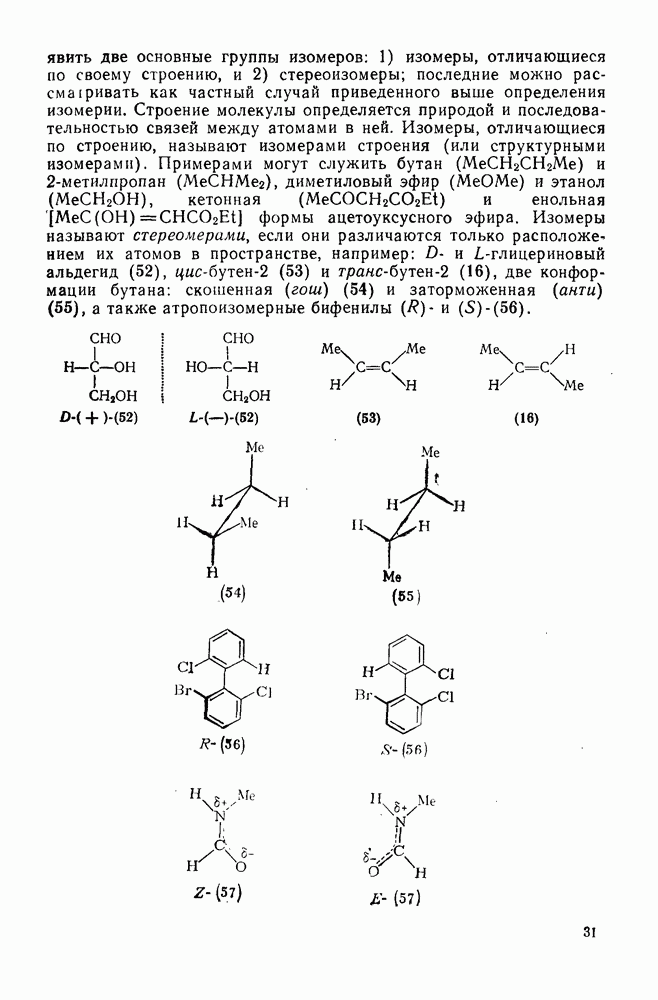
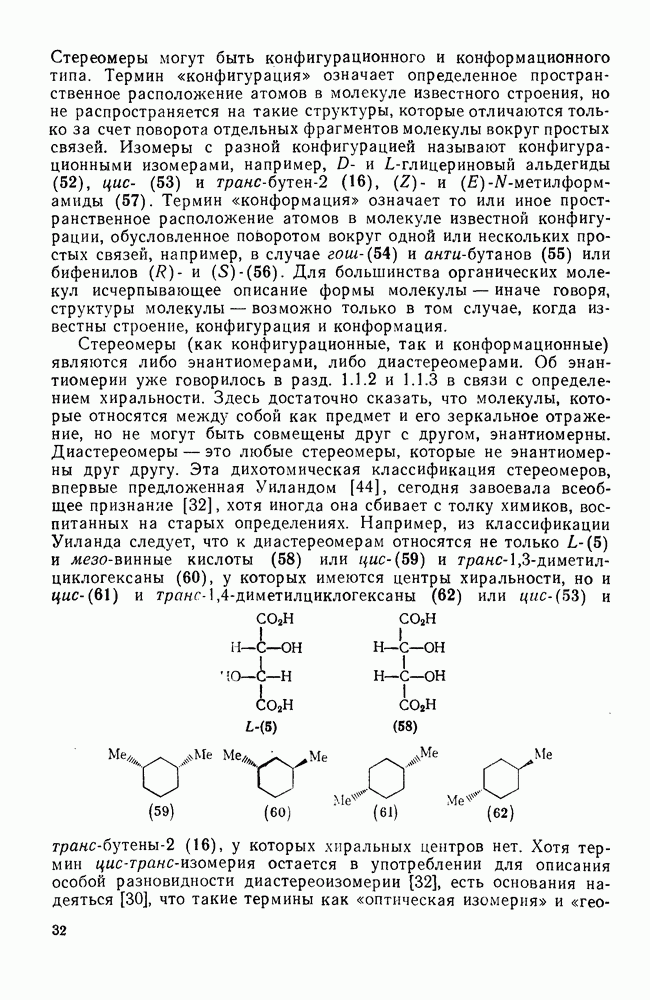
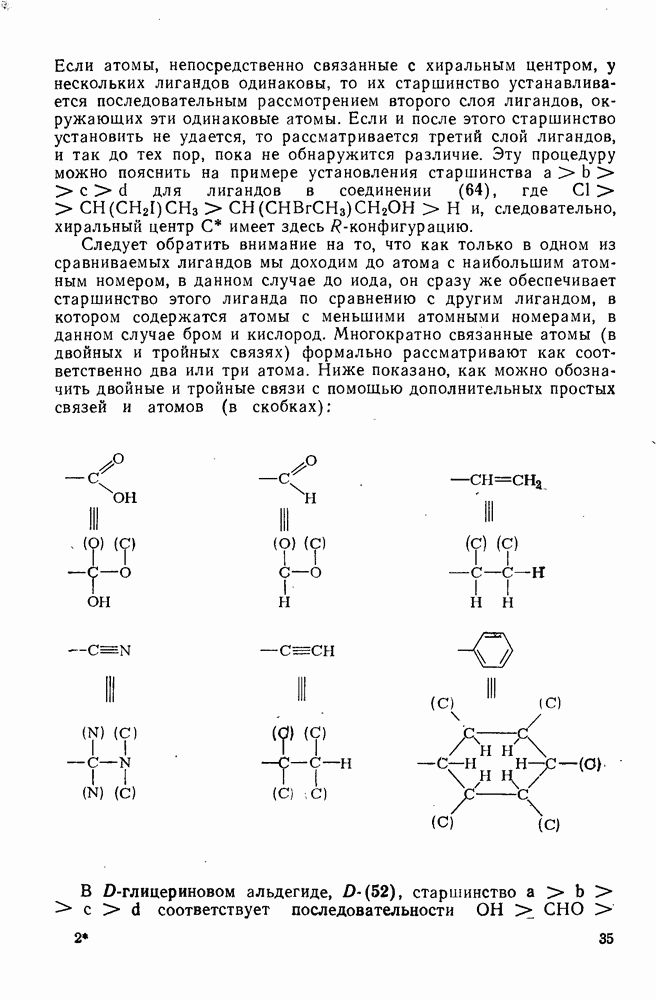
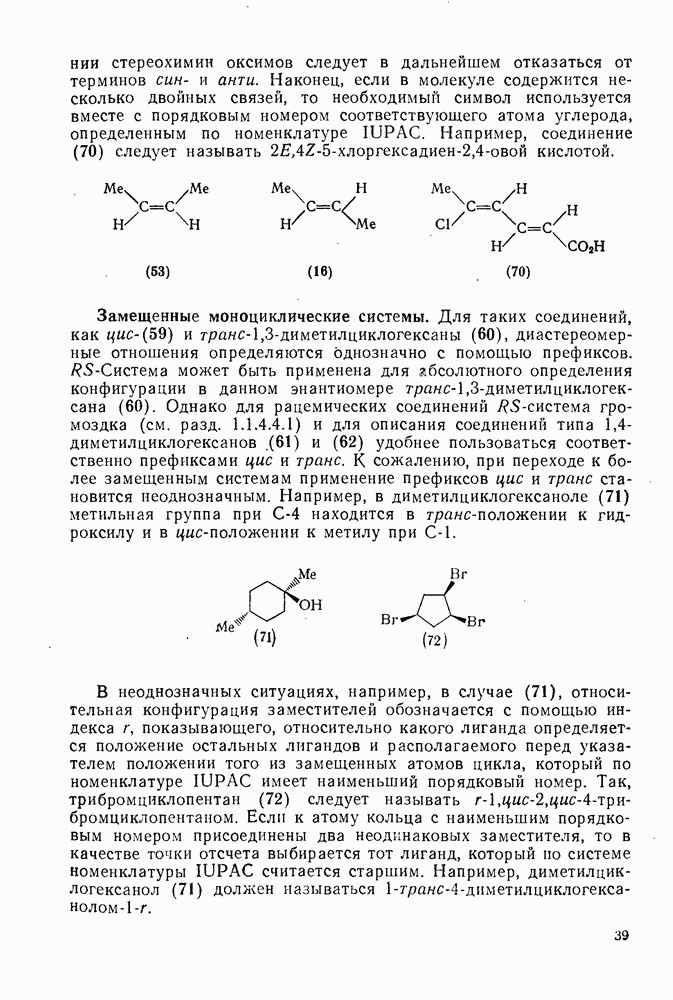
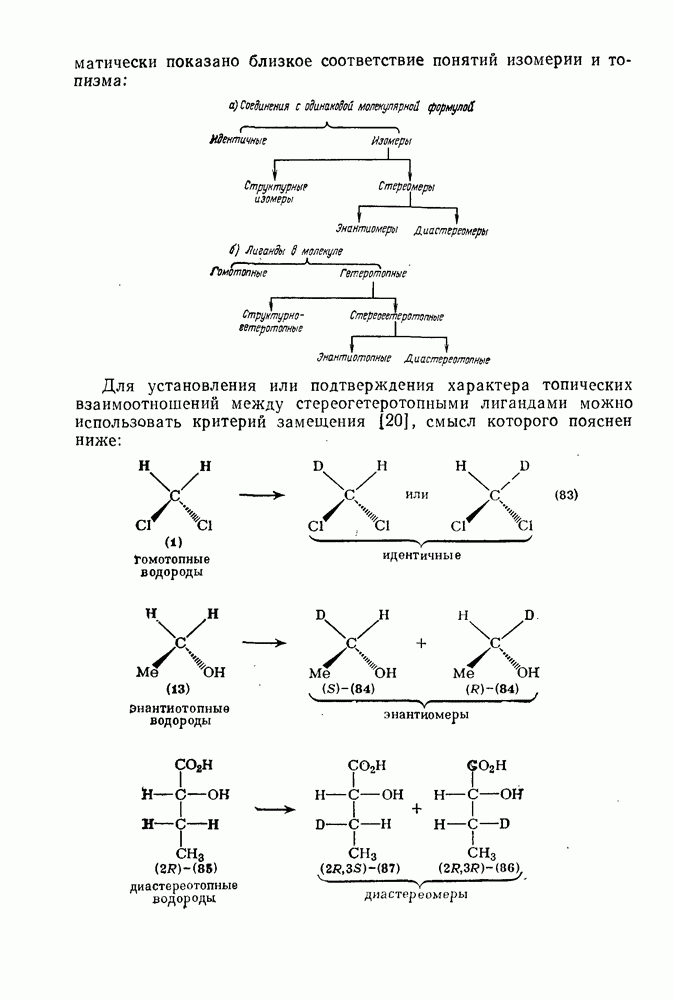
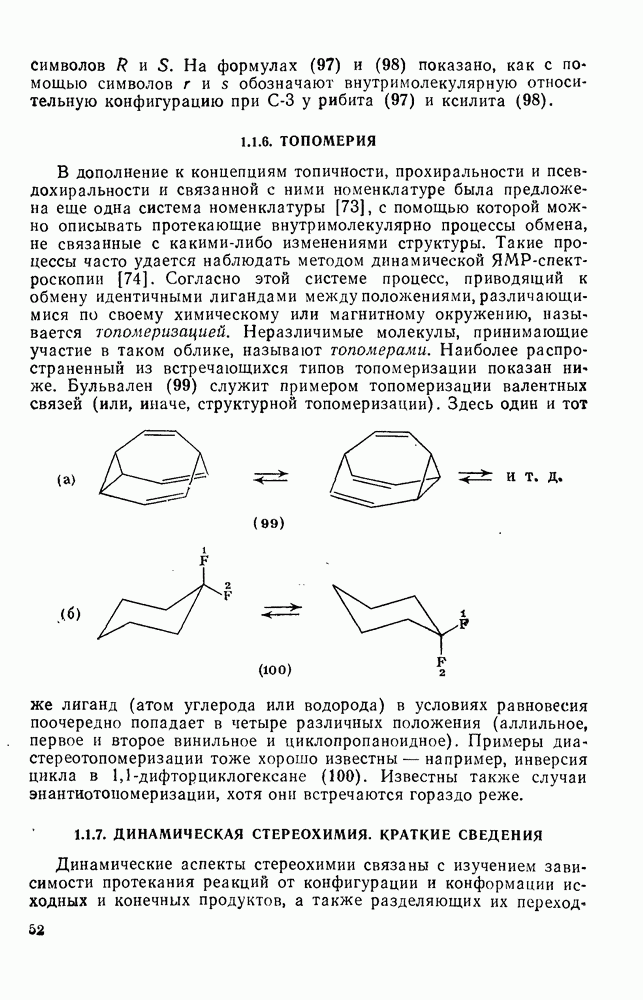
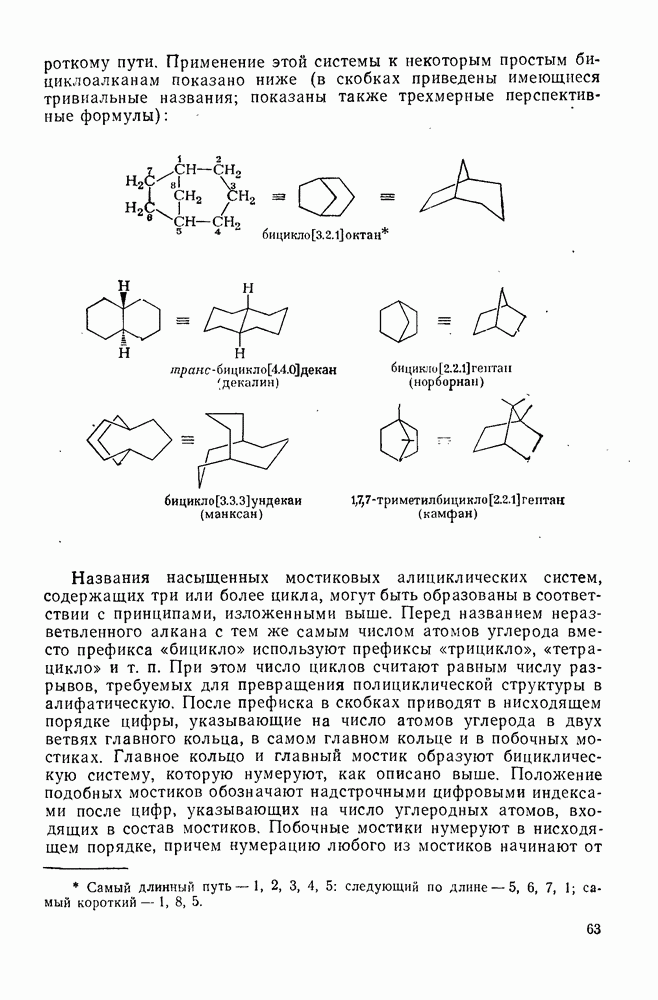
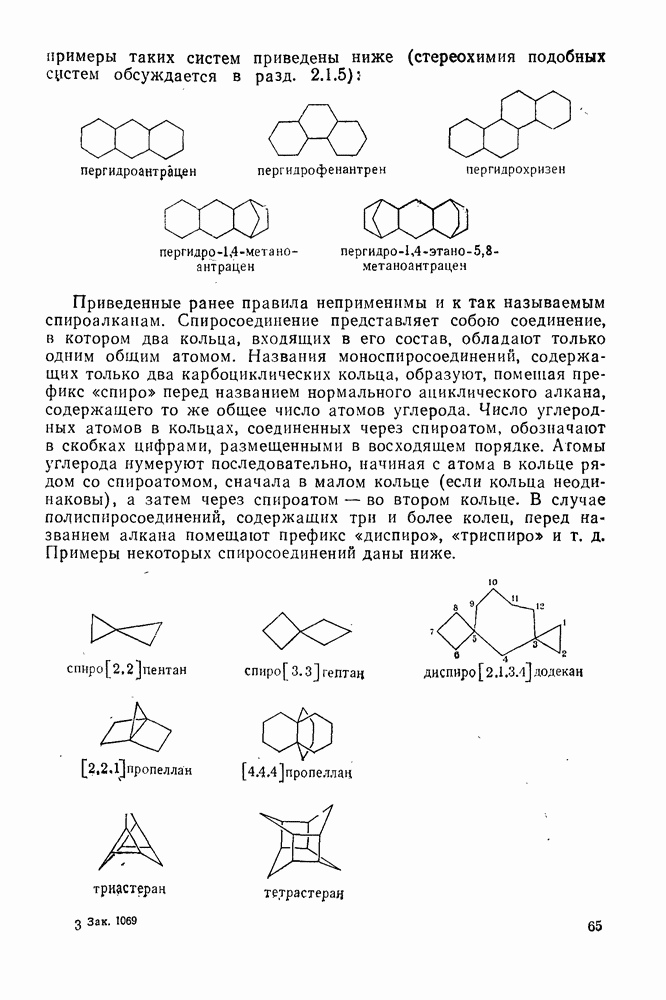
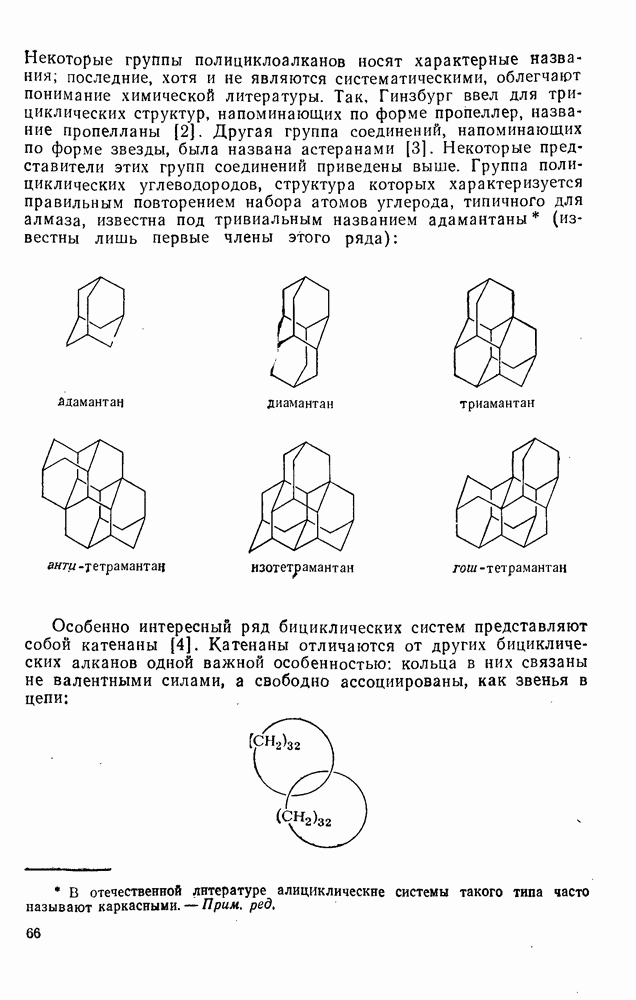
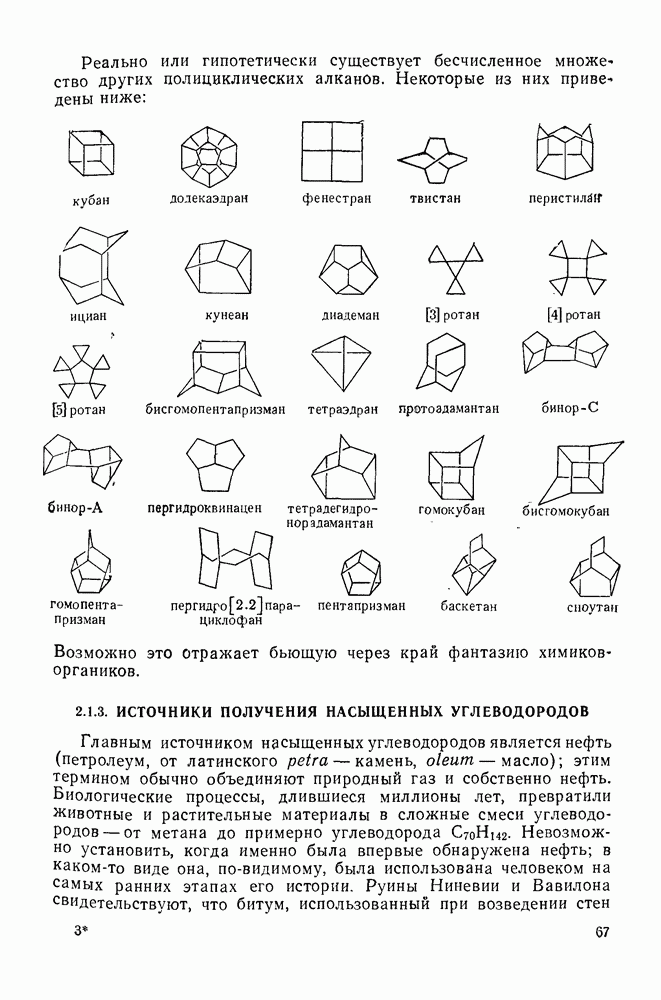
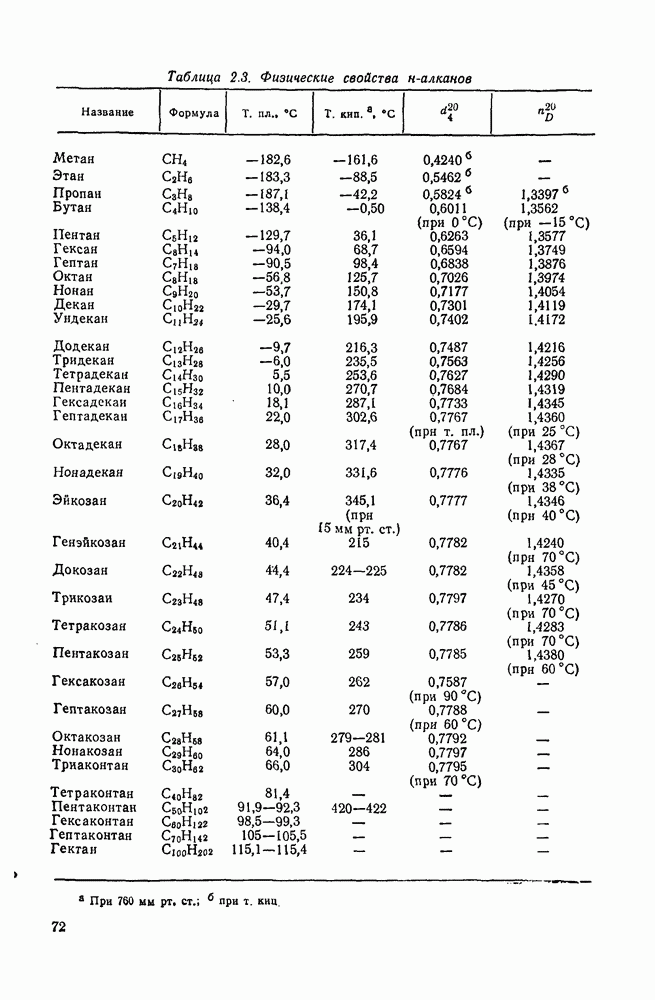
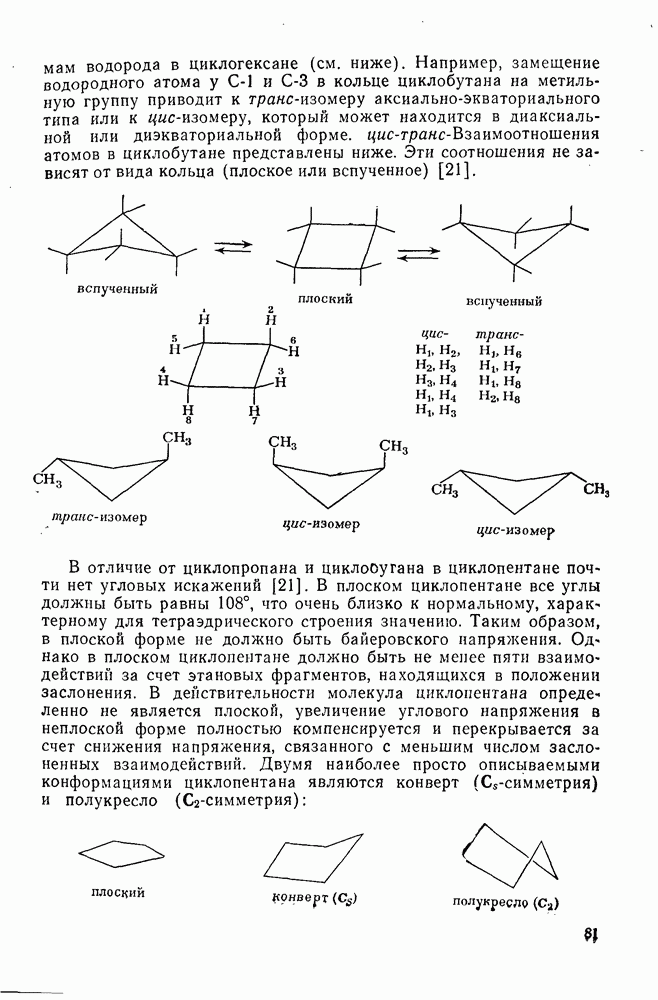
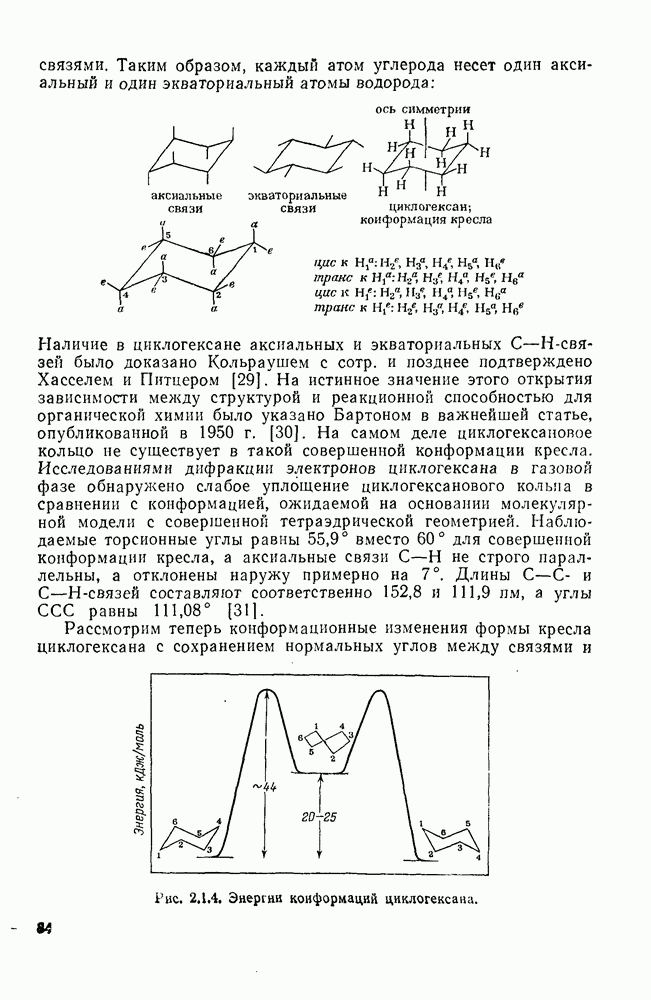
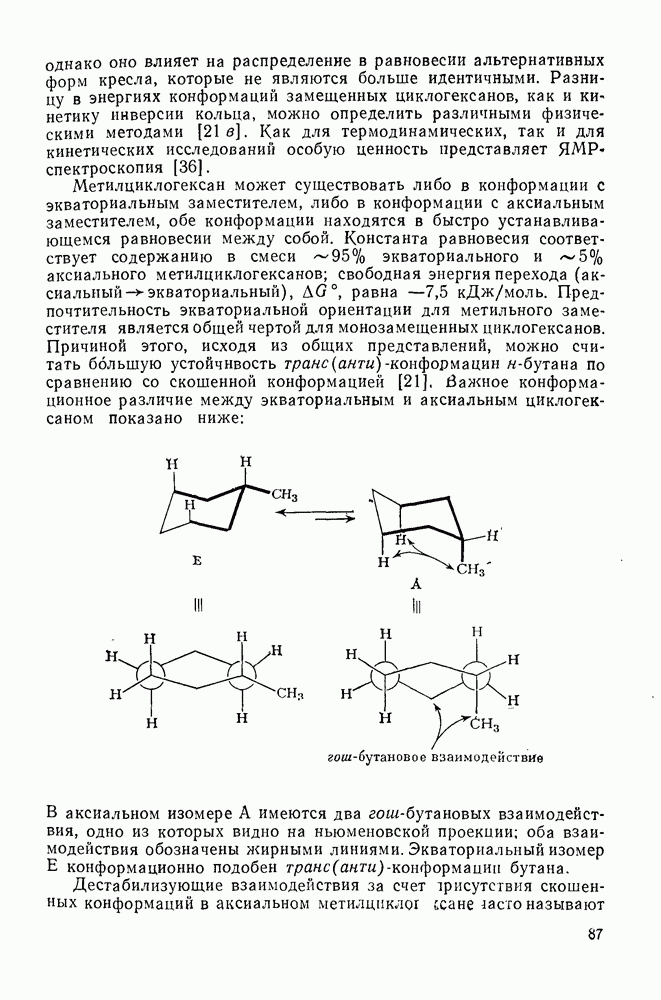
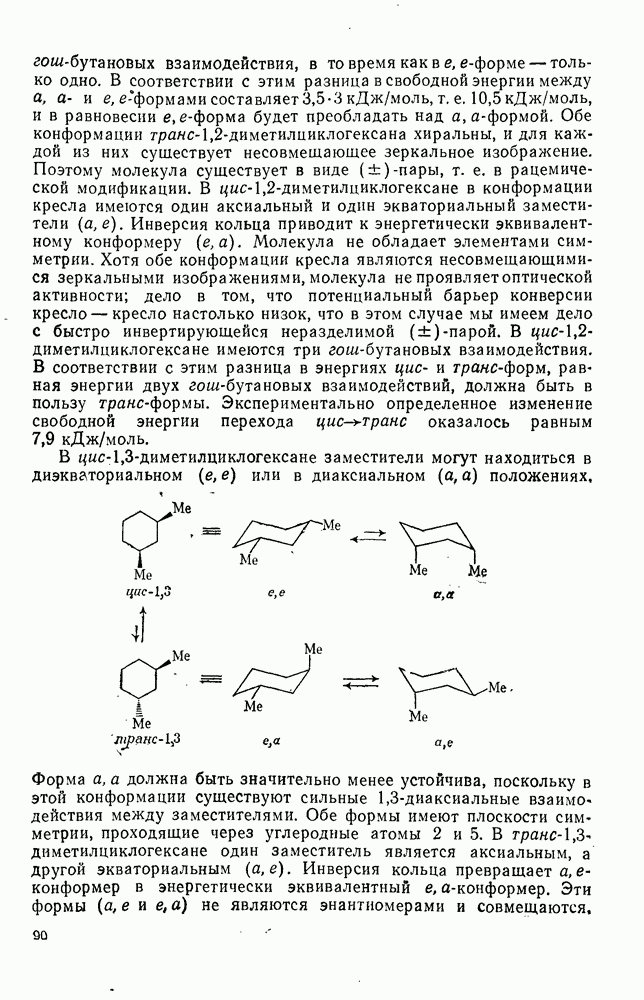
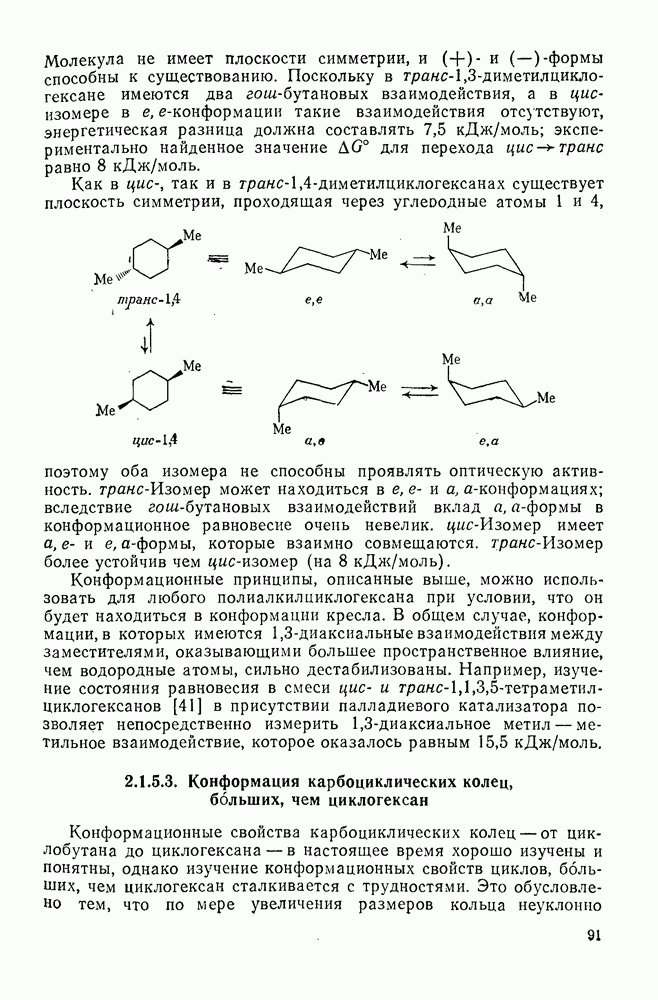
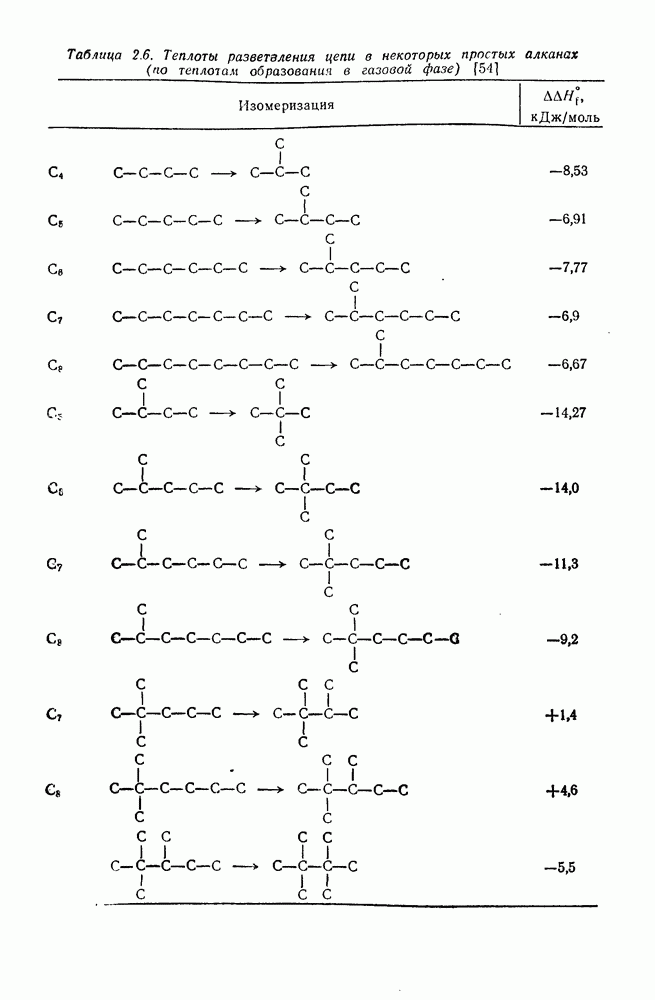
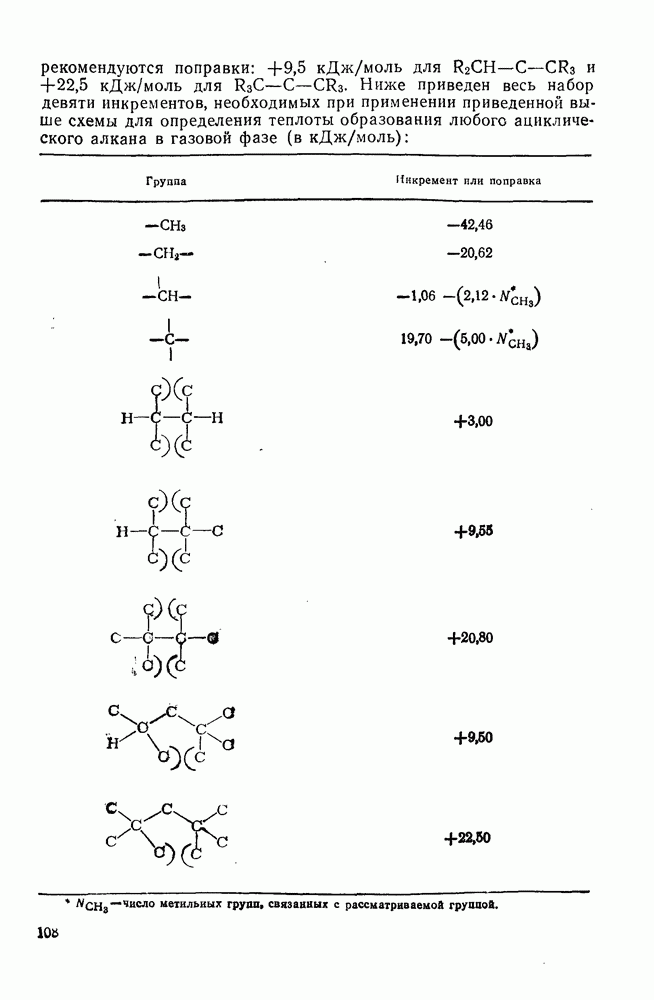
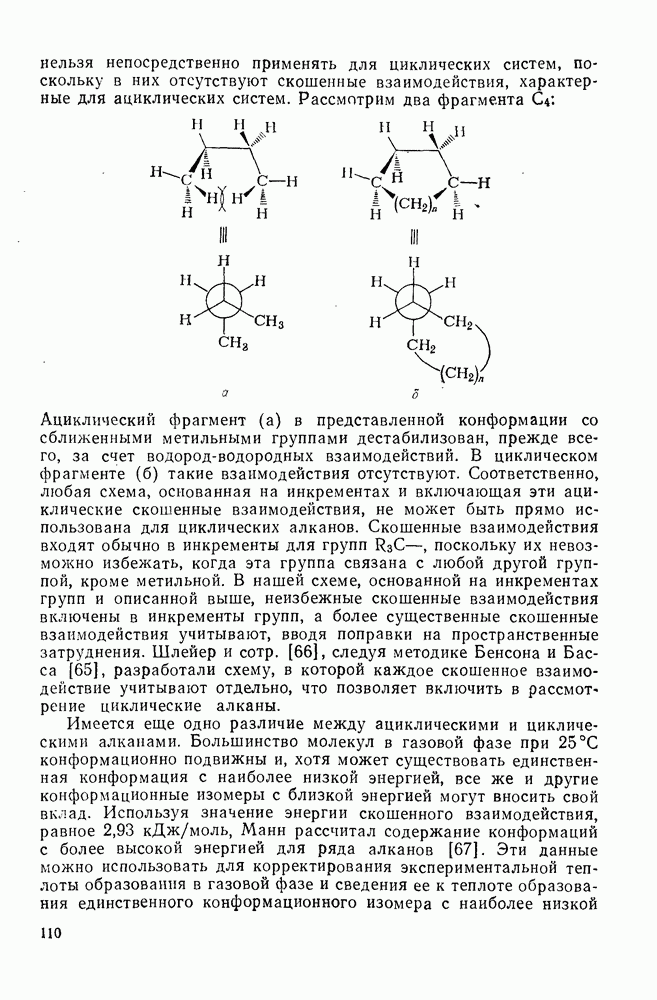
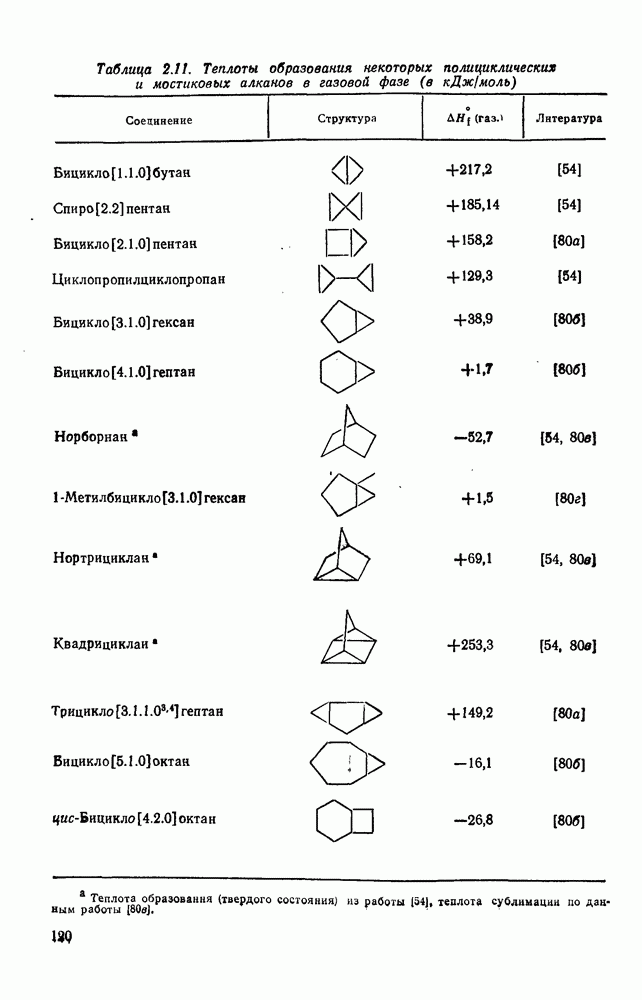
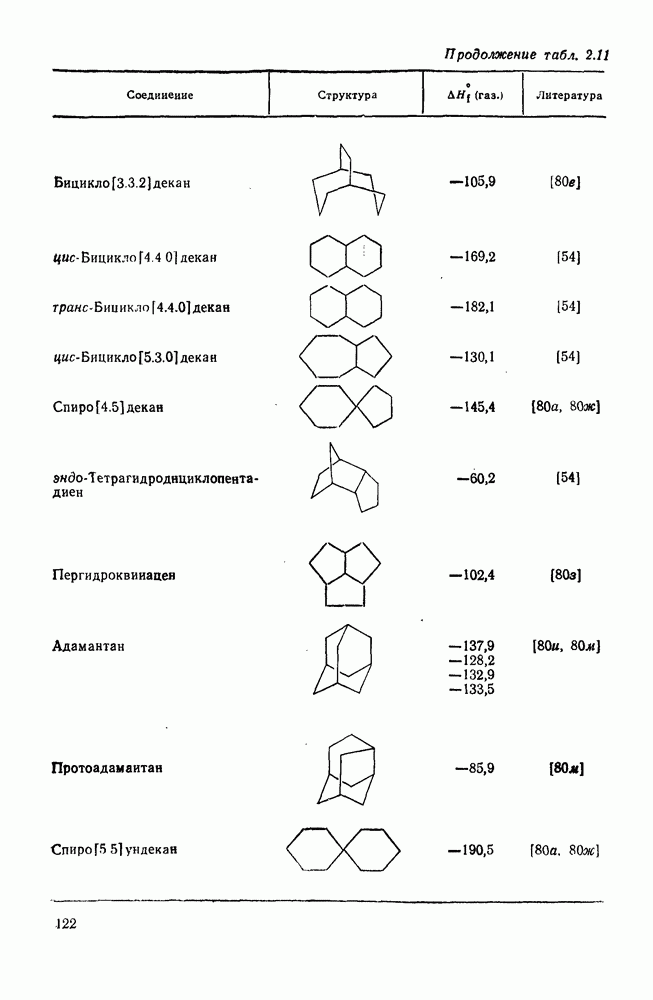
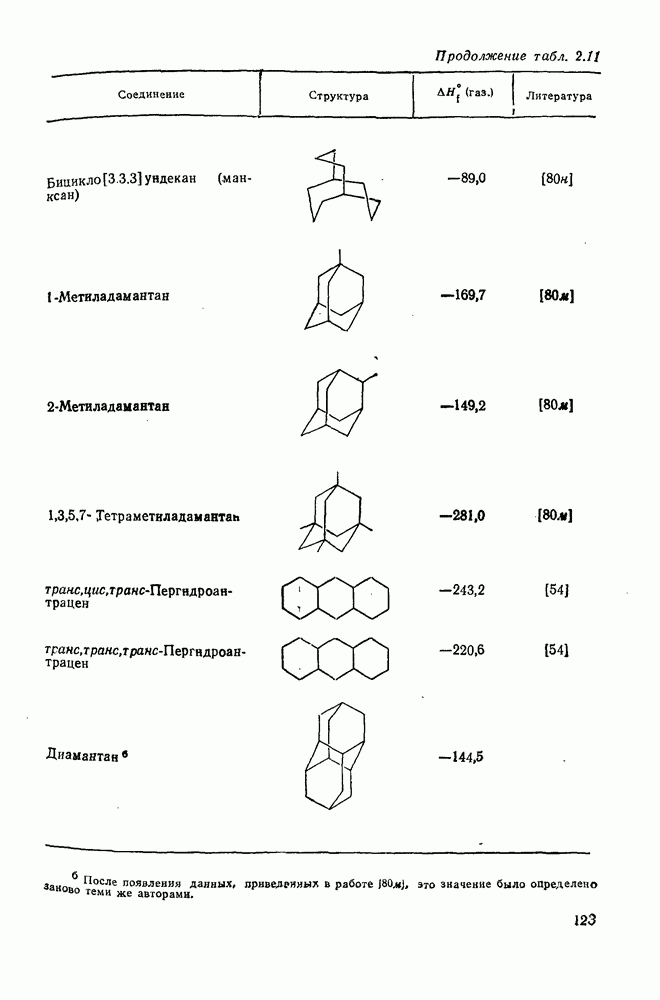
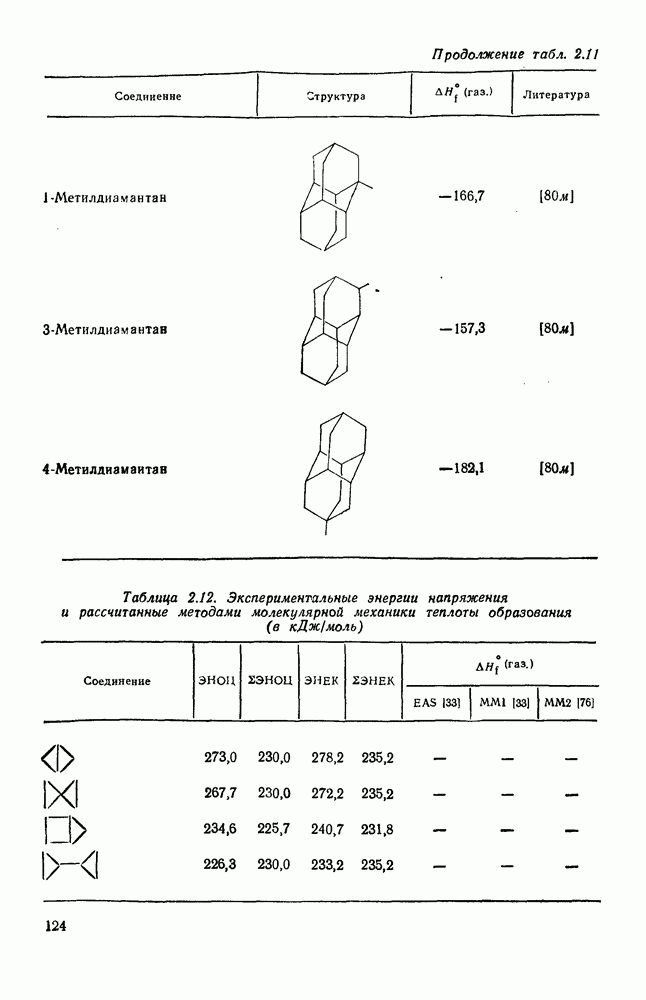
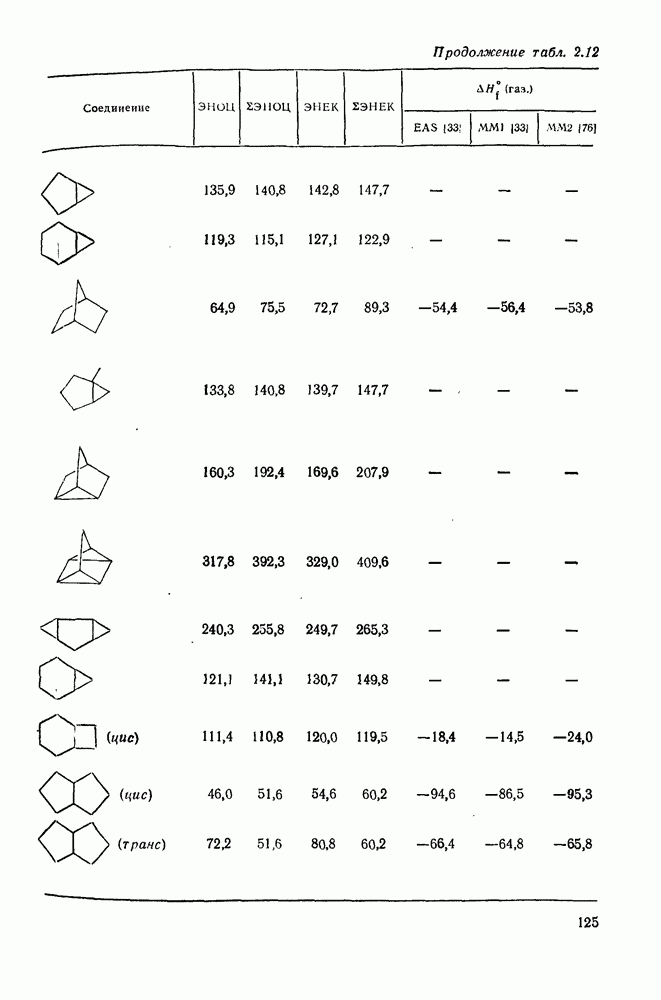
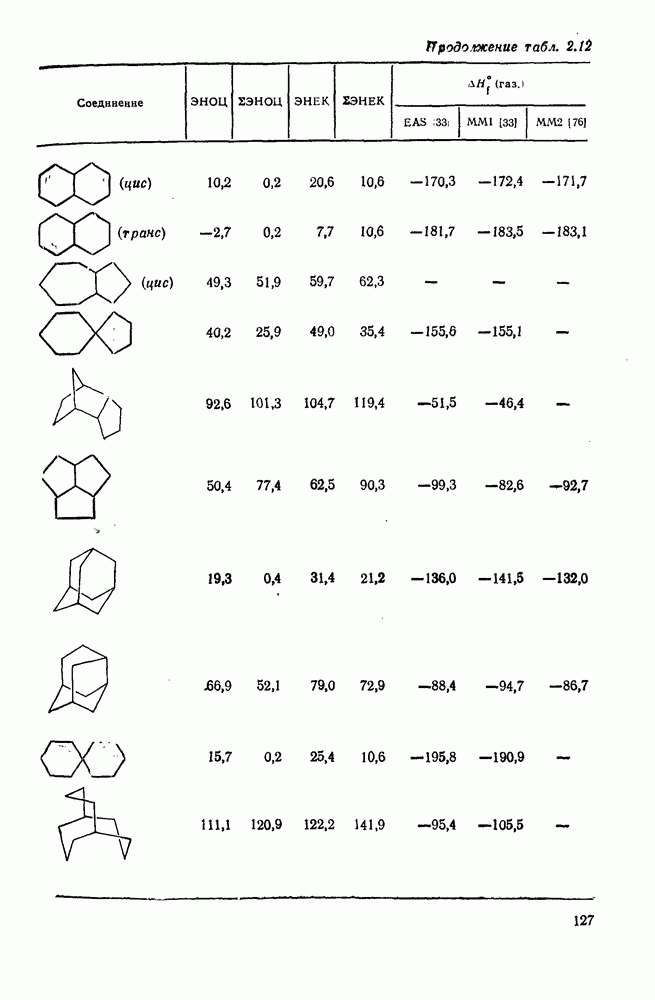
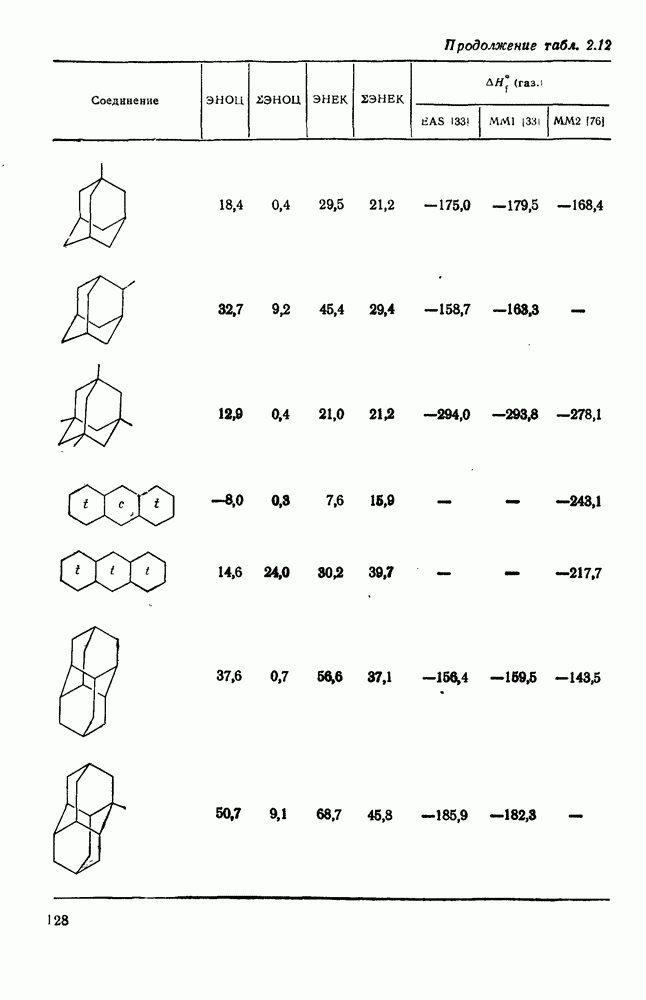
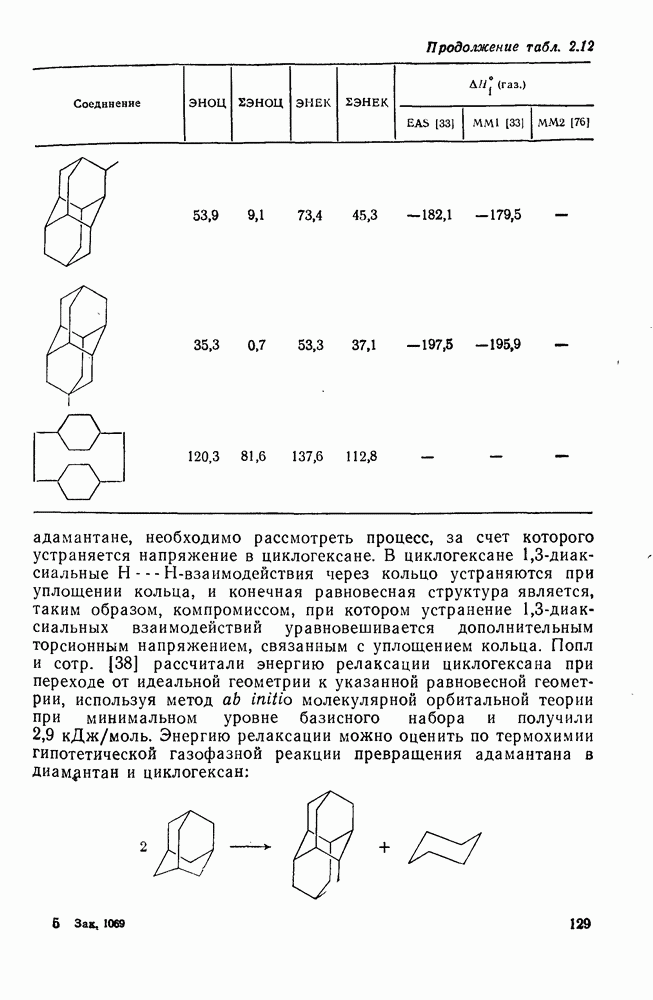
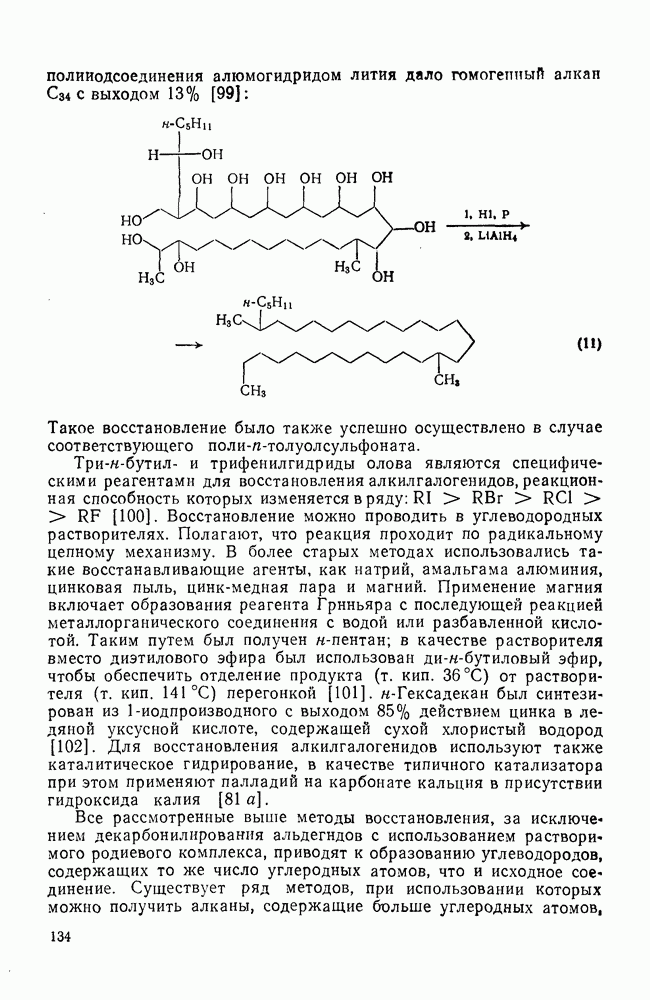
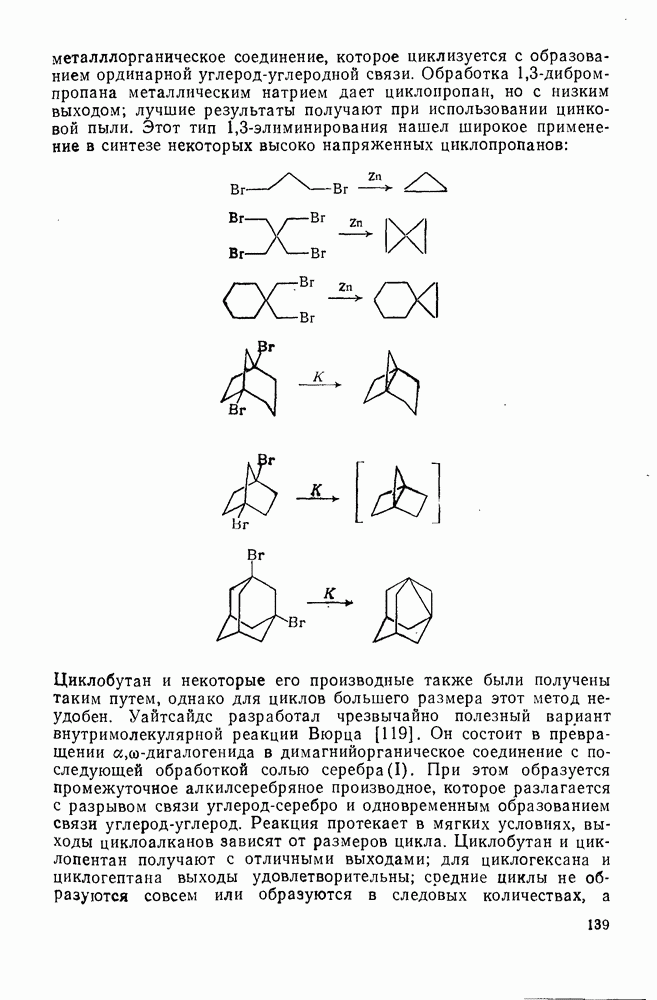
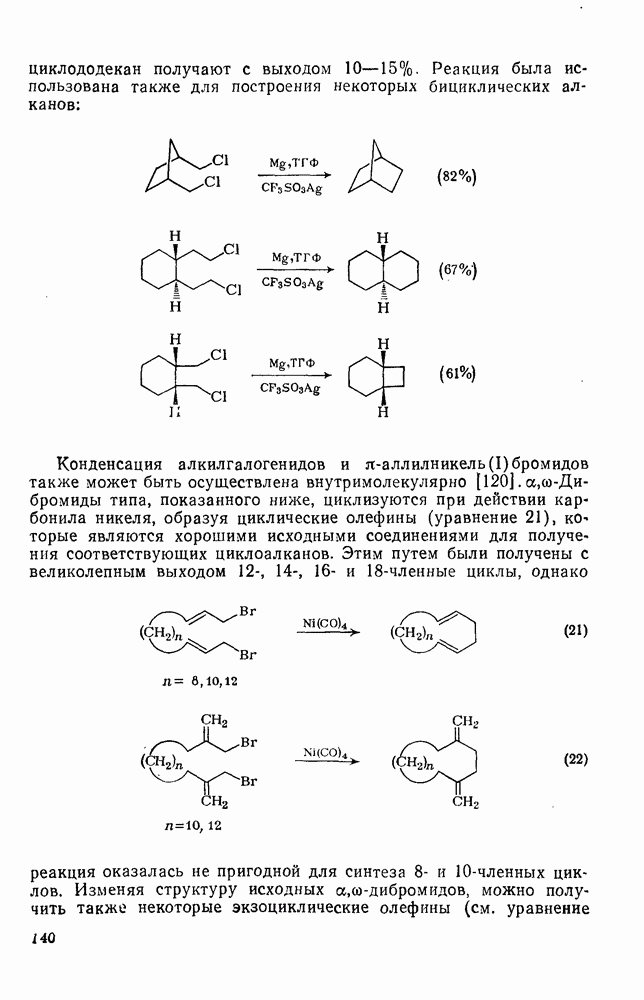
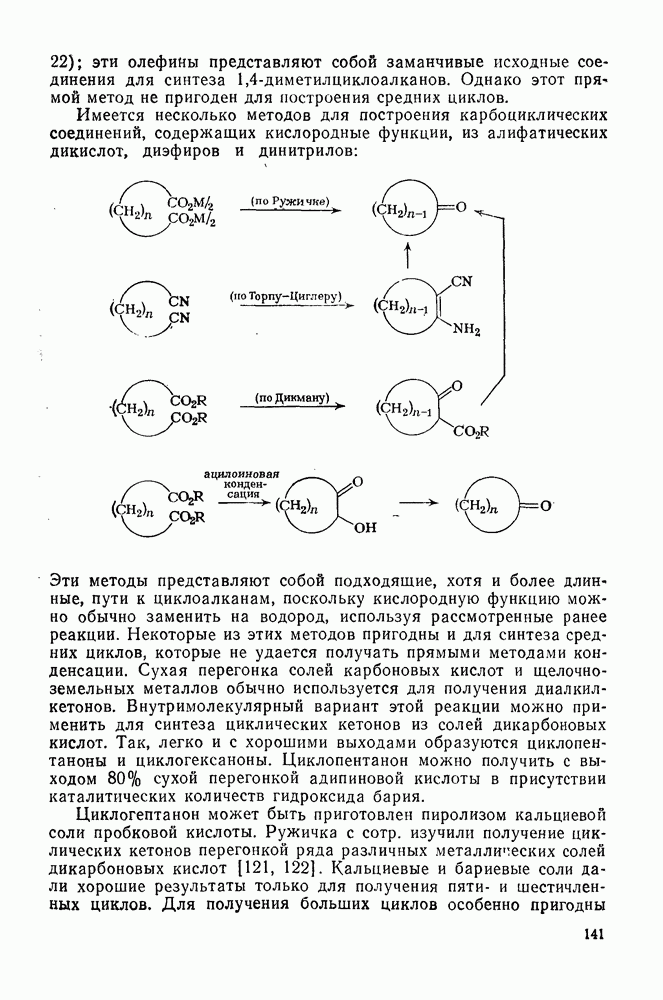
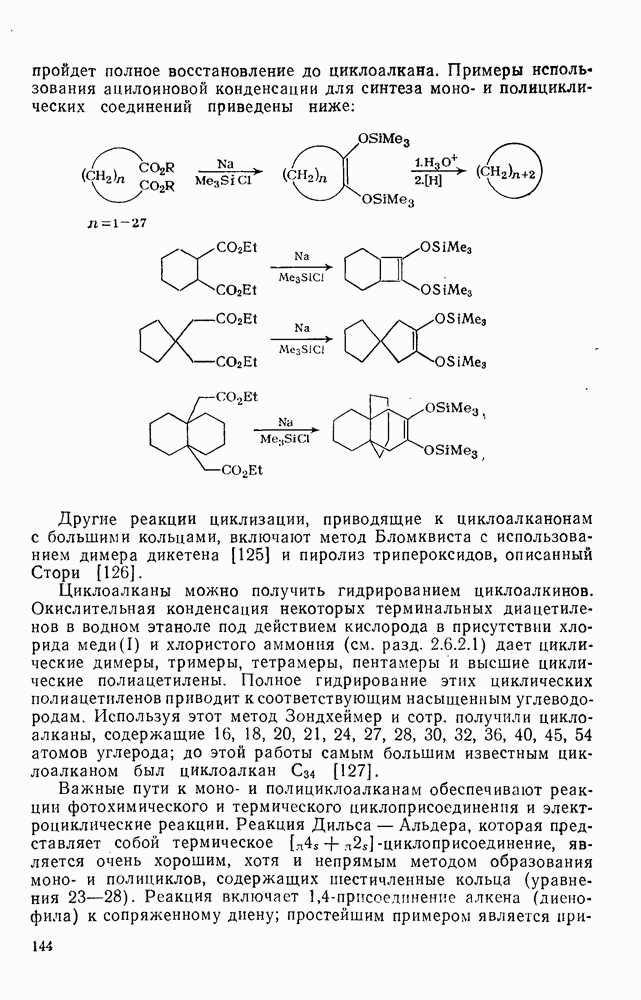
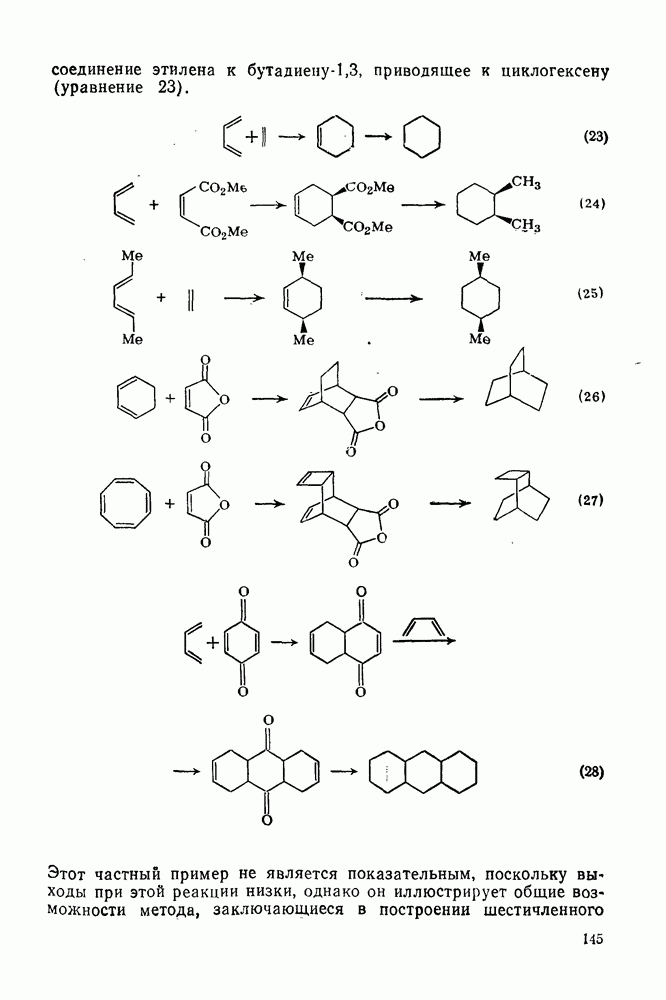
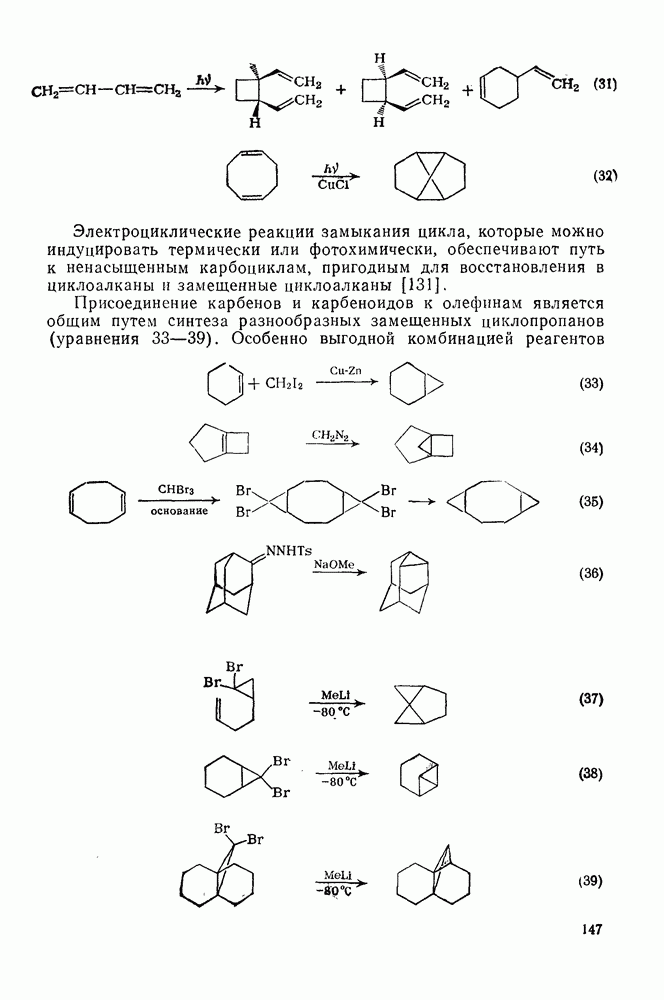
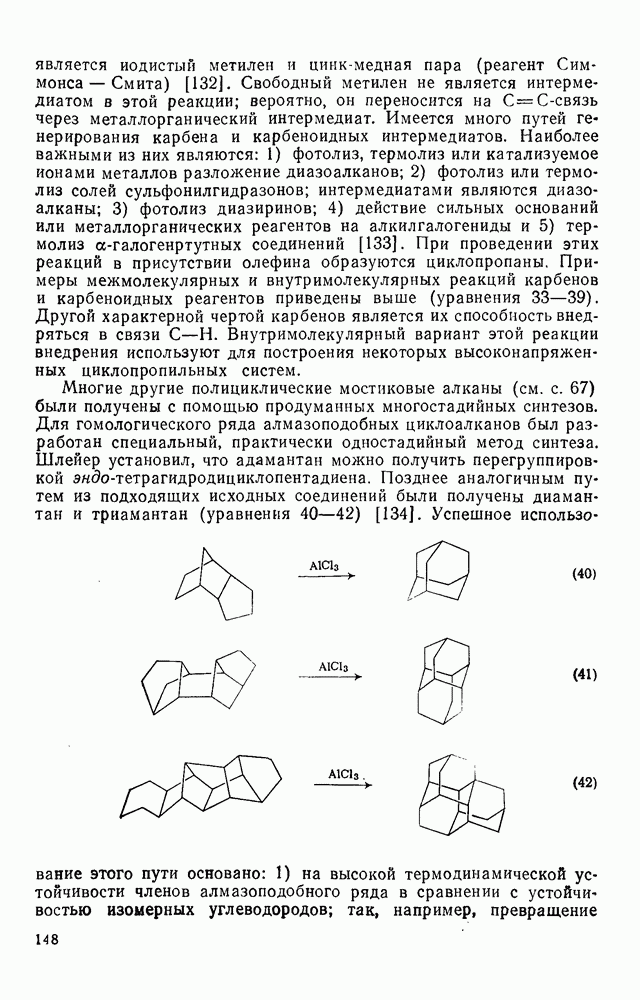
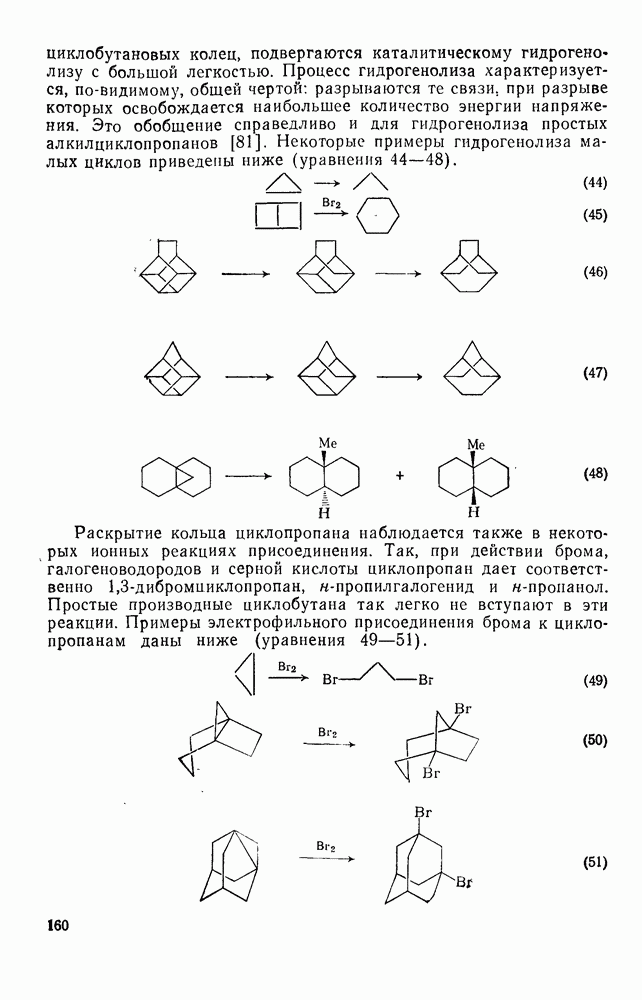
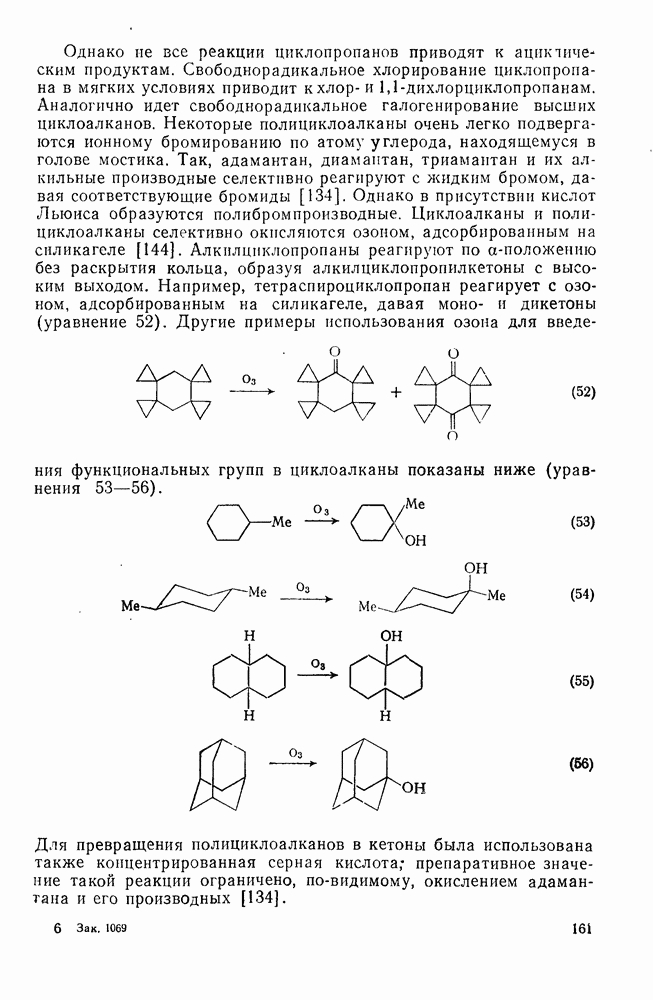
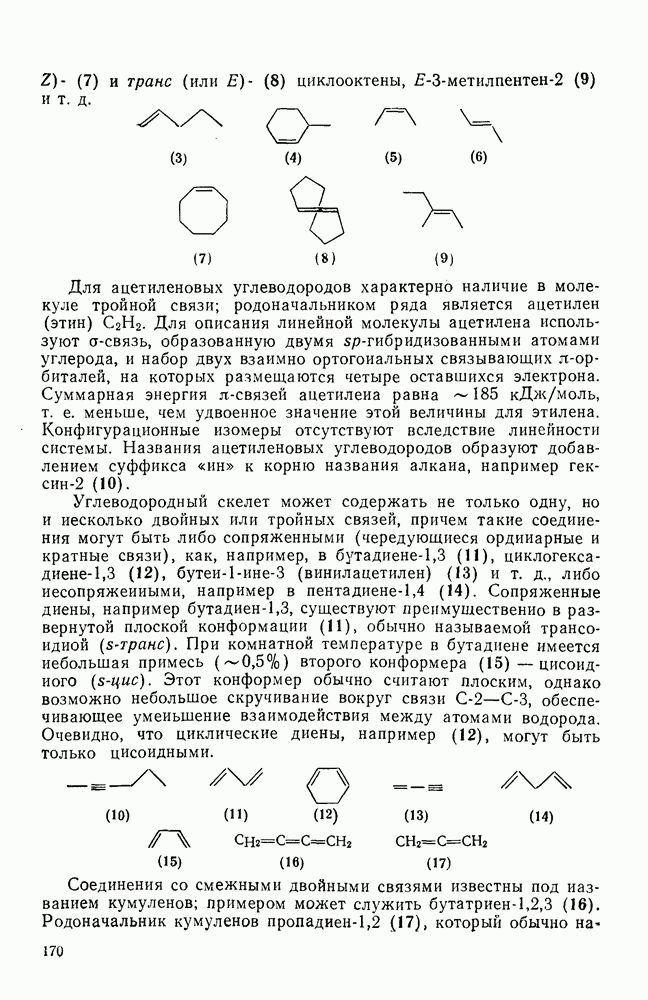
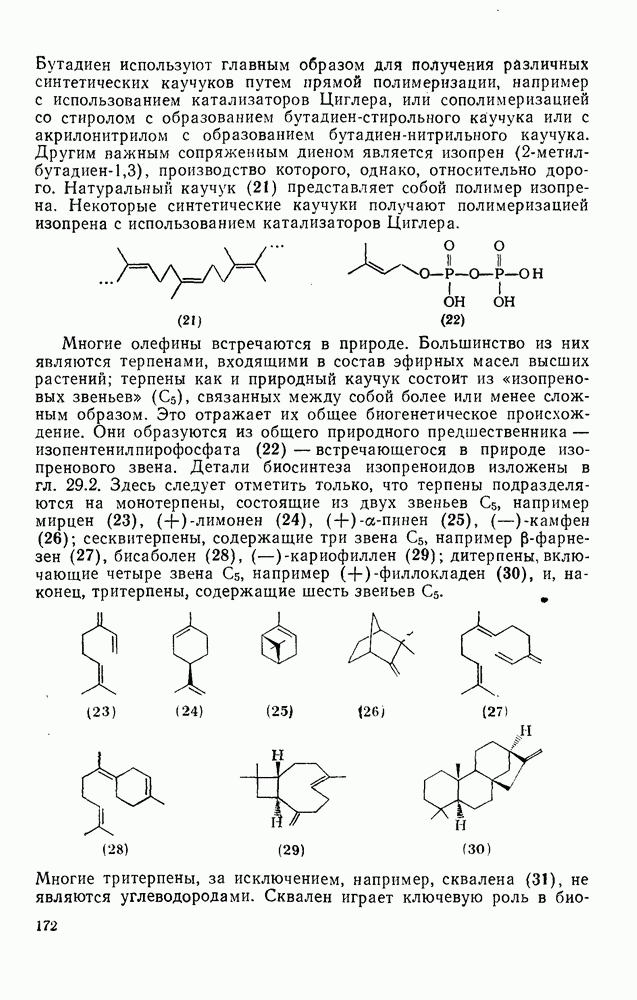
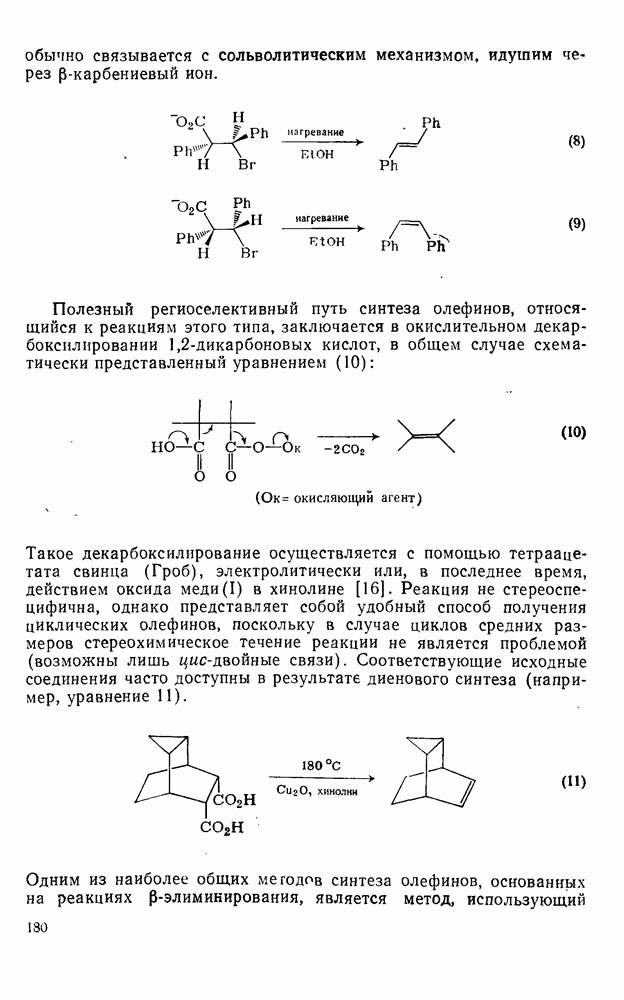
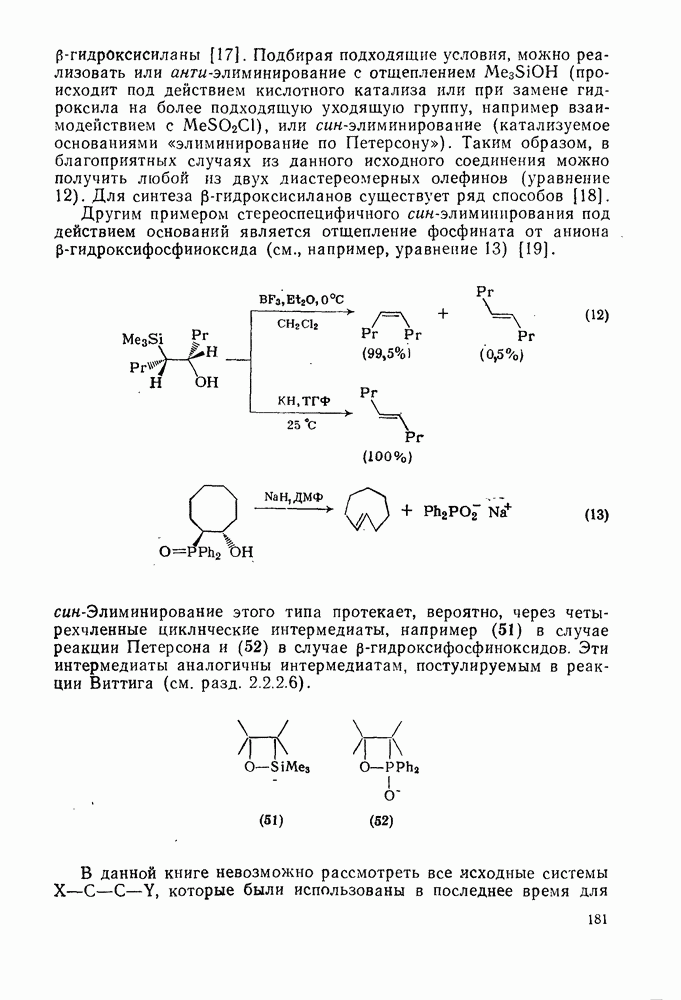
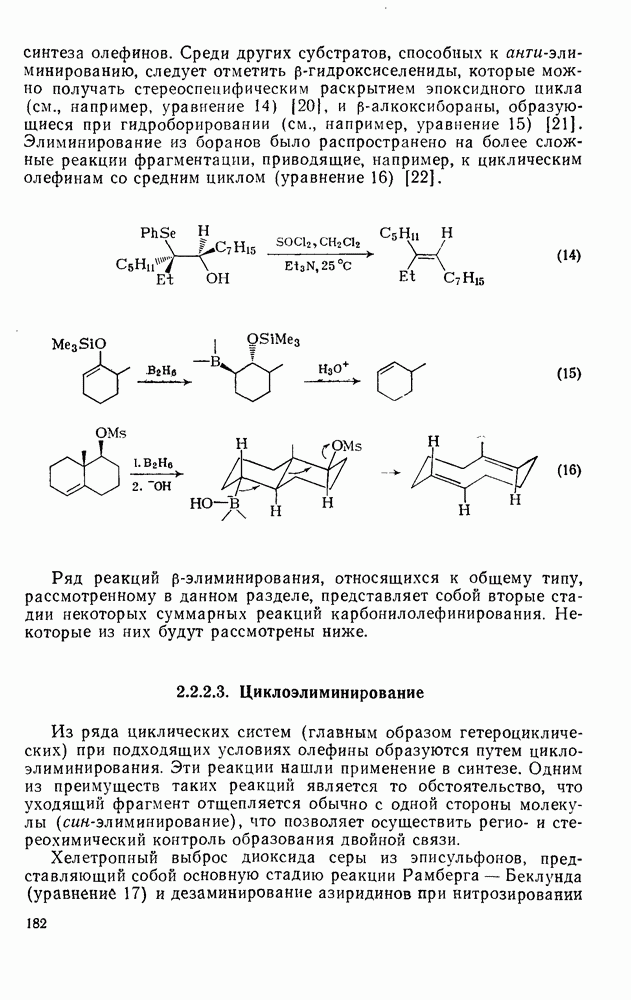
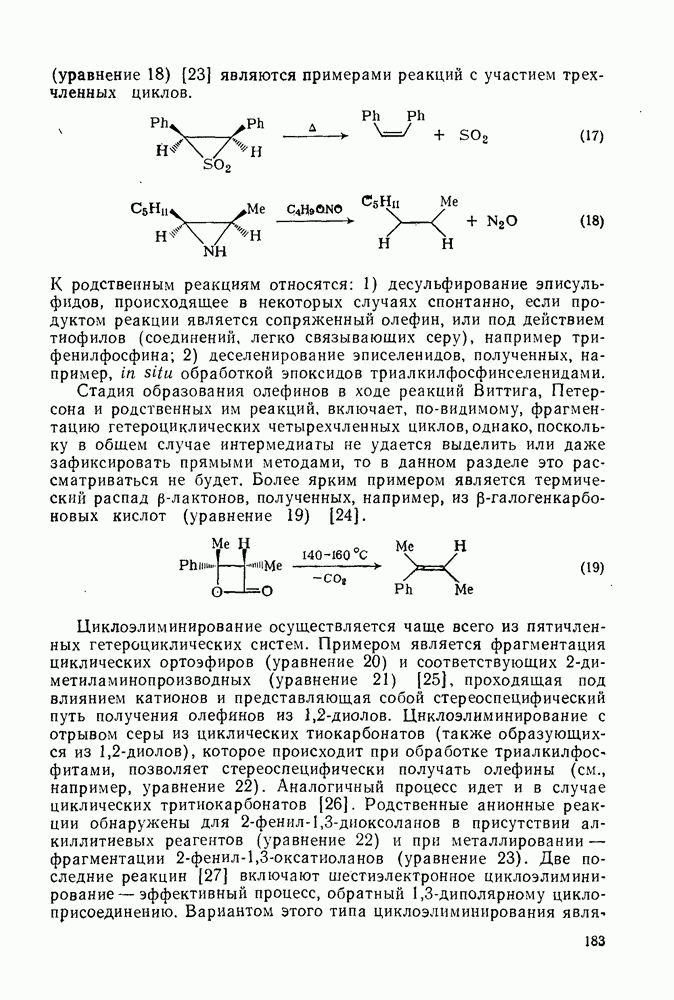
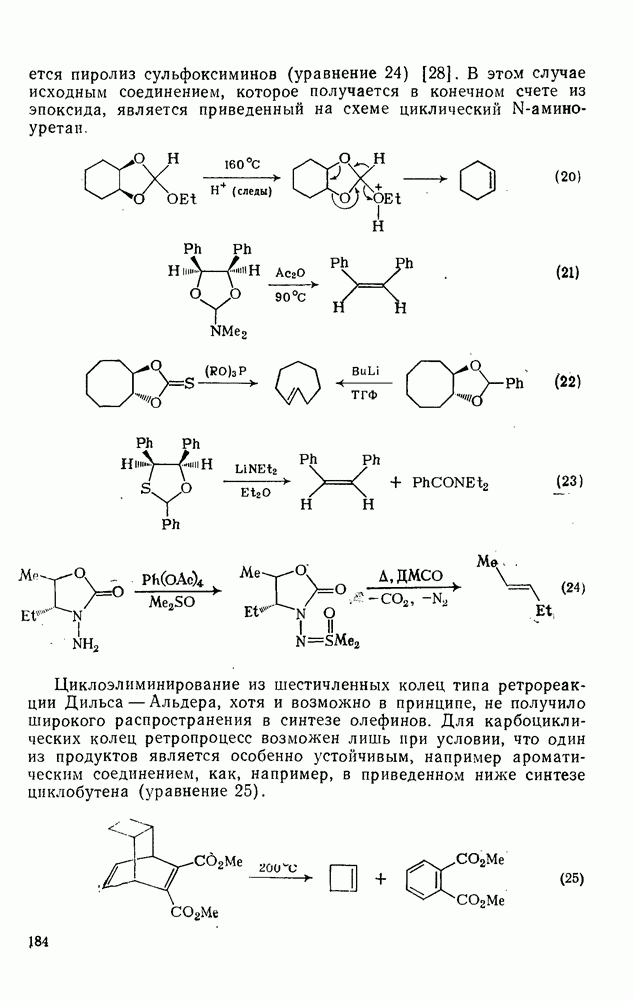
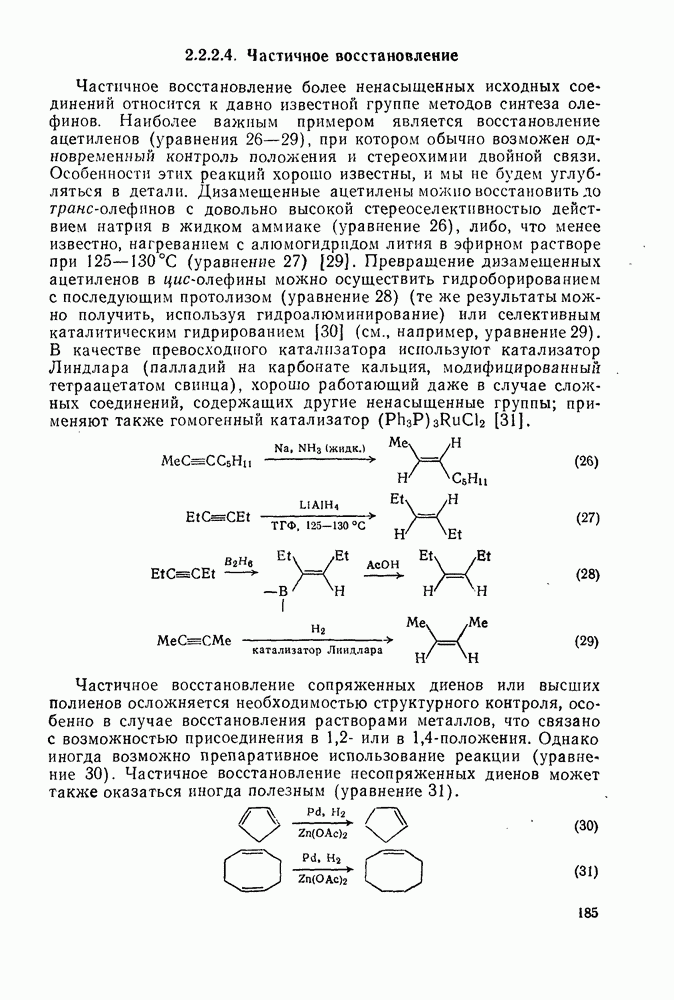
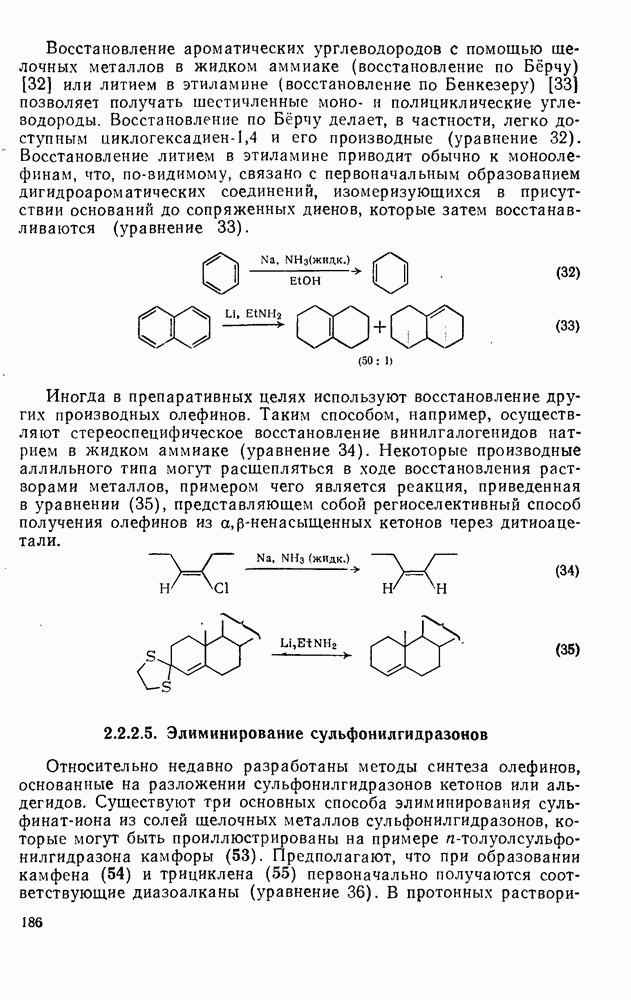
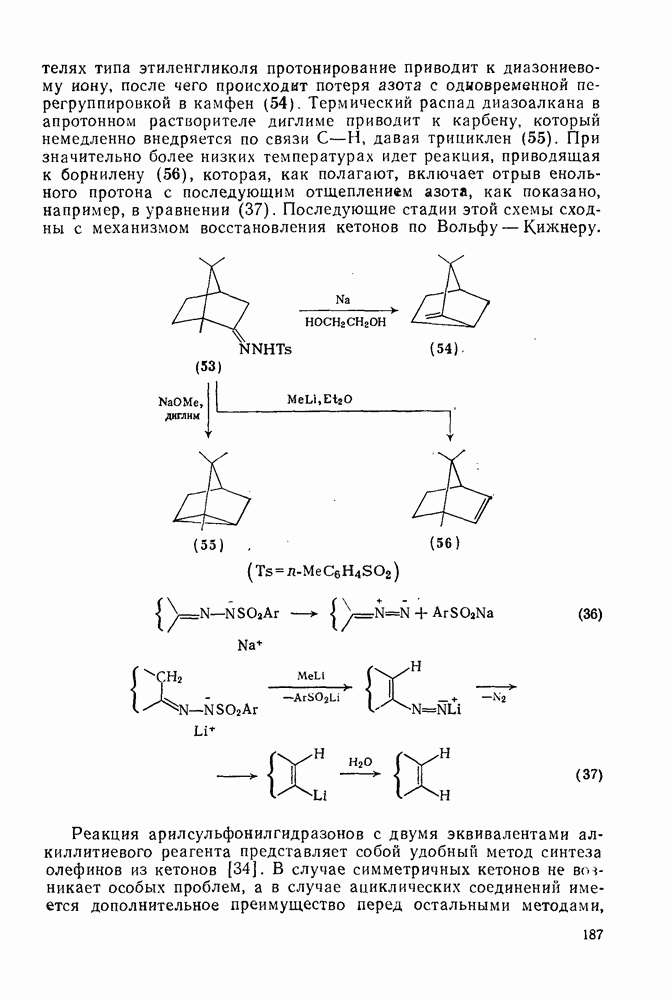
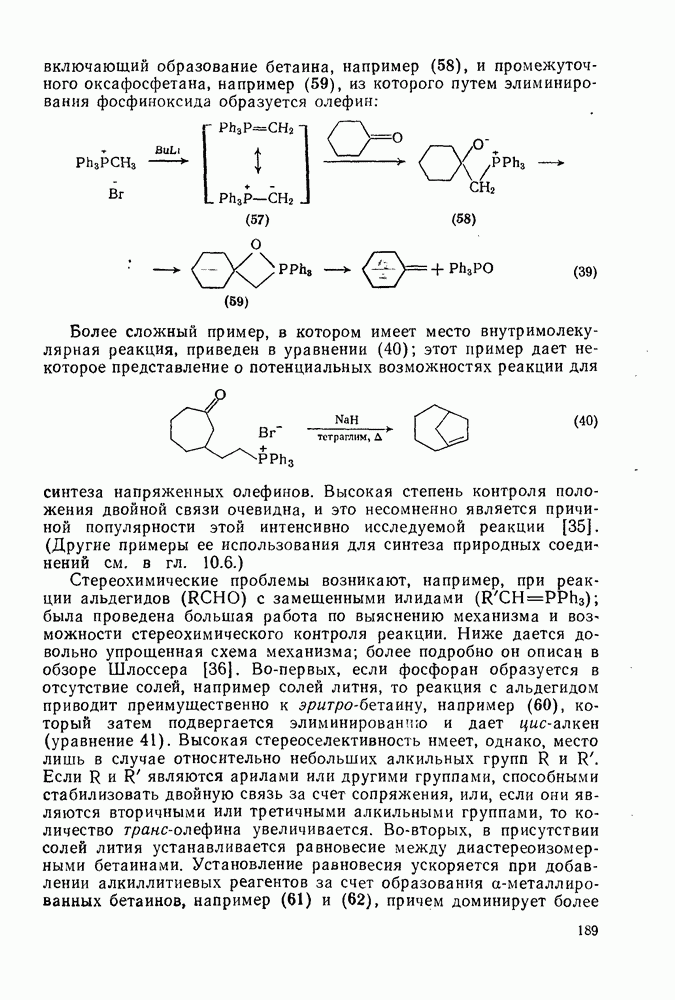
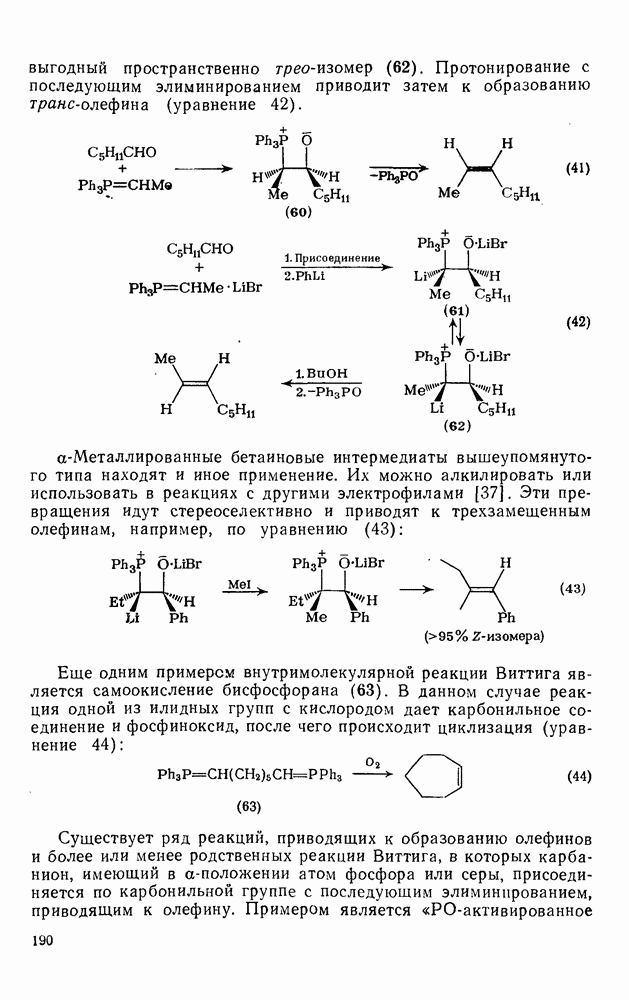
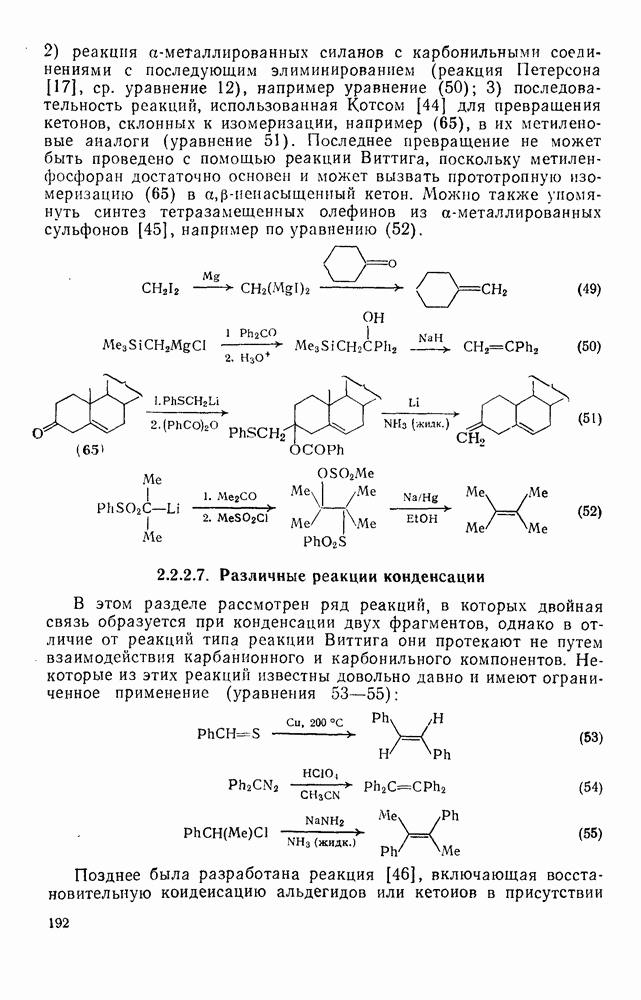
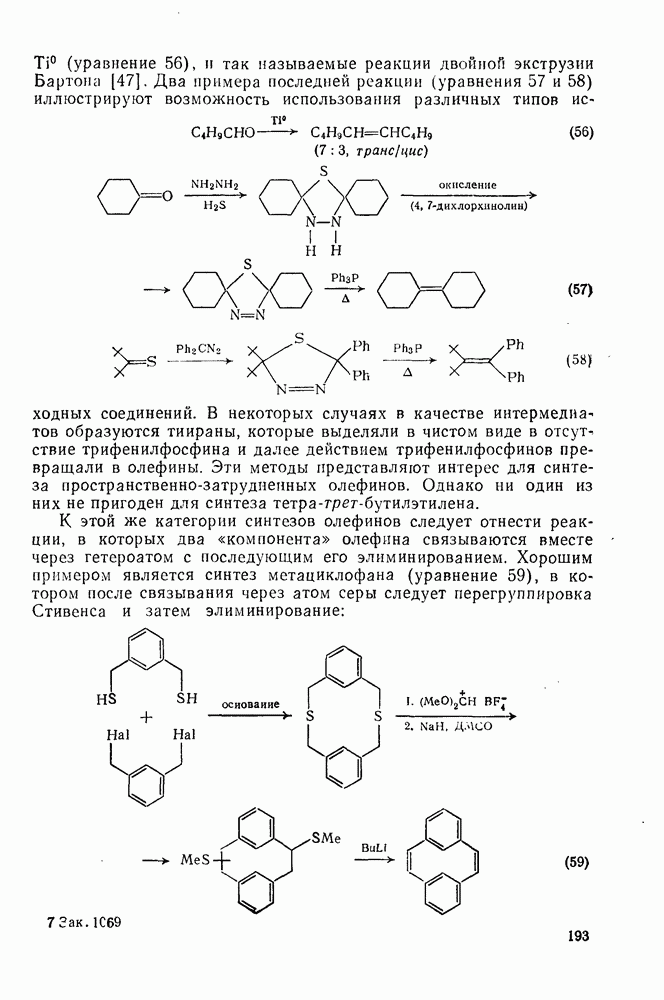
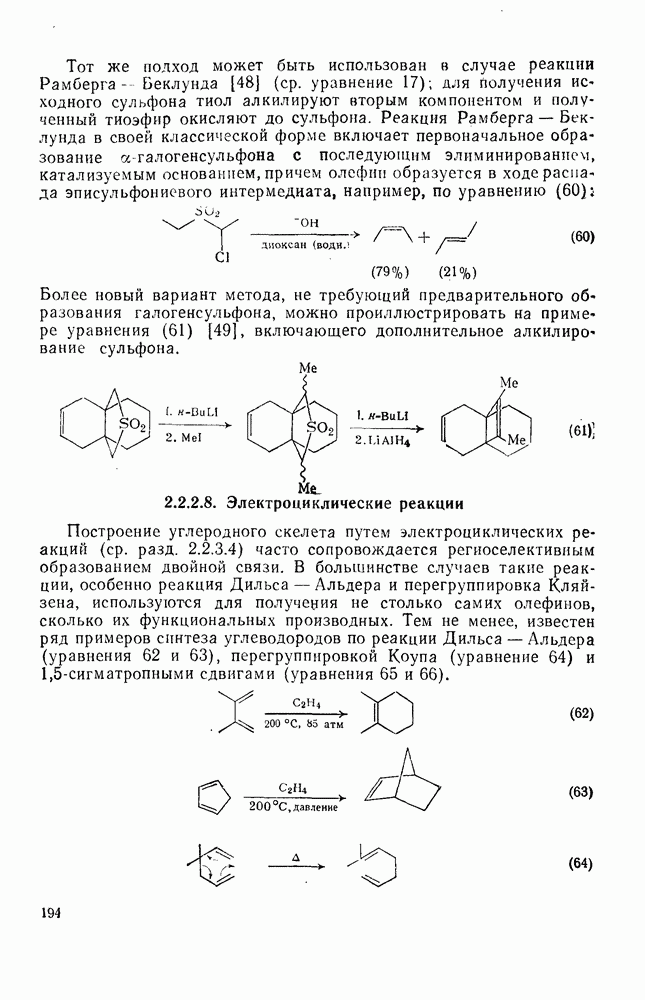
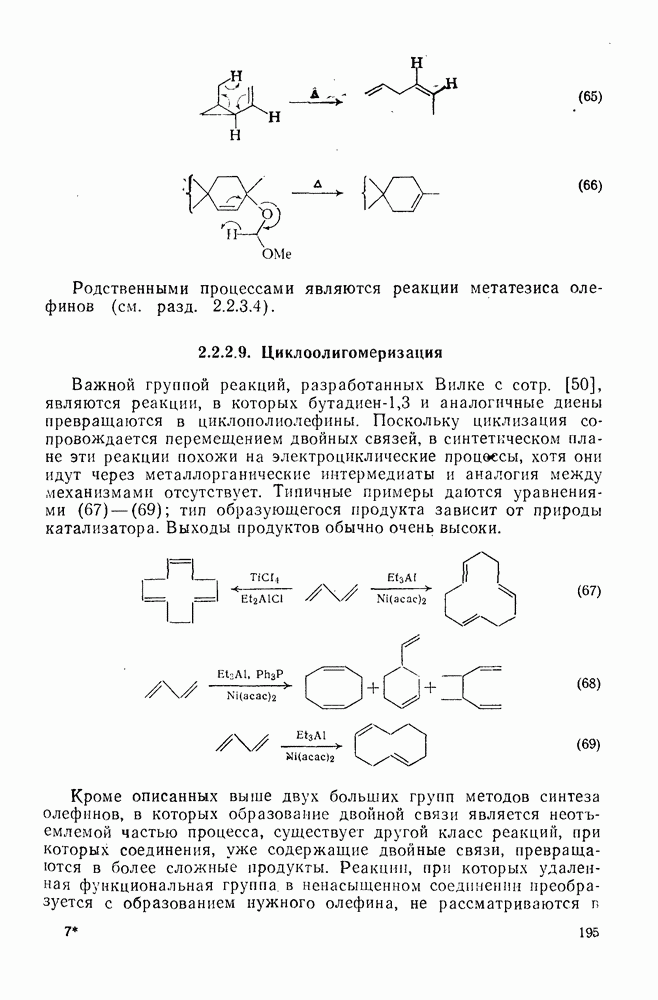
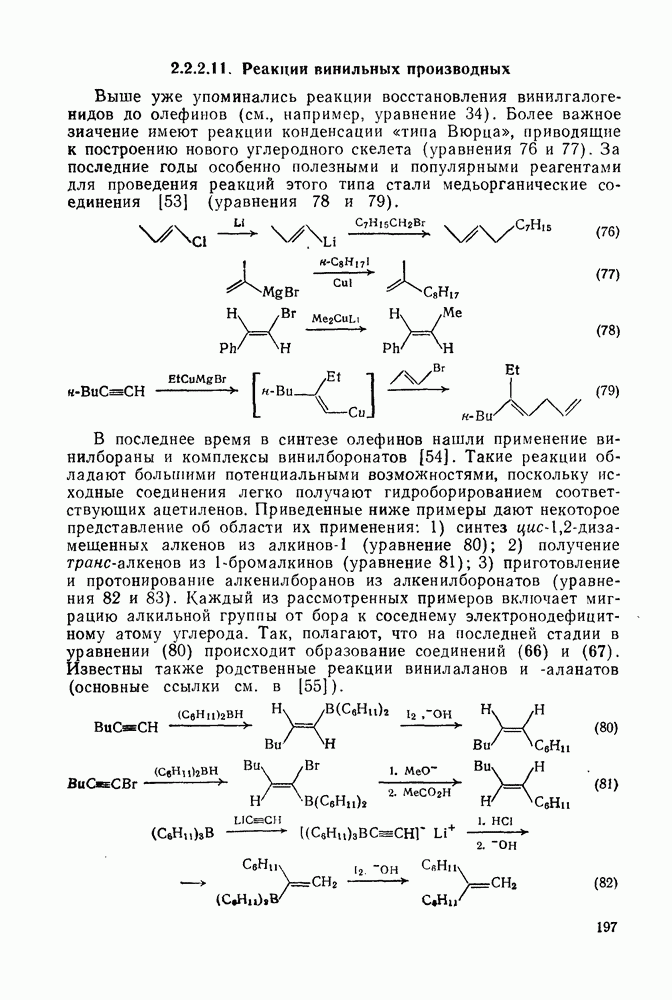
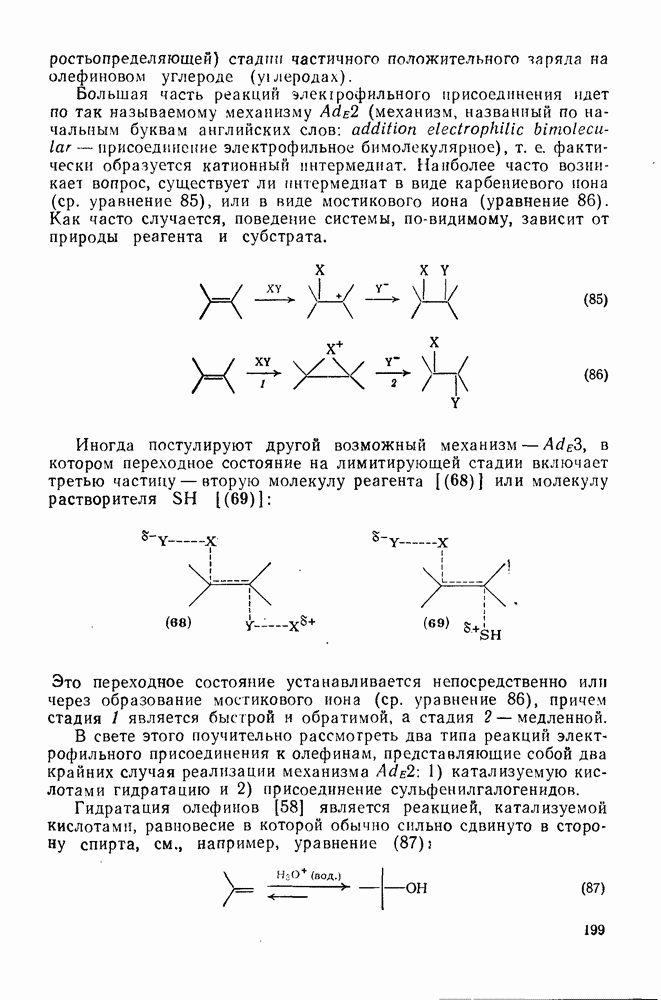
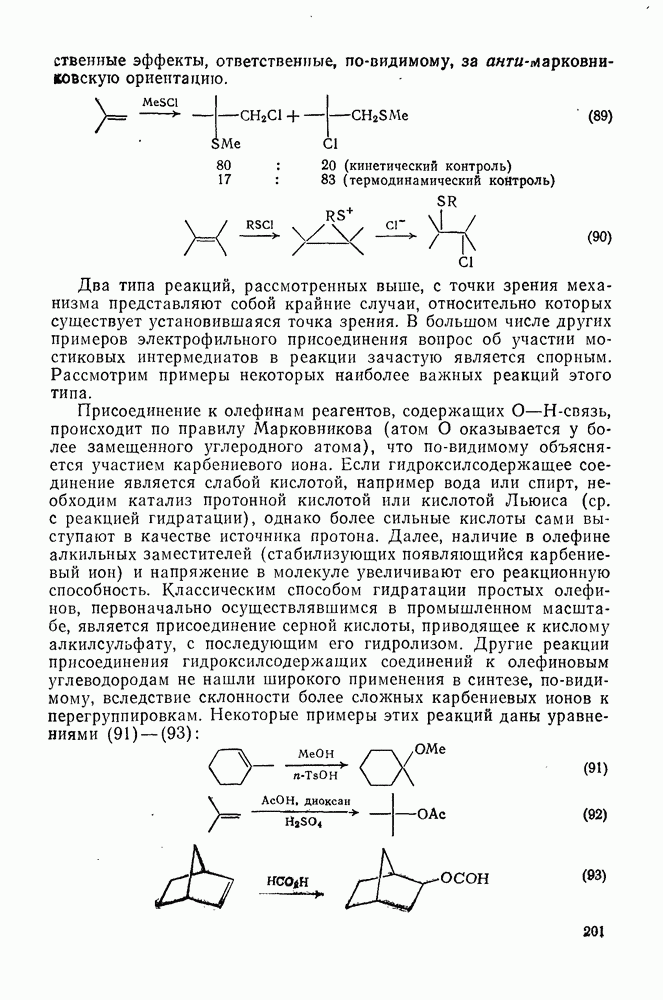
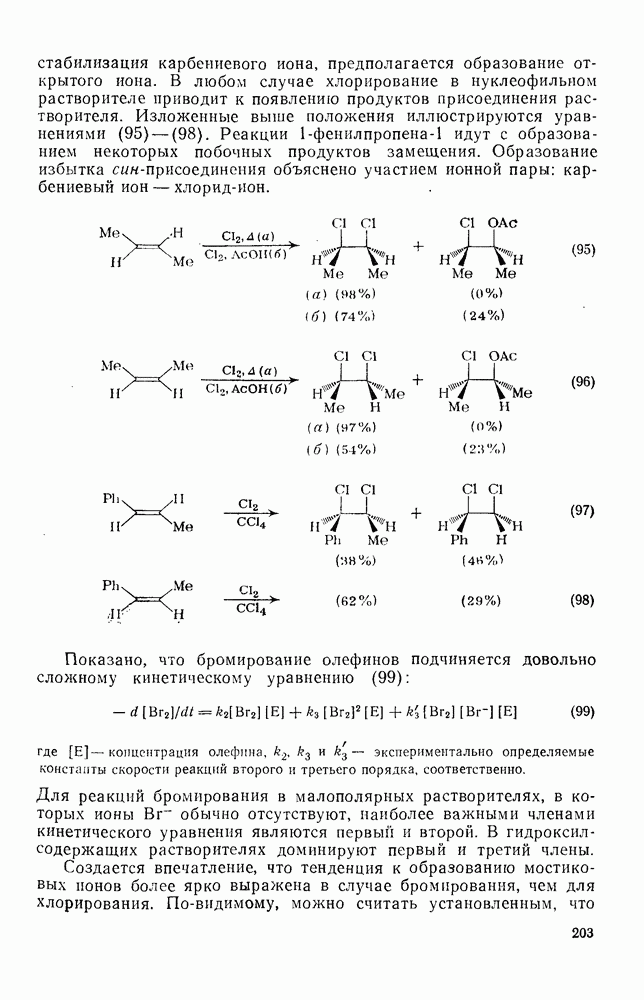
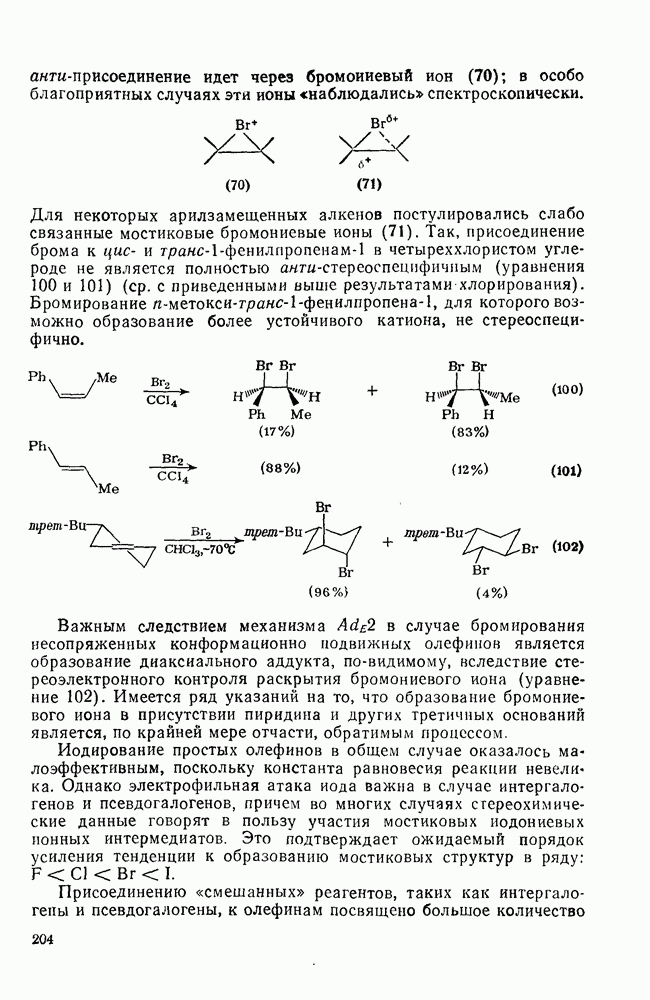
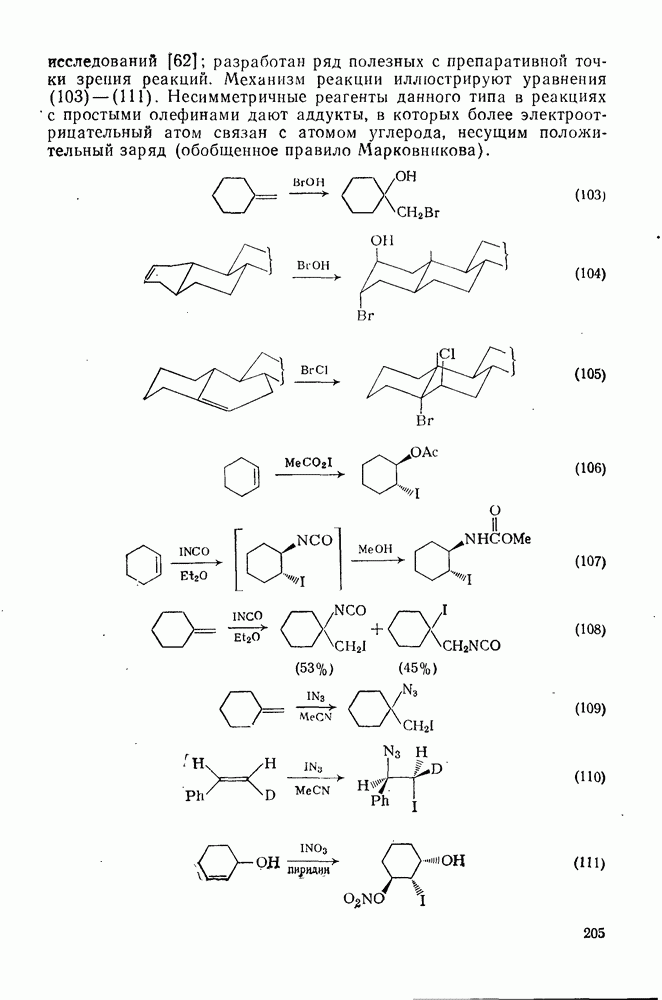
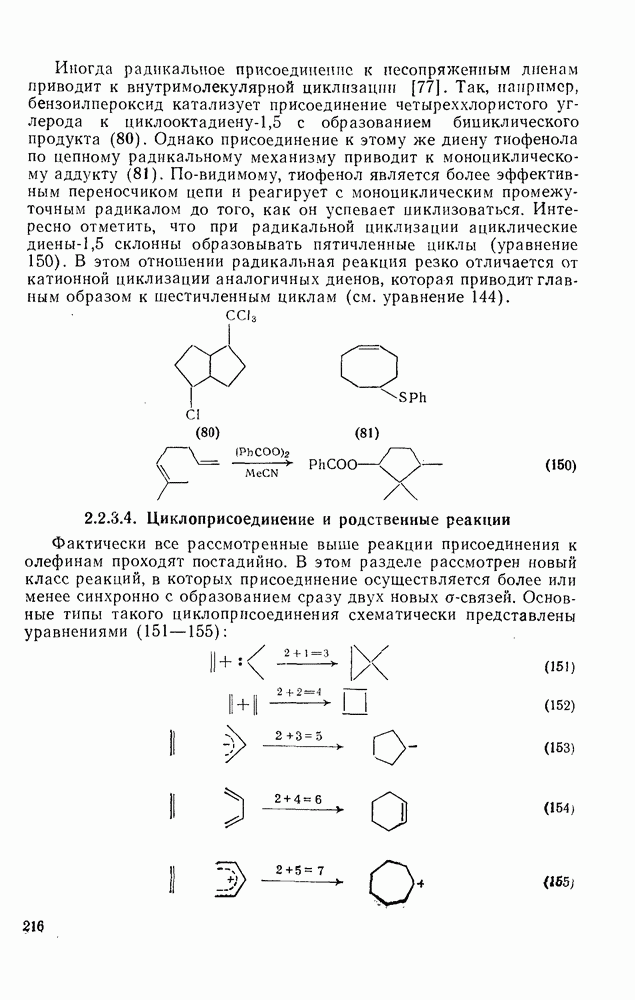
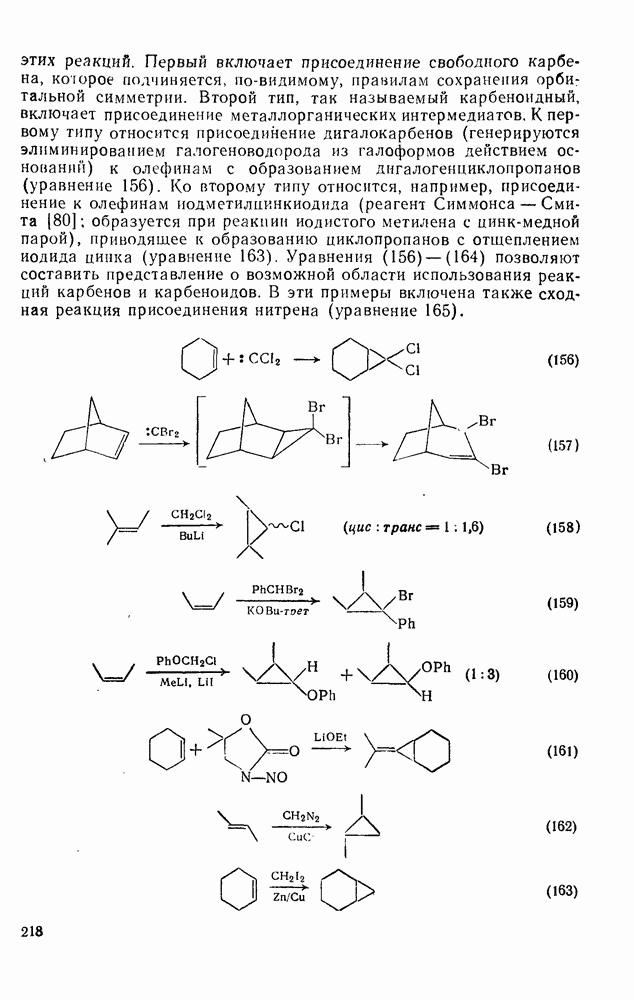
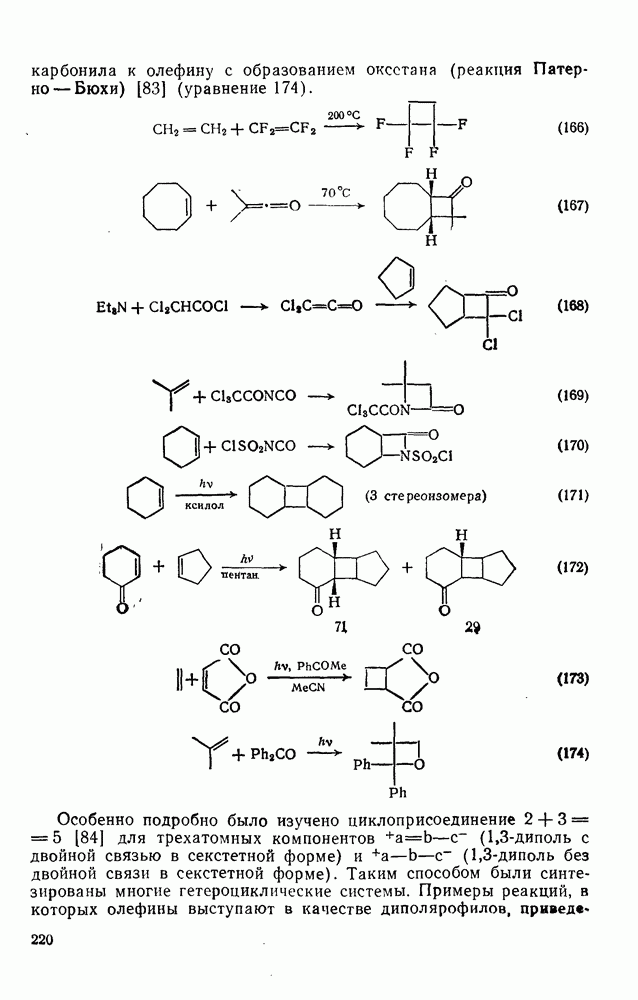
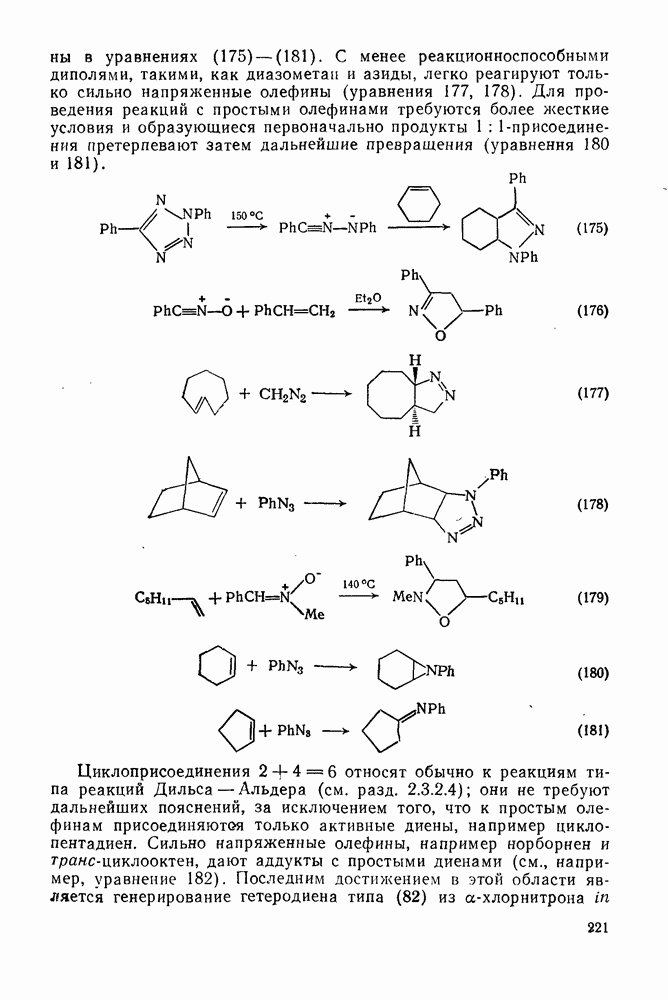
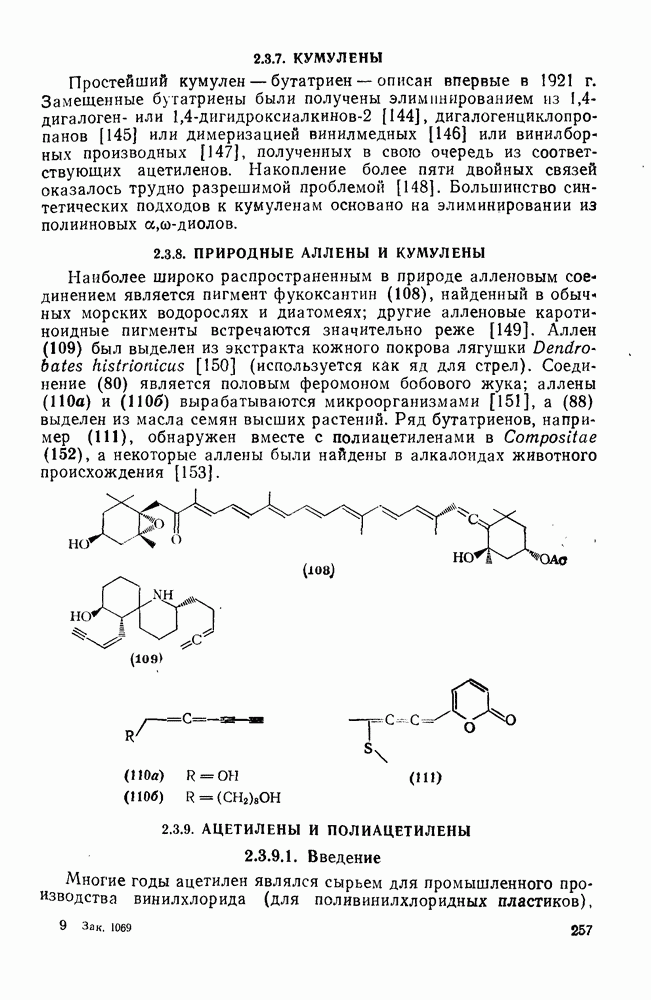
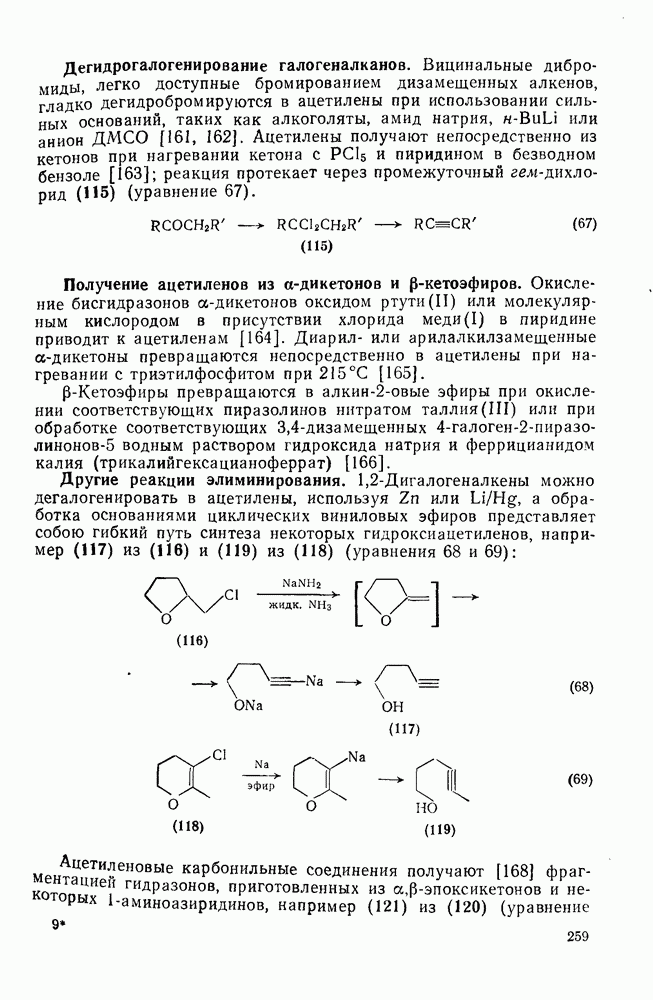
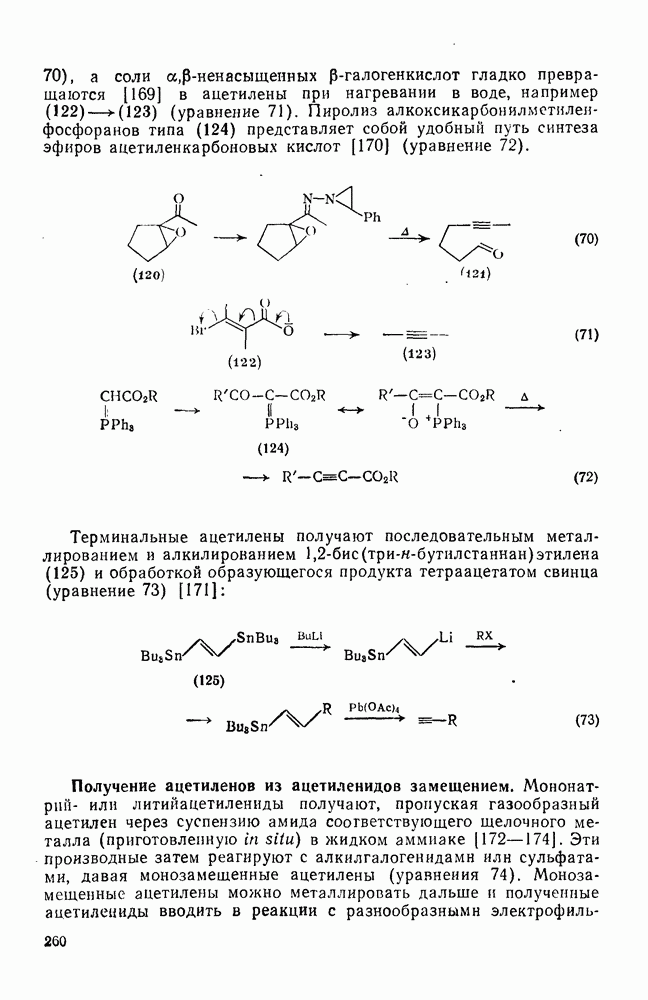
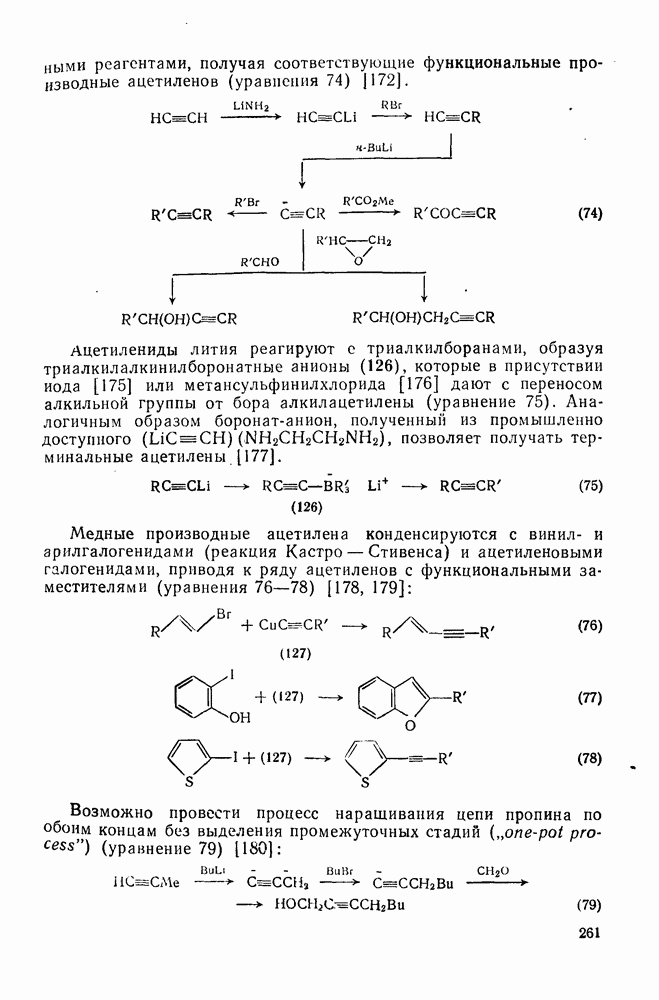
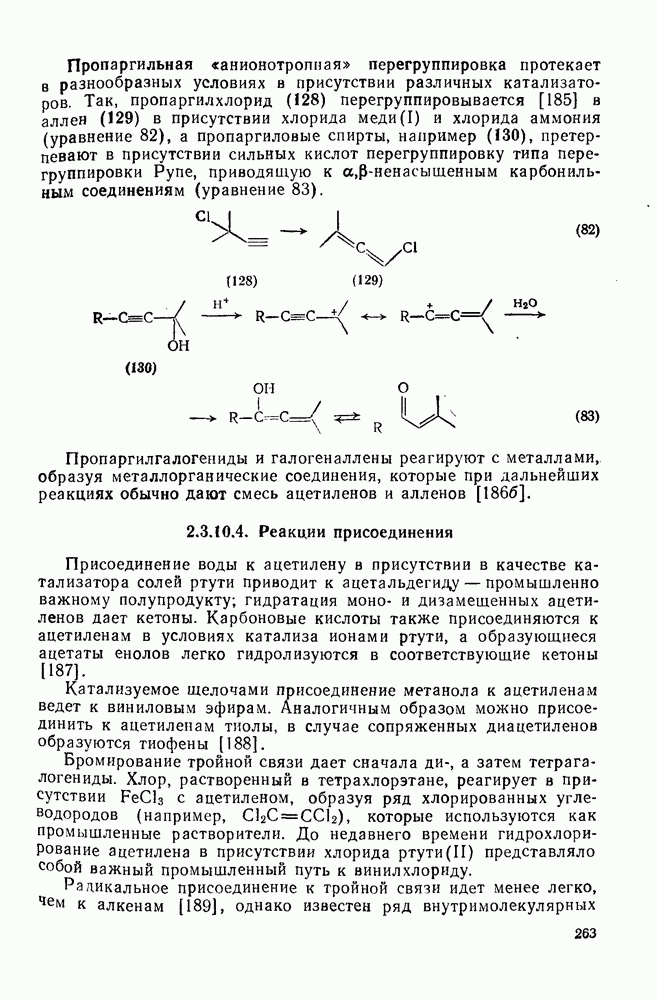
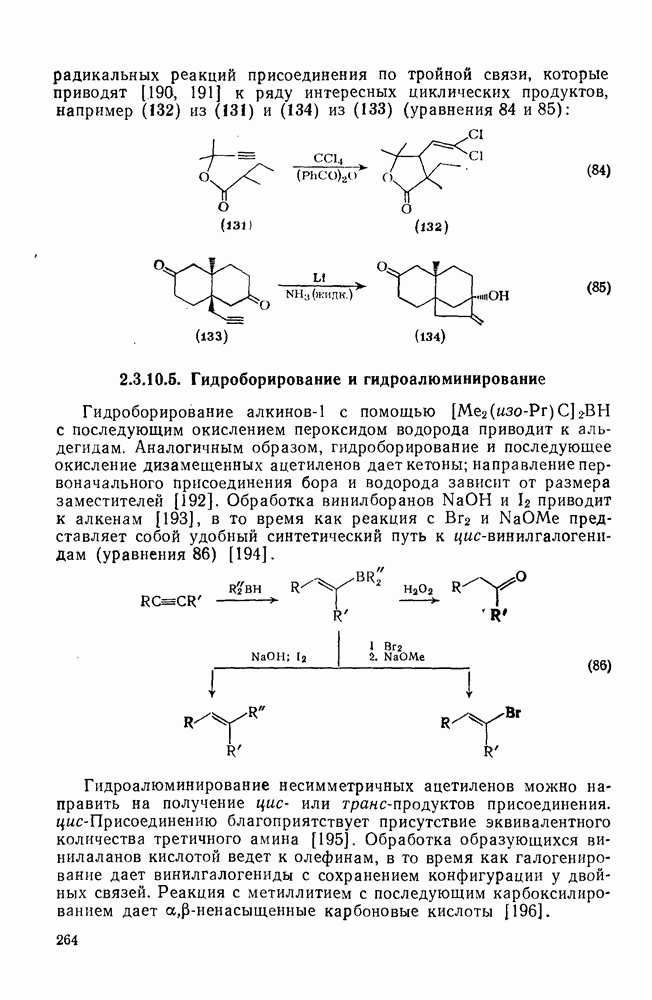
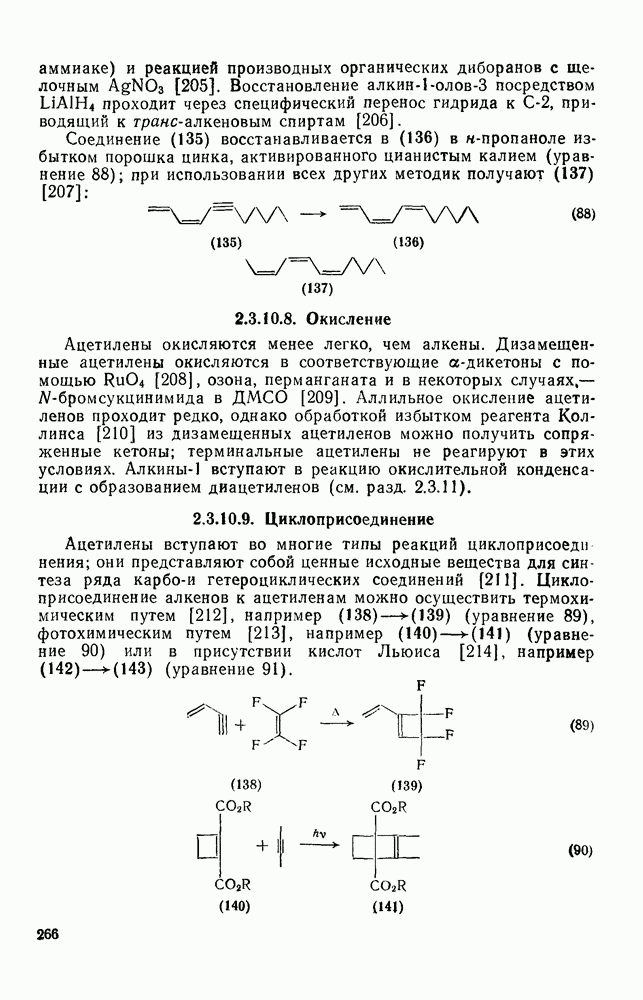
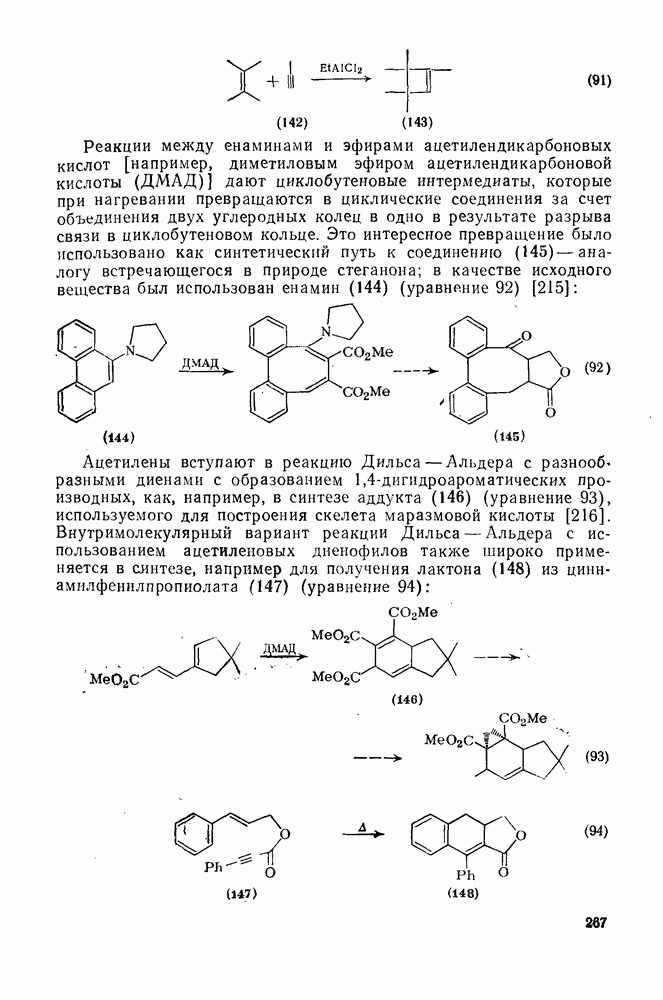
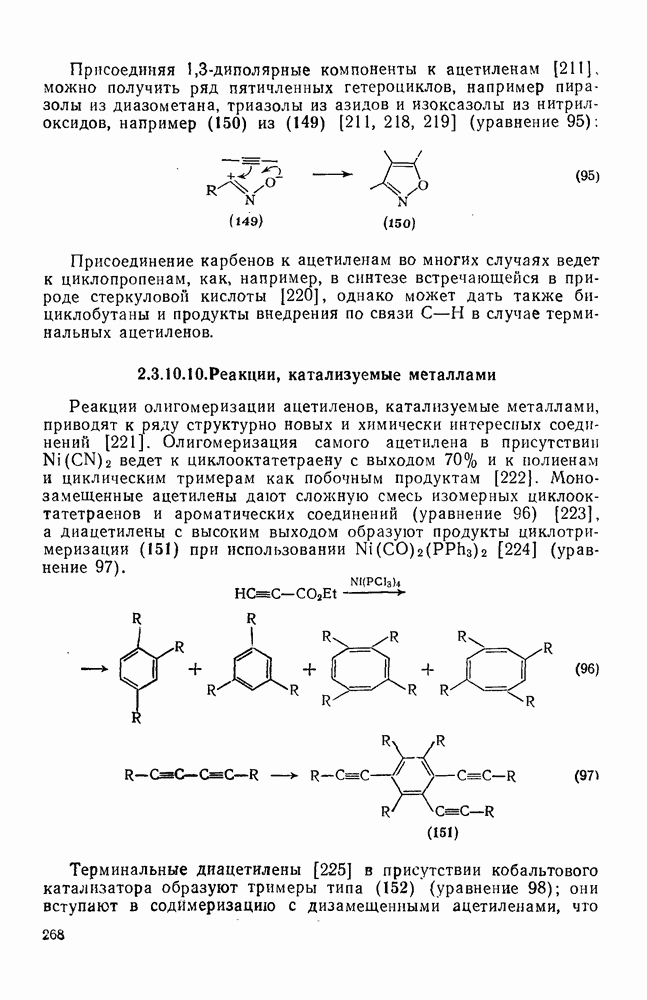
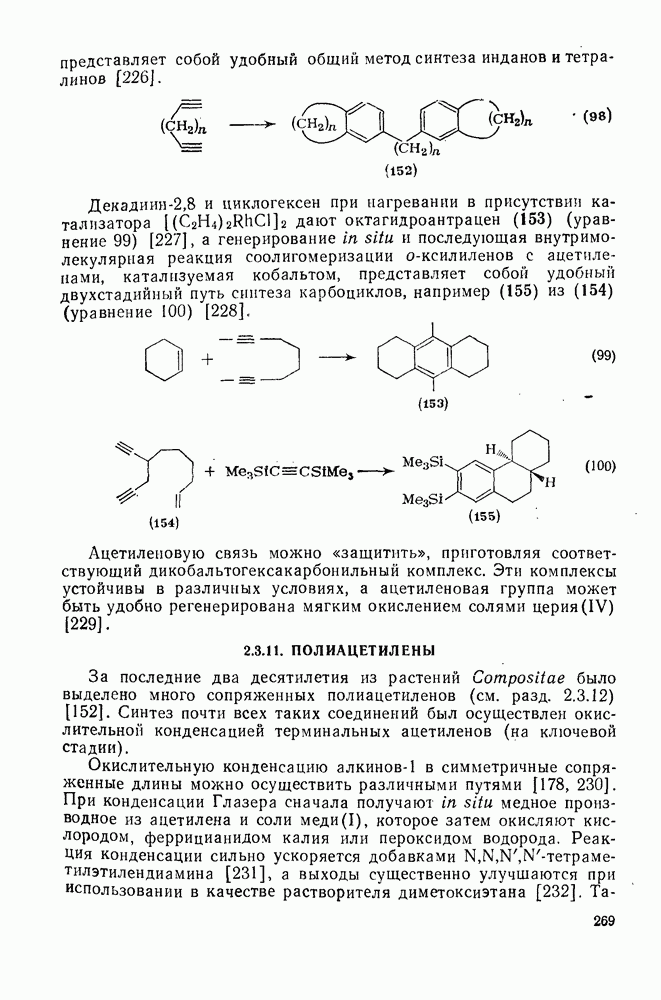
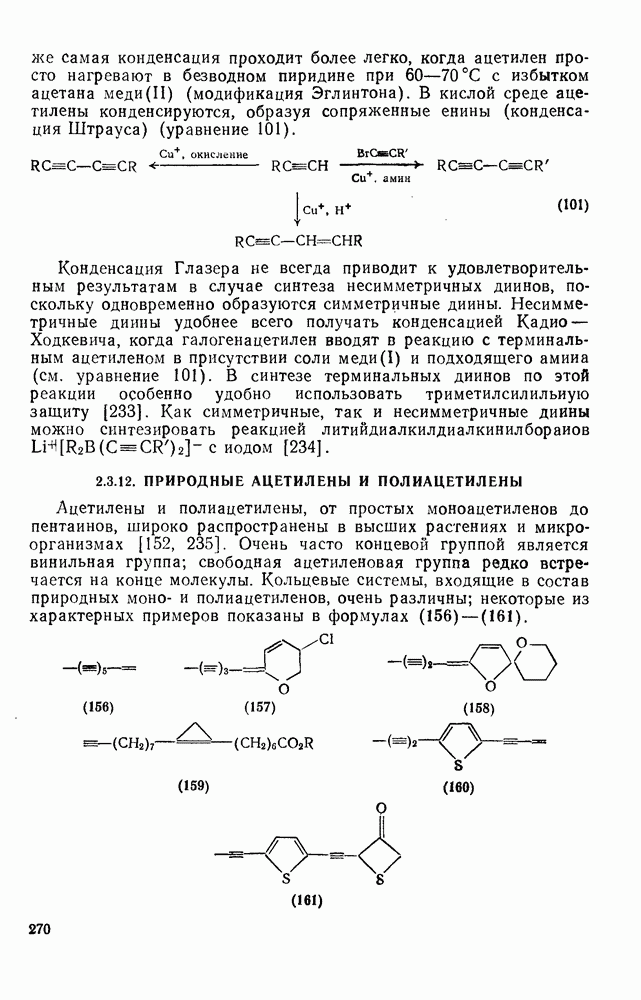
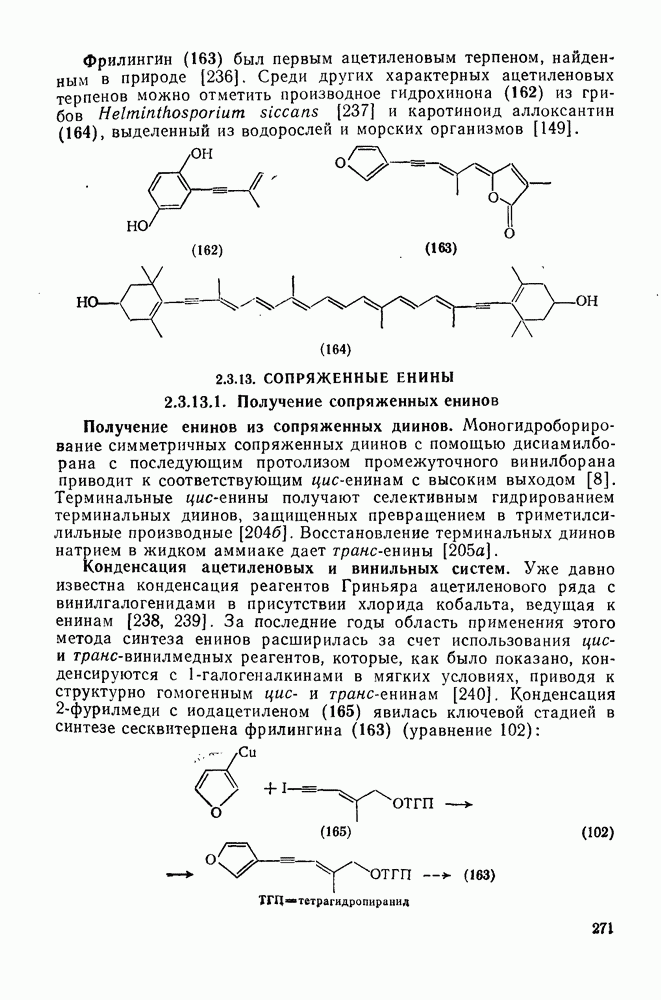
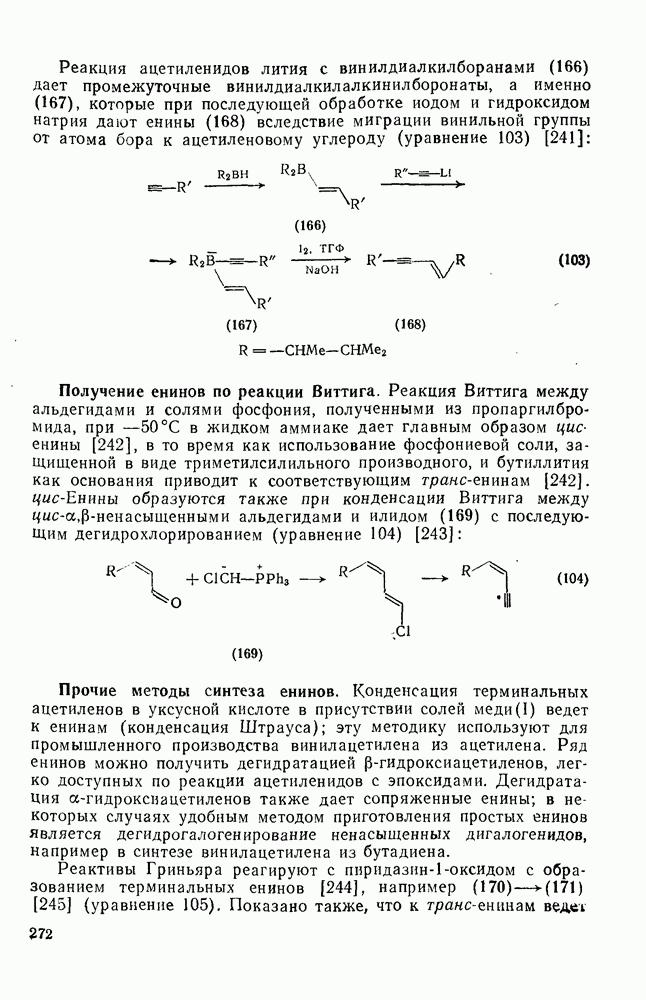
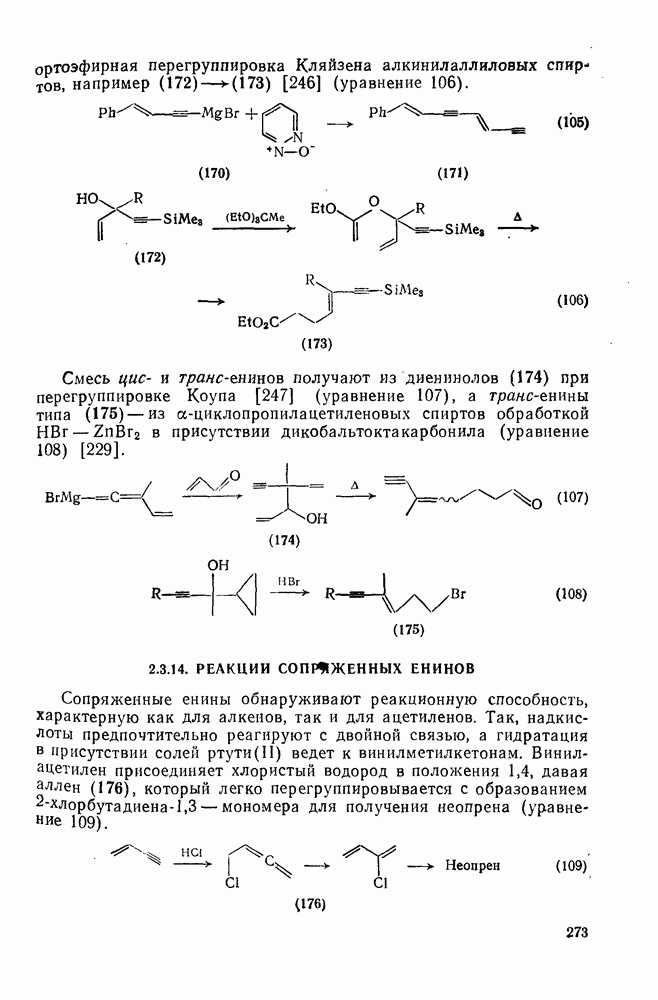
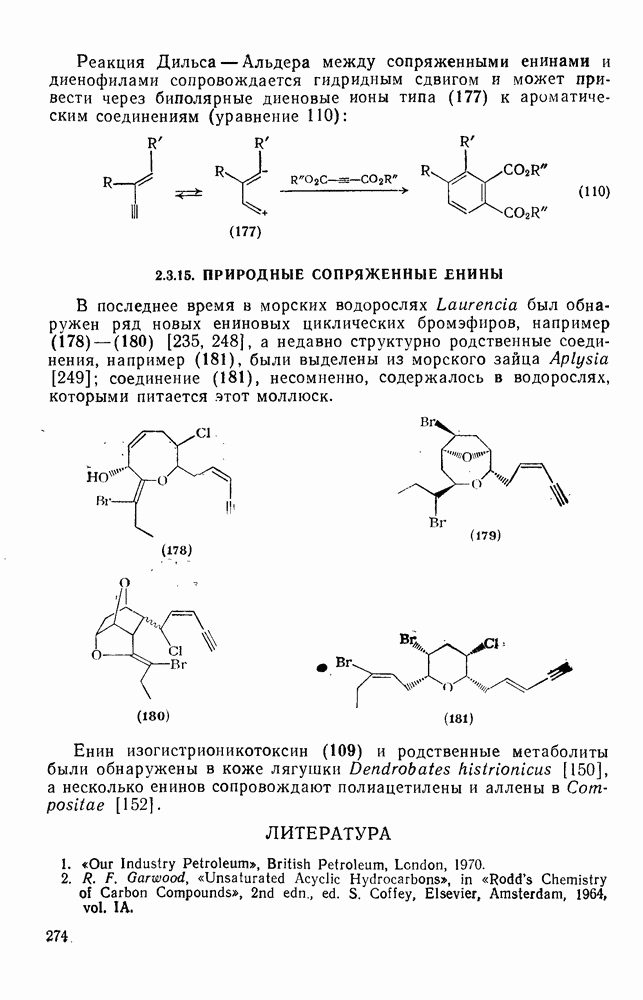
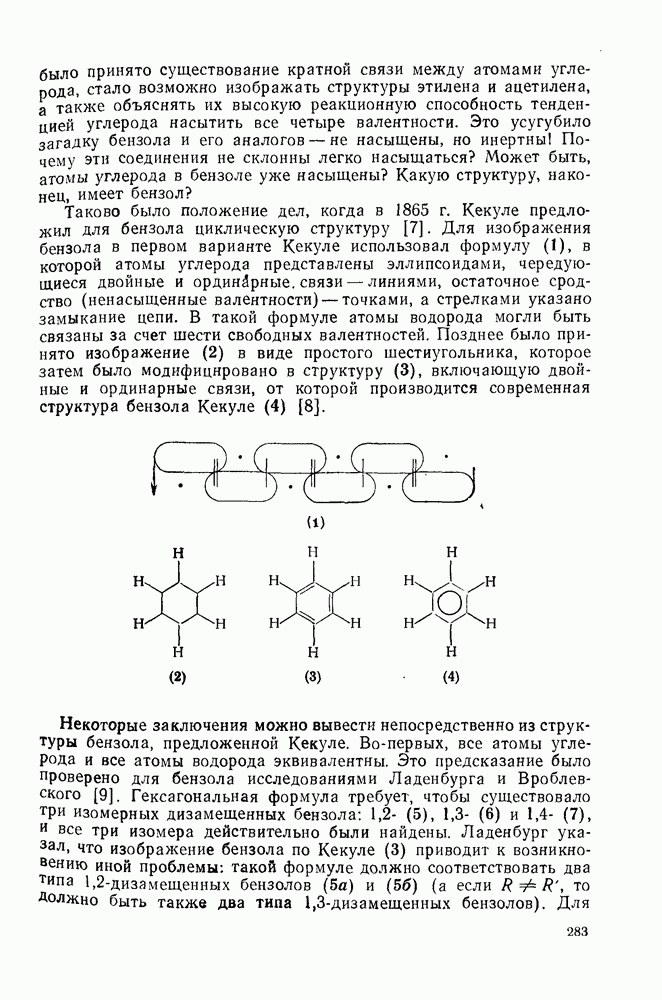
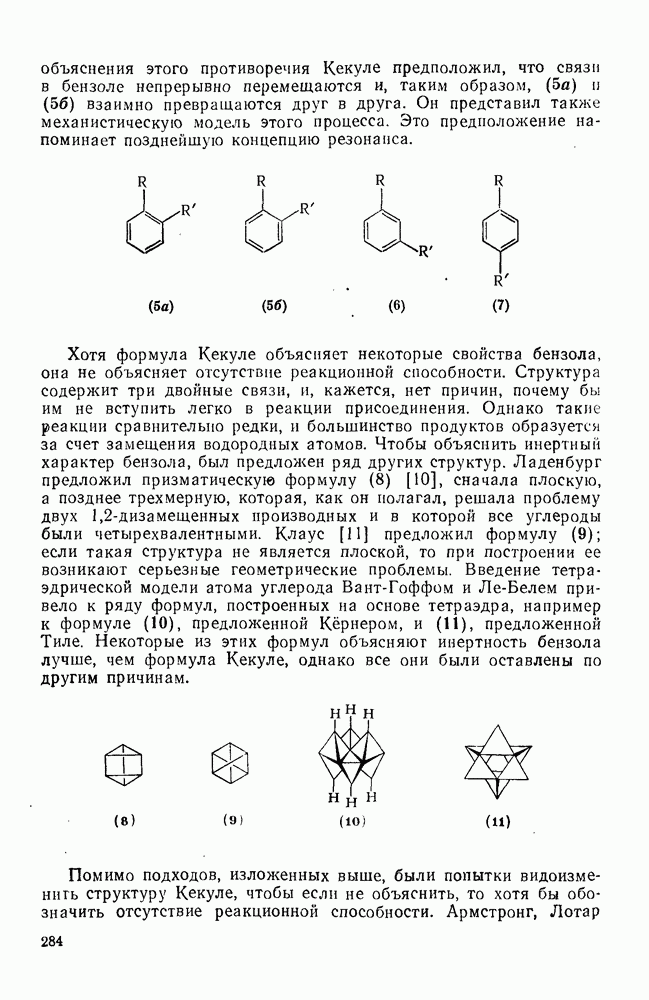
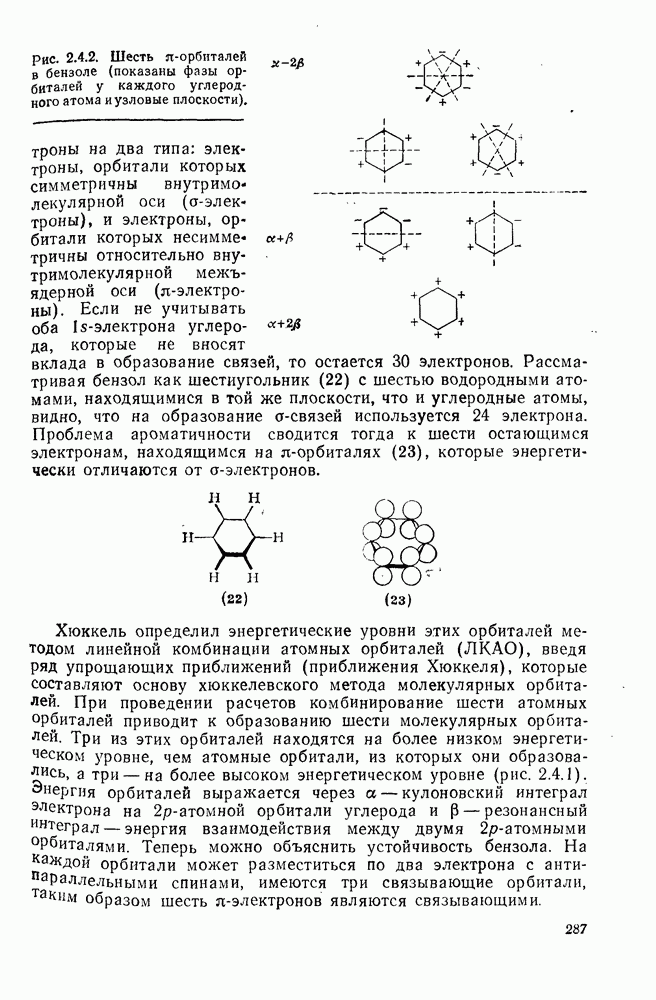
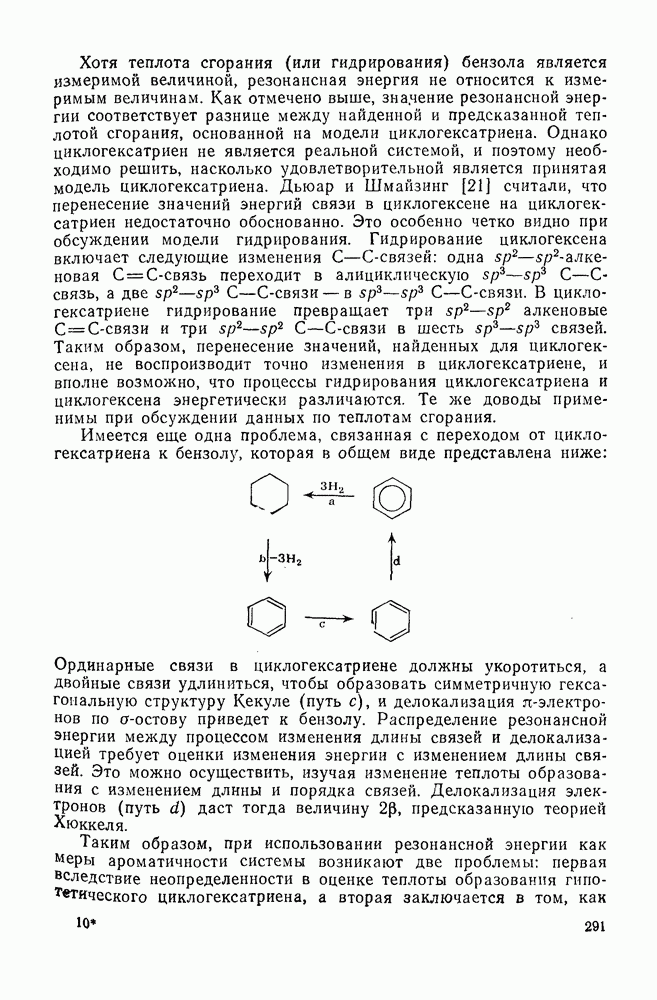
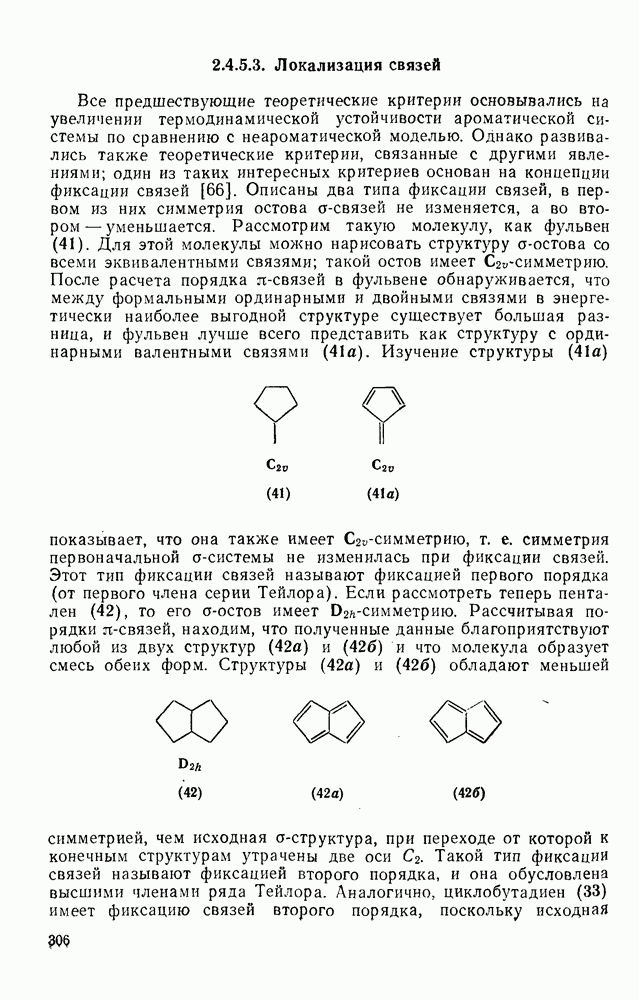
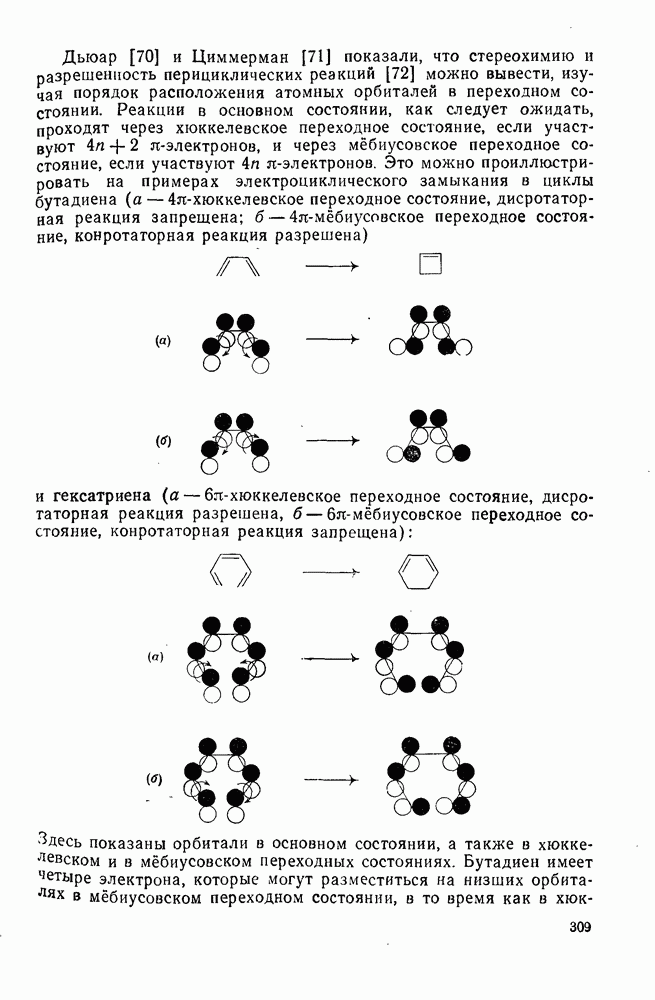
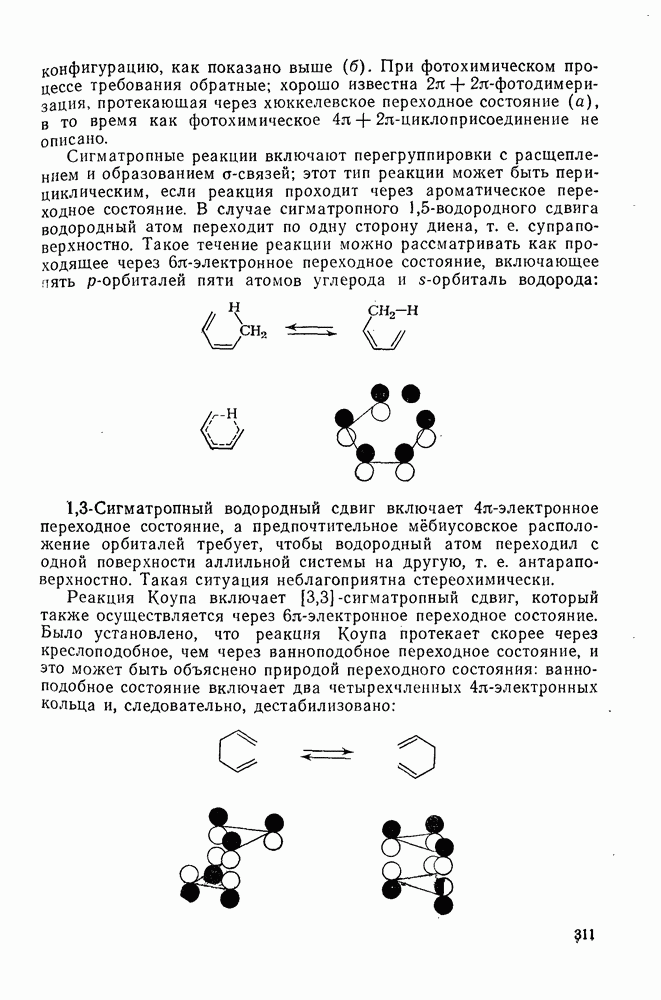
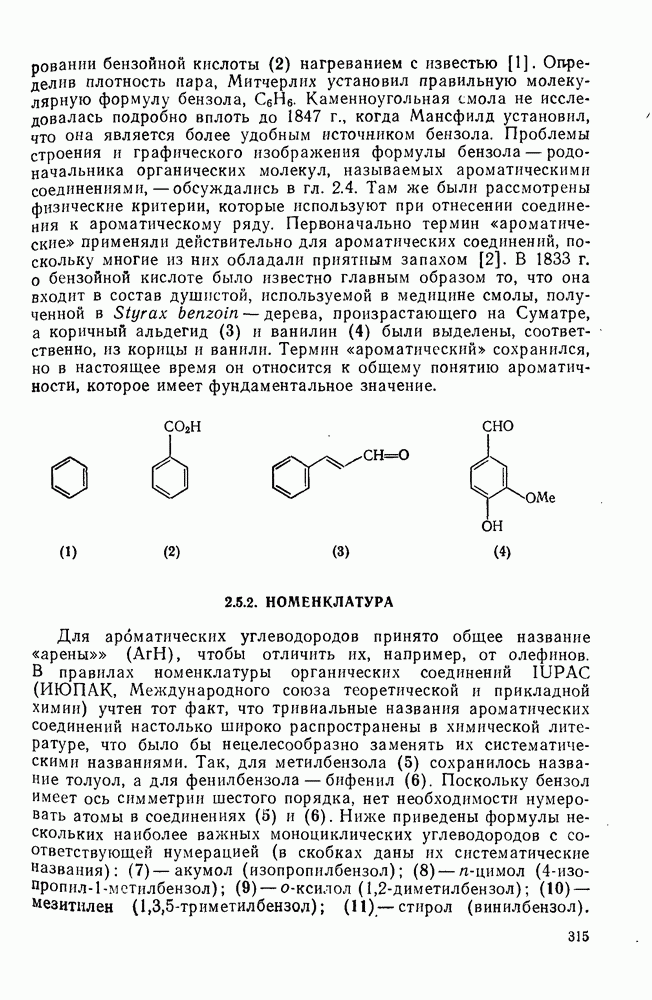
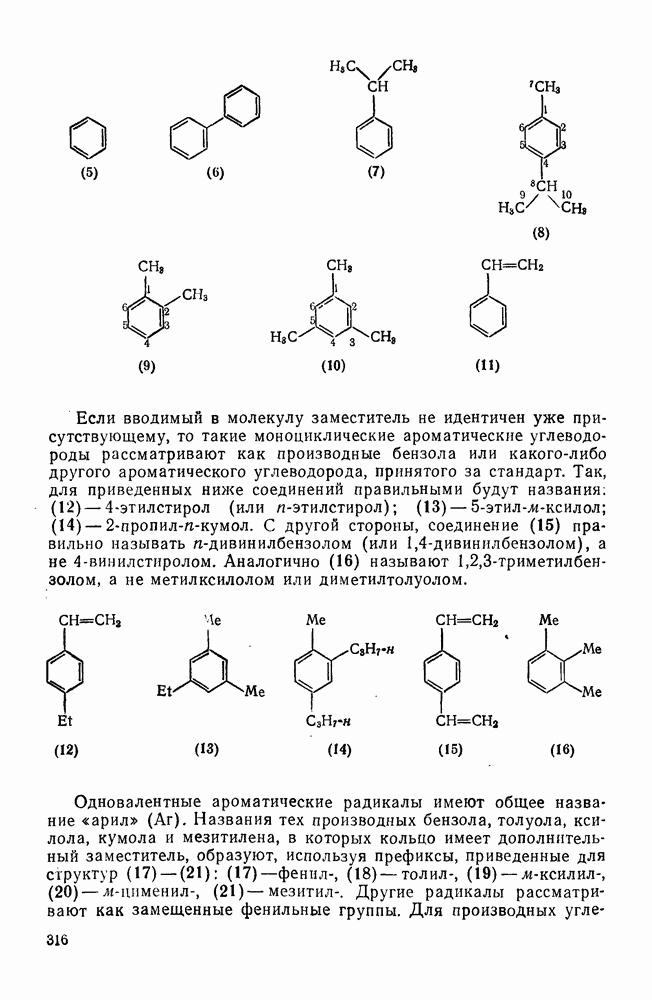
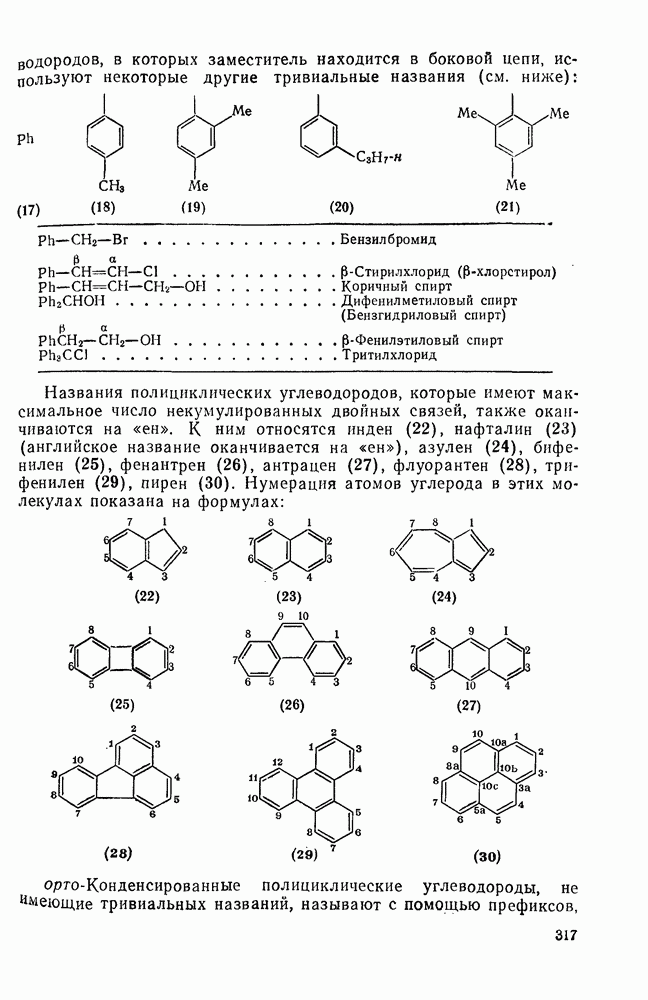
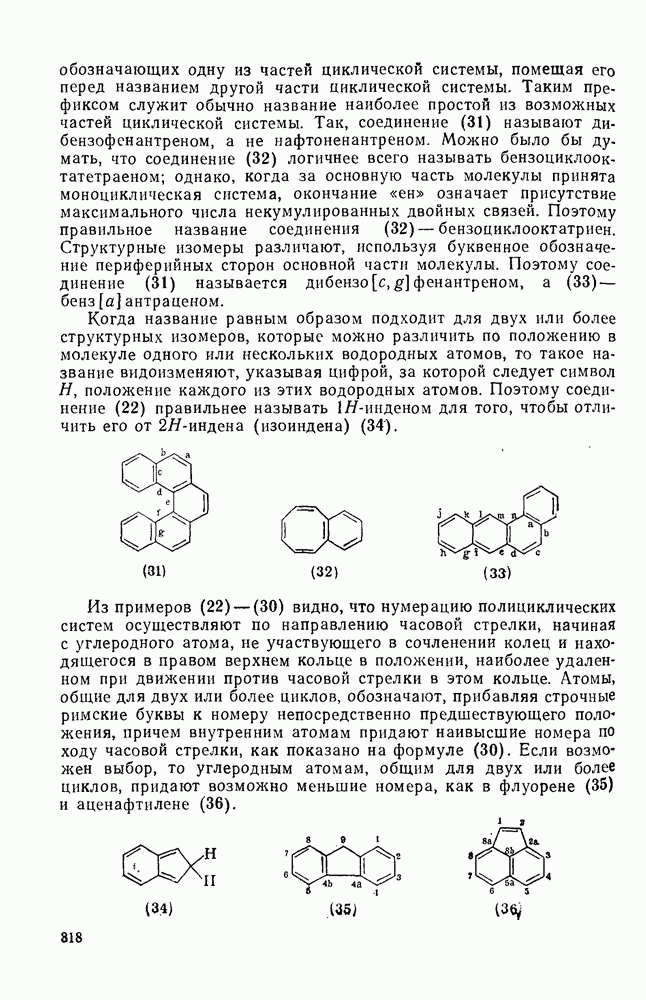
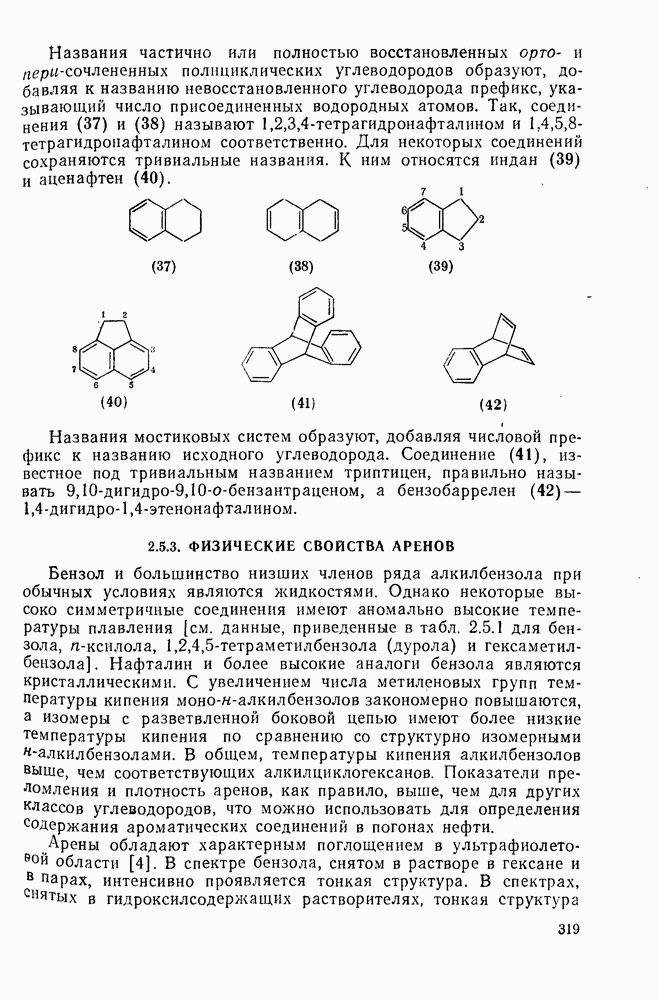
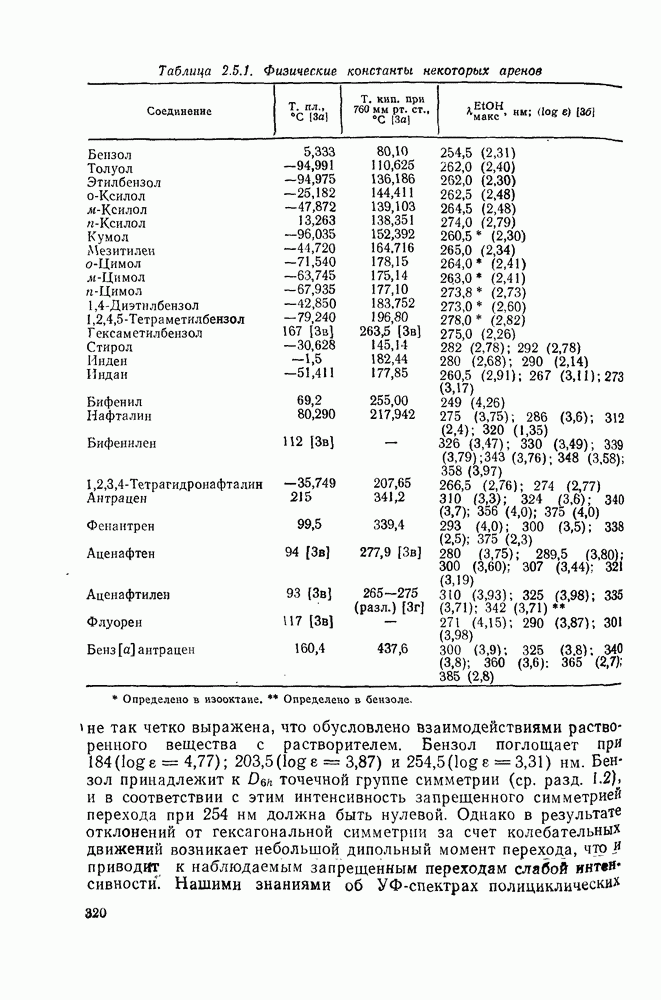
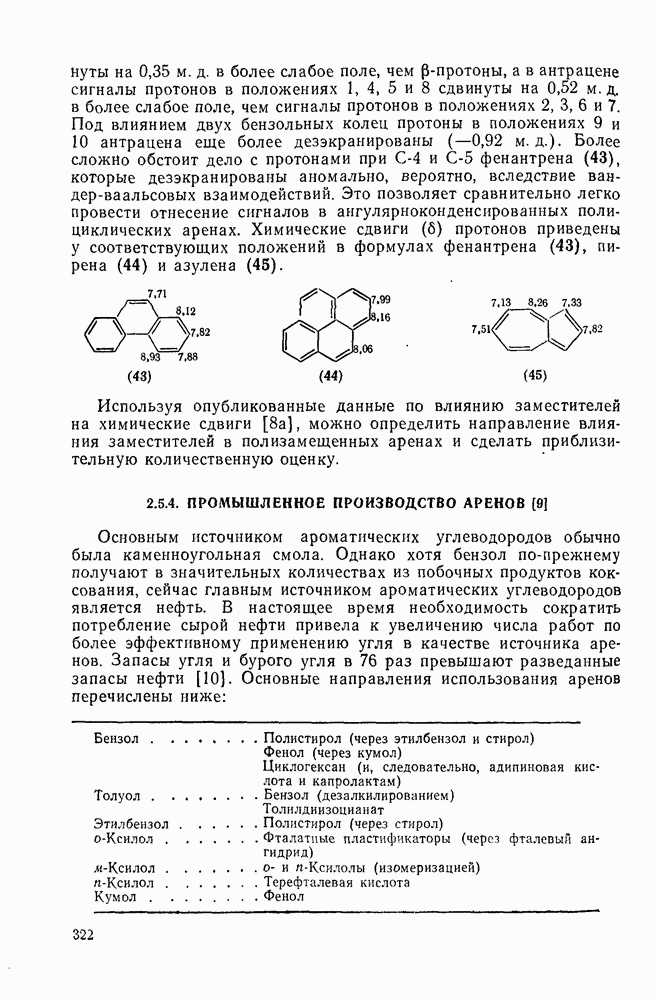
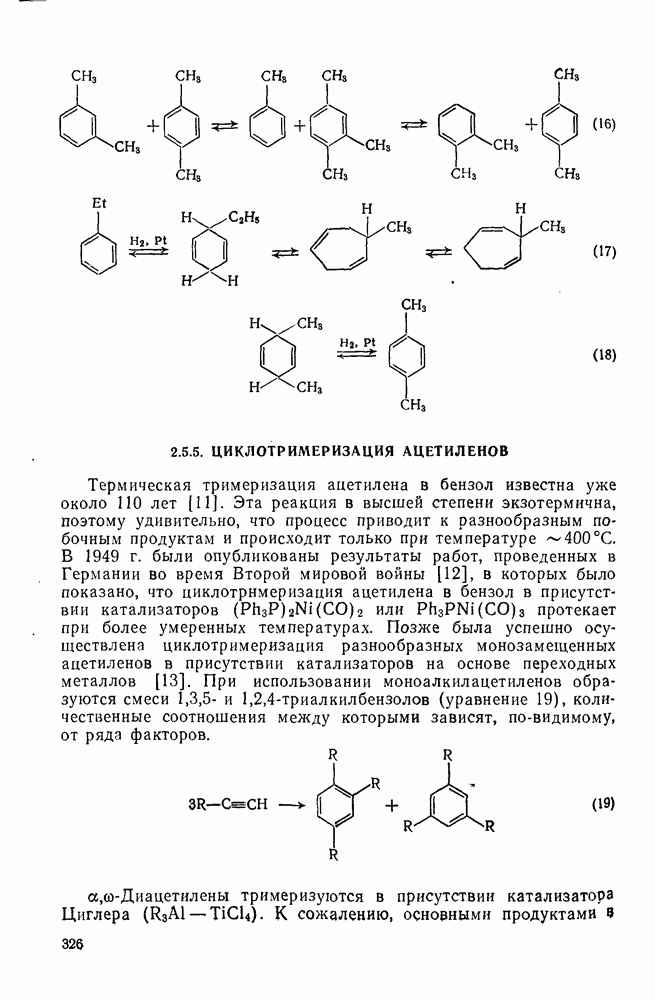
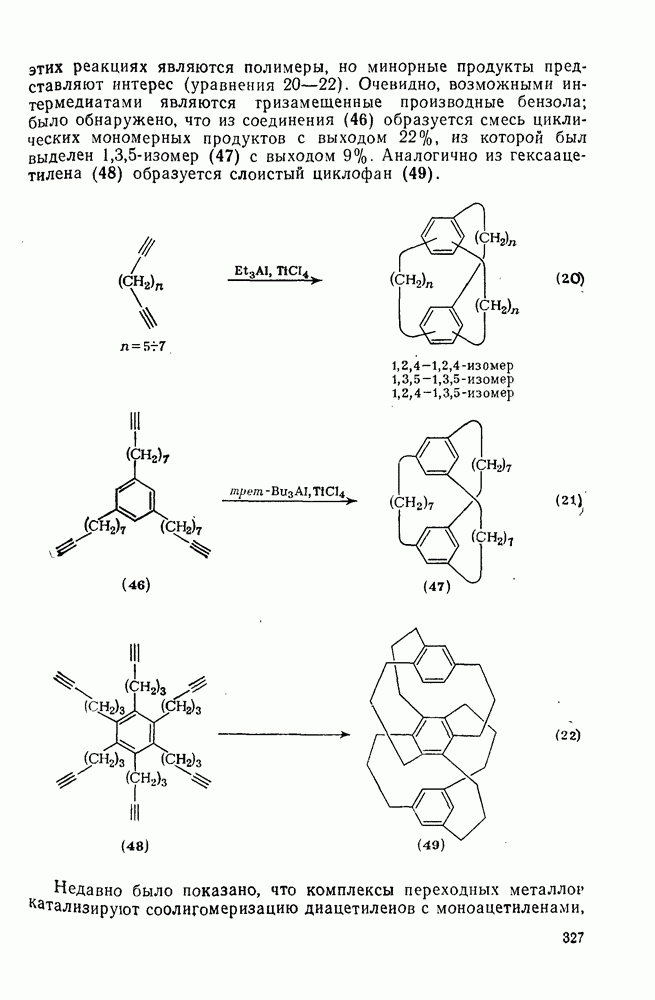
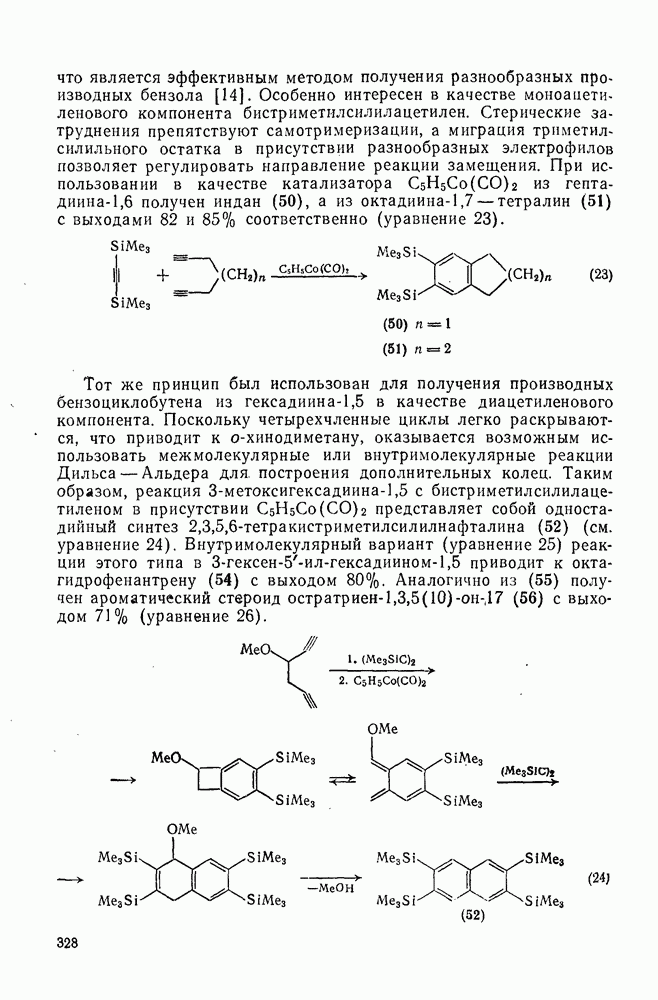
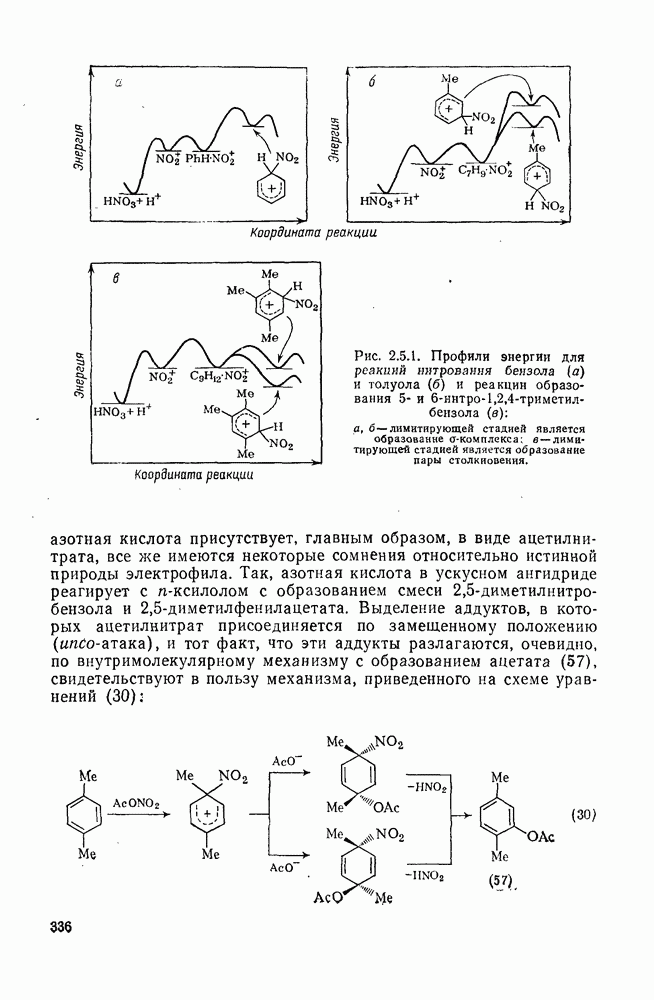
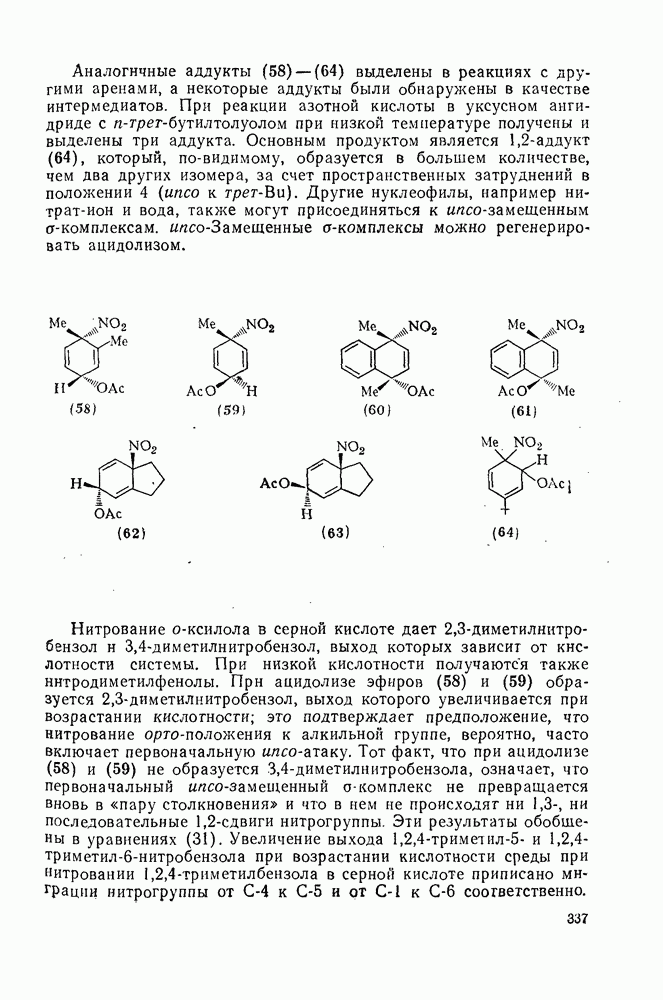
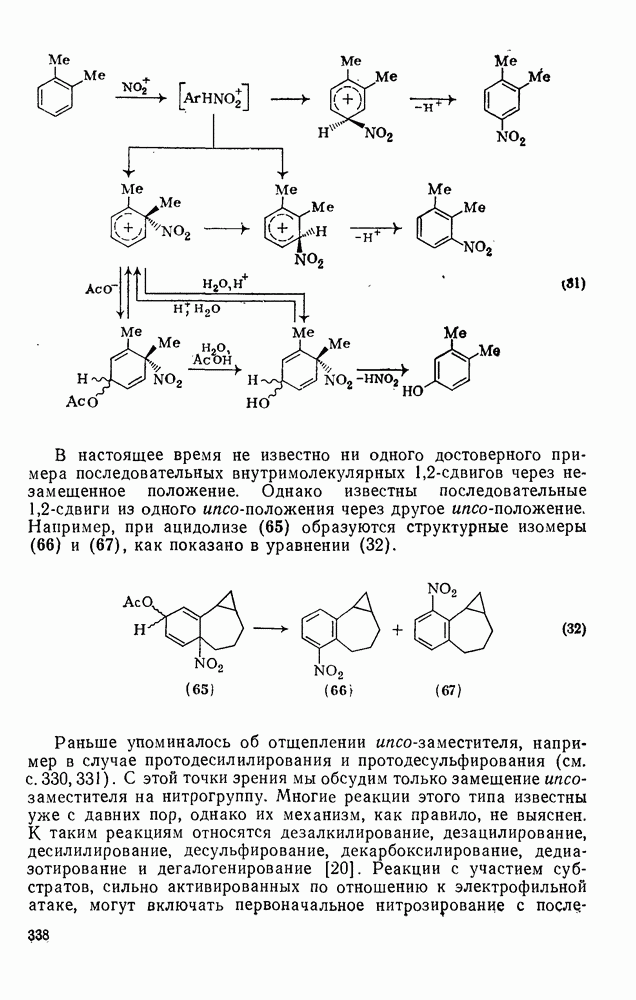
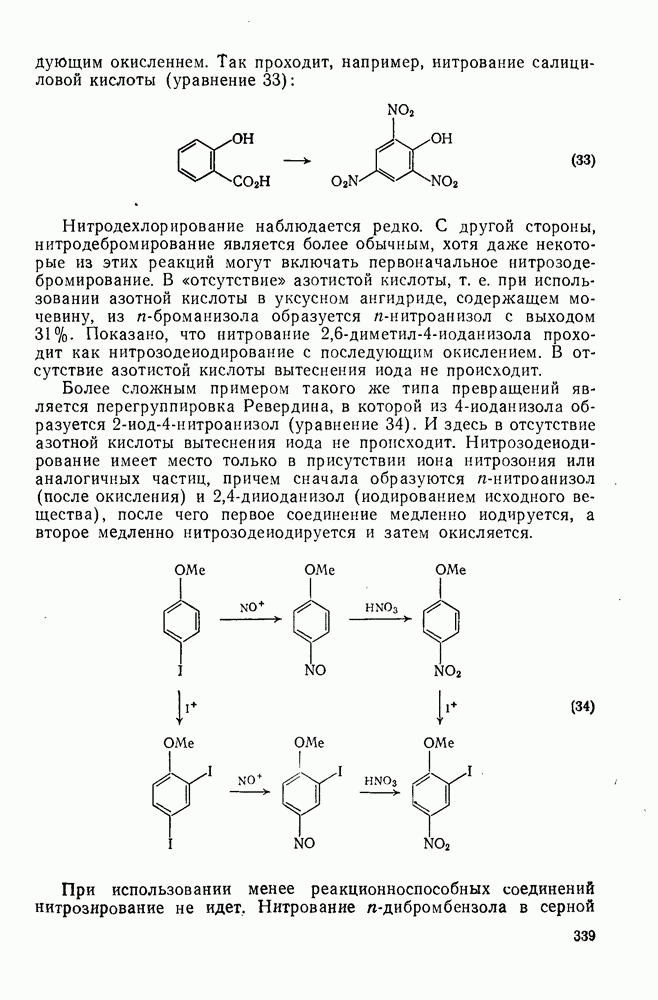
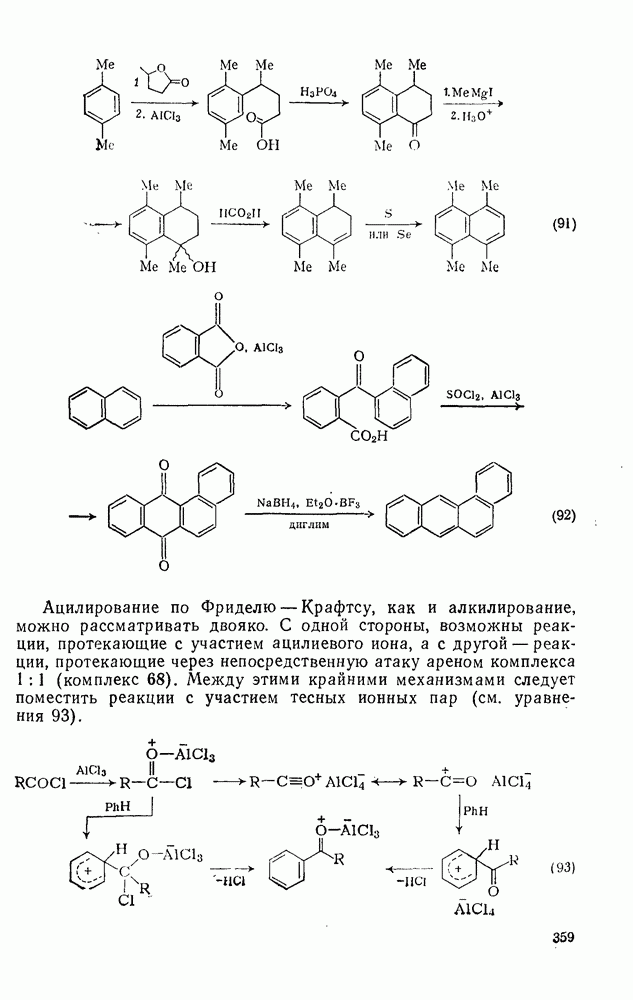
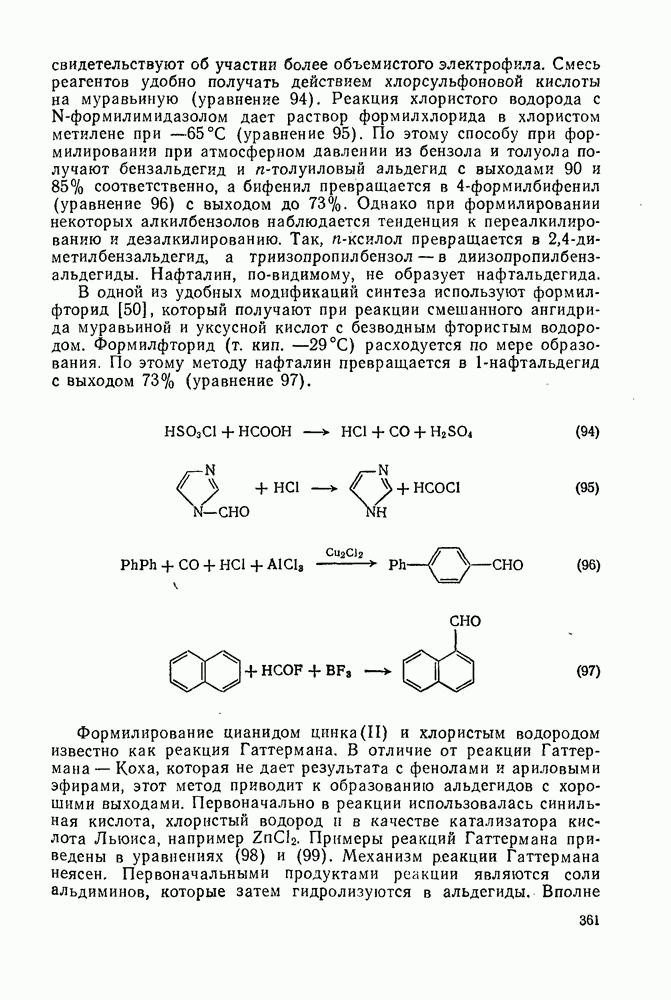
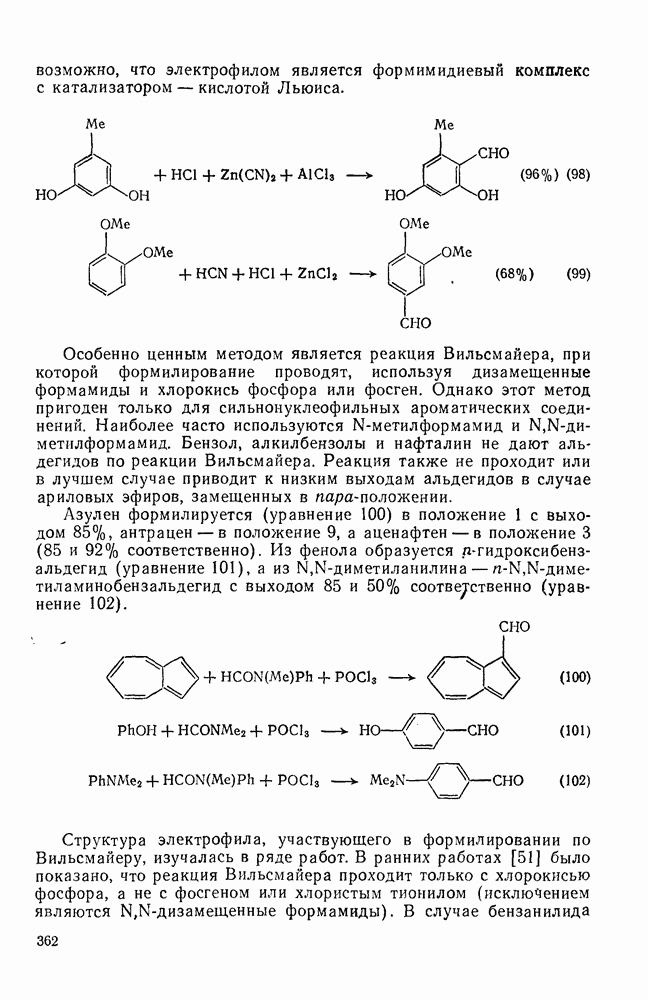
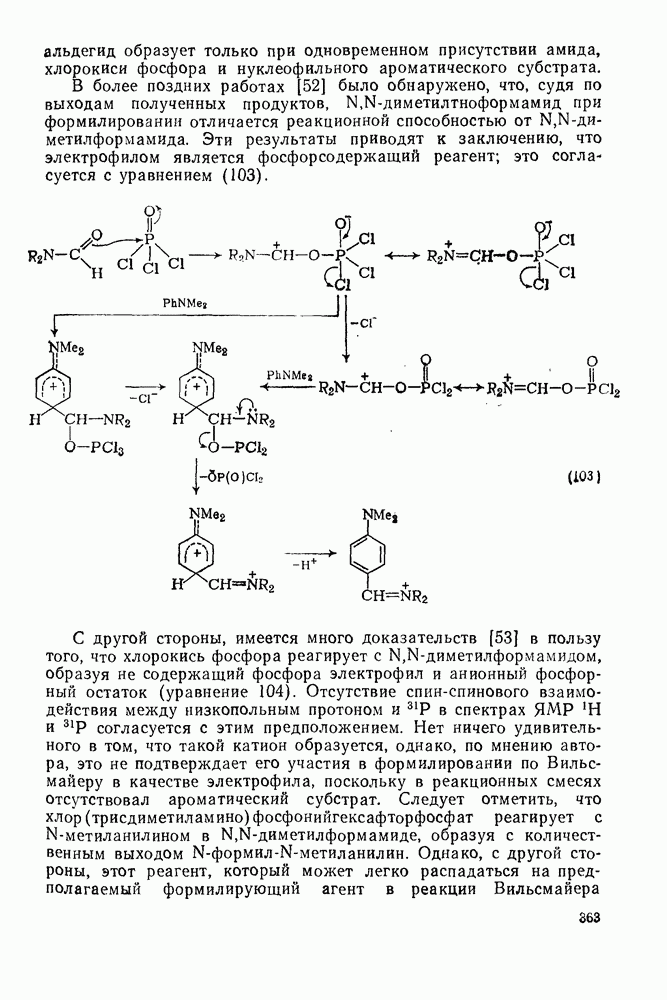
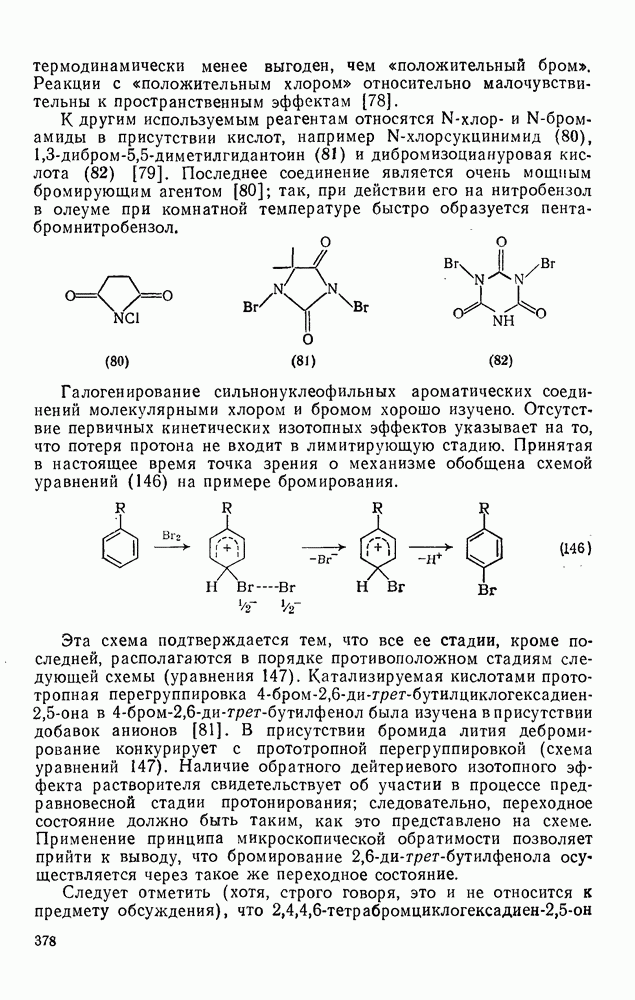
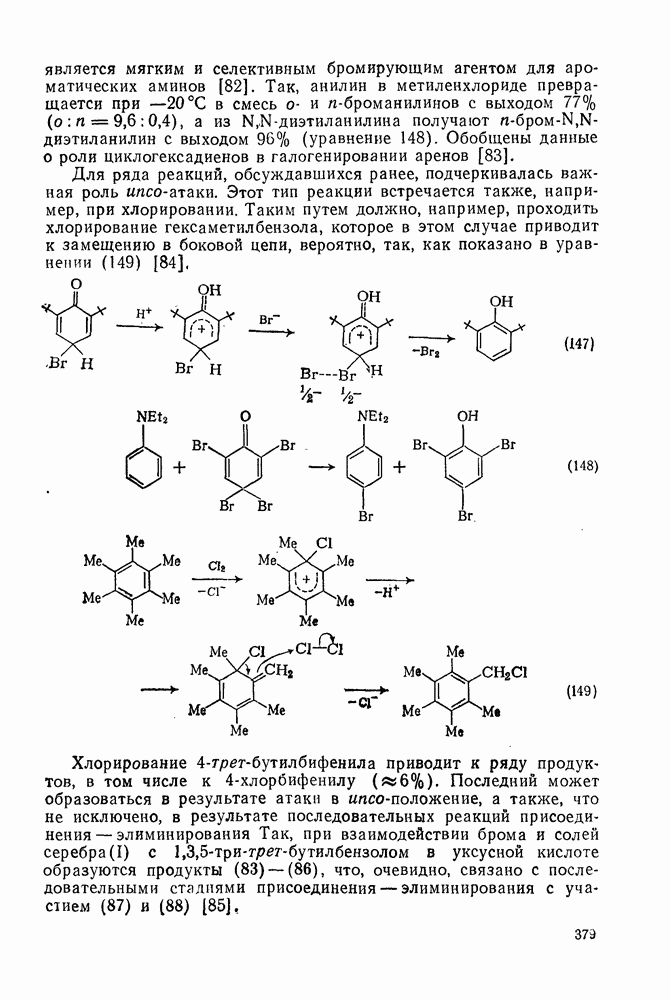
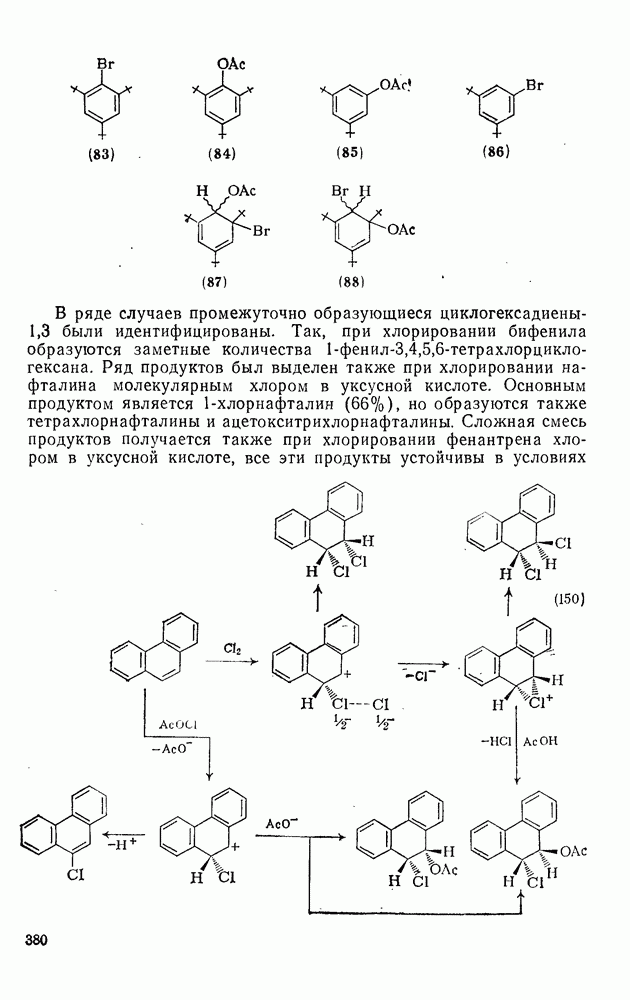
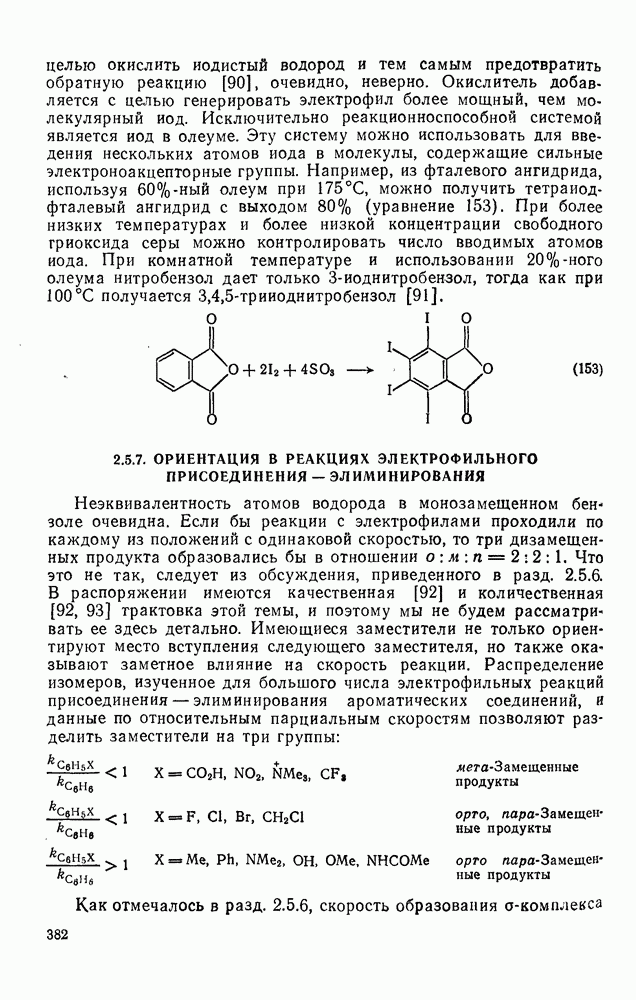
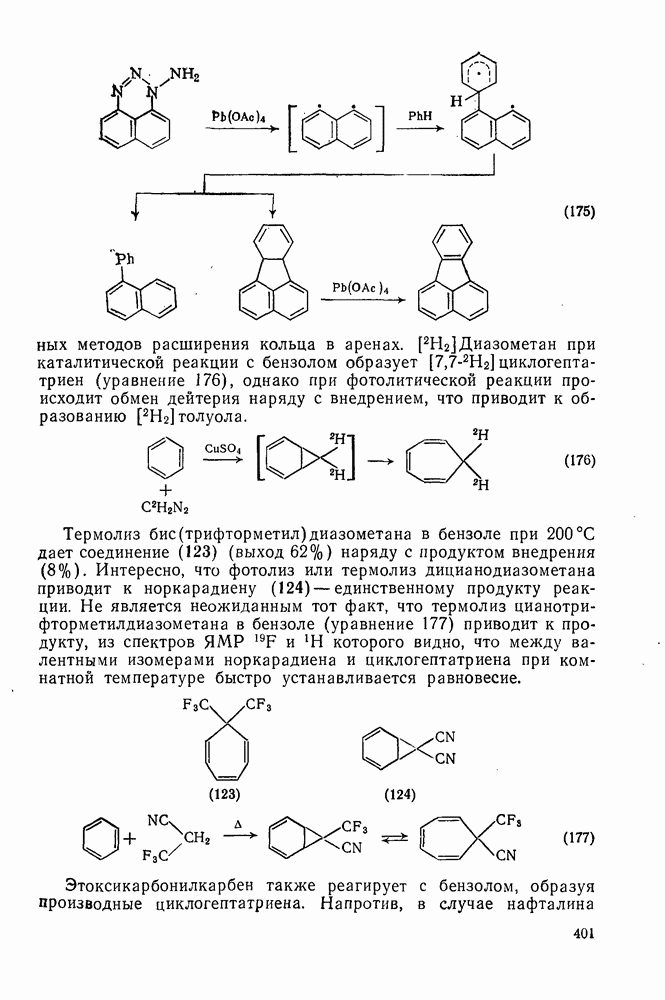
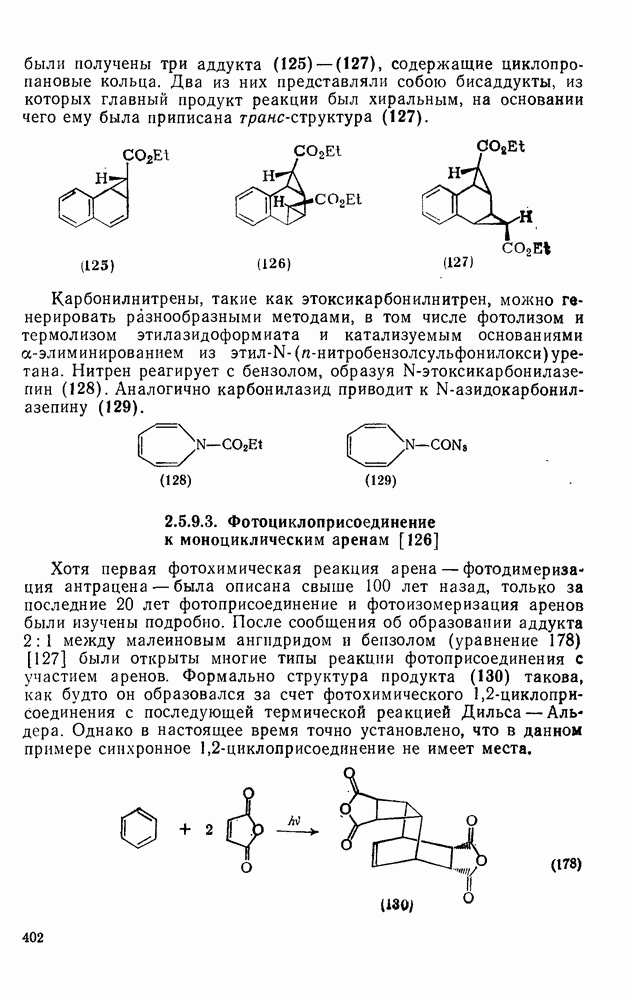
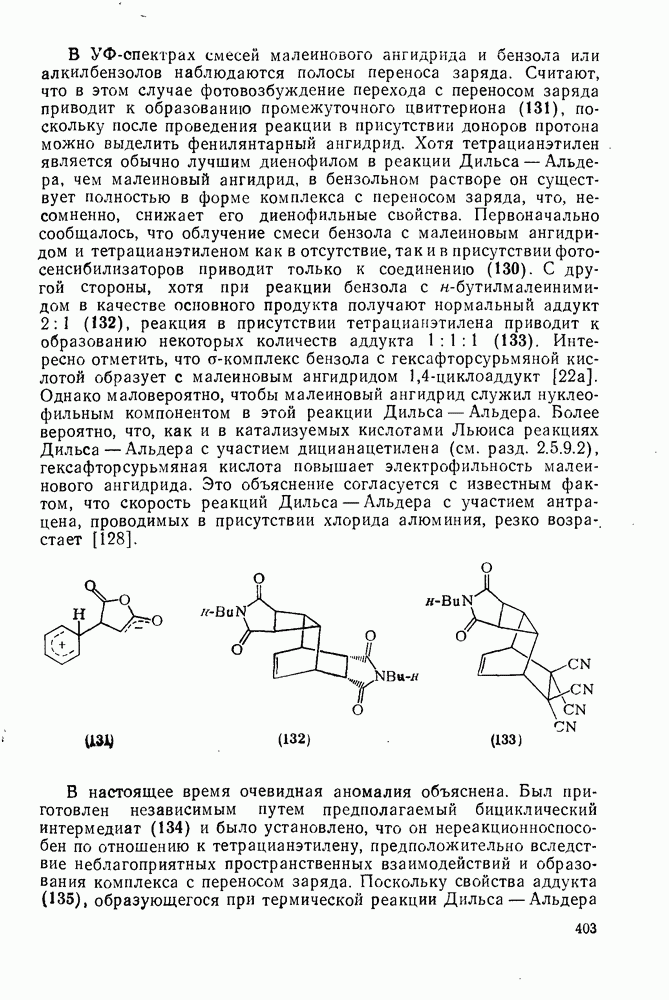
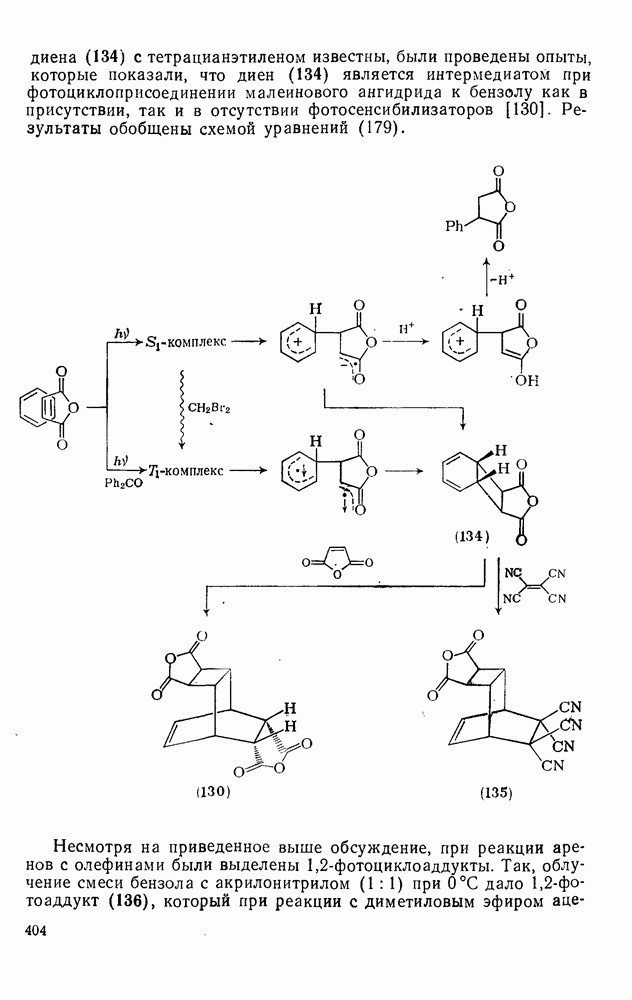
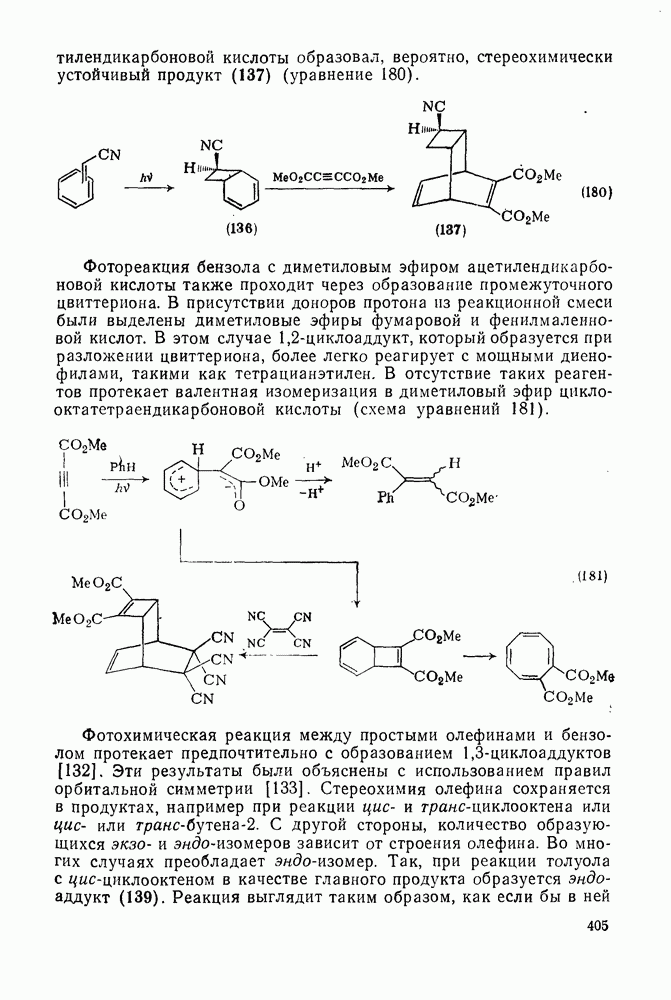
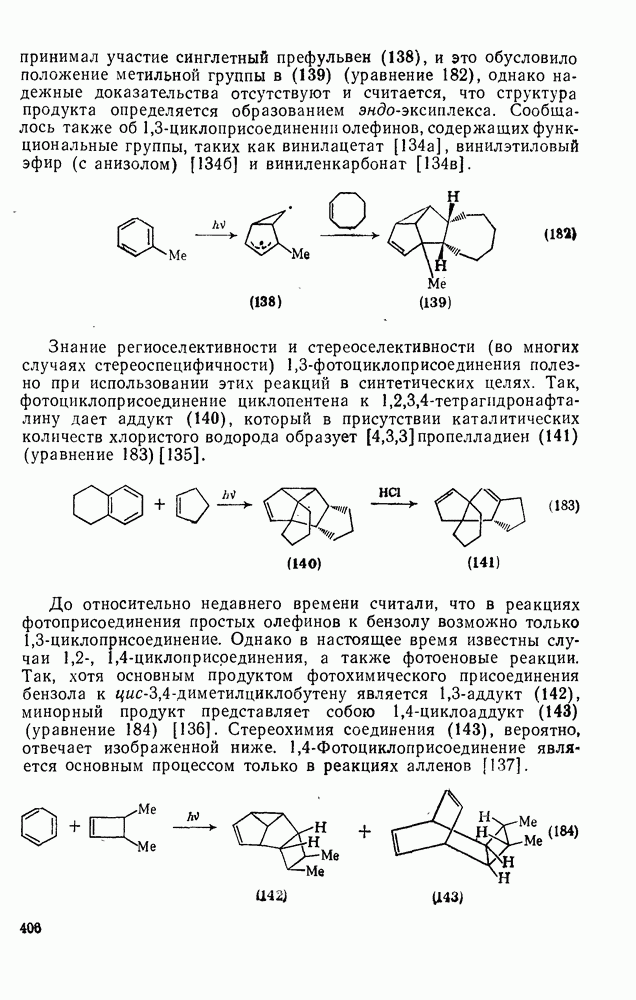
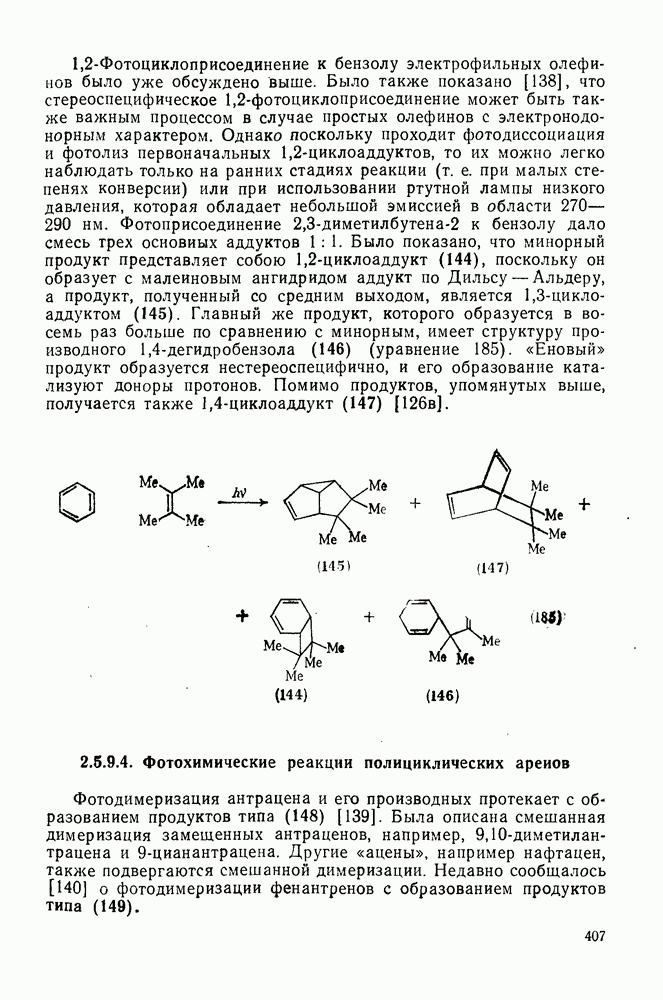
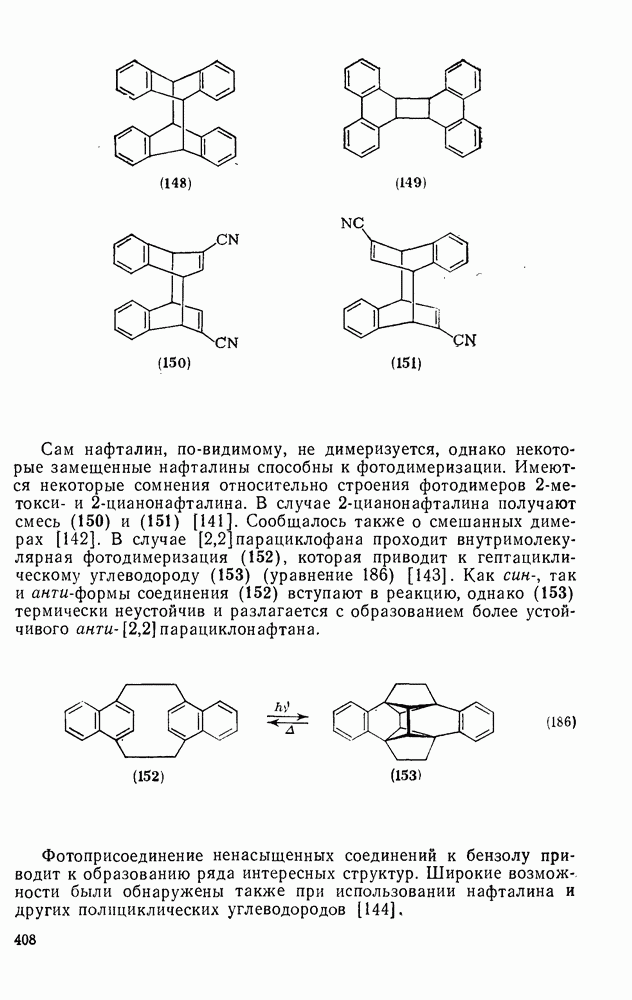
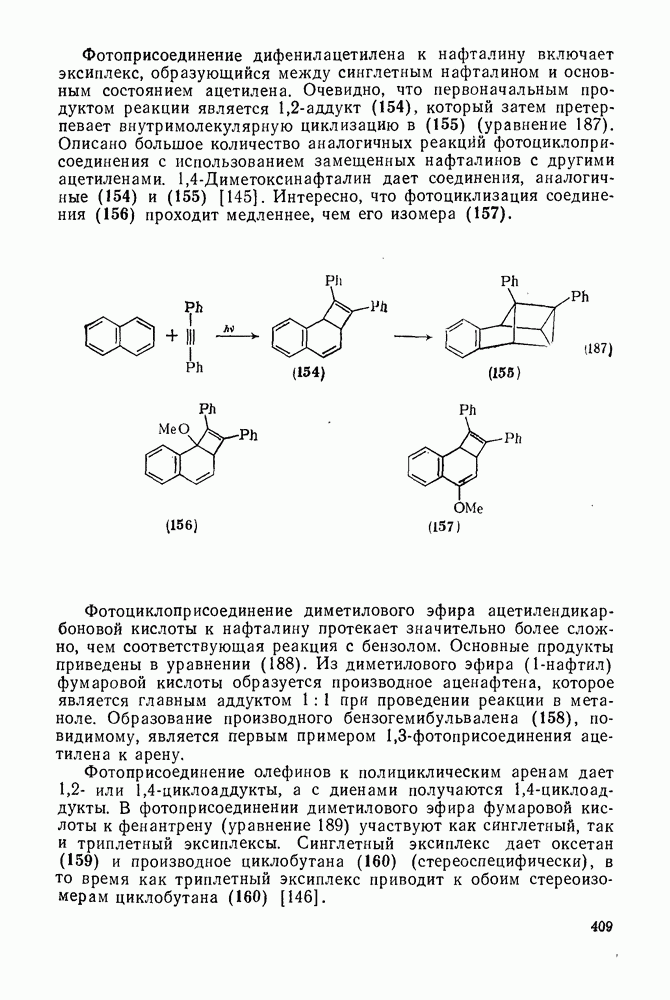
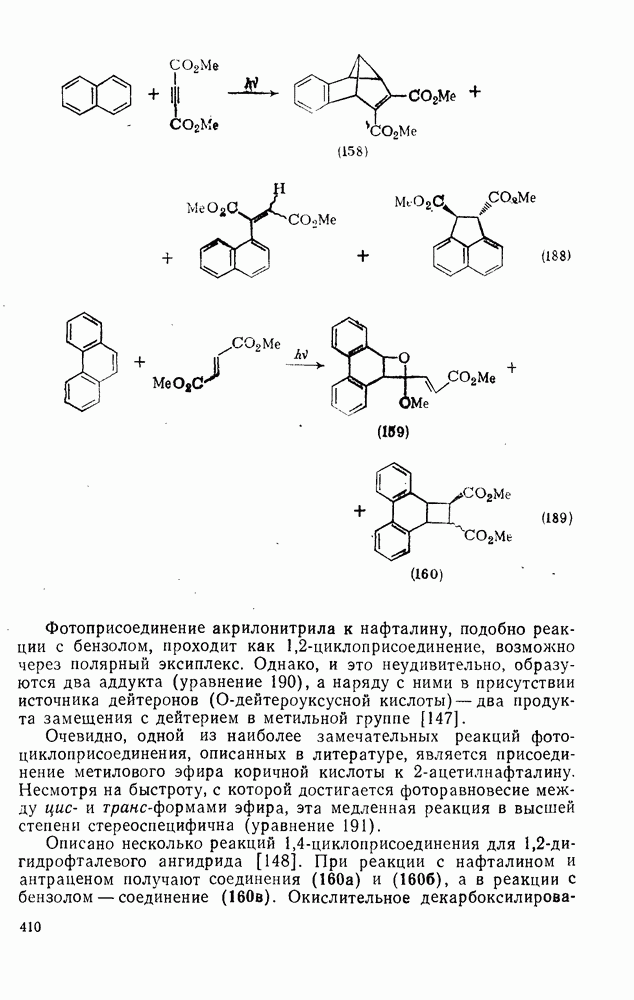
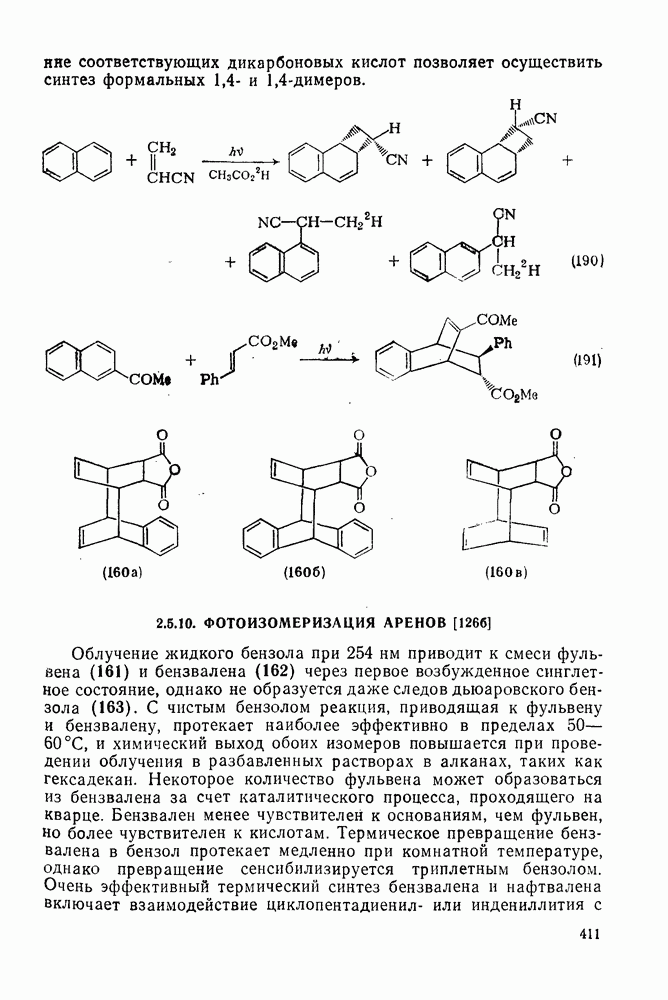
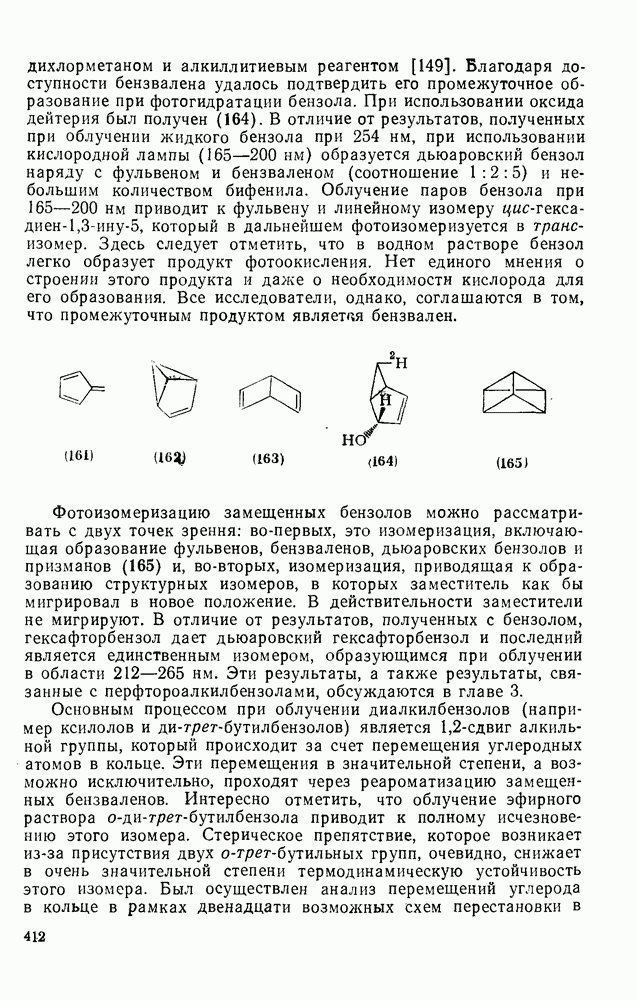
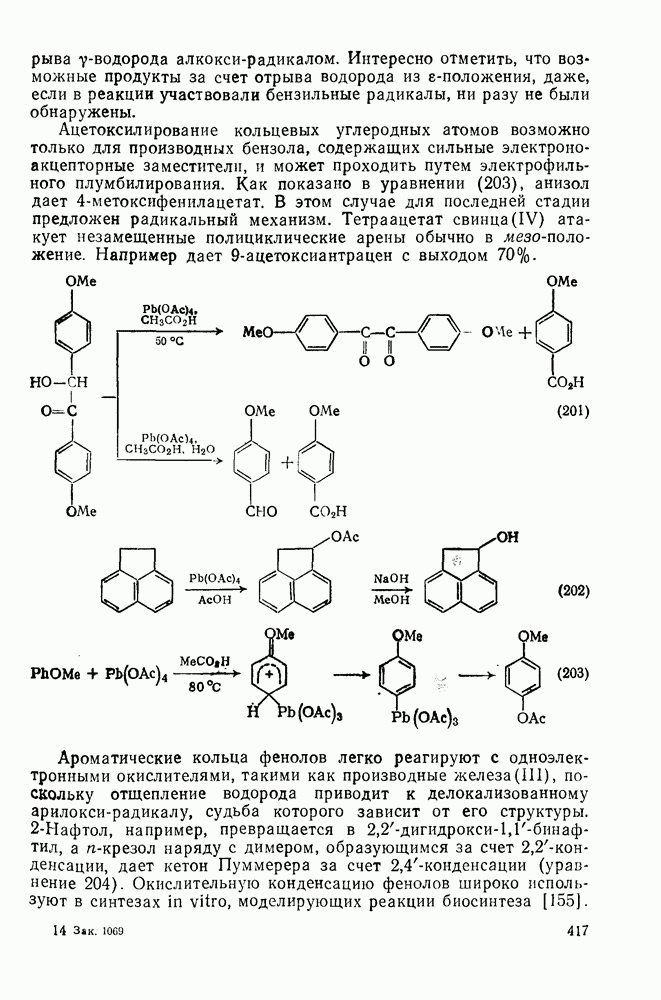
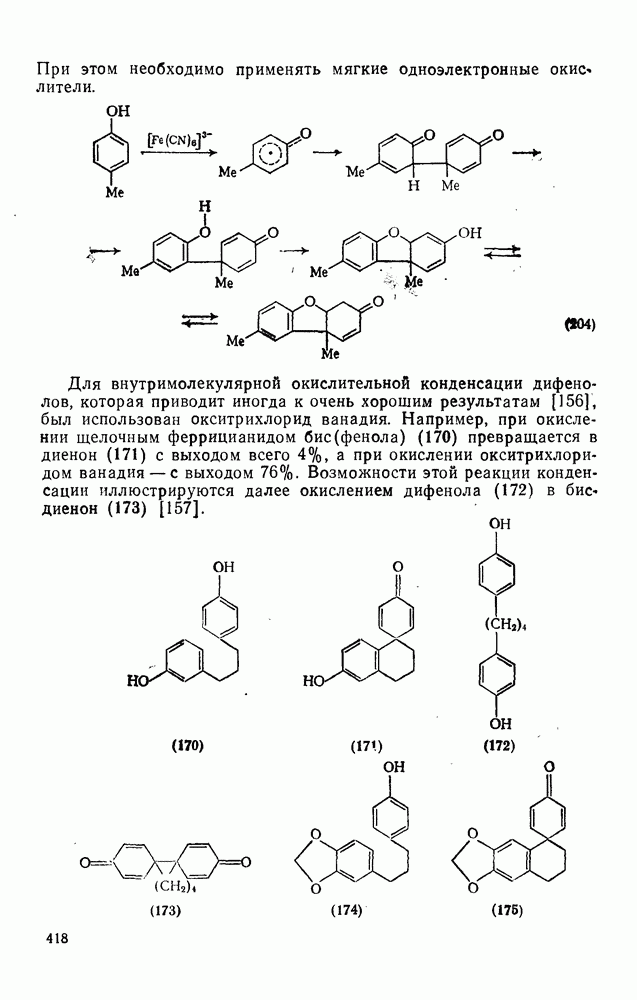
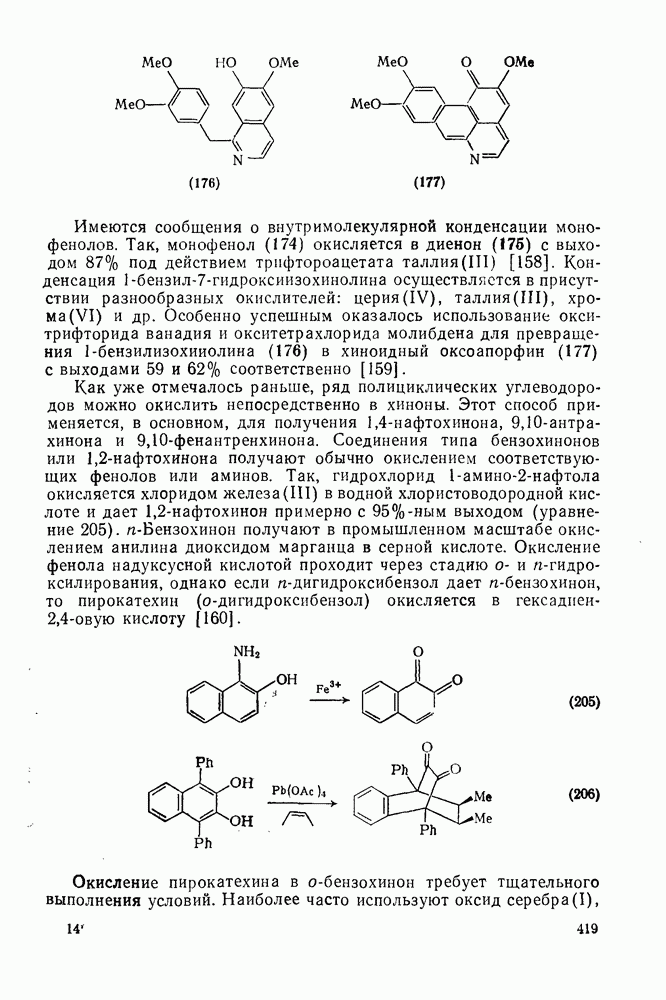
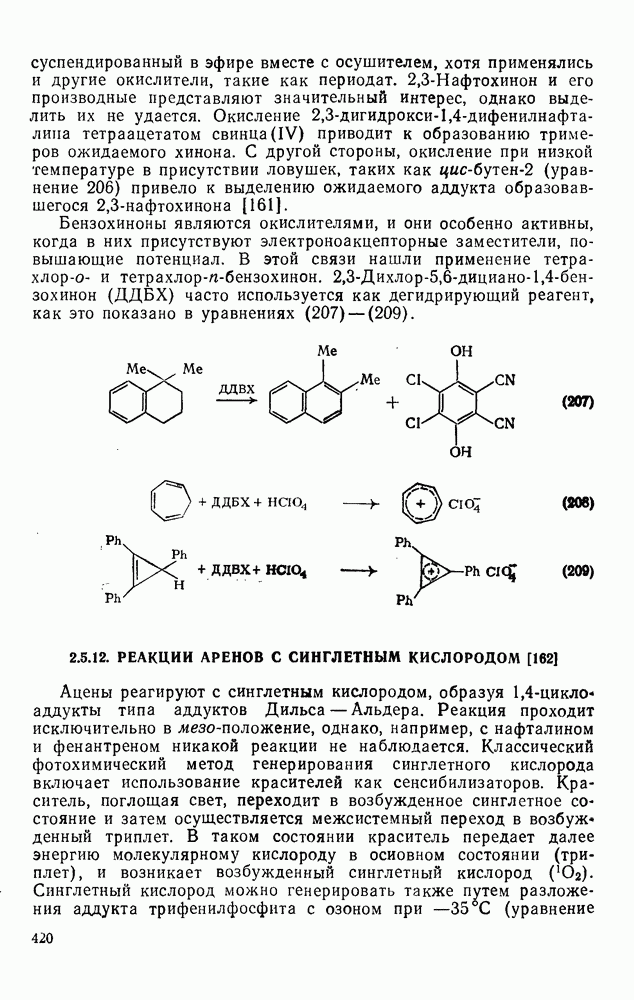
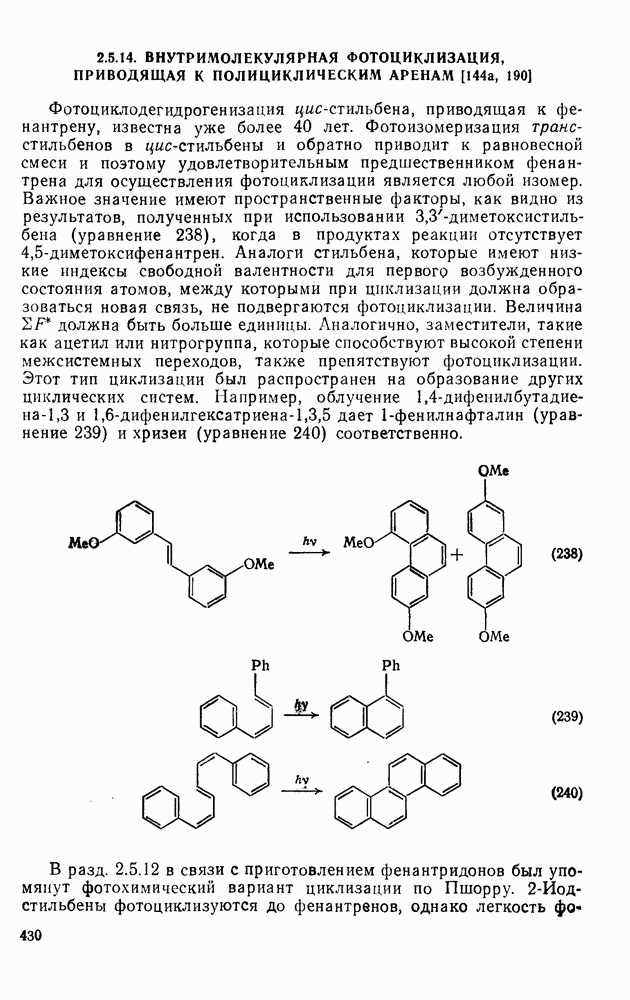
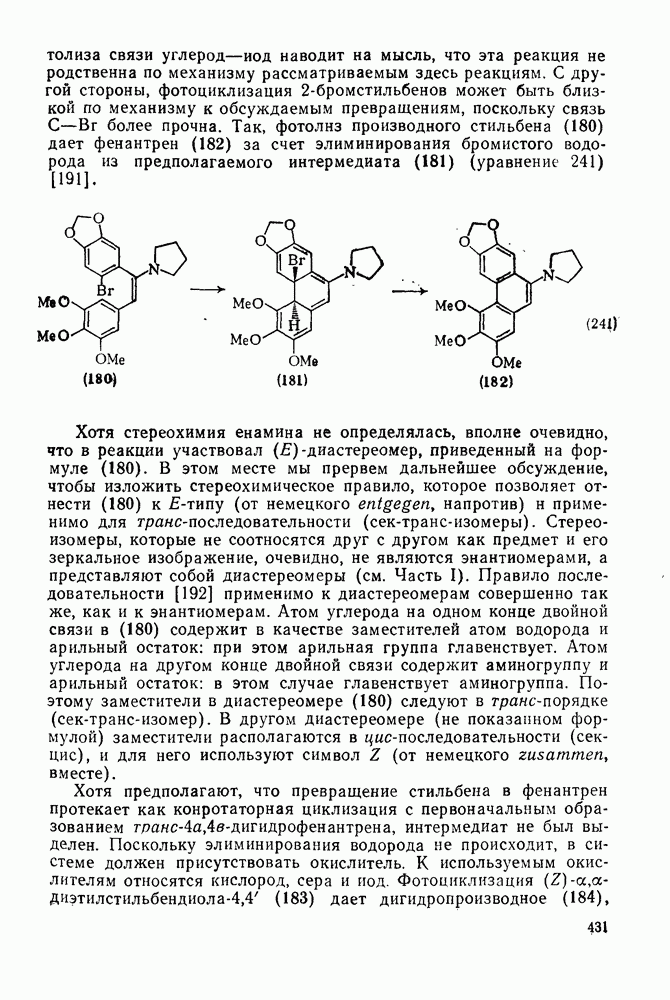
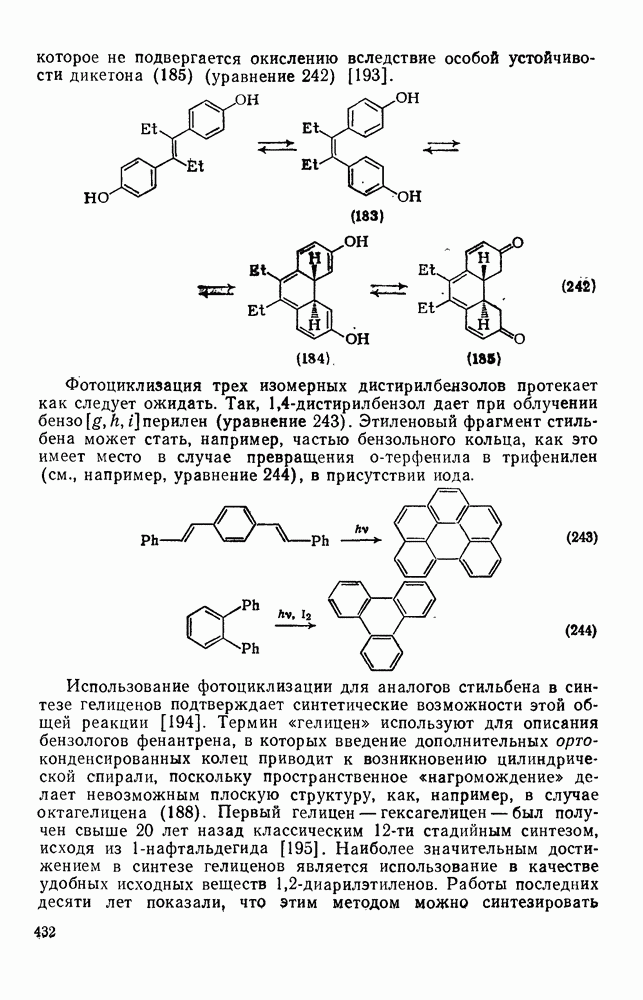
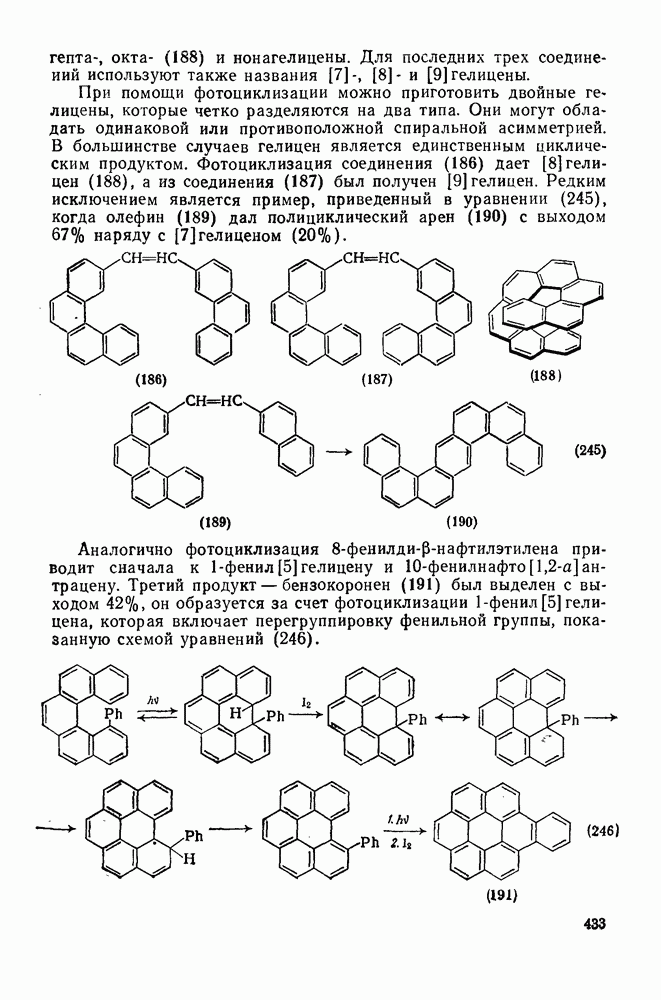
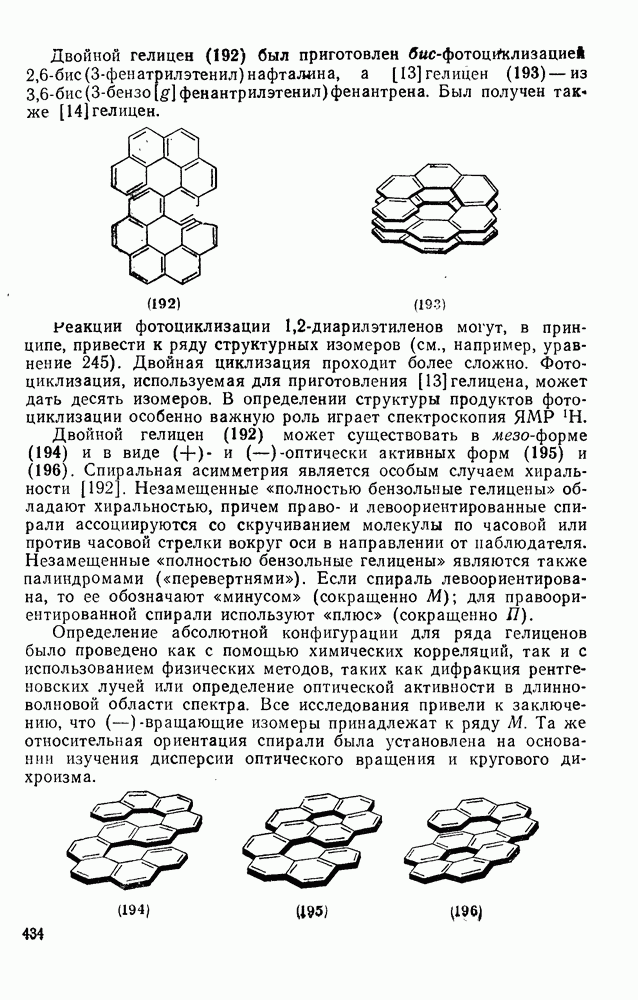
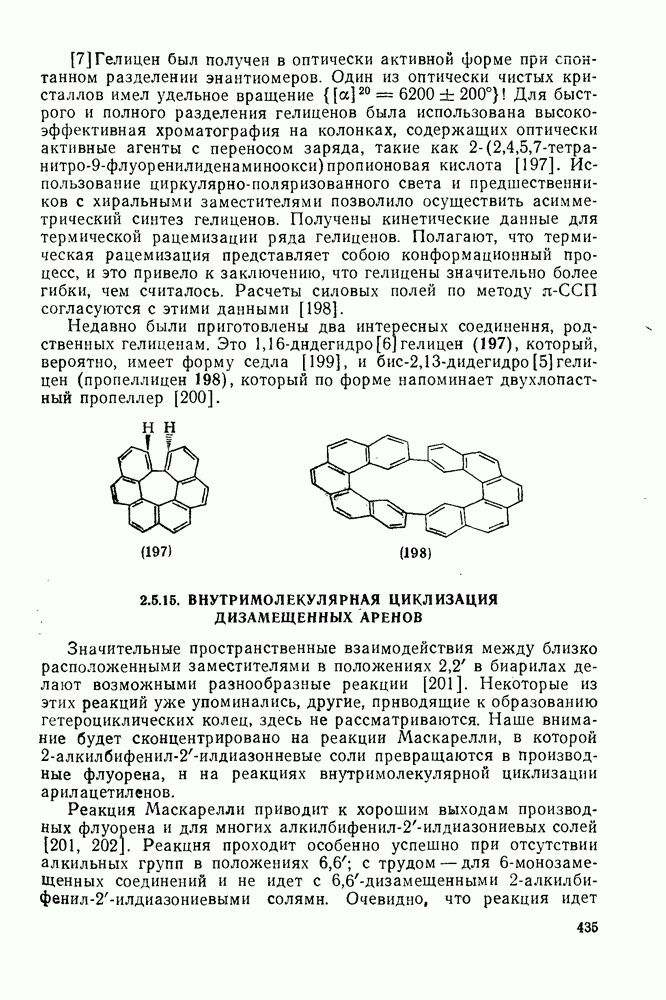
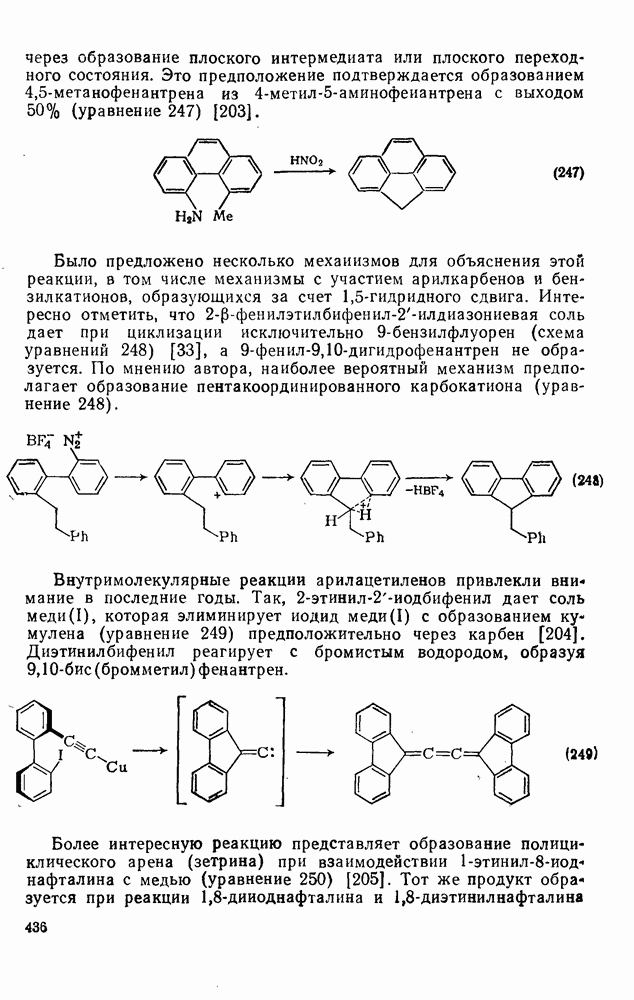
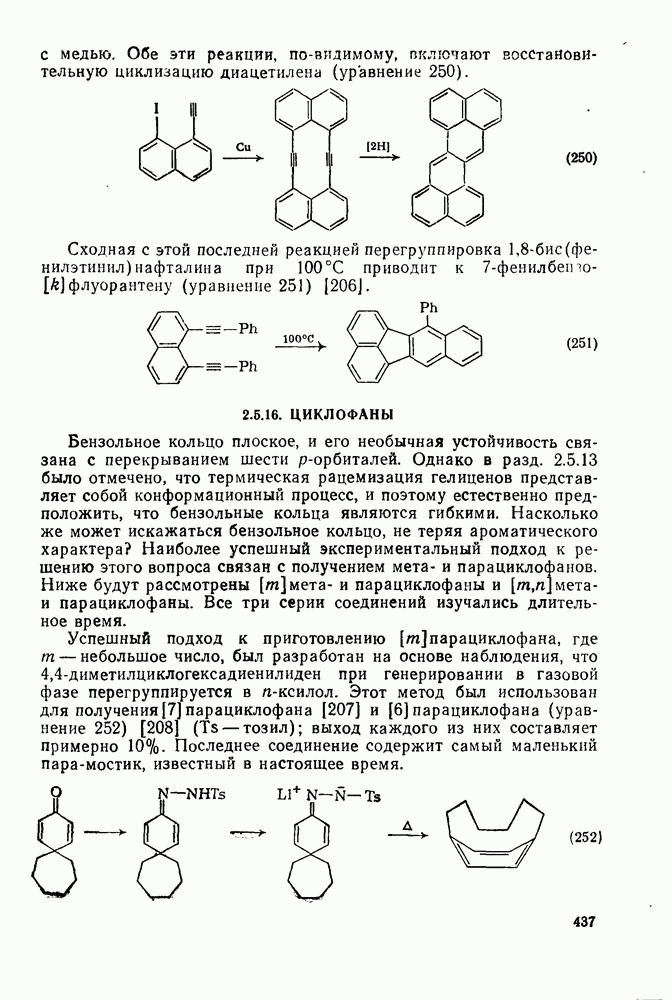
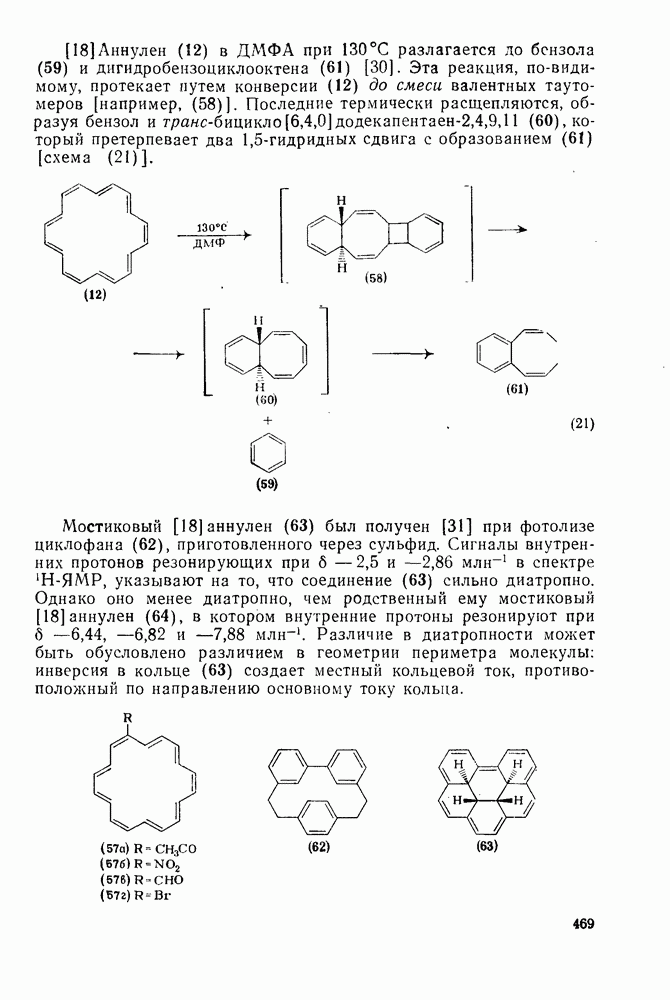
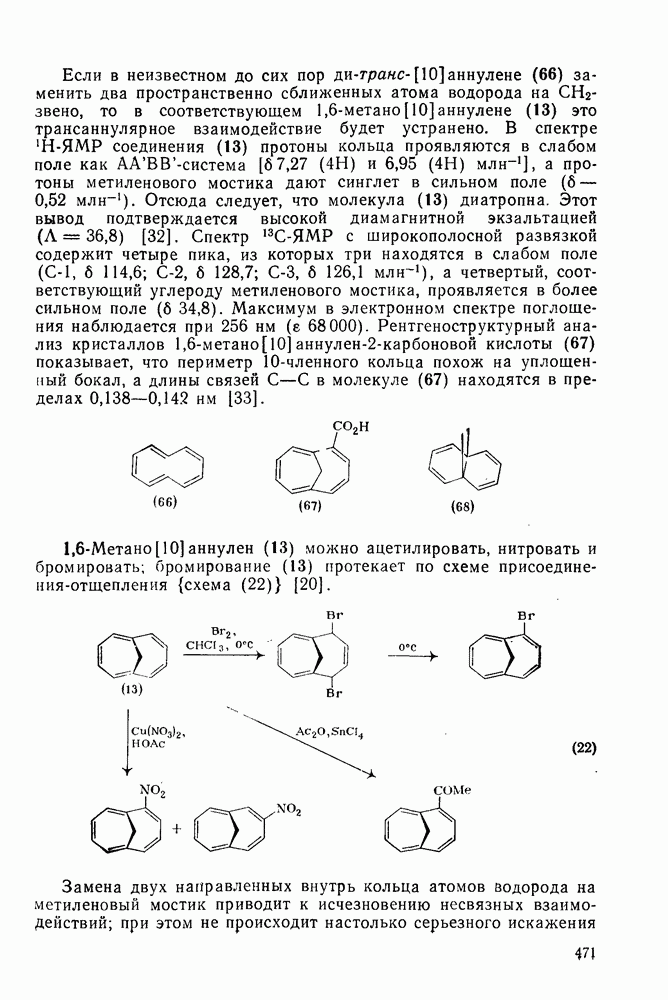
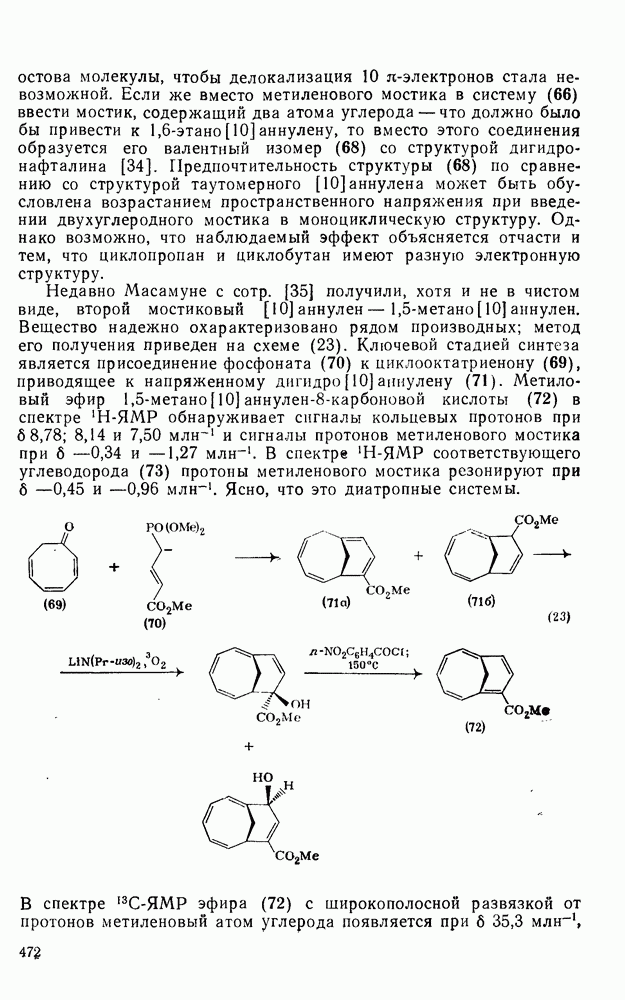
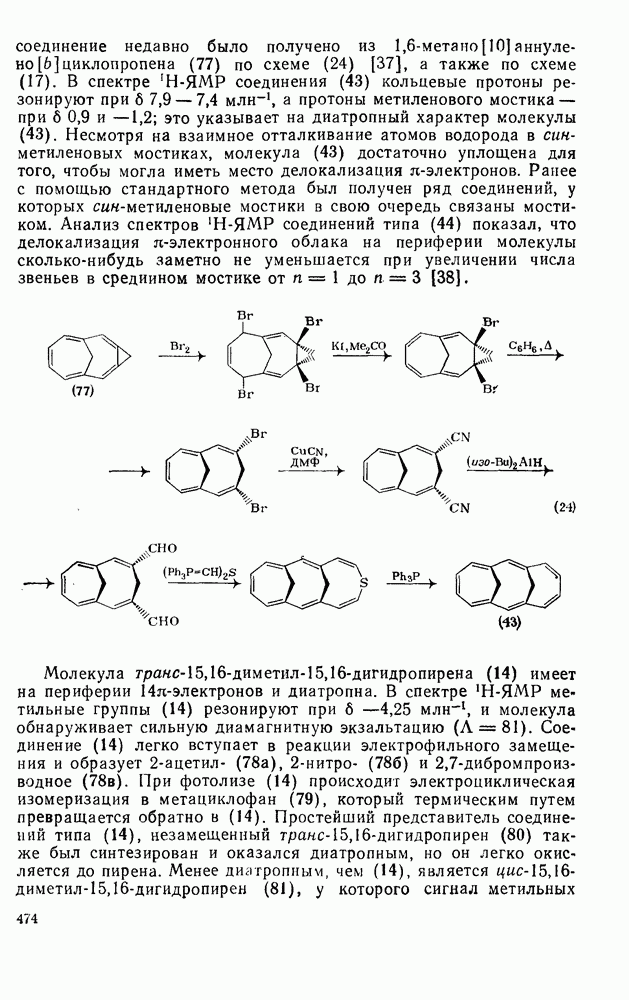
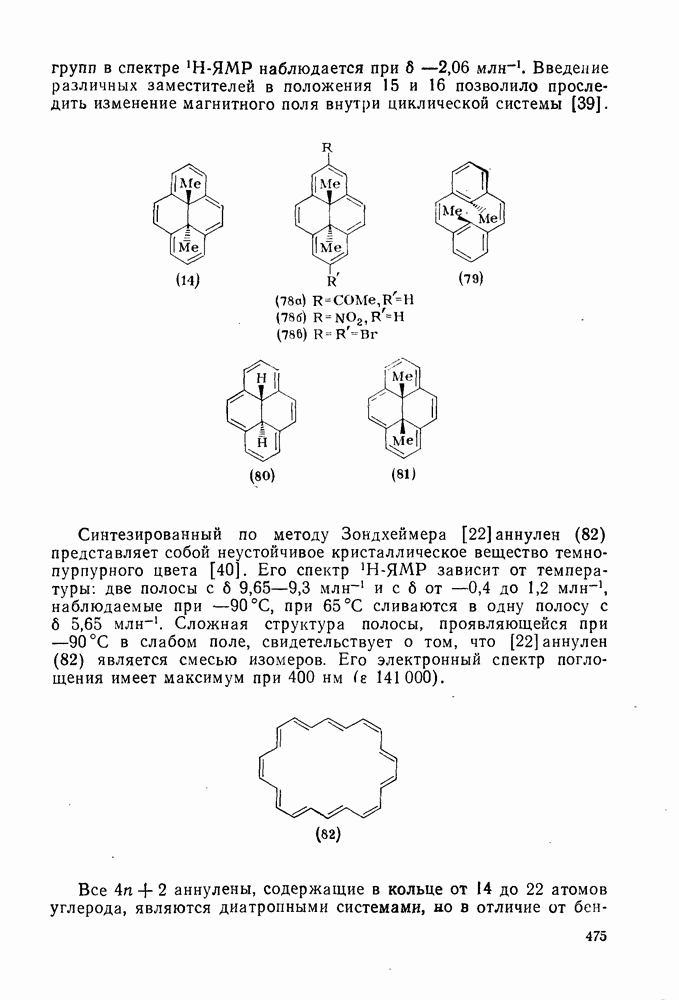
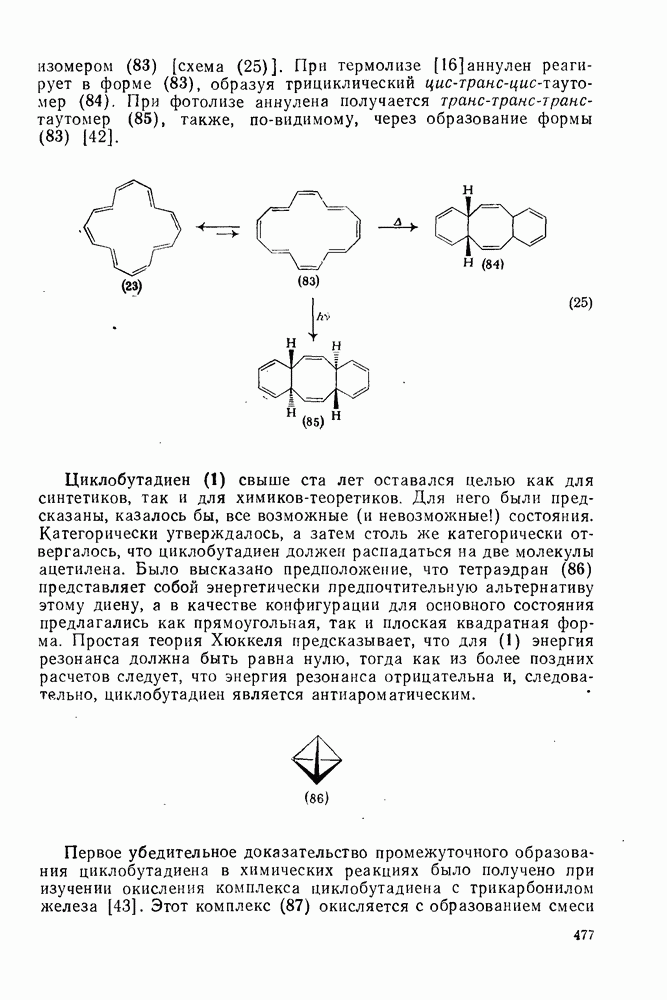
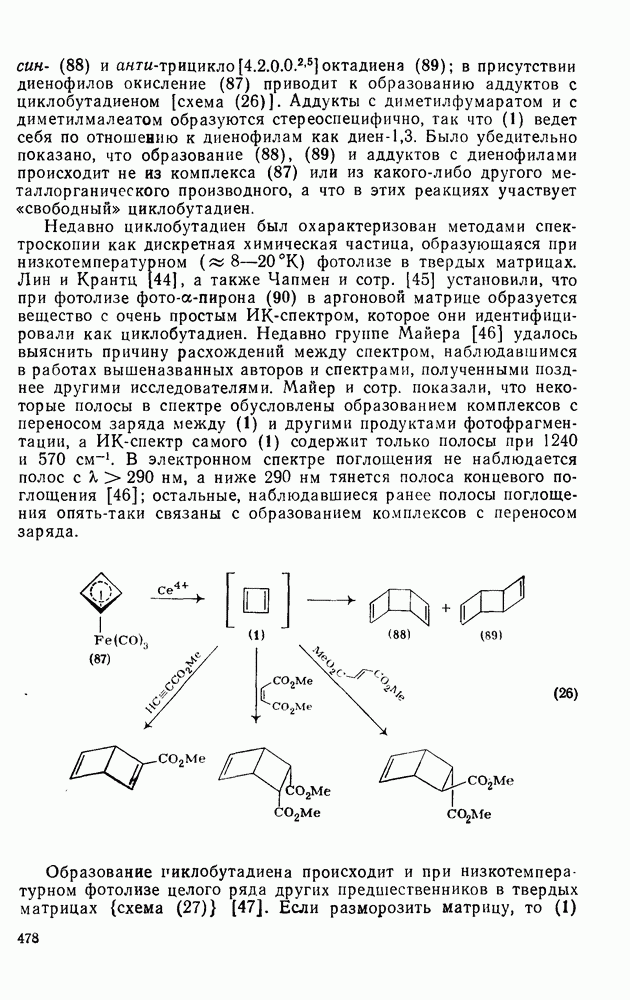
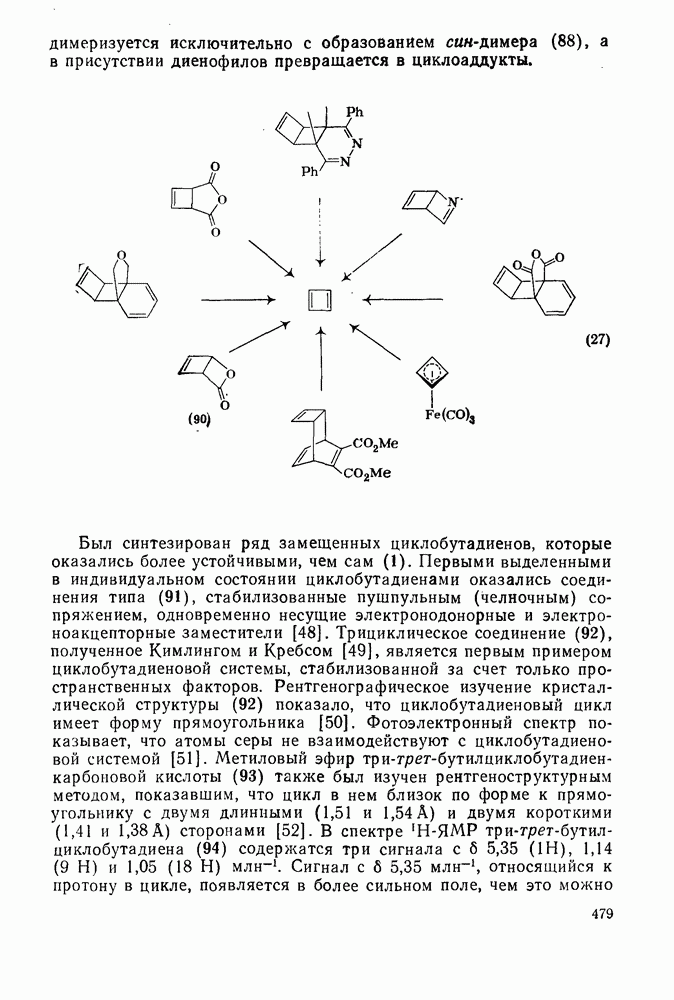
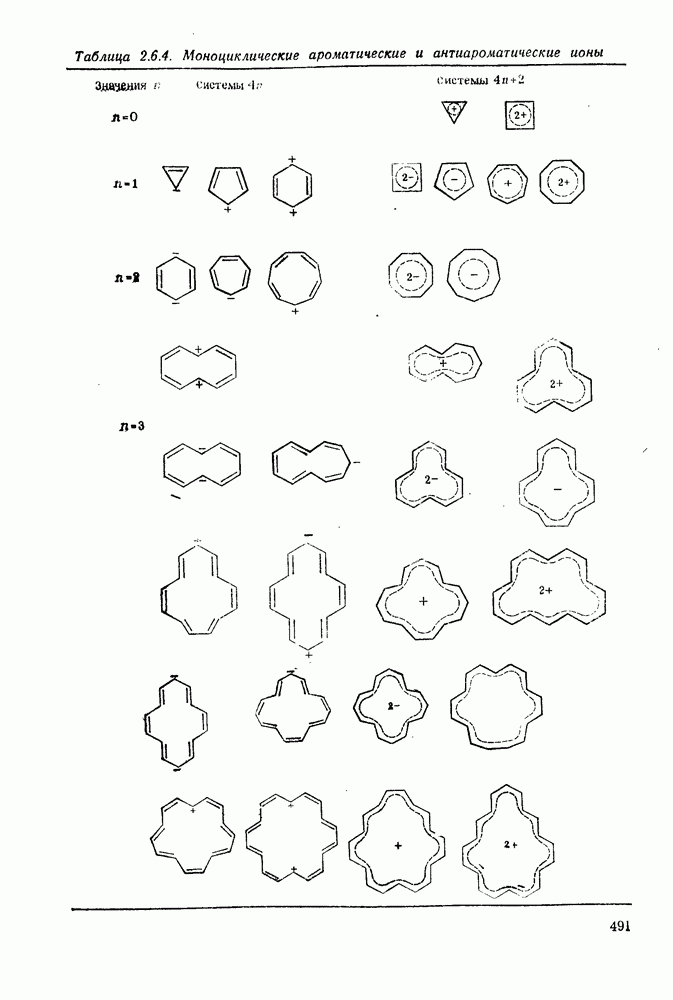
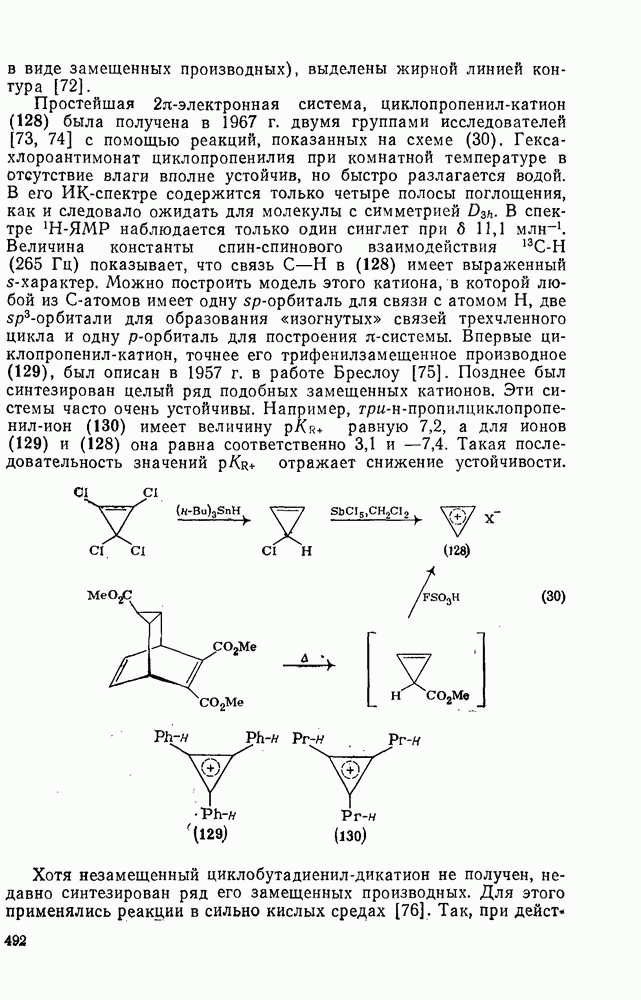
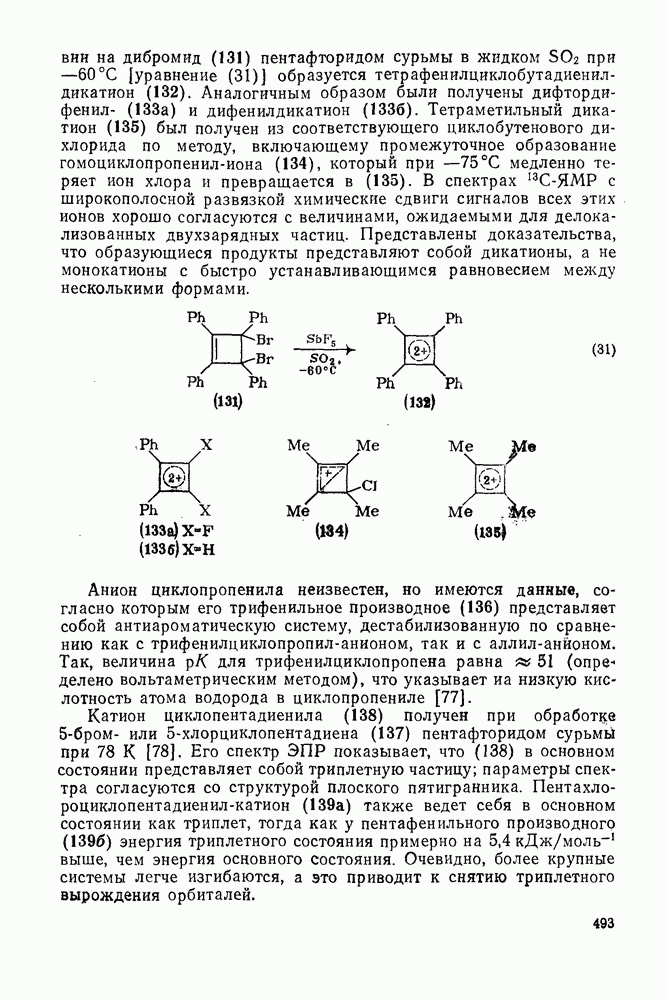
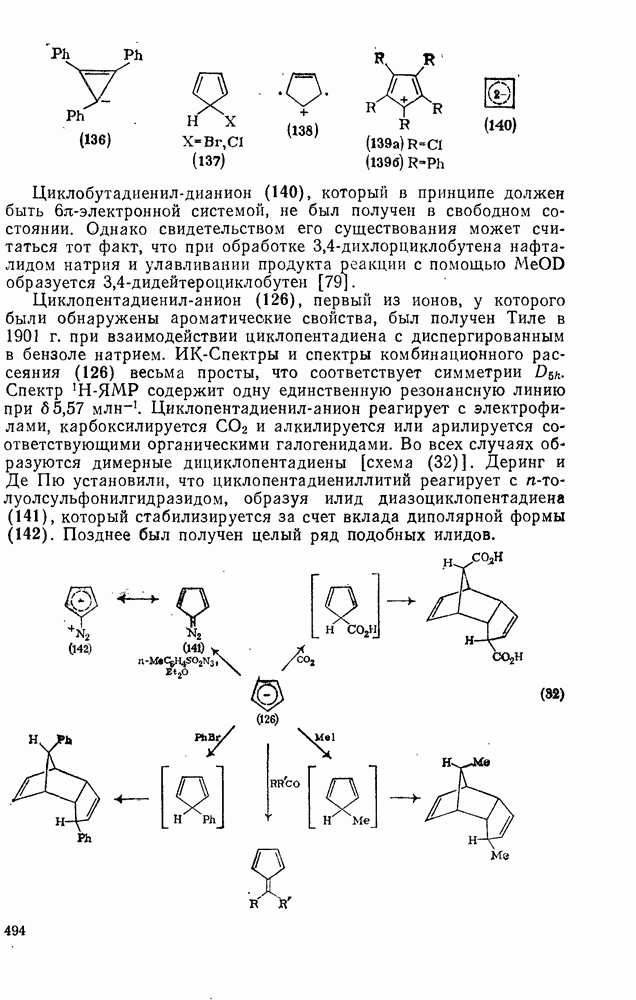
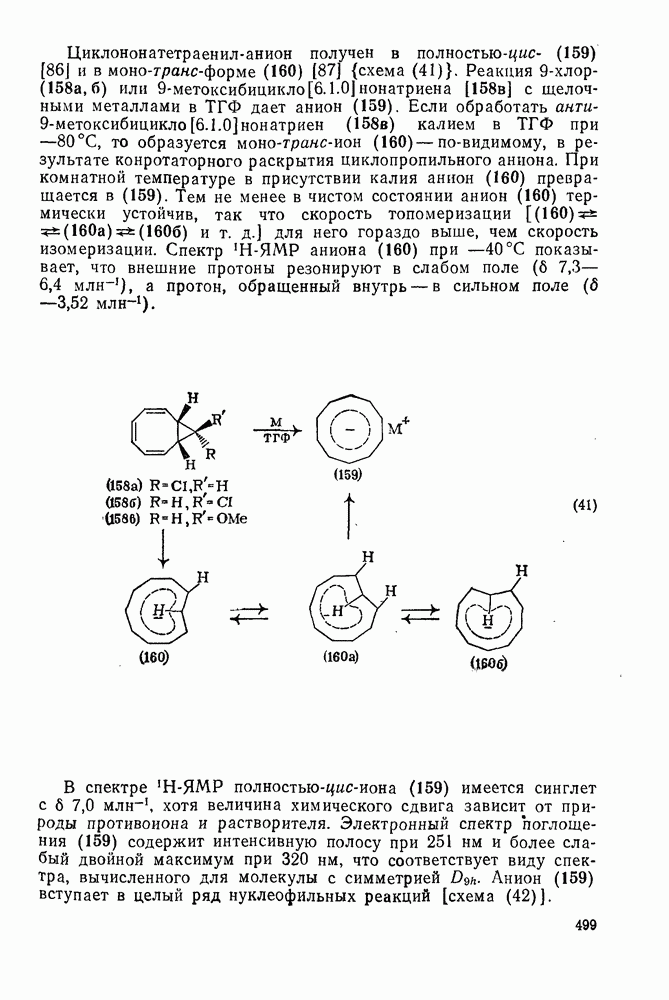
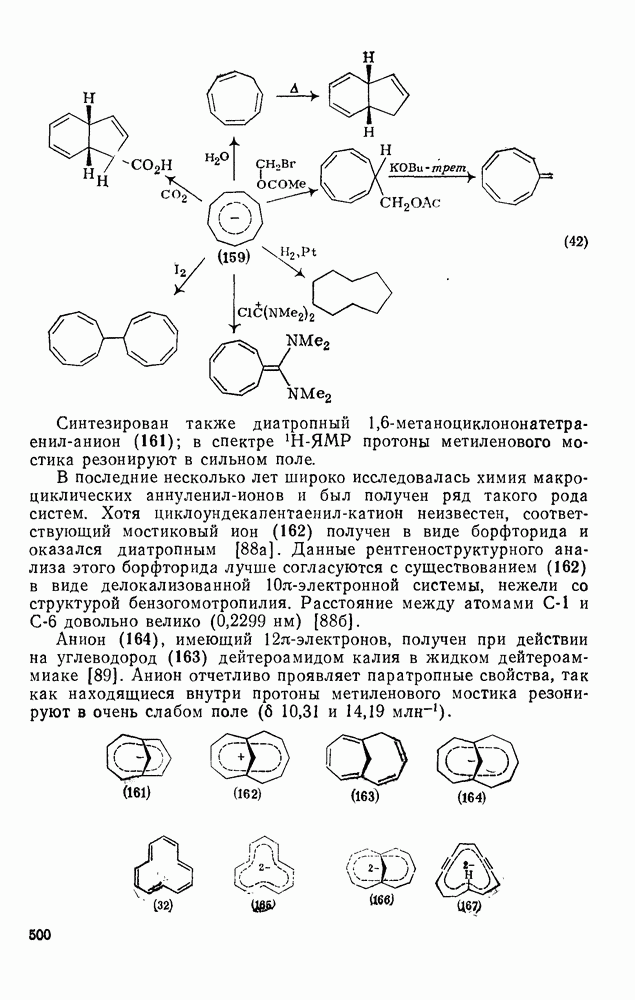
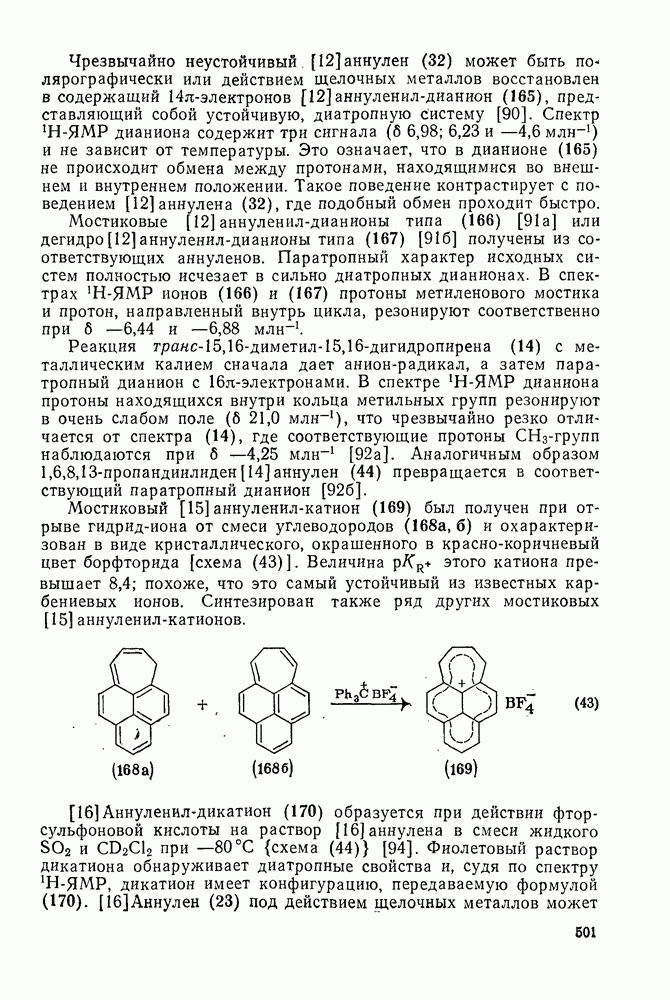
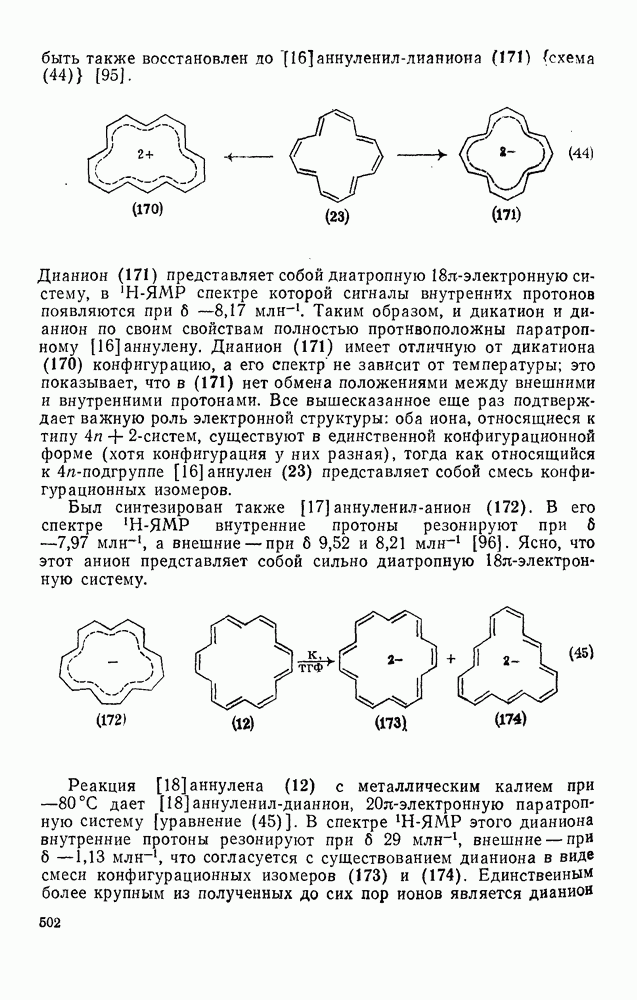
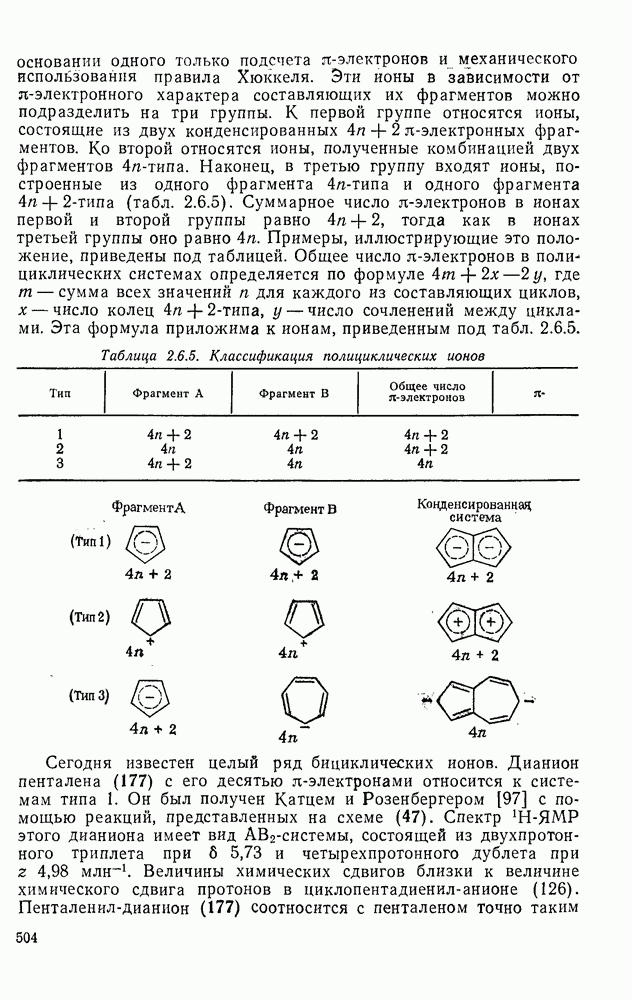
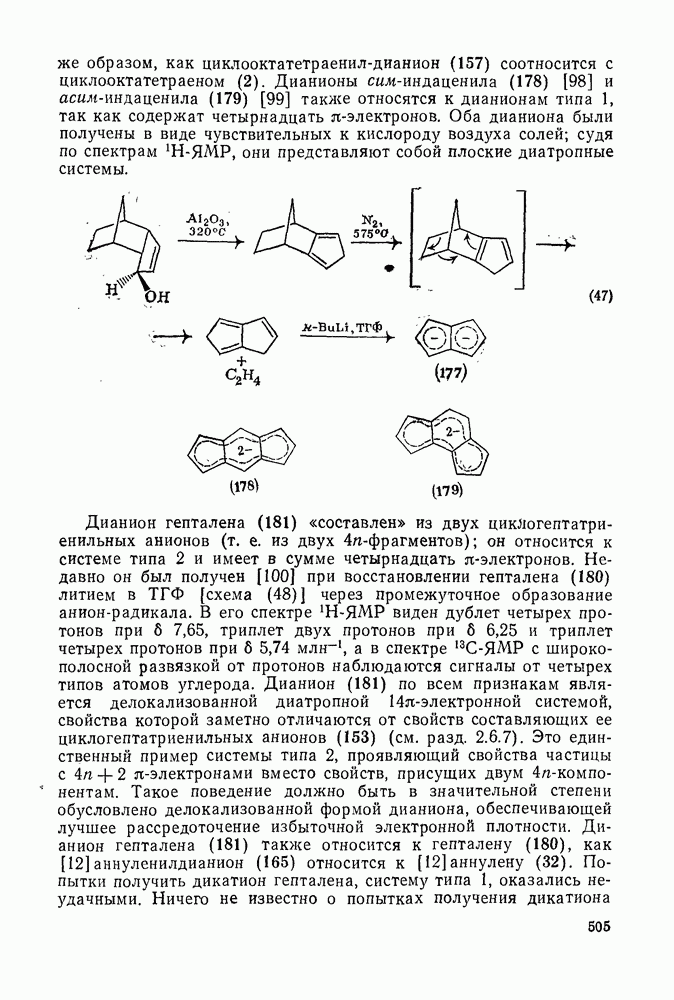
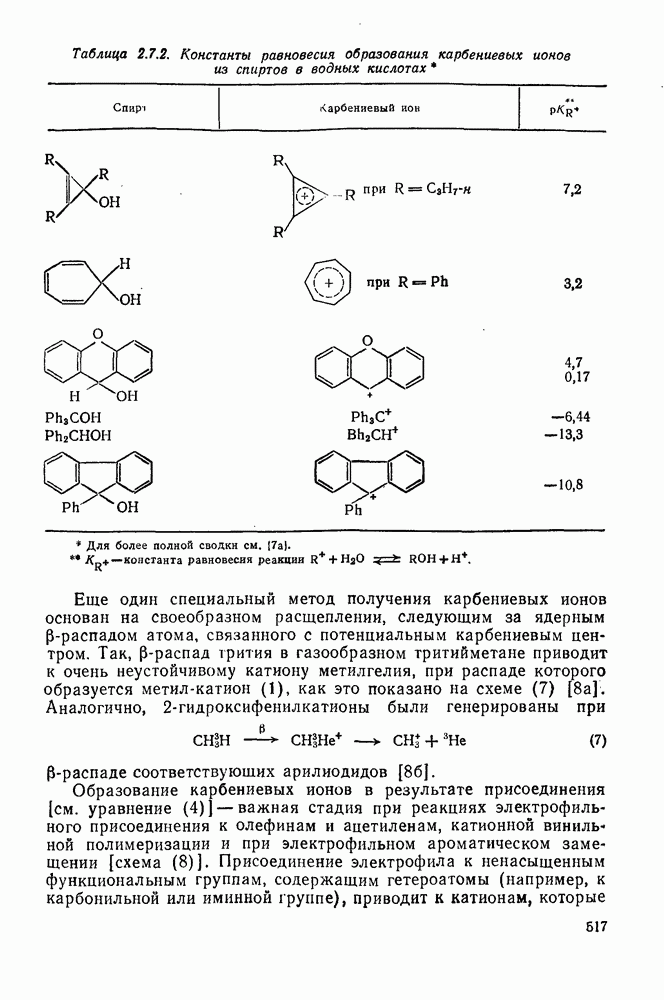
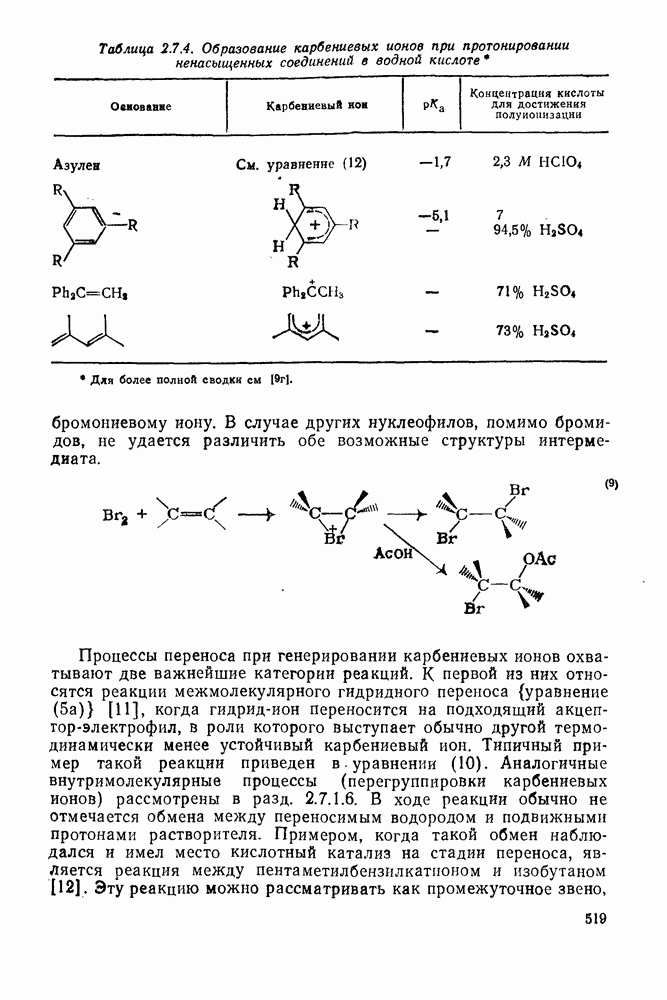
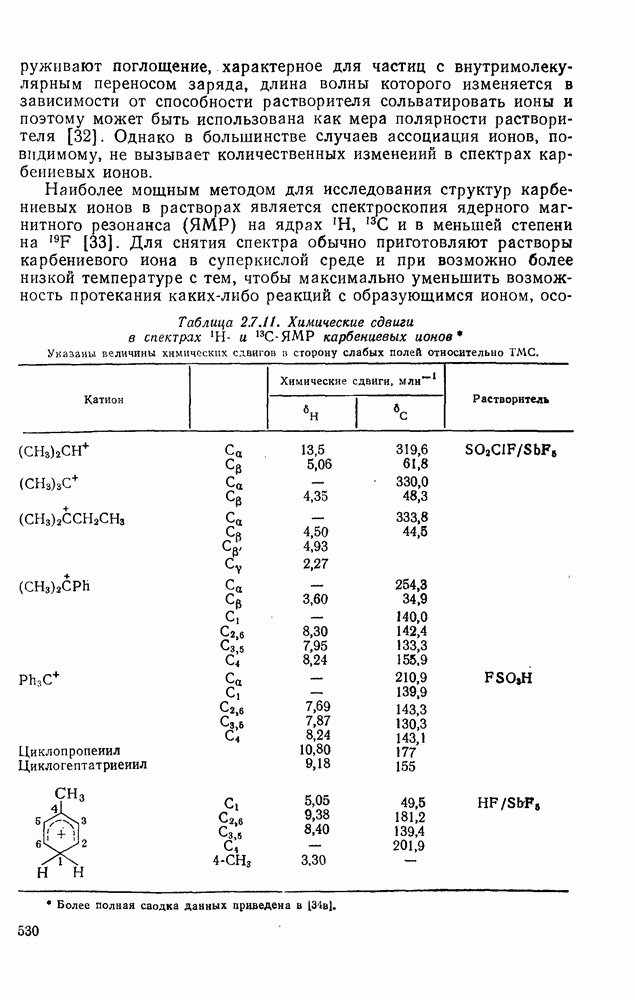
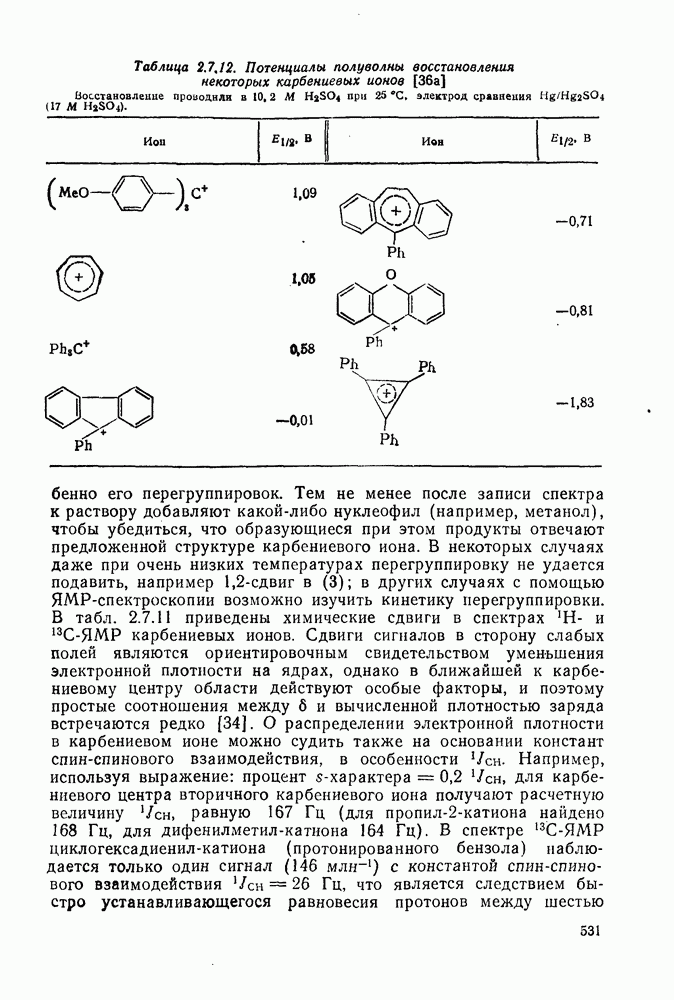
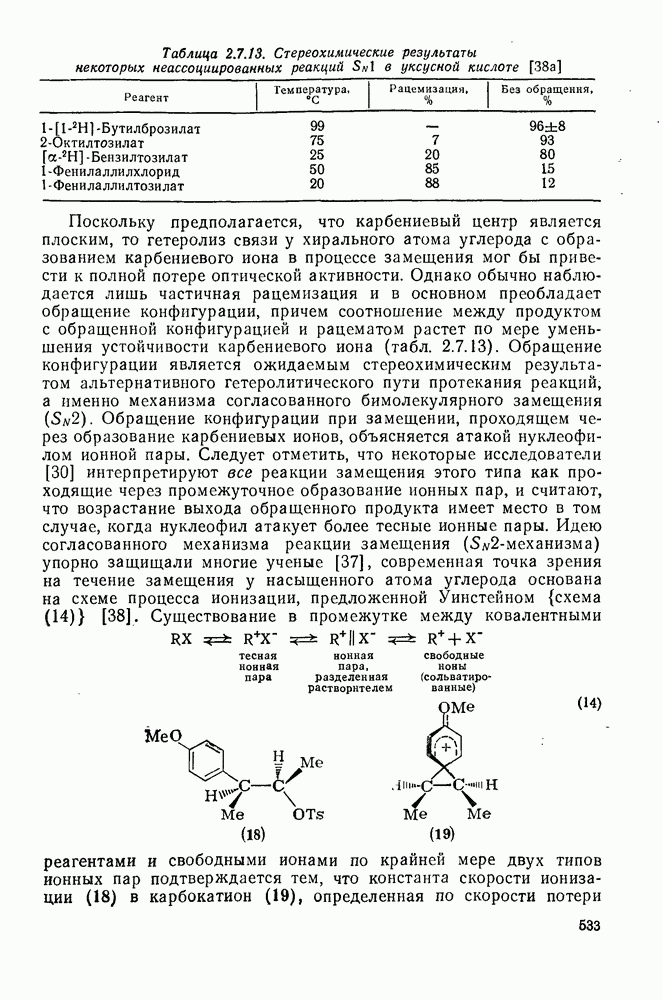
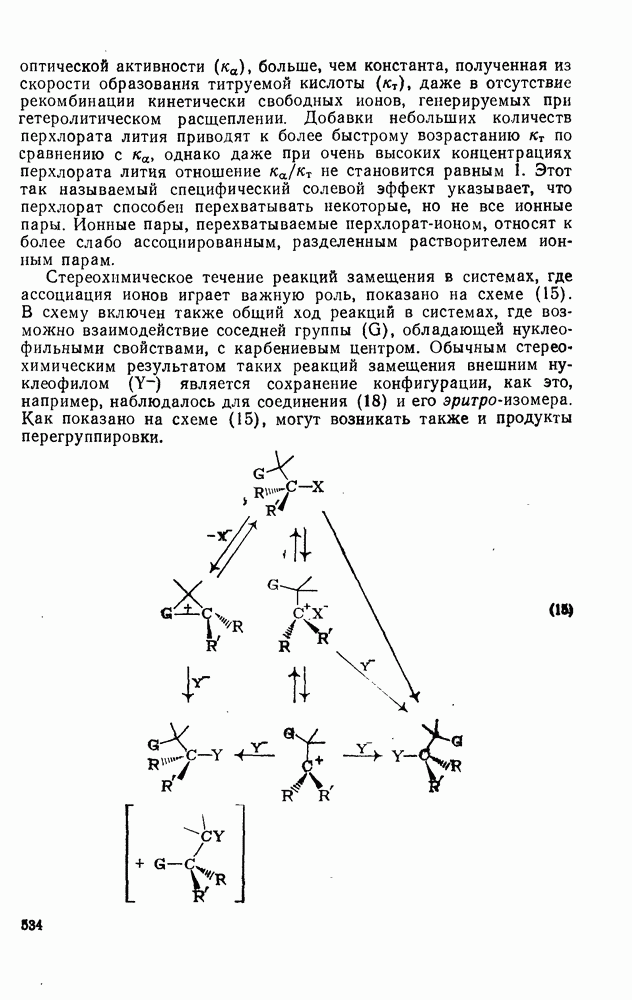
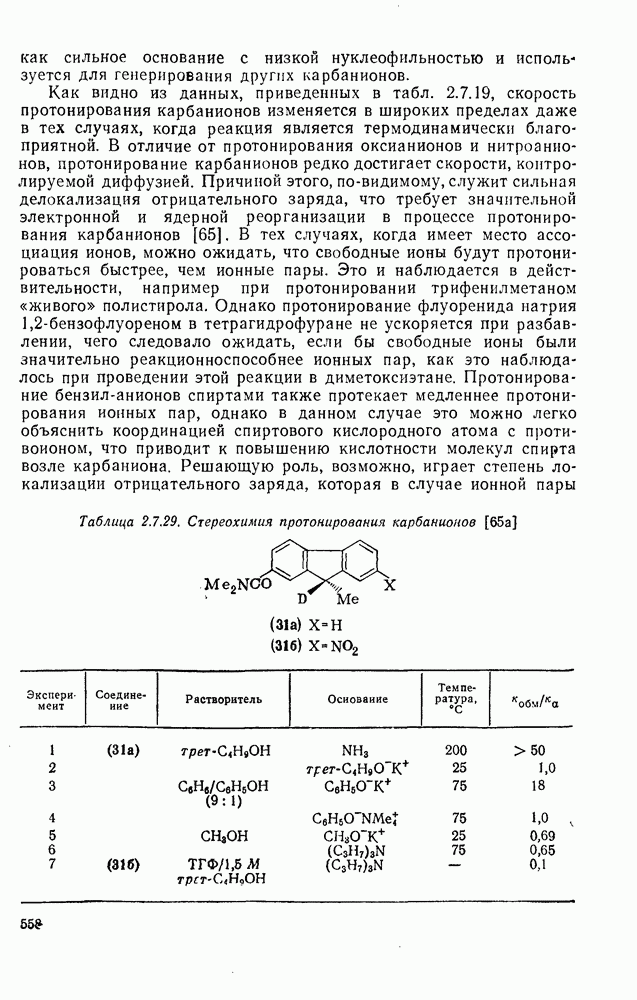
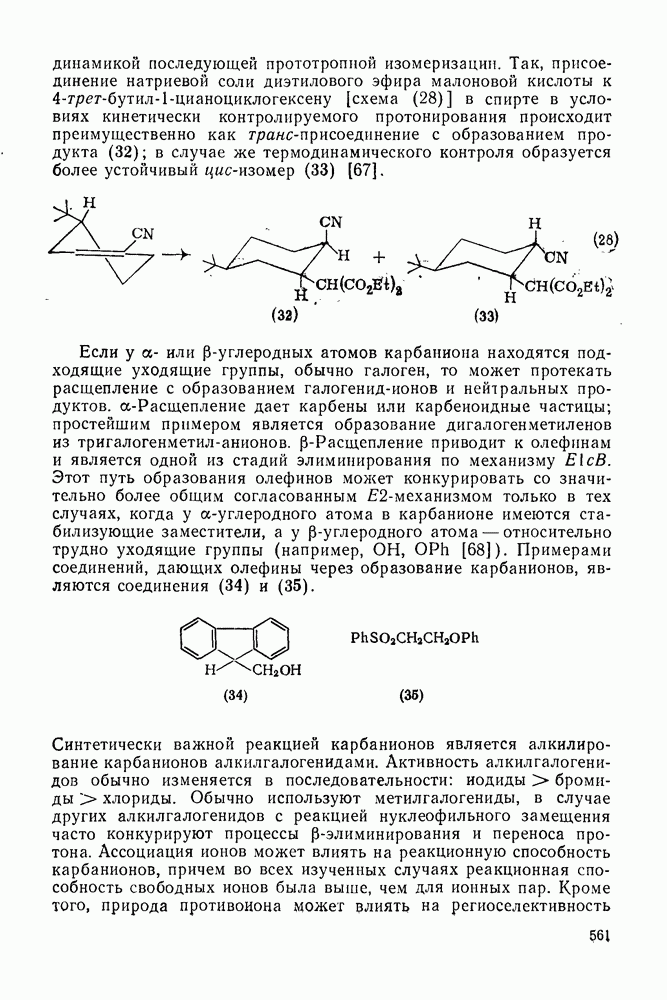
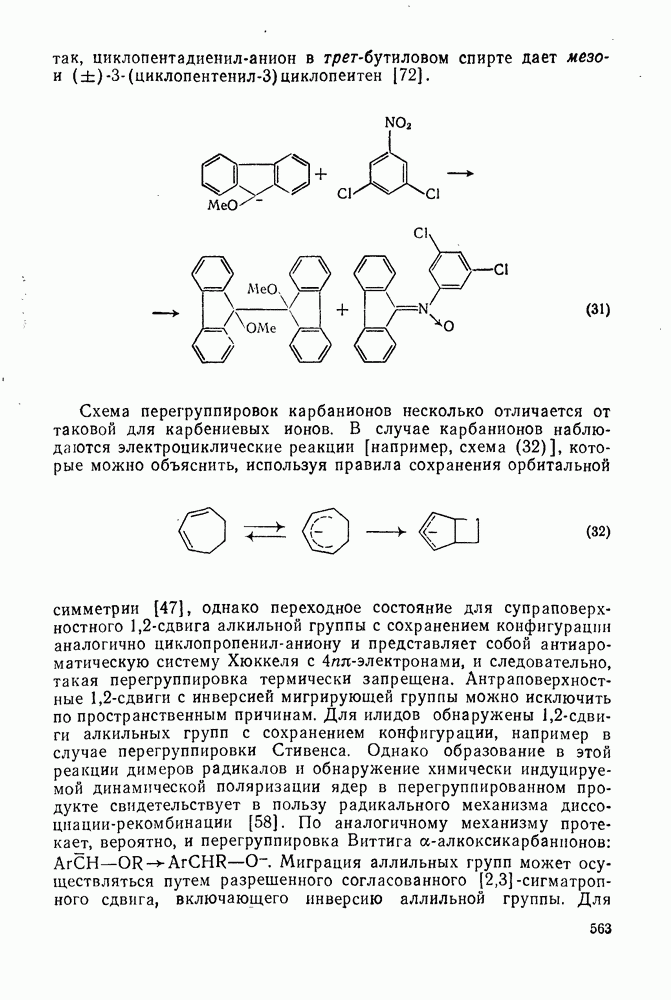
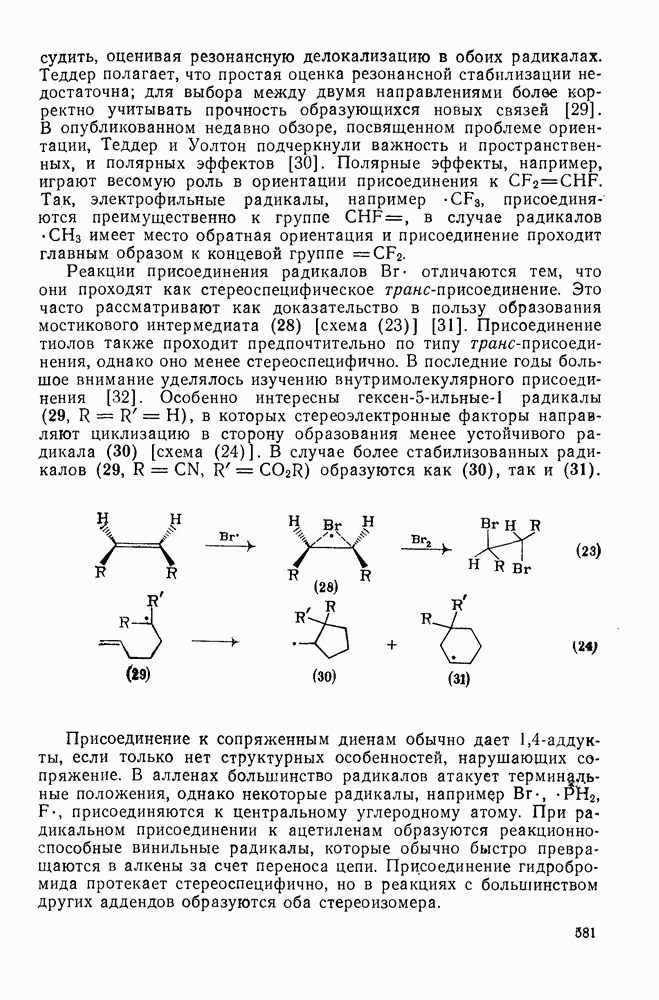
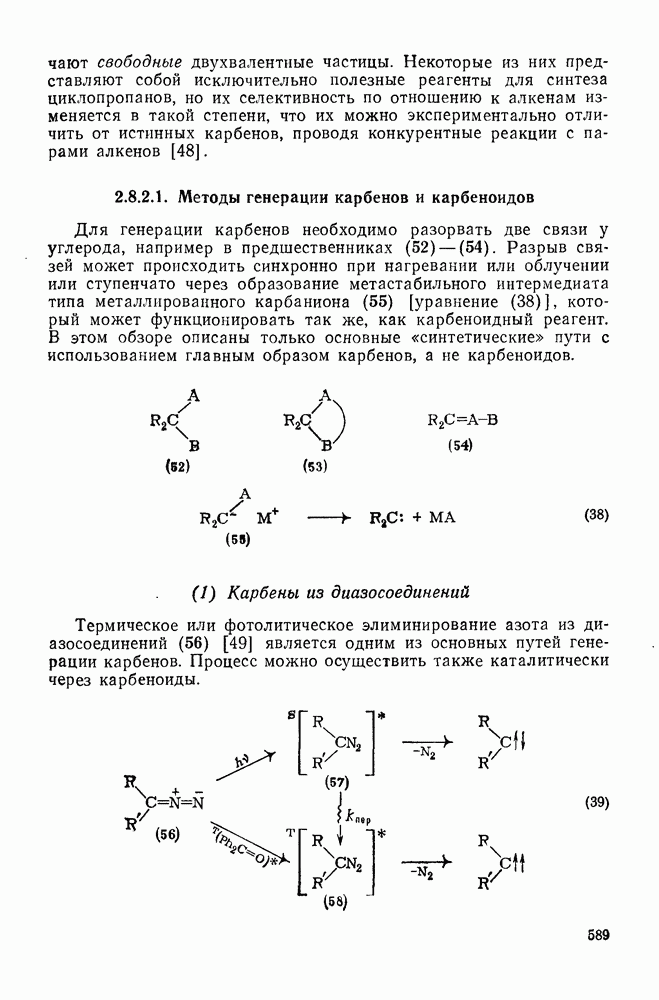
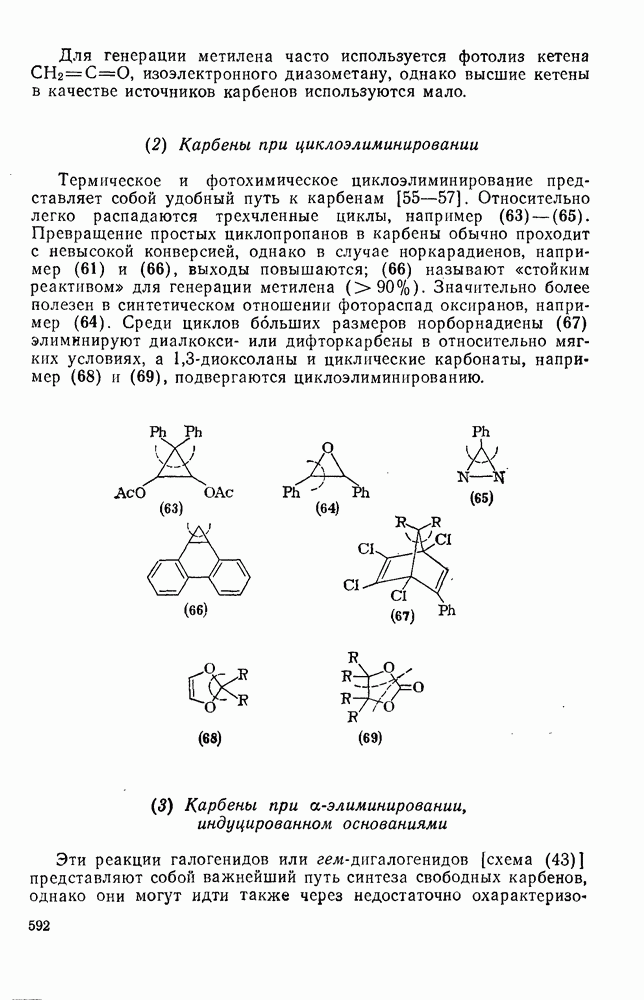
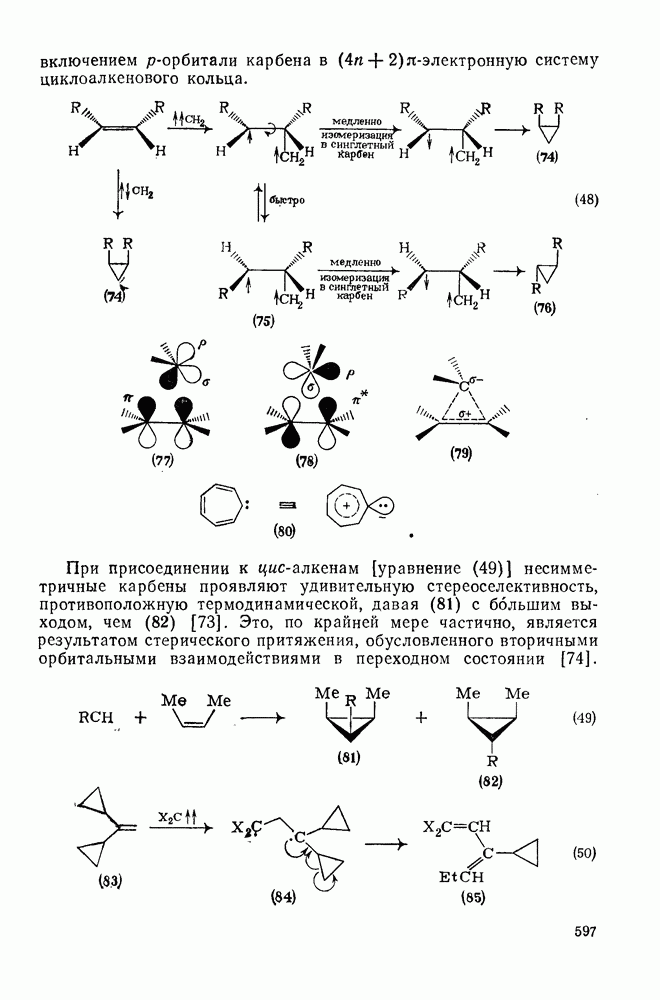
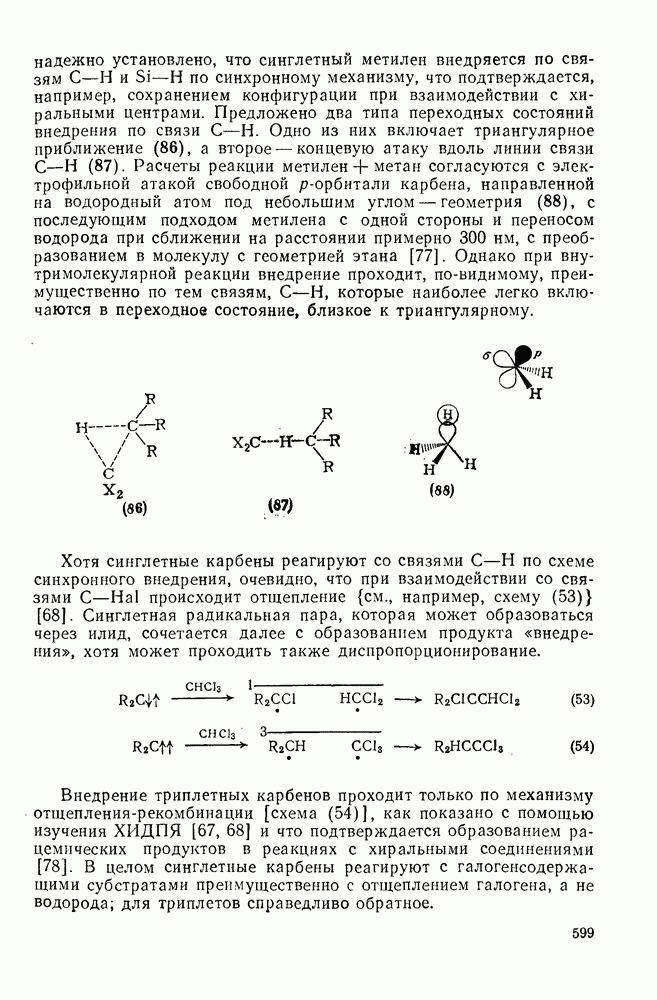
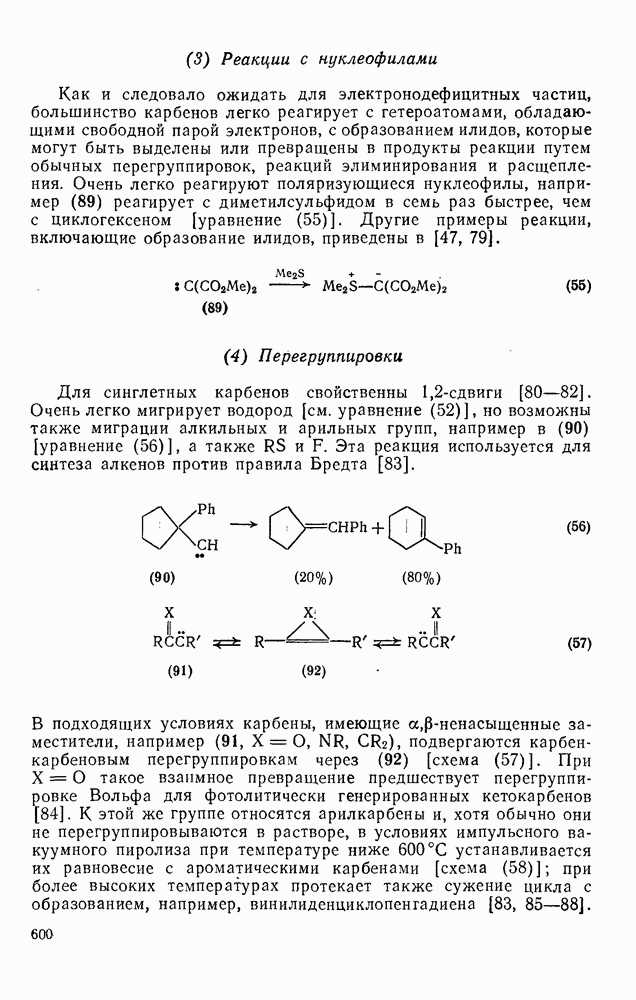
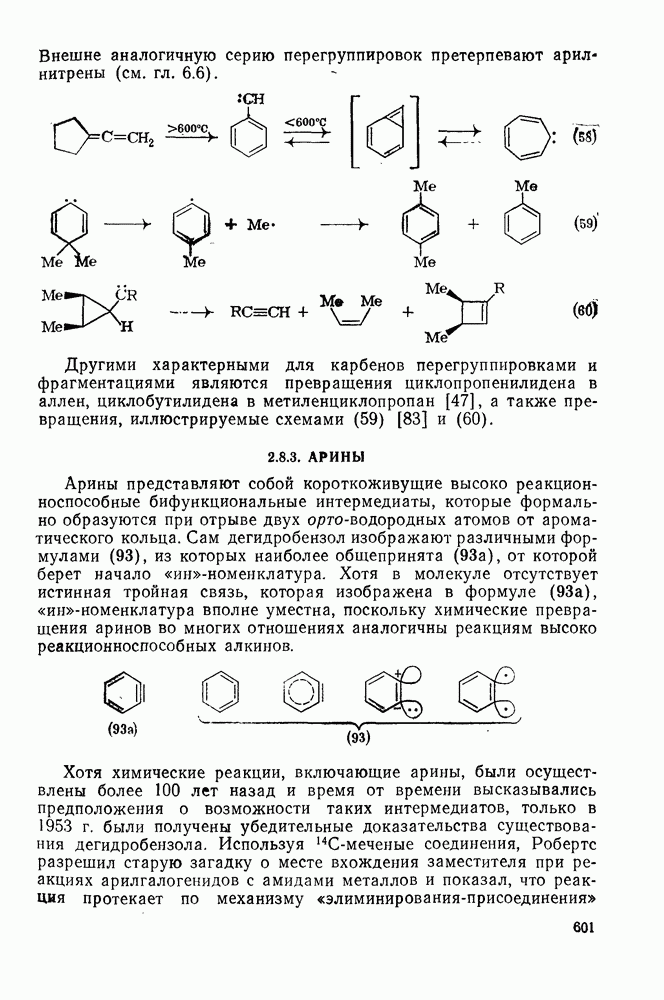
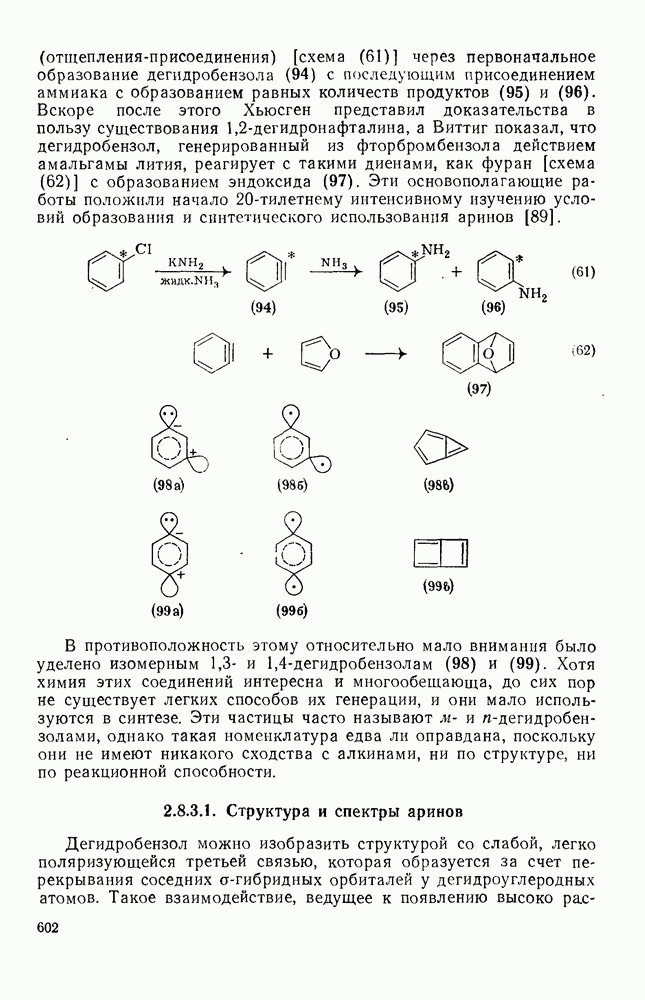
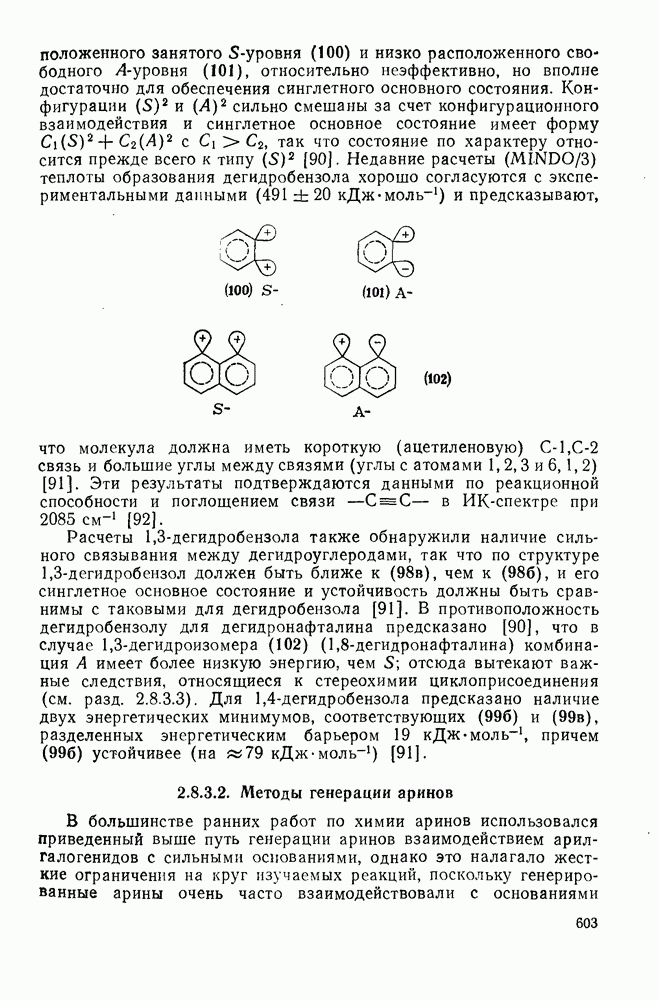
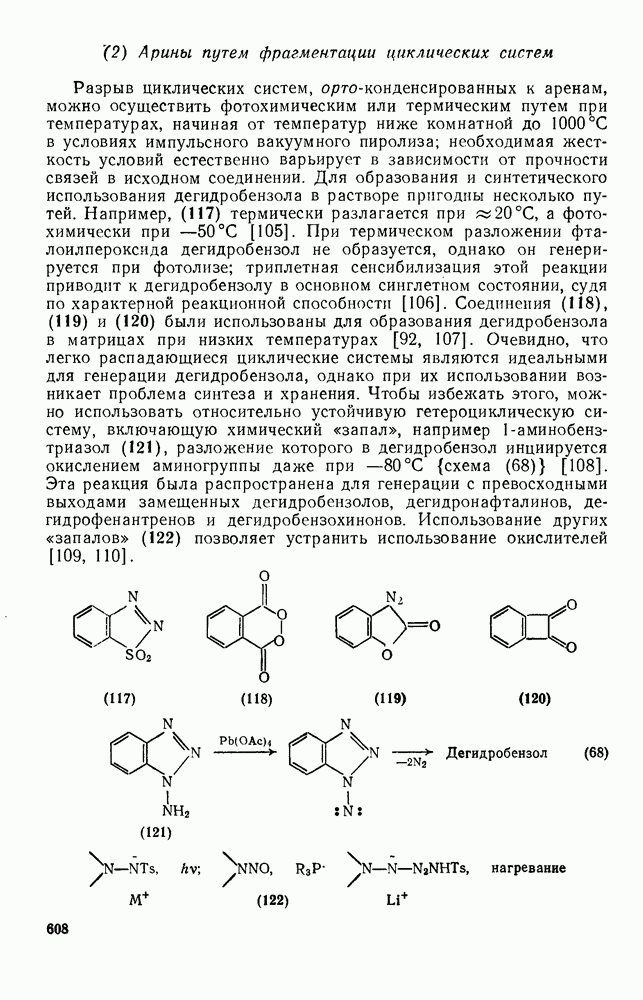
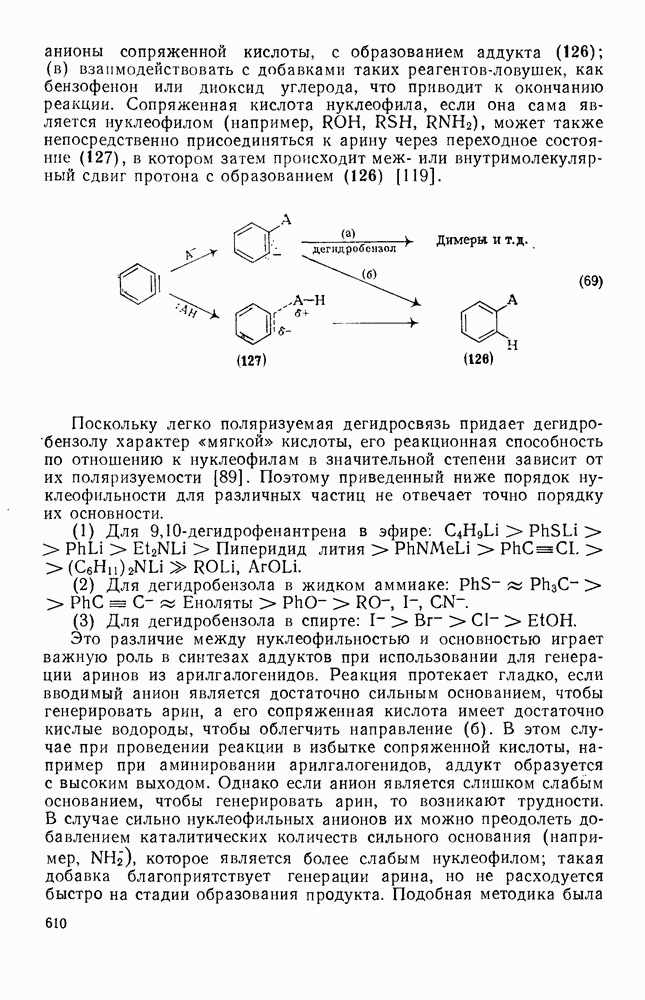
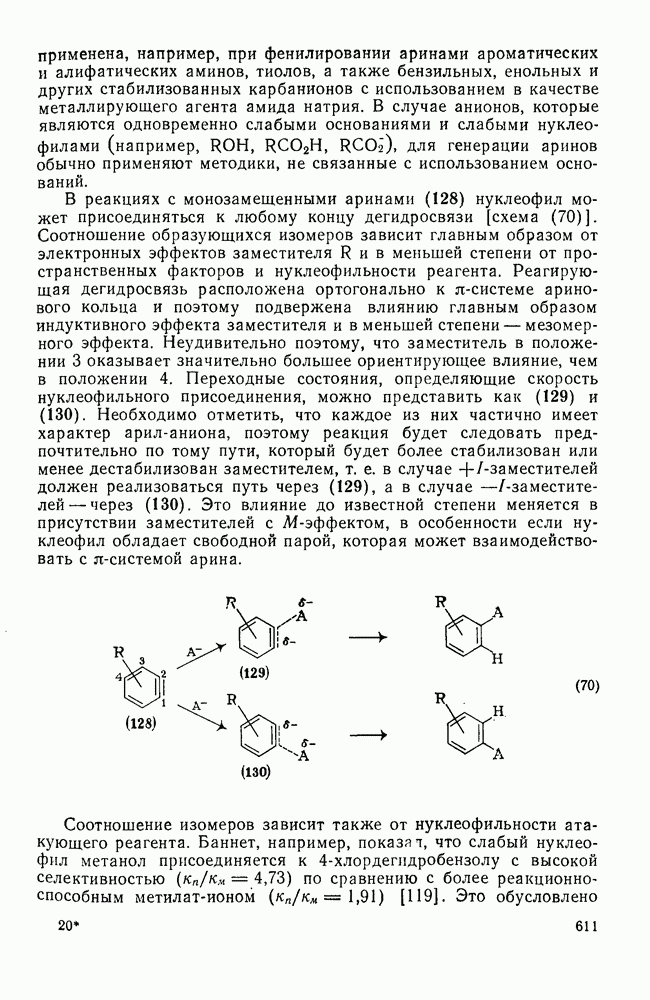
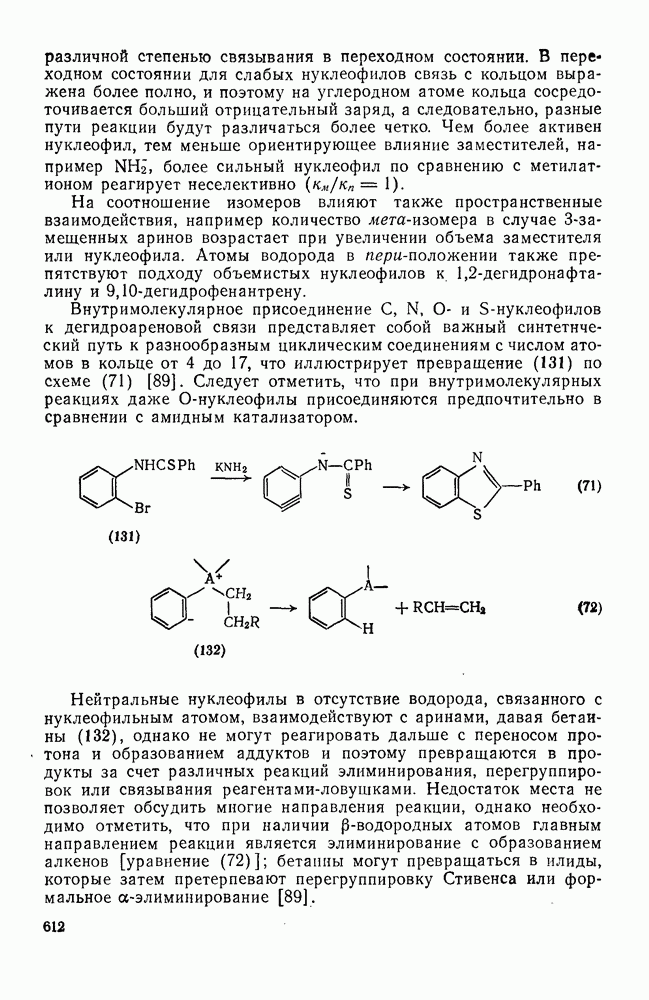
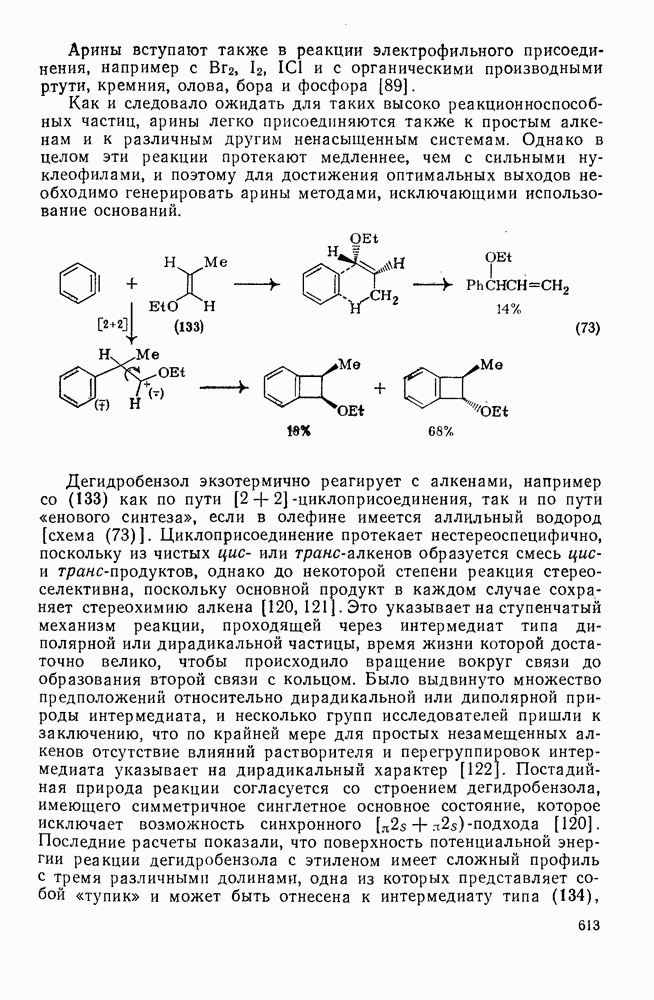
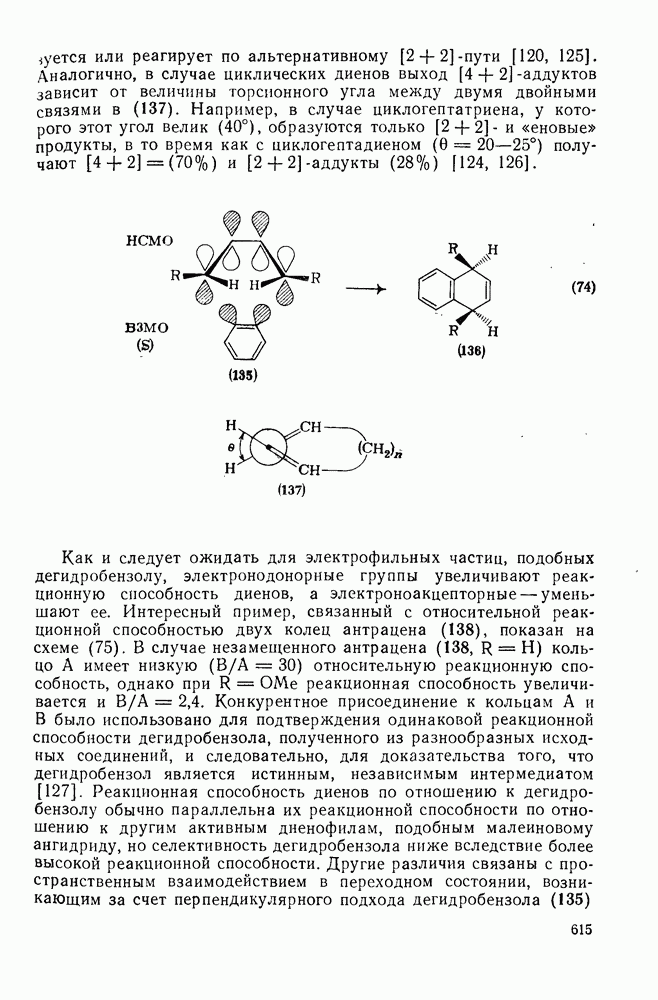
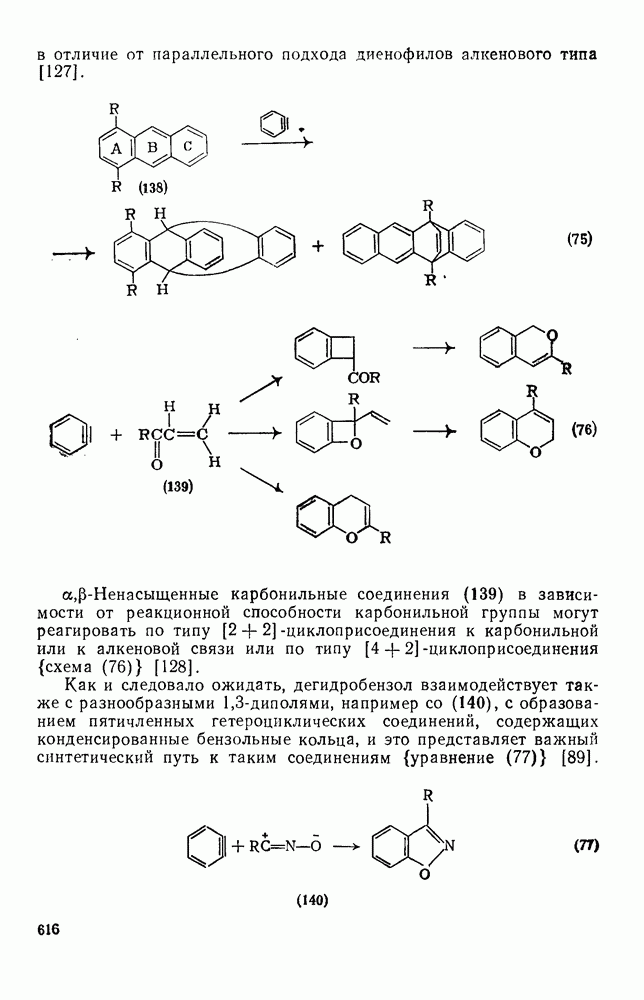
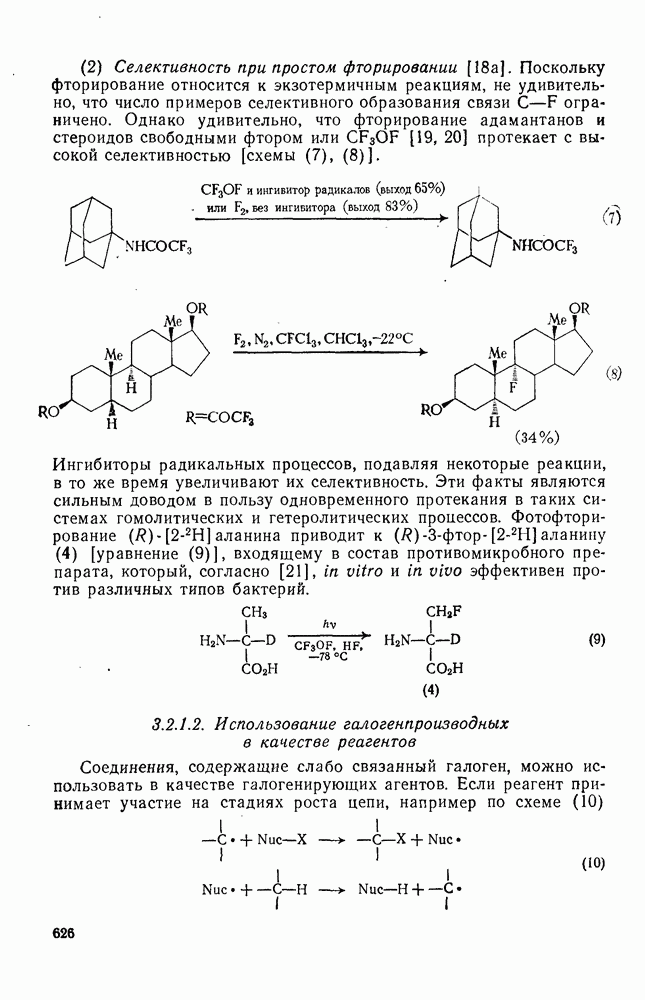
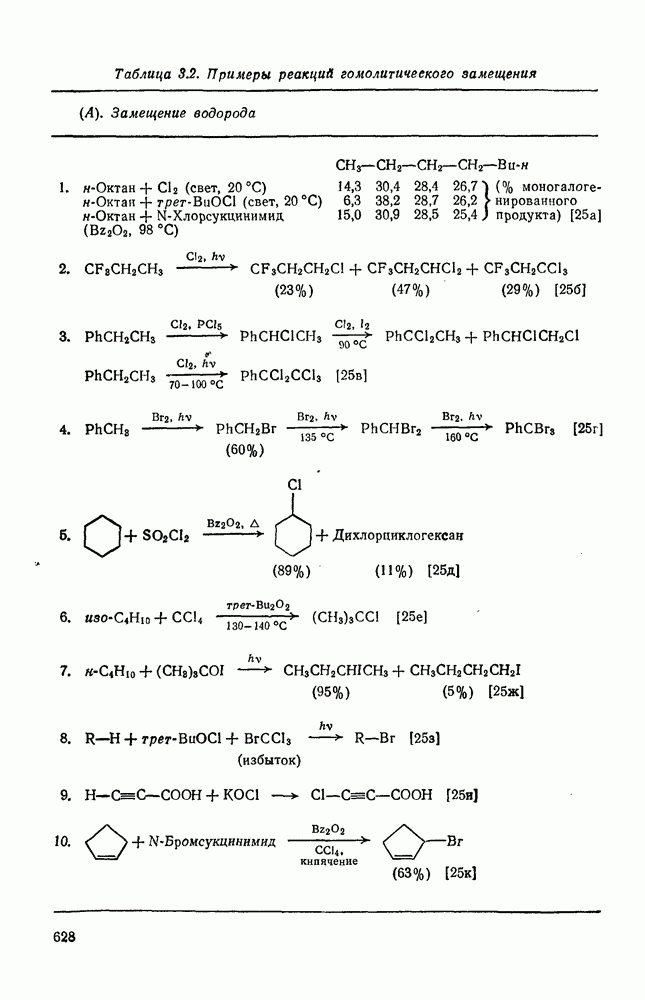
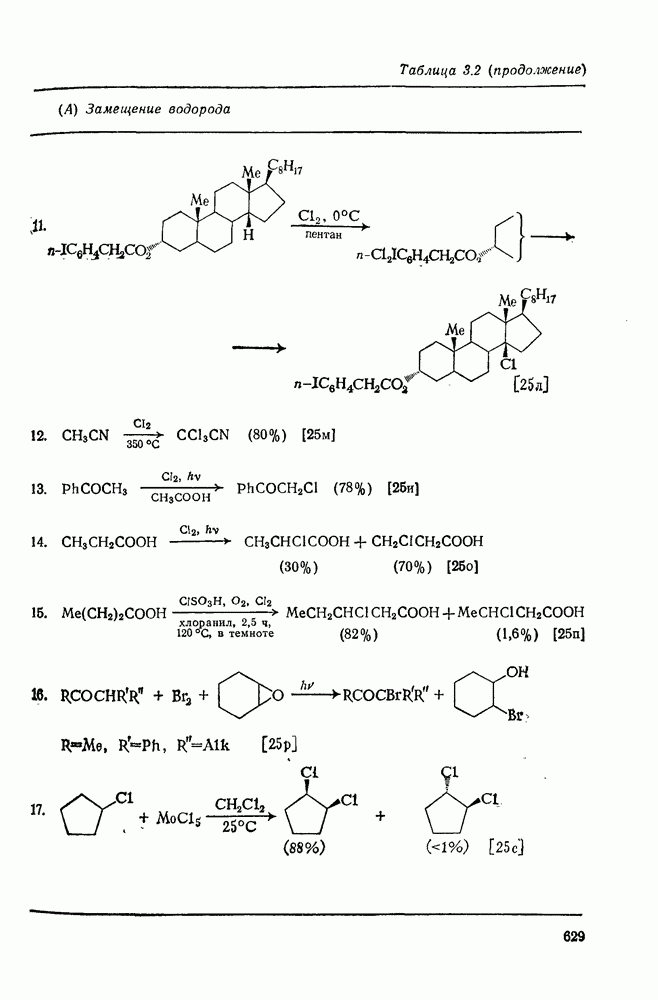
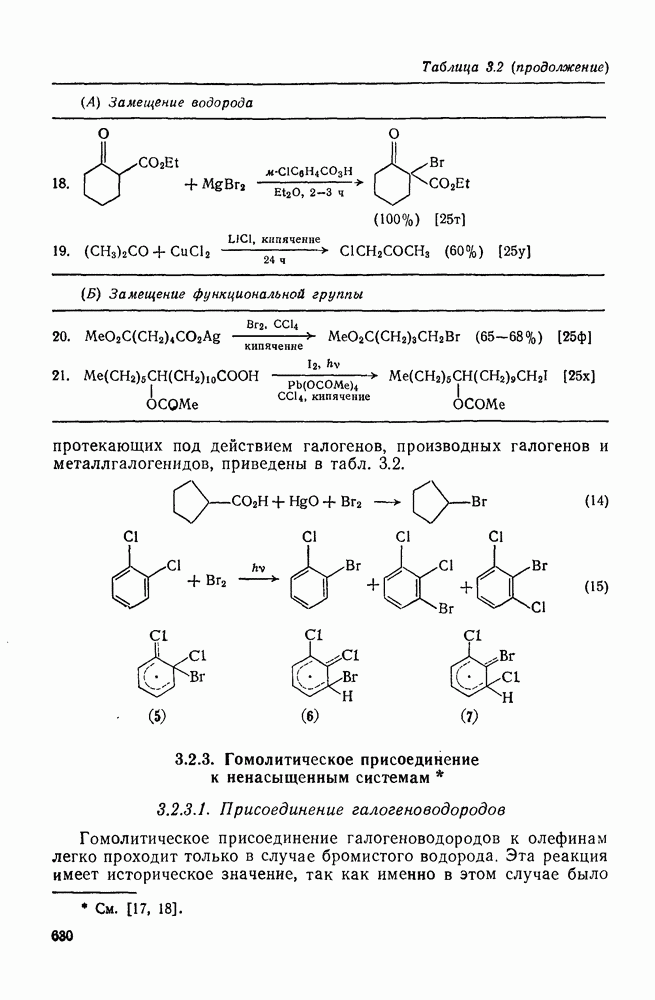
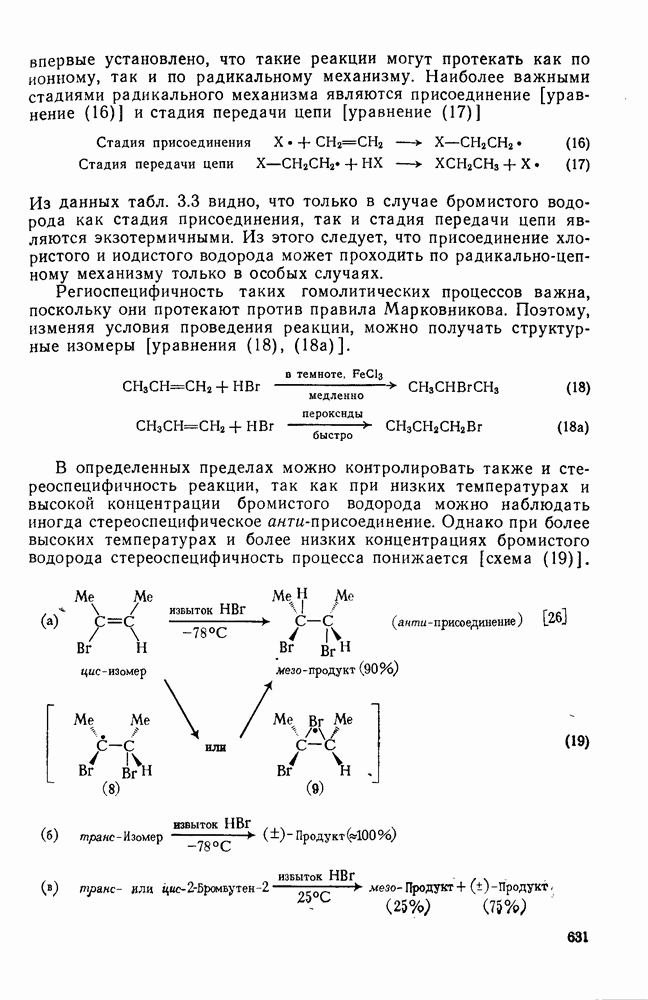
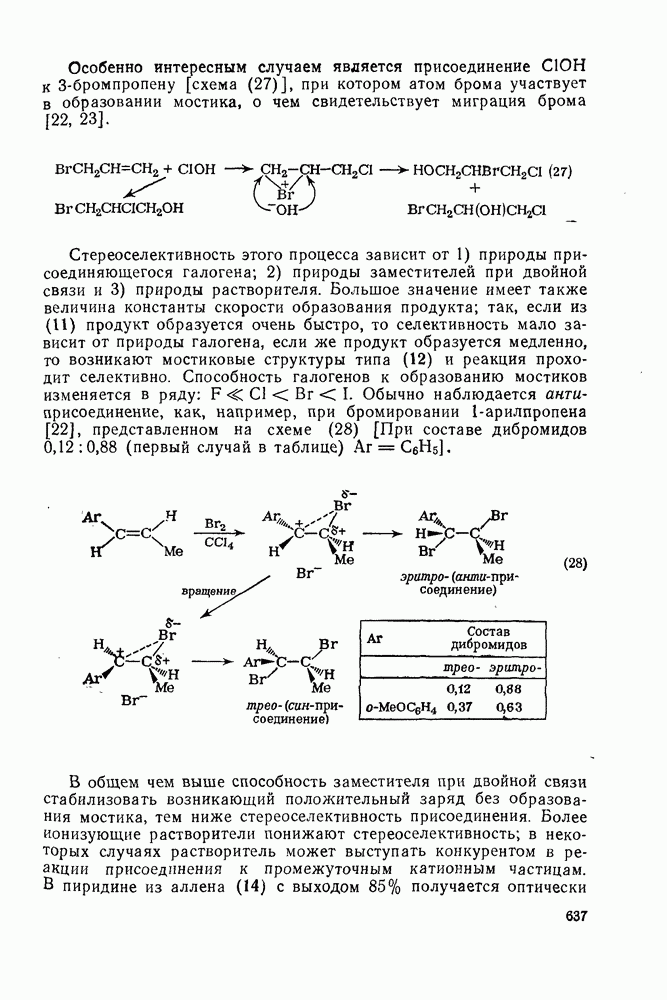
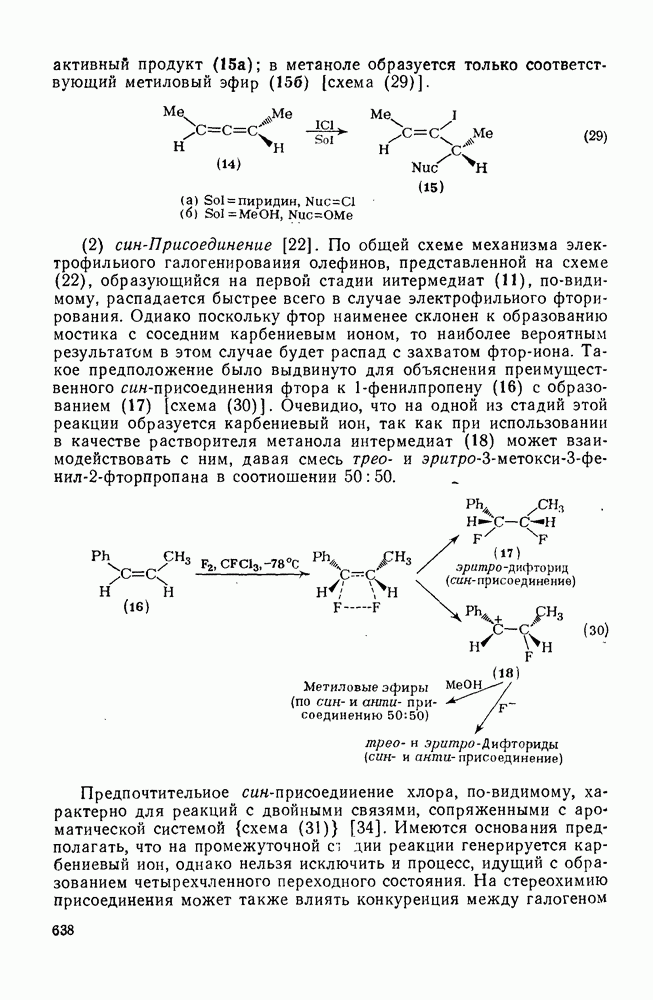
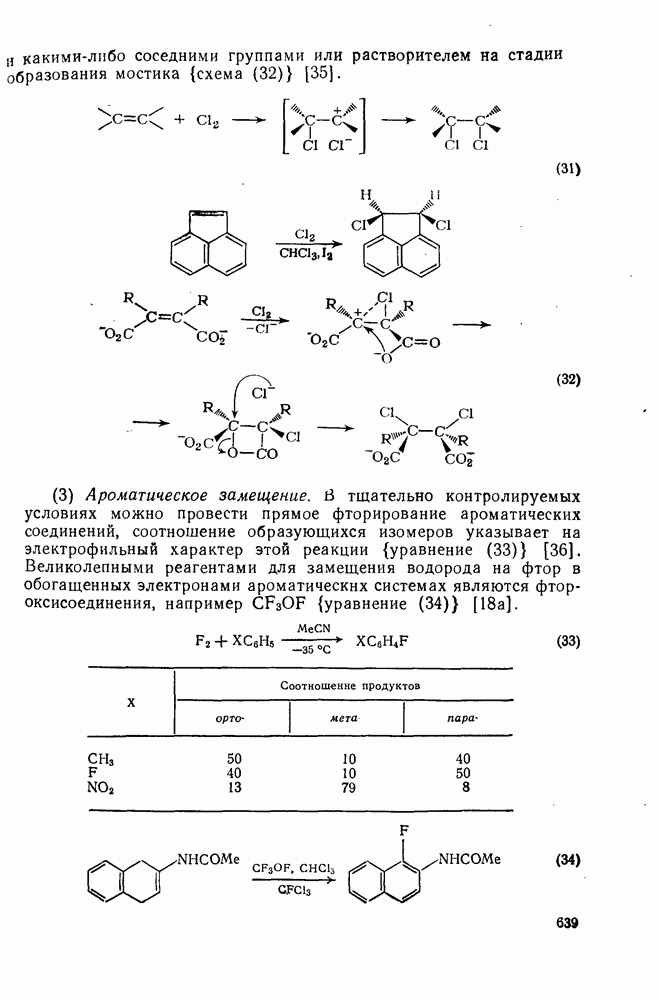
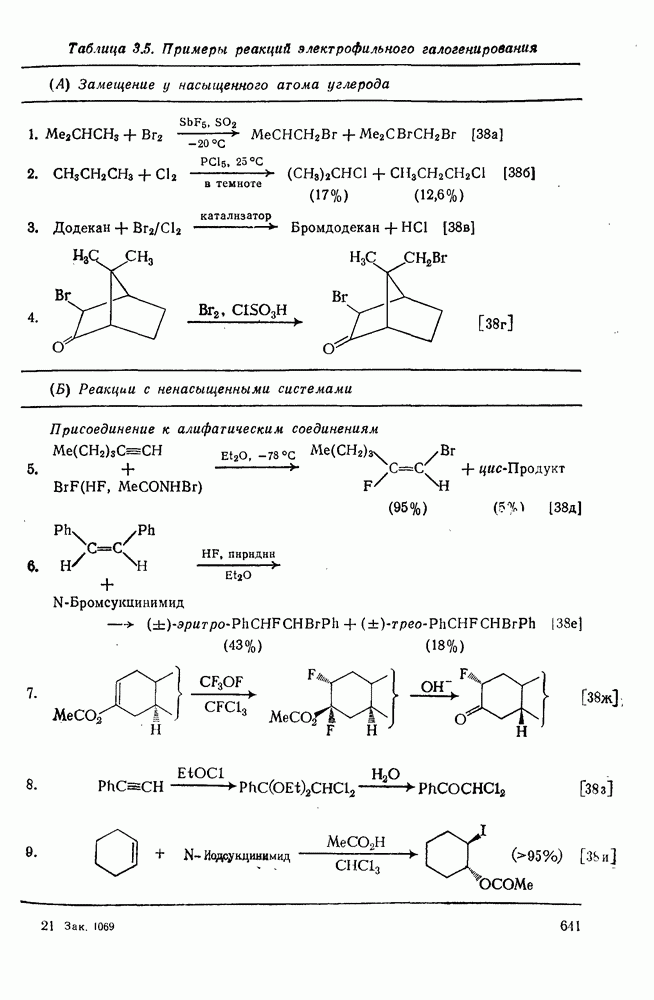
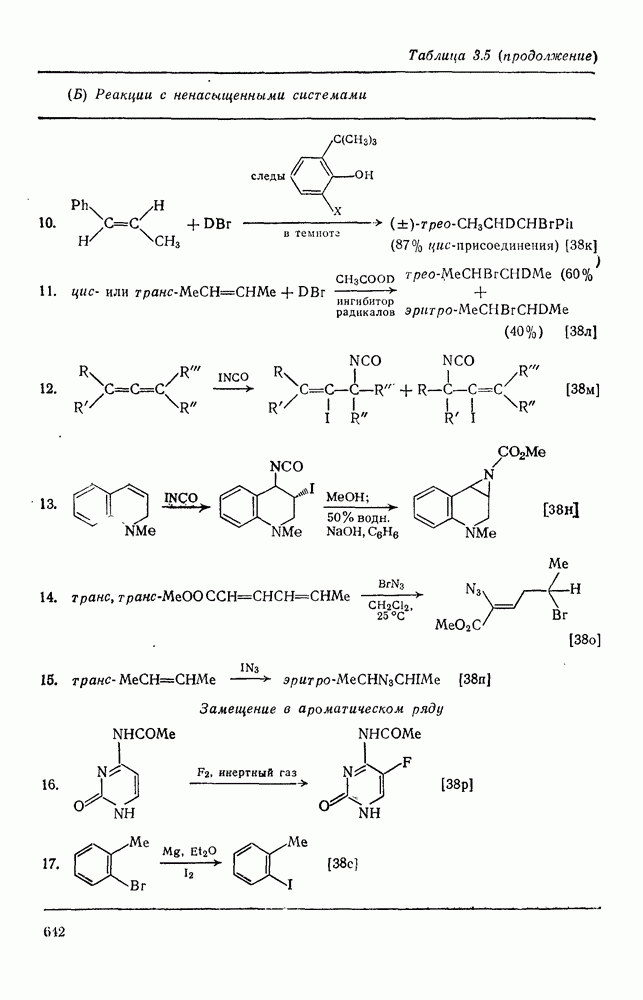
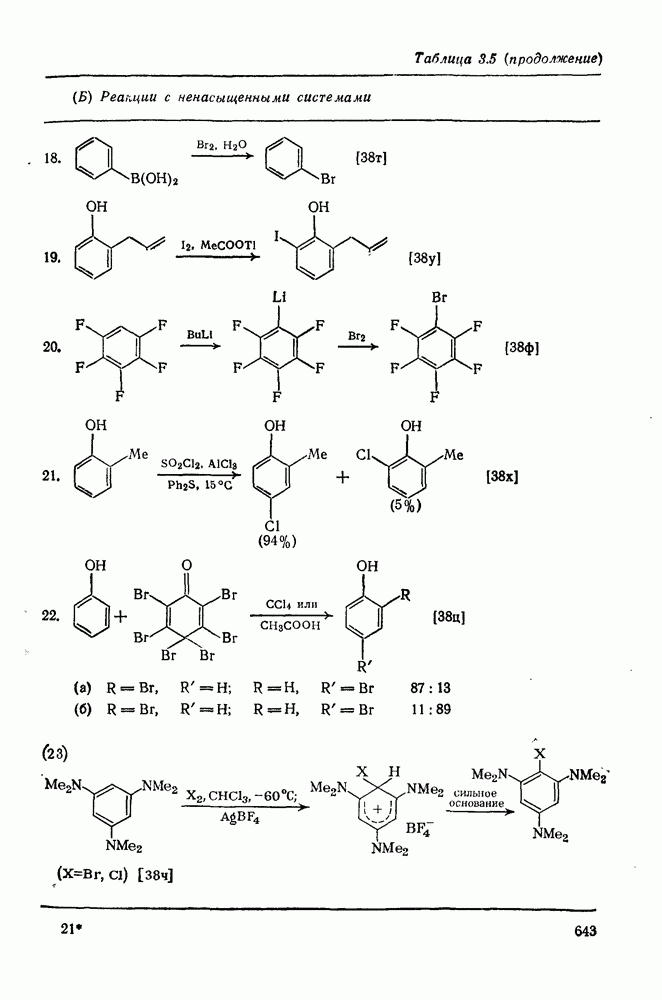
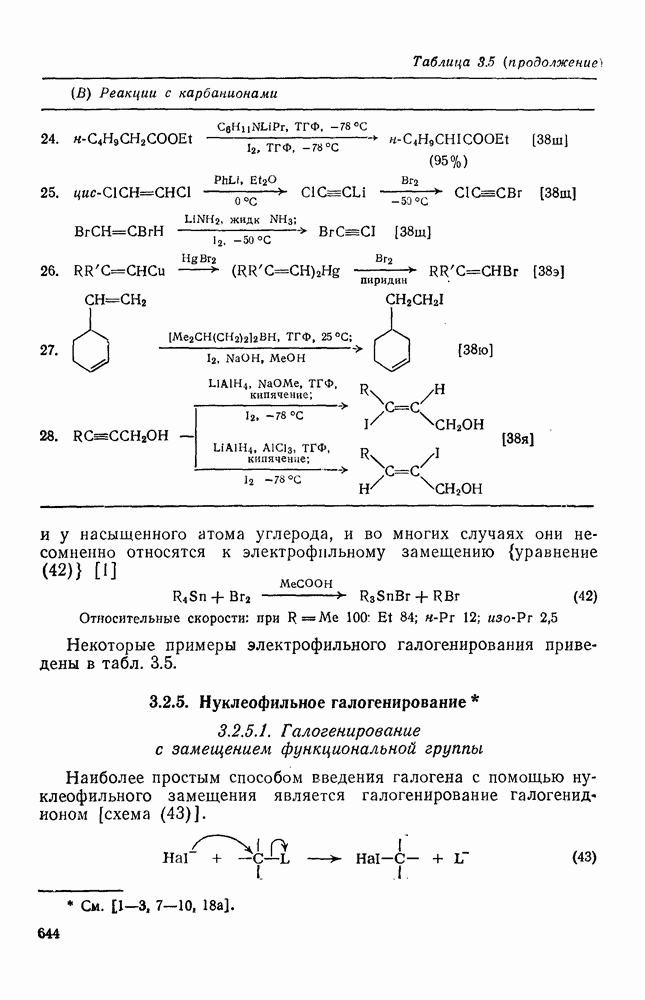
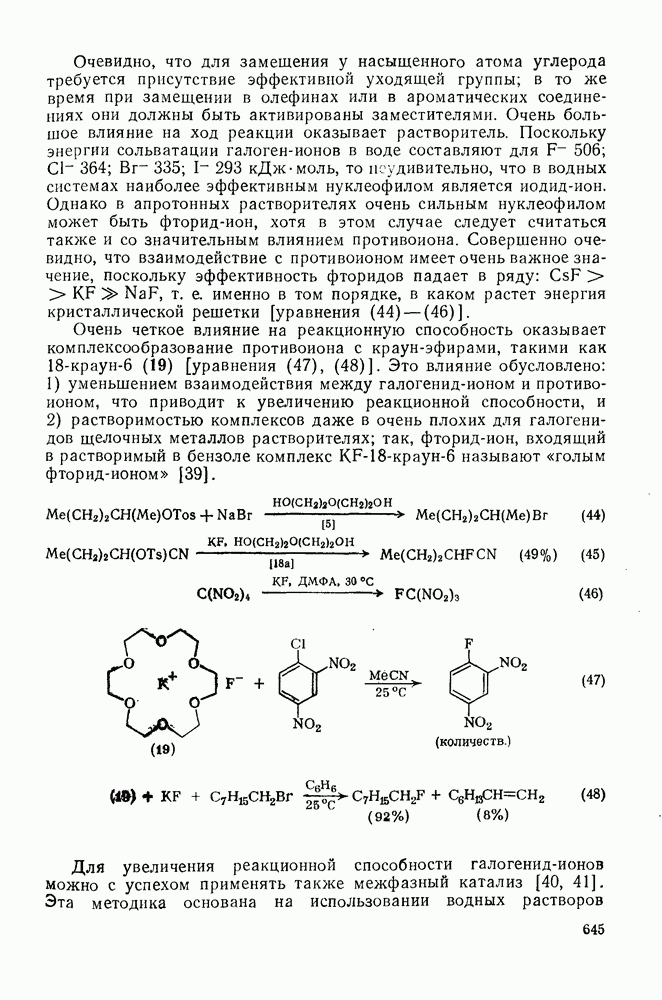
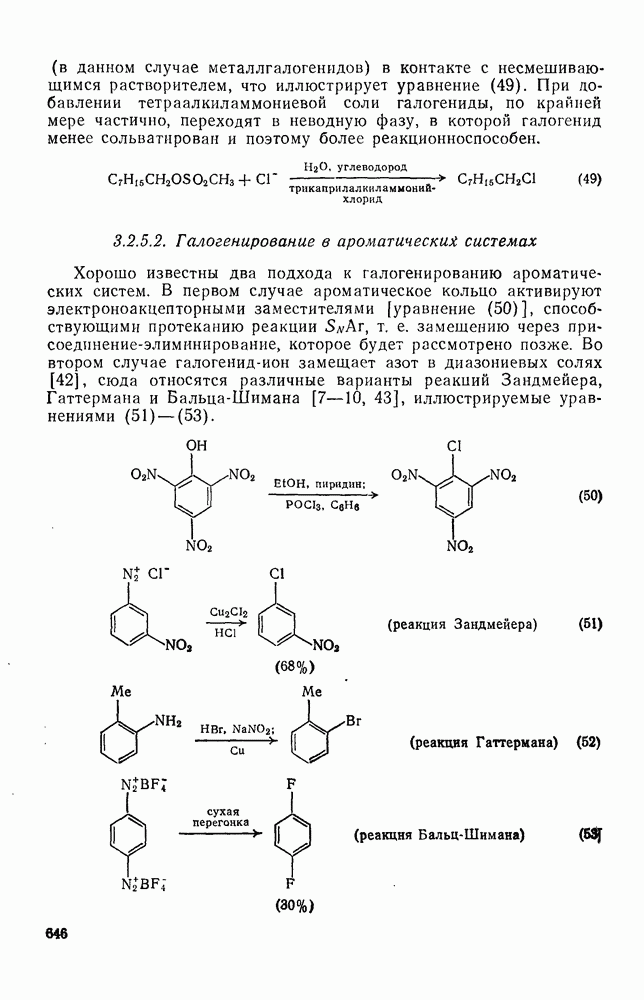
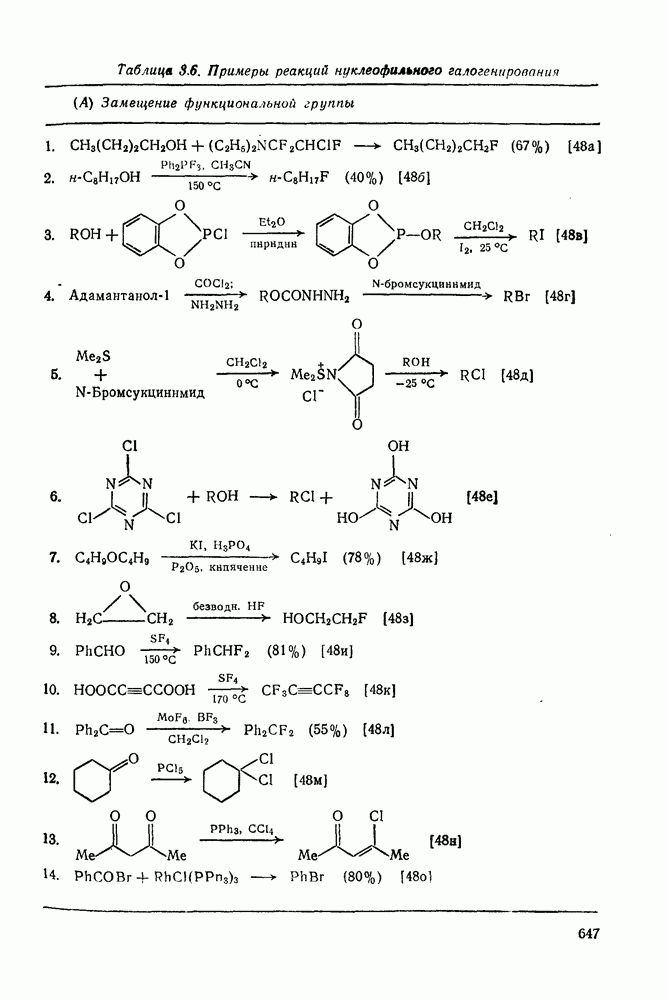
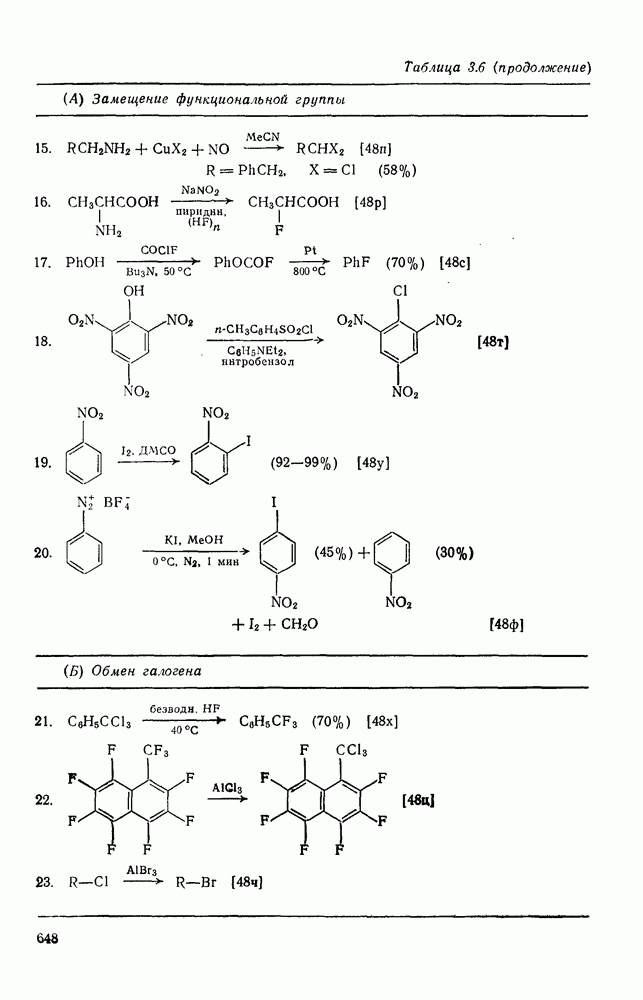
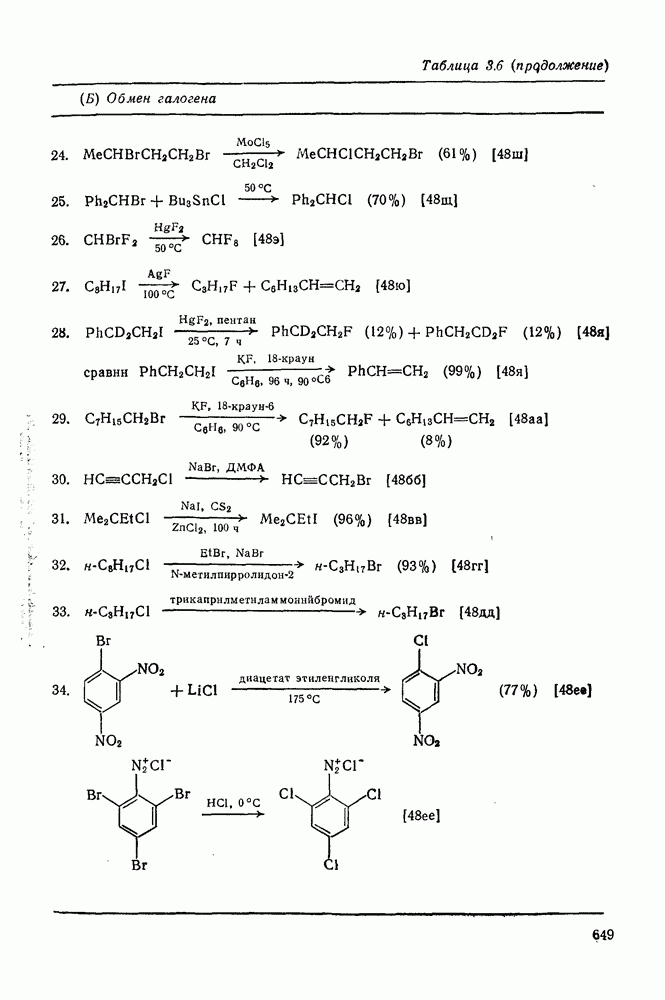
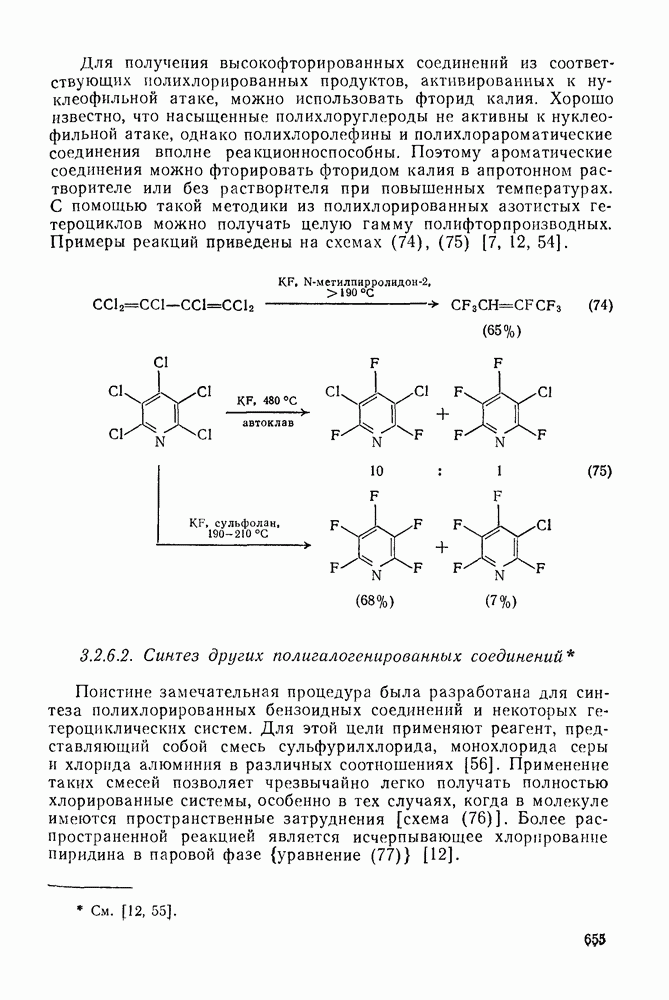
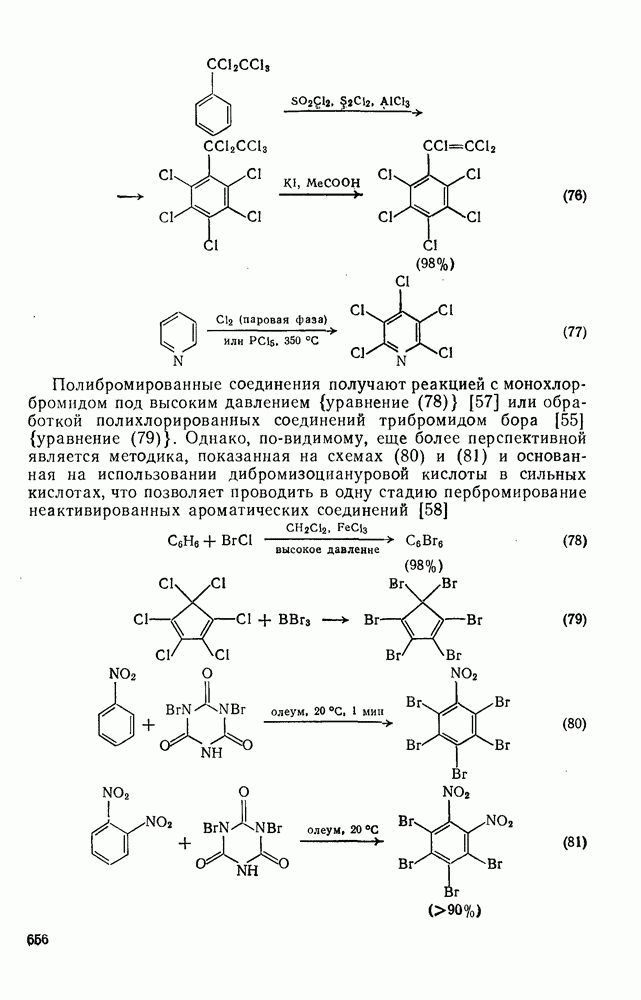
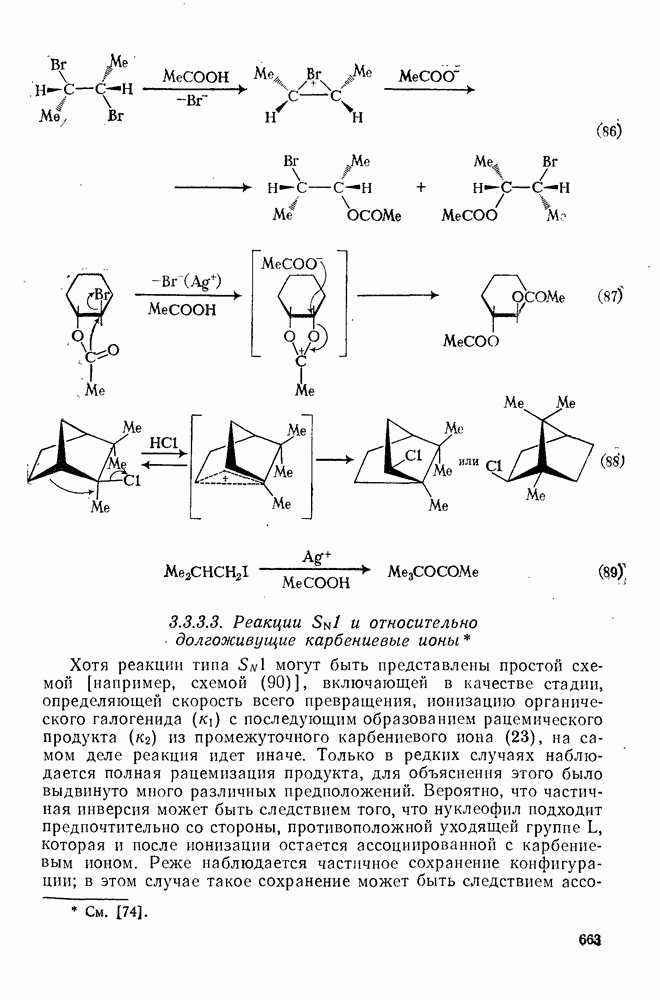
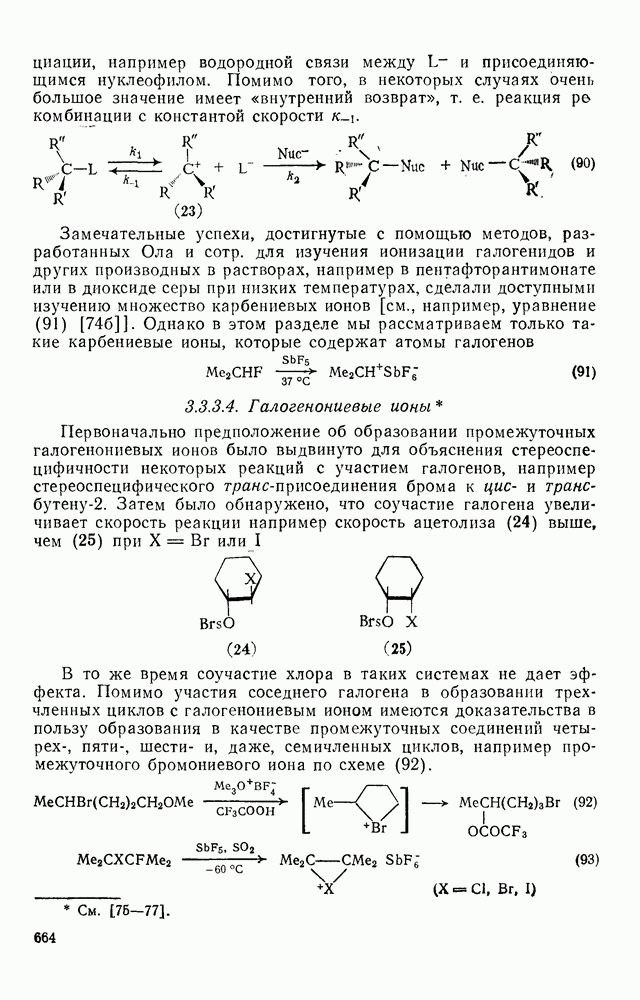
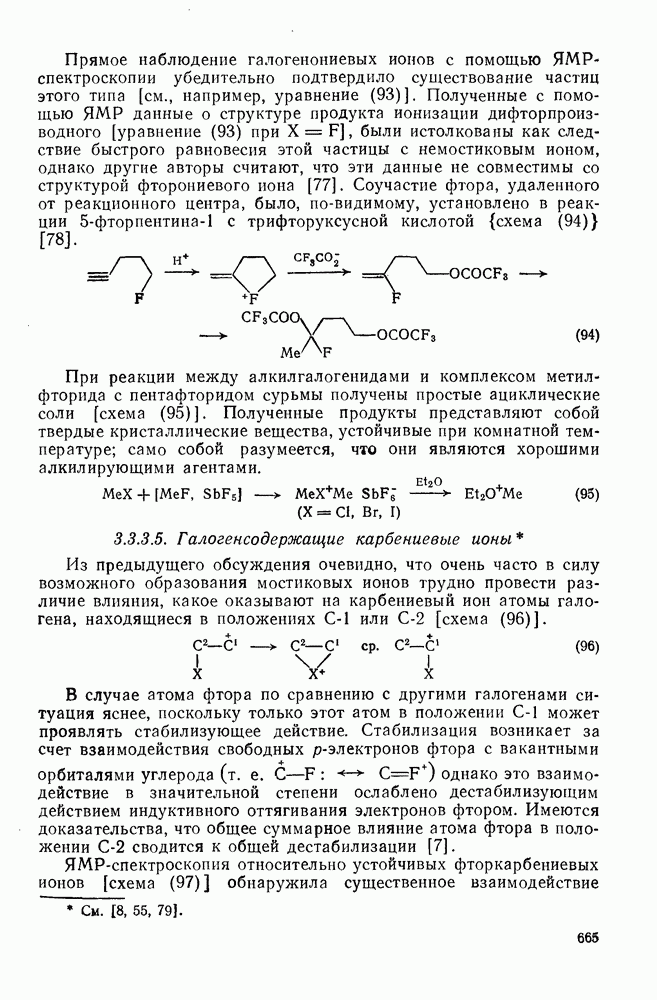
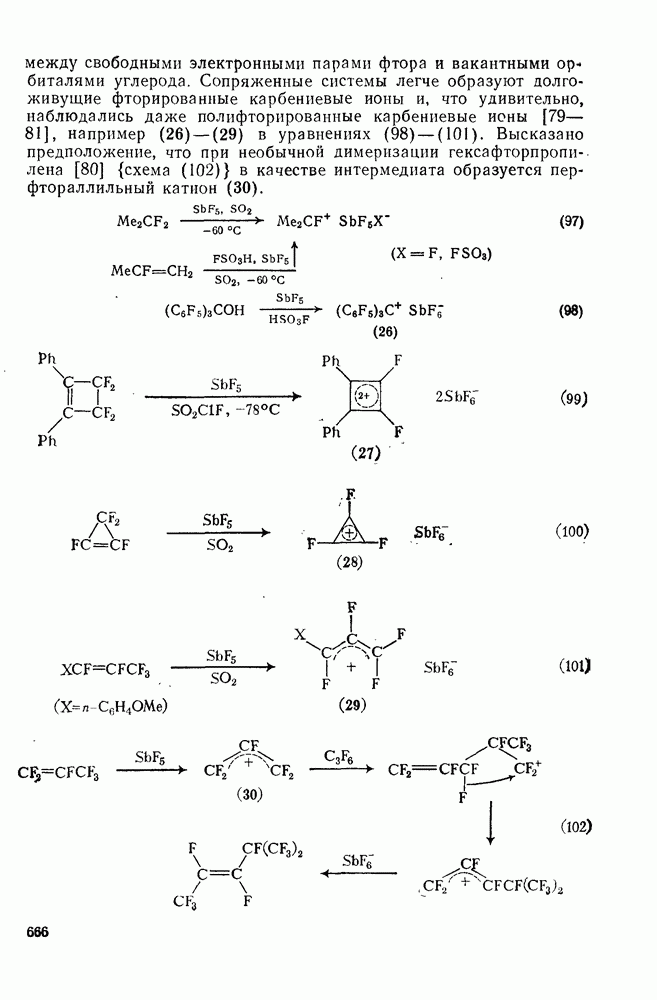
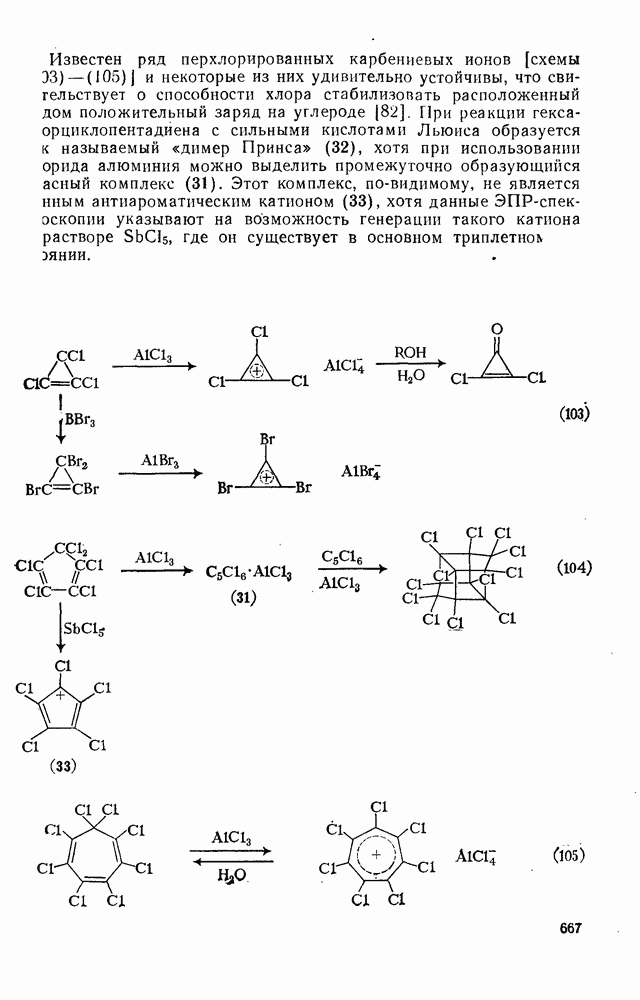
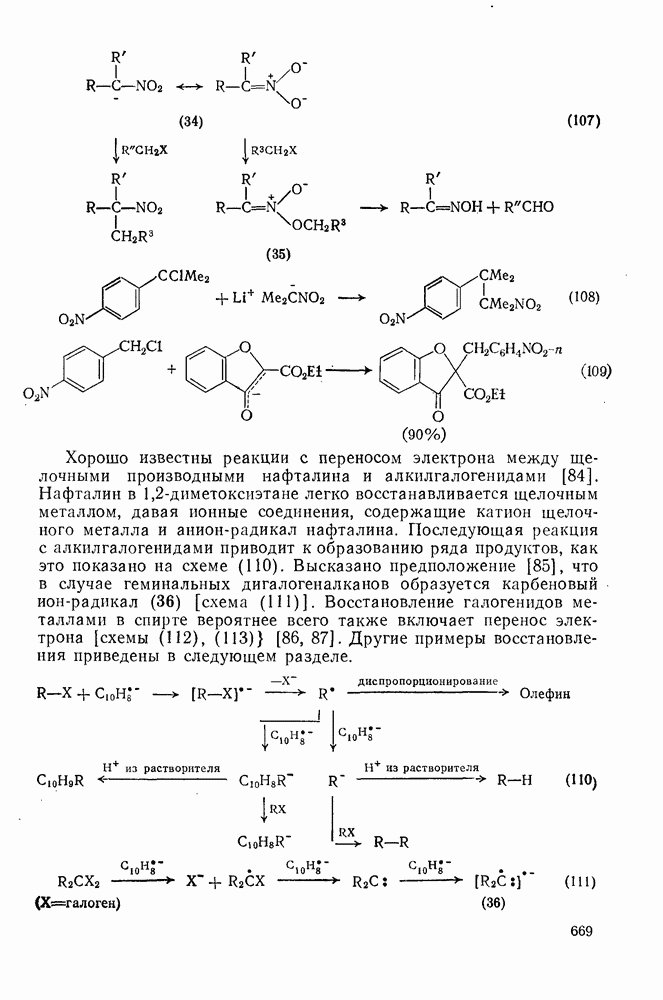
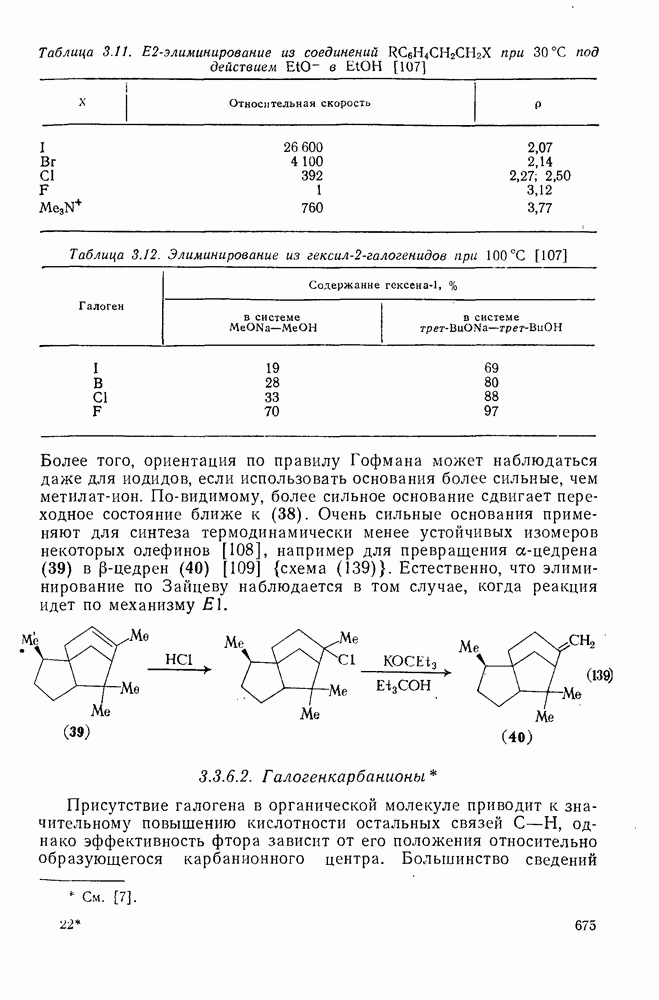
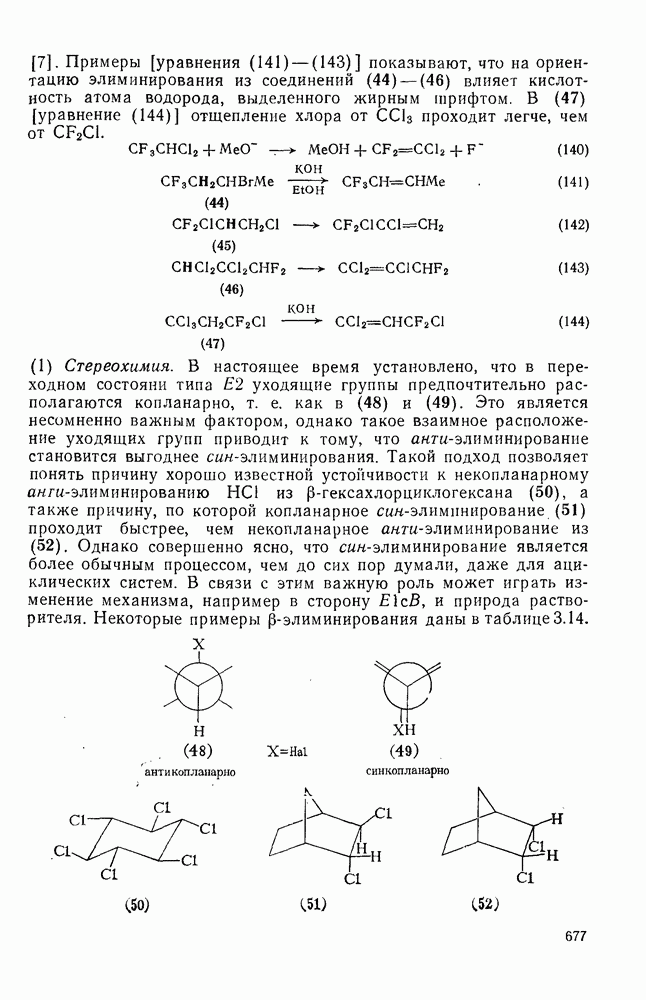
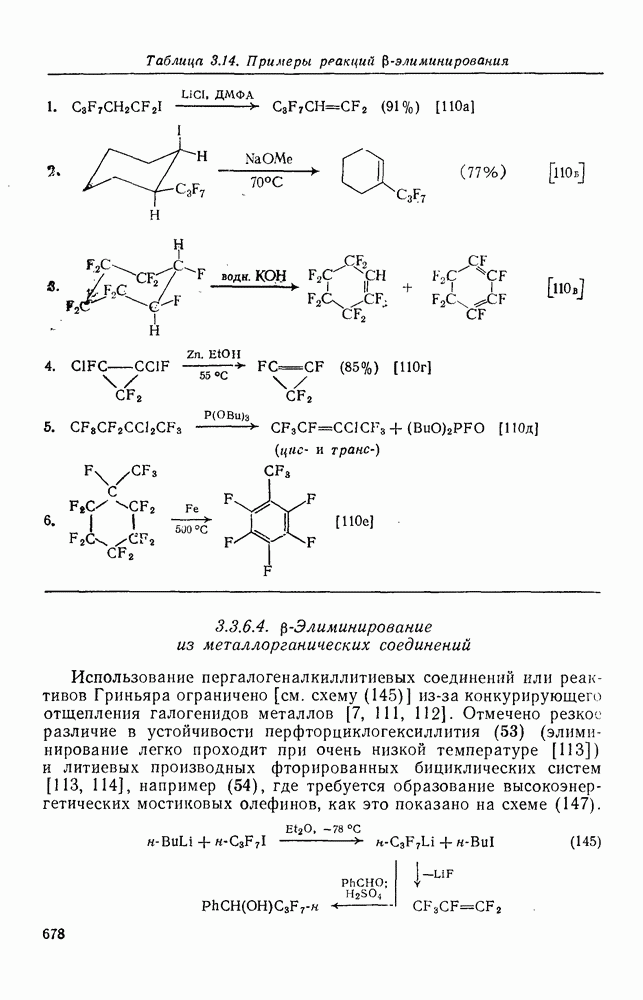
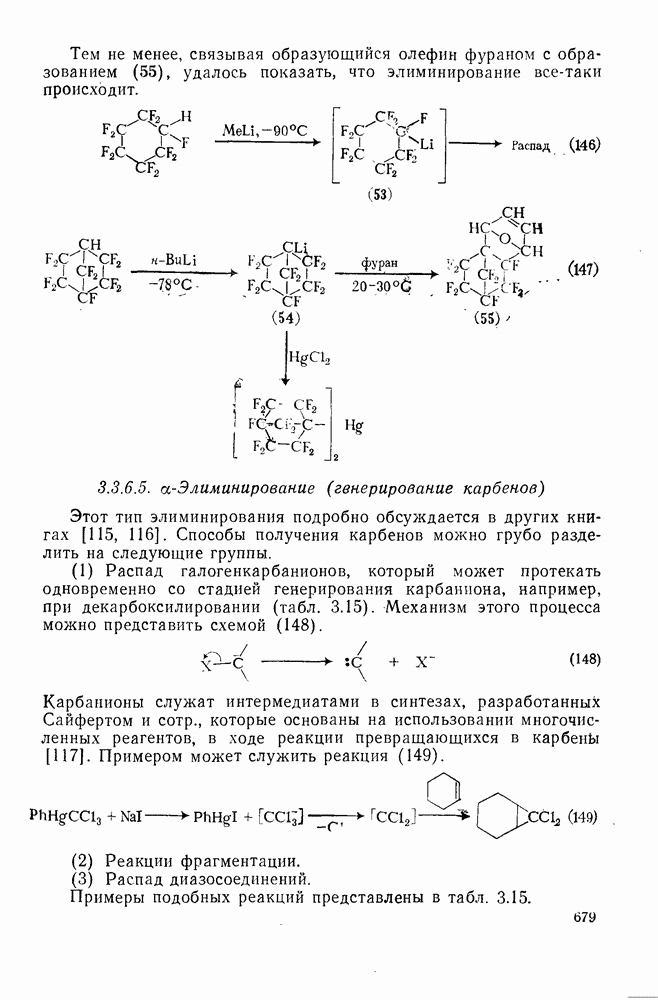
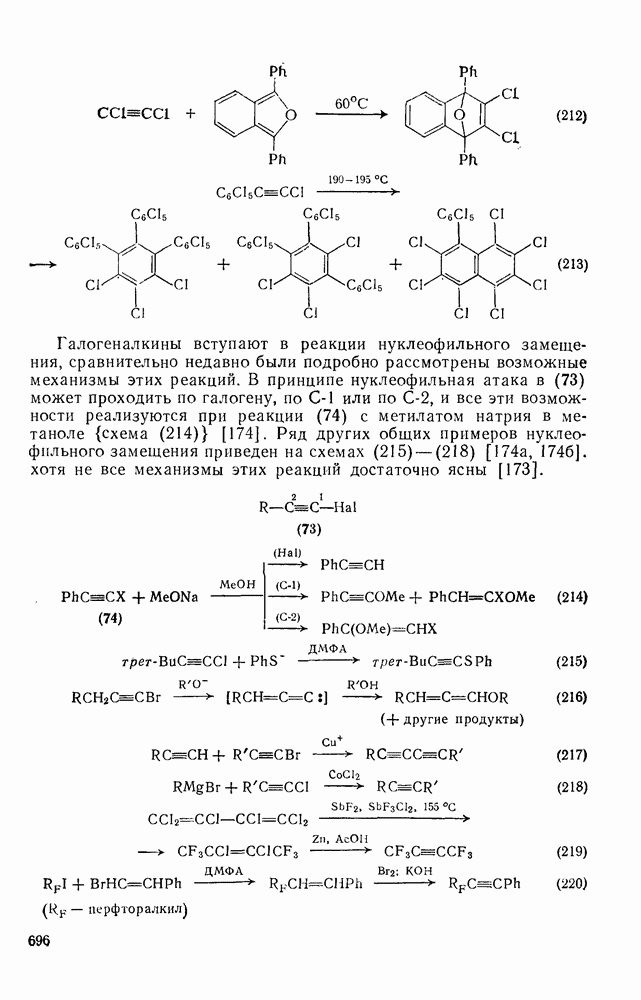
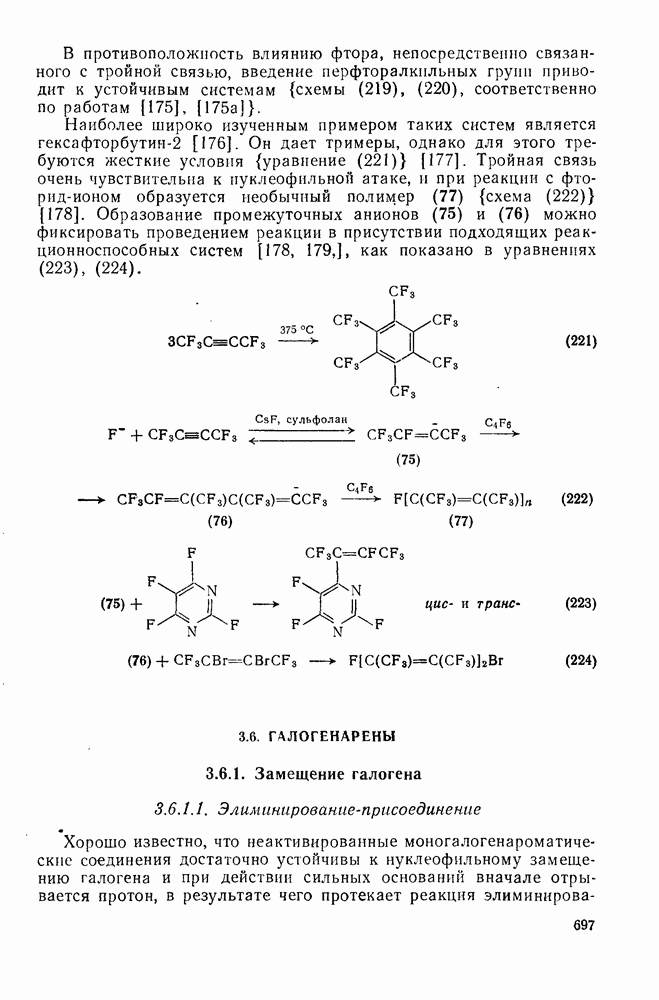
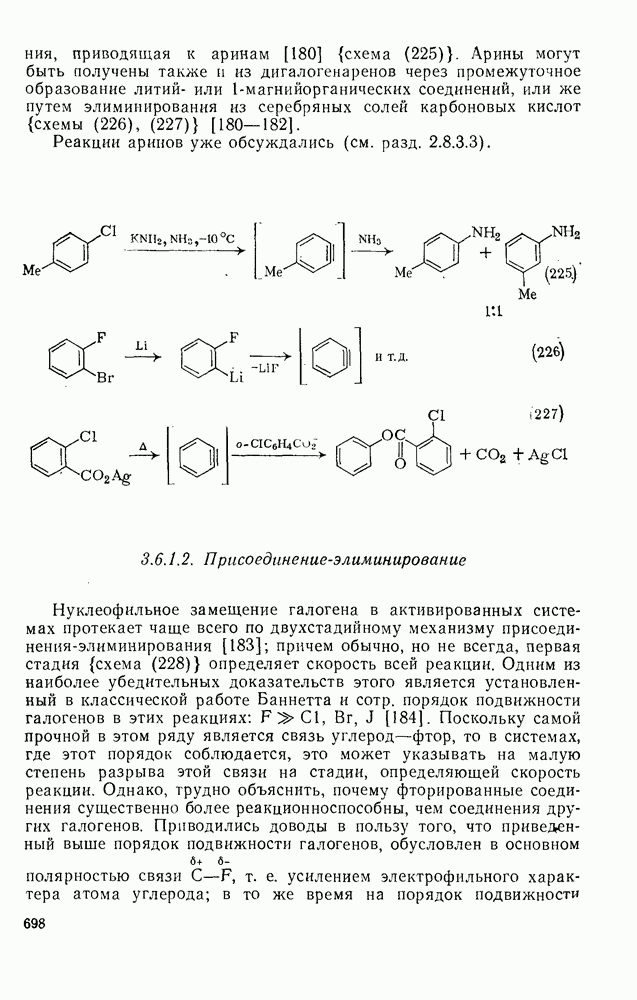
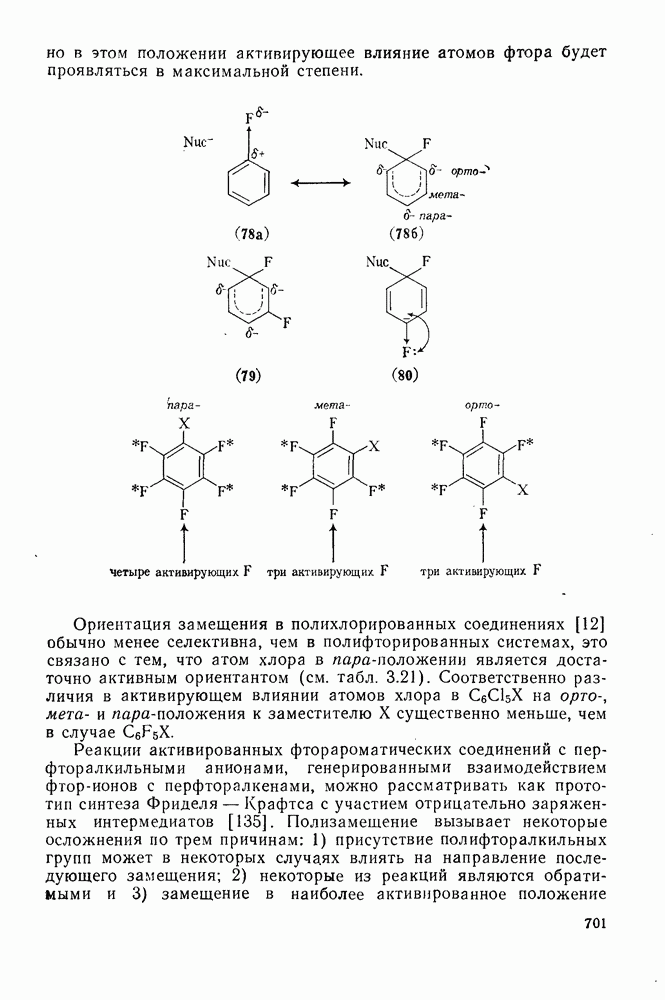
2.1.6.7. Молекулярно-механические расчеты теплот образования и энергии напряжения
Рассмотренные выше схемы, основанные на энергиях связей и инкрементах групп, хотя и могут быть доведены до высокой степени точности, не обладают гибкостью и широтой. Эти качества присущи схемам, включающим молекулярно-механические расчеты (иначе, расчеты, основанные на эмпирических силовых полях), которые в настоящее время широко используют в органической химии. В данном разделе обсуждение этих схем ограничено применением их для определения теплот образования, разницы энергий, энергий напряжения и молекулярной структуры насыщенных углеводородов. Однако достоинством подхода, основанного на молекулярной механике, является возможность его применения для функциональных групп, алкенов, ароматических и металлорганиче-ских соединений, конформаций полимеров и пептидов и для реакционноспособных интермедиатов. Подход на основе молекулярной механики был впервые развит Вестхаймером [68] и позднее расширен Хендриксоном [69] и Вайбергом [70]. В этом случае молекулу рассматривают как комбинацию частиц (атомов), удерживаемых вместе пружинками (связями), и проводят расчеты, применяя соответствующие типы молекулярных моделей. В этом случае электронную систему отдельно не рассматривают. Деформация модели растягиванием или сгибанием пружинок или смещением частиц друг относительно друга приводит к изменениям энергии, которые можно рассчитать, если известны необходимые силовые законы и константы.
Для определения потенциальных функций растяжения, сгибания, несвязанных взаимодействий и одновременного растягивания и сгибания (кручения) используют классические уравнения механики. Комбинирование этих функций приводит к установлению геометрии единственной конформации с минимальной энергией и полной пространственной энергии молекулы. Варьированием молекулярных параметров можно свести геометрию молекулярной модели к конформации с минимальной энергией. Технические детали минимизации энергии, которая проводится на быстродействующих вычислительных машинах, приведены в обзорах [71, 72]. Тщательный подбор потенциальных функций для каждого типа взаимодействия внутри молекулы позволяет получить силовое поле, отвечающее экспериментальным данным.
Пространственную энергию можно использовать непосредственно для определения разницы в энергиях между стереоизомерными и изологичными молекулами (т. е. молекулами, различающимися последовательностью связей, но содержащими одинаковое число групп  , СН и С). Примерами непосредственного применения энергий напряжения являются определение разницы в энергиях между формами ванны и кресла циклогексана, диастереомерами
, СН и С). Примерами непосредственного применения энергий напряжения являются определение разницы в энергиях между формами ванны и кресла циклогексана, диастереомерами  -диметилциклогексана или этилциклопентаном и метил-циклогексаном. Теплоты образования, необходимые для сравнения других типов энергии, рассчитывают, используя пространственные энергии с учетом инкрементов групп или инкрементов энергии связи, полученных при оптимизации пространственных энергий соотнесением с набором экспериментально известных теплот образования. Методом проб и ошибок можно успешно модифицировать параметры таким образом, чтобы достигнуть приемлемого соответствия для сопоставляемого ряда углеводородов различного структурного типа.
-диметилциклогексана или этилциклопентаном и метил-циклогексаном. Теплоты образования, необходимые для сравнения других типов энергии, рассчитывают, используя пространственные энергии с учетом инкрементов групп или инкрементов энергии связи, полученных при оптимизации пространственных энергий соотнесением с набором экспериментально известных теплот образования. Методом проб и ошибок можно успешно модифицировать параметры таким образом, чтобы достигнуть приемлемого соответствия для сопоставляемого ряда углеводородов различного структурного типа.
Одной из наиболее трудных задач при определении силового поля является выбор потенциальной функции для несвязанных атомов. Тар и Тенпас [73] отметили, что расчеты теплот образования не позволяют учесть с необходимой точностью ван-дер-ваальсовы взаимодействия в алканах. Это подтверждается обычно хорошим совпадением силовых полей при использовании сильно различающихся потенциальных функций для несвязанных взаимодействий. Параметризация силовых полей проводится обычно до согласования с энергетическими и структурными данными, однако имеются доказательства того, что по мере усовершенствования методов различные потенциалы несвязанных взаимодействий сводятся к единственному набору величин. Аллинджер [72] подчеркивал, что нельзя смешивать молекулярную модель с физической реальностью; если силовые поля не накладываются одно на другое, то это означает просто, что мы не можем получить информацию о физическом смысле того или иного явления, используя данную модель. Интерпретация явлений, которые резко изменяются от одного силового поля к другому, имеет сомнительный физический смысл.
В настоящее время, в рутинной работе используют несколько силовых полей. Наиболее широко используемыми моделями для углеводородов являются модели Бартелла [74), Бойда [75], Аллинджера (силовые поля  и
и  [76]) и Шлейера (силовое поле EAS) [33]. Силовое поле
[76]) и Шлейера (силовое поле EAS) [33]. Силовое поле  Аллинджера было введено для сведения к минимуму некоторых недостатков более раннего варианта
Аллинджера было введено для сведения к минимуму некоторых недостатков более раннего варианта  . Ниже сравнение экспериментальных и рассчитанных по методам молекулярной механики данных для циклоалканов и полициклоалканов проведено на основе результатов, полученных на моделях
. Ниже сравнение экспериментальных и рассчитанных по методам молекулярной механики данных для циклоалканов и полициклоалканов проведено на основе результатов, полученных на моделях  и EAS, поскольку именно на этих моделях было получено наибольшее количество данных. Преимущество подхода, основанного на молекулярной механике, заключается в том, что его можно применять для систем, расчет которых невозможен обычными методами с использованием инкрементов. Метод, основанный на молекулярной механике, также представляет собой, конечно, метод интерполяции и экстраполяции существующих экспериментальных данных, и здесь отсутствие достаточного количества надежных экспериментальных данных затрудняет точную параметризацию. Вследствие этого сохраняется проблема распространения молекулярно-механических расчетов на структурные типы углеводородов, для которых в наборе, используемом для параметризации, не имеется данных. Проведение расчетов до некоторой степени ускоряет их усовершенствование, стимулируя новые экспериментальные исследования, на которых будет основано создание новых силовых полей.
и EAS, поскольку именно на этих моделях было получено наибольшее количество данных. Преимущество подхода, основанного на молекулярной механике, заключается в том, что его можно применять для систем, расчет которых невозможен обычными методами с использованием инкрементов. Метод, основанный на молекулярной механике, также представляет собой, конечно, метод интерполяции и экстраполяции существующих экспериментальных данных, и здесь отсутствие достаточного количества надежных экспериментальных данных затрудняет точную параметризацию. Вследствие этого сохраняется проблема распространения молекулярно-механических расчетов на структурные типы углеводородов, для которых в наборе, используемом для параметризации, не имеется данных. Проведение расчетов до некоторой степени ускоряет их усовершенствование, стимулируя новые экспериментальные исследования, на которых будет основано создание новых силовых полей.