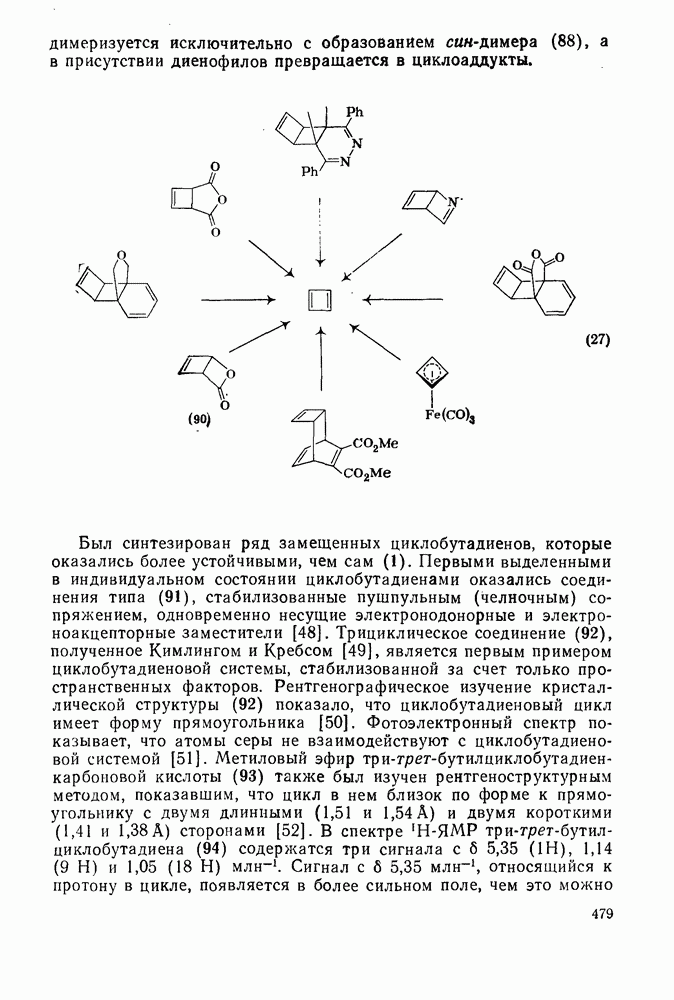
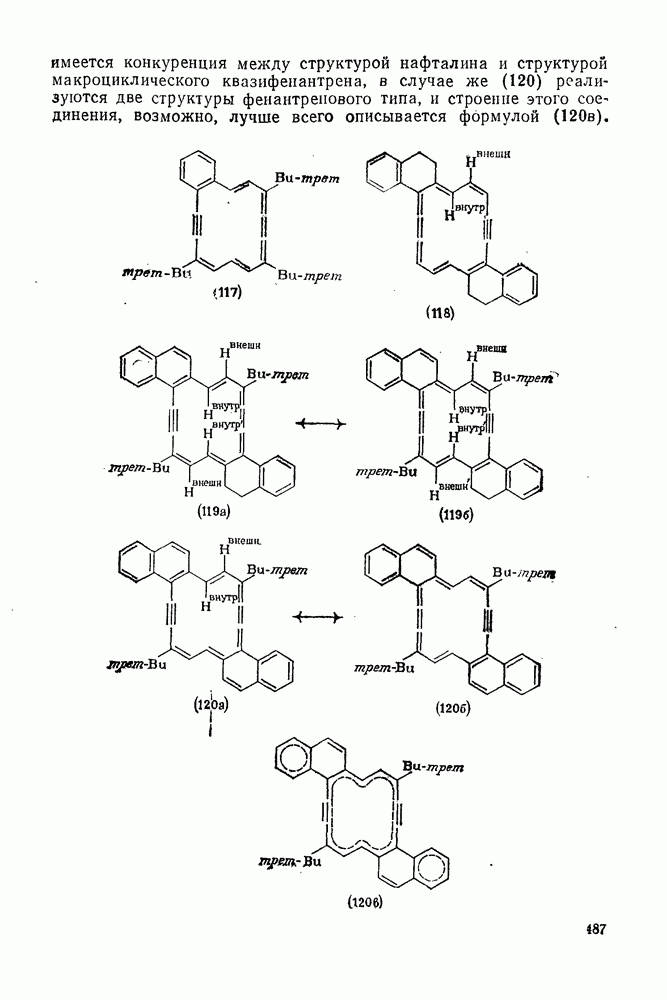
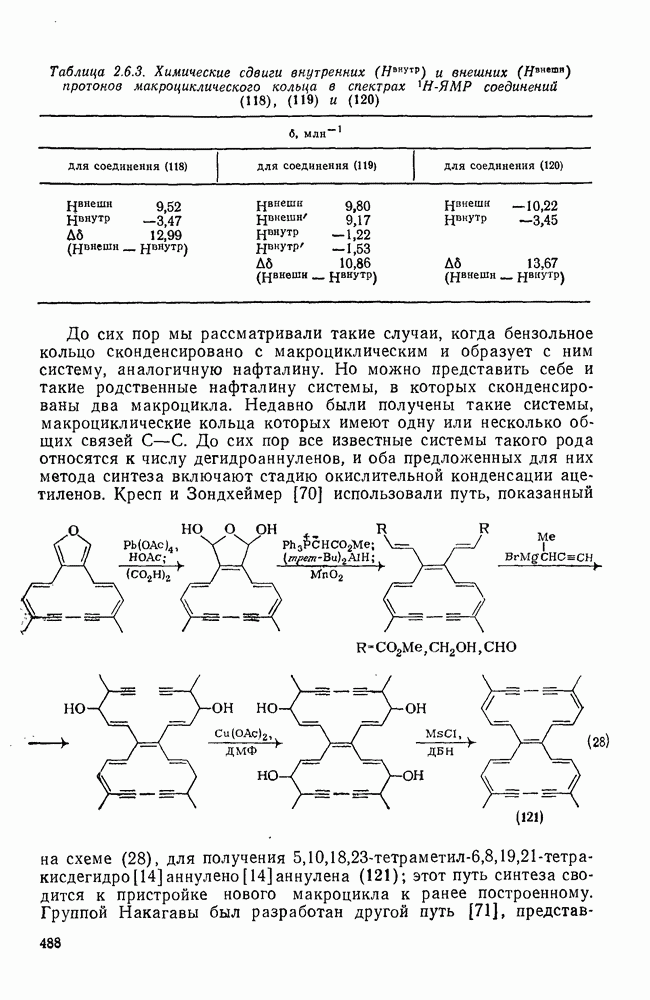
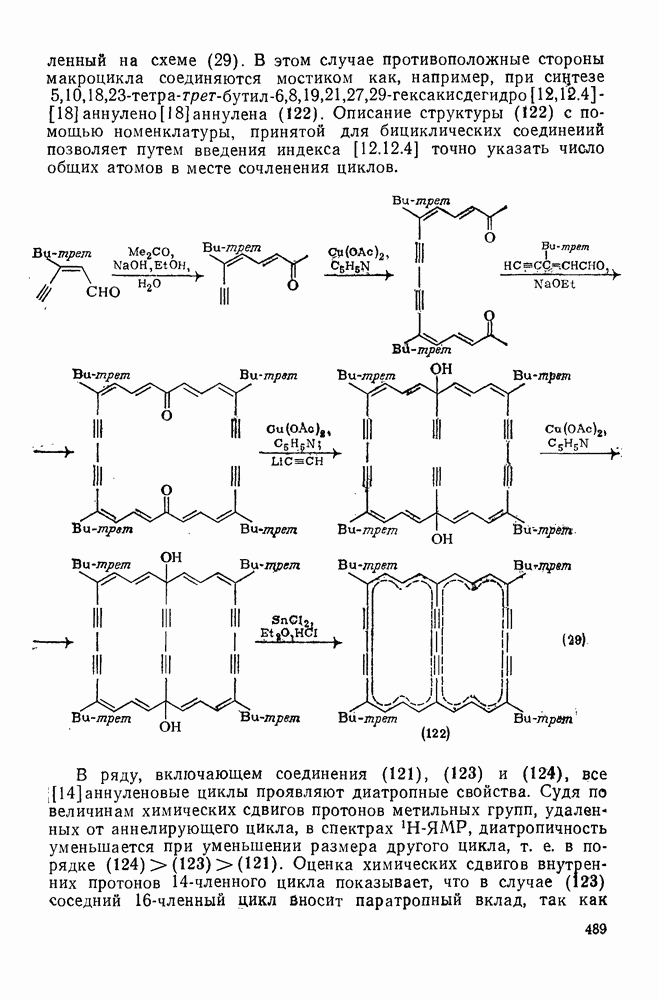
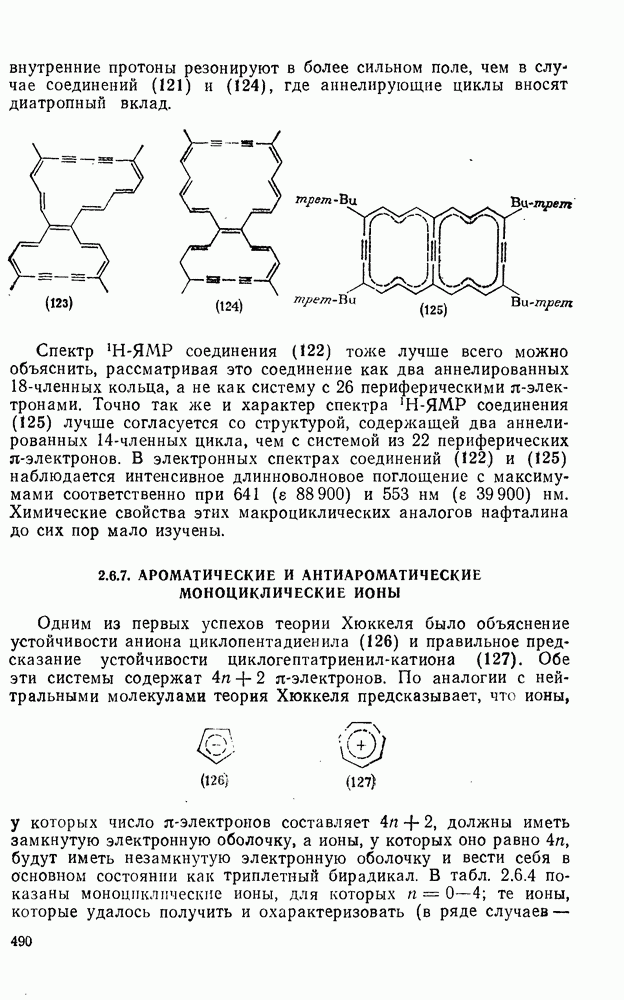
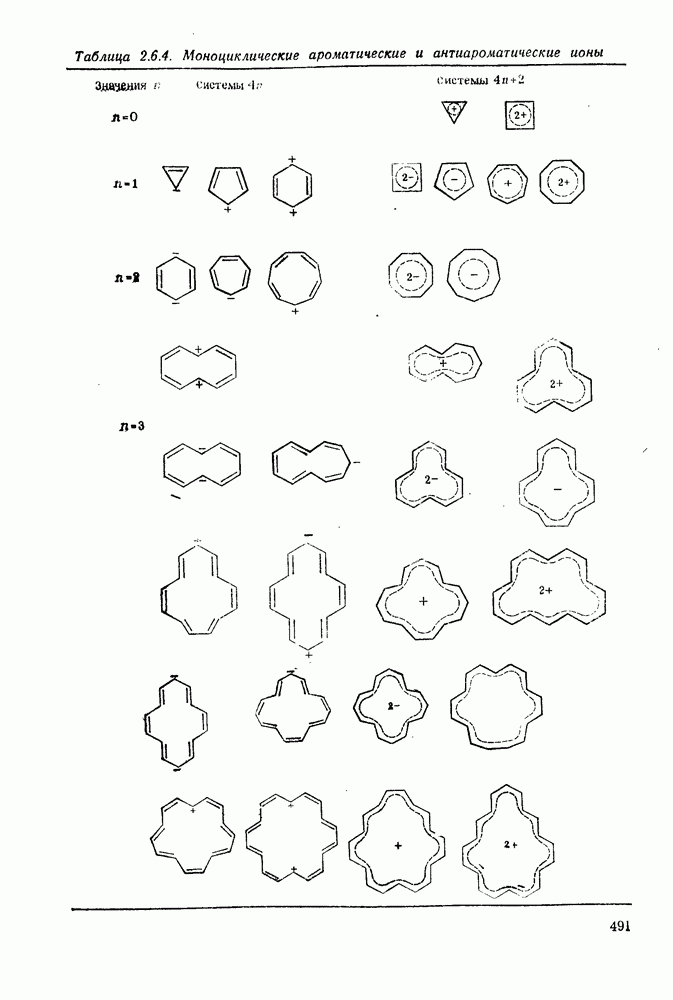
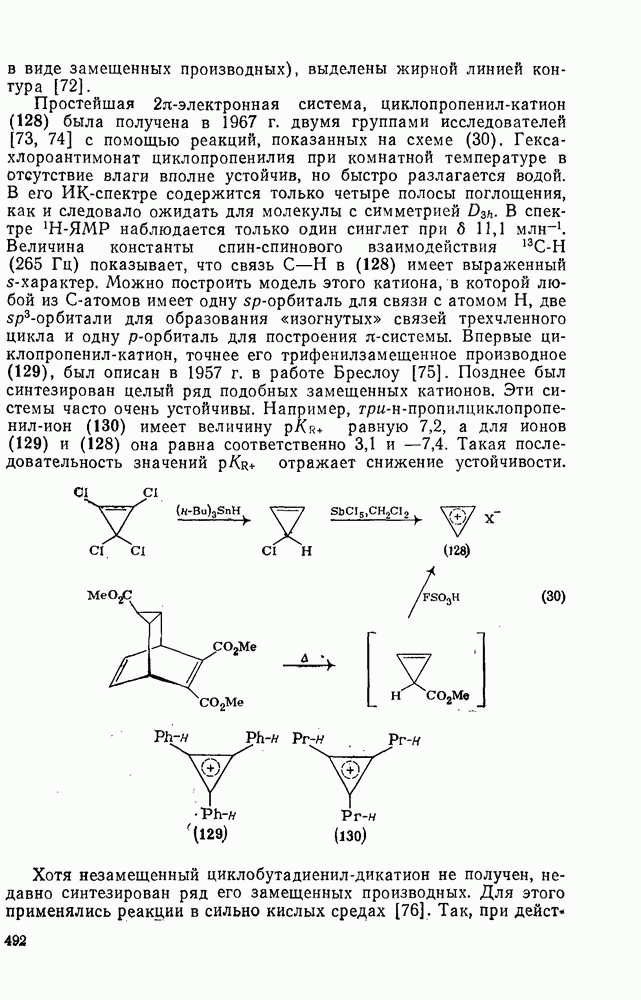
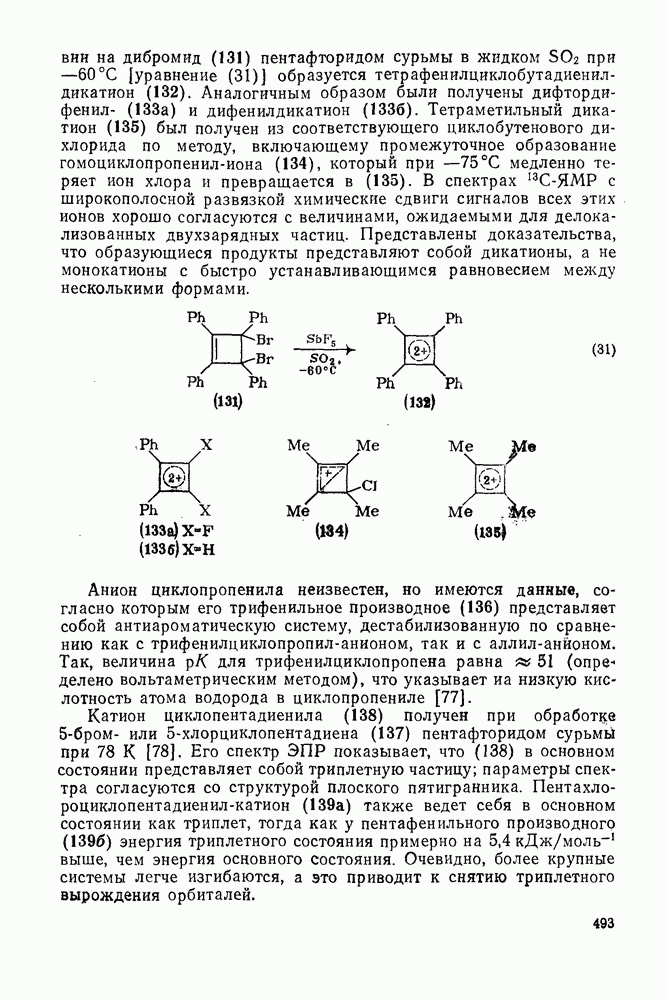
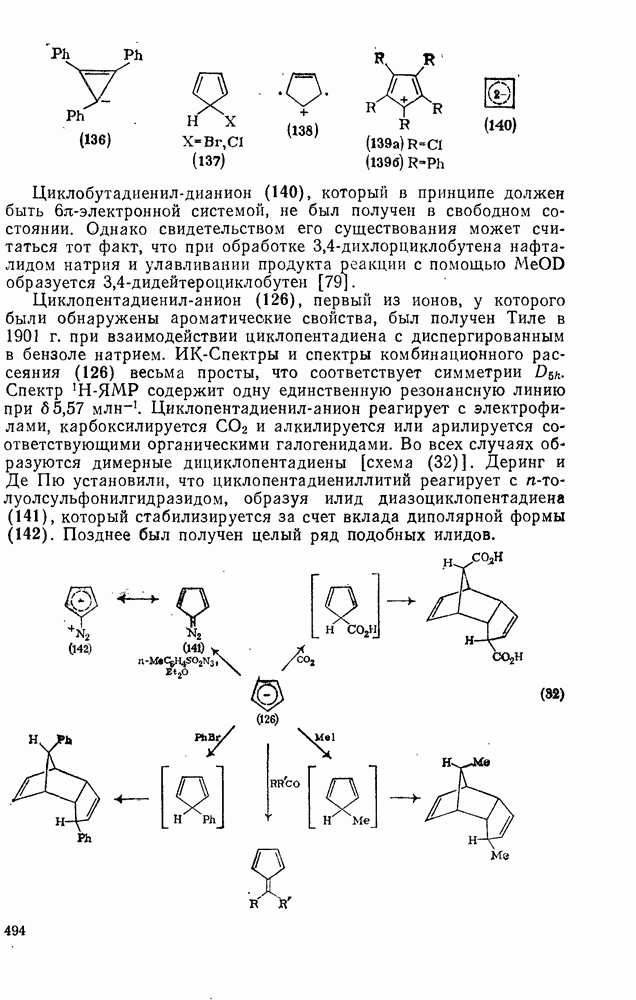
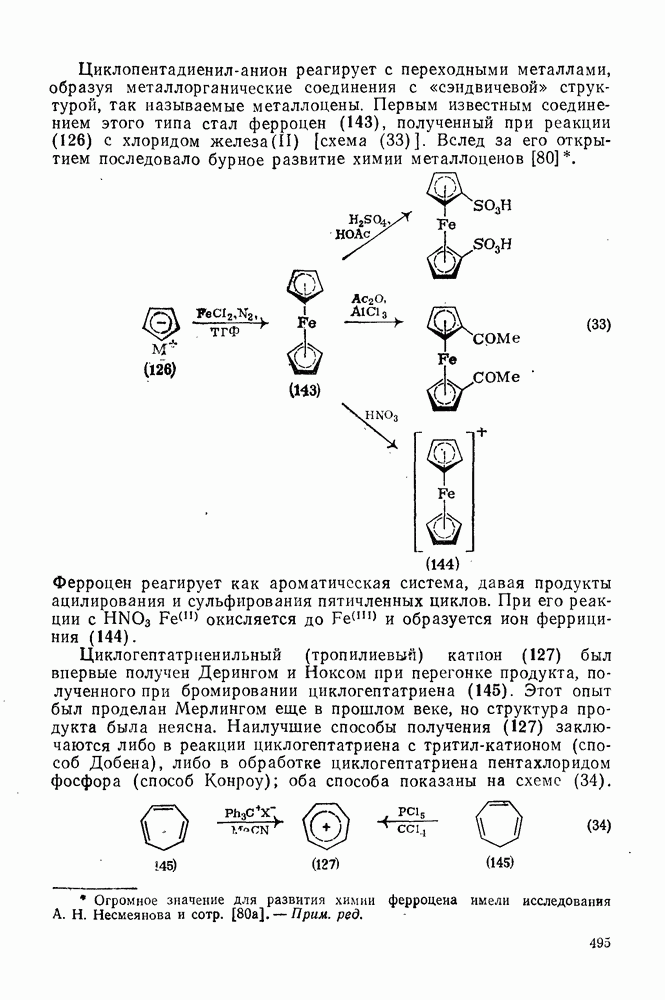
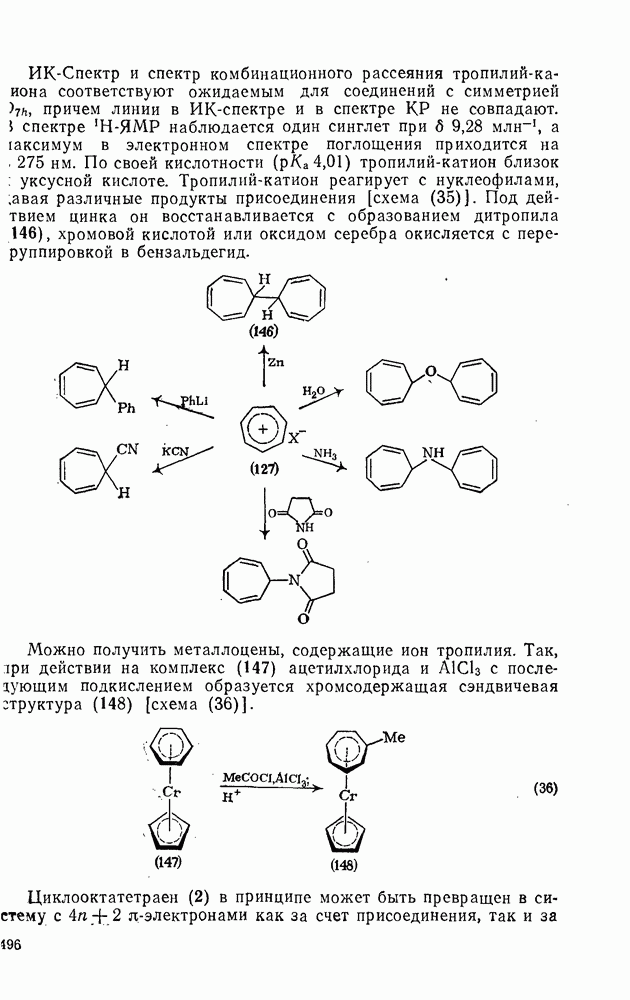
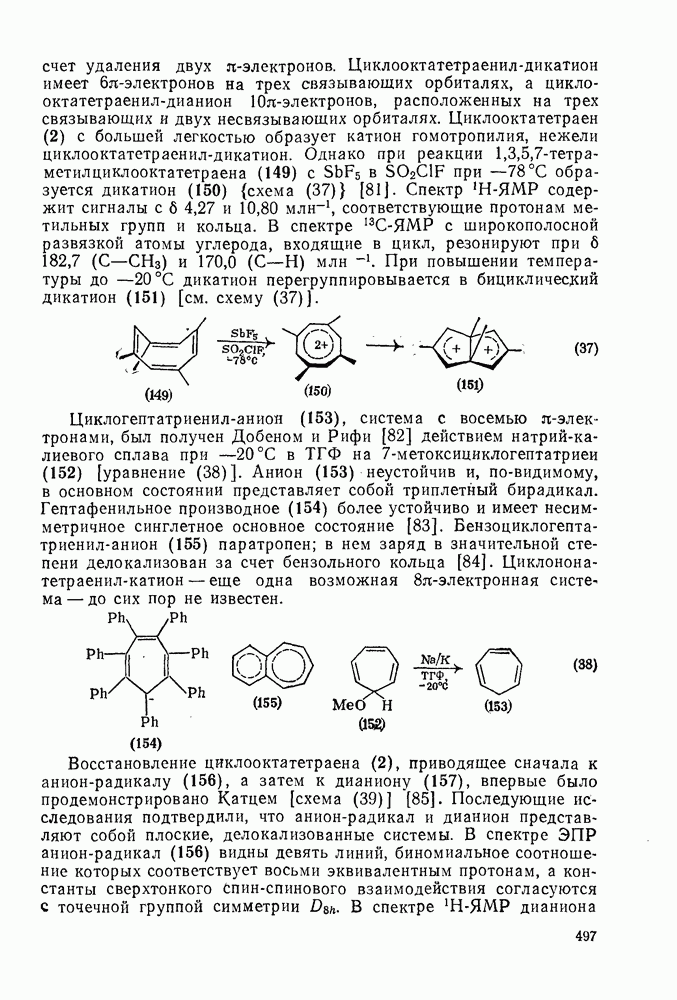
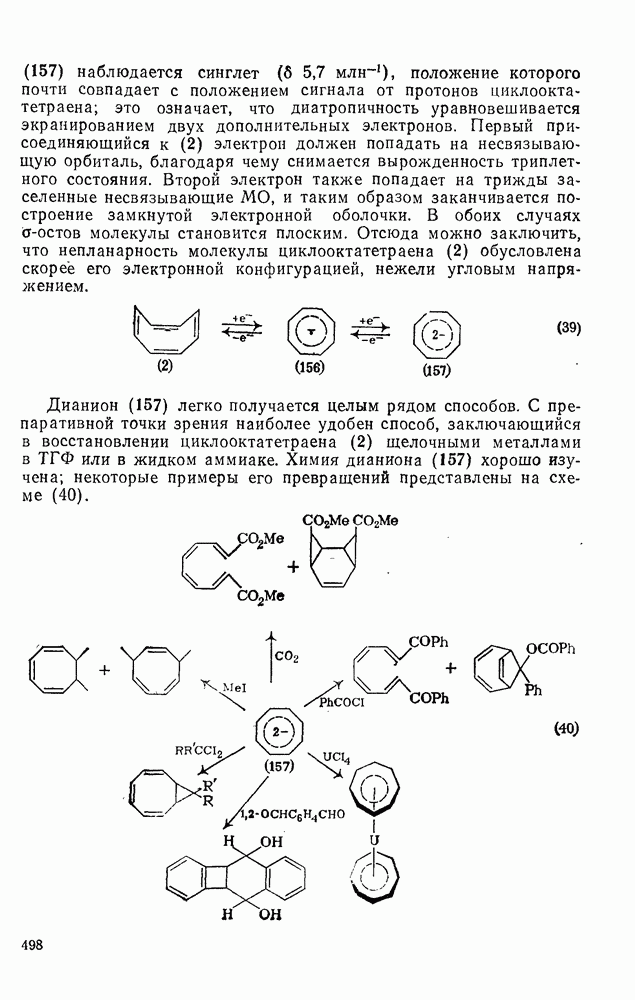
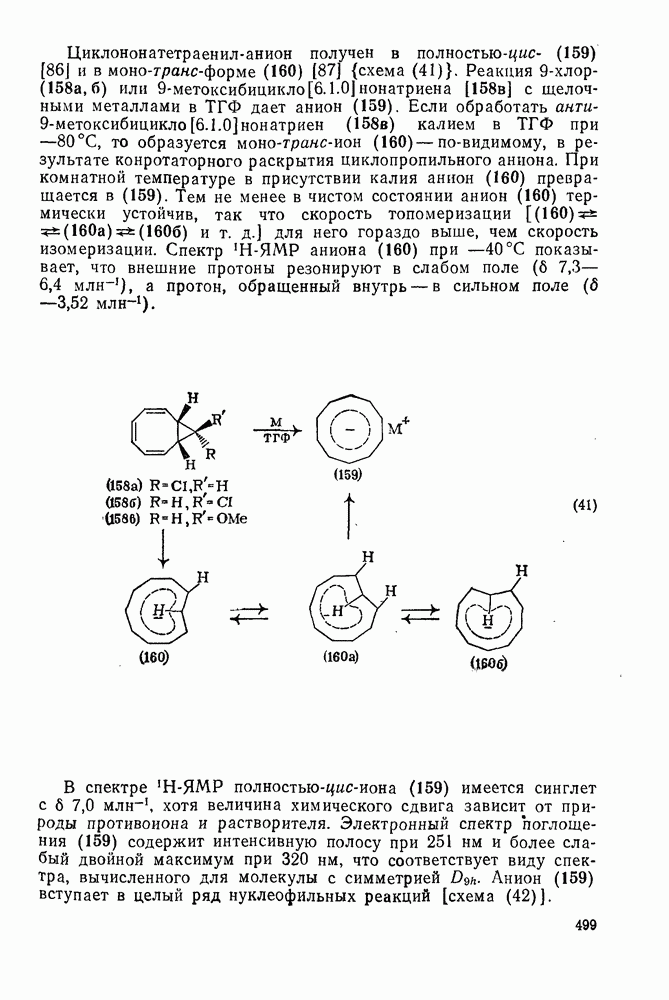
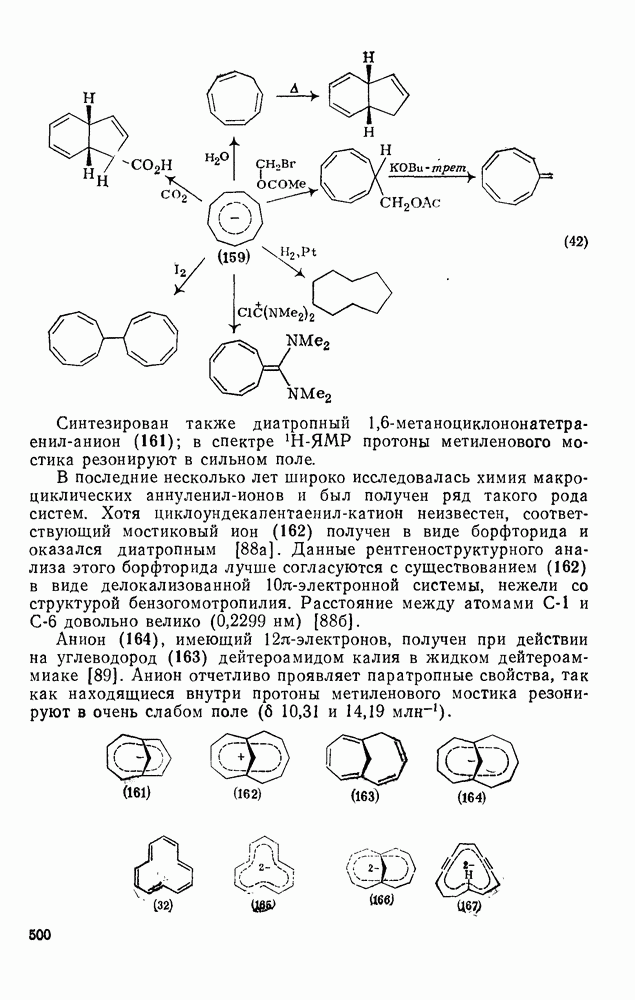
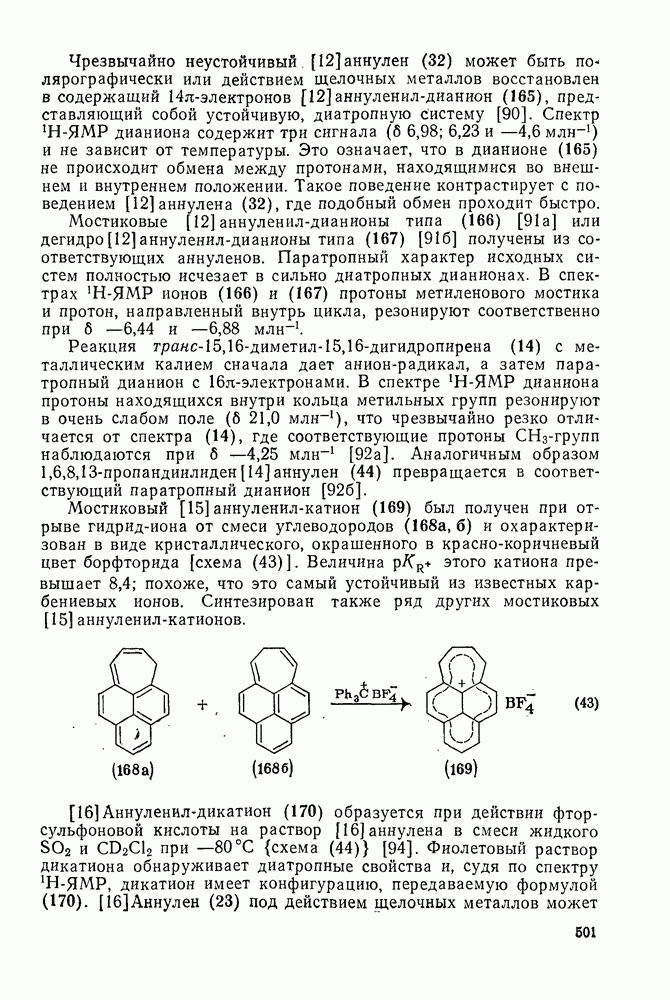
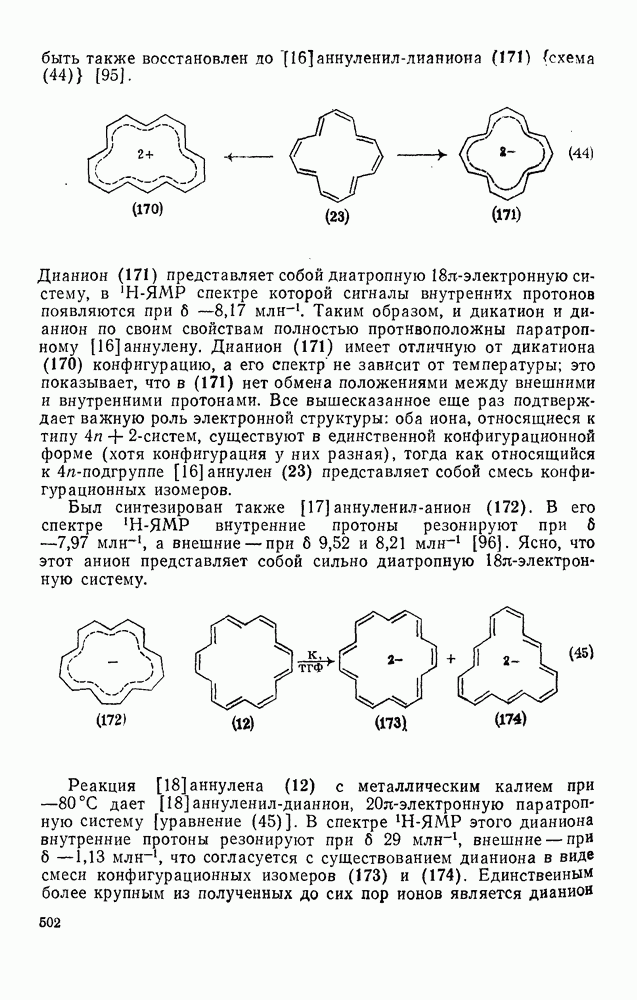
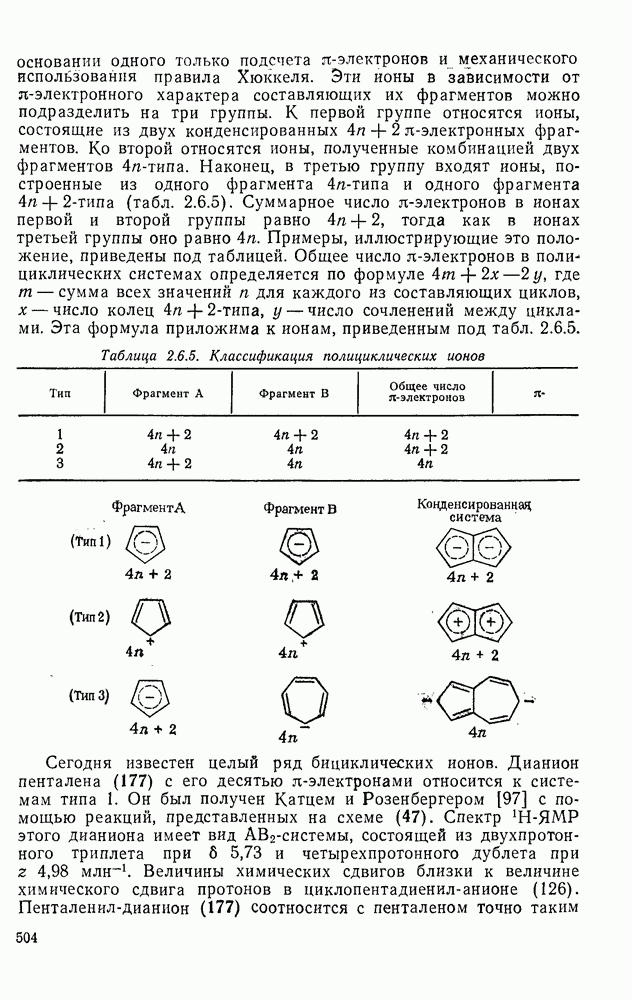
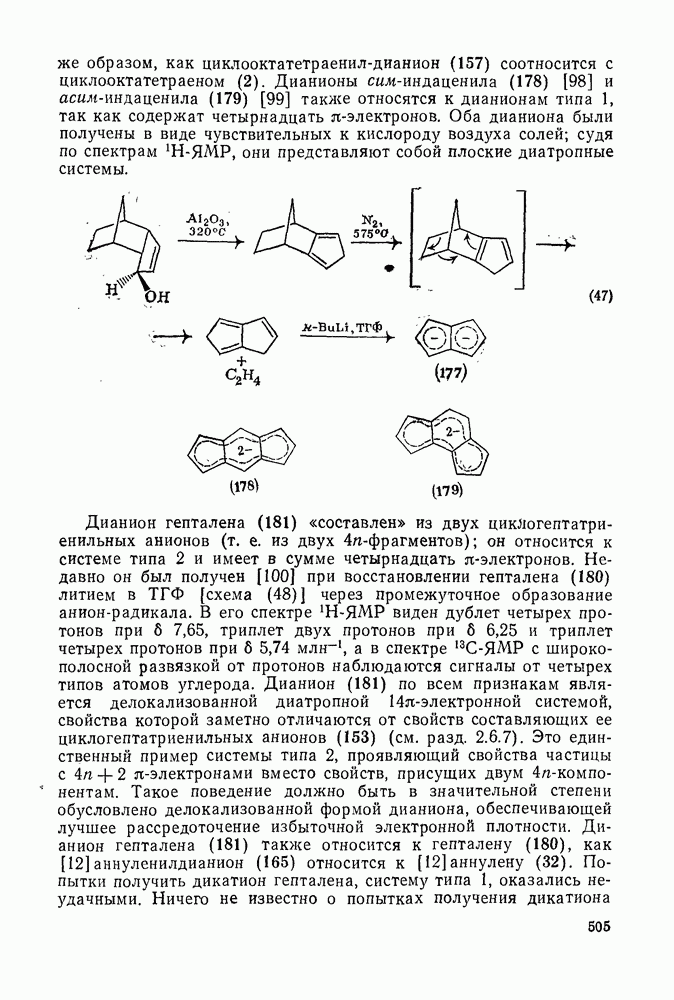
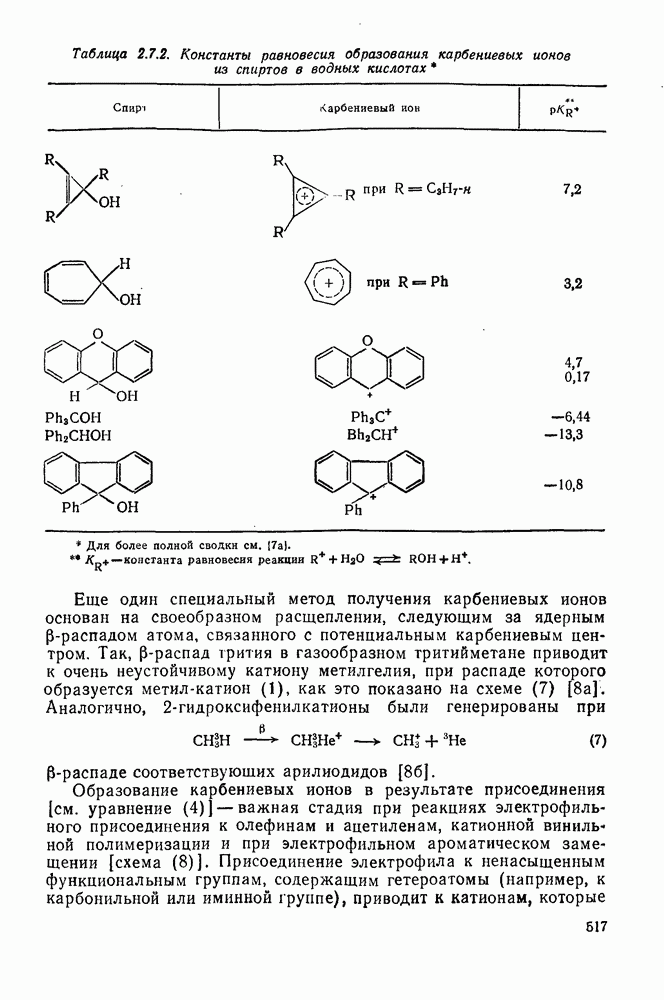
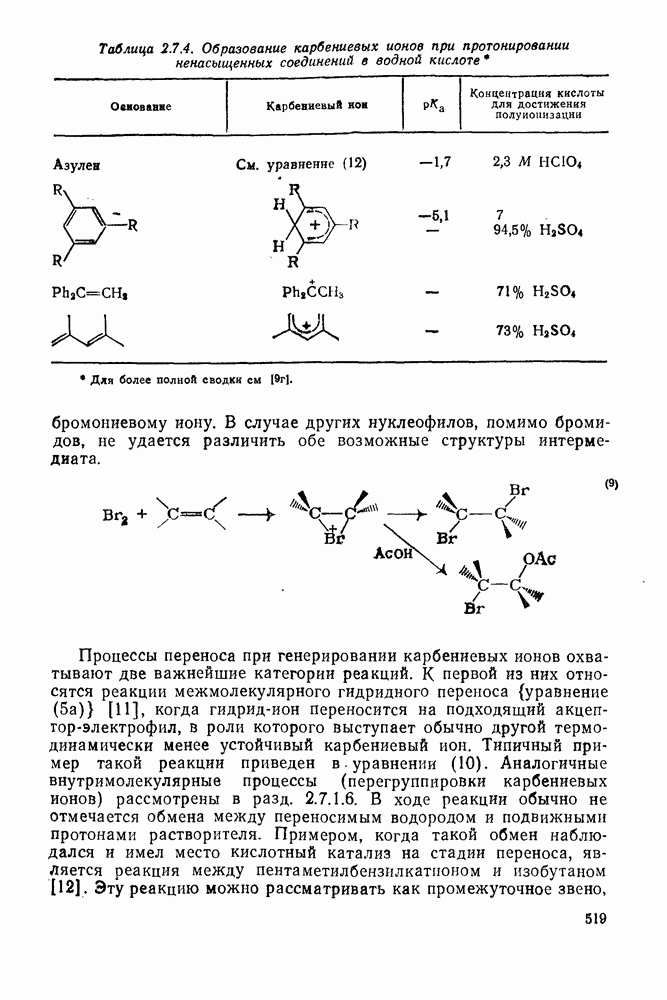
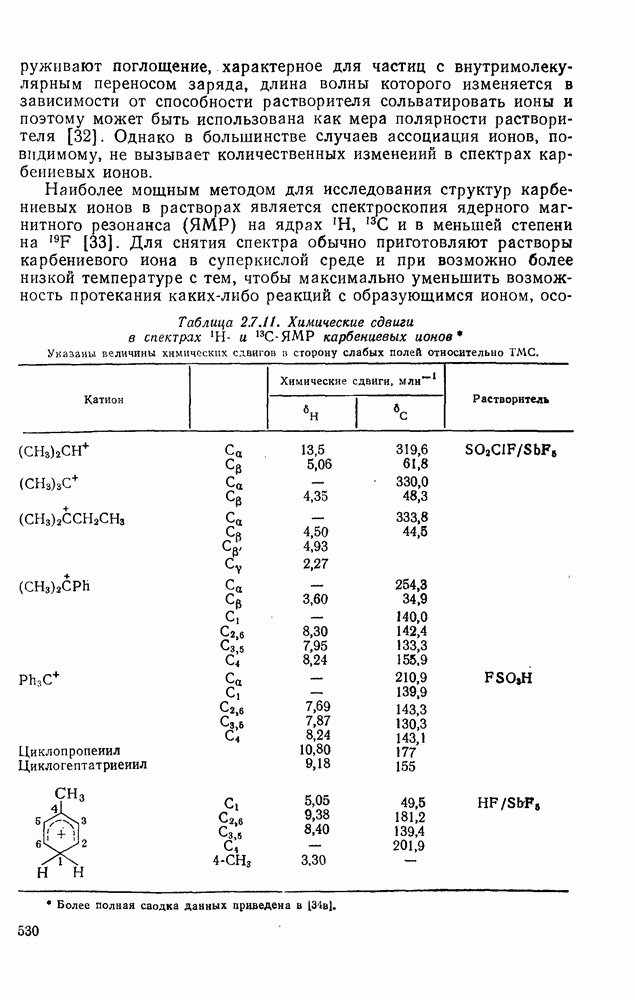
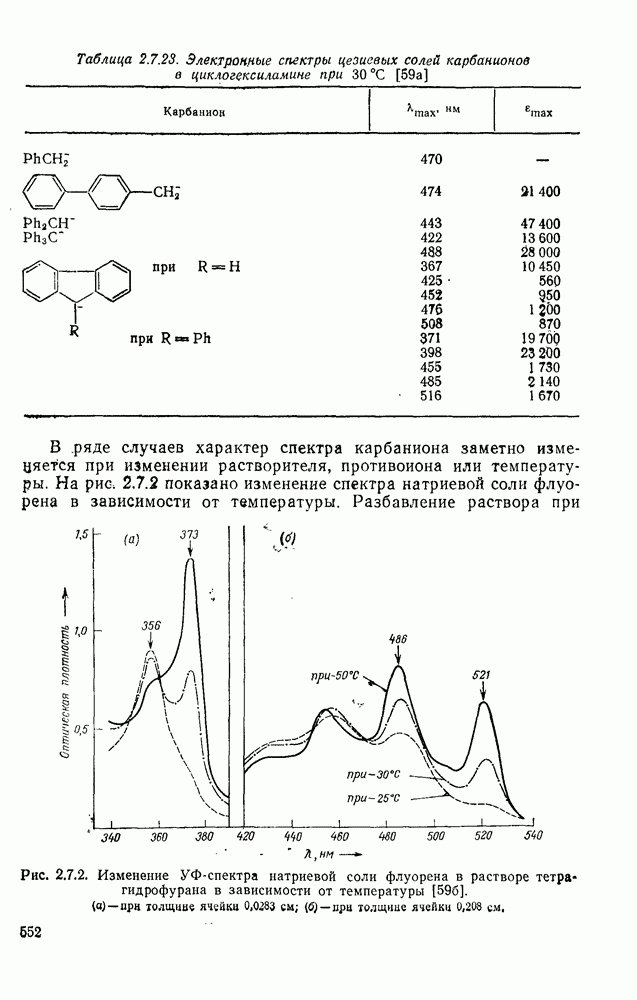
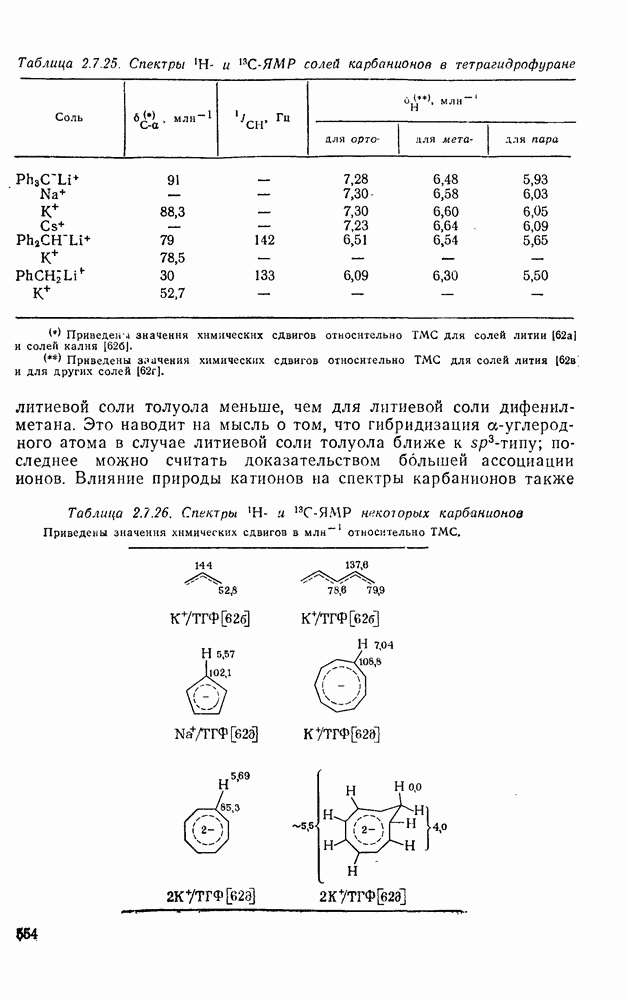
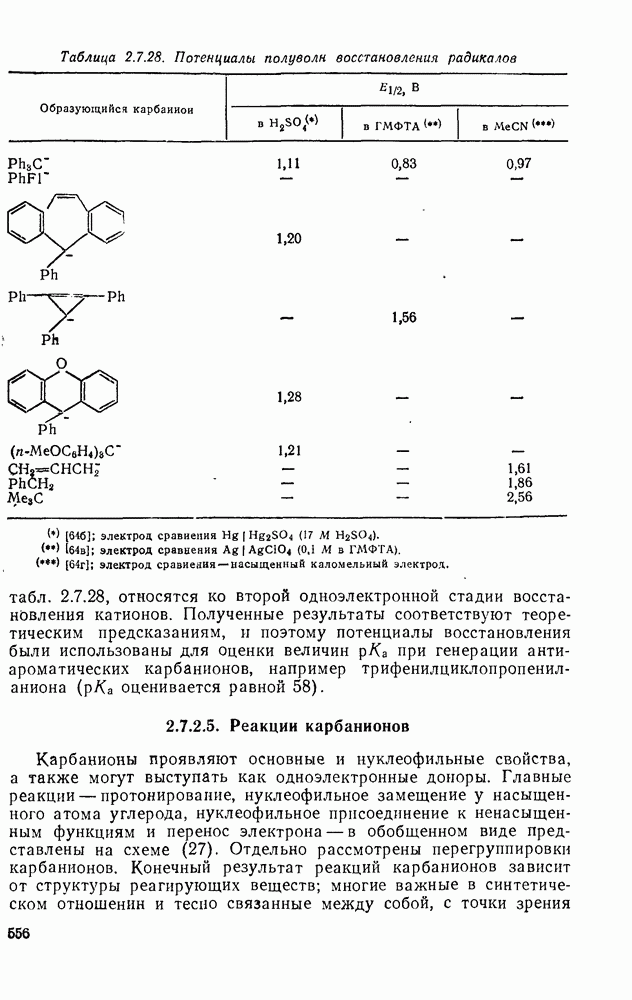
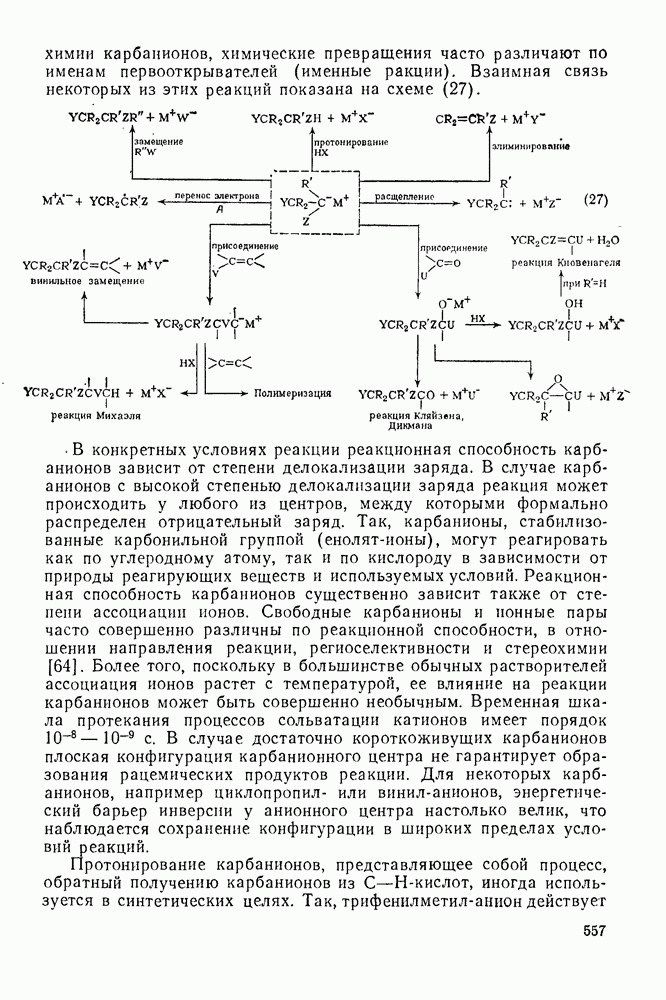
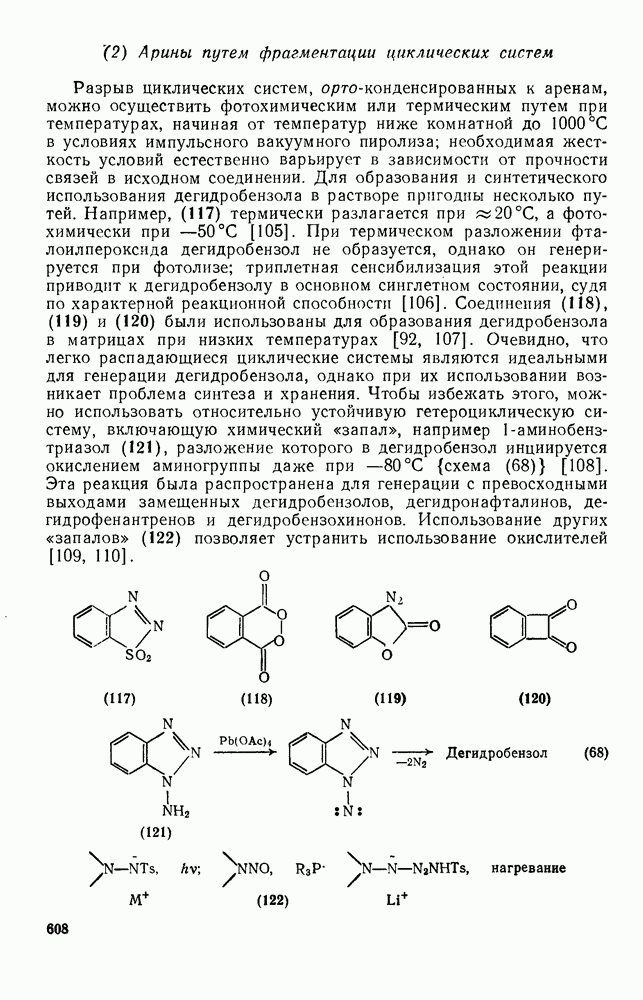
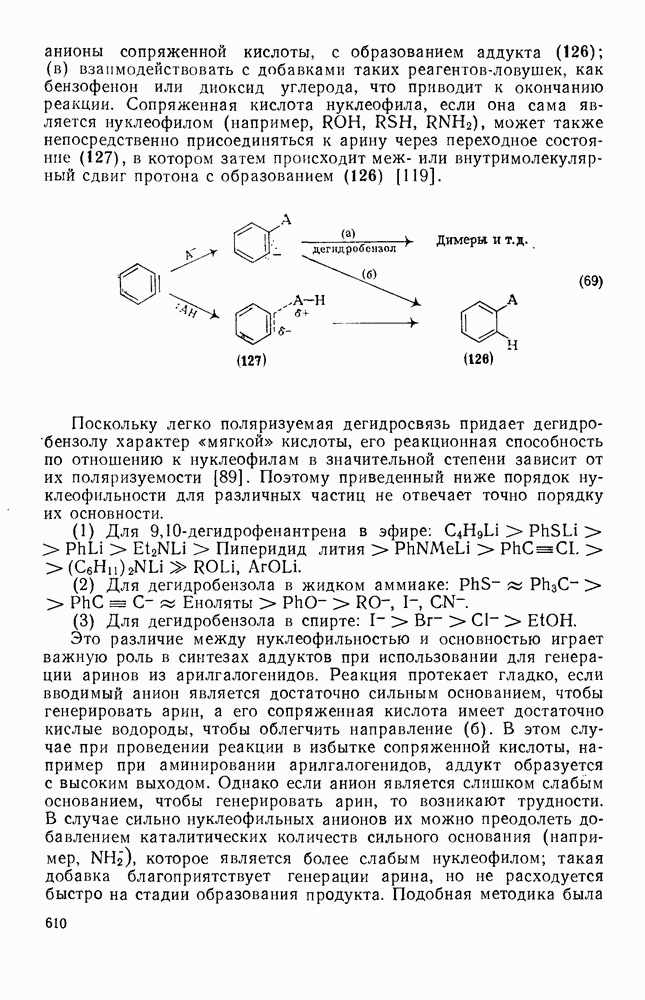
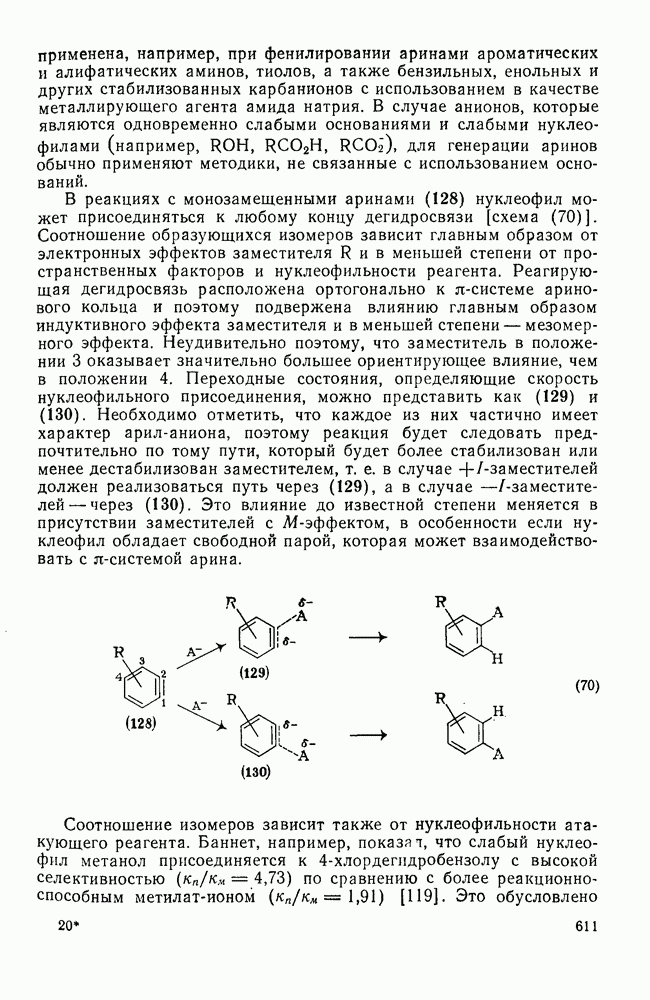
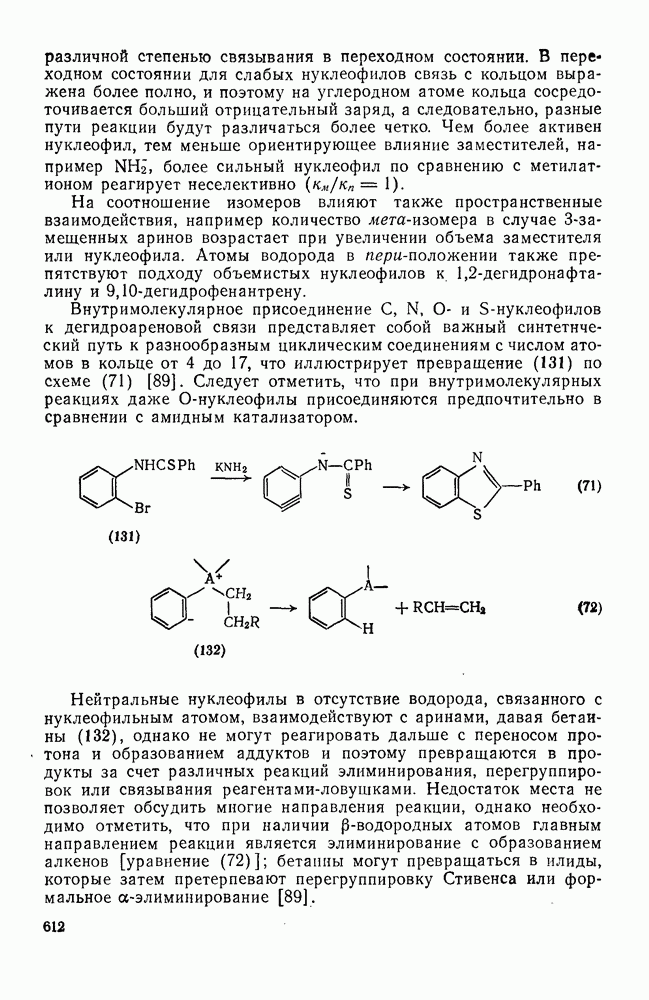
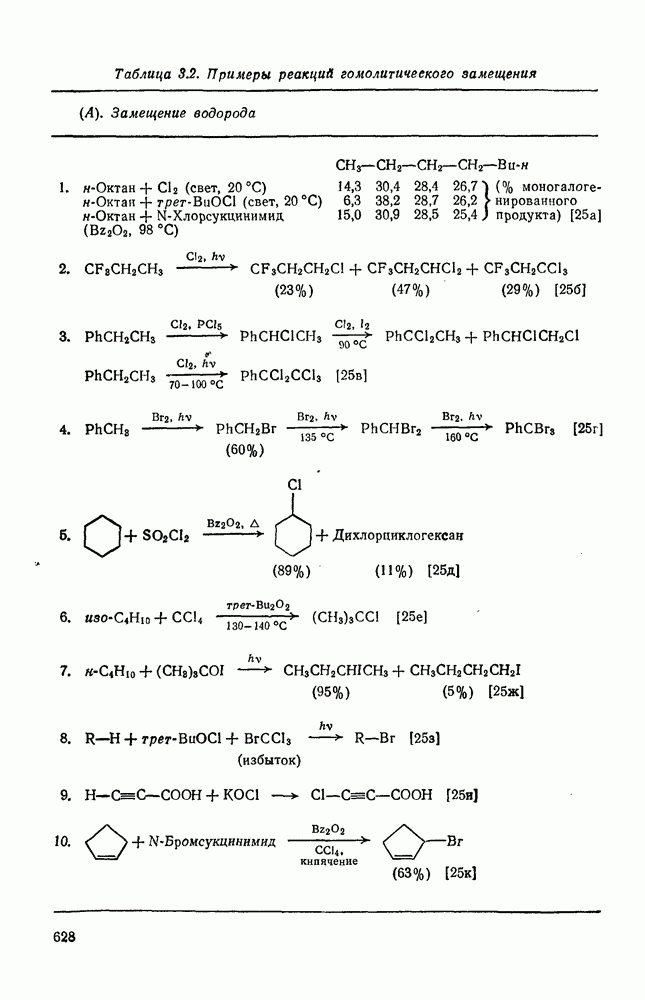
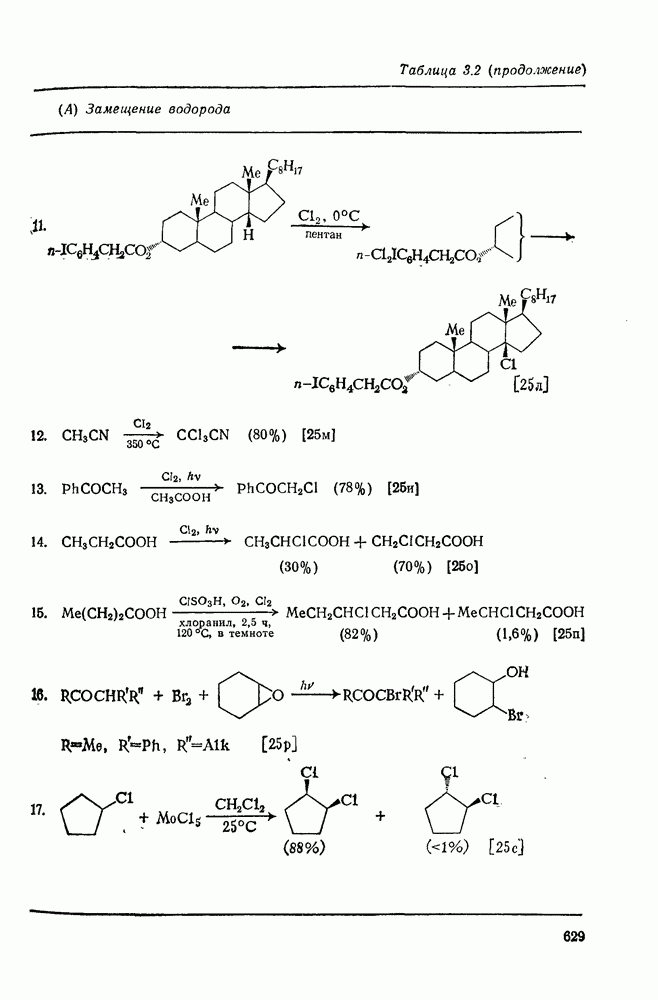
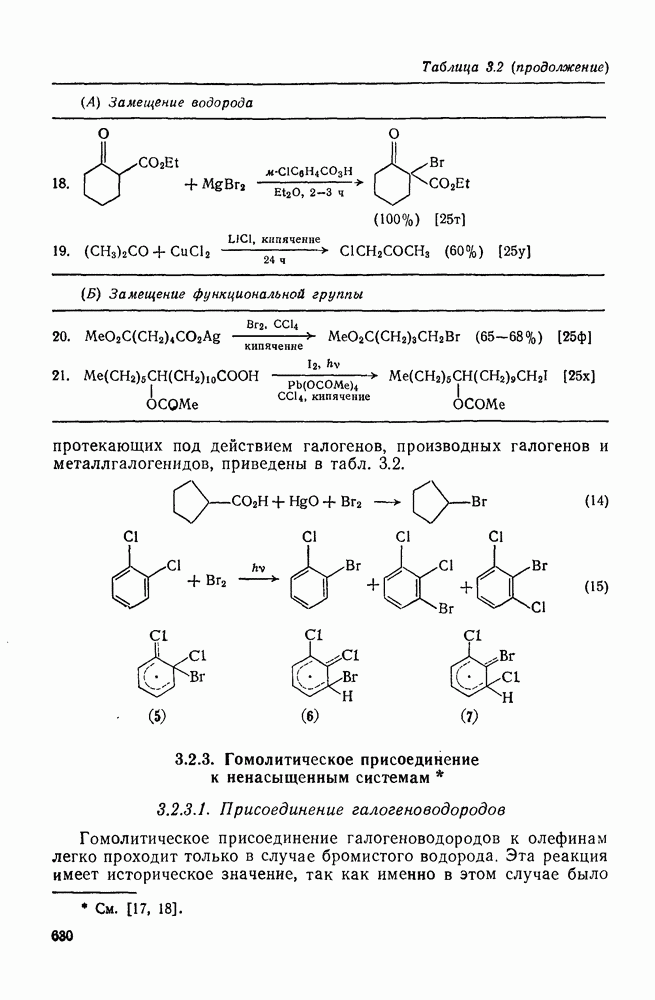
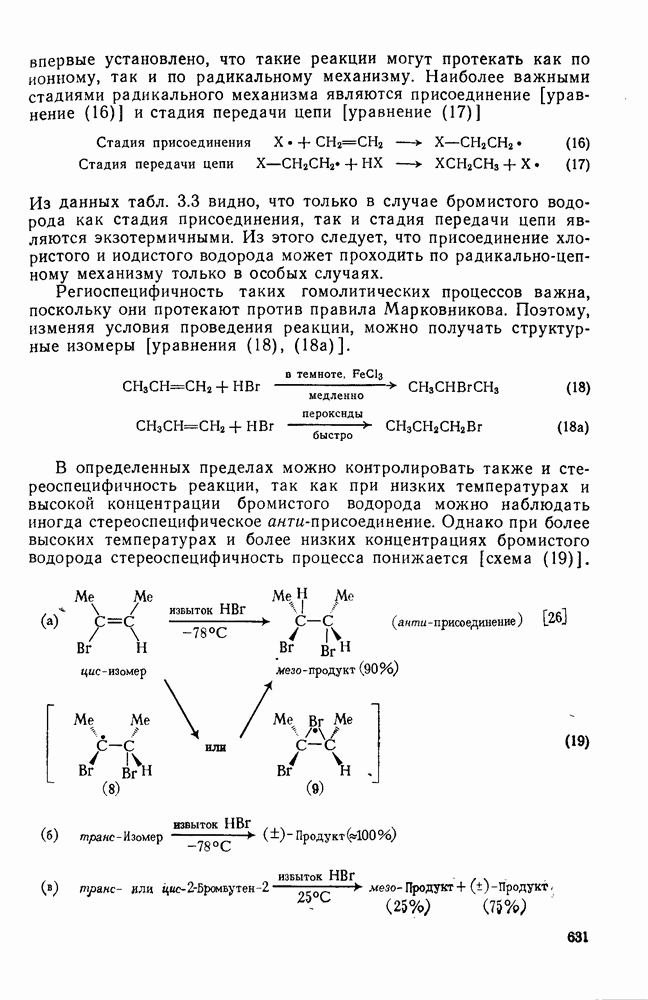
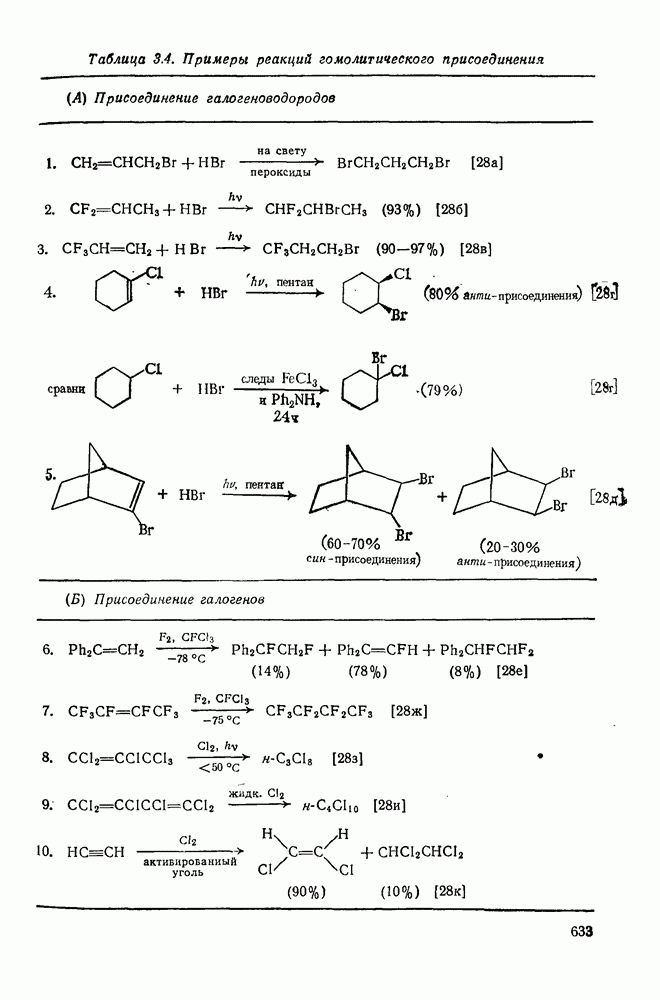
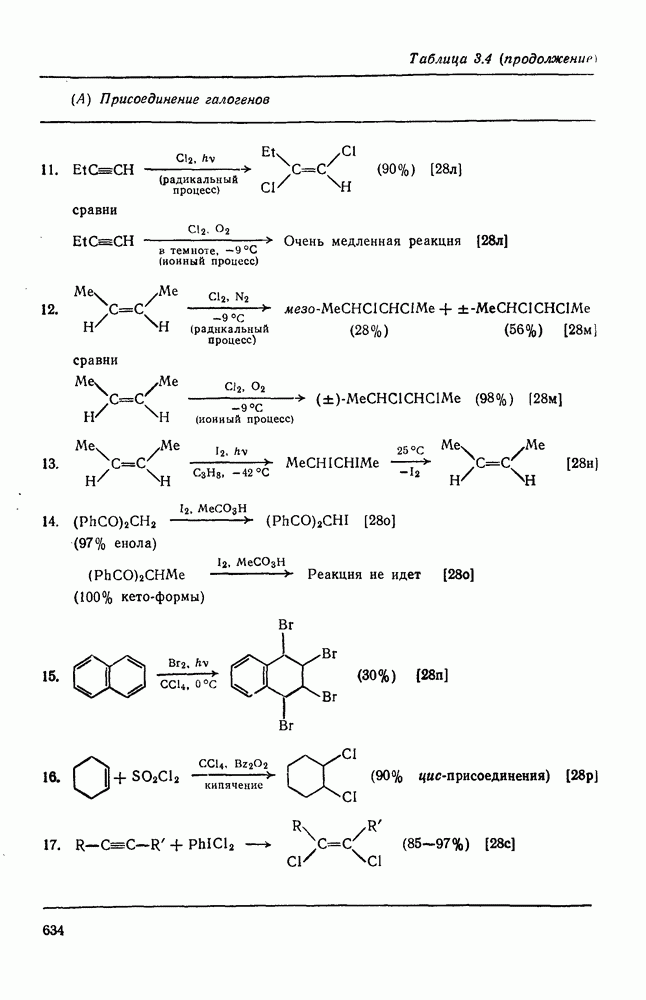
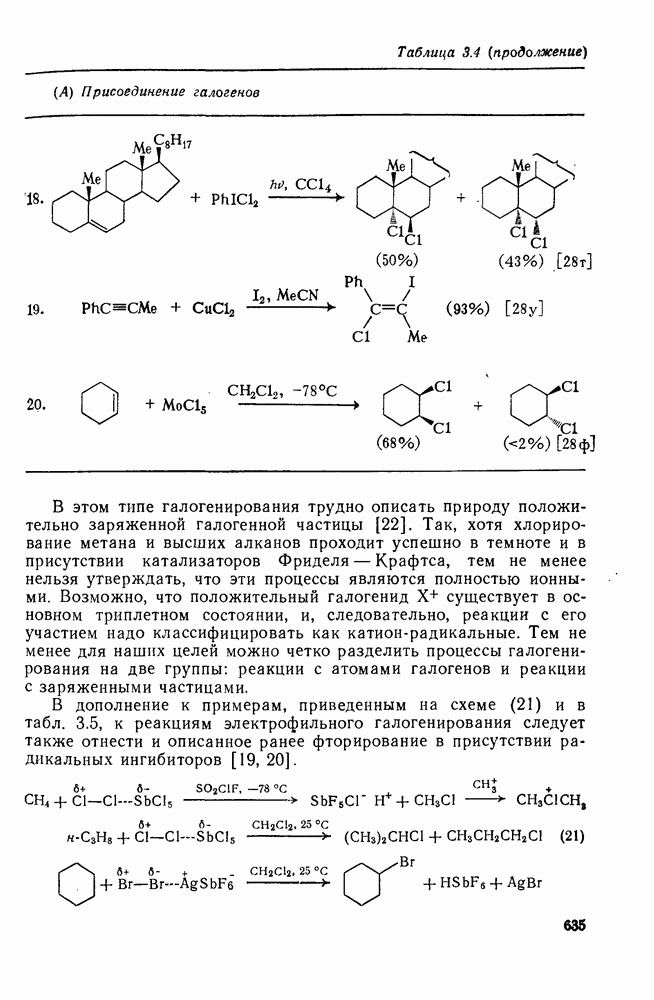
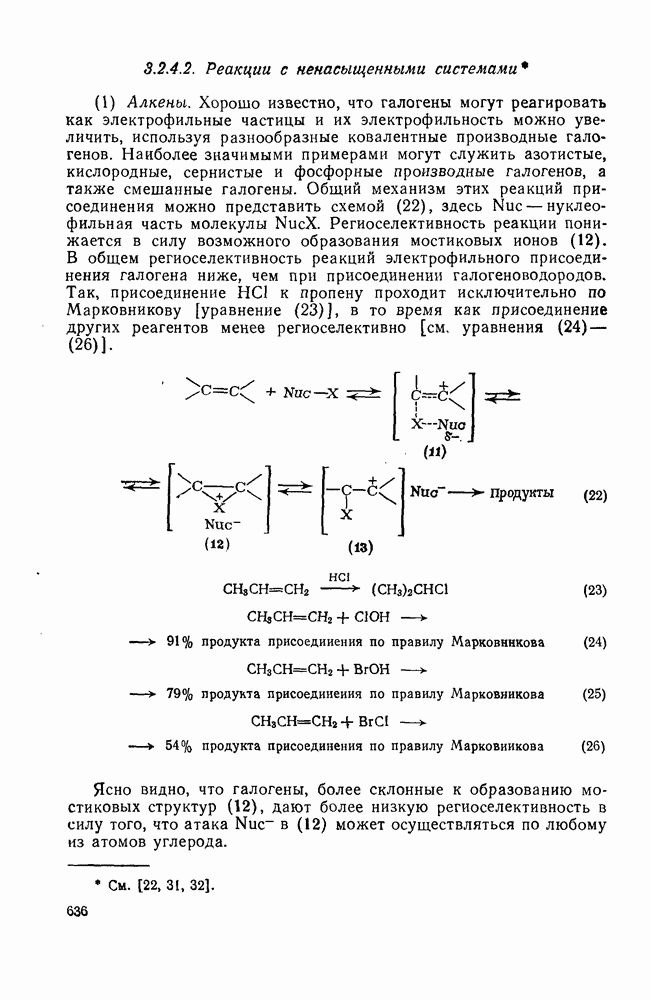
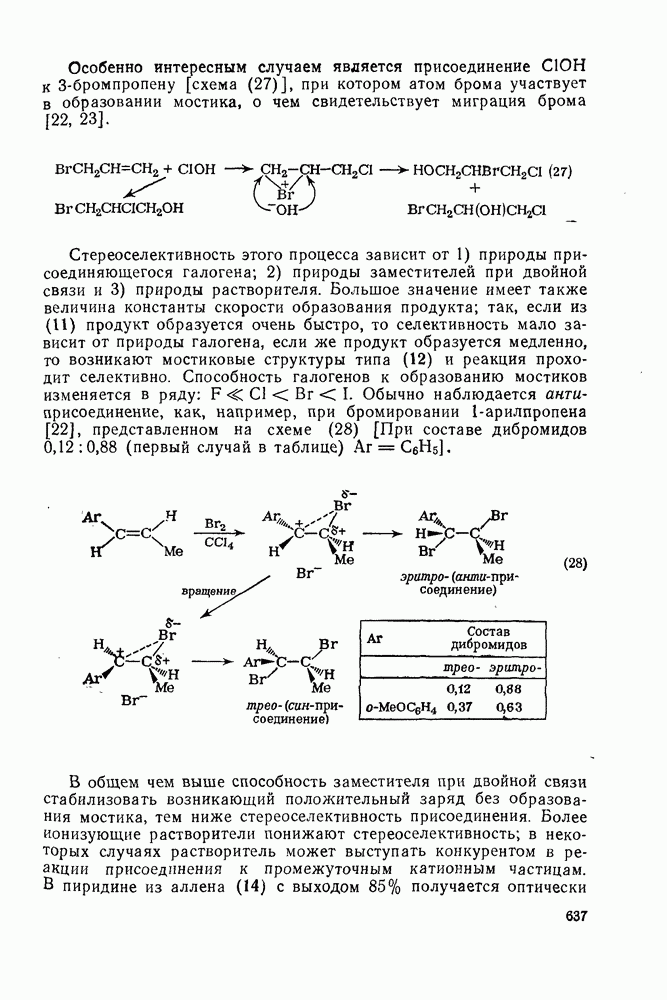
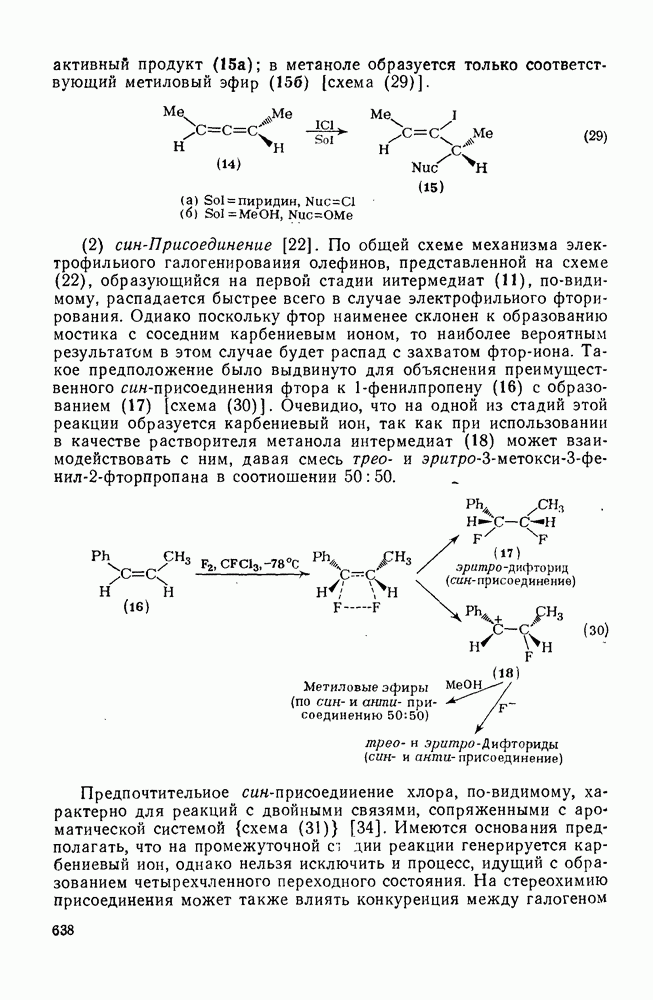
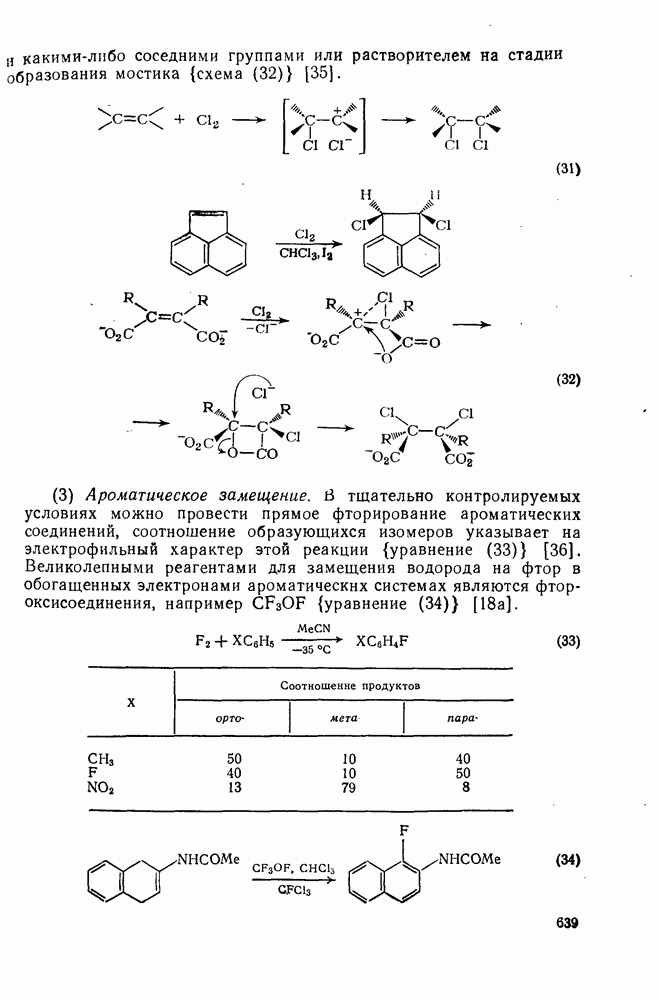
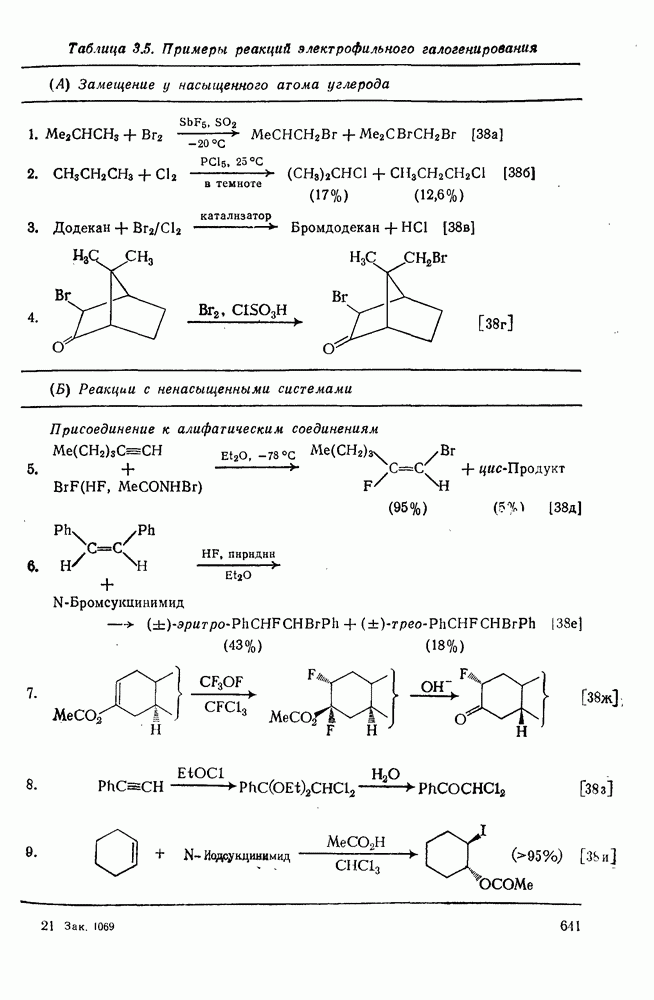
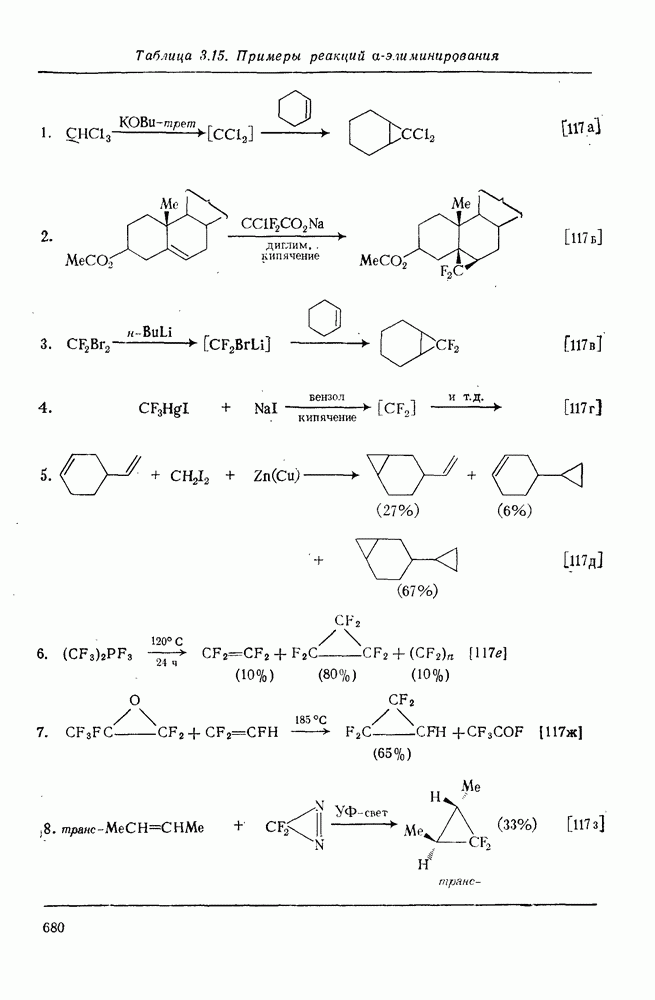
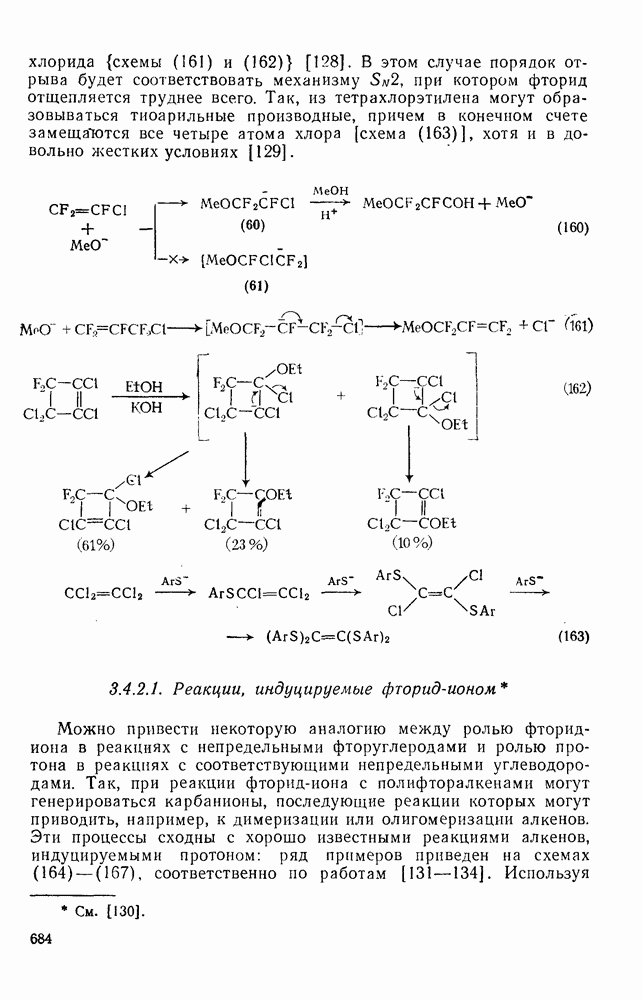
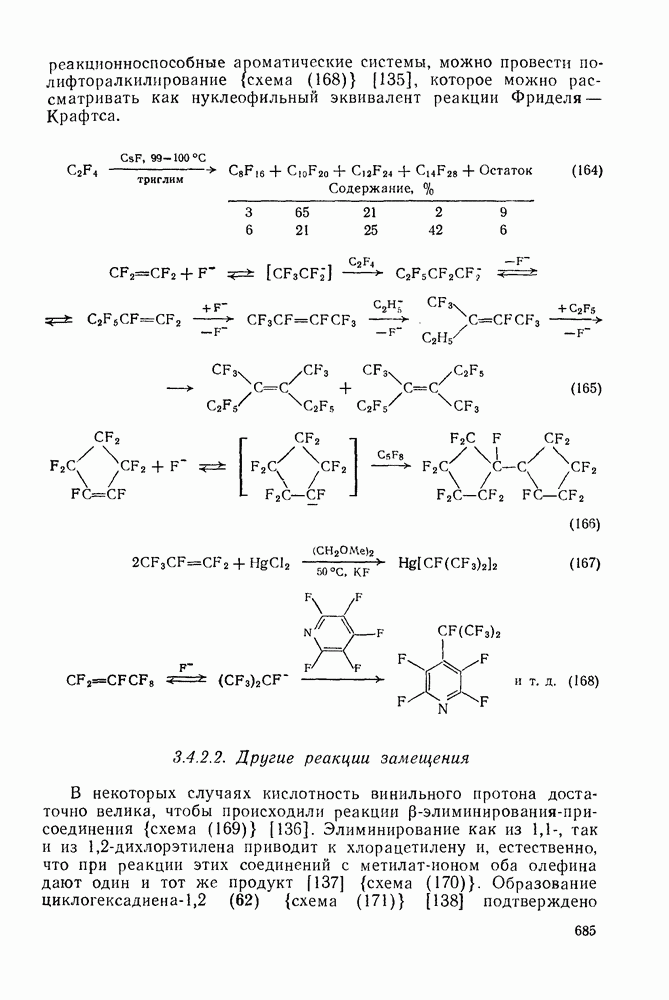
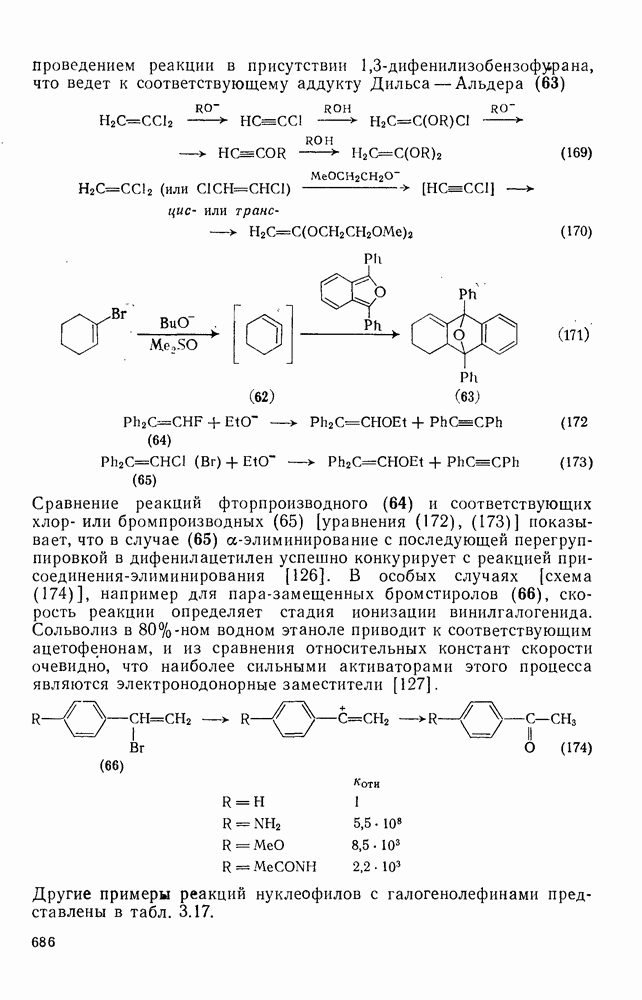
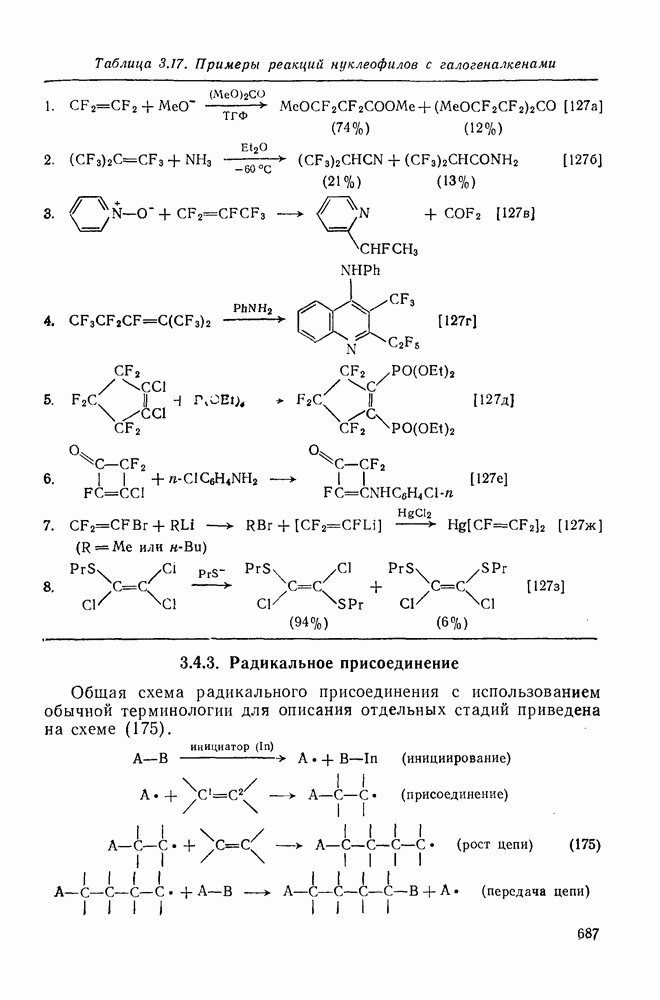
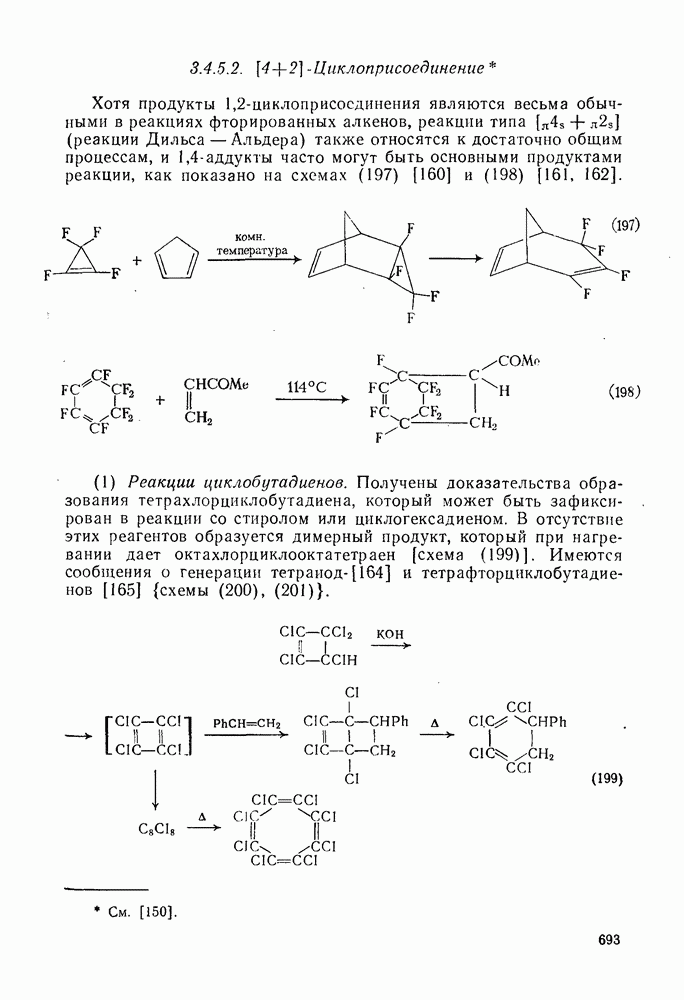
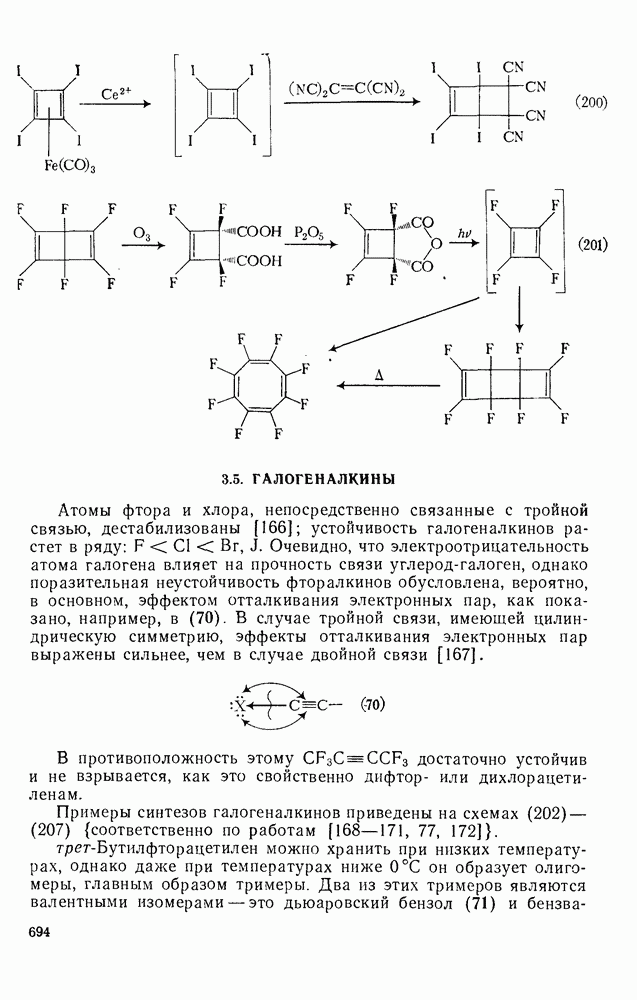
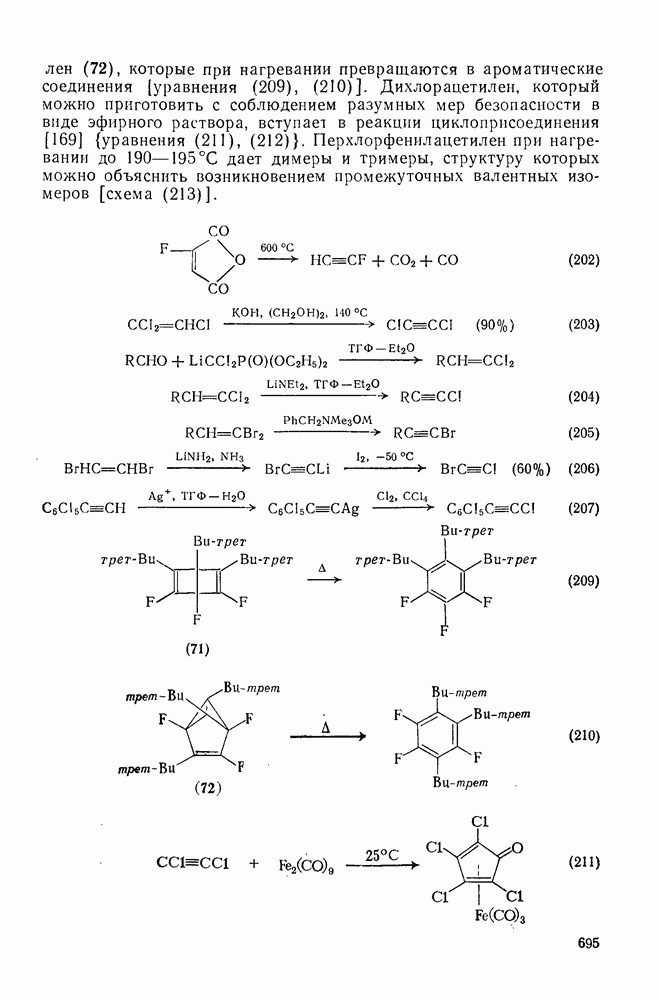
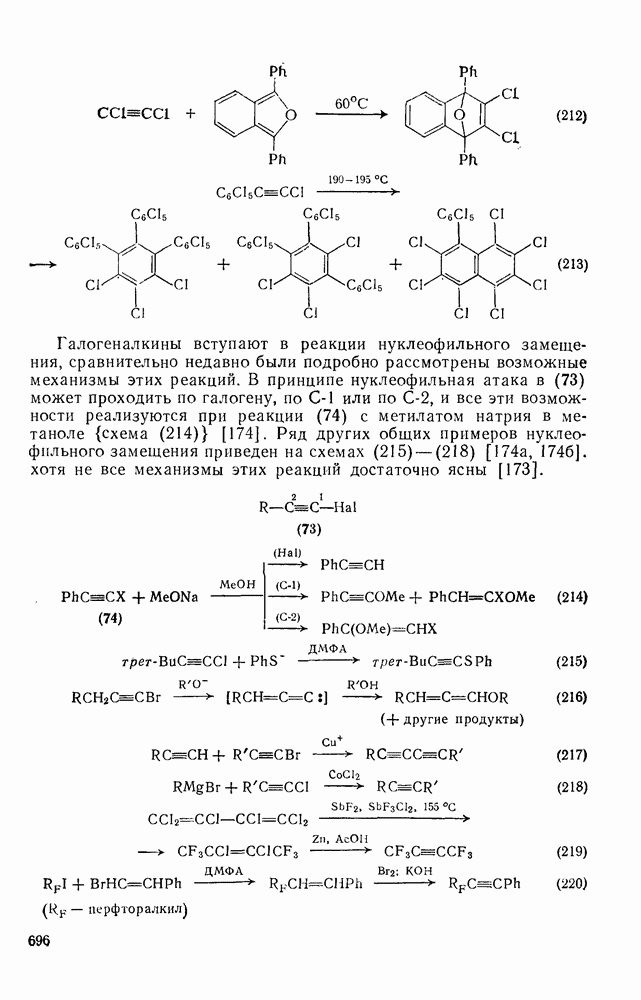
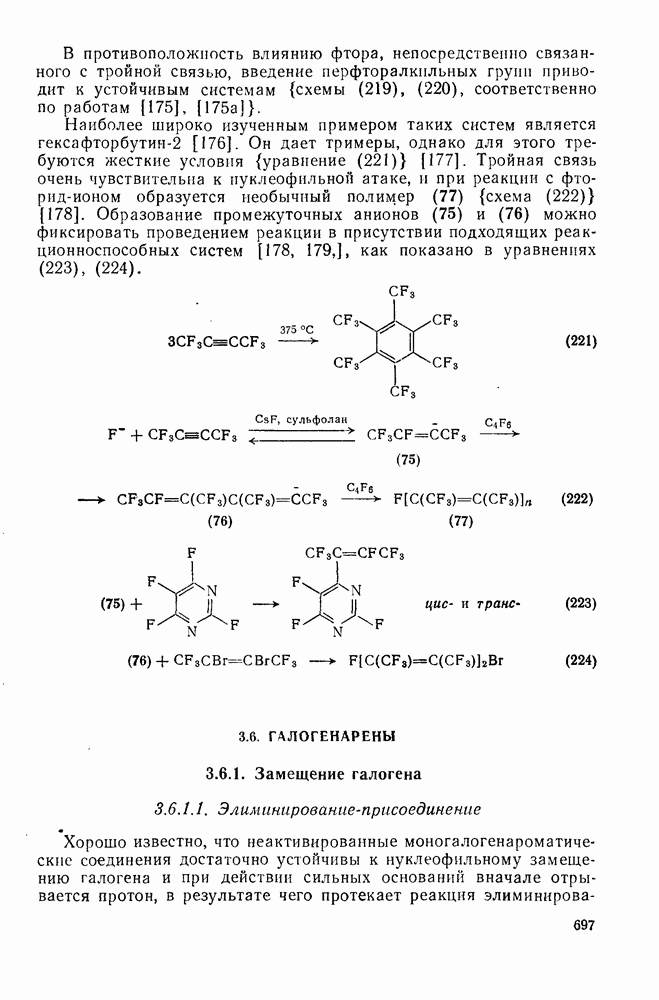
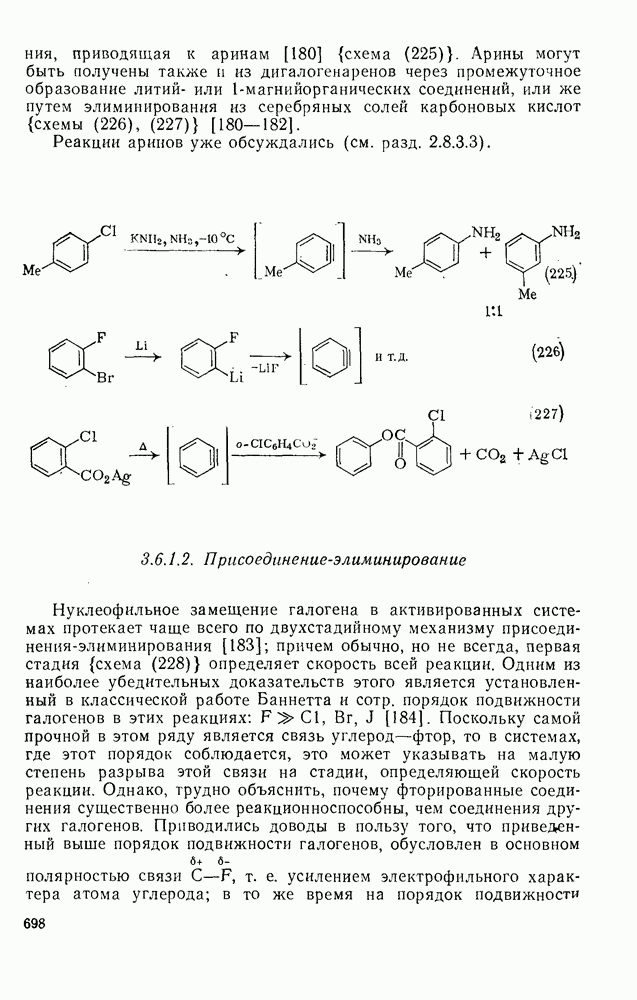
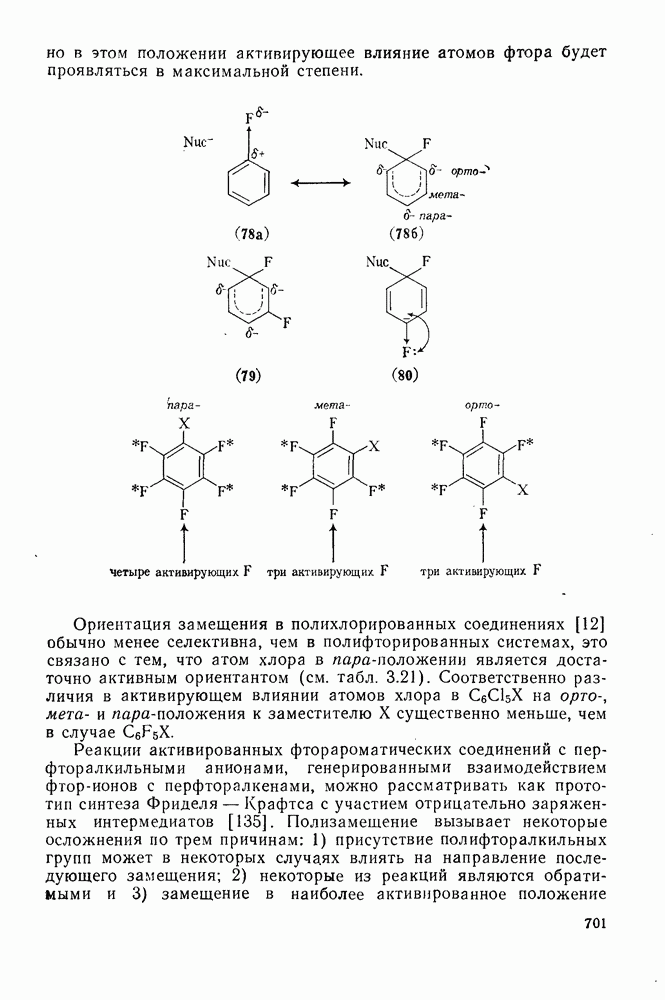
2.8.2.2. Реакции карбенов
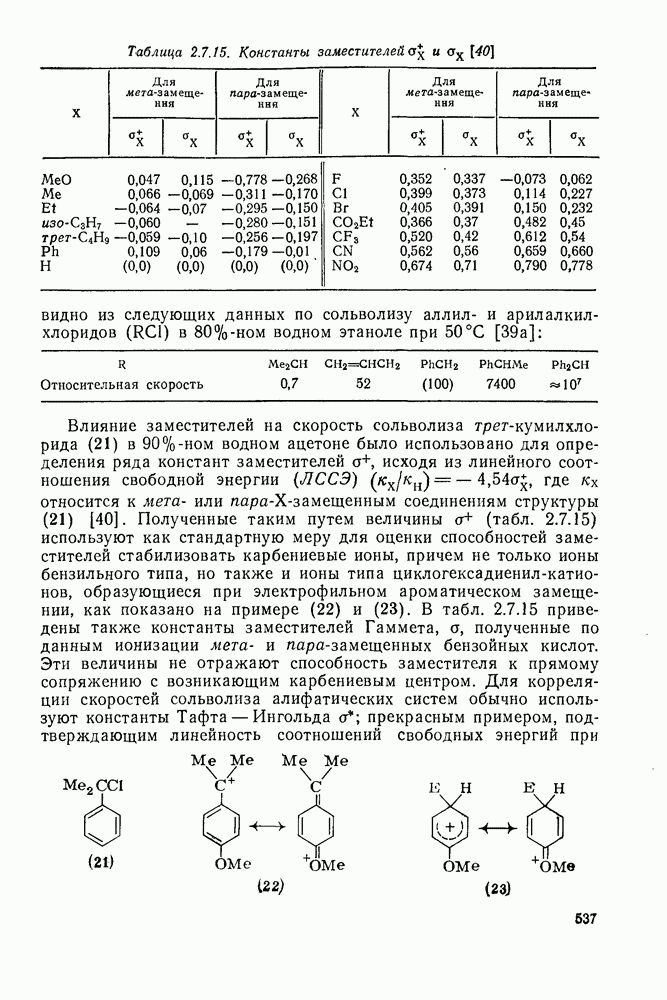
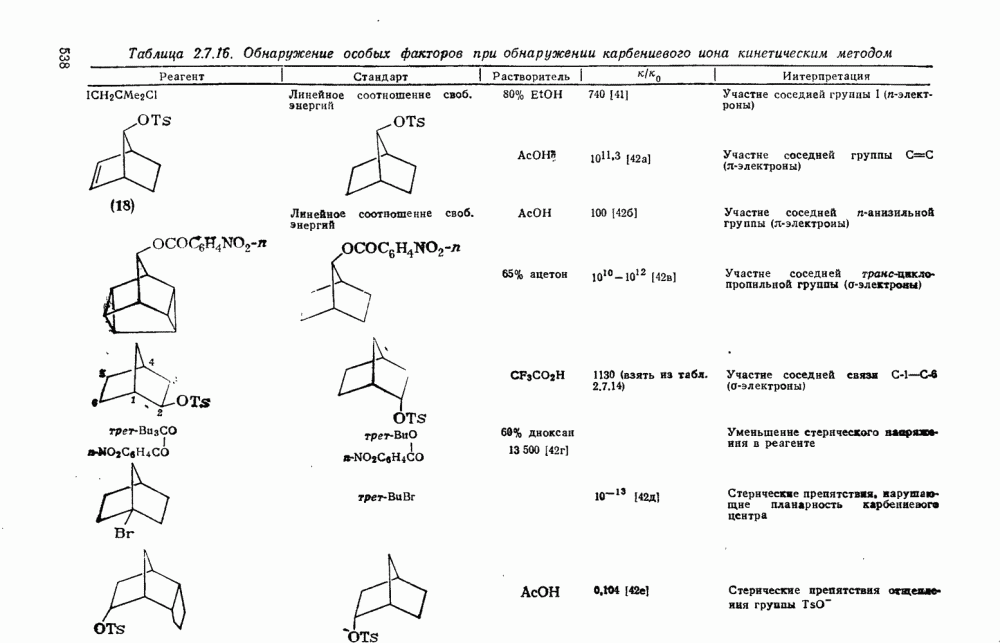
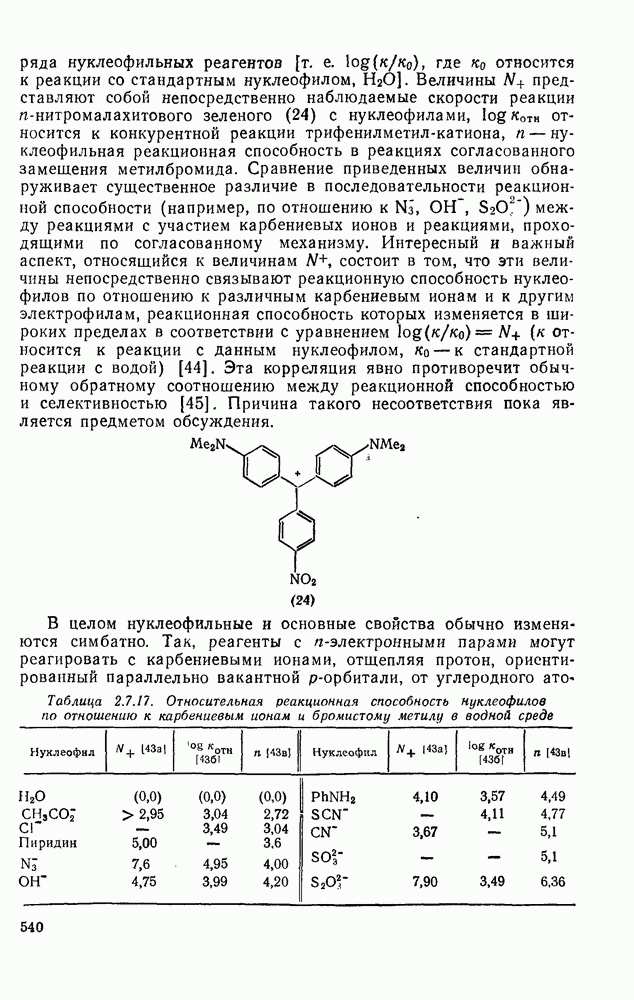
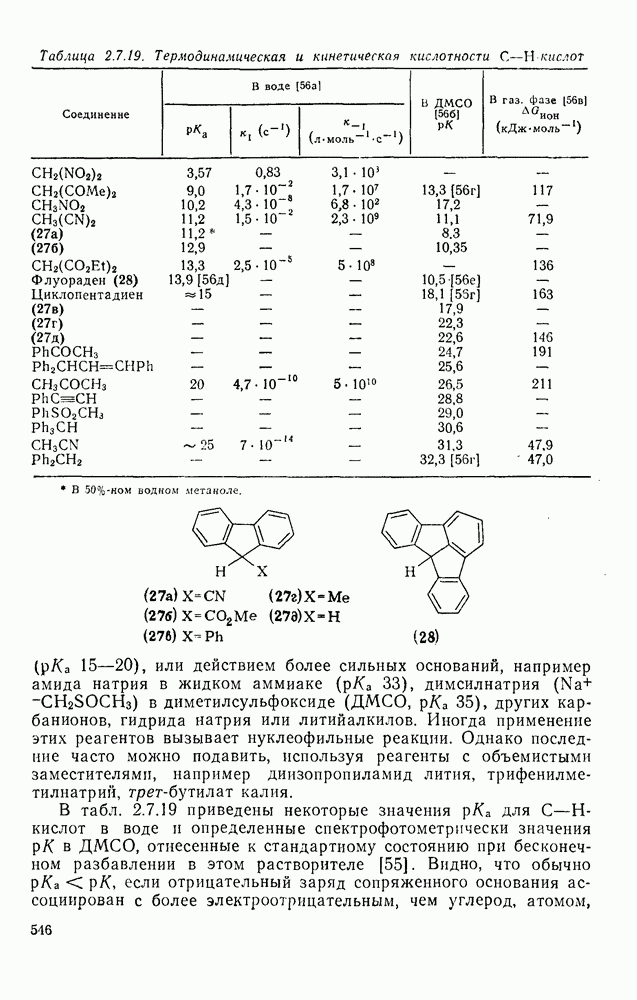
Наше знание химии карбенов в значительной мере стало возможным благодаря применению физических методов, таких как химически индуцнроваиная динамическая поляризация ядер, электронный парамагнитный резонанс и импульсный фотолиз для изучения реакций карбенов в растворе [66]. Обсуждение этих методов выходит за рамки данного раздела, поэтому мы отсылаем читателей к существующим превосходным обзорам [67, 68].
(1) Циклоприсоединение
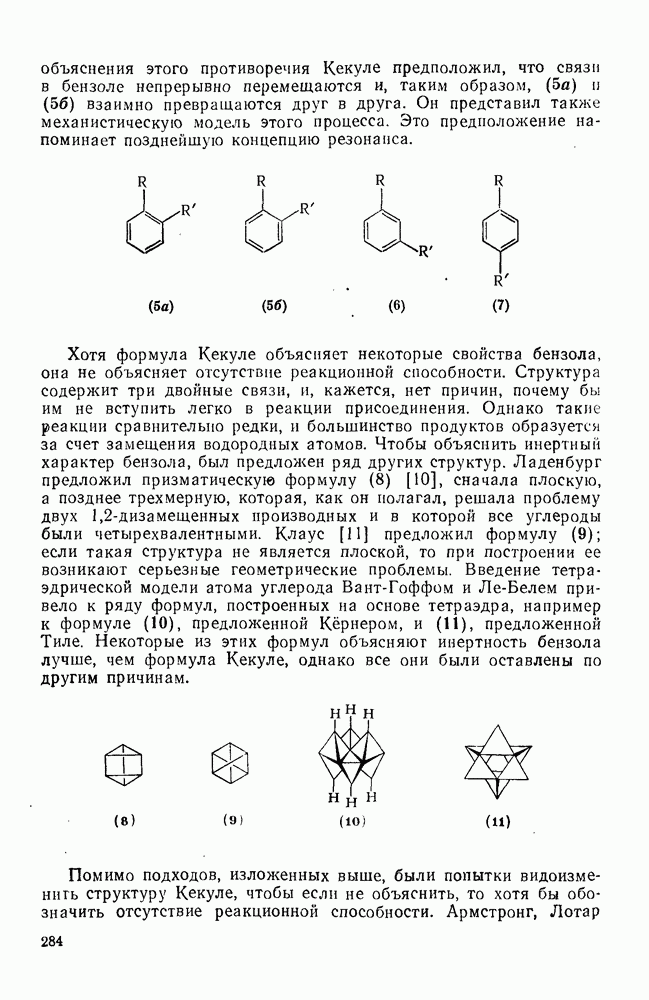
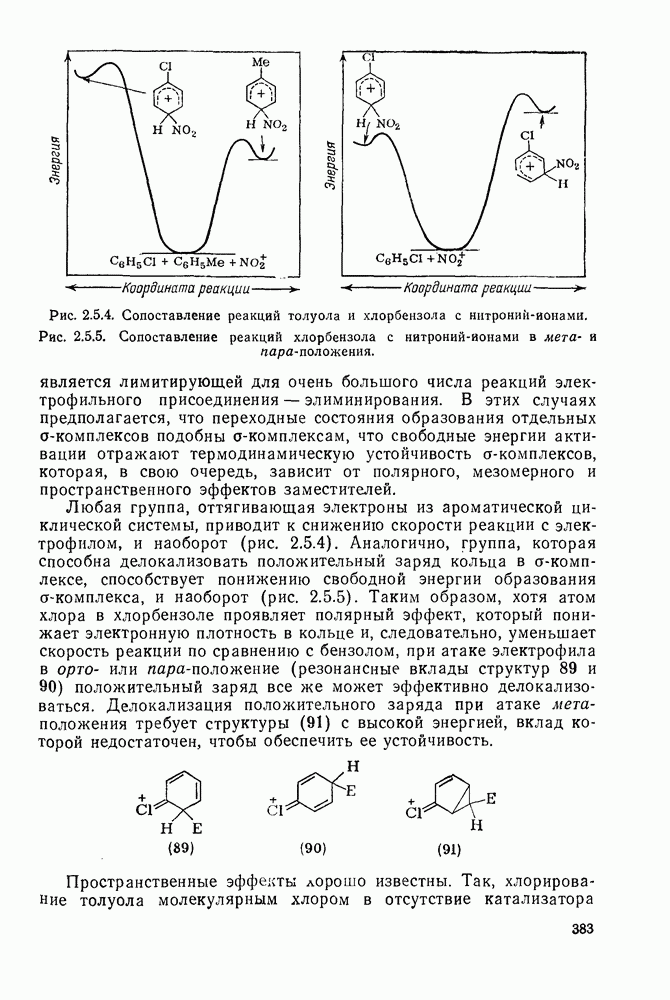
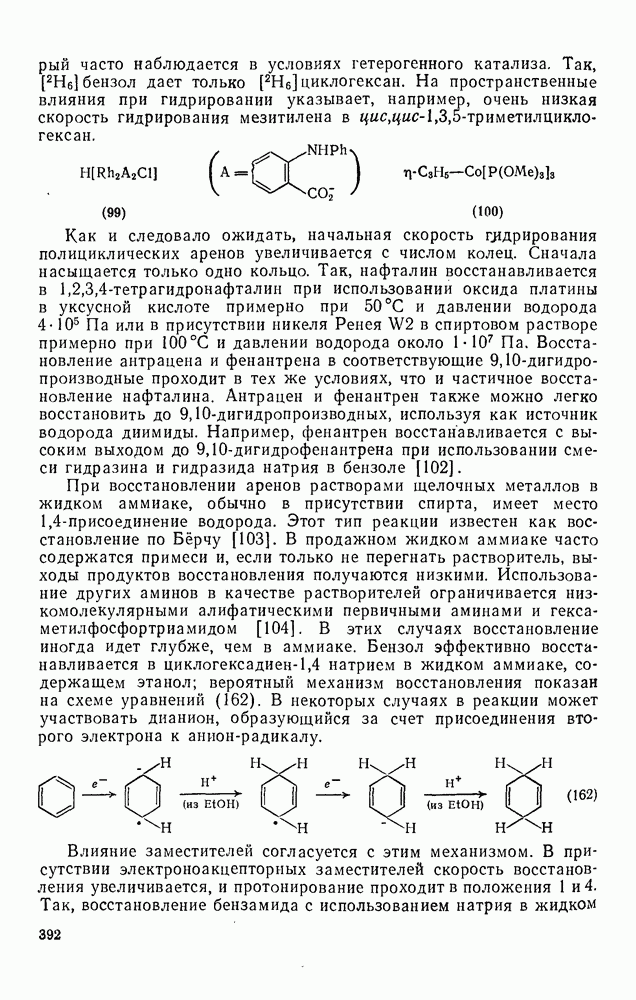
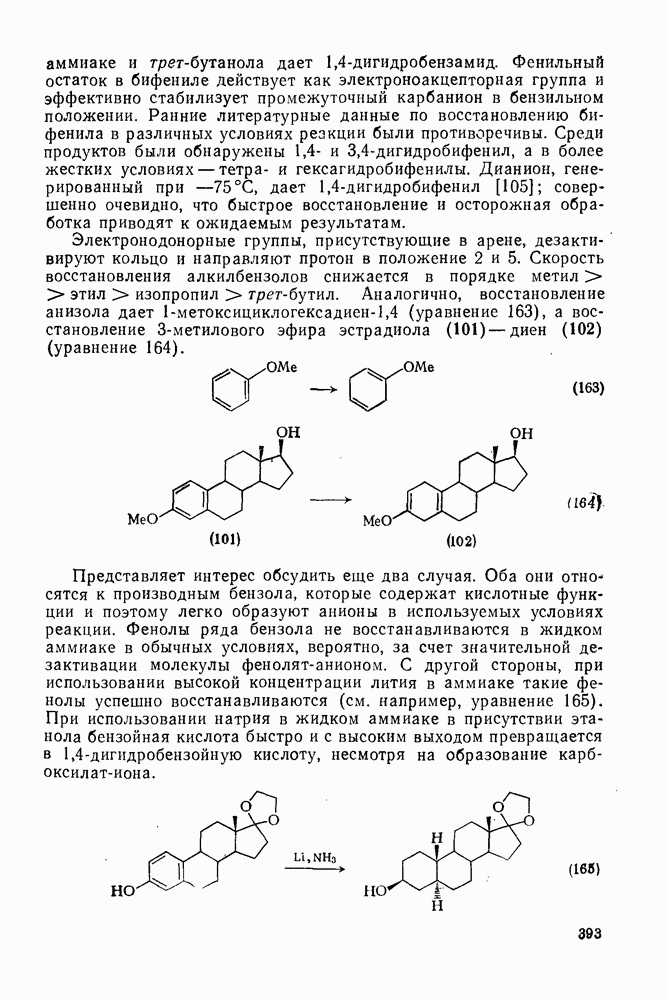
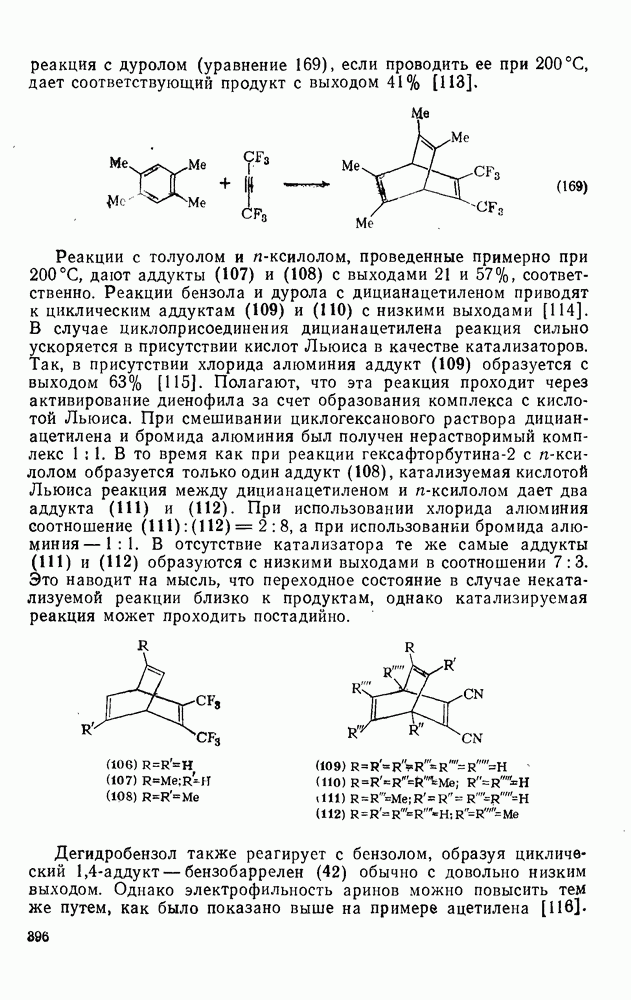
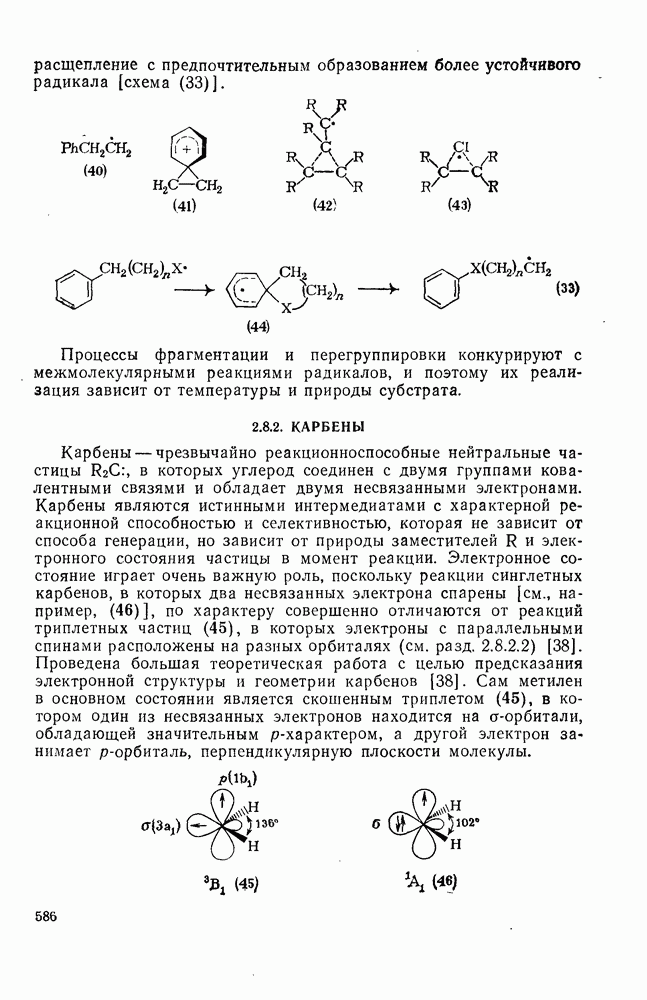
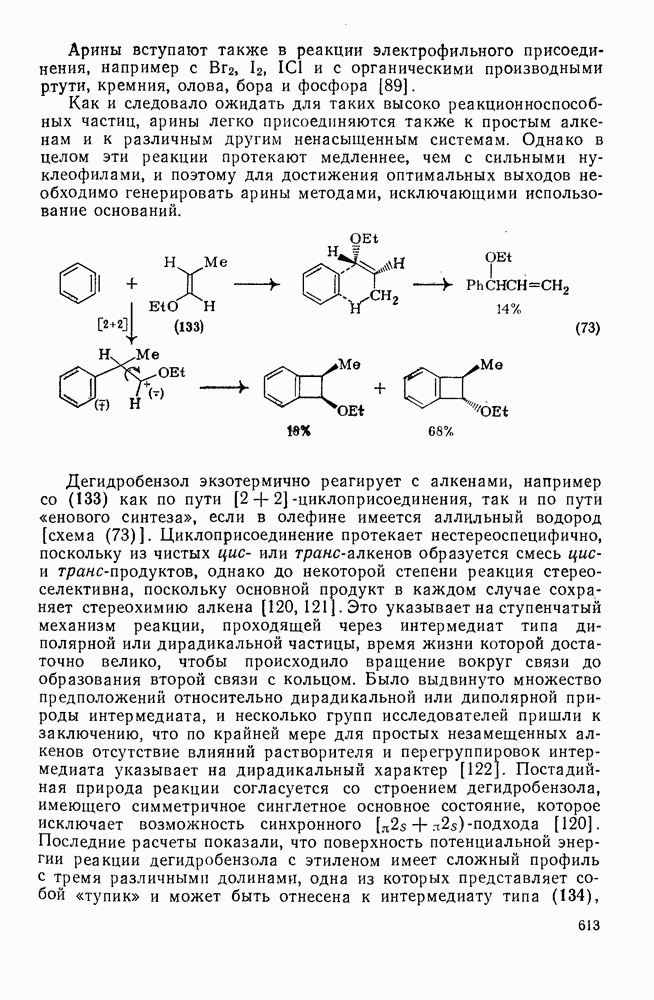
Присоединение карбенов к алкенам с образованием циклопропанов [схемы (35), (42) и (46)] является одной из наиболее характерных и синтетически полезных реакций [69]. Часто возможно успешно осуществить внутримолекулярные варианты этой реакции [см., например, уравнение  ], однако они зачастую проходят с низкими выходами и осложняются водородными сдвигами и другими конкурирующими реакциями. Расчеты молекул, аналогичных (73), в которых внутримолекулярное образование циклопропана стереохимически невозможно, показывают, что в таких соединениях электронное взаимодействие с двойной связью вполне достаточно для стабилизации синглетного основного состояния карбена. Такие карбены называют «листовидными» метиленами, и было затрачено много сил, чтобы обнаружить экспериментально влияние такой неклассической стабилизации на свойства карбена [69].
], однако они зачастую проходят с низкими выходами и осложняются водородными сдвигами и другими конкурирующими реакциями. Расчеты молекул, аналогичных (73), в которых внутримолекулярное образование циклопропана стереохимически невозможно, показывают, что в таких соединениях электронное взаимодействие с двойной связью вполне достаточно для стабилизации синглетного основного состояния карбена. Такие карбены называют «листовидными» метиленами, и было затрачено много сил, чтобы обнаружить экспериментально влияние такой неклассической стабилизации на свойства карбена [69].

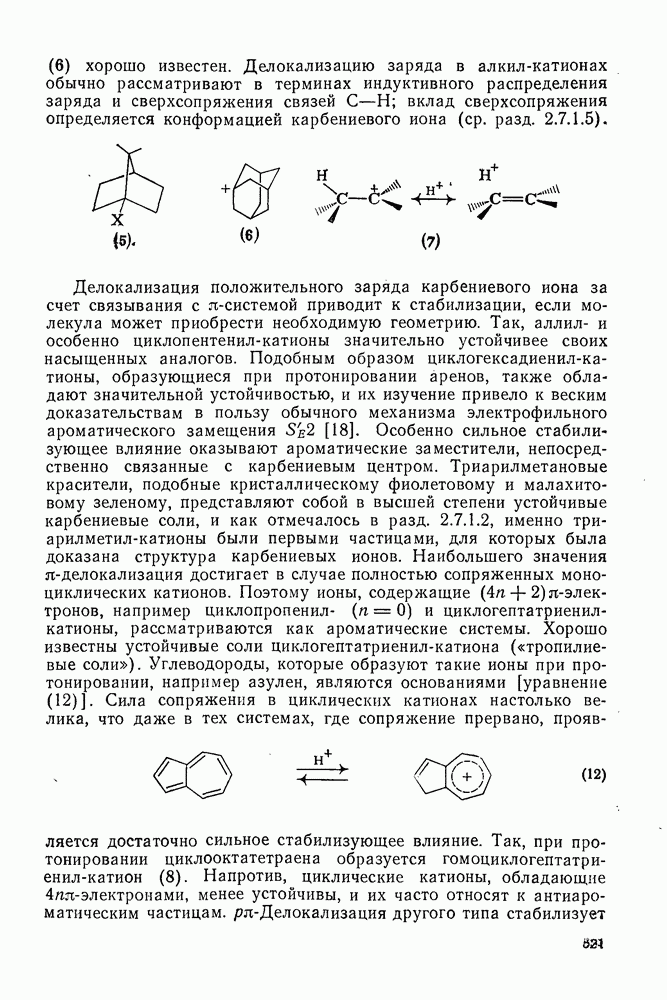
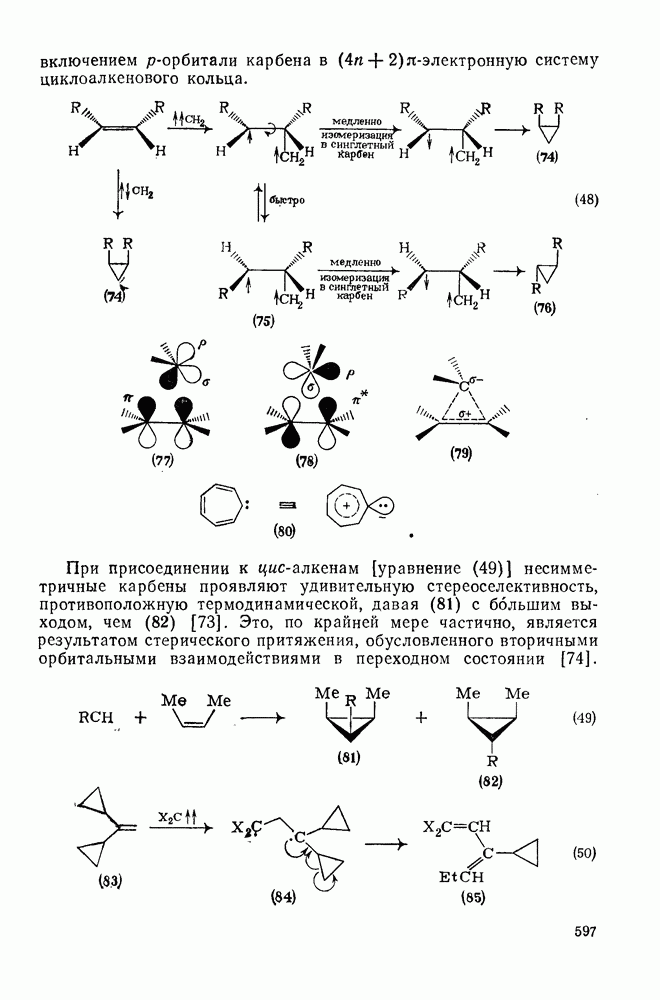
Механизм межмолекулярного присоединения карбенов к алкенам широко обсуждался в течение многих лет. С поразительной химической интуицией Скелл в 1956 г. предположил, что механизм и стереохимия реакции зависят от спинового состояния карбена.
Он привел доводы в пользу того, что синглетные карбены присоединяются путем синхронного образования обоих новых  -связей, давая только (74) и сохраняя таким образом стереохимию исходного алкена, в то время как триплетные карбены присоединяются по радикальному двухстадийному механизму с образованием в первую очередь бирадикала (75), в котором может происходить вращение вокруг связи до инверсии спина и замыкания кольца, что приводит к обоим диастереомерам (74) и (76). Несмотря на широкое обсуждение справедливости теоретических предпосылок, правило Скелла исключительно успешно объясняет многие экспериментальные данные, полученные для этих реакций присоединения. Однако при использовании правила следует соблюдать определенную осторожность, так как в его основе лежат некоторые предположения об относительных скоростях стадий схемы (48), которые могут соблюдаться не во всех случаях [38]. Таким образом, прежде чем однозначно приписать определенную реакционную способность одному из спиновых состояний карбена, следует выяснить свойства обоих состояний. В ряде случаев, когда это требование было точно соблюдено, например в случае метилена, бисметоксикарбонилкарбена, флуоренилидена и др., результаты всегда соответствовали предсказаниям Скелла. Расчет поверхности потенциальной энергии присоединения синглетного метилена к этилену [40, 70] подтверждает синхронность реакции и свидетельствует, что она осуществляется по принципу наименьшего движения через разрешенный орбитальной симметрией подход (77), при котором вакантная р-орбиталь (НСМО) карбена взаимодействует с занятой
-связей, давая только (74) и сохраняя таким образом стереохимию исходного алкена, в то время как триплетные карбены присоединяются по радикальному двухстадийному механизму с образованием в первую очередь бирадикала (75), в котором может происходить вращение вокруг связи до инверсии спина и замыкания кольца, что приводит к обоим диастереомерам (74) и (76). Несмотря на широкое обсуждение справедливости теоретических предпосылок, правило Скелла исключительно успешно объясняет многие экспериментальные данные, полученные для этих реакций присоединения. Однако при использовании правила следует соблюдать определенную осторожность, так как в его основе лежат некоторые предположения об относительных скоростях стадий схемы (48), которые могут соблюдаться не во всех случаях [38]. Таким образом, прежде чем однозначно приписать определенную реакционную способность одному из спиновых состояний карбена, следует выяснить свойства обоих состояний. В ряде случаев, когда это требование было точно соблюдено, например в случае метилена, бисметоксикарбонилкарбена, флуоренилидена и др., результаты всегда соответствовали предсказаниям Скелла. Расчет поверхности потенциальной энергии присоединения синглетного метилена к этилену [40, 70] подтверждает синхронность реакции и свидетельствует, что она осуществляется по принципу наименьшего движения через разрешенный орбитальной симметрией подход (77), при котором вакантная р-орбиталь (НСМО) карбена взаимодействует с занятой  -молекулярной орбиталью алкена, причем карбен расположен так, чтобы перекрывание было максимальным, а пространственные взаимодействия минимальны. Более симметричный подход (78), когда занятая
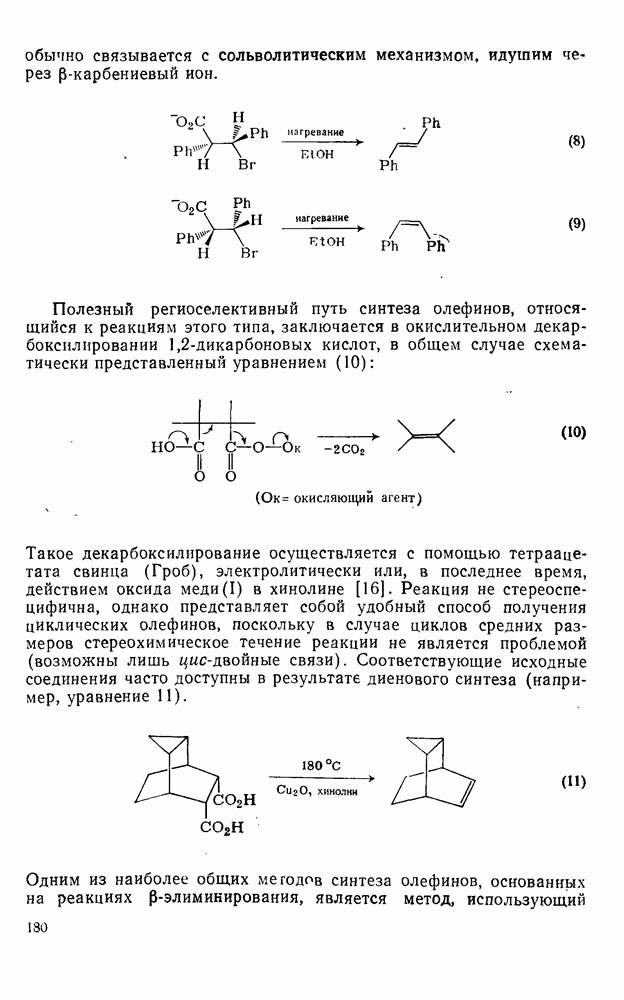
-молекулярной орбиталью алкена, причем карбен расположен так, чтобы перекрывание было максимальным, а пространственные взаимодействия минимальны. Более симметричный подход (78), когда занятая  -орбиталь карбена взаимодействует с
-орбиталь карбена взаимодействует с  -системой, запрещен орбитальной симметрией и по расчету обладает более высокой энергией, чем (77). Расчеты (77) указывают на наличие
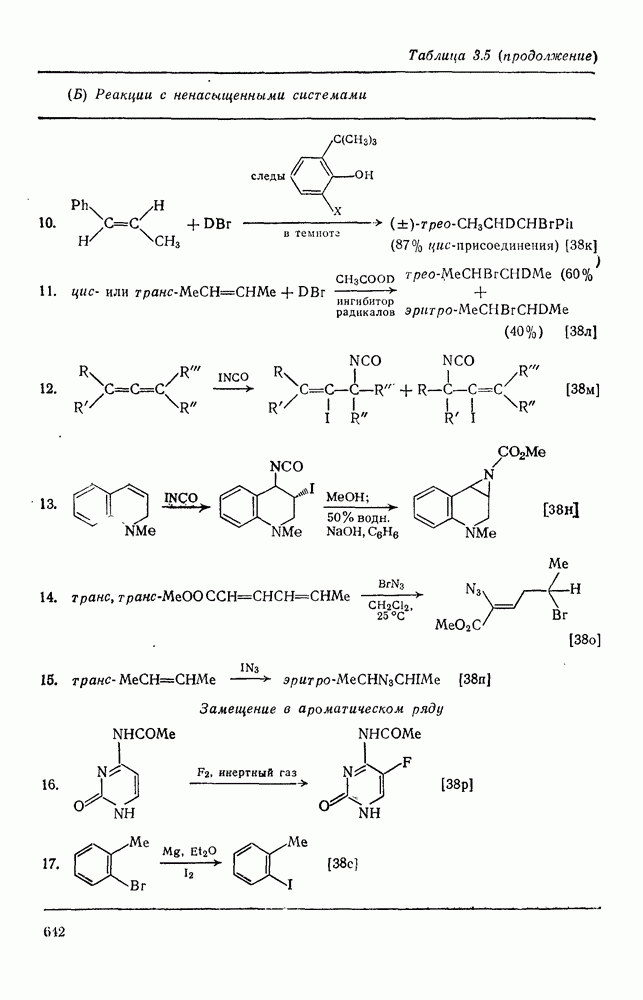
-системой, запрещен орбитальной симметрией и по расчету обладает более высокой энергией, чем (77). Расчеты (77) указывают на наличие  -переноса заряда в переходном состоянии (79), что согласуется с экспериментально наблюдаемым ускорением присоединения большинства карбенов к алкенам, содержащим электронодонорные заместители, и свидетельствует об электрофильной атаке карбена. Многочисленные исследования относительной реакционной способности карбенов с целью выяснения влияния пространственных и электронных эффектов различных заместителей в алкенах и карбенах критически оценены Моссом [48], который показал недавно, что селективность многих карбенов типа
-переноса заряда в переходном состоянии (79), что согласуется с экспериментально наблюдаемым ускорением присоединения большинства карбенов к алкенам, содержащим электронодонорные заместители, и свидетельствует об электрофильной атаке карбена. Многочисленные исследования относительной реакционной способности карбенов с целью выяснения влияния пространственных и электронных эффектов различных заместителей в алкенах и карбенах критически оценены Моссом [48], который показал недавно, что селективность многих карбенов типа  при реакции с олефинами коррелирует как с резонансными, так и с индуктивными параметрами X и Y [71]. Большинство карбенов, в том числе сильно
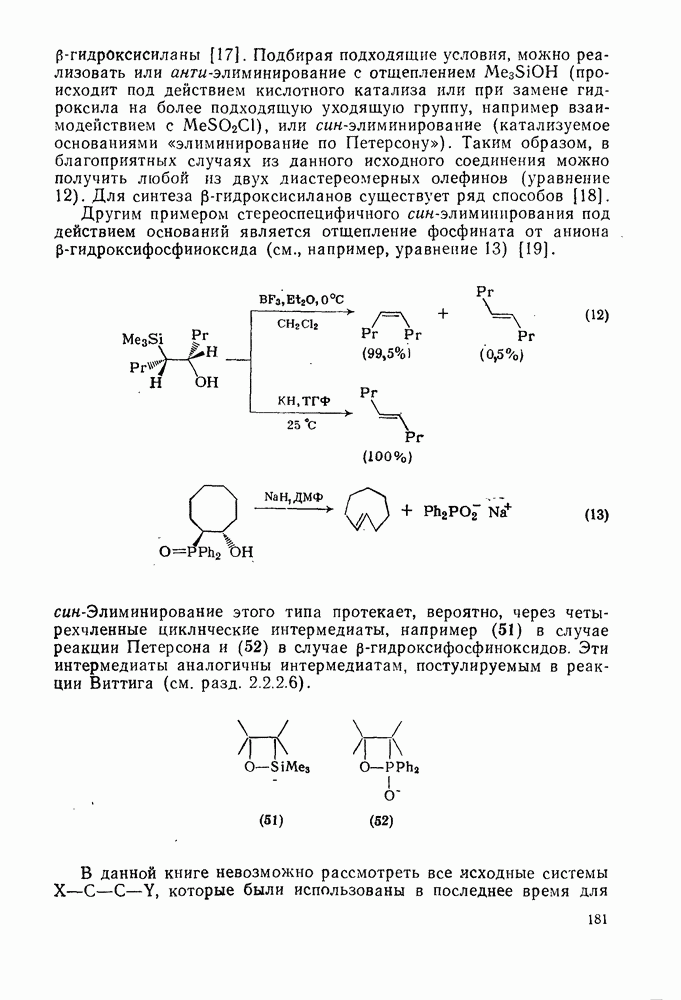
при реакции с олефинами коррелирует как с резонансными, так и с индуктивными параметрами X и Y [71]. Большинство карбенов, в том числе сильно  -стабилизованный
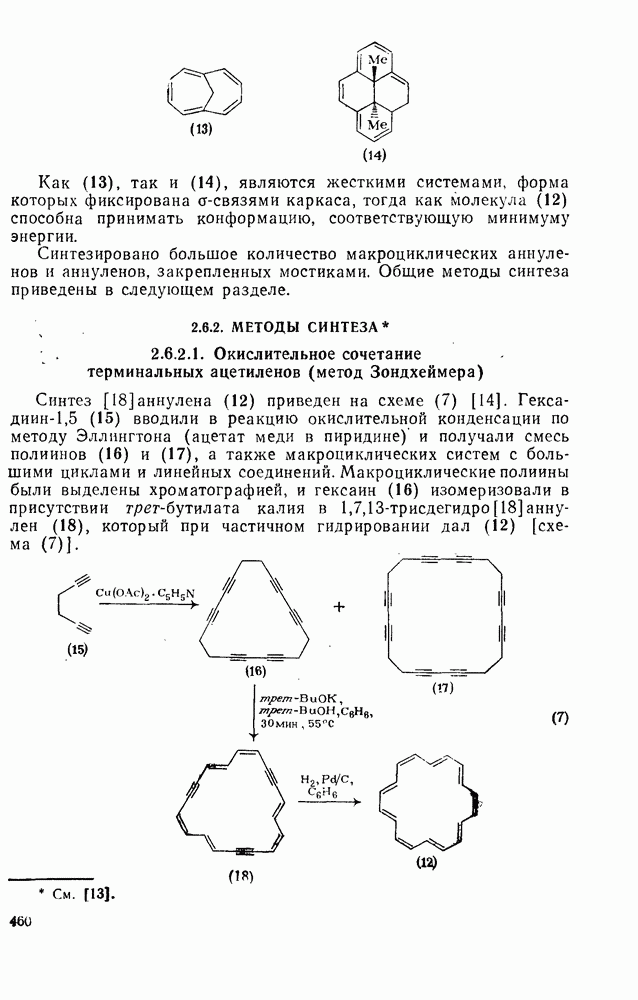
-стабилизованный  (49), ведут себя как типичные электрофилы, однако «ароматические» карбены, такие как (80) и (47), проявляют нуклеофильные свойства, например (80) присоединяется через переходное состояние, поляризованное противоположно (79) [72].
(49), ведут себя как типичные электрофилы, однако «ароматические» карбены, такие как (80) и (47), проявляют нуклеофильные свойства, например (80) присоединяется через переходное состояние, поляризованное противоположно (79) [72].
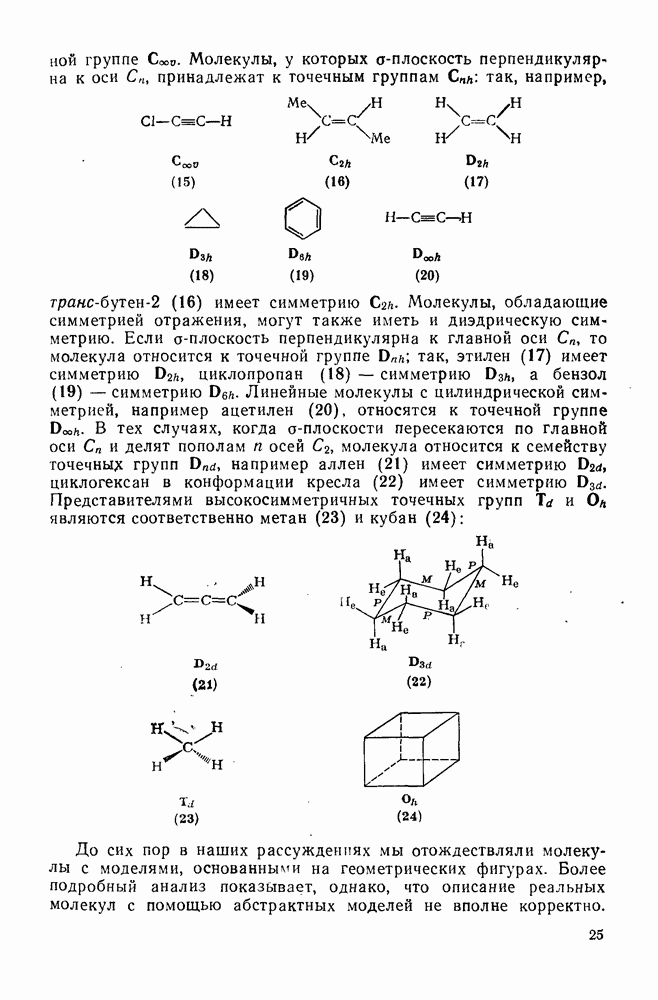
Полагают, что это обусловлено включением р-орбитали карбена в  -электронную систему циклоалкенового кольца.
-электронную систему циклоалкенового кольца.

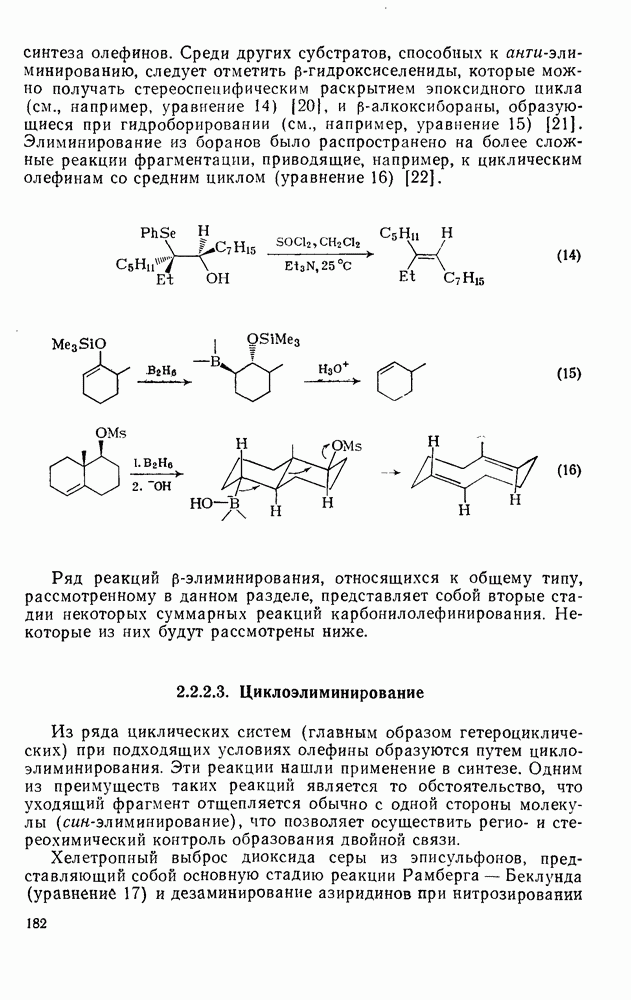
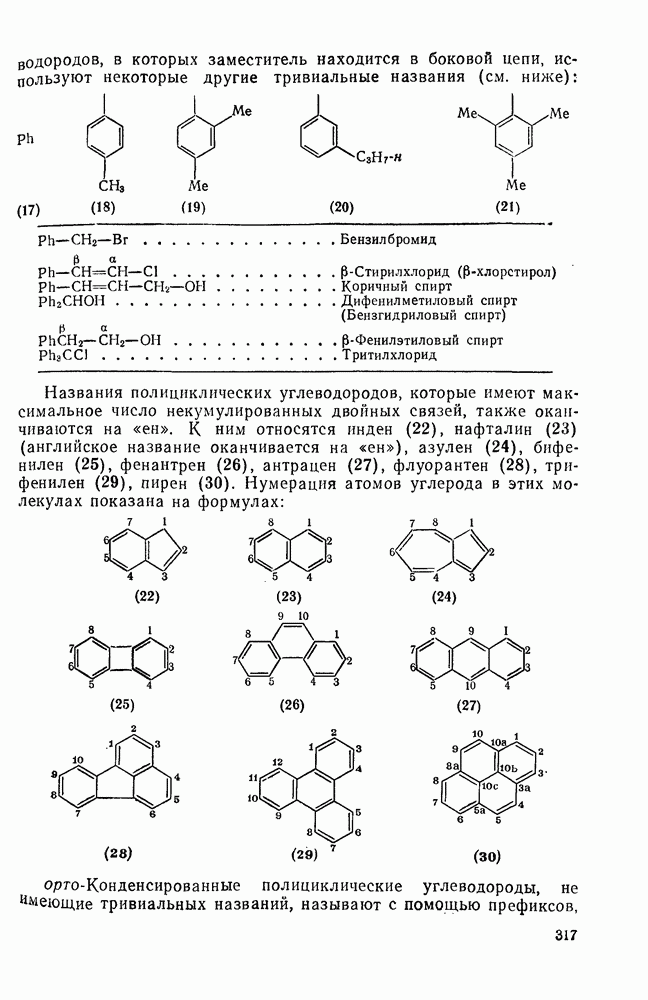
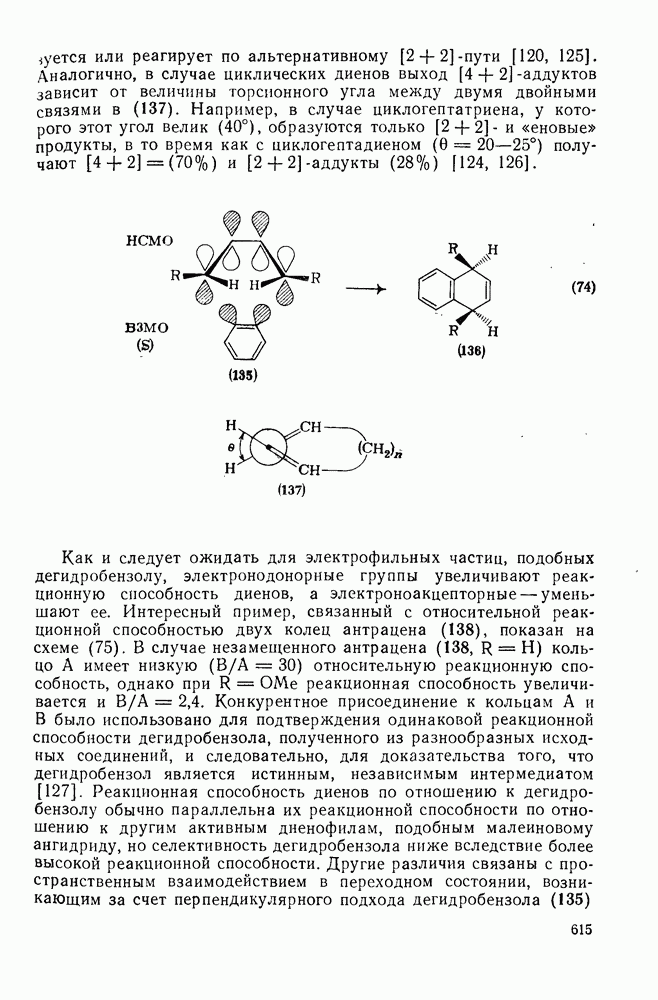
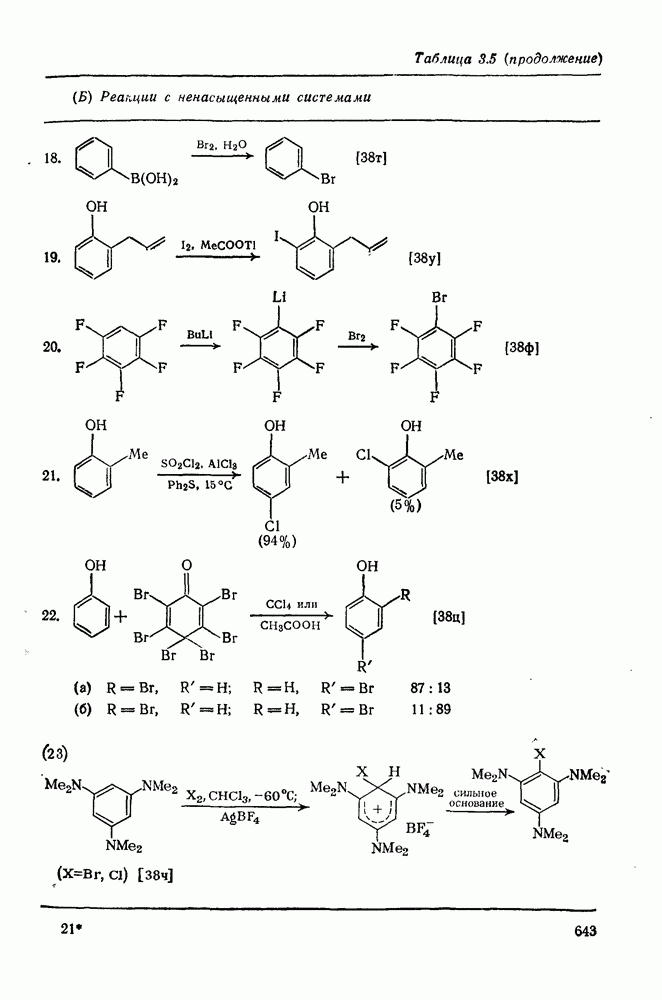
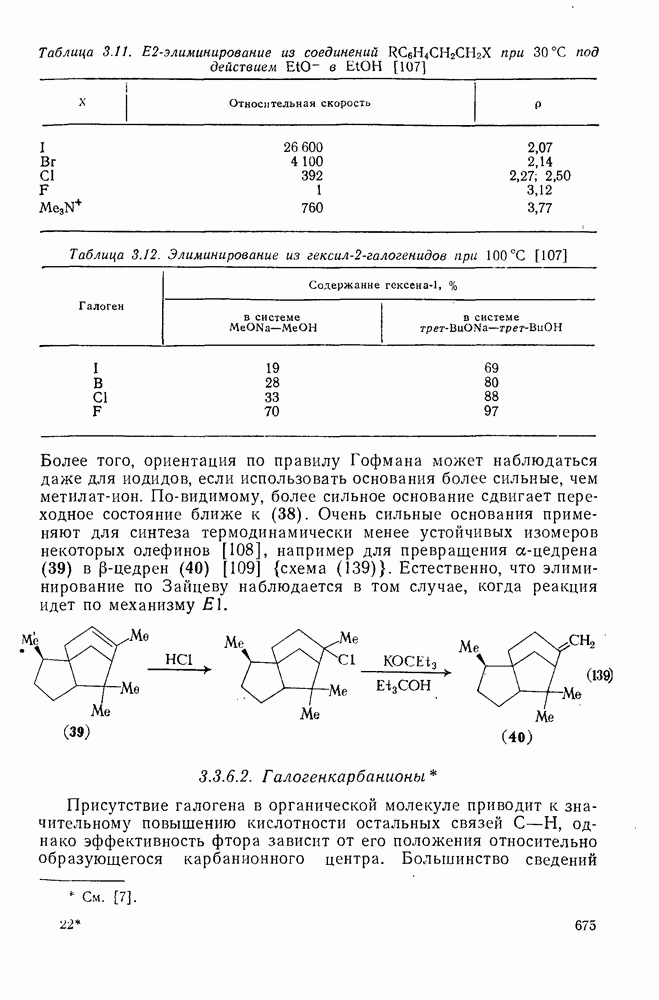
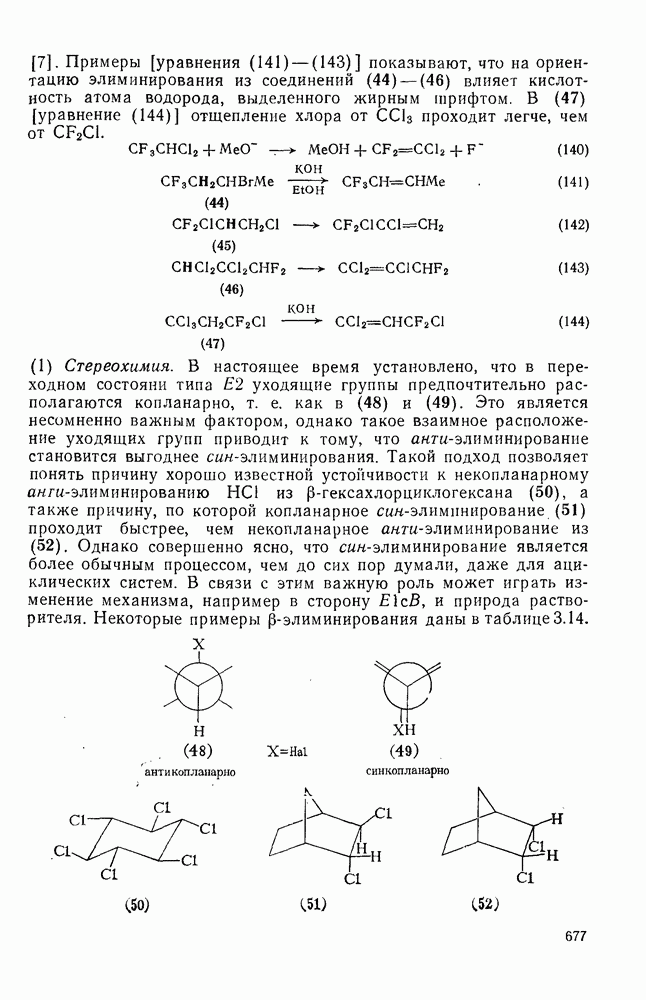
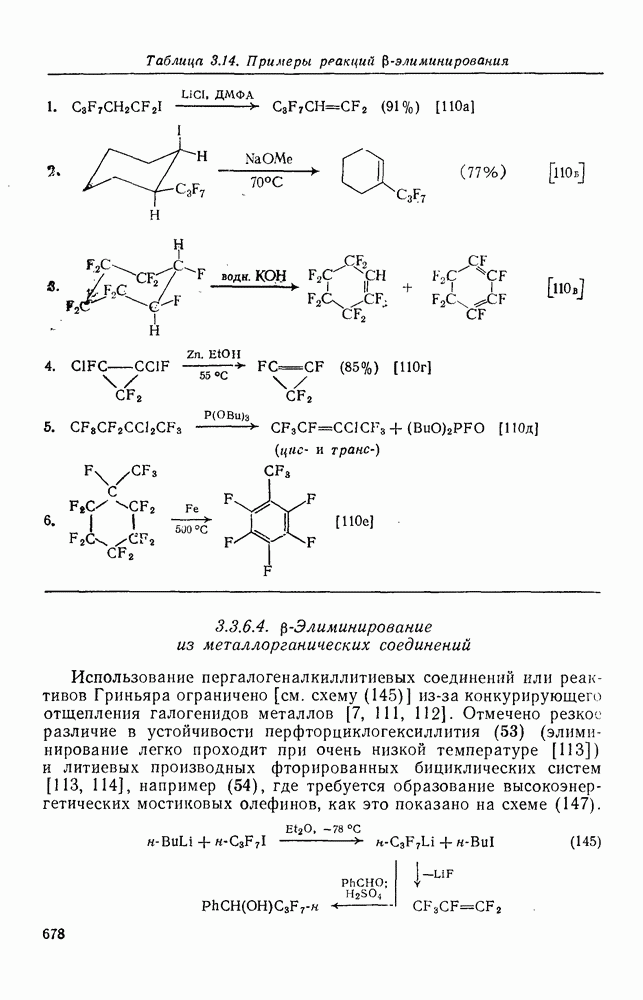
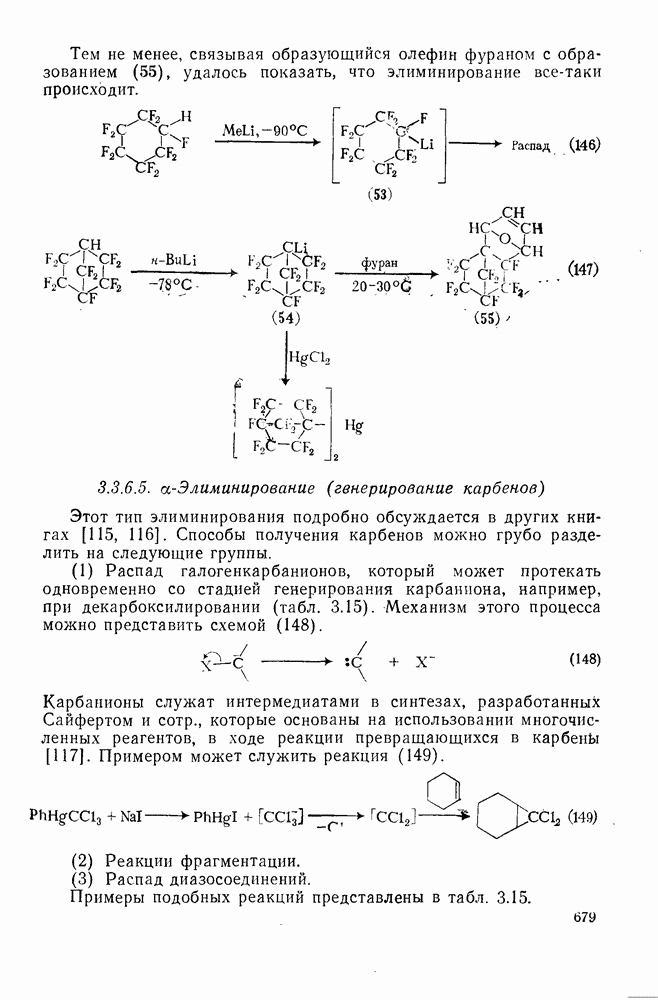
При присоединении к цис-алкенам [уравнение (49)] несимметричные карбены проявляют удивительную стереоселективность, противоположную термодинамической, давая (81) с большим выходом, чем (82) [73]. Это, по крайней мере частично, является результатом стерического притяжения, обусловленного вторичными орбитальными взаимодействиями в переходном состоянии [74].

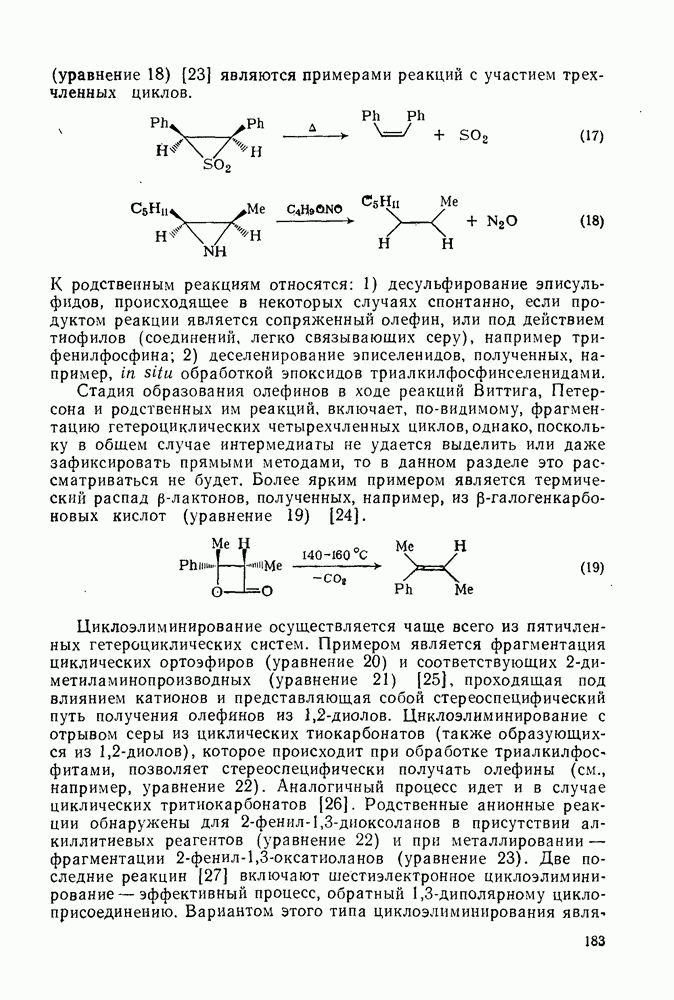
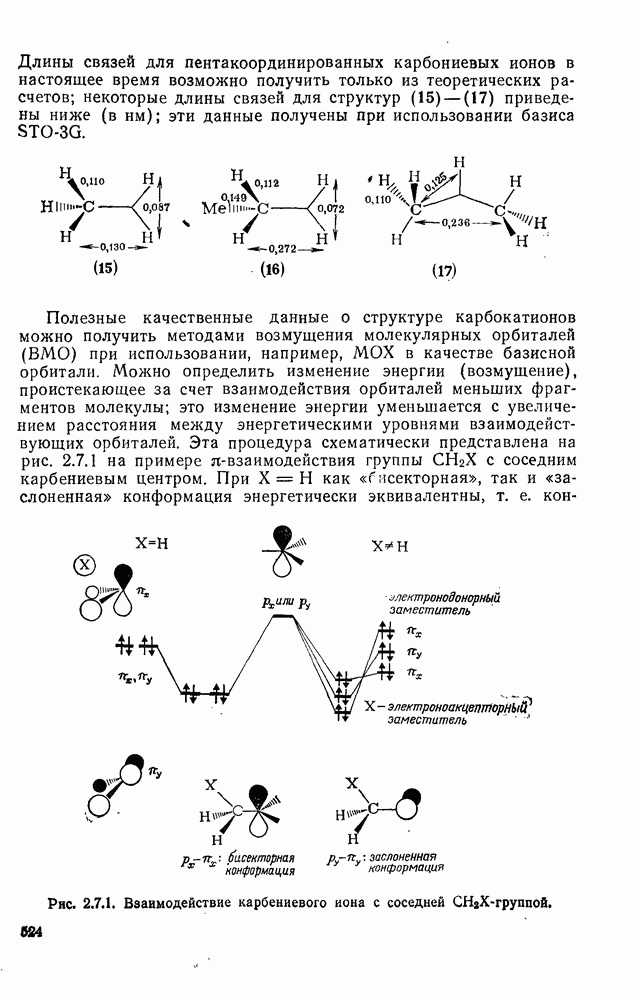
Расчеты присоединения триплетного метилена к этилену указывают на первоначально  -подобный подход с последующим связыванием карбена с одной стороны с образованием триплетного триметиленового бирадикала [50]. Триплетные карбены обычно реагируют с алкенами медленнее синглетных, для диенов справедливо обратное соотношение. Алкен (83) служит полезным индикатором мультиплетности карбена, поскольку синглетные карбены присоединяются к нему без перегруппировки, в то время как присоединение триплета приводит к (84), который быстро перегруппировывается с образованием (85) [50], как показано на схеме (50). Правила орбитальной симметрии разрешают как
-подобный подход с последующим связыванием карбена с одной стороны с образованием триплетного триметиленового бирадикала [50]. Триплетные карбены обычно реагируют с алкенами медленнее синглетных, для диенов справедливо обратное соотношение. Алкен (83) служит полезным индикатором мультиплетности карбена, поскольку синглетные карбены присоединяются к нему без перегруппировки, в то время как присоединение триплета приводит к (84), который быстро перегруппировывается с образованием (85) [50], как показано на схеме (50). Правила орбитальной симметрии разрешают как  , так и
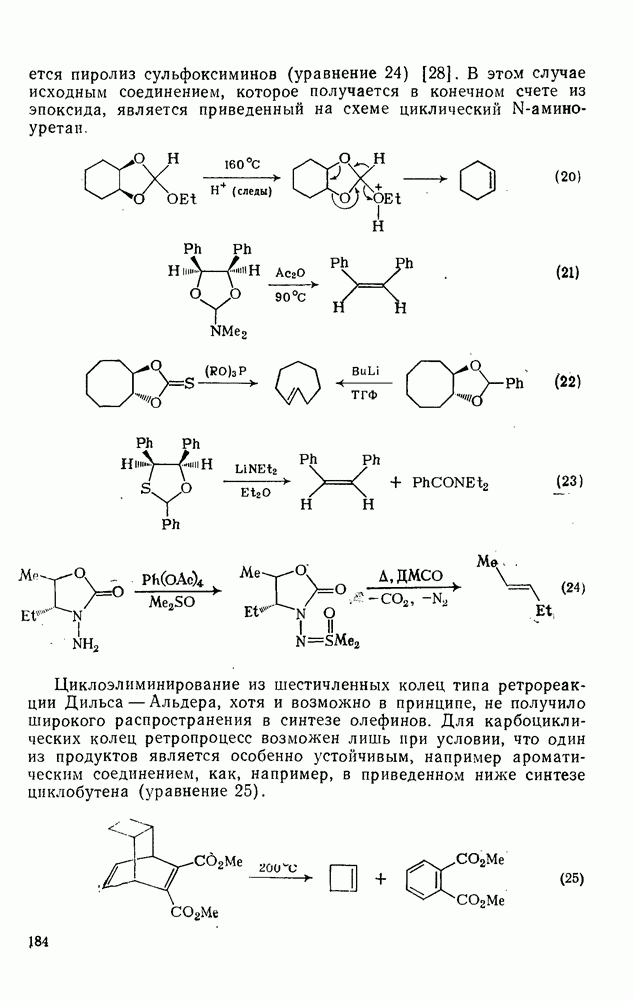
, так и  -присоединение к сопряженным диенам, однако отталкивание за счет замкнутой оболочки делает невыгодным
-присоединение к сопряженным диенам, однако отталкивание за счет замкнутой оболочки делает невыгодным  -присоединение, и оно наблюдается очень редко [75]. Однако цикло-пропенилиден присоединяется в положение 1,4 к тетрациклонам, а дифторкарбен присоединяется к норборнадиену в
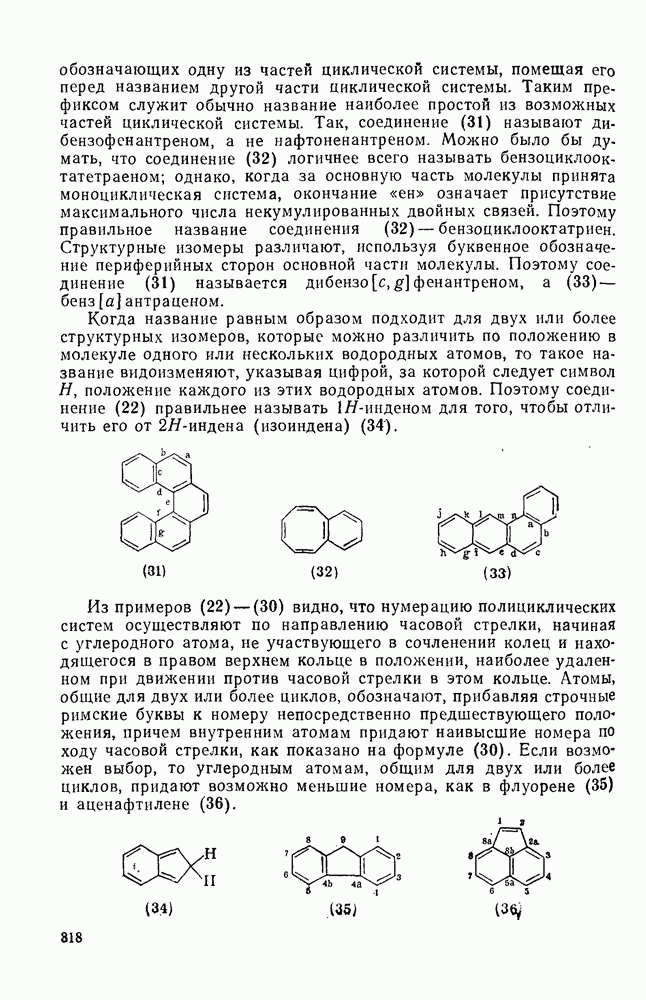
-присоединение, и оно наблюдается очень редко [75]. Однако цикло-пропенилиден присоединяется в положение 1,4 к тетрациклонам, а дифторкарбен присоединяется к норборнадиену в  [76].
[76].
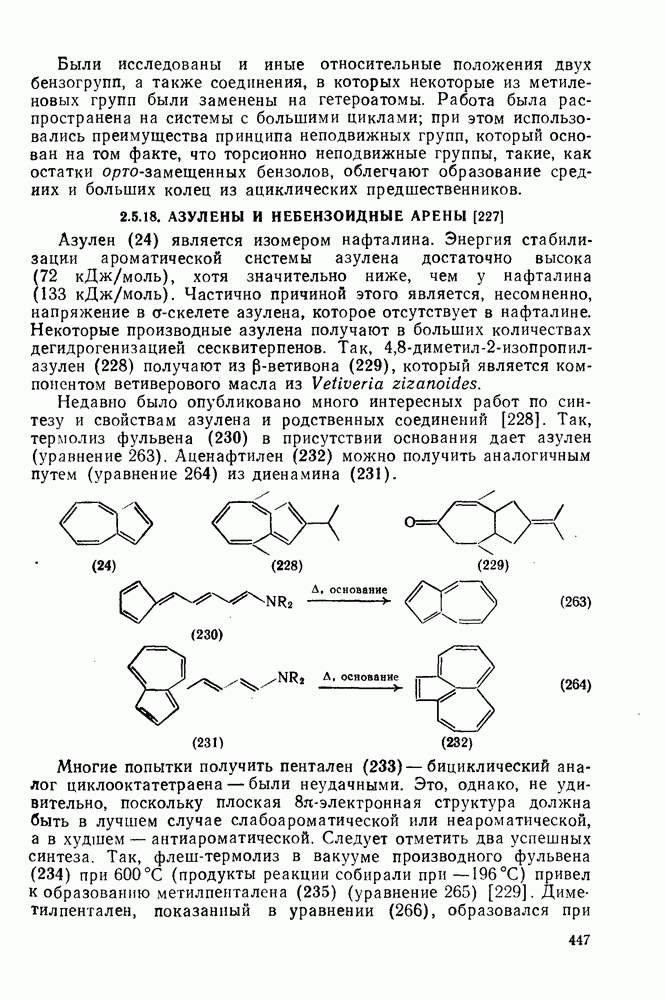
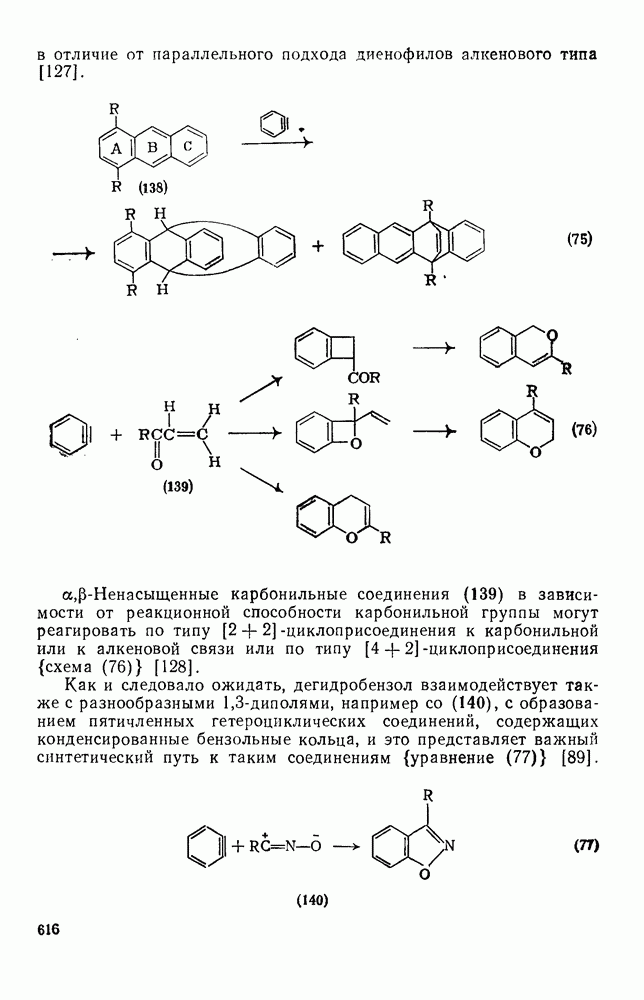
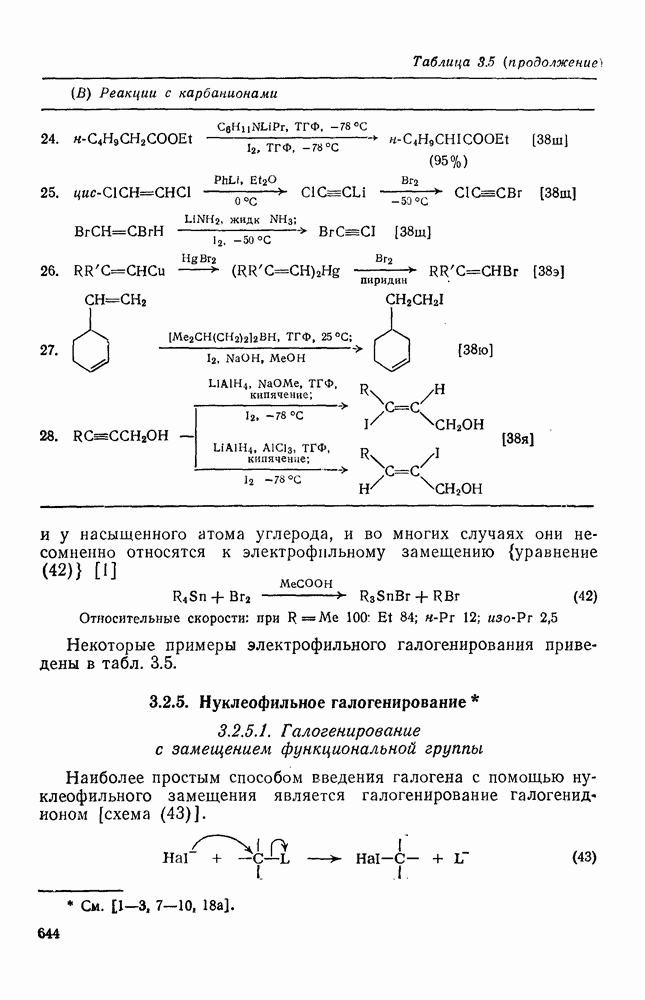
Карбены присоединяются также к алкинам, аренам, гетероаренам и различным другим ненасыщенным группам, таким как  и
и  [69].
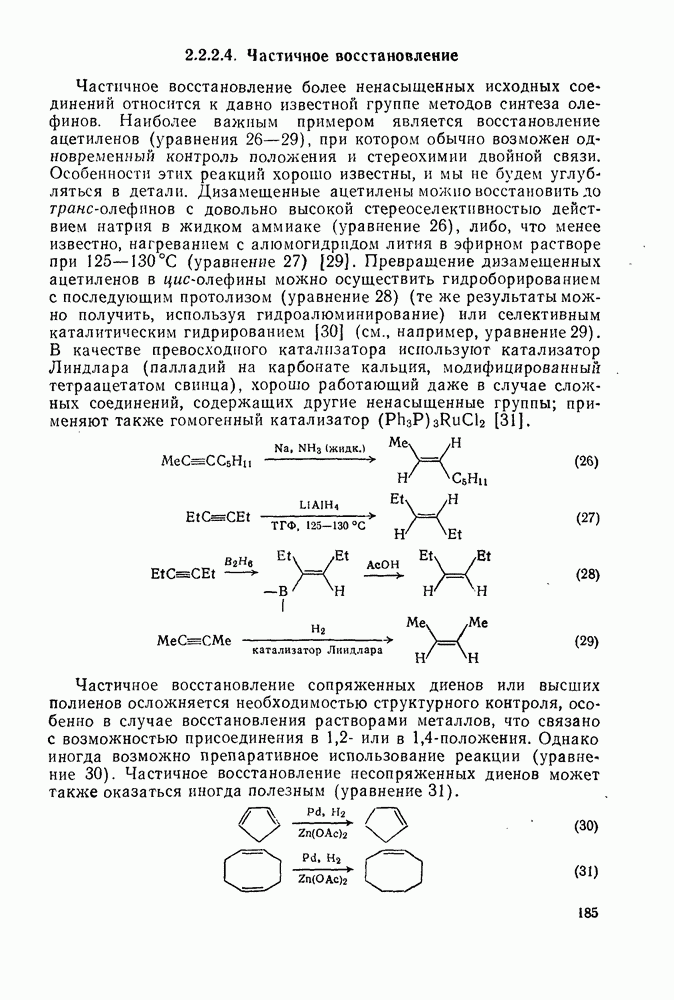
[69].
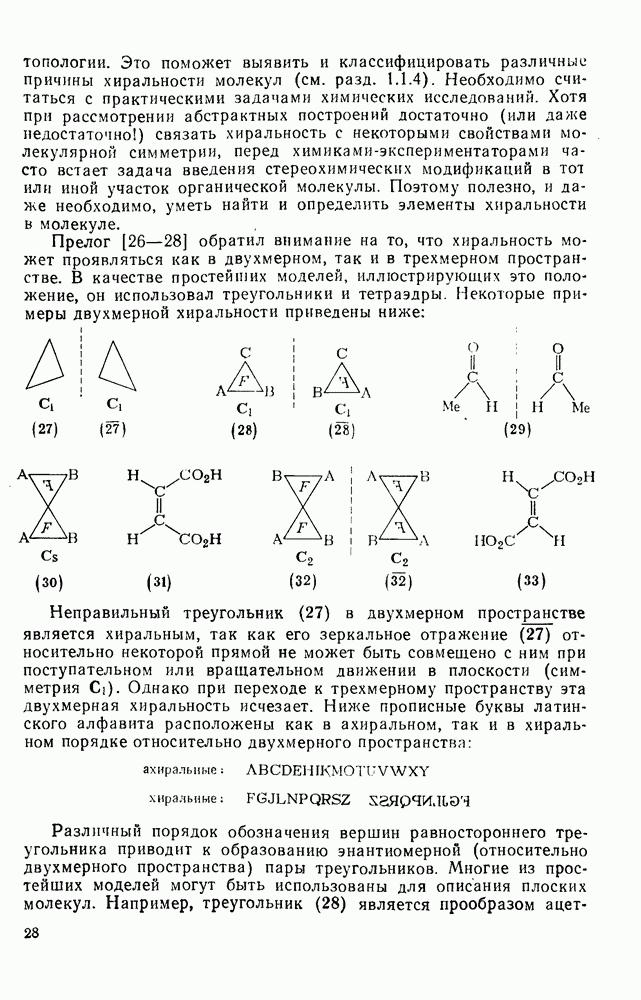
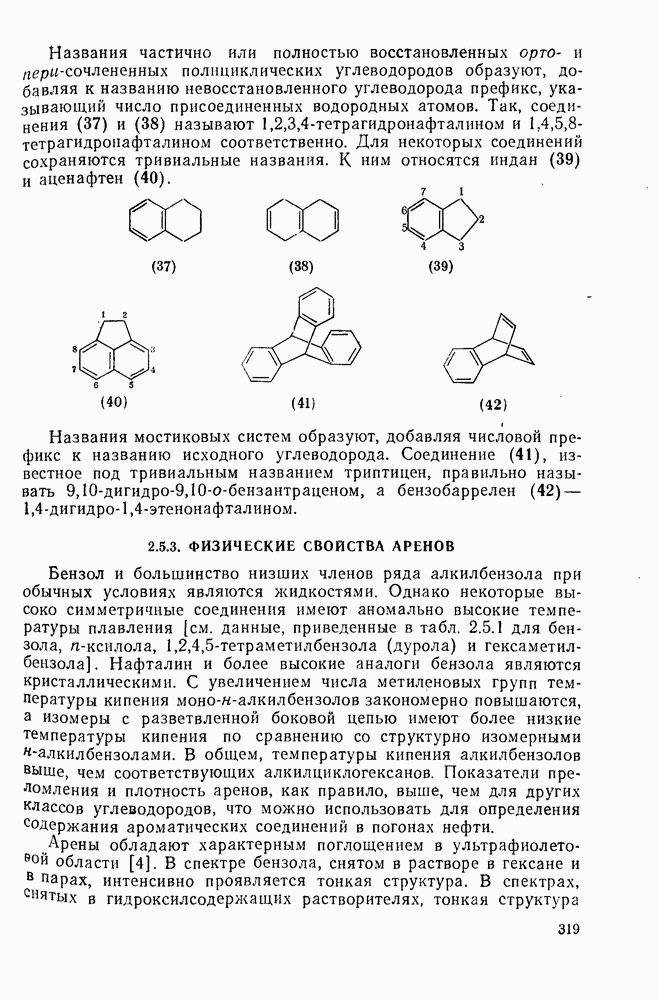
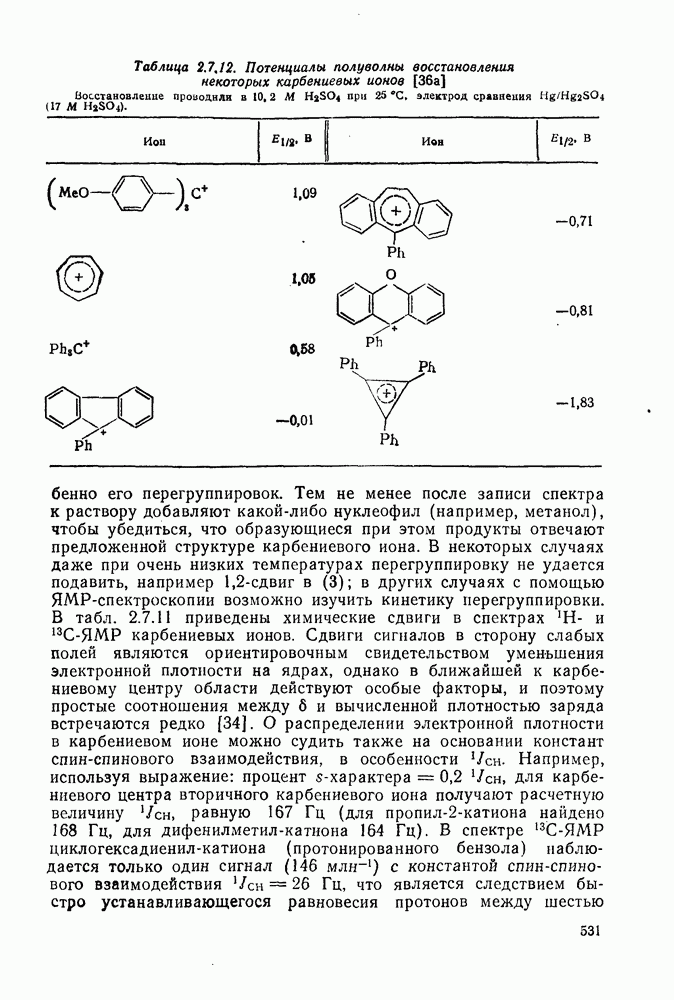
(2) Реакции внедрения и отщепления
Характерные межмолекулярные и внутримолекулярные реакции внедрения по связи [см., например, уравнения (51) и (52)], присущи большинству карбенов и многим типам связей, например  [47]. При внедрении самого метилена по связи
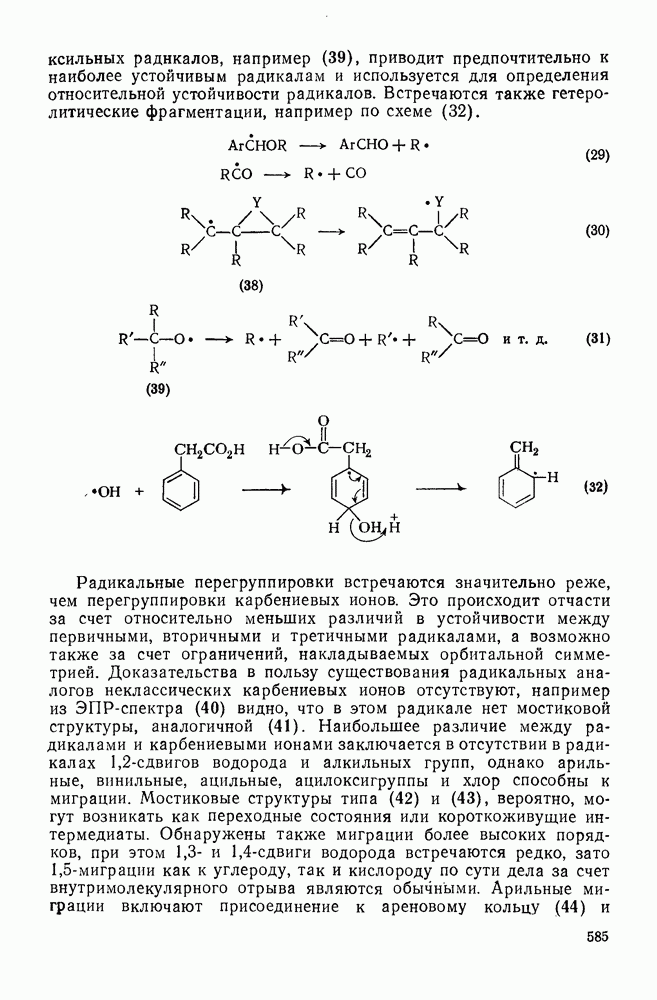
[47]. При внедрении самого метилена по связи  в растворе фактически не обнаруживается различий между первичными, вторичными и третичными связями; в газофазных реакциях такие различия не слишком велики, например в случае изопентана относительная скорость внедрения следующая: первичная связь 1,0; вторичная связь 1,2; третичная связь 1,4. При наличии в карбене арильных групп, галогенов или электроноакцепторных заместителей селективность возрастает. Пр и взаимодействии с алкенами основной реакцией является присоединение по двойной связи; внедрение по связи
в растворе фактически не обнаруживается различий между первичными, вторичными и третичными связями; в газофазных реакциях такие различия не слишком велики, например в случае изопентана относительная скорость внедрения следующая: первичная связь 1,0; вторичная связь 1,2; третичная связь 1,4. При наличии в карбене арильных групп, галогенов или электроноакцепторных заместителей селективность возрастает. Пр и взаимодействии с алкенами основной реакцией является присоединение по двойной связи; внедрение по связи  лишь слабо конкурирует с ним и только в случае более реакционноспособных карбенов.
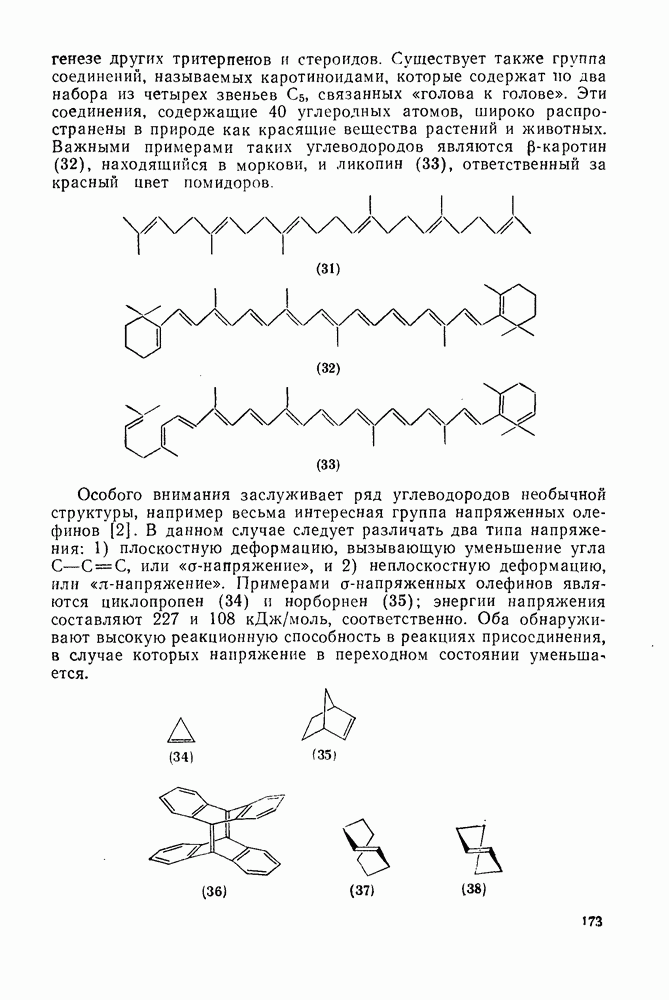
лишь слабо конкурирует с ним и только в случае более реакционноспособных карбенов.

Как и в случае реакции присоединения, возможен одностадийный синхронный или двухстадийный механизмы [38].
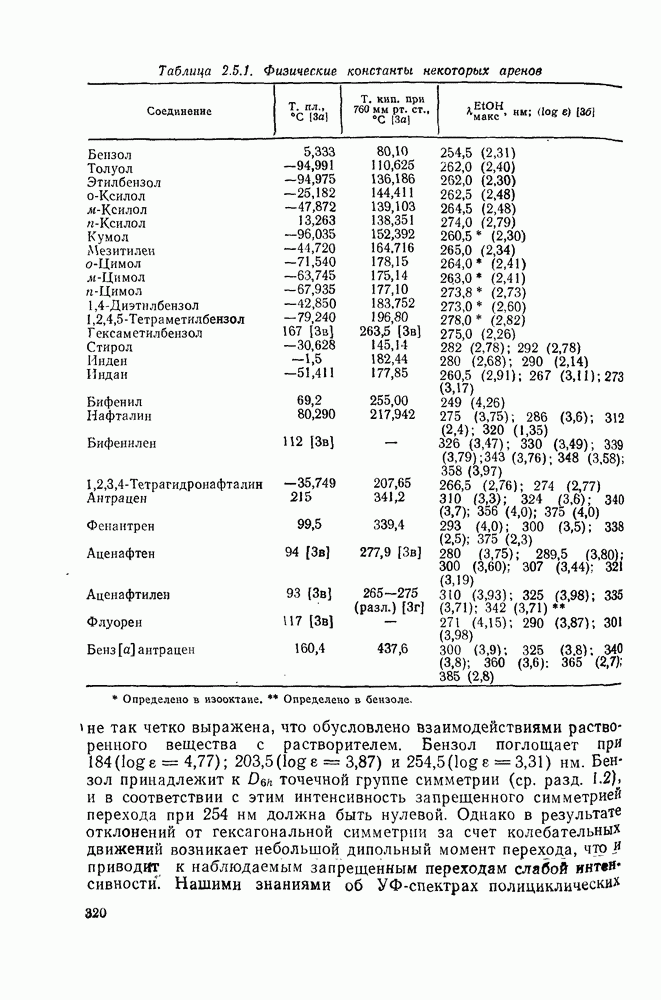
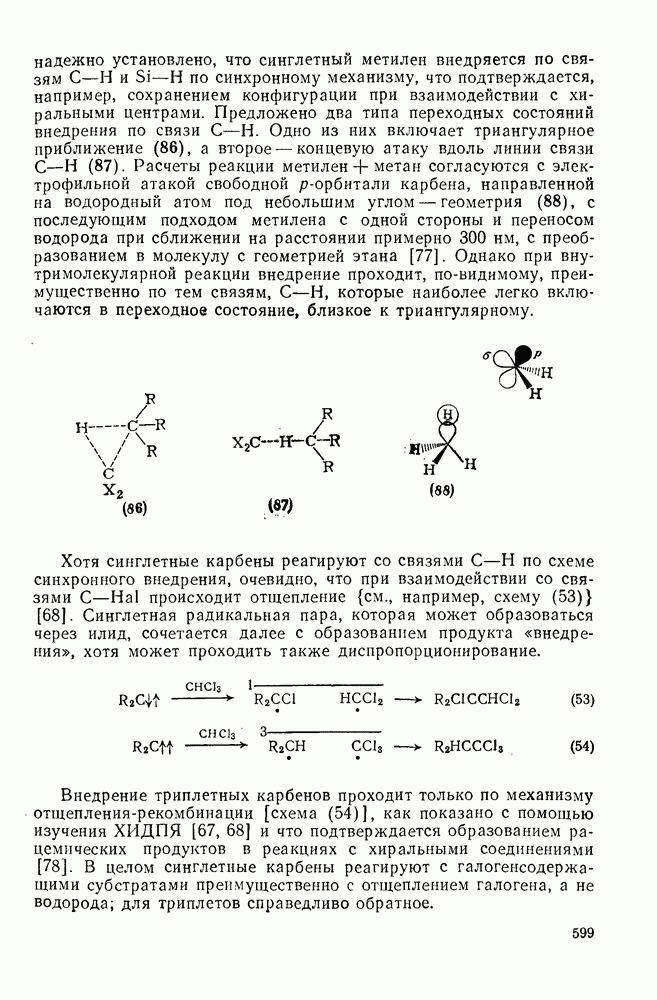
Достаточно надежно установлено, что синглетный метилен внедряется по связям  и
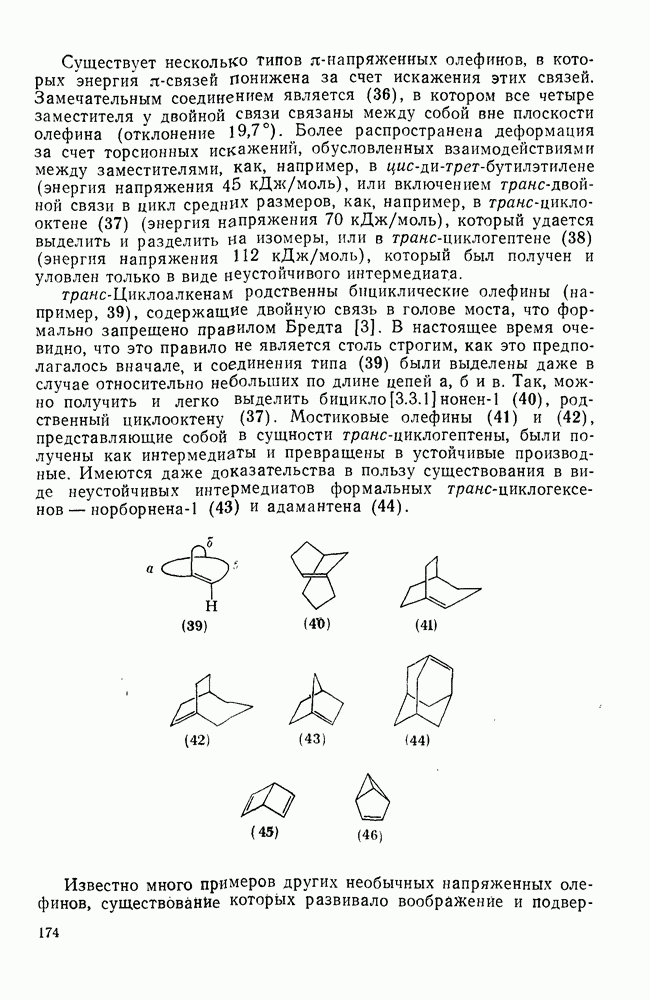
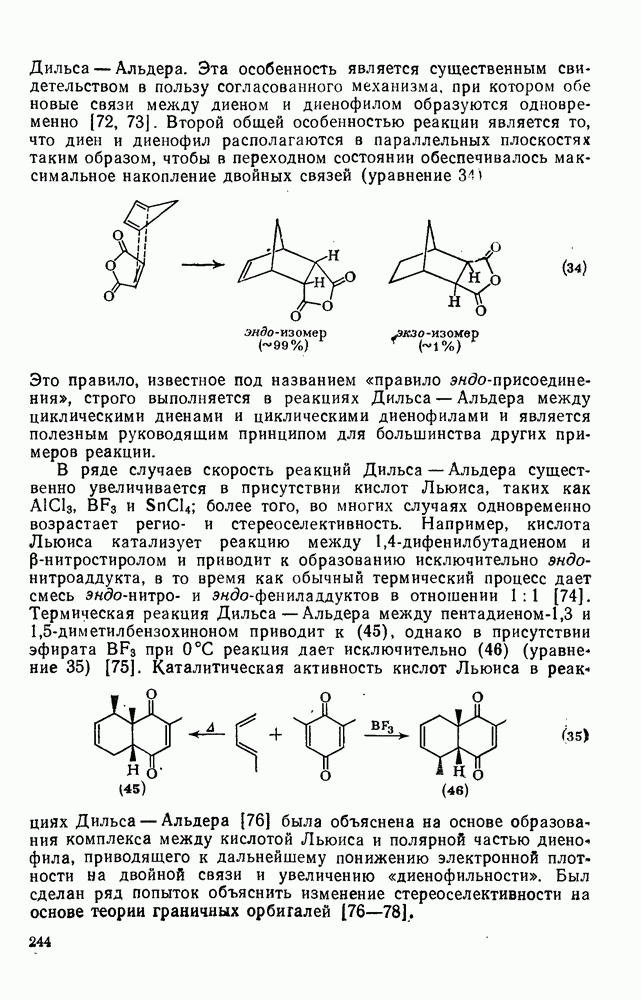
и  по синхронному механизму, что подтверждается, например, сохранением конфигурации при взаимодействии с хиральными центрами. Предложено два типа переходных состояний внедрения по связи
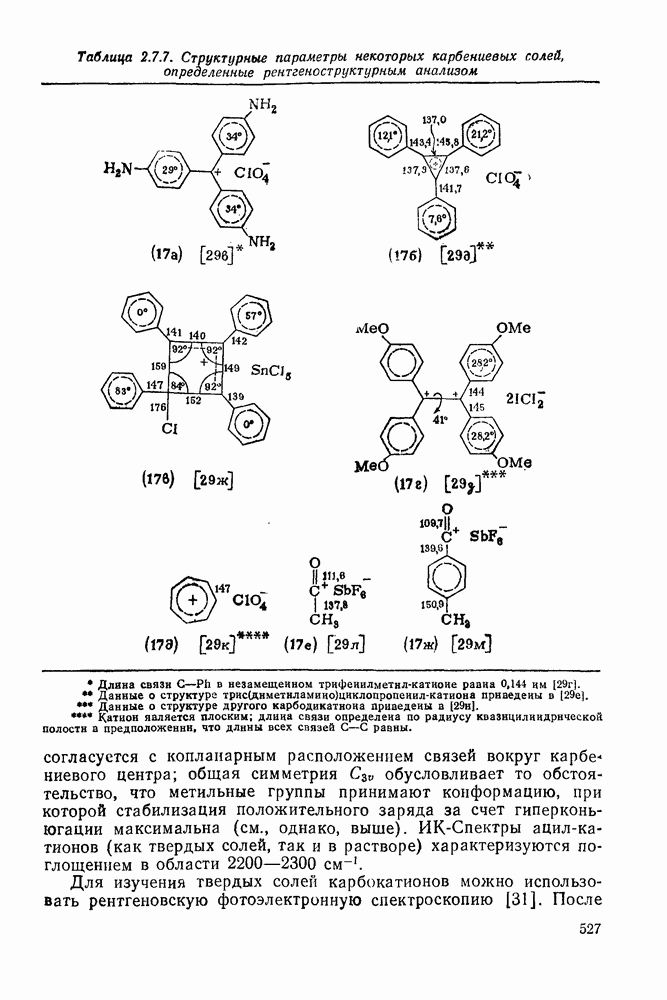
по синхронному механизму, что подтверждается, например, сохранением конфигурации при взаимодействии с хиральными центрами. Предложено два типа переходных состояний внедрения по связи  . Одно из них включает триангулярное приближение (86), а второе — концевую атаку вдоль линии связи
. Одно из них включает триангулярное приближение (86), а второе — концевую атаку вдоль линии связи  (87). Расчеты реакции метилен+метан согласуются с электрофильной атакой свободной р-орбитали карбена, направленной на водородный атом под небольшим углом — геометрия (88), с последующим подходом метилена с одной стороны и переносом водорода при сближении на расстоянии примерно 300 нм, с преобразованием в молекулу с геометрией этана [77]. Однако при внутримолекулярной реакции внедрение проходит, по-видимому, преимущественно по тем связям,
(87). Расчеты реакции метилен+метан согласуются с электрофильной атакой свободной р-орбитали карбена, направленной на водородный атом под небольшим углом — геометрия (88), с последующим подходом метилена с одной стороны и переносом водорода при сближении на расстоянии примерно 300 нм, с преобразованием в молекулу с геометрией этана [77]. Однако при внутримолекулярной реакции внедрение проходит, по-видимому, преимущественно по тем связям,  , которые наиболее легко включаются в переходное состояние, близкое к триангулярному.
, которые наиболее легко включаются в переходное состояние, близкое к триангулярному.

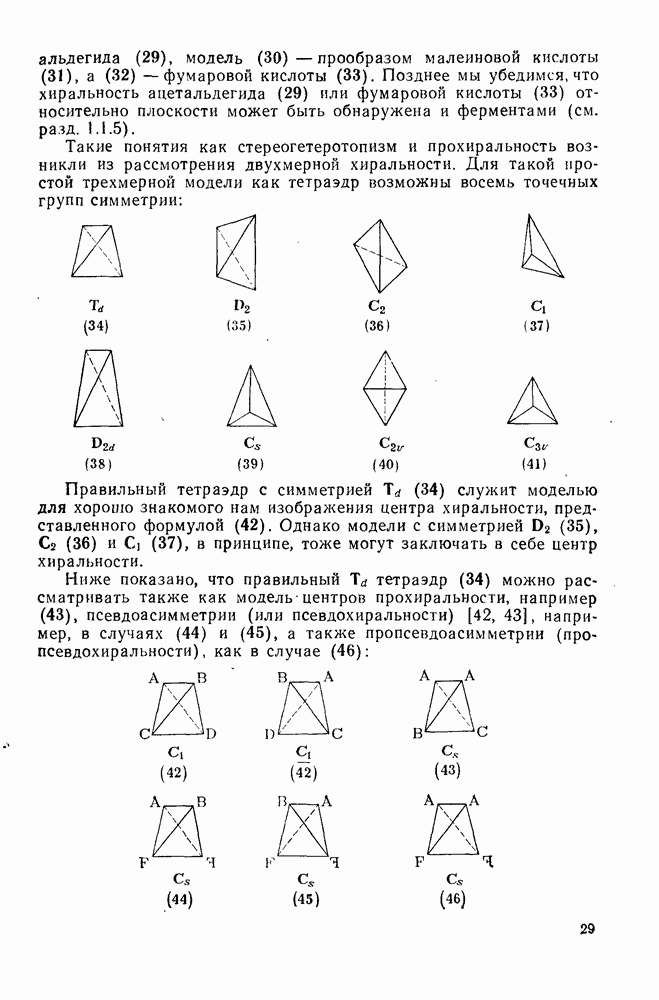
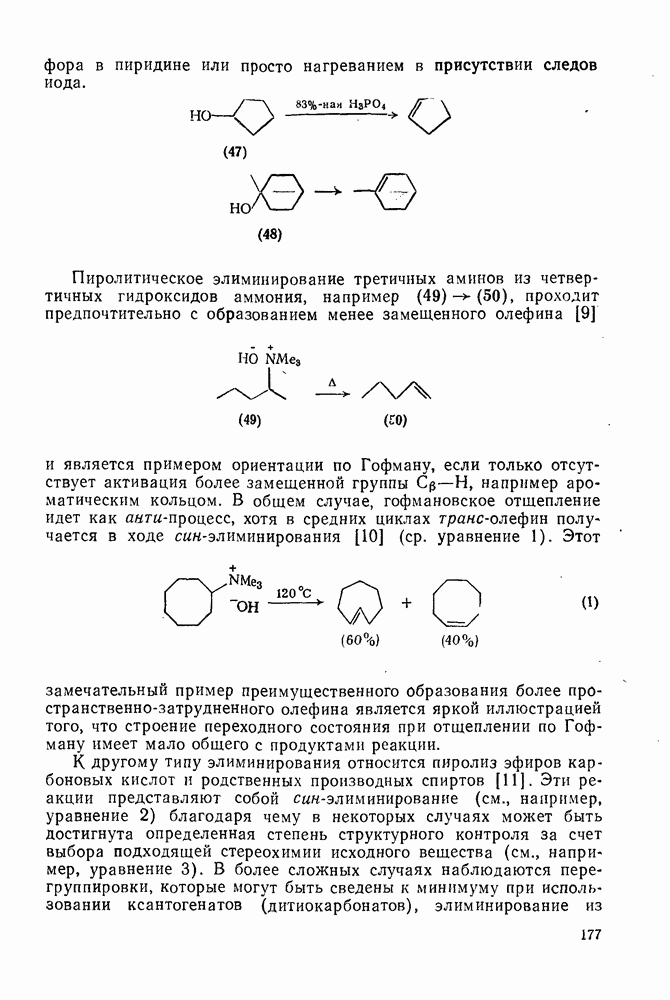
Хотя синглетные карбены реагируют со связями  по схеме синхронного внедрения, очевидно, что при взаимодействии со связями
по схеме синхронного внедрения, очевидно, что при взаимодействии со связями  происходит отщепление {см., например, схему
происходит отщепление {см., например, схему  ] [68]. Синглетная радикальная пара, которая может образоваться через илид, сочетается далее с образованием продукта «внедрения», хотя может проходить также диспропорционирование.
] [68]. Синглетная радикальная пара, которая может образоваться через илид, сочетается далее с образованием продукта «внедрения», хотя может проходить также диспропорционирование.

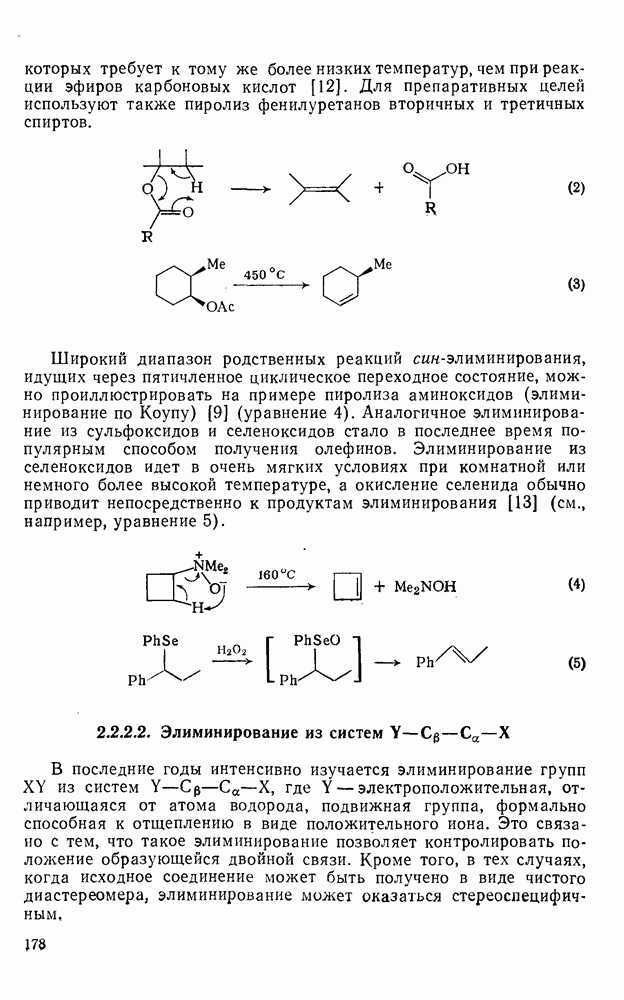
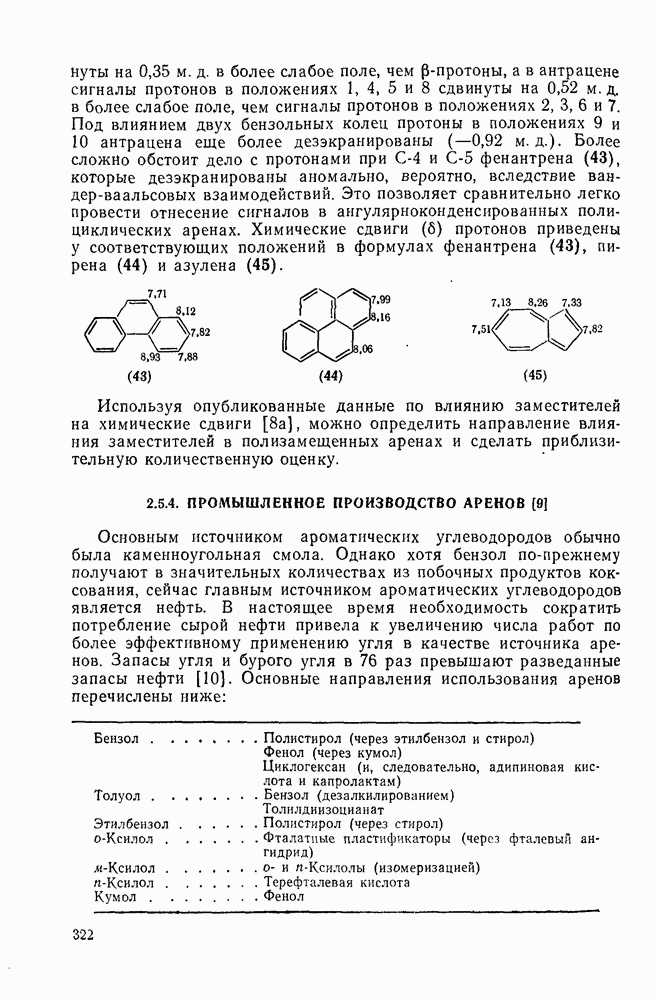
Внедрение триплетных карбенов проходит только по механизму отщепления-рекомбинации [схема  ], как показано с помощью изучения ХИДПЯ [67, 68] и что подтверждается образованием рацемических продуктов в реакциях с хиральными соединениями [78]. В целом синглетные карбены реагируют с галогенсодержащими субстратами преимущественно с отщеплением галогена, а не водорода; для триплетов справедливо обратное.
], как показано с помощью изучения ХИДПЯ [67, 68] и что подтверждается образованием рацемических продуктов в реакциях с хиральными соединениями [78]. В целом синглетные карбены реагируют с галогенсодержащими субстратами преимущественно с отщеплением галогена, а не водорода; для триплетов справедливо обратное.
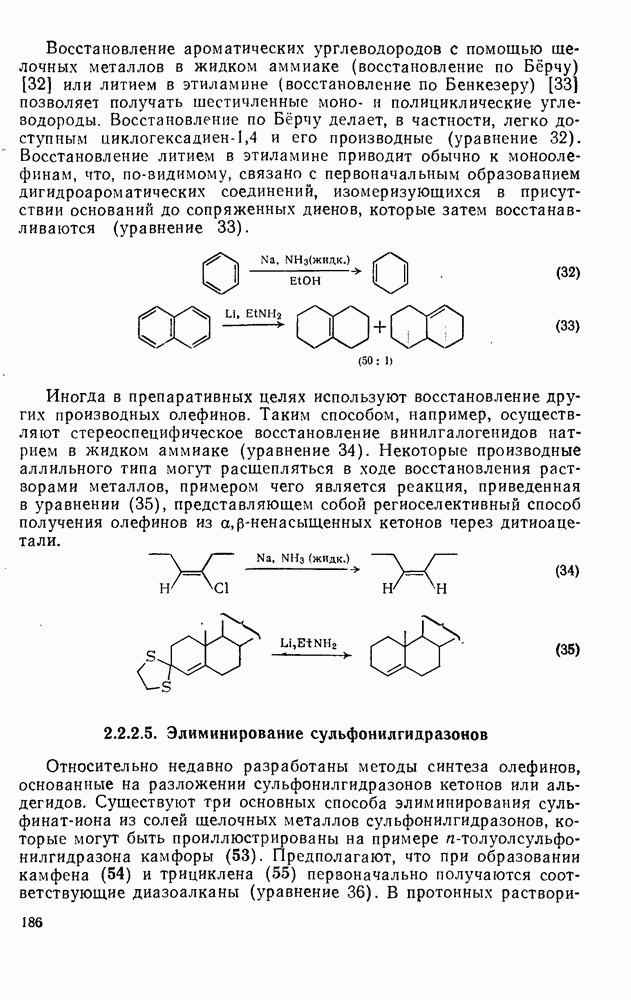
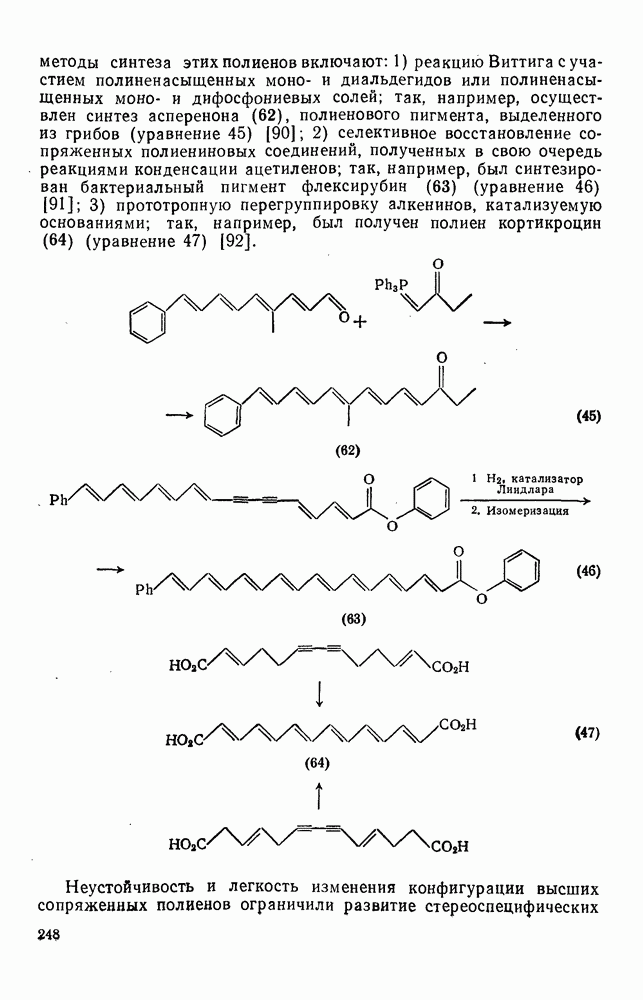
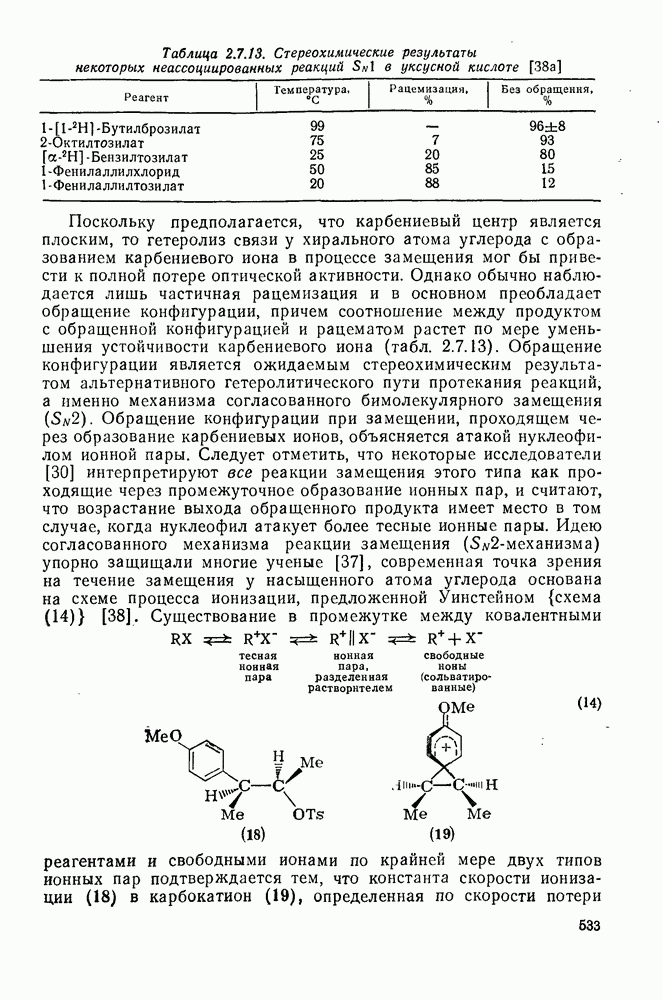
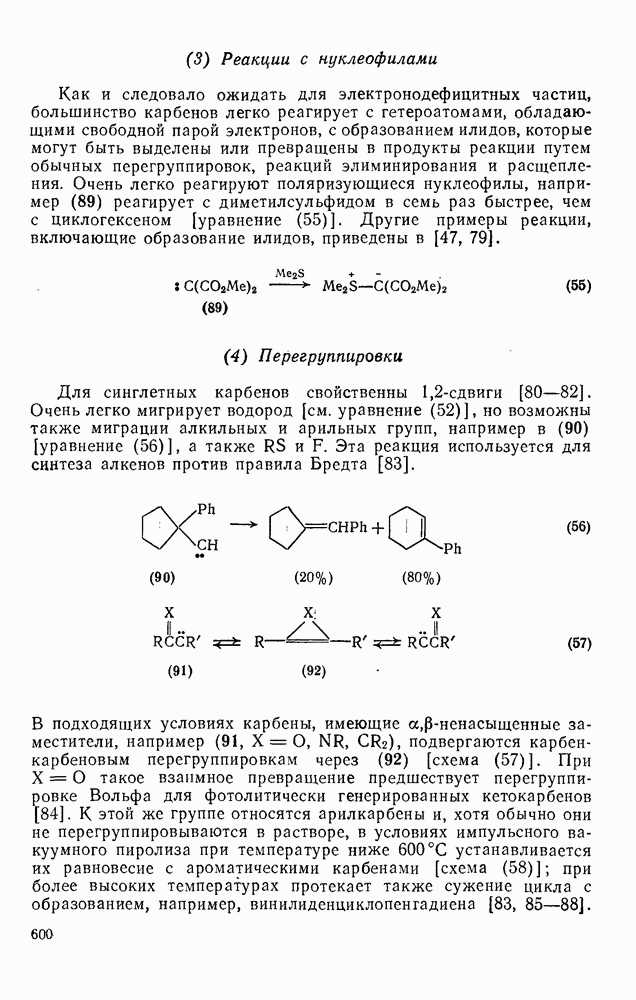
(3) Реакции с нуклеофилами
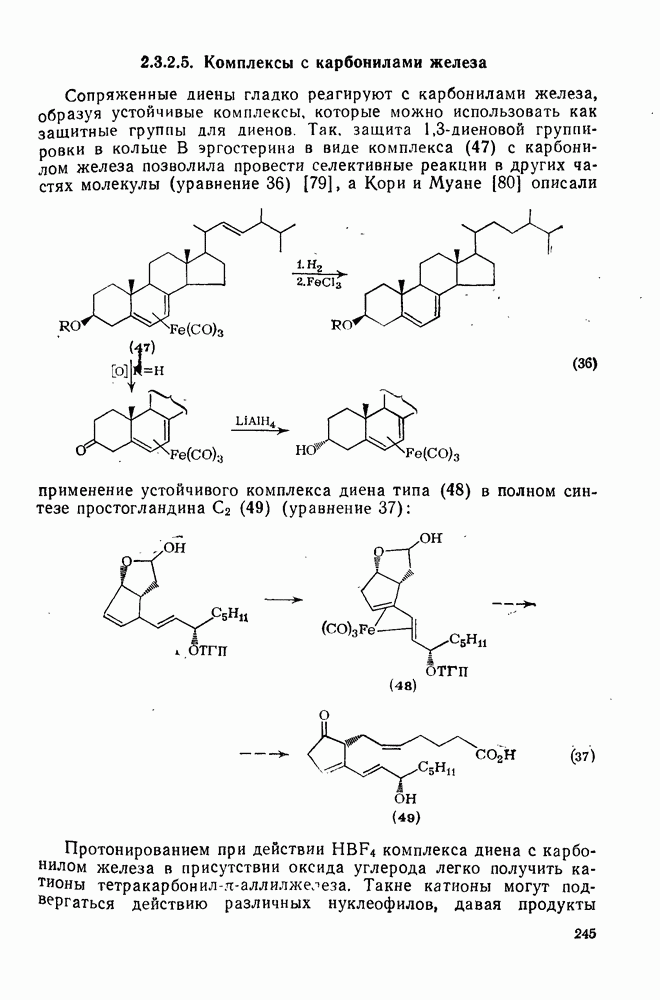
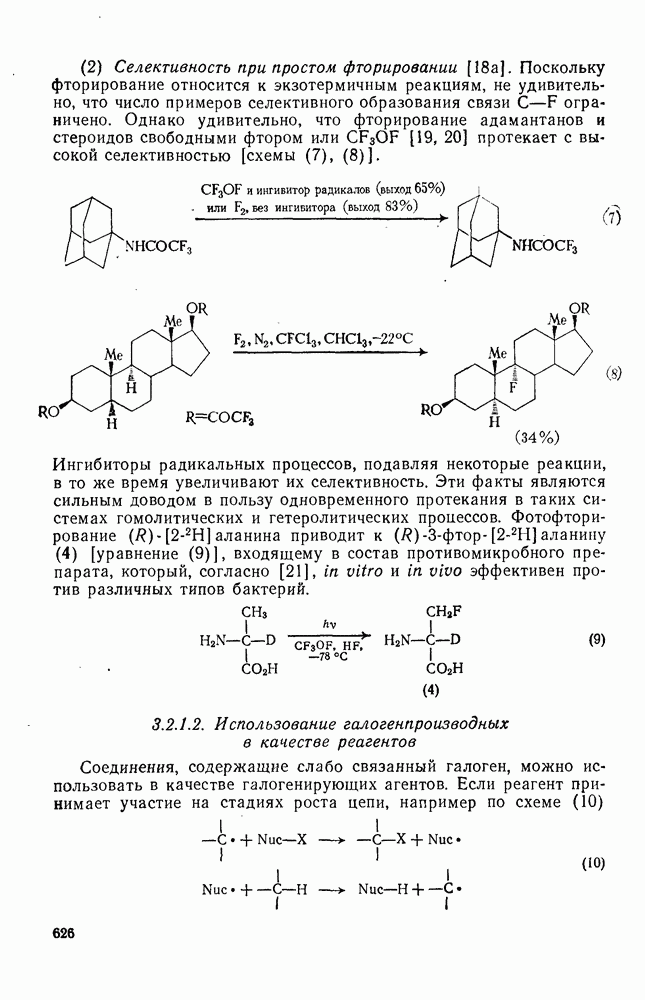
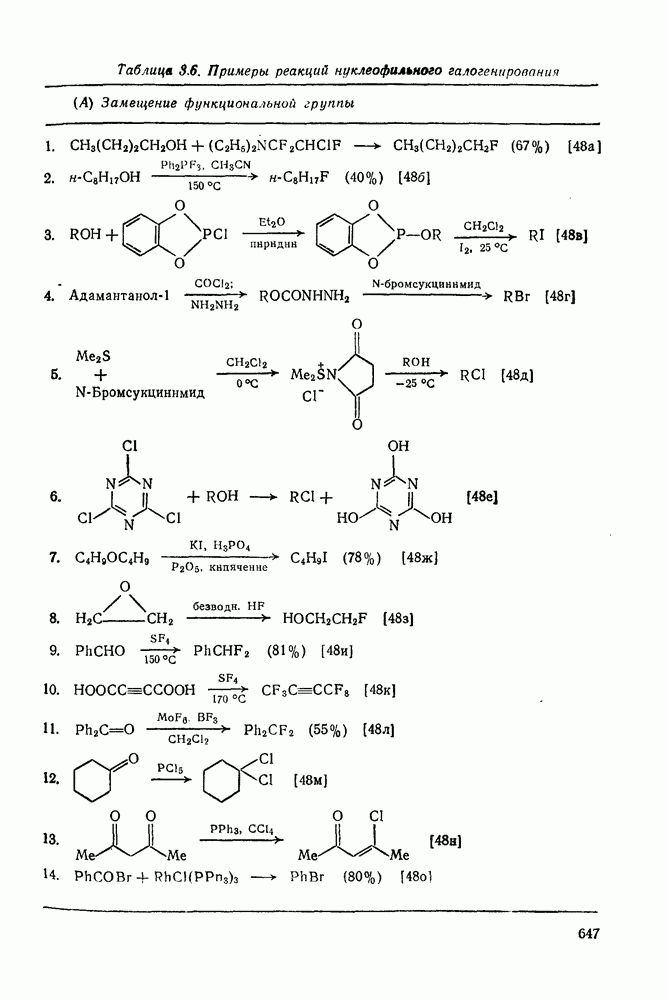
Как и следовало ожидать для электронодефицитных частиц, большинство карбенов легко реагирует с гетероатомами, обладающими свободной парой электронов, с образованием илидов, которые могут быть выделены или превращены в продукты реакции путем обычных перегруппировок, реакций элиминирования и расщепления. Очень легко реагируют поляризующиеся нуклеофилы, например (89) реагирует с диметилсульфидом в семь раз быстрее, чем с циклогексеном [уравнение (55)]. Другие примеры реакции, включающие образование илидов, приведены в [47, 79].

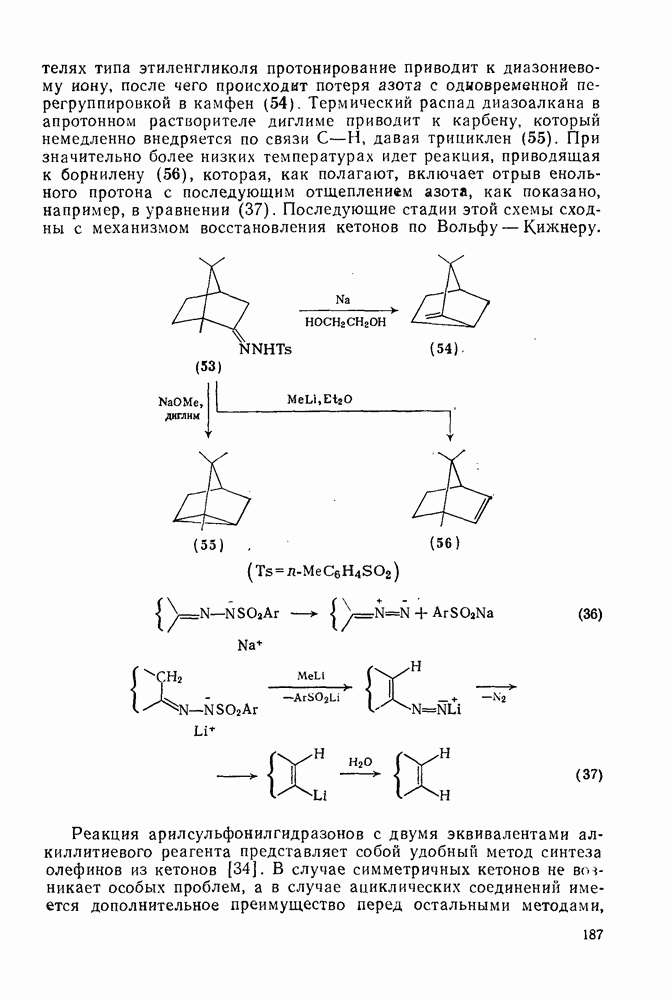
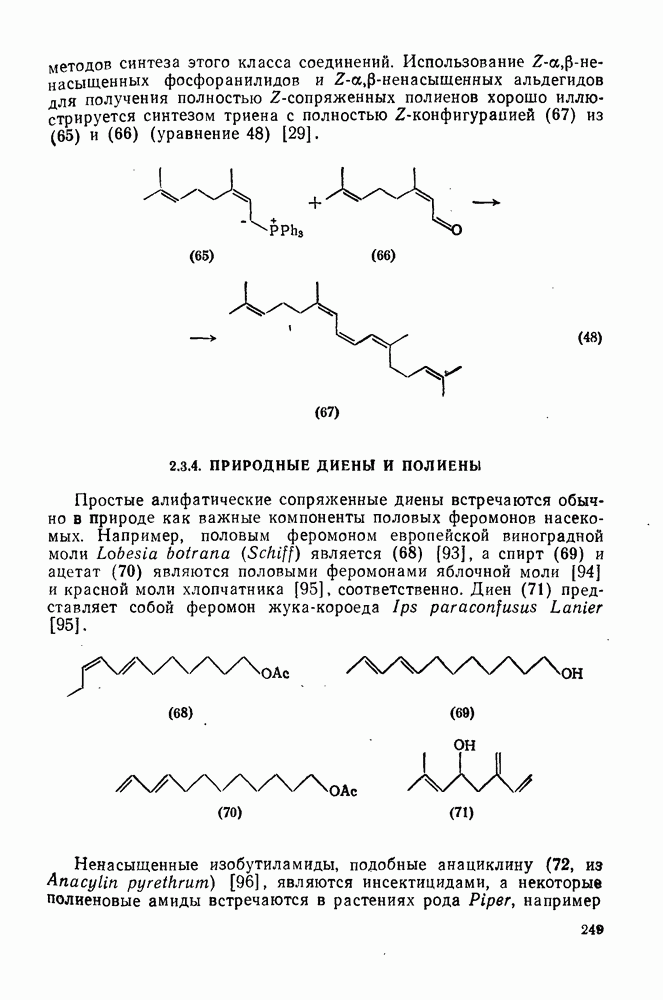
(4) Перегруппировки
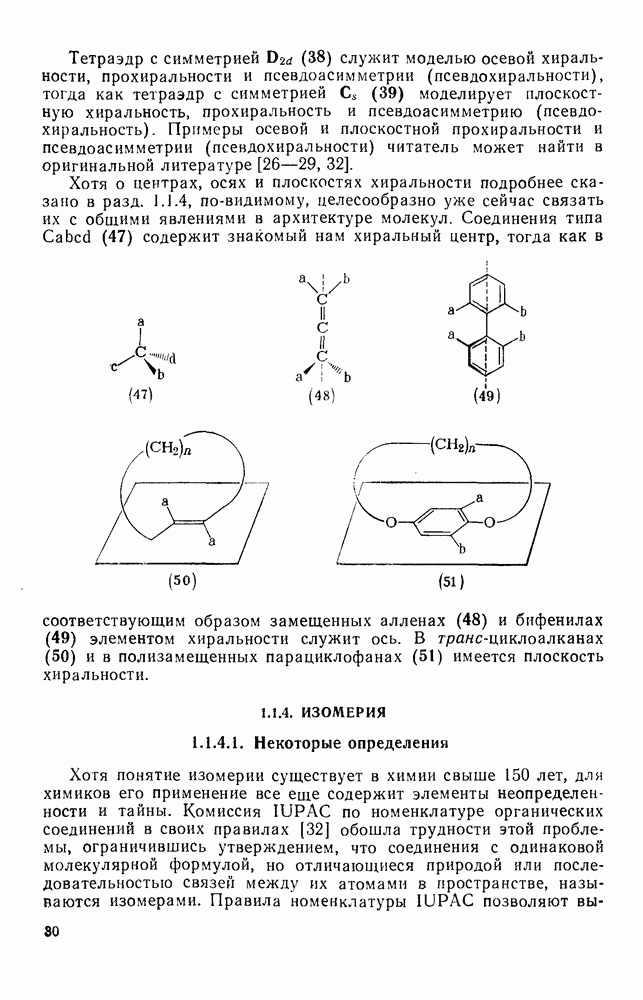
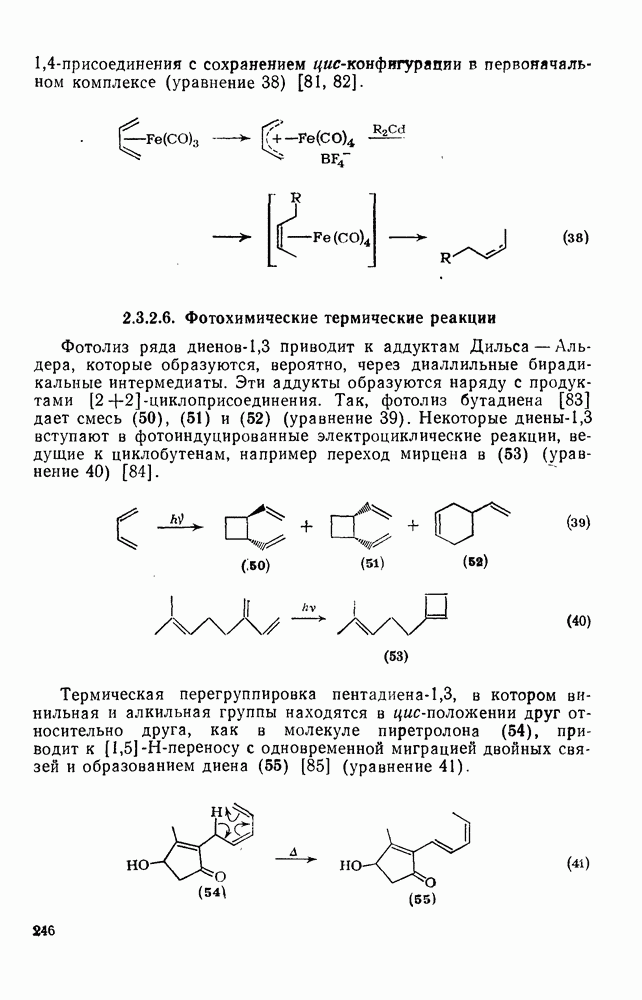
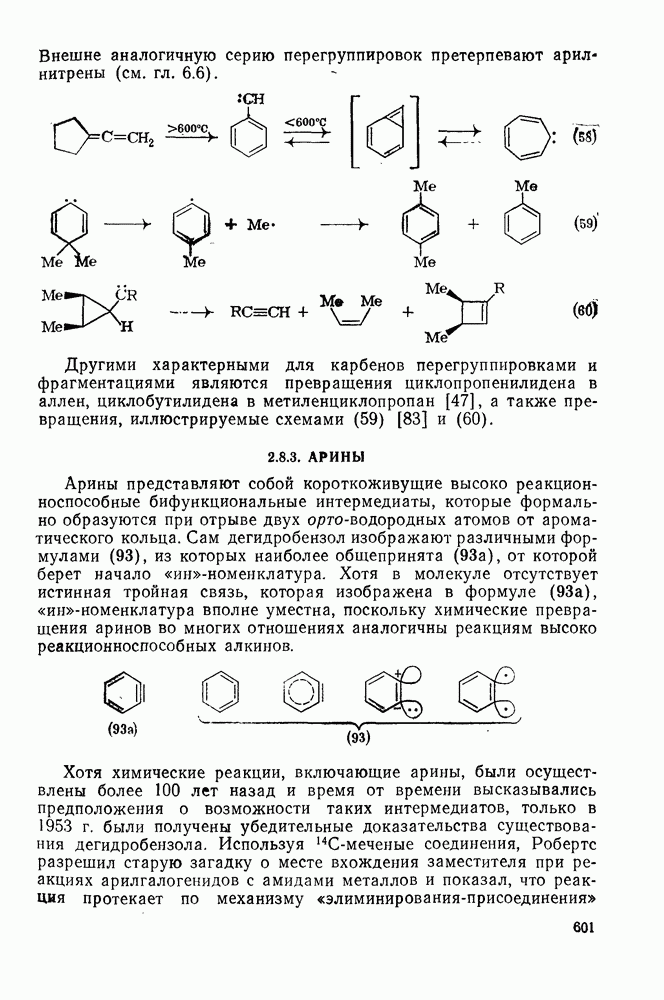
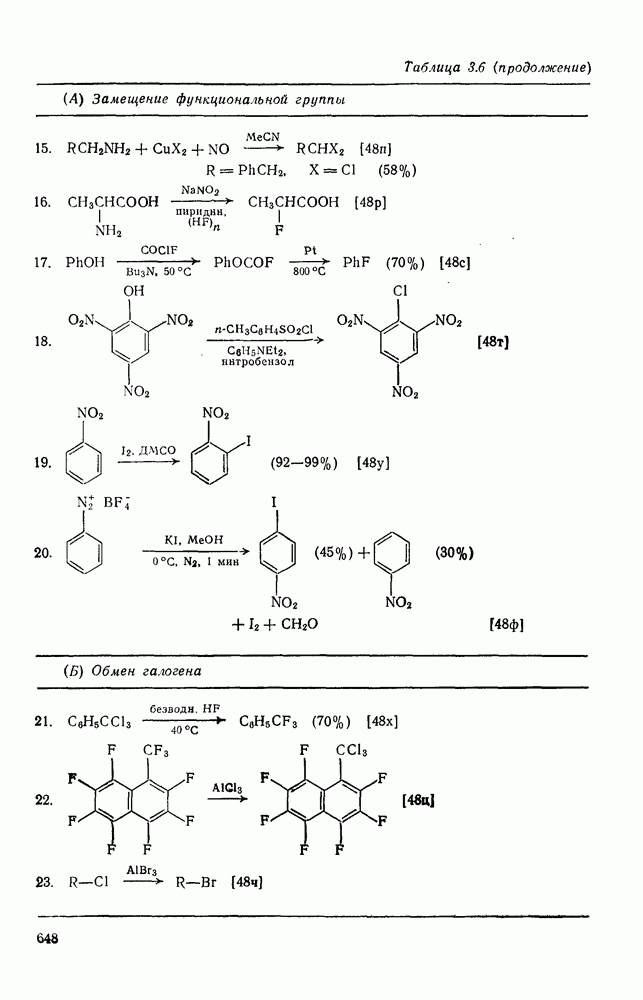
Для синглетных карбенов свойственны  -сдвиги [80—82]. Очень легко мигрирует водород [см. уравнение
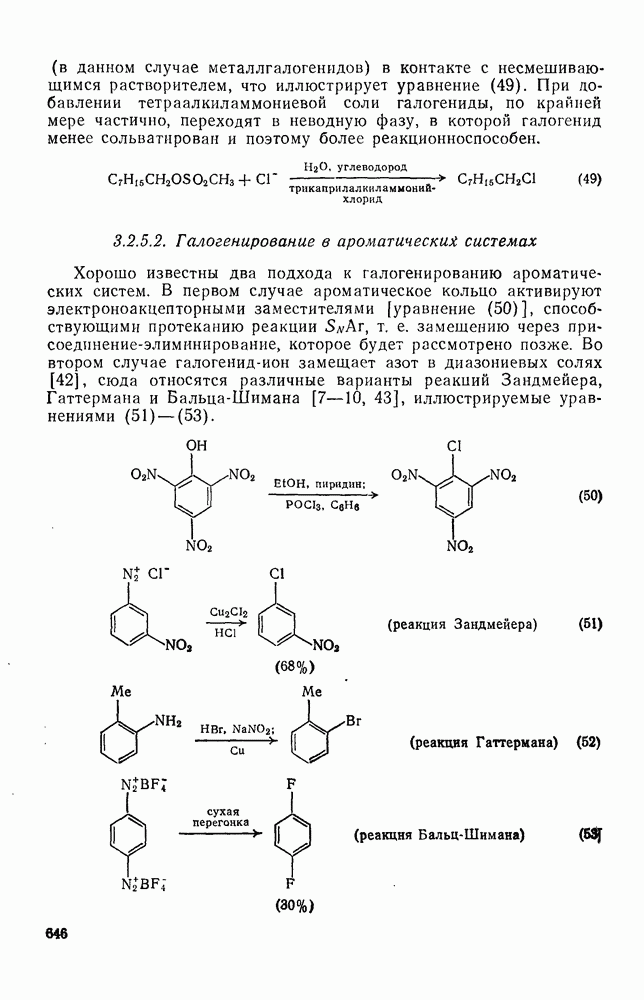
-сдвиги [80—82]. Очень легко мигрирует водород [см. уравнение  ], но возможны также миграции алкильных и арильных групп, например в (90) [уравнение
], но возможны также миграции алкильных и арильных групп, например в (90) [уравнение  ], а также RS и F. Эта реакция используется для синтеза алкенов против правила Бредта [83].
], а также RS и F. Эта реакция используется для синтеза алкенов против правила Бредта [83].

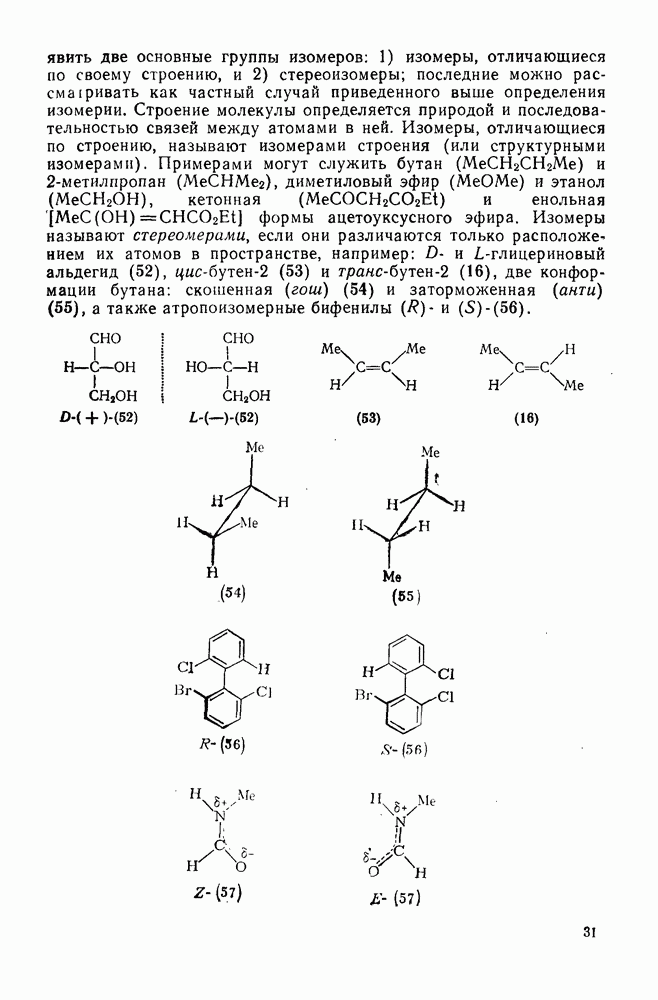
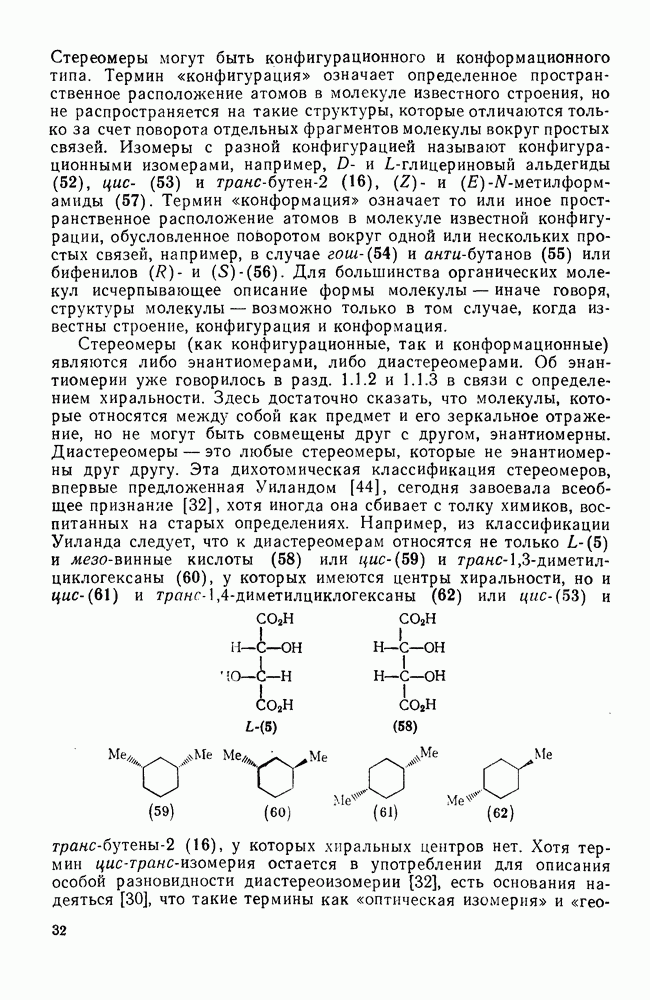
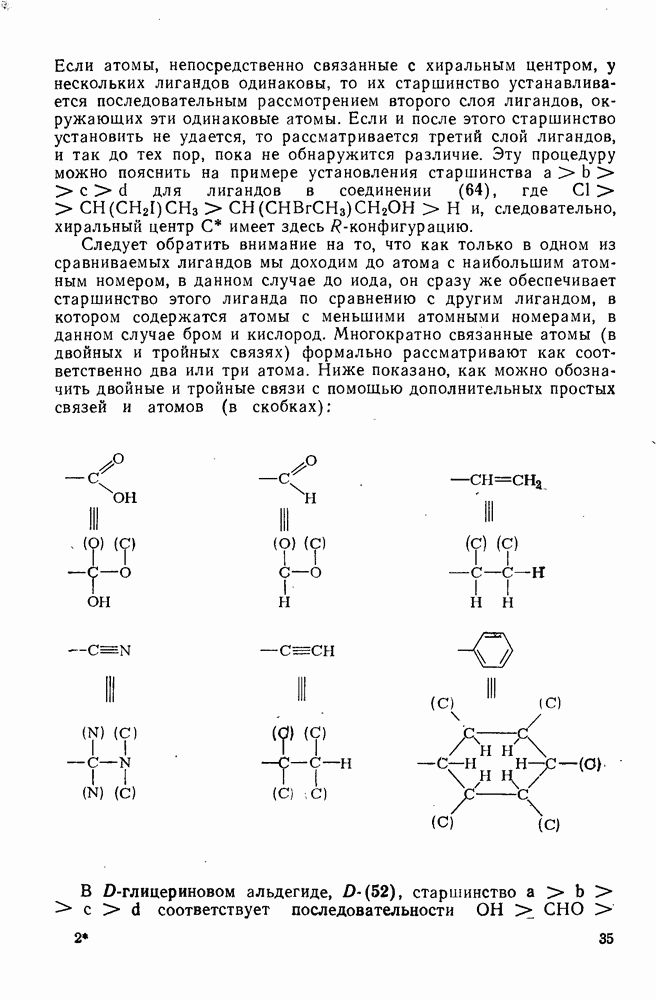
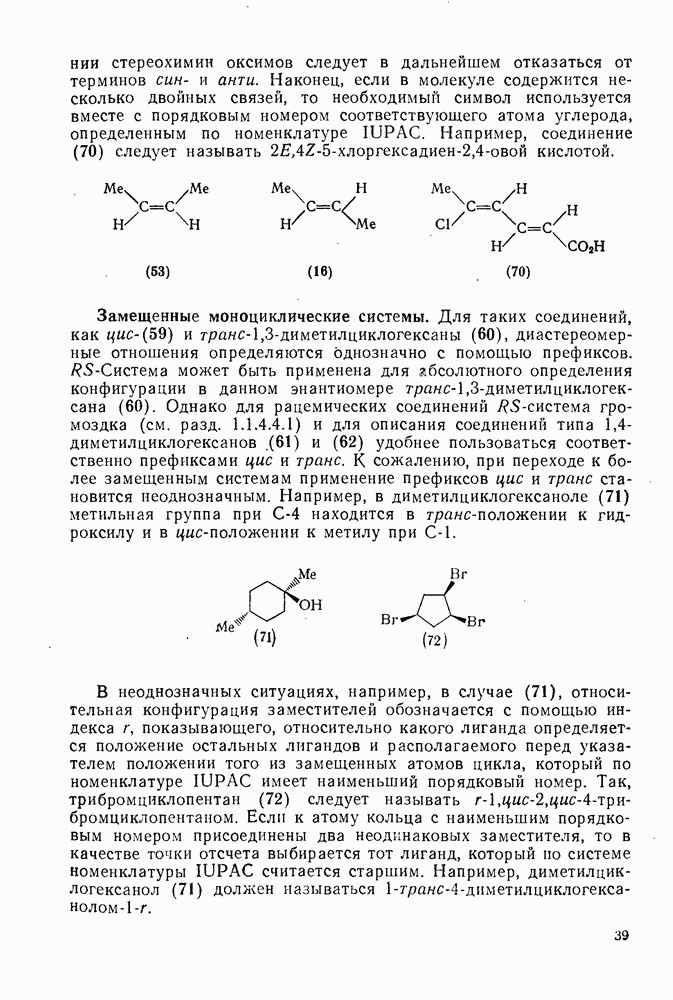
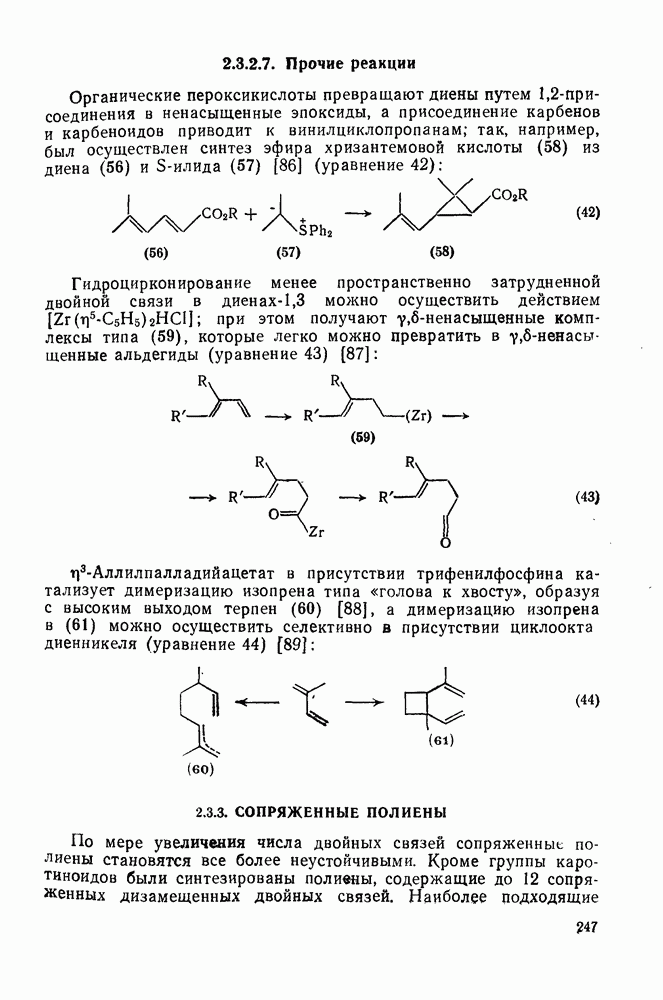
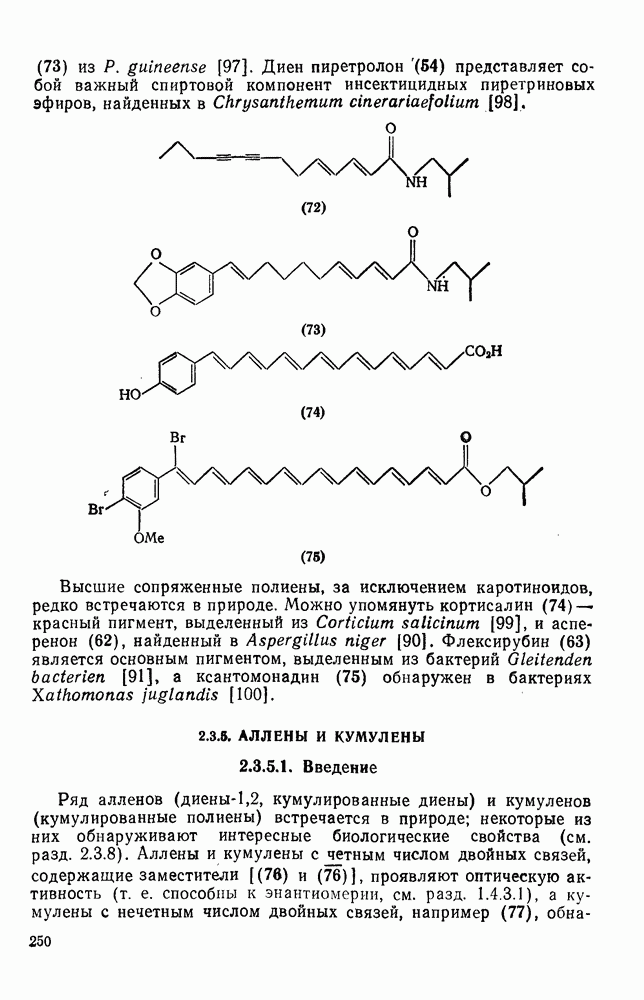
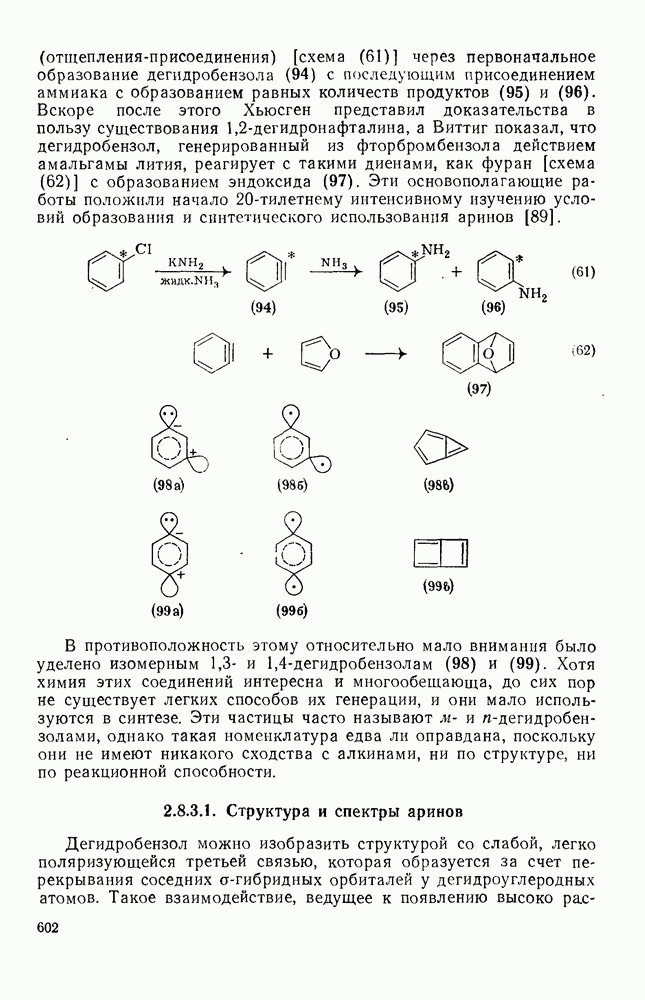
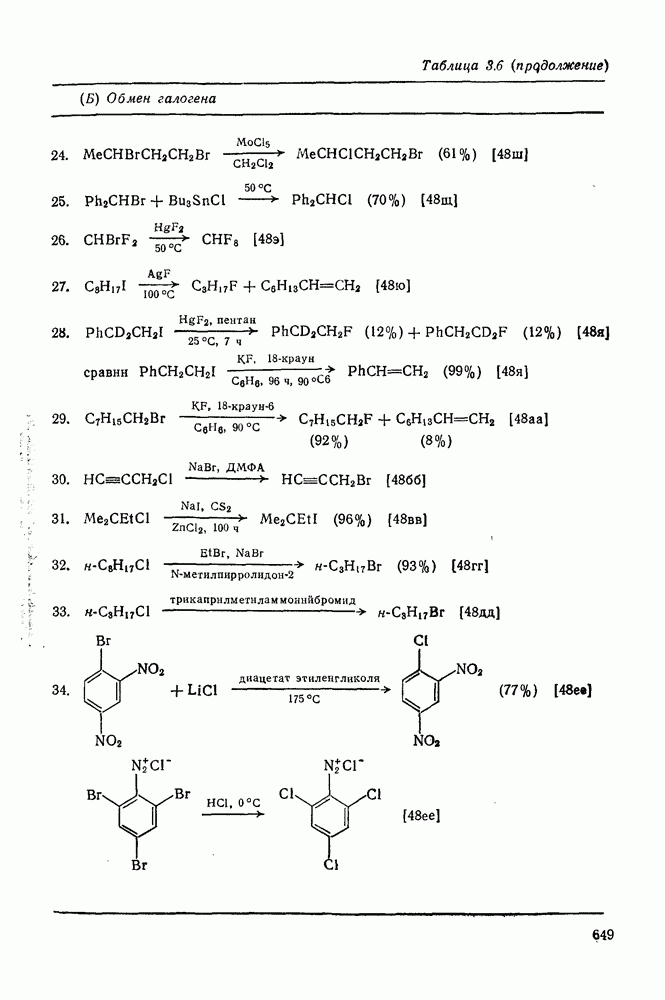
В подходящих условиях карбены, имеющие  -ненасыщенные заместители, например (
-ненасыщенные заместители, например ( ), подвергаются карбен-карбеновым перегруппировкам через (92) [схема (57)]. При X=0 такое взаимное превращение предшествует перегруппировке Вольфа для фотолитически генерированных кетокарбенов [84]. К этой же группе относятся арилкарбены и, хотя обычно они не перегруппировываются в растворе, в условиях импульсного вакуумного пиролиза при температуре ниже
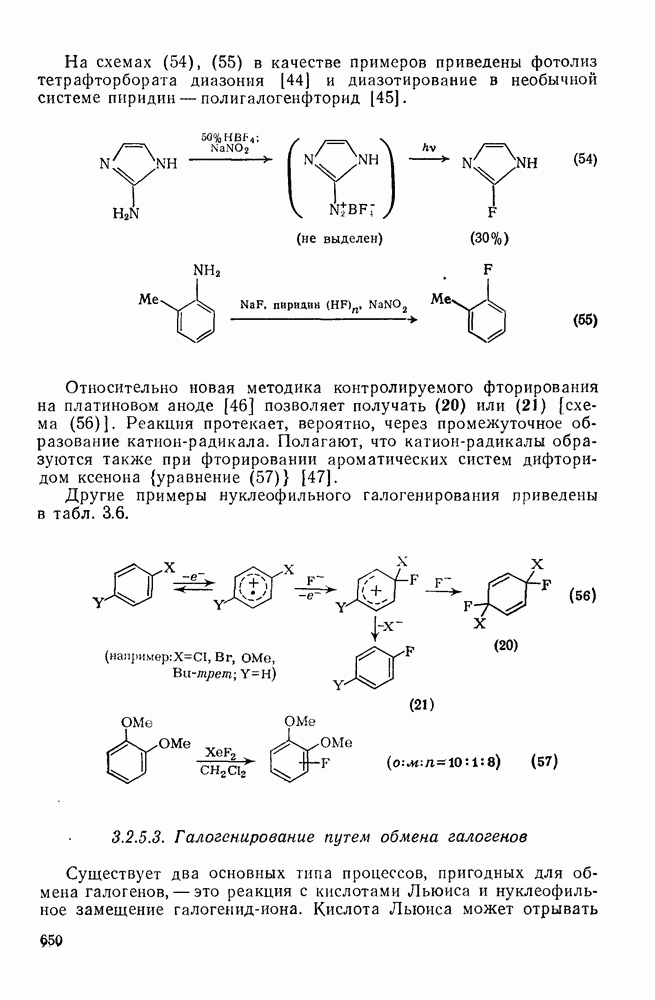
), подвергаются карбен-карбеновым перегруппировкам через (92) [схема (57)]. При X=0 такое взаимное превращение предшествует перегруппировке Вольфа для фотолитически генерированных кетокарбенов [84]. К этой же группе относятся арилкарбены и, хотя обычно они не перегруппировываются в растворе, в условиях импульсного вакуумного пиролиза при температуре ниже  устанавливается их равновесие с ароматическими карбенами [схема
устанавливается их равновесие с ароматическими карбенами [схема  ] при более высоких температурах протекает также сужение цикла с образованием, например, винилиденциклопенгадиена [83, 85—88].
] при более высоких температурах протекает также сужение цикла с образованием, например, винилиденциклопенгадиена [83, 85—88].
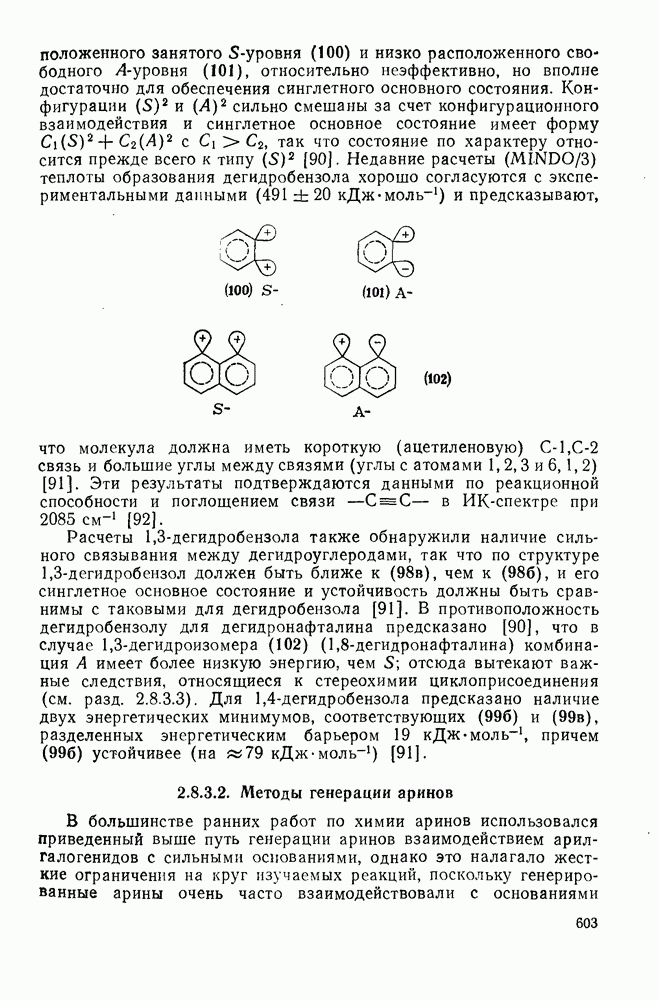
Внешне аналогичную серию перегруппировок претерпевают арил-нитрены (см. гл. 6.6).

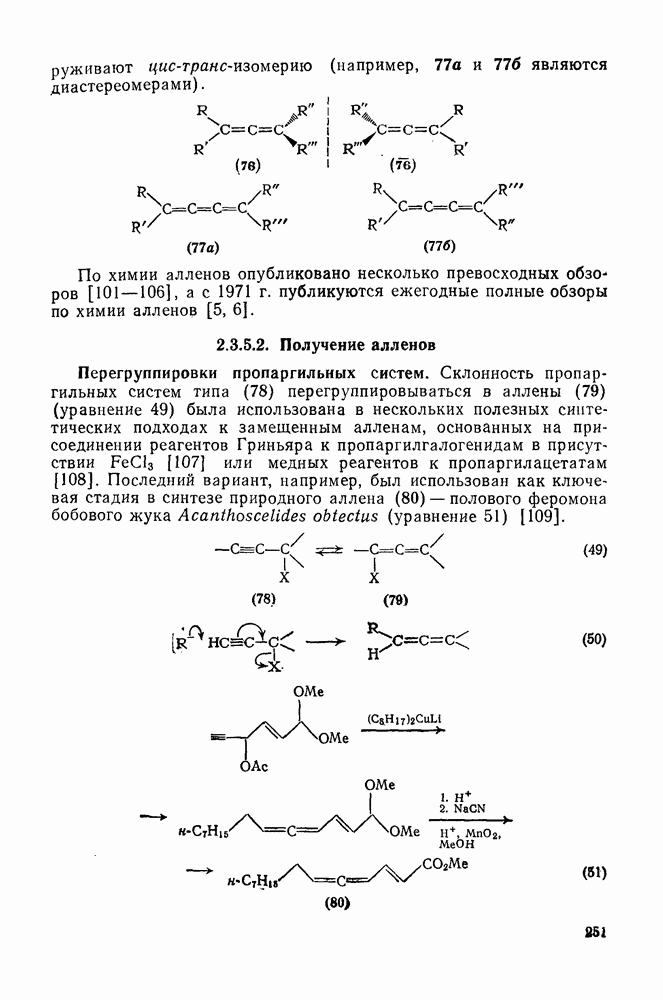
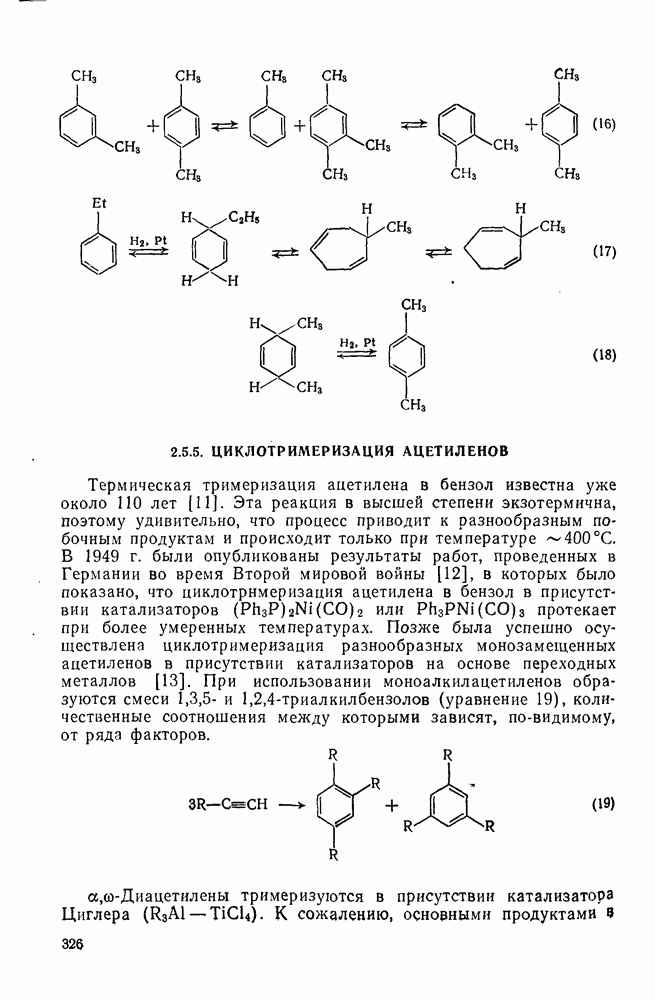
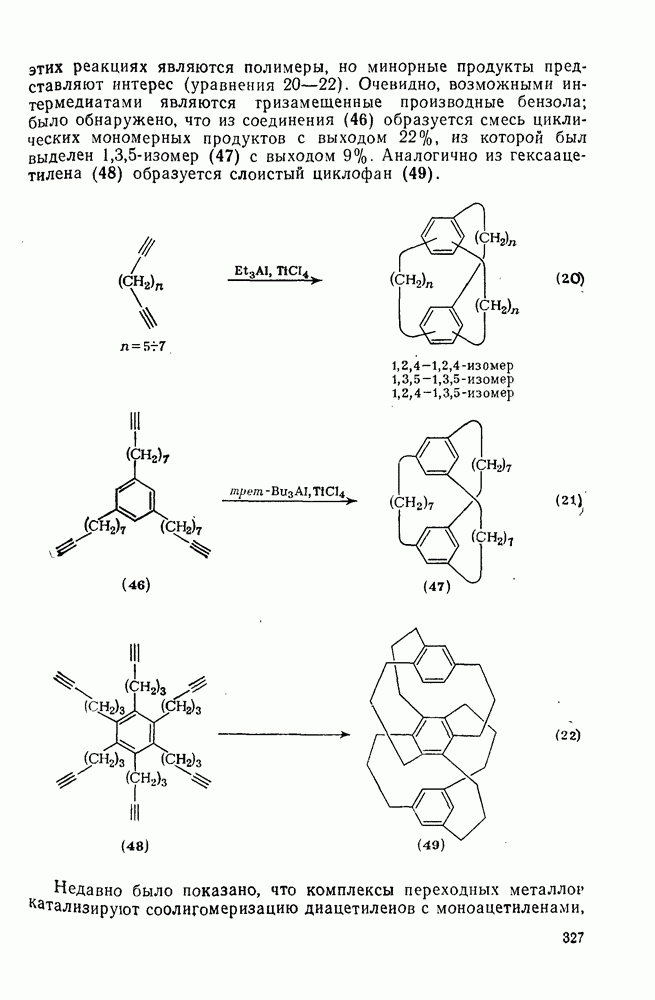
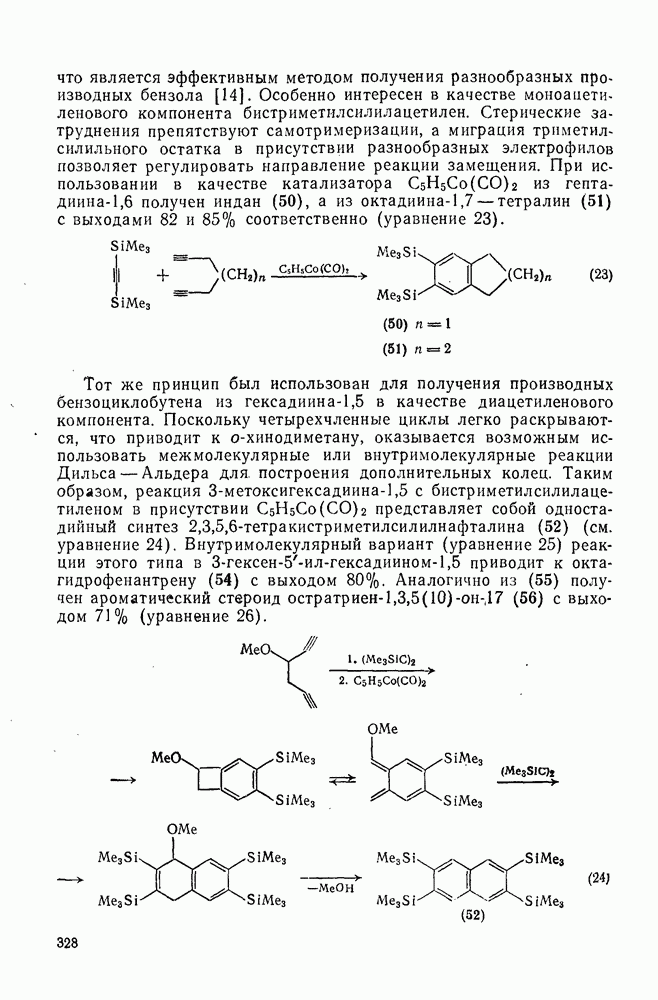
Другими характерными для карбенов перегруппировками и фрагментациями являются превращения циклопропенилидена в аллен, циклобутилидена в метиленциклопропан [47], а также превращения, иллюстрируемые схемами (59) [83] и (60).