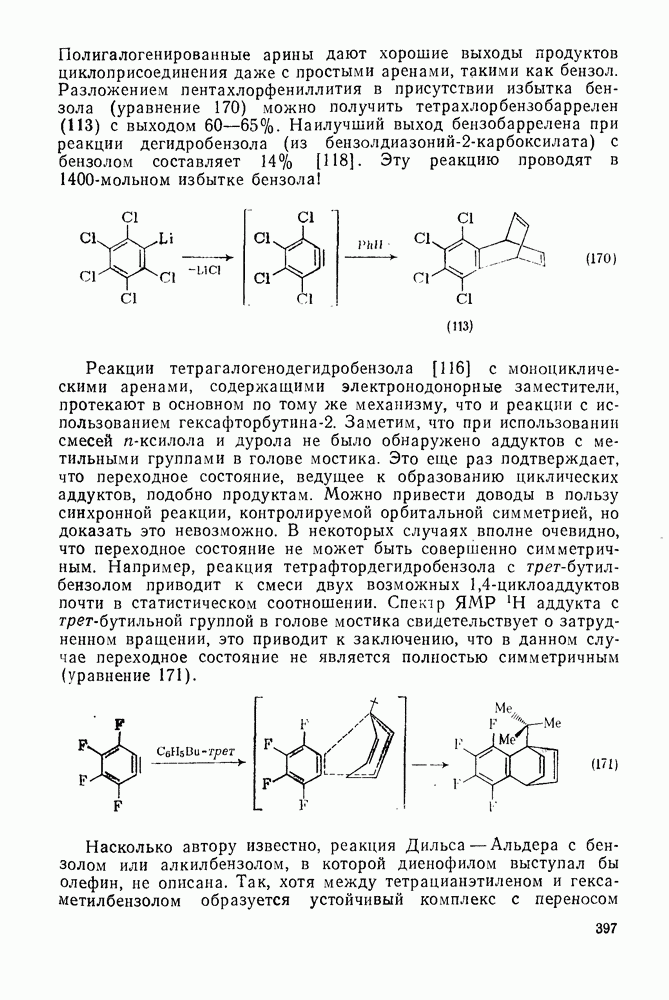
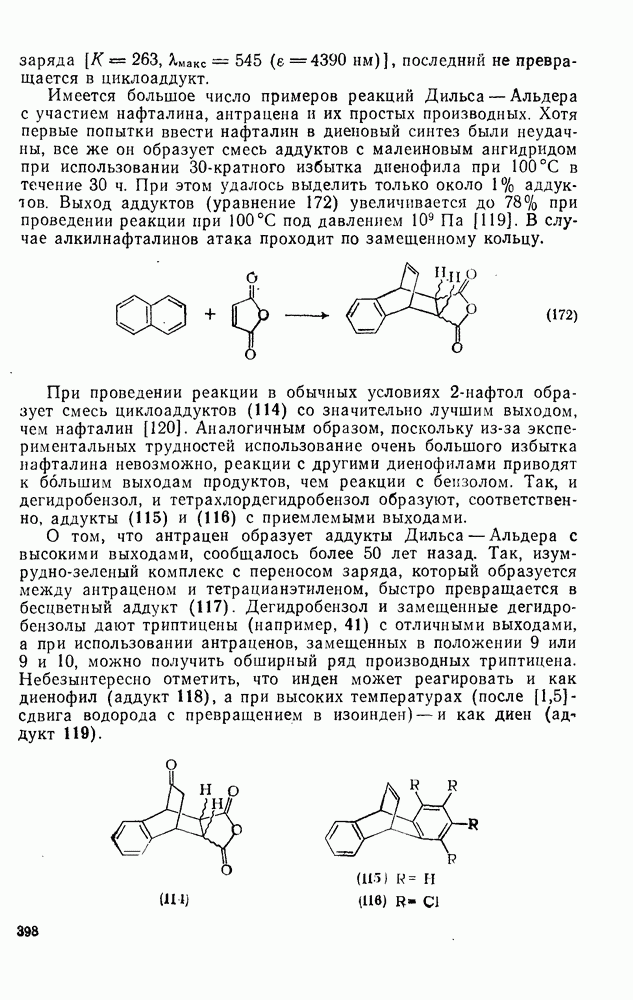
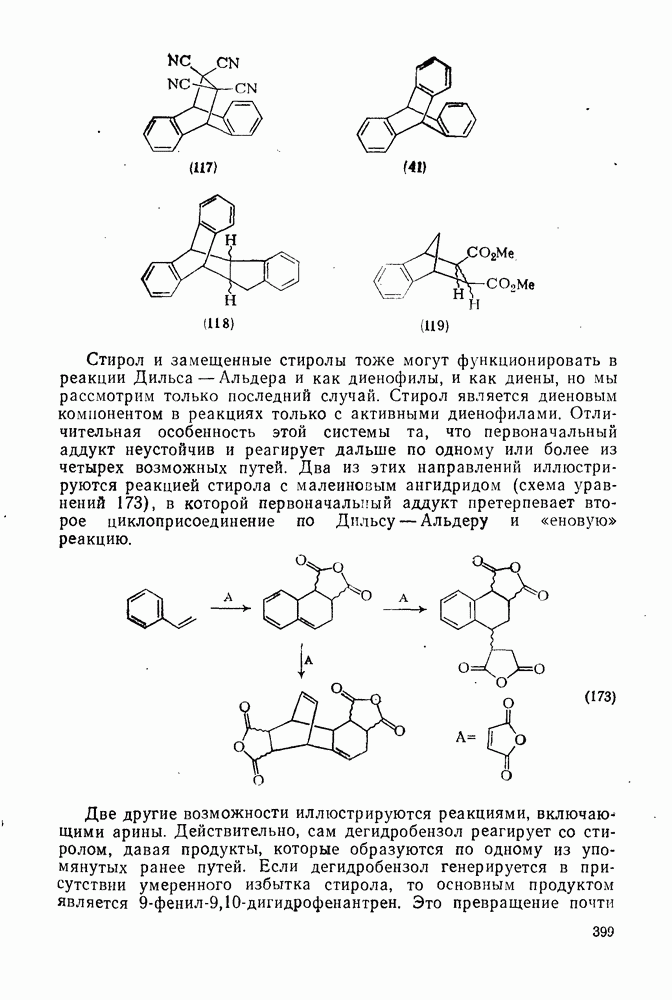
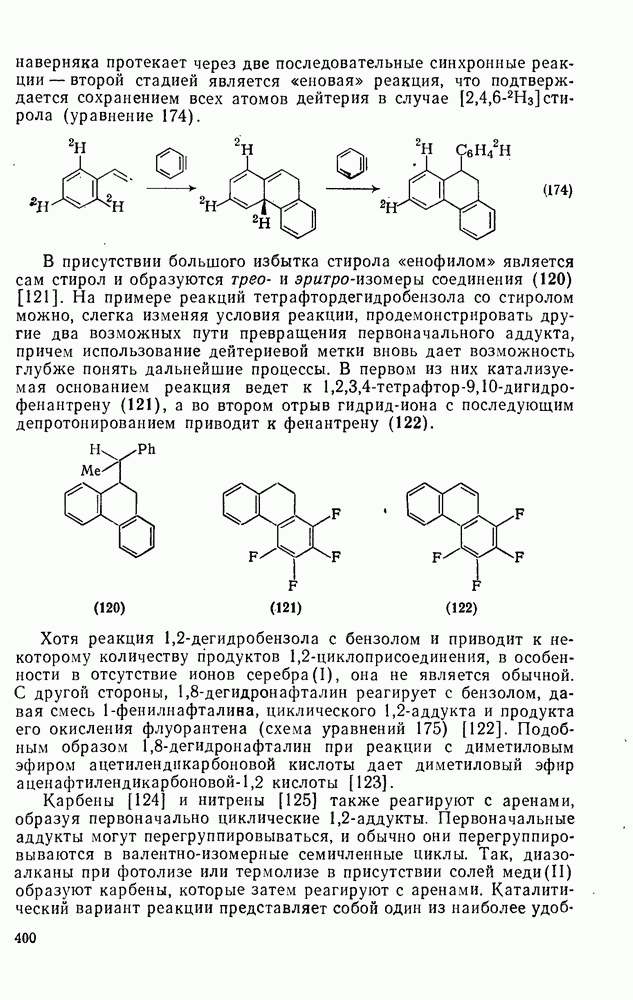
Пред.
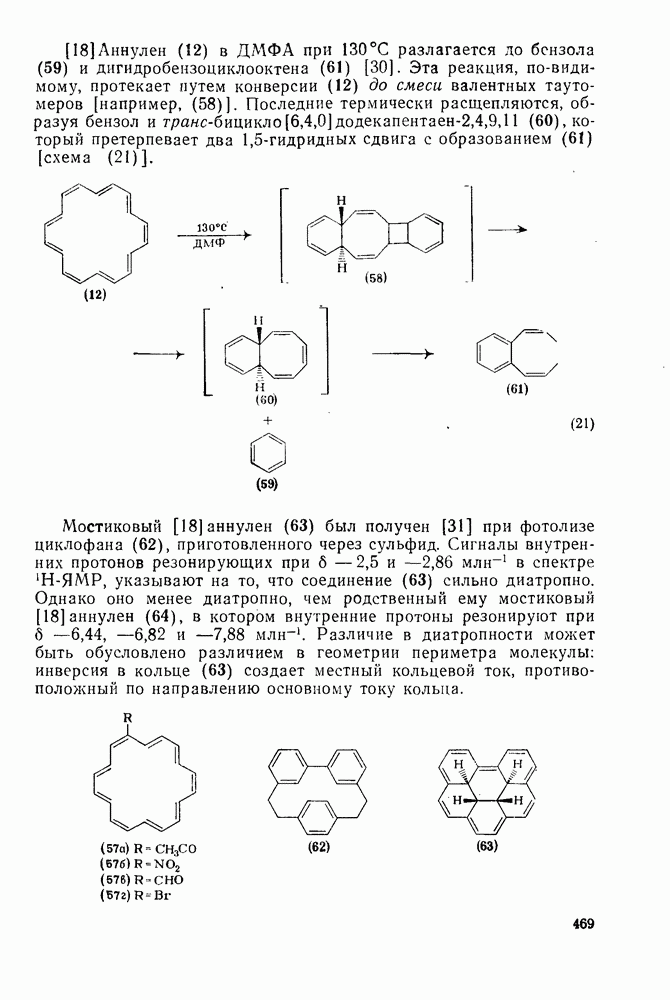
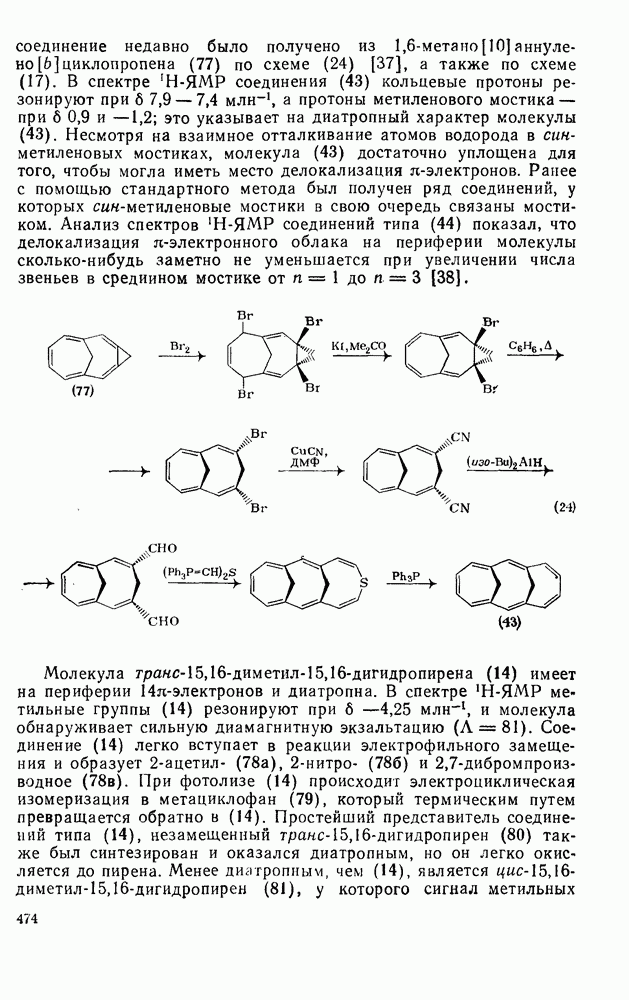
След.
Макеты страниц
Распознанный текст, спецсимволы и формулы могут содержать ошибки, поэтому с корректным вариантом рекомендуем ознакомиться на отсканированных изображениях учебника выше Также, советуем воспользоваться поиском по сайту, мы уверены, что вы сможете найти больше информации по нужной Вам тематике ДЛЯ СТУДЕНТОВ И ШКОЛЬНИКОВ ЕСТЬ
ZADANIA.TO
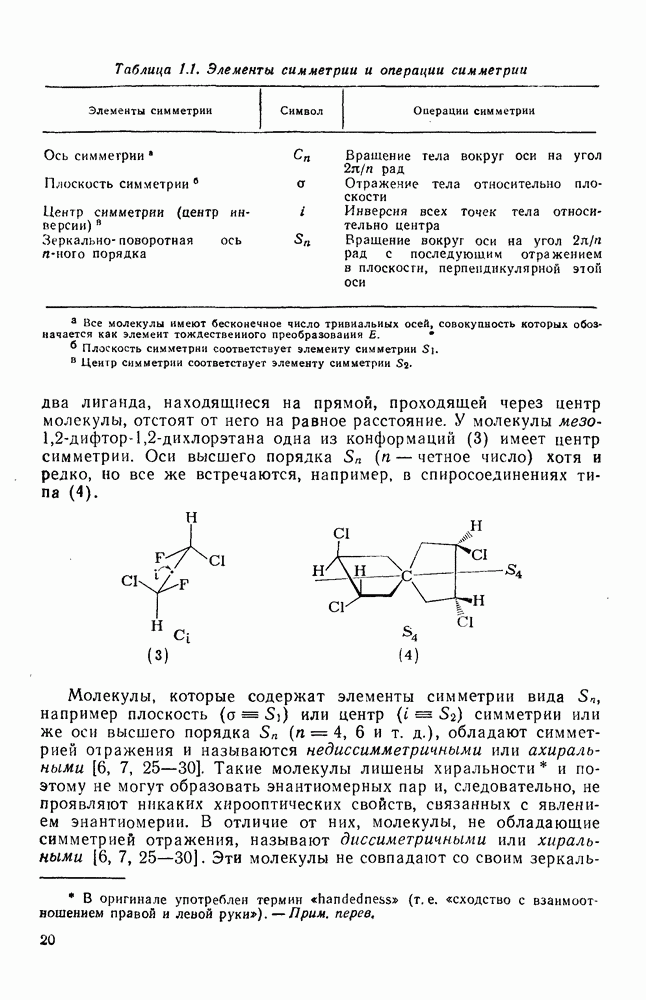
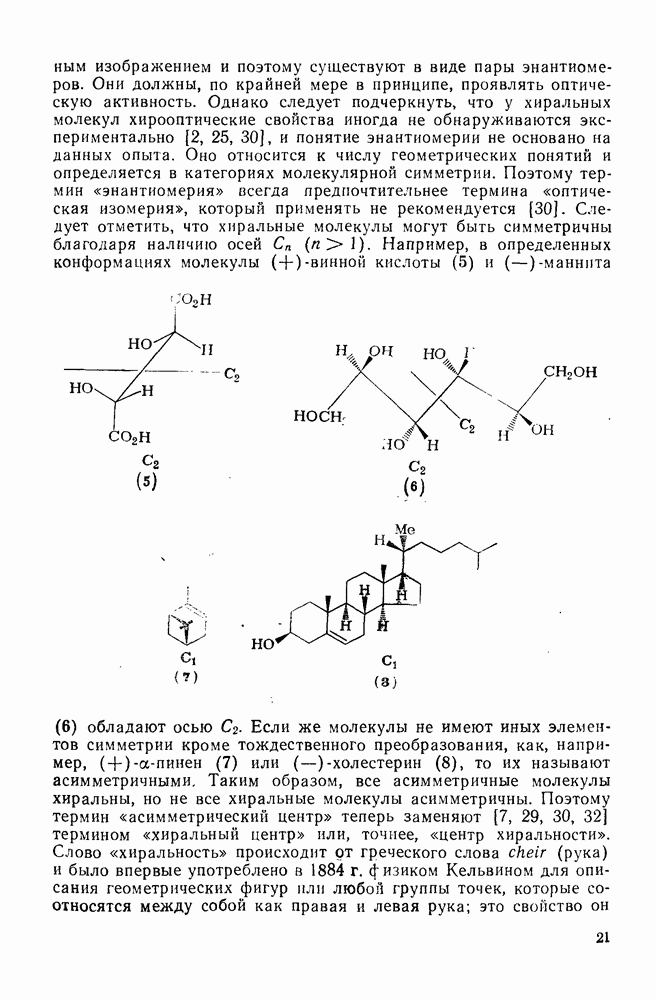
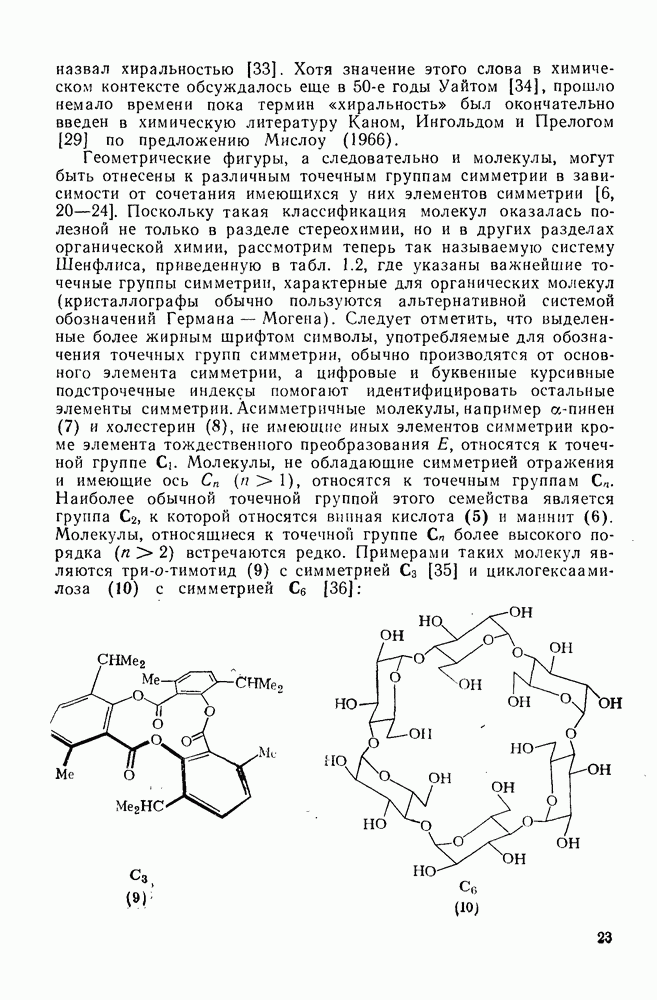
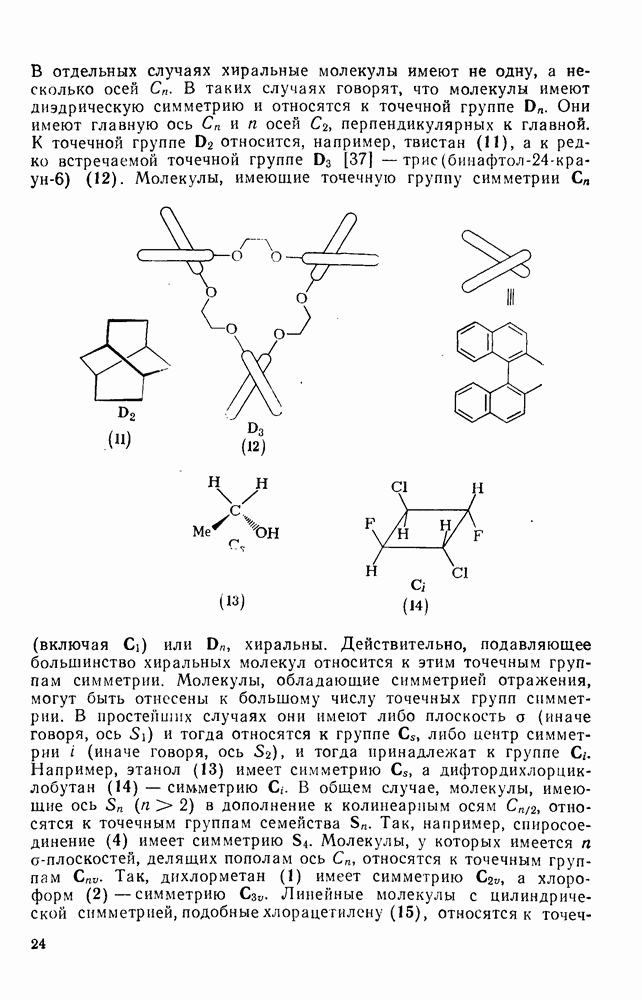
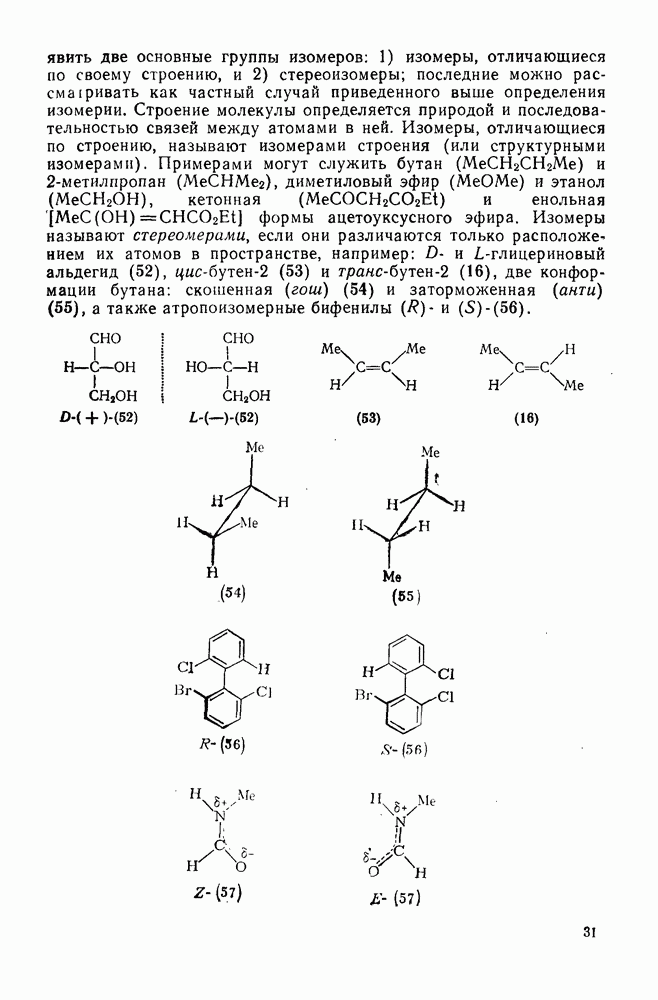
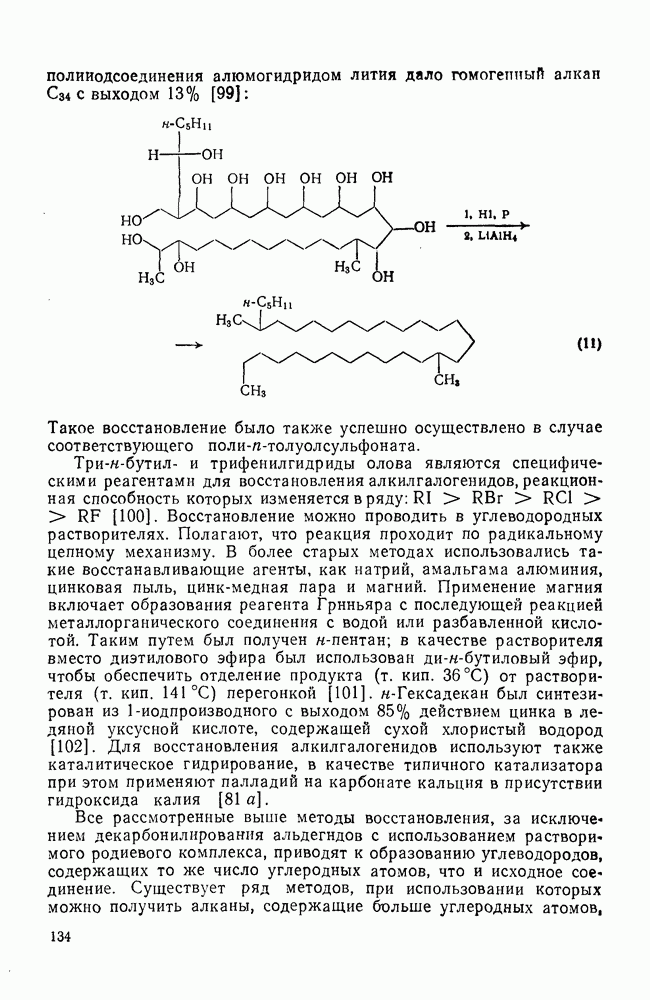
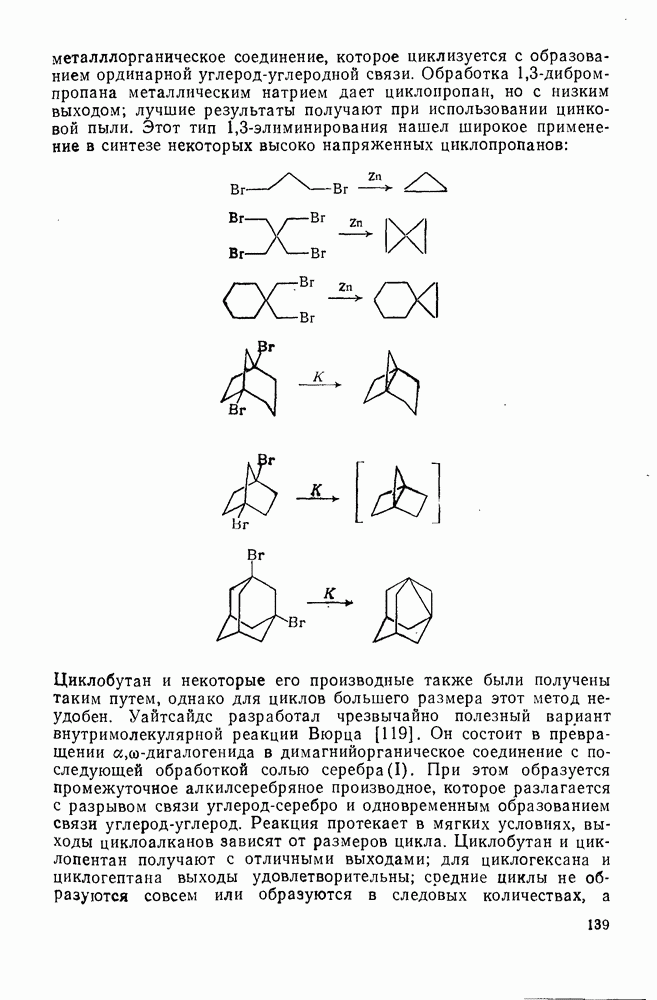
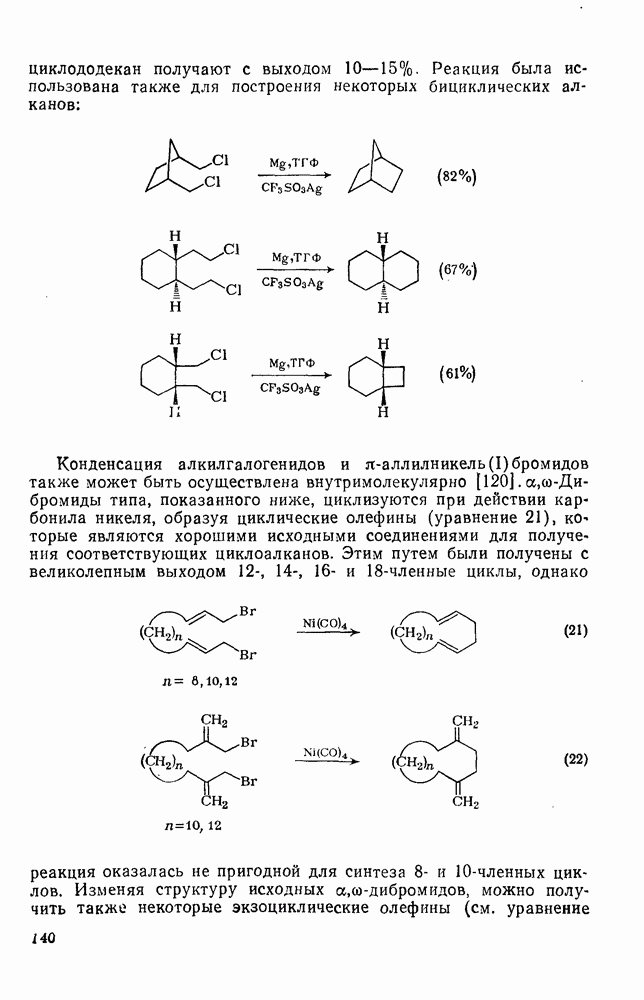
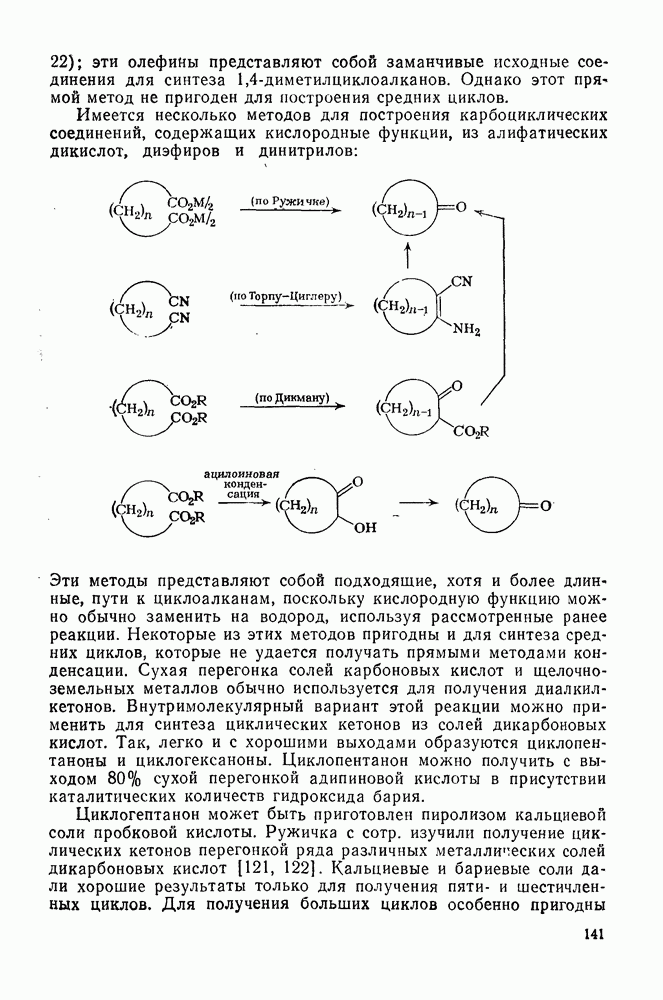
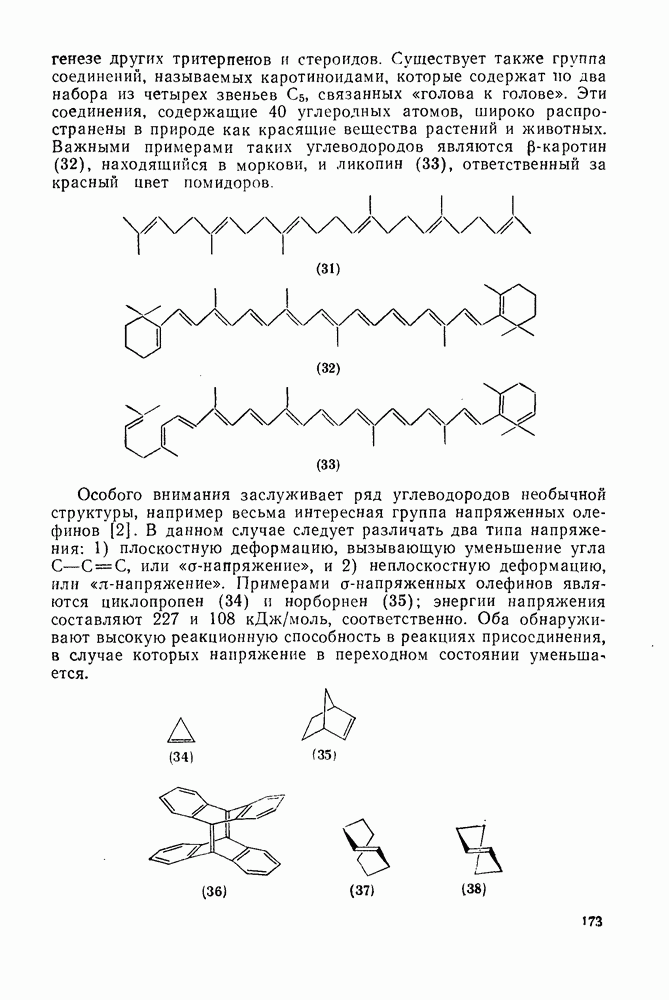
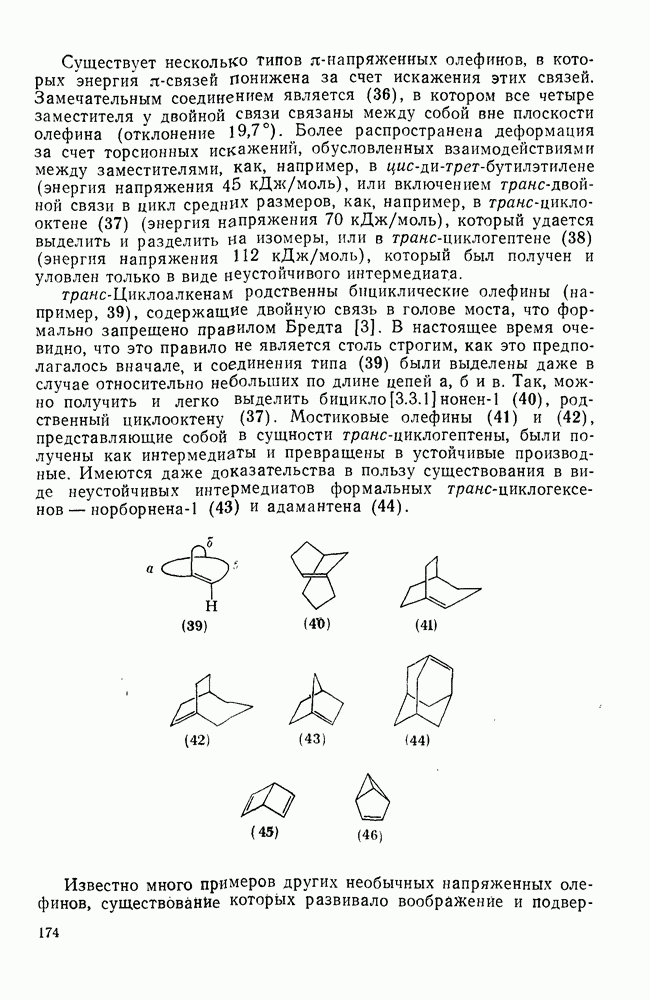
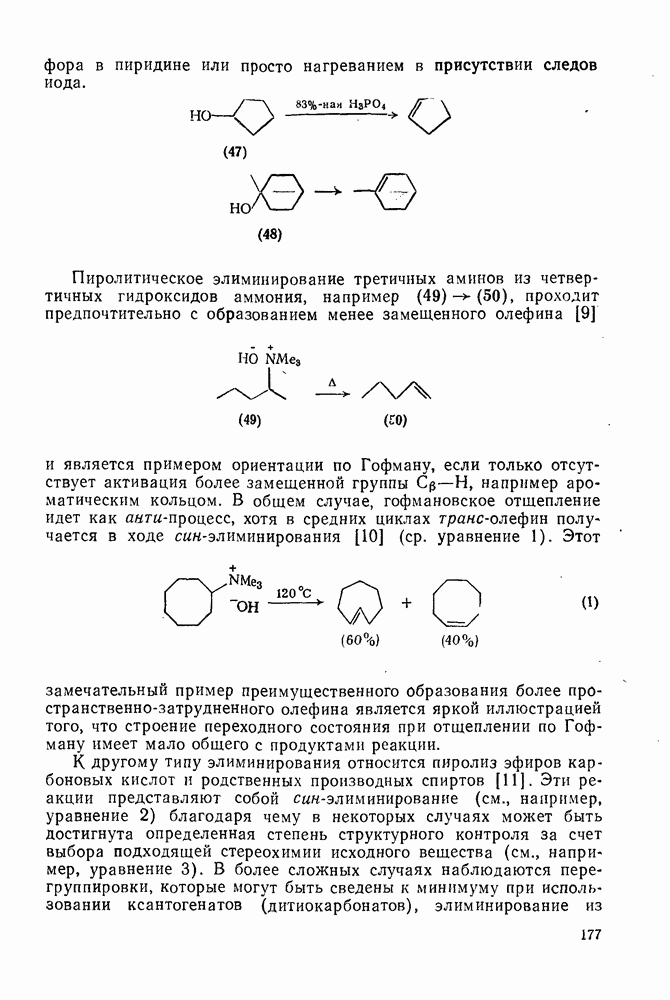
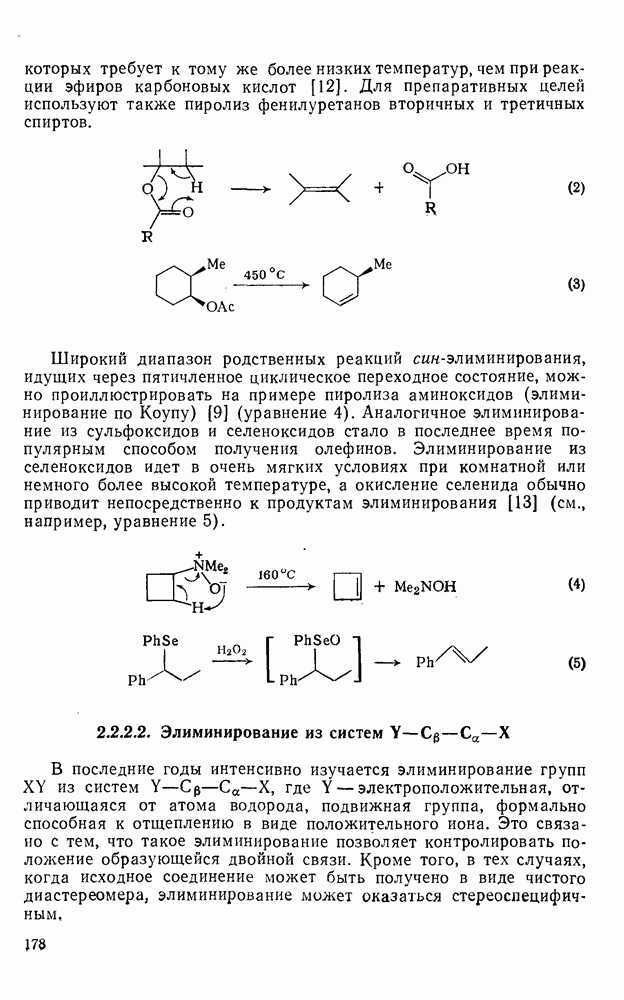
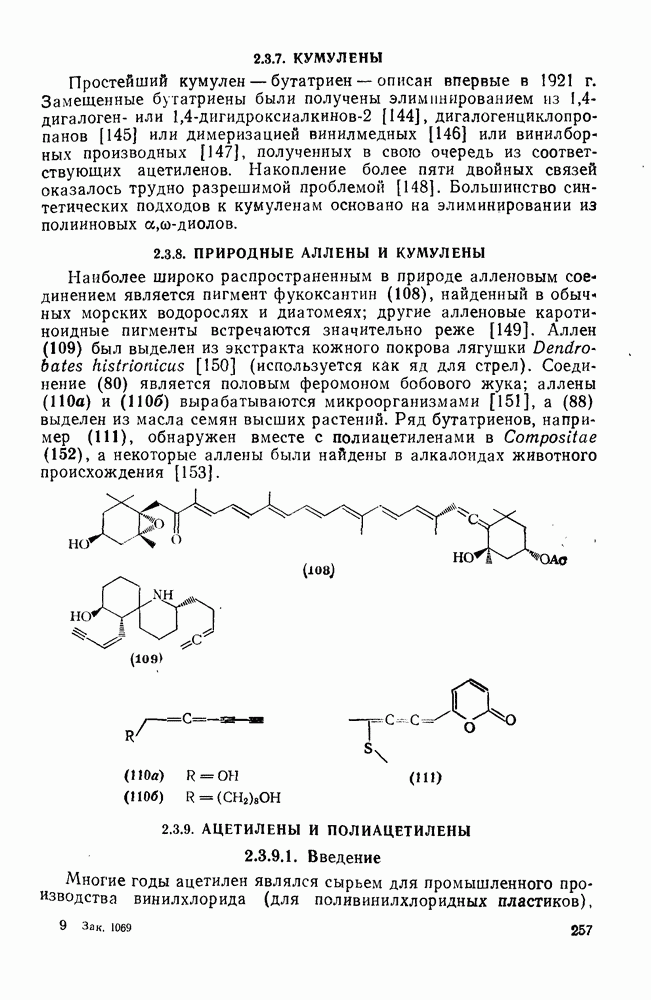
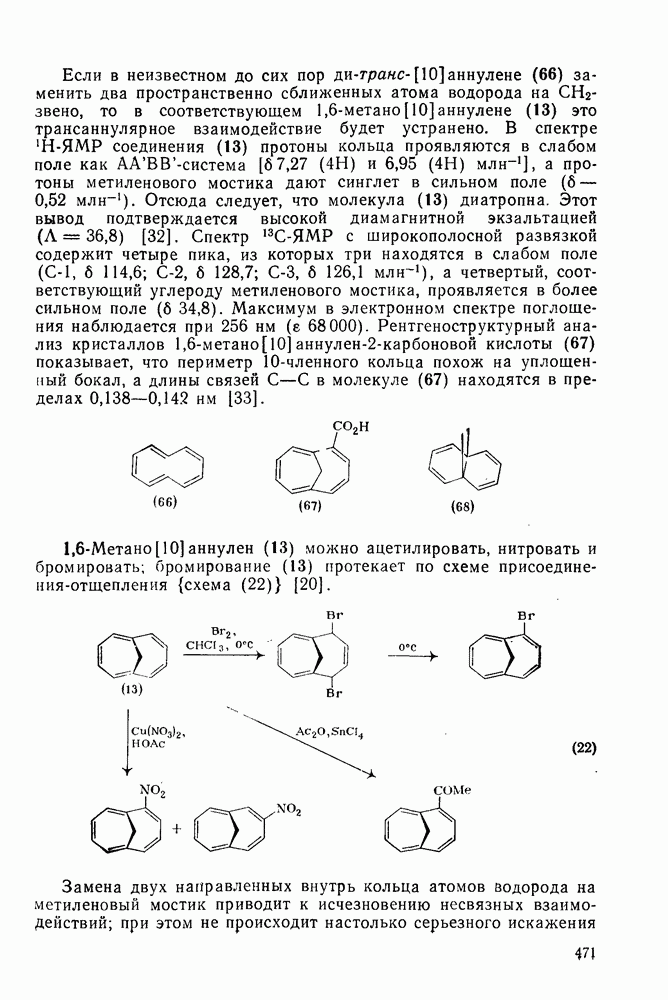
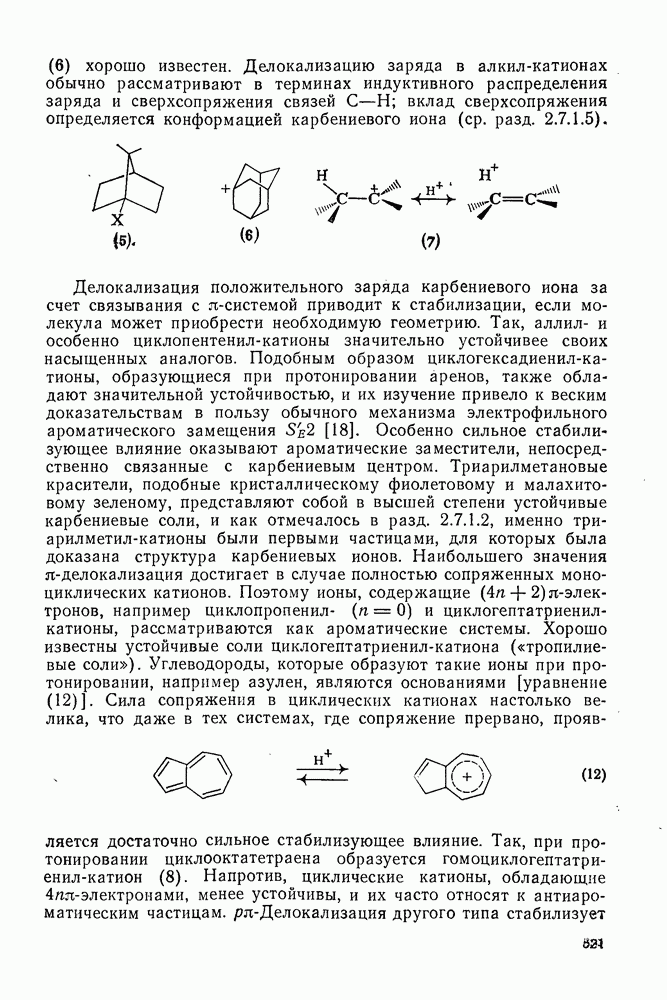
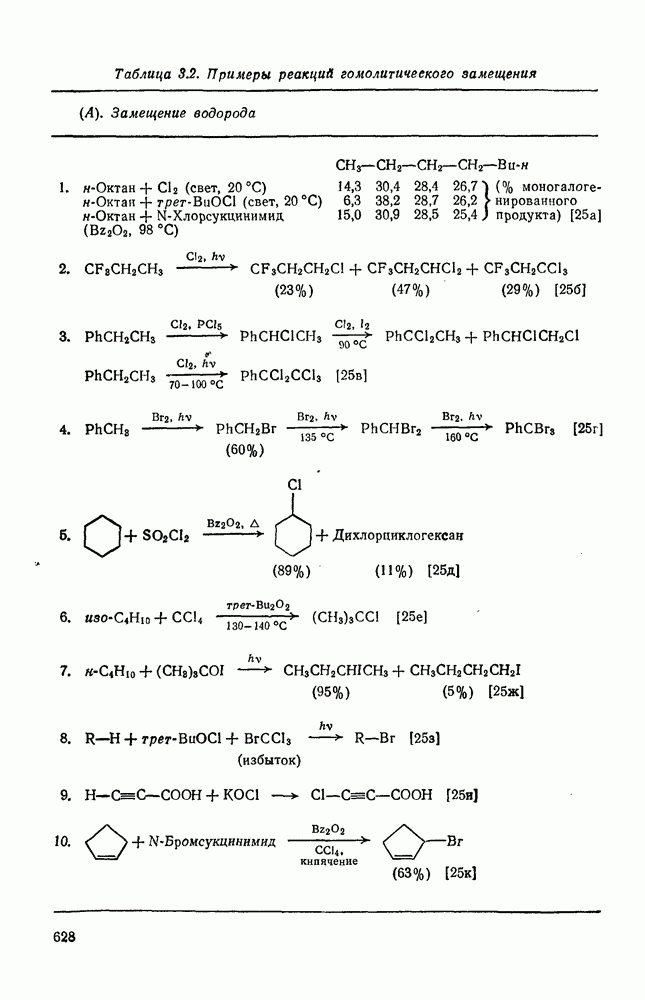
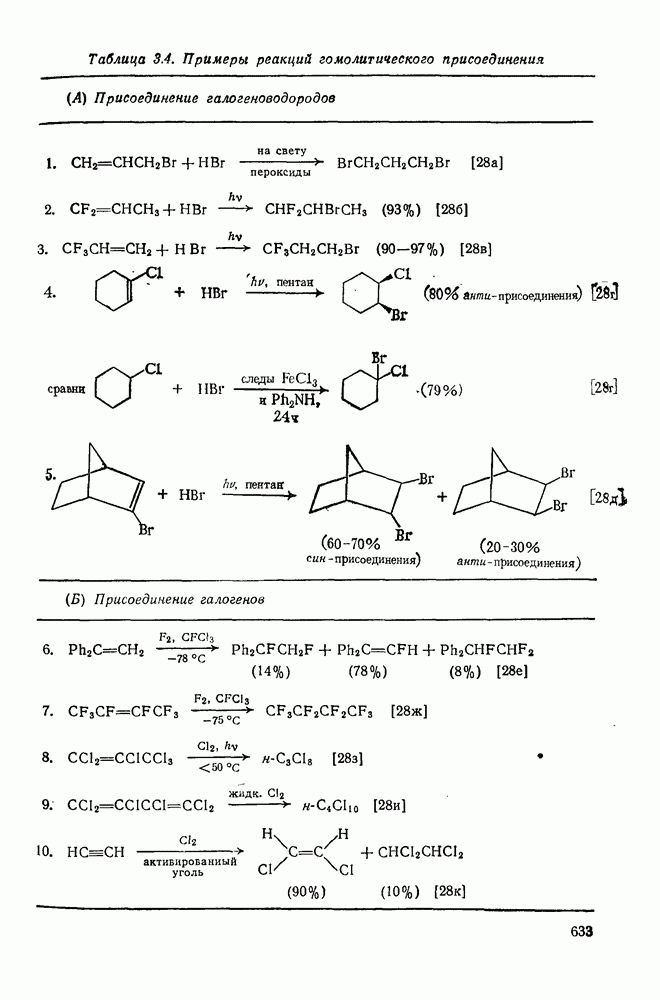
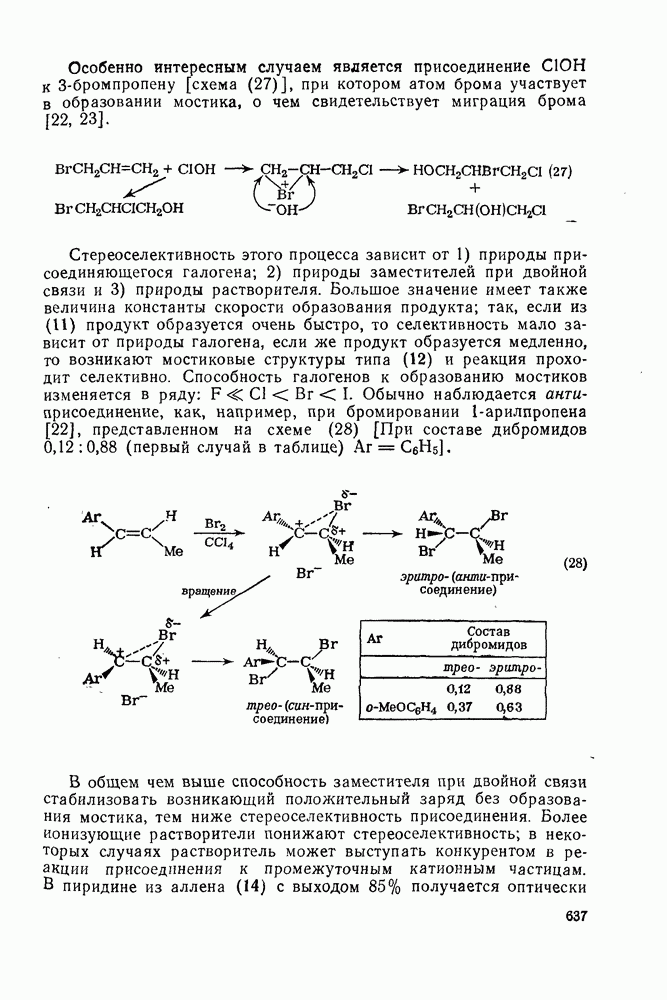
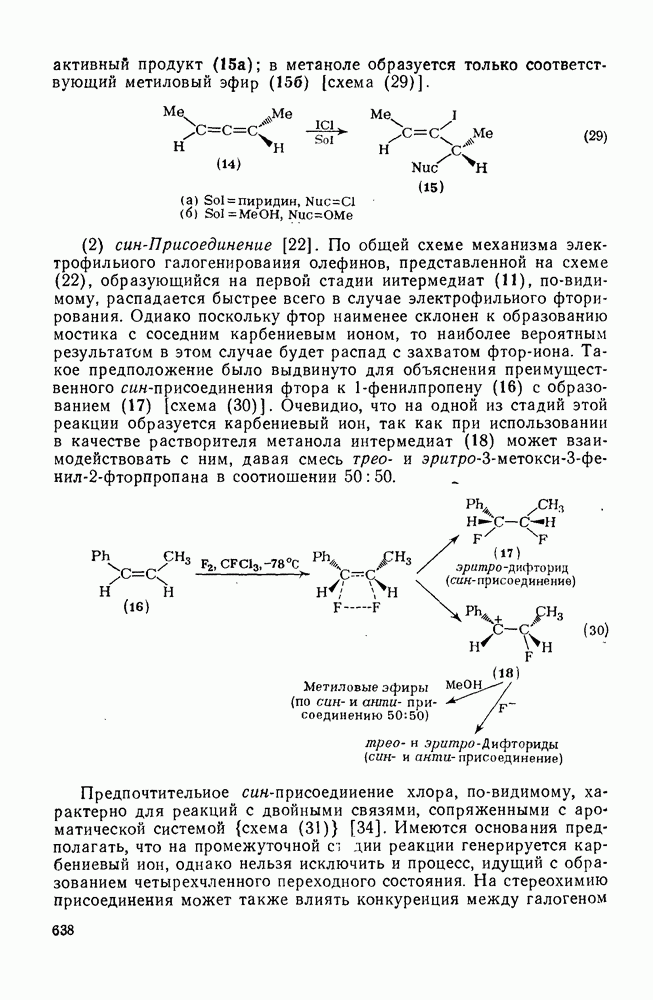
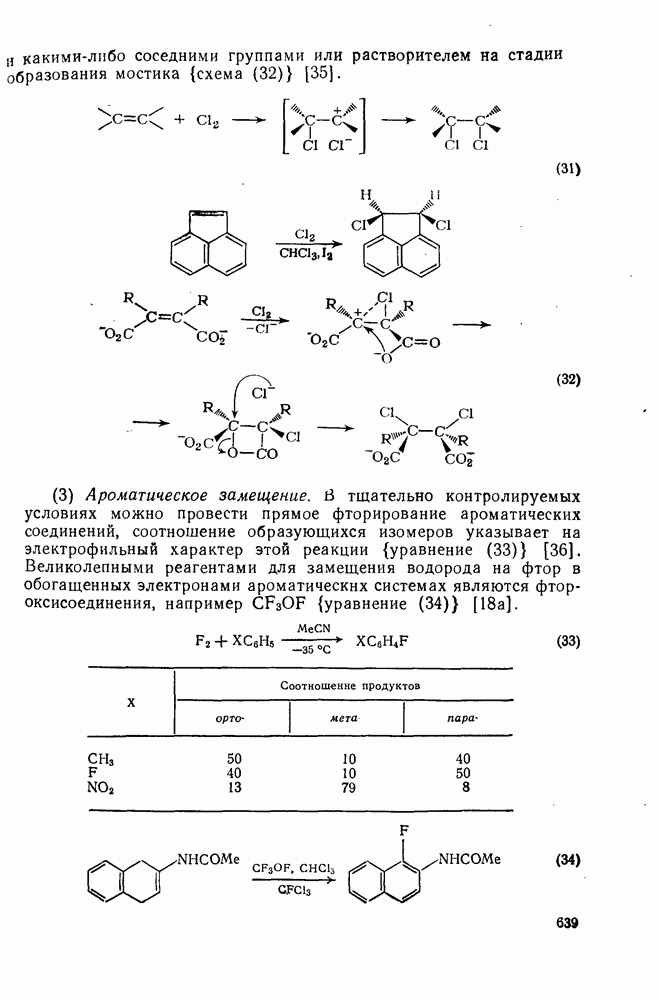
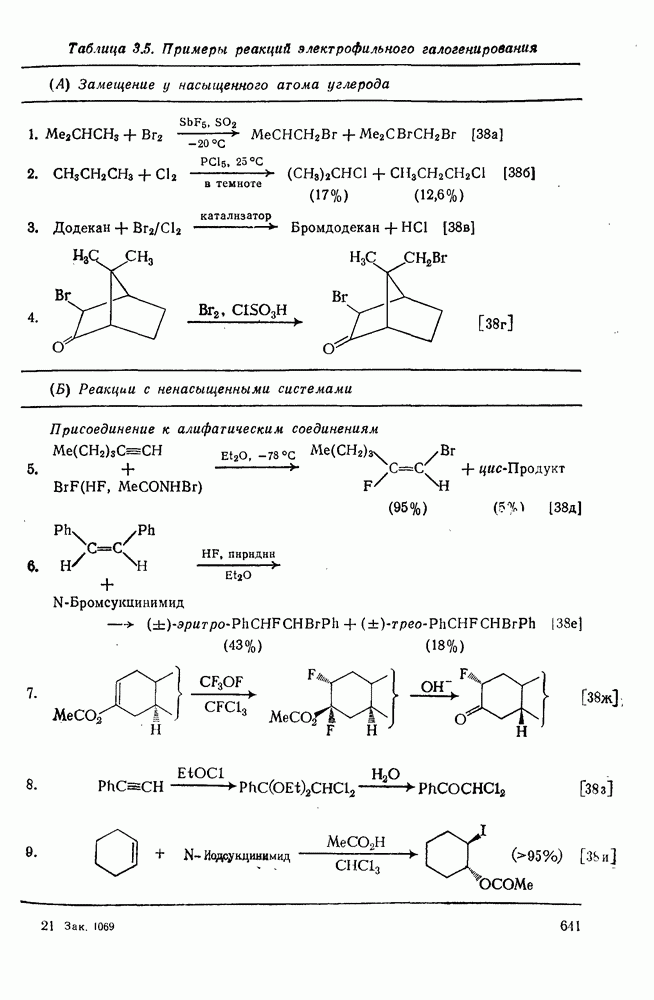
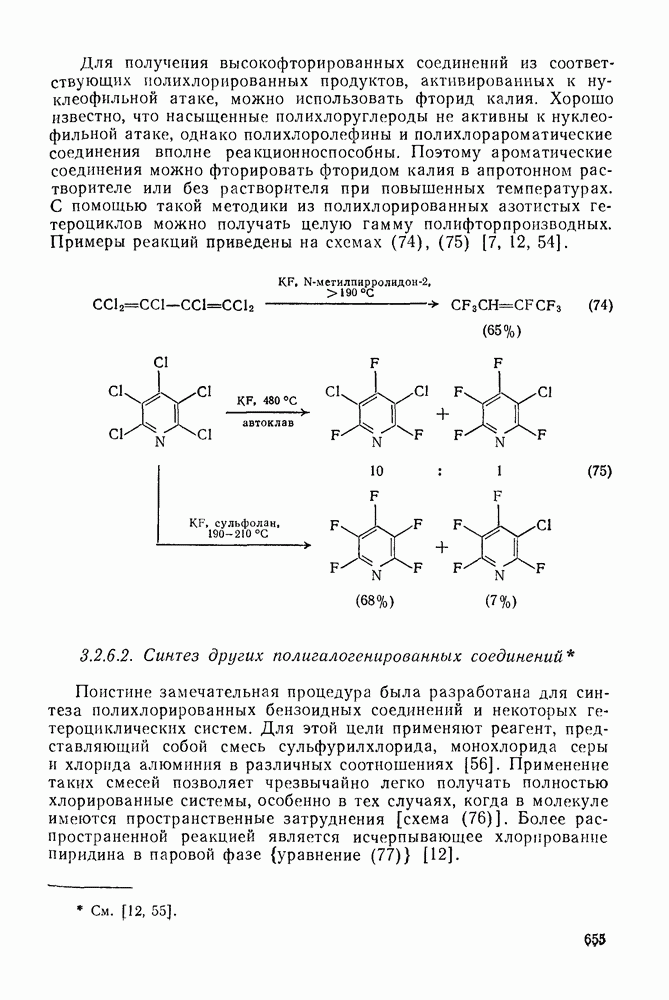
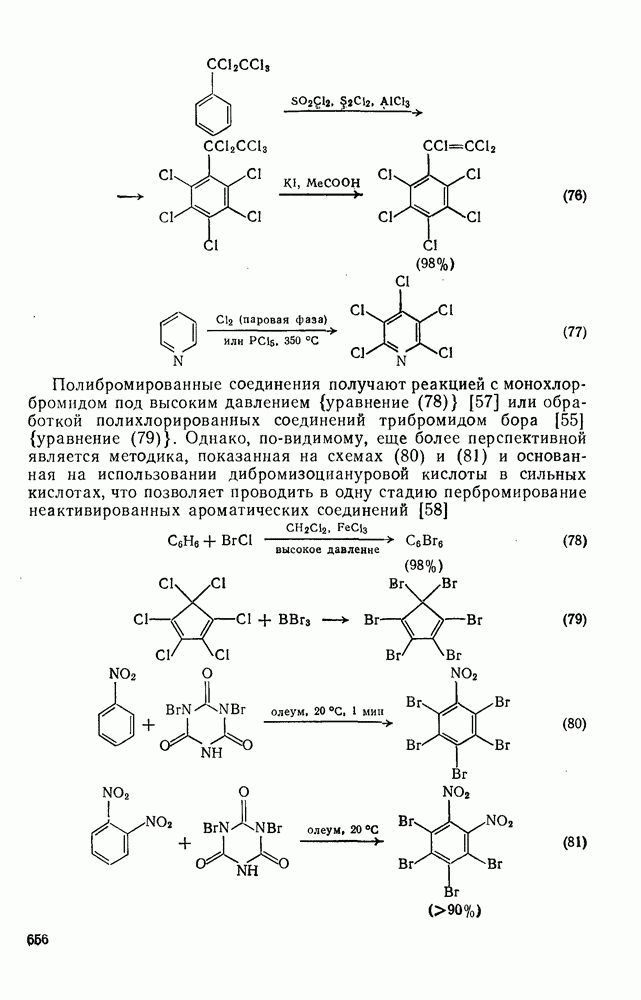
2.2.3. РЕАКЦИИ ОЛЕФИНОВВажное значение олефинов и большое разнообразие их реакций способствовали широкому изучению химии олефинов. Для того чтобы иметь возможность наряду с хорошо известными реакциями включить последние достижения в этой области, пришлось ограничить материал и в значительной степени изложить его схематично. Получение и свойства комплексов переходных металлов с олефинами не освещены, поскольку они рассматриваются в гл. 15.6. 2.2.3.1. Электрофильное присоединение
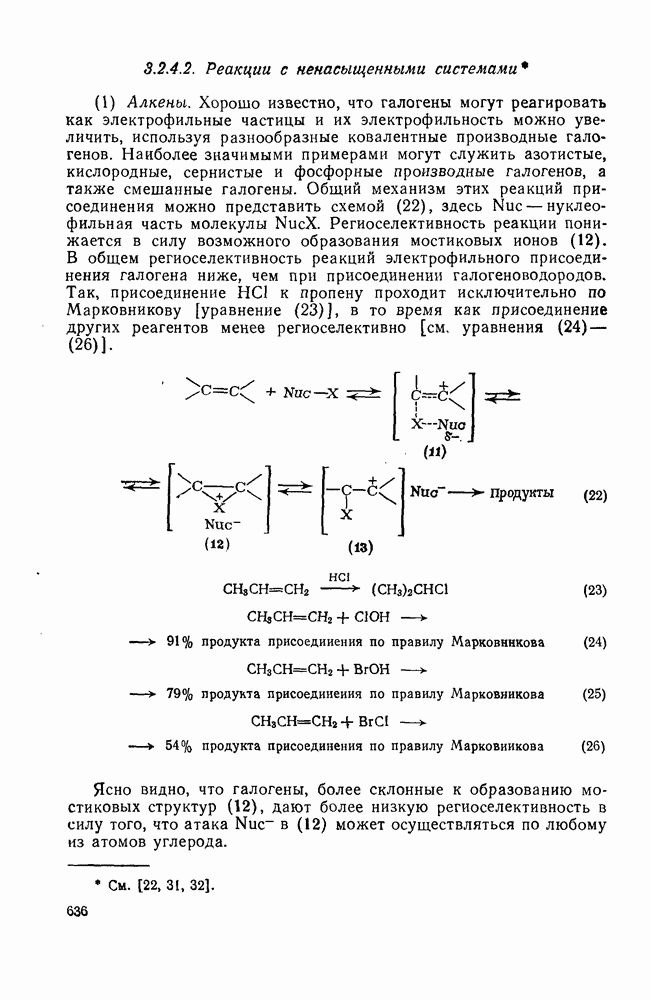
Большинство реакций присоединения реагентов типа XY к простым олефинам по своей природе являются электрофильными [типичный реагент
Большая часть реакций элекфофильного присоединения идет по так называемому механизму
Иногда постулируют другой возможный механизм —
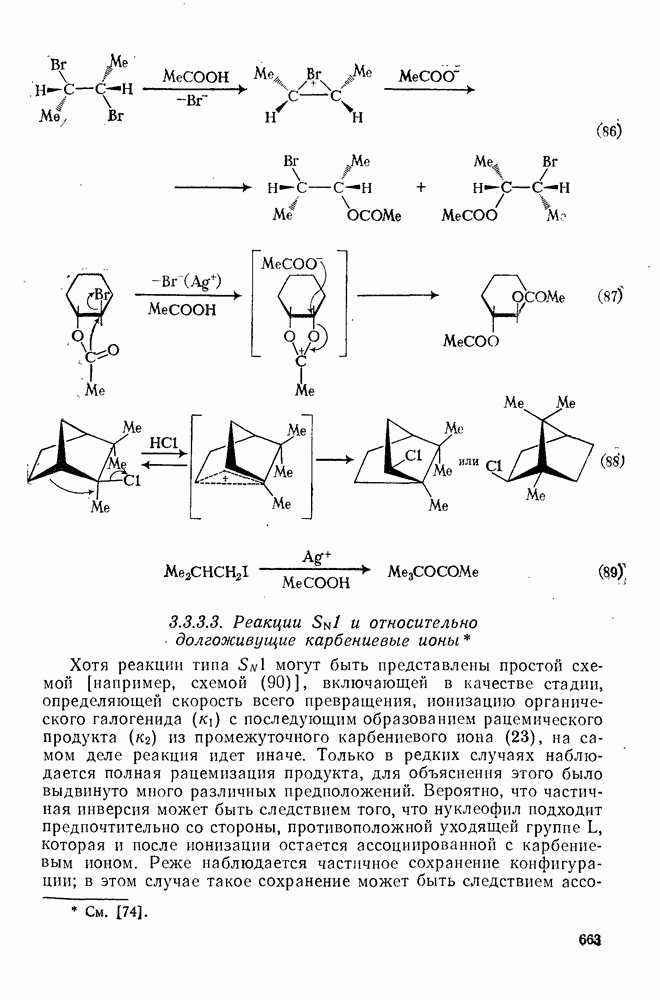
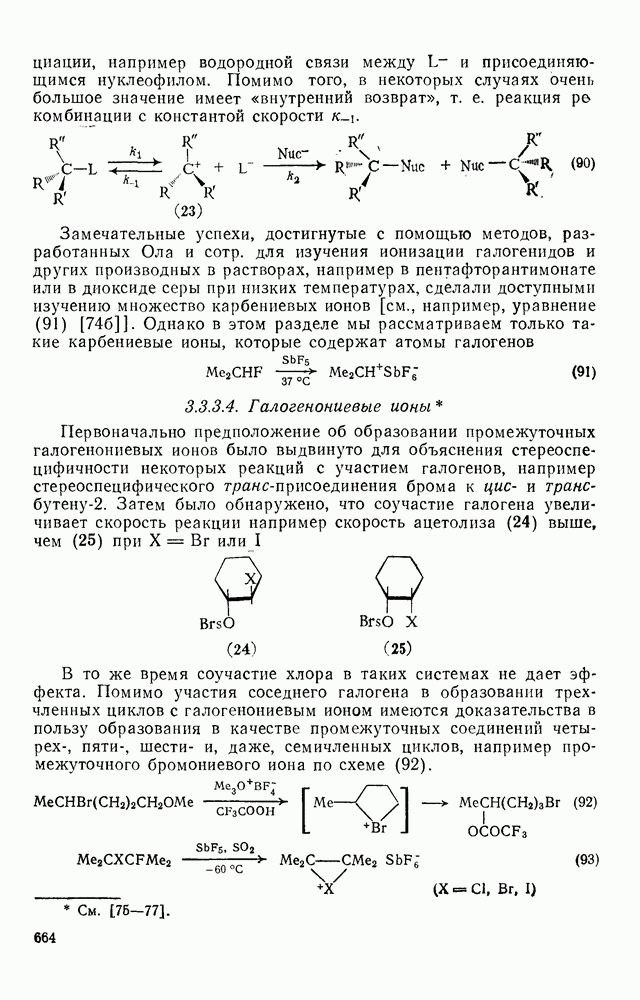
Это переходное состояние устанавливается непосредственно или через образование мостикового иона (ср. уравнение 86), причем стадия 1 является быстрой и обратимой, а стадия 2 — медленной. В свете этого поучительно рассмотреть два типа реакций электрофильного присоединения к олефинам, представляющие собой два крайних случая реализации механизма Гидратация олефинов [58] является реакцией, катализуемой кислотами, равновесие в которой обычно сильно сдвинуто в сторону спирта, см., например, уравнение (87):
Наблюдается ориентация по Марковникову (т. е. протон в общем случае присоединяется к менее замещенному атому углерода, что приводит к образованию более устойчивого карбениевого иона);
Все это, очевидно, свидетельствует в пользу карбкатионного механизма, причем близкие скорости присоединения к
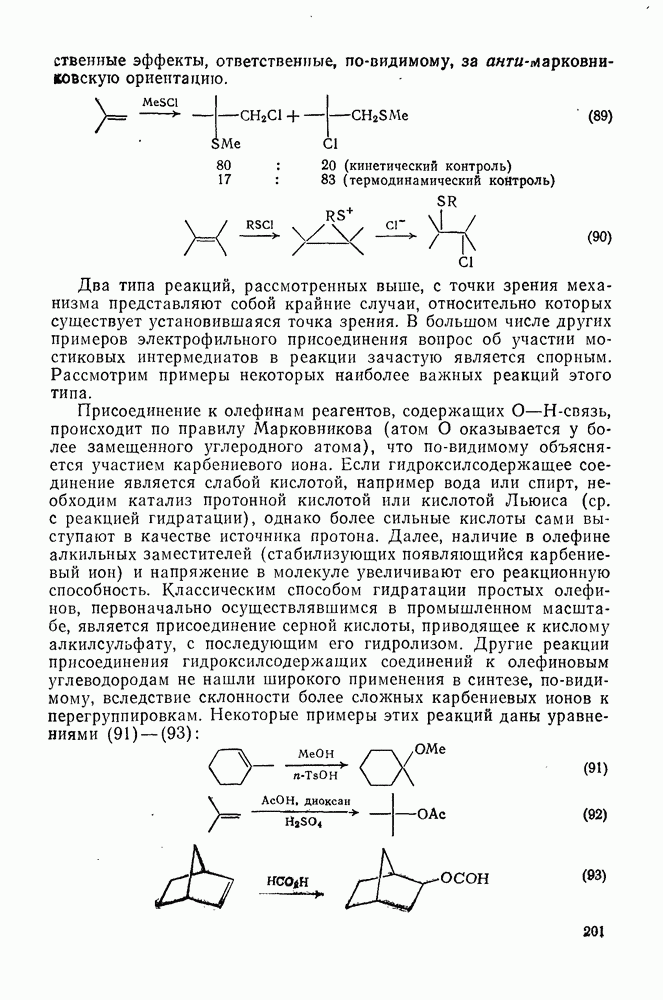
Картина, наблюдаемая при присоединении сульфенилгалогени-дов к олефинам [59], резко отличается от таковой для гидратации. В условиях кинетического контроля присоединение часто идет «против правила Марковникова»: преобладает продукт присоединения галогена (более электроотрицательного фрагмента) к менее замещенному атому углерода (уравнение 89). Последовательное введение в олефин алкильных заместителей влияет слабо. Так, например, относительные скорости присоединения метансульфенилхлорида к приведенным ниже олефинам изменяются следующим образом:
Даже для арилзамещенных олефинов наблюдается четкое анти-присоединение. Все имеющиеся данные говорят в пользу механизма
Два типа реакций, рассмотренных выше, с точки зрения механизма представляют собой крайние случаи, относительно которых существует установившаяся точка зрения. В большом числе других примеров электрофильного присоединения вопрос об участии мостиковых интермедиатов в реакции зачастую является спорным. Рассмотрим примеры некоторых наиболее важных реакций этого типа. Присоединение к олефинам реагентов, содержащих О—Н-связь, происходит по правилу Марковникова (атом О оказывается у более замещенного углеродного атома), что по-видимому объясняется участием карбениевого иона. Если гидроксилсодержащее соединение является слабой кислотой, например вода или спирт, необходим катализ протонной кислотой или кислотой Льюиса (ср. с реакцией гидратации), однако более сильные кислоты сами выступают в качестве источника протона. Далее, наличие в олефине алкильных заместителей (стабилизующих появляющийся карбениевый ион) и напряжение в молекуле увеличивают его реакционную способность. Классическим способом гидратации простых олефинов, первоначально осуществлявшимся в промышленном масштабе, является присоединение серной кислоты, приводящее к кислому алкилсульфату, с последующим его гидролизом. Другие реакции присоединения гидроксилсодержащих соединений к олефиновым углеводородам не нашли широкого применения в синтезе, по-видимому, вследствие склонности более сложных карбениевых ионов к перегруппировкам. Некоторые примеры этих реакций даны уравнениями (91) — (93):
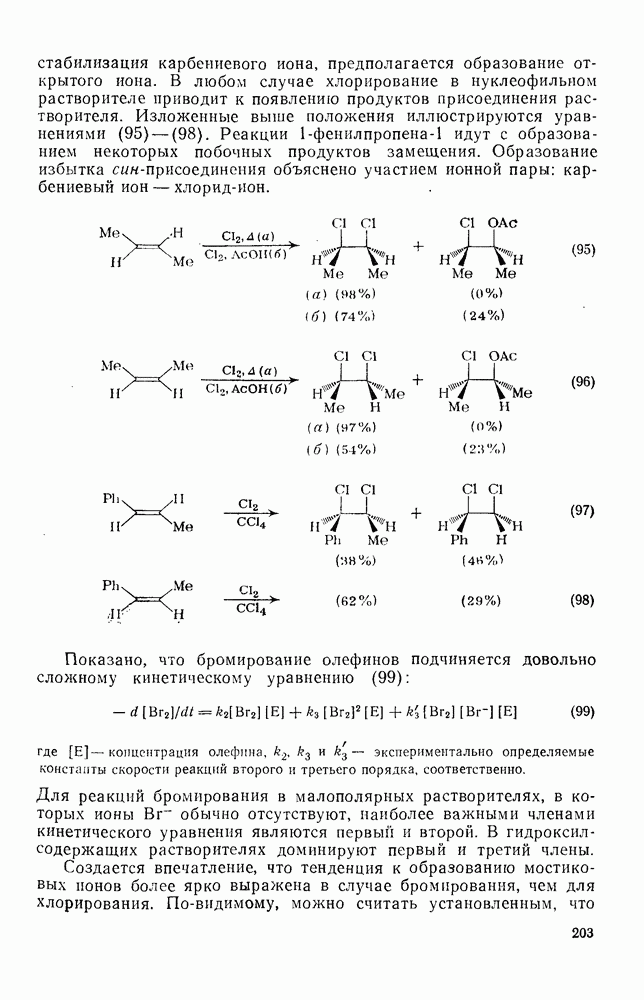
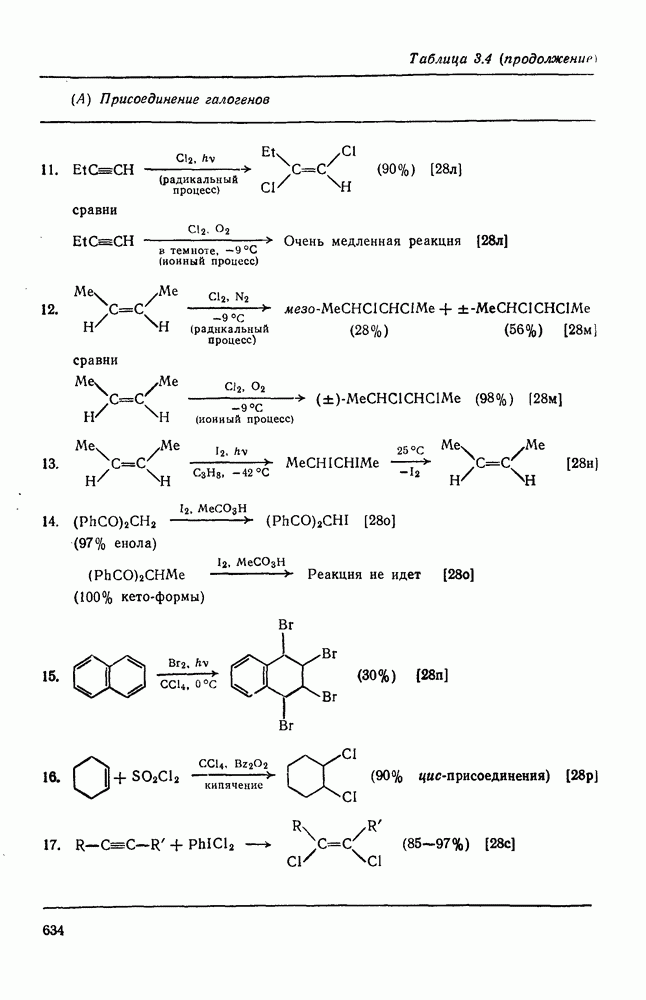
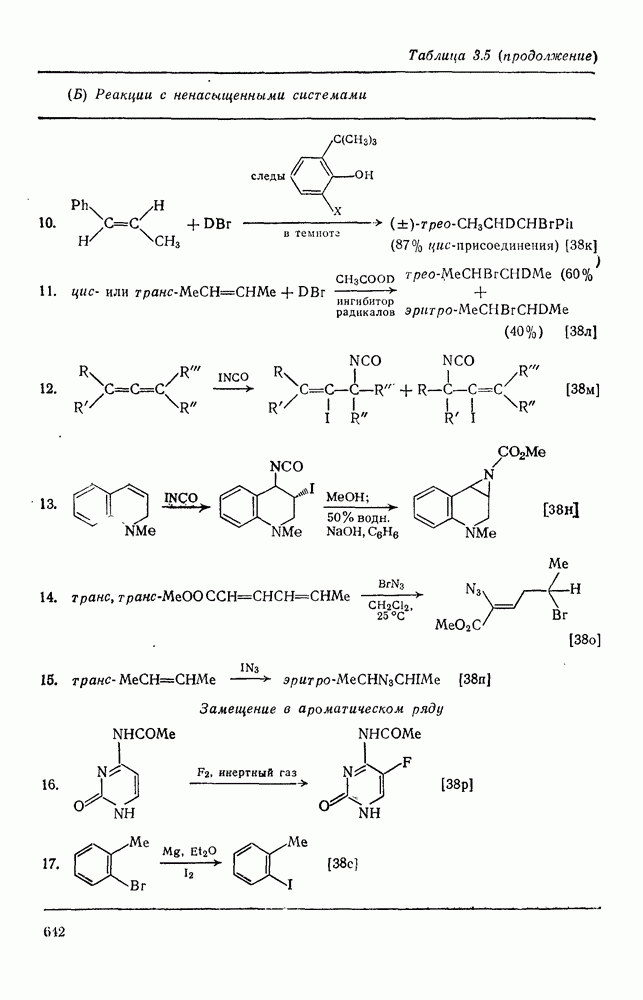
Присоединение галогеноводородов к олефинам [60] представляет собой другую давно известную электрофильную реакцию, которая в полярных средах проходит в соответствии с правилом Марковникова (галоген присоединяется к более замещенному углеродному атому). Эта реакция применяется в случае простых олефинов, но менее распространена для сложных систем из-за возможности перегруппировок. Механизм и стереохимия реакции широко исследованы. Для несопряженных олефинов, например при присоединении хлористого или бромистого водорода в уксусной кислоте к циклогексену, полученные данные говорят в пользу Присоединение галогенов является, вероятно, наиболее характерной реакцией олефинов, механизм и препаративные возможности которой были подробно исследованы [61]. Имеются данные, что при Если устранить возможность радикального цепного присоединения (проведение реакции в темноте, в присутствии ингибиторов радикалов), то возможно гетеролитическое присоединение молекулярного хлора к олефинам. Данные кинетических исследований, в особенности влияния заместителей, согласуются с предположением о бимолекулярном электрофильном механизме. Свидетельством образования промежуточного карбениевого иона является появление продуктов перегруппировок при реакциях с достаточно чувствительными системами (уравнение 94):
Стереохимия реакции наводит на мысль, что в случае простых олефинов происходит анти-присоединение хлора по механизму В любом случае хлорирование в нуклеофильном растворителе приводит к появлению продуктов присоединения растворителя. Изложенные выше положения иллюстрируются уравнениями
Показано, что бромирование олефинов подчиняется довольно сложному кинетическому уравнению (99):
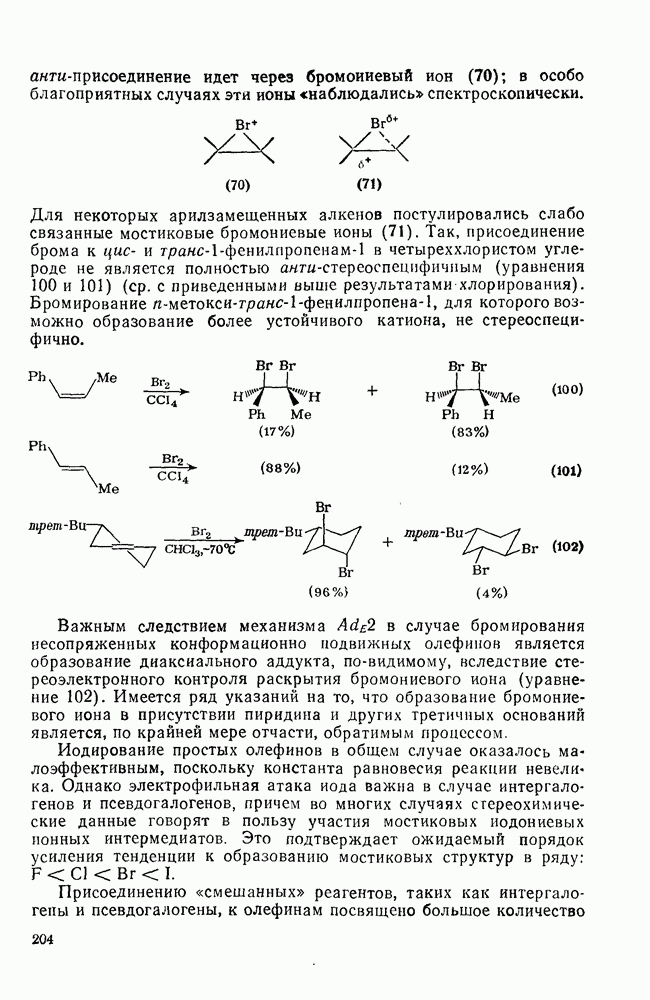
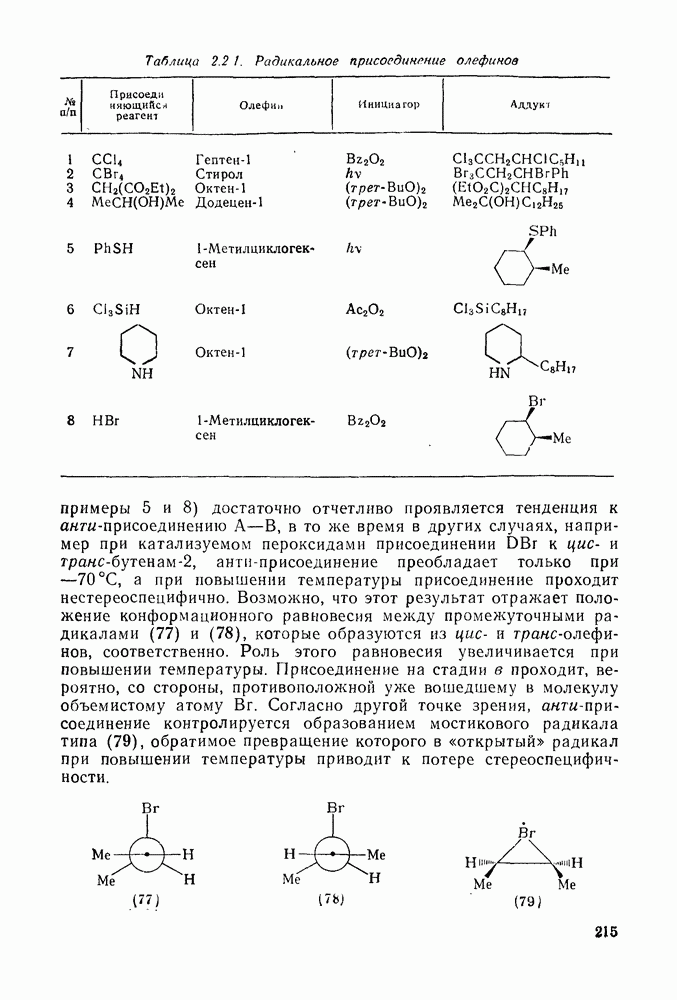
где [ Для реакций бромирования в малополярных растворителях, в которых ионы Создается впечатление, что тенденция к образованию мостико-вых ионов более ярко выражена в случае бромирования, чем для хлорирования. По-видимому, можно считать установленным, что анти-присоединение идет через бромоиневый ион (70); в особо благоприятных случаях эти ионы «наблюдались» спектроскопически.
Для некоторых арилзамещенных алкенов постулировались слабо связанные мостиковые бромониевые ионы (71). Так, присоединение брома к
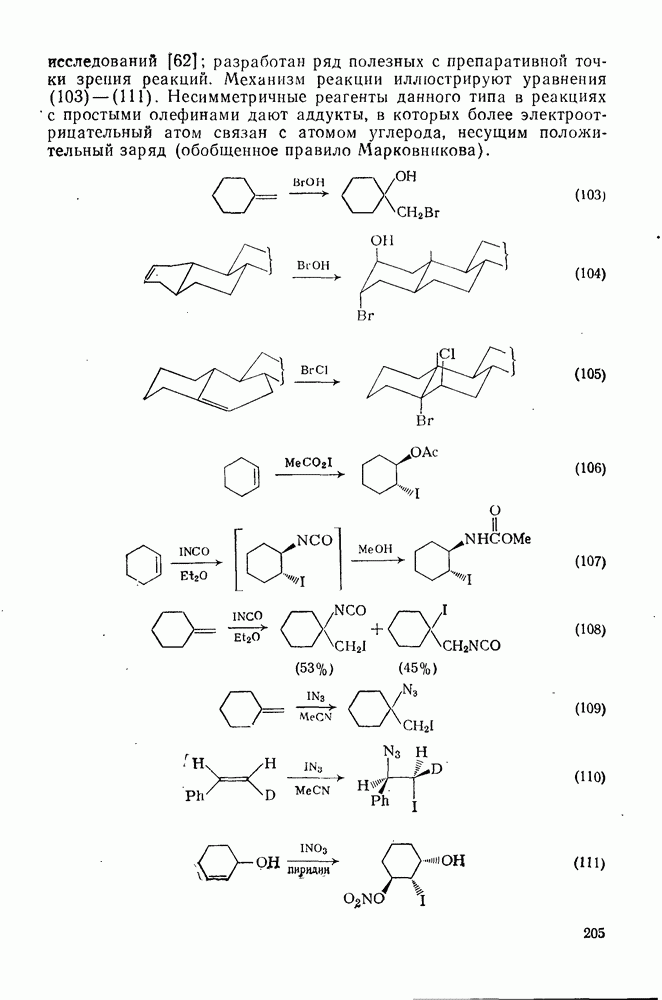
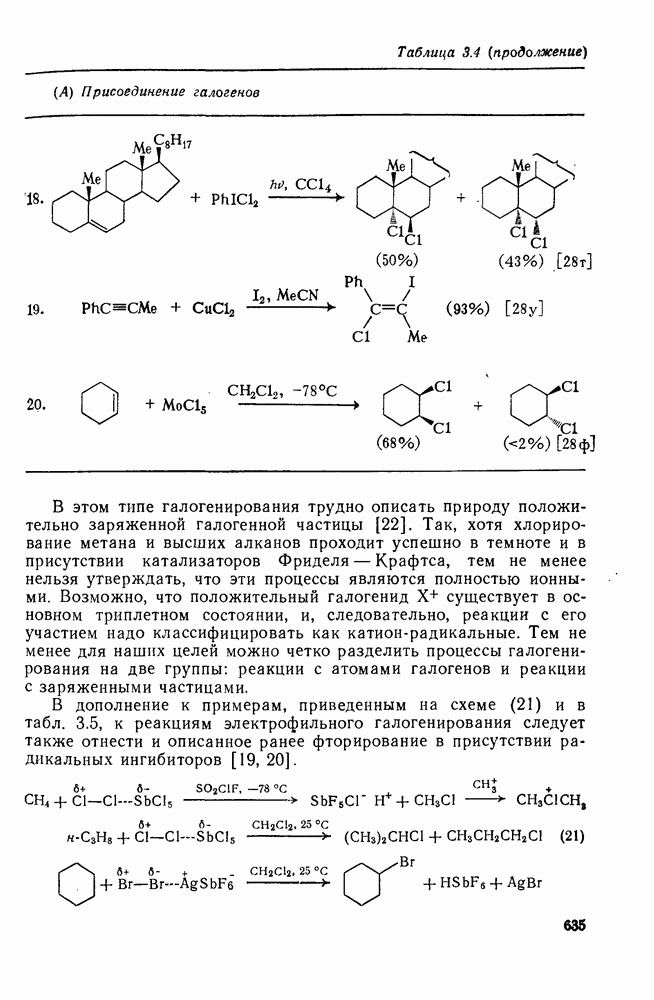
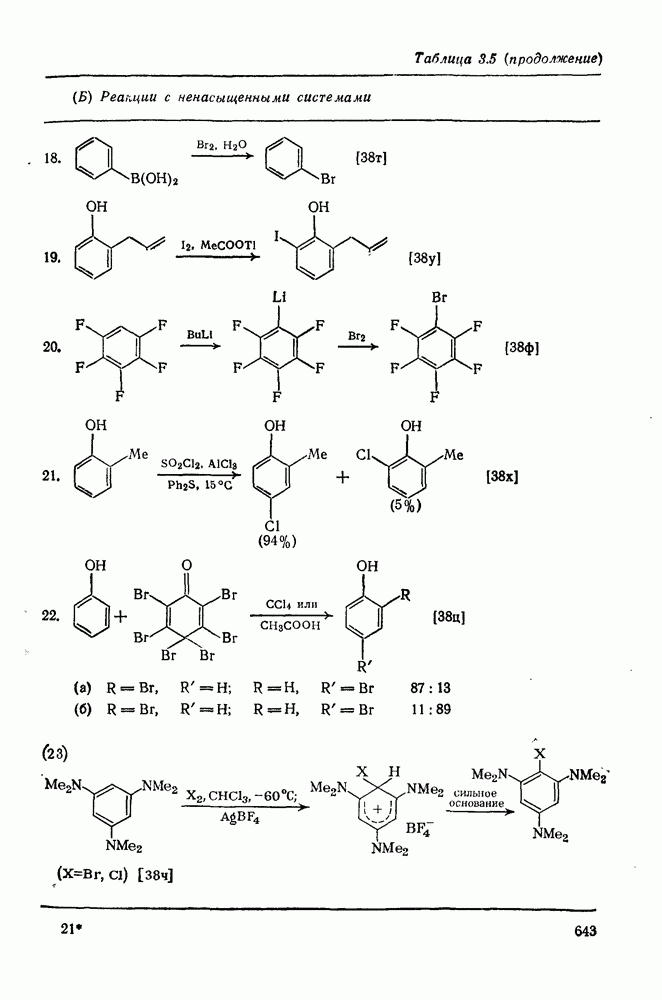
Важным следствием механизма Иодирование простых олефинов в общем случае оказалось малоэффективным, поскольку константа равновесия реакции невелика. Однако электрофильная атака иода важна в случае интергало-генов и псевдогалогенов, причем во многих случаях стереохимические данные говорят в пользу участия мостиковых иодониевых ионных интермедиатов. Это подтверждает ожидаемый порядок усиления тенденции к образованию мостиковых структур в ряду: Присоединению «смешанных» реагентов, таких как интергалогены и псевдогалогены, к олефинам посвящено большое количество исследований [62]; разработан ряд полезных с препаративной точки зрения реакций. Механизм реакции иллюстрируют уравнения (103) — (111). Несимметричные реагенты данного типа в реакциях с простыми олефинами дают аддукты, в которых более электроотрицательный атом связан с атомом углерода, несущим положительный заряд (обобщенное правило Марковникова).
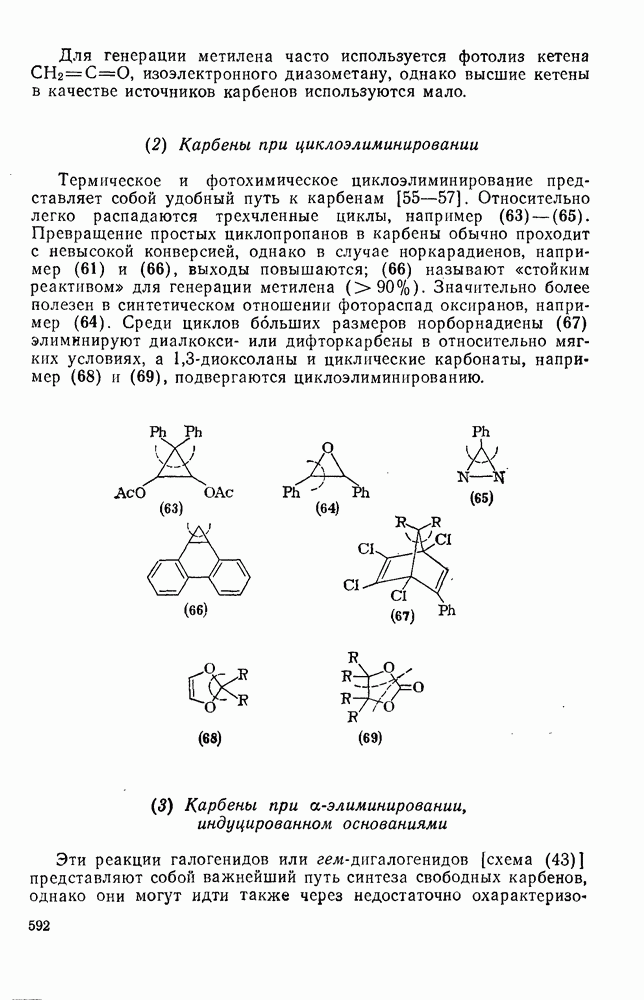
Для более сложных циклических систем (уравнение 105) наблюдаемую ориентацию вероятнее всего можно объяснить диаксиальным раскрытием первоначально образующегося Присоединение сульфенилгалогенидов как прототип электро-фильного присоединения к олефинам, включающего образование мостиковых ионов, уже рассмотрено ранее. Гпдроборирование олефинов дибораном и моно- или диалкил-боранамн [63] представляет собой электрофильное присоединение, которое, однако, во многом отличается от реакций, рассмотренных выше. Во всех случаях происходит син-присоединение, причем атом бора связывается с менее замещенным атомом углерода (уравнение 112). В эту реакцию вступают все олефины, даже сильно пространственно-затрудненные, однако три- и тетраметилэтилены, быстро присоединяя диборан, дают соответственно только диалкил- или моноалкилбораны (уравнения 113 и 114)
Большое значение реакции гидроборирования заключается в возможности дальнейшего широкого использования образующихся алкилборанов [64]; так, окисление их щелочным раствором пероксида водорода дает спирты. Относительные скорости гидроборирования не отвечают известной зависимости скорости электрофильного присоединения от степени замещения; так, относительные скорости для Можно думать, что этот результат отражает существенные черты первоначального переходного состояния, в котором небольшие пространственные и электронные эффекты достаточно хорошо сбалансированы. Пространственные эффекты начинают доминировать при гидроборировании с использованием замещенных боранов (уравнение 115); эта реакция может иметь полезные синтетические применения. Хотя до сих пор еще нет четких представленией о переходном состоянии при гидроборировании, однако наиболее подходящей гипотезой, объясняющей как ориентацию, так и стереохимию, может служить четырехнентровая модель (72).
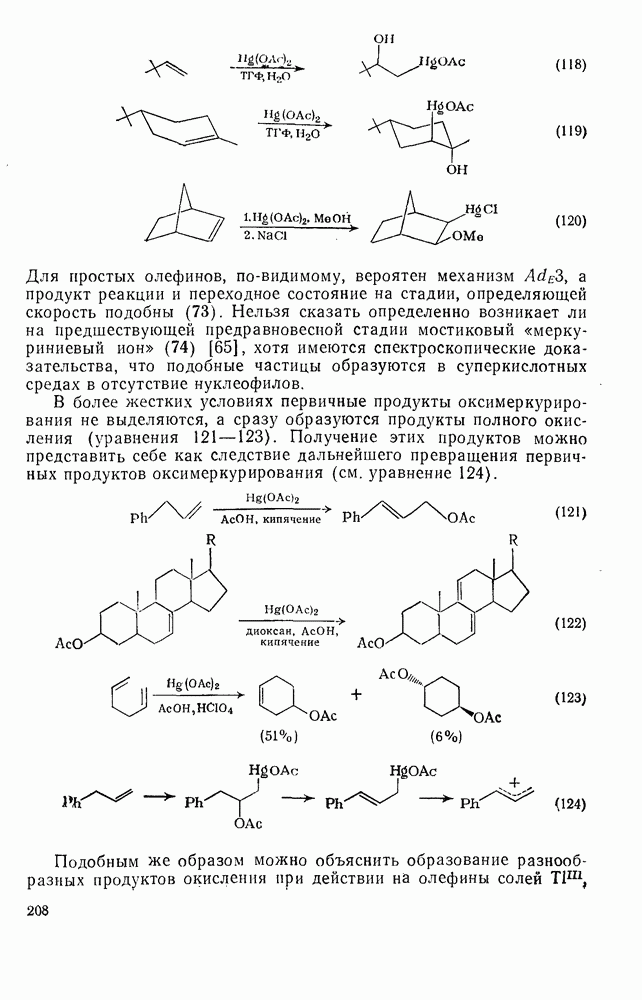
Иной тип электрофильного присоединения с образованием продуктов оксиметаллирования (уравнение 116) наблюдался при присоединении солей типа
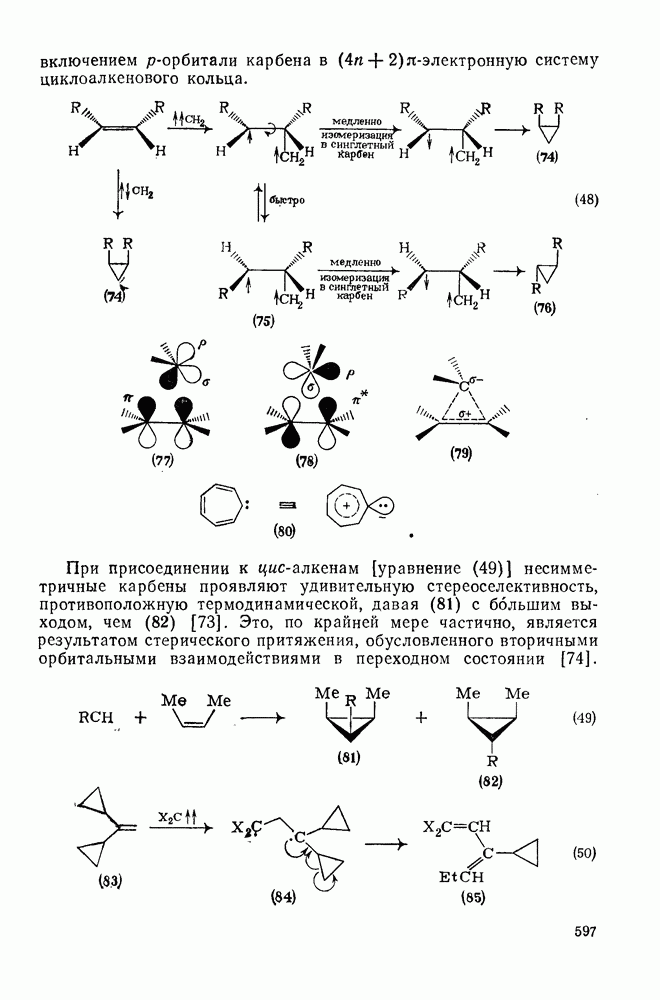
Характерными чертами реакции оксимеркурирования (уравнения 117—120) являются: строгая ориентация по правилу Марковникова; отсутствие заметной тенденции к перегруппировкам в подходящих для этого системах (уравнение 118); анти-стереоспецифичность для нормальных олефинов, что ведет к диаксиальному присоединению в случае конформационно подвижных олефинов (уравнение 119), но к син-присоединению для норборнеиа, поскольку здесь анти-копланарное присоединение затруднено (уравнение 120).
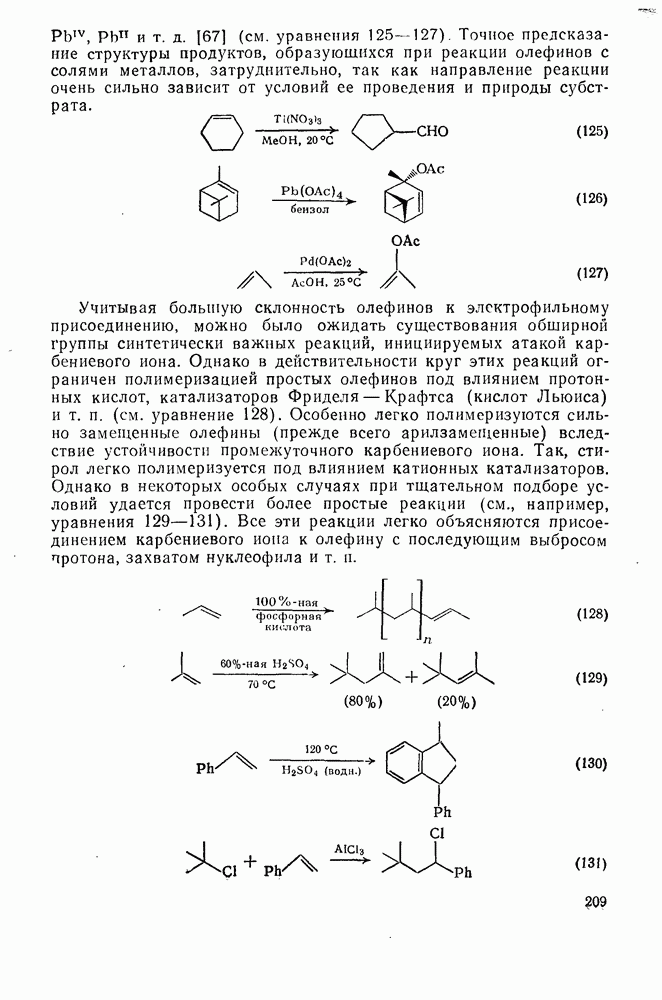
Для простых олефинов, по-видимому, вероятен механизм В более жестких условиях первичные продукты оксимеркуриро-вания не выделяются, а сразу образуются продукты полного окисления (уравнения 121 —123). Получение этих продуктов можно представить себе как следствие дальнейшего превращения первичных продуктов оксимеркурирования (см. уравнение 124).
Подобным же образом можно объяснить образование разнообразных продуктов окисления при действии на олефины солей Точное предсказание структуры продуктов, образующихся при реакции олефинов с солями металлов, затруднительно, так как направление реакции очень сильно зависит от условий ее проведения и природы субстрата.
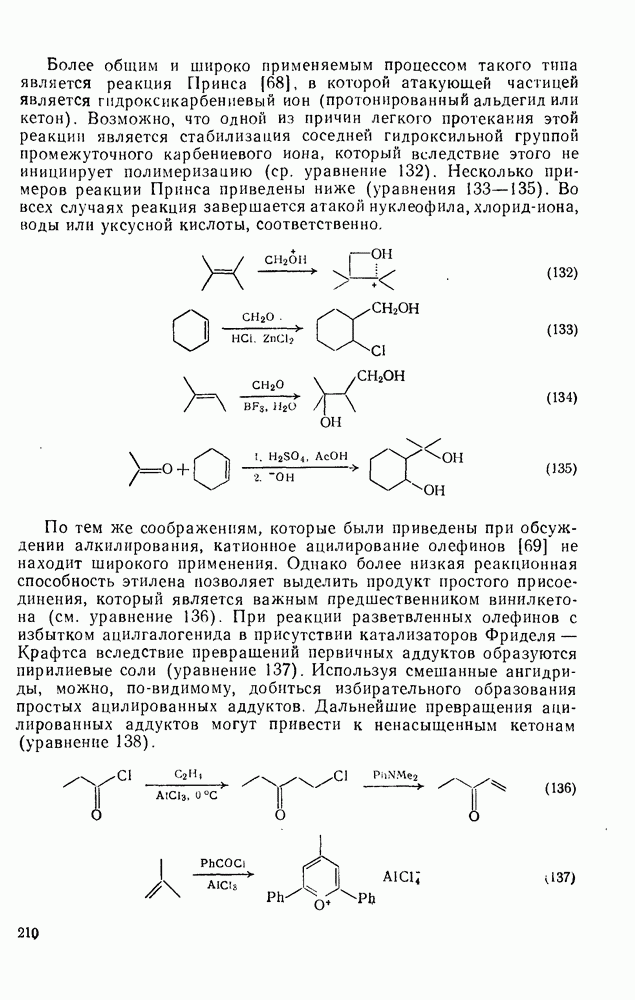
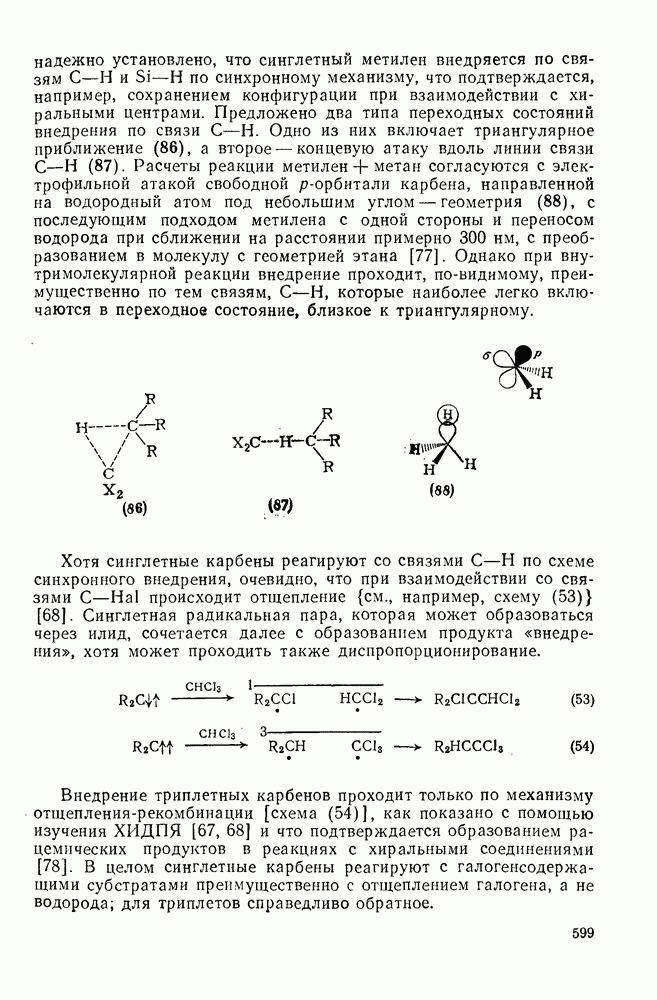
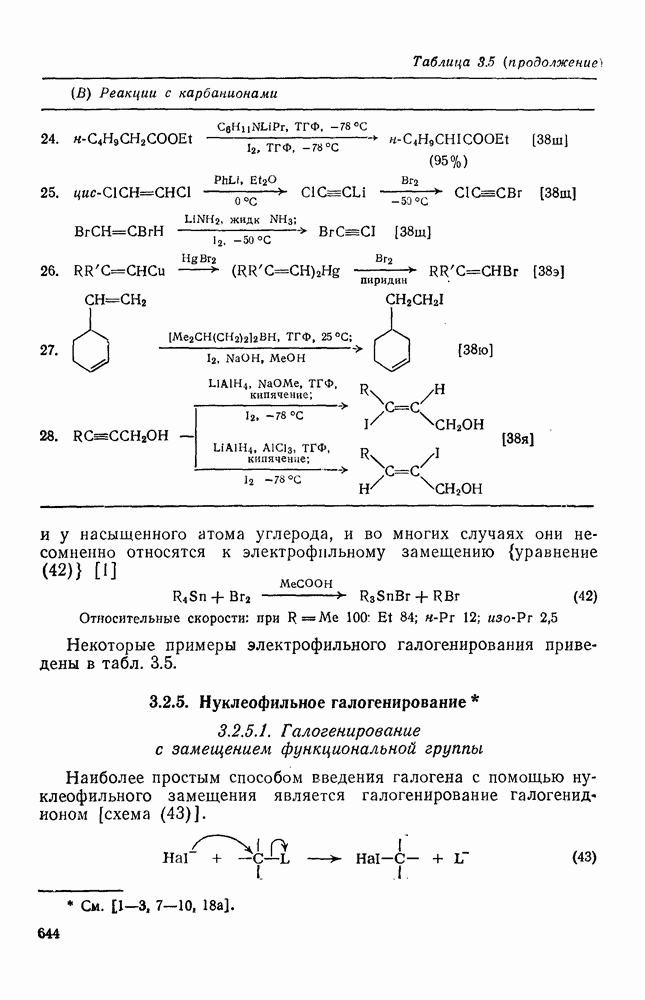
Учитывая большую склонность олефинов к электрофильному присоединению, можно было ожидать существования обширной группы синтетически важных реакций, инициируемых атакой карбениевого иона. Однако в действительности круг этих реакций ограничен полимеризацией простых олефинов под влиянием протонных кислот, катализаторов Фриделя — Крафтса (кислот Льюиса) и т. п. (см. уравнение 128). Особенно легко полимеризуются сильно замещенные олефины (прежде всего арилзамегценные) вследствие устойчивости промежуточного карбениевого иона. Так, стирол легко полимеризуется под влиянием катионных катализаторов. Однако в некоторых особых случаях при тщательном подборе условий удается провести более простые реакции (см., например, уравнения 129—131). Все эти реакции легко объясняются присоединением карбениевого иона к олефину с последующим выбросом протона, захватом нуклеофила и т. п.
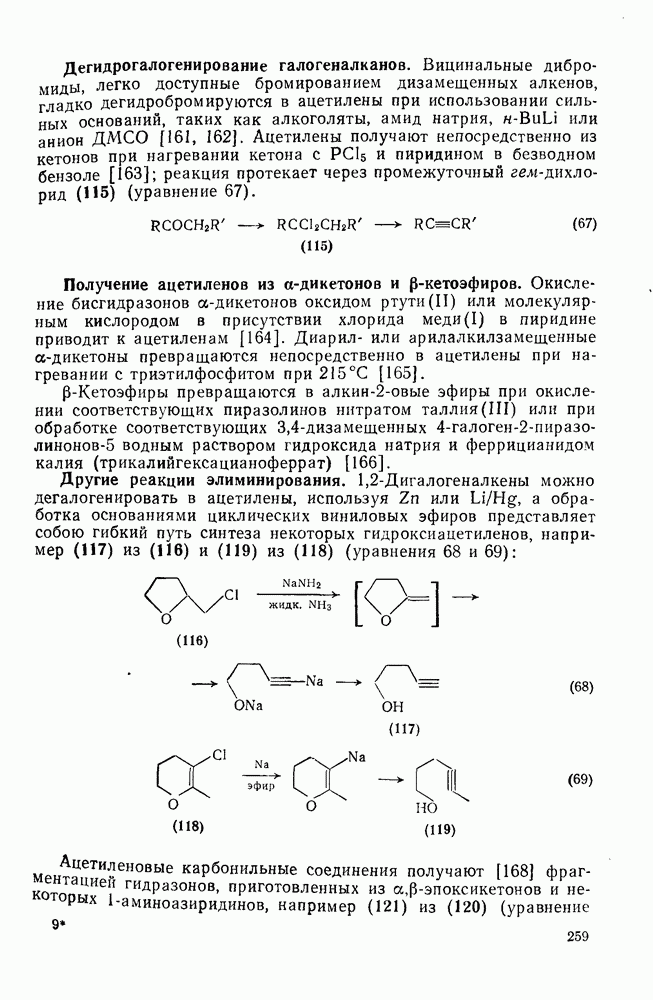
Более общим и широко применяемым процессом такого типа является реакция Принса [68], в которой атакующей частицей является гидроксикарбениевый ион (протонированный альдегид или кетон). Возможно, что одной из причин легкого протекания этой реакции является стабилизация соседней гидроксильной группой промежуточного карбениевого иона, который вследствие этого не инициирует полимеризацию (ср. уравнение 132). Несколько примеров реакции Принса приведены ниже (уравнения 133—135). Во всех случаях реакция завершается атакой нуклеофила, хлорид-иона, воды или уксусной кислоты, соответственно.
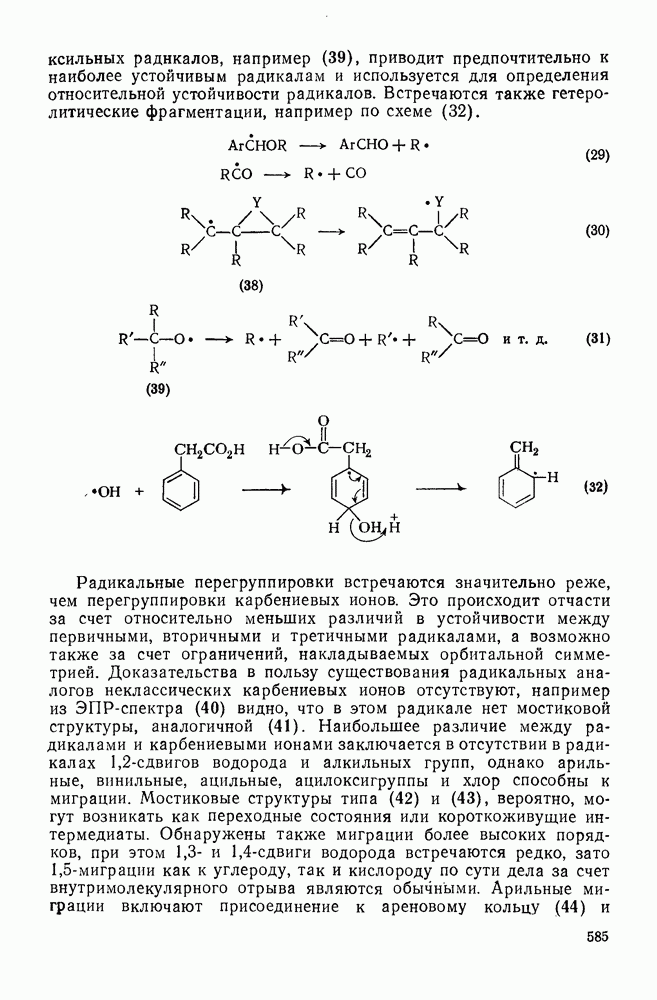
По тем же соображениям, которые были приведены при обсуждении алкилирования, катионное ацилирование олефинов [69] не находит широкого применения. Однако более низкая реакционная способность этилена позволяет выделить продукт простого присоединения, который является важным предшественником винилкегона (см. уравнение 136). При реакции разветвленных олефинов с избытком ацилгалогенида в присутствии катализаторов Фриделя — Крафтса вследствие превращений первичных аддуктов образуются пирилиевые соли (уравнение 137). Используя смешанные ангидриды, можно, по-видимому, добиться избирательного образования простых ацилированных аддуктов. Дальнейшие превращения аци-лированных аддуктов могут привести к ненасыщенным кетонам (уравнение 138).
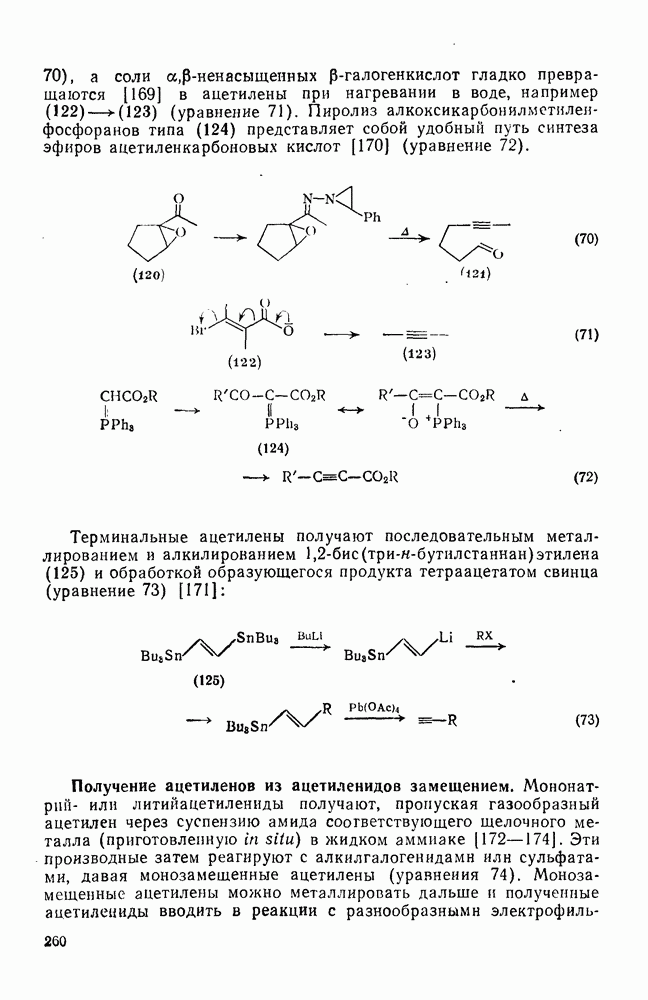
Заканчивая обзор реакций электрофильного присоединения к олефинам следует выделить группу реакций, в которых первой стадией является протонирование олефина с образованием карбениевого иона с последующей атакой нуклеофилом. Эти реакции типичны не только для олефинов, поскольку промежуточные карбениевые ионы могут быть получены и из других предшественников, поэтому в этом разделе такие реакции будут рассмотрены очень кратко. Приведенные ниже примеры включают: 1) алкилирование оксида углерода (генерированного из муравьиной кислоты) — карбоксилирование по Коху — Хаафу [70] (уравнения 139, 140); 2) алкилирование нитрилов с образованием амидов — реакция Риттера [71] (уравнение 141); 3) алкилирование тиолов (уравнение 142); 4) гидридный перенос от силилгидридов, ведущий к полному восстановлению [72] (уравнение 143). Однако такие карбениевые ионы склонны к перегруппировкам (см. уравнение 140).
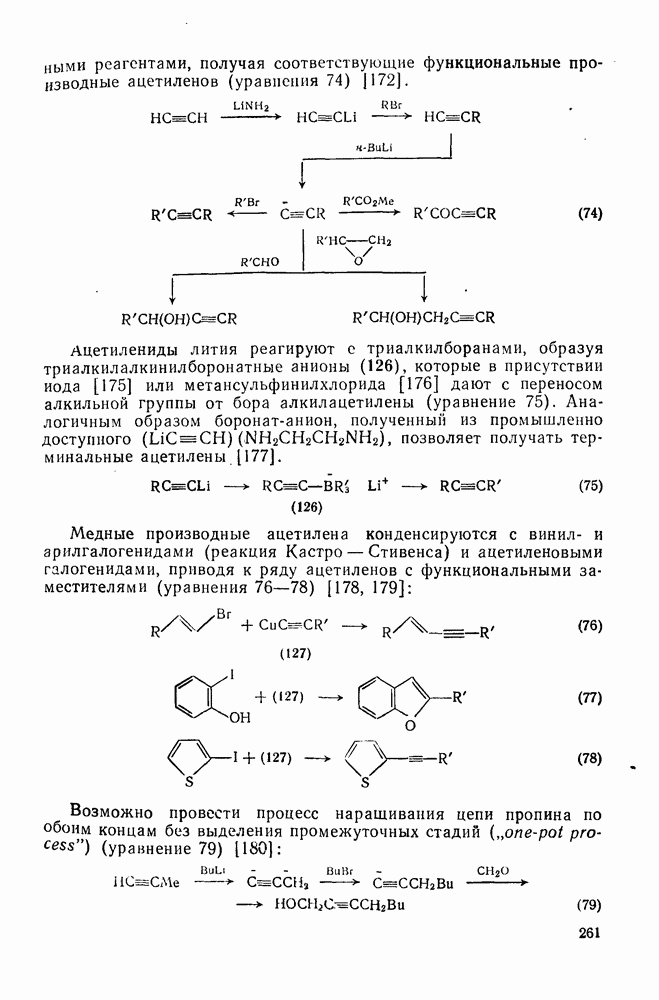
В полиенах образование карбениевого иона при протонировании одной двойной связи приводит иногда к внутримолекулярному присоединению по другой двойной связи. Такие реакции являются прототипом биогенетических циклизаций, приводящих к терпенам и т. п. (см. гл. 29.2). Хорошим примером, иллюстрирующим преимущественное образование шестичленных циклов с экваториальным вхождением протона и нуклеофила (анти-присоединение), является катализуемая кислотами циклизация сесквитерпенового углеводорода гермакрена (уравнение 144):
|
 |
Оглавление
|





















































































































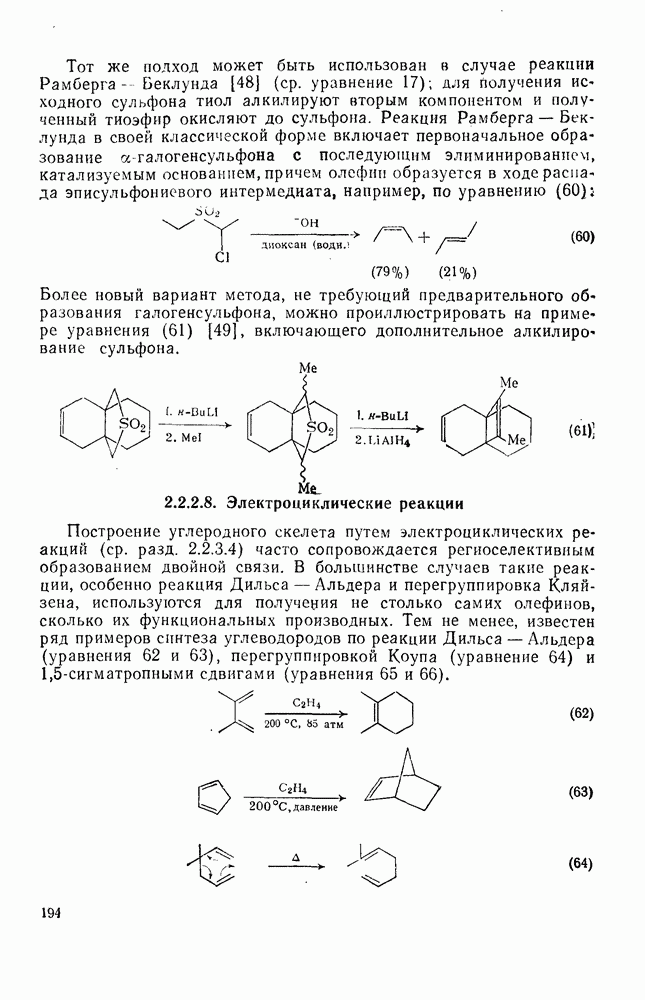









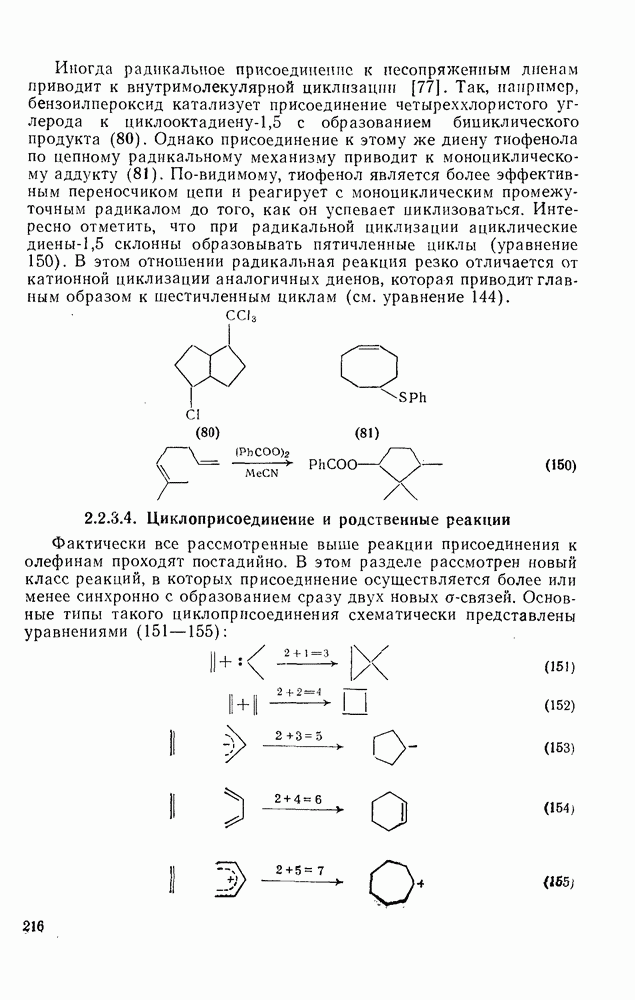

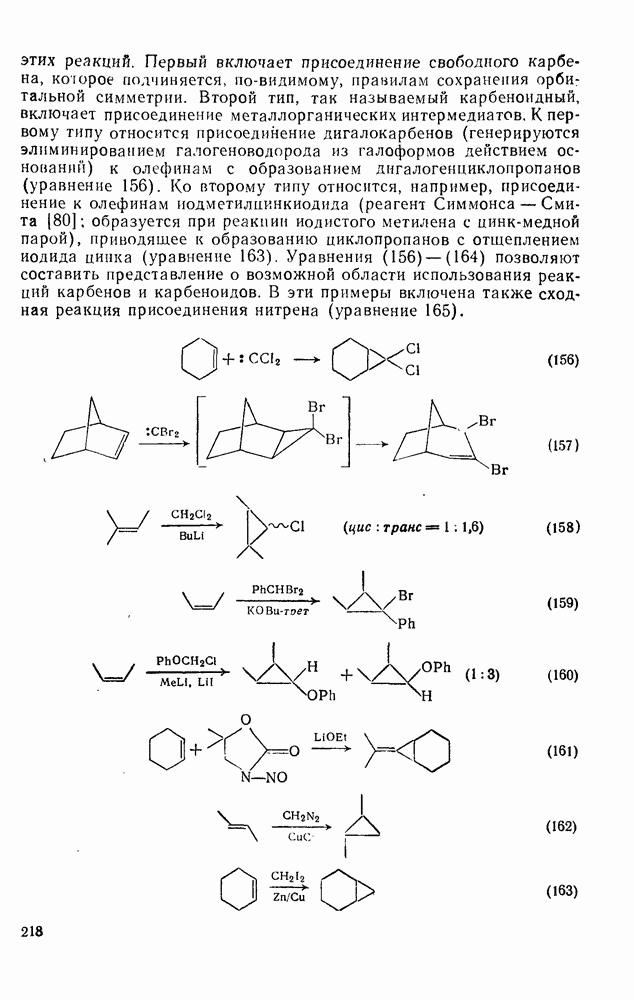

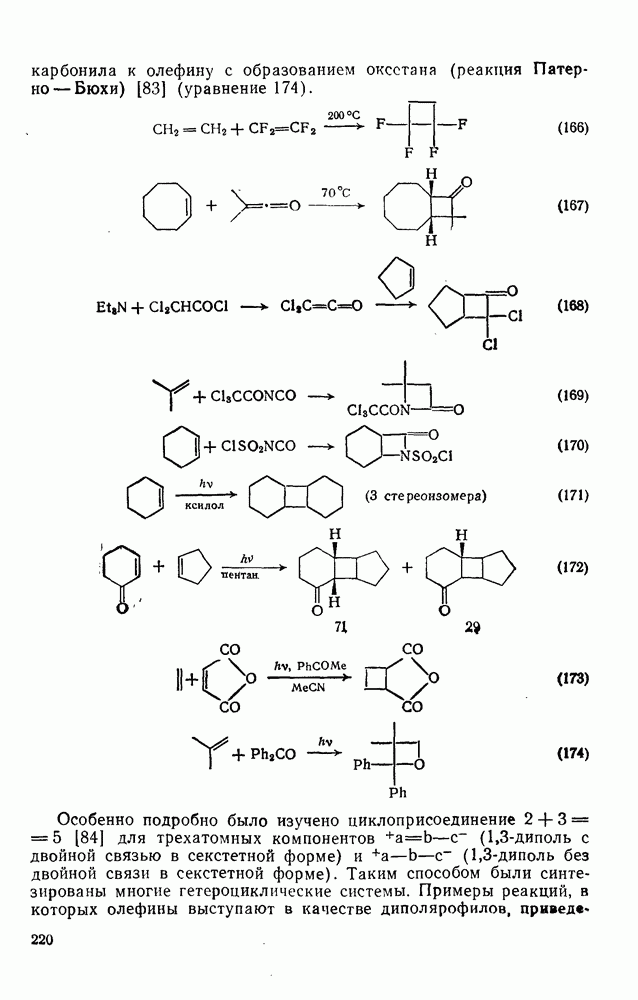
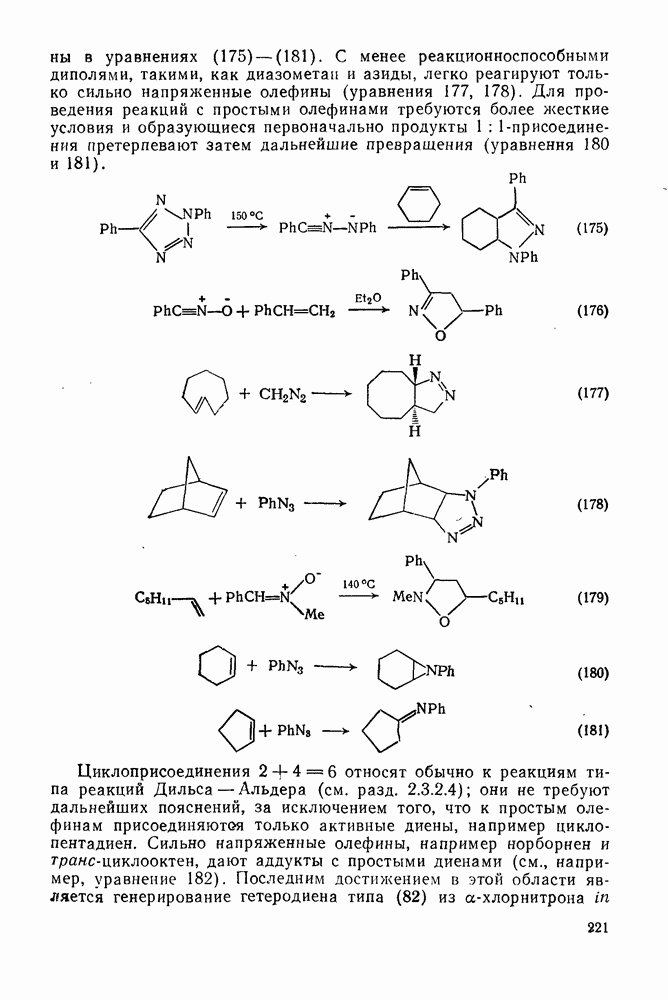


























































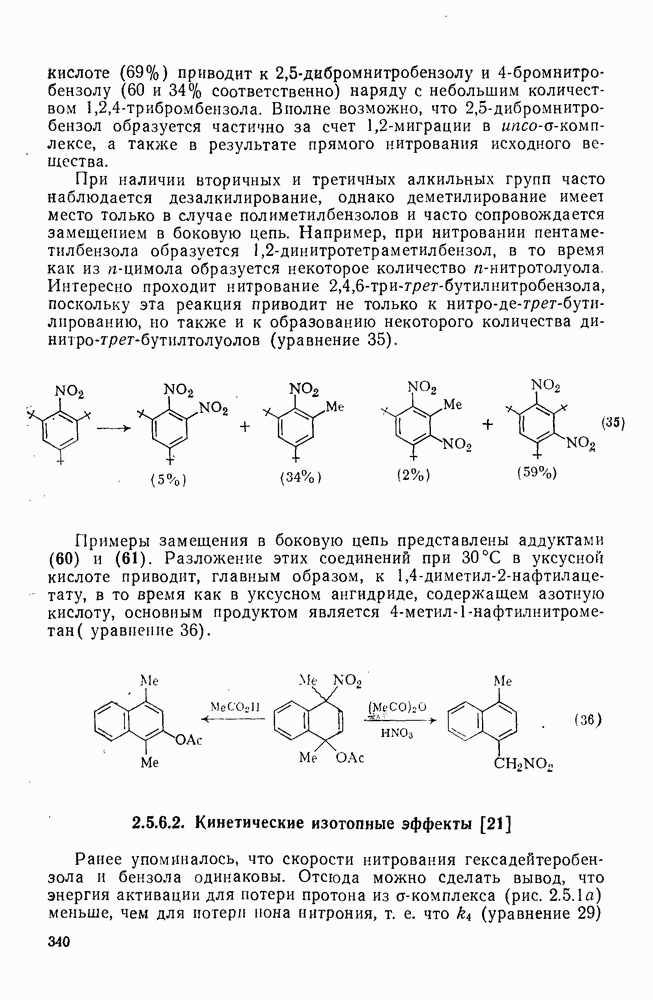










































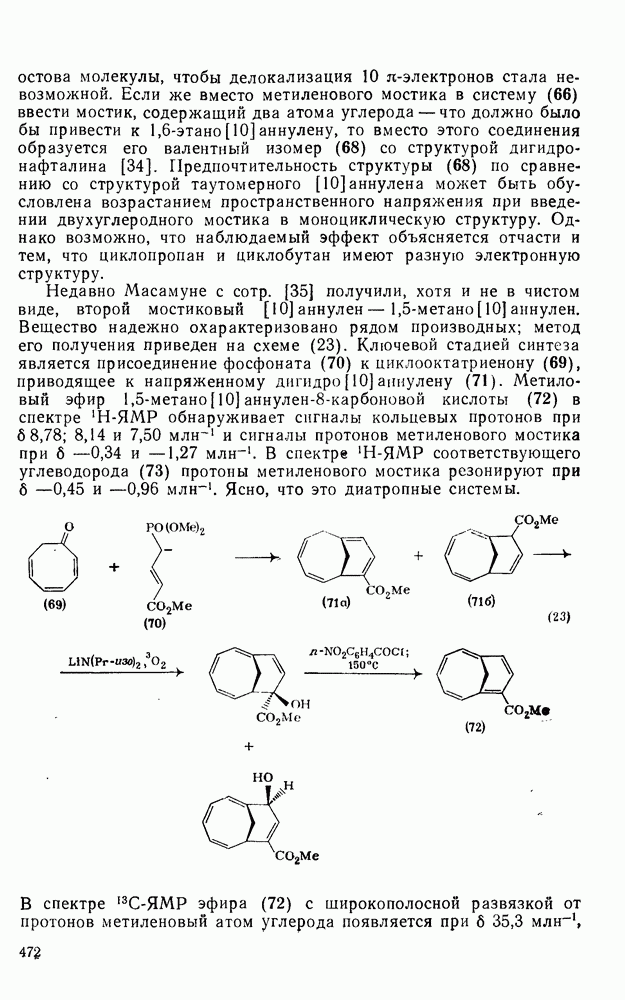





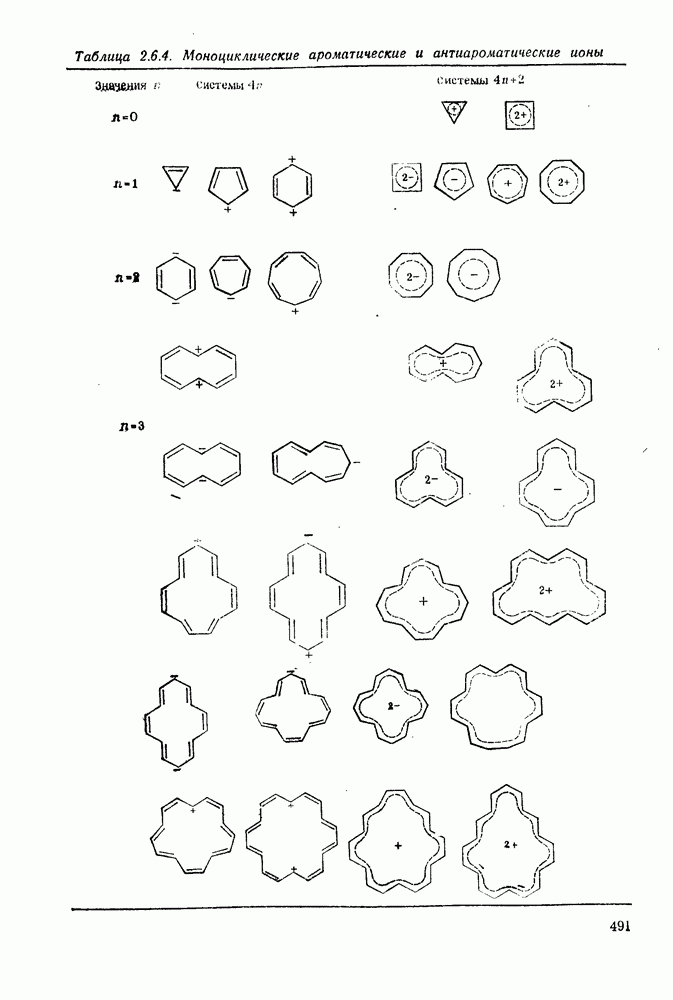
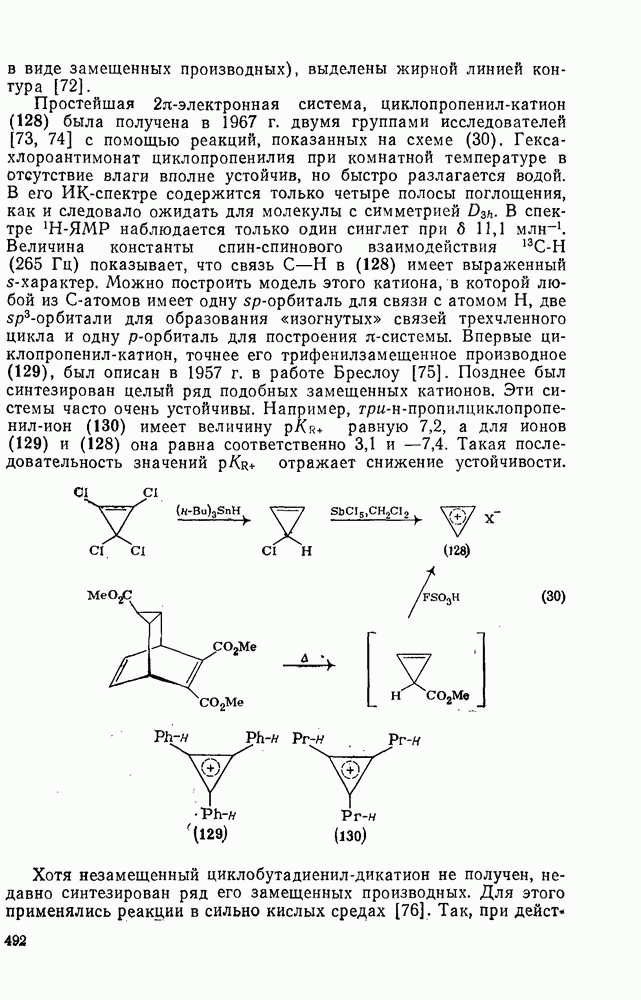
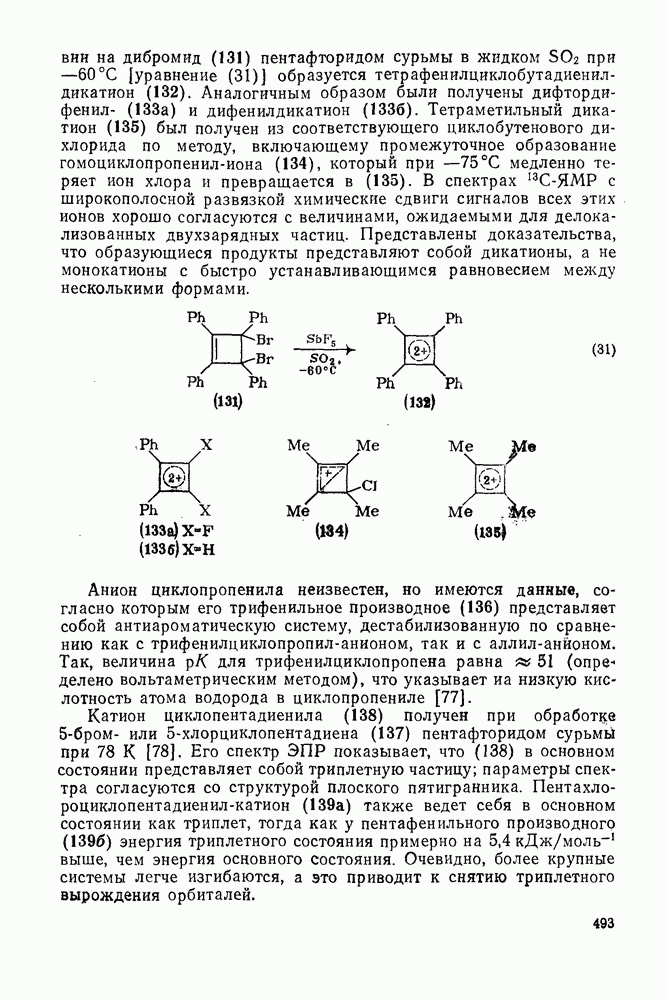
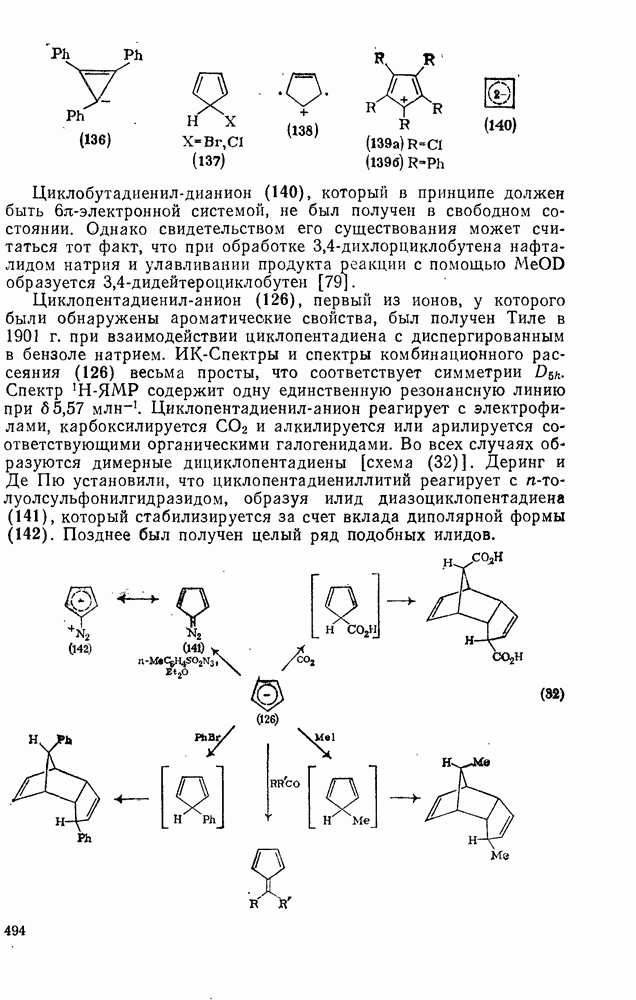



















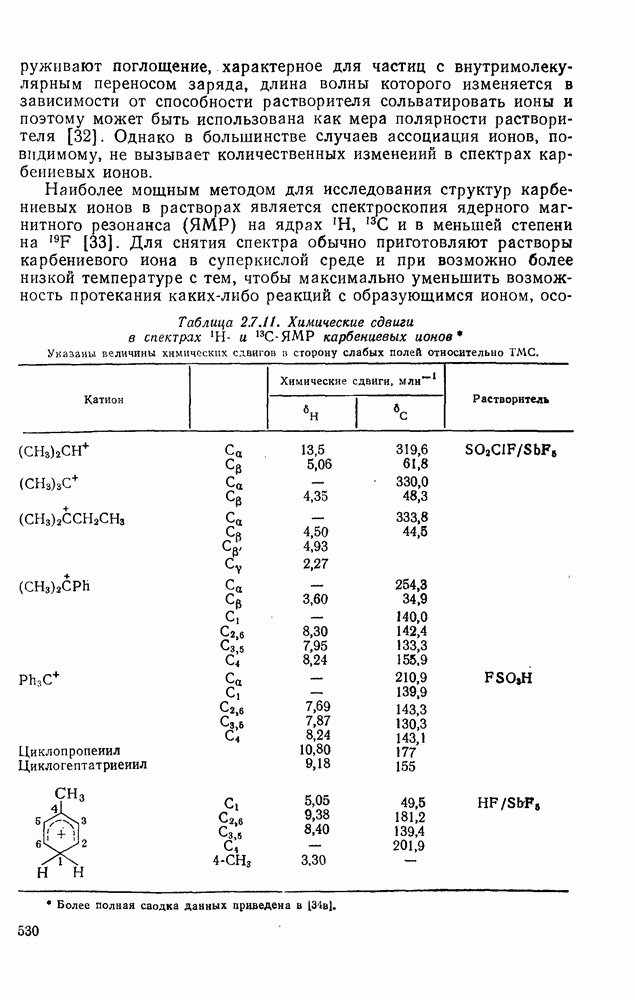
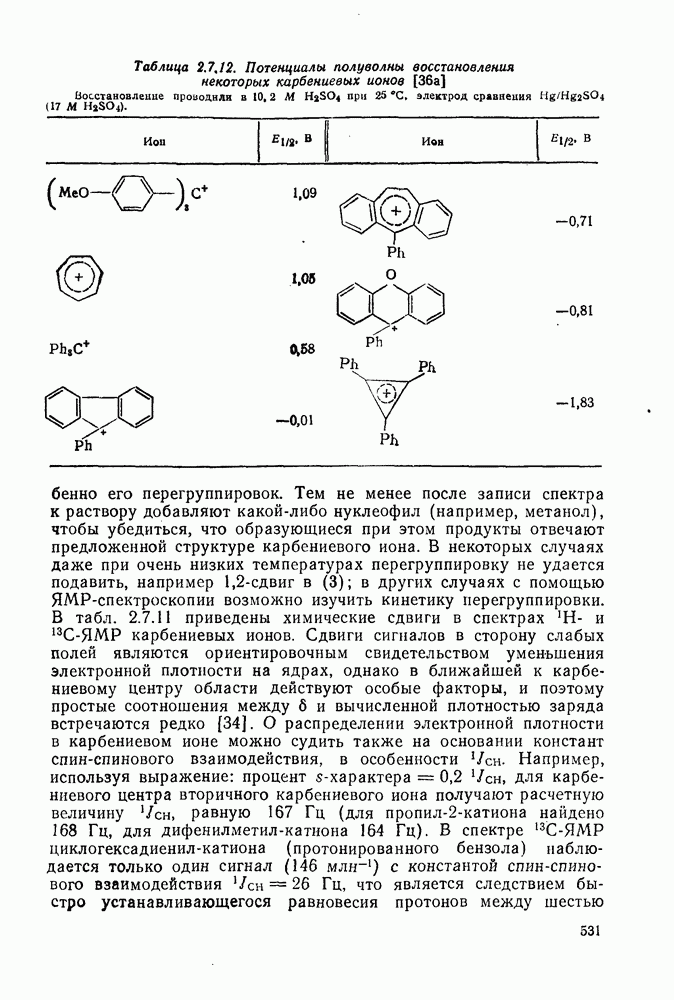

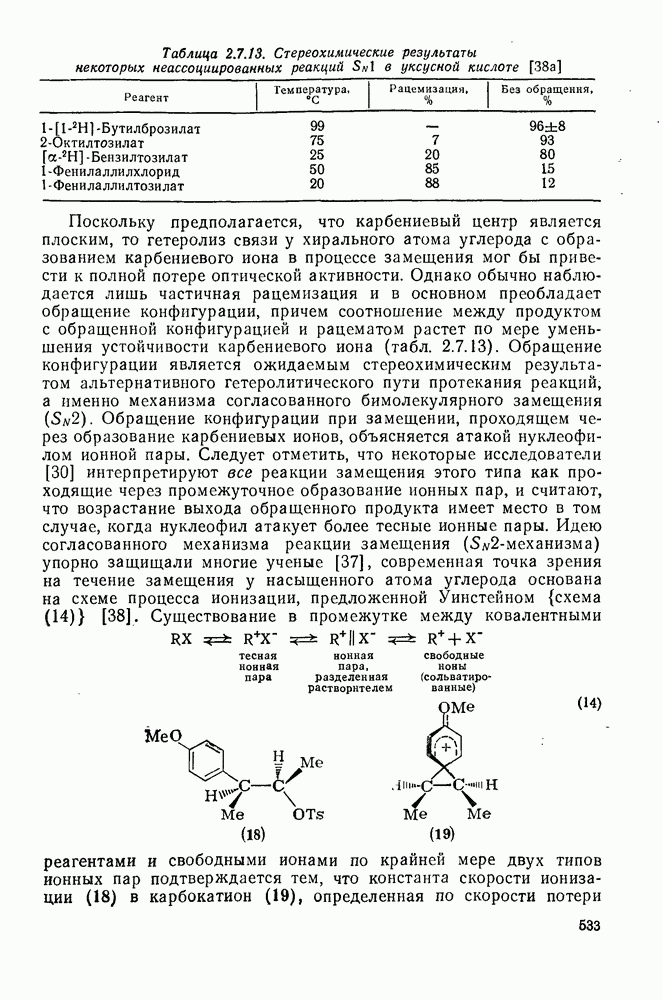
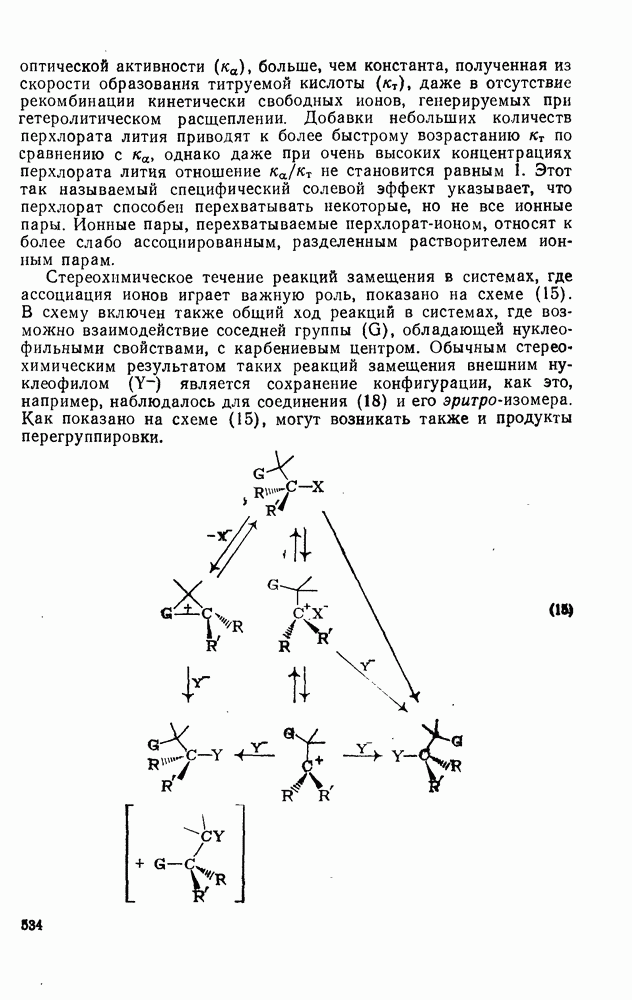


































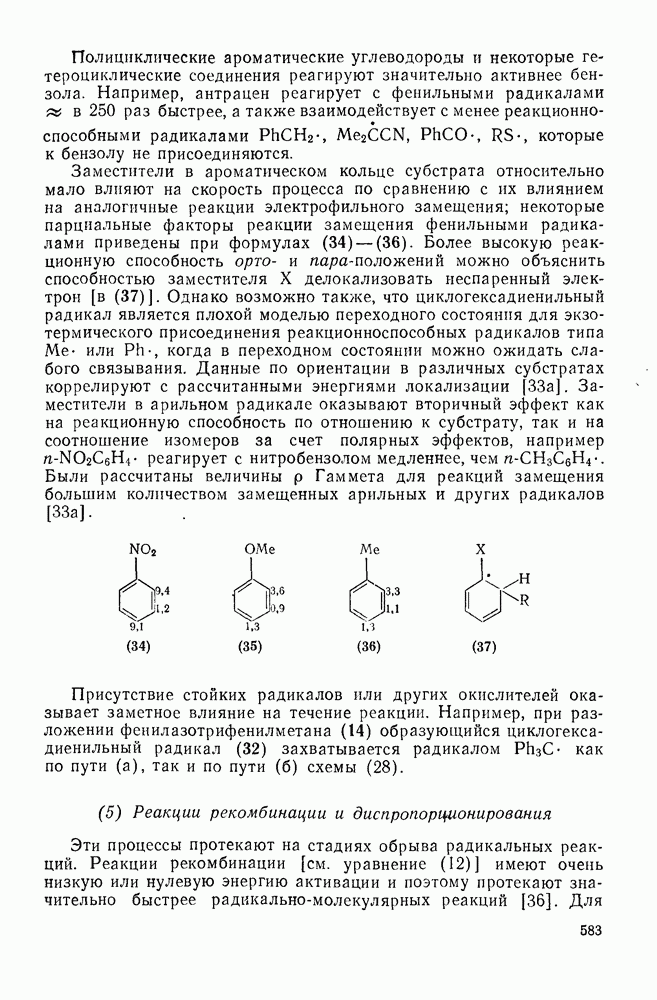









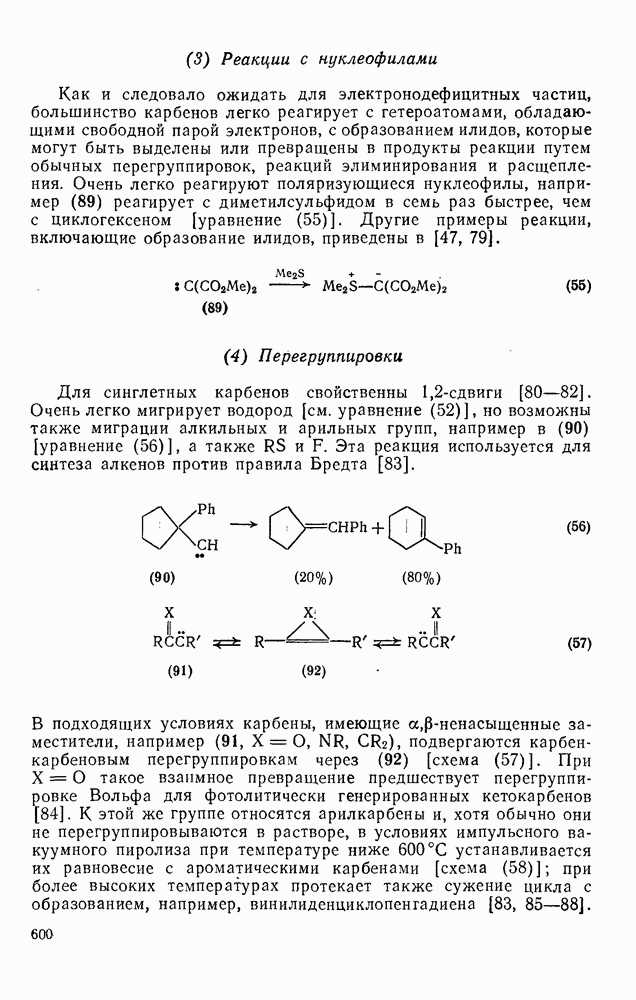
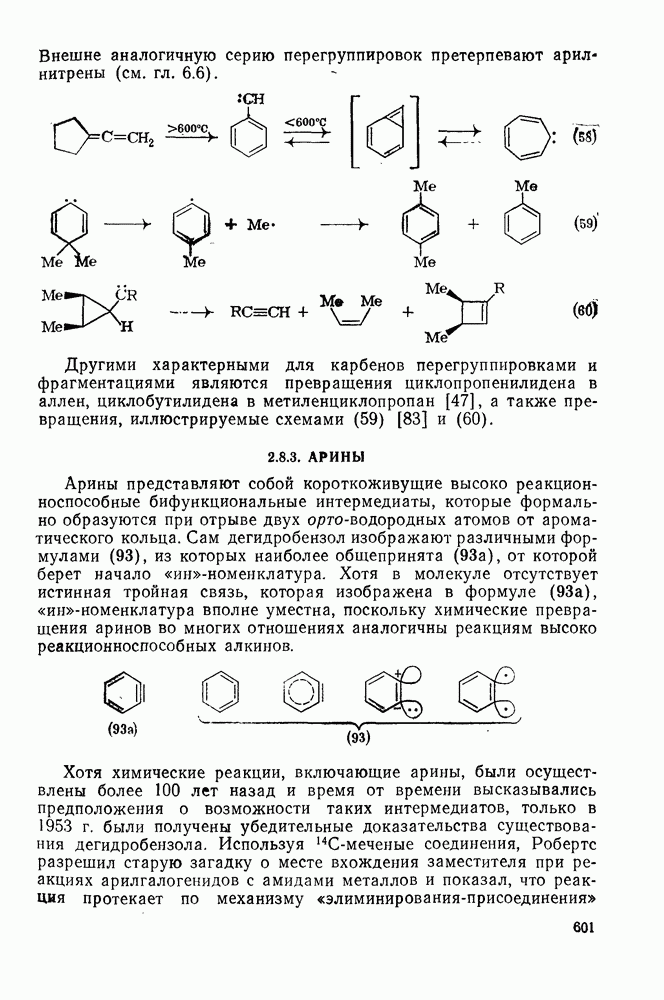
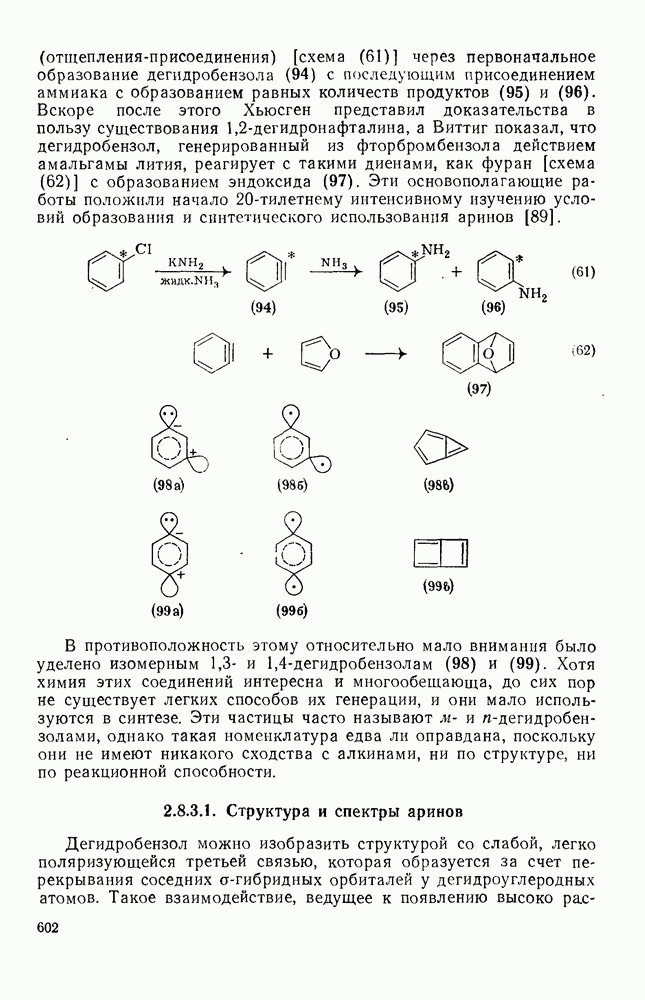
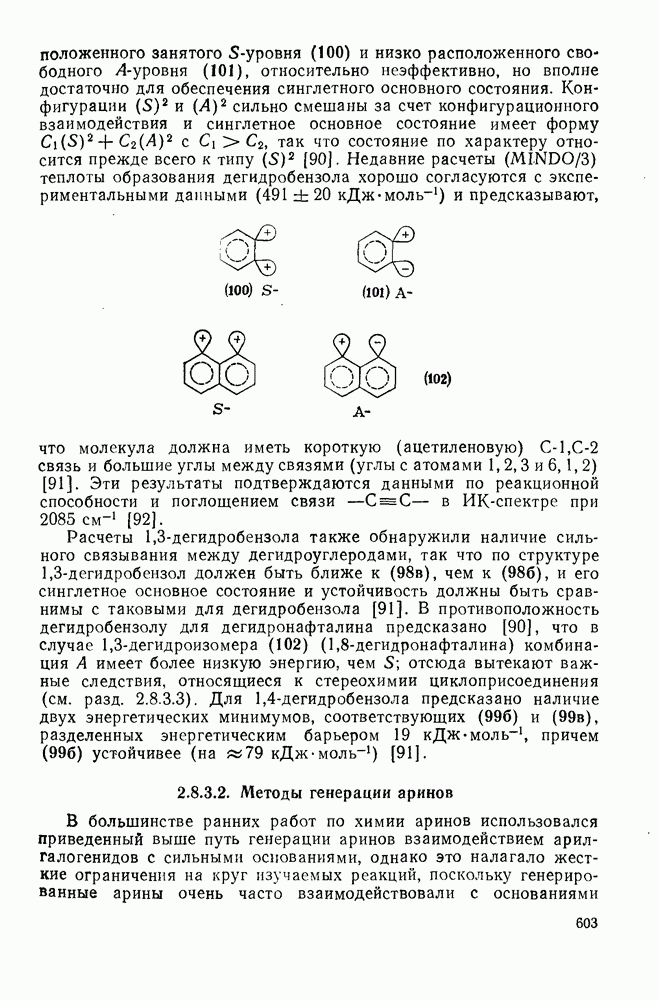

























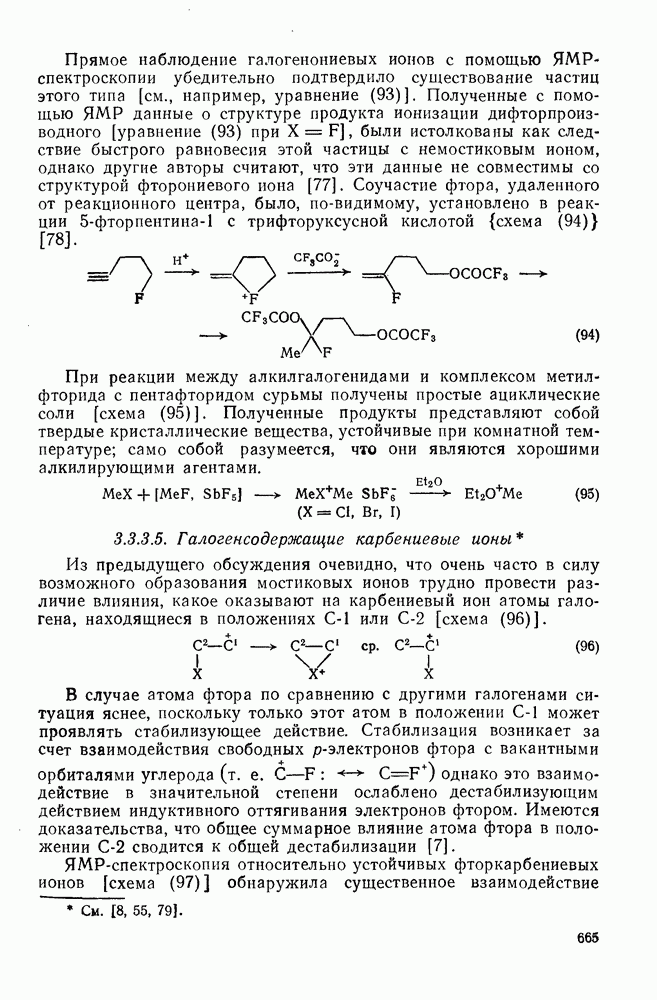
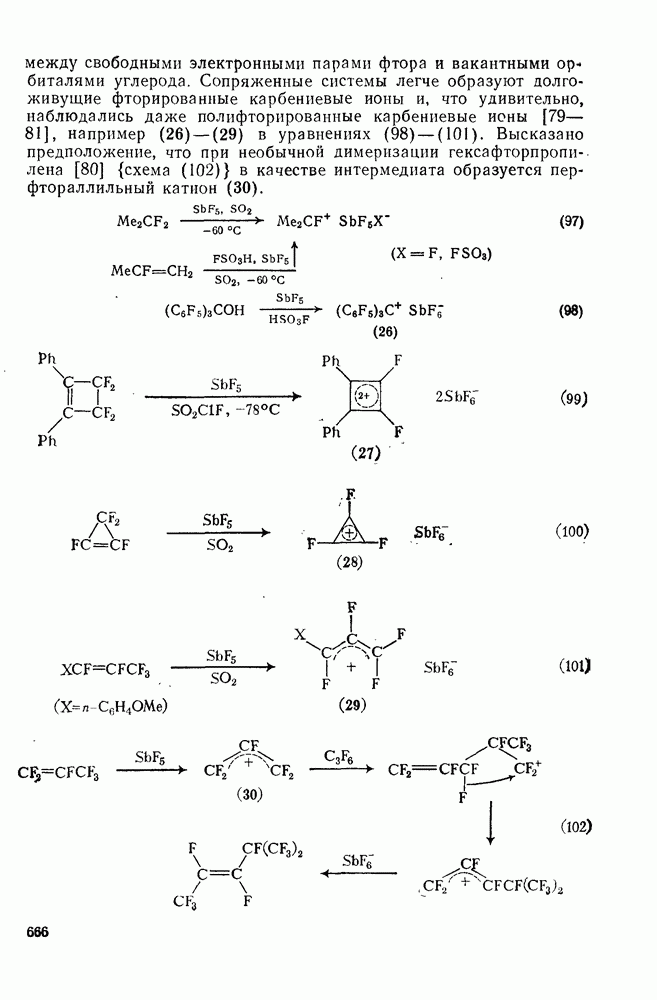
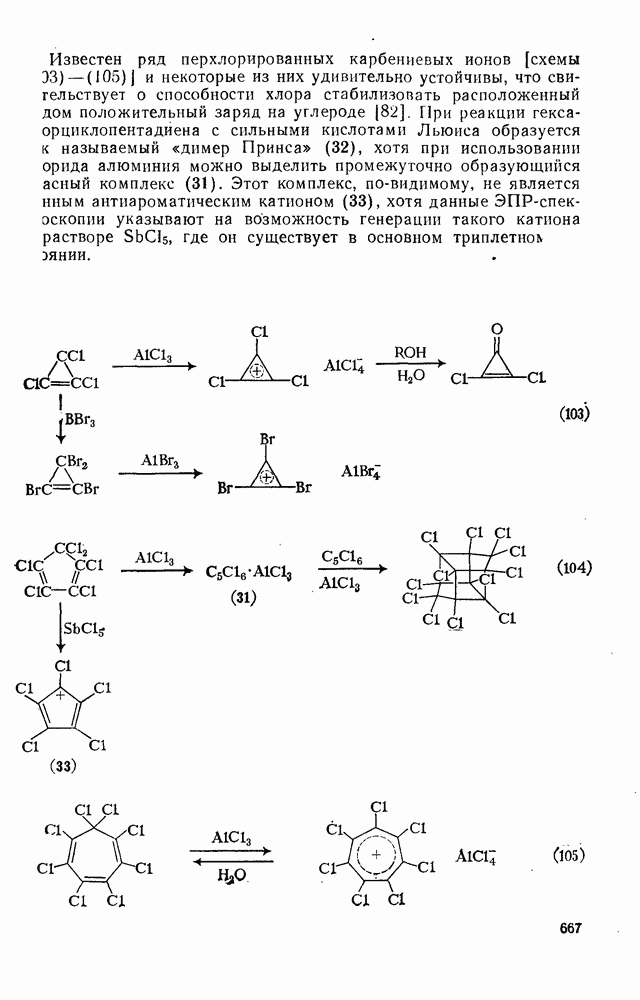

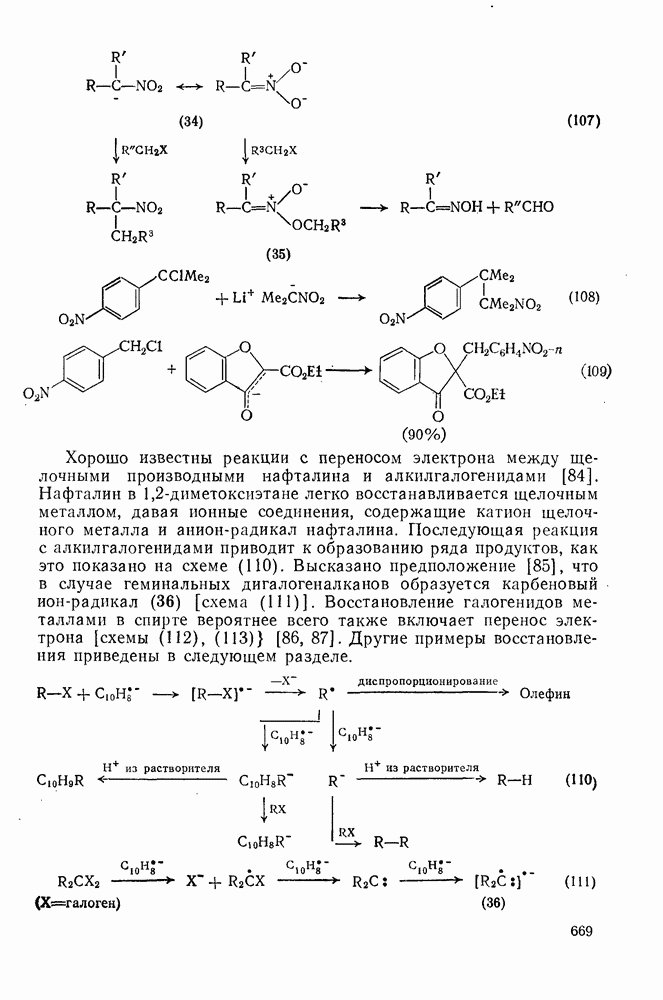


























 поляризован
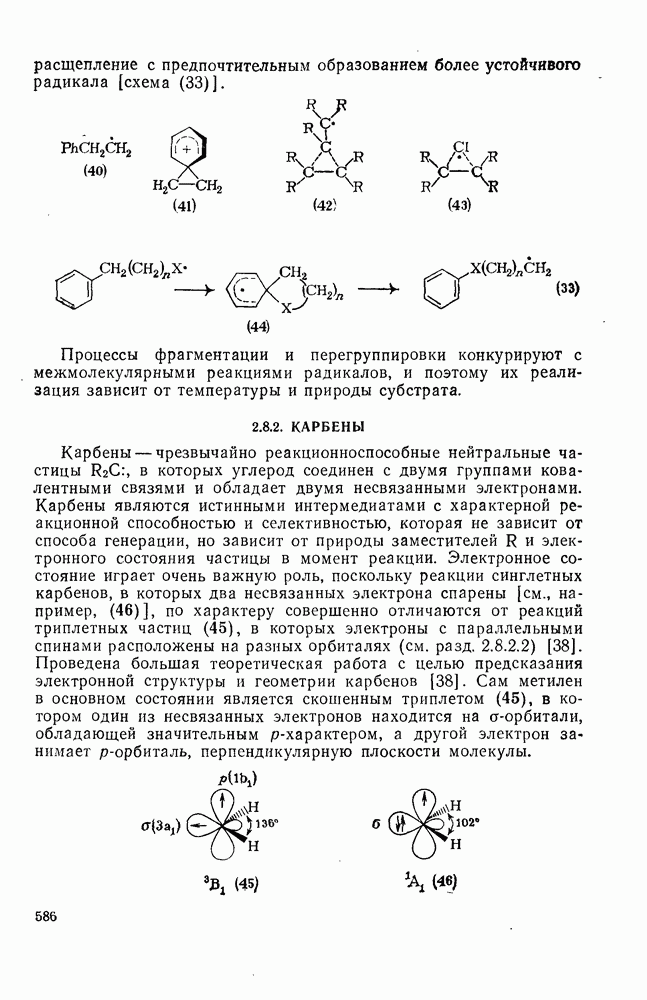
поляризован  , где Y более электроотрицателен, чем X)] [57]. Так, влияние замены заместителей указывает на появление в переходном состоянии лимитирующей (скоростьопределяющей) стадии частичного положительного чаряда на олефиновом углероде (углеродах).
, где Y более электроотрицателен, чем X)] [57]. Так, влияние замены заместителей указывает на появление в переходном состоянии лимитирующей (скоростьопределяющей) стадии частичного положительного чаряда на олефиновом углероде (углеродах).
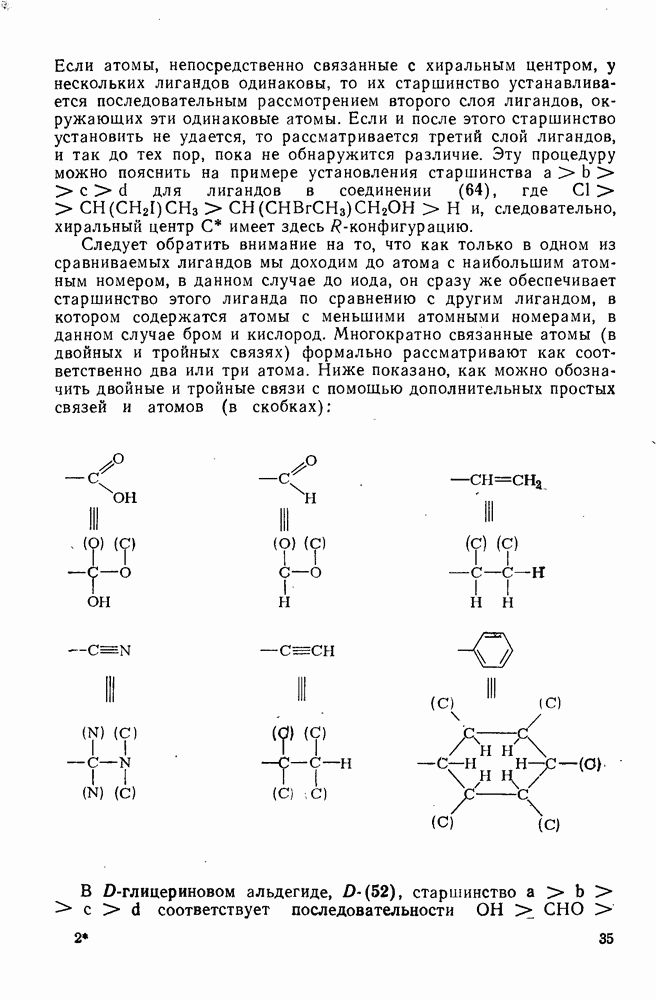
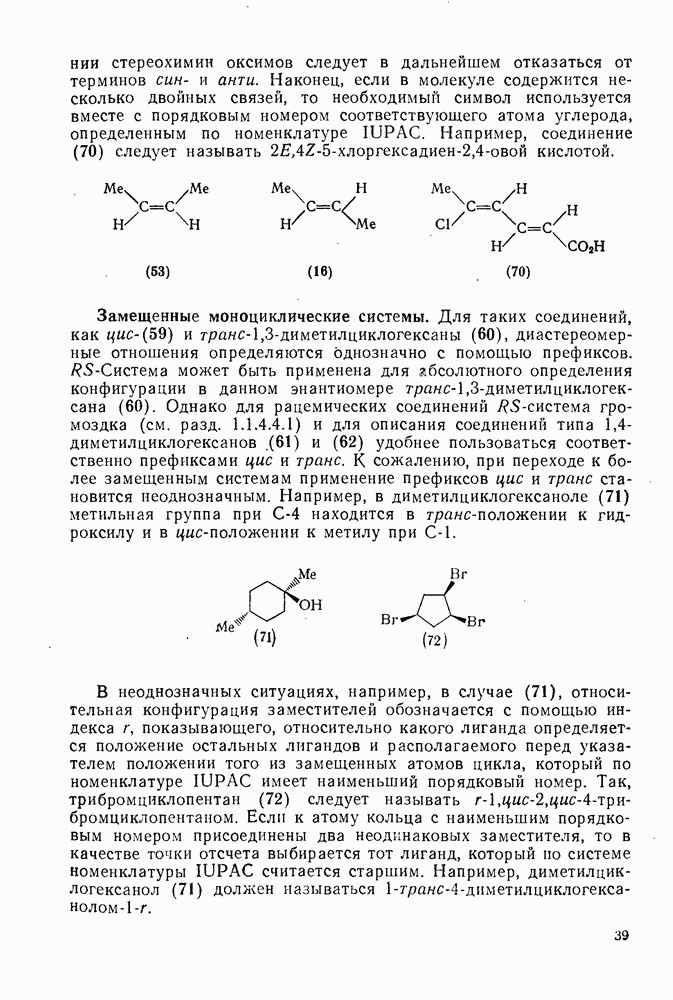
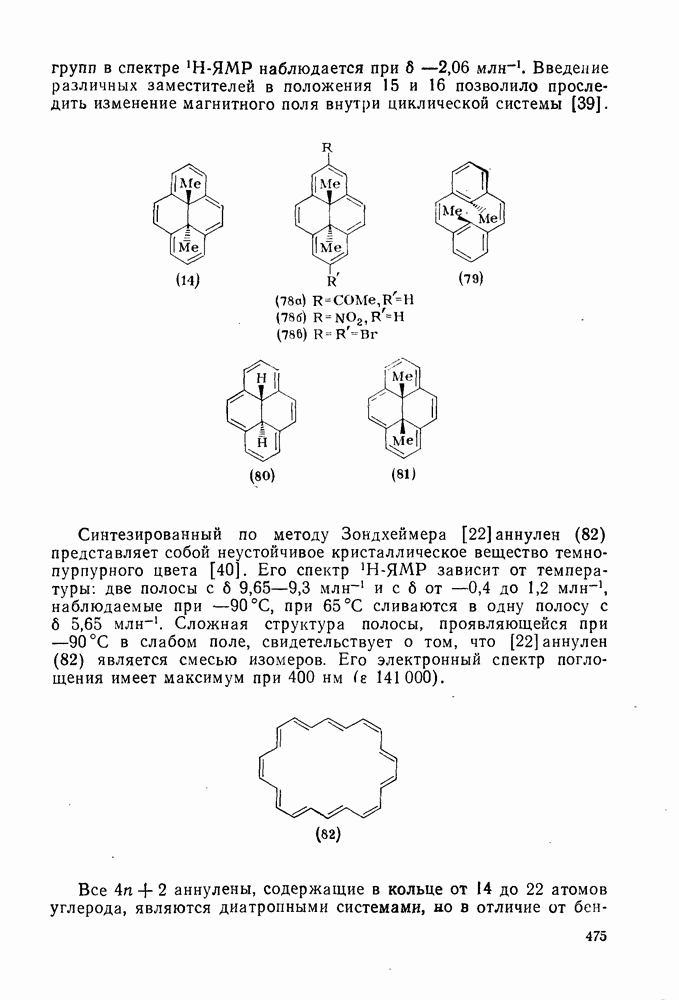
 (механизм, названный по начальным буквам английских слов: addition electrophilic bimolecular — присоединение электрофильное бимолекулярное), т. е. фактически образуется катионный интермедиат. Наиболее часто возникает вопрос, существует ли интермедиат в виде карбениевого иона (ср. уравнение 85), или в виде мостикового иона (уравнение 86). Как часто случается, поведение системы, по-видимому, зависит от природы реагента и субстрата.
(механизм, названный по начальным буквам английских слов: addition electrophilic bimolecular — присоединение электрофильное бимолекулярное), т. е. фактически образуется катионный интермедиат. Наиболее часто возникает вопрос, существует ли интермедиат в виде карбениевого иона (ср. уравнение 85), или в виде мостикового иона (уравнение 86). Как часто случается, поведение системы, по-видимому, зависит от природы реагента и субстрата.

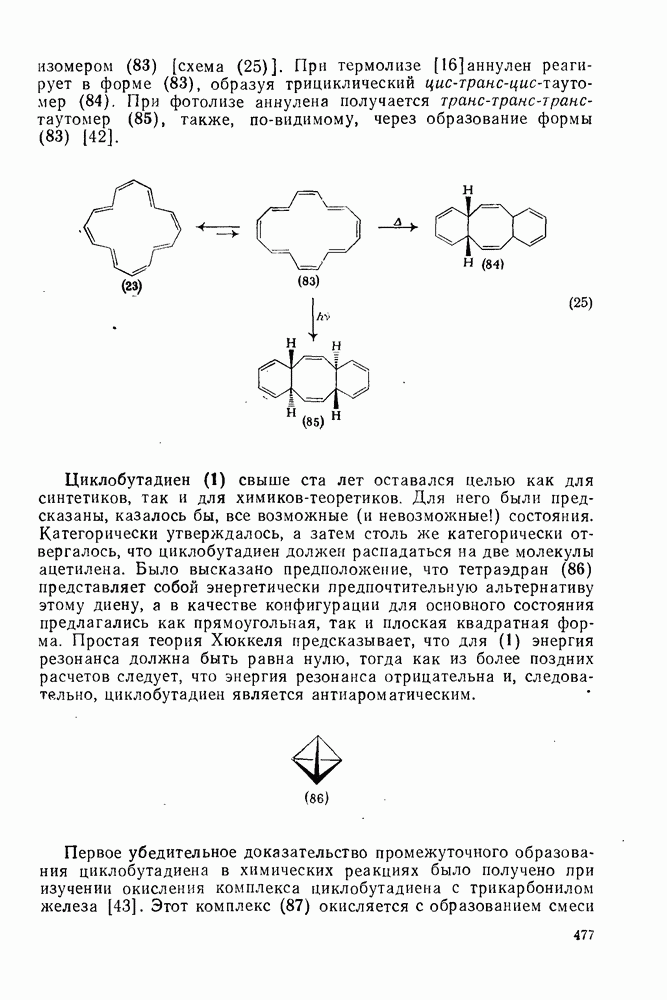
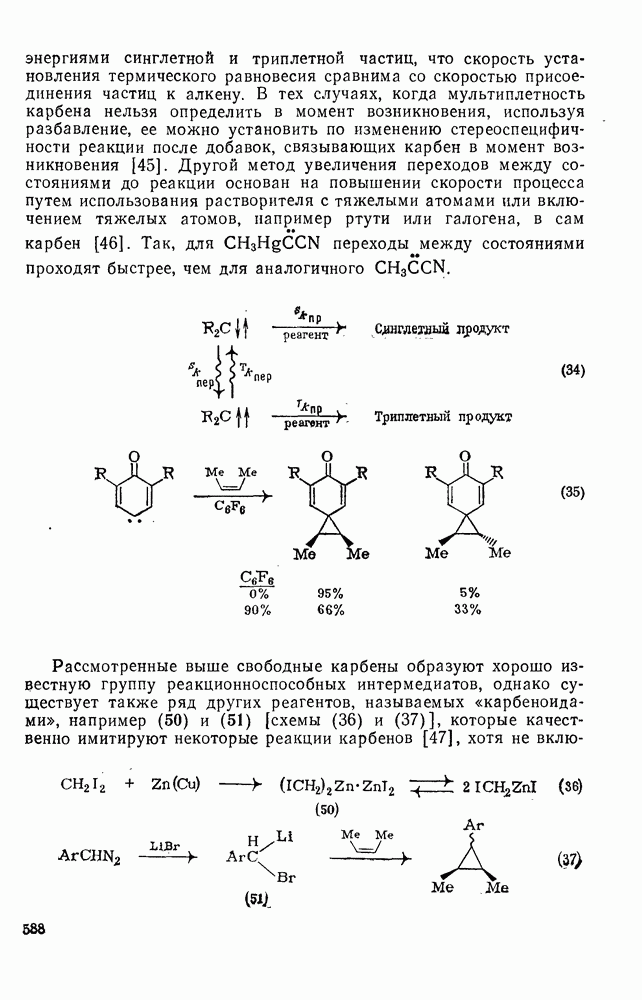
 , в котором переходное состояние на лимитирующей стадии включает третью частицу — вторую молекулу реагента
, в котором переходное состояние на лимитирующей стадии включает третью частицу — вторую молекулу реагента  или молекулу растворителя
или молекулу растворителя  :
:

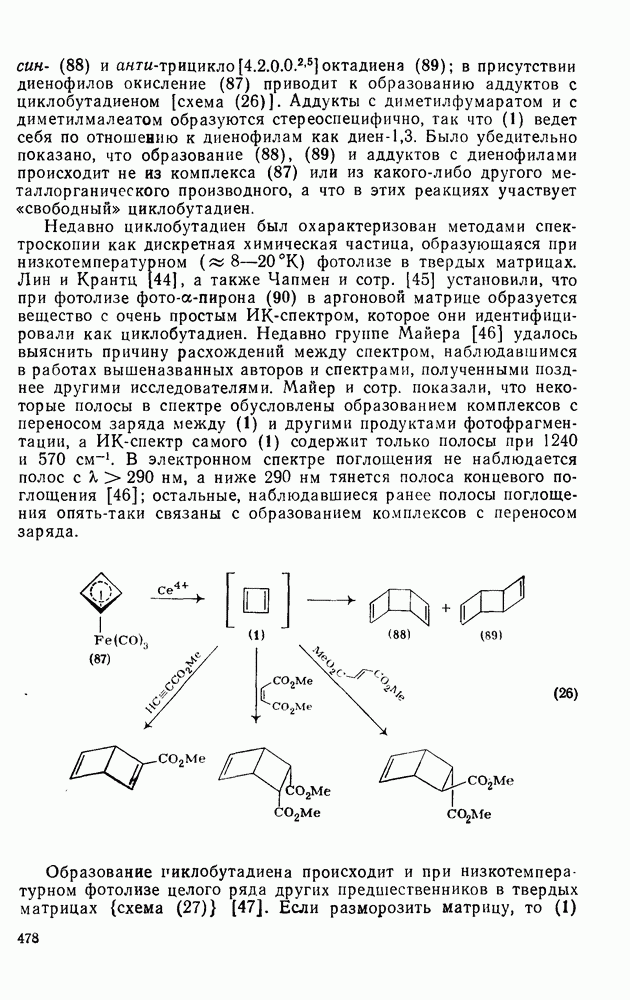
 : 1) катализуемую кислотами гидратацию и 2) присоединение сульфенилгалогенидов.
: 1) катализуемую кислотами гидратацию и 2) присоединение сульфенилгалогенидов.

 реакции гидратации замещенных стиролов прекрасно коррелирует со значениями
реакции гидратации замещенных стиролов прекрасно коррелирует со значениями  , при этом
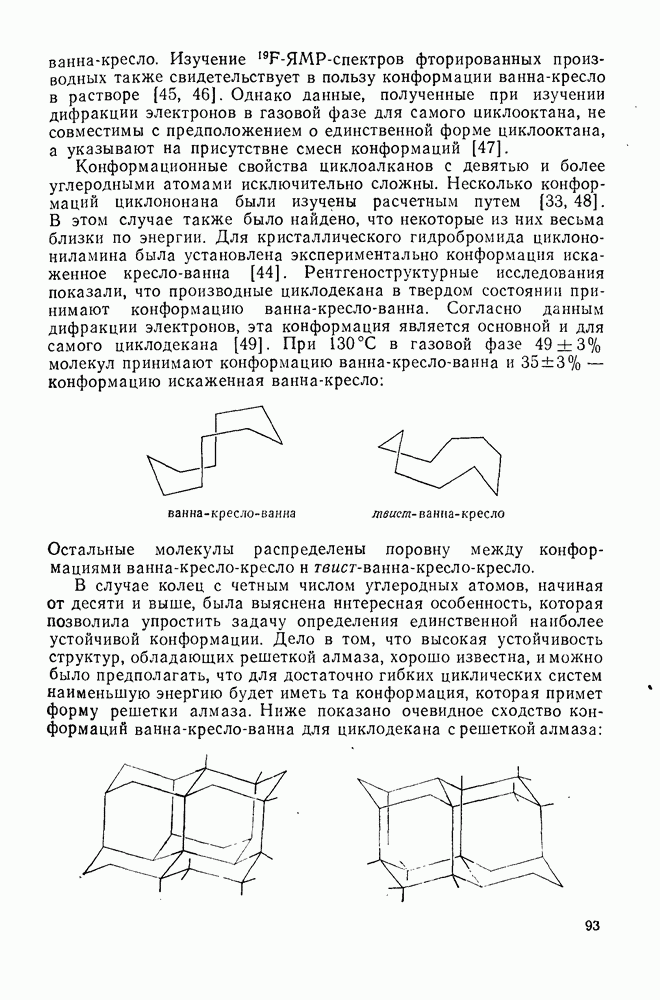
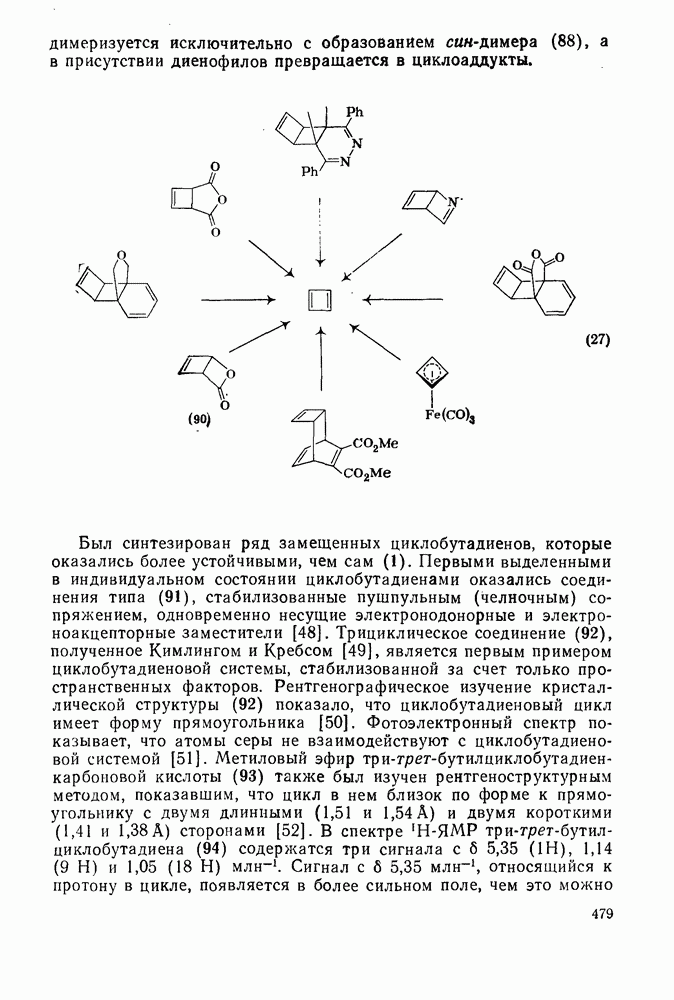
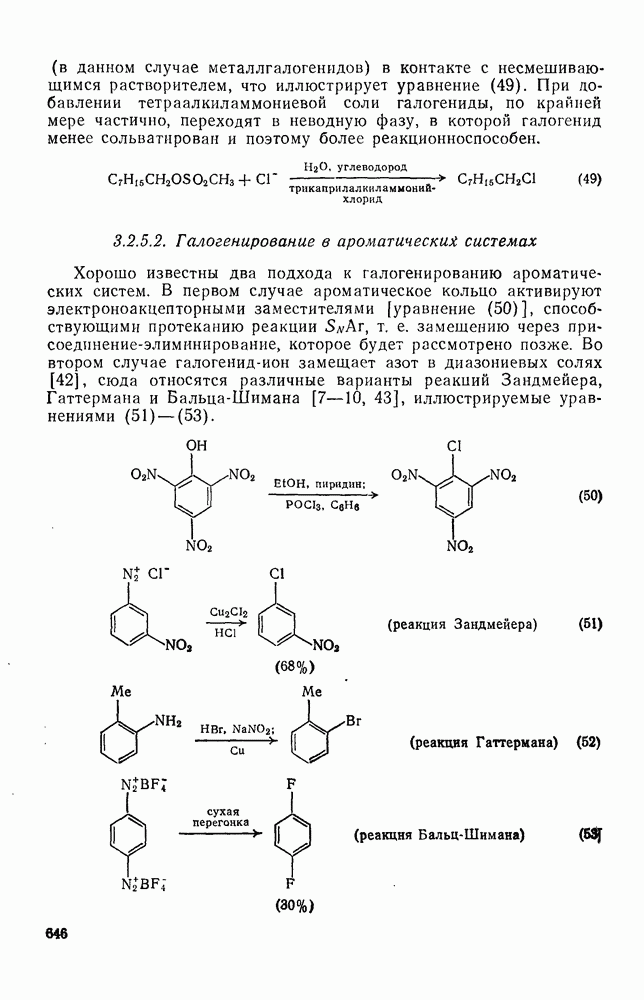
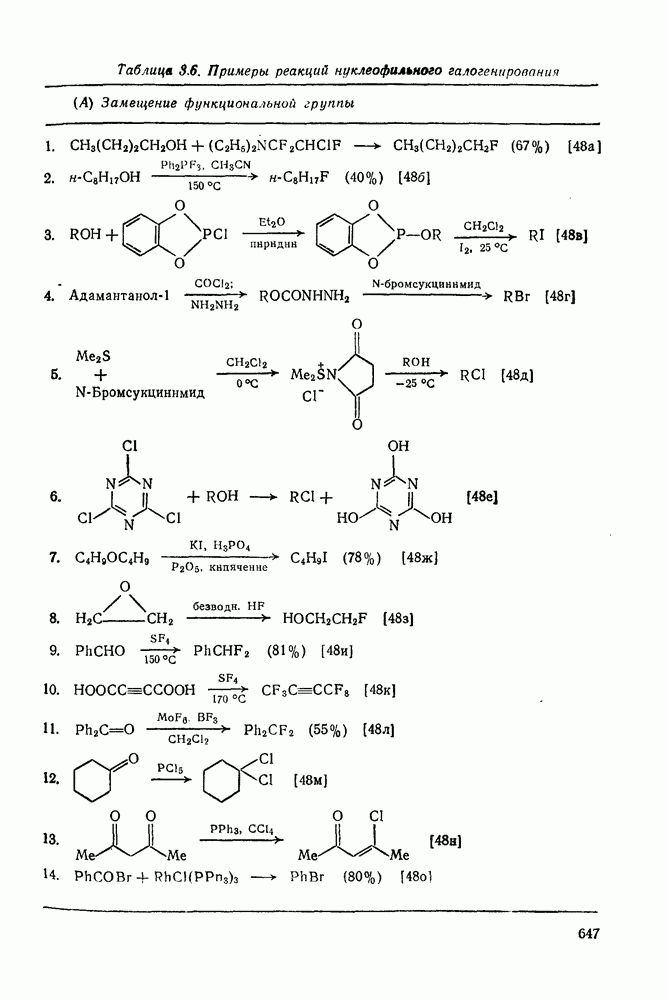
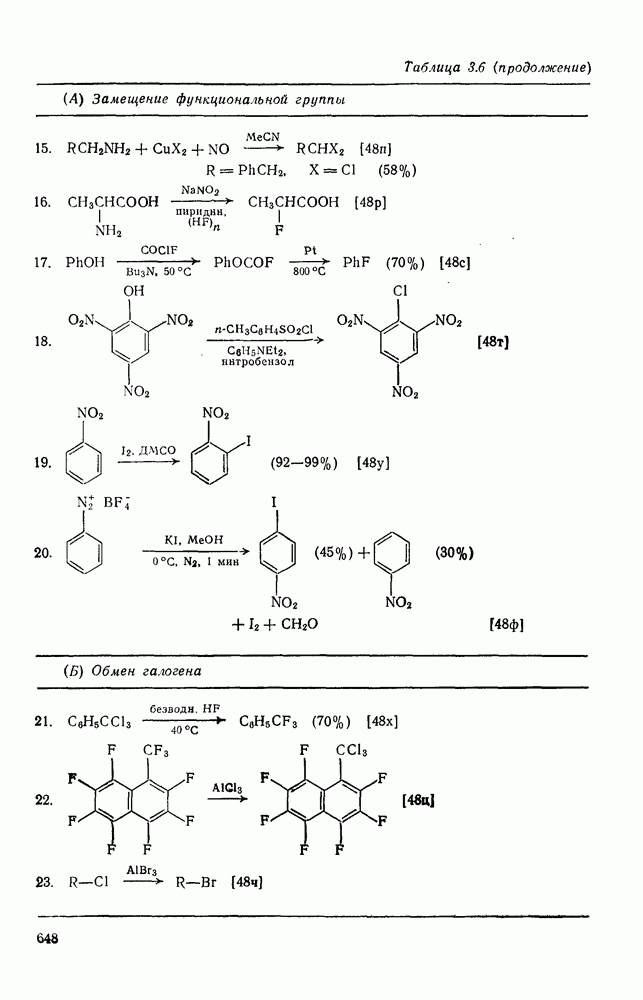
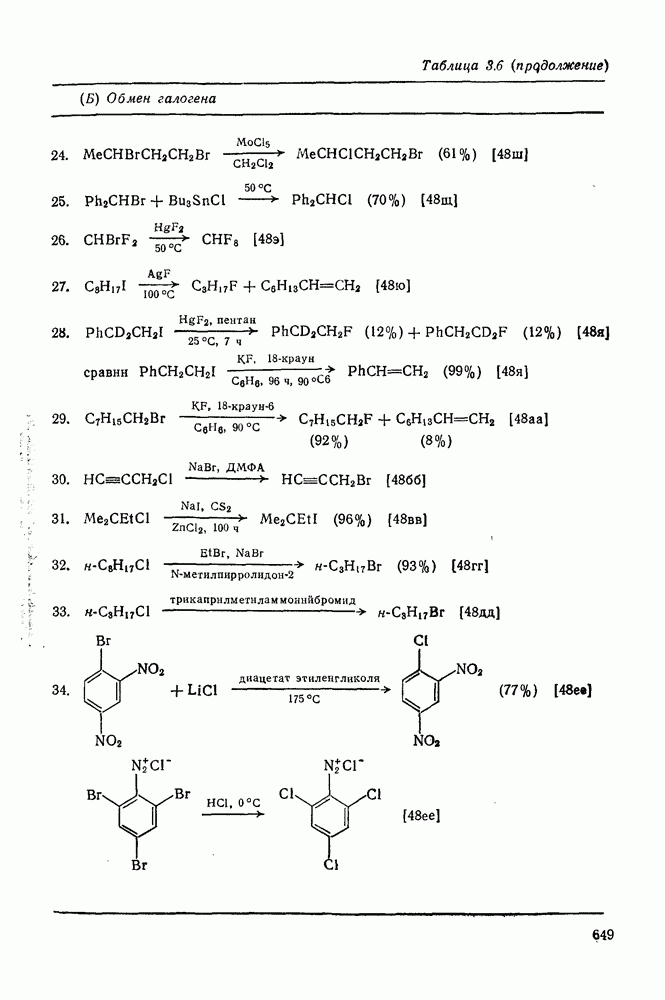
, при этом  имеет большое отрицательное значение (
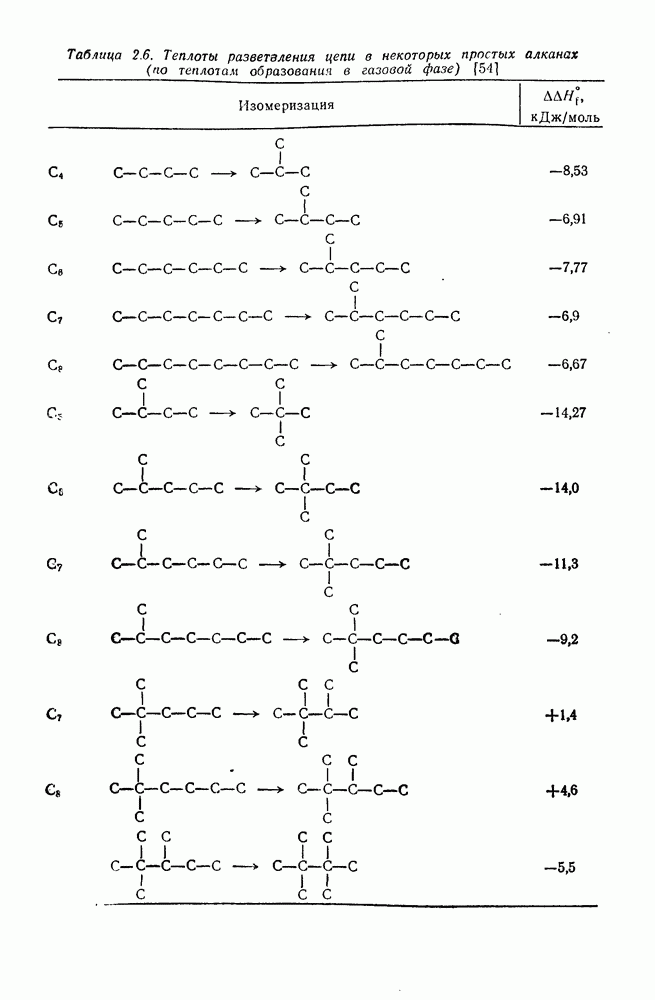
имеет большое отрицательное значение ( -наблюдаемая константа скорости реакции первого порядка, а
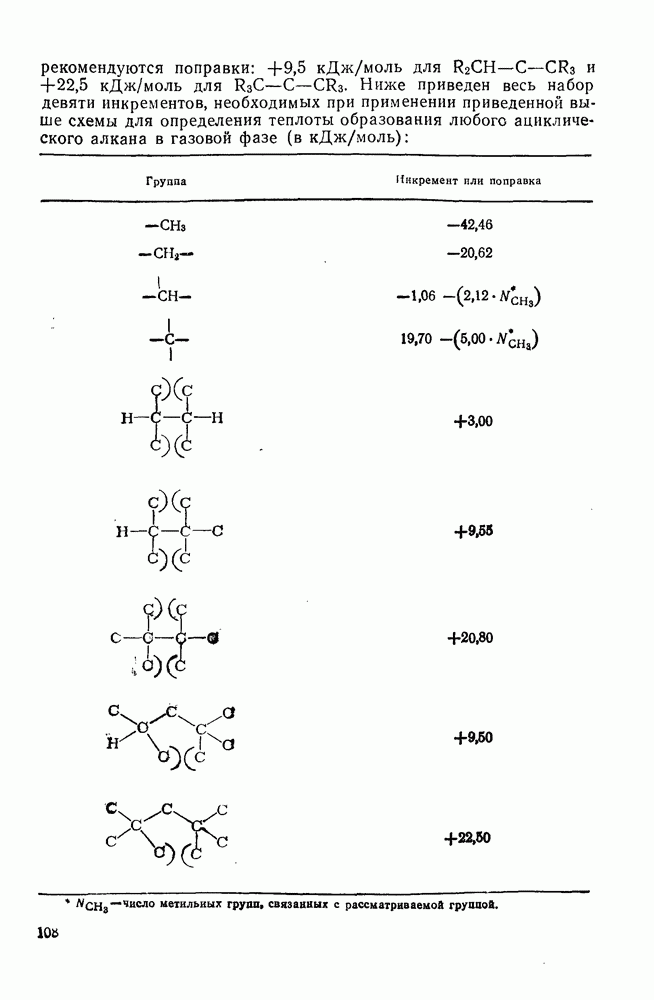
-наблюдаемая константа скорости реакции первого порядка, а  и
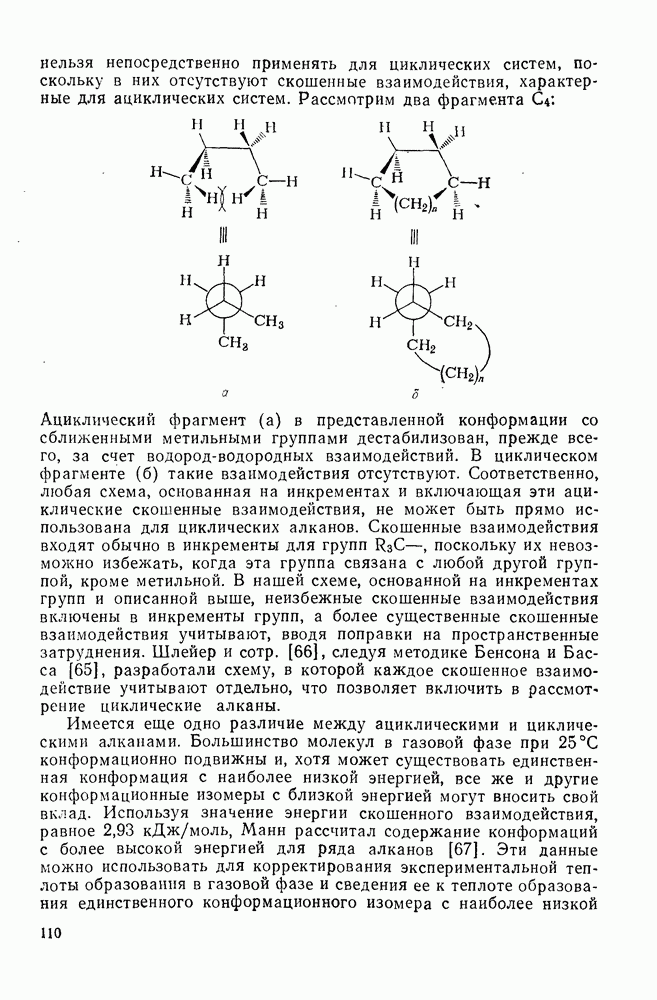
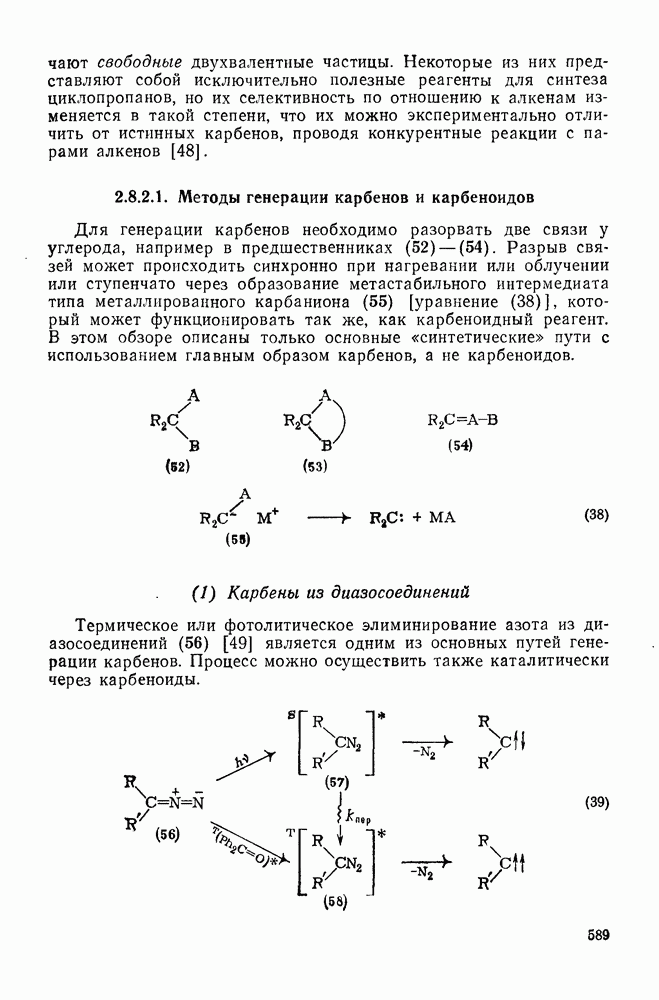
и  — соответствующие постоянные в уравнении Гаммета), а в подходящих системах наблюдаются перегруппировки, характерные для карбениевых ионов. Скорость гидратации увеличивается в последовательности:
— соответствующие постоянные в уравнении Гаммета), а в подходящих системах наблюдаются перегруппировки, характерные для карбениевых ионов. Скорость гидратации увеличивается в последовательности:

 и
и  лучше всего согласуются с промежуточным образованием карбениевого иона, а не мостикового иона. Более того, было показано, что в реакции гидратации напряженных олефинов имеет место общий кислотный катализ. Для всех олефинов, кроме сильно дезактивированных, эти данные согласуются с простым механизмом, представленным уравнением (88):
лучше всего согласуются с промежуточным образованием карбениевого иона, а не мостикового иона. Более того, было показано, что в реакции гидратации напряженных олефинов имеет место общий кислотный катализ. Для всех олефинов, кроме сильно дезактивированных, эти данные согласуются с простым механизмом, представленным уравнением (88):


 , идущего через образование мостикового интермедиата (эписульфониевого иона), олефиновые атомы углерода которого обладают в переходном состоянии слабо выраженным катионным характером (уравнение 90). На стадию, определяющую характер продукта, - раскрытие эписульфониевого иона — влияют пространственные эффекты, ответственные, по-видимому, за антимарковнивскую ориентацию.
, идущего через образование мостикового интермедиата (эписульфониевого иона), олефиновые атомы углерода которого обладают в переходном состоянии слабо выраженным катионным характером (уравнение 90). На стадию, определяющую характер продукта, - раскрытие эписульфониевого иона — влияют пространственные эффекты, ответственные, по-видимому, за антимарковнивскую ориентацию.


 -механизма, который объясняет наблюдаемое анти-присоединение. В случае сопряженных олефинов таких, как аценафтилен, инден и другие, происходит предпочтительно син-присоединение (которое можно наблюдать, используя бромистый дейтерий), что объясняют образованием промежуточной ионной пары—ион карбення — галогенид-нон, которому благоприятствует сопряжение с заместителем (заместителями), приводящее к стабилизации карбениевого иона.
-механизма, который объясняет наблюдаемое анти-присоединение. В случае сопряженных олефинов таких, как аценафтилен, инден и другие, происходит предпочтительно син-присоединение (которое можно наблюдать, используя бромистый дейтерий), что объясняют образованием промежуточной ионной пары—ион карбення — галогенид-нон, которому благоприятствует сопряжение с заместителем (заместителями), приводящее к стабилизации карбениевого иона.
 происходит электрофильное присоединение молекулярного фтора к простым олефинам. Преобладает син-присоединение, однако в этом случае участие мостиковых ионов в реакции не постулируется, и экспериментальные данные в пользу их образования в процессе реакции также отсутствуют.
происходит электрофильное присоединение молекулярного фтора к простым олефинам. Преобладает син-присоединение, однако в этом случае участие мостиковых ионов в реакции не постулируется, и экспериментальные данные в пользу их образования в процессе реакции также отсутствуют.

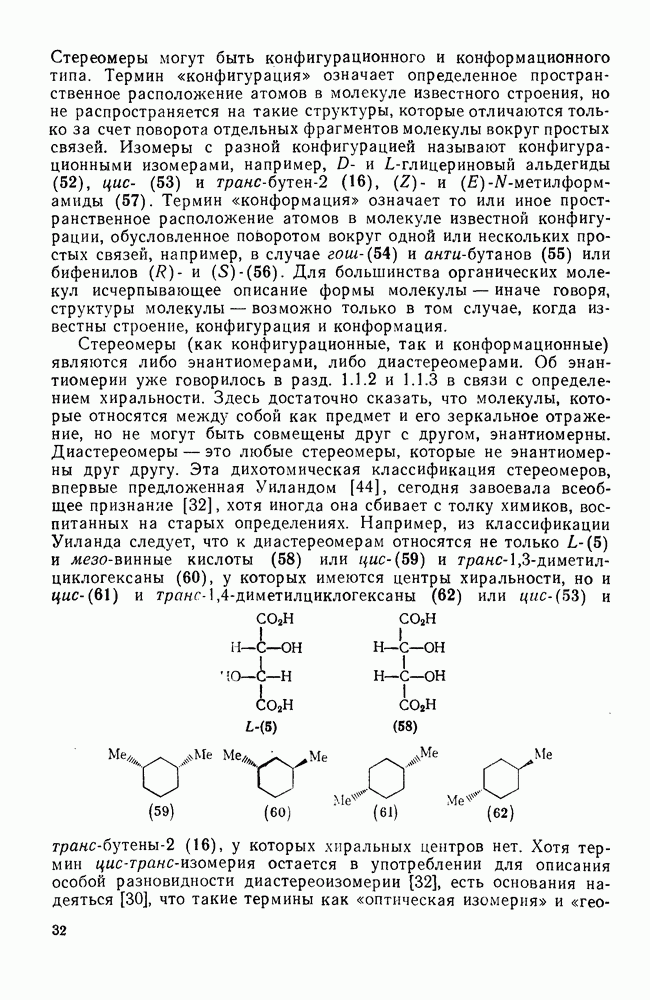
 с образованием промежуточного мостикового иона, в то время как для арилзамещенных олефинов, в которых возможна бензильная стабилизация карбениевого иона, предполагается образование открытого иона.
с образованием промежуточного мостикового иона, в то время как для арилзамещенных олефинов, в которых возможна бензильная стабилизация карбениевого иона, предполагается образование открытого иона.
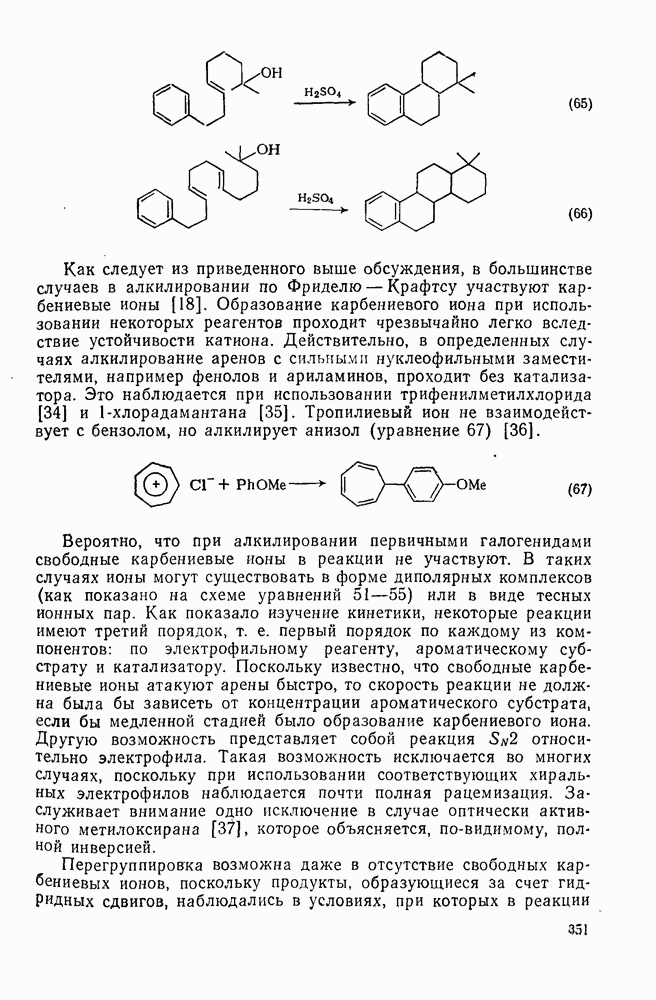
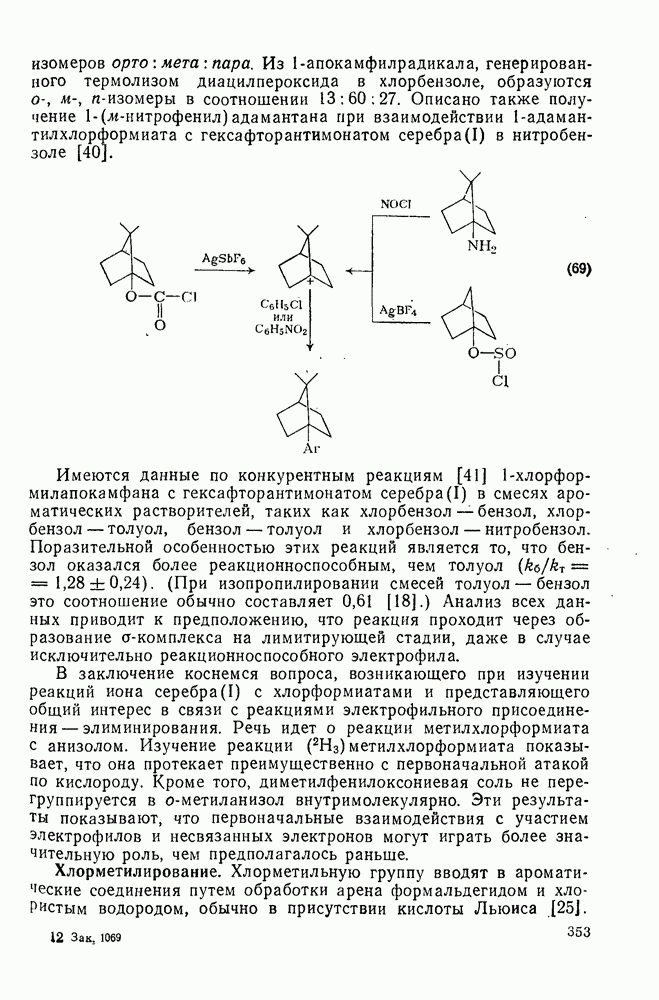
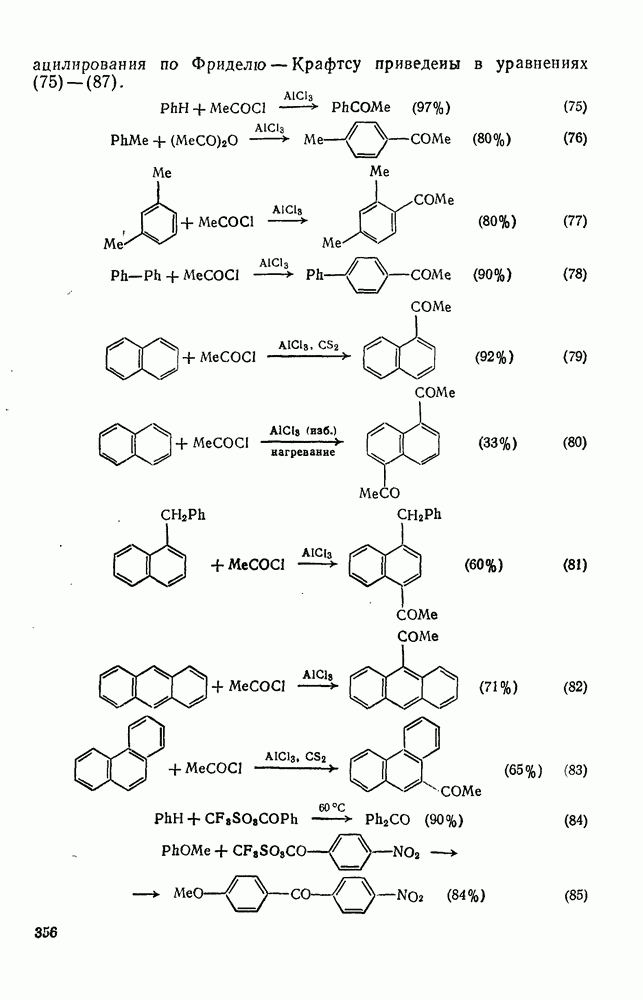
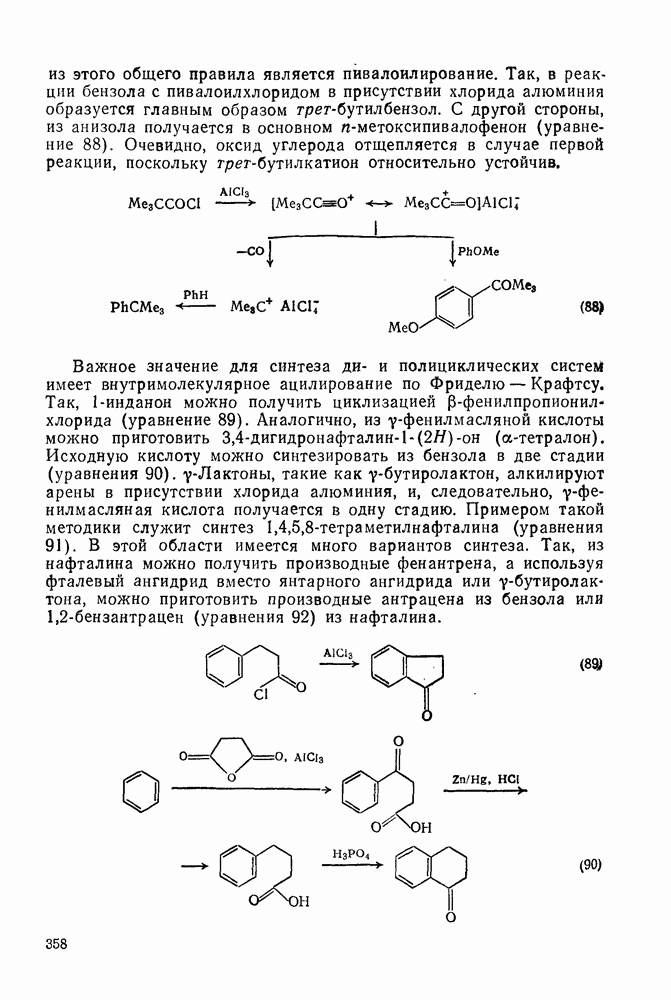
 . Реакции
. Реакции  идут с образованием некоторых побочных продуктов замещения. Образование избытка син-присоединения объяснено участием ионной пары: карбениевый ион — хлорид-ион.
идут с образованием некоторых побочных продуктов замещения. Образование избытка син-присоединения объяснено участием ионной пары: карбениевый ион — хлорид-ион.


 -концентрация олефина,
-концентрация олефина,  и
и  экспериментально определяемые константы скорости реакций второго и третьего порядка, соответственно.
экспериментально определяемые константы скорости реакций второго и третьего порядка, соответственно.
 обычно отсутствуют, наиболее важными членами кинетического уравнения являются первый и второй. В гидроксилсодержащих растворителях доминируют первый и третий члены.
обычно отсутствуют, наиболее важными членами кинетического уравнения являются первый и второй. В гидроксилсодержащих растворителях доминируют первый и третий члены.

 и
и  в четыреххлористом углероде не является полностью анти-стереоспецефичным (уравнения 100 и 101) (ср. с приведенными выше результатами хлорирования). Бромирование
в четыреххлористом углероде не является полностью анти-стереоспецефичным (уравнения 100 и 101) (ср. с приведенными выше результатами хлорирования). Бромирование  , для которого возможно образование более устойчивого катиона, не стереоспецифично.
, для которого возможно образование более устойчивого катиона, не стереоспецифично.

 в случае бромирования несопряженных конформационно подвижных олефинов является образование диаксиального аддукта, по-видимому, вследствие стереоэлектронного контроля раскрытия бромониевого иона (уравнение 102). Имеется ряд указаний на то, что образование бромониевого иона в присутствии пиридина и других третичных оснований является, по крайней мере отчасти, обратимым процессом.
в случае бромирования несопряженных конформационно подвижных олефинов является образование диаксиального аддукта, по-видимому, вследствие стереоэлектронного контроля раскрытия бромониевого иона (уравнение 102). Имеется ряд указаний на то, что образование бромониевого иона в присутствии пиридина и других третичных оснований является, по крайней мере отчасти, обратимым процессом.
 .
.

 -бромониевого иона. Некоторые из реагентов с «положительным иодом», обычно генерируемые действием иода на серебряные соли, приведены в уравнениях
-бромониевого иона. Некоторые из реагентов с «положительным иодом», обычно генерируемые действием иода на серебряные соли, приведены в уравнениях  . В этом случае ориентация зависит от природы реагента (уравнения 108 и 109). Азид иода обнаруживает выраженную тенденцию к анты-присоединепию даже в случае арилзамещенных олефинов (уравнение 110), что указывает, по-видимому, на промежуточное образование иодониевого иона. Аналогично, обратимое образование иона иодония объясняет стереоселективность протекания реакции, представленной уравнением (111); имеются доказательства, что такое равновесие с ионом иодония наблюдается и в других случаях.
. В этом случае ориентация зависит от природы реагента (уравнения 108 и 109). Азид иода обнаруживает выраженную тенденцию к анты-присоединепию даже в случае арилзамещенных олефинов (уравнение 110), что указывает, по-видимому, на промежуточное образование иодониевого иона. Аналогично, обратимое образование иона иодония объясняет стереоселективность протекания реакции, представленной уравнением (111); имеются доказательства, что такое равновесие с ионом иодония наблюдается и в других случаях.

 , циклогексена и
, циклогексена и  -диметил
-диметил  составляют соответственно 1,00, 0,5, 0,30 и 0,07.
составляют соответственно 1,00, 0,5, 0,30 и 0,07.

 к олефинам (обычно в присутствии гидроксилсодержащих растворителей). Аддукты могут быть выделены, как, например, в случае
к олефинам (обычно в присутствии гидроксилсодержащих растворителей). Аддукты могут быть выделены, как, например, в случае  , а иногда они претерпевают дальнейшие превращения, образуя продукты полного окисления олефинов, например при применении
, а иногда они претерпевают дальнейшие превращения, образуя продукты полного окисления олефинов, например при применении  или
или  .
.



 , а продукт реакции и переходное состояние на стадии, определяющей скорость подобны (73). Нельзя сказать определенно возникает ли на предшествующей предравновесной стадии мостиковый «меркуриниевый ион» (74) [65], хотя имеются спектроскопические доказательства, что подобные частицы образуются в суперкислотных средах в отсутствие нуклеофилов.
, а продукт реакции и переходное состояние на стадии, определяющей скорость подобны (73). Нельзя сказать определенно возникает ли на предшествующей предравновесной стадии мостиковый «меркуриниевый ион» (74) [65], хотя имеются спектроскопические доказательства, что подобные частицы образуются в суперкислотных средах в отсутствие нуклеофилов.

 ,
,  ,
,  и т. д. [67] (см. уравнения 125-127).
и т. д. [67] (см. уравнения 125-127).





 (141)
(141)
