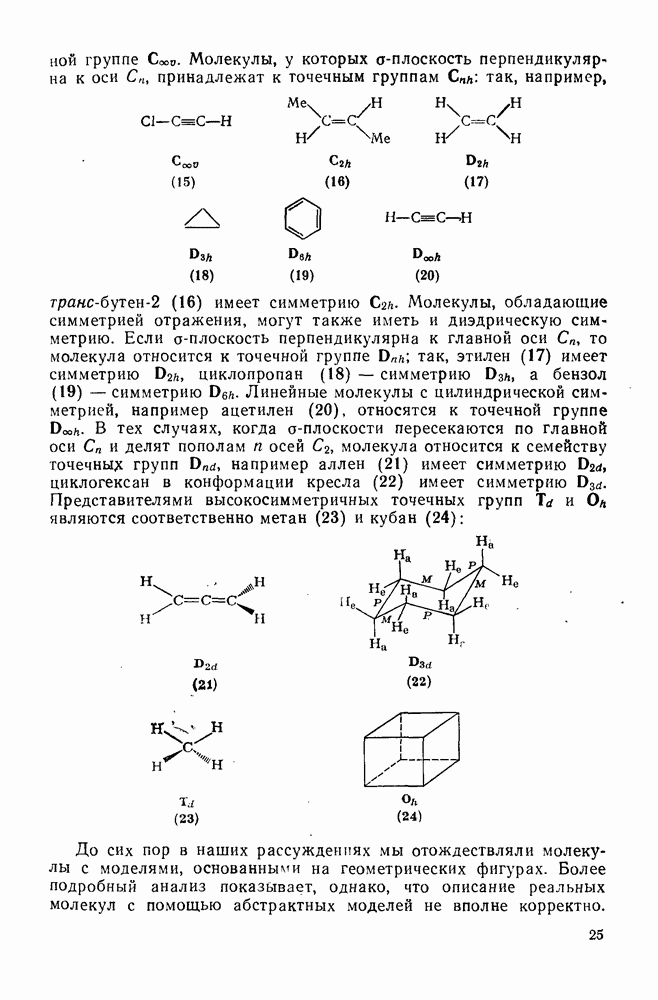
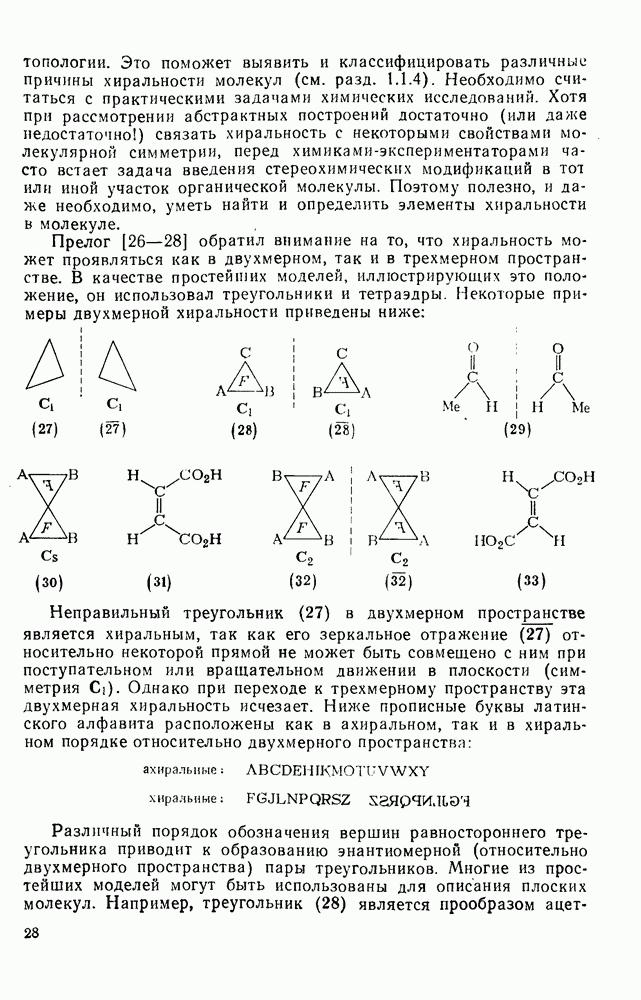
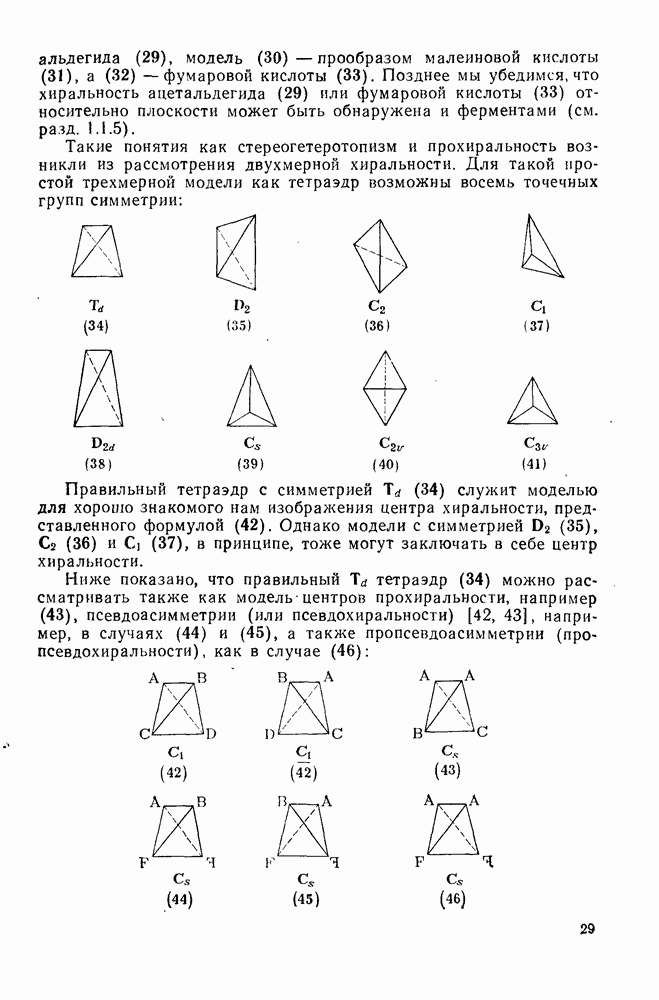
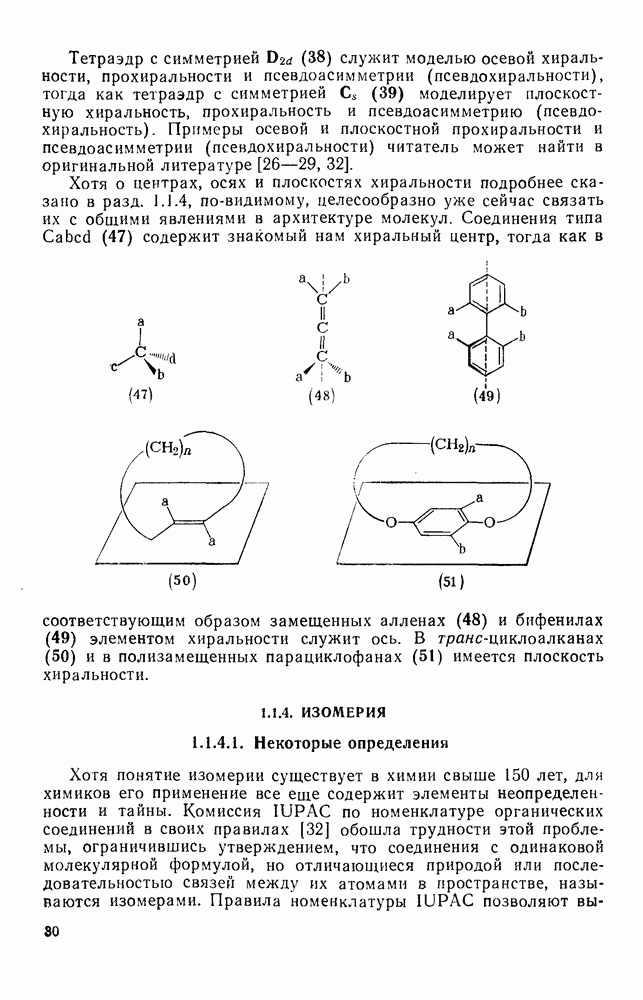
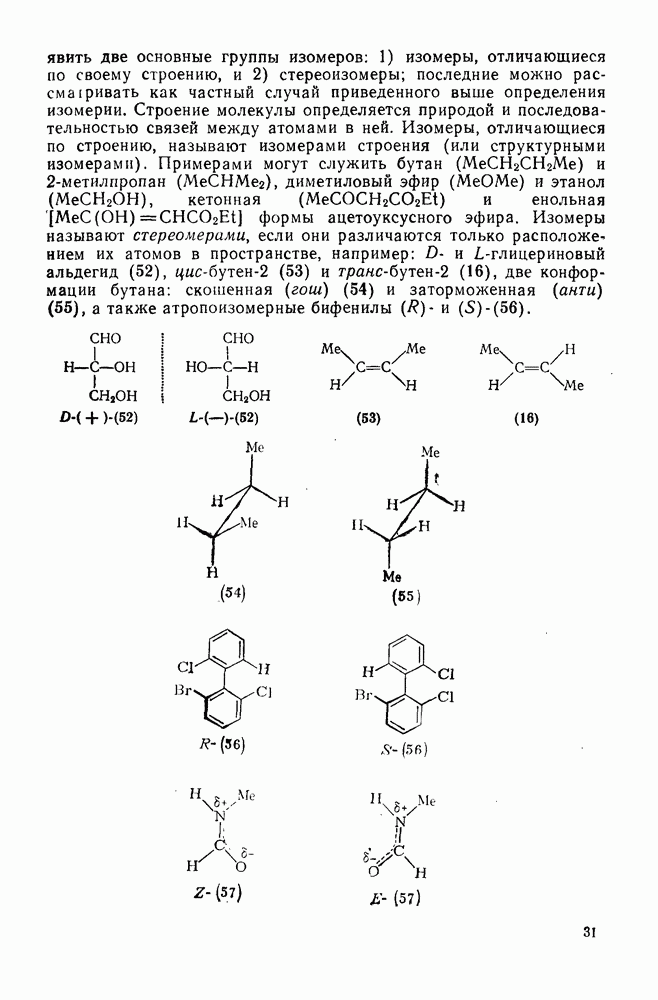
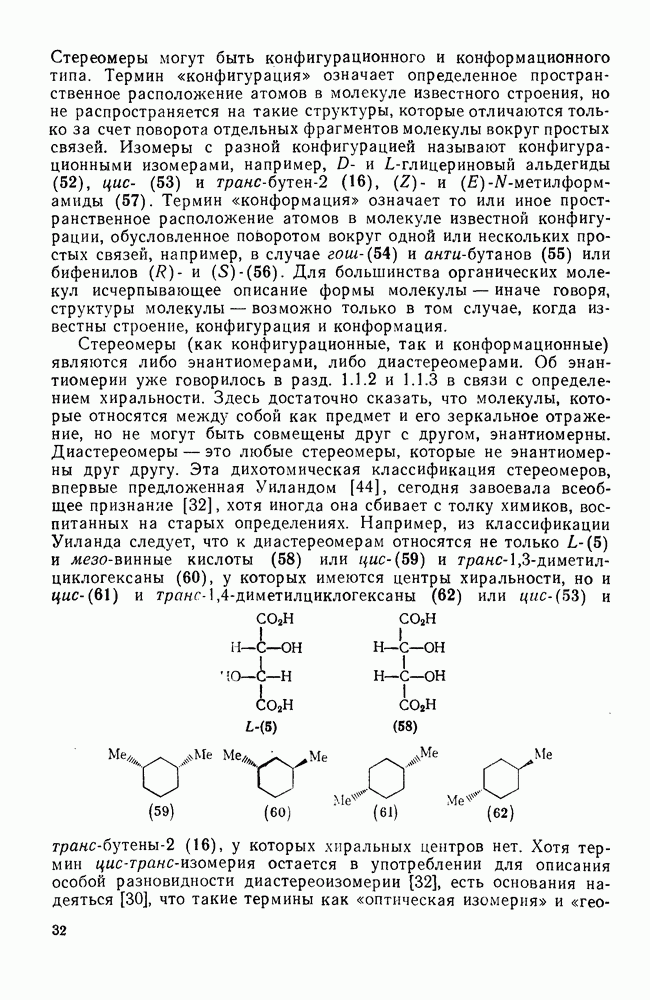
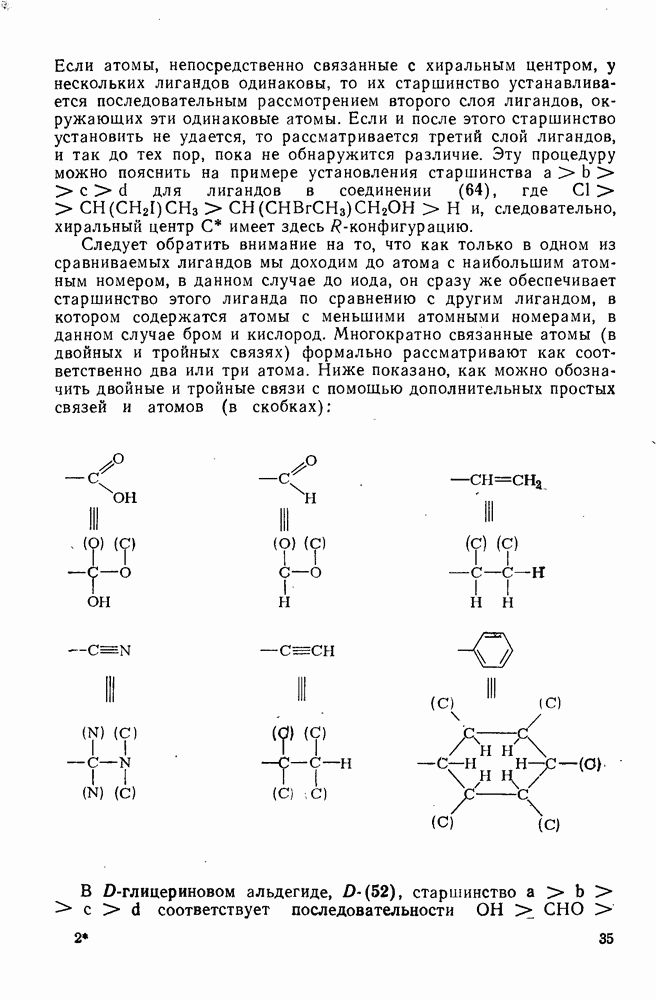
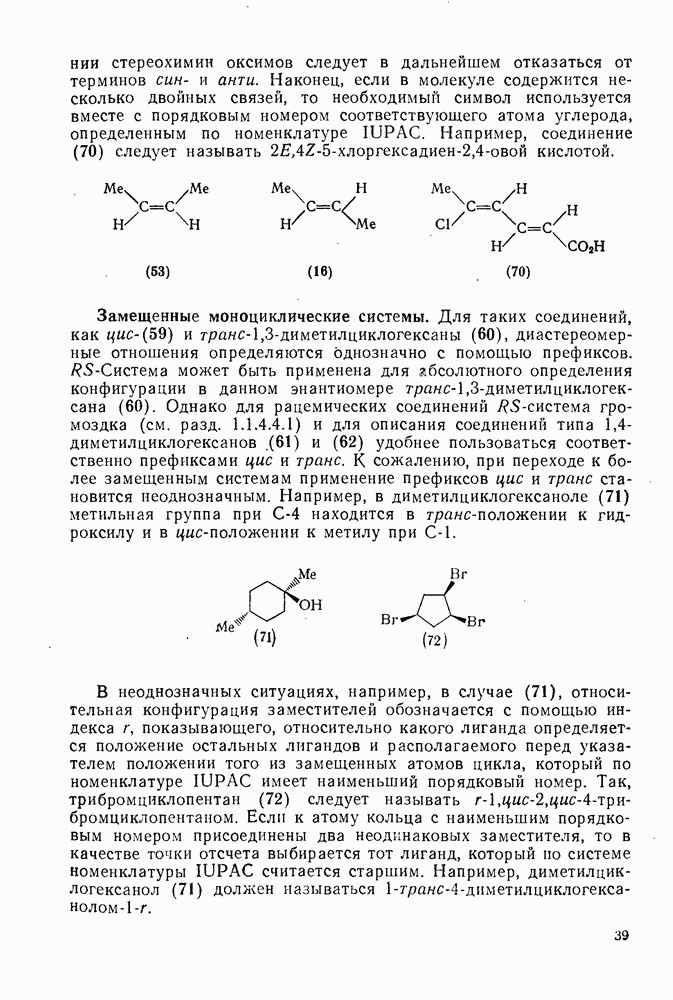
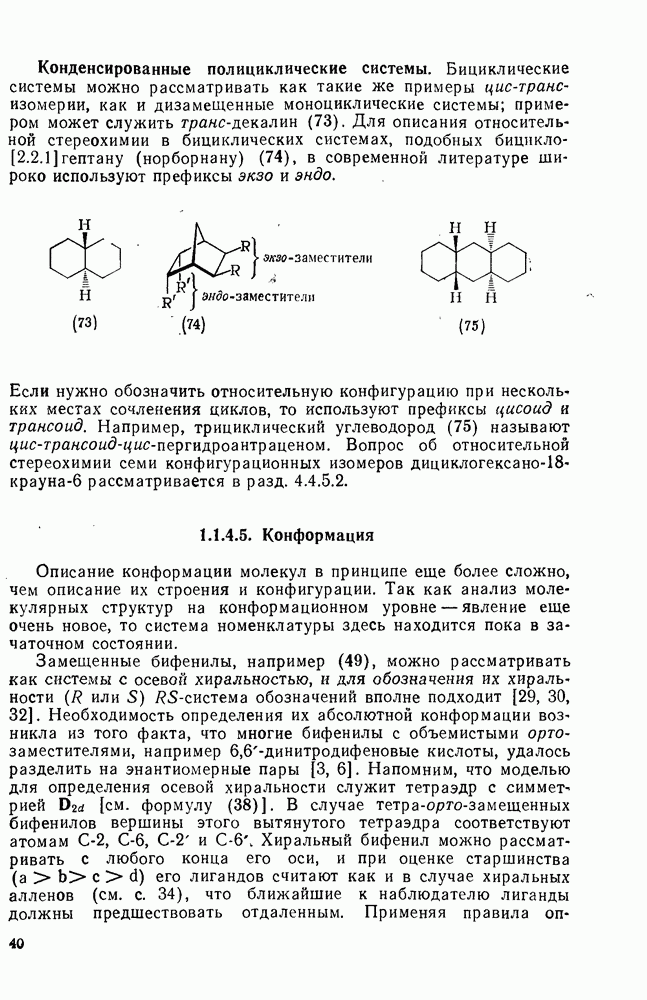
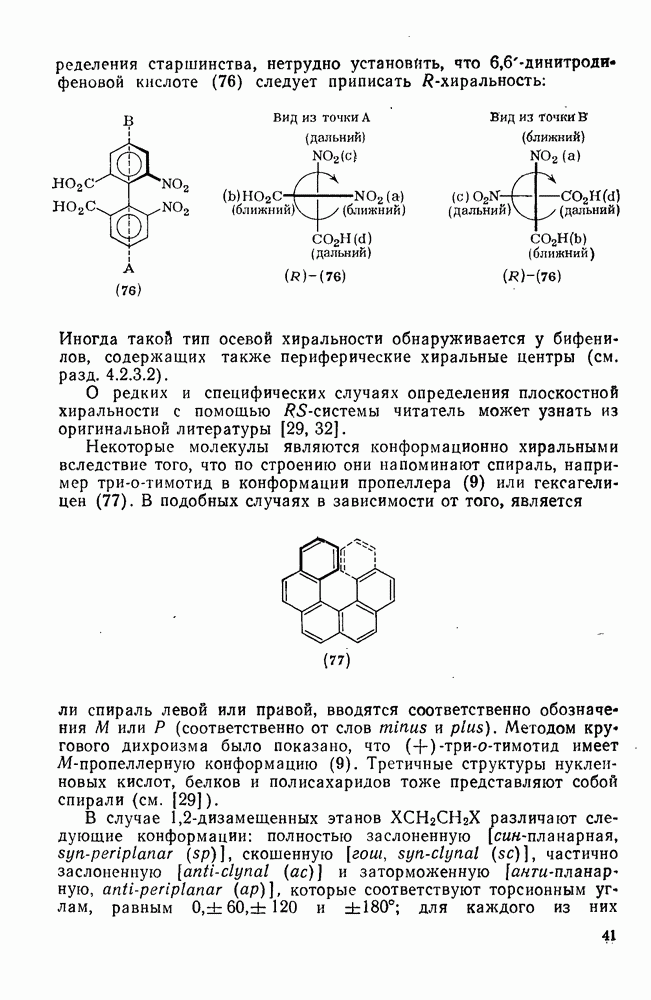
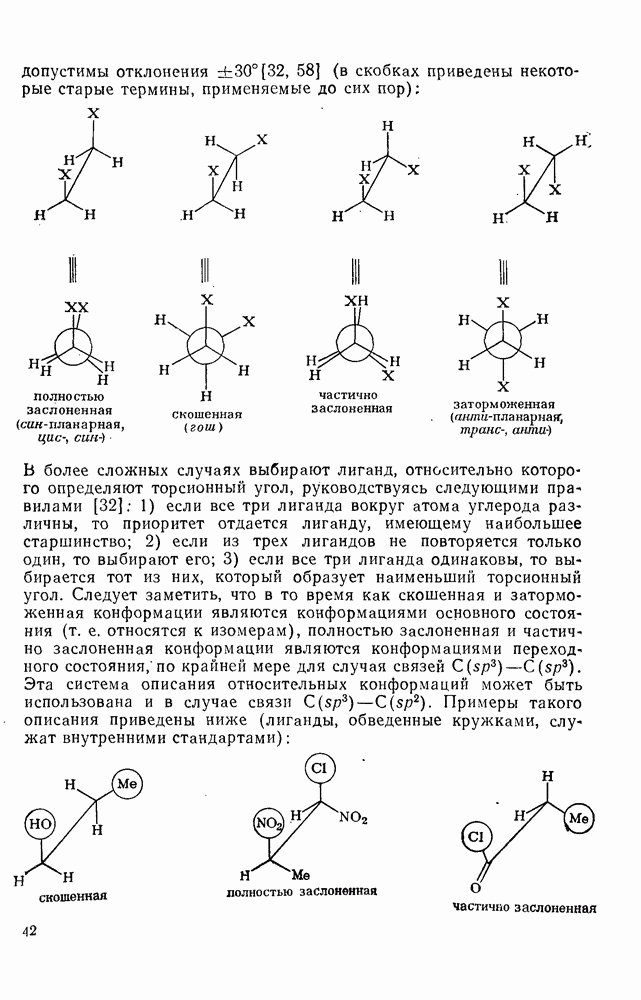
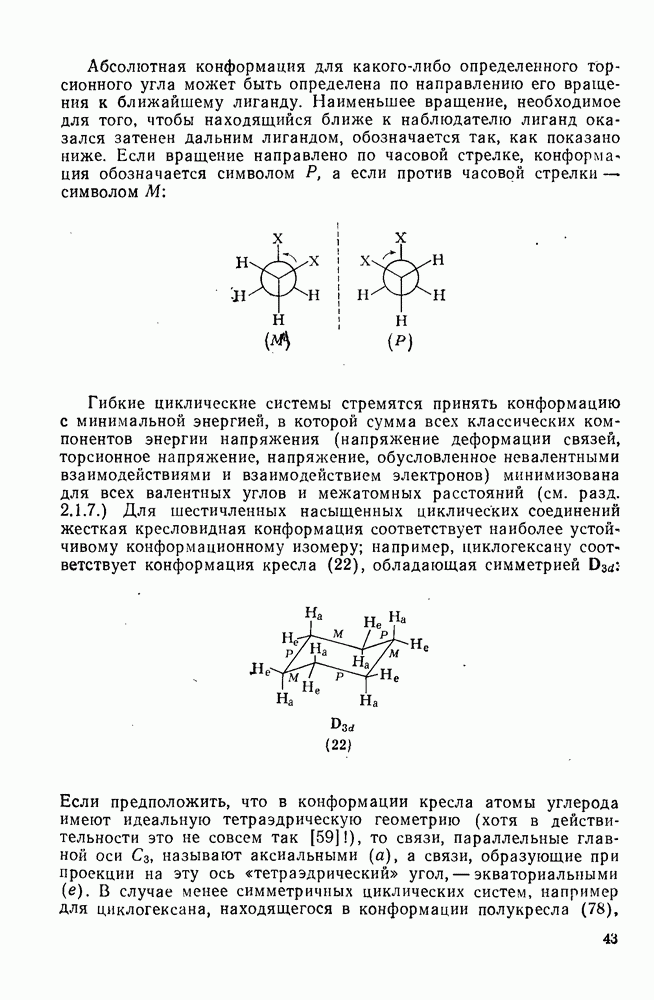
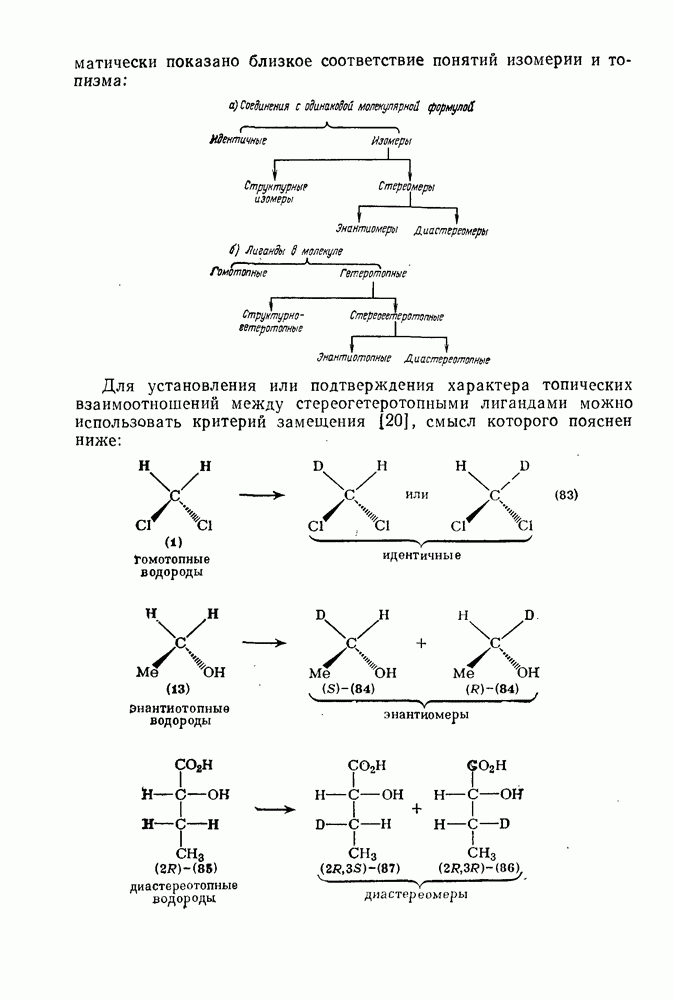
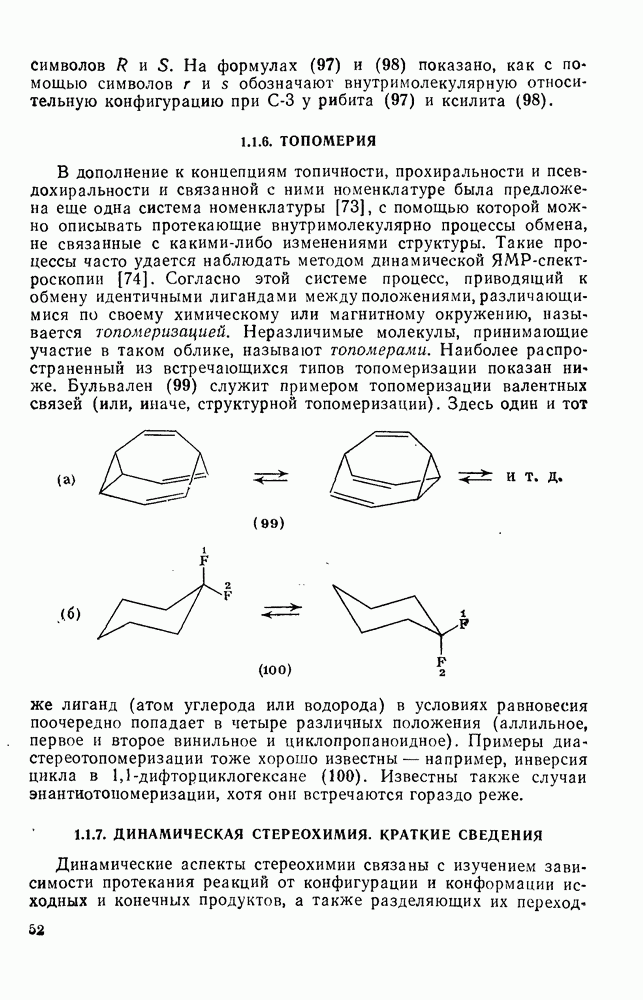
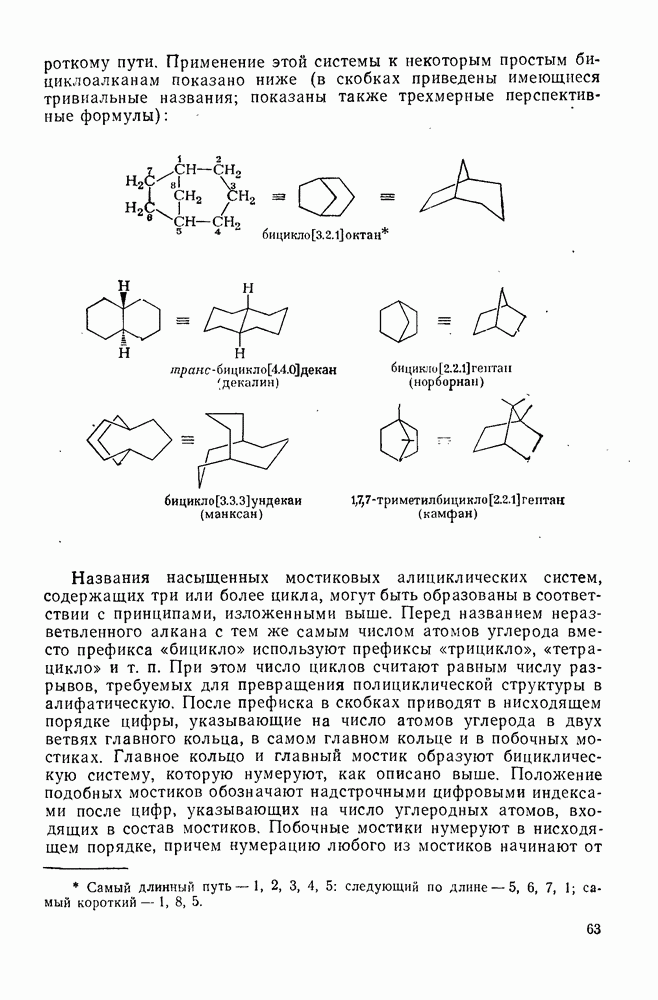
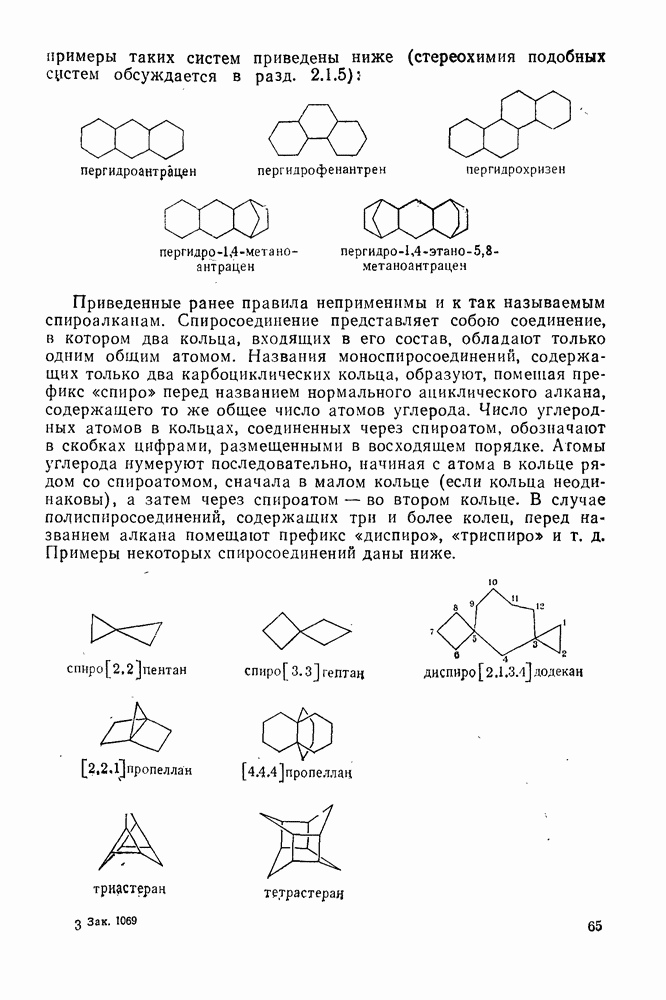
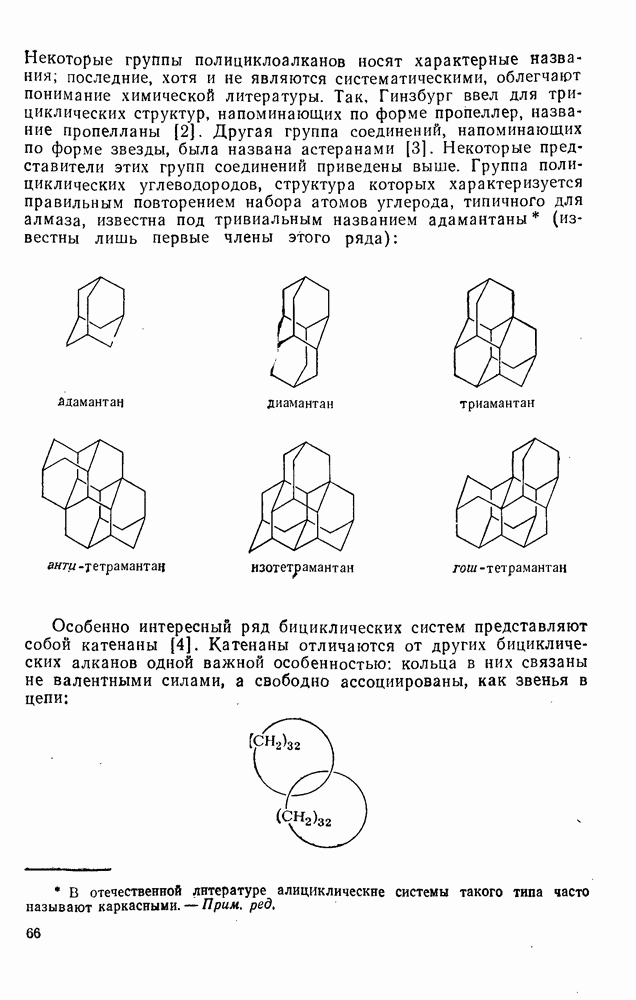
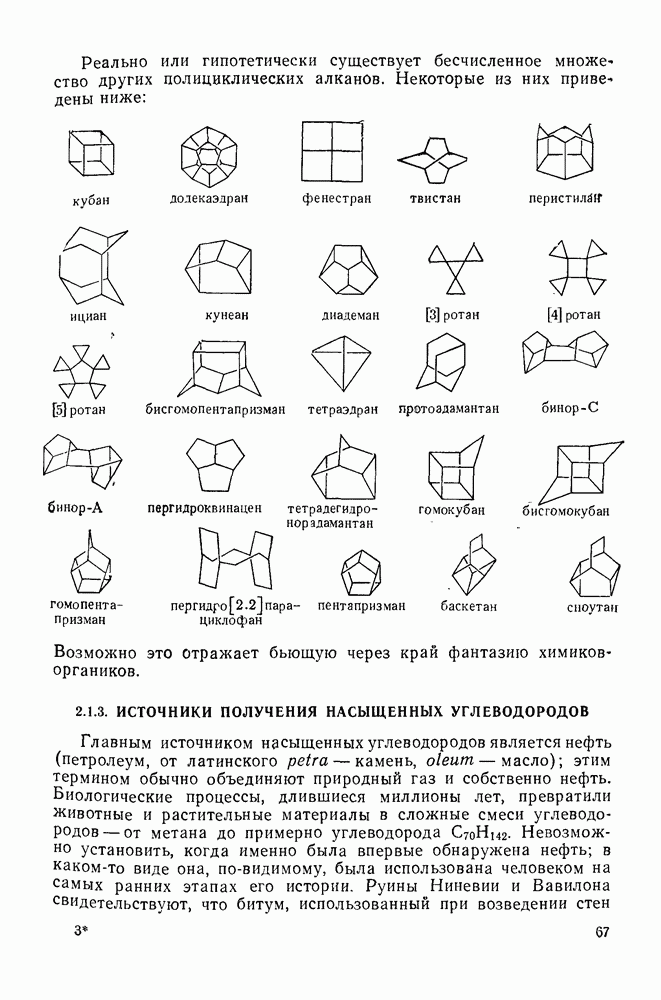
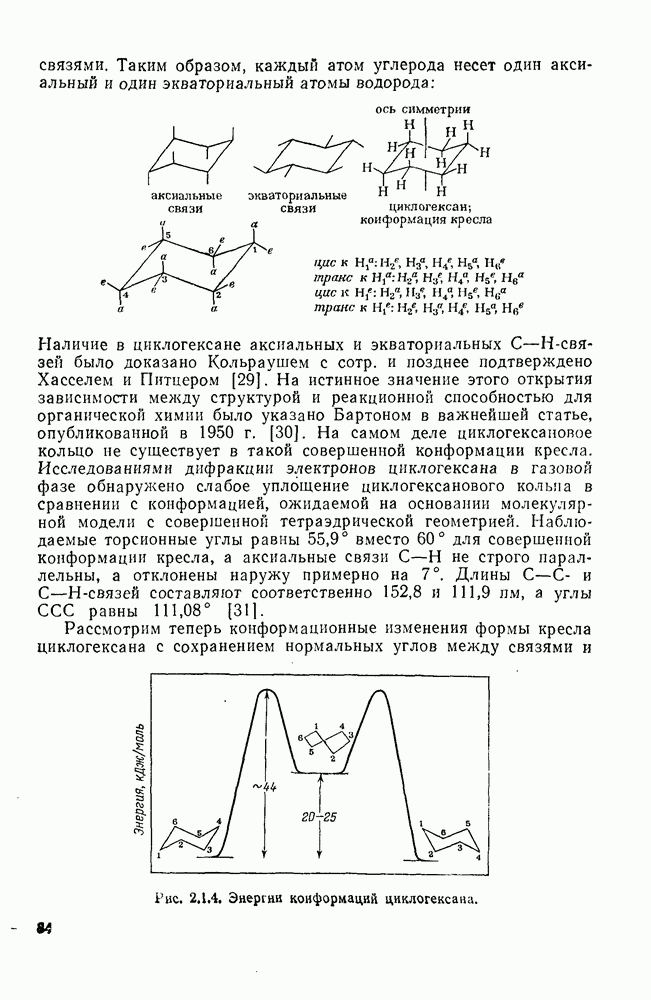
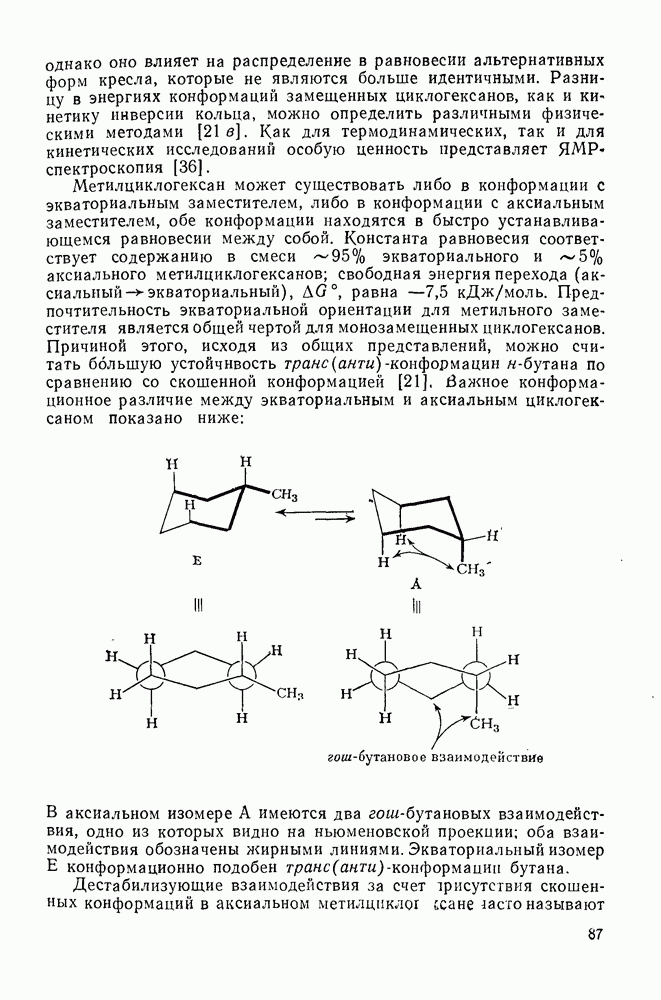
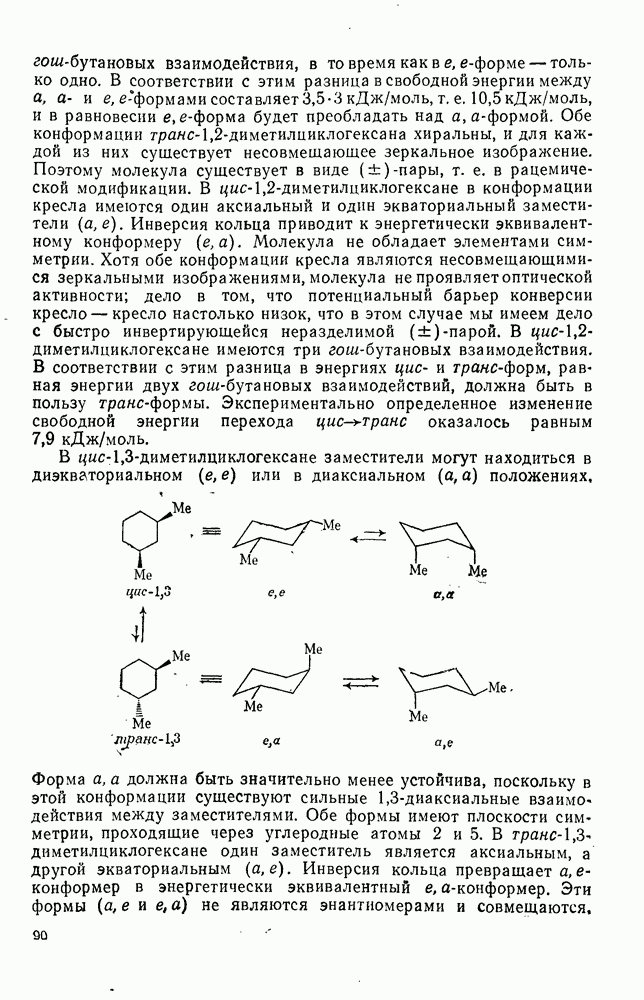
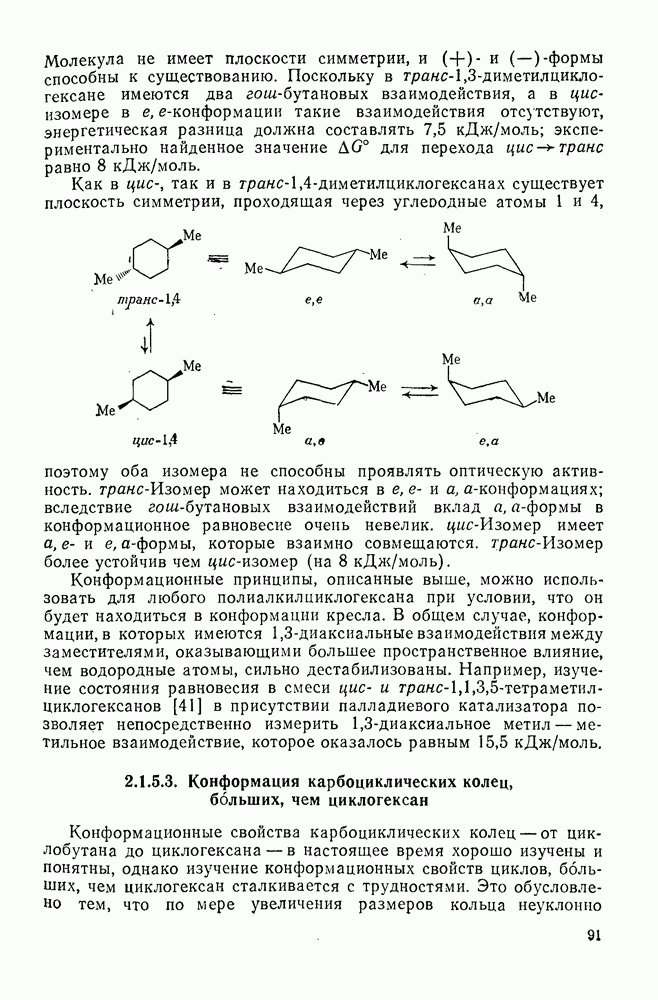
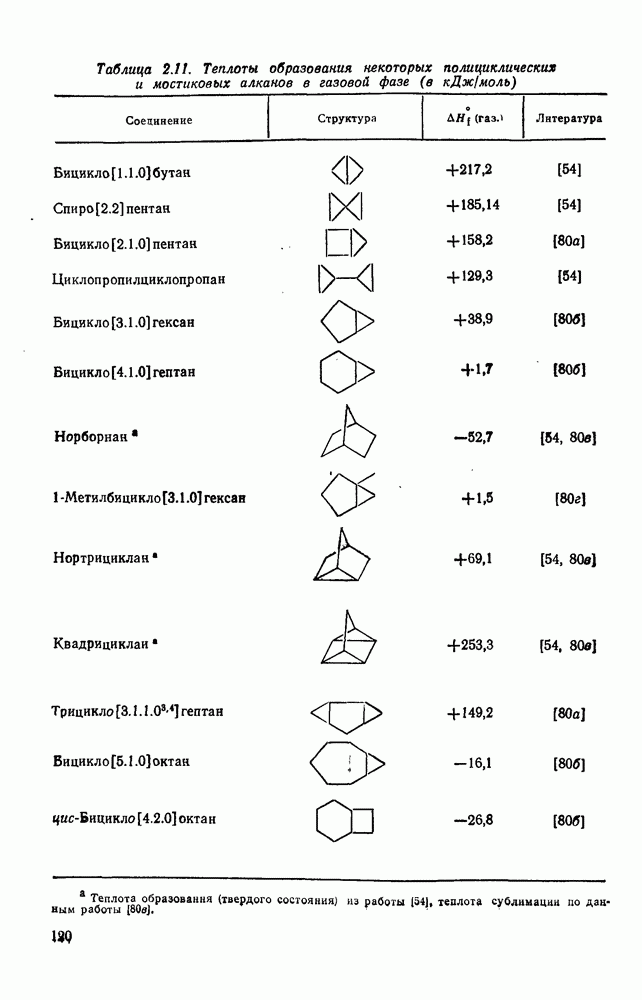
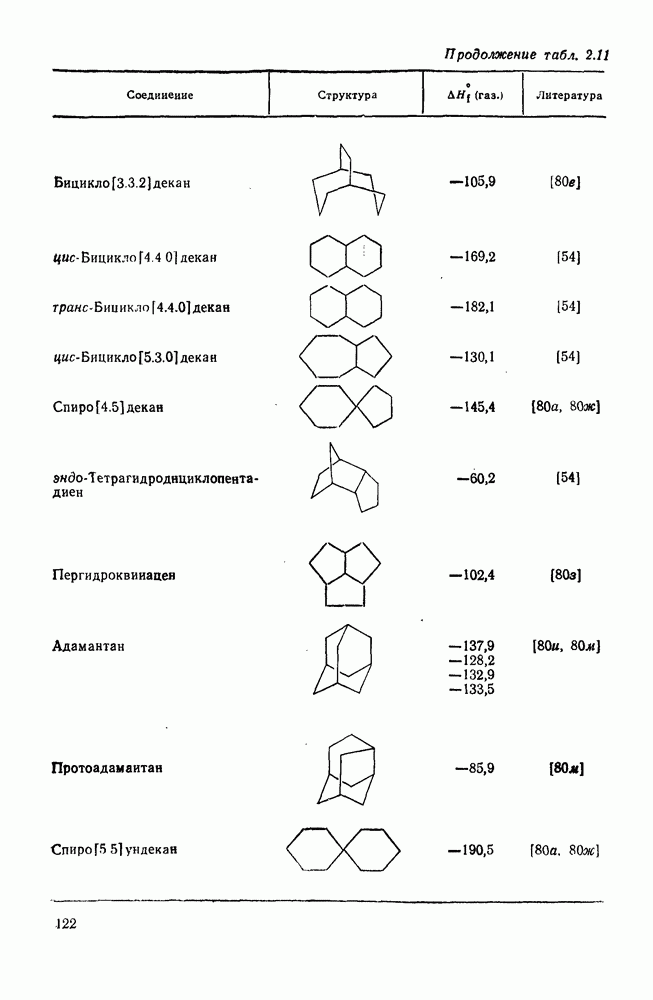
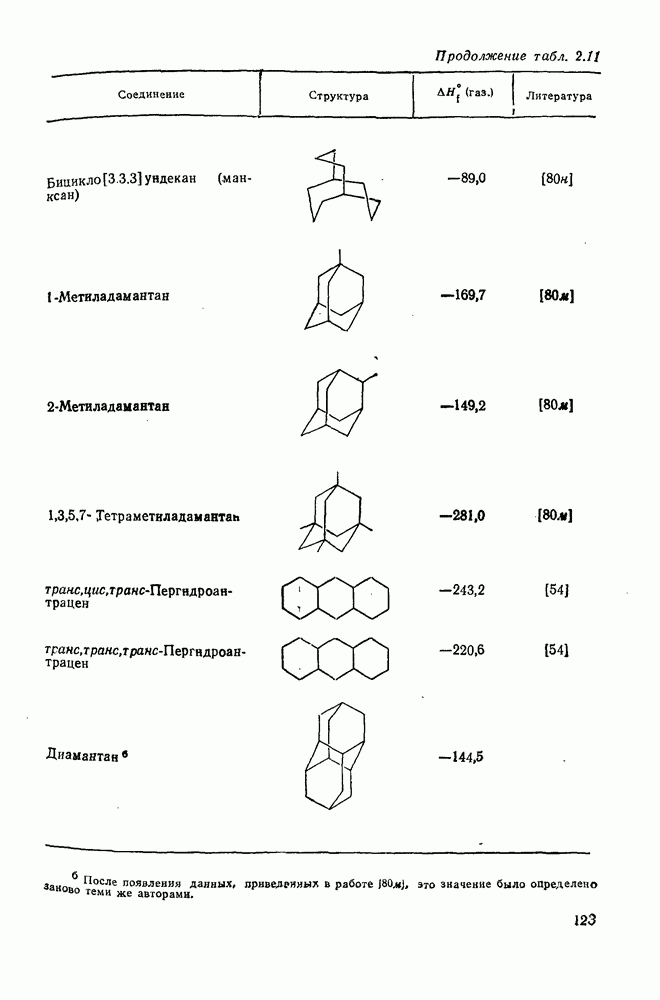
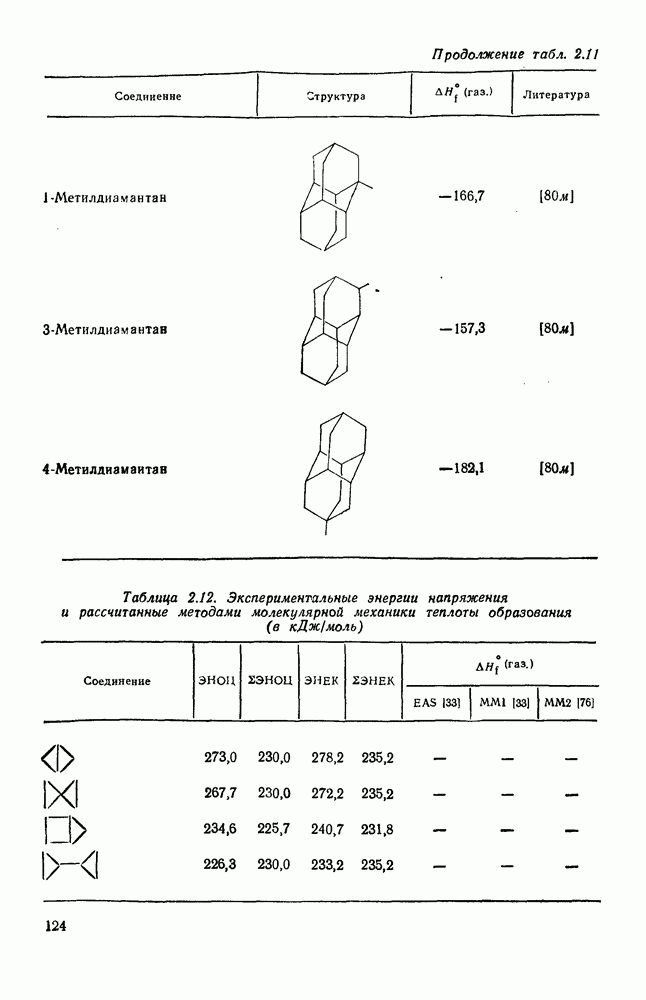
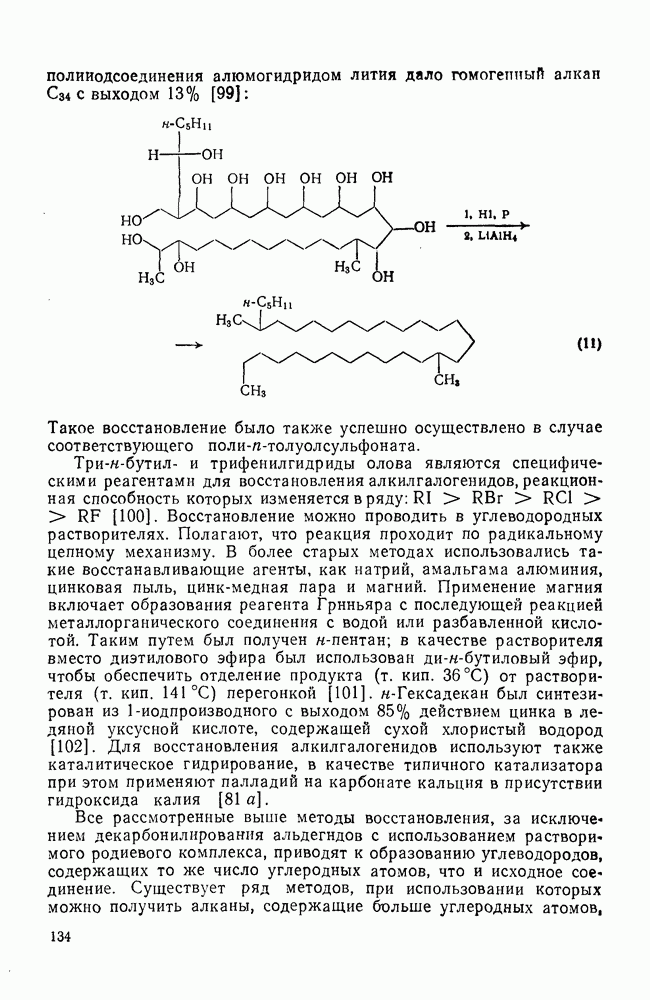
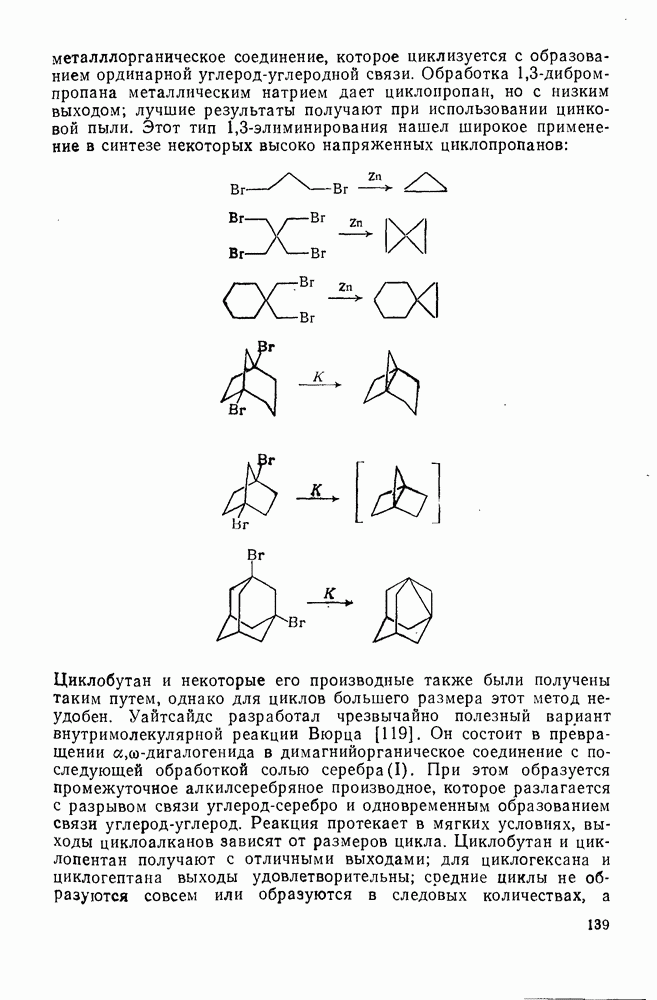
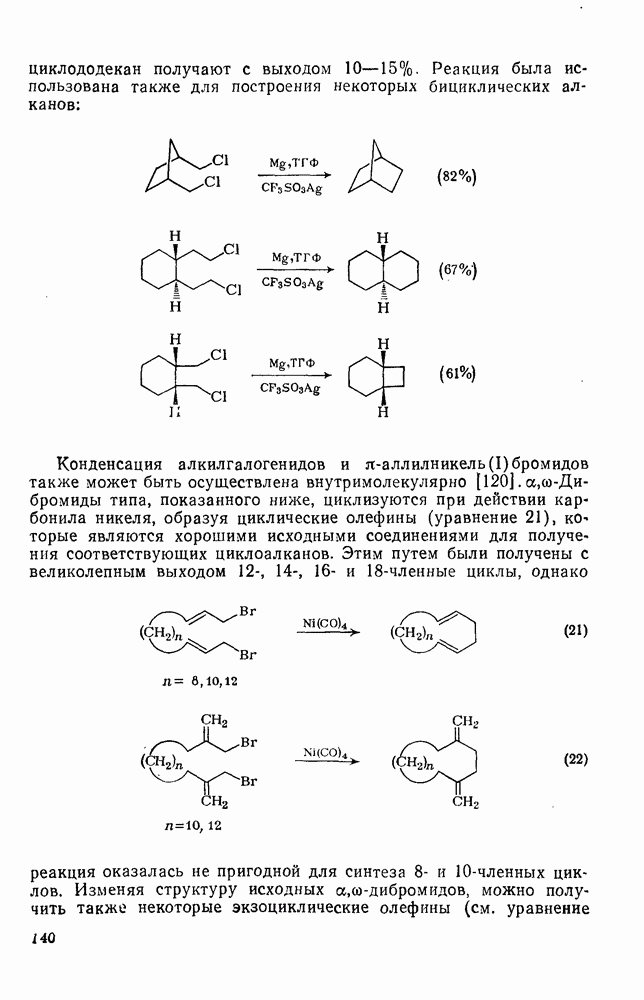
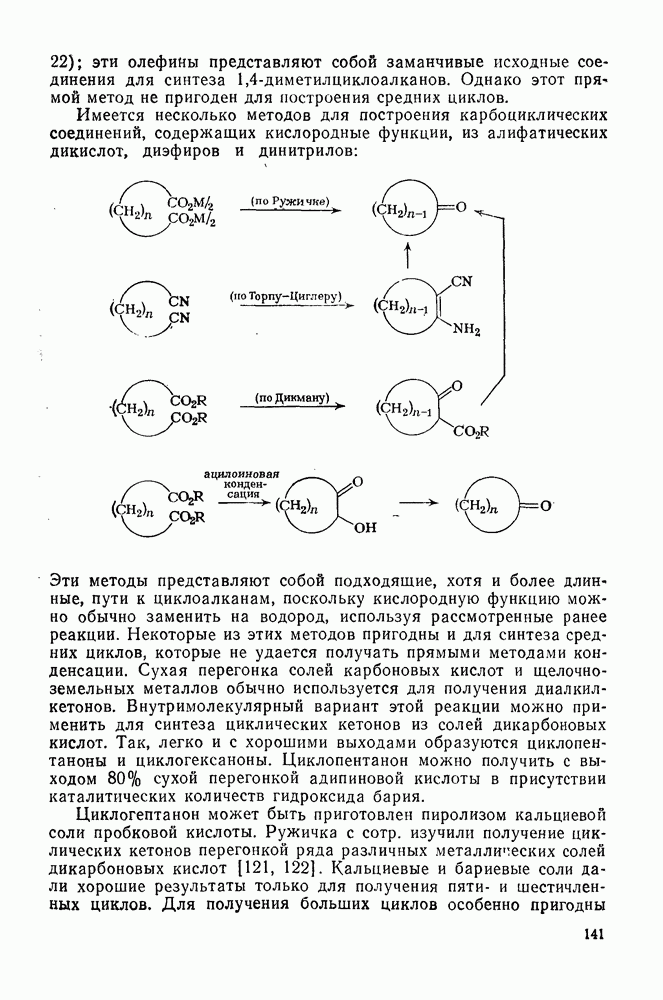
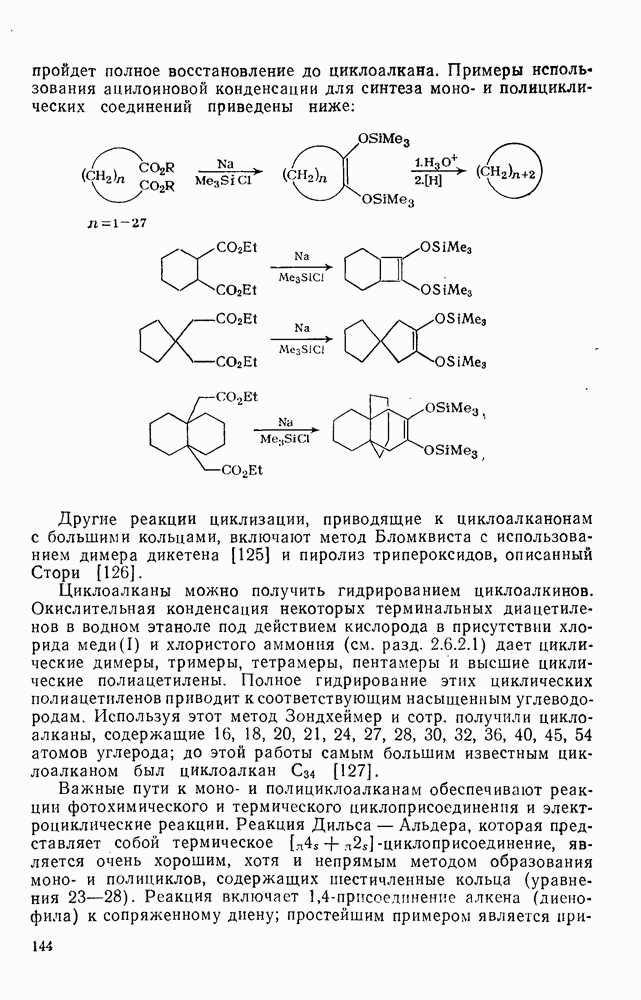
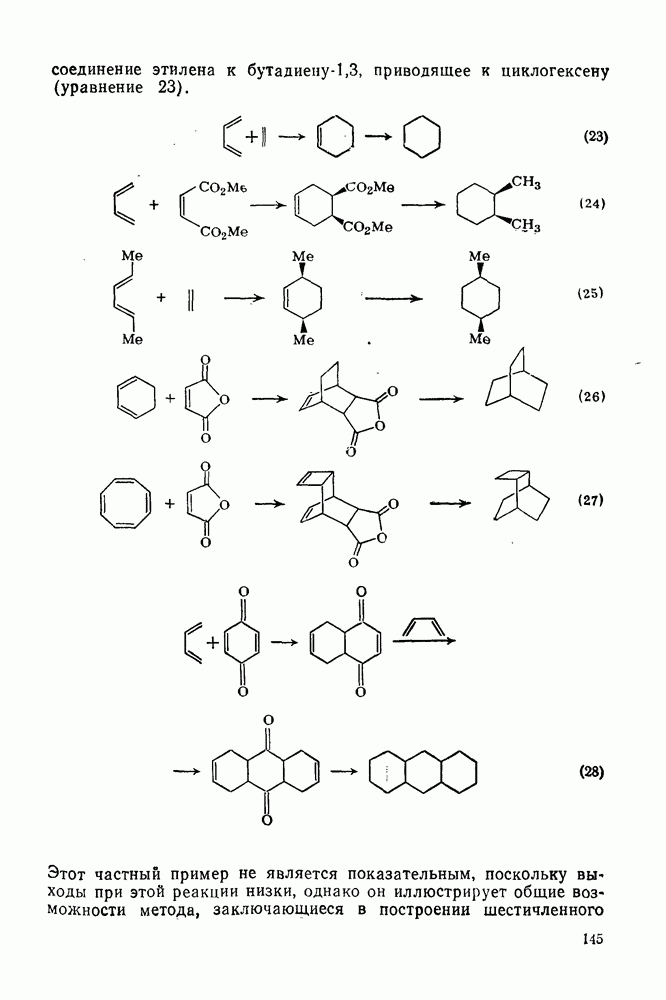
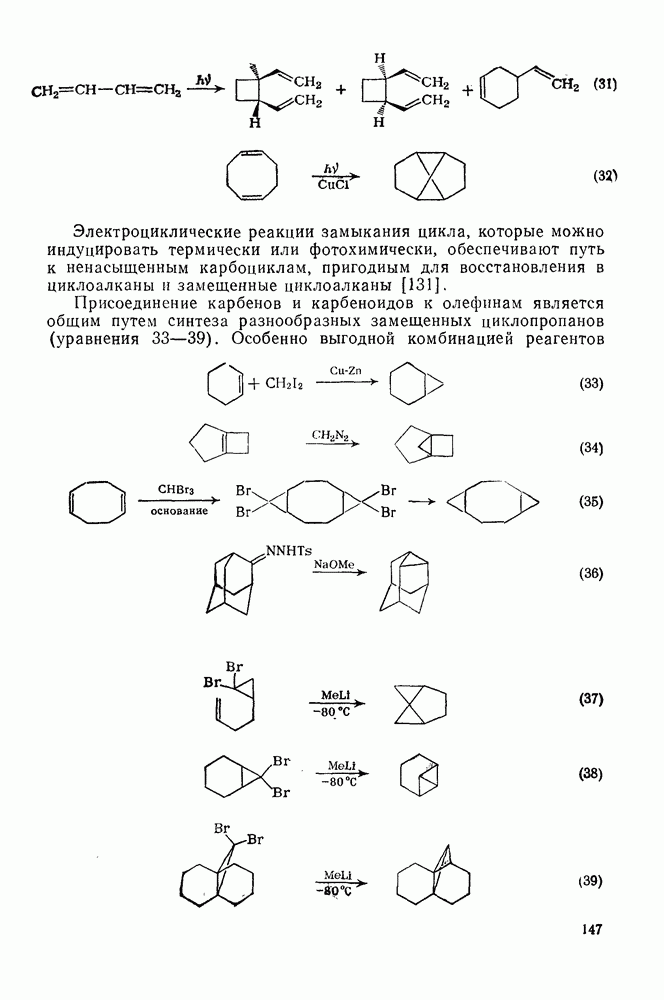
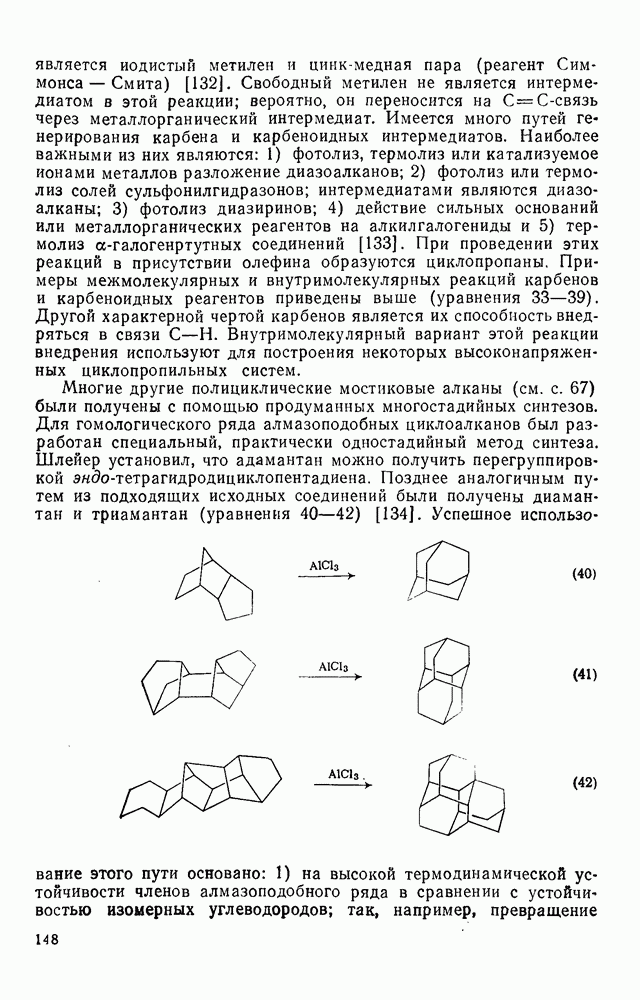
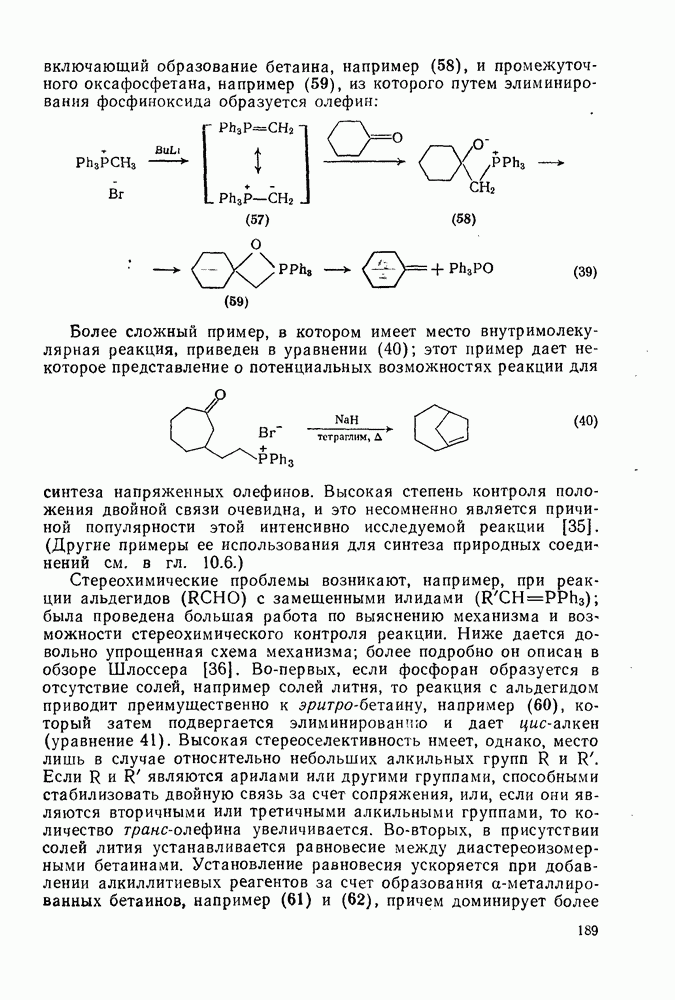
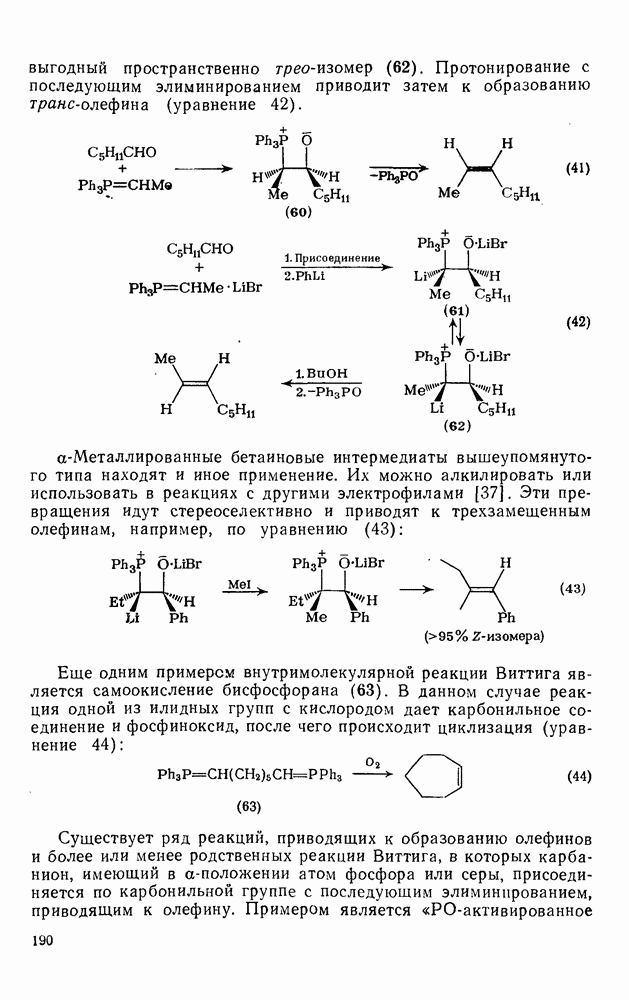
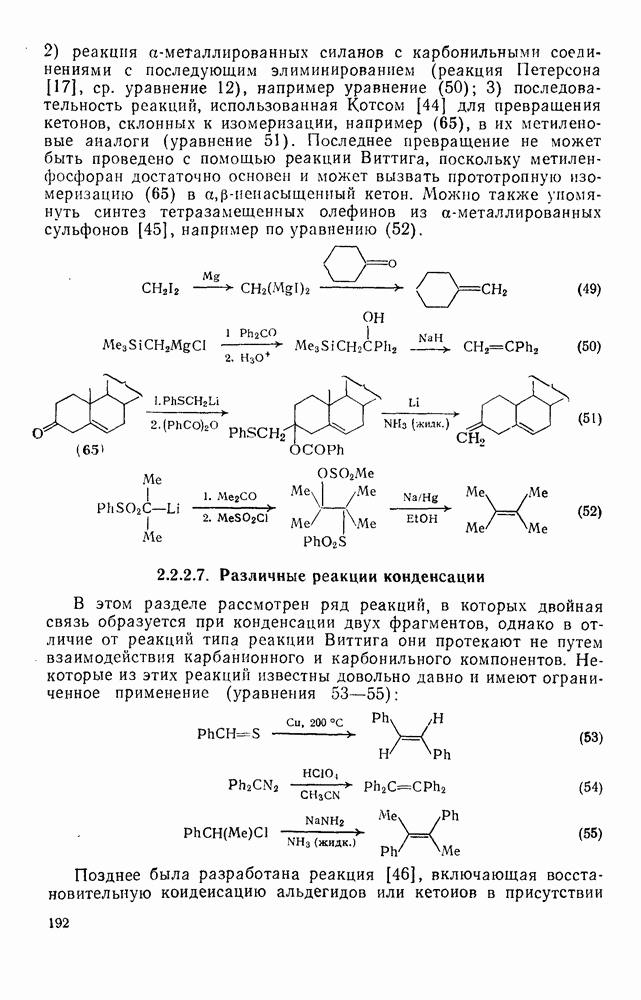
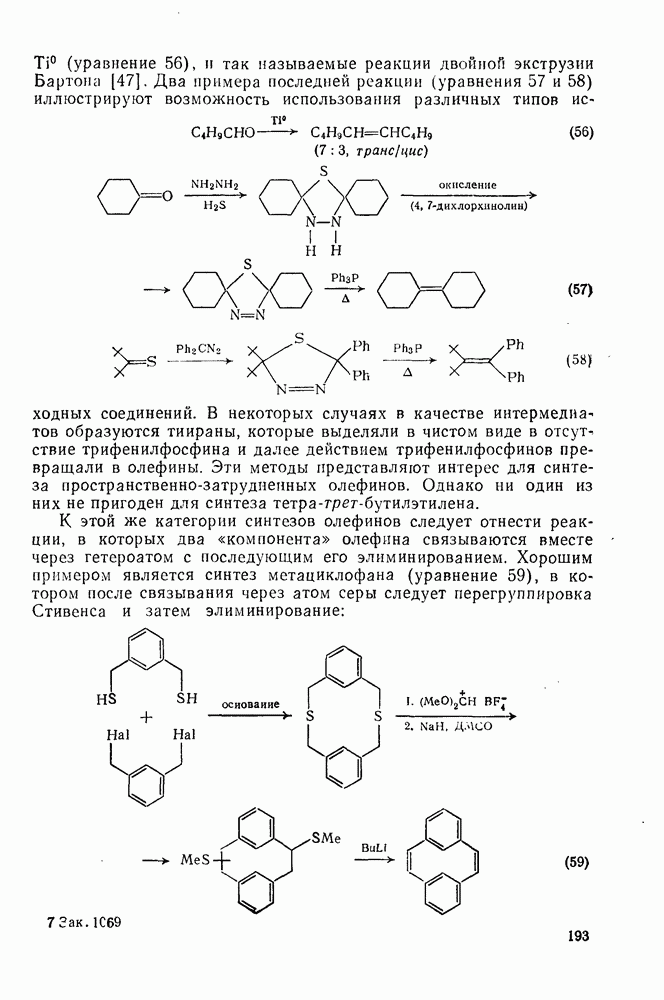
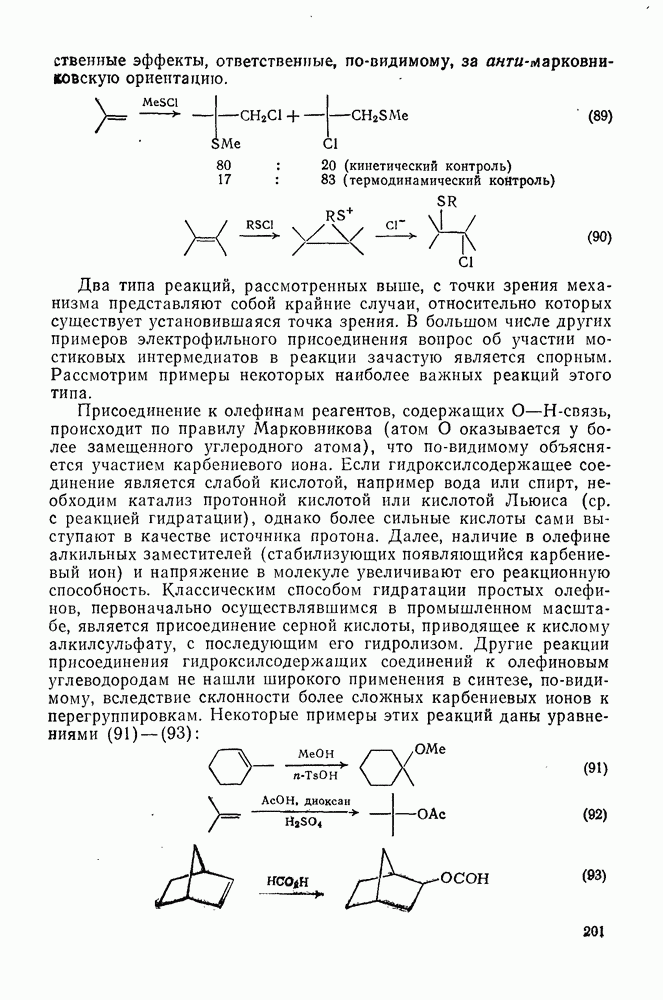
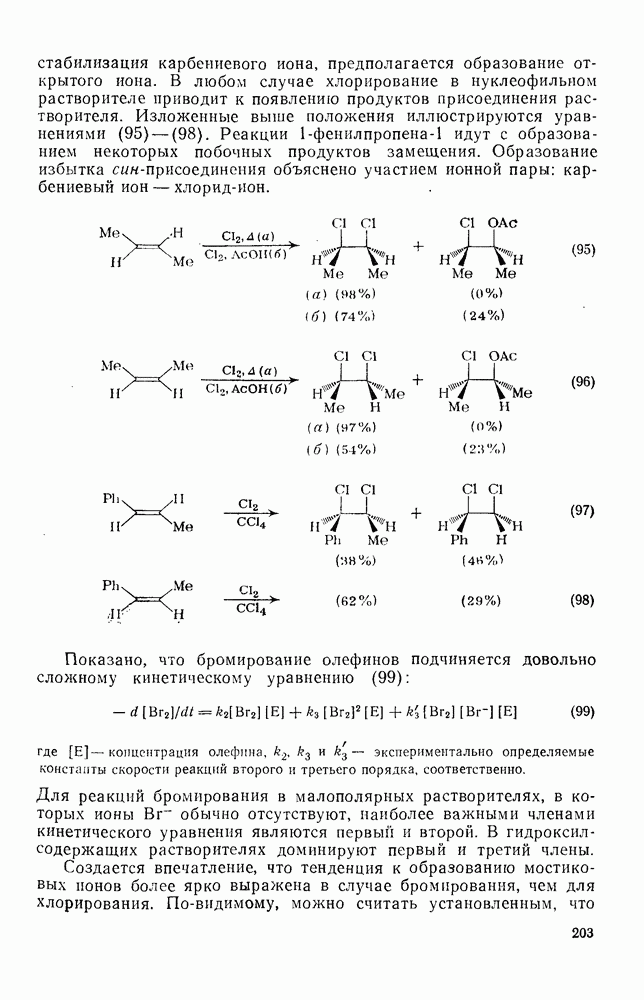
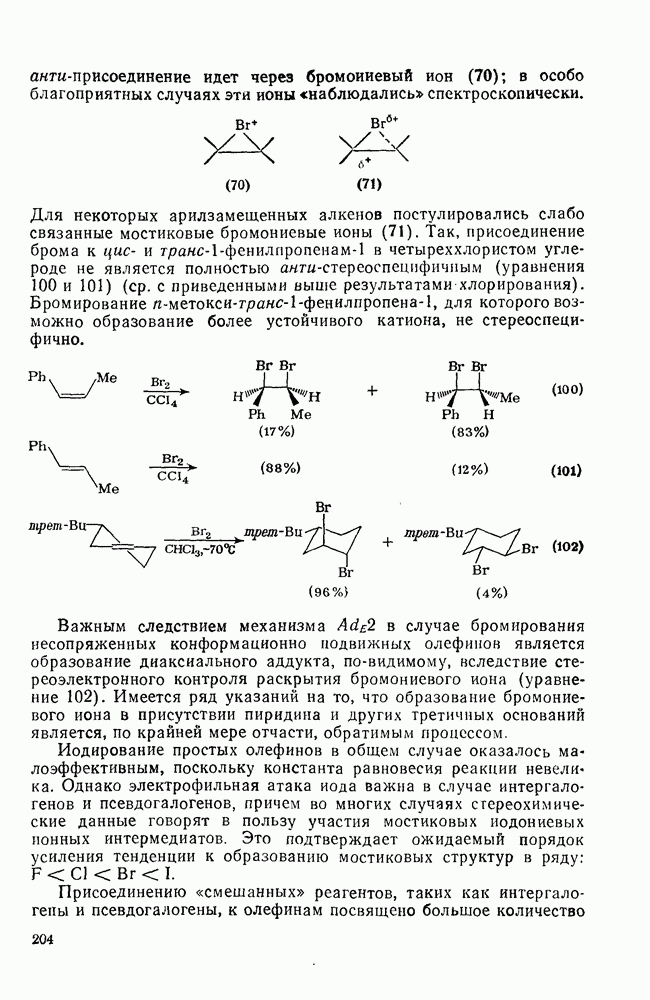
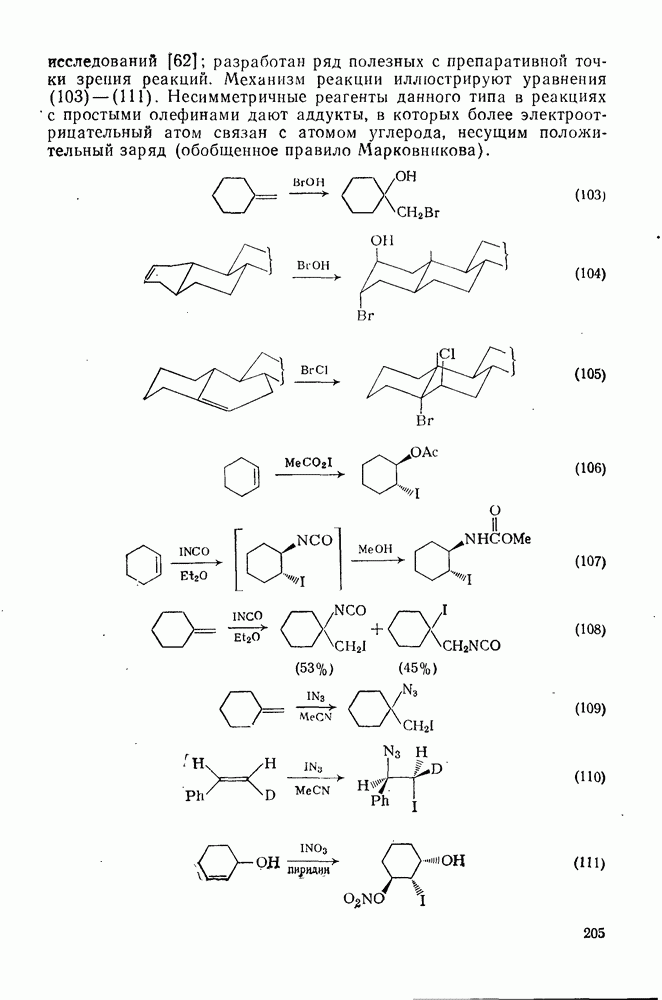
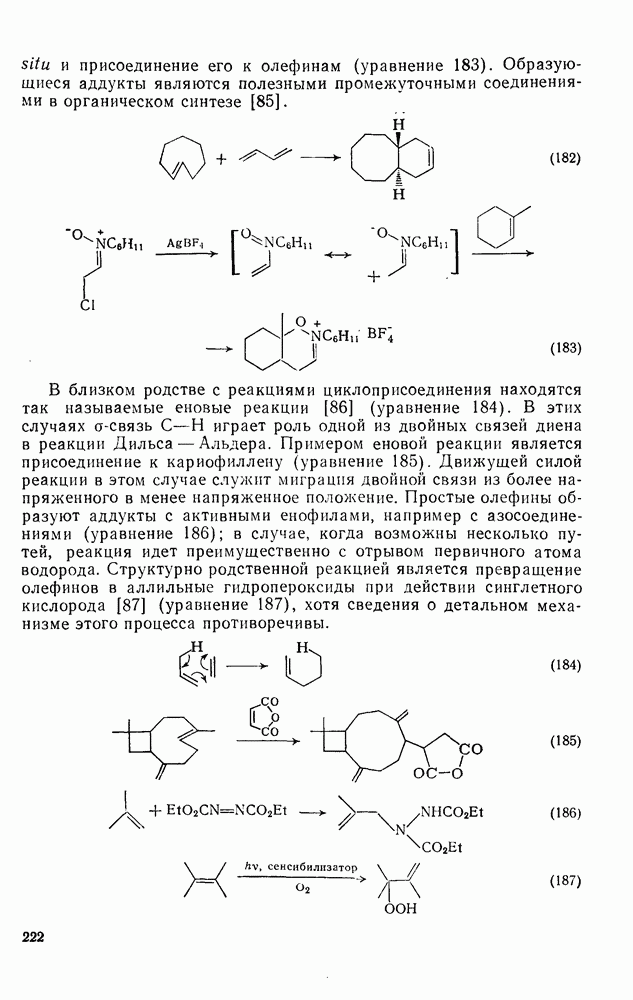
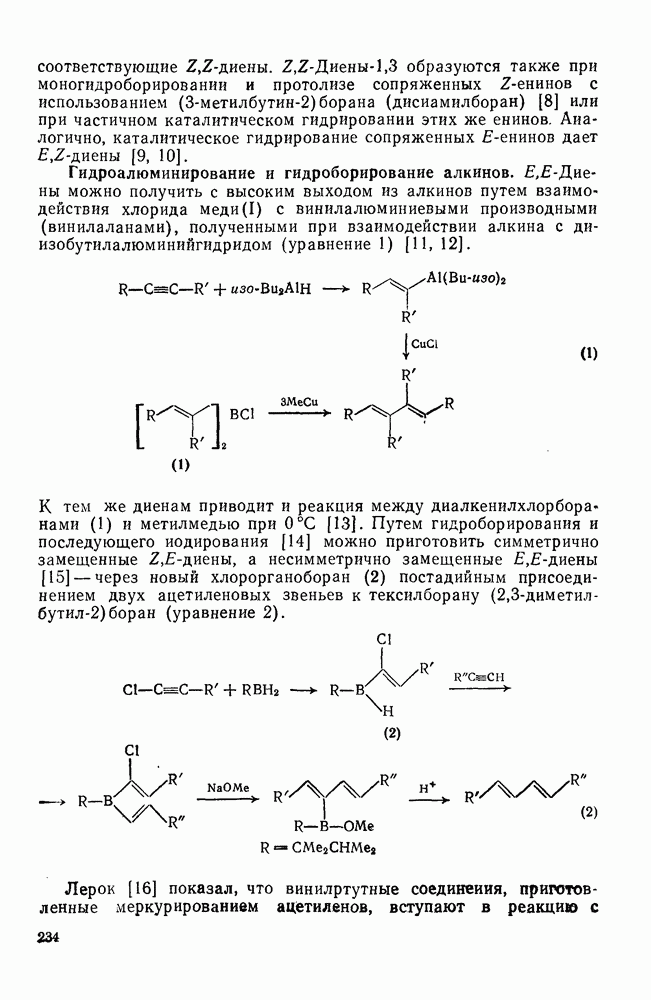
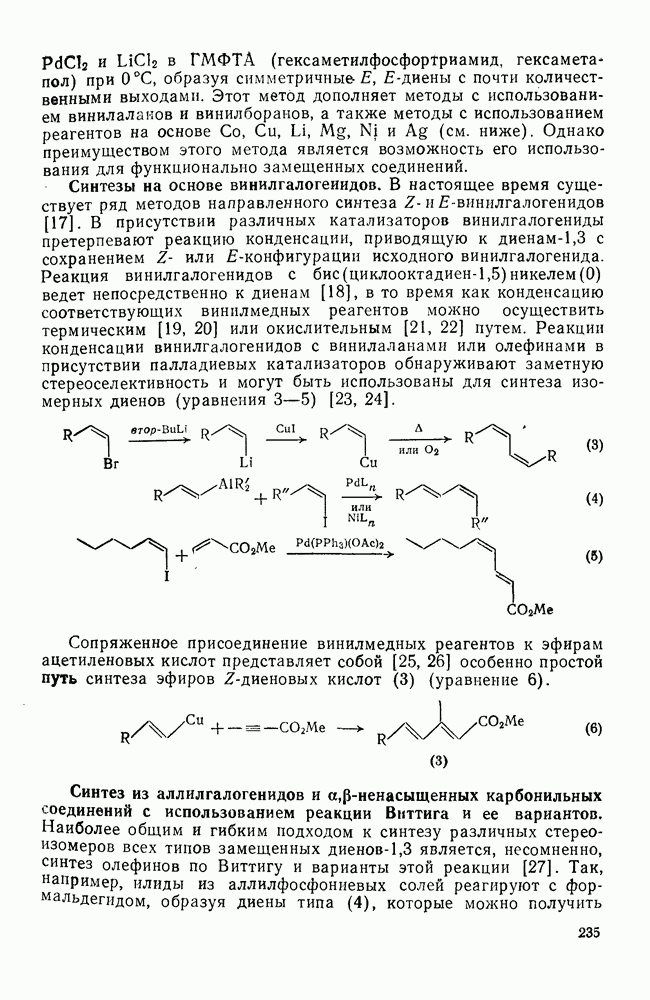
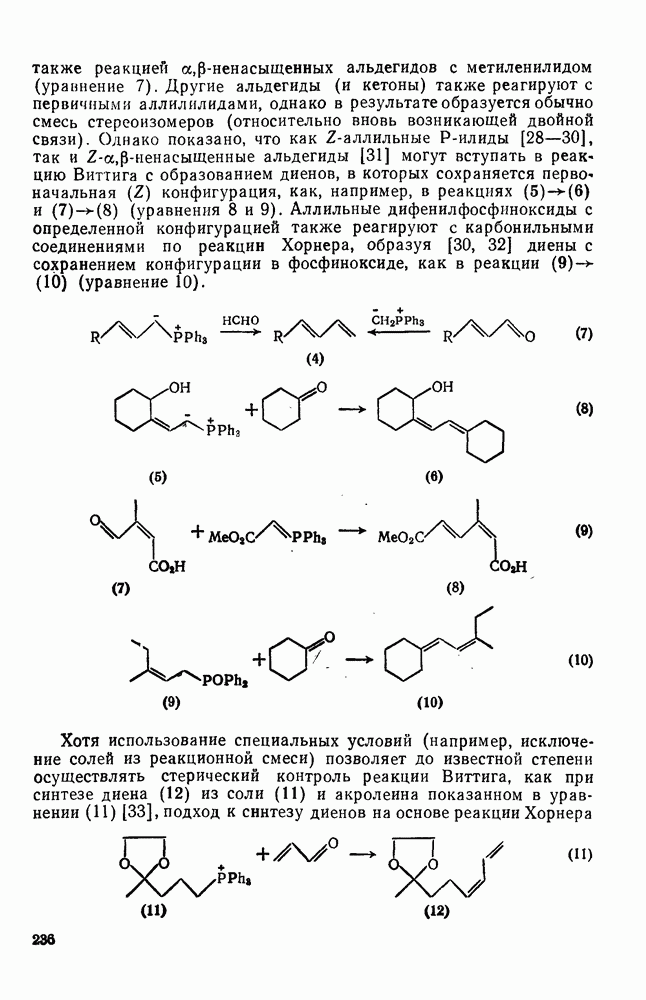
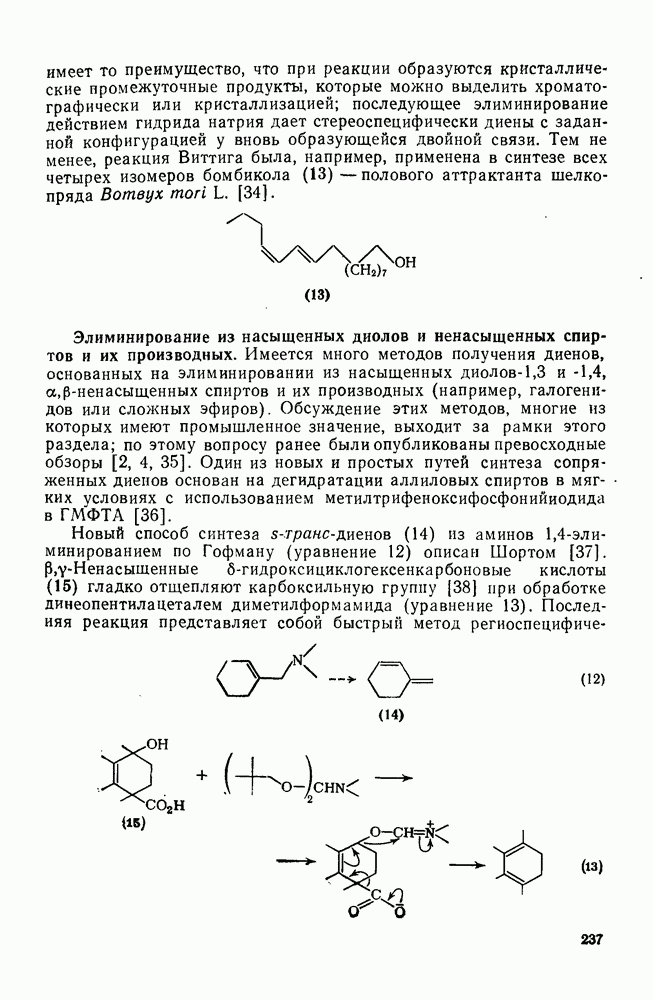
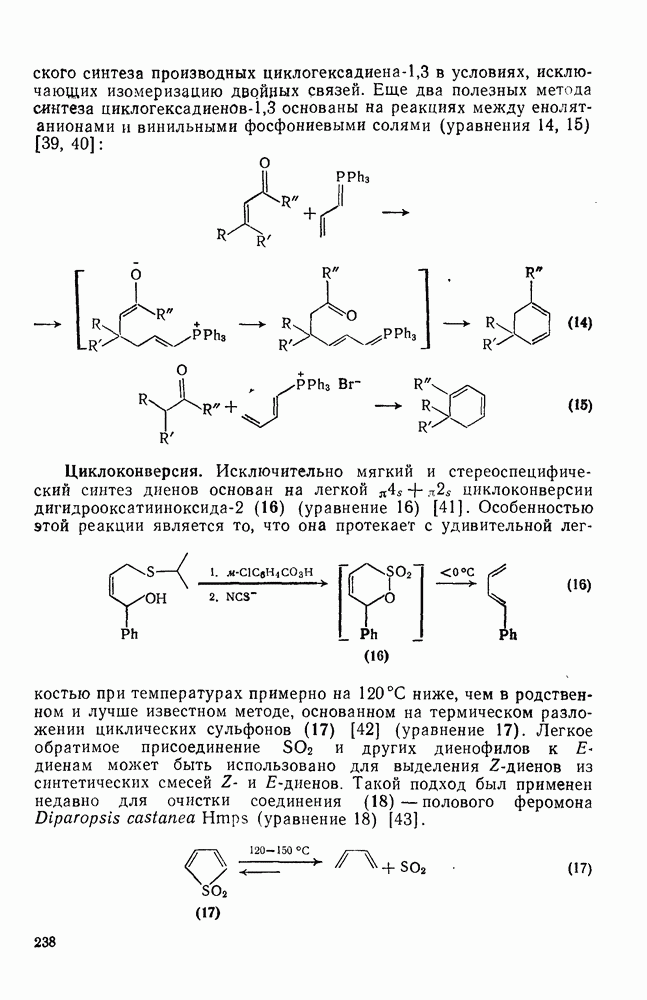
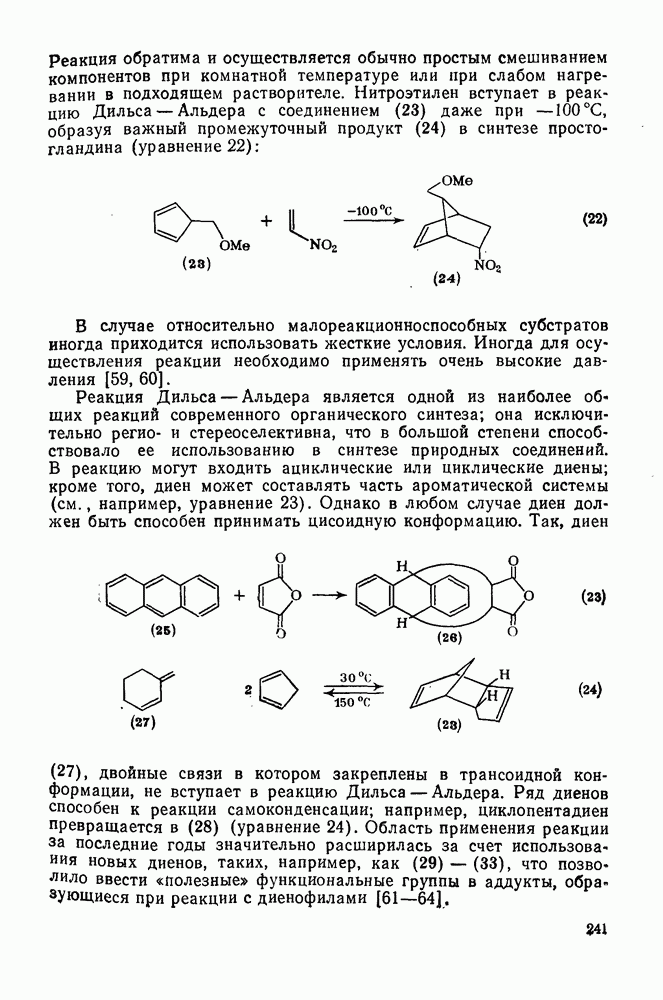
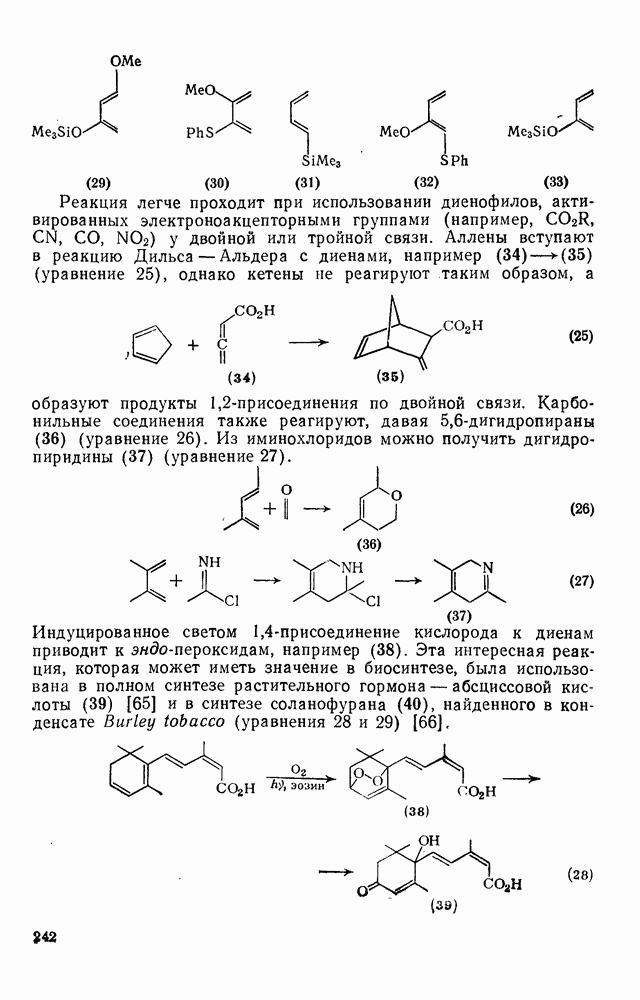
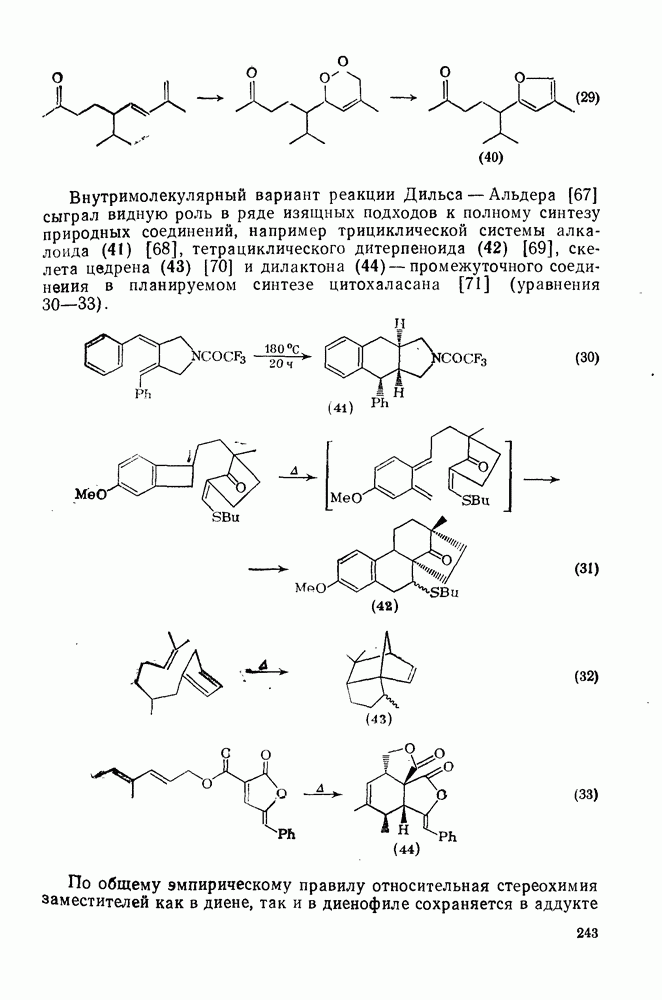
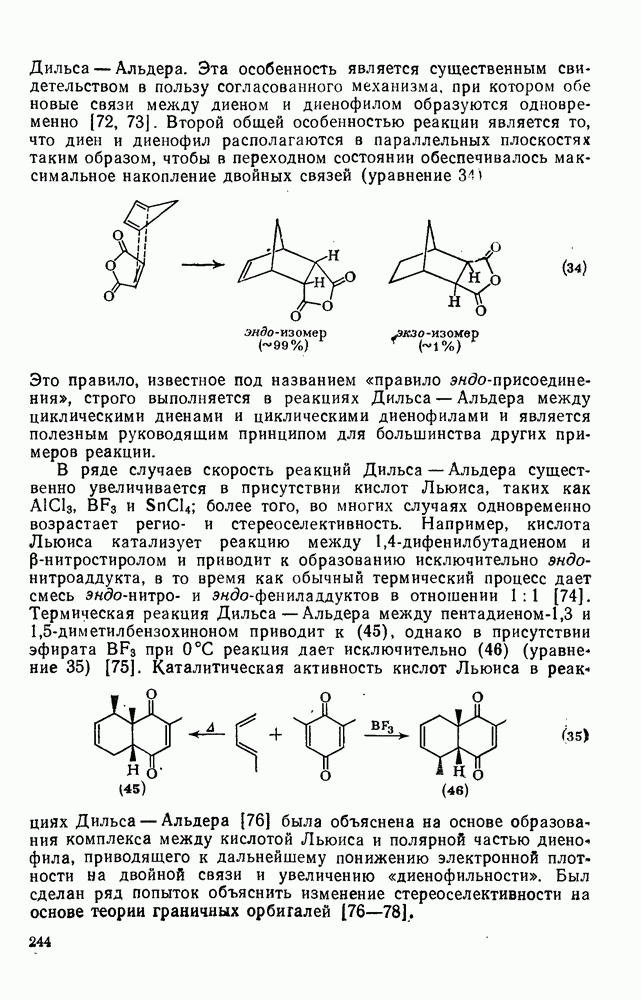
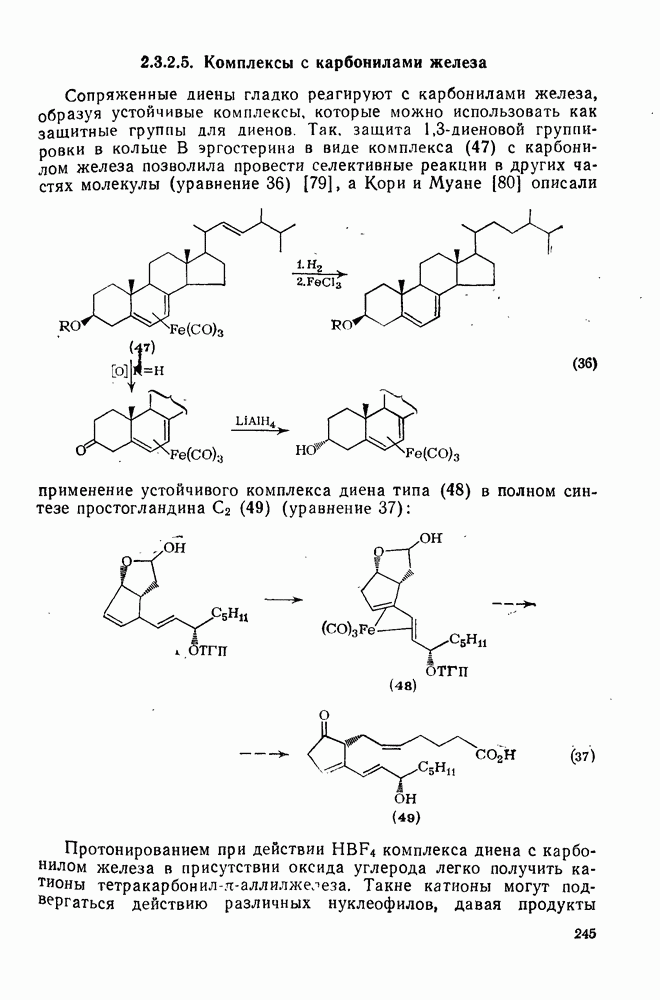
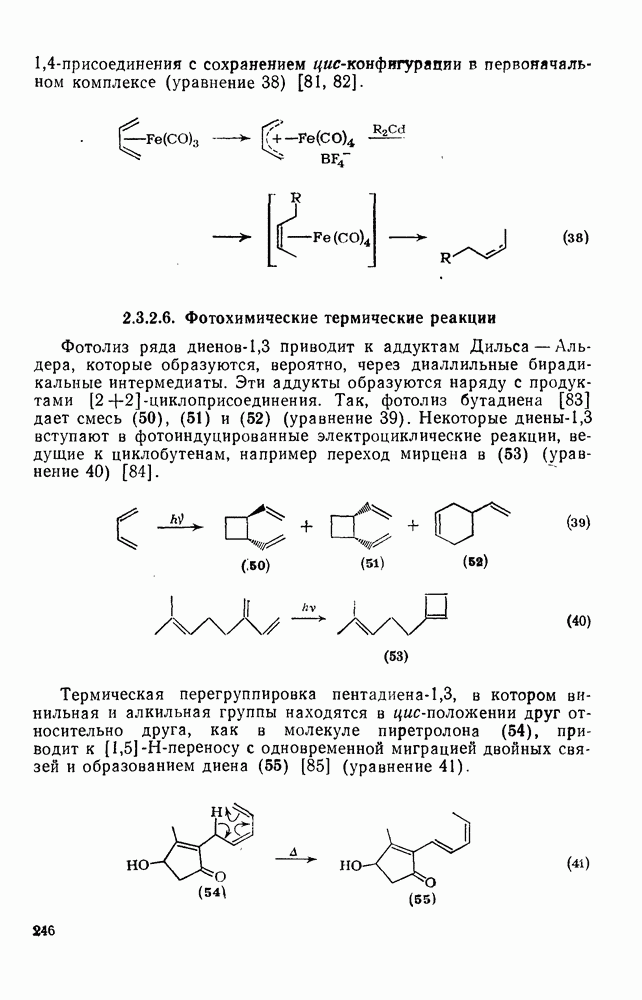
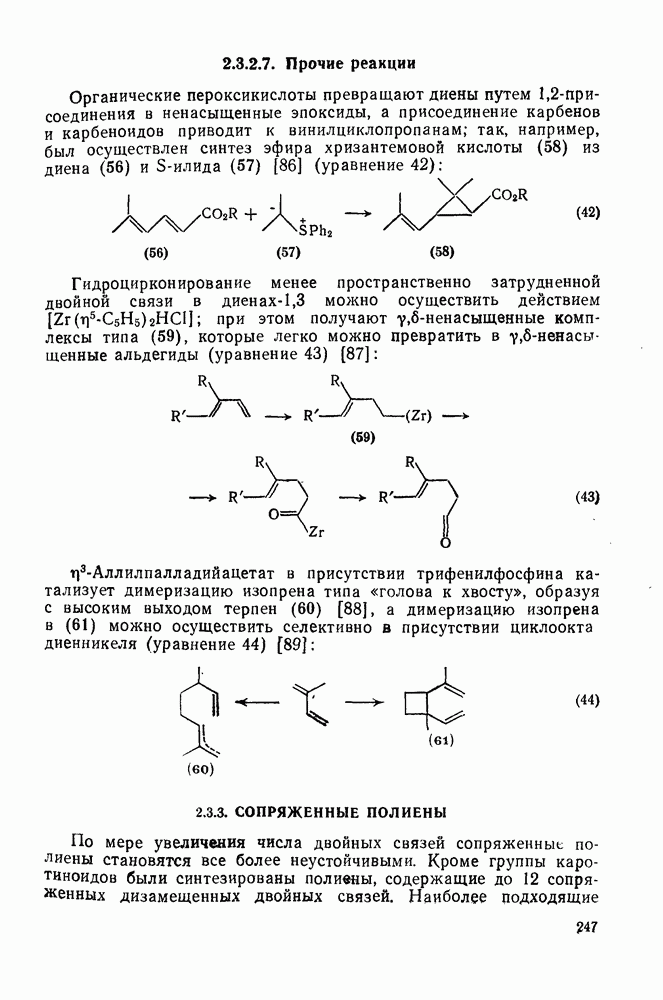
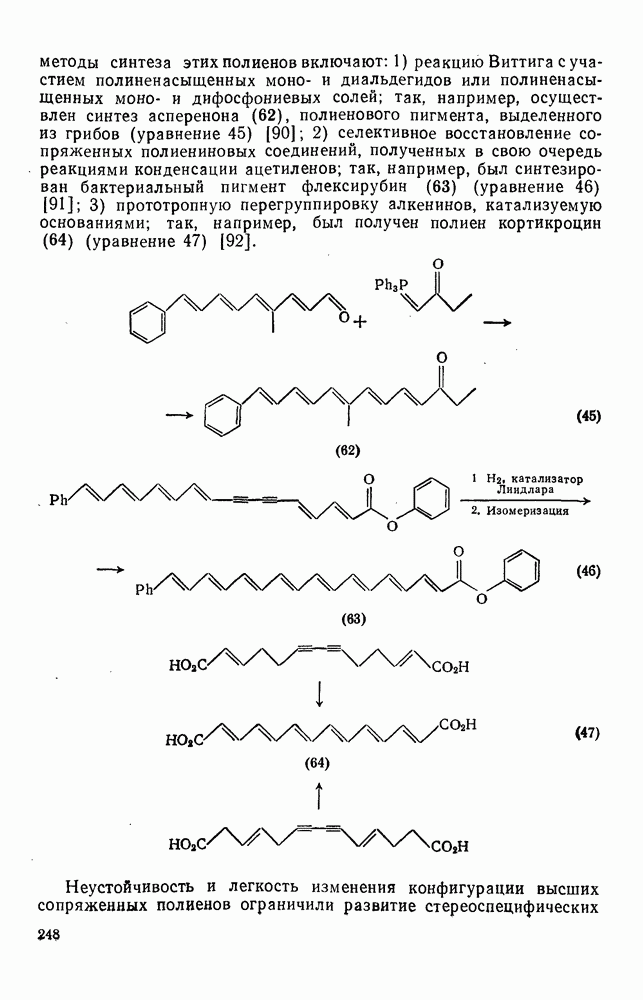
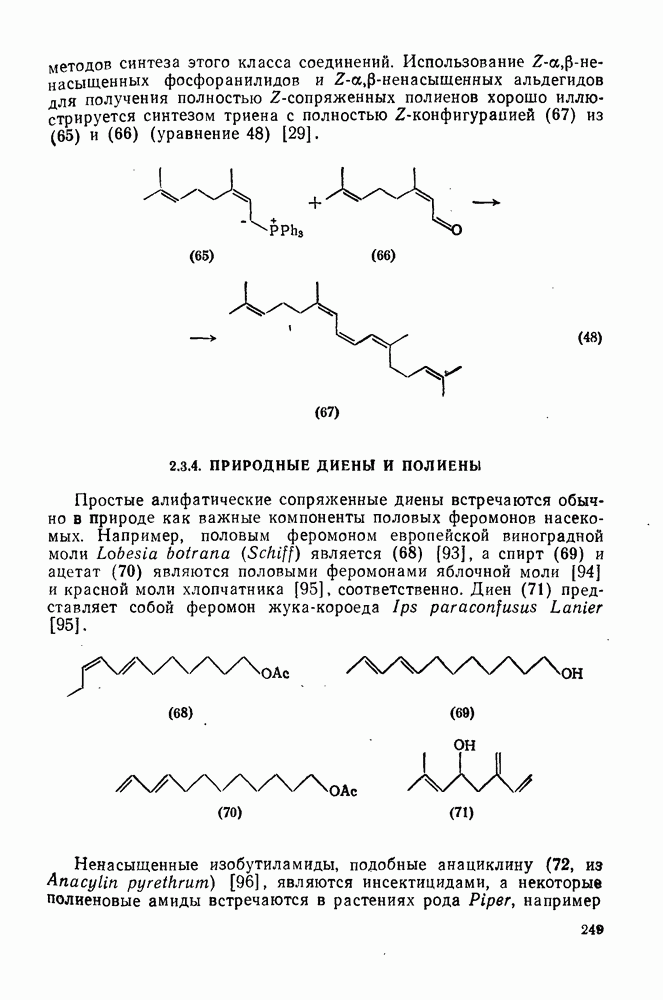
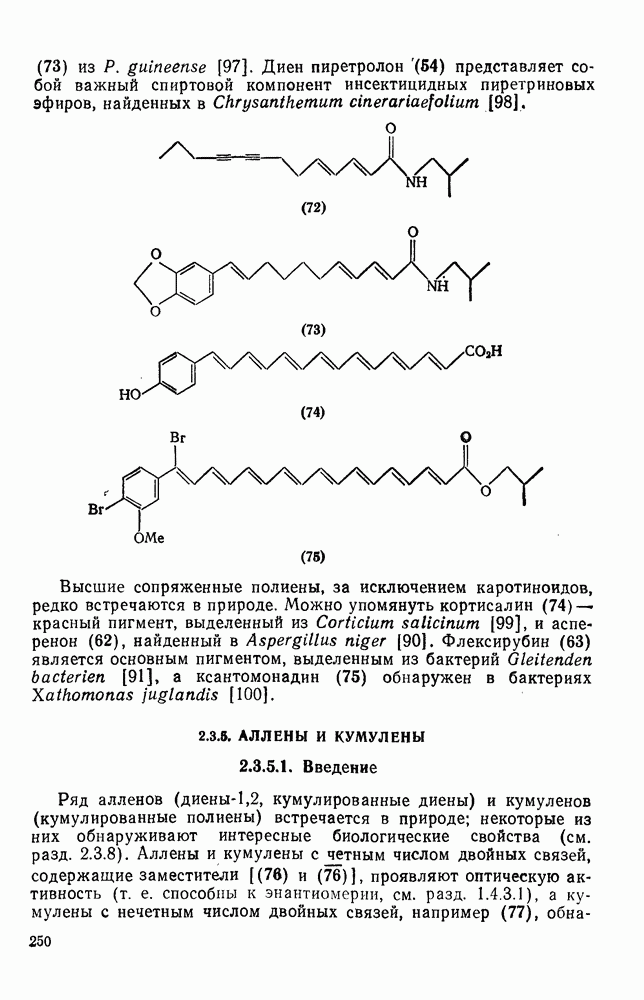
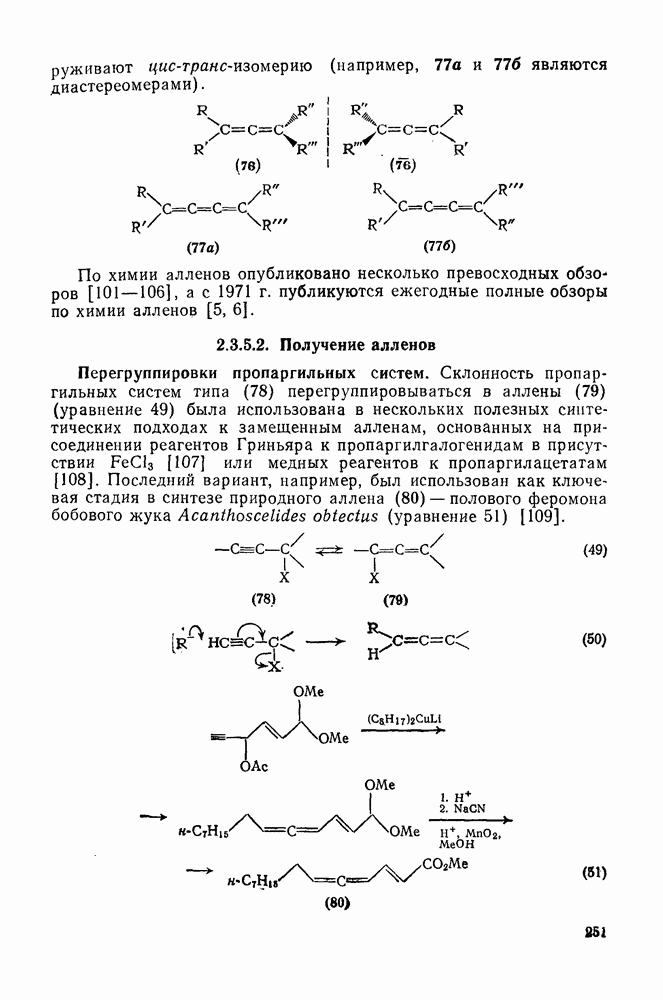
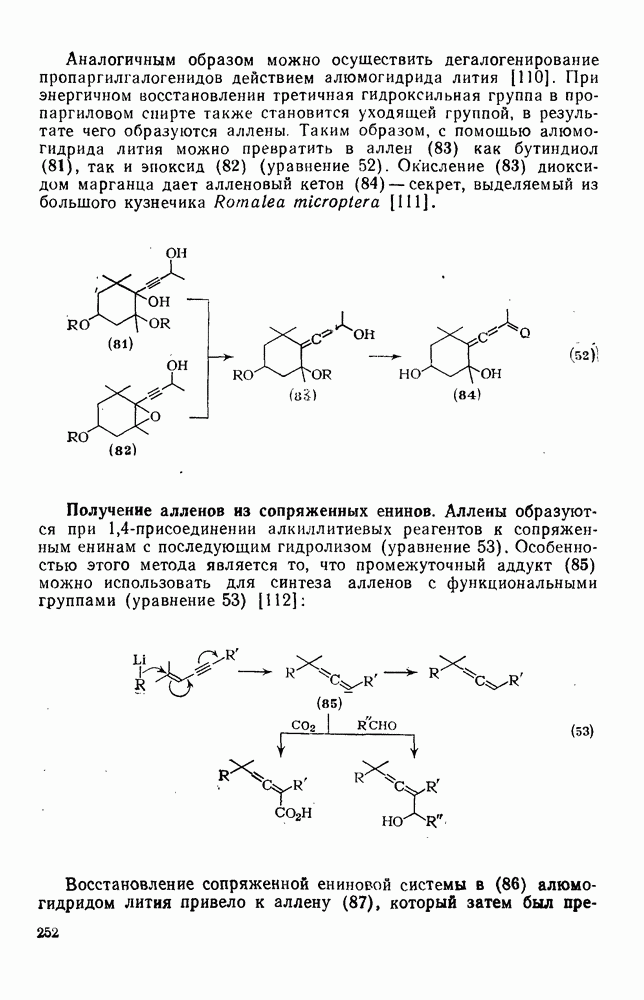
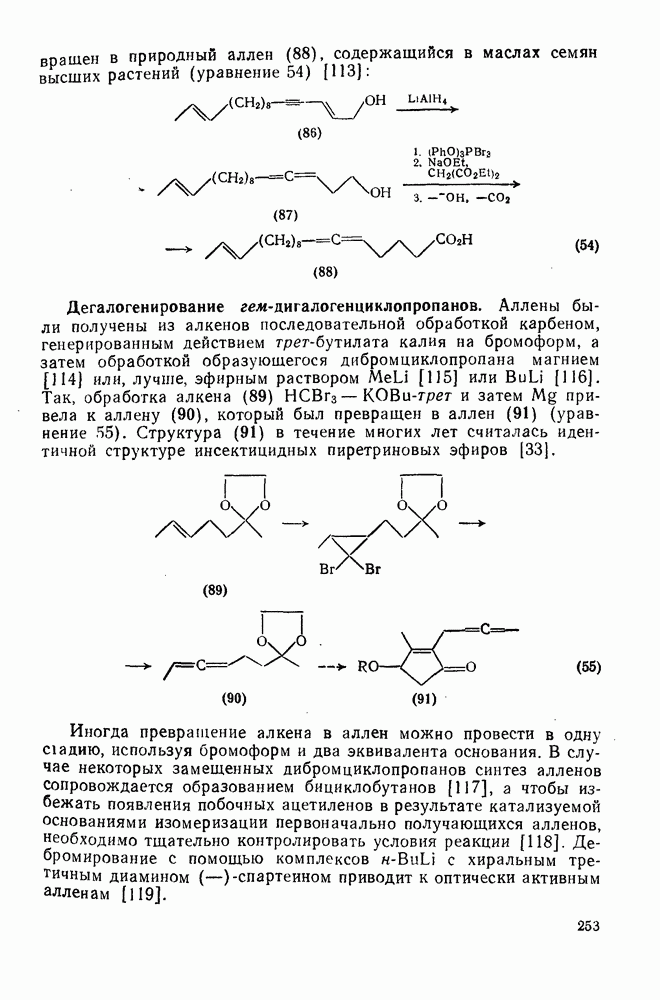
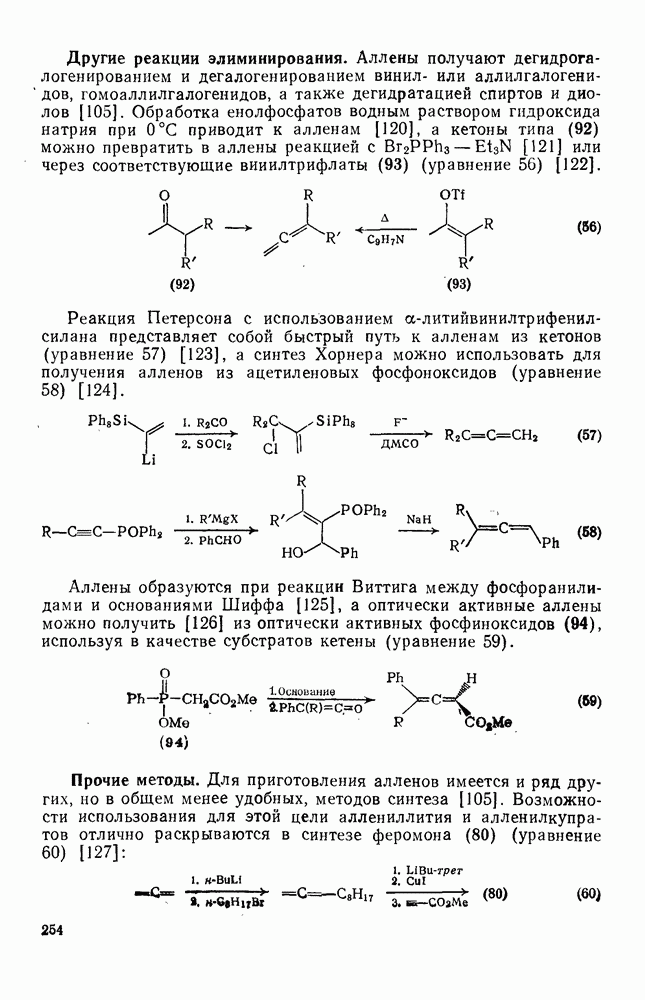
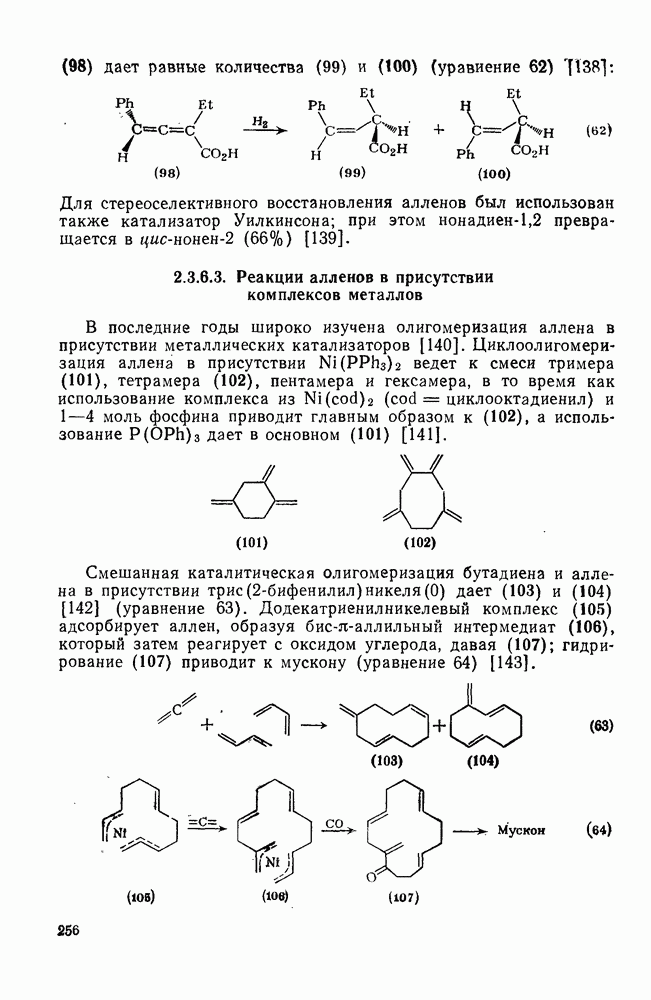
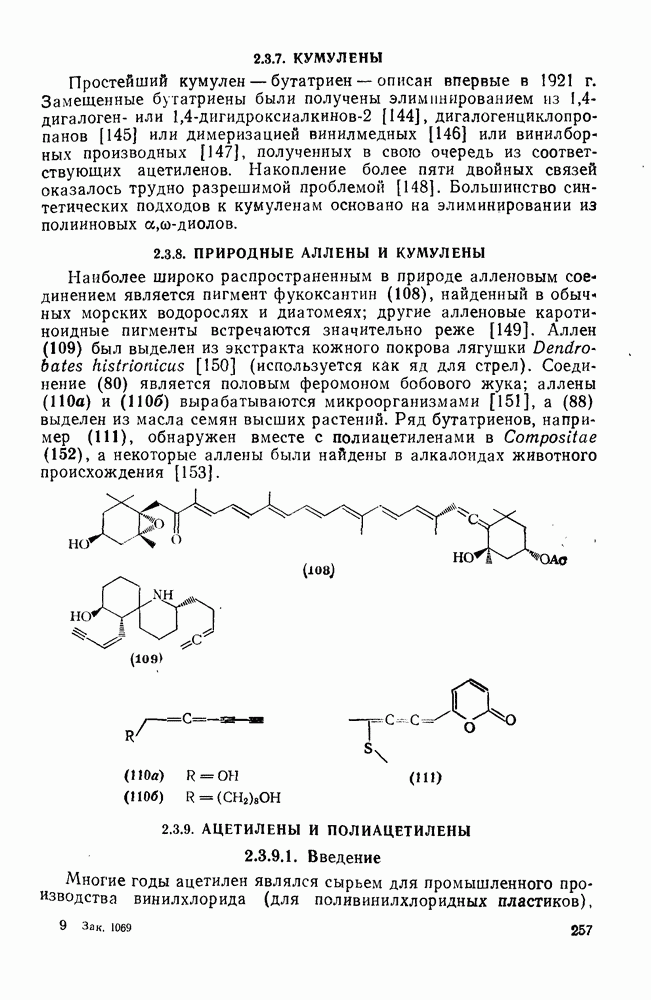
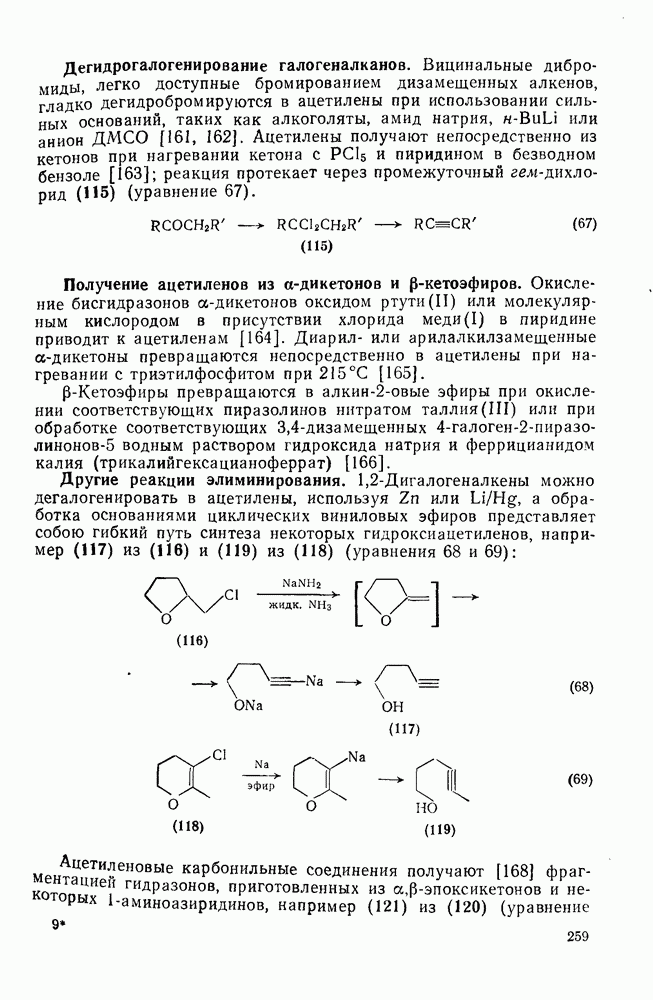
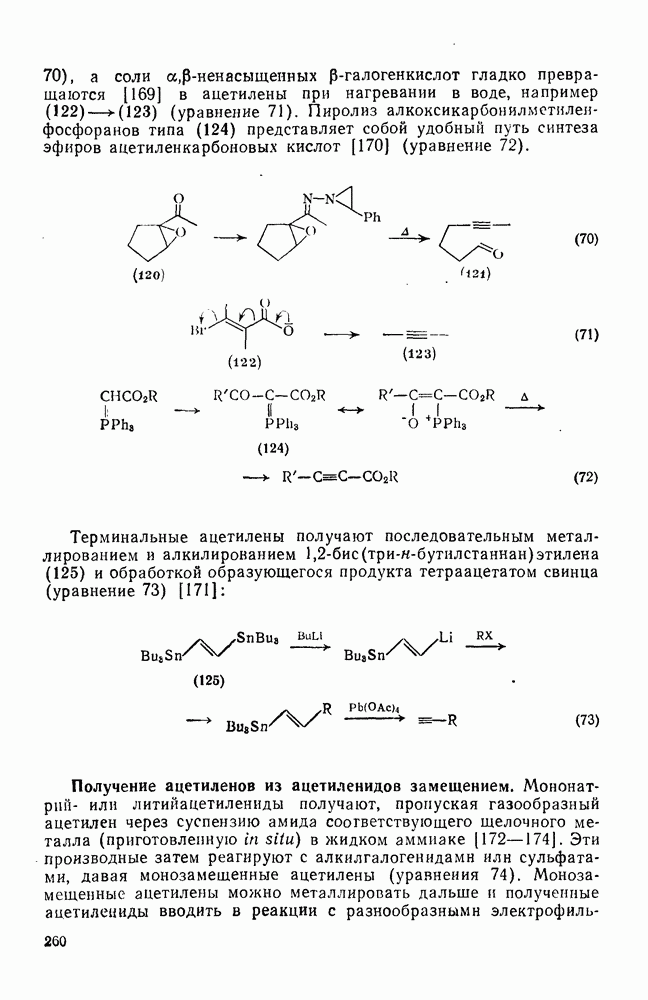
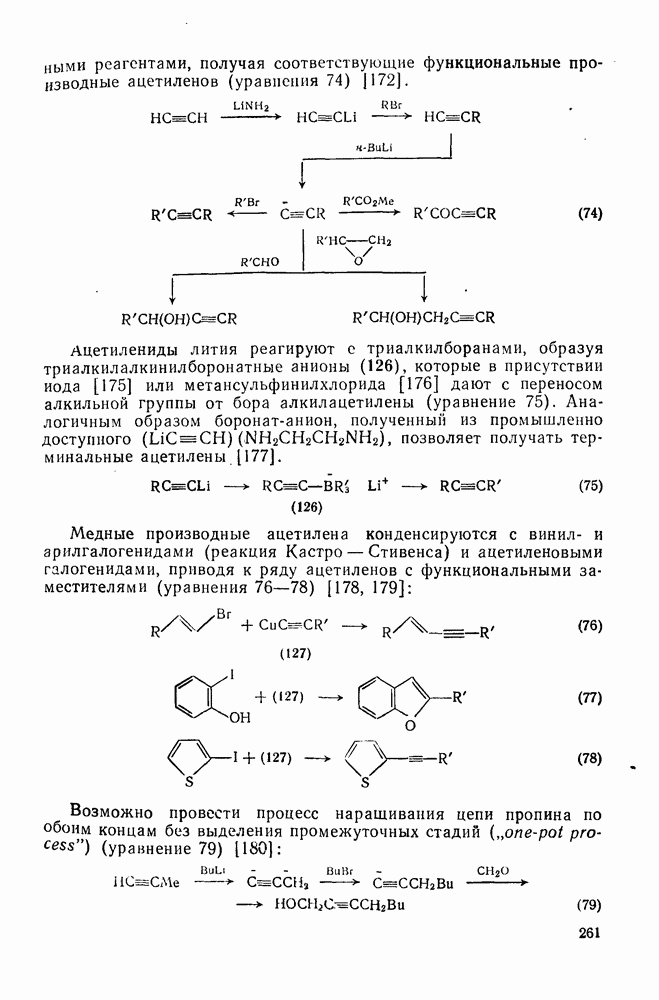
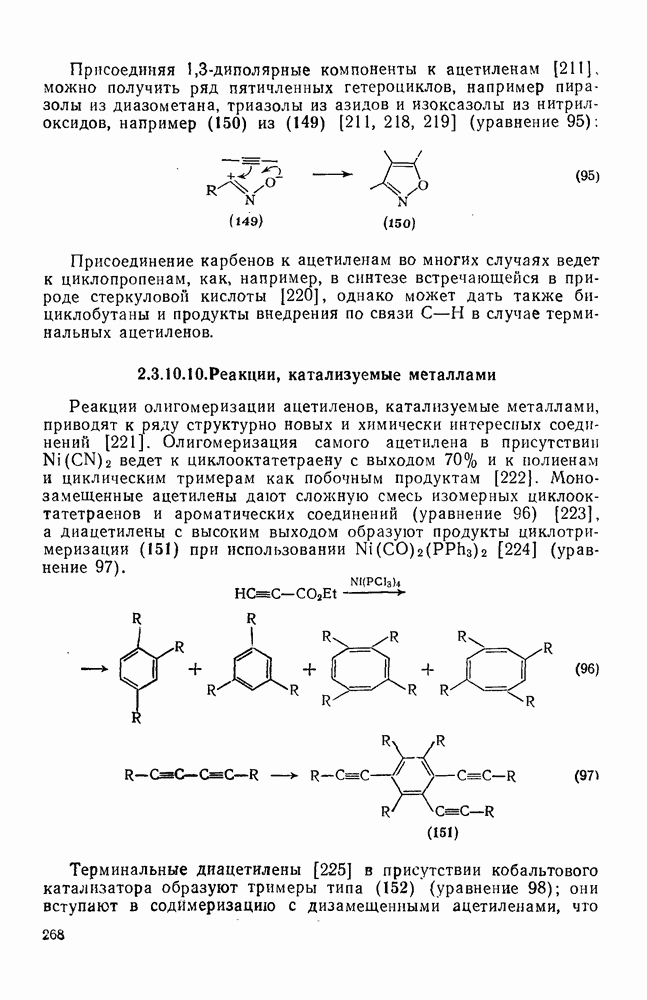
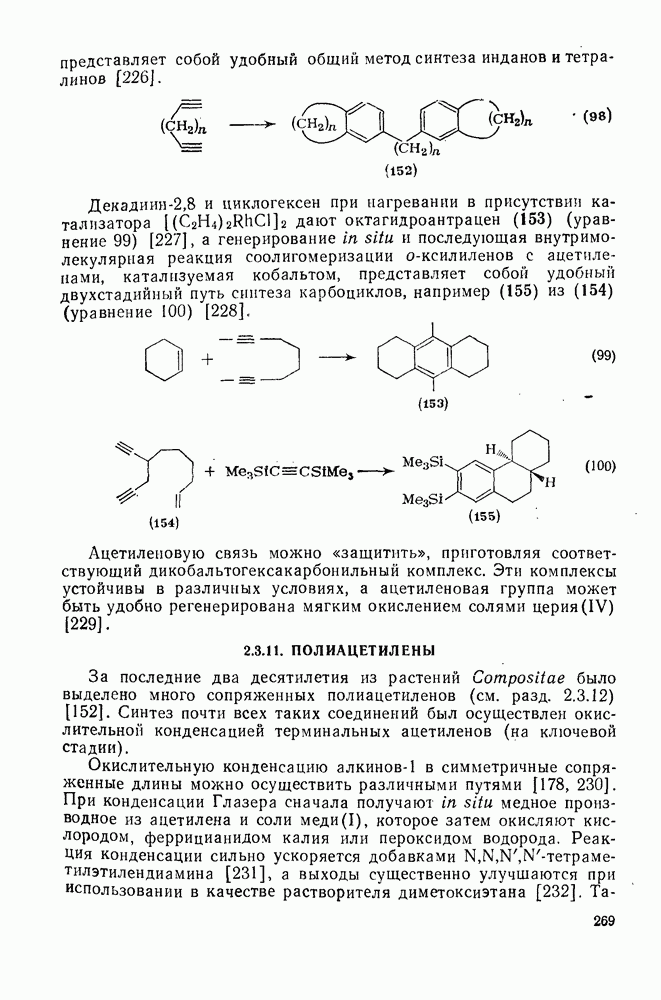
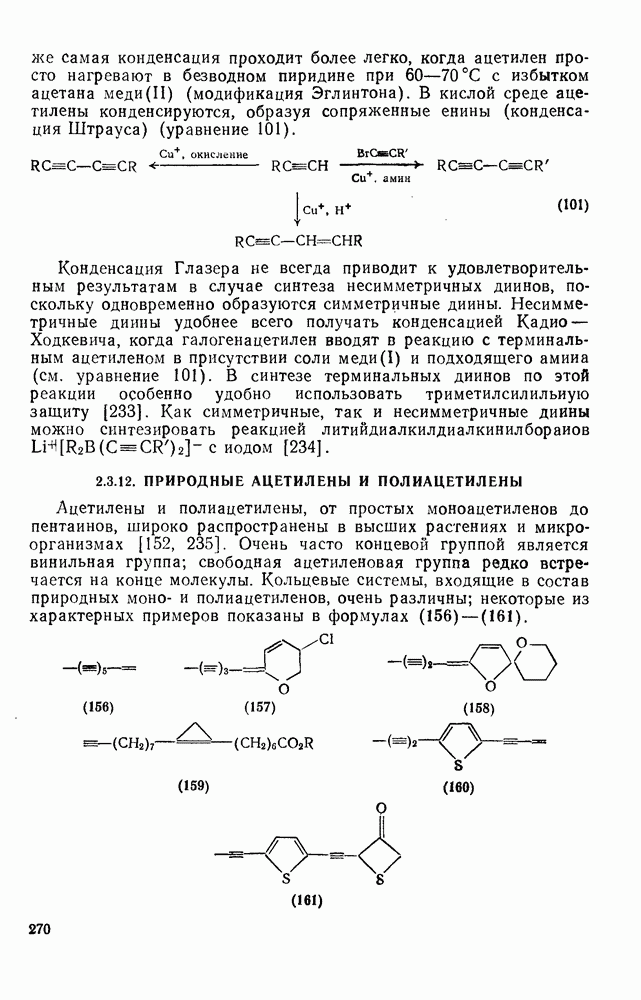
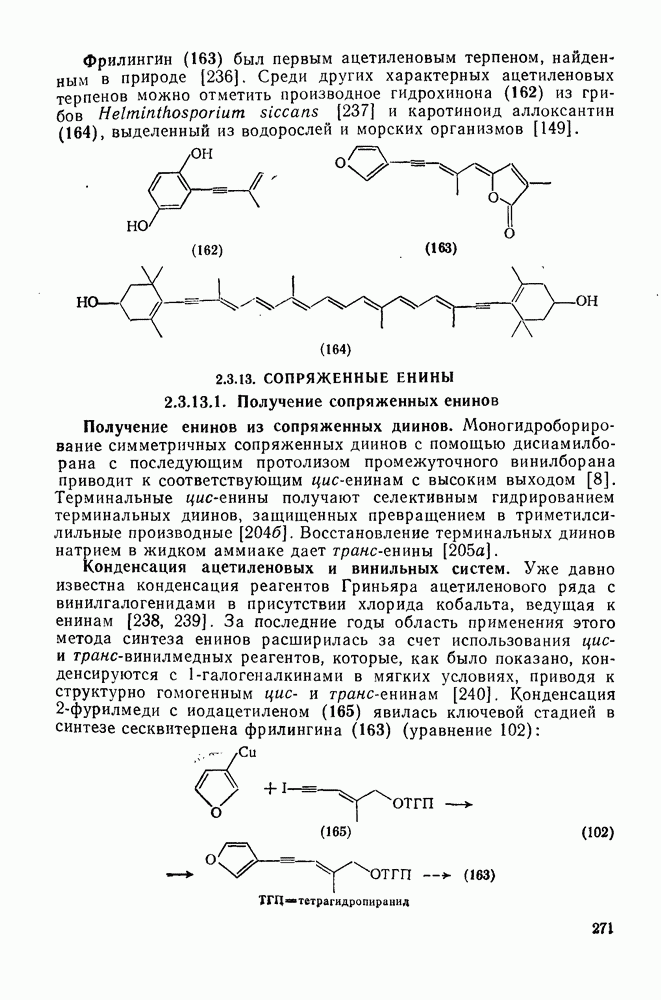
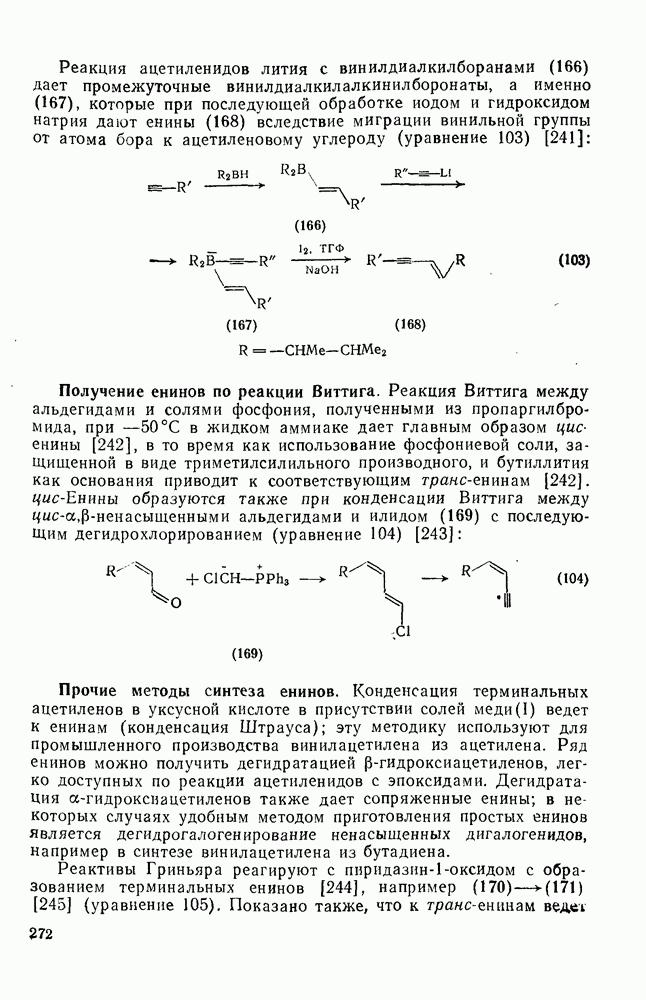
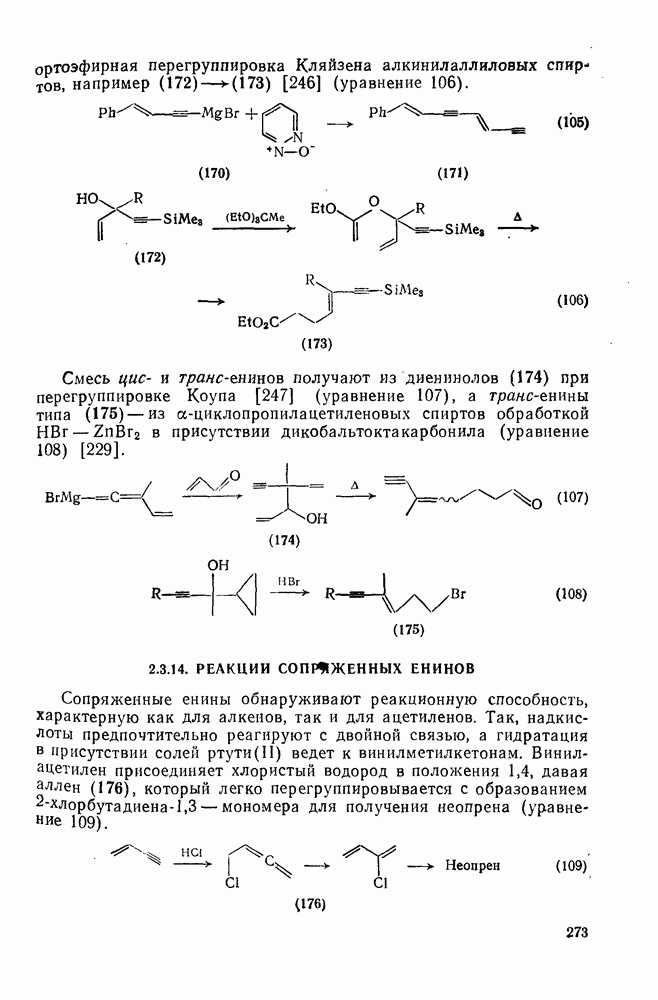
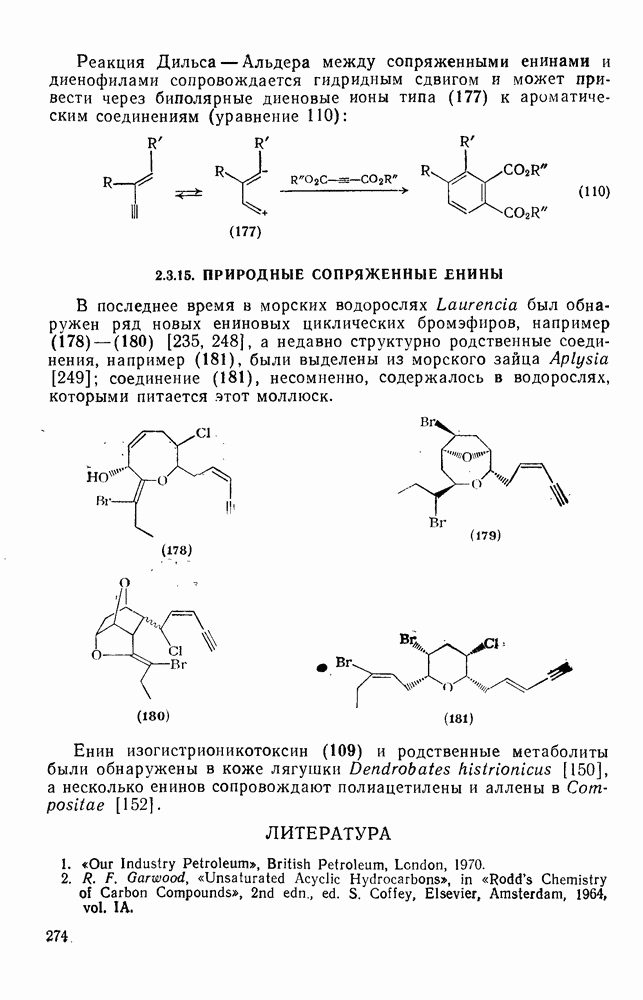
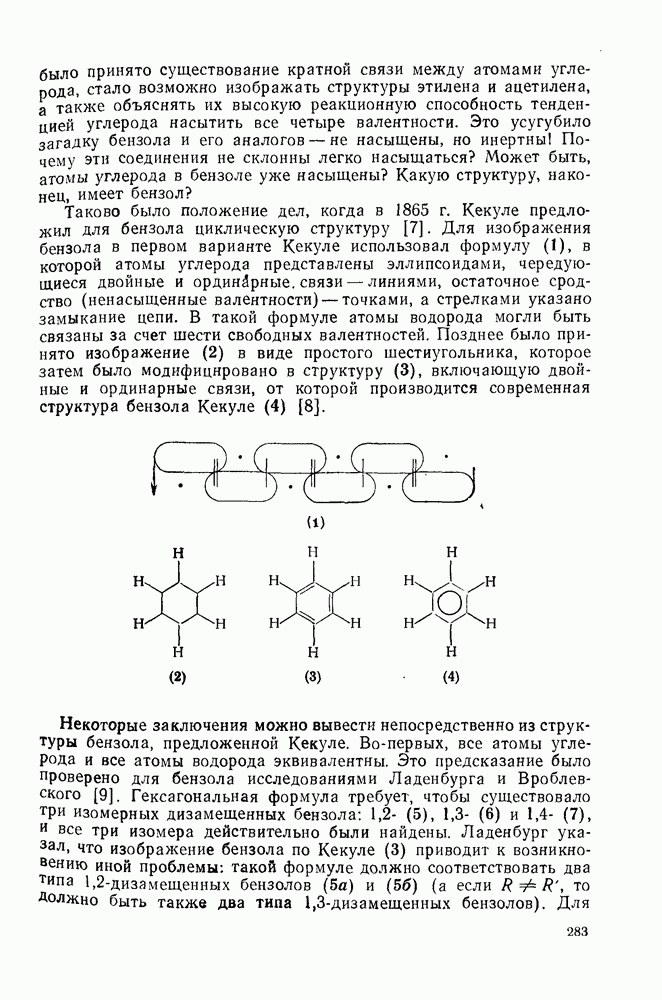
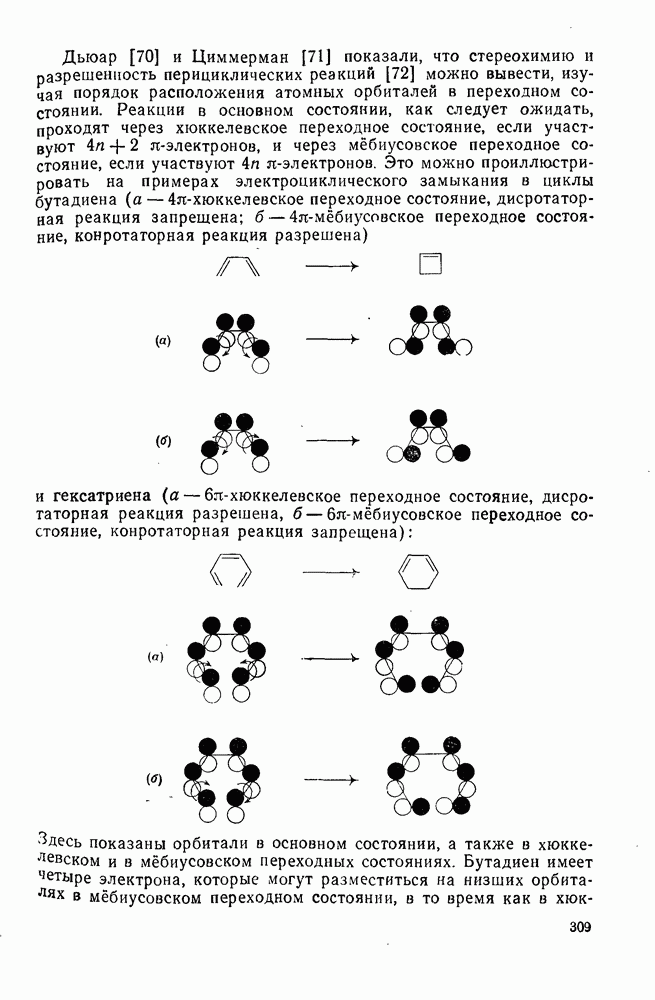
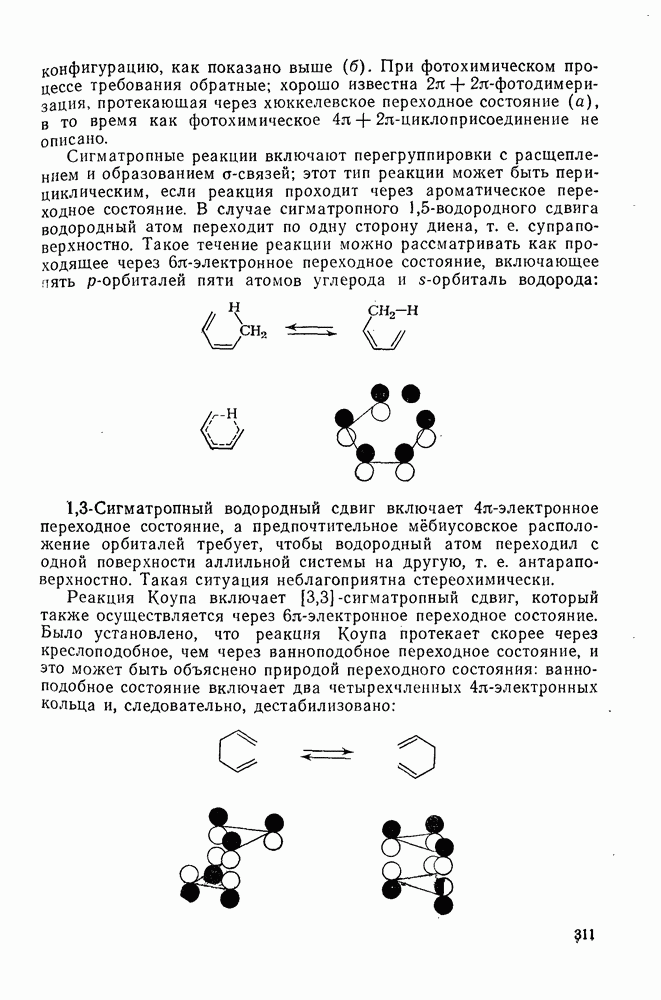
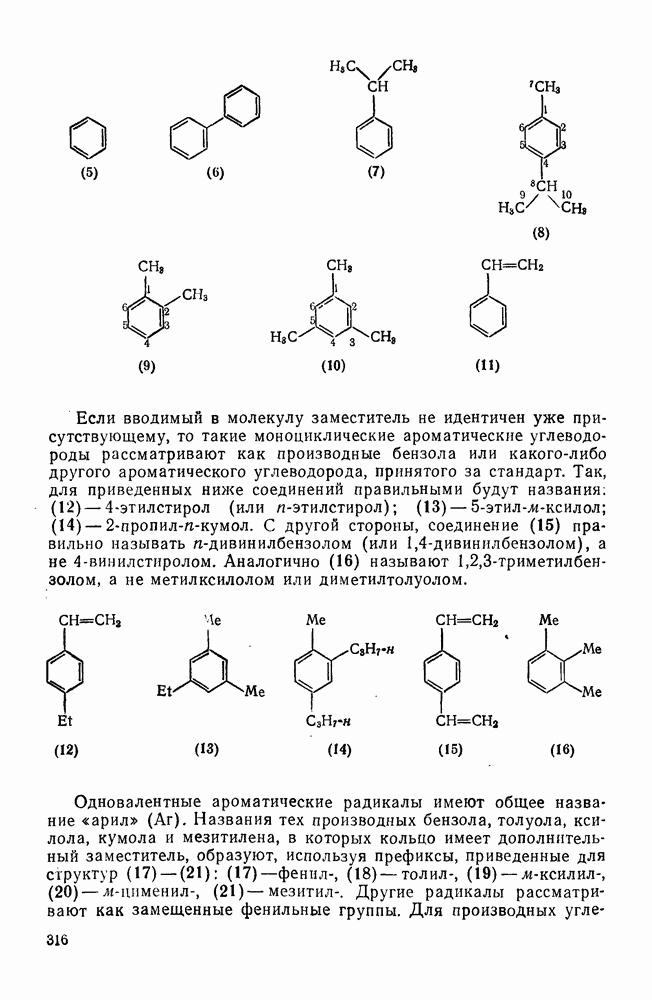
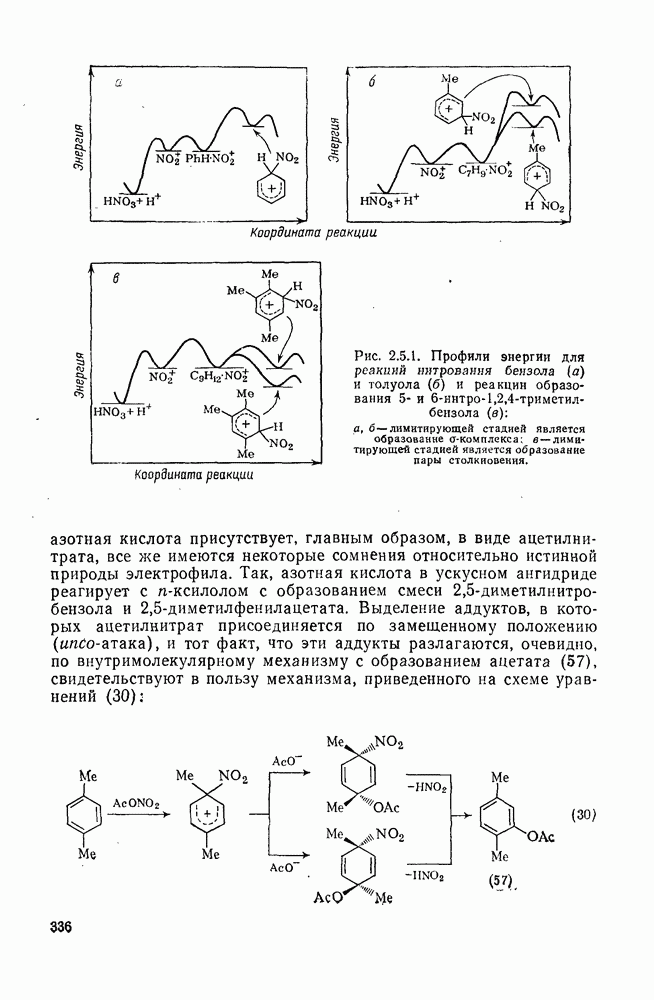
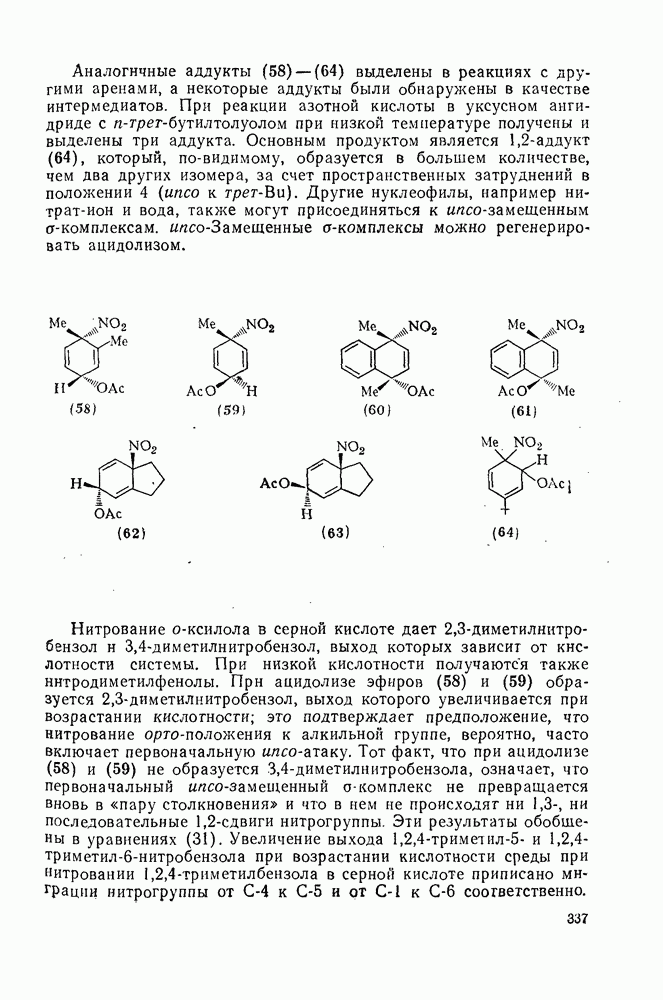
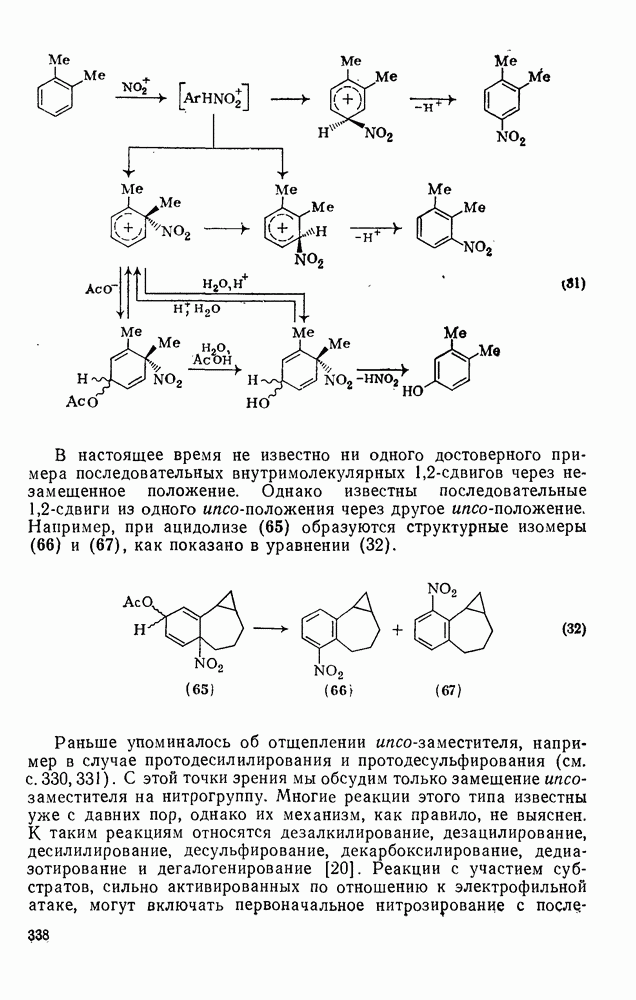
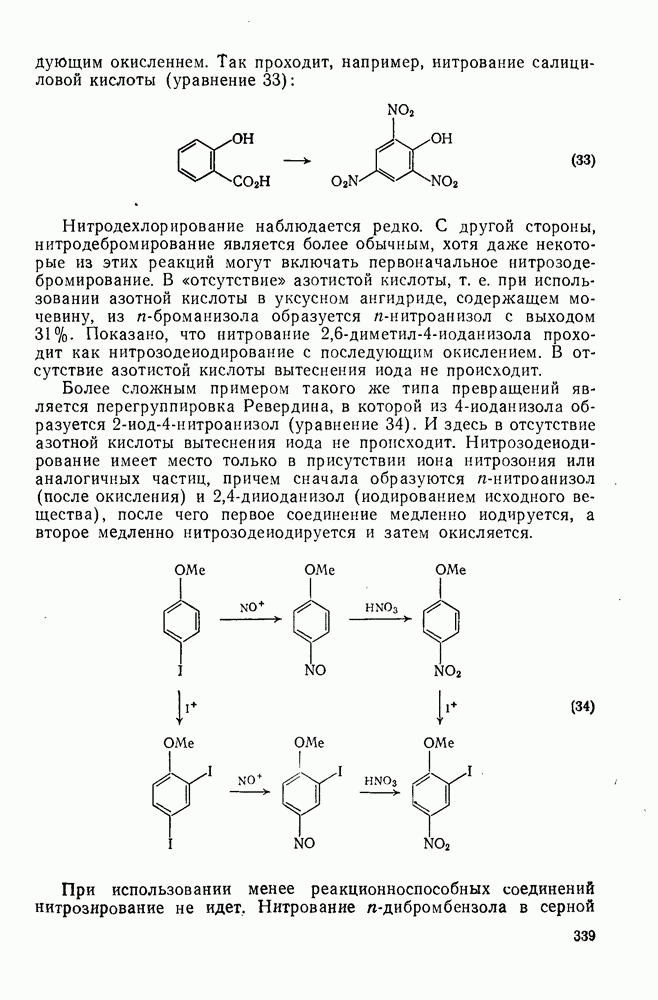
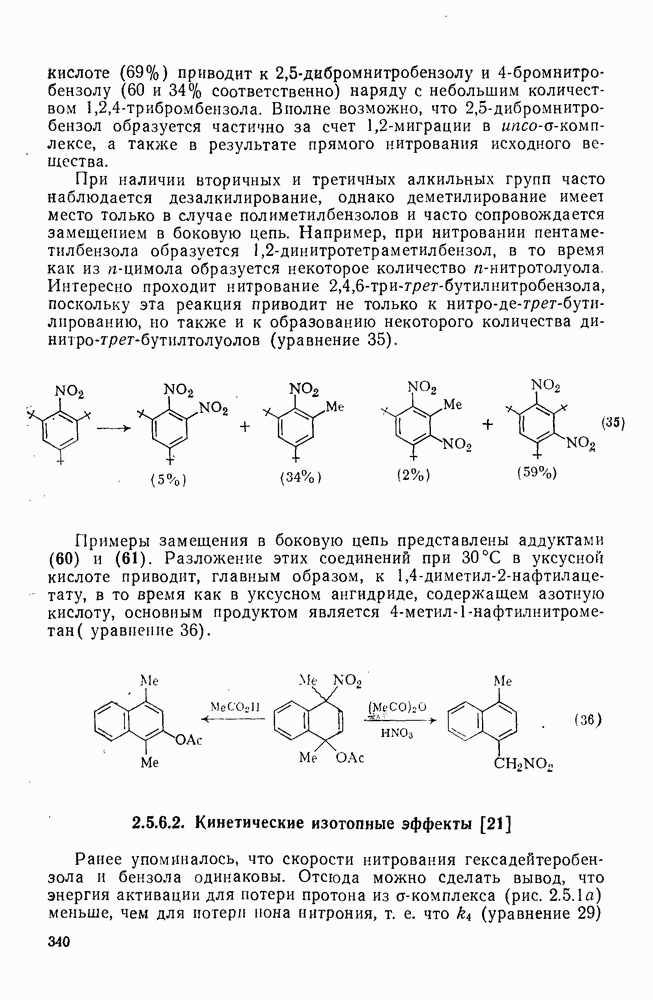
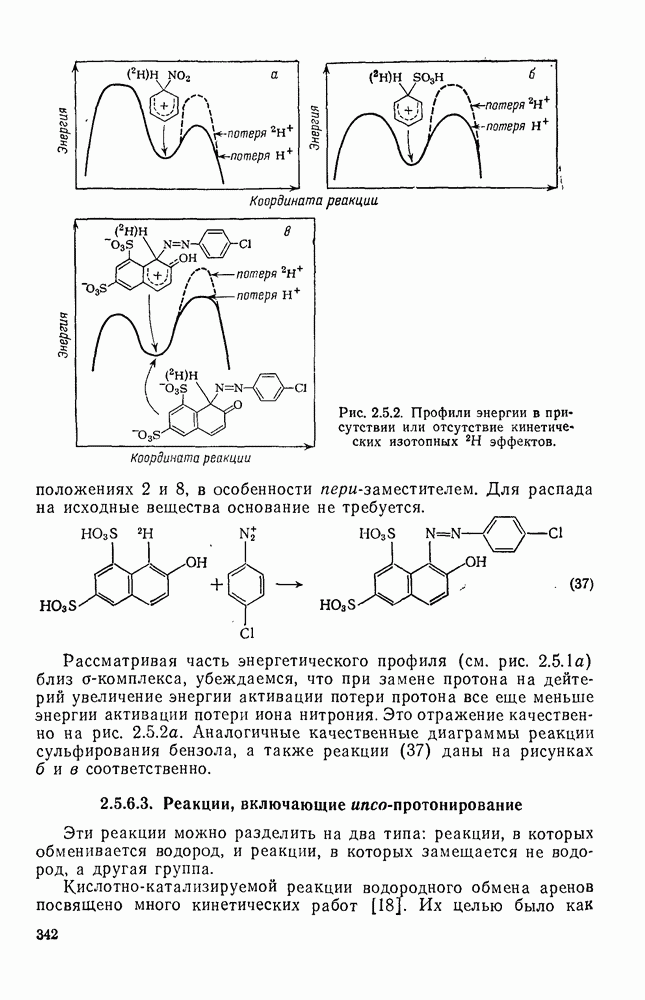
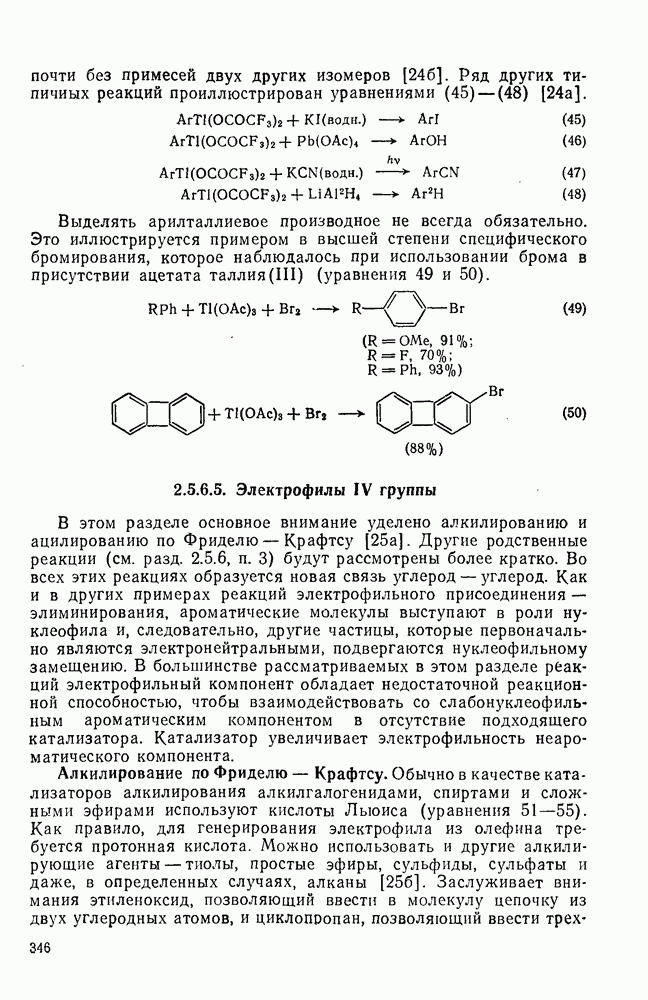
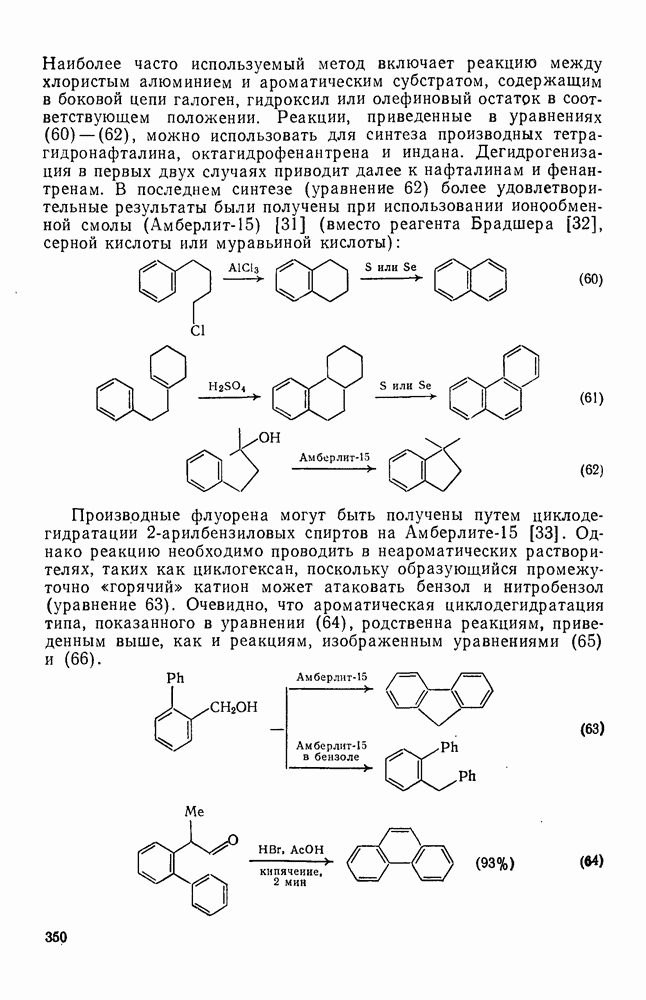
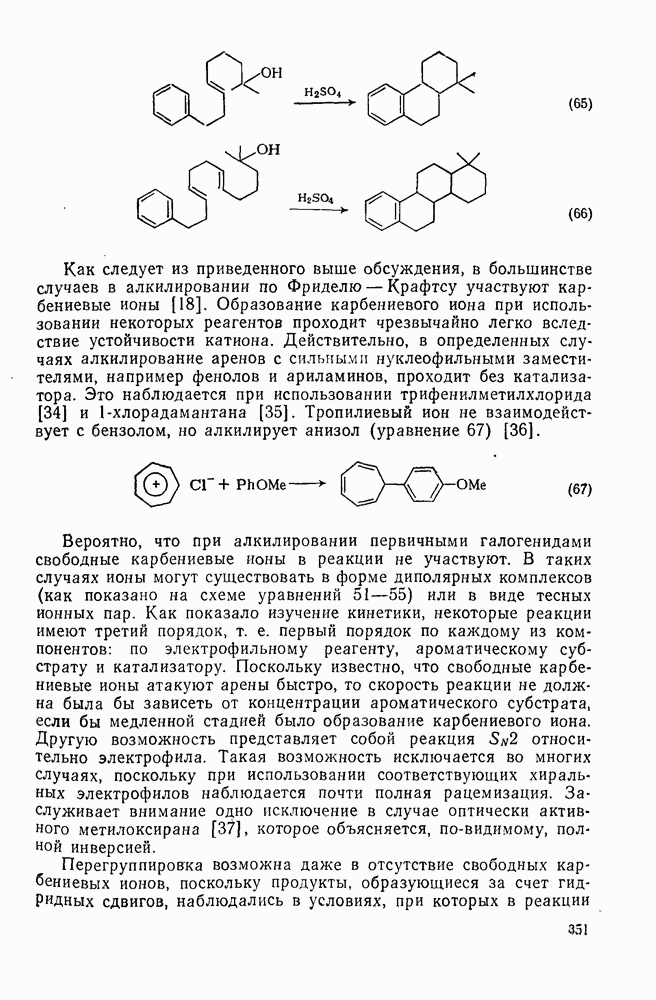
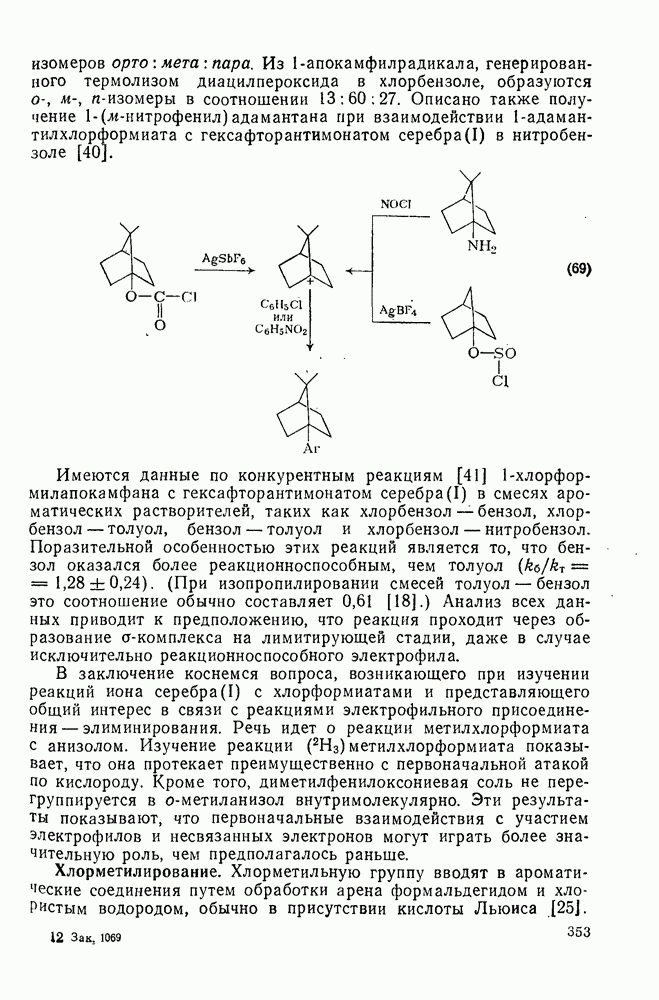
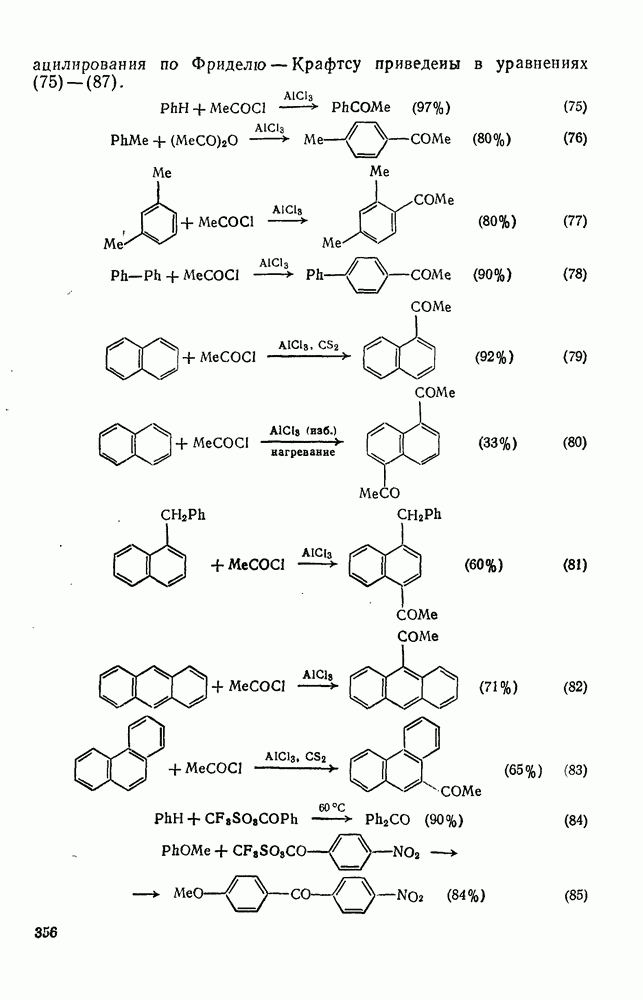
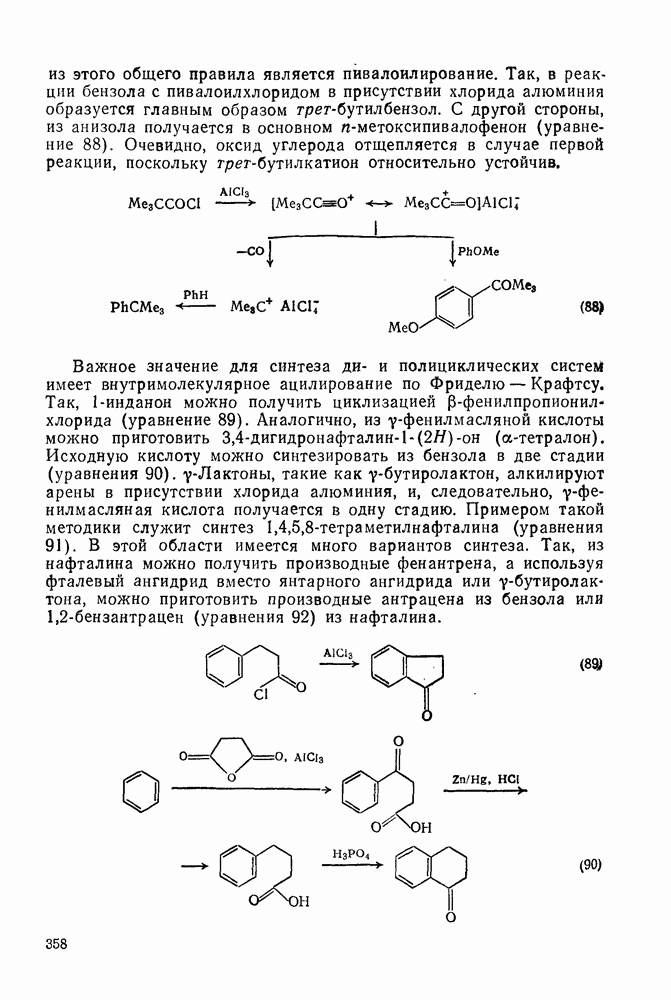
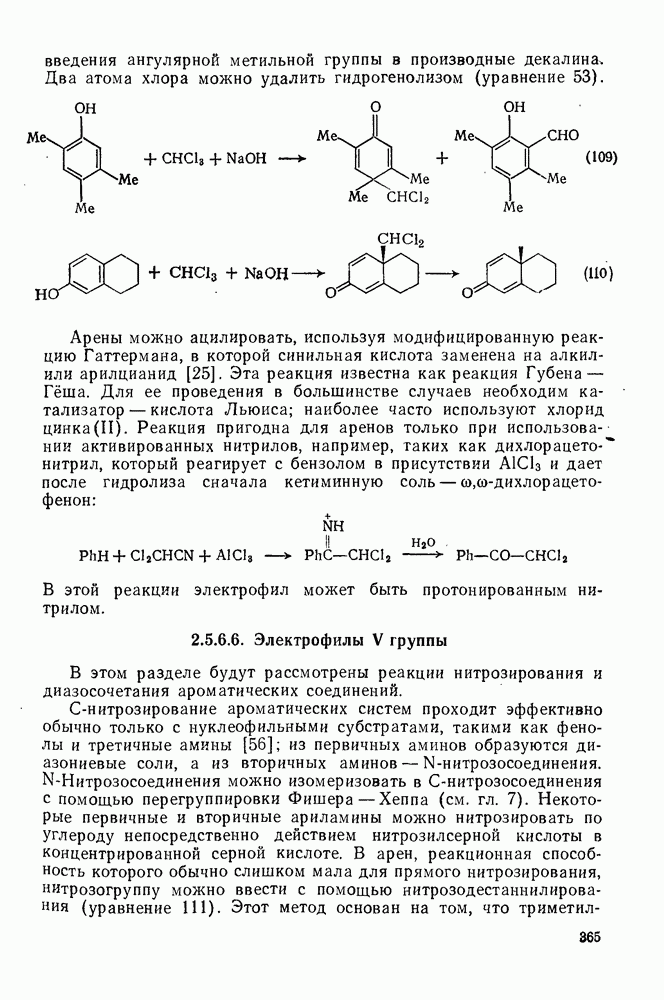
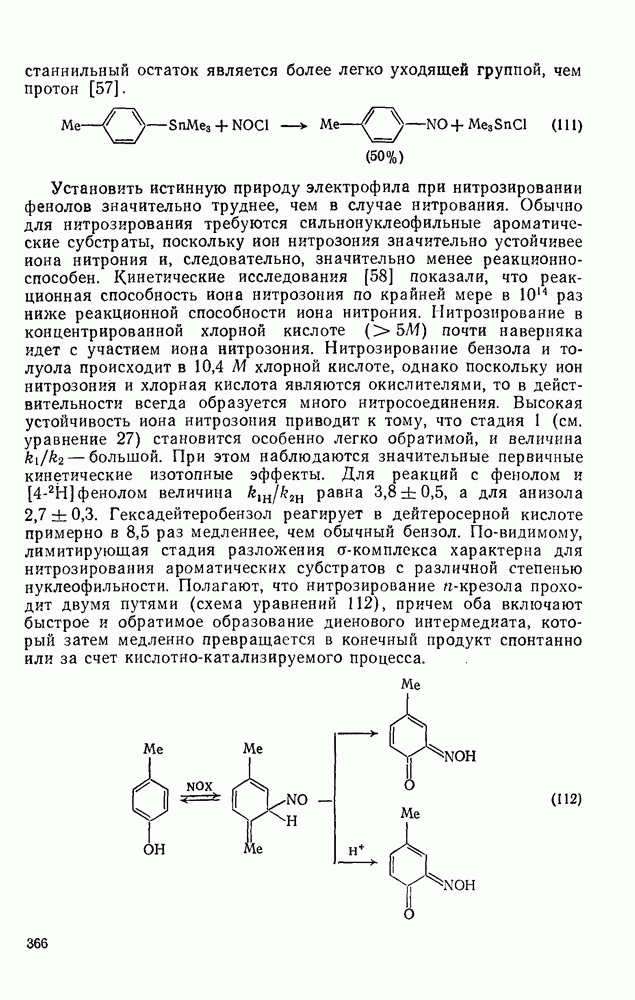
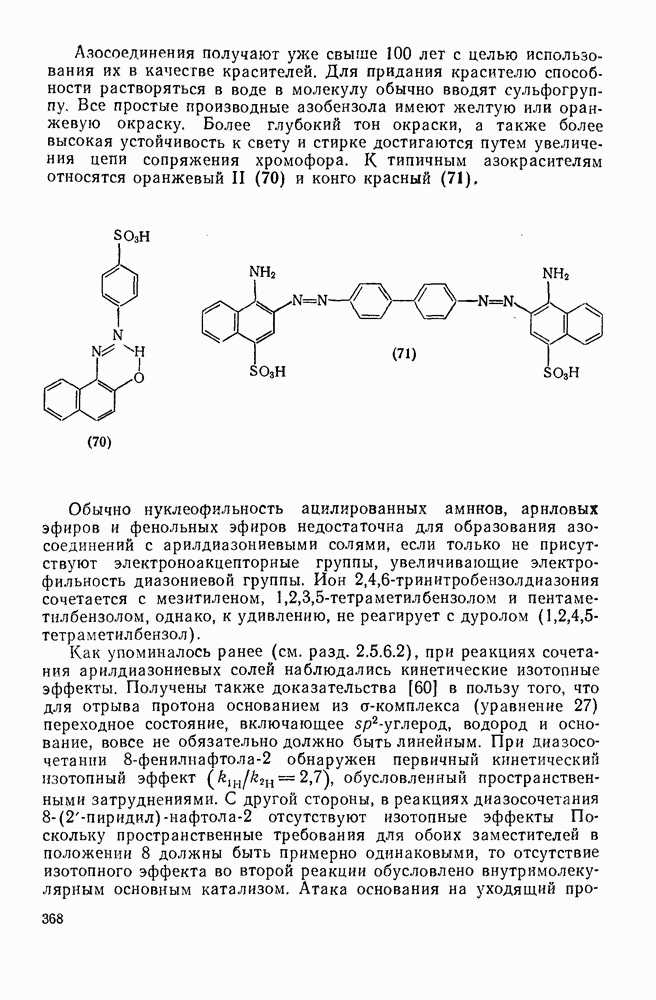
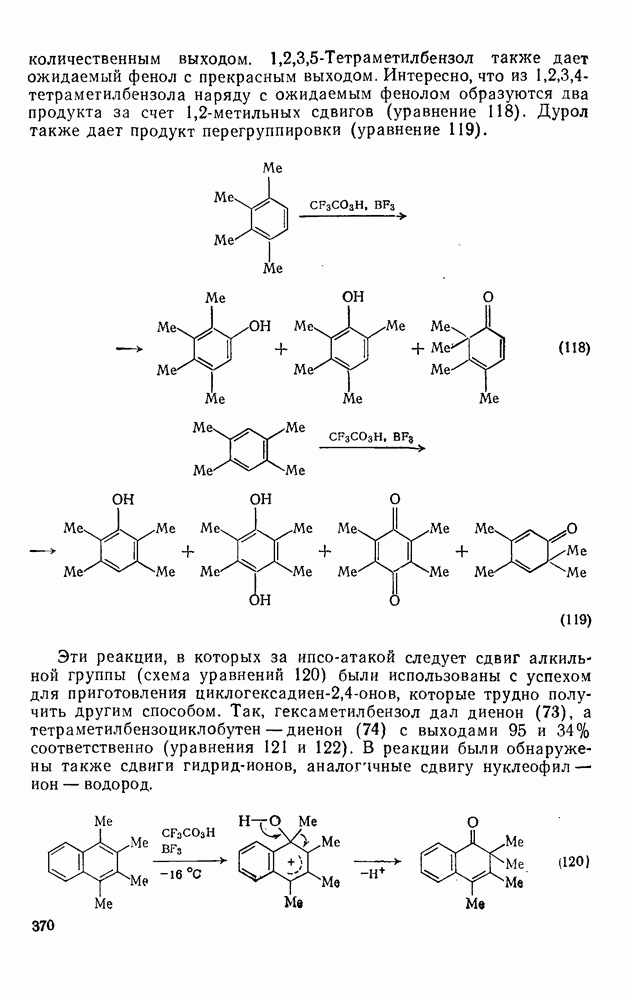
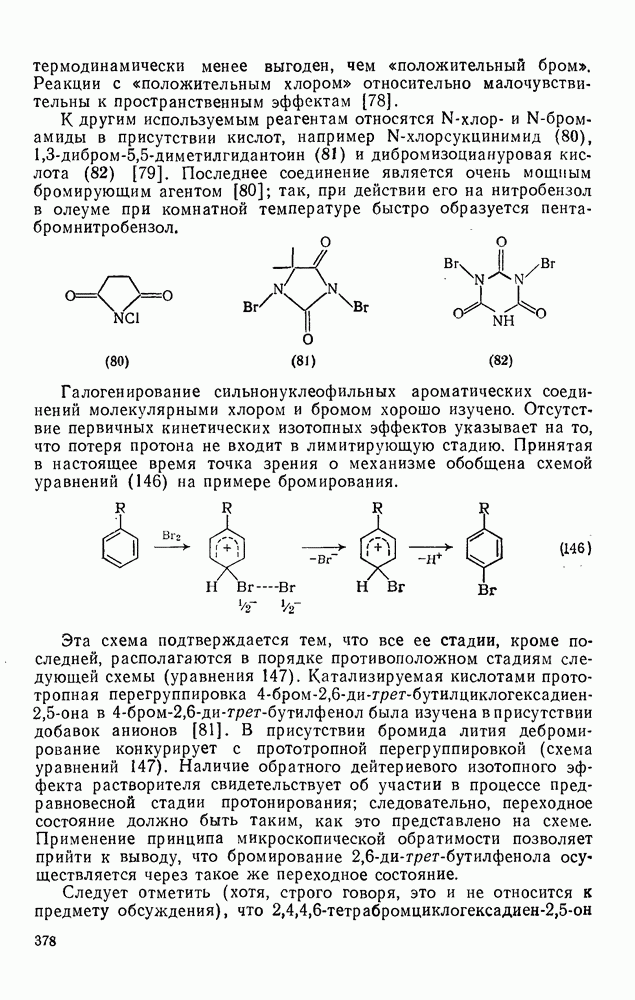
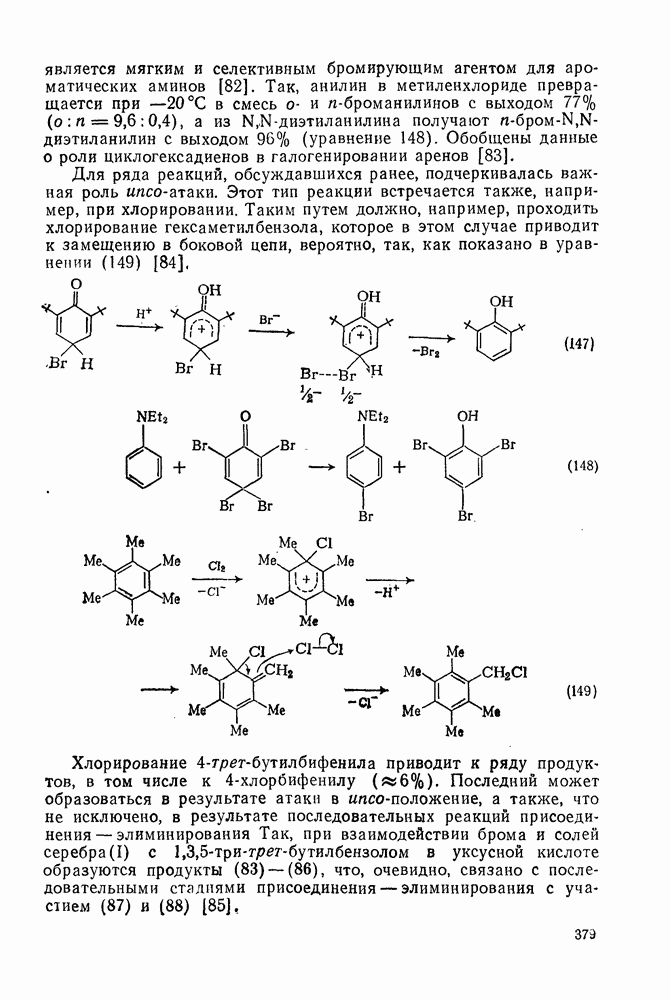
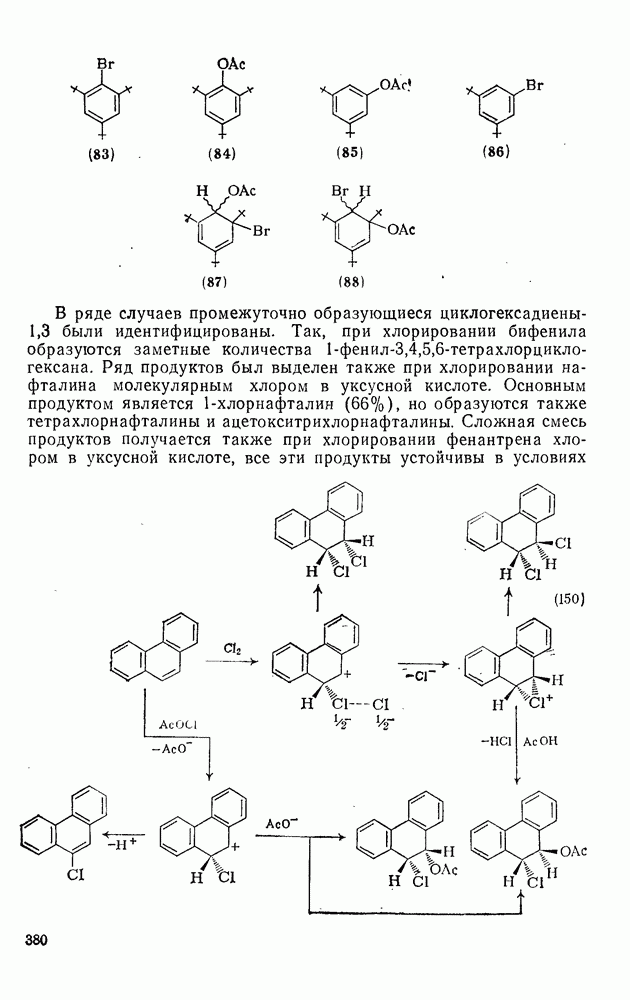
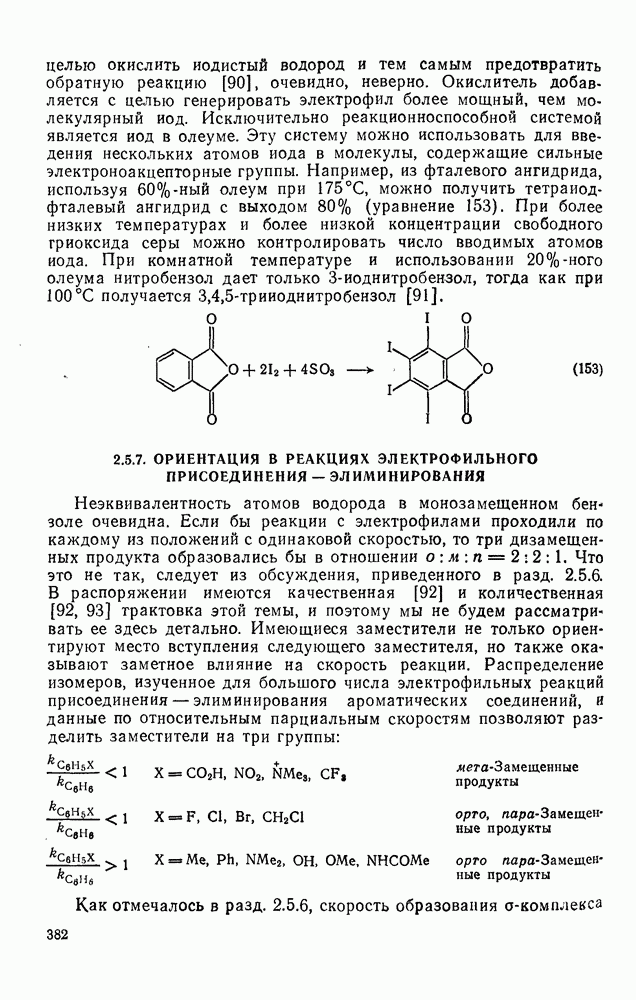
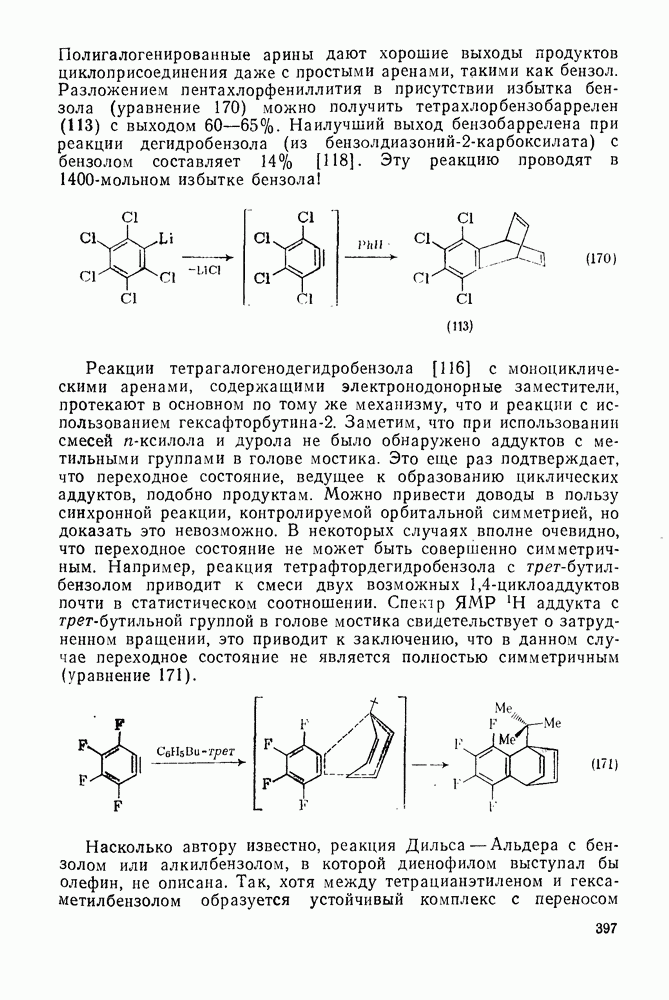
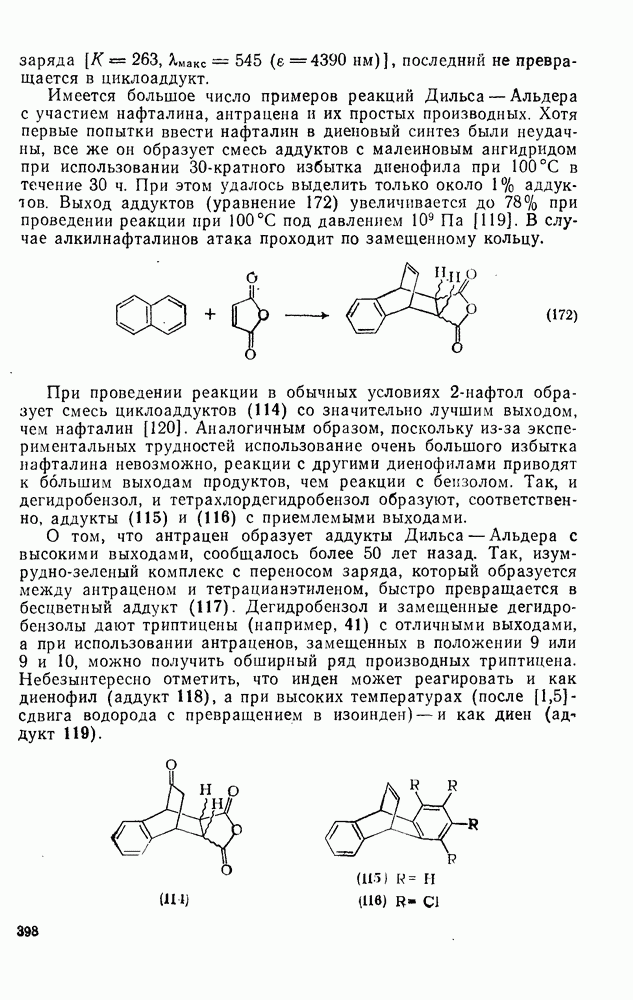
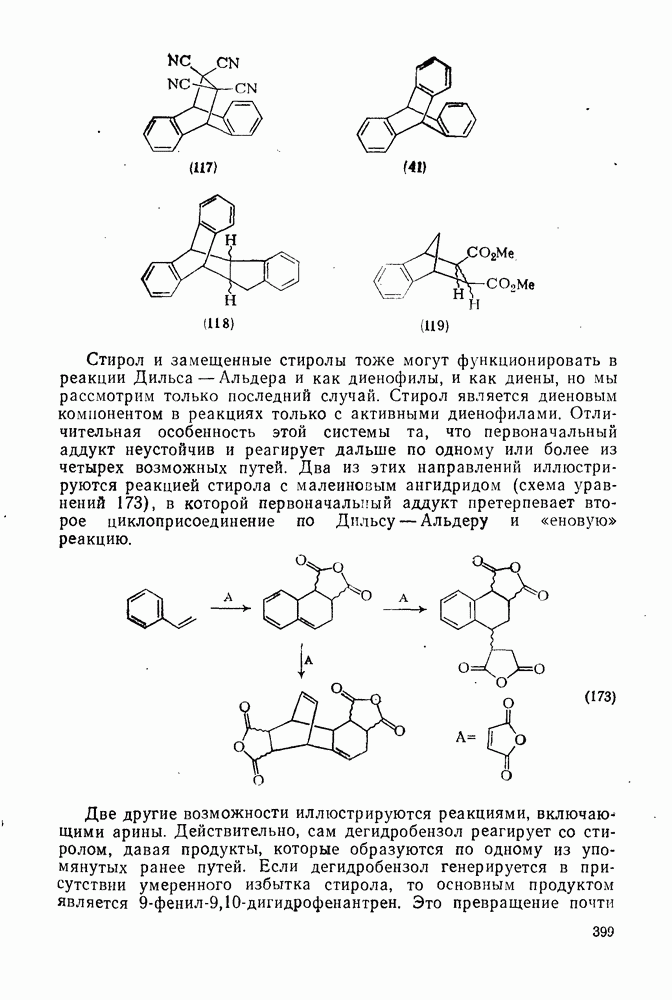
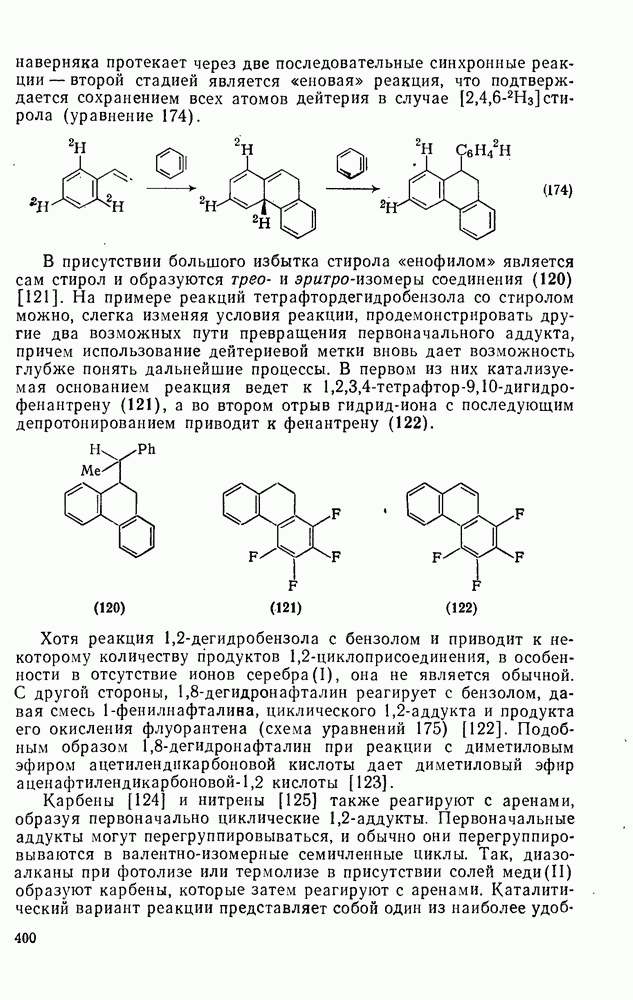
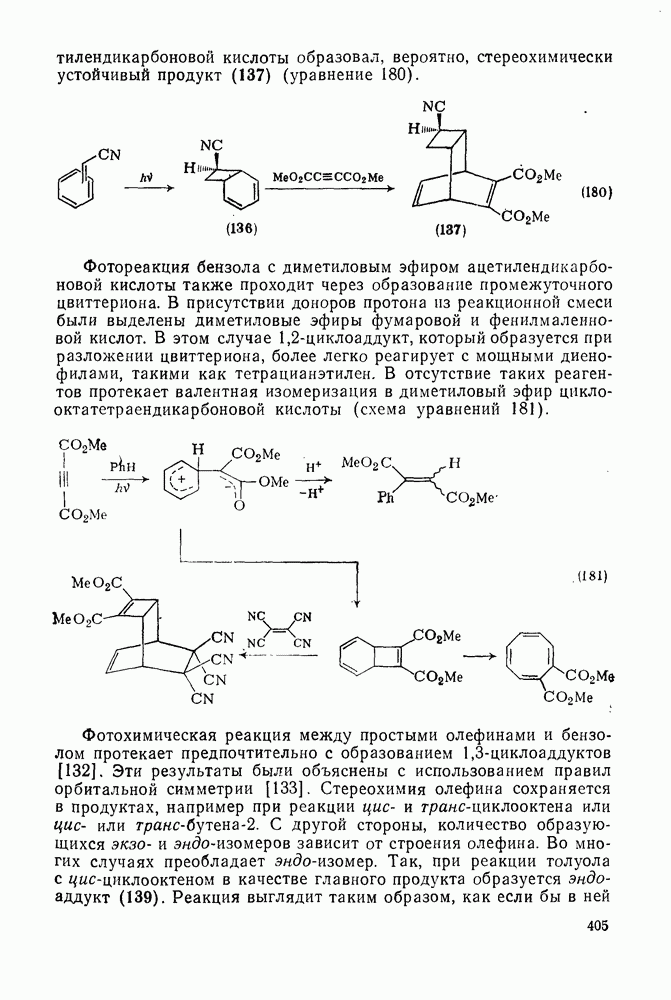
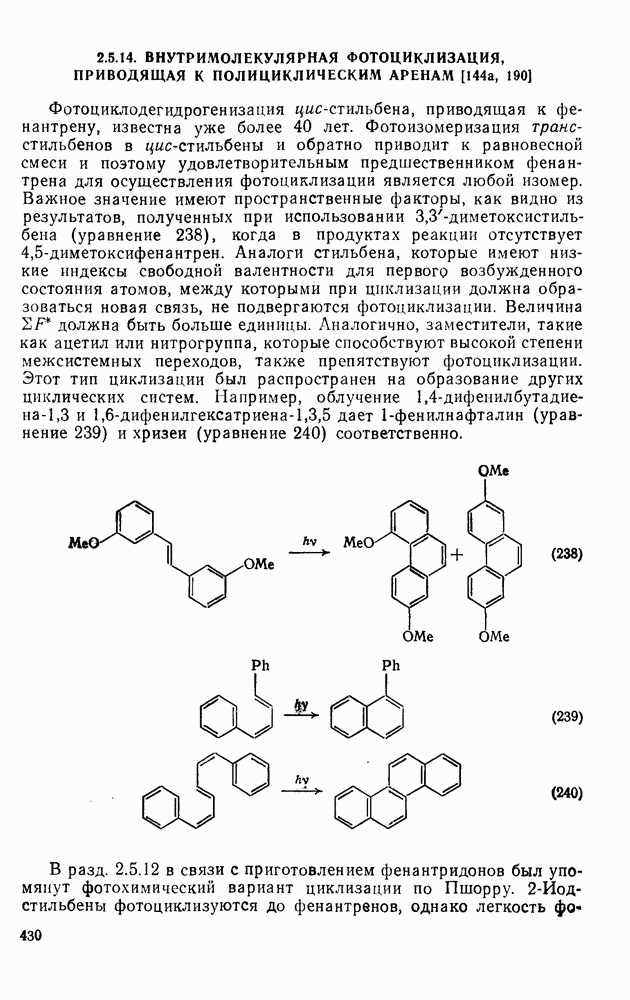
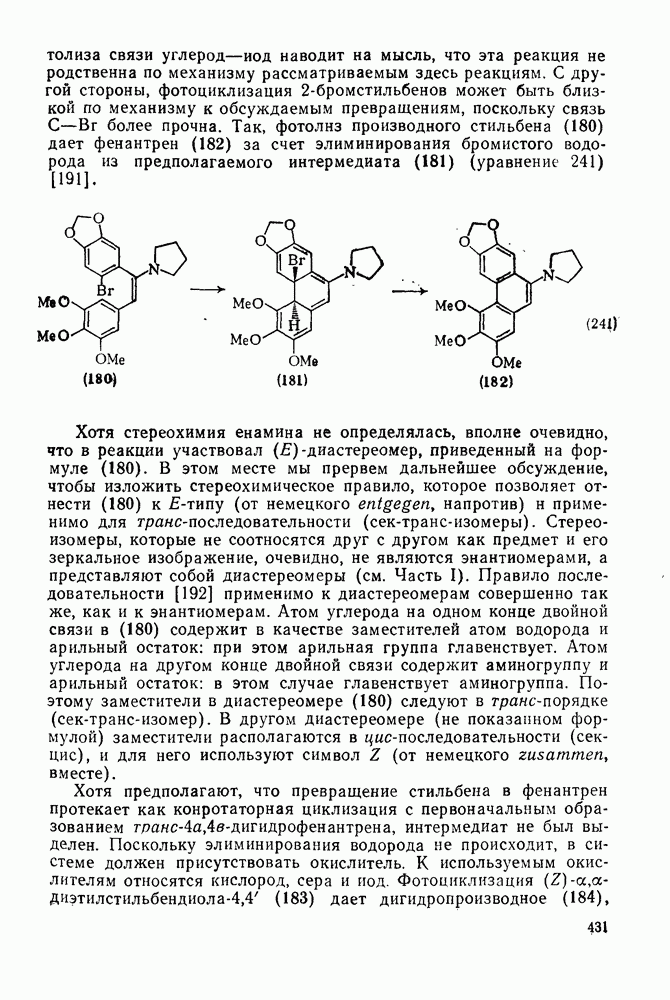
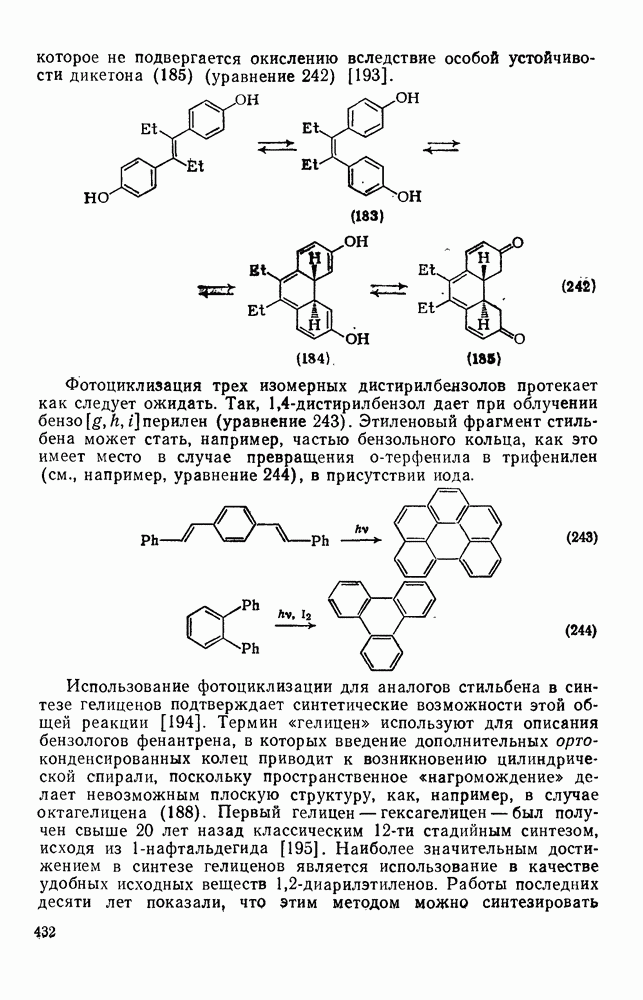
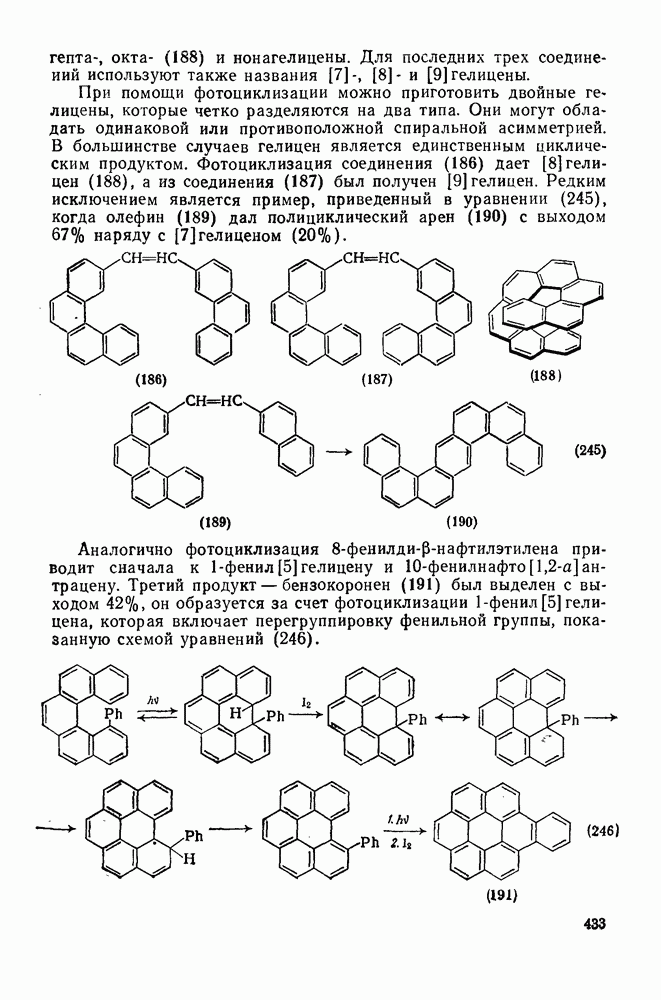
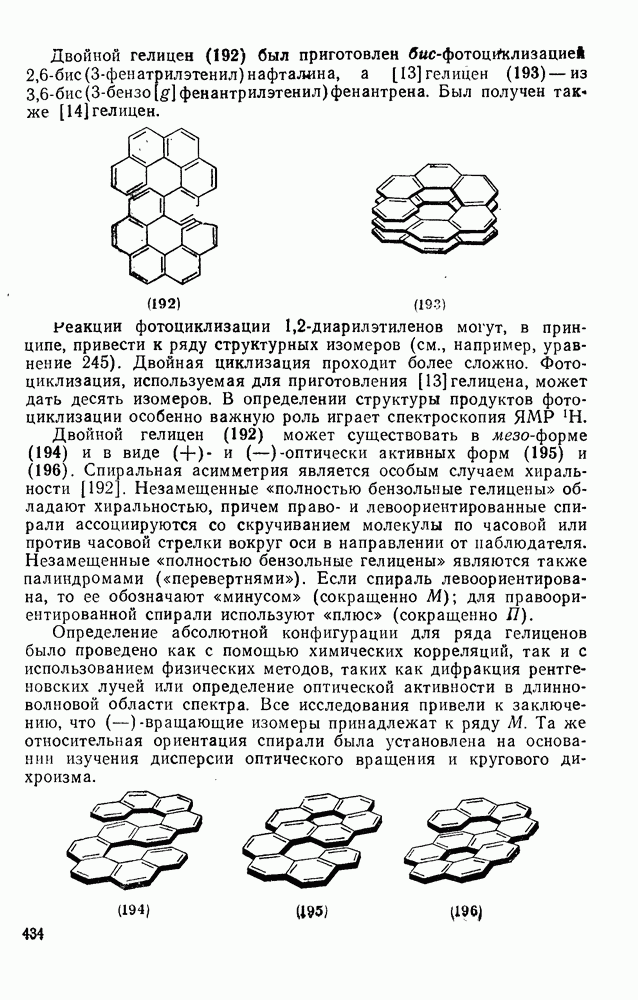
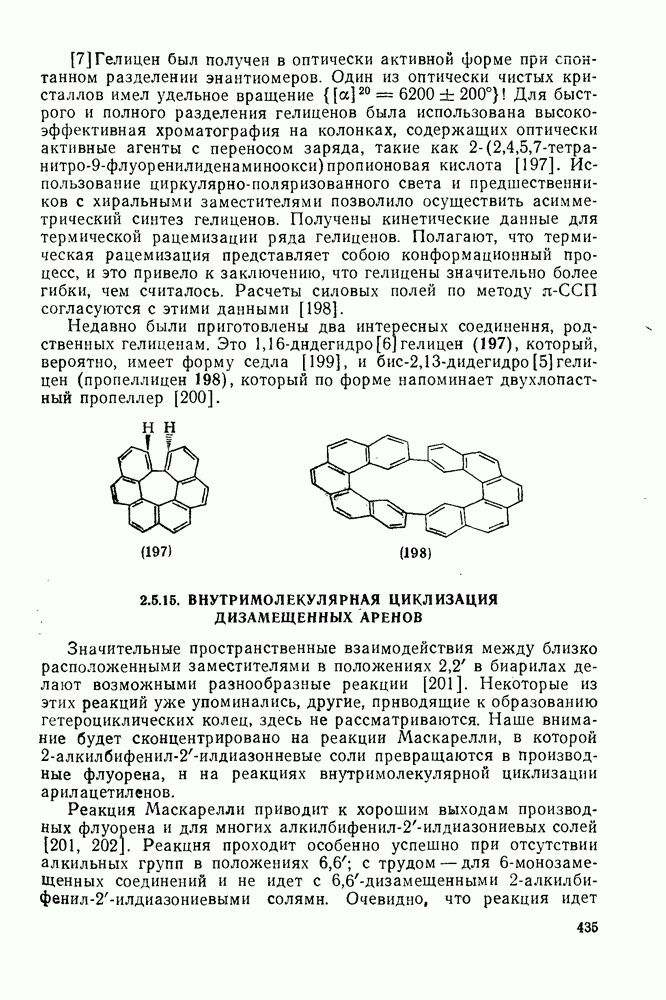
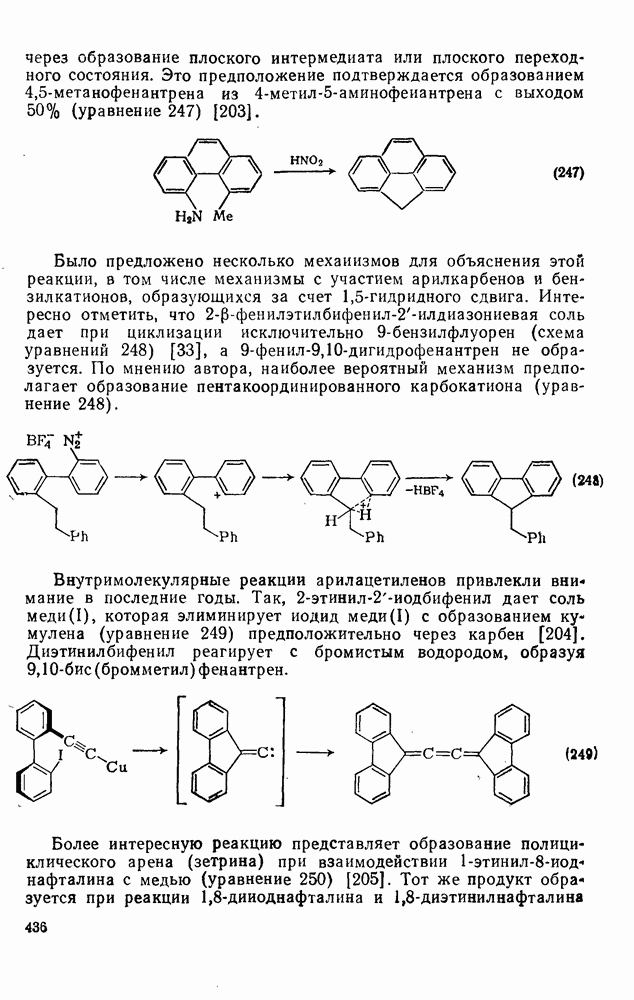
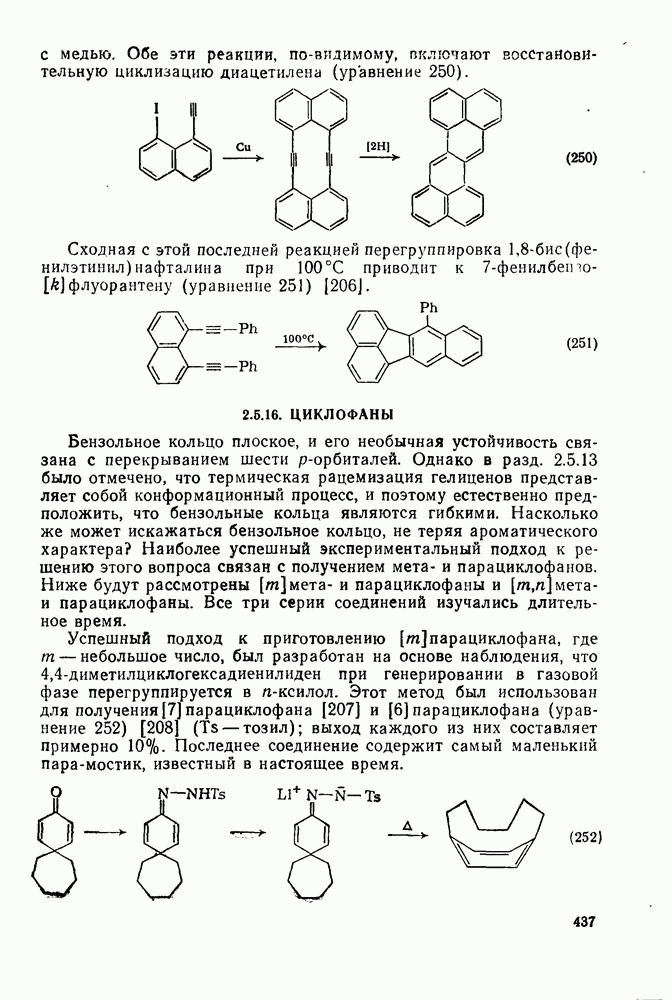
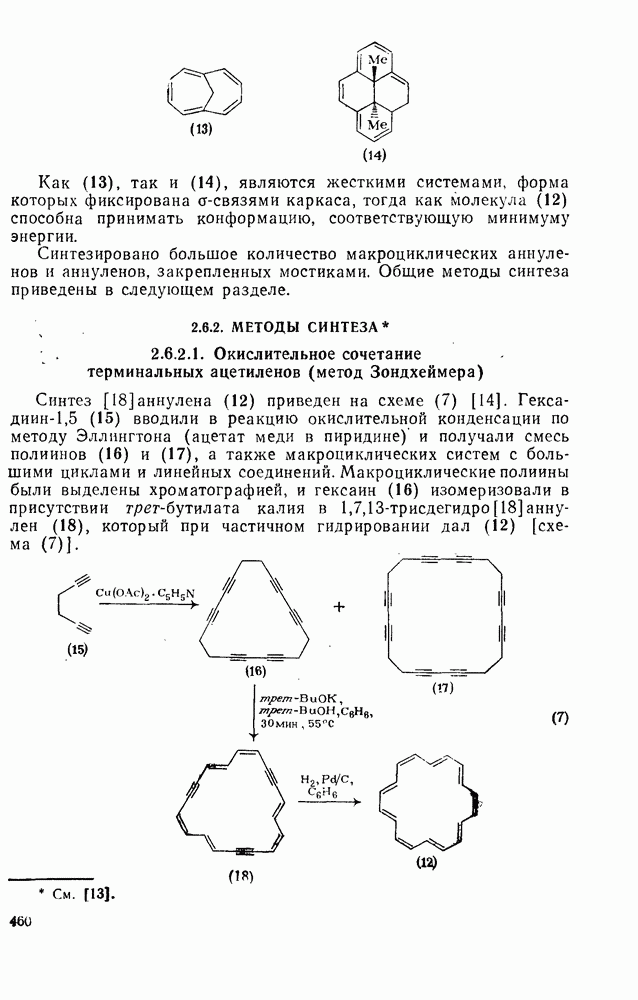
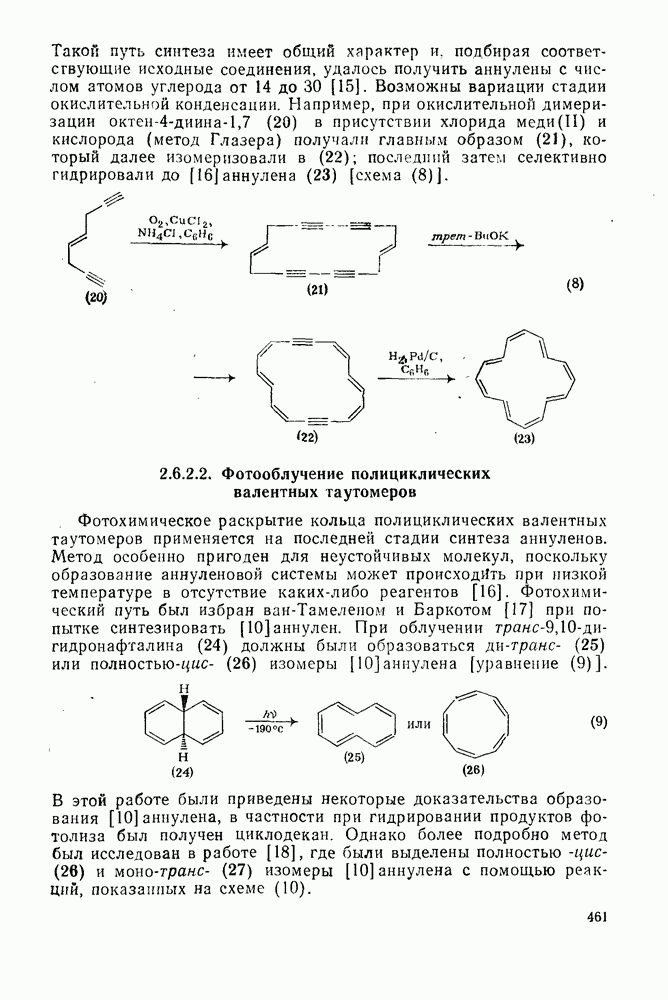
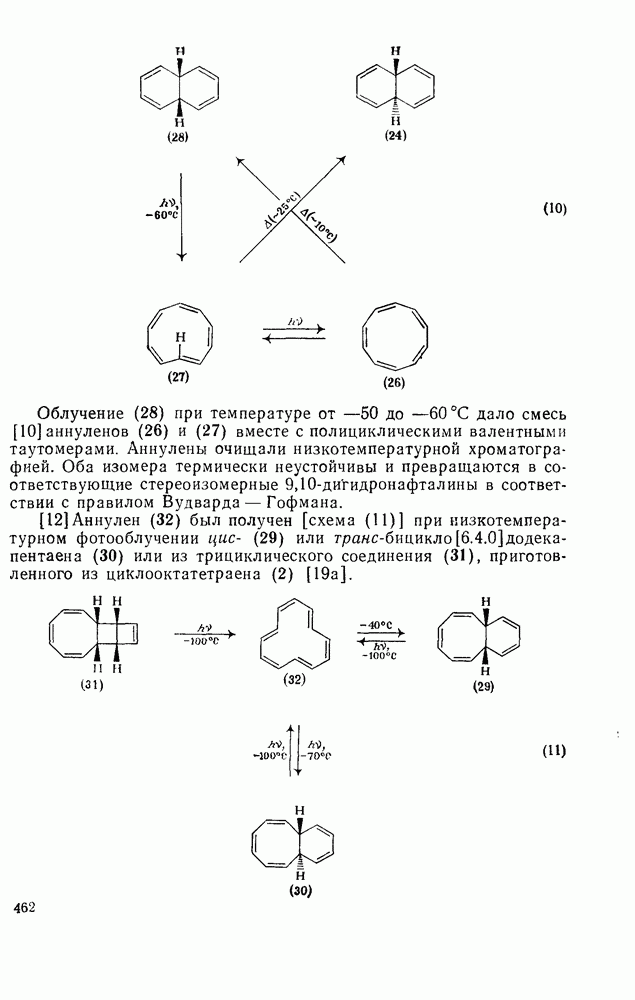
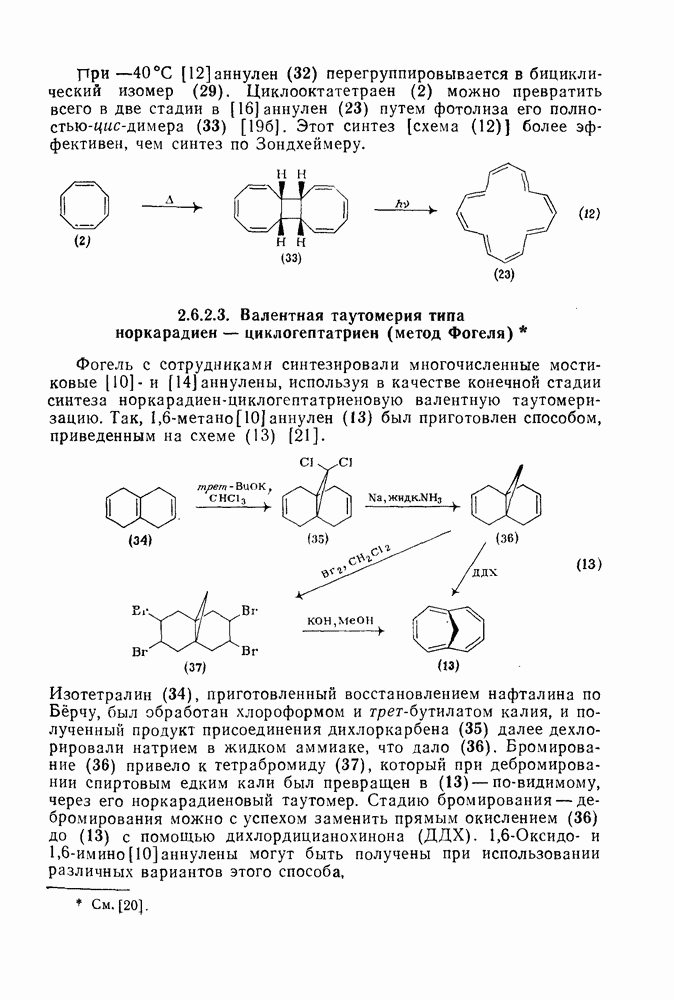
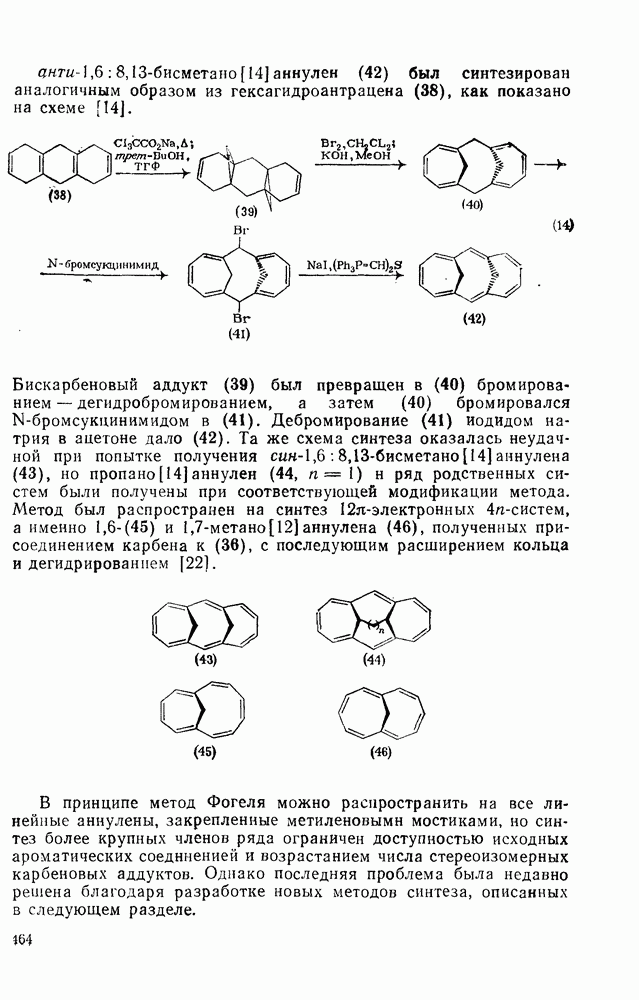
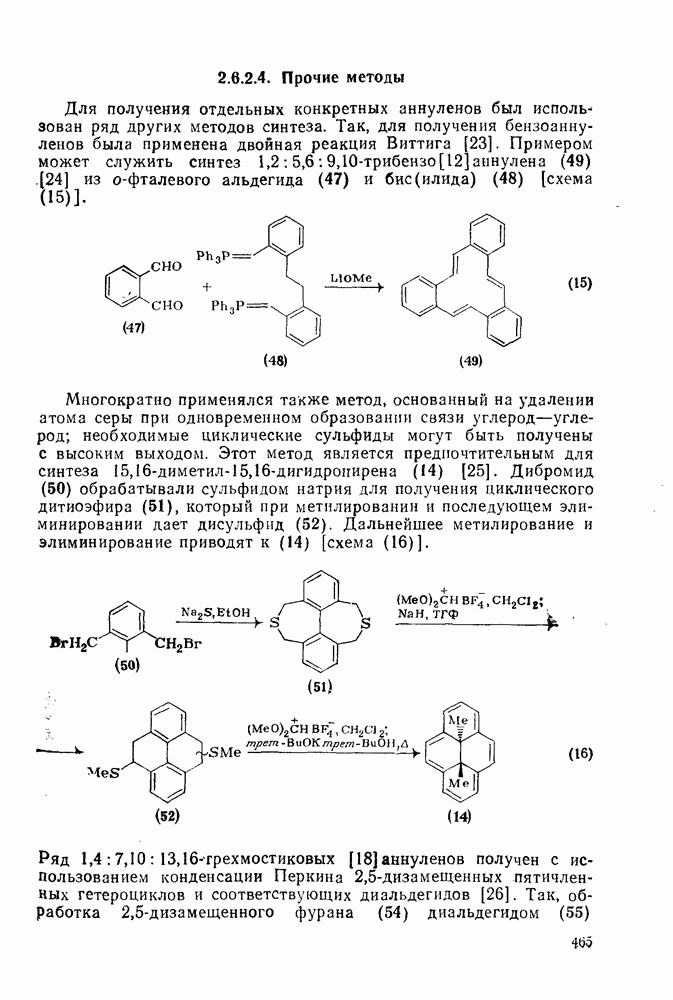
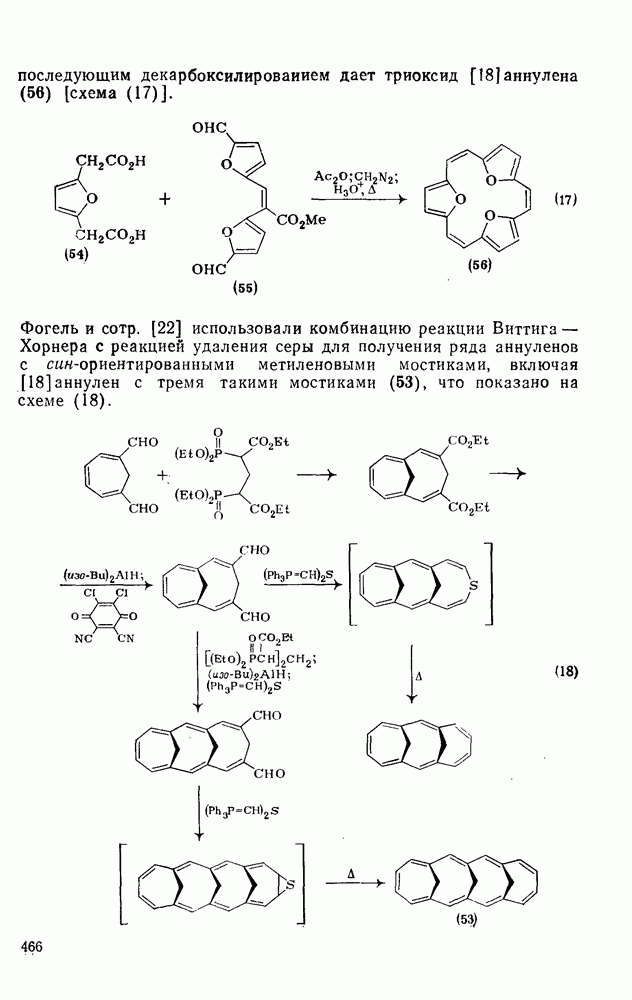
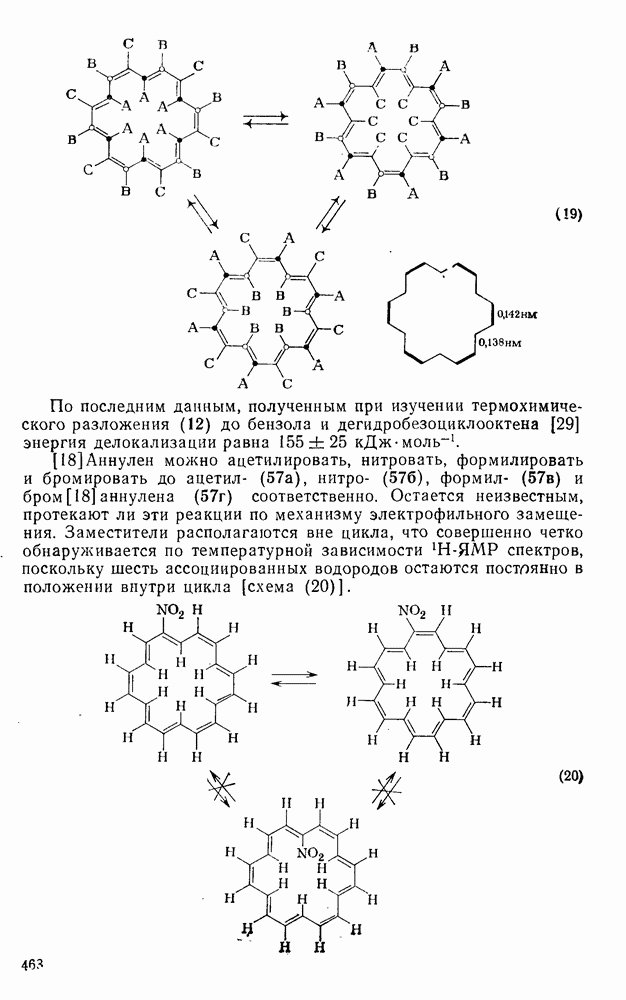
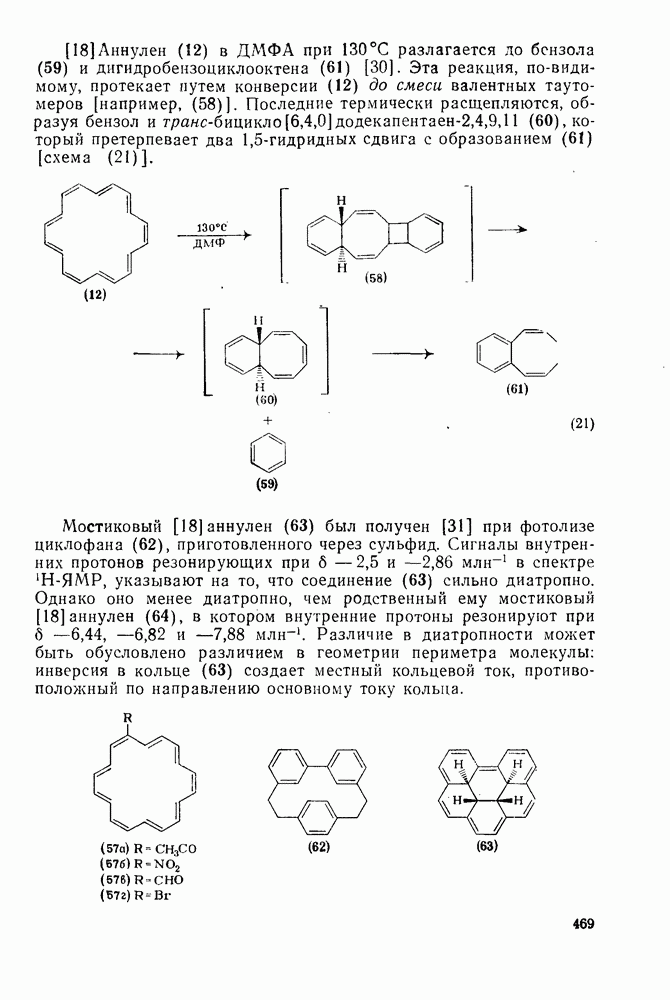
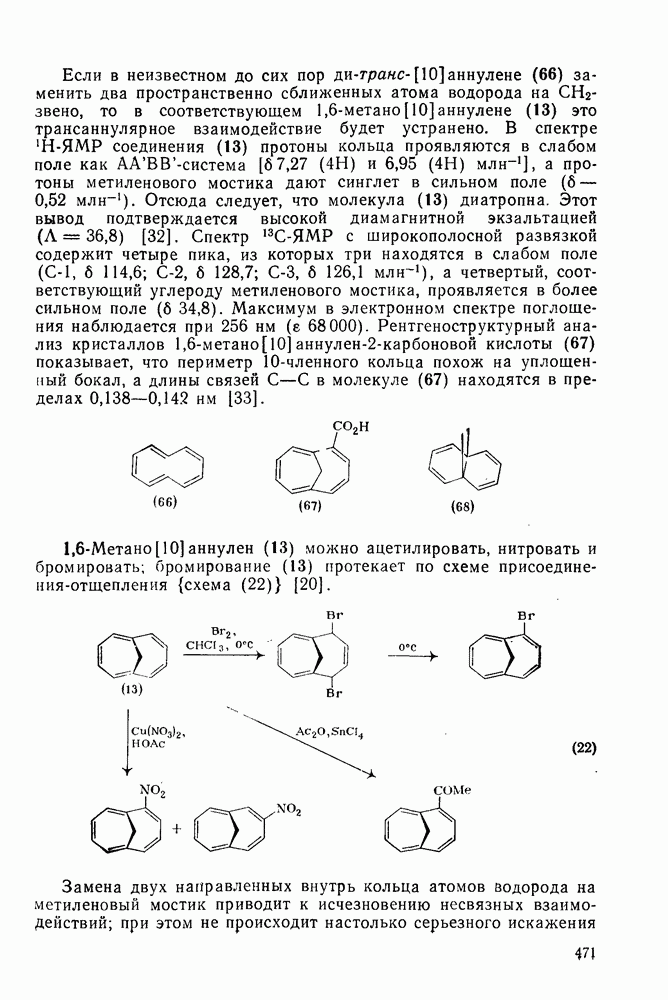
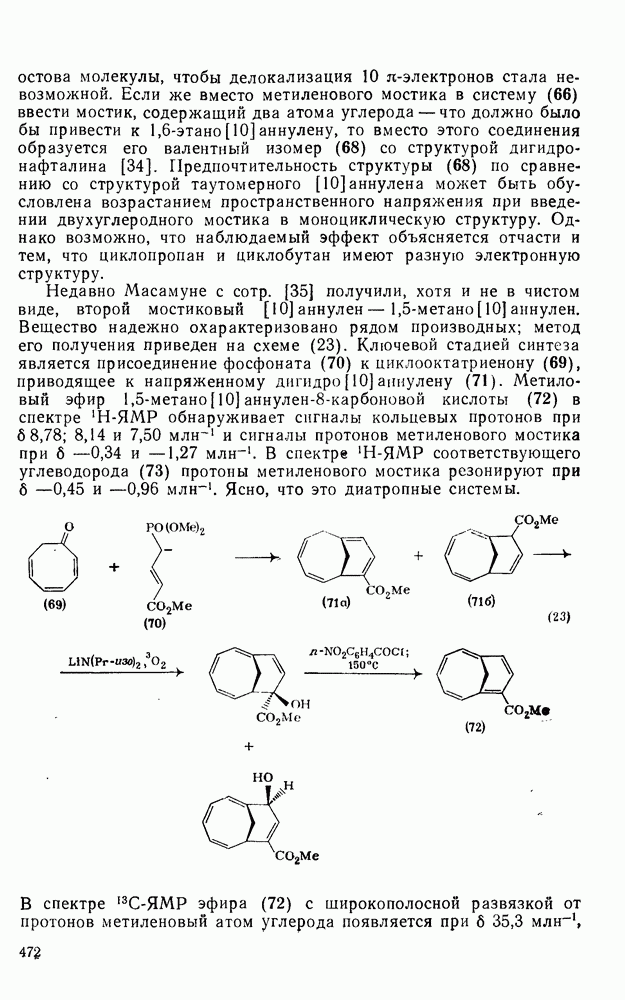
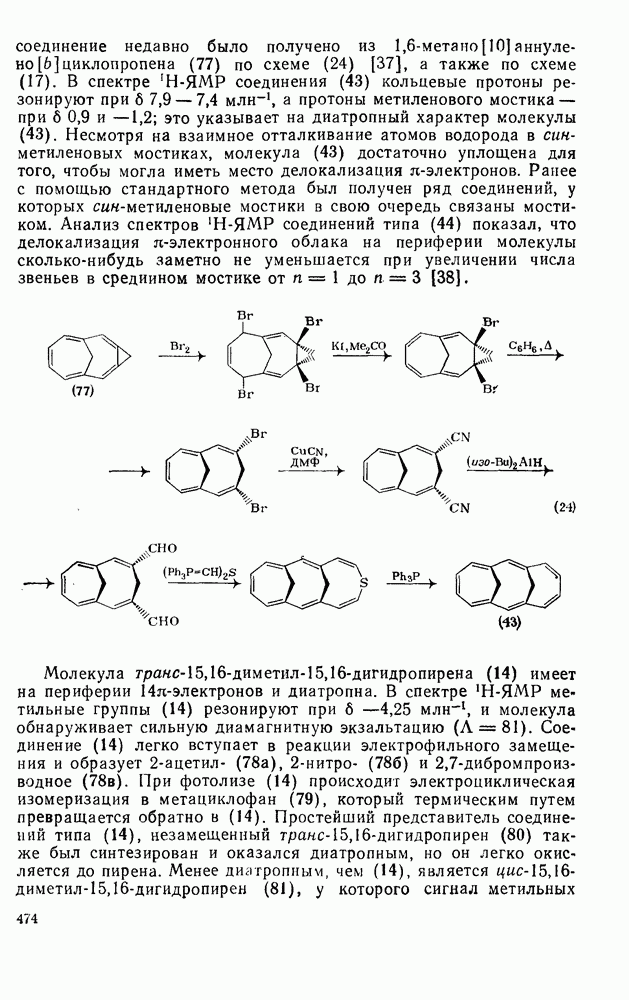
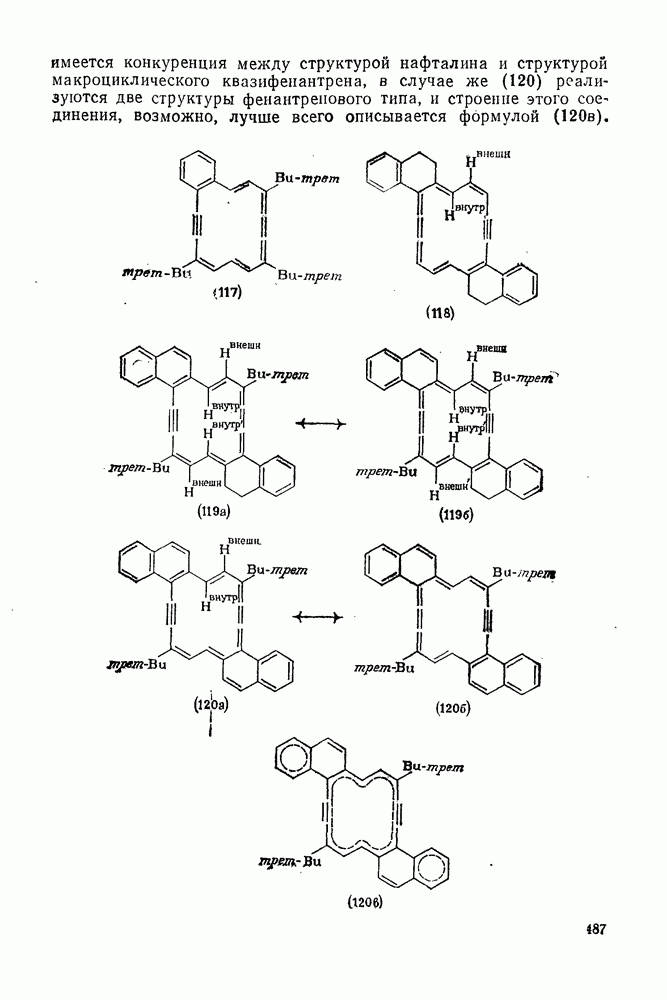
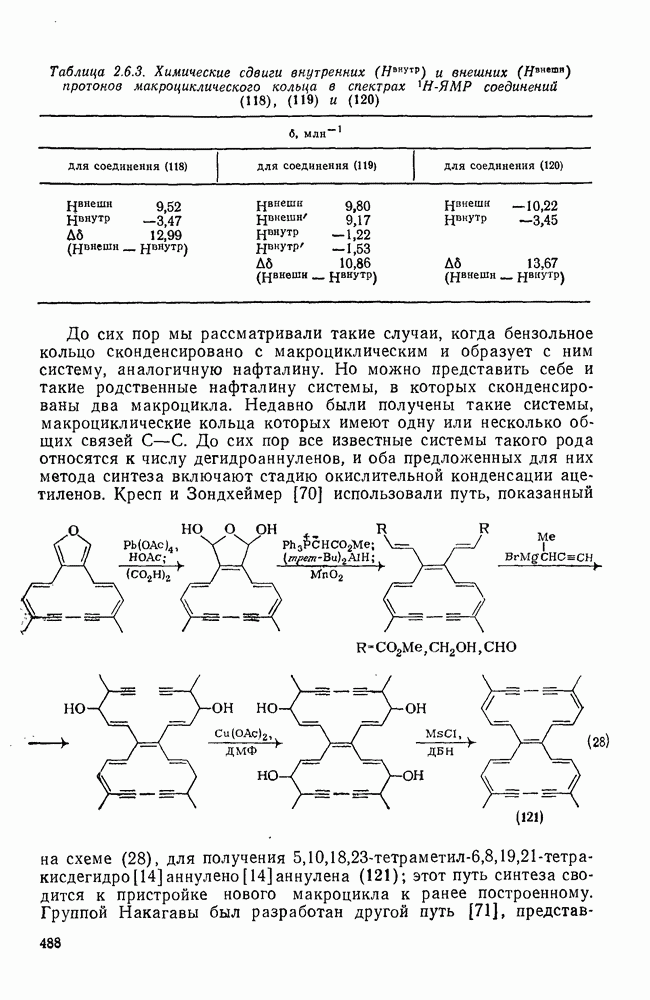
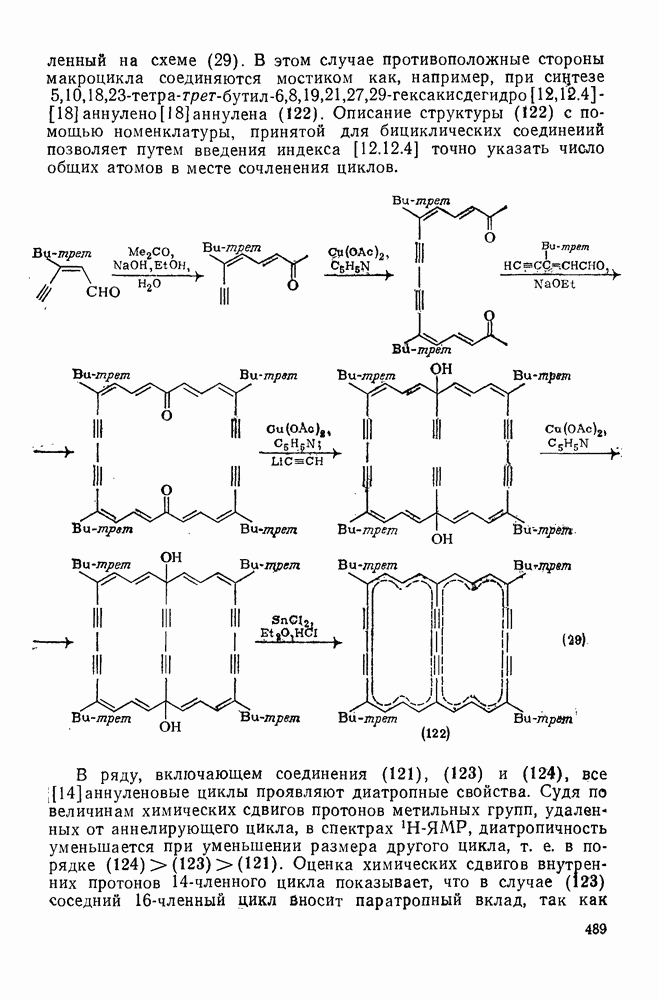
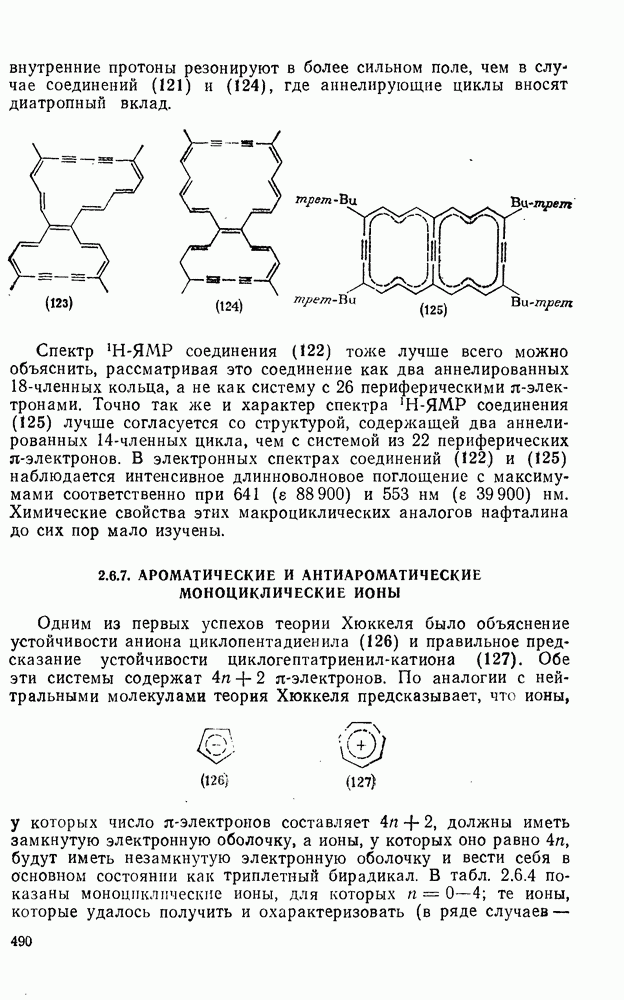
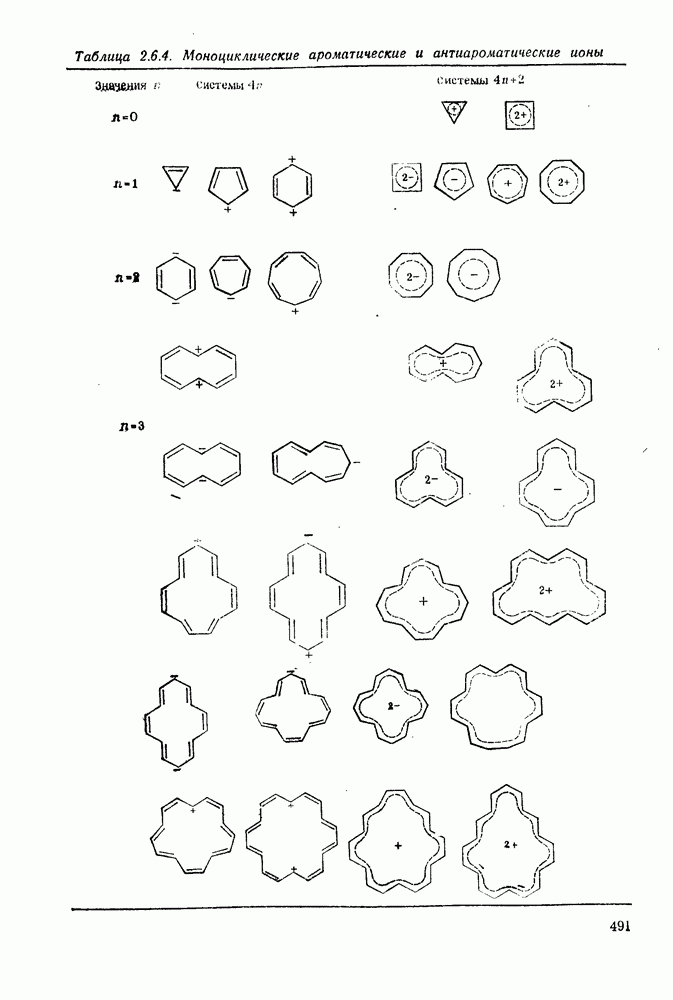
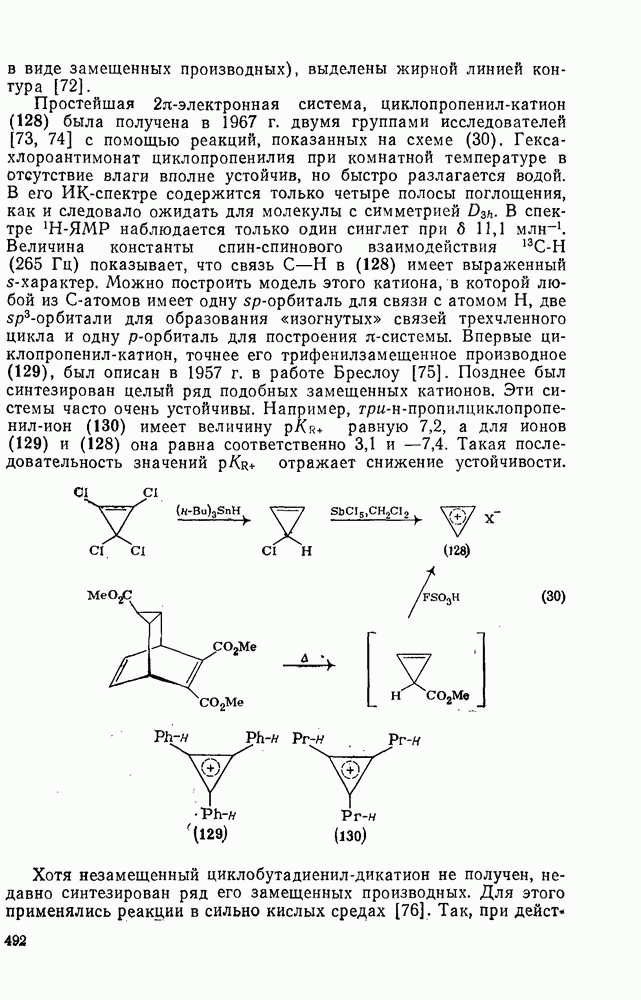
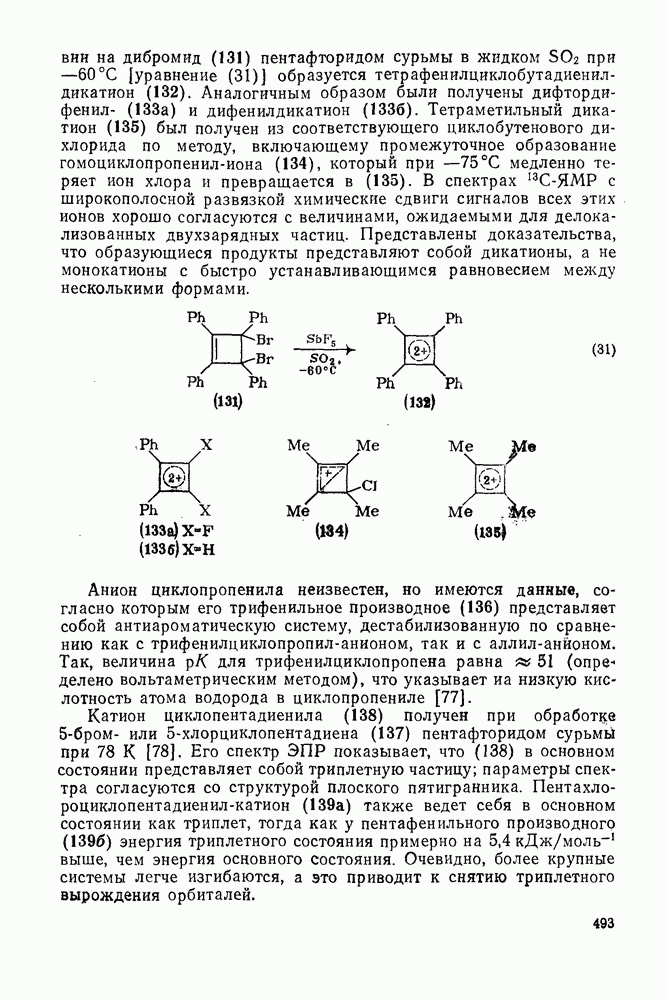
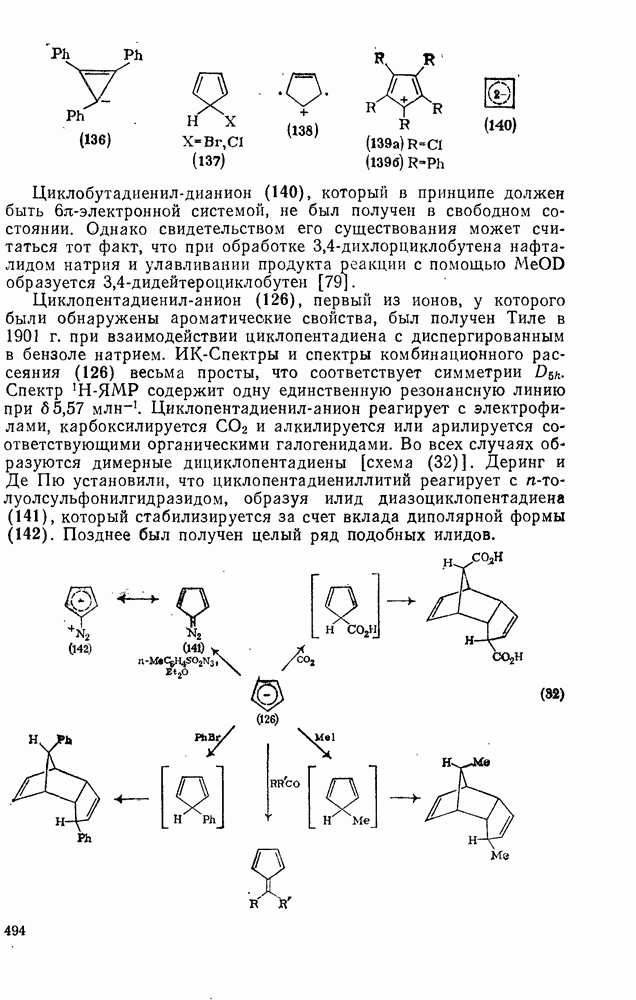
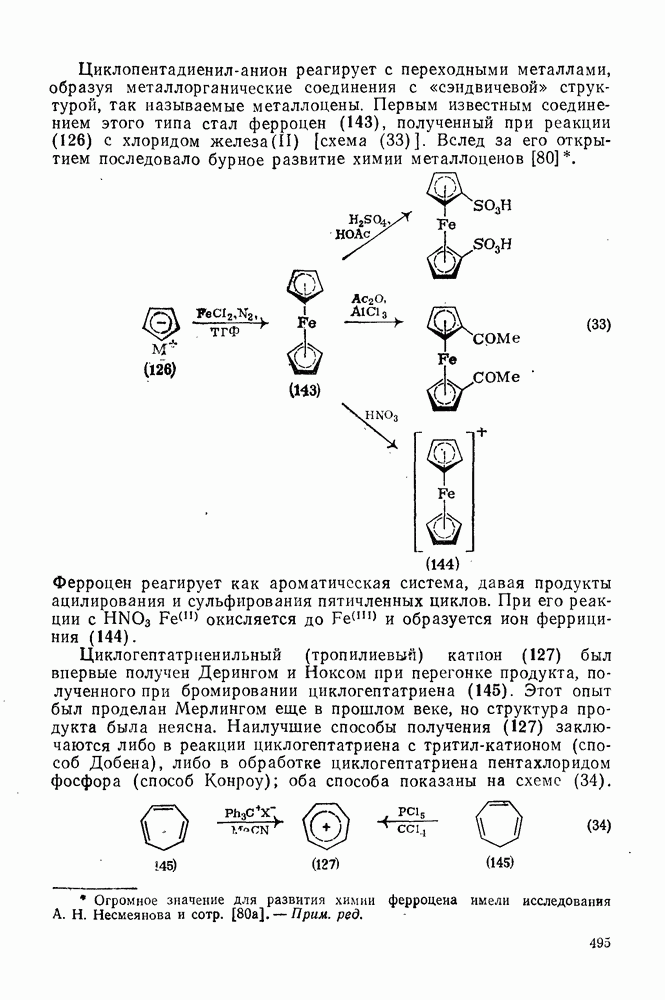
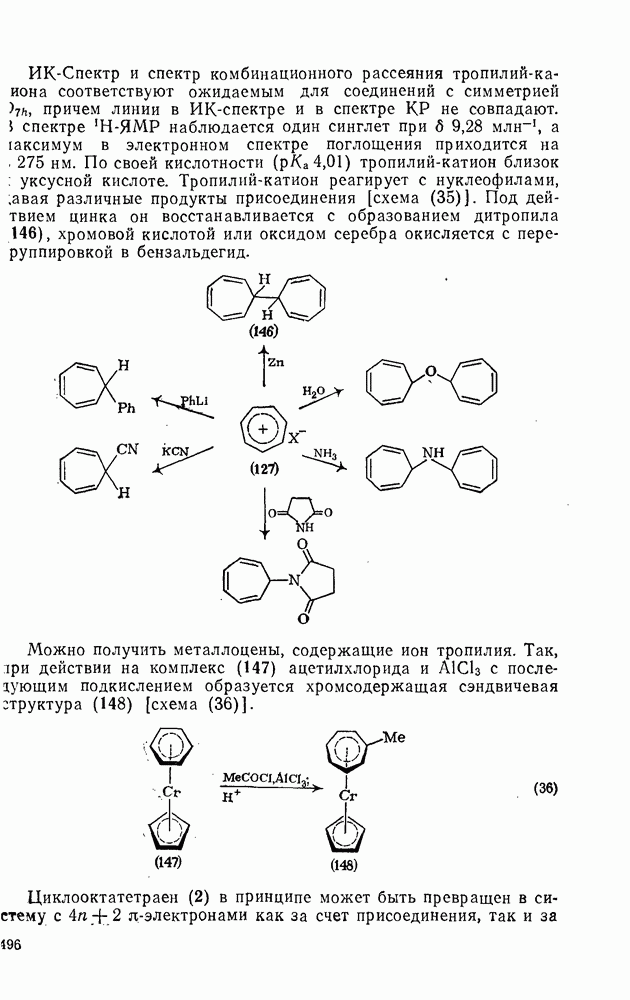
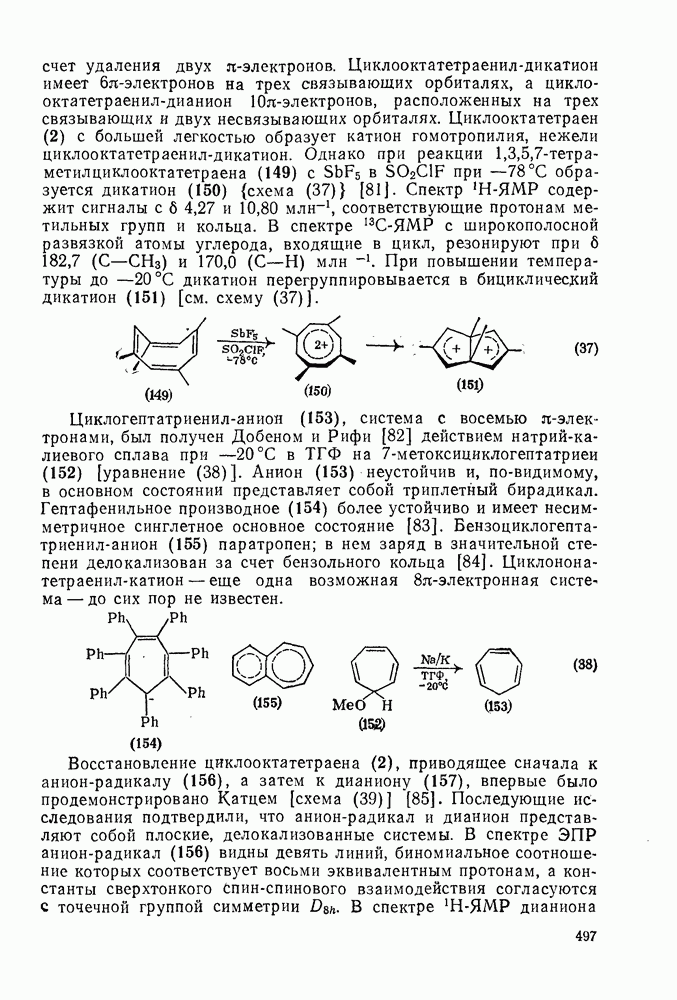
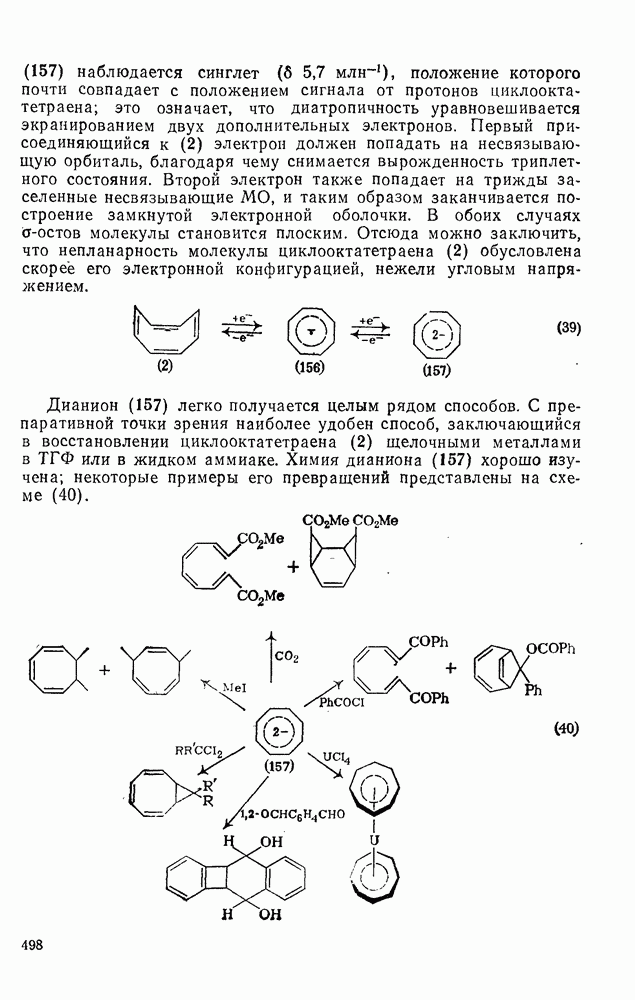
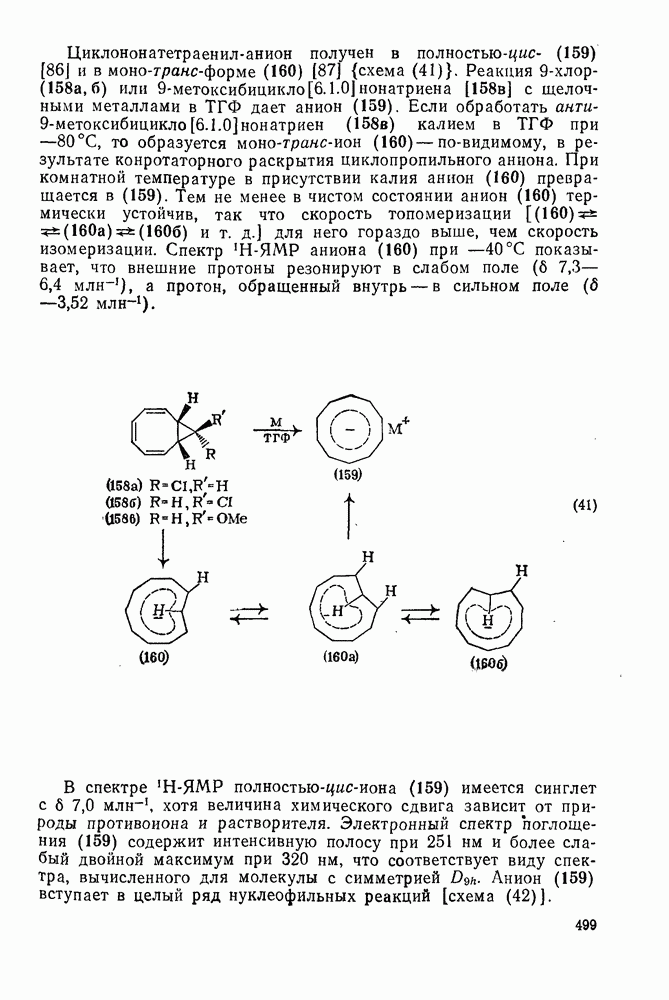
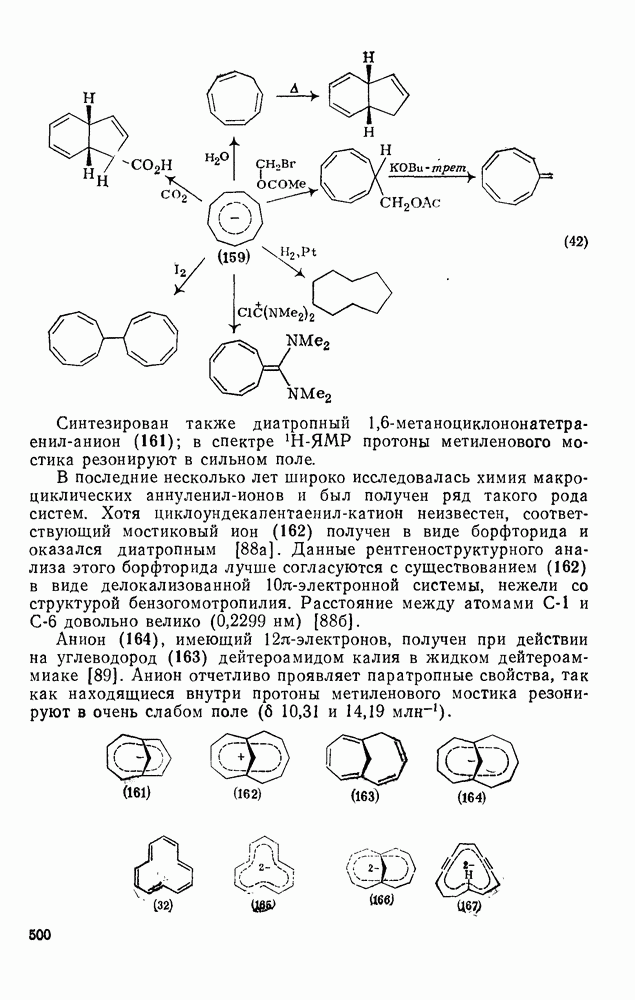
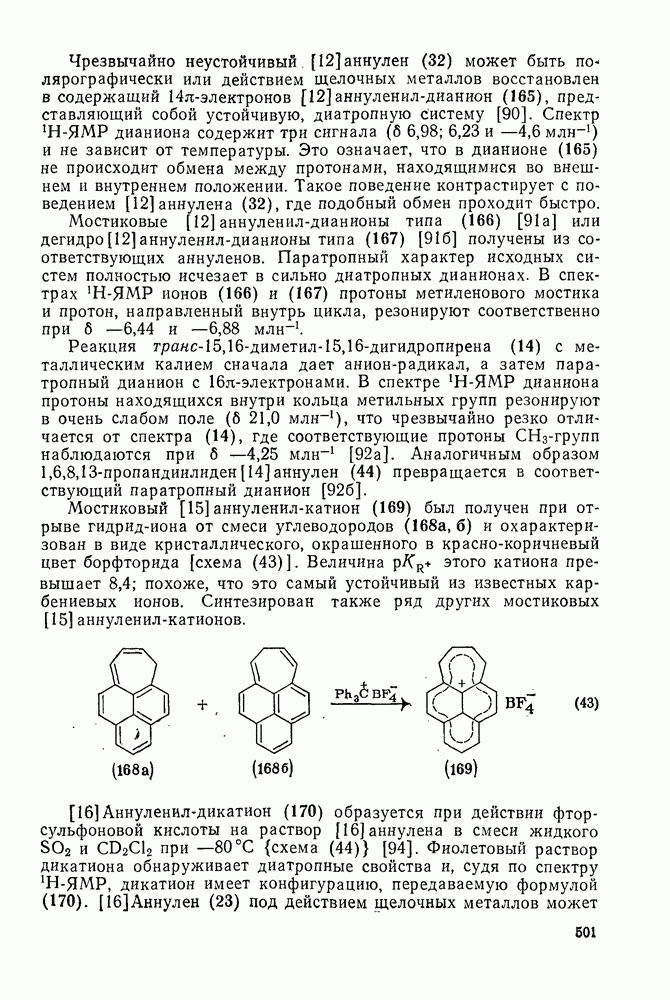
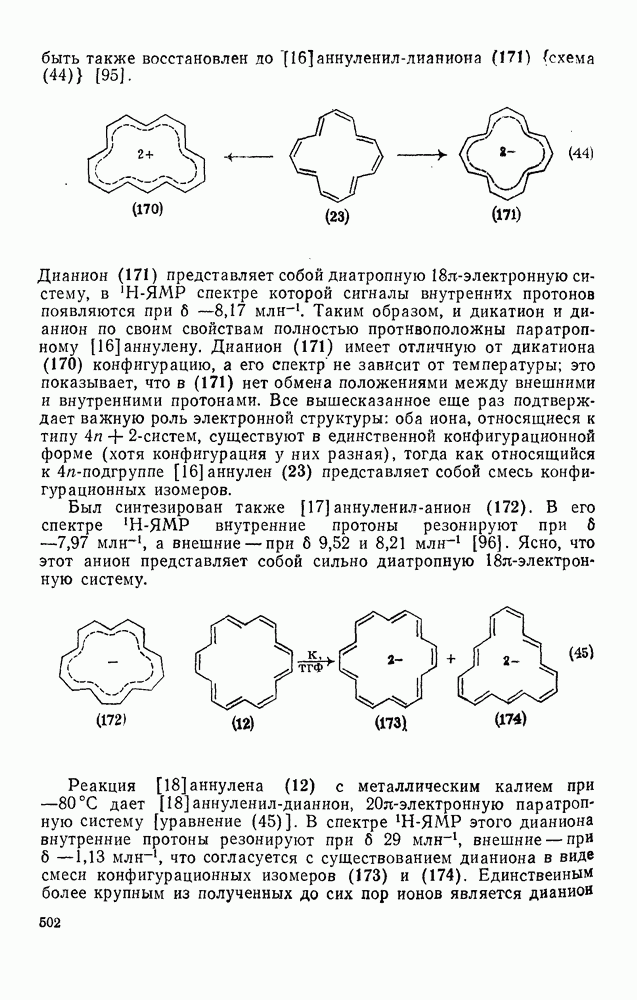
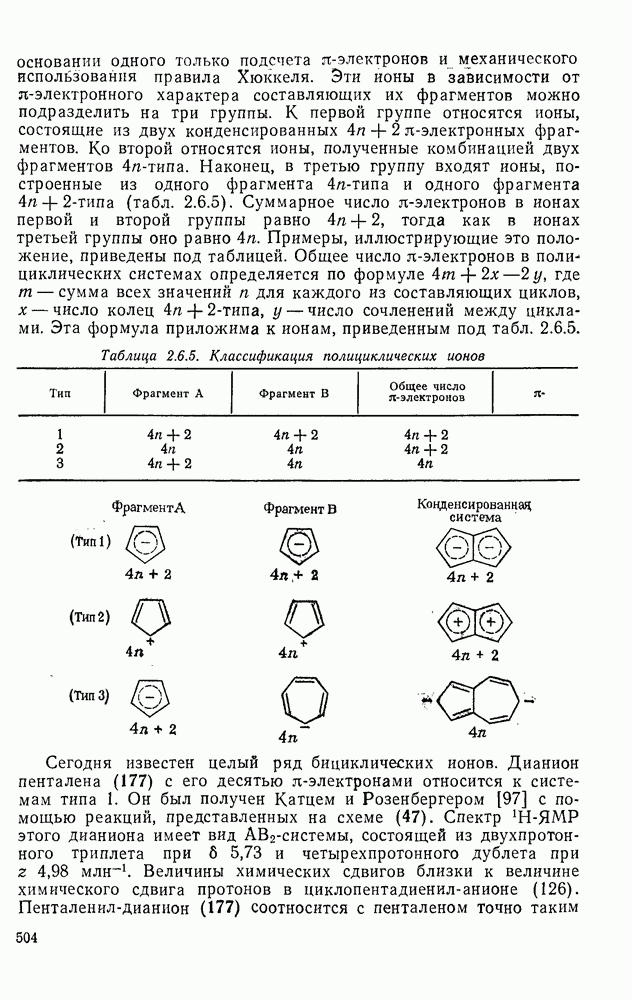
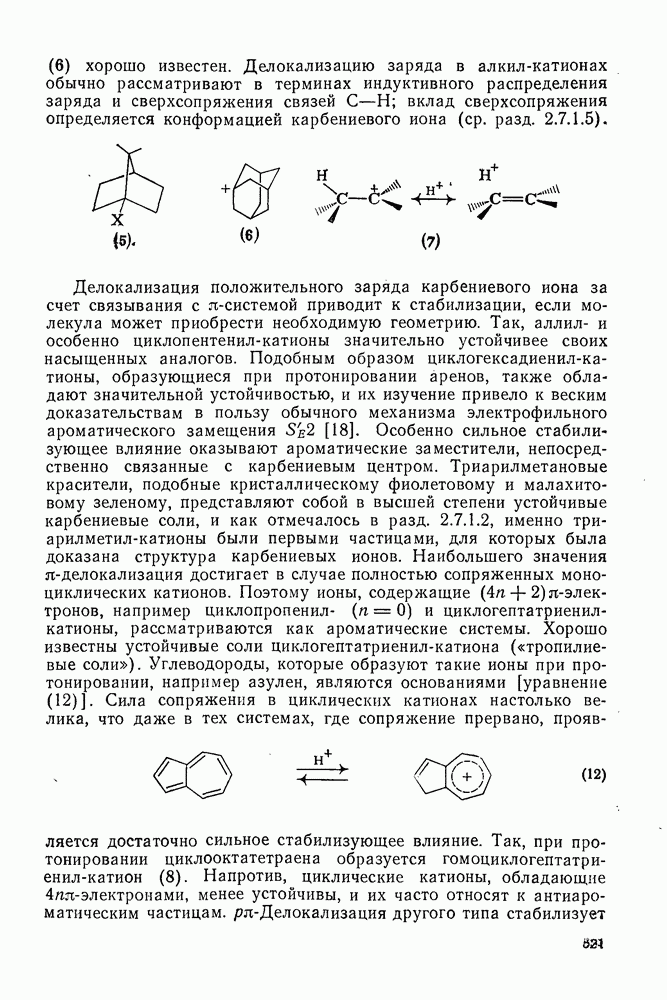
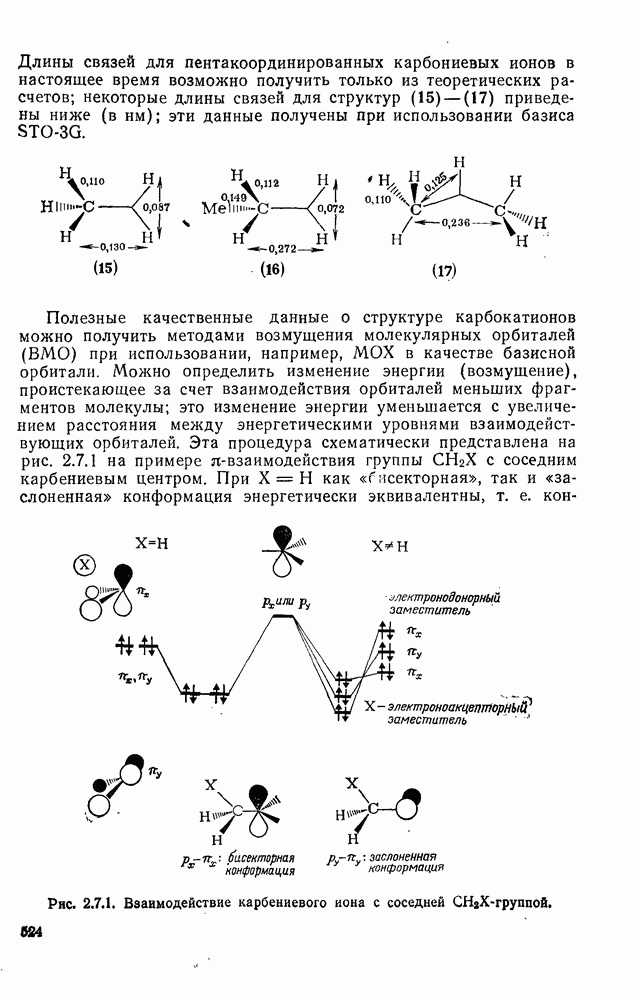
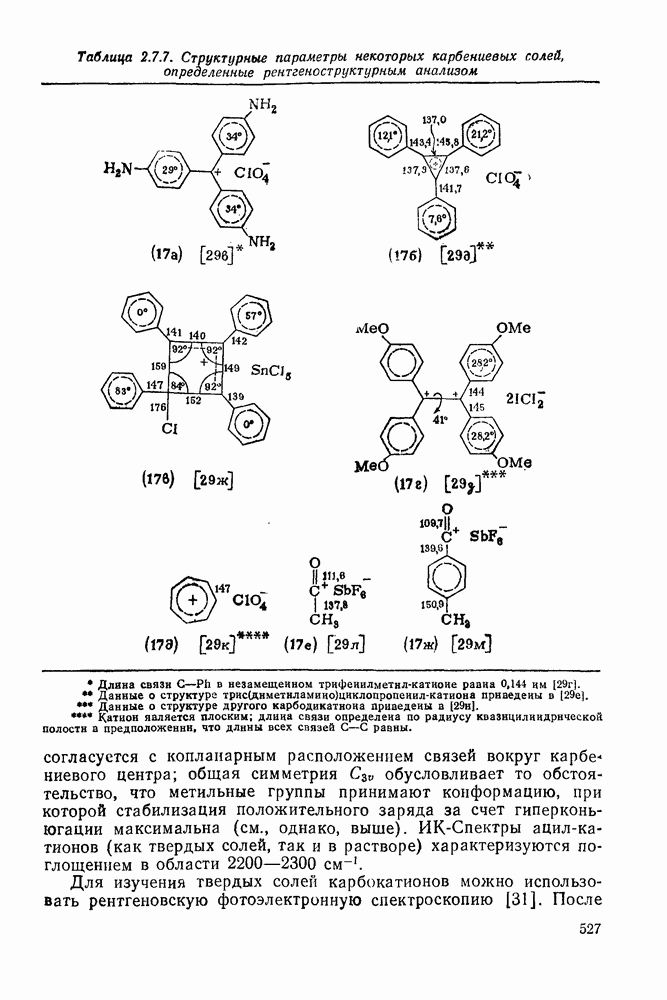
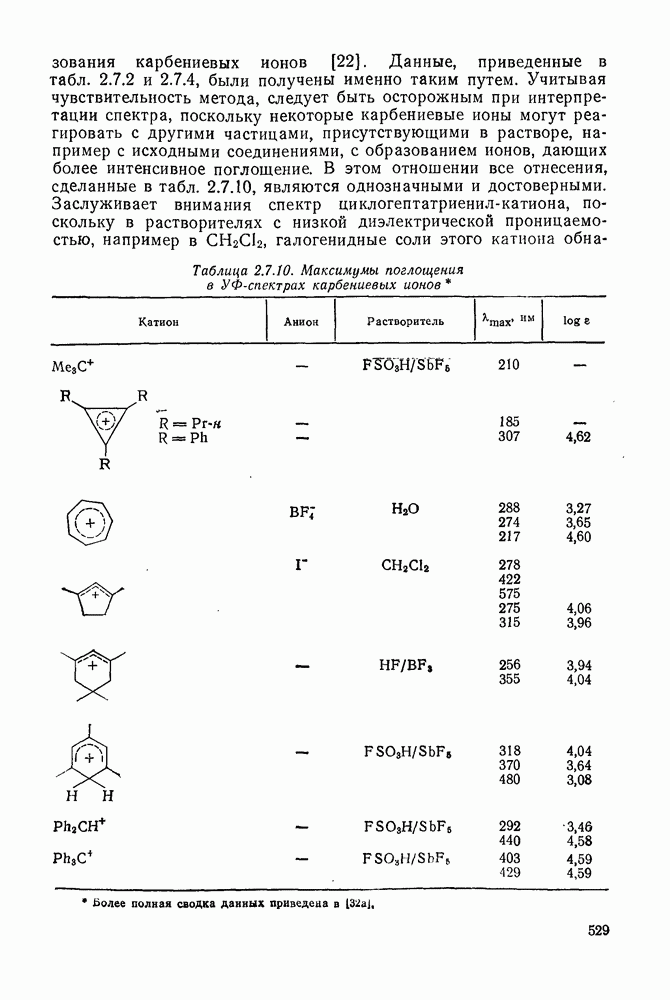
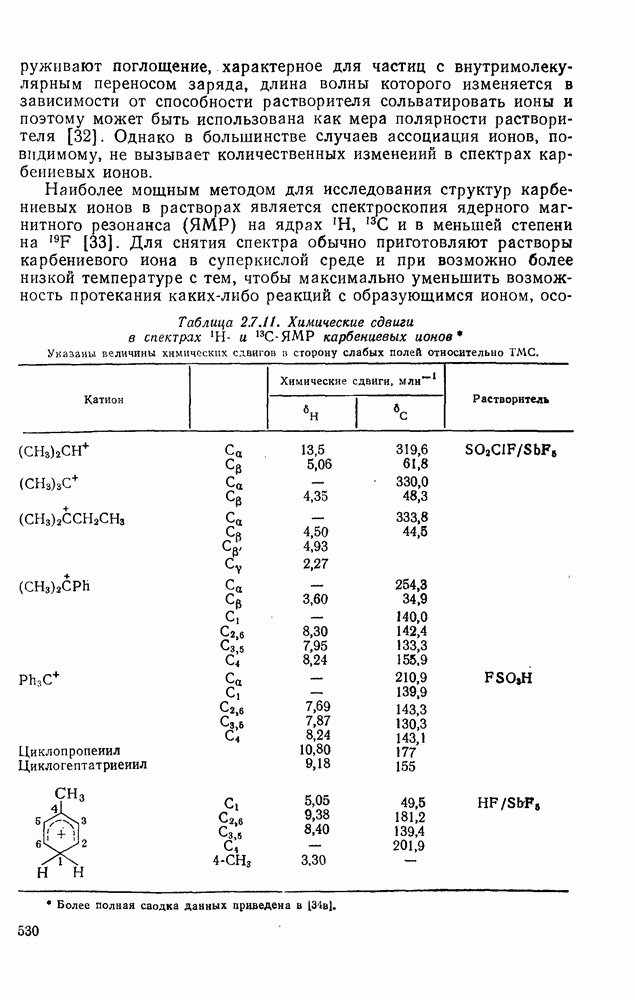
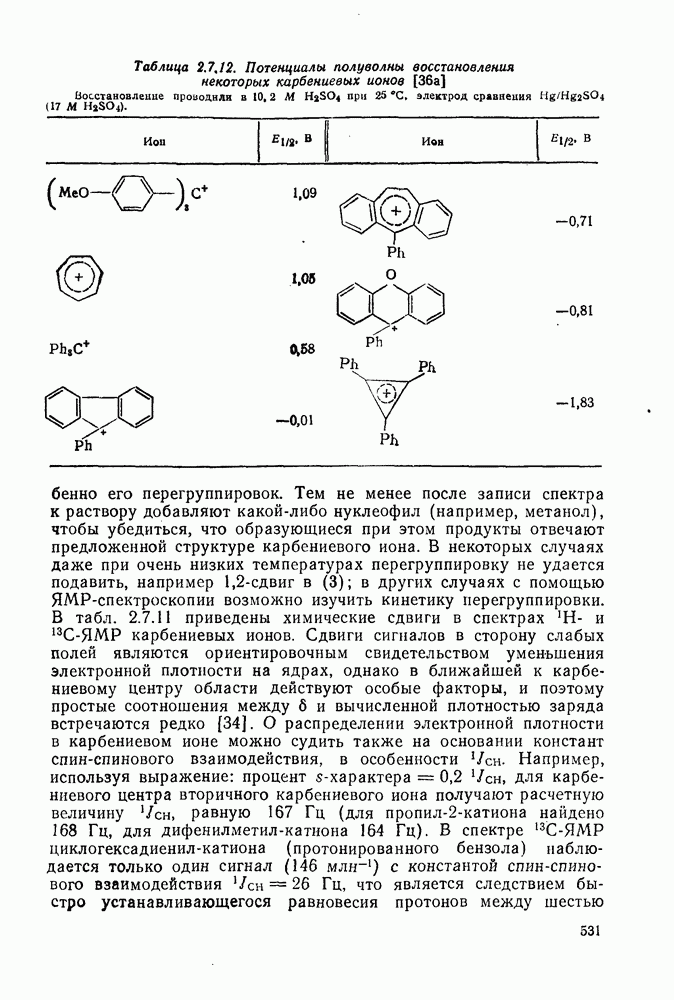
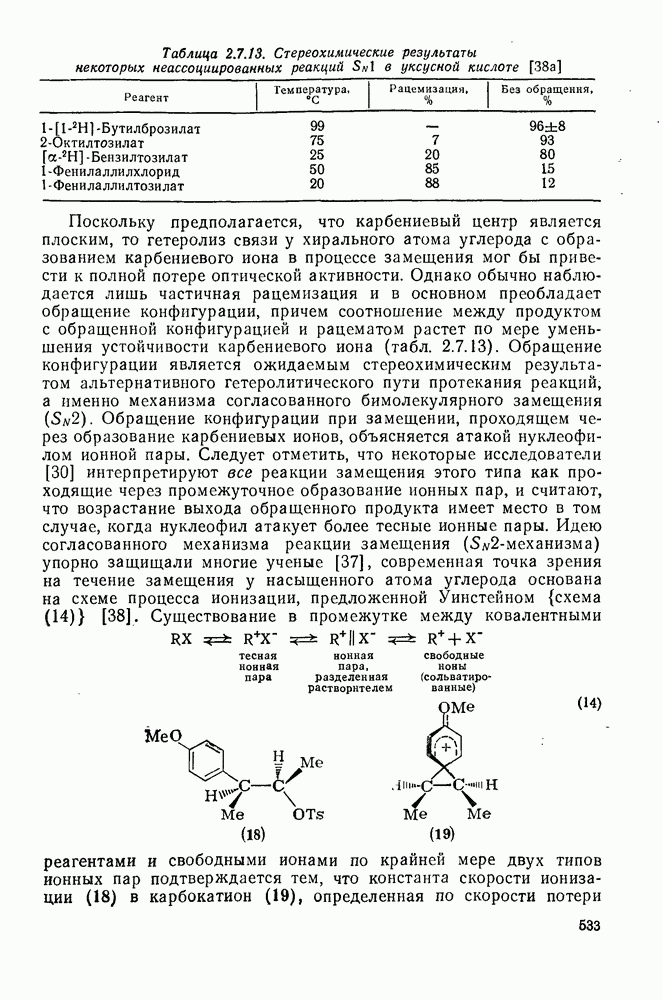
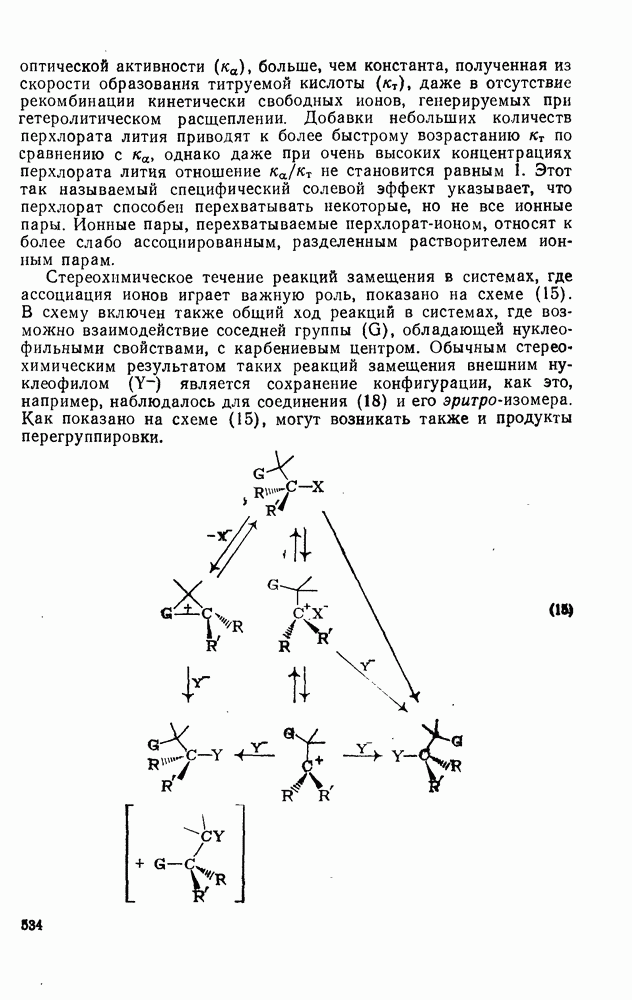
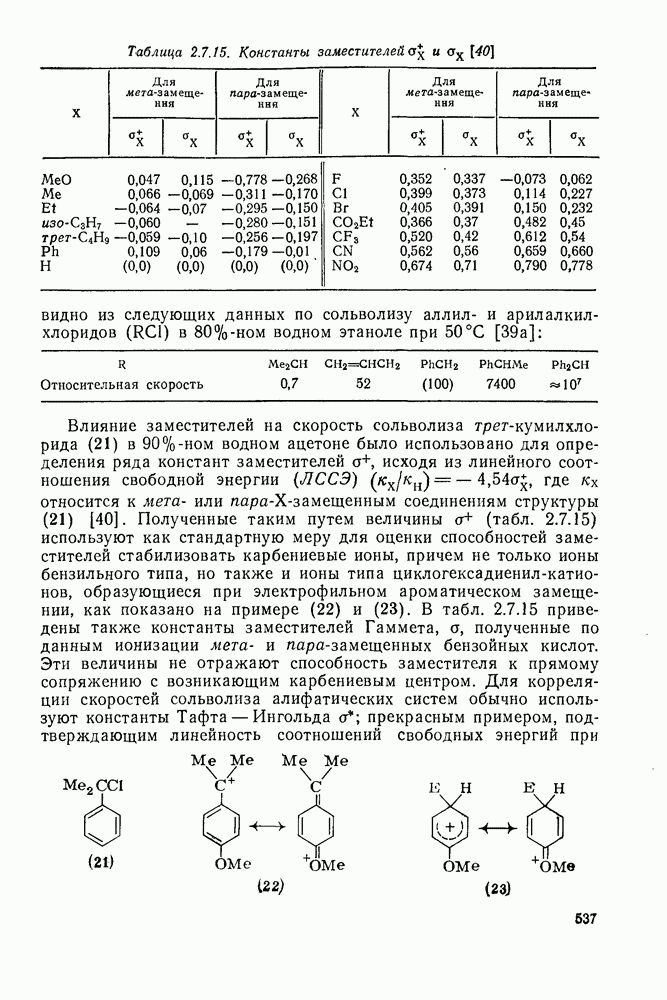
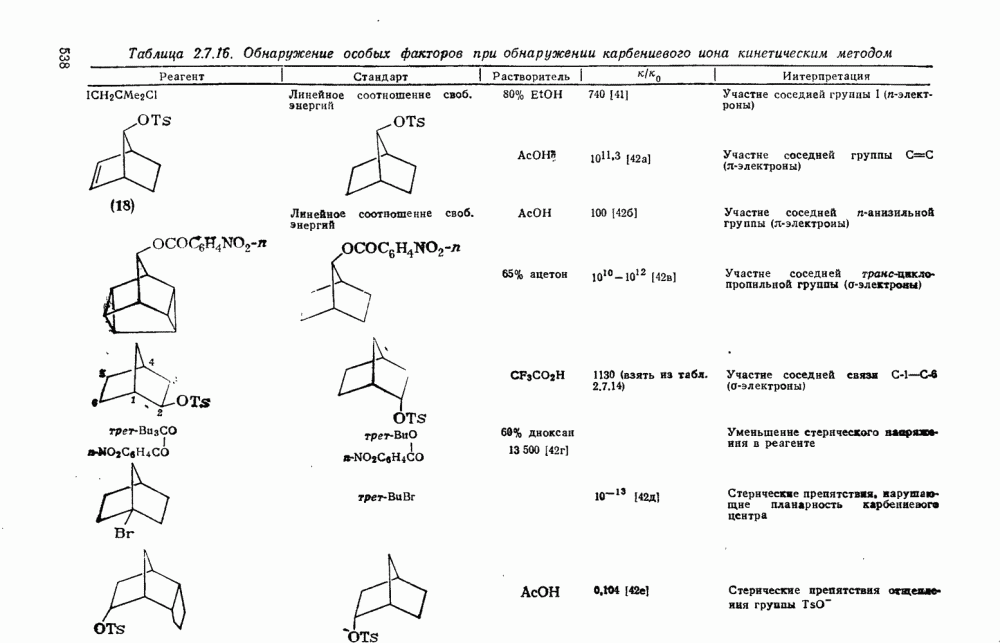
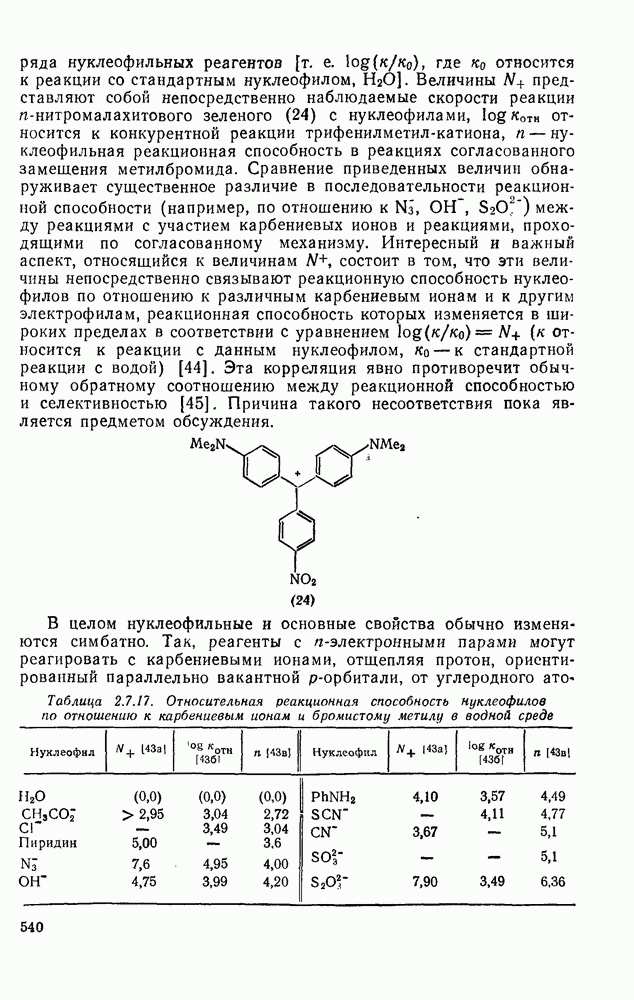
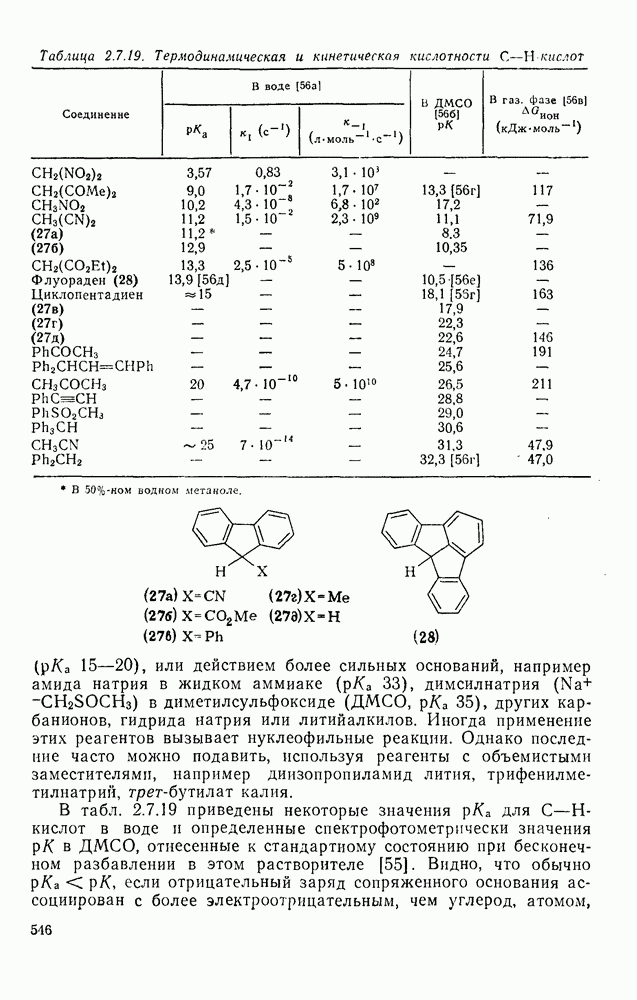
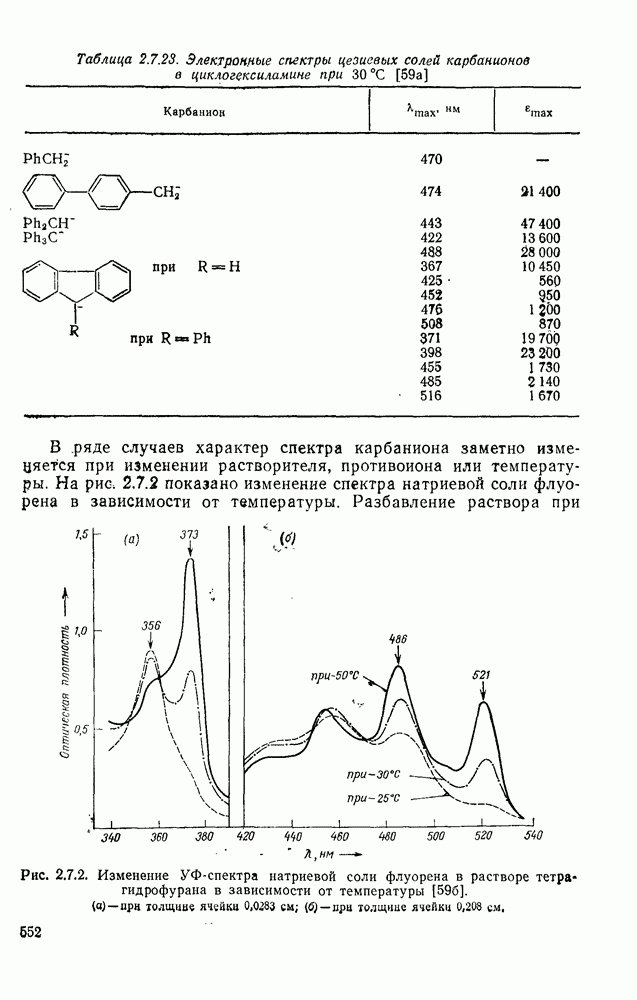
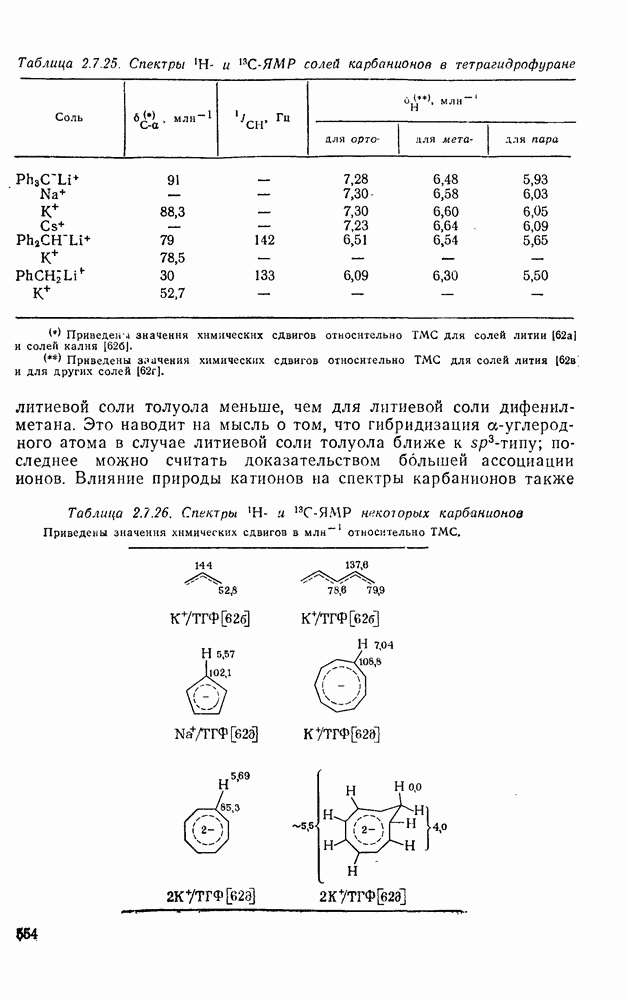
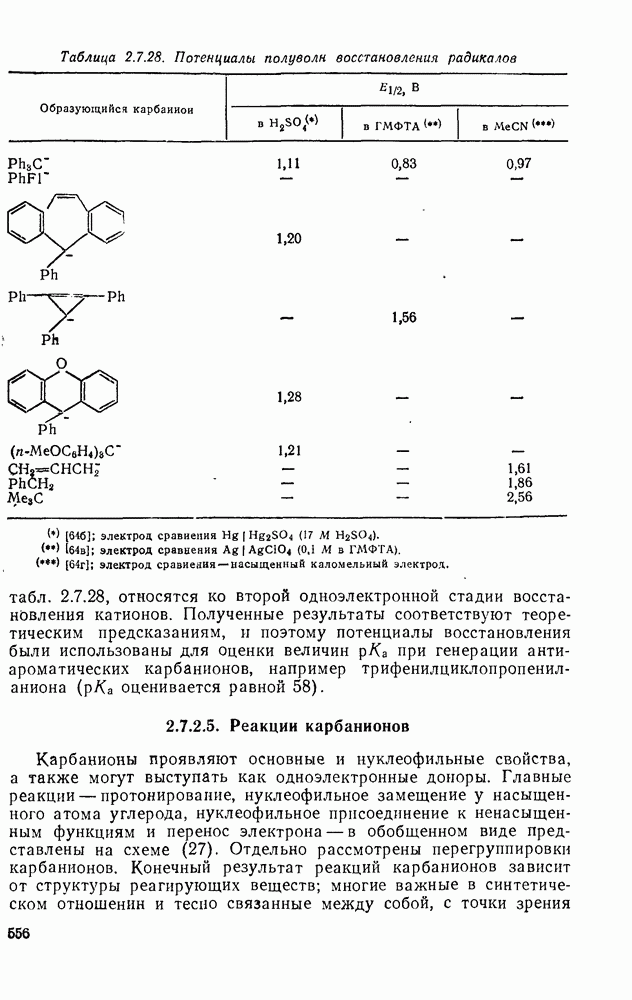
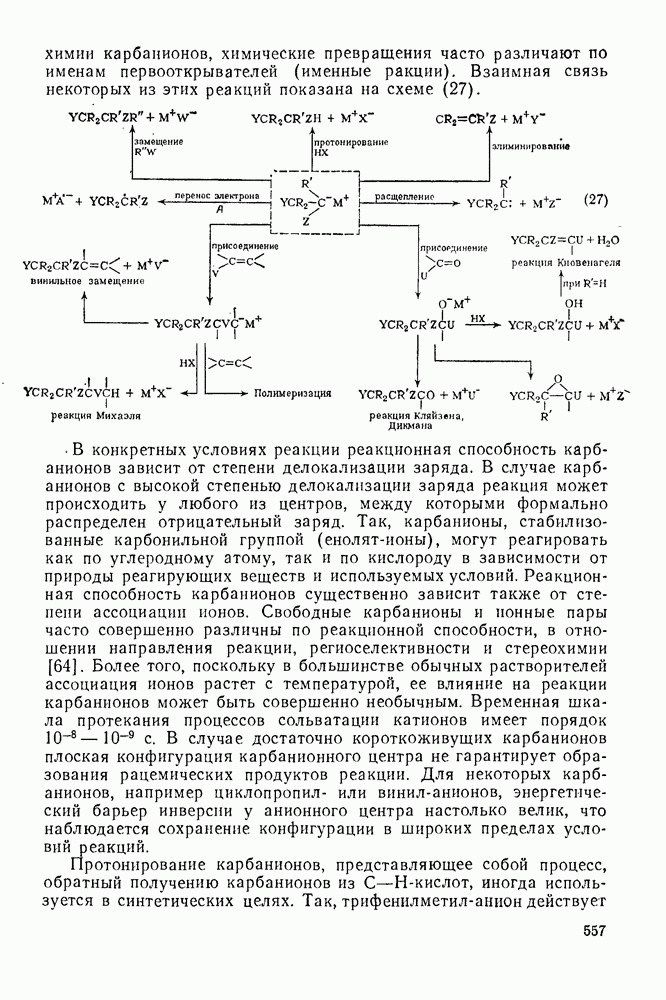
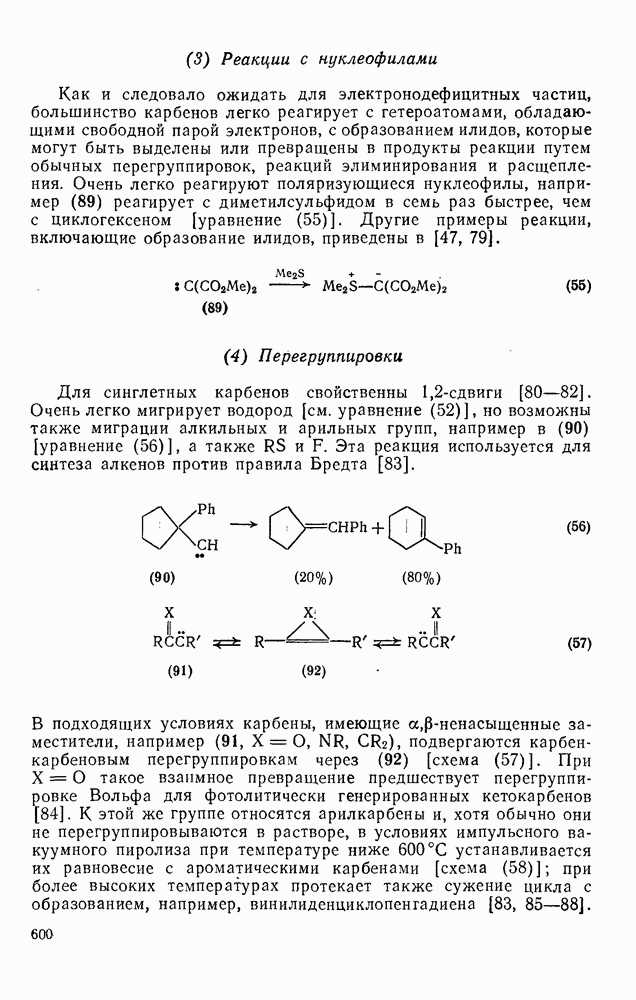
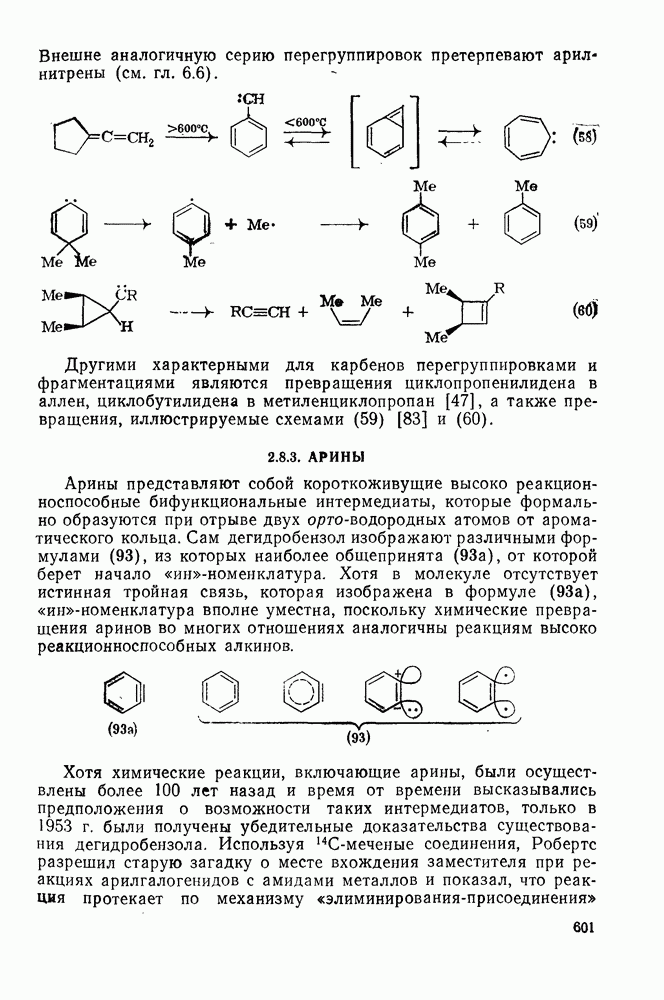
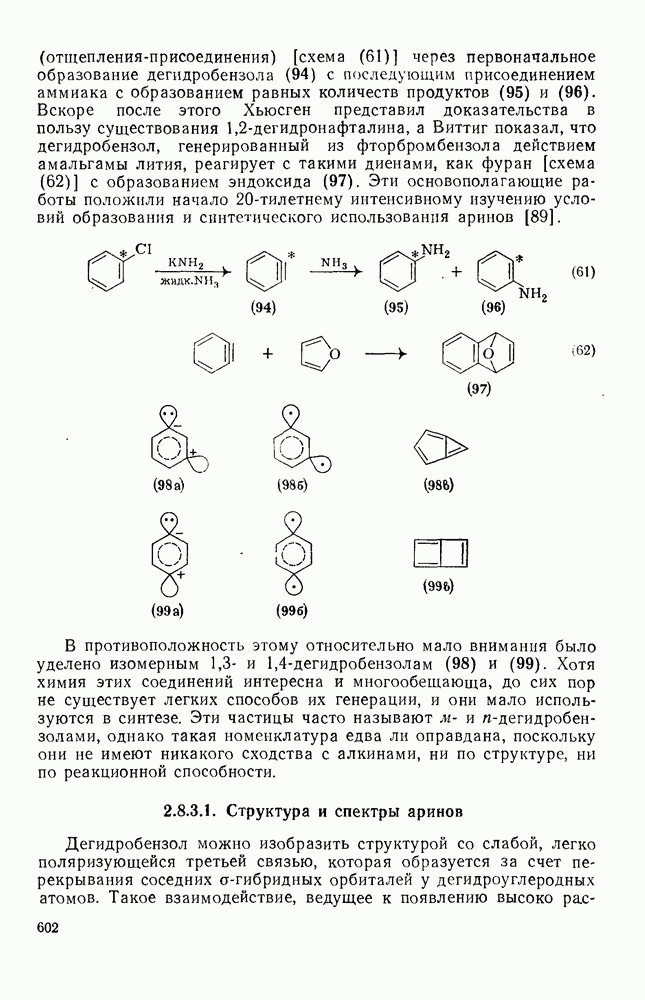
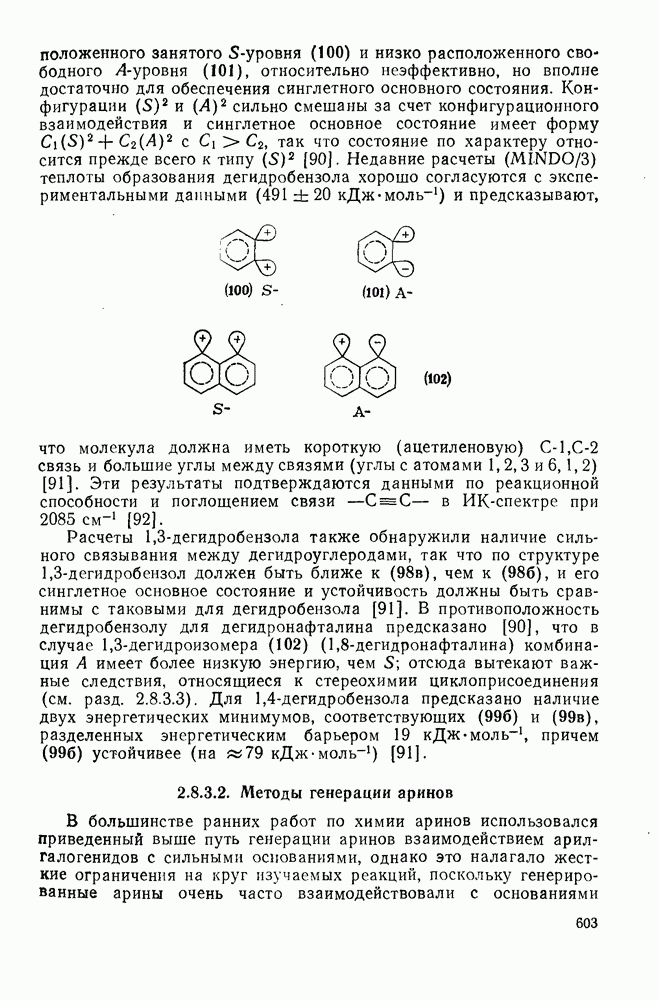
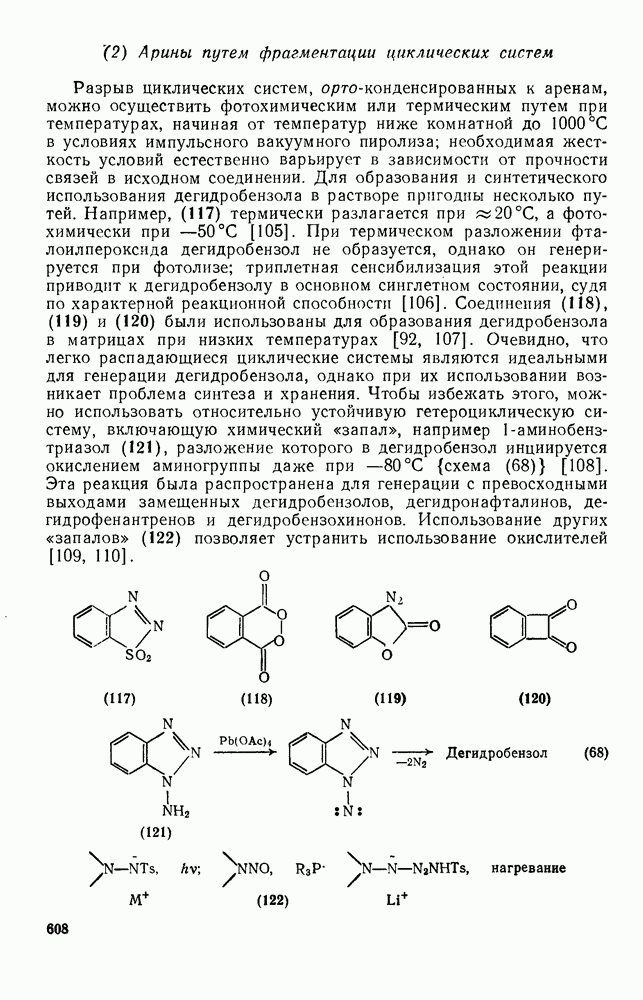
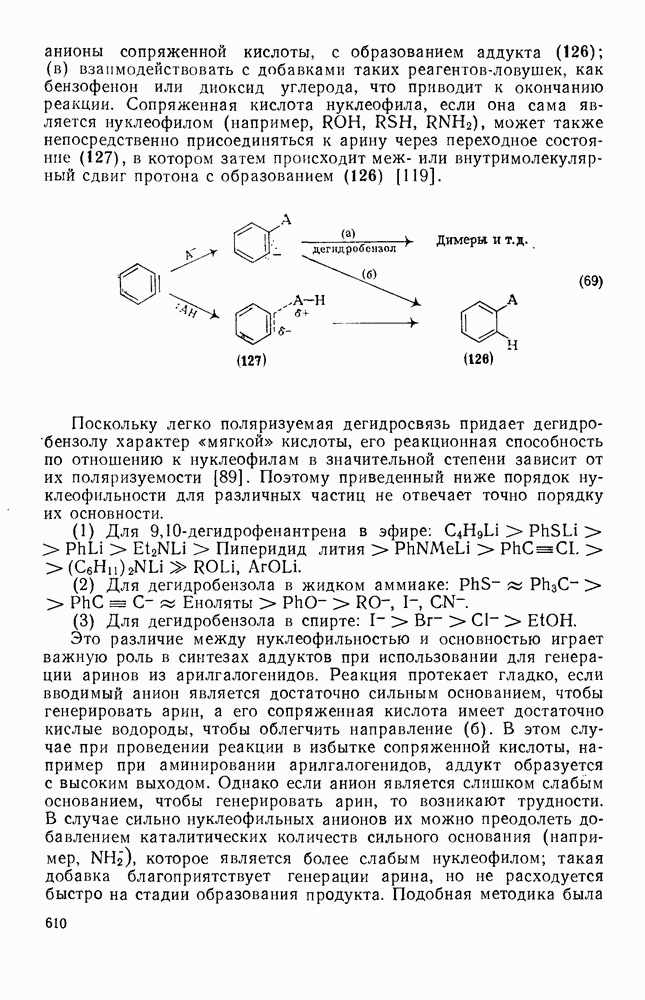
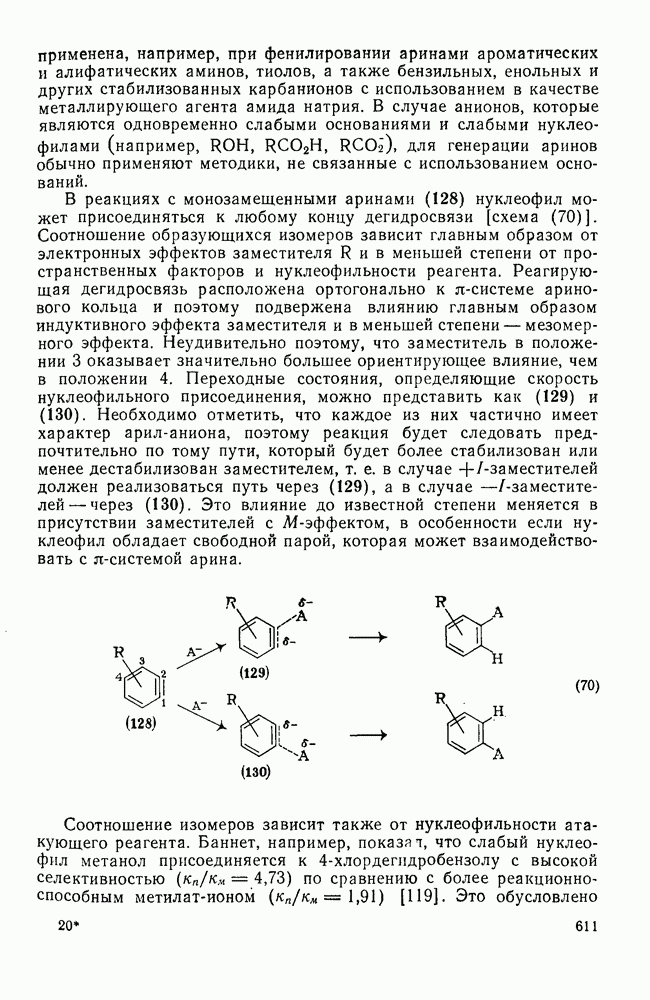
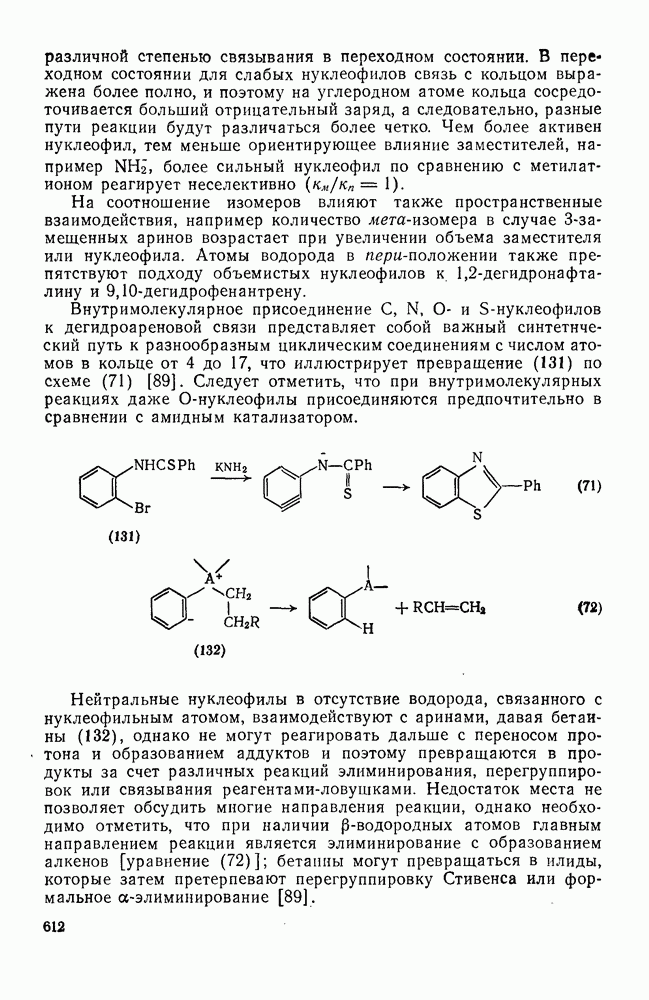
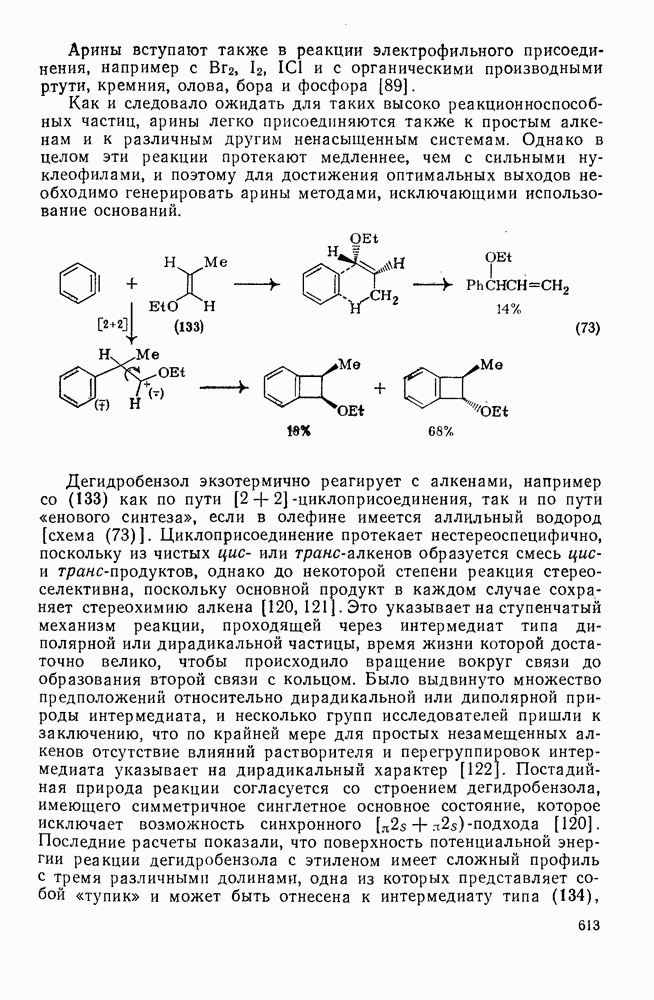
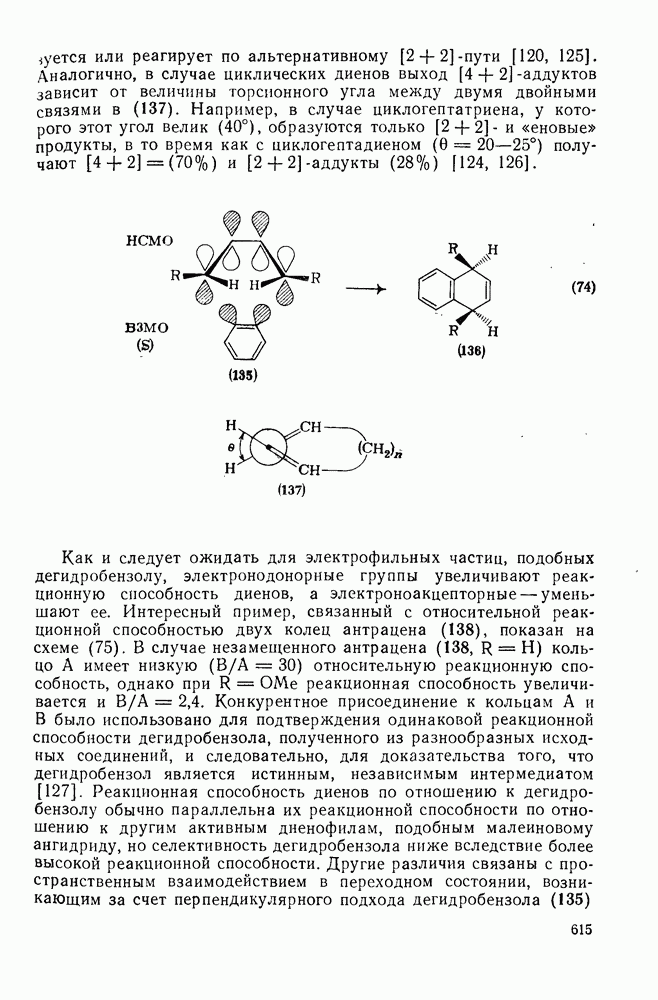
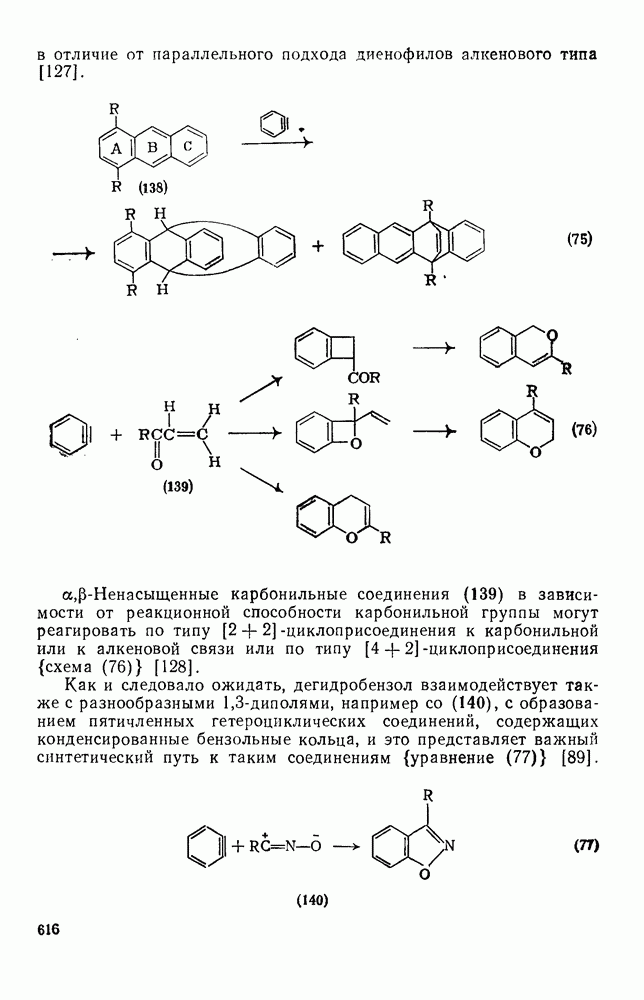
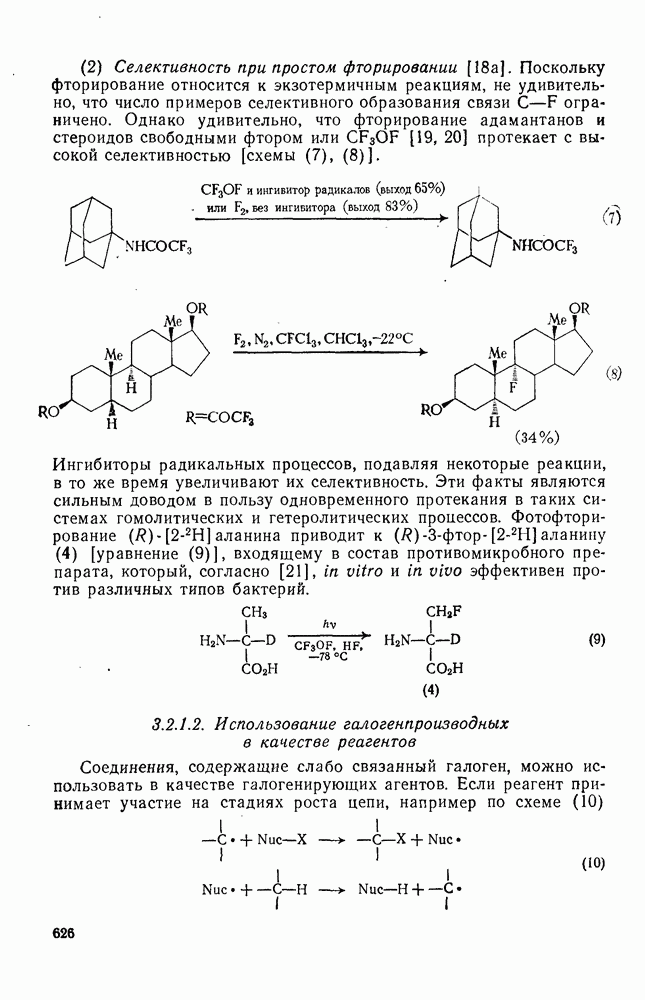
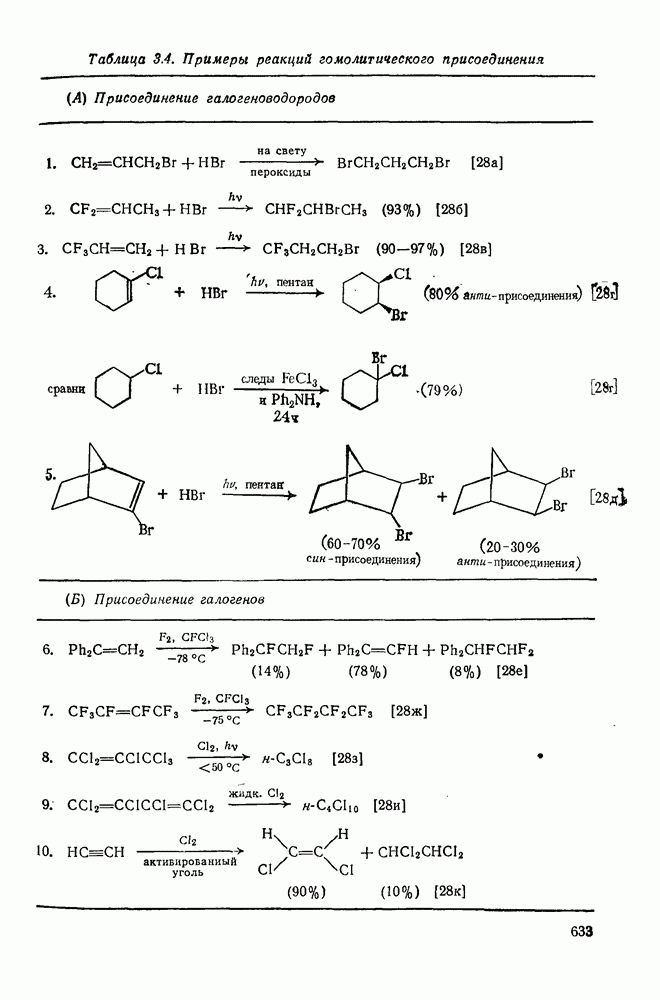
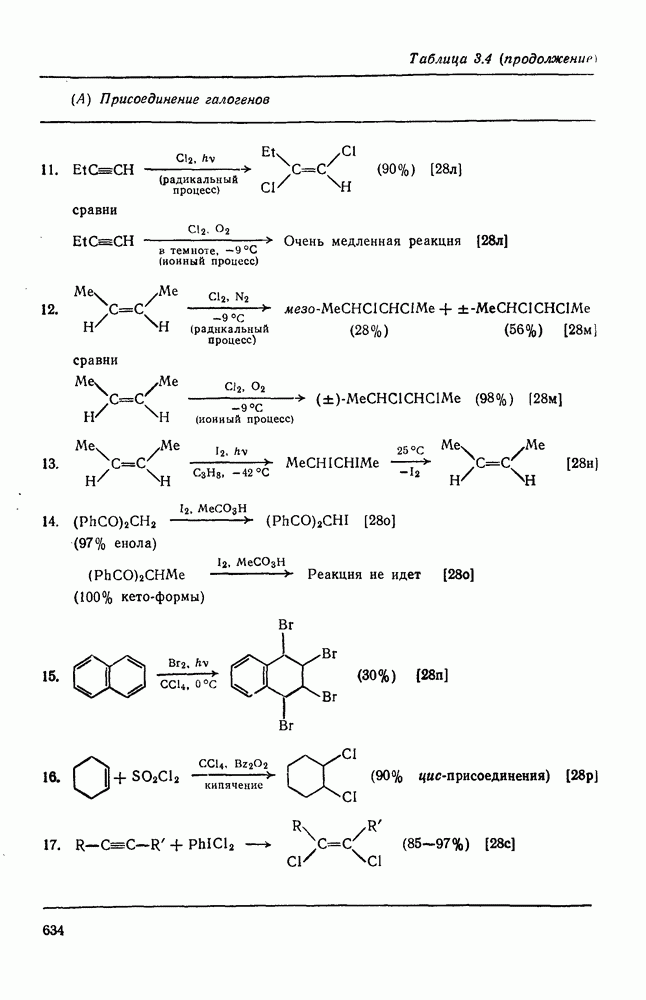
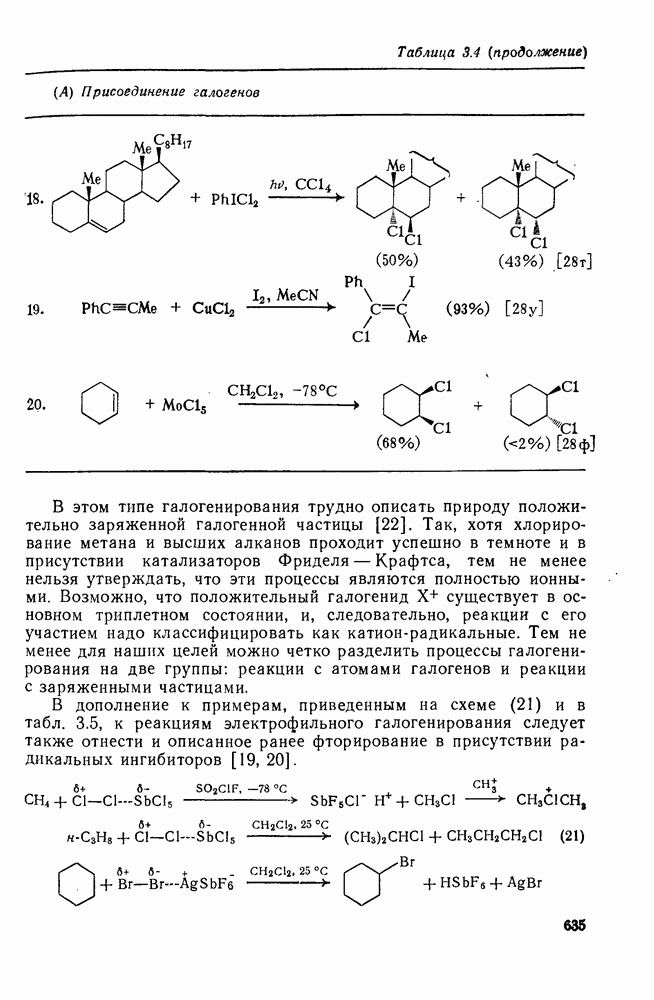
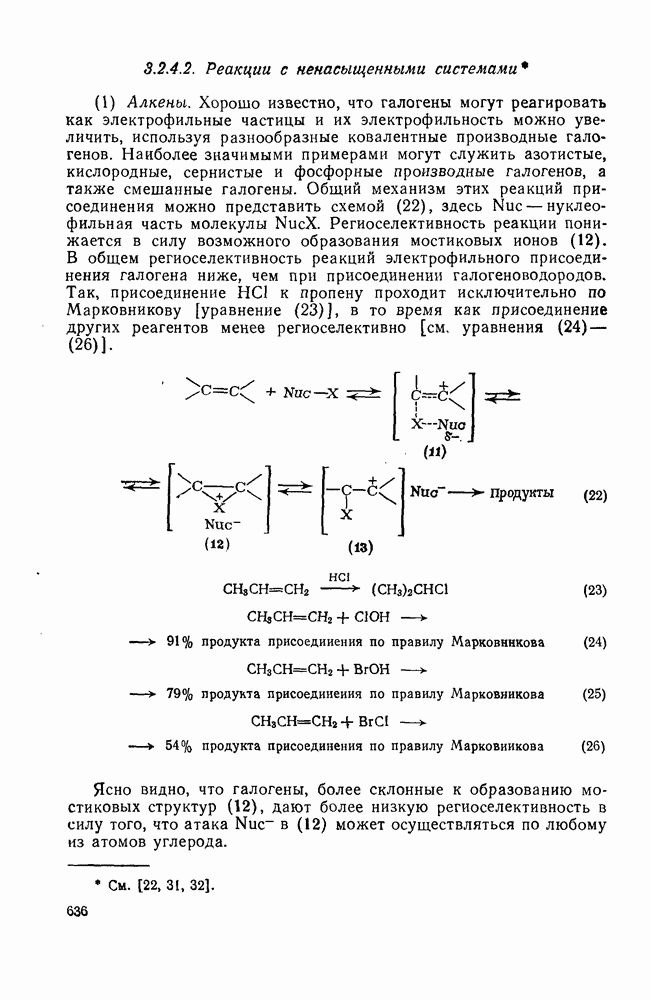
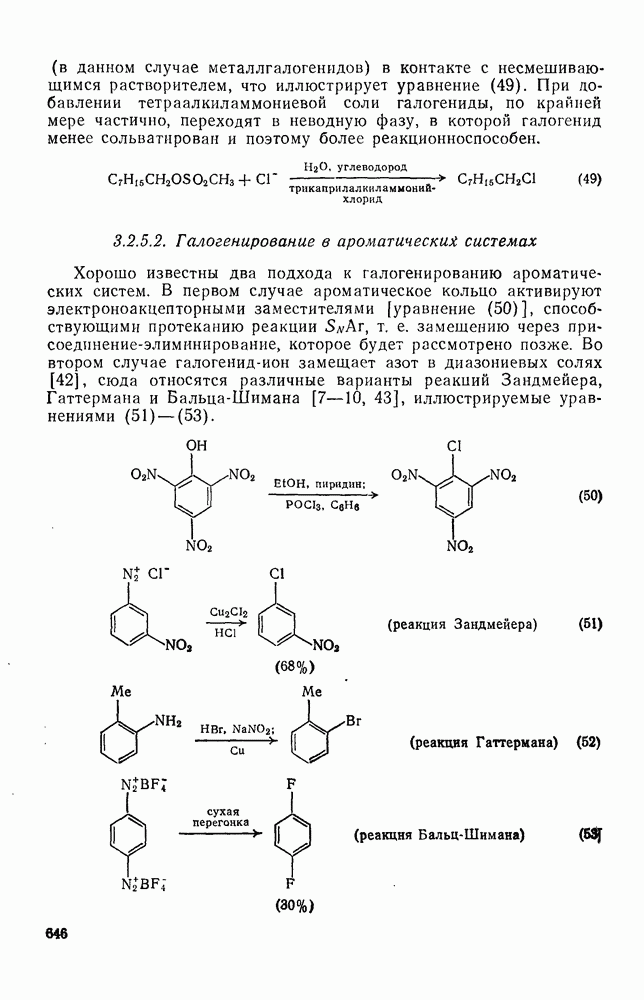
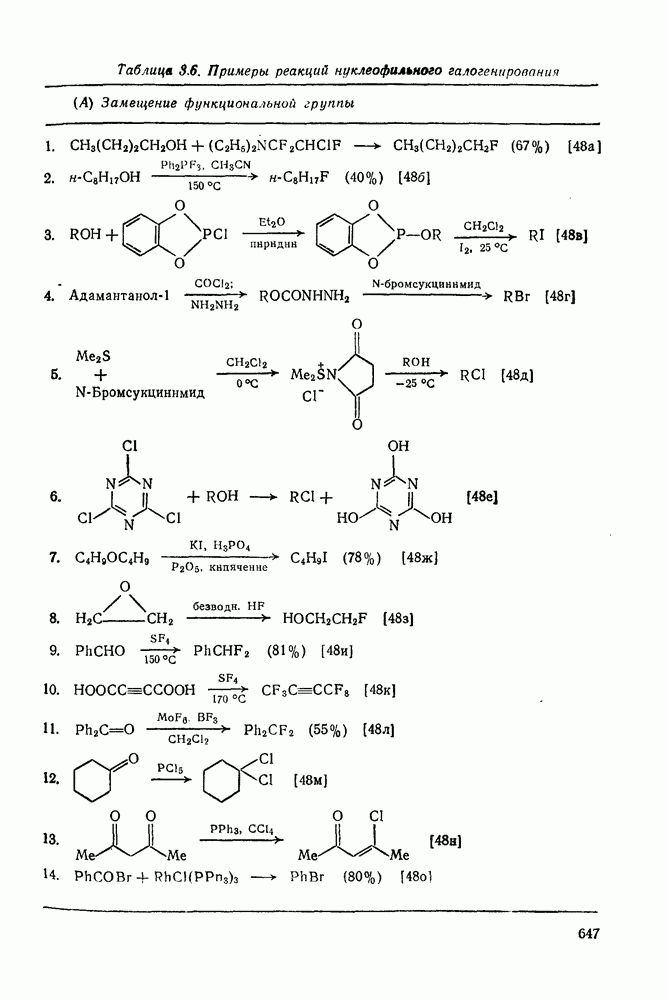
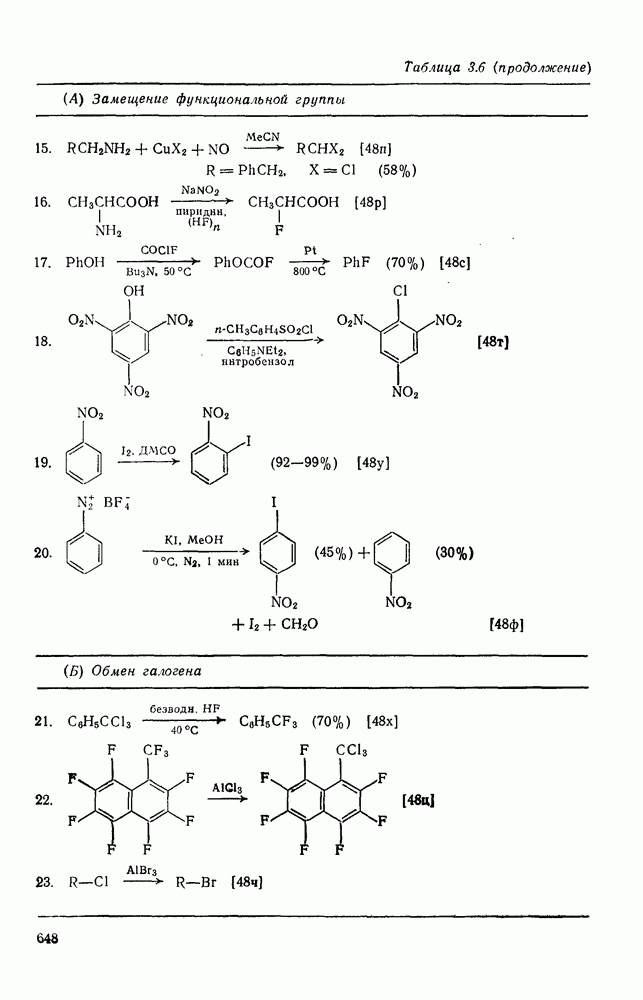
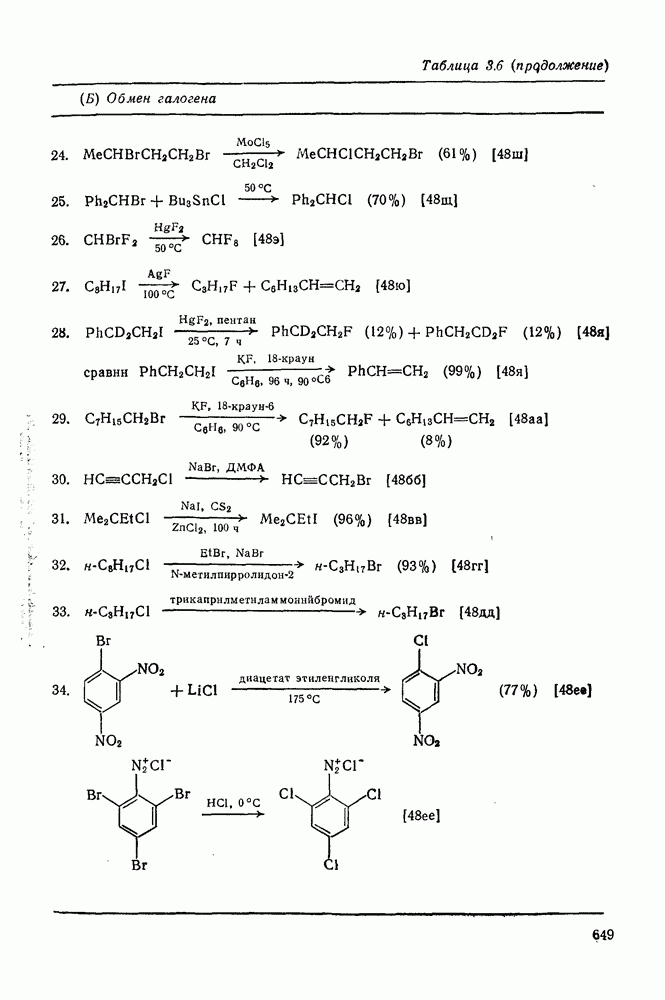
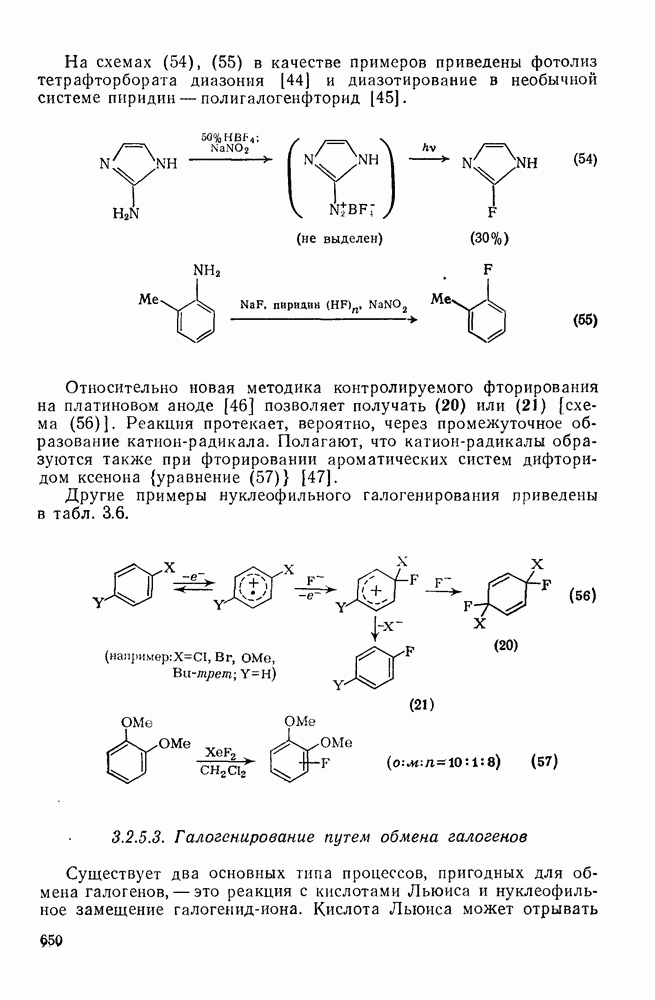
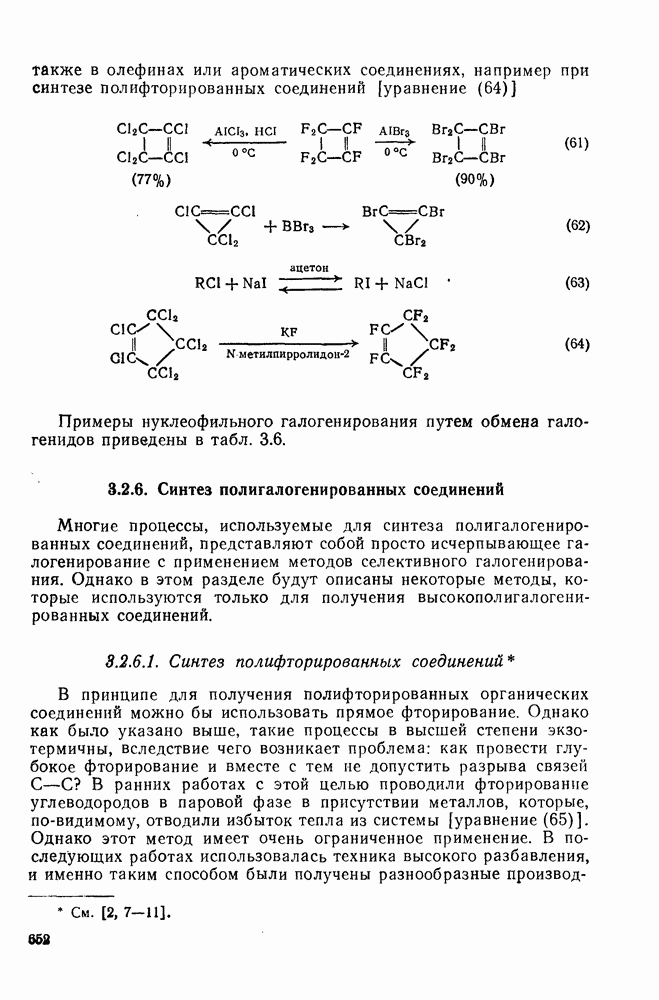
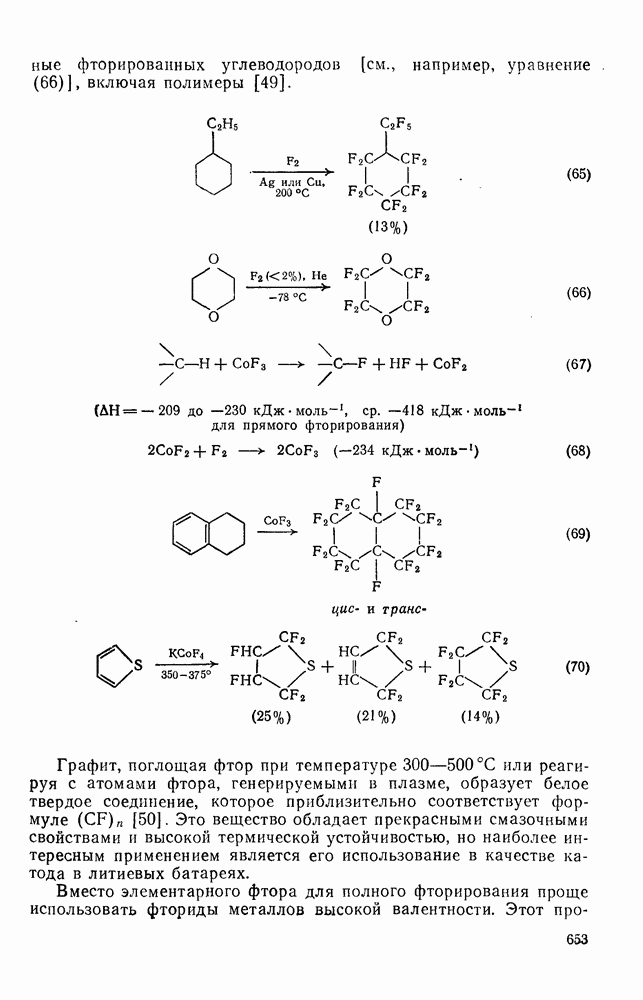
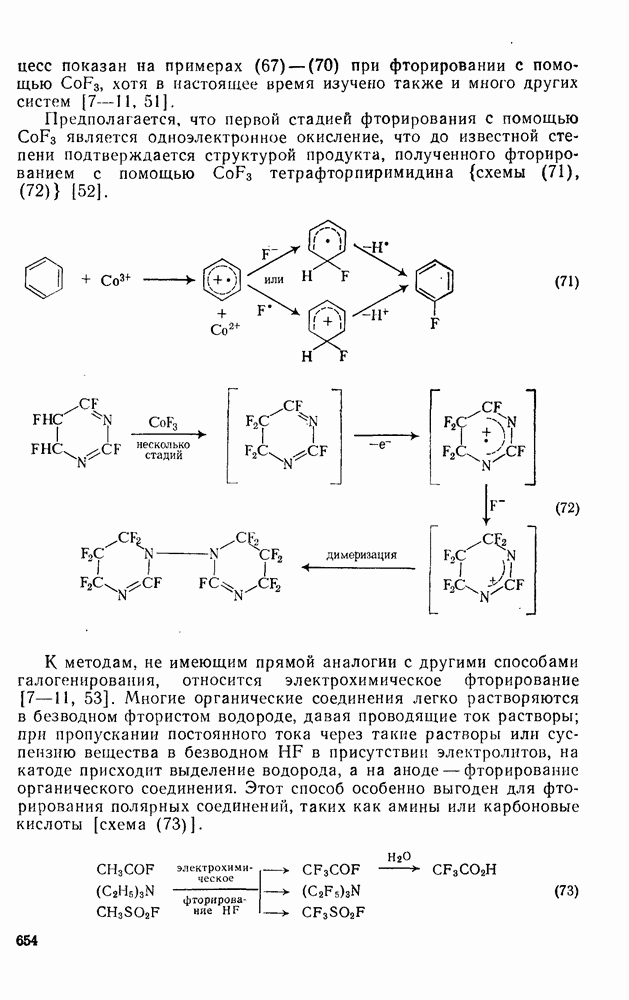
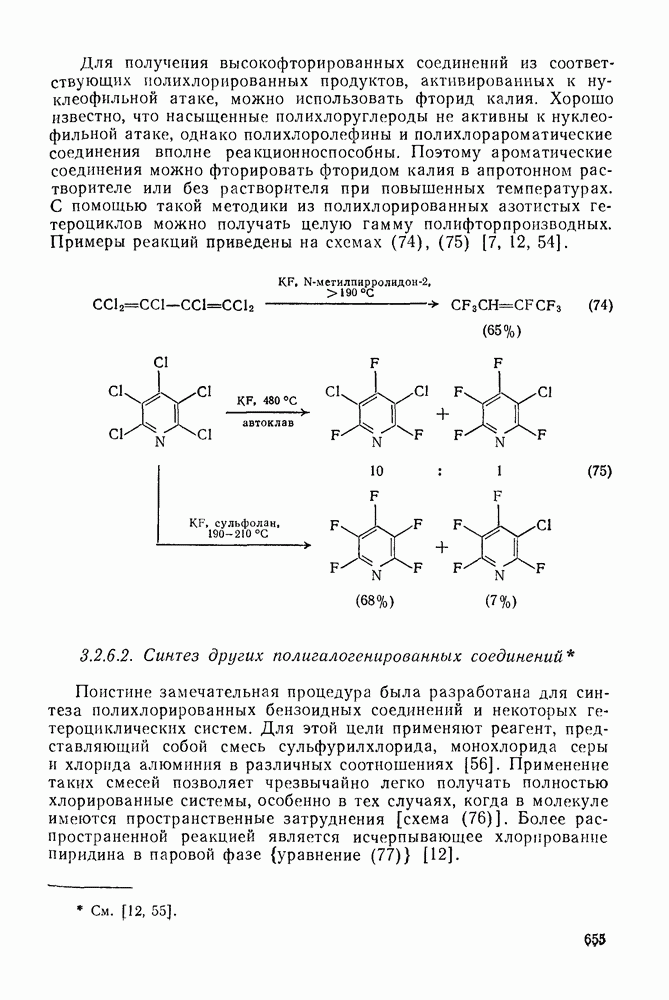
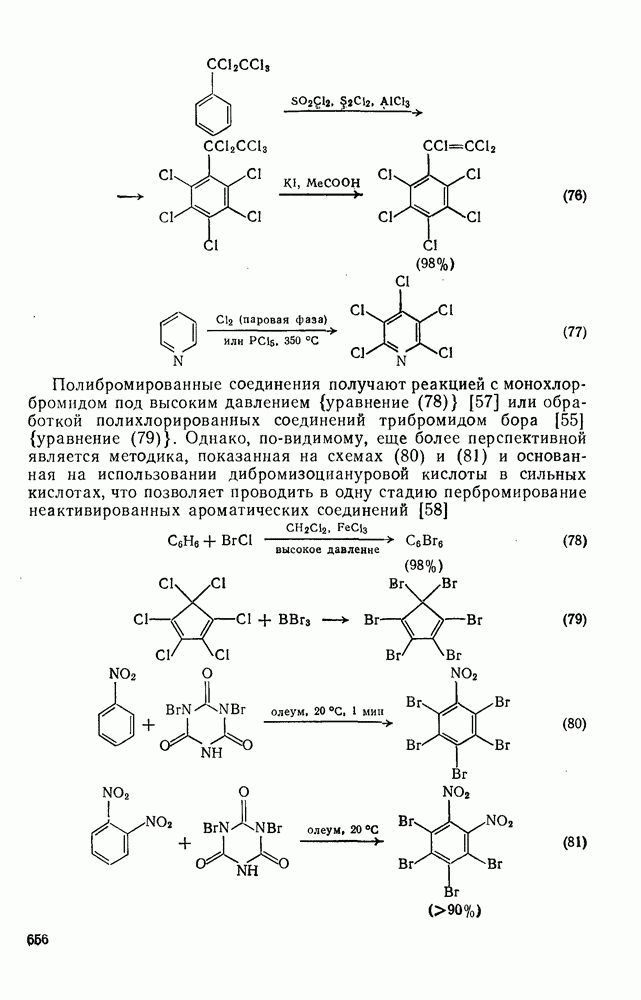
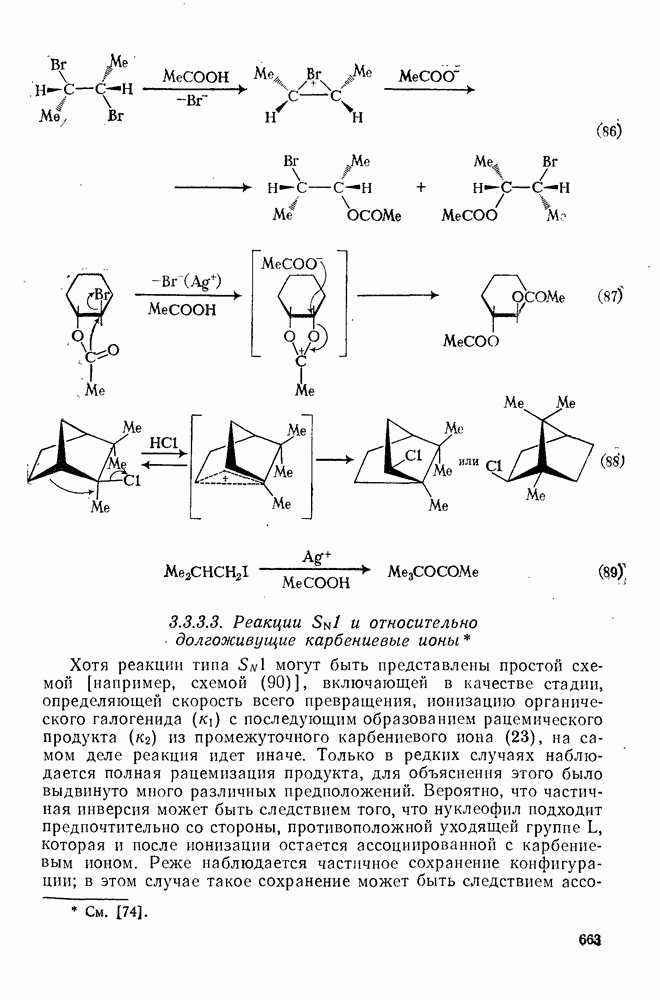
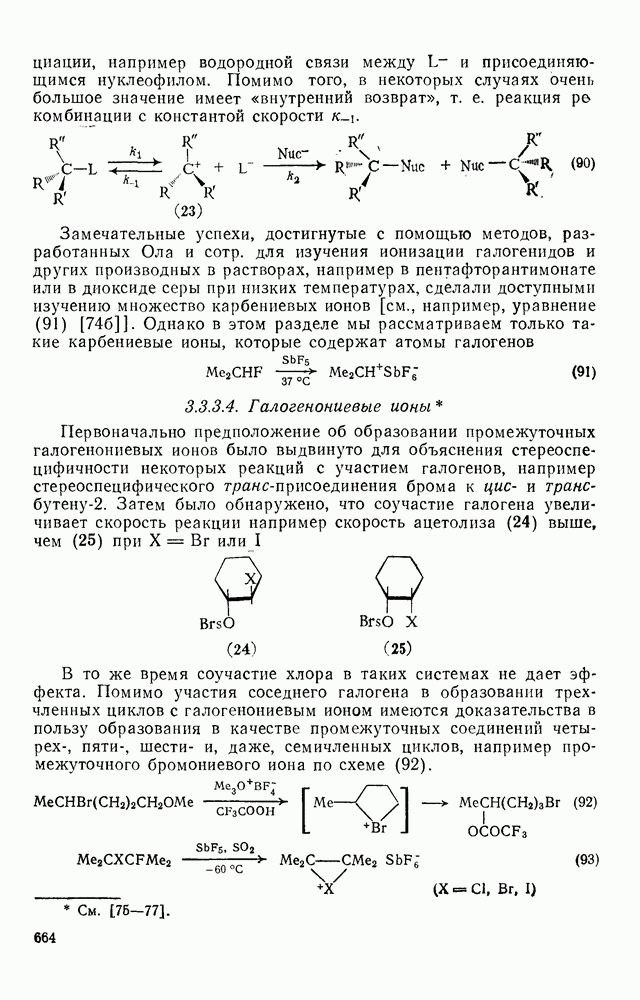
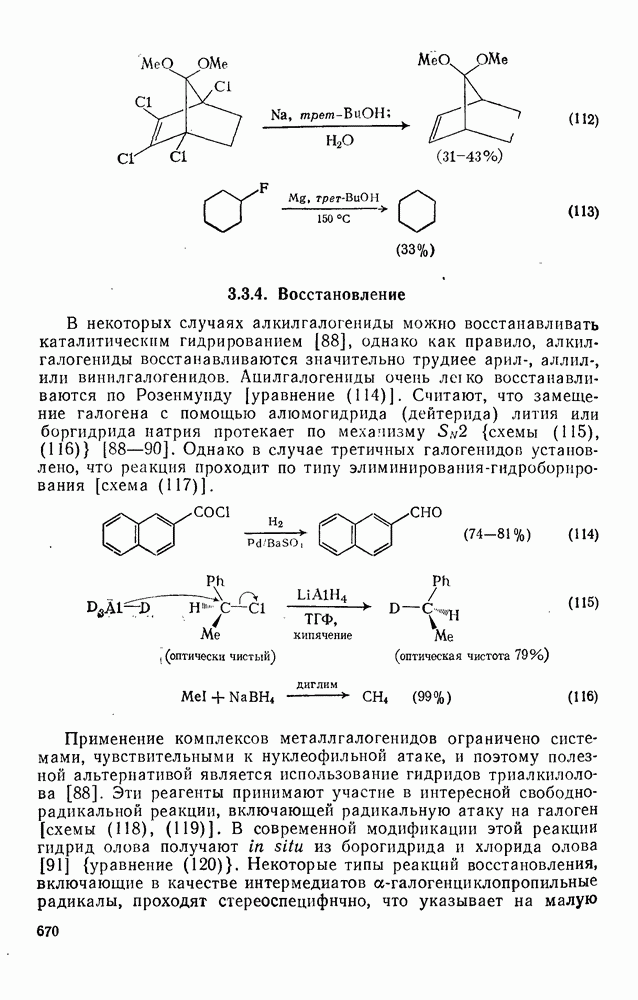
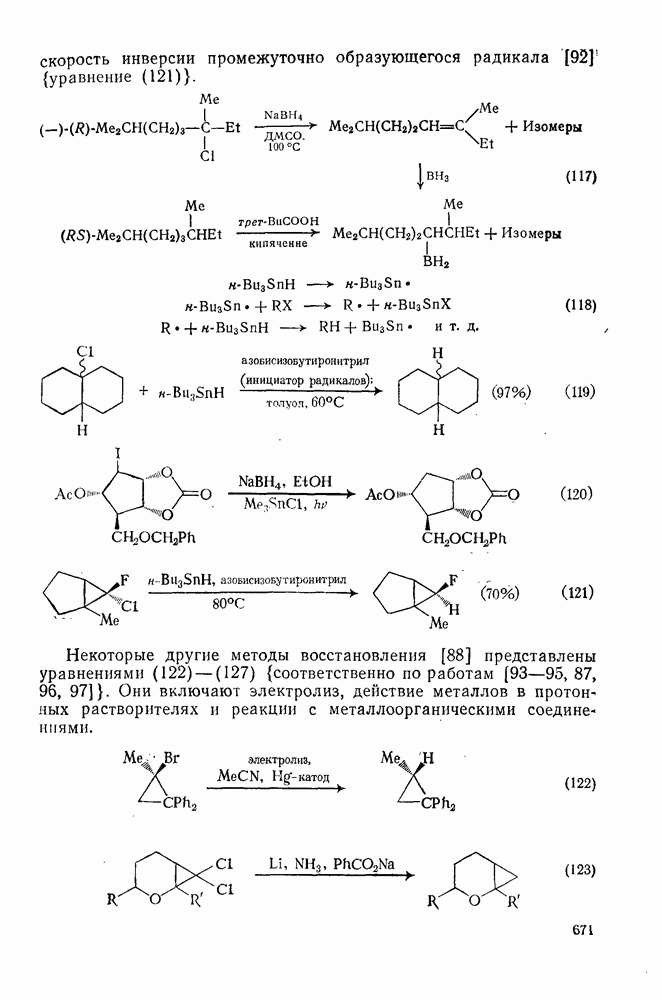
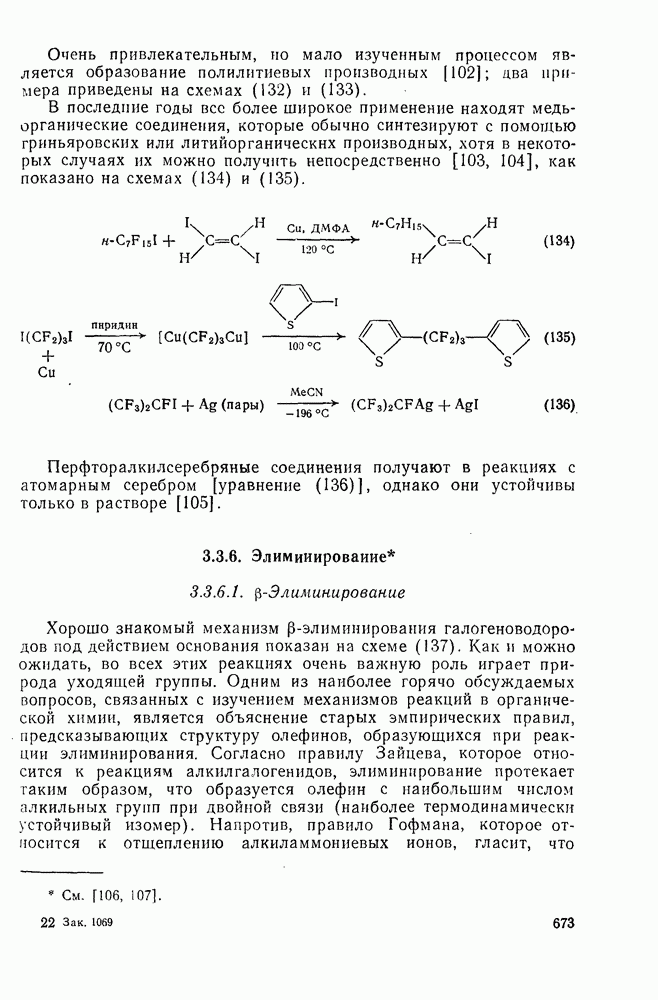
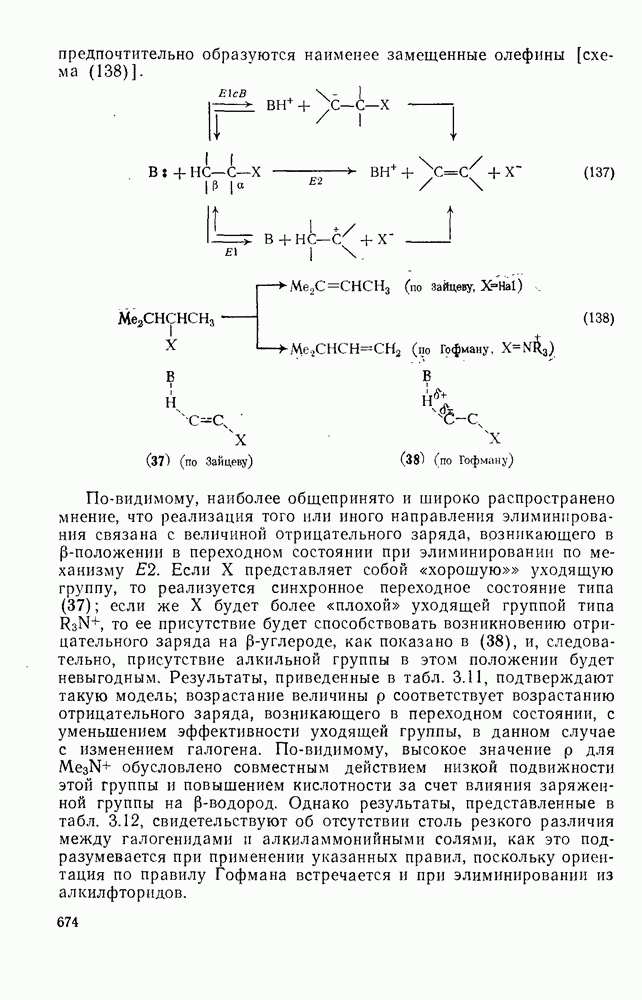
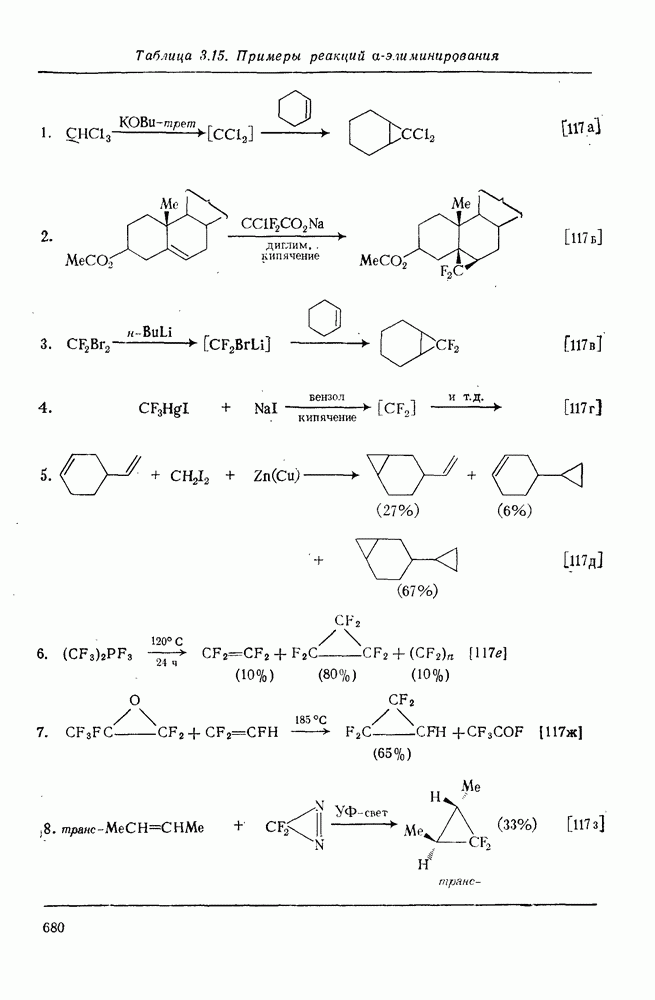
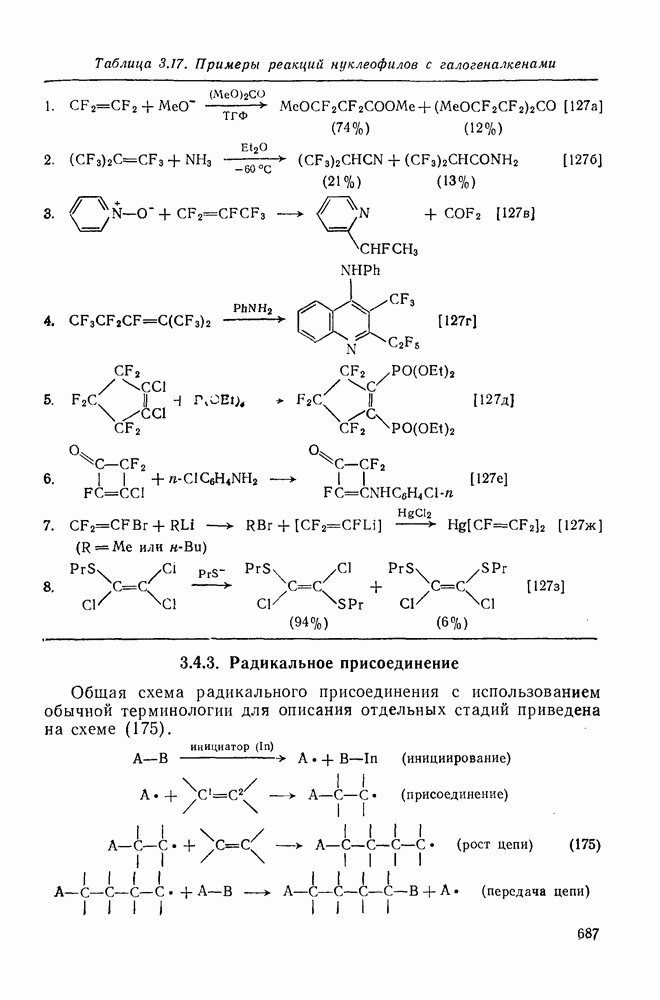
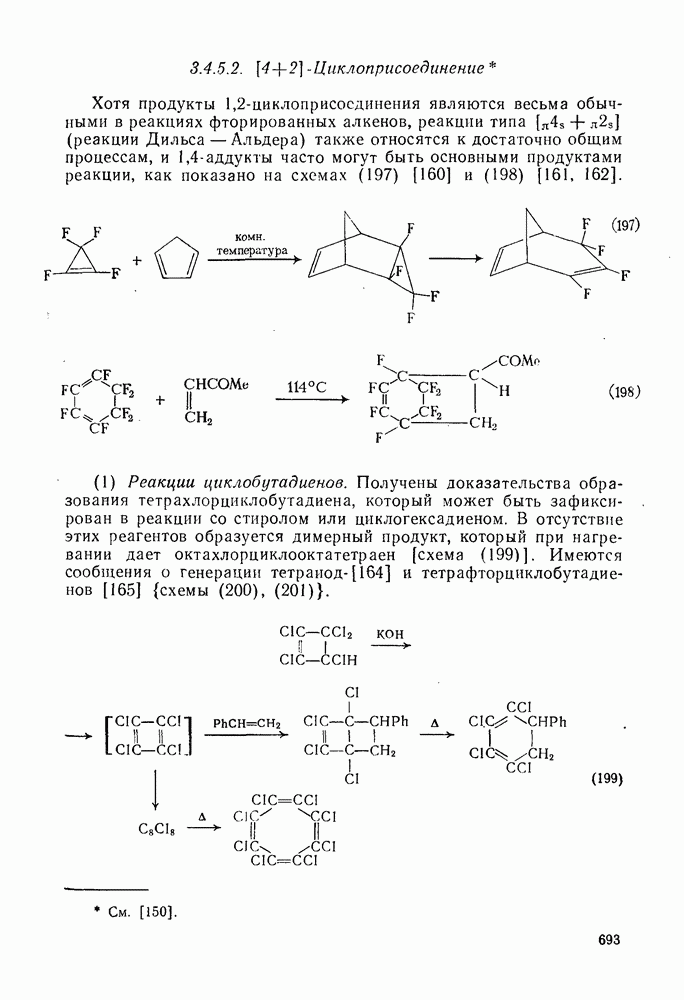
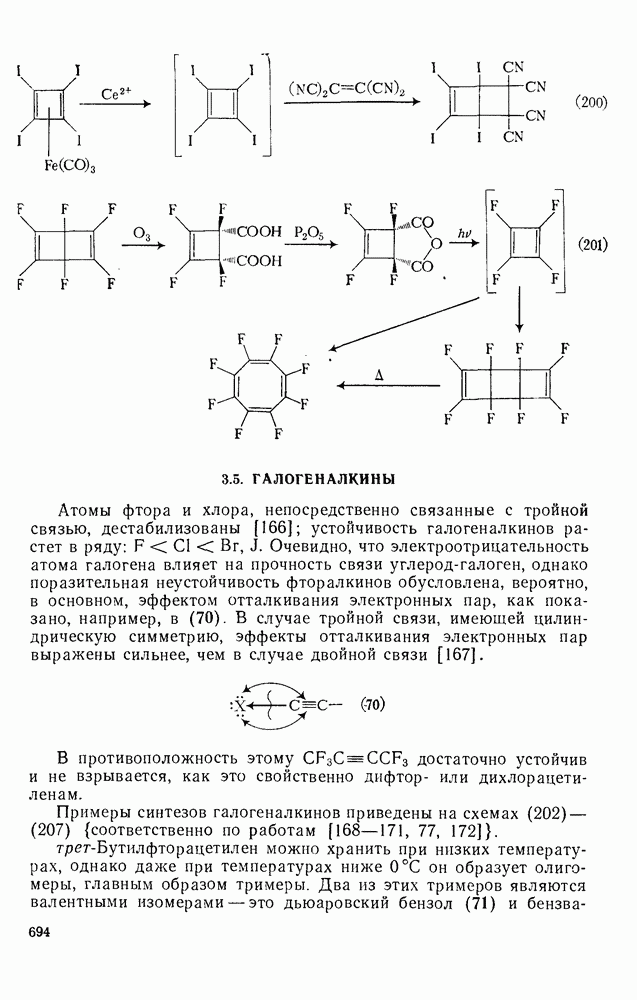
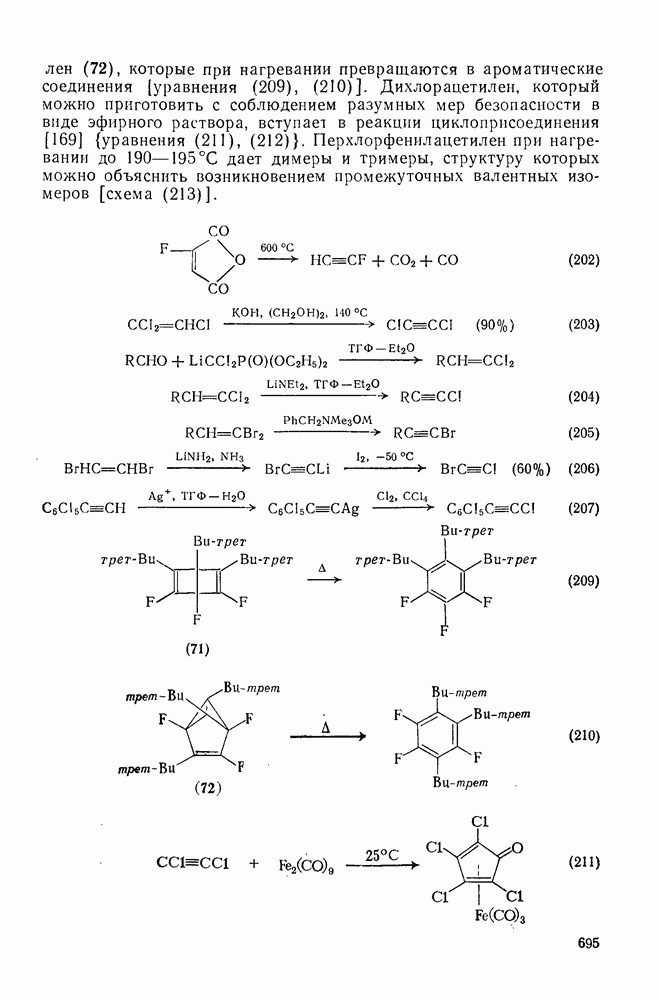
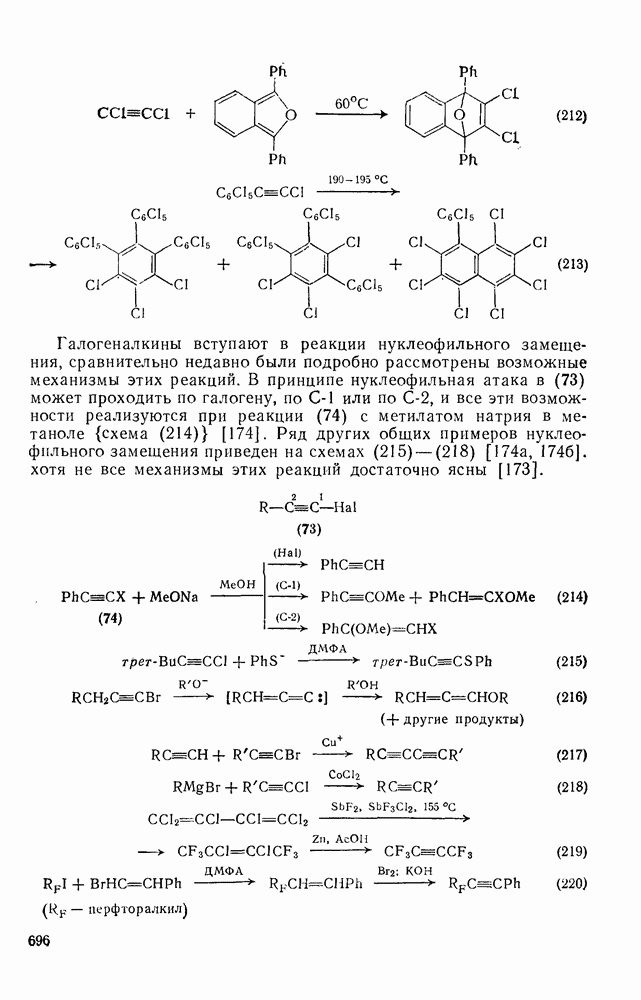
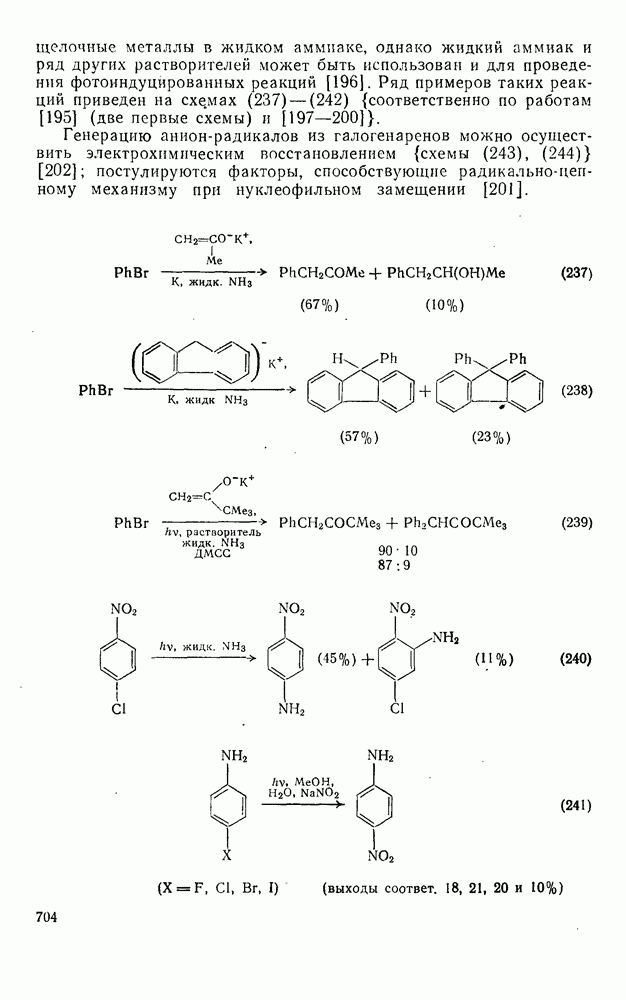
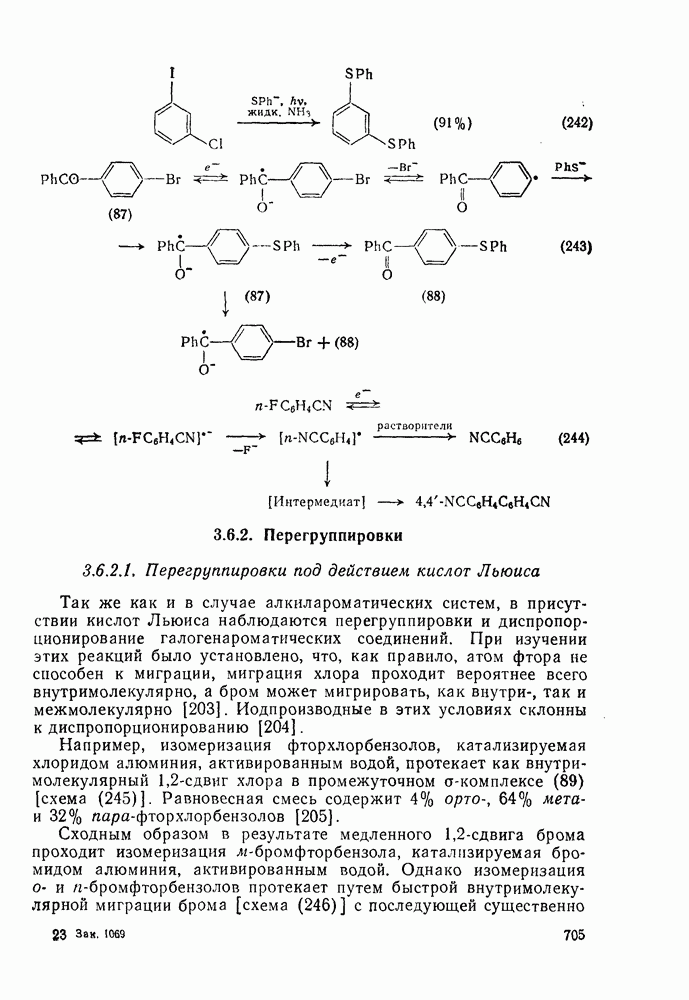
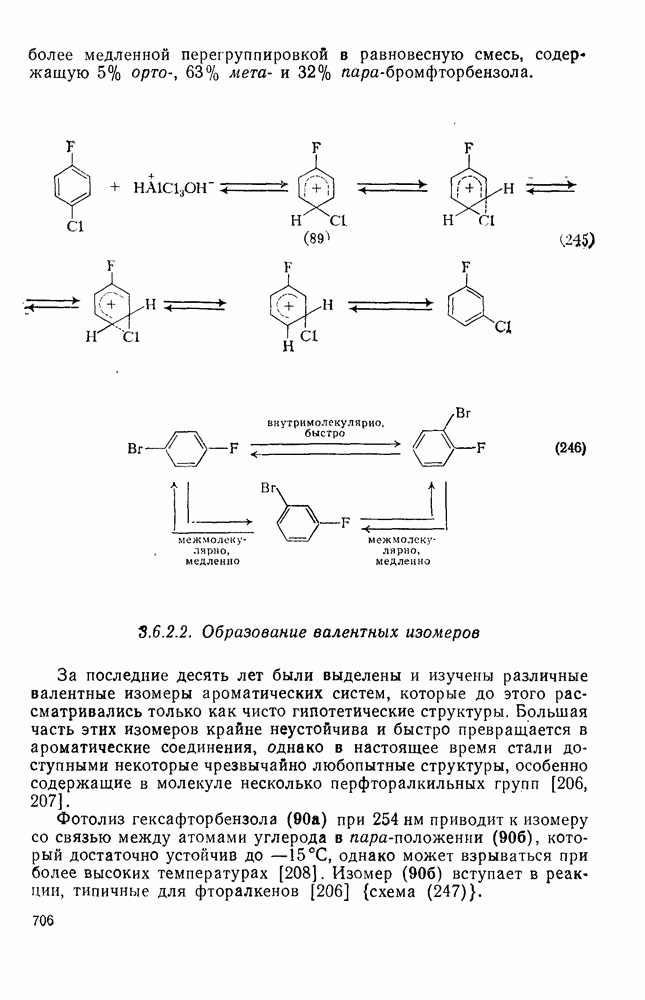
3.6.1.2. Присоединение-элиминирование
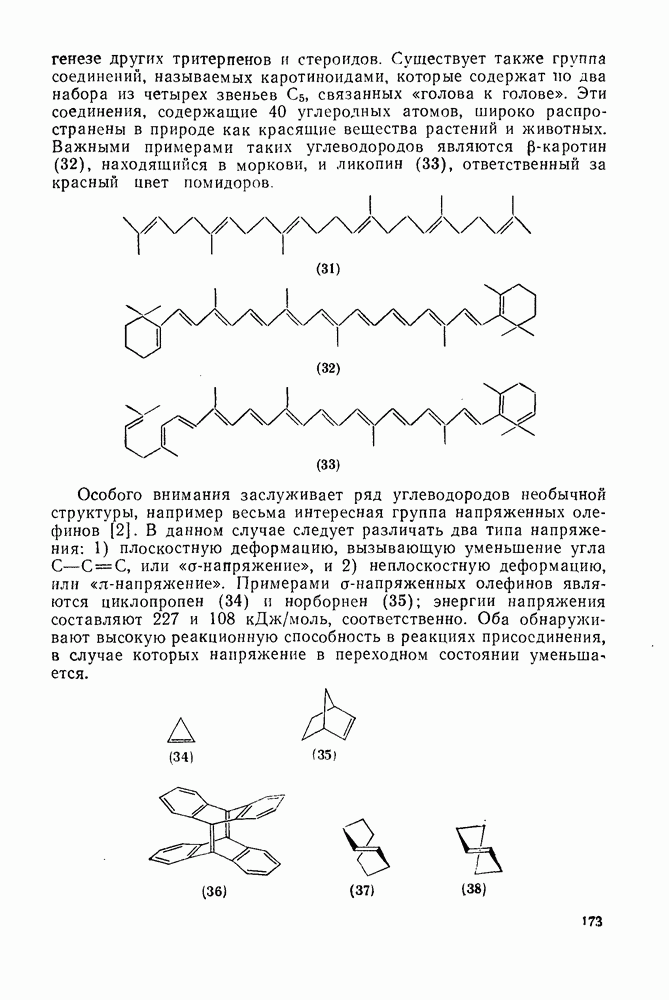
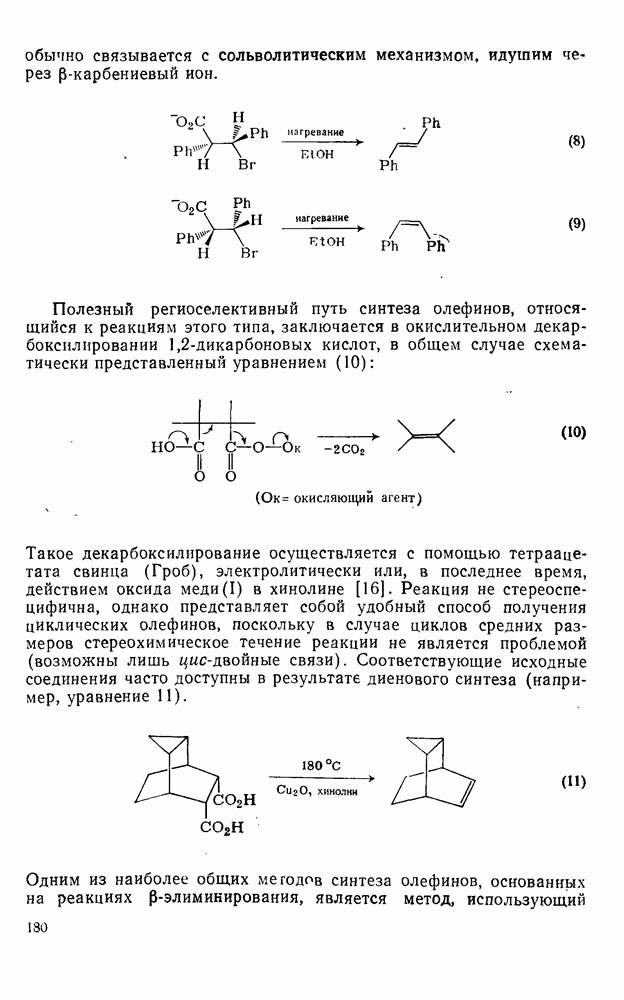
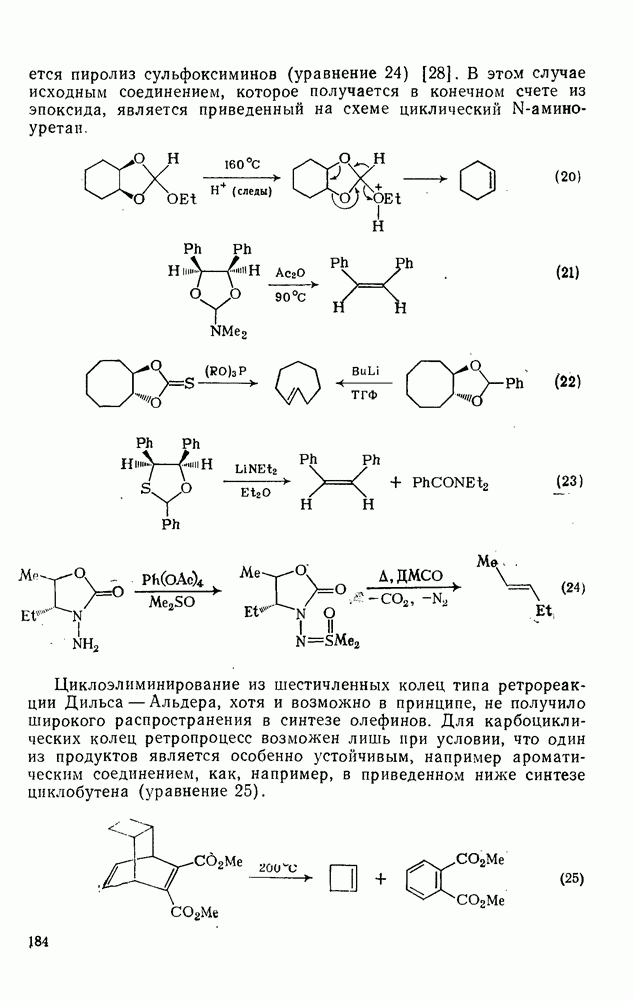
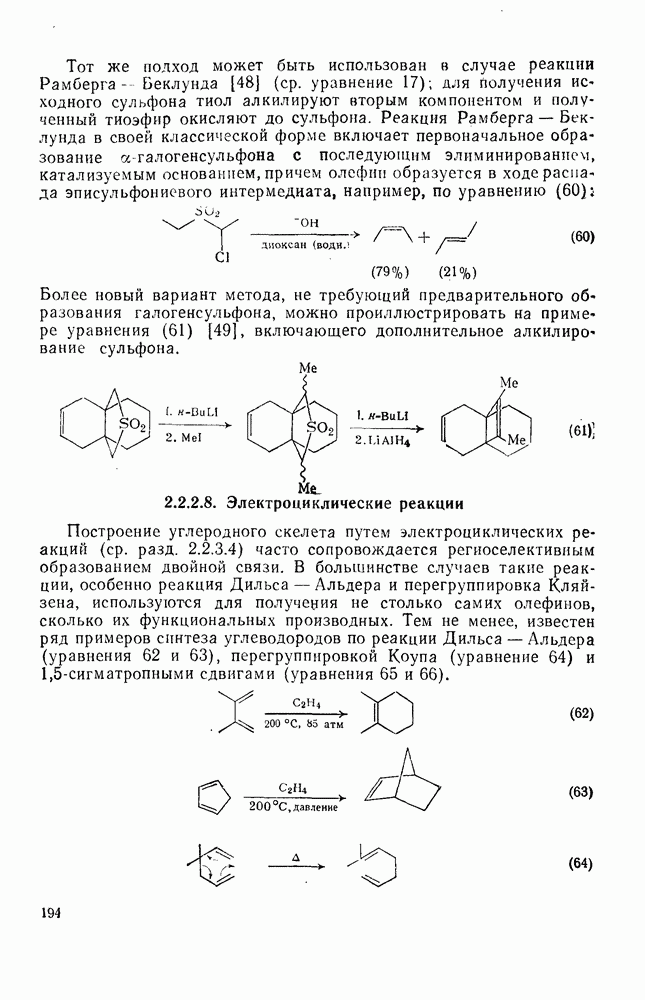
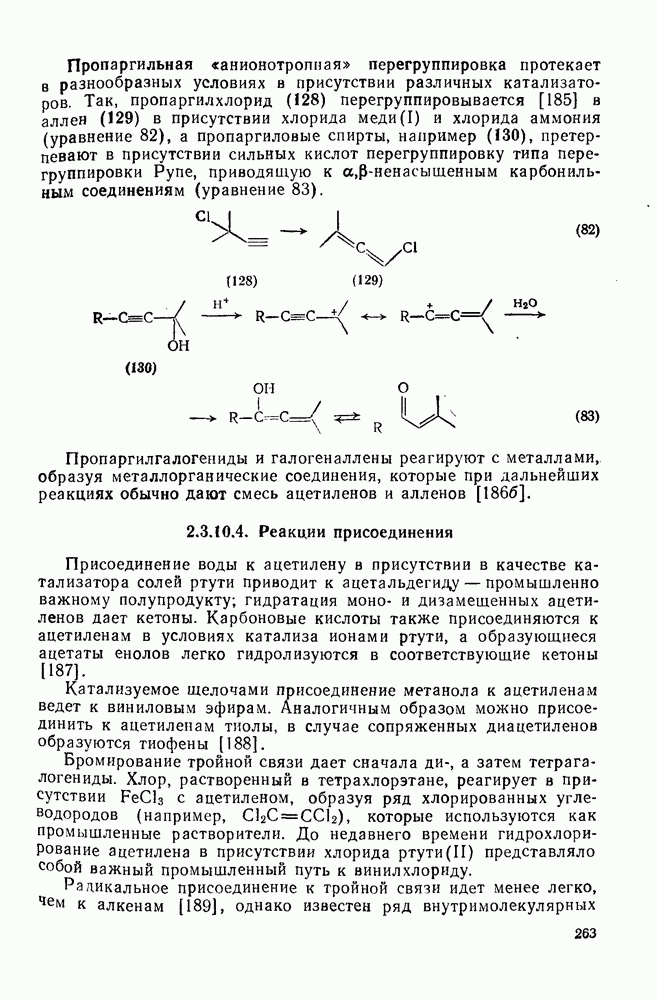
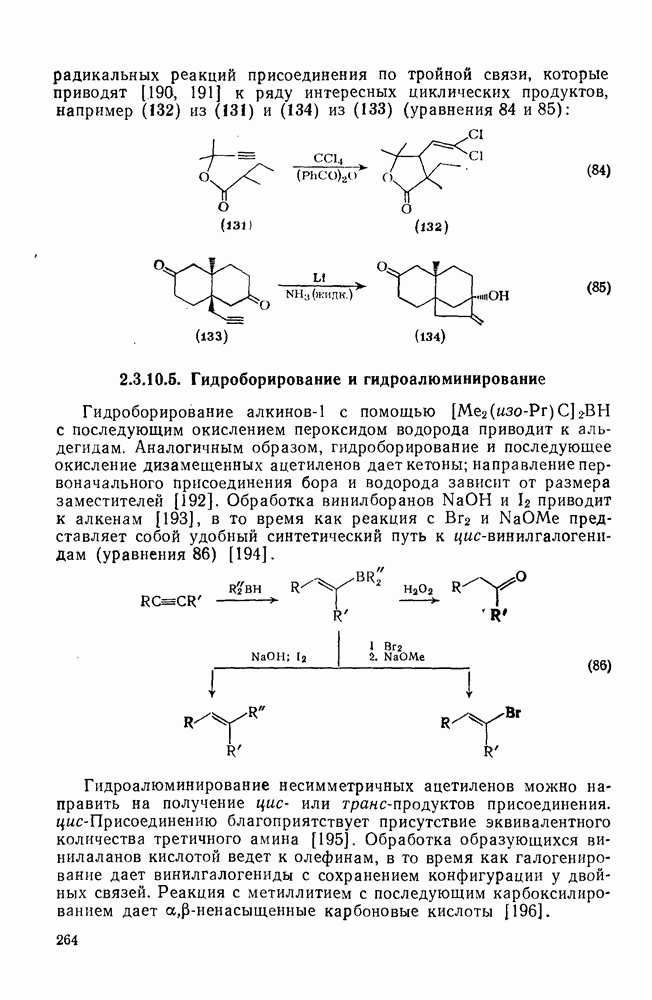
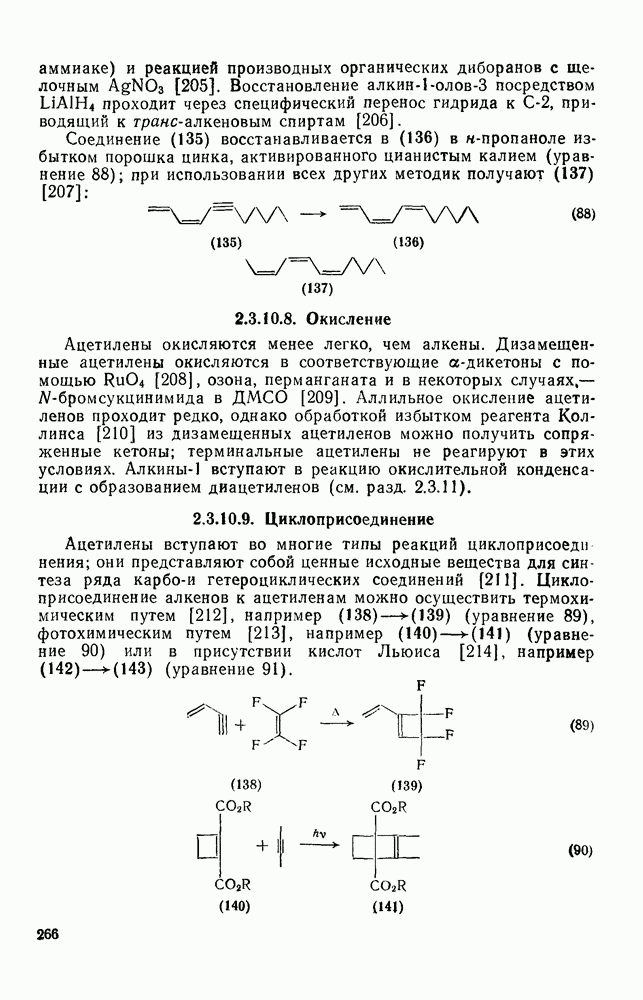
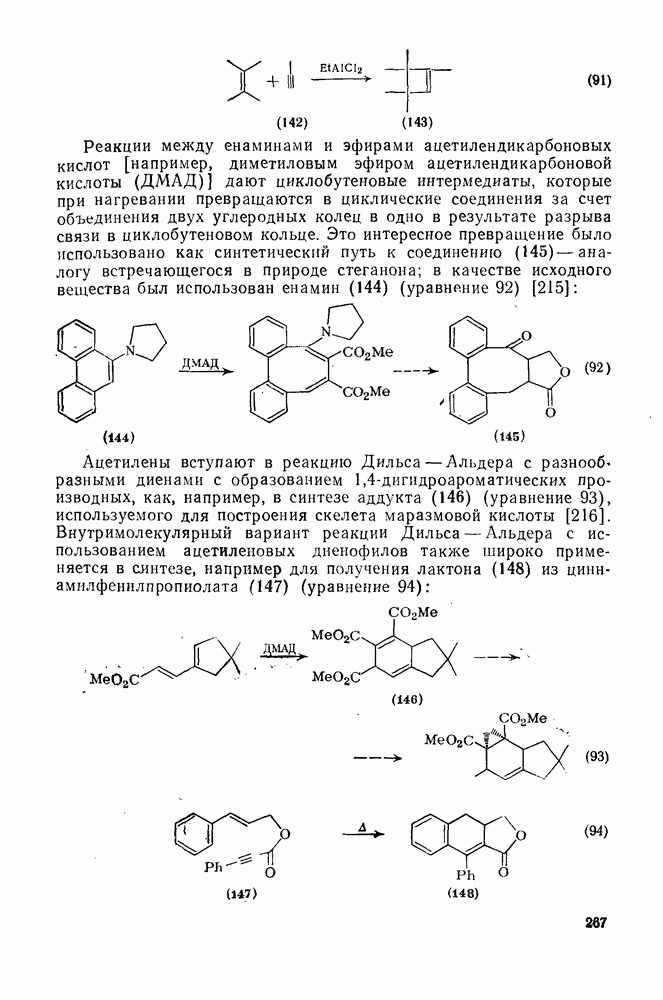
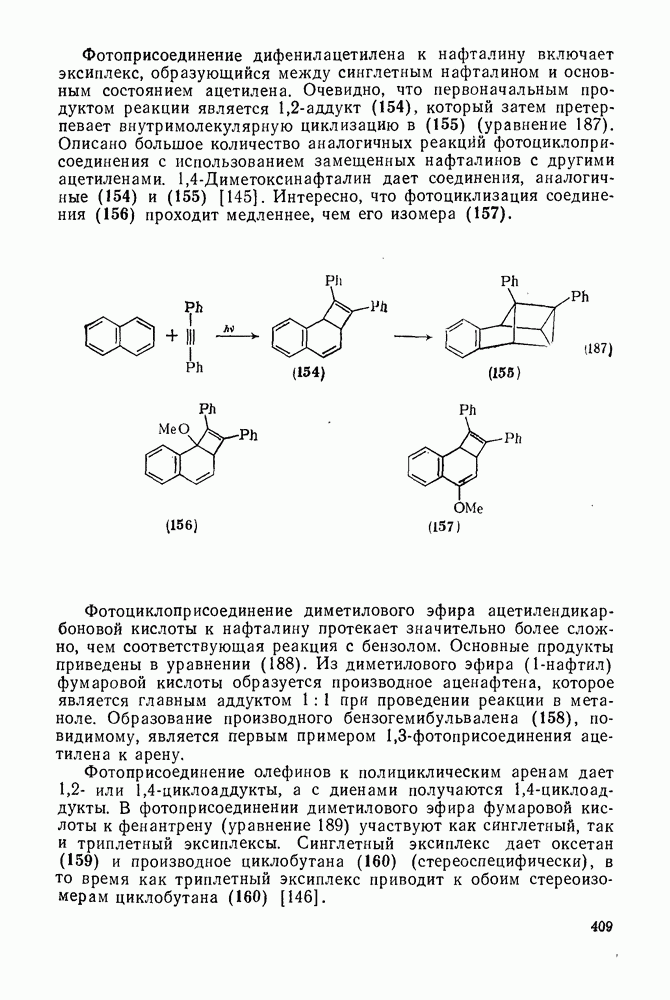
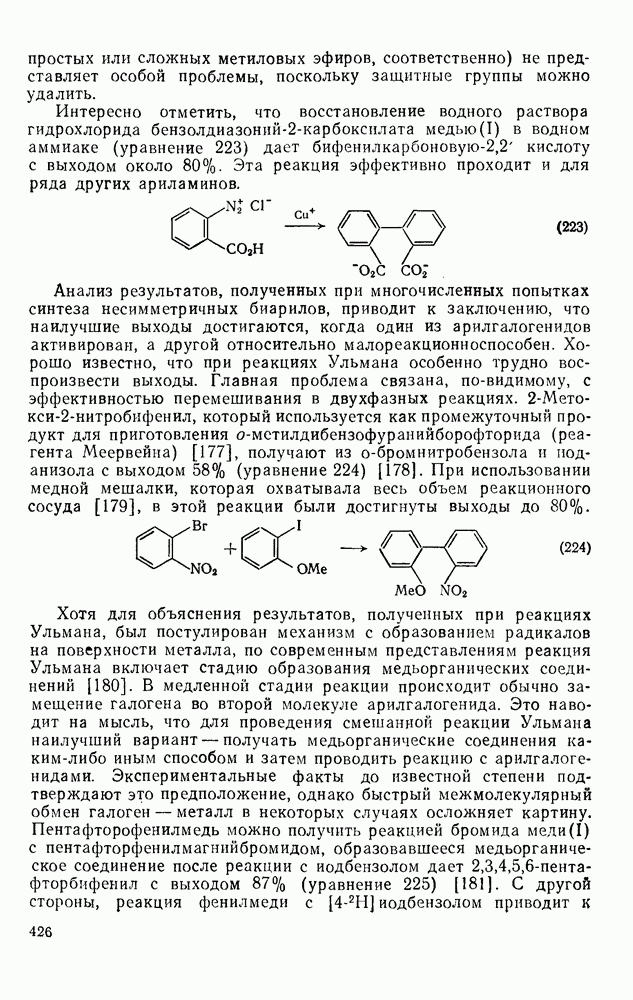
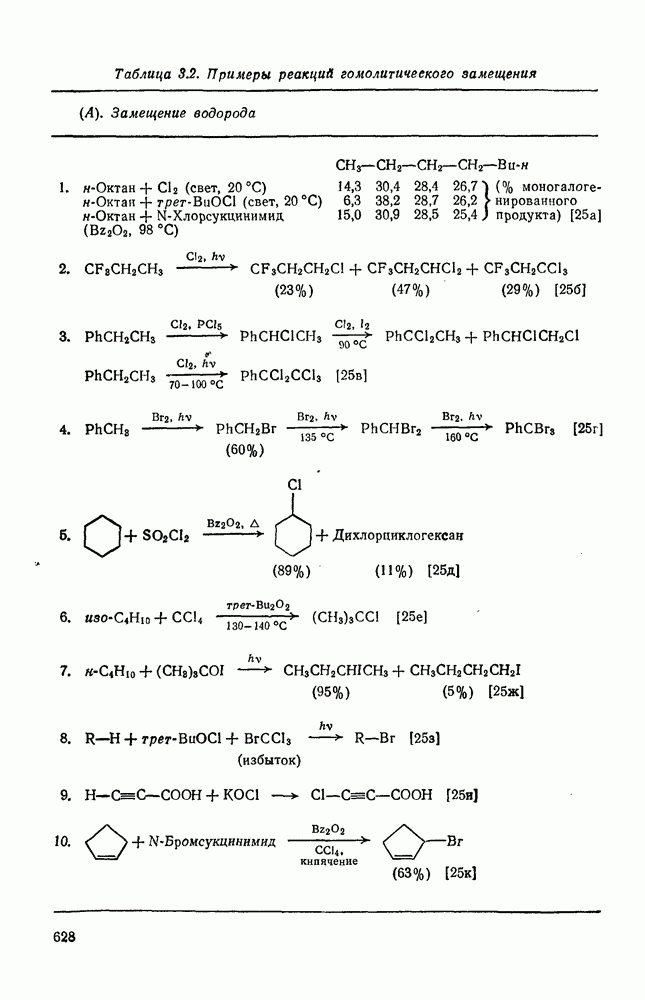
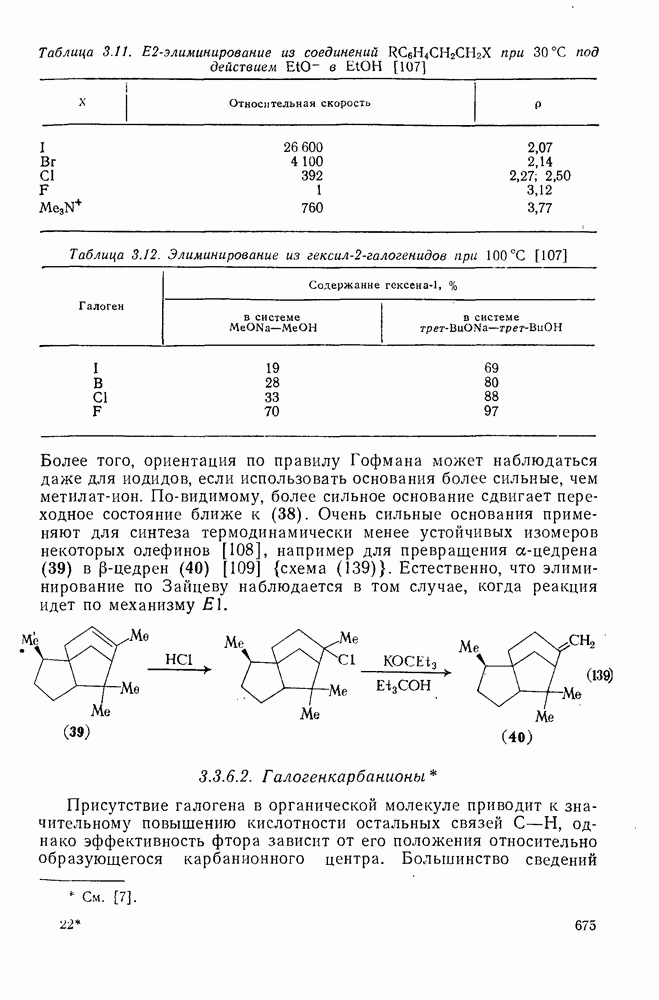
Нуклеофильное замещение галогена в активированных системах протекает чаще всего по двухстадийному механизму присоединения-элиминирования [183]; причем обычно, но не всегда, первая стадия  определяет скорость всей реакции. Одним из наиболее убедительных доказательств этого является установленный в классической работе Баннетта и сотр. порядок подвижности галогенов в этих реакциях:
определяет скорость всей реакции. Одним из наиболее убедительных доказательств этого является установленный в классической работе Баннетта и сотр. порядок подвижности галогенов в этих реакциях:  [184]. Поскольку самой прочной в этом ряду является связь углерод—фтор, то в системах, где этот порядок соблюдается, это может указывать на малую степень разрыва этой связи на стадии, определяющей скорость реакции. Однако, трудно объяснить, почему фторированные соединения существенно более реакционноспособны, чем соединения других галогенов. Приводились доводы в пользу того, что приведенный выше порядок подвижности галогенов, обусловлен в основном
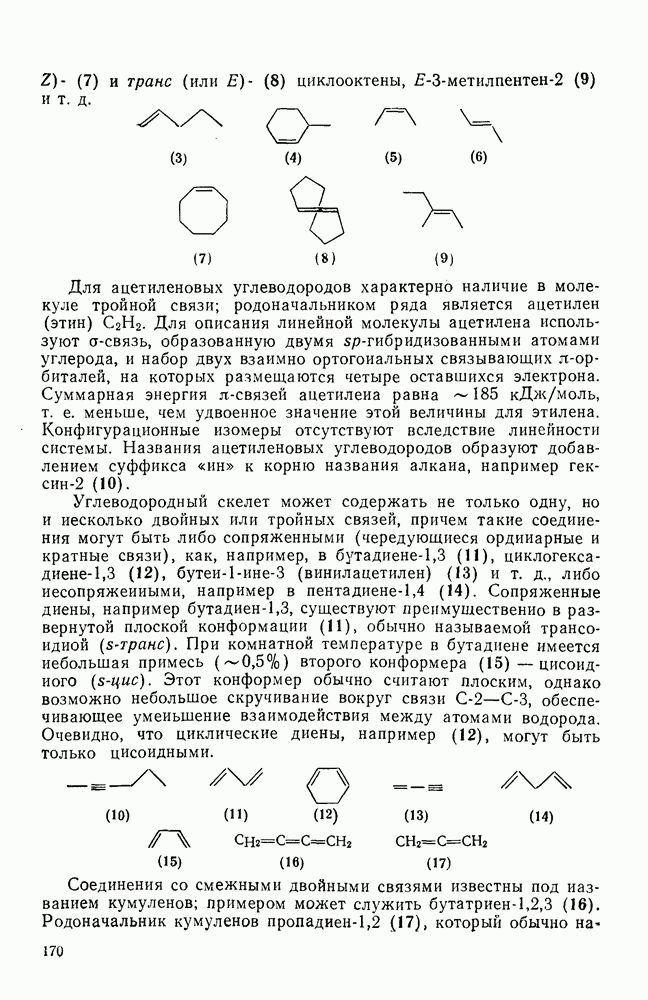
[184]. Поскольку самой прочной в этом ряду является связь углерод—фтор, то в системах, где этот порядок соблюдается, это может указывать на малую степень разрыва этой связи на стадии, определяющей скорость реакции. Однако, трудно объяснить, почему фторированные соединения существенно более реакционноспособны, чем соединения других галогенов. Приводились доводы в пользу того, что приведенный выше порядок подвижности галогенов, обусловлен в основном  полярностью связи
полярностью связи  , т. е. усилением электрофильного характера атома углерода; в то же время на порядок подвижности влияют, вероятно, также и различия в пространственных факторах для атомов галогенов.
, т. е. усилением электрофильного характера атома углерода; в то же время на порядок подвижности влияют, вероятно, также и различия в пространственных факторах для атомов галогенов.

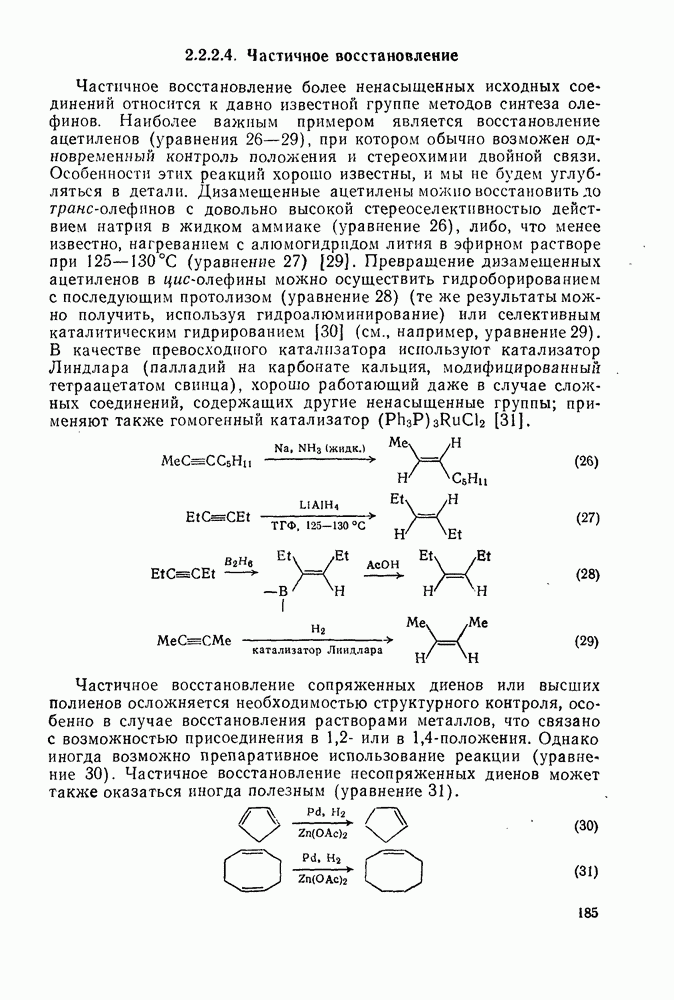
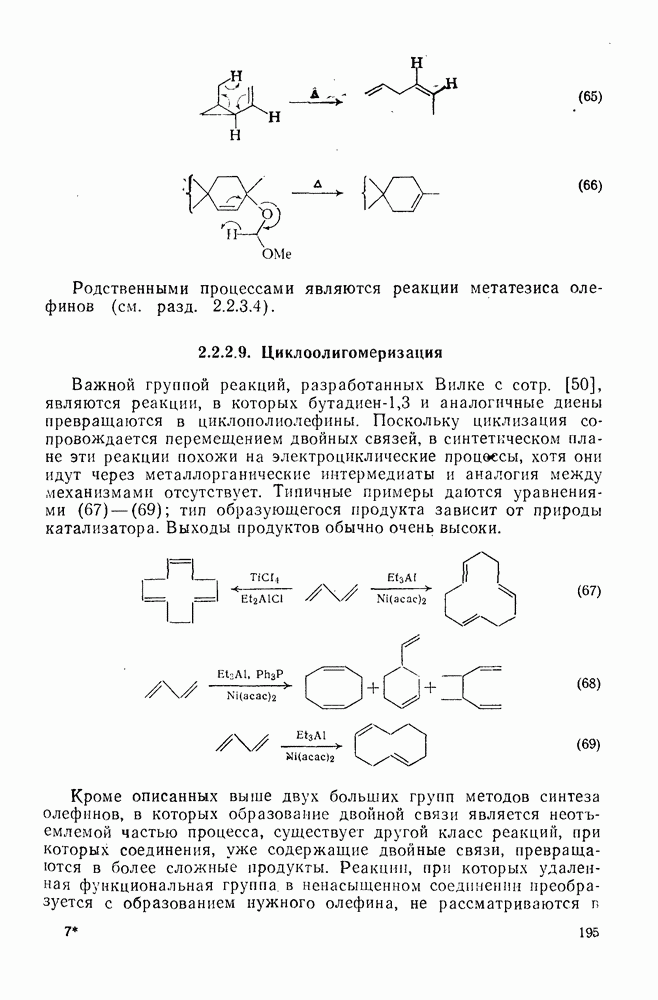
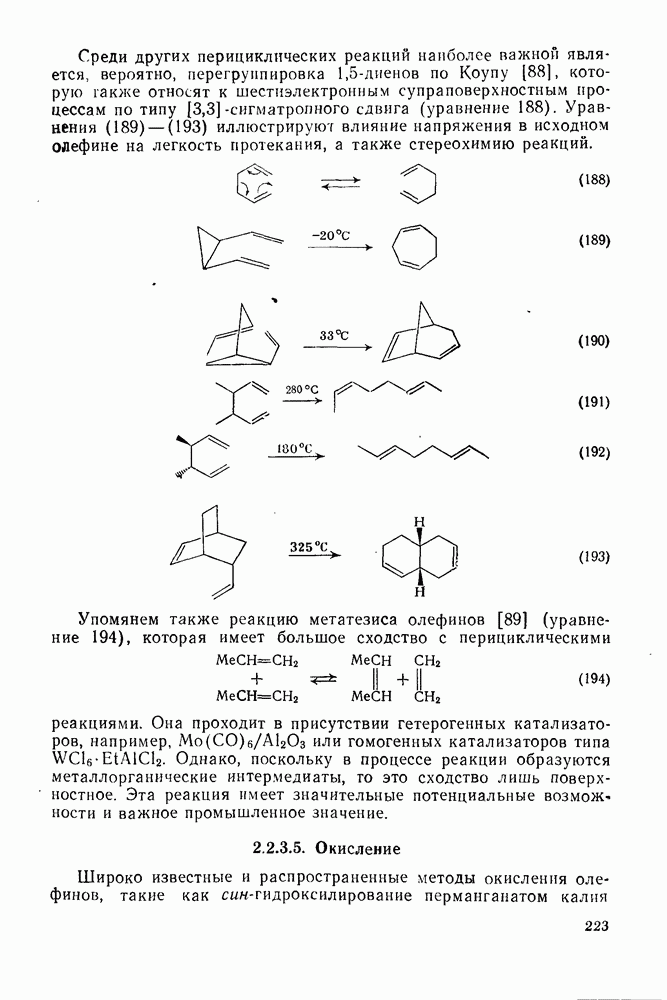
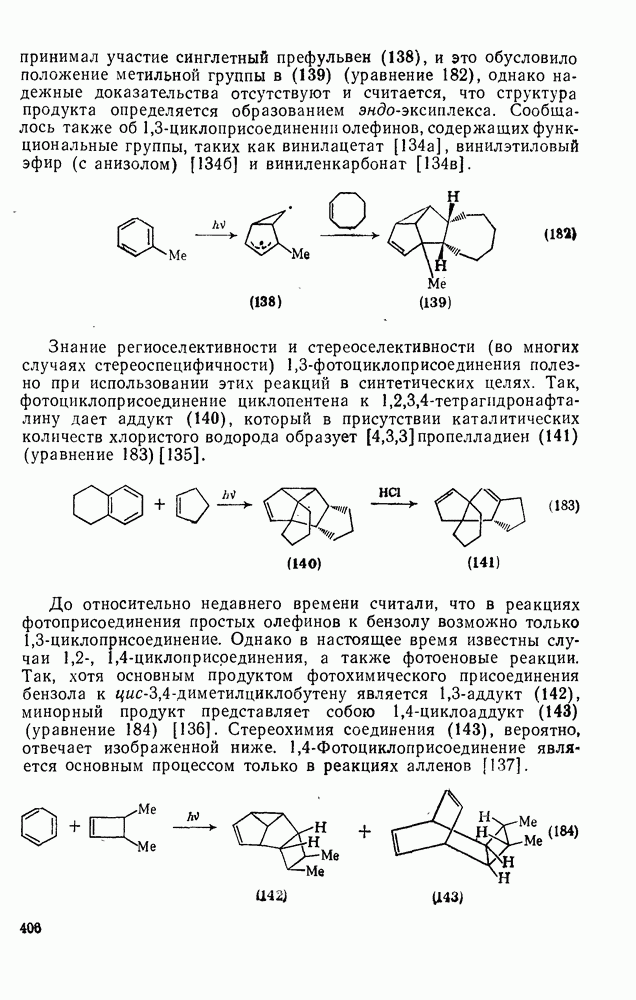
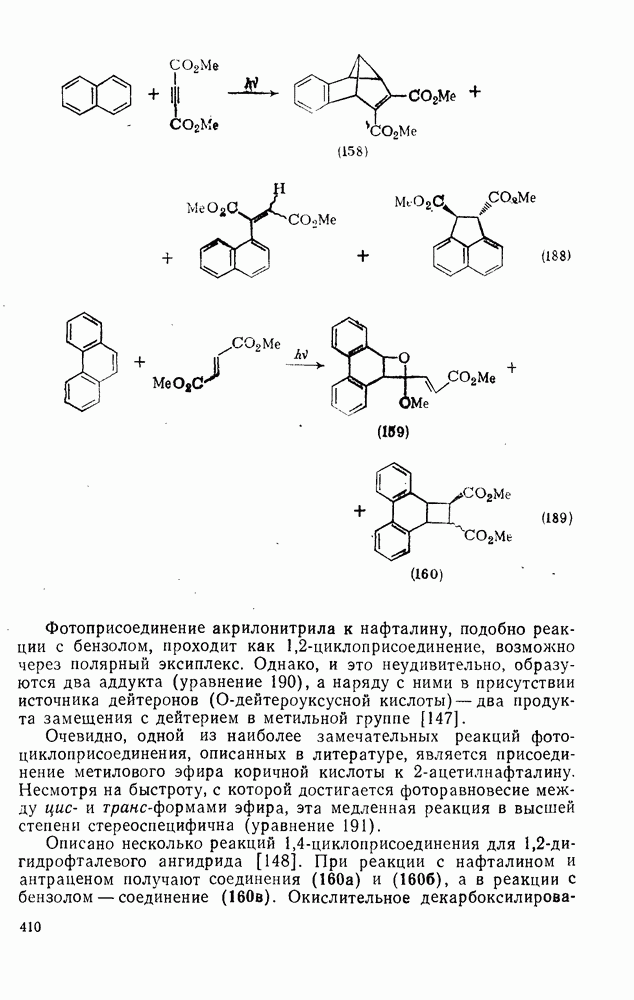
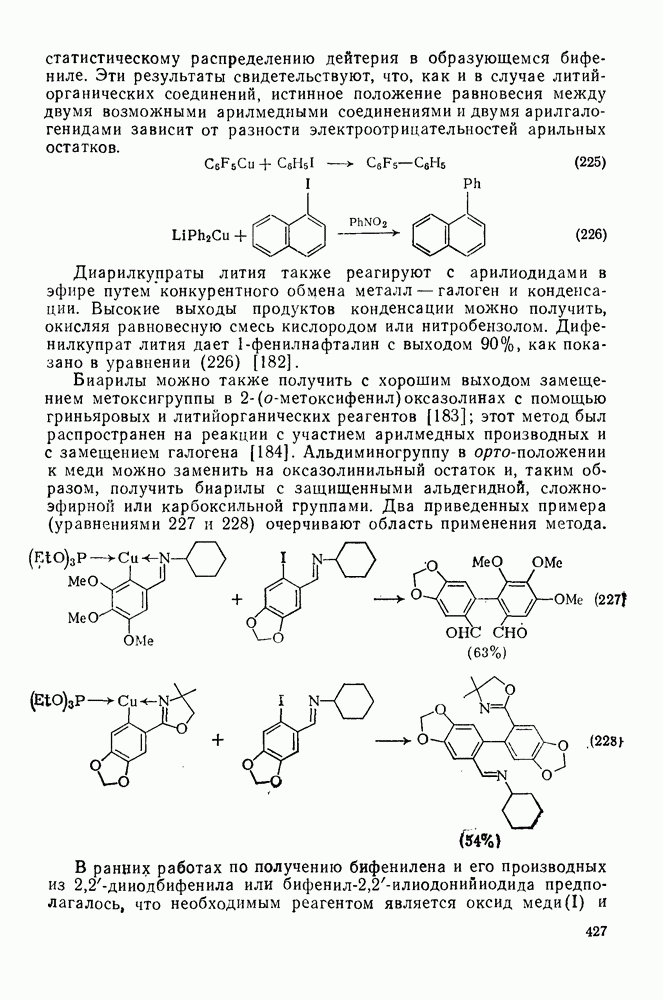
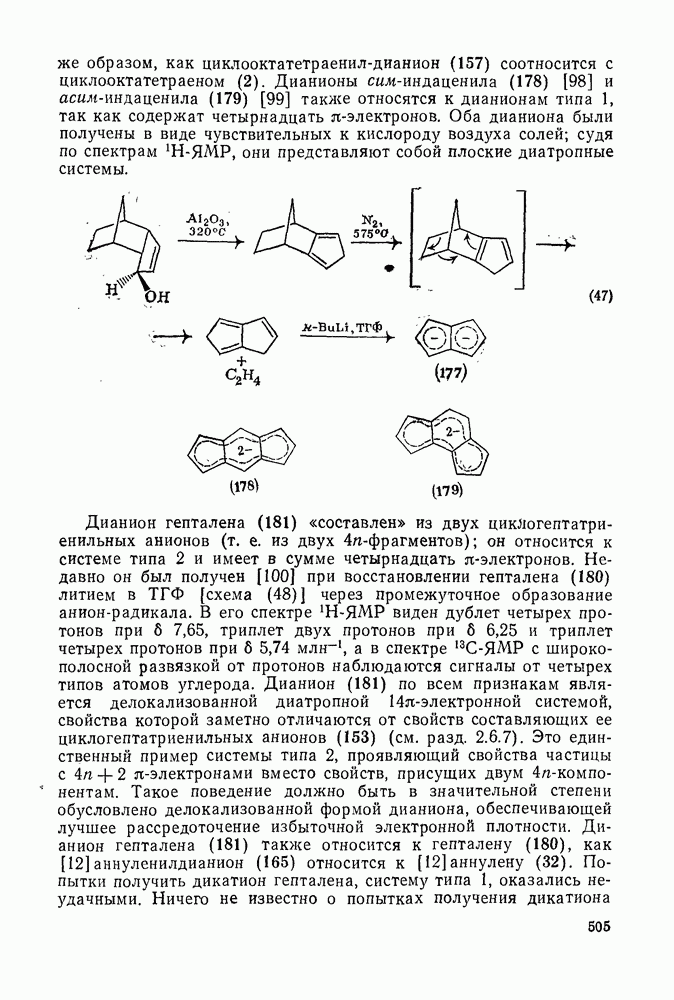
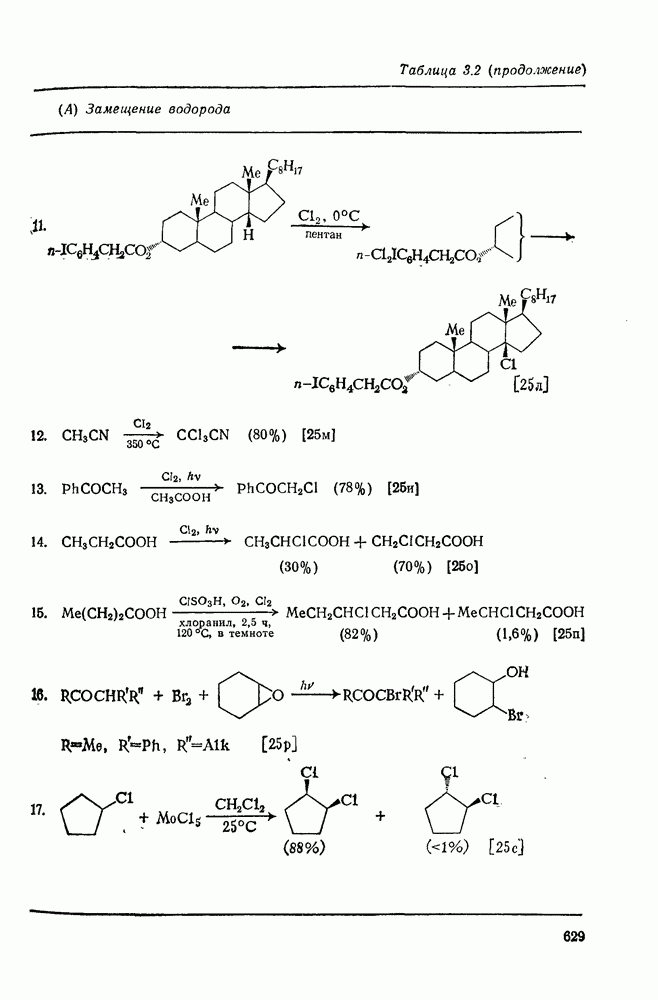
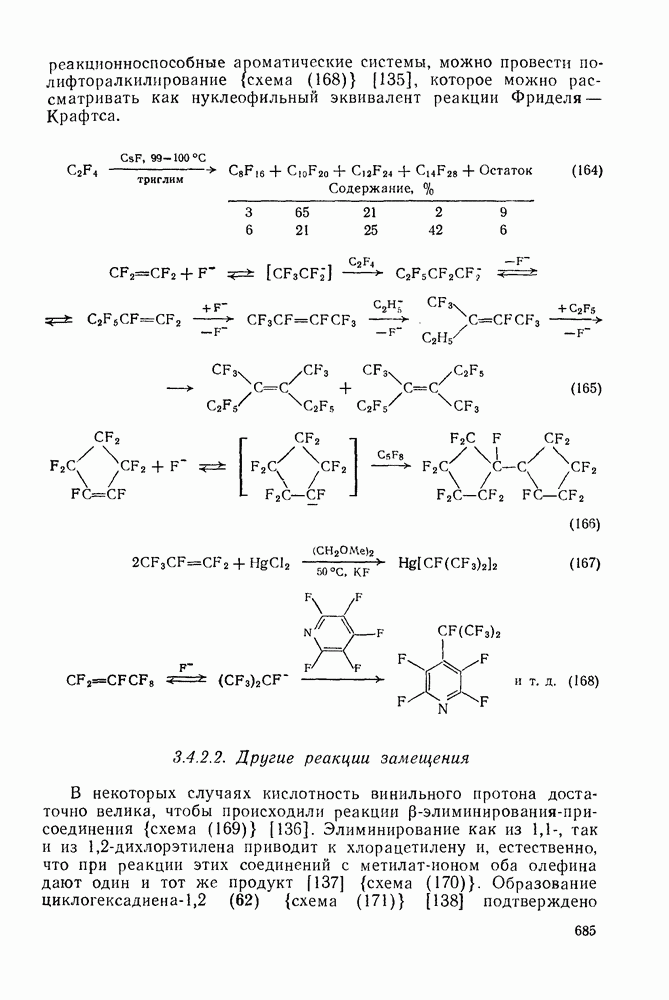
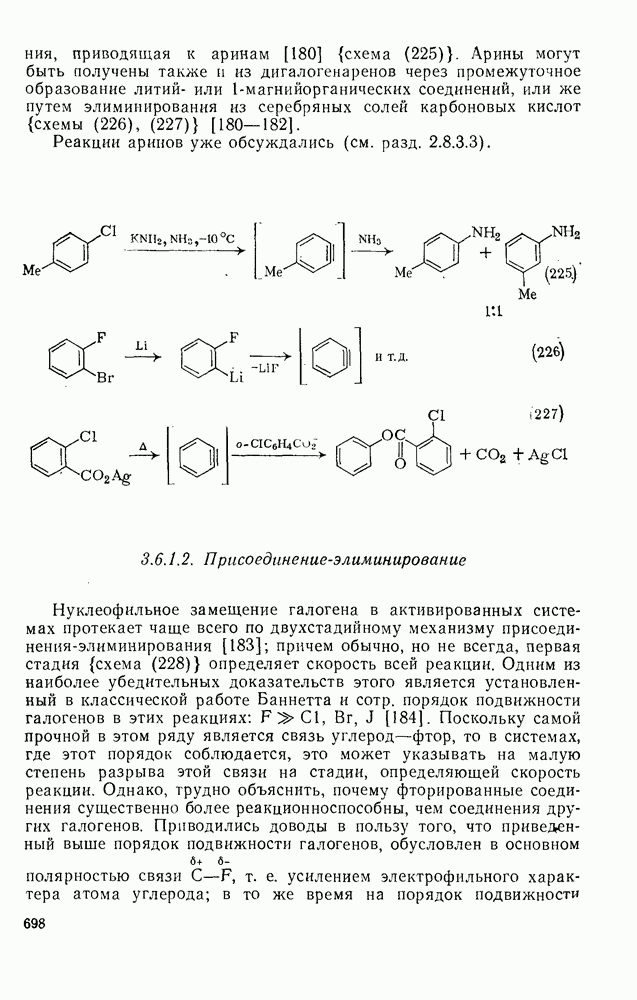
Проблемы, связанные с направлением замещения, которые возникают в случае полигалогенированных систем, сходны с классическими проблемами ориентации при электрофильном ароматическом замещении. Ориентация при замещении и реакционная способность полигалогенароматических соединений была предметом многочисленных дискуссий, и только в последнее время картина стала достаточно ясной [185,186]  . В большинстве случаев замещение в перфторбензолах
. В большинстве случаев замещение в перфторбензолах  приводит к замене атома фтора в пара-положении к заместителю X (
приводит к замене атома фтора в пара-положении к заместителю X ( ) (
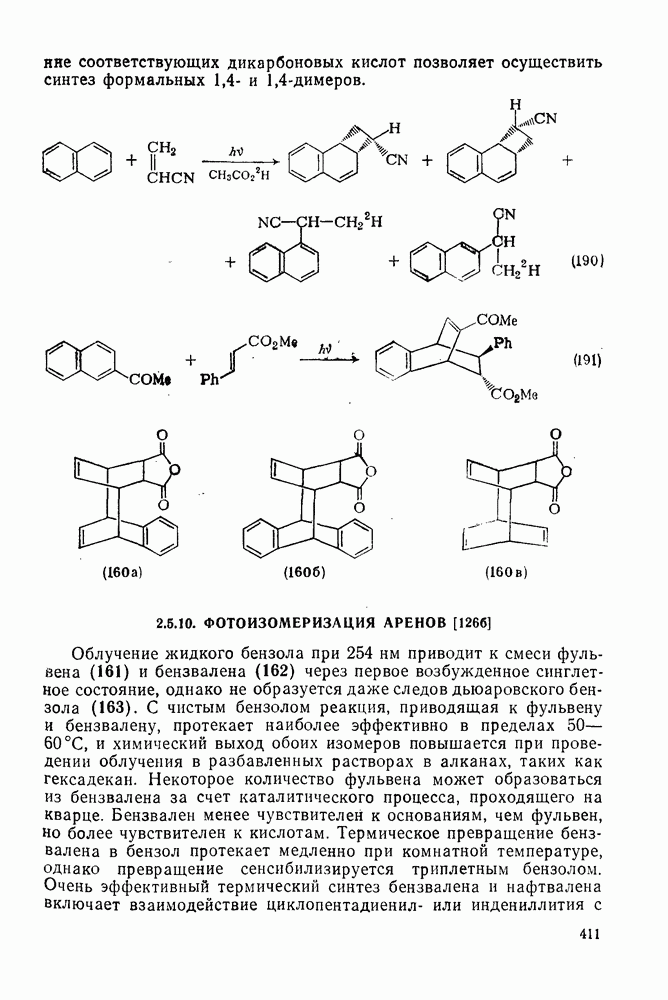
) ( и т. д.) [187, 188]. В отдельных случаях преобладает мета-замещение (X
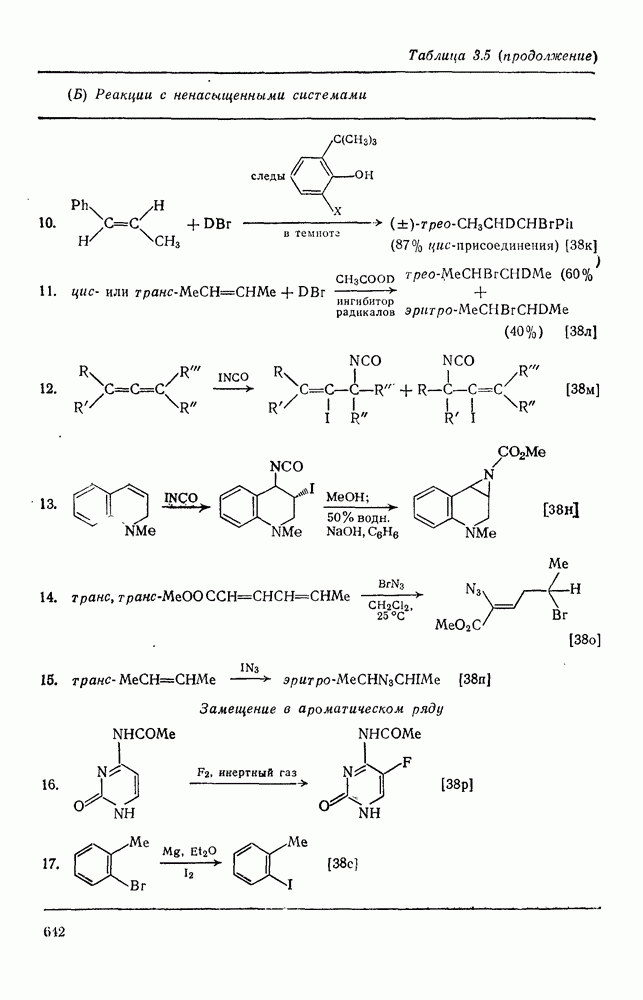
и т. д.) [187, 188]. В отдельных случаях преобладает мета-замещение (X  ,
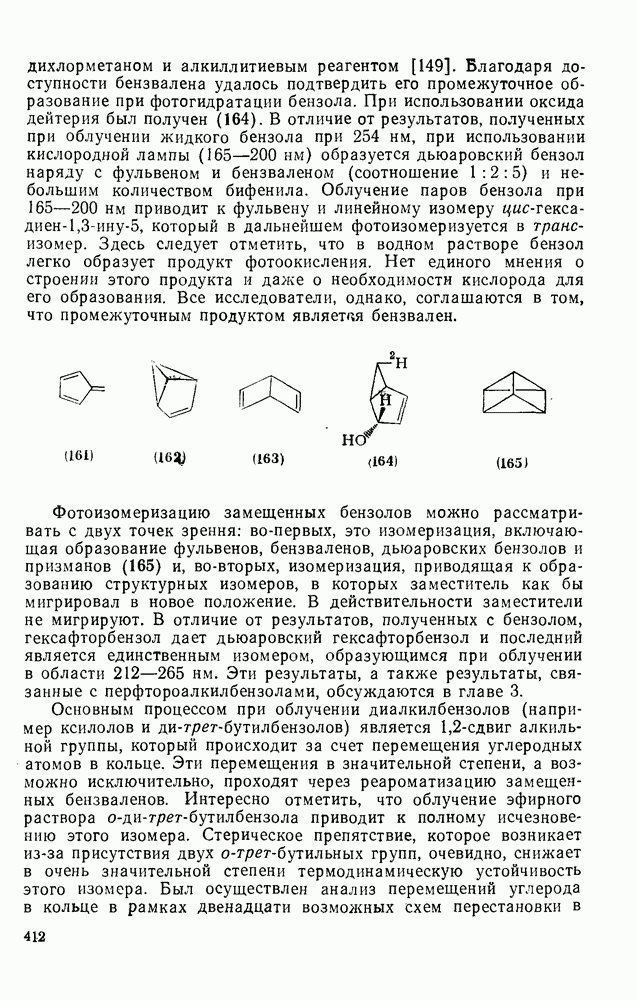
,  ). Однако на константу скорости заместители влияют в том же направлении, что при нуклеофильном ароматическом замещении, т. е. электронодонорные группы понижают (например,
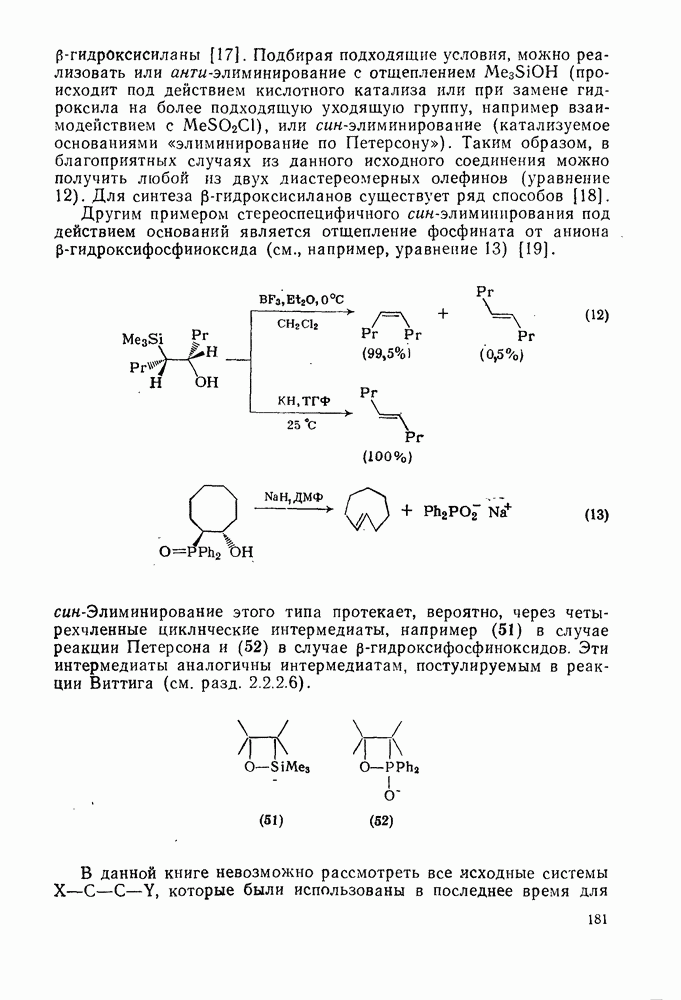
). Однако на константу скорости заместители влияют в том же направлении, что при нуклеофильном ароматическом замещении, т. е. электронодонорные группы понижают (например,  и
и  сильно дезактивируют), а электроноакцепторные группы повышают скорость реакции. Относительные константы скорости [189], приведенные в табл. 3.21, показывают, что, как и в случае электрофильного ароматического замещения, эти величины очень сильно зависят от типа заместителя. Напротив, направление замещения мало чувствительно к типу заместителя.
сильно дезактивируют), а электроноакцепторные группы повышают скорость реакции. Относительные константы скорости [189], приведенные в табл. 3.21, показывают, что, как и в случае электрофильного ароматического замещения, эти величины очень сильно зависят от типа заместителя. Напротив, направление замещения мало чувствительно к типу заместителя.
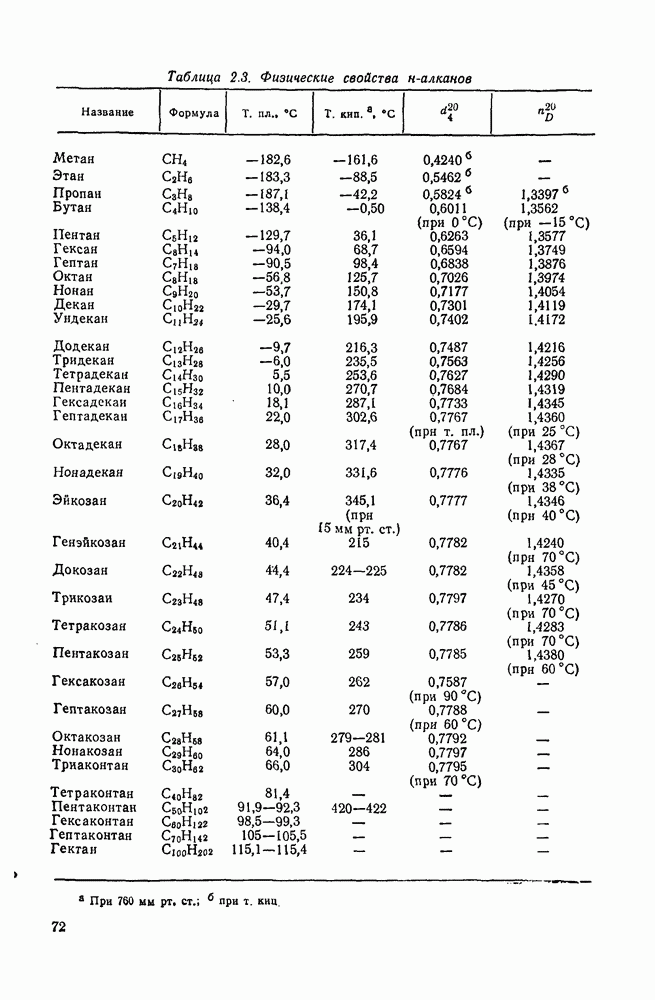
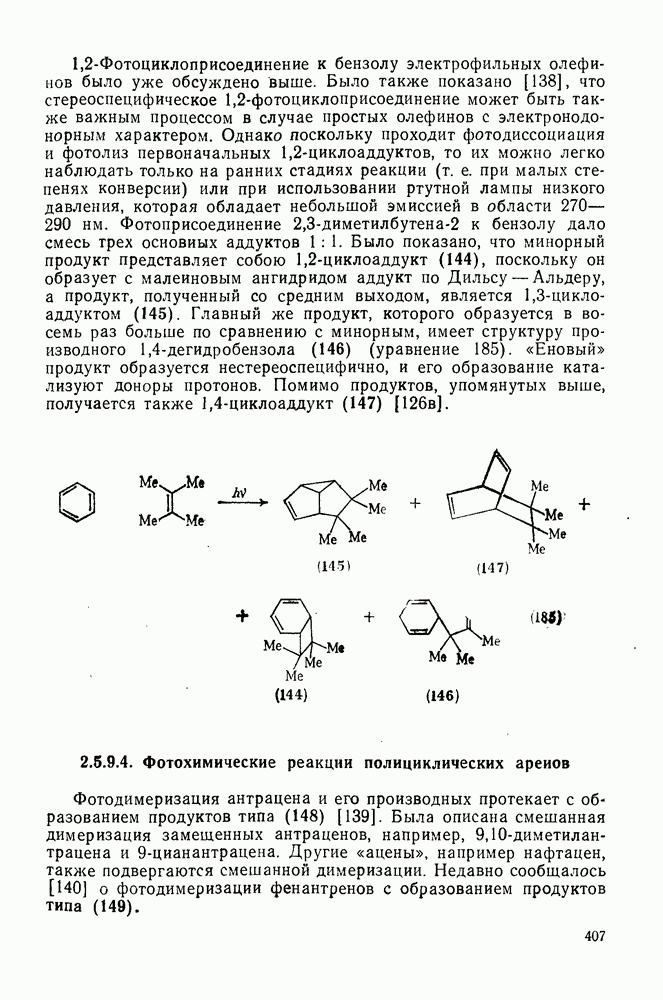
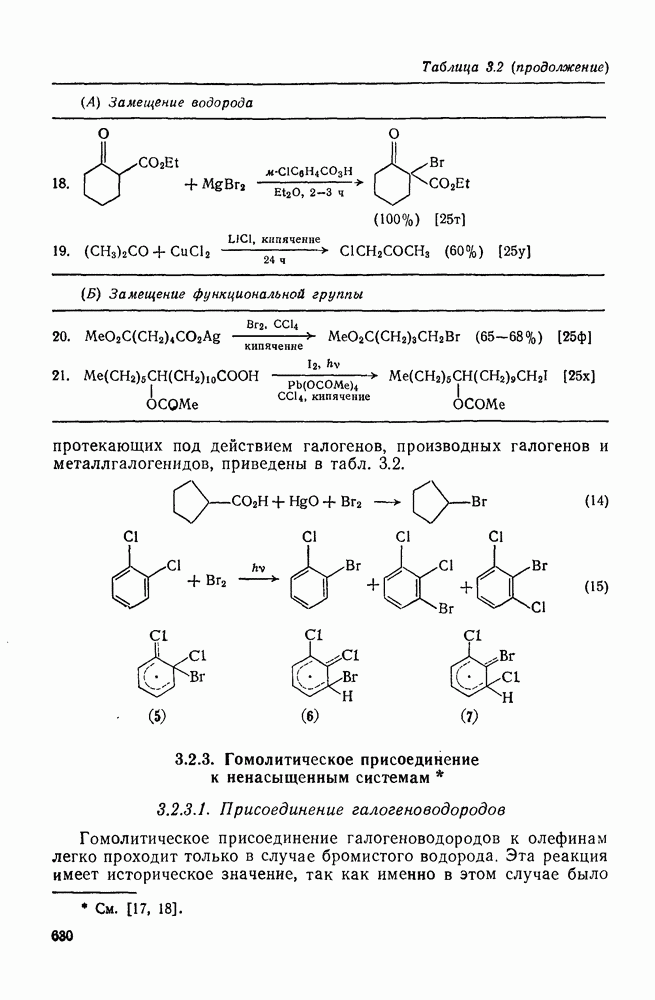
Таблица 3.21. Относительные константы скорости реакции  с
с  в
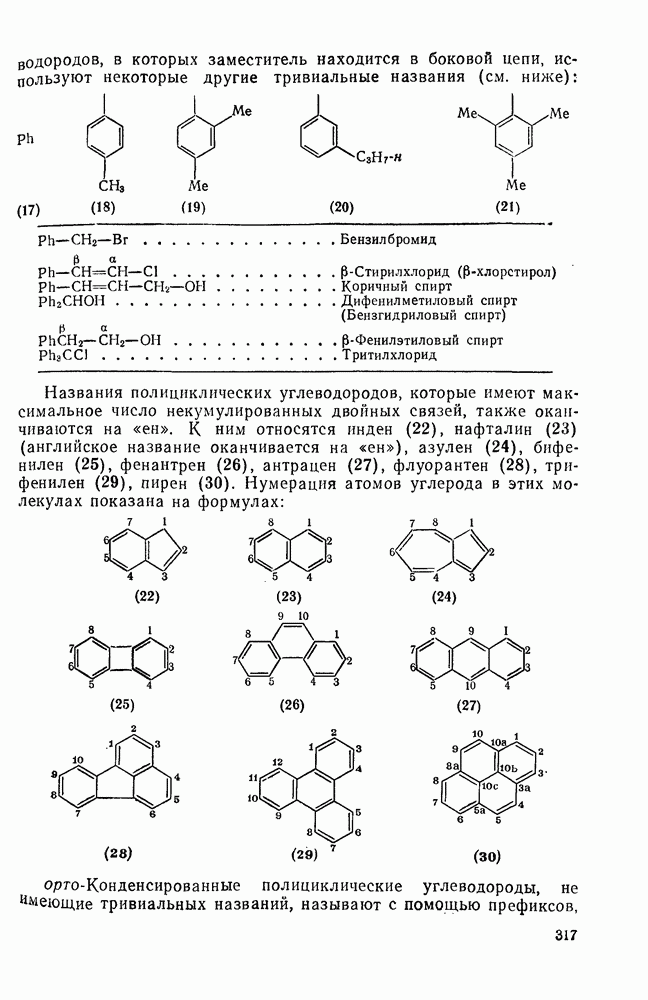
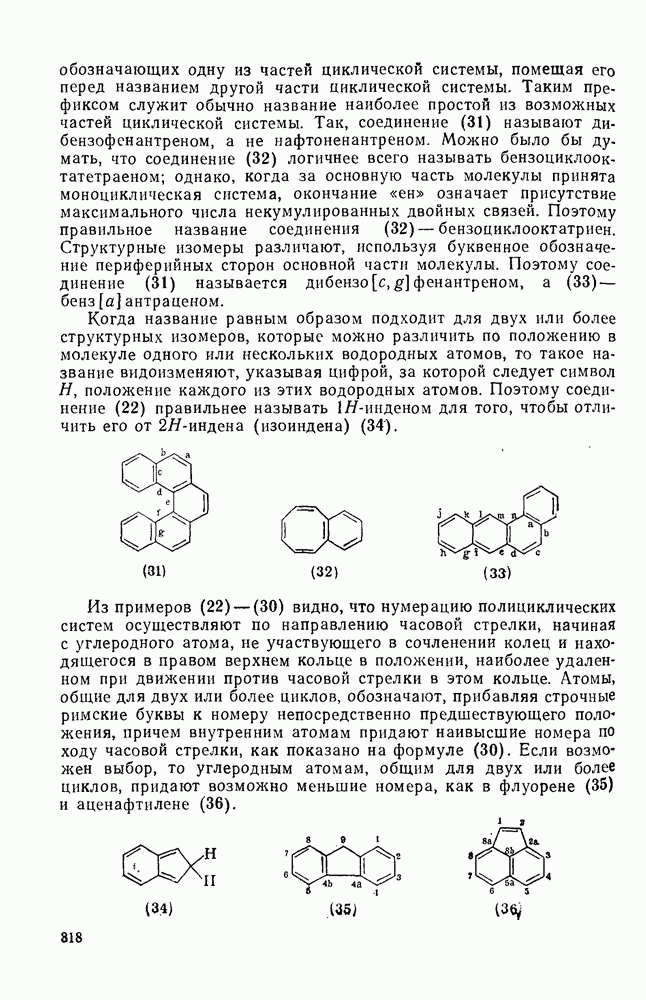
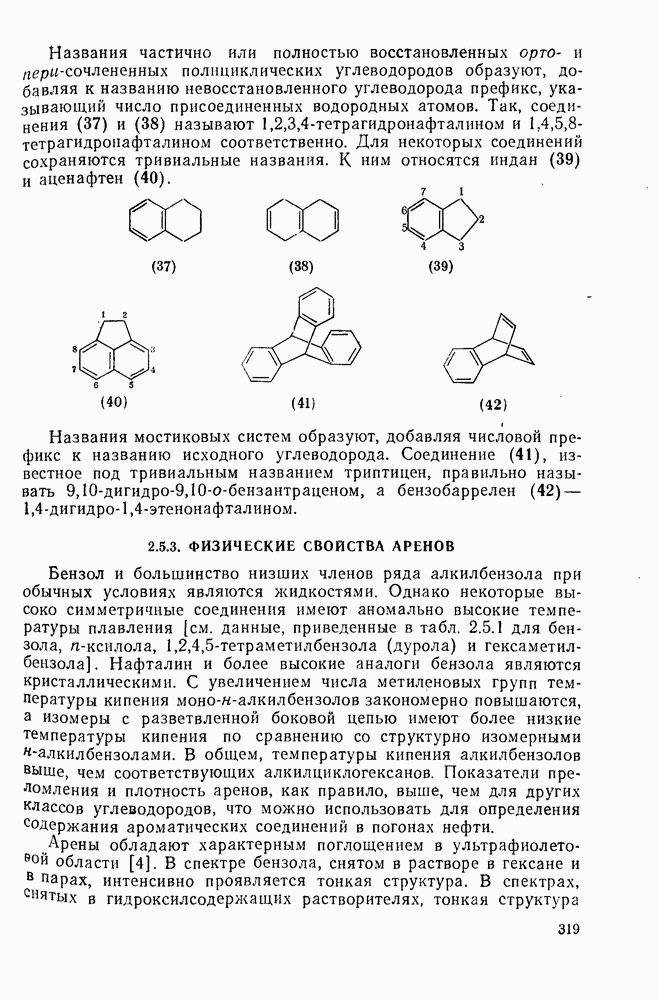
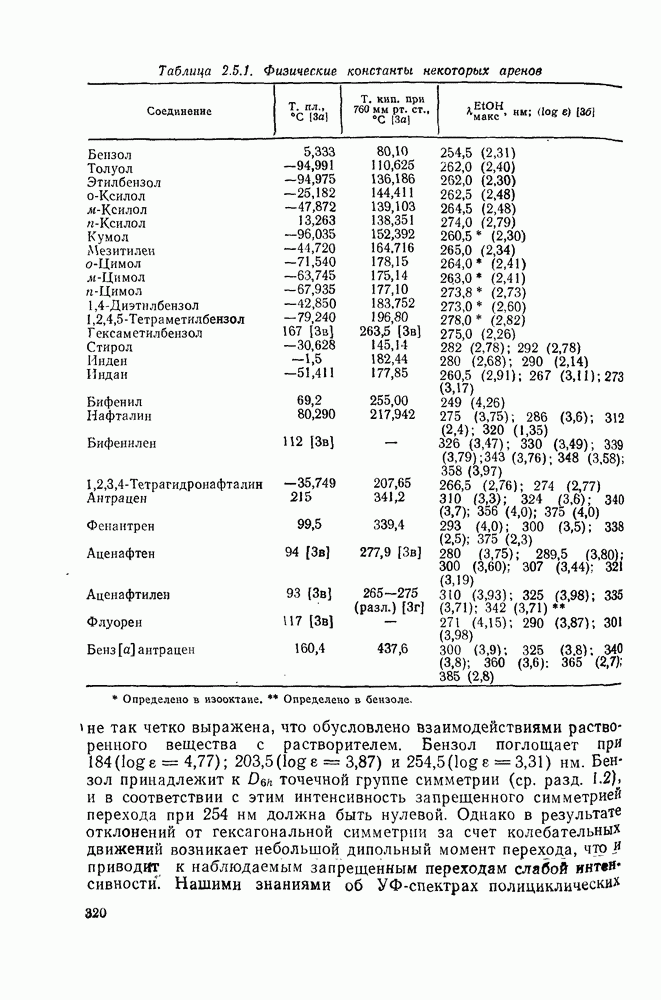
в  -диметилацетамиде при
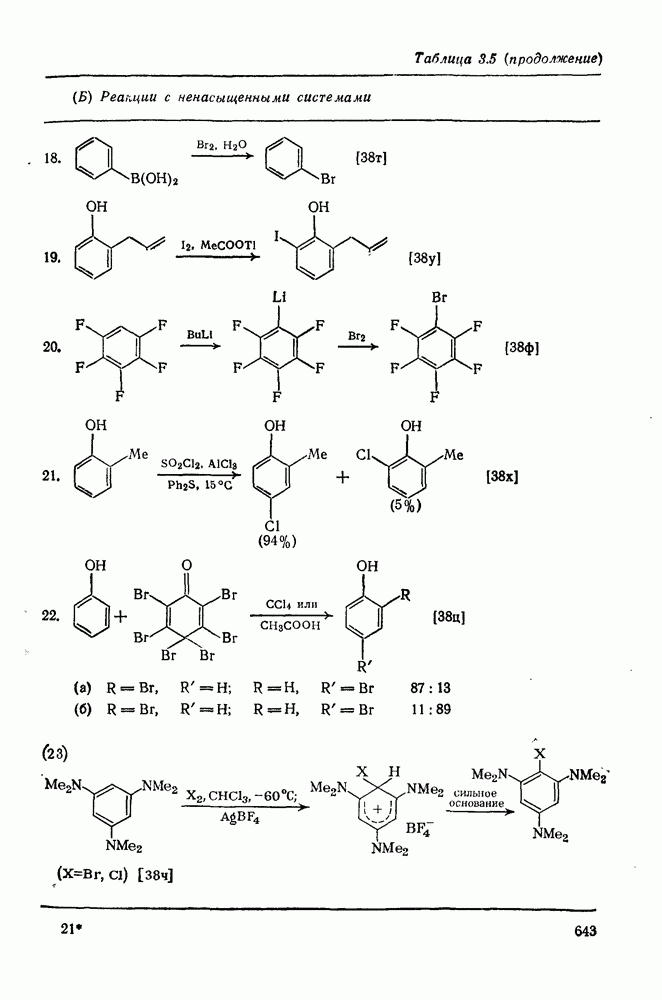
-диметилацетамиде при 


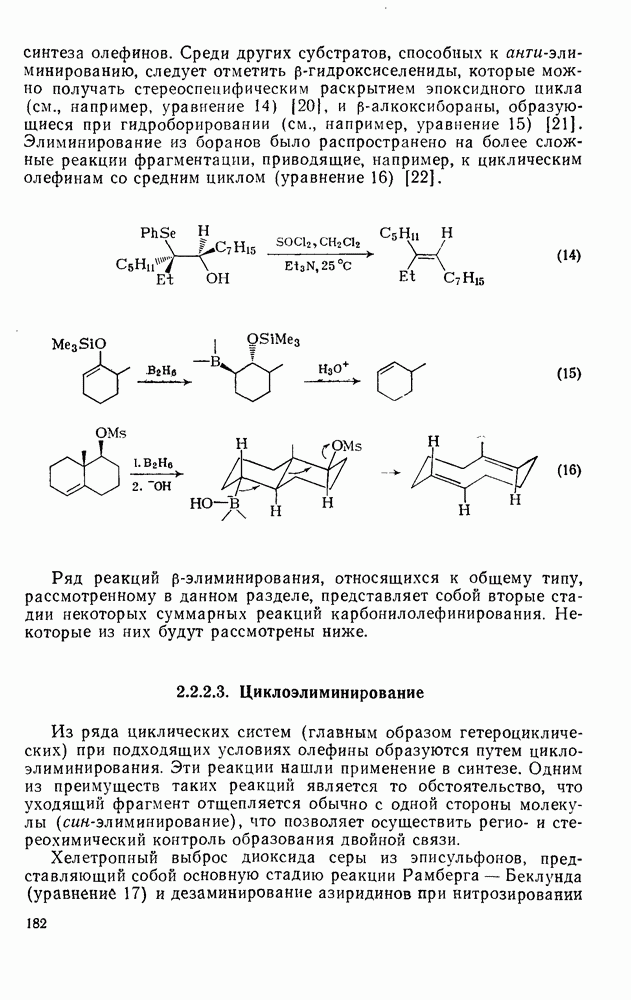
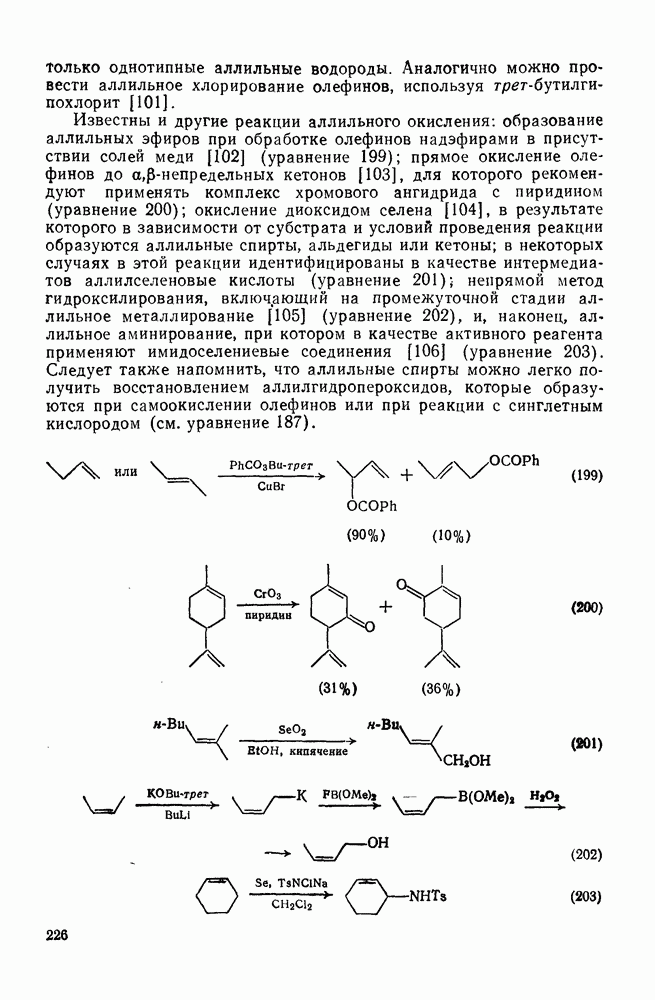
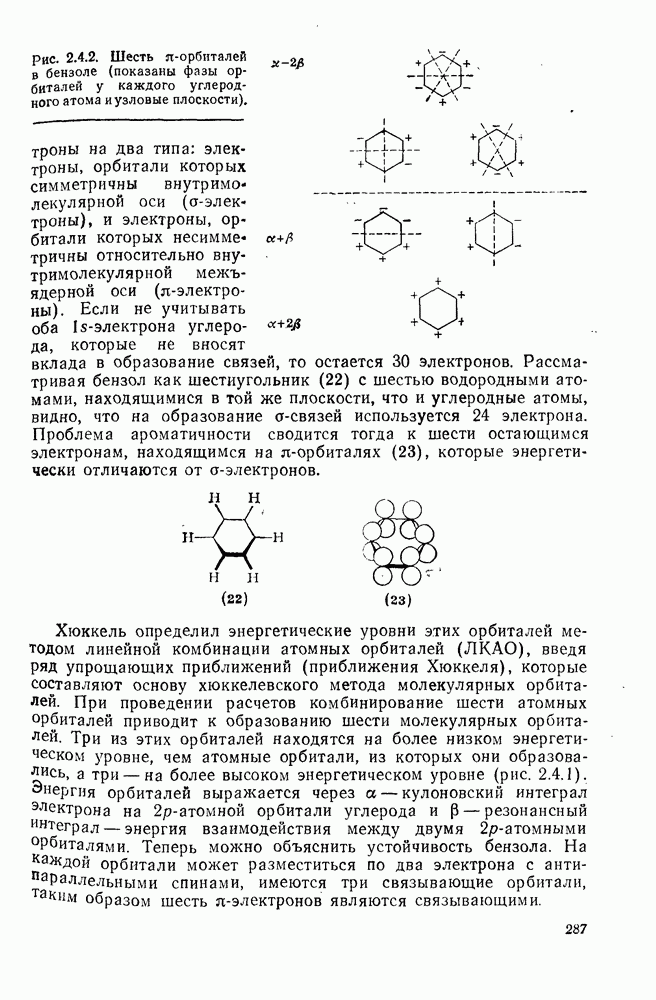
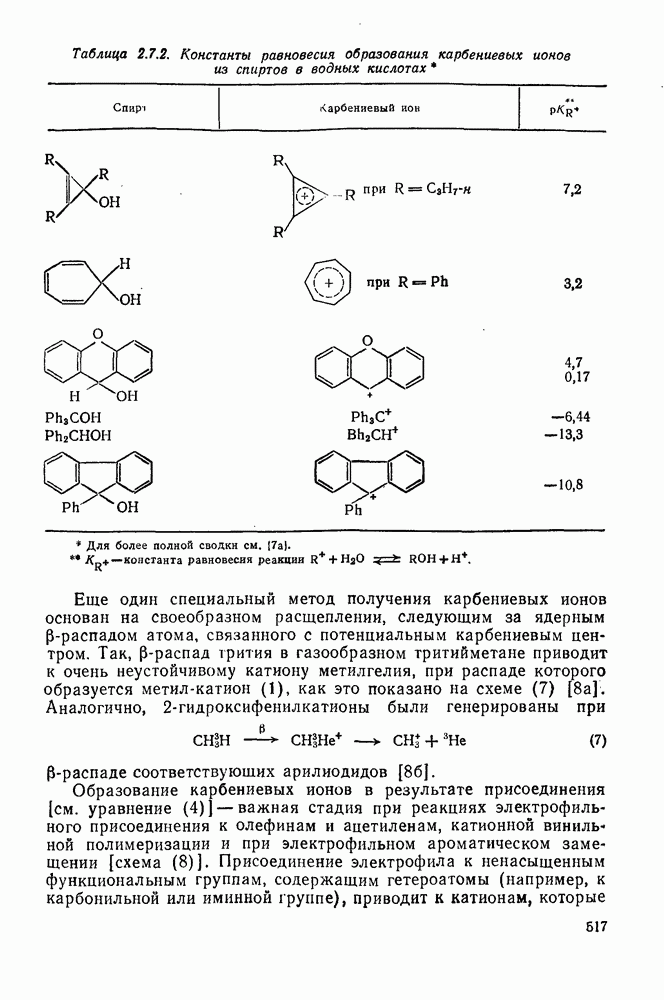
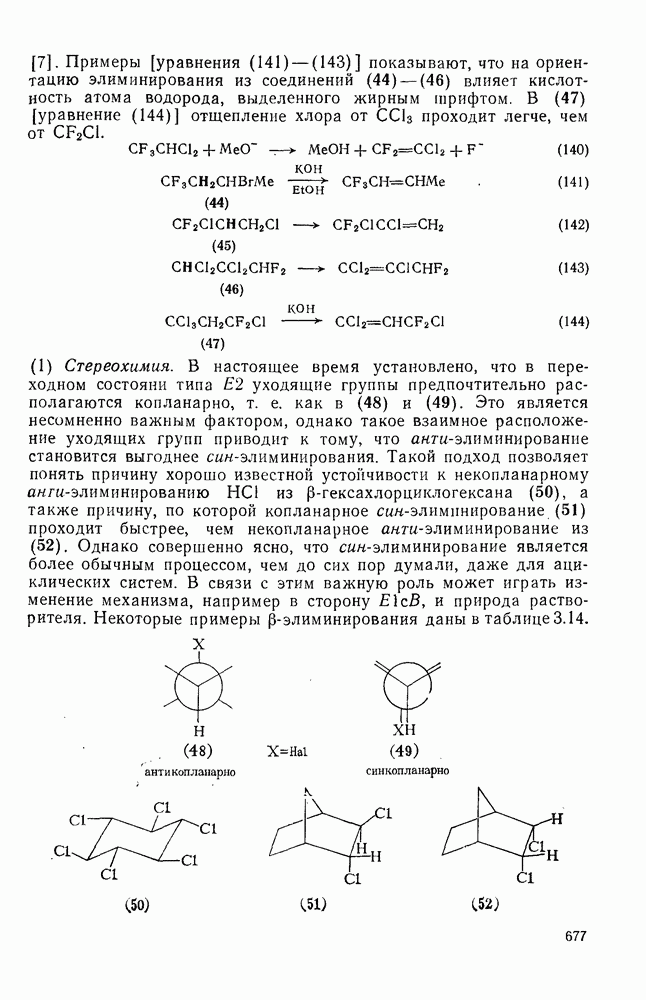
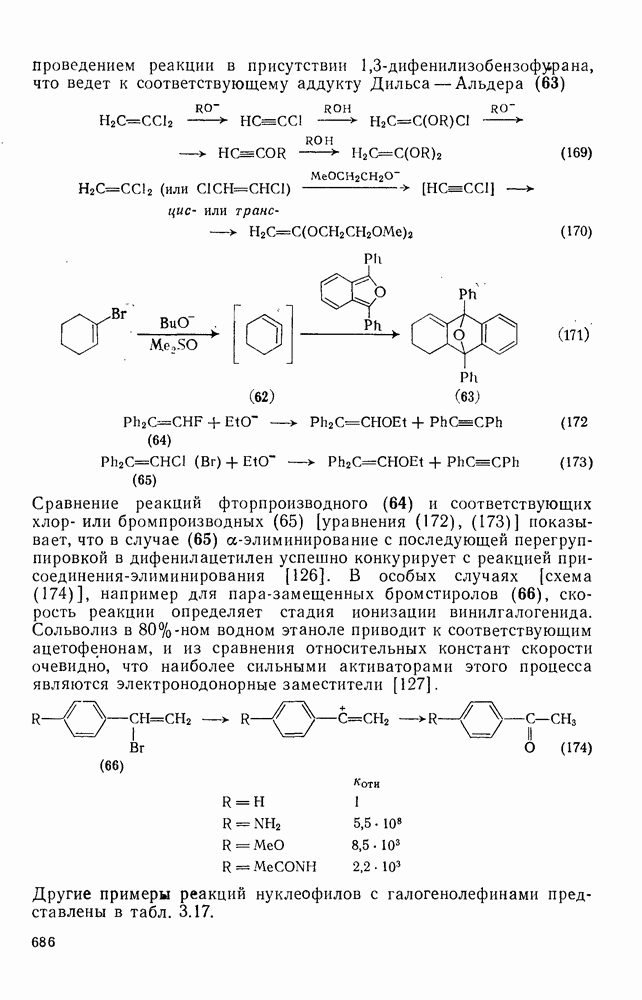
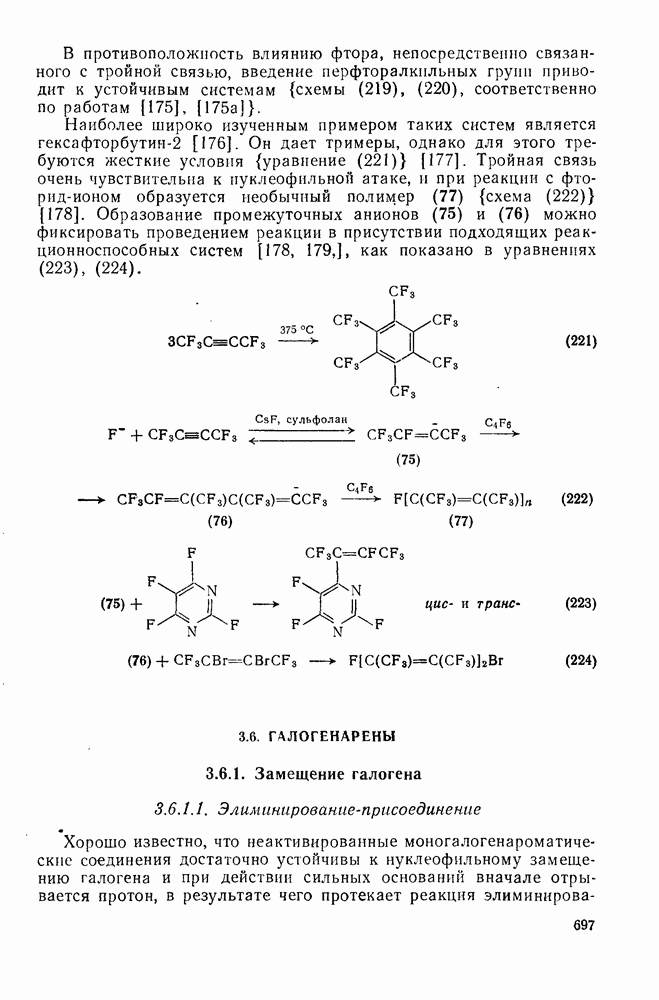
Причиной необычного ориентирующего влияния галогенов является различие в активирующем влиянии атомов фтора, находящихся в орто-, пара- и мета-положении относительно места нуклеофильной атаки.

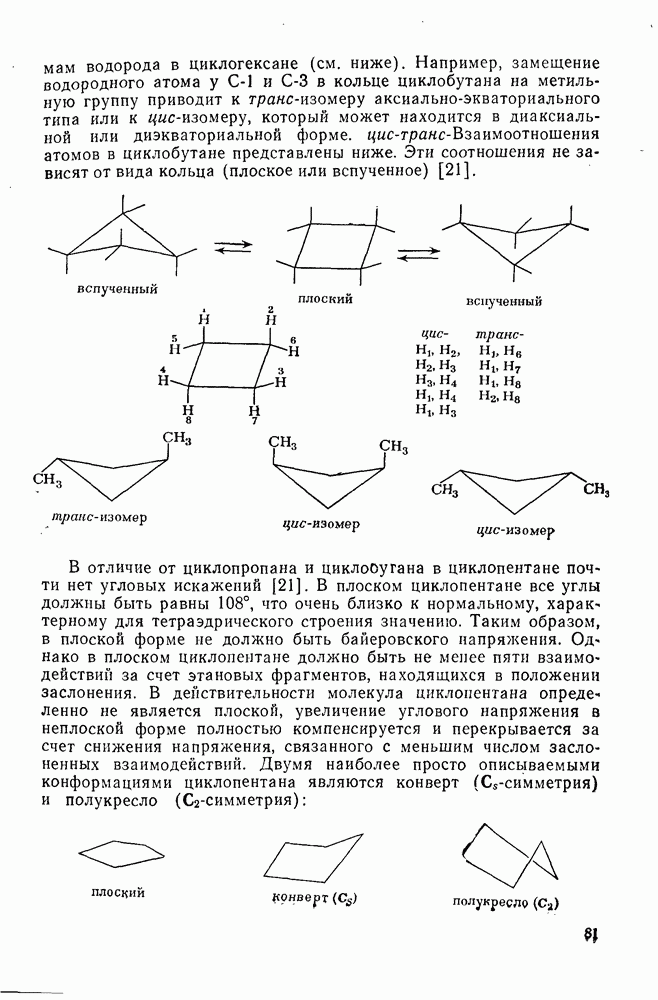
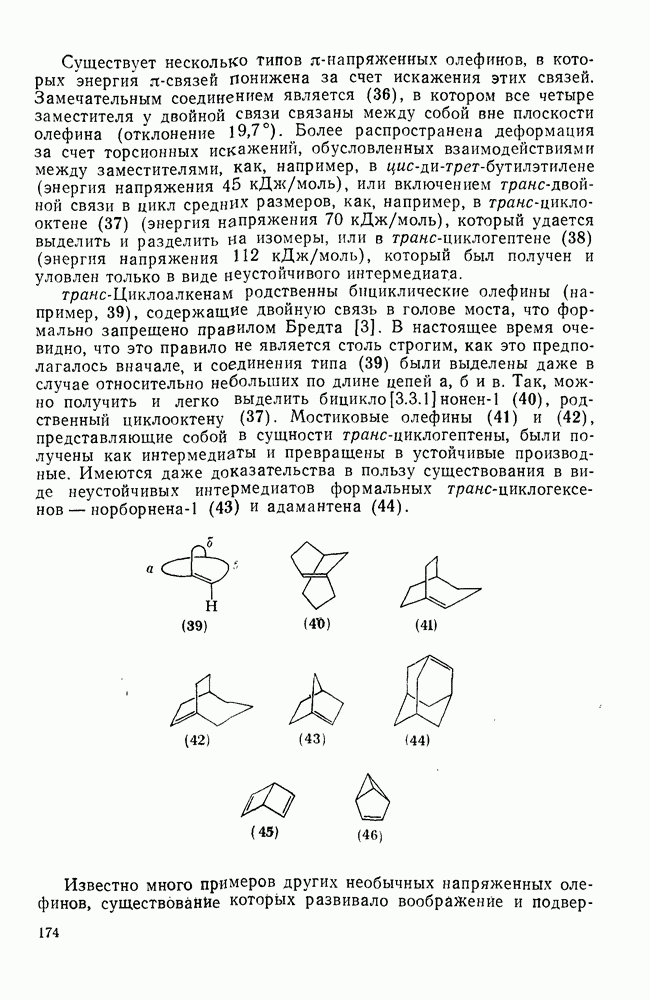
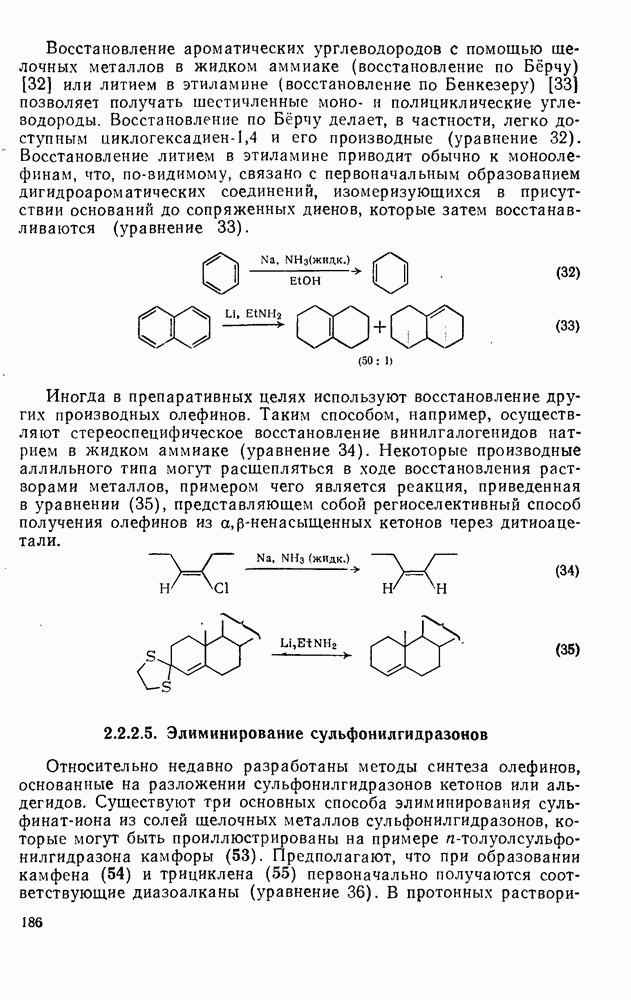
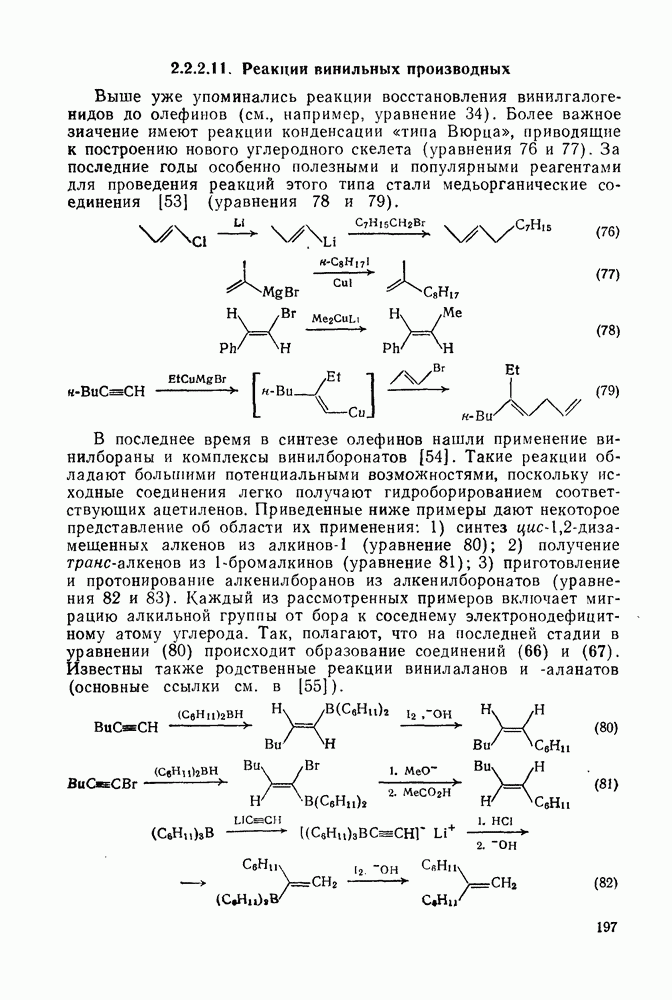
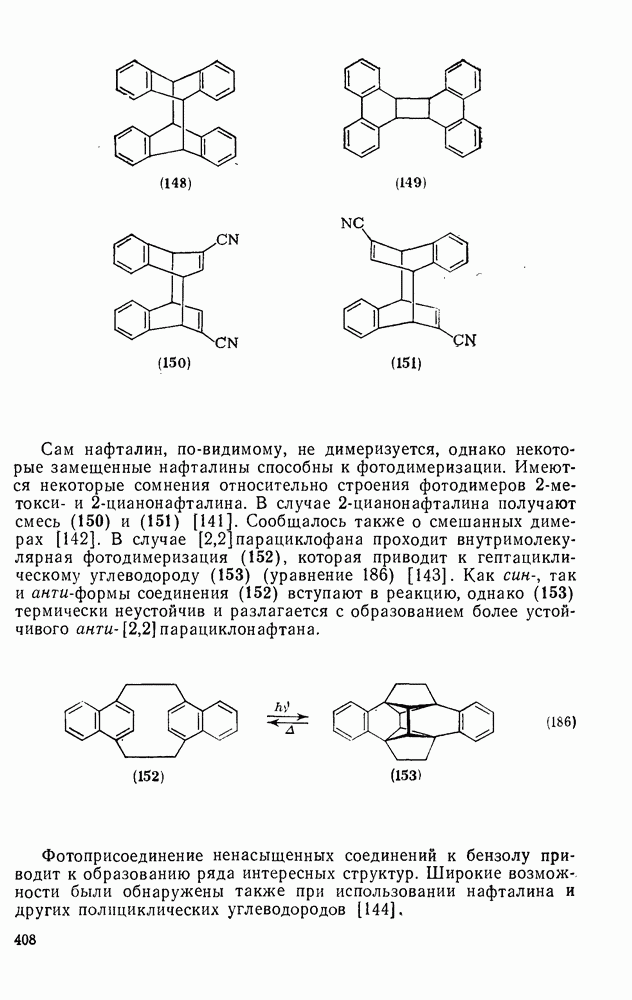
Такого рода различия в активирующем влиянии были установлены для реакций с метилат-ионом в метаноле [186]; влияние атома фтора в пара-положении к реакционному центру мало чем отличается от влияния атома водорода в том же положении, однако атомы фтора в мета- и орто-положениях очень сильно активируют реакцию. Предложенное объяснение этих эффектов основано на принятии для переходного состояния на стадии, определяющей скорость процесса, модели типа (78) [185, 186]. В этом случае становится понятным активирующее влияние фтора в лега-положе-нии, где он оказывается соседним с центром максимальной дело-кализации заряда в кольце [79]. Аналогично, малое влияние фтора в пара-положении является следствием конкуренции между индуктивным эффектом и отталкиванием электронных пар в (80). Однако принятие в качестве модели переходного состояния структуры (786) приводит к трудностям при объяснении сильного активирующего влияния фтора в орго-положении, поскольку модель подразумевает сходное влияние фтора, находящегося как в орто-, так и в пара-положениях. Можно предположить, что атом фтора в орто-положении дополнительно активирует молекулу за счет усиления электрофильности углеродного атома, участвующего в реакции, при атаке первоначальной структуры (78а). Тем не менее, независимо от причины активирующего влияния фтора, очевидно, что нуклеофильная атака в  будет приводить, главным образом, к замещению в пара-положение к заместителю X, поскольку именно в этом положении активирующее влияние атомов фтора будет проявляться в максимальной степени.
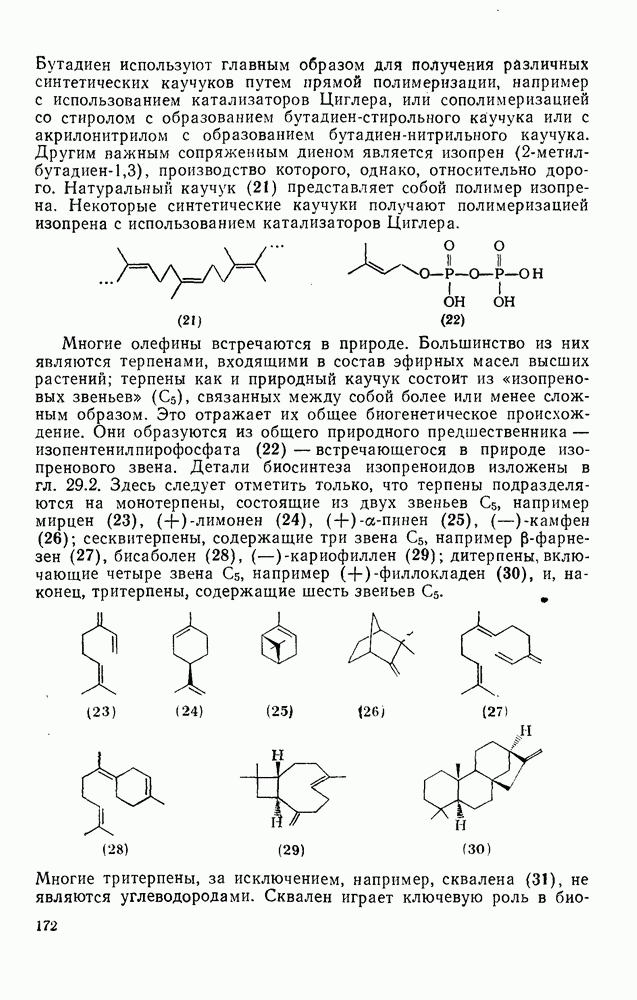
будет приводить, главным образом, к замещению в пара-положение к заместителю X, поскольку именно в этом положении активирующее влияние атомов фтора будет проявляться в максимальной степени.

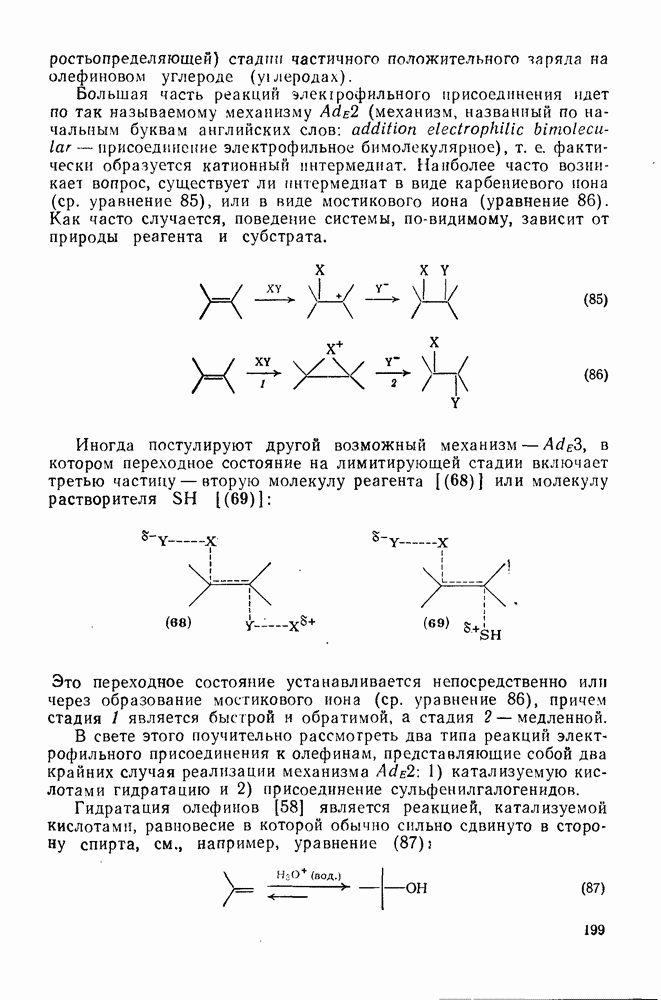
Ориентация замещения в полихлорированных соединениях [12] обычно менее селективна, чем в полифторированных системах, это связано с тем, что атом хлора в пара-положении является достаточно активным ориентантом (см. табл. 3.21). Соответственно различия в активирующем влиянии атомов хлора в  на орто-, мета- и пара-положения к заместителю X существенно меньше, чем в случае
на орто-, мета- и пара-положения к заместителю X существенно меньше, чем в случае  .
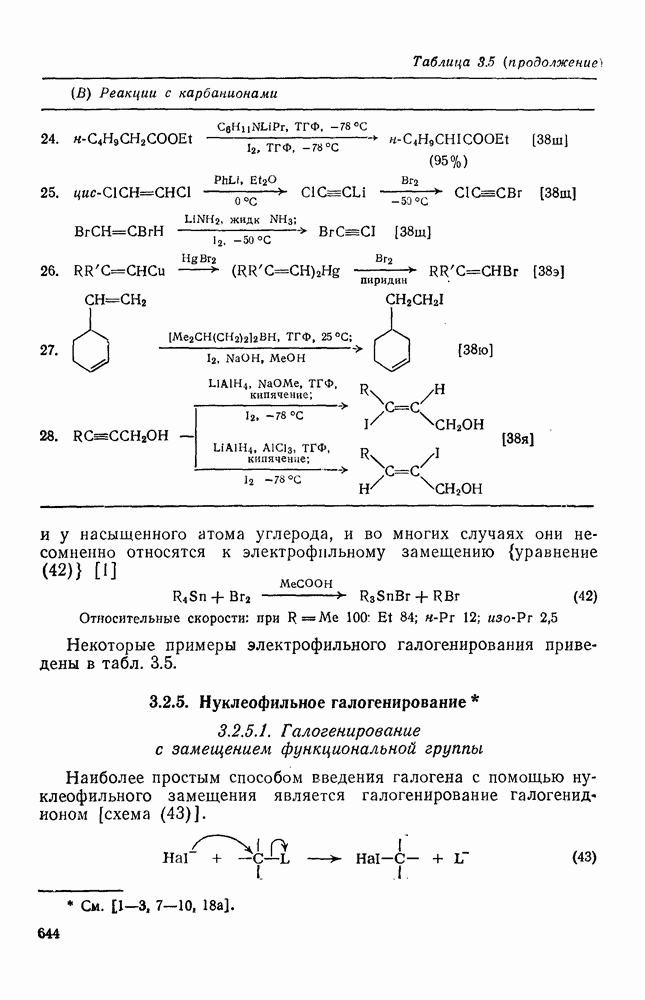
.
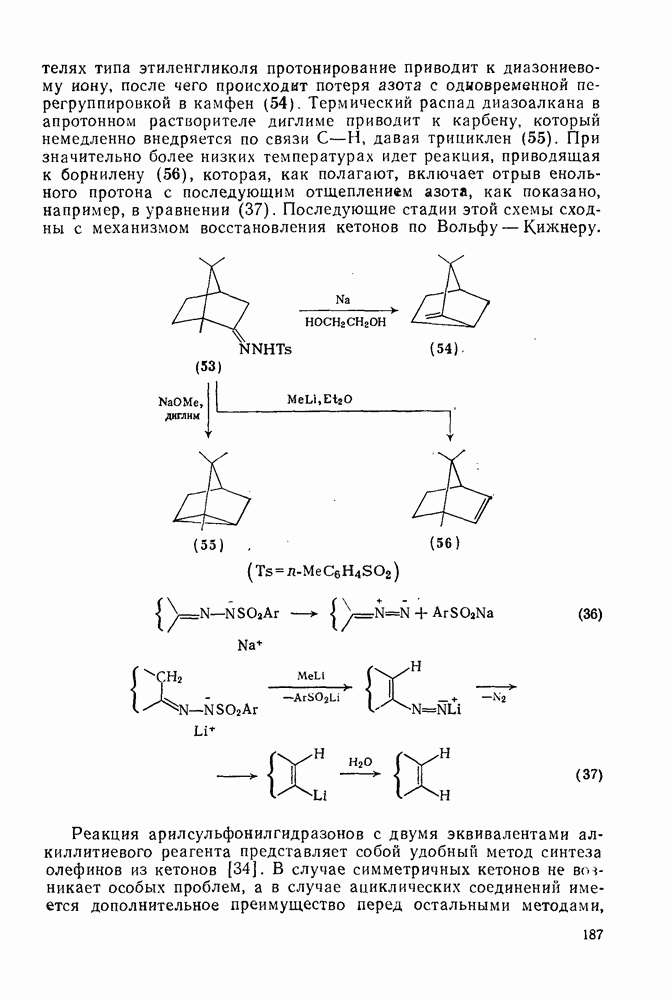
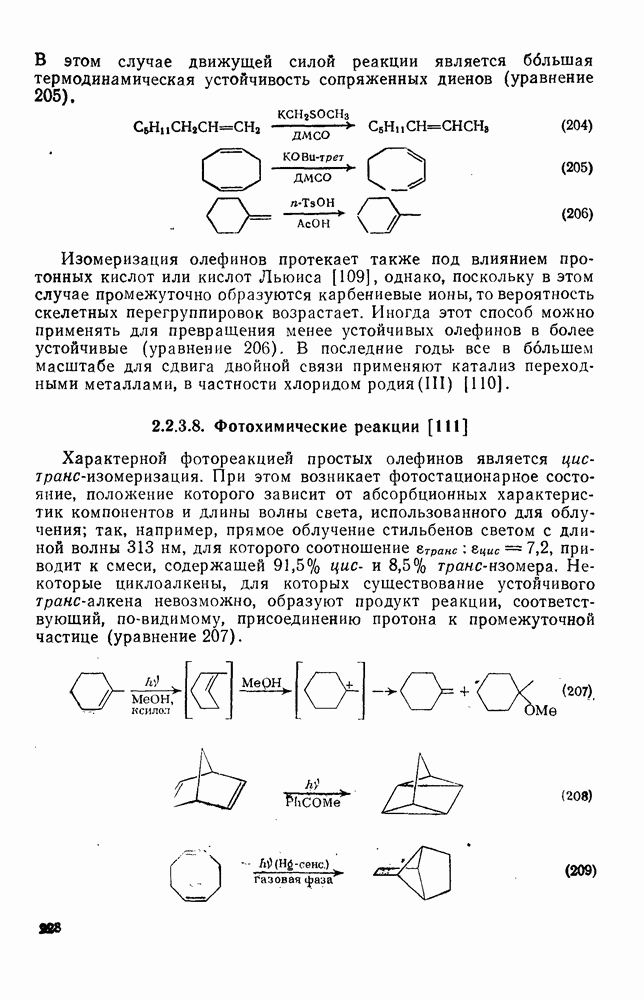
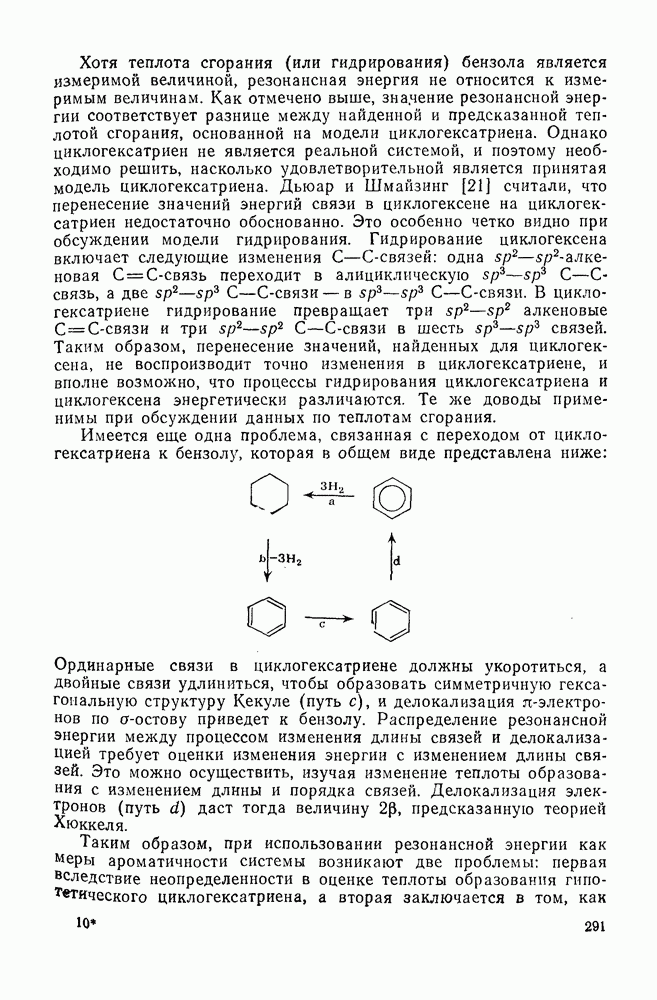
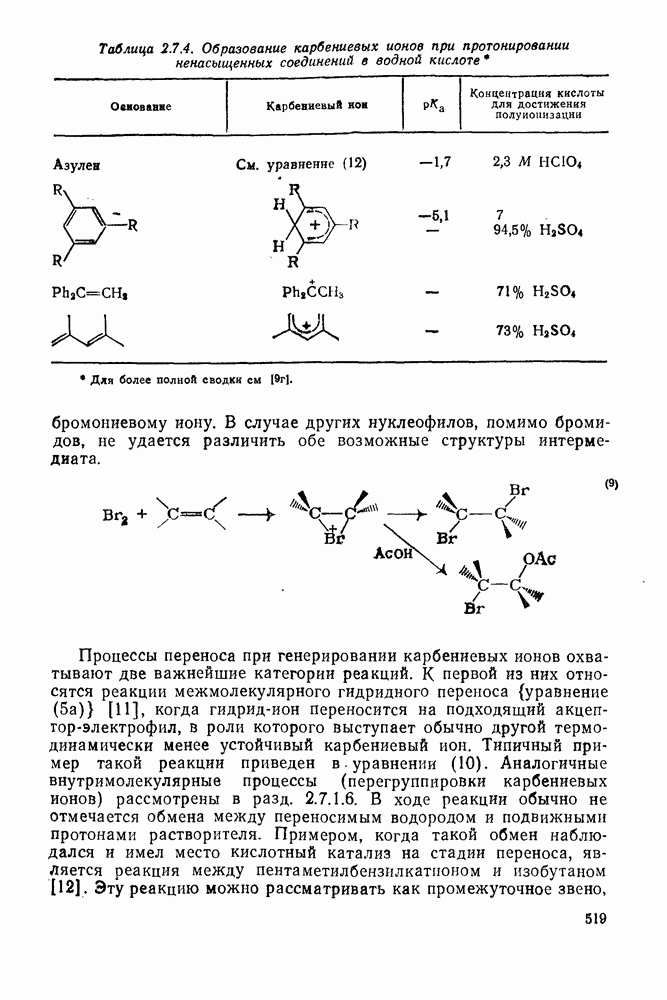
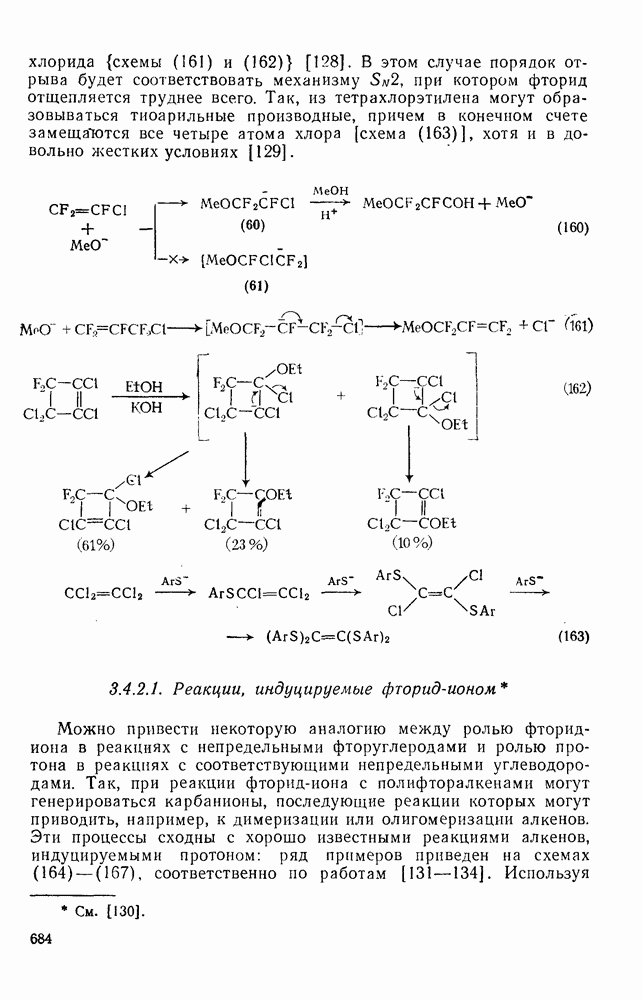
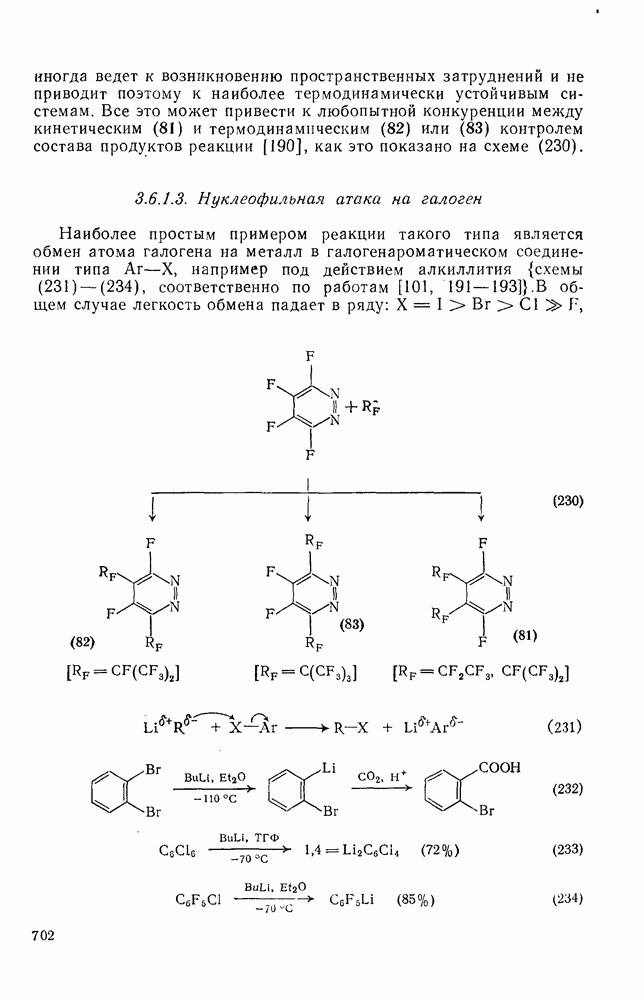
Реакции активированных фторароматических соединений с перфторалкильными анионами, генерированными взаимодействием фтор-ионов с перфторалкенами, можно рассматривать как прототип синтеза Фриделя — Крафтса с участием отрицательно заряженных интермедиатов [135]. Полизамещение вызывает некоторые осложнения по трем причинам: 1) присутствие полифторалкильных групп может в некоторых случаях влиять на направление последующего замещения; 2) некоторые из реакций являются обратимыми и 3) замещение в наиболее активированное положение иногда ведет к возникновению пространственных затруднений и не приводит поэтому к наиболее термодинамически устойчивым системам.
Все это может привести к любопытной конкуренции между кинетическим (81) и термодинамическим (82) или (83) контролем состава продуктов реакции [190], как это показано на схеме (230).